Изобретение относится к новым производным колхицина, имеющим антипролиферативную, противоопухолевую, противовоспалительную и релаксирующую мышцы активность, способам их получения и содержащим их фармацевтическим готовым препаративным формам.
Колхицин является известным псевдоалкалоидом, широко используемым в течение очень долгого времени в терапии для лечения подагры, - патологии, на которую он действует очень быстро и специфически, несмотря на то, что его следует использовать в течение короткого времени вследствие его токсичности. Производное колхицина, а именно тиоколхикозид, широко применяют для лечения контрактур и воспалительных состояний скелетных мышц. Кроме того, колхицин является очень сильным, препятствующим росту опухолевых клеток средством, которое действует, блокируя образование митотической веретенообразной структуры во время деления клеток; этот последний аспект был исследован тщательно для любой антинеопластической (противоопухолевой) активности, и для этой цели было получено большое число производных колхицина. Колхицин, как таковой, и ряд его производных нельзя использовать клинически вследствие их высокой токсичности и, следовательно, их неприемлемого соотношения риск/польза. Только одно производное колхицина, демеколцин, используют до некоторой степени в онкологии для лечения некоторых форм лейкемии. Продукты по данному изобретению отличаются от продуктов известного уровня техники высокой активностью, более низкой токсичностью и более высоким терапевтическим индексом. Более конкретно, в исследования неоплазии были сосредоточены на поиске продуктов, имеющих, кроме обычной цитотоксичности цитотоксичность, нацеленную на клеточные линии, устойчивые к известным, обычным антибластическим лекарственным средствам, препятствующим росту опухолевых клеток.
Производные по настоящему изобретению имеют формулу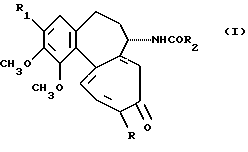
где R представляет метоксильную или тиометильную группу; R представляет гидрокси; β-D-глюкопиранозилокси - остаток; β-D-глюкопиранозилокси-остаток, кетализированный у гидроксилов 4' и 6' алифатическими или ароматическими альдегидами; 6-дезоксигалактопиранозилокси - остаток; ацилоксигруппа полиненасыщенных жирных кислот от C16 до C22; неразветвленный, разветвленный или циклический O-алкил C1-C6, насыщенный или ненасыщенный; и R2 представляет C1-C6-галогеналкильную группу.
Особенно предпочтительными соединениями формулы I являются соединения, у которых R1 представляет метоксигруппу, β-D-глюкопиранилокси-остаток, необязательно кетализирован у 4'- и 6'-гидроксилов ароматическими или алифатическими альдегидами, например 2- или 3-тиеналем, или ксименоилоксигруппа.
R2 предпочтительно представляет трифторметил, пентафторэтил или гептафторпропил.
Соединения I получают из природных соединений колхицина и тиоколхицина (формула I, R1 = -OCH3, R2 = CH3 R = -OCH3 или -SCH3 соответственно) или из их соответствующих производных, гликозилированных по гидроксилу в положении 3, а также из их N-формил-N-деацетилпроизводных.
Гидролиз этих природных соединений водными растворами сильных минеральных кислот делает возможным селективное получение, при изменении температуры и времени реакции, соответствующих N-деацетил- и 3-деметил-N-деацетилпроизводных, которые можно затем подвергнуть обычным реакциям N-ацилирования и алкилирования или ацилирования по гидроксигруппе в положении 3.
В случае тиоколхицина гидролиз галогеноводородными кислотами или, более предпочтительно, серной кислотой (20% H2SO4- 120 час) позволяет получить N-деацетилтио-колхицин и 3-деметил-N-деацетилтиоколхицин почти с количественными выходами.
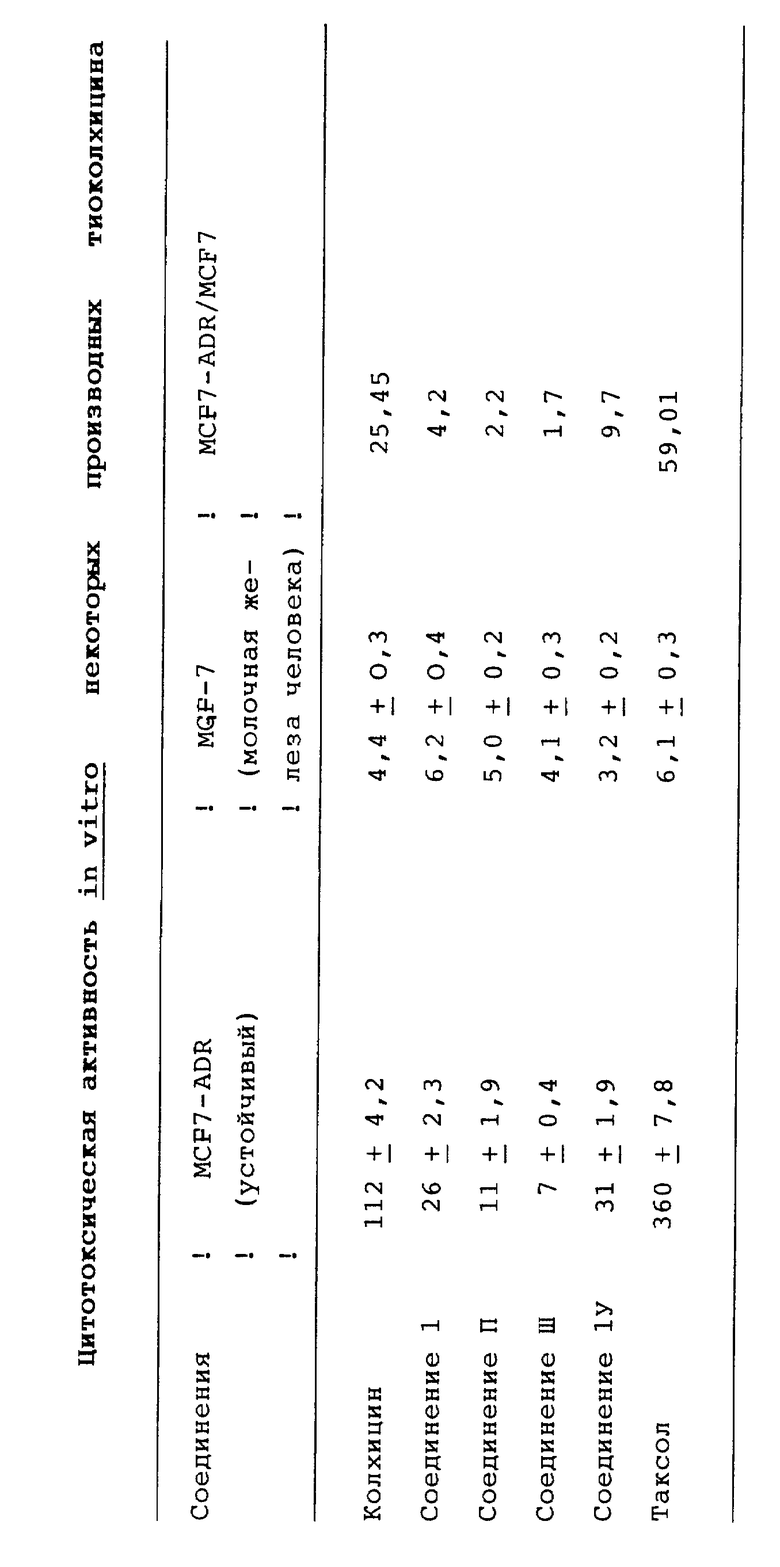
Соединения по данному изобретению имеют необычную противоопухолевую активность.
Таблица показывает антимитотическую активность соединений по настоящему изобретению, на культивированном эксплантате опухоли молочной железы по сравнению с колхицином и Таксолом.
Эта таблица свидетельствует о том, что соединение по данному изобретению имеет существенные преимущества по воздействию на устойчивые клеточные линии, которые в настоящее время рассматриваются в качестве основной мишени для цитотоксических лекарственных средств.
Кроме того, продукты по данному изобретению имеют противовоспалительную и релаксирующую мышцы активность, и их можно вводить в фармацевтические готовые препаративные формы, которые можно использовать для введения лекарственного средства для указанной патологии. Готовые препаративные формы для внутривенного, перорального, чрескожного, накожного введений можно получить подходящим способом.
Доказано, что среди наполнителей, которые можно использовать для получения этих готовых препаративных форм, природные и синтетические фосфолипиды особенно полезны для получения липосомных форм для парентеральных, внутривенных и/или местных путей введения. Выяснилось, что такие же готовые препаративные формы можно использовать при местном лечении кожных эпителиом и кожных гиперпролиферативных состояний, таких как псориаз. Оказывается, что в специфической противоопухолевой области кроме фосфолипидов, которые позволяют вводить лекарственное средство в липосомной форме, особенно полезны некоторые поверхностно-активные вещества, такие как полиэтоксилированные касторовые масла или полисорбаты, действующие синергитически с активным ингредиентом. Действующее начало предпочтительно измельчают до микронных размеров, пока соединение не будет растворяться в воде. В онкологии эти продукты используют при дозах от 1 до 100 мг/м2.
Следующие примеры далее иллюстрируют настоящее изобретение.
Пример I
Получение N-деацетил-N-пентафторпропионилтиоколхицина (Соединение I; R = -SCH3, R1 = -OCH3, R2= -CF2-CF3).
20 г тиоколхицина растворяют в 300 мл 20% серной кислоты и нагревают в атмосфере азота в течение 36 час при 100oC, реакционную смесь подщелачивают до pH 8, для выделения 15 г N-деацетилтиоколхицина. Этот продукт растворяют в ацетоне и в присутствии безводного Na2CO3 подвергают реакции с 1,5 эквивалента перфторпропионового ангидрида при сильном перемешивании; через 2 часа реакционную смесь фильтруют и растворитель выпаривают. Масляный остаток растворяют в метаноле, из которого N-деацетил-N-пентафторпропионилтиоколхицин выделяют кристаллизацией.
Пример II
Получение N-деацетил-N-пентафторпропионил-3-O-кси-меноилтиоколхицина. (Соединение II, R = -SCH3; R1 = -O-ксименоил, R2 = -CF2-CF3)
20 г тиоколхикозида растворяют в 300 мл 20% серной кислоты и весь раствор нагревают в атмосфере азота 36 час при 100oC; из реакционной смеси выделяют 12 г N-деацетил- 3-O-деметилтиоколхицина.
Этот продукт растворяют в ацетоне и в присутствии безводного Na2CO3 подвергают реакции с 3 эквивалентами перфторпропионового ангидрида при сильном перемешивании; через 2 часа реакционную смесь фильтруют и растворитель и избыток реагента удаляют в вакууме. Остаток, состоящий из N-деацетил-N-пентафторпропионил-3-O-деметил-3-O-пентафторпропионата, растворяют в метаноле, содержащем NH4Cl, контролируя гидролиз сложного эфира фенола тонкослойной хроматографией (смесь толуол/этилацетат, 1:1); растворитель выпаривают в вакууме досуха и остаток растворяют в ацетоне, отделяя нерастворимые компоненты фильтрованием. Раствор в ацетоне концентрируют досуха и остаток растворяют в 100 мл пиридина; этот раствор охлаждают до 0oC и добавляют 2 экв. хлорангидрида ксимениновой кислоты при сильном перемешивании. Реакционную смесь оставляют стоять в течение ночи и затем выливают в 500 г льда. Образованную водную суспензию экстрагируют три раза 500 мл метиленхлорида. Органическую фазу промывают водой, затем разбавленным раствором соляной кислоты и снова водой. Органическую фазу сушат над Na2SO4 и концентрируют досуха. Остаток кристаллизуют из смеси этилацетат/изопропиловый эфир, получая 27 г N-деацетил-N-пентафторпропионил-3-O-ксименоил-тиоколхицина.
Пример III
Получение N-деацетил-N-пентафторпропионил-3-O-деметил-3-O-циклопентилтиоколхицина. (Соединение III, R = -SCH3, R1 =-O-циклопентенил, R2 = CF2-CF3).
20 г тиоколхикозида растворяют в 300 мл 20% серной кислоты и смесь нагревают в атмосфере азота в течение 36 часов при 100oC; из реакционной смеси отделяется 12 г N-деацетил-3-O-деметилтиоколхицина.
Этот продукт растворяют в ацетоне в присутствии безводного Na2CO3, подвергают реакции с 3 эквивалентами перфторпропионового ангидрида при сильном перемешивании; через 2 часа реакционную смесь фильтруют и растворитель и избыток реагента удаляют в вакууме. Остаток, состоящий из N-деацетил-N-пентафторпропионил-3-O-деметил-3-O-пентафторпропионата растворяют в метаноле, содержащем NH4Cl, контролируя гидролиз сложного эфира фенола тонкослойной хроматографией (смесь толуол/этилацетат, 1:1); растворитель выпаривают в вакууме досуха и остаток растворяют в ацетоне, отфильтровывая нерастворимую часть. К раствору в ацетоне - добавляют Na2CO3 и 5 эквивалентов (относительно исходного продукта) циклопентилбромида. Реакционную смесь перемешивают в течение 6 час, контролируя алкилирование тонкослойной хроматографией. Когда реакция завершается, соль отделяют фильтрованием и растворитель перегоняют в вакууме. Остаток хроматографируют на колонке с сидикагелем, используя этилацетат в качестве элюента. Фракции, содержащие целевой продукт, собирают, растворитель удаляют и продукт кристаллизуют из смеси ацетон/гексан. Получают 9,2 г N-деацетил-N-пентафторпропионил-3- O-циклопентенилтиоколхицина.
Пример IV
Получение N-деацетил-N-гептафторбутироилтиоколхицина
(Соединение IV R =-SCH3, R1 =-OCH3, R2 =-CF2-CF2-CF3).
10 г N-деацетилтиоколхицина растворяют в 150 мл безводного ацетона в присутствии Na2CO3 и обрабатывают при комнатной температуре 1,5 экв ангидрида гептафтормасляной кислоты. Na2CO3 и растворитель удаляют и остаток очищают изопропиловым эфиром, получая 12,5 г N-деацетил-N-гептафторбутироилтиоколхицина.
Пример V
Получение N-деацетил-N-пентафторпропионил-3-O- изопропилтиоколхицина. (R =-SCH3, R1 =-O-изопропил, R2 =-CF2-CF3).
Для получения этого производного повторяют методики примера III, используя изопропилбромид в качестве реагента. После очистки сырого продукта реакции на силикагеле и кристаллизации получают 7,6 г N-деацетил-N-пентафторпропионил-3-O-изопропилтиоколхицина, имеющего спектроскопические характеристики, соответствующие целевой молекуле.
Производные колхицина формулы I, где R - метоксильная или тиометильная группа; R1 - ацилоксигруппа полиненасыщенных жирных кислот от C16 до C22, неразветвленный, разветвленный или циклический O-алкил C1-6, насыщенный или ненасыщенный; R2 - C1-6-галогеналкильная группа, при условии, что когда R2 - C1-галогеналкильная группа, R1 отличается от метокси, проявляют цитотоксическую активность. Продукты по данному изобретению отличаются от продуктов известного уровня техники высокой активностью, более низкой токсичностью и более высоким терапевтическим индексом. 2 с. и 3 з.п. ф-лы. 1 табл.
где R - метоксильная или тиометильная группа;
R1 - ацилоксигруппа полиненасыщенных жирных кислот от C16 до C22, неразветвленный, разветвленный или циклический О-алкил C1 - C6, насыщенный или ненасыщенный;
R2 - C1 - C6-галогеналкильная группа при условии, что когда R2 - C1-галогеналкильная группа, R1 отличается от метокси.
| Способ получения геминальных 2,2диаминопроизводных индандиона-1,3 | 1972 |
|
SU449039A1 |
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| J | |||
| Med | |||
| Chem | |||
| Vol | |||
| Нивелир для отсчетов без перемещения наблюдателя при нивелировании из средины | 1921 |
|
SU34A1 |
| LIEBIGS ANNALEN DER CHEMIE | |||
| Устройство для поддержания напряжения сети постоянным при переменном числе оборотов генератора переменного тока | 1924 |
|
SU758A1 |

