ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способу получения С-4′′-замещенных производных 9-деоксо-9а-аза-9а-гомоэритромицина А (здесь и далее, "азалид(ы)"), которые могут использоваться в качестве антибактериальных агентов и агентов против простейших у млекопитающих, включая человека, а также рыб и птиц. Данное изобретение также относится к способу получения стабильных промежуточных продуктов заявленных азалидов, а также к кристаллической соли промежуточного соединения в способе получения заявленных азалидов. Данное изобретение также относится к фармацевтическим композициям, содержащим новые соединения, полученные заявленным способом, и к способам лечения бактериальных инфекций и инфекций, вызванных простейшими, у млекопитающих, рыб и птиц путем введения новых соединений, полученных заявленными способами, млекопитающим, рыбам и птицам, нуждающимся в таком лечении.
При лечении широкого спектра бактериальных инфекций и инфекций, вызванных простейшими, у млекопитающих, рыб и птиц известно использование антибиотиков макролидного ряда. Такие антибиотики включают в себя различные производные эритромицина А, такие как азитромицин, который является коммерчески доступным и описан в патентах США 4474768 и 4517359, оба приведены здесь в качестве ссылки в своем полном объеме. Как азитромицин, так и другие антибиотики макролидкого ряда, макролидные соединения по настоящему изобретению обладают сильным действием против различных бактериальных инфекций и инфекций, вызванных простейшими, как описано далее.
Производство заявленных азалидов в коммерческом масштабе имеет некоторые трудности, включая, но не ограничиваясь ими, небольшой выход и неустойчивость некоторых синтетических промежуточных продуктов, а также присутствие нежелательных примесей.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
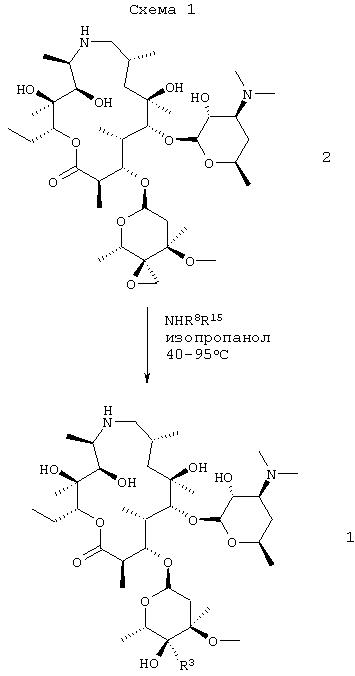
Настоящее изобретение относится к способу получения соединения формулы 1
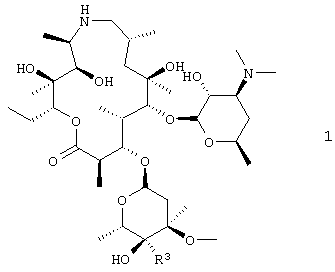
или его фармацевтически приемлемой соли, который включает: взаимодействие соединения формулы 2
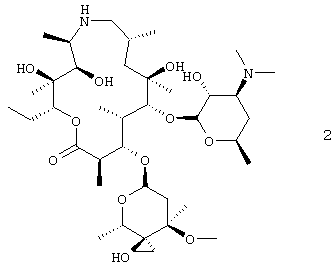
с амином формулы HNR8R15 в органическом растворителе, содержащем изопропанол;
где взаимодействие проводят при температуре, по крайней мере, около 40°С;
где:
R3 представляет собой -CH2NR8R15;
R8 представляет собой C1-С10 алкил; и
R15 представляет собой Н или C1-С10 алкил.
В предпочтительном воплощении способа R8 представляет собой пропил и R15 представляет собой Н. В особенно предпочтительном воплощении R8 представляет собой н-пропил и R15 представляет собой Н.
В особенно предпочтительном воплощении органическим растворителем является изопропанол.
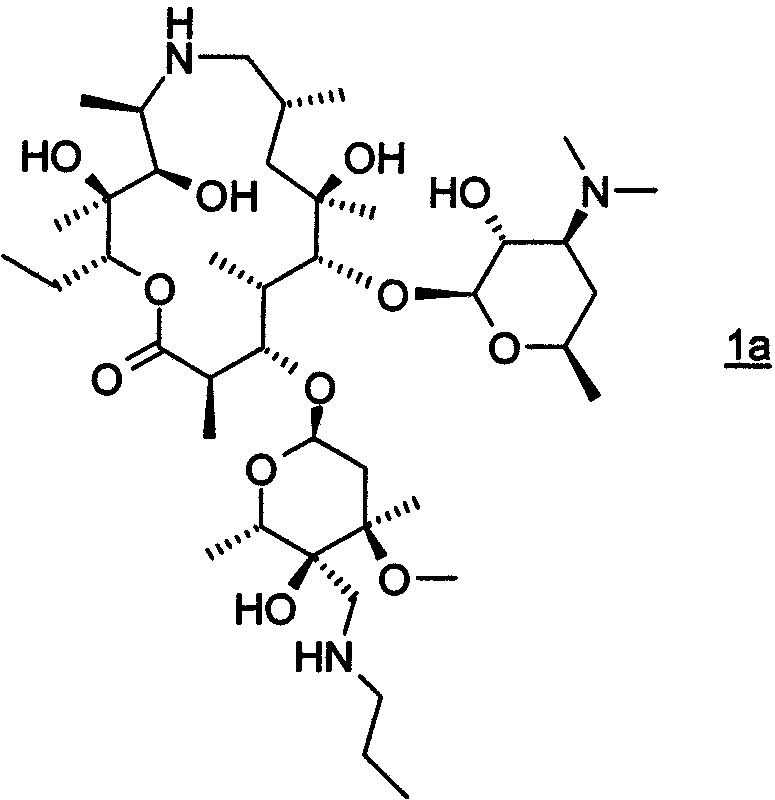
В другом предпочтительном воплощении изобретение относится к способу получения соединения формулы 1а или его фармацевтически приемлемой соли
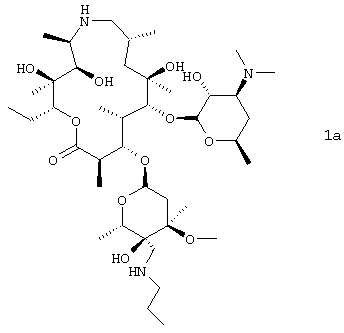
путем взаимодействия соединения формулы 2 с н-пропиламином в органическом растворителе, включая изопропанол; где взаимодействие проводят при температуре, по крайней мере, около 40°С. В особенно предпочтительном воплощении органическим растворителем является изопропанол.
Необходимо отметить, что термины "раствор" и "смесь", как используется здесь, если не указано иного, используются взаимозаменяемо без указания структуры дисперсии их компонентов. Фраза "органический растворитель, включая изопропанол", как используется здесь, если не указано иного, означает безводный растворитель или смесь безводных растворителей, где, по крайней мере, один растворитель является изопропанолом. В этой заявке термин "соединение формулы 1" включает в себя как соединение формулы 1, так и соединение формулы 1а. Соединение формулы 1а представляет собой особенно предпочтительное воплощение соединения формулы 1, где все воплощения и предпочтительные воплощения способа описаны здесь далее.
В воплощении описанного здесь способа температура, по крайней мере, ниже около 95°С и в предпочтительном воплощении температура, по крайней мере, ниже около 80°С. В более предпочтительном воплощении температура находится в области от около 50°С до около 76°С. В особенно предпочтительном воплощении температура находится в области от около 50°С до около 55°С.
В предпочтительном воплощении описанного здесь способа взаимодействие проводят при атмосферном давлении. В этой заявке термин "атмосферное давление" означает давление в пределах нормальных значений метеорологического атмосферного давления на высоте над уровнем моря, тогда как термин "повышенное давление" означает давление выше атмосферного давления. В другом воплощении описанного здесь способа взаимодействие проводят при повышенном давлении. В другом воплощении изобретения кроме изопропанола может присутствовать триэтиламин.
В дополнение к заявляемым предпочтительным воплощениям взаимодействие соединения формулы 2 с амином с получением соединения формулы 1 успешно осуществляли в растворителях, иных чем те, которые включают изопропанол. Соответственно настоящее изобретение также относится к способу получения соединения формулы 1 при взаимодействии соединения формулы 2 с амином формулы HNR8R15 в органическом растворителе, где растворитель выбран из группы, включающей бензиловый спирт, ацетон, метилизобутилкетон, ДМСО, трет-бутанол, н-бутанол, диизопропиловый эфир, смесь МТВЕ и ДМФ и их сочетания, где взаимодействие проводят при температуре, по крайней мере, около 40°С. Взаимодействие можно проводить при повышенном давлении, но предпочтительно проводить при атмосферном давлении. В другом воплощении взаимодействие ускоряется путем добавления каталитического количества кислоты Льюиса. В таком воплощении кислотой Льюиса является реагент, такой как бромид магния, йодид калия, перхлорат лития, перхлорат магния, тетрафторборат лития, пиридин гидрохлорид или йодид тетрабутиламмония. Кислотой Льюиса предпочтительно является бромид магния.
В воплощении описанного здесь способа молярное количество амина, по меньшей мере, приблизительно в пять раз больше молярного количества соединения формулы 2. В другом воплощении описанного здесь способа концентрация амина в изопропаноле составляет, по крайней мере, около 5 моль. В особенно предпочтительном воплощении концентрация н-пропиламина в изопропаноле равна приблизительно 6-7 моль.
В воплощении вышеуказанного способа соединение формулы 2 взаимодействует с амином, по крайней мере, в течение около 24 часов. В предпочтительном воплощении молярное количество амина, по меньшей мере, приблизительно в пять раз больше молярного количества соединения формулы 2 и соединение формулы 2 взаимодействует с амином, по крайней мере, в течение около 24 часов. В более предпочтительном воплощении температура составляет от около 50°С до около 80°С. В еще более предпочтительном воплощении молярное количество амина приблизительно в двадцать раз больше молярного количества соединения формулы 2, концентрация амина в изопропаноле составляет около 6 моль, и соединение формулы 2 взаимодействует с амином в течение, по крайней мере, около 24 часов при температуре от около 50°С до около 55°С.
Другое воплощение описанного здесь способа, кроме того, включает кристаллизацию соединения формулы 1 в виде свободного основания. В воплощении соединение формулы 1 в виде свободного основания кристаллизовали из водной смеси растворителя. В предпочтительном воплощении водная смесь растворителя включает воду и неводный растворитель, выбранный из группы, включающей метанол, этанол, изопропанол и ацетон. В другом воплощении соединение формулы 1 в виде свободного основания кристаллизовали из органического (С6-С10) алканового растворителя или смеси таких органических алкановых растворителей. В предпочтительном воплощении соединение формулы 1 кристаллизуется путем нагревания соединения с алкановым растворителем с последующим охлаждением для осуществления кристаллизации. В предпочтительном воплощении органический (С6-С10) алкановый растворитель выбран из гептана или октана, более предпочтительно, из гептана. В другом воплощении, как описано далее, свободное основание получали из соли добавлением кислоты соединения формулы 1. Понятно, что "алкан", как используется здесь, если не указано иного, включает насыщенные одновалентные углеводороды с прямой, циклической или разветвленной цепью или их смеси.
В другом воплощении описанного здесь способа соль добавления кислоты соединения формулы 1 получали путем обработки соединения формулы 1 раствором, содержащим кислоту в смеси растворителем в воде. В предпочтительном воплощении раствор кислоты добавляли к раствору, содержащему соединение формулы 1 и воду. В более предпочтительном воплощении кислота представляет собой фосфорную кислоту, L-винную кислоту или дибензоил-D-винную кислоту. В особенно предпочтительном воплощении кислота представляет собой фосфорную кислоту. В другом, более предпочтительном воплощении, растворитель включает этанол. В другом предпочтительном воплощении вышеуказанный способ, кроме того, включает в себя выделение соли добавления кислоты соединения формулы 1.
В воплощении описываемого здесь способа образуется соединение формулы 1, которое является чистым, по крайней мере, на 90%, более предпочтительно, по крайней мере, на 95% и наиболее предпочтительно, по крайней мере, на 98%. В частности, способ по изобретению дает соединение формулы 1 с подходящими параметрами очистки для использования соединения формулы 1 при получении композиции для парентерального введения. Требуемые условия для парентеральных составов хорошо известны в данной области, например: исключительная чистота и малый размер частиц в растворе, и стерильное получение составов, и удаление пирогенов (см., Remington′s Pharmaceutical Sciences, Mack Publishing Company, Easton, Pa., 18th Edition, Gennaro, ed. (1990), с. 1545-1580).
В другом предпочтительном воплощении вышеуказанный способ, кроме того, включает в себя обработку соли добавления кислоты соединения формулы 1 основанием в смеси с водой и неполярным растворителем с получением соединения формулы 1 в виде свободного основания. В более предпочтительном воплощении основанием является двухосновная карбонатная соль, и в особенно предпочтительном воплощении двухосновной карбонатной солью является карбонат калия. В другом более предпочтительном воплощении неполярный растворитель представляет собой дихлорметан. Еще в другом воплощении способ, кроме того, включает в себя кристаллизацию соединения формулы 1 в виде свободного основания, как описано выше, и другие воплощения, относящиеся к этому, описаны выше.
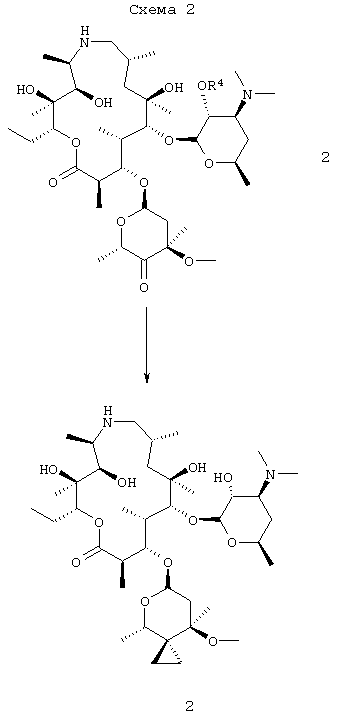
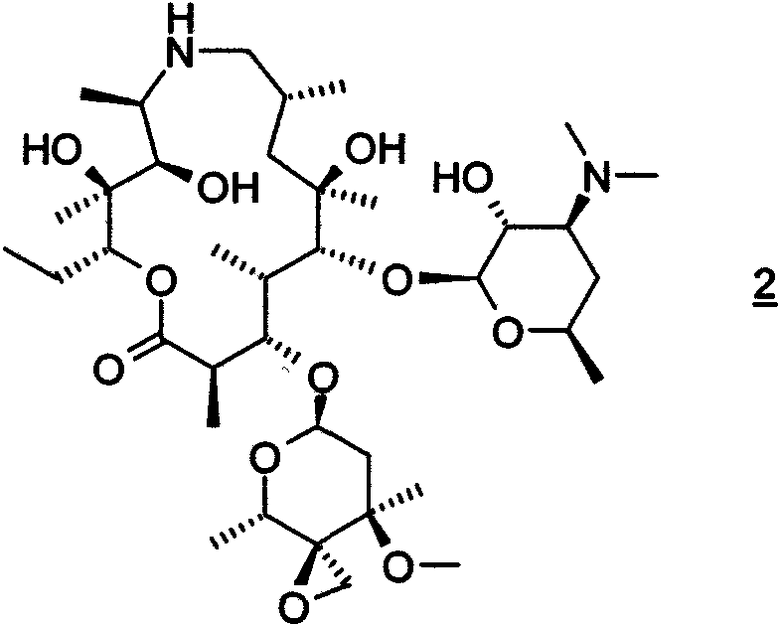
Данное изобретение также относится к получению соединения формулы 2, которое включает в себя:
(а) взаимодействие свободного основания соединения формулы 3
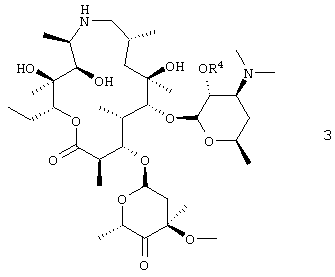
с ионом метилида сульфония;
(b) гашение реакции на стадии (а) водным раствором слабой кислоты и распределение продукта в неводный раствор; и
(c) снятие защиты у продукта со стадии (b) с получением соединения формулы 2;
где R4 представляет собой гидроксизащитную группу.
В воплощении вышеуказанный способ, кроме того, включает выделение соединения формулы 2. В предпочтительном воплощении соединение формулы 2 выделяют в виде гидрата, более предпочтительно, в виде моногидрата. В воплощении содержание воды определяется по методу Карла-Фишера (Karl-Fischer). В воплощении гидрат получают из смеси, содержащей соединение формулы 2 и растворитель или смесь растворителей, выбранного(ых) из ацетона, ацетон/вода, ацетон/гептан и МТВЕ/гептан. В других воплощениях соединение формулы 2 выделяли в виде ацетатной соли, соли L-винной кислоты или соли дибензоил-D-винной кислоты.
Данное изобретение относится к моногидрату соединения формулы 2. В предпочтительном воплощении вышеуказанного способа R4 представляет собой безилоксикарбонил.
В другом предпочтительном воплощении вышеуказанного способа стадию (а) проводили при температуре от около -80°С до около -45°С.
В другом воплощении вышеуказанного способа соединение формулы 3 в виде свободного основания получали из соли добавления кислоты соединения формулы 3. В предпочтительном воплощении соль добавления кислоты представляет собой соль добавления трифторуксусной кислоты. В других воплощениях вышеуказанного способа соль добавления кислоты соединения формулы 3 выбрана из дибензоил-D-тартратной соли, L-тартратной соли или фосфатной соли. Соли добавления кислот описанных здесь соединений легко получали обычными способами.
В воплощении вышеуказанного способа метилид сульфония представляет собой метилид диметилсульфония. В предпочтительном воплощении метилид диметилсульфония получали взаимодействием галогенида или сульфоната триметилсульфония с сильным основанием. В более предпочтительном воплощении использовали галогенид триметилсульфония, который предпочтительно представлял собой бромид триметилсульфония. В другом, более предпочтительном воплощении, галогенид триметилсульфония взаимодействовал с сильным основанием в инертном органическом растворителе или его смесях. В особенно предпочтительном воплощении инертный органический растворитель представлял собой эфирный растворитель, который, предпочтительно, представляет собой тетрагидрофуран или смесь тетрагидрофурана и дихлорметана.
В воплощении стадия (с) включает каталитическое гидрирование, где R4 представляет собой бензилоксикарбонил. В предпочтительном воплощении катализатором является палладий на угле. В особенно предпочтительном воплощении катализатор палладий на угле представляет собой 10% Pd/C (Johnson-Matthey type A 402028-10). В другом воплощении стадии (с) у продукта со стадии (b) удаляют защиту путем каталитического гидрирования, предпочтительно с формиатом аммония, Pd/C в метаноле. В другом воплощении перед гидрированием продукт со стадии (b) обрабатывают фуллеровой землей. Подходящими растворителями для процедуры гидрирования являются ацетон, этилацетат, ТГФ, МТВЕ, изопропанол, этанол и метанол. Предпочтительным растворителем является ацетон.
Данное изобретение также относится к 2′-бензилоксикарбонилзащищенному соединению II
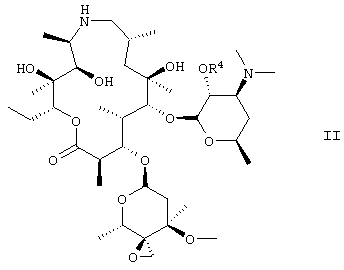
которое получают, минуя стадию (с) вышеуказанного способа.
Данное изобретение относится к получению соединения формулы 3
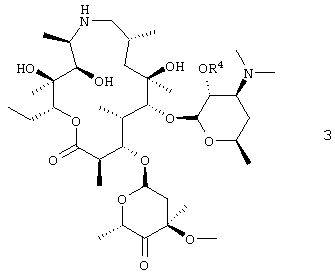
путем окисления С-4′′ гидроксигруппы соединения формулы 4
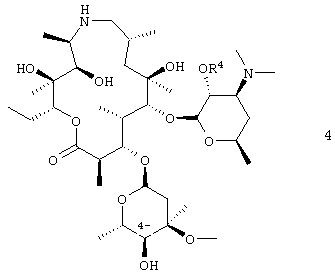
где R4 представляет собой гидроксизащитную группу.
В воплощении окисление проводят путем добавления диметилсульфоксида (ДМСО) в раствор, содержащий соединение формулы 4 и растворитель, охлаждением смеси до около -70°С, последующим добавлением трифторуксусного ангидрида, а затем добавлением триэтиламина. В других воплощениях ДМСО активируется при использовании оксалилхлорида (в присутствии или в отсутствие триметилсилилацетамида), полифосфорной кислоты, пиридина-SO3 или ангидрида уксусной кислоты. В другом воплощении в процессе добавления трифторуксусного ангидрида температуру поддерживали в интервале между -70°С и -60°С. В другом воплощении растворитель представляет собой дихлорметан. Частный случай вышеуказанного способа представляет собой активацию ДМСО in situ в присутствии реакционного спирта, который сводит к минимуму образование примесей, обычно встречаемых при активируемых ДМСО окислениях, и обычно включает в себя введение спирта в раствор, содержащий активированный ДМСО.
В воплощении вышеуказанный способ, кроме того, включает выделение соли добавления кислоты соединения формулы 3. В предпочтительном воплощении соль добавления кислоты представляет собой дибензоил-D-тартратную соль или фосфатную соль. В особенно предпочтительном воплощении данное изобретение относится к способу получения соли добавления трифторуксусной кислоты соединения формулы 3, который включает в себя обработку соединения формулы 3 трифторуксусной кислотой; и кристаллизации полученной соли добавления кислоты;
где R4 представляет собой гидроксизащитную группу.
В предпочтительном воплощении вышеуказанного способа R4 представляет собой бензилоксикарбонил.
В другом предпочтительном воплощении вышеуказанного способа соль добавления кислоты кристаллизовали из изопропанола.
Еще в другом предпочтительном воплощении вышеуказанного способа соль добавления кислоты кристаллизовали из смеси метиленхлорида и метил трет-бутилового эфира.
Соли добавления трифторуксусной кислоты, полученные способом по данному изобретению, не являются фармацевтически приемлемыми, но являются прекрасно очищенными и стабильными, что дает возможность для хранения и транспортировки соответствующих исходных продуктов для коммерческого получение соединений формулы 1.
В воплощении вышеуказанного способа соединение формулы 4 получают путем защиты 2′-гидроксигруппы соединения формулы 5
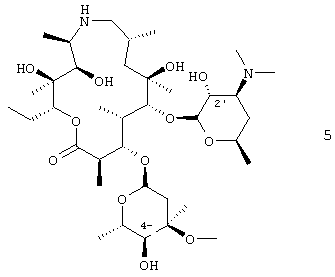
В предпочтительном воплощении 2′-гидроксигруппу защищали бензилоксикарбонилом. В другом предпочтительном воплощении соединение формулы 5 подвергали взаимодействию, по крайней мере, с двумя молярными эквивалентами бензилхлорформиата. В более предпочтительном воплощении взаимодействие проводили в дихлорметане. В еще более предпочтительном воплощении дихлорметан был, по крайней мере, в 15-кратном избытке по объему относительно объема исходного продукта. Данное изобретение также относится к соли добавления трифторуксусной кислоты соединения формулы 3, где R4 представляет собой бензилоксикарбонил:
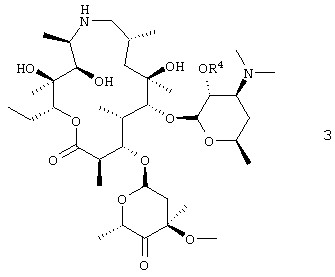
В предпочтительном воплощении соль имеет структуру, показанную формулой 3а
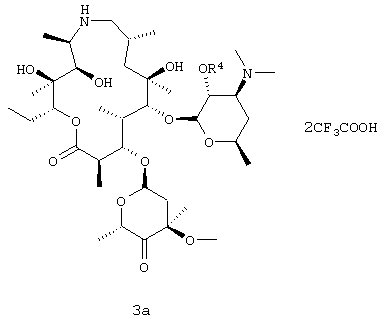
где R4 представляет собой бензилоксикарбонил. Данное изобретение также относится к дибензоил-D-тартратной соли соединения формулы 3, где R4 представляет собой бензилоксикарбонил.
Термин "гидроксизащитная группа", как используется здесь, если не указано иного, включает ацетильные, бенилоксикарбонильные и различные гидроксизащитные группы, хорошо известные специалистам в данной области, включая группы, перечисленные в Т.W. Greene, P.G. М. Wuts, "Protective Groups In Organic Synthesis," (J. Wiley & Sons, 1991). Предпочтительно гидроксизащитная группа R4 представляет собой бензилоксикарбонил ("CBZ").
Термин "галоген", как используется здесь, если не указано иного, включает фтор, хлор или бром, а термин "галогенид" относится к соответствующим одновалентным анионам, F-, Сl- или Вr- соответственно.
Термин "алкил", как используется здесь, если не указано иного, включает насыщенные одновалентные углеводородные радикалы с прямой, циклической или разветвленной цепью или их смеси.
Фраза "фармацевтически приемлемая(ые) соль(и)", как используется здесь, если не указано иного, включает соли кислотных или основных групп, которые могут находиться в соединениях по настоящему изобретению. Полученные способом по настоящему изобретению соединения, которые по своей природе являются основными, в частности, например, соединения формулы 1 в виде свободного основания, способны образовывать большое разнообразие солей с различными неорганическими и органическими кислотами. К кислотам, которые можно использовать для получения фармацевтически приемлемых солей добавления кислот таких основных соединений по настоящему изобретению, относятся такие, которые образуют нетоксичные соли добавления кислот, то есть соли, содержащие фармакологически приемлемые анионы, такие как гидрохлорид, гидробромид, гидройодид, нитрат, сульфат, бисульфат, фосфат, кислый фосфат, изоникотинат, ацетат, лактат, салицилат, цитрат, кислый цитрат, тартрат, пантотенат, битартрат, аскорбат, сукцинат, малеат, гентизинат, фумарат, глюконат, глюкаронат, сахарат, формиат, бензоат, глютамат, метансульфонат, этансульфонат, бензолсульфонат, п-туолуол- сульфонал и памоат [то есть 1,1′-метилен-бис-(2-гидрокси-3-нафтоат)]. Соединения, полученные способом по настоящему изобретению, которые включают в себя аминогруппу, могут образовывать фармацевтически приемлемые соли с различными аминокислотами, дополнительно к кислотам, указанным выше.
Термин "лечение", как используется здесь, если не указано иного, включает в себя лечение или предотвращение бактериальной инфекции или инфекции, вызванной простейшими, в соответствии со способом по настоящему изобретению.
Настоящее изобретение включает соединения по настоящему изобретению и их фармацевтически приемлемые соли, где один или несколько водородных, углеродных, азотных или других атомов замещены их изотопами. Такие соединения могут использоваться в качестве исследовательских и диагностических средств при изучении фармакокинетики метаболизма и при анализе связывания.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Способ по настоящему изобретению можно осуществлять в соответствии со схемами 1-4 ниже и последующими описаниями. В следующих схемах, если не указано иного, заместители R3, R4, R8 и R15 такие, как определено выше.
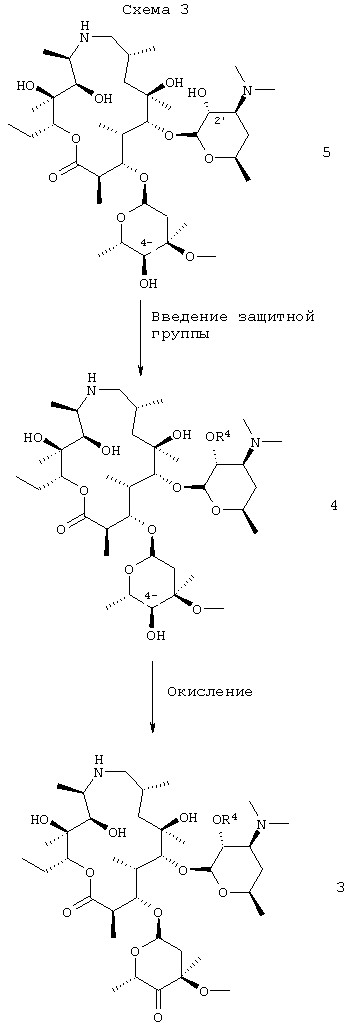
Соединение формулы 4, используемое в качестве исходного продукта в способе по настоящему изобретению, легко получали из соединения 5, то есть в котором R4 представляет собой водород, смотри WO 98/56802 и патенты Соединенных Штатов 4328334, 4474768 и 4517359, все приведены здесь в качестве ссылки в своем полном объеме.
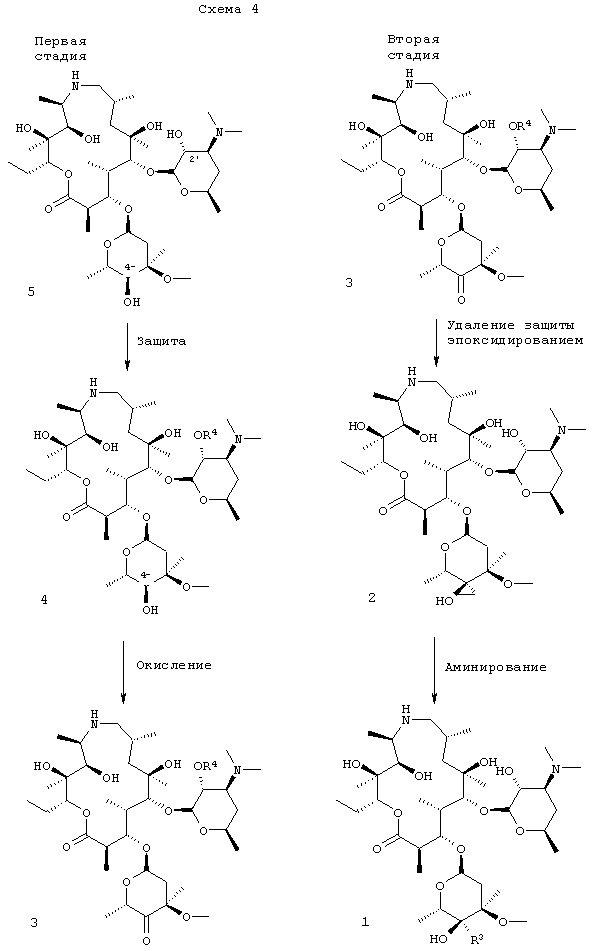
Схемы, приведенные выше, даны лишь для иллюстрации и более подробно описываются ниже и, кроме того, в примерах ниже здесь. На схеме 1 эпоксид формулы 2 преобразовывали в амин формулы 1, где R3 представляет собой –CH2NR15R8, где R15 и R8 такие, как описано выше. В наиболее предпочтительном воплощении изобретения амин представляет собой н-пропиламин, то есть R8 представляет собой н-пропил и R15 представляет собой Н.
Для получения соединения формулы 1 соединение формулы 2 предпочтительно подвергали взаимодействию с соединением формулы HNR15R8, где R15 и R8 такие, как описано здесь выше, в присутствии соответствующего растворителя, такого как изопропанол, или смеси органических растворителей, содержащей изопропанол предпочтительно при температуре от около 40°С до около 95°С. Наиболее предпочтительная температура для проведения взаимодействия составляет от около 50°С до около 55°С, хотя также можно использовать более высокие температуры, например 76°С. Наиболее предпочтительным давлением для проведения взаимодействия является атмосферное давление; однако взаимодействие также может проводиться при повышенном давлении.
В одном из предыдущих способов раскрытия эпоксидного кольца соединения формулы 2 (смотри WO 98/56802, примеры 48, 50, 51 и 70) 2′-гидроксигруппу защищали, и для получения соединения формулы 1 (или формулы 1а соответственно) требовались одновременные гидролиз защитной группы и аминирование эпоксида. Этот способ не был предпочтительным, так как проведение гидролиза в процессе стадии раскрытия эпоксида является неэффективным, и выделение соединения формулы 1 становилось более сложным из-за присутствия негидролизированной защитной группы и других примесей. В другом предыдущем методе соединение формулы 2 (в котором 2′-гидрокси не защищали) взаимодействовало с чистым алкиламином, то есть без органического растворителя. В этом случае взаимодействие протекало медленно при обычной температуре кипения н-пропиламина (около 48°С). Соответственно для того чтобы повысить температуру, взаимодействие проводили при повышенном давлении, что в меньшей степени подходит при получении в промышленном масштабе. (Смотри WO 98/56802, пример 8 (получение 2), с выходом 11%). Кроме того, в реакции использовали катализатор. Заявители обнаружили, что смесь н-пропиламина и изопропанола имеет точку кипения при атмосферном давлении, равную около 76°С, что позволяет проводить реакцию с большим выходом (свыше 85%) при температуре приблизительно 50°С-55°С без использования герметичного реакционного сосуда или катализатора(ов). Способ, предложенный заявителями, дает высокий выход (85%) и более лучшие параметры чистоты по сравнению с прежними способами, и делает возможными применить различные способы кристаллизации соединения формулы 1 как в виде свободного основания, так и в виде солей кислот с получением соединения формулы 1 в высокоочищенном виде, таком как желательно для использования в парентеральных составах.
На схеме 2 соединение формулы 2 может быть получено при взаимодействии соединения формулы 3 с метилидом серы при температуре от около -80°С до около -45°С с последующим удалением обычными способами 2′-защитной группы с получением соединения формулы 2. Исходным продуктом в способе, представленном на схеме 1, предпочтительно является соль добавления трифторуксусной кислоты соединения формулы 3, которую сначала преобразуют в форму свободного основания, охлаждают до низкой температуры, около -70°С, и затем проводят взаимодействие с низкотемпературным раствором метилида. Метилид серы представляет собой, предпочтительно, метилид диметилсульфония, например (CH3)2S+CH
На схеме 3 кетон 4′′ получали из соединения формулы 5 в одном сосуде продолжительным процессом. На первой стадии процесса гидроксигруппу 2′ избирательно защищали обычными способами, предпочтительно путем обработки 2′-гидрокси формулы 5, где R4 представляет собой водород, бензилхлорформиатом в дихлорметане с получением соединения формулы 4, где R4 представляет собой бензилоксикарбонил (CBZ). Предпочтительно использовали, по крайней мере, 2 молярных эквивалента бензилхлорформиата с тем, чтобы обеспечить полное преобразование 2′-гидроксигруппы в ее защищенную форму. В качестве растворителя предпочтительным является дихлорметан в том случае, если взаимодействие проводят, используя, по крайней мере, 15 объемов дихлорметана к объему исходного продукта, таким образом сводя к минимуму образование бис-CBZ примесей. Соединение формулы 4, где R4 представляет собой CBZ, может быть выделено в виде дибензоил-D-тартратной соли, что позволяет очистить от возможных бис-CBZ примесей. Однако водный экстракт соединения формулы 4 не является предпочтительным, так как выделение продукта является нестабильным из-за присутствия бензиламина, полученного при аминоалкилировании соединения формулы 4 бензилхлоридом (образованный при замещении бензил-хлорформиата). Соответственно после стадии защиты реакционную смесь предпочтительно использовать непосредственно на второй стадии без выделения соединения формулы 4. Вторая стадия, которую можно проводить в том же сосуде, который использовался в первой части, включая окисление 4′′-гидроксильной группы с получением 4′′-кетона формулы 3. Окисление предпочтительно является окислением, активированным ДМСО, как описано выше, то есть его проводят при пониженной температуре, например при от -60-70°С, и включает в себя активацию ДМСО in situ путем добавления трифторуксусного ангидрида к охлажденному раствору соединения в ДМСО с последующим добавлением триэтиламина. Реакционную смесь затем добавляли в воду и постепенно нагревали до температуры окружающей среды. Смесь предпочтительно промывали водой с получением раствора соединения формулы 3.
Соль трифторуксусной кислоты соединения формулы 3 может быть получена промывкой реакционной смеси водой на стадии окисления с последующим добавлением трифторуксусной кислоты и затем подходящего для кристаллизации соли растворителя, например изопропанола или смеси метиленхлорида и метил трет-бутилового эфира (МТВЕ). Другие соли добавления кислот, такие как соль дибензоил-D-винной кислоты и фосфатная соль, также могут быть получены обычным способом. В способе настоящего изобретения используются дибензоил-D-тартратная и фосфатная соли, но они в меньшей степени предпочтительны по сравнению с трифторуксусной кислотой.
Как показано на схеме 4, изобретение в целом относится к способу получения соединения формулы 1 в две стадии: на первой стадии получают соединение формулы 3 в одном сосуде, включая защиту 2′-гидроксигруппы соединения формулы 5 бензилоксикарбонилом, с получением соединения формулы 4, с последующим непосредственным окислением 4′′-гидроксигруппы 4 с выходом кетона формулы 3, который предпочтительно выделяли в виде его соли добавления трифторуксусной кислоты. На второй стадии соединение формулы 3 в виде свободного основания (предпочтительно полученного из его соли трифторуксусной кислоты) преобразовывали в 4′′-эпоксид формулы 2, 2′-защищенную группу удаляли с получением 2′-гидроксигруппы опять и эпоксид раскрывали с помощью амина путем нагревания в смеси, содержащей изопропанол, с получением соединения формулы 1.
Соединения, полученные способом по настоящему изобретению, который составляет его сущность, способны образовывать широкое разнообразие различных солей с различными неорганическими и органическими кислотами. Хотя такие соли могут быть фармацевтически приемлемыми для введения млекопитающим, на практике часто желательно на начальных стадиях выделить соединение, полученное способом по настоящему изобретению, из реакционной смеси в виде фармацевтически неприемлемой соли, а затем легко преобразовать последнюю снова в соединение в виде свободного основания обработкой щелочным реагентом для использования в последующих взаимодействиях или для получения фармацевтически приемлемой соли добавления кислоты. Соли добавления кислот основных соединений, полученные способом по настоящему изобретению, без труда получали обработкой основного соединения соответствующим эквивалентным количеством выбранной неорганической или органической кислоты в среде водного растворителя или в подходящем органическом растворителе. Желаемую соль легко получали при осторожном упаривании растворителя. Желаемая соль также может выпадать в осадок из раствора свободного основания в органическом растворителе при добавлении раствора соответствующей неорганической или органической кислоты. Соединения формулы 1, полученные способом по данному изобретению, и их фармацевтически приемлемые соли (здесь и далее "активные соединения") при лечении бактериальной инфекции или инфекции, вызванной простейшими, могут вводиться пероральным, парентеральным или ректальным путями.
Обычно активные соединения наиболее желательно вводить в дозах, находящихся в области от около 0,2 мг на кг веса тела в день (мг/кг/день) до около 200 мг/кг/день единой или раздельными дозами (то есть от 1 до 4 доз в день), хотя в зависимости от вида, массы или состояния субъекта, подвергаемого лечению, и определенного выбранного пути введения, необязательно, могут встречаться вариации. Однако наиболее желательно использовать уровень дозы, который находится в области от около 4 мг/кг/день до около 50 мг/кг/день. Вариации могут все-таки встречаться в зависимости от вида млекопитающего, рыбы или птицы, который подвергается лечению, и его индивидуальной реакции на указанный препарат, а также от типа выбранного фармацевтического состава и временного периода и интервала, с которым проводят введение. В некоторых случаях уровень доз, находящийся за пределами вышеуказанного нижнего уровня, может быть более адекватным, хотя в других случаях могут использоваться большие дозы без каких-либо побочных эффектов, при условии, что при введении в течение дня такие большие дозы сначала разделяют на несколько малых доз.
Активные соединения могут вводиться отдельно или в сочетании с фармацевтически приемлемыми носителями или разбавителями указанными выше путями, и такое введение может проводиться в виде единой или раздельных доз. Более предпочтительно, активные соединения могут вводиться в виде широкого разнообразия различных форм дозировки, то есть они могут быть объединены с различными фармацевтически инертными носителями в таблетки, капсулы, пилюли, дроже, твердые желатиновые капсулы, порошки, спреи, кремы, бальзамы, суппозитории, желе, гели, пасты, лосьоны, мази, водные суспензии, растворы для инъекций, эликсиры, сиропы и тому подобное. Такие носители включают твердые разбавители или наполнители, стерильную водную среду, различные нетоксические органические растворители и тому подобное. Кроме того, фармацевтические композиции для перорального введения могут быть подходящим образом подслащены и/или ароматизированы. Обычно активные соединения присутствуют в таких дозируемых формах с уровнями концентраций, находящихся в области от около 5,0 до около 70% по массе.
Для перорального введения таблетки, содержащие различные эксципиенты, такие как микрокристаллическая целлюлоза, цитрат натрия, карбонат кальция, дикальцийфосфат и глицин, могут быть использованы с различными разрыхлителями, такими как крахмал (и предпочтительно кукурузный, картофельный крахмал или крахмал из тапиоки), альгиновую кислоту и известный комплекс силикатов, вместе со связующим в гранулы агентом, таким как поливинилпирролидон, сахароза, желатин и аравийская камедь. Кроме того, для целей таблетирования также часто используют смазывающие агенты, такие как стеарат магния, лаурилсульфат натрия и тальк. Твердые композиции подобного типа также могут использоваться в качестве наполнителей в желатиновых капсулах; предпочтительные вещества в этом смысле включают лактозу или молочный сахар, а также высокомолекулярные полиэтиленгликоли. Если для перорального введения предпочтительными являются водные суспензии и/или эликсиры, то активное соединение может быть в сочетании с различными подсластителями и ароматизаторами, придающими окраску или красителями, и, если желательно, также эмульгаторами и/или суспендирующими агентами, вместе с такими растворителями, как вода, этанол, пропиленгликоль, глицерин и тому подобными их сочетаниями.
Для парентерального введения можно использовать растворы активного соединения либо в кунжутном или арахисовом масле, либо в водном пропиленгликоле. Если необходимо, водные растворы можно подходящим образом забуферировать, и жидкий разбавитель сначала сделать изотоничным. Эти водные растворы подходят для внутривенных инъекций. Масляные растворы подходят для внутриартериальных, внутримышечных и подкожных инъекций. Получение всех этих растворов в стерильных условиях легко осуществляют с помощью обычных способов, хорошо известных специалисту в данной области.
Кроме того, также возможно вводить активные соединения по настоящему изобретению местно, и это можно производить с помощью кремов, желе, гелей, пластырей, мазей и тому подобное в соответствии со стандартными фармацевтическими способами. Для введения животным, исключая людей, таким как крупный рогатый скот или домашние животные, активные соединения могут вводиться либо в пище для животных, либо пероральным вливанием.
Активные соединения могут также вводиться в виде систем липосомной доставки, таких как маленькие однослойные липосомальные везикулы, большие однослойные липосомальные везикулы и многослойные липосомальные везикулы. Липосомы могут быть образованы из различных фосфолипидов, таких как холестерол, стериламин или фосфотидилхолины.
Активные вещества можно также присоединить к растворимым полимерам в качестве носителей целевых лекарств. Такие полимеры могут включать поливинилпирролидон, сополимер пирана, фенилполигидроксипропилметакриламид, фенолполигидроксиэтиласпартамид или полилизинполиэтиленоксид, замещенный польмитоиловыми остатками. Более того, активные соединения можно присоединить к классу биодеградируемых полимеров, используемых для контролируемого высвобождения лекарства, например, к полимолочной кислоте, полигликолевой кислоте, сополимерам полимолочной и полигликолевой кислоты, полиэпсилонкапролактону, полигидроксимасляной кислоте, полиортоэфирам, полиацеталам, полидигидропиранам, полицианоакрилатам и перекрестносвязанным или амфипатическим блок-сополимерам гидрогелей.
Следующие примеры дополнительно иллюстрируют способ и промежуточные вещества по настоящему изобретению. Должно быть понятно, что настоящее изобретение не ограничивается этими конкретными подробностями приведенных ниже примеров.
Пример 1
Получение (2R,3S,4R,5R,8R,10R,11R,12S,13S,14R)-13-[(2,6-дидезокси-3-С-метил-3-О-метил-α-L-рибо-гексопиранозил)окси]-2-этил-3,4,10-тригидрокси-3,5,8,10,12,14-гексаметил-11-[[3,4,6-тридезокси-3-(диметиламино)-2-0-[(фенилметокси)-карбонил]-β-D-ксилогексопиранозил]окси]-1-окса-6-азациклопентадекан-15-она.
К охлажденному до 0-5°С раствору 25 кг (2R,3S,4R,5R,8R,10R,11R,12S,13S,14R)-13-[(2,6-дидезокси-3-С-метил-3-О-метил-α-L-рибо-гексопиранозил)окси]-2-этил-3,4,10-тригидрокси-3,5,8,10,12,14-гексаметил-11[[3,4,6-тридезокси-3-(диметиламино)-β-D-ксило-гексопиранозил]окси]-1-окса-6-азациклопентадекан-15-она в 425 л метиленхлорида добавляли раствор 13,7 кг бензилхлорформиата в 25 л метиленхлориде, поддерживая температуру ниже 5°С. Полученную смесь взбалтывали при этой температуре в течение трех часов и затем концентрировали до 148 л с получением сухого раствора, содержащего приблизительно 26,6 кг (90%) продукта (с помощью ВЭЖХ-Waters Symmetry C8, I.D. колонка 15 см × 3,9 мм, подвижная фаза 25 мМ буфер фосфат калия (рН 7,5):ацетонитрил:метанол (35:50:15), скорость потока 2,0 мл/мин, электрохимическое определение. Время удерживания = 8,2 минут). Эту смесь непосредственно использовали в примере 2.
Пример 2
Получение (2R,3S,4R,5R,8R,10R,11R,12S,13S,14R)-13-[(2,6-дедезокси-3-С-метил-3-О-метил-α-L-рибо-гексопиранозил)окси]-2-этил-3,4,10-тригидрокси-3,5,8,10,12,14-гексаметил-11-[[3,4,6-тридезокси-3-(диметиламино)-2-0-[(фенилметокси)-карбонил]-β-D-ксило-гексопиранозил]окси]-1-окса-6-азациклопентадекан-15-оновой соли добавления бис трифторуксусной кислоты.
В раствор, полученный в примере 1, добавляли 58,6 кг диметилсульфоксида ("ДМСО"), затем охлаждали до -70°С. Поддерживая температуру между -70 и -60°С, добавляли 16 кг трифторуксусного ангидрида и смесь перемешивали в течение 30 минут, затем добавляли 17,2 кг триэтиламина и полученную смесь перемешивали еще 30 минут. Реакционную смесь добавляли к 175 л воды и затем, постепенно нагревая до температуры окружающей среды, разделяли слои. Органический слой дважды промывали 170 л воды и концентрировали приблизительно до 100 л. После чего добавляли 7,8 кг трифторуксусной кислоты, а затем 236 л изопропанола, и смесь концентрировали до образования кристаллов 29,5 кг (87,9%) продукта, который был на 98% очищен ВЭЖХ.
Аналитические данные: т. пл. = 187-192°С. Элементный анализ для C49H76F6N2Ol8:
Вычислено: С 53,74; Н 6,99; F 10,41; N 2,56.
Найдено: С 53,87; Н 6,99; F 10,12; N 2,59.
Система ВЭЖХ такая же, как в примере 1; время удерживания = 9,5 минут. Рентгеновская дифракция на порошке (d интервал): 6,3, 8,3, 8,8, 9,4, 10,8, 11,8, 12,6, 13,0, 14,3, 15,4, 15,9, 16,4, 17,1, 17,4, 17,8, 18,1, 19,1, 19,8, 20,4, 21,1, 21,5, 21,7, 22,8, 23,4, 24,0.
Пример 3
Получение (2R,3S,4R,5R,8R,10R,11R,12S,13S,14R)-2-этил-3,4,10-тригидрокси-13-[[(3S,4S,6R,8R)-8-метокси-4,8-диметил-1,5-диоксаспиро[2.5]окт-6-ил]окси]-3,5,8,10,12,14-гексаметил-11-[[3,4,6-тридезокси-3-(диметиламино)-2-O-[(фенилметокси)-карбонил]-β-D-ксило-гексопиранозил]окси]-1-окса-6-азациклопентадекан-15-она.
(a) Раствор 109 кг продукта по примеру 2 в 327 л метиленхлорида обрабатывали раствором 27,5 кг карбоната калия в 327 л воды. Слои разделяли, водный слой промывали 327 л метиленхлорида и объединенные органические слои сушили и упаривали до около 327 л и охлаждали до -70°С.
(b) В отдельном сосуде упаривали суспензию 29,7 кг бромида триметилсульфония в 436 л тетрагидрофурана ("ТГФ") до приблизительно 170 л, охлаждали до -12°С и обрабатывали 36,8 кг трет-бутоксида калия в течение 75 минут при -10-15°С. Эту смесь затем добавляли в раствор метилен-хлорида со стадии (а) в течение около 30 минут, поддерживая при этом температуру при -70-80°С, и полученную смесь оставляли нагреваться до -65°С и перемешивали, по крайней мере, 1 час. Смесь затем добавляли в раствор 55,4 кг хлорида аммония в 469 л воды. После перемешивания при 15-25°С в течение 15 минут разделяли слои, водный слой промывали 360 л метиленхлорида и объединенные органические слои упаривали до приблизительно 227 л. К полученной смеси добавляли 750 л ацетона. Наконец, упаривали смесь до 227 л раствора, содержащего приблизительно 70,1 кг (80%) указанного в заголовке продукта (с помощью ВЭЖХ, система ВЭЖХ: колонка MetaSil AQ C18 (от MetaChem, номер партии 0520-250×046), подвижная фаза 50 мМ буфер фосфата калия (рН 8,0):ацетонитрил:метанол (30:60:10), скорость потока 1,0 мл/мин, электрохимическое определение. Время удерживания = 31,1 минут). Эту смесь непосредственно использовали в примере 4.
Пример 4
Получение (2R,3S,4R,5R,8R,10R,11R,12S,13S,14R)-2-этил-3,4,10-тригидрокси-13[[(3S,4S,6R,8R)-8-метокси-4,8-диметил-1,5-диоксаспиро[2.5]окт-6-ил]окси]-3,5,8,10,12,14-гексаметил-11-[[3,4,6-тридезокси-3-(диметиламино)-(3-D-ксило-гексопиранозил]окси]-1-окса-6-азациклопентадекан-15-она.
Раствор, содержащий продукт по примеру 3, объединяли с 11 кг активированного угля, 17,5 кг 10% палладия на угле (Johnson-Matthey, тип А 402028-10) и 637 л ацетона. Полученную смесь обрабатывали водородом при 50 фунт на кв. дюйм, 20-25°С до полного протекания реакции, и затем фильтровали. Фильтрат концентрировали до приблизительно 350 л и затем в течение 90 минут добавляли 1055 л воды. Кристаллизованный продукт собирали фильтрацией, промывали раствором 132 л воды и 45 л ацетона и сушили с выходом 57,5 кг (94,4%) указанного в заголовке эпоксида в виде моногидрата (содержание воды по методу Карла-Фишера). Аналитические данные: система ВЭЖХ: такая же как в примере 3; время задержки = 13,3 минут. Рентгеновская дифракция на порошке (d интервал): 6,0, 8,5, 9,4, 11,9, 12,7, 13,4, 15,2, 16,9, 17,5, 18,0, 18,9, 19,4, 19,9, 20,7, 21,2, 21,6, 22,8.
Пример 5
Получение (2R,3S,4R,5R,8R,10R,11R,12S,13S,14R)-13-[(2,6-дидезокси-3-С-метил-3-О-метил-4-С-[(пропиламино)метил]-α-L-рибо-гексопиранозил)окси-2-этил-3,4,10-тригидрокси-3,5,8,10,12,14-гексаметил-11-[[3,4,6-тридезокси-3-(диметиламино)-β-D-ксило-гексопиранозил]окси]-1-окса-6-азациклопентадекан-15-оновой соли добавления бисфосфорной кислоты.
С 280 л изопропанола и 108,2 кг н-пропиламина объединяли 56 кг эпоксида моногидрата по примеру 4. Смесь нагревали при 50-55°С в течение трех часов и затем концентрировали в вакууме до приблизительно 112 л. К концентрату добавляли 560 л этанола и 44/8 л воды. Для кристаллизации продукта в течение более двух часов к полученной смеси добавляли 16,8 кг фосфорной кислоты в 252 л этанола. После перемешивания полученной суспензии в течение 18 часов смесь фильтровали, твердое вещество промывали 28 л этанола и продукт сушили с выходом 64,6 кг (88%) указанного в заголовке соединения (с помощью ВЭЖХ, система ВЭЖХ: YMC-Pack Pro C18 (YMC Inc. Part #AS-12 SO 3-1546 WT), подвижная фаза 50 мМ буфера фосфата калия (рН 8,0):ацетонитрил:метанол 61:21:18, скорость потока 1,0 мл/мин, электрохимическое определение. Время удерживания = 26,4 минут).
Пример 6
Получение (2R,3S,4R,5R,8R,10R,11R,12S,13S,14R)-13-[(2,6-дидезокси-3-С-метил-3-О-метил-4-С[(пропиламино)метил]-α-L-рибо-гексопиранозил)окси-2-этил-3,4,10-тригидрокси-3,5,8,10,12,14-гексаметил-11[[3,4,6-тридезокси-3-(диметиламино)-β-D-ксило-гексопиранозил]окси]-1-окса-6-азациклопентадекан-15-она в виде свободного основания.
С 433 л метиленхлорида, 433 л воды и 27,6 кг карбоната калия объединяли 64,6 кг продукта по примеру 5. После перемешивания смеси в течение тринадцати минут слои разделяли и водный слой промывали 32 л метиленхлорида. Объединенные органические слои объединяли путем фильтрации и упаривали до приблизительно 155 л. К концентрату добавляли 386 л гептана, раствор упаривали до приблизительно 155 л и охлаждали до 20-25°С для эффекта кристаллизации. После перемешивания смеси в течение шести часов твердые продукты собирали фильтрацией, промывали 110 л гептана и сушили с выходом 40,3 кг (77%) указанного в заголовке соединения (с помощью ВЭЖХ; такая же система, как в примере 5; время удерживания = 26,4 минут).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 4"-ЗАМЕЩЕННЫХ ПРОИЗВОДНЫХ 9-ДЕОКСО-9А-АЗА-9А-ГОМОЭРИТРОМИЦИНА А | 2002 |
|
RU2263117C2 |
| Иммуномодулирующие азалиды на основе мочевины | 2021 |
|
RU2811591C1 |
| КОМПОЗИЦИИ И СПОСОБ ДЛЯ ЛЕЧЕНИЯ МИКРОБНЫХ И ПАРАЗИТАРНЫХ ИНФЕКЦИЙ У КРУПНОГО РОГАТОГО СКОТА И ДРУГИХ ЖИВОТНЫХ | 2004 |
|
RU2359667C2 |
| β, β-ДИЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ 9-ДЕЗОКСО-9А-N-ЭТЕНИЛ-9А-АЗА-9А-ГОМОЭРИТРОМИЦИНА А | 1998 |
|
RU2205185C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА И ИХ СОЛИ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ С АНТАГОНИСТИЧЕСКОЙ В ОТНОШЕНИИ АНГИОТЕНЗИНА АКТИВНОСТЬЮ НА ИХ ОСНОВЕ | 1995 |
|
RU2139869C1 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2014 |
|
RU2695219C2 |
| ЗАМЕЩЕННЫЕ ИЗОХИНОЛИНОВЫЕ И ИЗОХИНОЛИНОНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ Rho-КИНАЗЫ | 2007 |
|
RU2455302C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПИПЕРИДИНИЛАМИНОМЕТИЛ-ТРИФТОРМЕТИЛОВЫХ ЦИКЛИЧЕСКИХ ЭФИРОВ | 2000 |
|
RU2184116C1 |
| ХИНОЛИНОНКАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ АГОНИСТОВ 5-HT РЕЦЕПТОРОВ | 2005 |
|
RU2394033C2 |
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ПРОИЗВОДНОГО ДИАЗАБИЦИКЛООКТАНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2014 |
|
RU2801220C2 |
Изобретение относится к способу получения производного 9-деоксо-9a-аза-9a-гомоэритромицина A формулы 1а или его фармацевтически приемлемой соли, включающему взаимодействие моногидрата соединения формулы 2 с н-пропиламином в изопропаноле при температуре 50-55єC и атмосферном давлении. Способ получения соединения формулы 2 включает: (a) преобразование трифторуксуснокислой соли соединения формулы 3 в свободное основание соединения формулы 3, где R4 представляет собой гидроксизащитную группу; (b) взаимодействие указанного свободного основания соединения формулы 3 с ионом метилида сульфония; (c) гашение реакционной смеси со стадии (b) водным раствором слабой кислоты и распределение продукта в неводном растворе; и (d) удаление защитных групп продукта со стадии (c). Технический результат - повышение выхода и улучшение чистоты целевых продуктов. 3 с. и 2 з.п. ф-лы.

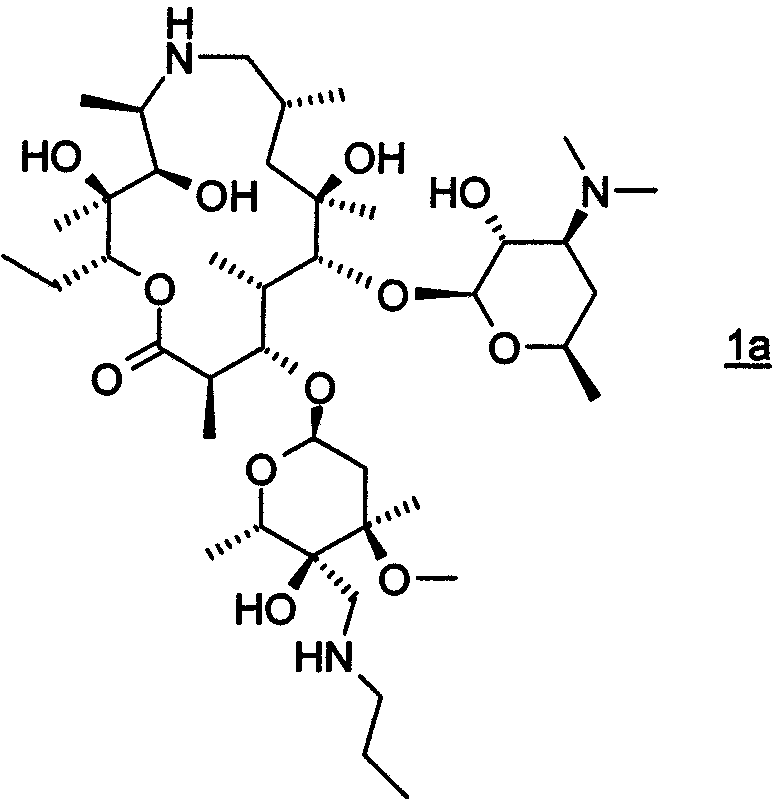
или его фармацевтически приемлемой соли, включающий взаимодействие моногидрата соединения формулы 2
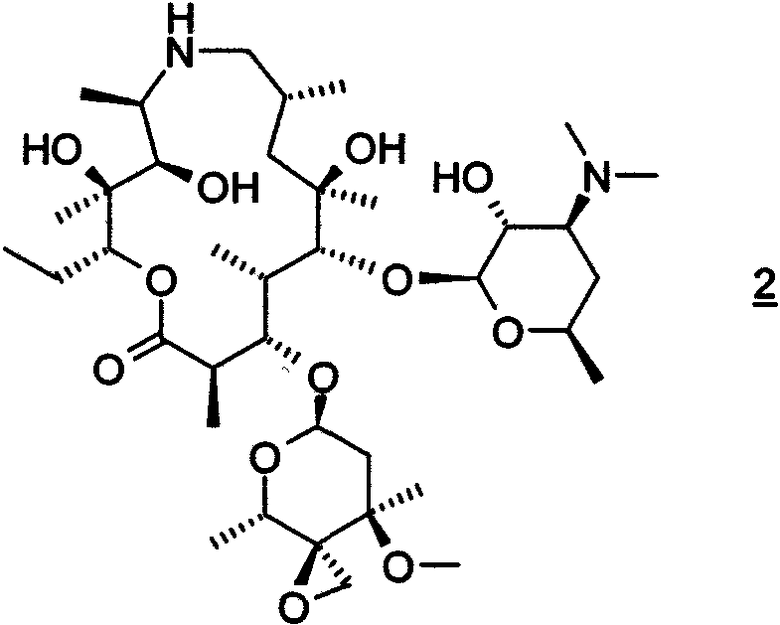
с н-пропиламином в изопропаноле при температуре 50-55°C и атмосферном давлении.
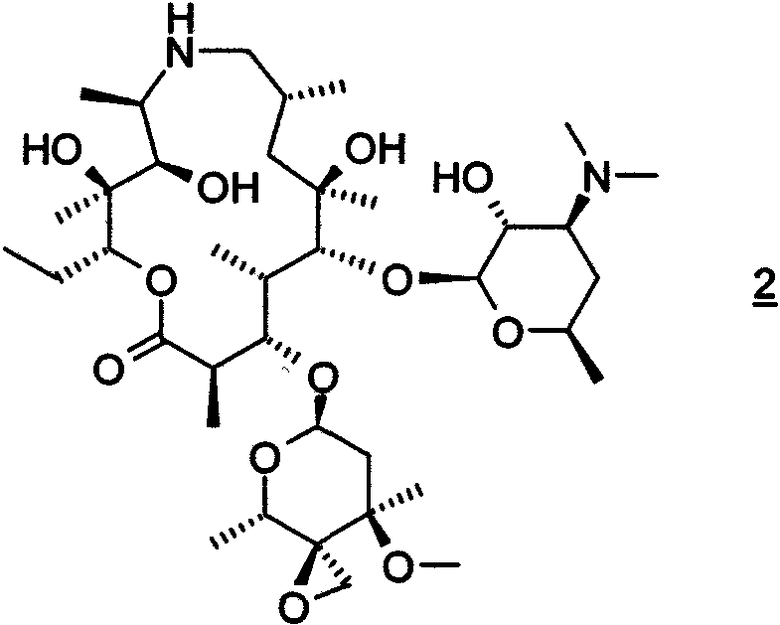
включающий (a) преобразование трифторуксуснокислой соли соединения формулы 3 в свободное основание соединения формулы 3
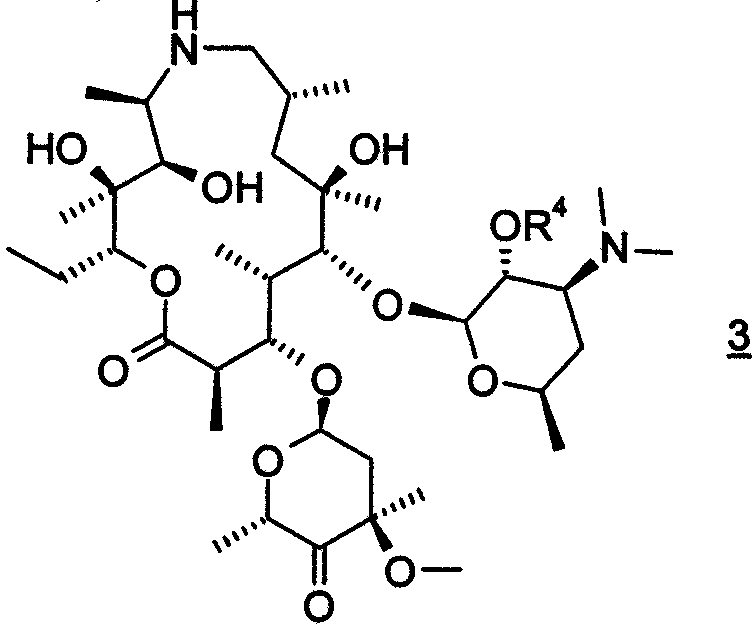
где R4 представляет собой гидроксизащитную группу,
(b) взаимодействие указанного свободного основания соединения формулы 3 с ионом метилида сульфония; (c) гашение реакционной смеси со стадии (b) водным раствором слабой кислоты и распределение продукта в неводном растворе и (d) удаление защитных групп продукта со стадии (c) с получением соединения формулы 2.
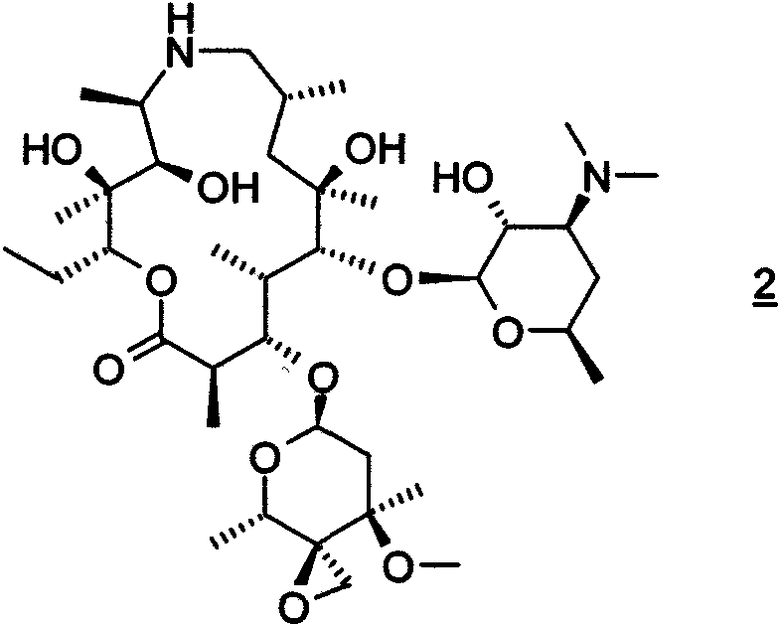
в виде моногидрата.
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| Общая органическая химия | |||
| - М.: Химия, 1982, т | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| с | |||
| Передвижная комнатная печь | 1922 |
|
SU383A1 |

