РАСКРЫТИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к производным N-деацетилтиоколхицина формулы I

в которой:
- линкер представляет собой двухвалентный линейный или разветвленный С1-С8 алкильный остаток, С3-С8 циклоалкил, фенилен или С4-С6 гетероциклическое кольцо;
- соединяющие группы G1 и G2, которые могут быть одинаковыми или различными, представляют собой группы -CO-, -CONH-, -CR2-, где R2 является водородом или линейным C1-C4 алкильным остатком,
или группа G1-линкер-G2 представляет собой группу -CO-.
Соединения формулы I обладают антипролиферативной, противовоспалительной, антиартритической и противовирусной активностью.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
С некоторого времени в медицинской практике известны колхицин и тиоколхицин. Колхицин применяют в терапии подагры и состояний, связанных с воспалительными процессами. 3-О-Деметилтиоколхицинглюкозид применяют в качестве миорелаксанта при спастичности и мышечных болях вследствие контрактуры (стойкого ограничения подвижности поврежденного сустава). Однако в обоих случаях применение этих соединений ограничено из-за их высокой токсичности.
Колхицин и тиоколхицин также известны как соединения, препятствующие росту опухолевых клеток, то есть соединения, которые могут дестабилизировать микротрубочки посредством взаимодействия с тубулином. Было изучено возможное применение целого ряда производных колхицина и тиоколхицина в качестве противоопухолевых лекарственных средств. Вследствие их низкого терапевтического индекса ни одно из них не имело успеха, за единственным исключением демеколхицина(деацетил-N-метилколхицина), введенного в терапию в шестидесятые годы для лечения лейкемических форм и впоследствии замененного более эффективными алкалоидами Винке (Vinca).
WO 01/68597 раскрывает димеры тиоколхицина, где остатки тиоколхицина связаны посредством линейного алифатического амидного или амидо-уреидного мостика.
РАСКРЫТИЕ ИЗОБРЕТЕНИЯ
В настоящее время обнаружено, что соединения формулы I обладают более высокой антипролиферативной активностью, чем колхицин и тиоколхицин, в частности, на клетках, экспрессирующих фенотип MDR (Multi-Drug Resistance - множественная лекарственная устойчивость).
Соединения изобретения также являются более предпочтительными, чем димеры, раскрытые в ссылке WO 01/68597. Вместе с тем, было обнаружено, что введение остатка ароматического основания в линкер повышает на порядок цитотоксичность соединений. Введение линкеров, способных должным образом сориентировать остатки тиоколхицина в пространстве, многократно увеличивает спектр активности в отношении устойчивых опухолей. Цитотоксичность соединений формулы I оказалась сравнимой с цитотоксичностью наиболее эффективных противоопухолевых лекарственных средств, тогда как спектр действия в отношении устойчивых опухолей значительно шире.
ПОДРОБНОЕ РАСКРЫТИЕ ИЗОБРЕТЕНИЯ
Раскрытыми являются производные N-деацетилтиоколхицина формулы
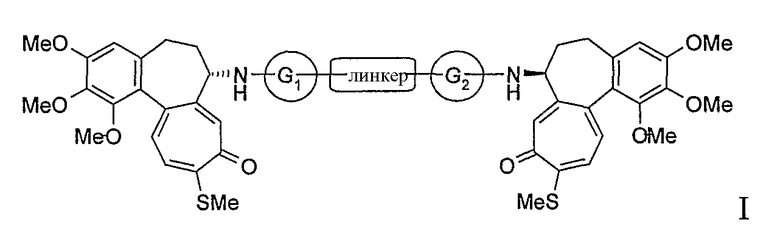
в которой:
- линкер представляет собой двухвалентный линейный или разветвленный С1-С8 алкильный остаток, С3-С8 циклоалкил, фенилен или С4-С6 гетероциклическое кольцо;
- соединяющие группы G1 и G2, которые могут быть одинаковыми или различными, представляют собой группы -CO-, -CONH-, -CR2-, где R2 является водородом или линейным C1-C4 алкильным остатком,
или группа G1-линкер-G2 представляет собой группу -CO-
с тем условием, что, когда G1 и G2 оба представляют собой группу -CO- или когда G1 является группой -CONH- и G2 является группой -CO-, линкер отличается от алкильного остатка.
Примеры алкильных двухвалентных остатков включают линейные остатки с двумя, тремя, четырьмя, пятью или шестью атомами углерода.
Примеры циклоалкильных групп включают 1,3-циклогексилен и 1,4-циклогексилен.
Примеры фениленовых групп включают 1,2-, 1,3- или 1,4-фенилен.
Примеры гетероциклических групп включают пиридил, пиразинил, пиримидинил, пиперидинил, пиперазинил, связанные с группами G1 и G2 посредством двух атомов углерода кольца, например, в положениях 3,5 или 2,5, или 2,6.
Предпочтительно G1 и G2 оба представляют собой группу -CO- или группу -CONH-.
Линкер предпочтительно представляет собой фениленовую группу, циклоалкиленовую группу или гетероциклическую группу, определенные выше, при этом предпочтительно гетероциклическая группа включает, по меньшей мере, один основный атом азота (пиридинил, пиримидинил, пиразинил, пиперидинил).
Формулы некоторых конкретных соединений формулы I представлены ниже:
Таблица 1

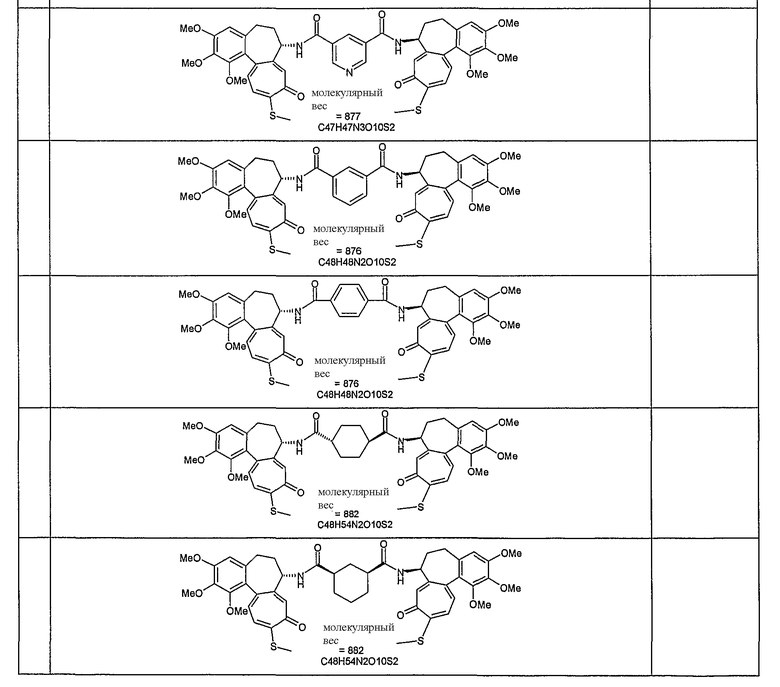

Соединения формулы I обладают антипролиферативной, противовоспалительной, антиартритической и противовирусной активностью.
Соединения настоящего изобретения получены таким образом, как описано ниже.
Соединения изобретения формулы I, в которой G1=G2=CO, (бис-амиды) получают реакцией N-деацетилтиоколхицина с активированными производными желаемых дикарбоновых кислот в инертных растворителях. Среди активированных производных дикарбоновых кислот особенно предпочтительными являются хлорангидриды и смешанные ангидриды, особенно с трифторуксусной кислотой. Среди инертных растворителей особенно предпочтительными являются хлорированные растворители. В качестве альтернативы N-деацетилтиоколхицин подвергают взаимодействию с желательной дикарбоновой кислотой в присутствии DMAP (4-N,N-диметиламинопиридин) и конденсирующего агента, DCC (дициклогексилкарбодиимид), при комнатной температуре или при нагревании и при тщательном перемешивании до исчезновения исходных продуктов.
Соединения изобретения формулы I, в которой G1=G2=CONH, (бис-мочевины) получают взаимодействием N-деацетилтиоколхицина с желательным бис-изоцианатом в инертном растворителе. Когда выбранный изоцианат коммерчески недоступен, его получают in situ путем обработки соответствующей гидроксамовой кислоты карбодиимидом и основанием (перегруппировка Лоссена (Lossen)). Альтернативно изоцианат получают обработкой соответствующего амида тетраацетатом свинца.
Соединения, где G1 и G2 представляют собой группу -CR2-, (бис-амины) получают восстановительным аминированием N-деацетилтиоколхицина подходящими диальдегидами. Альтернативно бис-амины получают алкилированием азота тиоколхицина подходящими галоген- или тозил-производными. В редких случаях бис-амины получают из соответствующих бис-амидов восстановлением боранами.
Соединения с различными G1 и G2 получают в две стадии посредством комбинирования способов, приведенных выше.
Активность соединений формулы I оценивали по большому ряду клеток опухолей, экспрессирующих различную устойчивость. Наиболее интересную активность наблюдали в линиях опухоли яичника, толстой кишки, печени и поджелудочной железы.
Таблица 2 представляет значения IC50 (выраженные в наномолях) некоторых соединений изобретения в сравнении с димером, раскрытым в примере 3 ссылки WO 01/68597.

Цитотоксическую активность оценивали по экспериментальной методике, описанной M.C. Alley et al., Cancer Research 1998, 48, 589-601.
Оказалось, что все димеры тиоколхицина с заявленными спейсерами обладают антипролиферативной, противовоспалительной и противовирусной активностью. Эти соединения, следовательно, применимы в лечении неоплазии различного происхождения, деформирующего ревматоидного артрита и в лечении опухоли Капоши (Kaposi), чей ретровирусный компонент установлен. Ингибирующее действие вирусной репликации в сочетании с ингибированием клеточной пролиферации в активно разрастающихся тканях представляет особый интерес, учитывая происхождение некоторых опухолей человека.
Для этой цели соединения изобретения вводят в виде фармацевтических композиций, пригодных для внутривенного, парентерального, перорального, чрескожного введения. Особенно полезный аспект изобретения относится к получению комплексов с белком плазмы крови, в частности, генно-инженерным человеческим альбумином. Белковые комплексы этих производных получают путем добавления соединения, растворенного в диоксане, к концентрированному раствору альбумина за время, достаточное для реакции участвующих в процессе видов молекул. После реакционного взаимодействия двух видов молекул (альбумин и производное колхицина) в условиях физиологических pH и ионной силы получающийся в результате раствор подвергают лиофилизации (сублимационная сушка). Если лиофилизированный раствор приготавливают в условиях абсолютной стерильности, то он готов для внутривенной инъекции; лиофилизированный раствор после диспергирования в подходящих и совместимых эксципиентах (вспомогательные вещества) может быть сконцентрирован и введен перорально, обеспечивая плазму лекарственными веществами в концентрациях, близких к концентрациям, получаемым парентерально. В качестве альтернативы применения белков плазмы крови, приводящих к плохой водорастворимости соединений, могут быть преимущественно использованы различно функционализированные циклодекстрины или акриловые матрицы, пригодные для парентерального введения в человека.
В зависимости от способа введения дозировка соединений будет колебаться в диапазоне от 1 до 20 мг/м2 площади тела. Предпочтительными способами введения являются локальная (locoregional) инъекция, внутривенное и пероральное введение.
Примеры композиций включают лиофилизат в пузырьках (ампулах), лиофилизированные таблетки на носителе (с наполнителем) и питьевые растворы. Любые другие агенты, которые растворяют соединения в приемлемой для введения степени, такие как Tween, Cremophor, и их подходящие смеси с полиэтиленгликолем (PEG) или спиртами также включены в настоящее изобретение.
Следующие примеры иллюстрируют изобретение более подробно.
ПРИМЕРЫ
Пример 1 - бис-(N-Деацетилтиоколхицин)амид 3,5-пиридиндикарбоновой кислоты
10 г N-деацетилтиоколхицина растворяют в 60 мл метиленхлорида. Затем добавляют при тщательном перемешивании 2,24 г (0,5 экв.) 3,5-пиридинкарбоновой кислоты, 1,64 г (0,5 экв.) N,N-диметиламинопиридина (DMAP) и 8,3 г (1,5 экв.) дициклогексилкарбодиимида (DCC). Смесь оставляют перемешиваться до исчезновения реагентов (приблизительно 12 часов), контролируя реакцию посредством тонкослойной хроматографии (TLC) (AcOEt:MeOH 10:1). Затем раствор фильтруют через целит (Celite), промывая фильтровальную подушку метиленхлоридом (2×100 мл). Объединенные органические фазы промывают сначала равным объемом 1N раствора HCl и затем 1N раствором NaOH. Органическую фазу сушат над сульфатом натрия и упаривают в вакууме. Остаток очищают фильтрацией на силикагеле (AcOEt:MeOH 10:1). Получающийся в результате продукт сушат в течение ночи в статическом сушильном аппарате при 40°С в вакууме, что дает 7,5 г конечного продукта.
1H-ЯМР (300 МГц, CDCl3): 1,00-1,42 (м, 2Н), 1,50-2,00 (м, 2Н), 2,05-2,30 (м, 2Н), 2,30-2,50 (м, 2Н), 2,52 (с, 3Н, SMe), 3,71 (с, 3Н, МеО-1), 3,90 (с, 3Н, МеО-2), 3,96 (с, 3Н, МеО-3), 4,80-4,92 (м, 2Н, Н-7), 6,47 (с, 2Н, Н-8), 7,18 (д, 12,0 Гц, 2Н, Н-12), 7,38 (д, 12,0 Гц, 2Н, Н-11), 7,67 (с, 2Н, Н-4), 8,52 (шир. с, 2Н, NH), 9,28 (с, 2Н, Н-2'+H-6'), 9,60 (с, 1Н, Н-4').
13С-ЯМР (75 МГц, CDCl3): 182,64, 164,25, 158,88, 154,12, 152,59, 152,34, 151,33, 141,92, 139,33, 135,48, 134,47, 132,74, 129,04, 128,54, 127,48, 125,71, 107,74, 61,88, 61,61, 56,49, 53,03, 30,09, 15,41.
ИК (KBr): 2934, 2854, 1664, 1605, 1540, 1485, 1424, 1403, 1349, 1321, 1266, 1235, 1195, 1153, 1137, 1095, 1051, 1021, 978, 921, 842, 796, 703.
Пример 2 - бис-(N-Деацетилтиоколхицин)амид транс-1,4-циклогександикарбоновой кислоты
10 г N-деацетилтиоколхицина растворяют в 60 мл метиленхлорида. Затем добавляют при тщательном перемешивании 2,31 г (0,5 экв.) транс-1,4-циклогександикарбоновой кислоты, 1,64 г (0,5 экв.) N,N-диметиламинопиридина (DMAP) и 8,3 г (1,5 экв.) дициклогексилкарбодиимида (DCC). Смесь оставляют перемешиваться в течение 12 часов и контролируют посредством тонкослойной хроматографии (TLC) (AcOEt:MeOH 10:1), затем фильтруют через целит (Celite), промывая фильтровальную подушку метиленхлоридом (2×20 мл). Объединенные органические фазы промывают равным объемом 1N раствора HCl, 1N раствора NaOH и рассола (насыщенный раствор NaCl), сушат над сульфатом натрия. После упаривания растворителя при пониженном давлении, остаток очищают прямой колоночной хроматографией (AcOEt:MeOH 12:1). Продукт кристаллизуют из метанола (10 объемов/объем) и сушат в течение ночи в статическом сушильном аппарате при 40°С в вакууме, что дает 6,6 г чистого соединения.
1H-ЯМР (300 МГц, CDCl3): 1,30-1,45 (м), 1,45-1,70 (м, 2Н), 1,80-1,89 (м), 1,89-2,10 (м), 2,18-2,56 (м), 2,43 (с, 3Н, SMe), 3,69 (с, 3Н, МеО-1), 3,92 (с, 3Н, МеО-2), 3,97 (с, 3Н, МеО-3), 4,80-4,92 (м, 2Н, Н-7), 6,47 (с, 2Н, Н-8), 7,09 (д, 12,0 Гц, 2Н, Н-12), 7,34 (д, 12,0 Гц, 2Н, Н-11), 7,86 (с, 2Н, Н-4), 8,93 (шир. с, 2Н, NH).
13С-ЯМР (75 МГц, CDCl3): 182,44, 176,35, 158,52, 153,80, 152,96, 151,55, 141,97, 139,22, 135,04, 134,69, 129,93, 127,09, 126,37, 107,04, 62,39, 61,68, 56,45, 51,13, 44,59, 36,92, 30,63, 29,61, 27,84, 15,36.
ИК (KBr): 3442, 3285, 2934, 2855, 1674, 1602, 1532, 1484, 1454, 1424, 1403, 1390, 1348, 1321, 1281, 1256, 1236, 1195, 1153, 1136, 1094, 1022, 982, 942, 921, 841, 619, 582.
Пример 3 - бис-(N-Деацетилтиоколхицин)амид 3,5-бензолдикарбоновой кислоты
Следуют экспериментальной методике примера 1, исходя из 5 г изофталевой кислоты, с получением продукта в виде кристаллического сухого вещества (выход: 82%).
1H-ЯМР (300 МГц, CDCl3): 1,00-1,42 (м, 2Н), 1,50-2,00 (м, 2Н), 2,05-2,30 (м, 2Н), 2,30-2,50 (м, 2Н), 2,52 (с, 3Н, SMe), 3,71 (с, 3Н, МеО-1), 3,90 (с, 3Н, МеО-2), 3,96 (с, 3Н, МеО-3), 4,80-4,92 (м, 2Н, Н-7), 6,47 (с, 2Н, Н-8), 7,18 (д, 12,0 Гц, 2Н, Н-12), 7,38 (д, 12,0 Гц, 2Н, Н-11), 7,67 (с, 2Н, Н-4), 8,52 (шир. с, 2Н, NH), 8,05 (м, 2Н, Н-4'+Н-6'), 8,50 (м, 1Н, Н-2'), 7,46 (дд, 8,0 Гц, Н-5').
Пример 4 - N-Деацетилтиоколхицин-1,4-фенилендиамин-бис-мочевина
2 г N-деацетилтиоколхицина растворяют в 150 мл безводного тетрагидрофурана. Затем добавляют 0,5 эквивалента 1,4-фенилендиизоцианата (0,4 г). Реакционную смесь оставляют перемешиваться при комнатной температуре в течение двух дней, контролируя реакцию посредством тонкослойной хроматографии (TLC) (DCM:EtOH 95:5, Rf=0,30). Растворитель выпаривают и сырой продукт рекристаллизуют из этилацетата с получением 0,95 г чистого продукта (38%).
1H-ЯМР (300 МГц, DMSO-d6): 1,72-1,88 (м), 2,08-2,36 (м), 2,58-2,70 (м), 2,42 (с, 3Н, SMe), 3,58 (с, 3Н, МеО-1), 3,82 (с, 3Н, МеО-2), 3,87 (с, 3Н, МеО-3), 4,24-4,35 (м, 2Н, Н-7), 6,82 (с, 2Н, Н-8), 7,17 (д, 12,0 Гц, 2Н, Н-12), 7,27 (д, 12,0 Гц, 2Н, Н-11), 7,13 (с, 2Н, Н-4), 8,52 (шир. с, 2Н, NH).
Пример 5 - N-Деацетилтиоколхицин-1,3-фенилендиамин-бис-мочевина
Следуют экспериментальной методике примера IV, исходя из 2 г N-деацетилтиоколхицина и 0,4 г 1,3-фенилендиизоцианата, с получением целевого продукта (выход: 42%).
1H-ЯМР (300 МГц, DMSO-d6): 1,72-1,88 (м), 2,08-2,36 (м), 2,58-2,68 (м), 2,43 (с, 3Н, SMe), 3,58 (с, 3Н, МеО-1), 3,83 (с, 3Н, МеО-2), 3,87 (с, 3Н, МеО-3), 4,24-4,35 (м, 2Н, Н-7), 6,82 (с, 2Н, Н-8), 7,18 (д, 12,0 Гц, 2Н, Н-12), 7,28 (д, 12,0 Гц, 2Н, Н-11), 7,14 (с, 2Н, Н-4), 8,48 (шир. с, 2Н, NH).
Пример 6 - бис-(N-Деацетилтиоколхицин)амид 2,9-диазасебациновой кислоты
Следуют экспериментальной методике примера IV, исходя из 2 г N-деацетилтиоколхицина и 0,4 г 1,4-бутандиизоцианата, с получением целевого продукта (выход: 63%, 0,4 г). TLC (DCM:EtOH 95:5) Rf=0,38.
1H-ЯМР (300 МГц, СDCl3): 1,80-1,95 (м), 2,38-2,54 (м), 2,70-2,95 (м), 3,40-3,60 (м), 2,50 (с, 3Н, SMe), 3,69 (с, 3Н, МеО-1), 3,95 (с, 3Н, МеО-2), 3,98 (с, 3Н, МеО-3), 4,54-4,65 (м, 2Н, Н-7), 6,58 (с, 2Н, Н-8), 7,21 (д, 12,0 Гц, 2Н, Н-12), 7,43 (д, 12,0 Гц, 2Н, Н-11), 7,81 (с, 2Н, Н-4), 8,20 (шир. с, 2Н, NH).
Пример 7 - 1,3-Бензолметилен-бис-N-деацетилтиоколхицин
2,0 г N-деацетилтиоколхицина растворяют в 100 мл хлороформа. Добавляют 0,5 эквивалента диметилацеталя изофталевого альдегида и 0,01% тозилата пиридиния. Смесь кипятят с обратным холодильником в течение ночи, дают остыть до комнатной температуры и помещают в ледяную баню. Добавляют 8 эквивалентов триацетоксиборгидрида натрия и смесь оставляют перемешиваться в течение одного дня. Затем раствор фильтруют, промывают тем же самым объемом 0,1 N раствора HCl и затем насыщенным водным раствором бикарбоната натрия. Органическую фазу сушат над сульфатом натрия, и растворитель выпаривают. Сырой продукт очищают флэш-хроматографией, что дает 0,49 г продукта.
1H-ЯМР (300 МГц, СDCl3): 1,00-1,42 (м, 2Н), 1,50-2,00 (м, 2Н), 2,05-2,30 (м, 2Н), 2,30-2,50 (м, 2Н), 2,52 (с, 3Н, SMe), 3,71 (с, 3Н, МеО-1), 3,90 (с, 3Н, МеО-2), 3,96 (с, 3Н, МеО-3), 4,80-4,92 (м, 2Н, Н-7), 6,47 (с, 2Н, Н-8), 7,18 (д, 12,0 Гц, 2Н, Н-12), 7,38 (д, 12,0 Гц, 2Н, Н-11), 7,67 (с, 2Н, Н-4), 8,52 (шир. с, 2Н, NH), 7,02 (м, 5Н).
Пример 8 - Состав для инъекций, представляющий собой комплекс бис-(N-деацетилтиоколхицин)амида 3,5-пиридиндикарбоновой кислоты с человеческим альбумином
1,0 г бис-(N-деацетилтиоколхицин)амида 3,5-пиридиндикарбоновой кислоты растворяют в 20 мл диоксана. Получающийся в результате раствор медленно добавляют по каплям в 5% физиологический раствор альбумина до получения гомогенной молочной суспензии. Смесь оставляют перемешиваться в стерильных условиях в течение двух часов, затем лиофилизируют.
Лиофилизированный продукт готов для введения путем инъекции.
| название | год | авторы | номер документа |
|---|---|---|---|
| АНАЛОГИ РЕБАУДИОЗИДА | 2017 |
|
RU2752317C2 |
| КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ ТИОКОЛХИЦИНА И БАККАТИНА В КАЧЕСТВЕ ПРОТИВООПУХОЛЕВЫХ АГЕНТОВ | 2001 |
|
RU2264393C2 |
| ВОДОРАСТВОРИМОЕ ПРОИЗВОДНОЕ ХЛОРОФИЛЛА α, МОДИФИЦИРОВАННОЕ ФРАГМЕНТОМ МИРИСТИНОВОЙ КИСЛОТЫ | 2017 |
|
RU2680523C1 |
| ОТБЕЛИВАЮЩАЯ КОМПОЗИЦИЯ И СПОСОБ ОТБЕЛИВАНИЯ СУБСТРАТА | 1999 |
|
RU2235125C2 |
| ПРОИЗВОДНЫЕ ГАЛАНТАМИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2001 |
|
RU2241001C2 |
| ПРОИЗВОДНЫЕ КОЛХИЦИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ИХ ОСНОВЕ | 1997 |
|
RU2181354C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПАКЛИТАКСЕЛА | 2001 |
|
RU2276147C2 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2010 |
|
RU2581367C2 |
| ИНГИБИТОРЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЕ | 2019 |
|
RU2806033C2 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 1997 |
|
RU2193029C2 |
Настоящее изобретение относится к производным N-деацетилтиоколхицина формулы (I) и фармацевтическим композициям на их основе, которые могут быть применены в качестве противоопухолевых средств:

где линкер представляет собой циклоалкил, фенилен или гетероциклическое кольцо; обе соединяющие группы G представляют собой -СО- или -CONH-. Технический результат - получение новых биологически активных соединений и фармацевтических композиций. 2 н. и 4 з.п. ф-лы.
1. Соединения формулы I

в которой линкер представляет собой С3-С8 циклоалкил, фенилен или С4-С6 гетероциклическое кольцо; и
обе соединяющие группы G представляют собой группу -СО- или группу -CONH-.
2. Соединения по п.1, где линкер выбирают из 1,3-циклогексилена и 1,4-циклогексилена.
3. Соединения по п.1 или 2, где линкер выбирают из 1,2-, 1,3- или 1,4-фенилена.
4. Соединения по п.1, где линкер выбирают из пиридила, пиперидинила, пиперазинила, связанных с группами G в положениях 3,5 или 2,5 или 2,6.
5. Соединения формулы I по п.1, предназначенные для противоопухолевого применения.
6. Фармацевтическая композиция для противоопухолевого применения, содержащая соединение формулы I по п.1 в качестве активного ингредиента в смеси с подходящими носителями и/или эксципиентами.
| ПРОИЗВОДНЫЕ КОЛХИЦИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1996 |
|
RU2163241C2 |
| БКИРЛ-ГСКЛИ '^Ь^"-^^' rV:.»В. И. Гинзбург | 0 |
|
SU168597A1 |
| US 4533675, 06.08.1985. | |||

