Изобретение относится к новым производным колхицина, обладающим антипролиферативной, противоопухолевой и противовоспалительной активностями, способам их получения и содержащим их фармацевтическим составам. Колхицин представляет собой известный псевдоалкалоид, широко применявшийся в течение длительного времени в терапии для лечения подагры; он действует очень быстро и специфично, даже несмотря на то, что его следует применять в течение коротких промежутков времени из-за его токсичности. Кроме того, колхицин является очень сильным средством, препятствующим клеточному росту, чье действие связано с механизмом, блокирующим образование митотического веретена во время клеточного деления; этот последний аспект тщательно исследовался на предмет какой-либо противоопухолевой активности, и было получено значительное количество производных колхицина данного назначения.
Колхицин как таковой и ряд его полученных производных не могут применяться из-за их высокой токсичности с точки зрения отношения риск/выгода. Лишь одно производное колхицина, демеколцин, применяют в некоторой степени в онкологии для лечения некоторых форм лейкоза. Что касается применения с целью оказания противовоспалительного эффекта, единственным продажным производным колхицина является тиоколхикозид, несущий тиометильный радикал в C10 и молекулу глюкозы при гидроксиле в C3; к терапевтическим применениям данного производного относят эффект мышечной релаксации и противовоспалительный эффект. Продукты по изобретению отличаются от таковых, существовавших ранее в данной области, высоким терапевтическим индексом. В области противоопухолевого действия исследования были сосредоточены на разработке продуктов, обладающих, помимо нормальной цитотоксичности, цитотоксичностью, направленной на клеточные линии, устойчивые к обычным лекарственным средствам, препятствующим клеточному росту.
Производные по настоящему изобретению имеют общую формулу I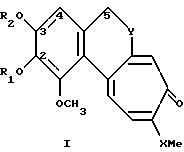
где X может представлять собой атом кислорода или серы; R1 и R2, которые могут быть одинаковыми или различными, представляют собой неразветвленные или разветвленные алкильные группы или циклоалкильные группы, содержащие от 1 до 6 атомов углерода; или насыщенные или ненасыщенные ацильные группы, содержащие от 16 до 22 атомов углерода, или остаток β- D-глюкозы как таковой или остаток β- D-глюкозы, где гидроксилы в положениях 4 и 6 защищены по типу кеталей алифатическими, или ароматическими, или гетероароматическими альдегидами; Y представляет собой группу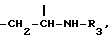
метиленовая группа которой присоединена к углероду в положении 5, а метиновая группа, имеющая ту же абсолютную конфигурацию, что и колхицин, присоединена к трополоновому кольцу, или группу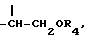
метиновая группа которой присоединена к углероду в положении 5 и к трополоновому кольцу и имеет абсолютную конфигурацию S; R3 представляет собой ацильную группу, содержащую от 2 до 6 атомов углерода, несущих от одного до трех атомов галогена, предпочтительно фтор или хлор, или ацильную группу природной аминокислоты, где аминогруппа может быть свободной или защищенной по типу трифторацетамида или бензамида;
R4 представляет собой ацильный остаток дикарбоновой кислоты, содержащий от 4 до 6 атомов углерода, или ацильный остаток природной аминокислоты, где аминогруппа может быть свободной или защищенной по типу трифторацетамида или бензамида, или гликозидный остаток, содержащий D-глюкозу, D-галактозу, L-фукозу или L-рамнозу. В данном случае сахара играют роль хемотактического агента для меланоцитов, гепатоцитов, фибробластов или они могут давать производное, характеризующее пролекарство.
Предпочтительными соединениями формулы I являются такие, где X представляет собой серу.
R1 и R2, которые являются одинаковыми или различными, предпочтительно представляют собой водород или C1-C6-алкил.
Y предпочтительно представляет собой группу формулы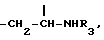
как указано выше.
Используемыми при получении продуктов по изобретению исходными продуктами являются природные вещества колхицин (1; X = 0; R1=R2=Me; Y=CH2-CH-NHAc), 2-O-деметилколхицин (1; X=O; R1=H; R2=Me; Y=CH2-CH-NHAc), 3-O-деметилколхицин (1; X=O; R1=Me; R2=H; Y=CH2-CH-NHAc), колхикозид (1; X=O; R1=Me; R2 = β- D-глюкоза; Y=CH2-CH-NHAc), которые могут быть выделены из растительных материалов в соответствии с методиками, известными в литературе. Данные природные вещества, будучи обработаны метилмеркаптаном в щелочном растворе в соответствии с методиками, также известными в литературе, приводят к получению соответствующих тиопроизводных, которые используют в качестве синтонов для получения производных общей формулы I, где X представляет собой серу.
Для получения производных общей формулы I, где R1 и R2 представляют собой алкильные и ацильные группы, используемыми исходными продуктами являются колхициновый или тиоколхициновый синтоны, деметилированные в положении 2 или 3. Данные синтоны подвергают алкилированию или ацилированию, применяя широко известные способы получения фенольных производных. Аналогично производные общей формулы I, где R1 или R2 представляют собой β- D-глюкозный остаток или β- D-глюкозный остаток, где гидроксилы в положениях 4 и 6 защищены по типу кеталей алифатическими, или ароматическими, или гетероароматическими альдегидами, получают из колхициновых или тиоколхициновых синтонов, деметилированных по положению 2 или 3. Данные синтоны подвергают взаимодействию с α- бромтетраацетил-D-глюкозой или с 2,3-ди-O-дихлорацетил -β- D-глюкозой, содержащей кеталевую группу, с гидроксилами в положениях 4 и 6 (сравни с Патентом Канады N 956939). После удаления защитных ацильных групп известными способами, описанными в литературе, получают производные, глюкозидированные по положению 2 или 3 по настоящему изобретению.
Производные формулы I, где Y представляет собой группу 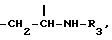 получают, подвергая колхициновые или тиоколхициновые синтоны, имеющие метокси- или гидроксигруппы в положениях 2 и 3, N-дезацетилированию в условиях кислотного катализа с последующим ацилированием первичного аминорадикала реакционноспособным производным кислоты, содержащим от одного до трех атомов фтора или хлора, или природную аминокислоту, аминогруппа которой может быть свободной или защищенной по типу трифторацетамида или бензамида. Таким образом получают производные формулы I, где R3 имеет значение, указанное выше. Производные формулы I, где Y представляет собой группу
получают, подвергая колхициновые или тиоколхициновые синтоны, имеющие метокси- или гидроксигруппы в положениях 2 и 3, N-дезацетилированию в условиях кислотного катализа с последующим ацилированием первичного аминорадикала реакционноспособным производным кислоты, содержащим от одного до трех атомов фтора или хлора, или природную аминокислоту, аминогруппа которой может быть свободной или защищенной по типу трифторацетамида или бензамида. Таким образом получают производные формулы I, где R3 имеет значение, указанное выше. Производные формулы I, где Y представляет собой группу 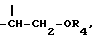 получают, подвергая колхициновые или тиоколхициновые синтоны, имеющие метокси- или гидроксигруппы в положениях 2 и 3, N-дезацетилированию с последующей обработкой нитритом натрия и уксусной кислотой, которые взаимодействуют с циклогептановым кольцом с образованием синтонов формулы I, где
получают, подвергая колхициновые или тиоколхициновые синтоны, имеющие метокси- или гидроксигруппы в положениях 2 и 3, N-дезацетилированию с последующей обработкой нитритом натрия и уксусной кислотой, которые взаимодействуют с циклогептановым кольцом с образованием синтонов формулы I, где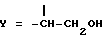
(J. Med. Chem. 36, 544, 1993). Полученный радикал первичного спирта путем взаимодействия с подходящими активированными дикарбоновыми кислотами или с активированными природными аминокислотами, где аминогруппа может быть свободной или защищенной по типу трифторацетамида или бензамида, или с реакционноспособной формой сахаров D-глюкозы, D-галактозы, L-фукозы или L-рамнозы приводит к получению производных общей формулы I, где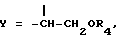
причем R4 имеет значение, указанное выше.
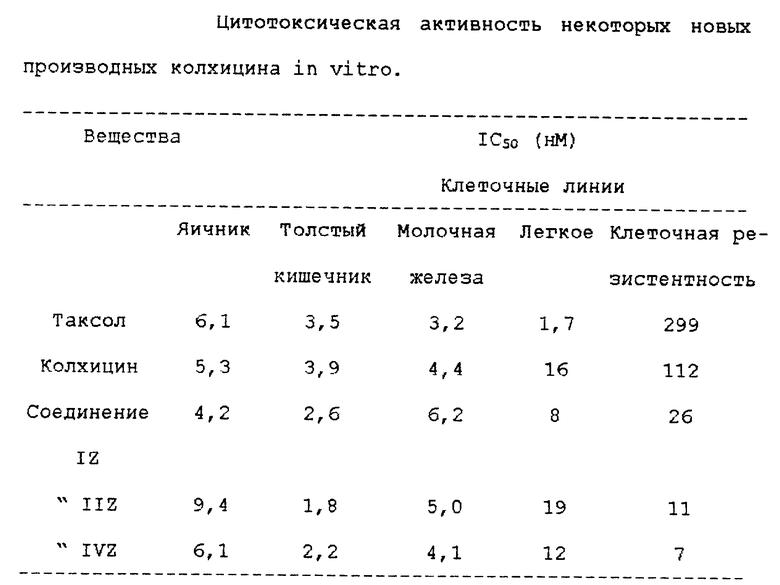
В таблице показано антимитотическая активность некоторых производных по изобретению на опухолевых клеточных линиях. Таксол и колхицин являются веществами сравнения.
Данная таблица свидетельствует, что новые производные имеют значительные преимущества на резистентных клеточных линиях, которые являются основной мишенью цитотоксических лекарственных средств. Продукты по настоящему изобретению могут входить в состав фармацевтических препаратов, используемых для введения лекарственного средства. Препараты для парентерального, перорального, чрескожного, накожного введения могут быть легко приготовлены.
Среди наполнителей, используемых для приготовления указанных препаратов, природные и синтетические фосфолипиды особенно применимы для приготовления липосомальных форм для парентерального, чрескожного или накожного путей введения; причем последние два препарата особенно получены при лечении воспалительных состояний суставов и периферических вен; причем указанные составы также применимы при местном лечении кожных эпителиом и в случае кожных гиперпролиферативных состояний, таких как псориаз. Оказалось, что в специфичной противоопухолевой области, помимо фосфолипидов, позволяющих вводить лекарственное средство в липосомальной форме, особенно применимы некоторые поверхностно-активные вещества, такие как полиэтоксилированное касторовое масло, как, например, Хемоформ L50, или полисорбат, как, например, Твин, действующие синергично с активным ингредиентом. В онкологии продукты применяют в дозах от 1 до 100 мг/м2, тогда как в качестве противовоспалительного средства доза изменяется от 1 до 20 мг на стандартную дозу, один или несколько раз в день. Все фармацевтические препараты, такие как ампулы, капсулы, кремы и так далее, могут быть приготовлены с большей частью указанных производных.
Следующие примеры дополнительно иллюстрируют изобретение.
Пример I. Получение N-дезацетил-N-трифторацетил-3-O-деметил- 3-O-циклопентилтиоколхицина, соединение IZ (1; X=S; R1=Me; R2=C5H9; Y=CH2CHNH-COCF3).
20 г 3-O-Деметилтиоколхицина (1; X=S; R1=Me; R2=H; Y=CH2CHNHAc) растворяют в 300 мл 20% серной кислоты и обрабатывают при 100oC в атмосфере азота в течение 36 ч. Реакционную смесь нейтрализуют, выделяя таким образом 12 г N-дезацетил-3-O-деметилтиоколхицина (1; X=S; R1=Me; R2=H; Y=CH2CHNH2). Данный продукт растворяют в ацетоне и подвергают взаимодействию с 3 эквивалентами трифторуксусного ангидрида при сильном перемешивании в присутствии безводного Na2CO3. Через 2 ч реакционную смесь фильтруют и раствор выпаривают досуха. Остаток, содержащий 3-O-деметил-N,3-O- бистрифторацетилтиоколхицин, гидролизуют в метаноле, содержащем NH4Cl. Реакционную смесь выпаривают досуха в вакууме и остаток обрабатывают ацетоном. Ацетоновый раствор фильтруют и нагревают с обратным холодильником в течение восьми часов с 5 эквивалентами циклопентилбромида в присутствии карбоната натрия. Соли отфильтровывают, раствор выпаривают досуха и остаток очищают с помощью хроматографии на колонке с силикагелем, используя в качестве элюента этилацетат. Путем кристаллизации из ацетона/гексана получают 8,6 г продукта, M+a m/z 523.
Пример II. Получение N-дезацетил-N-трифторацетил-3-O-деметил- 3-O-изопропилтиоколхицина, соединение IIZ (1; X= S; R1=Me; R2=изо-Pr; Y=CH2-CH-NHCOCF3).
Для получения данного производного повторяют методику примера I, используя изопропилбромид вместо циклопентилбромида. После очистки сырого продукта реакции на силикагеле и кристаллизации получают 7,6 г продукта, M+a m/z 497.
Пример Ill. Получение N-дезацетил-N-трифторацетилтиоколхикозида, соединение IIIZ (1; X=S; R1=Me; R2= β-D-глюкозил; Y=CH2-CH-NHCOCF3).
10 г N-Дезацетилтиоколхикозида (1; X= S; R1=Me; R2= β-D-глюкоза; Y= CH2-CH-NH2) растворяют в ацетоне и обрабатывают в течение двух часов при 10oC тремя эквивалентами трифторуксусного ангидрида. Смесь выпаривают досуха и остаток кристаллизуют из изопропанола и вслед за этим из этанола, получая 8,5 г продукта, M+a m/z 617.
Пример IV. Получение N-(N-трифторацетил- α-фенилглицил) дезацетилтиоколхицина, соединение IVZ (1; X=S; R1=R2=Me; Y=CH2-CH-NH-COCH(NHCOCF3)Ph).
400 мг N-Дезацетилтиоколхицина (1; X=S; R1=R2=Me; Y=CH2-CH-NH2) (1,07 ммоль) растворяют вместе с 265 мг (1,07 ммоль) L-N-трифторацетил -α- фенилглицина в 10 мл метиленхлорида в атмосфере азота. К раствору добавляют 221 мг (1,07 ммоль) N, N-дициклогексилкарбодиимида, перемешивая до исчезновения реагентов. Реакционную смесь охлаждают до -30oC и фильтруют для удаления выпавшей в осадок мочевины. Хлорметиленовый раствор концентрируют и очищают путем фильтрования через силикагель, элюируя смесью метиленхлорида/метанола 98:2. После кристаллизации из метиленхлорида/этилового эфира получают 350 мг продукта, M+a m/z 602.
Пример V. Получение N-(N-трифторацетил-L-аланил)-дезацетилтиоколхицина, соединение VZ (1; X=S; R1=R2=Me; Y=CH2-CH-NH-COCH(NHCOCF3)CH3).
400 мг N-Дезацетилтиоколхицина (1,07 ммоль) обрабатывают одним эквивалентом N-трифторацетил-L-аланина и одним эквивалентом N,N-дициклогексилкарбодиимида в 10 мл метиленхлорида и в атмосфере азота до исчезновения реагентов. Реакционную смесь охлаждают до -30oC и фильтруют для удаления выпавшей в осадок мочевины. Хлорметиленовый раствор концентрируют и очищают путем фильтрования через силикагель, элюируя смесью метиленхлорида/метанола 98: 2. После кристаллизации из метиленхлорида/этилового эфира получают 94 мг продукта, M+a m/z 540.
Пример VI. Получение N-N-трифторацетилметионил)-дезацетилтиоколхицина, соединение VIZ (1; X=S; R1=R2=Me; Y=CH2-CH-NHCO-CH(NHCOCF3)CH2-CH2-SMe).
Повторяют методику из примера IV, осуществляя взаимодействие с N-трифторацетилметионином. По завершении хроматографической очистки реакционного остатка методом фракционированной кристаллизации из 400 мг N-дезацетилтиоколхицина получают 84 мг продукта, M+a m/z 600.
Пример VII. Получение N-( α-фенилглицил)дезацетилтиоколхицина, соединение VIIZ (1; X=S; R1=R2=Me; Y=CH2-CH-NHCO-CHNH2-Ph).
400 мг Продукта, полученного в примере IV, растворяют в 5 мл 50% ацетона в присутствии 120 мг карбоната калия и нагревают до 60oC при перемешивании в течение 5 часов. Реакционную смесь охлаждают, насыщают NaCl и экстрагируют добавлением хлороформа. Органическую фазу сушат над безводным сульфатом натрия, затем концентрируют досуха и остаток хроматографируют на силикагеле смесью метиленхлорида/метанола 98:2. Получают 160 мг продукта, M+a m/z 506.
Пример VIII. Получение N-дезацетил-N-трифторацетил-3-O-деметил- 3-O-ксименинилтиоколхицина, соединение VIIIZ (1; X=S; R1=Me; R2=CO(CH2)7C ≡ C-CH=CH-(CH2)5CH3; Y=CH2-CH-NH-CO-CF3).
500 мг N-Дезацетил-N-трифторацетил-3-O-деметилтиоколхицина (1; X=S; R1= Me; R2= H; Y=CH2-CH-NHCOCF3) растворяют в 2,5 мл пиридина и к нему при 0oC добавляют 500 мг хлорангидрида ксимениновой кислоты. Реакционную смесь оставляют стоять в течение ночи при комнатной температуре, затем выливают на лед. Полученный осадок выделяют и кристаллизуют из ацетона/гексана, M+a m/z 715.
Пример IX. Получение 5,6-дигидро-6(S)-[( β-D-глюкопиранозилокси) метил] -1,2,3-триметокси-9-(метилтио)-8H-циклогепта[a] нафталин-8-она, соединение IXZ (1; X=S; R1=R2=Me; Y=CH-CH2 -β- D-глюкоза).
10 г N-Дезацетилтиоколхицина обрабатывают нитритом натрия, что дает 4 г 5,6-дигидро-6(S)-(гидроксиметил)-1,2,3-триметокси-9-метилтио- 8H-циклогепта[a]нафталин-8-она в соответствии со способом, описанным в J. Med. Chem., 36, 544, 1993. Полученный продукт обрабатывают в течение 12 часов при нагревании с обратным холодильником в ацетонитриле 26 г α-бромтетраацетилглюкозы в присутствии 85 г цианида ртути. Соли отфильтровывают, раствор выпаривают досуха, помещают в 70% ацетон и обрабатывают в течение двух часов 15% карбонатом натрия. Смесь нейтрализуют, экстрагируют добавлением этилацетата и хроматографируют на силикагеле, элюируя смесью метиленхлорида-этанола 9:1. Получают 2,1 г продукта, M+a m/z 536.
Пример X. Получение сукцинилового эфира 5,6-дигидро-6(S)- (гидроксиметил)-1,2,3-триметокси-9-(метилтио)-8H-циклогепта[a] нафталин-8-она, соединение XZ (1; X=S; R1=R2=Me; Y=CH-CH2-OCOCH2CH2CO2H).
10 г N-Дезацетилтиоколхицина обрабатывают, как в примере IX. Полученный нафталин-8-он растворяют в пиридине и обрабатывают при нагревании с обратным холодильником в течение 24 часов с избытком янтарного ангидрида. Реакционную смесь охлаждают, выливают в достаточное количество воды и экстрагируют добавлением метиленхлорида. Органическую фазу концентрируют до небольшого объема и очищают на силикагеле, элюируя смесью метиленхлорида-воды-метанола 70: 30: 5. После кристаллизации из метанола получают 7 г продукта, M+a m/z 474.
Пример XI. Фармацевтический препарат: таблетки
Композиция на 100 мг-овую таблетку, мг:
Соединение IIZ (по примеру II) - 10
Лактоза - 50
Микрокристаллическая целлюлоза - 32
Поперечносшитая натрий карбоксиметилцеллюлоза - 5
Коллоидный диоксид кальция - 1
Тальк - 1
Стеарат магния - 1д
Описываются новые производные колхицина формулы (I), где значения R1, R2, X, Me, Y указаны в п.1 формулы, обладающие антипролиферативной, противоопухолевой и противовоспалительной активностями. Новые соединения обладают цитотоксичностью по отношению к опухолевым клеточным линиям человека, сравнимой с таковой колхицина, но по сравнению с последним они являются менее токсичными и более избирательными, особенно по отношению к клеткам, устойчивым к обычным лекарственным средствам. Некоторые соединения обладают выраженной активностью по отношению к ФНО и интерлейкину-2 и, таким образом, являются очень мощными противовоспалительными средствами. Они могут быть включены в фармацевтические составы, применимые для парентерального, перорального и местного введения. Описывается также способ их получения и фармацевтическая композиция на основе соединений формулы (I). 3 с. и 3 з.п. ф-лы, 1 табл.
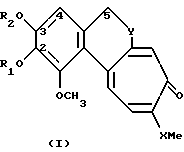
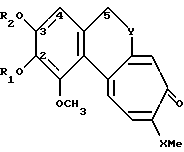
где R1 и R2 одинаковые или различные, представляют собой неразветвленную или разветвленную алкильную группу C1 - C6, циклоалкильную группу C5, ненасыщенную ацильную группу C16 - C22 или остаток β-D-глюкозы;
X - атом серы;
Y - группа формулы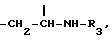
метиленовая группа которой присоединена к углероду в положении 5, а метиновая группа, имеющая ту же абсолютную конфигурацию, что и колхицин, присоединена к трополоновому кольцу, или группа =CH-CH2-OR4, метиновая группа которой присоединена к углероду в положении 5 или к трополоновому кольцу и имеет абсолютную конфигурацию s; R3 - ацильная группа C2 - C6, имеющая 1 - 3 атома галогена, предпочтительно фтора, или ацильная группа природной аминокислоты, где аминогруппа может быть защищена в виде трифторацетамида, при условии, что когда R1 и R2 - метил, R3 не является -COCH2F; R4 - ацильный остаток дикарбоновой кислоты C4 - C6 или гликозидный остаток, содержащий D-глюкозу.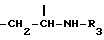
как указано выше.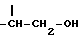
и последующим взаимодействием с производным дикарбоновой кислоты или реакционноспособным производных D-глюкозы.
| Уравнивающий мостик | 1984 |
|
SU1221142A1 |
| УСТРОЙСТВО для ПЕРЕКЛАДКИ ИЗДЕЛИЙ | 0 |
|
SU356137A1 |
| Орехов А.П | |||
| Химия алкалоидов | |||
| Устройство для охраны помещений, хранилищ и т.п. | 1925 |
|
SU1938A1 |
| J of Med | |||
| Chem | |||
| Коридорная многокамерная вагонеточная углевыжигательная печь | 1921 |
|
SU36A1 |

