Изобретение относится к газовой хроматографии и может быть использовано для анализа сложных смесей веществ природного и техногенного происхождения в различных отраслях промышленности: химической, нефтяной, газовой, нефтехимической, металлургии, медицине, биологии, экологии и др.
Известны различные способы хроматографического анализа с использованием капиллярных колонок, при которых дозируют малые количества жидких веществ в колонку, заключающиеся в том, что пробу сначала испаряют в предварительно вакуумированной камере, после чего, малый объем образовавшегося пара переводят в капиллярную колонку для разделения и измеряют их концентрации в потоке газа-носителя на выходе из колонки с помощью детектора [Руденко Б.А. Капиллярная хроматография. М.: Наука, 1978. С. 129-143].
Известен также способ получения калибровочной смеси для газовых хроматографов с малыми концентрациями исходного вещества в потоке газа-разбавителя, заключающийся в том, что исходное вещество подают в поток путем барботирования дополнительного потока газа-разбавителя через нелетучую жидкость, в которой растворено это исходное вещество [Панков А.Г., Трубин А.М., Березкин В.Г. и др. Авт. свид. СССР. 603898. Бюл. изобр. 15 от 25.04.78].
Однако известные способы имеют относительно невысокую точность хроматографических измерений, так как невозможно одновременно хроматографировать на капиллярных колонках газообразные и жидкие пробы в период одного цикла анализа, а также вводить пробы анализируемых смесей для разделения в виде узкой полосы на входе колонки.
Наиболее близким к изобретению по совокупности существенных признаков является способ одновременного дозирования и последующего совместного анализа на хроматографической капиллярной колонке фиксированного количества трех различных по объему веществ, при этом одна из проб для вещества в газовой фазе, а две другие - для веществ в жидкой фазе [Арутюнов Ю.И., Платонов И.А. , Лобачев А.Л. Патент РФ 2069365. Бюл. изобр. 32 от 20.11.96].
Недостатком известного способа является невозможность ввода пробы в виде узкой полосы, кроме того, величина вводимого объема паро-газовой пробы зависит от летучести анализируемой жидкости, поскольку фиксированное количество вводимой жидкости испаряется в процессе переноса ее в колонку потоком газа-носителя, поэтому не обеспечивается необходимая точность измерения и эффективность хроматографического разделения на капиллярных колонках.
Задачей изобретения является повышение точности хроматографических измерений и эффективности разделения.
Эта задача решается за счет того, что в способе хроматографического анализа с использованием капиллярных колонок, заключающегося в одновременном дозировании фиксированного количества несорбирующегося газообразного вещества и исследованных жидких веществ в хроматографическую колонку для разделения и измерения их концентраций в потоке газа-носителя на выходе из колонки, при этом исследуемые жидкие вещества растворяют в нелетучей жидкости, через которую барботируют несорбирующееся газообразное вещество, а паро-газовый поток на выходе барботера разбавляют газом-носителем в определенном соотношении и заполняют дозировочную петлю фиксированного объема при температуре, исключающей конденсацию паров исследуемых жидких веществ, переводят содержимое петли в капиллярную колонку потоком газа-носителя и проводят анализ в условиях программирования температуры колонки с начальной температурой, обеспечивающей конденсацию исследуемых жидких веществ.
При решении поставленной задачи создается технический результат, который заключается в одновременном вводе малых количеств газообразного несорбирующегося вещества и исследуемых жидких веществ в капиллярную колонку в виде паро-газовой смеси фиксированного объема, а также в возможности сжатия хроматографической полосы пробы на начальном участке колонки, за счет понижения температуры начала анализа в режиме линейного программирования до температуры, обеспечивающей конденсацию исследуемых жидких веществ, что позволяет повысить эффективность разделения и точность хроматографического измерения.
Пример конкретного выполнения способа.
Предлагаемый способ поясняется чертежом, на котором изображено устройство для хроматографического анализа.
На схеме устройства показаны: линия несорбирующего газообразного вещества (метана) 1, соединенная с барботером 2, капиллярная колонка 3, дроссели 4, линия газовой смеси 5, соединенная с дозирующим устройством 6, снабженным дозировочной петлей 7, линия сброса 8, пламенно-ионизационный детектор 9, термостаты 10, 11.
Способ осуществляется следующим образом.
Несорбирующееся газообразное вещество метан с заданной скоростью 1÷2 см3/мин по линии 1 поступает в барботер 2, в который помещают 1÷2 см3 раствора исследуемых жидких веществ в нелетучей жидкости - апиезоне L, одновременно используемой в качестве неподвижной жидкой фазы в капиллярной колонке 3. Паро-газовую смесь на выходе барботера 2 с помощью дросселей 4 разбавляют азотом, используемым в качестве газа-носителя, в соотношении, обеспечивающем скорость газа в линии 5 на выходе дозирующего устройства 6 25÷50 см3/мин. Дозирующее устройство 6 снабжено дозировочной петлей 7 объемом 0,125 см3. В одном положении дозирующего устройства дозировочная петля 7 продувается паро-газовой смесью в линию сброса 8 (операция "набор"). В другом положении газ-носитель - азот вымывает анализируемую паро-газовую смесь из дозировочной петли 7 в капиллярную колонку 3 для разделения. Измерение изменения концентраций компонентов в газе-носителе на выходе колонки производится пламенно-ионизационным детектором 9. Барботер 2 и дозирующее устройство 6 помещают в специальный термостат 10, в котором поддерживают температуру Т=373 К, исключающую конденсацию паров исследуемых жидких веществ. Хроматографирование проводят в режиме программирования температуры в термостате колонки 11. Начальная температура колонки Т0=308 К, при которой обеспечивается конденсация исследуемых жидких веществ. Линейная скорость подъема температуры 25oС в мин. Конечная температура колонки 393 К выдерживается постоянной в течение всего времени элюирования анализируемых компонентов пробы.
Экспериментальная оценка выполнения предлагаемого и известного способов хроматографического анализа с использованием капиллярных колонок проводилась на примере анализа трехкомпонентной смеси декана (0,15 мольных долей), додекана (0,39 мольных долей) и октанола-1 (0,46 мольных долей).
Методом взвешивания в барботер 2 вводилась нелетучая жидкость апиезон L и смесь исследуемых жидких веществ в количестве 5÷8 % от массы нелетучей жидкости.
Паро-газовая смесь, содержащая несорбирующийся газ-метан и пары декана, додекана, октанола-1, разбавленная азотом, хроматографируется в режиме программирования температуры на капиллярной кварцевой колонке (длина 30 м, внутренний диаметр 0,25 мм), на внутренние стенки которой нанесена пленка неподвижной жидкой фазы - апиезон L.
Температура колонки при программировании определяется по уравнению
Тс = Т0 + bt,
где Тс = 393 К - изотермическая площадка, выдерживаемая до окончания анализа; t = 3 мин 24 с - время линейного подъема температуры колонки от Т0 до Тс; Т0 = 308 К - начальная температура колонки при t = 0; b = 25 oС/мин - линейная скорость подъема температуры.
Анализ исследуемой жидкой смеси (декан, додекан, октанол-1) и несорбирующегося газа (метан) известным способом проводился на хроматографе. Цвет 500 с пламенно-ионизационым детектором. Дозатор с объемом пробы Vпp=2•10-3 см3 для жидких веществ и Vnp=0,125 см3 для метана устанавливался над испарителем хроматографа. На выходе испарителя часть газового потока сбрасывалась с помощью делителя. Объемная скорость газа-носителя - азота при среднем (по длине) колонки давлении  и температуре колонки Тс,
и температуре колонки Тс,  Скорость газа-носителя в линии сброса Fсброса=95,7 см3/мин. Давление на входе в колонку 0,56 кгс/см2. Температура в термостате колонки Тс = 393 К.
Скорость газа-носителя в линии сброса Fсброса=95,7 см3/мин. Давление на входе в колонку 0,56 кгс/см2. Температура в термостате колонки Тс = 393 К.
Хроматографирование анализируемой смеси известным и предлагаемым способами проводилось на одной и той же капиллярной колонке.
Для исследуемых сорбатов были рассчитаны чистый объем удерживания VN, фактор TZ, характеризующий максимальное число пиков, которые могут уместиться на хроматограмме между двумя гомологами н-алканов, разделенными со степенью RS,Г и эффективное число теоретических тарелок Neff по следующим уравнениям: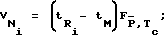
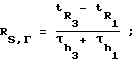
TZ = RS,Г-1;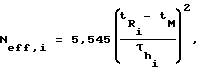
где 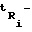 время удерживания i-го компонента; tM - время удерживания несорбирующегося вещества - метана;
время удерживания i-го компонента; tM - время удерживания несорбирующегося вещества - метана; 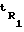 и
и 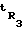 - время удерживания н-алканов декана и додекана;
- время удерживания н-алканов декана и додекана; 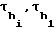 и
и 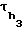 - ширина полосы, измеренная на середине высоты пика соответственно i-го компонента и н-алканов декана и додекана в единицах времени.
- ширина полосы, измеренная на середине высоты пика соответственно i-го компонента и н-алканов декана и додекана в единицах времени.
В связи с тем, что определение массы неподвижной жидкой фазы в капиллярной колонке wL вызывает определенные трудности, удельный объем удерживания октанола-1 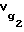 и додекана
и додекана 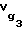 определяли по известному справочному значению
определяли по известному справочному значению 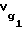 для декана по уравнениям:
для декана по уравнениям: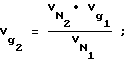
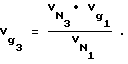
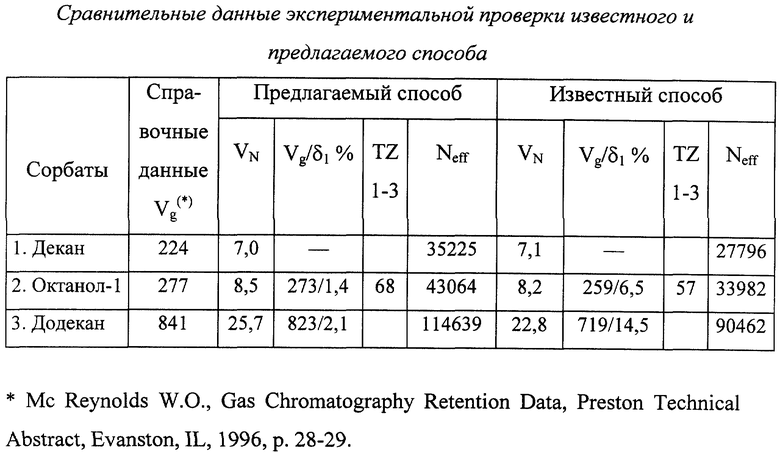
Результаты эксперимента сведены в таблицу "Сравнительные данные экспериментальной проверки известного и предлагаемого способа".
Таким образом, из приведенных в таблице данных видно, что предлагаемый способ обеспечивает значительное повышение точности определения удельного объема удерживания Vg по сравнению с известным способом, а также обеспечивает большую эффективность и разделительную способность колонки, например, фактор TZ увеличивается в 1,2 раза, а число эффективных теоретических тарелок Neff для додекана увеличивается в 1,3 раза.
Использование предлагаемого способа хроматографического анализа позволяет:
1. Одновременно вводить исследуемые жидкие вещества и несорбирующийся газ в капиллярную колонку без деления потока в виде паро-газовой смеси, разбавленной газом-носителем.
2. Повысить эффективность и разделительную способность колонки за счет уменьшения ширины полосы, занимаемой пробой в начале колонки.
3. Значительно повысить точность измерения удельного объема удерживания за счет исключения системы деления потока газа-носителя на входе колонки и измерения разности величин удерживания сорбатов и несорбирующегося вещества.
Изобретение относится к газовой хроматографии. В способе хроматографического анализа фиксированные количества несорбирующегося газообразного вещества и исследуемых жидких веществ дозируют в хроматографическую колонку для разделения и измеряют изменение их концентраций в потоке газа-носителя на выходе из колонки. Исследуемые жидкие вещества растворяют в нелетучей жидкости, через которую барботируют несорбирующееся газообразное вещество, а парогазовый поток на выходе барботера разбавляют газом-носителем в определенном соотношении и заполняют дозировочную петлю фиксированного объема при температуре, исключающей конденсацию паров исследуемых жидких веществ. Переводят содержимое петли в капиллярную колонку потоком газа-носителя и проводят анализ в условиях программирования температуры колонки с начальной температурой, обеспечивающей конденсацию исследуемых жидких веществ. Технический результат - повышение точности хроматографических измерений и эффективности разделения. 1 ил., 1 табл.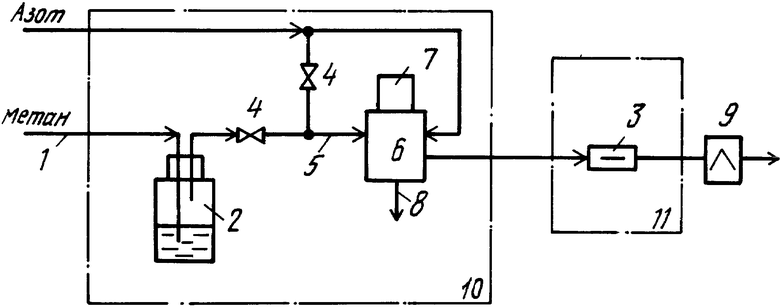
Способ хроматографического анализа с использованием капиллярных колонок, при котором фиксированные количества несорбирующегося газообразного вещества и исследуемых жидких веществ дозируют в хроматографическую колонку для разделения и измеряют изменение их концентраций в потоке газа-носителя на выходе из колонки, отличающийся тем, что исследуемые жидкие вещества растворяют в нелетучей жидкости, через которую барботируют несорбирующееся газообразное вещество, а парогазовый поток на выходе барботера разбавляют газом-носителем в соотношении, обеспечивающем скорость газа в линии на выходе дозирующего устройства 25-50 см3/мин, и заполняют дозировочную петлю фиксированного объема при температуре, исключающей конденсацию паров исследуемых жидких веществ, переводят содержимое петли в капиллярную колонку потоком газа-носителя и проводят анализ в условиях программирования температуры колонки с начальной температурой, обеспечивающей конденсацию исследуемых жидких веществ.
| ДОЗИРУЮЩИЙ КРАН ДЛЯ ХРОМАТОГРАФА | 1992 |
|
RU2069365C1 |
| Способ получения калибровочных смесей для газовых хроматографов | 1974 |
|
SU603898A1 |
| ГРАНУЛЯТОР КОРМОВ | 1972 |
|
SU427671A1 |
| Способ размножения копий рисунков, текста и т.п. | 1921 |
|
SU89A1 |

