ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Изобретение относится к способам получения С-4"-замещенных производных 9-деоксо-9а-аза-9а-гомоэритромицина А (в дальнейшем «азалид(ы)»), которые полезны в качестве антибактериальных и антипротозойных агентов для млекопитающих, в том числе человека, а также рыб и птиц. Данное изобретение относится также к способам получения стабильных промежуточных продуктов рассматриваемых азалидов, а также к кристаллической соли промежуточного продукта в способе получения рассматриваемых азалидов. Данное изобретение относится также к фармацевтическим композициям, содержащим новые соединения, полученные рассматриваемыми способами, и к способам лечения бактериальных инфекций и протозойных инфекций у млекопитающих, рыб и птиц введением новых соединений, полученных рассматриваемыми способами, млекопитающим, рыбам и птицам, нуждающимся в таком лечении.
Известно, что макролидные антибиотики являются полезными при лечении широкого спектра бактериальных инфекций и протозойных инфекций у млекопитающих, рыб и птиц. Такие антибиотики включают различные производные эритромицина А, такие как азитромицин, который является коммерчески доступным и упоминается в патентах США 4474768 и 4517359, оба из которых включены в описание в качестве ссылки во всей их полноте. Подобно азитромицину и другим макролидным антибиотикам, макролидные соединения настоящего изобретения обладают сильной активностью против различных бактериальных инфекций и протозойных инфекций, как описано ниже.
Получение рассматриваемых азалидов в коммерческом масштабе имеет некоторые трудности, включающие, но не ограничивающиеся ими, низкие выходы и нестабильность некоторых синтетических промежуточных продуктов, а также присутствие нежелательных примесей.
КРАТКОЕ ИЗЛОЖЕНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к способу получения соединения формулы 1
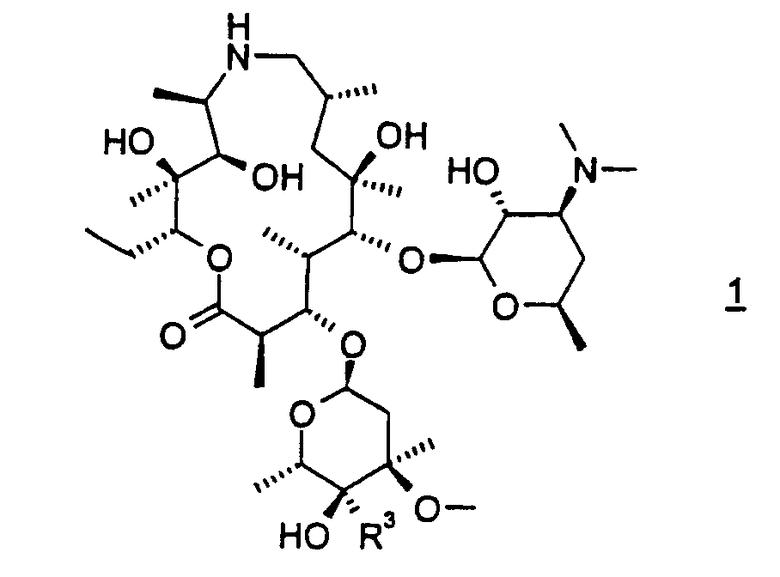
или его фармацевтически приемлемой соли, который включает:
взаимодействие соединения формулы 2
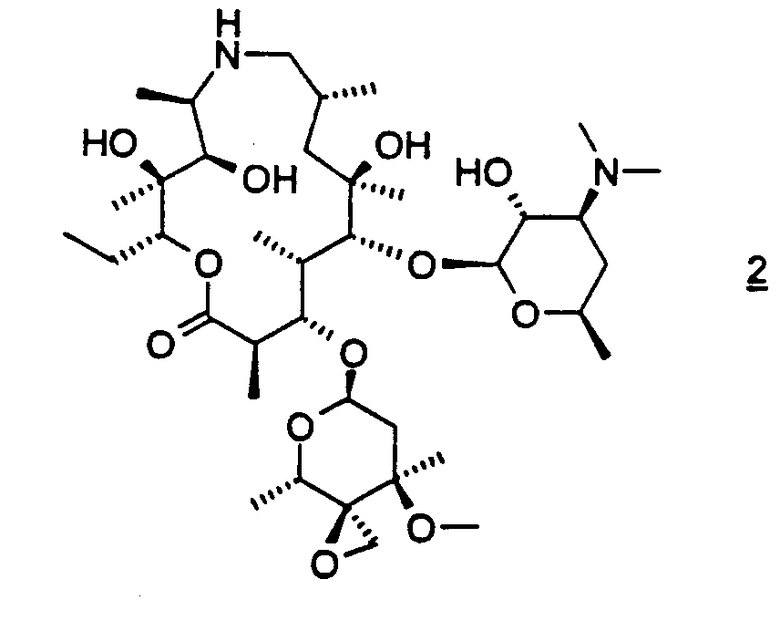 с амином формулы HNR8R15 в органическом растворителе, содержащем изопропанол; где взаимодействие проводят при температуре, по меньшей мере, приблизительно 40°С;
с амином формулы HNR8R15 в органическом растворителе, содержащем изопропанол; где взаимодействие проводят при температуре, по меньшей мере, приблизительно 40°С;
где:
R3 представляет СН2NR8R15;
R8 представляет С1-С10-алкил и
R15 представляет Н или С1-С10-алкил.
В предпочтительном варианте осуществления способа R8 представляет пропил и R15 представляет Н. В особенно предпочтительном варианте осуществления R8 представляет н-пропил и R15 представляет Н. В особенно предпочтительном варианте осуществления органическим растворителем является изопропанол.
Другой предпочтительный вариант осуществления изобретение относится к способу получения соединения формулы 1а или его фармацевтически приемлемой соли
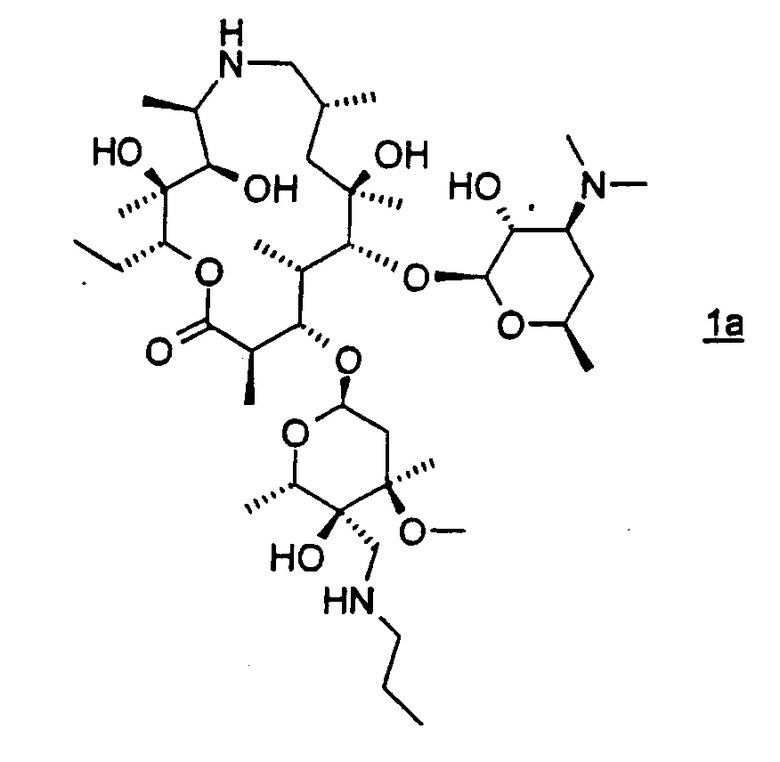
взаимодействием соединения формулы 2 с н-пропиламином в органическом растворителе, содержащем изопропанол; где взаимодействие проводят при температуре, по меньшей мере, приблизительно 40°С. В особенно предпочтительном варианте его осуществления органическим растворителем является изопропанол.
Следует отметить, что используемые здесь термины «раствор» и «смесь», если не оговорено особо, используются взаимозаменяемым образом без учета состояния дисперсии их компонентов. Фраза «органический растворитель, содержащий изопропанол», используемая в описании, если не оговорено особо, означает неводный растворитель или смесь неводных растворителей, где, по меньшей мере, одним растворителем является изопропанол. В данной заявке термин «соединение формулы 1» включает как соединение формулы 1, так и соединение формулы 1а. Соединение формулы 1а является особенно предпочтительным вариантом соединения формулы 1, для которого применяют все из вариантов осуществления и предпочтительных вариантов осуществления описанных здесь способов.
В варианте осуществления описанных здесь способов температура составляет ниже чем приблизительно 95°С, и в предпочтительном варианте осуществления температура составляет ниже чем приблизительно 80°С. В более предпочтительном варианте осуществления температура составляет от приблизительно 50°С до приблизительно 76°С. В особенно предпочтительном варианте осуществления температура составляет от приблизительно 50°С до приблизительно 55°С.
В предпочтительном варианте осуществления описанных здесь способов взаимодействие проводят при приблизительно атмосферном давлении. В данной заявке термин «атмосферное давление» означает давление в пределах нормального диапазона метеорологического атмосферного давления для определенной высоты, тогда как термин «повышенное давление» означает давление выше атмосферного давления. В другом варианте осуществления описанных здесь способов взаимодействие проводят при повышенном давлении. В другом варианте осуществления изобретения кроме изопропанола может присутствовать триэтиламин.
В дополнение к предпочтительным вариантам осуществления взаимодействие соединения формулы 2 с амином для получения соединения формулы 1 успешно проводили в растворителях, отличных от растворителей, содержащих изопропанол. Соответственно, изобретение относится также к способу получения соединения формулы 1 взаимодействием соединения формулы 2 с амином формулы NHR8R15 в органическом растворителе, где растворитель выбирают из группы, состоящей из бензилового спирта, ацетона, метилизобутилкетона, диметилсульфоксида (ДМСО), трет-бутанола, н-бутанола, диизопропилового эфира, смеси метилтретбутилового эфира (МТВЕ) и диметилформамида (ДМФ) и их комбинаций, где взаимодействие проводят при температуре, по меньшей мере, приблизительно 40°С. Взаимодействие можно проводить при повышенных давлениях, но предпочтительно его проводят при приблизительно атмосферном давлении. В следующем варианте осуществления взаимодействие ускоряют добавлением каталитического количества кислоты Льюиса. В варианте осуществления кислота Льюиса представляет собой реагент, такой как бромид магния, иодид калия, перхлорат лития, перхлорат магния, тетрафторборат лития, гидрохлорид пиридиния или иодид тетрабутиламмония. Предпочтительной кислотой Льюиса является бромид магния.
В варианте осуществления описанных здесь способов молярное количество амина составляет, по меньшей мере, приблизительно пятикратное молярное количество соединения формулы 2. В другом варианте осуществления описанных здесь способов, концентрация амина в изопропаноле является, по меньшей мере, приблизительно 5 моляльной. В особенно предпочтительном варианте осуществления концентрация н-пропиламина является приблизительно 6-7 моляльной в изопропаноле.
В варианте осуществления вышеуказанных способов соединение формулы 2 подвергают взаимодействию с амином в течение, по меньшей мере, приблизительно 24 часов. В предпочтительном варианте осуществления молярное количество амина составляет, по меньшей мере, пятикратное молярное количество соединения формулы 2, и соединение формулы 2 подвергают взаимодействию с амином в течение, по меньшей мере, 24 часов. В более предпочтительном варианте осуществления температура составляет от приблизительно 50°С до приблизительно 80°С. В еще более предпочтительном варианте осуществления молярное количество амина составляет приблизительно двенадцатикратное количество молярного количества соединения формулы 2, концентрация амина в изопропаноле является приблизительно 6 моляльной, и соединение формулы 2 взаимодействует с амином в течение, по меньшей мере, приблизительно 24 часов при температуре от приблизительно 50°С до приблизительно 55°С.
Другой вариант осуществления описанных здесь способов дополнительно включает кристаллизацию формы свободного основания соединения формулы 1. В варианте осуществления форму свободного основания соединения формулы 1 кристаллизуют из водной смеси растворителей. В предпочтительном варианте осуществления водная смесь растворителей содержит воду и неводный растворитель, выбранный из группы, состоящей из метанола, этанола, изопропанола и ацетона. В другом варианте осуществления форму свободного основания соединения формулы 1 кристаллизуют из органического (С6-с10)алканового растворителя или смеси таких органических алкановых растворителей. В предпочтительном варианте осуществления соединение формулы 1 кристаллизуют нагреванием соединения вместе с алкановым растворителем с последующим охлаждением для осуществления кристаллизации. В предпочтительном варианте осуществления органический (С6-с10)алкановый растворитель выбирают из гептана или октана, более предпочтительно гептана. В другом варианте осуществления, описанном ниже, свободное основание получают из кислотно-аддитивной соли соединения формулы 1. Следует понимать, что «алкан», используемый здесь, если не оговорено особо, включает насыщенные одновалентные углеводороды, имеющие неразветвленные, циклические или разветвленные фрагменты или их смеси.
В следующем варианте осуществления описанных здесь способов кислотно-аддитивную соль соединения формулы 1 получают обработкой соединения формулы 1 раствором, содержащим кислоту в смешиваемом с водой растворителе. В предпочтительном варианте осуществления раствор кислоты добавляют к раствору, содержащему соединение формулы 1 и воду. В более предпочтительном варианте осуществления кислотой является фосфорная кислота, L-винная кислота или дибензоил-D-винная кислота. В особенно предпочтительном варианте осуществления кислота является фосфорной кислотой. В другом более предпочтительном варианте осуществления растворитель содержит этанол. В другом предпочтительном варианте осуществления вышеуказанные способы дополнительно включают выделение кислотно-аддитивной соли соединения формулы 1.
В варианте осуществления описанные здесь способы дают соединение формулы 1, которое имеет чистоту, по меньшей мере, 90%, более предпочтительно чистоту, по меньшей мере, 95% и наиболее предпочтительно чистоту, по меньшей мере, 98%. В частности, способы изобретения дают соединение формулы 1, имеющее профиль чистоты, подходящий для использования соединения формулы 1 при получении препаративных форм для парентерального введения. Требования для парентеральных препаративных форм хорошо известны в данной области, например исключительная чистота и небольшой размер частиц в растворе и стерильность при изготовлении препаративных форм и исключение пирогенов (см. Remington,s Pharmaceutical Sciences, Mack Publishing Company, Easton, Pa., 18th Edition, Gennaro, ed. (1990), pages 1545-1580.
В другом предпочтительном варианте осуществления вышеуказанные способы дополнительно включают обработку кислотно-аддитивной соли соединения формулы 1 основанием в смеси воды и неполярного растворителя с образованием формы свободного основания соединения формулы 1. В более предпочтительном варианте осуществления основанием является двухосновная карбонатная соль и в особенно предпочтительном варианте осуществления двухосновной карбонатной солью является карбонат калия. В другом более предпочтительном варианте осуществления неполярным растворителем является дихлорметан. Еще в одном предпочтительном варианте осуществления способ дополнительно включает кристаллизацию формы свободного основания соединения формулы 1, как описано выше, и дополнительные варианты осуществления, относящиеся к нему, которые описаны выше.
Изобретение относится также к способу получения соединения формулы 2, который включает:
(а) взаимодействие формы свободного основания соединения формулы

с ионом метилида сульфония;
(b) гашение реакционной смеси стадии (а) водной слабой кислотой и распределение продукта в неводном растворе и
(с) снятие защиты у продукта стадии (b) с образованием соединения формулы 2, где R4 представляет гидроксизащитную группу.
В варианте осуществления вышеуказанный способ дополнительно включает выделение соединения формулы 2. В предпочтительном варианте осуществления соединение формулы 2 выделяют в форме гидрата, более предпочтительно моногидрата. В варианте осуществления содержание воды определяют способом Карл-Фишера. В варианте осуществления гидрат получают из смеси, содержащей соединение формулы 2 и растворитель или смесь растворителей, выбранных из ацетона, смеси ацетон/вода, ацетон/гептан и МТВЕ/гептан. В других вариантах осуществления соединение формулы 2 выделяют в виде его ацетатной соли, L-тартратной соли или дибензоил-D-тартратной соли.
Изобретение относится к моногидрату соединения формулы 2. В предпочтительном варианте осуществления вышеуказанного способа R4 представляет бензилоксикарбонил.
В другом предпочтительном варианте осуществления вышеуказанного способа стадию (а) проводят при температуре от приблизительно -80°С до приблизительно -45°С.
В другом варианте осуществления вышеуказанного способа форму свободного основания соединения формулы 3 получают из кислотно-аддитивной соли соединения формулы 3. В предпочтительном варианте осуществления кислотно-аддитивной солью является аддитивная соль трифторуксусной кислоты. В других вариантах осуществления вышеуказанного способа кислотно-аддитивную соль соединения формулы 3 выбирают из дибензоил-D-тартратной соли, L-тартратной соли или фосфатной соли. Кислотно-аддитивные соли описанных здесь соединений легко получают общепринятыми способами.
В варианте осуществления вышеуказанного способа метилидом сульфония является метилид диметилсульфония. В предпочтительном варианте осуществления метилид диметилсульфония получают взаимодействием галогенида или сульфоната триметилсульфония с сильным основанием. В более предпочтительном варианте осуществления используют галогенид триметилсульфония, который, предпочтительно, является бромидом триметилсульфония. В другом, более предпочтительном варианте осуществления галогенид триметилсульфония подвергают взаимодействию с сильным основанием в инертном органическом растворителе или их смеси. В особенно предпочтительном варианте осуществления инертным органическим растворителем является эфирный растворитель, который, наиболее предпочтительно, представляет собой тетрагидрофуран, или смесь тетрагидрофурана и дихлорметана.
В варианте осуществления стадия (с) включает каталитическое гидрирование, где R4 представляет бензилоксикарбонил. В предпочтительном варианте осуществления катализатором для гидрирования является катализатор палладий/углерод. В особенно предпочтительном варианте осуществления катализатором палладий/углерод является 10% Pd/C (тип А402028-10 Johnson-Matthey). В другом варианте осуществления стадии (с) у продукта стадии (b) снимают защиту каталитическим гидрированием с переносом, предпочтительно, формиатом аммония, Pd/С в метаноле. В следующем варианте осуществления продукт стадии (b) перед гидрированием обрабатывают фуллеровой землей. Подходящими растворителями для способа гидрирования являются ацетон, этилацетат, ТГФ, МТВЕ, изопропанол, этанол и метанол. Предпочтительным растворителем является ацетон.
Изобретение относится также к 2'-бензилоксикарбонилзащищенному соединению II:
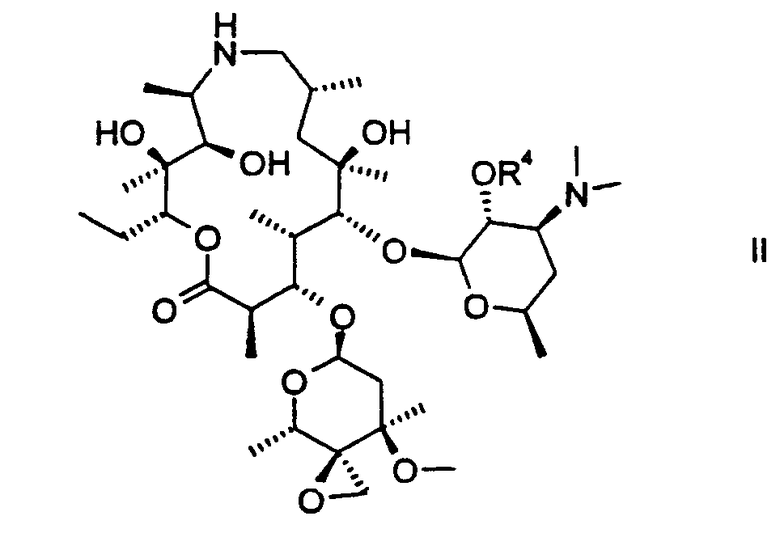
которое получают исключением стадии (с) вышеуказанных способов.
Данное изобретение относится к способу получения соединения формулы 3

окислением С-4"-гидроксигруппы соединения формулы 4

где R4 представляет гидроксизащитную группу.
В варианте осуществления окисление проводят добавлением диметилсульфоксида («ДМСО») к раствору, содержащему соединение формулы 4 и растворитель, охлаждением смеси до приблизительно -70°С и затем добавлением трифторуксусного ангидрида с последующим добавлением триэтиламина. В другом варианте осуществления ДМСО активируют с использованием оксалилхлорида (с триметилсилилацетамидом или без него), полифосфорной кислоты, пиридин-SO3 или уксусного ангидрида. В следующем варианте осуществления температуру поддерживают между -70°С и -60°С во время добавления трифторуксусного ангидрида. В другом варианте осуществления растворителем является дихлорметан. Особым преимуществом вышеуказанного способа является активация ДМСО in situ в присутствии взаимодействующего спирта, которая устраняет образование примесей, в типичном случае встречающихся при окислениях активированного ДМСО, которая обычно включает введение спирта в раствор, содержащей активированный ДМСО.
В варианте осуществления вышеуказанный способ дополнительно включает выделение кислотно-аддитивной соли соединения формулы 3. В предпочтительном варианте осуществления кислотно-аддитивной солью является дибензоил-D-тартратная соль или фосфатная соль. В особенно предпочтительном варианте осуществления данное изобретение относится к способу получения аддитивной соли трифторуксусной кислоты соединения формулы 3, который включает обработку соединения формулы 3 трифторуксусной кислотой и кристаллизацию образовавшейся кислотно-аддитивной соли; где R4 представляет гидроксизащитную группу.
В предпочтительном варианте осуществления вышеуказанного способа R4 представляет бензилоксикарбонил.
В другом предпочтительном варианте осуществления вышеуказанного способа кислотно-аддитивную соль кристаллизуют из изопропанола.
Еще в одном предпочтительном варианте осуществления вышеуказанного способа кислотно-аддитивную соль кристаллизуют из смеси метиленхлорида и метил-трет-бутилового эфира.
Аддитивные соли трифторуксусной кислоты, полученные способами настоящего изобретения, не являются фармацевтически приемлемыми, но обеспечивают прекрасную очистку и стабильность, что позволяет проводить хранение и транспортировку подходящих исходных материалов при коммерческом получении соединений формулы 1.
В варианте осуществления вышеуказанного способа соединение формулы 4 получают защитой 2'-гидроксигруппы соединения формулы 5
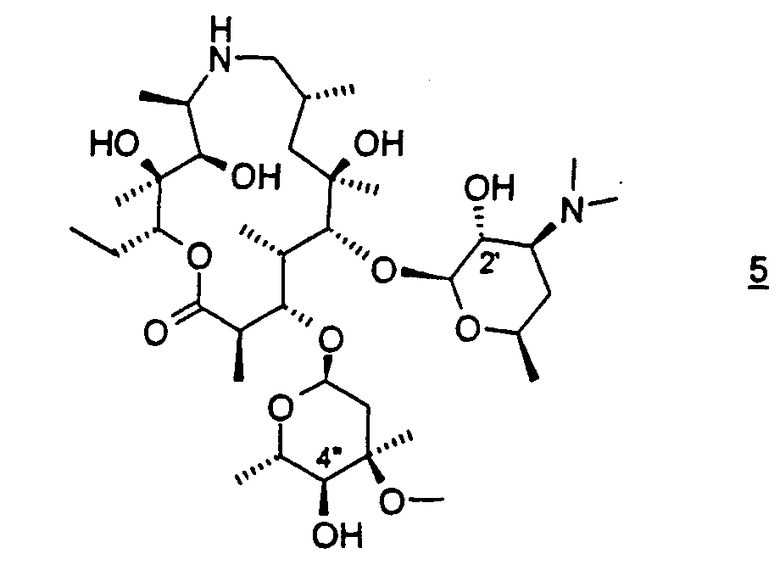
В предпочтительном варианте осуществления 2'-гидроксигруппу защищают бензилоксикарбонилом. В другом предпочтительном варианте осуществления соединение формулы 5 подвергают взаимодействию с, по меньшей мере, двумя молярными эквивалентами бензилхлорформиата. В более предпочтительном варианте осуществления взаимодействие проводят в дихлорметане. В еще более предпочтительном варианте осуществления дихлорметан присутствует, по меньшей мере, в 15-кратном избыточном объеме относительно объема исходного материала. Данное изобретение относится также к аддитивной соли трифторуксусной кислоты соединения формулы 3, где R4 представляет бензилоксикарбонил:
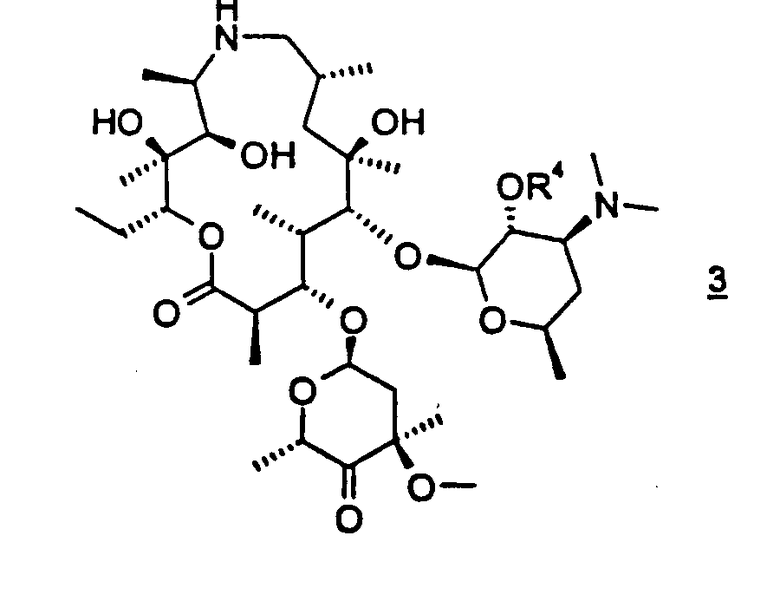
В предпочтительном варианте осуществления соль имеет структуру, показанную в формуле 3а,

где R4 представляет бензилоксикарбонил.
Данное изобретение относится также к дибензоил-D-тартратной соли соединения формулы 3, где R4 представляет бензилоксикарбонил.
Термин «гидроксизащитная группа», используемый здесь, если не оговорено особо, включает ацетил, бензилоксикарбонил, и различные гидроксизащитные группы, подобные группам, известным специалисту в данной области, включают группы, указанные в T.W.Greene, P.G.M.Wuts, «Protective Groups in Organic Synthesis», (J.Wiley & Sons, 1991).
Гидроксизащитной группой R4 предпочтительно является бензилоксикарбонил («CBZ»).
Термин «галоген», используемый здесь, если не оговорено особо, включает фтор, хлор или бром, и термин «галогенид» относится к соответствующим моноанионам, F-, Cl- или Br- соответственно.
Термин «алкил», используемый здесь, если не оговорено особо, включает насыщенные одновалентные углеводородные радикалы, имеющие неразветвленные, циклические или разветвленные фрагменты или их смеси.
Фраза «фармацевтически приемлемая соль(и)», используемая здесь, если не оговорено особо, включает соли кислотных или основных групп, которые могут присутствовать в соединениях настоящего изобретения. Соединения, полученные способами настоящего изобретения, которые являются основными по природе, особенно, например, форма свободного основания соединений формулы 1, способны образовывать большое число солей с различными неорганическими и органическими кислотами. Кислотами, которые можно использовать для получения фармацевтически приемлемых кислотно-аддитивных солей таких основных соединений настоящего изобретения, являются кислоты, которые образуют нетоксичные кислотно-аддитивные соли, т.е. соли, содержащие фармакологически приемлемые анионы, такие соли как гидрохлорид, гидробромид, гидроиодид, нитрат, сульфат, бисульфат, фосфат, кислый фосфат, изоникотинат, ацетат, лактат, салицилат, цитрат, кислый цитрат, тартрат, пантотенат, битартрат, аскорбат, сукцинат, малеат, гентизинат, фумарат, глюконат, глюкаронат, сахарат, формиат, бензоат, глутамат, метансульфонат, этансульфонат, бензолсульфонат, п-толуолсульфонат и памоат [т.е. 1,1'-метилен-бис-(2-гидрокси-3-нафтоат)]. Соединения, полученные способами настоящего изобретения, которые включают аминогруппу, могут образовывать фармацевтически приемлемые соли с различными аминокислотами, в дополнении к указанным выше кислотам.
Термин «лечение», используемый здесь, если не оговорено особо, включает лечение или профилактику бактериальной инфекции или протозойной инфекции, как предлагается в способе настоящего изобретения.
Настоящее изобретение включает соединения настоящего изобретения и их фармацевтически приемлемые соли, в которых один или несколько атомов водорода, углерода, азота или других атомов заменены их изотопами. Такие соединения могут быть полезными в качестве исследовательских и диагностических средств при фармакокинетических изучениях метаболизма и в анализах связывания.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Способ настоящего изобретения можно проводить по приведенным ниже схемам 1-4 и описанию, которое следует за этим. В следующих схемах, если не оговорено особо, заместители R3, R4, R8 и R15 имеют указанные выше значения.

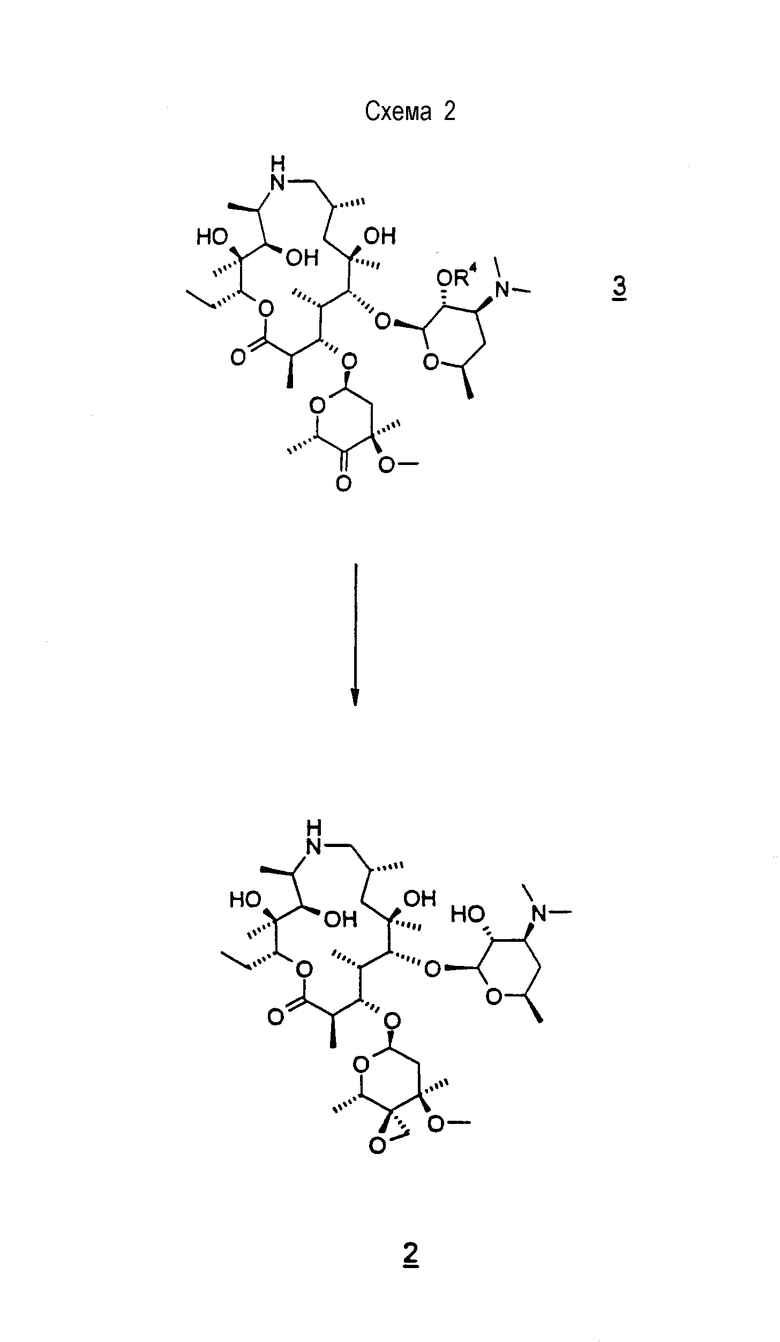
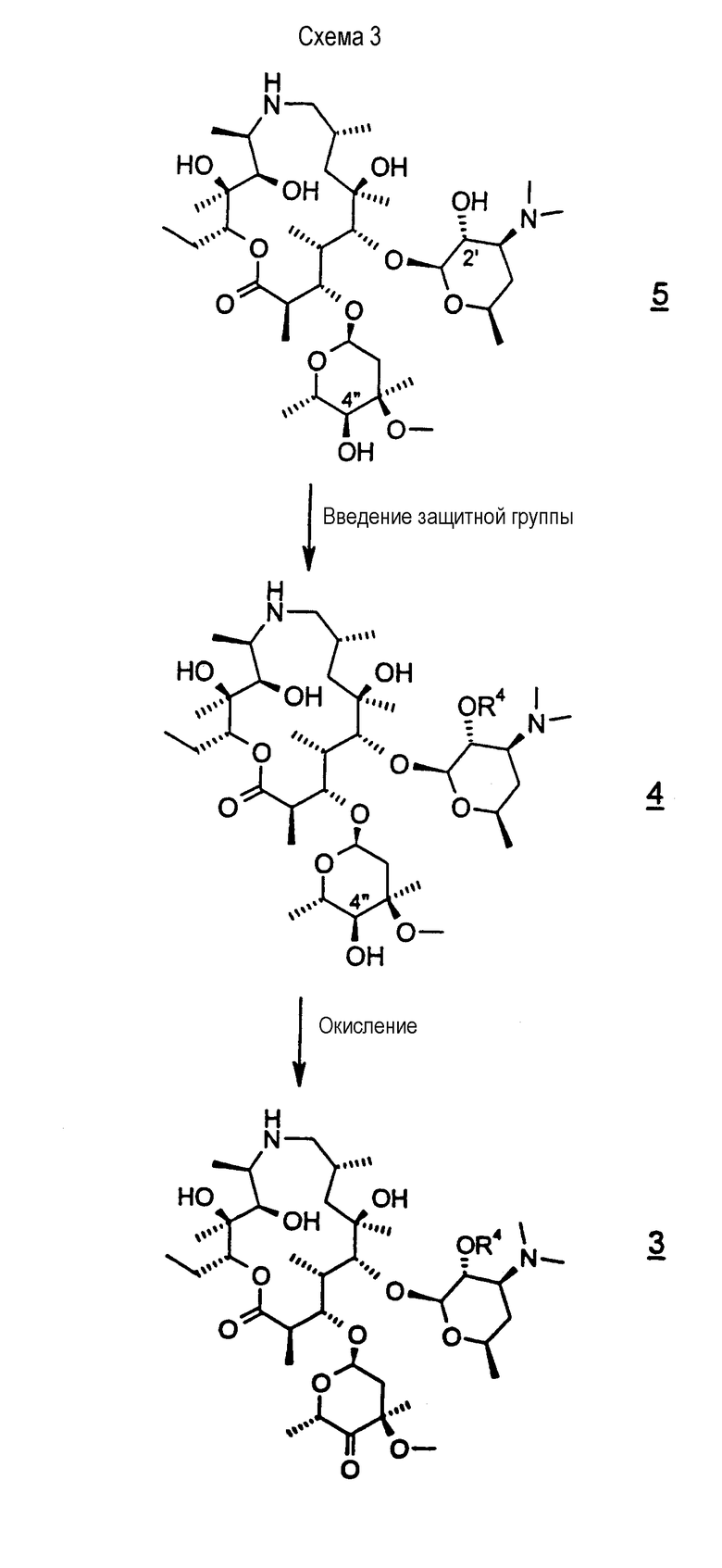
Соединение формулы 4, используемое в качестве исходного материала для способов настоящего изобретения, легко получают из соединения 5, т.е. соединения, у которого R4 представляет водород, см. WO 98/56802 и патенты США 4328334, 4474768 и 4517359, все из которых включены в описание в качестве ссылки во всей их полноте.
Приведенные выше схемы являются только иллюстративными и описываются более подробно ниже и в примерах, дополнительно приведенных ниже. На схеме 1 эпоксид формулы 2 превращают в амин формулы 1, где R3 представляет -CH2NR15R8, где R15 и R8 имеют значения, указанные выше. В наиболее предпочтительном варианте осуществления изобретения амином является н-пропиламин, т.е. R8 представляет н-пропил и R15 представляет Н.
Для получения соединения формулы 1 соединение формулы 2 предпочтительно обрабатывают соединением формулы HNR15R8, где R15 и R8 имеют значения, указанные выше, в присутствии подходящего растворителя, такого как изопропанол, или смеси органических растворителей, содержащей изопропанол, предпочтительно при температуре от приблизительно 40°С до приблизительно 95°С. Наиболее предпочтительной температурой для проведения взаимодействия является диапазон от приблизительно 50°С до 55°С, но можно также использовать более высокие температуры, например 76°С. Наиболее предпочтительным давлением для проведения взаимодействия является приблизительно атмосферное давление; однако взаимодействие можно также проводить при повышенных давлениях.
В одном способе раскрытия кольца эпоксида формулы 2 (см. WO 98/56802, примеры 48, 50, 51 и 70) 2'-гидроксигруппу защищают и получение соединения формулы 1 (или формулы 1а соответственно) требует одновременный гидролиз защитной группы и аминирование эпоксида. Этот способ не был предпочтительным, поскольку проведение гидролиза во время стадии раскрытия кольца эпоксида было неэффективным, и выделение соединения формулы 1 становилось более трудным вследствие присутствия негидролизованной защитной группы и других примесей. В другом предыдущем способе соединение формулы 2 (у которого 2'-гидроксигруппа является незащищенной) подвергали взаимодействию с чистым алкиламином, т.е. без органического растворителя. В этом случае взаимодействие протекало медленно при нормальной температуре кипения н-пропиламина (приблизительно 48°С). Следовательно, чтобы создать более высокую температуру взаимодействие проводили при повышенном давлении, менее предпочтительной характеристике при коммерческом масштабе. (См. WO 98/56802, пример 8 (получение 2), имеющий выход 11%.) Кроме того, в реакции использовали катализатор. Заявителями было сделано открытие, что смесь н-пропиламина и изопропанола имеет точку кипения при атмосферном давлении окружающей среды приблизительно 76°С, которая позволяет реакции протекать с высоким выходом (выше 85%) при температуре приблизительно 50-55°С без использования реакционного работающего при повышенном давлении сосуда или катализатора(ов). Способ заявителей обеспечивает высокий выход (85%) и лучший профиль чистоты, чем более ранние способы, и позволяет использовать различные процедуры кристаллизации как для формы свободного основания, так и кислотных солей соединения формулы 1, чтобы получить соединение формулы 1 в форме соединения с такой высокой чистотой, какая требуется для использования в парентеральных препаративных формах.
На схеме 2 соединение формулы 2 можно получить обработкой соединения формулы 3 метилидом серы при температуре от приблизительно -80°С до приблизительно -45°С с последующим удалением 2'-защитной группы общепринятыми способами с образованием соединения формулы 2. Исходным материалом для способа схемы 2 является предпочтительно аддитивная соль трифторуксусной кислоты соединения формулы 3, которую сначала превращают в форму свободного основания, охлаждают до низкой температуры, приблизительно -70°С, и затем подвергают взаимодействию с охлажденным до низкой температуры раствором метилида серы. Метилидом серы предпочтительно является метилид диметилсульфония, например (СН3)2S+CH2 -, полученный общепринятыми способами, например обработкой триметилсульфониевой соли, например (СН3)3SX, где Х представляет галоген, предпочтительно бром, или сульфонат, более предпочтительно бромид триметилсульфония, активирующим агентом, таким как гидроксид калия, трет-бутоксид калия, трет-бутоксид натрия, этоксид калия, этоксид натрия, гексаметилдисилазид калия (KHMDS) или метоксид натрия, предпочтительно трет-бутоксид калия, в эфирном растворителе, таком как ТГФ, или в СН2Cl2, ДМФ или ДМСО или смеси двух или более указанных выше растворителей. Защитную группу удаляют общепринятыми способами, например каталитическим гидрированием, когда R4 представляет CBZ.
На схеме 3 4"-кетон получают из соединения формулы 5 непрерывным способом, проводимом в одном сосуде. В первой стадии способа 2'-гидроксигруппу селективно защищают общепринятыми способами, предпочтительно обработкой 2'-гидрокси формулы 5, где R4 представляет водород, бензилхлорформиатом в дихлорметане с получением соединения формулы 4, где R4 представляет бензилоксикарбонил («CBZ»). Используют предпочтительно, по меньшей мере, 2 молярных эквивалента бензилхлорформиата, чтобы обеспечить полное превращение 2'-гидроксигруппы в ее защищенную форму. В качестве растворителя предпочтительным является дихлорметан, когда взаимодействие проводят с использованием, по меньшей мере, 15 объемов дихлорметана относительно объема исходного материала, что, таким образом, минимизирует образование примесей бис-CBZ. Соединение формулы 4, где R4 представляет CBZ, можно выделить в виде его дибензоил-D-тартратной соли, которая позволяет очистить соединение от возможной примеси бис-CBZ. Однако водная экстракционная обработка соединения формулы 4 не является предпочтительной, поскольку выделенный продукт является нестабильным из-за присутствия бензиламина, образованного аминным алкилированием соединения формулы 4 бензилхлоридом (образованным разложением бензилхлорформиата). В соответствии с этим после стадии защиты реакционную смесь, предпочтительно, переводят непосредственно на вторую стадию без выделения соединения формулы 4. Вторая стадия, которую можно проводить в том же сосуде, что и первую стадию, включает окисление 4"-гидроксильной группы с образованием 4"-кетона формулы 3. Окислением, предпочтительно, является окисление активированным-ДМСО, как описано выше, т.е. проведенное при пониженной температуре, например, при температуре от -60 до -70°С, причем оно включает активацию ДМСО in situ добавлением трифторуксусного ангидрида к охлажденному раствору соединения в ДМСО с последующим добавлением триэтиламина. Реакционную смесь затем добавляют к воде и постепенно нагревают до температуры окружающей среды. Смесь, предпочтительно, промывают в воде, получая при этом раствор соединения формулы 3.
Соль трифторуксусной кислоты соединения формулы 3 можно получить промыванием реакционной смеси стадии окисления водой с последующим добавлением трифторуксусной кислоты и затем растворителя, подходящего для кристаллизации соли, например изопропанола или смеси метиленхлорида и метил-трет-бутилового эфира («MTBE»"). Общепринятым способом можно также получить другие кислотно-аддитивные соли, такие как дибензоил-D-тартратная соль и фосфатная соль. Дибензоил-D-тартратная и фосфатная соли являются полезными в способах изобретения, но менее предпочтительными по сравнению с трифторуксусной кислотой.
Как показано на схеме 4, в общем изобретение относится к способу получения соединения формулы 1 в две стадии: на первой стадии соединение формулы 3 получают проводимом в одном сосуде способом, включающим защиту бензилоксикарбонилом 2'-гидроксигруппы соединения формулы 5 с образованием соединения формулы 4 с последующим непосредственным окислением 4"-гидроксигруппы соединения 4 с образованием кетона формулы 3, который, предпочтительно, выделяют в виде его аддитивной соли с трифторуксусной кислотой. На второй стадии форму свободного основания соединения формулы 3 (предпочтительно, полученного из его соли трифторуксусной кислоты) превращают в 4"-эпоксид формулы 2, 2'-защитную группу удаляют для возвращения 2'-гидроксигруппы и эпоксидное кольцо размыкают амином нагреванием в смеси, содержащей изопропанол, получая при этом соединение формулы 1.

Соединения, полученные способами настоящего изобретения, которые являются основными по природе, способны образовывать большое число различных солей с различными неорганическими и органическими кислотами. Хотя такие соли должны быть фармацевтически приемлемыми для введения млекопитающим, часто желательно на практике сначала выделить соединение, полученное способами настоящего изобретения, из реакционной смеси в виде фармацевтически неприемлемой соли и затем просто превратить последнюю обратно в соединение в виде свободного основания обработкой щелочным реагентом для использования в последующих реакциях или для получения фармацевтически приемлемой кислотно-аддитивной соли. Кислотно-аддитивные соли основных соединений, полученных способами данного изобретения, легко получают обработкой основного соединения по существу эквивалентным количеством выбранной минеральной или органической кислоты в среде водного растворителя или в подходящем органическом растворителе. Требуемую твердую соль легко получают после осторожного выпаривания растворителя. Требуемую соль можно также осадить из раствора свободного основания в органическом растворителе добавлением к раствору подходящей минеральной или органической кислоты. Соединения формулы 1, полученные способами данного изобретения, и их фармацевтически приемлемые соли (далее «активные соединения») можно вводить через пероральные, парентеральные, местные или ректальные пути при лечении бактериальных и протозойных инфекций.
В общем, активные соединения наиболее желательно вводить в дозах, составляющих от приблизительно 0,2 мг на кг массы тела в день (мг/кг/день) до приблизительно 200 мг/кг/день в виде разовой дозы или разделенных доз (т.е. от 1 до 4 доз в день), хотя, безусловно, могут иметь место варианты в зависимости от видов, массы и состояния подвергаемого лечению субъекта и конкретного выбранного пути введения. Однако уровень дозы, который находится в диапазоне от приблизительно 4 мг/кг/день до приблизительно 50 мг/кг/день, является наиболее желательно используемым. Тем не менее могут иметь место варианты в зависимости от вида подвергаемого лечению млекопитающего, рыбы или птицы и его индивидуальной реакции на указанное лекарственное средство, а также от типа выбранной фармацевтической композиции и периода времени и интервала, при котором проводят такое введение. В некоторых случаях уровни доз ниже низшего предела вышеуказанного диапазона могут быть более чем достаточными, тогда как в других случаях могут быть использованы еще более высокие дозы, не вызывающие какие-либо вредные побочные действия, при условии, что такие более высокие дозы сначала делят на несколько меньших доз для введения на протяжении всего дня.
Активные соединения можно вводить отдельно или в комбинации с фармацевтически приемлемыми носителями или разбавителями ранее указанными путями, и такое введение можно проводить в виде разовой дозы или множественных доз. Более конкретно, активные соединения можно вводить в виде большого числа различных лекарственных форм, т.е. их можно комбинировать с различными фармацевтически приемлемыми инертными носителями в форме таблеток, капсул, лепешек, пастилок, твердых леденцов, порошков, спреев, кремов, мазей, суппозиториев, желе, гелей, паст, лосьонов, линиментов, водных суспензий, инъецируемых растворов, эликсиров, сиропов и тому подобное. Такие носители включают твердые разбавители или наполнители, стерильную водную среду и различные нетоксичные органические растворители и т.д. Кроме того, пероральные фармацевтические композиции можно подходящим образом подсластить и/или ароматизировать. В общем, активные соединения присутствуют в таких лекарственных формах при уровнях концентраций, составляющих от приблизительно 5,0 до приблизительно 70 мас.%.
Для перорального введения можно использовать таблетки, содержащие различные эксципиенты, такие как микрокристаллическая целлюлоза, цитрат натрия, карбонат кальция, дикальцийфосфат и глицин вместе с различными дезинтегрантами, такими как крахмал (предпочтительно кукурузный, картофельный или тапиока), альгиновая кислота и некоторые комплексные силикаты, вместе со связующими грануляции, подобными поливинилпирролидону, сахарозе, желатину и аравийской камеди. Кроме того, смазывающие агенты, такие как стеарат магния, лаурилсульфат натрия и тальк, часто являются очень полезными для целей таблетирования. Твердые композиции подобного типа можно также использовать в качестве наполнителей в желатиновых капсулах; предпочтительные материалы в этой связи также включают лактозу или молочный сахар, а также высокомолекулярные полиэтиленгликоли. Когда водные суспензии и/или эликсиры желательны для перорального введения, активное соединение можно комбинировать с различными подслащивающими агентами или ароматизаторами, красящим веществом или красителями и, при желании, эмульгирующими и/или суспендирующими агентами, кроме того, вместе с такими разбавителями, как вода, этанол, пропиленгликоль, глицерин и различные, подобные им комбинации.
Для парентерального введения можно использовать растворы активного соединения либо в кунжутном, либо в арахисовом масле или в водном пропиленгликоле. Водные растворы, если необходимо, должны быть подходящим образом забуферены и жидкому разбавителю сначала должна быть придана изотоничность. Эти водные растворы являются подходящими для целей внутривенной инъекции. Масляные растворы являются подходящими для целей внутрисуставной, внутримышечной и подкожной инъекции. Получение всех этих растворов при стерильных условиях легко выполняют стандартными фармацевтическими способами, хорошо известными специалистам в данной области.
Кроме того, активные соединения настоящего изобретения можно также вводить местным способом и это можно сделать посредством кремов, желе, гелей, паст, пластырей, мазей и тому подобное в соответствии со стандартной фармацевтической практикой.
Для введения животным, отличным от людей, таким как кошки или домашние животные, активные соединения можно вводить в корма животных или вводить перорально в виде композиции для вливания животному.
Активные соединения могут быть также введены в форме липосомных систем доставки, таких как небольшие однослойные везикулы, большие однослойные везикулы и многослойные везикулы. Лимосомы можно получить из различных фосфолипидов, таких как холестерин, стеариламин или фосфатидилхолины. Активные соединения можно также сочетать с растворимыми полимерами в качестве носителей, доставляющих лекарственное средство к цели. Такие полимеры могут включать поливинилпирролидон, сополимер пирана, полигидроксипропилметакриламидфенил, полигидроксиэтиласпартамидфенол или полиэтиленоксидполилизин, замещенный пальмитоильными остатками. Кроме того, активные соединения можно сочетать с классом биоразлагаемых полимеров, полезных в достижении контролируемого высвобождения лекарственного средства, например полимолочной кислотой, полигликолевой кислотой, сополимерами полимолочной кислоты и полигликолевой кислоты, полиэпсилонкапролактоном, полигидроксимасляной кислотой, полиортоэфирами, полиацеталями, полидигидропиранами, полицианоакрилатами и сшитыми или амфипатическими блок-сополимерами гидрогелей.
Следующие примеры дополнительно иллюстрируют способ и промежуточные продукты настоящего изобретения. Должно быть понятно, что настоящее изобретение не ограничивается конкретными деталями примеров, представленных ниже.
Пример 1
Получение (2R,3S,4R,5R,8R,10R,11R,12S,13S,14R)-13-[(2,6-дидеокси-3-С-метил-3-О-метил-α-L-рибогексопиранозил)окси]-2-этил-3,4,10-тригидрокси-3,5,8,10,12,14-гексаметил-11-[[3,4,6-тридеокси-3-(диметиламино)-2-О-[(фенилметокси)карбонил]-β-D-ксилогексопиранозил]окси]-1-окса-6-азациклопентадекан-15-она
К раствору 25 кг (2R,3S,4R,5R,8R,10R,11R,12S,13S,14R)-13-[(2,6-дидеокси-3-С-метил-3-О-метил-α-L-рибогексопиранозил)окси]-2-этил-3,4,10-тригидрокси-3,5,8,10,12,14-гексаметил-11-[[3,4,6-тридеокси-3-(диметиламино)-β-D-ксилогексопиранозил]окси]-1-окса-6-азациклопентадекан-15-она в 425 л метиленхлорида, охлажденному до 0-5°С, добавляют раствор 13,7 кг бензилхлорформиата в 25 л метиленхлорида при скорости, позволяющей поддерживать температуру при 5°С. Образовавшуюся смесь перемешивают при этой температуре в течение трех часов и затем концентрируют до 148 л, получая при этом сухой раствор, содержащий приблизительно 26,6 кг (90%) продукта (определено ВЭЖХ - Waters Symmetry C8, колонка 15 см х 3,9 мм I.D., подвижная фаза 25 мМ фосфат калия в качестве буфера (рН 7,5):ацетонитрил:метанол (35:50:15), скорость потока 2,0 мл/мин, электрохимическое детектирование, время удерживания=8,2 минуты). Эту смесь непосредственно используют в примере 2.
Пример 2
Получение бис-трифторацетатной соли (2R,3S,4R,5R,8R,10R, 11R,12S,13S,14R)-13-[(2,6-дидеокси-3-С-метил-3-О-метил-α-L-рибогексопиранозил)окси]-2-этил-3,4,10-тригидрокси-3,5,8,10,12,14-гексаметил-11-[[3,4,6-тридеокси-3-(диметиламино)-2-О-[(фенилметокси)карбонил]-β-D-ксилогексопиранозил]окси]-1-окса-6-азациклопентадекан-15-она
К раствору, полученному в примере 1, добавляют 58,6 кг диметилсульфоксида («ДМСО») с последующим охлаждением до -70°С. При поддержании температуры между -70 и -60°С добавляют 16 кг трифторуксусного ангидрида и смесь перемешивают в течение 30 минут, затем добавляют 17,2 кг триэтиламина и образовавшуюся смесь перемешивают в течение дополнительных 30 минут. Реакционную смесь добавляют к 175 л воды и после постепенного нагревания до температуры окружающей среды слои разделяют. Органический слой промывают дважды 170 л воды и концентрируют приблизительно до 100 л. Затем добавляют 7,8 кг трифторуксусной кислоты с последующим добавлением 236 л изопропанола и смесь концентрируют для кристаллизации, получая при этом 29,5 кг (87,9%) продукта, который имеет чистоту по ВЭЖХ 98%. Аналитические данные: т.пл.=187-192°С. Элементный анализ. (Вычислено для С49Н76F6N2O18: С, 53,74; Н 6,99; F 10,41; N 2,56; найдено: С, 53,87; Н 6,99; F 10,12; N 2,59. Система ВЭЖХ: такая же, как в примере 1; время удерживания=9,5 мин. Порошковая дифракция рентгеновских лучей (d расстояние): 6,3, 8,3, 8,8, 9,4, 10,8, 11,8, 12,6, 13,0, 14,3, 15,4, 15,9, 16,4, 17,1, 17,4, 17,8, 18,1 19,1, 19,8, 20,4, 21,1, 21,5, 21,7, 22,8, 23,4, 24,0.
Пример 3
Получение (2R,3S,4R,5R,8R,10R,11R,12S,13S,14R)-2-этил-3,4,
10-тригидрокси-13-[[3S,4S,6R,8R)-8-метокси-4,8-диметил-1,5-диоксаспиро[2,5]окт-6-ил]окси]-3,5,8,10,12,14-гексаметил-11-[[3,4,6-тридеокси-3-(диметиламино)-2-О-[(фенилметокси)карбонил]-β-D-ксилогексопиранозил]окси]-1-окса-6-азациклопентадекан-15-она
(а) Раствор 109 кг продукта примера 2 в 327 л метиленхлорида обрабатывают раствором 27,5 кг карбоната калия в 327 л воды. Слои разделяют, водный слой промывают 327 л метиленхлорида и объединенные органические слои сушат и упаривают приблизительно до 327 л и охлаждают до -70°С.
(b) В отдельном сосуде суспензию 29,7 кг бромида триметилсульфония в 436 л тетрагидрофурана («ТГФ») упаривают приблизительно до 170 л, охлаждают до -12°С и обрабатывают 36,8 кг трет-бутоксида калия в течение 75 минут при температуре от -10 до -15°С. Данную смесь затем добавляют к раствору в метиленхлориде стадии (а) в течение периода приблизительно 30 минут при поддержании температуры при от -70 до -80°С и образовавшейся смеси дают возможность нагреться до -65°С и перемешивают в течение, по меньшей мере, 1 часа. Смесь затем добавляют к раствору 55,4 кг хлорида аммония в 469 л воды. После перемешивания смеси при 15-25°С в течение 15 минут слои разделяют и водный слой промывают 360 л метиленхлорида и объединенные органические слои упаривают приблизительно до 227 л. К образовавшейся смеси добавляют 750 л ацетона. Наконец, смесь упаривают до 227 л раствора, содержащего приблизительно 70,1 кг (80%) указанного в заголовке продукта (ВЭЖХ-системой ВЭЖХ: колонка С18 MetaSil AQ (от MetaChem, номер партии 0520-250х046), подвижная фаза 50 мМ фосфат калия в качестве буфера (рН 8,0):ацетонитрил:метанол (30:60:10), скорость потока 1,0 мл/мин, электрохимическое детектирование, время удерживания=31,1 минуты). Эту смесь непосредственно используют в примере 4.
Пример 4
Получение (2R,3S,4R,5R,8R,10R,11R,12S,13S,14R)-2-этил-3,4,10-тригидрокси-13-[[3S,4S,6R,8R)-8-метокси-4,8-диметил-1,5-диоксаспиро[2,5]окт-6-ил]окси]-3,5,8,10,12,14-гексаметил-11-[[3,4,6-тридеокси-3-(диметиламино)-β-D-ксилогексопиранозил]окси]-1-окса-6-азациклопентадекан-15-она
Раствор, содержащий продукт примера 3, смешивают с 11 кг активированного угля, 17,5 кг 10% палладия на углероде (Johnson-Matthey type A402028-10) и 637 л ацетона. Образовавшуюся смесь обрабатывают водородом при 50 фунт/кв. дюйм при 20-25°С до завершения реакции, и затем смесь фильтруют. Фильтрат концентрируют приблизительно до 350 л и затем в течение 90 минут добавляют 1055 л воды. Кристаллизованный продукт собирают фильтрованием, промывают смесью 132 л воды и 45 л ацетона и сушат, получая при этом 57,5 кг (94,4%) указанного в заголовке эпоксида в виде моногидрата (содержание воды по способу Карл-Фишера).
Аналитические данные: система ВЭЖХ: такая же, как в примере 3; время удерживания=13,3 минуты. Порошковая дифракция рентгеновских лучей (d расстояние): 6,0, 8,5, 9,4, 11,9, 12,7, 13,4, 15,2, 16,9, 17,5, 18,0, 18,9, 19,4, 19,9, 20,7, 21,2, 21,6, 22,8.
Пример 5
Получение бис-фосфатной соли (2R,3S,4R,5R,8R,10R,11R,12S,13S,14R)-13-[(2,6-дидеокси-3-С-метил-3-О-метил-4-С-[(пропиламино)метил]-α-L-рибогексопиранозил)окси-2-этил-3,4,10-тригидрокси-3,5,8,10,12,14-гексаметил-11-[[3,4,6-тридеокси-3-(диметиламино)-β-D-ксилогексопиранозил]окси]-1-окса-6-азациклопентадекан-15-она
Моногидрат эпоксида примера 4 в количестве 56 кг смешивают с 280 л изопропанола и 108,2 кг н-пропиламина. Смесь нагревают при 50-55°С в течение тридцати часов и затем концентрируют в вакууме приблизительно до 112 л. К концентрату добавляют 560 л этанола и 44,8 л воды. К образовавшейся смеси добавляют в течение приблизительно двух часов 16,8 кг фосфорной кислоты в 252 л этанола для кристаллизации продукта. После перемешивания образовавшейся суспензии в течение 18 часов смесь фильтруют, твердое вещество промывают 28 л этанола и продукт сушат, получая при этом 64,6 кг (88%) указанного в заголовке соединения (ВЭЖХ-система ВЭЖХ: YMC-PacK Pro C18 (YMC Inc. Part #AS-12S03-1546WT), подвижная фаза 50 мМ двухосновный фосфат калия в качестве буфера (рН 8,0):ацетонитрил:метанол (61:21:18), скорость потока 1,0 мл/мин, электрохимическое детектирование, время удерживания = 26,4 минуты).
Пример 6
Получение (2R,3S,4R,5R,8R,10R,11R,12S,13S,14R)-13-[(2,6-дидеокси-3-С-метил-3-О-метил-4-С-[(пропиламино)метил]-α-L-рибогексопиранозил)окси-2-этил-3,4,10-тригидрокси-3,5,8,10,12,
14-гексаметил-11-[[3,4,6-тридеокси-3-(диметиламино)-β-D-ксилогексопиранозил]окси]-1-окса-6-азациклопентадекан-15-она, свободного основания
Продукт примера 5 в количестве 64,6 кг смешивают с 433 л метиленхлорида, 433 л воды и 27,6 кг карбоната калия. После перемешивания смеси в течение тридцати минут слои разделяют и водный слой промывают 32 л метиленхлорида. Объединенные органические слои очищают фильтрованием и упаривают приблизительно до 155 л. К концентрату добавляют 386 л гептанов и раствор упаривают приблизительно до 155 л и охлаждают до 20-25°С для осуществления кристаллизации. После перемешивания смеси в течение шести часов твердое вещество собирают фильтрованием, промывают 110 л гептанов и сушат, получая при этом 40,3 кг (77%) указанного в заголовке соединения (ВЭЖХ: такая же система, как в примере 5; время удерживания 26,4 минуты).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 4''-ЗАМЕЩЕННЫХ ПРОИЗВОДНЫХ 9-ДЕОКСО-9А-АЗА-9А-ГОМОЭРИТРОМИЦИНА А | 2002 |
|
RU2233287C2 |
| Иммуномодулирующие азалиды на основе мочевины | 2021 |
|
RU2811591C1 |
| N-ЗАМЕЩЕННЫЕ 2-ЦИАНПИРРОЛИДИНЫ | 1999 |
|
RU2251544C2 |
| β, β-ДИЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ 9-ДЕЗОКСО-9А-N-ЭТЕНИЛ-9А-АЗА-9А-ГОМОЭРИТРОМИЦИНА А | 1998 |
|
RU2205185C2 |
| N-ЗАМЕЩЕННЫЕ 2-ЦИАНОПИРРОЛИДИНЫ | 1997 |
|
RU2180901C2 |
| 9А-N-(N'-КАРБАМОИЛ)- ИЛИ 9А-N-(N'-ТИОКАРБАМОИЛ) ПРОИЗВОДНЫЕ 9-ДЕОКСО-9А-АЗА-9А-ГОМОЭРИТРОМИЦИНА А, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1994 |
|
RU2131878C1 |
| ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ | 2008 |
|
RU2489439C2 |
| КОМБИНАЦИИ, СОДЕРЖАЩИЕ ИНГИБИТОР ТРАНСДУКЦИИ СИГНАЛА И ПРОИЗВОДНОЕ ЭПОТИЛОНА | 2002 |
|
RU2313345C2 |
| 8-ЦИАН-1-ЦИКЛОПРОПИЛ-7-(2,8-ДИАЗАБИЦИКЛО(4.3.0)-НОНАН-8-ИЛ)-6-ФТОР-1,4-ДИГИДРО-4-ОКСО-3-ХИНОЛИНКАРБОНОВЫЕ КИСЛОТЫ, ИХ ПРОИЗВОДНЫЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АНТИБАКТЕРИАЛЬНОЙ АКТИВНОСТЬЮ | 1997 |
|
RU2173318C2 |
| ПРОИЗВОДНЫЕ ХИНОЛОНКАРБОНОВОЙ КИСЛОТЫ, СМЕСЬ ИХ ИЗОМЕРОВ ИЛИ ОТДЕЛЬНЫЕ ИЗОМЕРЫ, ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ ГИДРАТЫ И СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2077533C1 |
Производное 9-деоксо-9а-аза-9а-гомоэритромицина А формулы 3, где R4 представляет собой гидроксильную защитную группу, получают путем защиты 2'-гидроксигруппы соединения формулы 5 с получением соединения формулы 4 и окисления С-4"-гидроксигруппы соединения формулы 4, которое проводят добавлением диметилсульфоксида к раствору, содержащему соединение формулы 4 и растворитель, охлаждением смеси до приблизительно -70°С, затем активацией диметилсульфоксида in situ и, наконец, гашением реакционной смеси. Соединение формулы 4 непосредственно переводят на стадию окисления без выделения. Также предлагается аддитивная соль трифторуксусной кислоты соединения формулы 3 и способ ее получения путем обработки соединения формулы 3 трифторуксусной кислотой. Технический результат - повышение выхода и улучшение чистоты продукта. 3 н. и 8 з.п. ф-лы.


где R4 представляет гидроксильную защитную группу, включающий стадии защиты 2' - гидроксигруппы соединения формулы 5
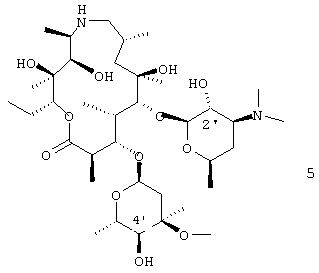
с образованием соединения формулы 4

где R представляет гидроксизащитную группу, окисления С-4"-гидроксигруппы соединения формулы 4, которую проводят добавлением диметилсульфоксида к раствору, содержащему соединение формулы 4 и растворитель, охлаждением смеси до приблизительно -70°С и затем активацией диметилсульфоксида in situ и, наконец, гашением реакционной смеси, отличающийся тем, что соединение формулы 4 непосредственно переводят на стадию окисления без выделения.
где R4 представляет гидроксизащитную группу, включающий обработку соединения формулы 3 трифторуксусной кислотой и кристаллизацию образовавшейся кислотно-аддитивной соли.
где R4 представляет бензилоксикарбонил.

где R4 представляет бензилоксикарбонил.
| WO 00/31097, A1, 02.06.2000 | |||
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| Способ получения 4"-дезокси-4"-OKCO-эРиТРОМициНА A | 1978 |
|
SU837326A3 |

