Данное изобретение относится к новым производным колхицина, обладающим антипролиферативной, противоопухолевой и противовоспалительной активностями, способам их получения и содержащим их фармацевтическим составам.
Колхицин является известным псевдоалкалоидом, широко использовавшимся в течение длительного времени в терапии для лечения подагры, патологии, на которую он воздействует очень быстро и специфически, несмотря на то, что он должен использоваться кратковременно из-за его токсичности. Производное колхицина, называемое тиоколхикозидом, широко используется для лечения контрактур и воспалительных состояний скелетных мышц. Кроме того, колхицин является очень сильным антибластическим агентом (препятствующим росту), оказывающим блокирующее действие на образование митотической веретенообразной структуры во время деления клеток; этот последний аспект глубоко исследован в отношении любой противоопухолевой активности, и с этой целью было получено большое количество производных колхицина. Колхицин сам по себе и ряд его производных не могут быть использованы клинически ввиду их высокой токсичности и, следовательно, их неприемлемого соотношения риск/польза. Только одно производное колхицина - демеколцин используется в некоторой степени в онкологии для лечения некоторых форм лейкоза.
Таким образом, существуют проблемы доступности противоопухолевых лекарственных препаратов, имеющих удовлетворительный коэффициент риск/польза, то есть высокую терапевтическую эффективность при слабых или отсутствующих побочных действиях.
Другая проблема в области противоопухолевых средств состоит в устойчивости к медицинским препаратам, имеющей место при специфических фенотипах.
Теперь неожиданно обнаружено, что некоторые производные колхицина оказывают высокое цитотоксическое действие как на нормальные раковые клетки, так и на соответствующий устойчивый фенотип (MDR).
Соединения по изобретению являются эффективными индукторами апоптоза, показавшими себя значительно лучше, чем соединения, известные из уровня техники. Благодаря их липофильным свойствам, соединения особенно биодоступны после перорального введения. Кроме того, соединения по данному изобретению могут быть введены с равным успехом парентерально или локально.
Описание изобретения
Данное изобретение относится к соединениям формулы I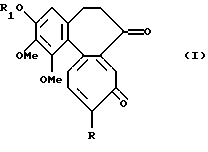
где R обозначает метокси- или метилтио- группу, a R1 обозначает линейную или разветвленную алкильную группу с 1-6 углеродными атомами.
Примерами алкильной группы служат метил, этил, пропил, изопропил, н-бутил, изо-бутил, трет-бутил, пентил, неопентил, гексил.
Соединения формулы I получают исходя из природных соединений колхицина или тиоколхицина или из их С3-производных, выпускаемых промышленностью или получаемых известными из литературы способами. Как описано в литературе, С3-производные могут быть получены реакцией 3-0-диметилпроизводного с алкил- или ацил-галогенидом. Гидролиз указанных соединений водными растворами сильной неорганической кислоты позволяет получать селективно, варьируя температуру и реакционное время, соответствующие N-деацетил-производные. В частности, деацетилирование тиоколхицина или его С3-производных может быть выполнено проведением кислотного гидролиза соединений; в случае тиоколхицина гидролиз галогенкислотами или, более предпочтительно, с серной кислотой (20% H2SO4 - 120 ч) позволяет получать N-деацетилтиоколхицин и 3-диметил-N-деацетилтиоколхицин с почти количественными выходами.
Для получения соединений формулы I проводят реакции N-деацетил-производных с 4-формил-1-метилпиридиний-п-толуол-сульфонатом и 1,8-диазабицикло[5.4.0]ундец-7-еном (DBU).
Альтернативно, реакцией N-деацетил-производных с 2,3-дитрет-бутил-1,2-бензохиноном получают соединения формулы II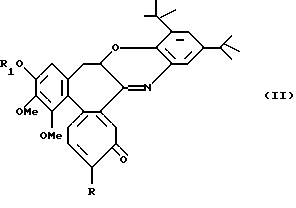
где R обозначает метилтио, R1 обозначает линейную или разветвленную C1-6 алкильную группу.
Следующей целью данного изобретения являются соединения формулы II.
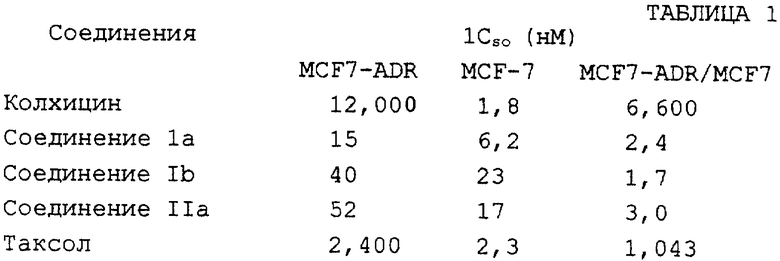
Предложенные данным изобретением соединения обладают значительной противоопухолевой активностью как in vitro, так и in vivo. Таблица 1 демонстрирует противомитотическое действие соединений по изобретению на эксплантаты культивированной опухоли молочной железы, нормальные (MCF-7) или устойчивые как к адриамицину, так и винбластину (MCF7-ADR), в сравнении с действием колхицина и таксола.
Таблица 1 показывает, что соединения по изобретению имеют значительные преимущества на резистентных линиях клеток, которые в настоящее время рассматриваются как основная мишень цитотоксических лекарственных средств.
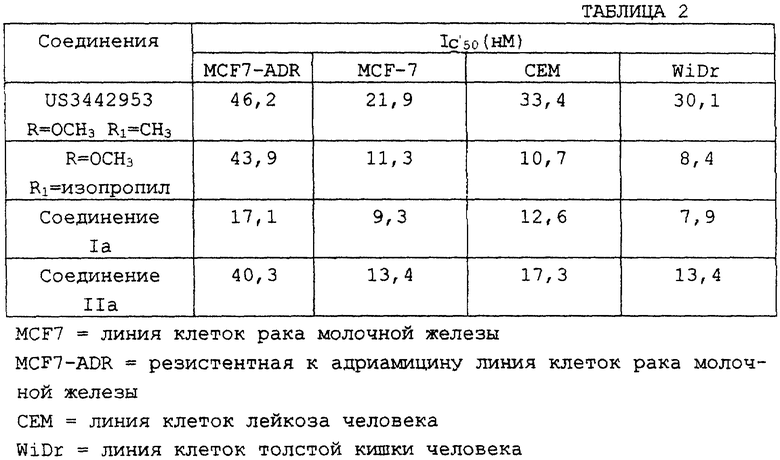
Также было проведено сравнение соединений настоящего изобретения (Iа, IIа и соединение, в котором R=OCH3 и R''=изопропил) с ближайшим структурным аналогом - соединением по патенту США 3442953. Процедура, используемая для анализа цитотоксичности, опубликована в Alley M.C. et al., Cancer Research 48, 589, 1988. Как показано в представленной ниже таблице 2, соединения по настоящему изобретению более эффективны в ингибировании клеточных линий опухоли.
Кроме того, соединения по данному изобретению обладают противовоспалительной и противоартритной активностями (дегенеративный ревматоидный артрит и подобные патологии) и могут быть включены в фармацевтические составы, используемые при введении лекарства от указанной патологии. Они пригодны для получения составов внутривенного, перорального, трансдермального и подкожного введений.
Среди используемых для получения указанных составов эксципиентов природные и синтетические фосфолипиды представляются особенно полезными для получения липосомных форм для парентерального и/или локального способов введения. Те же самые составы предложены для использования в локальной обработке кожных эпителий и в случаях кожных гиперпролиферативных состояний, таких как псориаз. В специфической противоопухолевой области, кроме фосфолипидов, которые позволяют введение лекарственного средства в липосомной форме, оказались особенно полезными некоторые поверхностно-активные соединения, такие как полиэтоксилированные касторовые масла или полисорбаты, взаимодействующие синергически с активным ингредиентом. Предпочтительно, активную составляющую микронизируют, чтобы растворить соединение в воде. Необычно активной, удобной формой является комплекс этих соединений с циклодекстринами.
В онкологии продукты используют в дозировках от 1 до 100 мг/м2.
Последующие примеры дополнительно иллюстрируют изобретение.
В 1ЯМР-спектрах:
Hz - Гц
ppm - м.д.
molt - вид
int - интерпретация
type - тип
eq. - равный
ах - аксиальный
s - синглет
t - триплет
d - дублет
ddd - тройной дублет
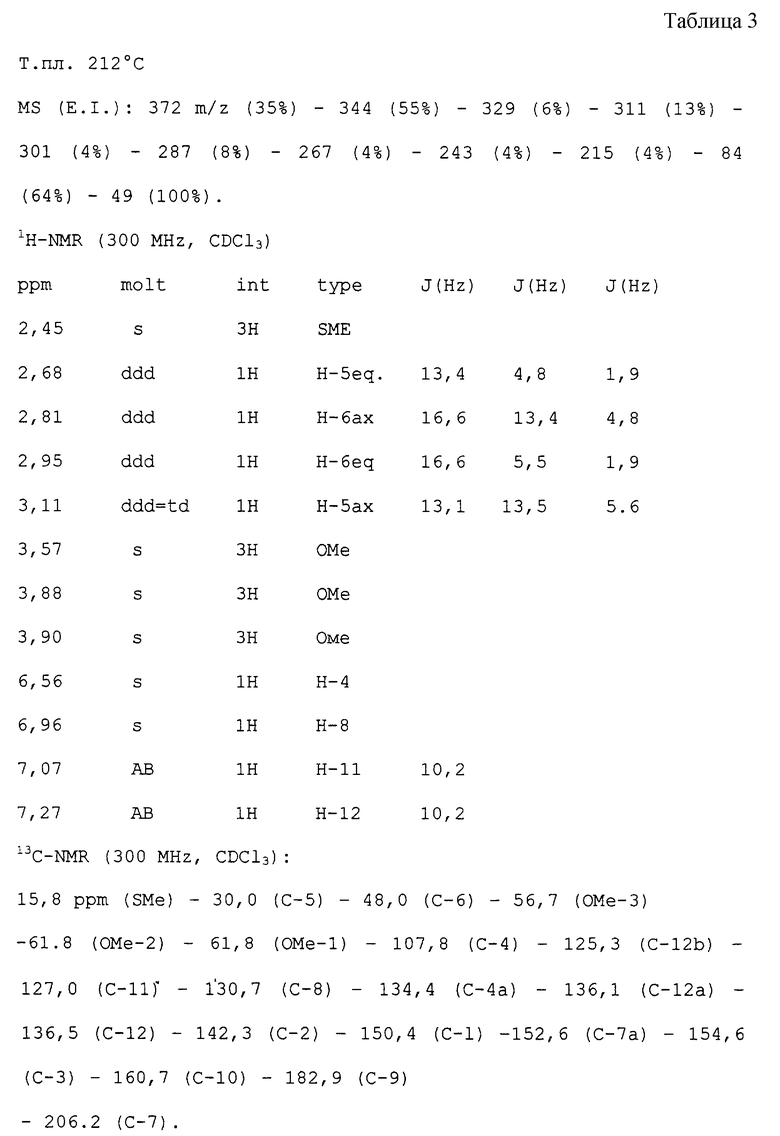
ПРИМЕР 1
Получение тиоколхикона из N-деацетилтиоколхицина.
(la:R=SMe; Ri=Me)
100 мл СН2Сl2 и 30 мл ДМФ смешивают в атмосфере азота, затем добавляют 4 г деацетилтиоколхицина (м. в. 373, 10,7 ммоль) и 24,2 г п-толуолсульфоната 4-формил-1-метилпиридиния (м. в. 279,15 ммоль); смесь нагревают до температуры кипения с обратным холодильником в течение 3 часов или в любом случае до исчезновения амина. Раствор охлаждают до 0oС и затем добавляют по каплям 1,94 г DBU (м.в. 152, 12,8 ммоль), получая темно-красный раствор. Через 15 минут добавляют 150 мл водного раствора щавелевой кислоты, смесь оставляют реагировать в течение ночи, затем неоднократно экстрагируют СН2Сl2, сушат над сульфатом натрия и растворитель упаривают досуха. Остаток кристаллизуют из этилацетата, что дает 78% выход. Тиоколхикон имеет следующие физико-химические и спектроскопические характеристики (см. табл. 3 в конце текста).
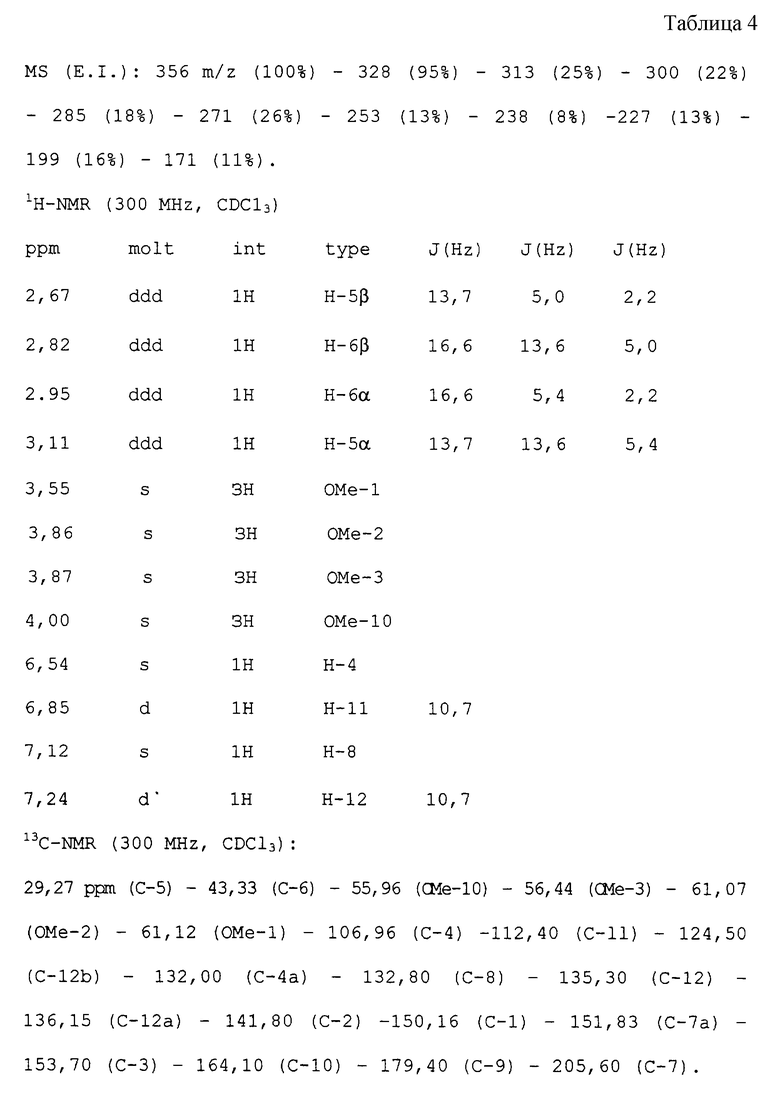
ПРИМЕР 2
Получение колхикона из N-деацетил-колхицина,
(Ib:R=Оме; R1=Me)
3,58 г N-деацетилколхицина обрабатывают по способу примера 1. Получают 2,6 г колхикона, имеющего следующие физико-химические и спектроскопические характеристики (см. табл. 4 в конце текста).
ПРИМЕР 3
Получение 3-0-изопропил-колхикона
3,8 г 3-0-изопропил-N-деацетилколхицина растворяют в 100 мл СН2Сl2 и добавляют 25 г 4-формил-1-метилпиридиний-п-толуолсульфоната; реакционную смесь кипятят с обратным холодильником в течение 2,5 часов. Раствор охлаждают до 0oС и затем добавляют по каплям 2 г DBU с получением темно-красного раствора.
Через 15 минут добавляют 150 мл водного раствора щавелевой кислоты, смесь оставляют реагировать в течение ночи, затем несколько раз экстрагируют CH2Cl2.
Растворитель выпаривают до сухости и остаток выкристаллизовывают из смеси этилацетат/ацетон с получением 3-0-изопропил-колхикона.
Т.пл.222o-4oС
МС(Е.1.): 384 m/z
ПРИМЕР 4
Получение 3-0-изопропил-тиоколхикона
3,8 г 3-0-изопропил-N-деацетилтиоколхицина растворяют в 100 мл СН2Сl2 и добавляют 25 г 4-формил-1-метилпиридиний-п-толуолсульфоната; реакционную смесь кипятят с обратным холодильником в течение 2,5 часов. Раствор охлаждают при 0oС и затем добавляют по каплям 2 г DBU с получением темно-красного раствора.
Через 15 минут добавляют 150 мл водного раствора щавелевой кислоты, смесь оставляют реагировать в течение ночи, затем несколько раз экстрагируют СН2Сl2.
Растворитель выпаривают досуха и остаток выкристаллизовывают из смеси этилацетат/ацетон с получением 3-0-изопропил-тиоколхикона.
МС(Е.I.): 396 m/z
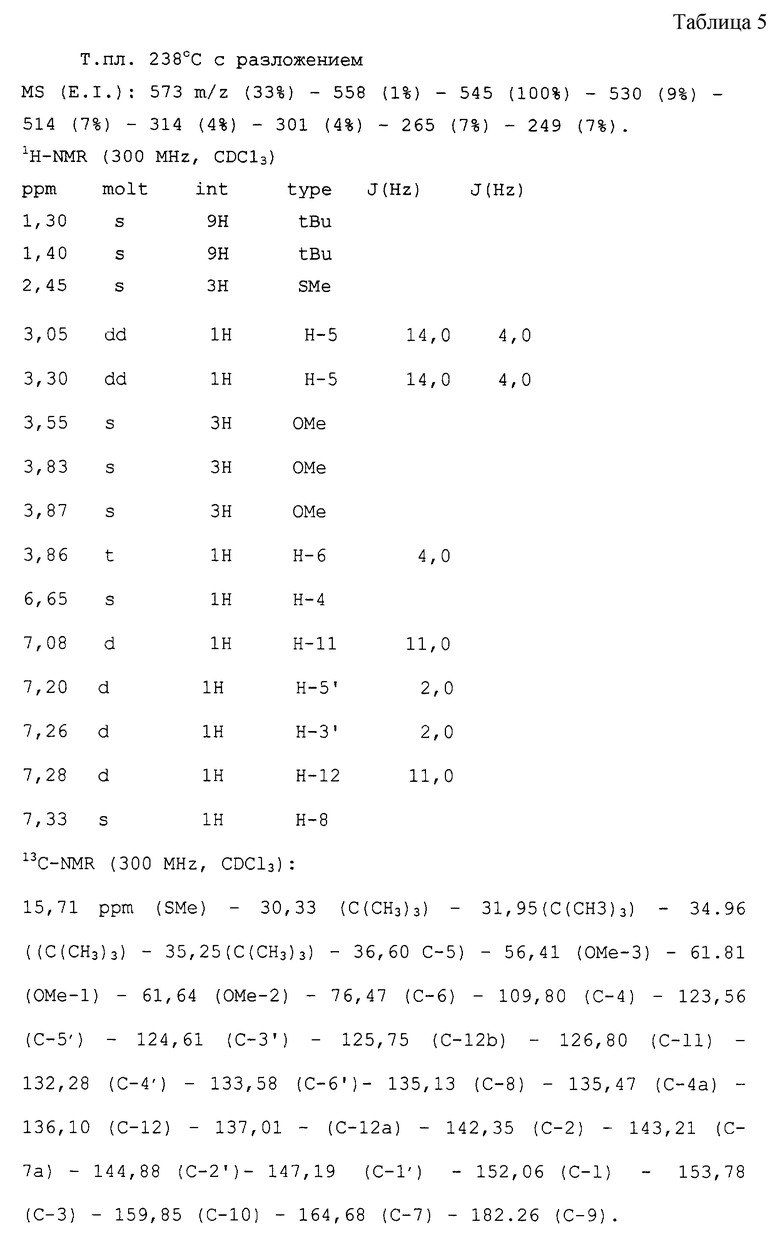
ПРИМЕР 5
Получение продукта конденсации тиоколхицина и 3,5-дитретбутил-1,2-бензохинона
(IIa:R=SMe; R1=Me)
500 мг деацетилтиоколхицина (м. в. 373, 1,34 ммоль) и 590 мг 3,5-дитрет-бутил-1,2-бензохинона (м.в. 220, 2,69 ммоль) растворяют в 50 мл метанола при нормальном атмосферном давлении.
После реакции проводят ТСХ (СН2Сl2:ацетон 30:1) и приблизительно через 18 часов растворитель упаривают в вакууме.
Теплый сырой продукт растворяют в 1 объеме этилацетата, добавляют 1-1,5 объема гексана и смесь охлаждают льдом. Реакционный продукт выделяют фильтрованием, выход составляет 70%. Это соединение имеет следующие физико-химические и спектроскопические характеристики (см. табл. 5 в конце текста).
ПРИМЕР 6
Состав таблеток, содержащих соединение (Iа)
Соединение (Iа) - 25 мг
Лактоза - 47 мг
Микрокристаллическая целлюлоза - 20 мг
Поперечно-сшитая натрий карбоксиметилцеллюлоза - 5 мг
Коллоидная двуокись кремния - 1 мг
Тальк - 1 мг
Стеарат магния - 1 мг
А. Смешать все вышеперечисленные ингредиенты за исключением натрий кроскармелозы, талька и стеарата магния.
В. Пропустить смесь А) через сито 60 меш.
С. Перенести В) в кубический смеситель и смешивать до получения гомогенной смеси.
D. Добавить натрий кроскармелозу к С) и смешивать в течение 5 минут.
Е. Пропустить стеарат магния и тальк через сито 60 меш.
F. Смешивать Е и D в кубическом смесителе в течение 2 минут.
G. Спрессовать 100 мг смеси F) в таблетки с использованием вогнутого штампа (диаметр 6 мм).
ПРИМЕР 7
Состав липосомного крема, содержащего соединение (IIа)
Соединение (IIа) - 0,20 г
Фосфатидилхолин - 20,00 г
Холестерол - 0,50 г
Бутилгидрокситолуол - 0,01 г
95% Этанол - 8,00 г
Динатрий эдетат - 0,15 г
Имидизолидинил-мочевина - 0,30 г
Дегидроацетат натрия - 0,20 г
Гидроксиэтилцеллюлоза (Natrosol 250 HHX-Aqualon) - 2,00 г
Дистиллированная вода - 67,75
А. Расплавить фосфатидилхолин, добавить 95% этанол и перемешивать до получения прозрачной жидкости.
В. Подогреть фазу А и добавлять при перемешивании до полного растворения холестерол, бутилгидрокситолуол.
С. К фазе В добавить соединение IIа и перемешивать до получения прозрачной вязкой жидкости. Нагреть до 40oС.
D. В дистиллированной воде растворить динатрий эдетат, имидазолидинмочевину и дегидроксиацетат натрия. Нагреть до 40oС.
Е. К фазе D добавить фазу С при интенсивном перемешивании в условиях вакуума.
F. Диспергировать гидроксиэтилцеллюлозу в фазе Е до образования геля.
ПРИМЕР 8
Получение раствора для инъекции, содержащего соединение (Iа)
Соединение (Iа) - 15 мг
PEG-660 12-гидроксистеарат - 2,500 мг
Пропиленгликоль - 1,000 мг
Спирт q.s. до - 5 мл
А. К пропиленгликолю при перемешивании добавить спирт.
В. При интенсивном перемешивании к фазе А добавить ПЭГ-660 12 гидроксистеарат.
С. К фазе В добавить соединение Iа и перемешивать до получения прозрачной жидкости.
D. Довести до нужного объема при помощи спирта и перемешивать до получения прозрачной жидкости.
ПРИМЕР 9
Получение раствора для инъекций, содержащего соединение (Ia)
Соединение Iа - 15 мг
Полиэтоксилированное касторовое масло - 2,500 мг
Пропиленгликоль - 1,000 мг
Спирт - достаточное количество до 5 мл
А. К пропиленгликолю при перемешивании добавить спирт.
В. При интенсивном перемешивании к фазе А добавить полиэтоксилированное касторовое масло.
С. К фазе В добавить соединение Iа и перемешивать до получения прозрачной жидкости.
D. Довести до нужного объема при помощи спирта и перемешивать до получения прозрачной жидкости.
Изобретение относится к производным колхицина формулы (I), где R означает метокси- или метилтиогруппу; R1 означает линейный или разветвленный C1 - C6 алкил, при условии, что, когда R означает метокси, R1 не может быть метил; и соединениям формулы II, где R означает метилтио; R1 означает линейный или разветвленный C1 - С6-алкил. Соединения I получают взаимодействием N-деацетилтио-колхицина или N-деацетилколхицина с 1,8-диазабицикло[5,4,0] ундец-7-еном. Соединения II получают взаимодействием N-деацетилтио-колхицина с 3,5-дитретбутил-1,2-бензохиноном. Соединения I и II проявляют противоопухолевую и антипролиферативную активность, что позволяет использовать их в фармацевтической композиции в качестве активного ингредиента, пригодной для локального применения. 6 с. и 7 з.п. ф-лы, 5 табл.
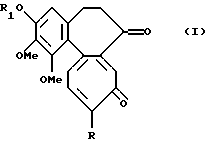
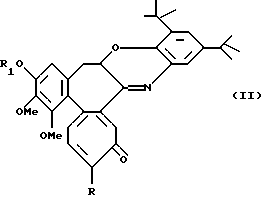
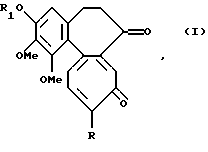
где R обозначает метокси- или метилтиогруппу;
R1 обозначает линейную или разветвленную алкильную группу с 1-6 атомами углерода, при условии, что когда R обозначает метокси, R1 не может обозначать метил.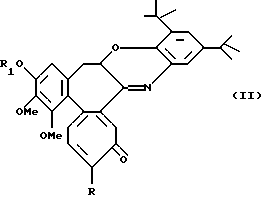
где R обозначает метилтио;
R1 обозначает линейную или разветвленную алкильную группу с 1-6 атомами углерода.
| US 3442953, 06.05.1969 | |||
| US 4349548, 14.09.1982 | |||
| ЕР 729933, 28.02.1996 | |||
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИЦИКЛИЧЕСКИХ АРОМАТИЧЕСКИХ СОЕДИНЕНИЙ | 0 |
|
SU198320A1 |

