Изобретение относится к области способов очистки органических соединений, включая но не ограничиваясь органическими соединениями, которые являются конечными продуктами и промежуточными веществами, особенно последними, получаемыми синтетическими методами органической химии. В частности, способы являются способами очистки алкиловых сложных эфиров органических соединений, которые представляют собой карбоновые кислоты. Настоящее изобретение относится к улучшенному способу очистки посредством фазового разделения ди(С1-С6 алкиловых) сложных эфиров (6-хлор-2-карбазолил) метилмалоновой кислоты, особенно диэтиловых сложных эфиров, которые иногда относят далее к "сложным эфирам карбазола", хотя этот термин также используют как общую ссылку ко всем ди(С1-С6 алкиловым) сложным эфирам, включенным в способ по настоящему изобретению.
Сложный эфир карбазола является исходным продуктом для способа производства карпрофена, высокоэффективного СОХ-2 селективного противовоспалительного препарата, одобренного Министерством по Пищевым и Лекарственным Продуктам, Комитетом по Ветеринарной Медицине (FDA/CVM) для лечения собак в Соединенных Штатах. Исходный продукт, представляющий собой сложный эфир карбазола, как известно, потенциально содержит по крайней мере одну примесь, которая образуется в течение одной стадии соответствующего способа производства и которая может составлять целых 0,9% по весу от исходного продукта, представляющего собой сложный эфир карбазола. Состав этой примеси обсуждается в деталях далее, а способ очистки по настоящему изобретению рассматривается с включением в его рамки не только этой примеси, но также и других примесей. Для того чтобы получить конечный продукт карпрофен в существенно чистом виде для использования в качестве лекарственного препарата для животных, все такие примеси должны быть снижены до минимума.
Звален в патенте США 4264500 описывает способ получения 6-хлор--α-метилкарбазол-2-уксусной кислоты. Последнее промежуточное соединение для целевого продукта представляет собой сложный диэтиловый эфир (6-хлор-2-карбазолил) метилмалоновой кислоты, который, согласно описанию Звалена превращают в целевое соединение посредством гидролиза и декарбоксилирования. Стадии реакции, как утверждается, альтернативно выполняют in situ или с последующим выделением указанного предпоследнего промежуточного продукта известным способом, например, кристаллизацией. Однако у Звалена нет указания на способ очистки такого промежуточного продукта, как тот, что обеспечивается настоящим изобретением, или на удивительно высокие выходы, полученные в соответствии со способом по настоящему изобретению.
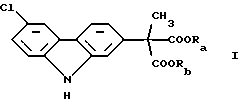
В соответствии с наиболее широкими аспектами по настоящему изобретению обеспечивается способ очистки ди(С1-С6 алкилового) сложного эфира (6-хлор-2-карбазолил) метилмалоновой кислоты формулы (I):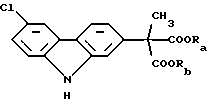 ,
,
где Ra и Rb должны быть одинаковыми и выбраны из группы, состоящей из C1-С6 алкила, предпочтительно C1-C4 алкила; включающий фазовое отделение одной или более примесей от указанного сложного эфира по крайней мере один раз, где растворитель, используемый для проведения упомянутого фазового отделения, представляет собой уксусную кислоту.
В соответствии с настоящим изобретением далее обеспечивается вышеописанный способ очистки указанного сложного эфира формулы (I), где упомянутый сложный эфир получают с чистотой по крайней мере 99,80% по весу, так что количество присутствующих в нем примесей равно 0,2% или менее по весу; и более того, где указанная уксусная кислота представляет собой ледяную уксусную кислоту, температуру которой поддерживают от примерно 30oС до примерно 110oС; и далее где указанное разделение фаз необязательно проводят два или более раз.
Более того, в соответствии с настоящим изобретением обеспечивается вышеописанный способ очистки указанного сложного эфира формулы (I), где упомянутый сложный эфир является диэтиловым сложным эфиром; и более того, где упомянутый сложный эфир формулы (I) получают с чистотой по крайней мере 99,90% по весу, так что количество присутствующих в нем примесей равно 0,1% или менее по весу; и далее, где указанная уксусная кислота представляет собой ледяную уксусную кислоту, температуру которой поддерживают от примерно 40oС до примерно 90oС, более предпочтительно от примерно 45oС до примерно 75oС и наиболее предпочтительно от примерно 50oС до примерно 70oС; и далее, где указанное разделение фаз необязательно проводят только один раз.
В соответствии с более узкими, но не менее предпочтительными реализациями настоящего изобретения, указанный ди(С1-С6 алкиловый) сложный эфир (6-хлор-2-карбазолил)метилмалоновой кислоты формулы (I), который необходимо очистить, присутствует в форме дисперсного твердого вещества, аморфного или кристаллического, которое образует преимущественно суспензию в своем растворе в ледяной уксусной кислоте.
Далее, обеспечивается, что упомянутые примеси могут образоваться прямо или косвенно в ходе способа получения упомянутого сложного эфира и могут состоять из одного или более исходных веществ, промежуточных продуктов синтеза, реагирующих веществ, побочных продуктов реакции, продуктов разложения, растворителей, в которых проводили различные реакционные стадии упомянутого способа получения, или нежелательные аналоги с близкой родственной химической структурой по отношению к указанному сложному эфиру формулы (I). В особенности обеспечивается, что указанные примеси могут возникать косвенно из указанного способа получения как результат неправильного его выполнения или осуществления его в условиях ниже оптимальных.
Также обеспечивается, что указанные примеси могут быть получены случайно из источников, которые не включены в число прямо или косвенно получаемых в течение указанного способа получения указанного сложного эфира формулы (I), например, из-за загрязнения оборудования, в котором проводят упомянутый способ получения, из-за загрязнения исходных веществ, растворителей или вспомогательных веществ для синтеза, используемых в упомянутом способе получения, из-за загрязняющих примесей в окружающей атмосфере, т.е. среде, окружающей указанный способ получения, которые абсорбируются в указанный способ, или от загрязнения указанного сложного эфира формулы (I), пока его хранят или затем обрабатывают для его подготовки.
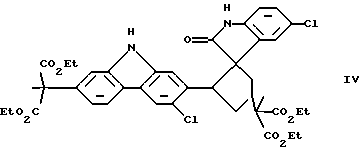
При особенно предпочтительной реализации способа очистки по настоящему изобретению, промежуточное соединение, которое необходимо очистить, представляет собой (диэтиловый) сложный эфир карбазола, и примесь, которую необходимо удалить, является димером формулы (IV):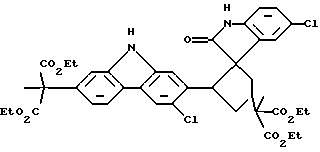
Вышеописанный ди(С1-С6 алкиловый) сложный эфир (6-хлор-2-карбазолил) метилмалоновой кислоты формулы (I):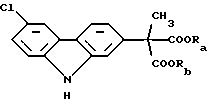
где Ra и Rb должны быть одинаковыми и выбраны из группы, состоящей из C1-С6 алкила, который необходимо очистить в соответствии со способами по настоящему изобретению, является конечным промежуточным веществом в синтезе карпрофена. Карпрофен, как уже было описано, представляет собой одобренный противовоспалительный лекарственный препарат, особенно полезный при лечении боли и воспаления у собак.
Требуется, чтобы Ra и Rb были одинаковыми и чтобы они были выбраны из группы, состоящей из C1-С6 алкила, предпочтительно C1-C4 алкила. Если Ra и Rb было бы позволено представлять различные алкильные группы, например метил и этил, что приводило бы к смешанным сложным диэфирам, тогда углерод малоновой кислоты становился бы хиральным центром, давая (S) и (R) энантиомеры сложного эфира формулы (I). Этот результат стал бы далее усложнять и возможно полностью уничтожил бы удовлетворительную очистку промежуточного продукта, представляющего собой сложный эфир формулы (I). Например, тогда было бы необходимо использовать известные способы фазового разделения диастереоизомеров, полученных из рацемической смеси посредством смешения с оптически чистой молекулой, например винной кислотой и ее производными.
Ra и Rb используют здесь как различные обозначения заместителей, несмотря на факт, что части, которые они представляют, должны обе быть одинаковыми. Цель этой отличающейся идентификации состоит в подчеркивании, что потенциальные примеси, от которых должен быть отделен сложный эфир формулы (I), включают смешанные сложные эфиры, которые могут быть получены из-за неправильного проведения, способа получения или из-за других, неизвестных или непредвиденных случаев. Ra и Rb предпочтительно выбирают из С1-С4 алкилов, которые могут быть линейными или разветвленными, и включать метил, этил, н-пропил, изопропил, н-бутил, втор-бутил и трет-бутил. Из этих характерных соединений метил и этил, и в особенности этил, являются предпочтительными.
Карпрофен, 6-хлор-α-метил-9Н-карбазол-2-уксусная кислота, которую получают из промежуточного продукта, представляющего собой сложный эфир формулы (I), можно представить формулой (II):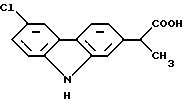
Необходимо заметить, что активное вещество карпрофен формулы (II) отличается от эфирного предшественника формулы (I) тем, что он подвергнут гидролизу и монодекарбоксилированию. В предпочтительном способе производства карпрофена являющийся промежуточным продуктом сложный эфир карбазола формулы (I) имеет свое собственное промежуточное соединение, показанное ниже формулой (III). Промежуточный продукт, представляющий собой сложный эфир карбазола формулы (I), в свою очередь, отличается от промежуточного соединения формулы (III), которое предшествует ему, тем, что он ароматизирован введением двух дополнительных двойных связей в фенильное кольцо, к которому присоединена часть молекулы α-метилуксусной кислоты. Это будет легко понято из изображения промежуточного продукта следующей формулы (III):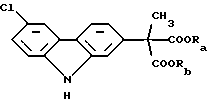
Вышеупомянутые модификации промежуточного продукта, представляющего собой сложный эфир формулы (I) и предшествующего ему промежуточного продукта формулы (III), имеют место в предпочтительном способе производства карпрофена, который проводят в соответствии со стадиями синтеза, описанными в вышеупомянутом патенте США 4264500 Звалена.
Первая стадия в синтезе Звалена представляет собой ароматизацию промежуточного соединения формулы (III) обработкой его хлором. Эту стадию предпочтительно проводят в апротонном растворителе, таком как толуол, метиленхлорид или этиленхлорид при повышенной температуре, как, например, при температуре перегонки реакционной смеси с дефлегмацией, в то время как хлор медленно добавляют к указанной смеси. Добавление хлора предпочтительно имеет место в течение периода времени от 2 до 8 ч. В типичном методе проведения этой стадии толуол используют в качестве растворителя и реакцию проводят при 75oС в течение 4 ч. Ароматизированное соединение, которое получают в результате, является промежуточным продуктом, представляющим собой сложный эфир карбазола формулы (I):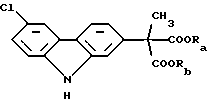
Ароматизация сложного эфира формулы (III) дает промежуточный продукт, представляющий собой сложный эфир карбазола, представленный формулой (I), показанной выше, который затем подвергают гидролизу и декарбоксилированию, чтобы получить конечный продукт карпрофен. При предпочтительном проведении этой последней упомянутой стадии синтеза промежуточное соединение формулы (I) подвергают гидролизу и декарбоксилированию в соответствии с известными методами, включающими обработку кислотами, например смесью ледяной уксусной кислоты и соляной кислоты.
Вышеописанные синтетические превращения могут быть представлены вместе в соответствии со следующей реакционной схемой: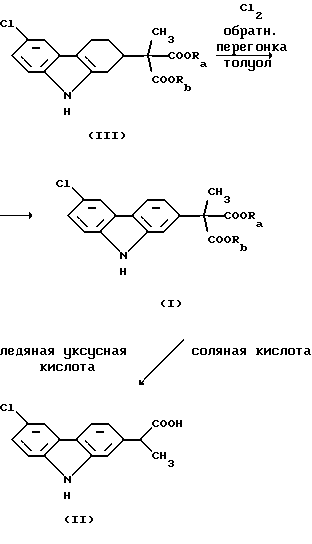
В соответствии с настоящим изобретением широко рассматривается, что примесь или примеси, которые отделяют от промежуточного продукта, представляющего собой сложный эфир формулы (I), могут значительно различаться по природе и могут происходить из различных источников. По своему существу, очистка, которую проводят по настоящему изобретению, опирается, как правило, на основной принцип способов очистки, который как предпочтительная реализация представляет собой процедуру разделения фаз. Такие известные способы могут достичь очень высоких уровней разделения, даже для соединений, которые являются очень близко родственными по структуре, как объяснено более подробно далее. Параметры способа очистки по настоящему изобретению были выбраны таким образом, что указанный способ не будет обязан своей работоспособностью и высокой селективностью структуре примесей, которые отделяют. Следовательно, не рассматривается, что настоящее изобретение каким-либо образом ограничивается характером таких примесей.
Было предпринято исследование одной из наиболее вызывающих затруднения встречающихся примесей по отношению к промежуточному продукту, представляющему собой сложный эфир карбазола формулы (I). Эта примесь появляется с течением времени как осадок в растворе упомянутого промежуточного продукта, представляющего собой сложный эфир, так же как в растворах конечного продукта карпрофена формулы (II). Эта примесь была идентифицирована рентгеновской кристаллографией и другими аналитическими данными как димерная спирооксиндольная форма промежуточного продукта, представляющего собой сложный эфир карбазола, которая получается во время стадии ароматизации, включающей хлорирование промежуточного соединения формулы (III), показанной выше на схеме синтеза. Структуру примеси димера спирооксиндола можно представить формулой (IV):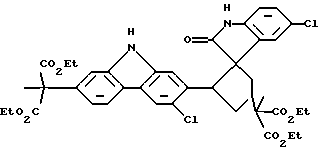
Примесь димера обладает кристаллизационными свойствами, которые ставят под сомнение традиционные процедуры очистки, которые могут быть сведены на нет соосаждением. Первые попытки достигнуть требуемых уровней очистки, полученные посредством способа по настоящему изобретению, с использованием обычных систем растворителей не были успешными. Ацетон, ацетонитрил, этанол, пропанол, бутанол, этилацетат, N,N-диметилфорамид, N,N-диметилацетамид, метилизобутилкетон и сочетания данных систем растворителей привели к увеличенным уровням примеси димера из-за вышеупомянутых кинетических эффектов кристаллизации этих систем растворителей. Более приемлемые результаты были достигнуты при использовании систем растворителей толуол/метансульфоновая кислота и толуол/бутанол. Выходы (75-85%) и качество продукта (<0,1% примеси димера), полученные с использованием системы толуол/метансульфоновая кислота, были удовлетворительными, тогда как выходы из толуол/бутанол системы были ниже. Затем система толуол/метансульфоновая кислота была испытана в стрессовых условиях, чтобы симулировать условия, которые встречаются в течение реального производства. Очистку проводили при повышенных температурах 60-65oС в течение длительного времени >2 ч. Эти стрессовые условия привели к возникновению продукта разложения, который было нельзя отделить и удалить.
Удовлетворительные результаты были также получены первоначально при перекристаллизации с этанолом/изопропиловым эфиром, которая дала высокий выход и хорошее удаление примесей. Однако когда эта система очистки была подвергнута стрессовым экспериментам с использованием длительных времен гранулирования, полученные результаты были неприемлемы. Кристаллизация была кинетической по природе с продуктом, выкристаллизовывающимся первым, затем в течение 1 ч следовала примесь димера. Этот временной интервал, в течение которого примесь димеpa выкристаллизовывается, является слишком коротким для производства в коммерческом масштабе.
Система растворителя, которая была успешной и на которой основано настоящее изобретение, оказалась системой, включающей теплую уксусную кислоту. Суспензия системы, включающей теплую уксусную кислоту, т.е. суспензия с разделением фаз, выдержала при стрессовых экспериментах, которые состояли из длительного времени гранулирования (> 36 ч), длительного времени нагрева (> 12 ч) и избыточного нагрева (> 70oС). Система растворителя, представляющего собой уксусную кислоту, была впоследствии постепенно масштабирована для производства 40 кг продукта, что включало в себя изменения во временах цикла, а также в оборудовании. Производственный цикл был чрезвычайно успешный, приведя к получению только 0,02% примеси димера, как было определено анализом ВЭЖХ.
Кроме конкретной вышеупомянутой примеси димера спирооксиндола, несомненно существуют многие другие потенциальные примеси. Эти примеси могут получаться прямо или косвенно в ходе способа получения указанного промежуточного продукта, представляющего собой сложный эфир карбазола формулы (I), и могут включать в себя одно или более из исходных веществ, промежуточных продуктов синтеза, реагирующих веществ, побочных продуктов реакции, продуктов разложения, растворителей, в которых проводили различные реакционные стадии упомянутого способа получения, или нежелательные аналоги с близкой родственной химической структурой по отношению к упомянутому сложному эфиру карбазола формулы (I). Упомянутые примеси наиболее типично возникают в течение обычных процедур, включенных в конкретный способ получения, который был применен, и являются по этой причине относимыми здесь к примесям, прямо относящимся к упомянутому способу получения.
Однако частым случаем является, когда способ получения является неправильно спланированным, что касается основной химической технологии, используя не отвечающие требованиям исходные материалы, реагирующие вещества или растворители или требуя не соответствующие параметры процесса, такие как время и температура для проведения реакции. С другой стороны, способ получения может базироваться на совершенно подходящей химической технологии, но в ходе ее выполнения случайно делаются какие-то ошибки. Например, могут быть использованы неправильный исходный материал или ненадлежащее количество реагирующего вещества; или температура, при которой проводят реакцию, может быть слишком высокой или слишком низкой. Такие ошибки исполнения процесса могут также приводить к образованию примесей вместе с желаемым конечным продуктом. Примеси этого типа появляются извне рамок процедур, включенных в применяемый способ получения, и по этой причине их относят здесь к примесям, "косвенно" относящимся к упомянутому способу получения.
Также возможно, что примеси могут быть не связаны ни прямо, ни косвенно со способом получения. Вместо этого такие примеси могут получаться случайно из различных источников, например от загрязнения оборудования, в котором проводят процесс получения, от загрязнения исходных веществ, растворителей или вспомогательных веществ для синтеза, используемых в способе получения, от загрязняющих примесей в окружающей атмосфере, т.е. среде, окружающей способ получения. Примеси из этих источников могут абсорбироваться в процедуры способа получения. После того как способ получения завершен, необходимо отделить конечный продукт и затем обрабатывать его или хранить его каким-то способом, подготовительным к составлению его рецептуры в фармацевтический состав в соответствии с известными процедурами. Таким образом, примеси могут возникать как результат загрязнения упомянутого сложного эфира формулы (I), пока он хранится или затем обрабатывается для его подготовки посредством контакта с источником упомянутых примесей.
Способ очистки по настоящему изобретению обеспечивает выход промежуточного продукта, представляющего собой сложный эфир карбазола формулы (I), достаточно высокий, так что чистота упомянутого конечного продукта промежуточного соединения, представляющего собой сложный эфир карбазола, равна по крайней мере 99,80% по весу так, что вес примесей в нем равен 0,20% или менее по весу. Показанный весовой процент основан на весе промежуточного продукта, представляющего собой сложный эфир, в конечном продукте, разделенном на вес упомянутого конечного продукта Х 100. Часто является более удобным, однако, вычислять процент чистоты из результатов количественного анализа конечного продукта, который определяет количество присутствующей примеси, из которых затем вычисляют процент примеси. Такие количественные аналитические процедуры хорошо известны, одна или более из которых могут быть адаптированы для нужд способа, описанного здесь.
В предпочтительном варианте реализации настоящего изобретения указанный промежуточный продукт, представляющий собой сложный эфир карбазола формулы (I), представляет собой диэтиловый сложный эфир и указанный промежуточный продукт, представляющий собой сложный эфир карбазола, получают с чистотой по крайней мере 99,90% по весу так, что количество присутствующих в нем примесей равно 0,10% или менее по весу. В еще более предпочтительном варианте реализации настоящего изобретения указанный промежуточный продукт, представляющий собой сложный эфир карбазола формулы (I), представляет собой диэтиловый сложный эфир и упомянутый промежуточный продукт, представляющий собой сложный эфир карбазола, получают с чистотой по крайней мере 99,95% по весу так, что количество присутствующих в нем примесей равно 0,05% или менее по весу.
Уксусная кислота, которую используют, может быть в форме высококонцентрированных неводных растворов, в которых уксусная кислота является в значительной степени преобладающим компонентом. Однако такие неводные растворы уксусной кислоты будут обычно связаны с низкими уровнями чистоты в конечном продукте промежуточного соединения, представляющего собой сложный эфир карбазола формулы (I). Соответственно в предпочтительных реализациях настоящего изобретения упомянутая уксусная кислота представляет собой ледяную уксусную кислоту.
Способ очистки по настоящему изобретению в его предпочтительной реализации использует горячую уксусную кислоту в качестве растворителя, который добавляют к твердому продукту, включающему в себя промежуточный продукт, представляющий собой сложный эфир карбазола формулы (I), и примеси, содержащиеся в нем. Примеси, которые необходимо удалить, хорошо растворимы в этом горячем растворителе, являющемся уксусной кислотой, но конечный продукт промежуточного соединения, представляющего собой сложный эфир карбазола, имеет очень низкую растворимость в горячем растворителе, являющемся уксусной кислотой. Уровень нерастворимости промежуточного продукта, представляющего собой сложный эфир карбазола формулы (I), в горячем растворителе, являющемся уксусной кислотой, по порядку равен примерно 85% по весу, т.е. только примерно 15% промежуточного продукта, представляющего собой сложный эфир карбазола, будет растворено в горячей уксусной кислоте. Остающийся промежуточный продукт, представляющий собой сложный эфир карбазола, присутствует в виде твердого вещества, которое диспергировано в горячей уксусной кислоте и поэтому может быть описано как суспензия или пульпа. После того как из растворителя, представляющего собой горячую уксусную кислоту, было осаждено так много промежуточного продукта, представляющего собой сложный эфир карбазола, как это возможно, он и уже диспергированный промежуточный продукт, представляющий собой сложный эфир карбазола, который не растворился в растворителе, отделяют от растворителя. Это разделение представляет собой разделение фаз, в котором твердую фазу промежуточного продукта, представляющего собой сложный эфир карбазола, отделяют от жидкой фазы, в которой растворяются примеси.
Температуру растворителя, являющегося уксусной кислотой, поддерживают при температуре от примерно 30oС до примерно 110oС; предпочтительно при температуре от примерно 35oС до примерно 90oС, более предпочтительно от примерно 40oС до примерно 75oС и наиболее предпочтительно от примерно 45oС до примерно 70oС. Процесс осаждения, т.е. процесс разделения фаз, который включает объемную фазу промежуточного продукта, представляющего собой сложный эфир карбазола в форме суспензии, можно проводить так много раз, как это требуется. В то время как каждый цикл фазового разделения будет давать более чистый продукт, но это будет достигнуто при стоимости дополнительной затраченной энергии и, следовательно, уменьшать эффективность. Однако одним из удивительных преимуществ настоящего изобретения является то, что чистота настолько высокая, как по крайней мере 99,90% по весу, и настолько высокая, как 99,95% по весу или выше, включая даже 99,98% по весу, может быть достигнута в единственном фазовом разделении. Проведение процесса фазового разделения дважды - это обычно все, что требуется для получения конечного продукта с высокой чистотой, требуемой для коммерческого распространения в качестве лекарственного препарата для животных.
Далее рассматривается, что способ очистки по настоящему изобретению можно выполнять в ряде различных вариантов реализации относительно характера и истории способа получения промежуточного продукта, представляющего собой сложный эфир карбазола формулы (I), который необходимо очистить. Например, рассматривается, что указанный промежуточный продукт, представляющий собой сложный эфир карбазола, может быть в виде выделенного твердого вещества в качестве промежуточного продукта из способа получения такого, как описан выше более детально. Упомянутое вещество промежуточного продукта, представляющего собой сложный эфир, может быть выделено в виде твердого вещества для того, чтобы позволить его хранить для последующей переработки на том же самом участке производства или транспортировать его для заключительной стадии на другом производственном оборудовании. Такой выделенный твердый промежуточный продукт представляет прекрасную возможность для удобного удаления присутствующих примесей, поскольку обработка промежуточного продукта, представляющего собой сложный эфир карбазола, по настоящему изобретению будет полностью совместима с последовательностью стадий производственного синтеза, которые используются. Упомянутое выделенное твердое промежуточное соединение промежуточного продукта, представляющего собой сложный эфир карбазола, можно непосредственно обработать горячим растворителем для разделения фаз, представляющим собой уксусную кислоту по способу настоящего изобретения. В менее предпочтительной реализации упомянутый твердый промежуточный продукт, представляющий собой сложный эфир карбазола, можно сначала растворить в каком-то неводном растворителе, который совместим с уксусной кислотой, которую необходимо затем добавить.
Способ очистки по настоящему изобретению необходимо проводить не только в соответствии с этим описанием, но также в соответствии с принципами процедур очистки, особенно процедур фазового разделения, которые хорошо известны. Эти принципы кратко описываются ниже для того, чтобы суммировать те соображения, которые наиболее часто играют роль при модификациях способа очистки по настоящему изобретению квалифицированными в этой области специалистами. Краткое обсуждение этих принципов также служит для того, чтобы осветить непредсказуемую природу результатов процессов фазового разделения в общем, и неожиданного успеха способа по настоящему изобретению в частности.
Таким образом, например, очистка посредством фазового разделения по настоящему изобретению включает в себя не только присутствие промежуточного продукта, представляющего собой сложный эфир, в диспергированной, суспендированной форме, но также и некоторое количество осадка промежуточного продукта, представляющего собой сложный эфир, который должен иметь место, в то время как примеси удерживают в растворенном состоянии в уксусной кислоте, являющейся растворителем. Осаждением обычно считают по существу процесс отделения частиц твердого вещества из первоначально прозрачного раствора посредством физических или химических изменений в нем. Это необходимо отличать тогда от присутствия промежуточного продукта, представляющего собой сложный эфир, в диспергированном состоянии с начального этапа процесса очистки по настоящему изобретению. Одним из важнейших применений фазового разделения является очистка твердых веществ, где оно может быть отнесено в общем случае к осаждению.
В своем наиболее простом аспекте разделение фаз включает в себя твердое вещество, содержащее примеси, которое растворяют в подходящем растворителе при повышенных температурах и при охлаждении основные примеси остаются растворенными, в то время как осажденный продукт отделяют оттуда и посредством этого очищают. В случае промежуточных продуктов, представляющих собой сложный эфир формулы (I), продукт имеет низкую растворимость даже в присутствии растворителя, представляющего собой уксусную кислоту, при высоких температурах, приводя к первоначальному образованию суспензии. Процесс фазового разделения по настоящему изобретению можно повторить несколько раз, если это желательно, и растворитель, представляющий собой уксусную кислоту, можно использовать при различных температурах.
Твердый промежуточный продукт, представляющий собой сложный эфир формулы (I), который является продуктом очистки посредством способа фазового разделения по настоящему изобретению, может быть аморфным, или в форме кристаллов, или в обоих формах. Если он находится в аморфной форме, твердый конечный продукт может включать в себя любую из ряда различных типов и размеров, и эти аморфные частицы могут также быть агломерированы или флокулированы друг с другом с образованием более значительных мас. Если он находится в кристаллической форме, твердый конечный продукт может включать в себя более чем одну кристаллическую форму, и эти формы могут также присутствовать в комбинации. Размер кристаллических частиц может различаться в широком диапазоне размеров.
В более конкретных определениях разделение фаз или кристаллизацию относят к получению твердой, однокомпонентной, аморфной или кристаллической фазы из многокомпонентной жидкой фазы, и в случае настоящего изобретения упомянутая жидкая фаза представляет собой раствор уксусной кислоты, в котором растворены нежелательные примеси. Там, где целью разделения фаз или кристаллизации является получение чистого сухого твердого вещества, что представляет собой случай некоторых реализации по настоящему изобретению, будет необходимо отделить твердое вещество от упомянутой жидкой фазы, и это обычно выполняют посредством центрифугирования или фильтрования, за чем следует сушка. Выгодные свойства такого сухого твердого аморфного или кристаллического продукта включают в себя легкость обращения, стабильность, хорошую текучесть и привлекательный внешний вид. Как правило, разделение фаз или кристаллизацию проводят в сосудах с рубашкой или с мешалкой, и условия, необходимые для получения подходящей чистоты, выхода и возможно кристаллической формы, должны быть определены посредством эксперимента.
Там, где разделение фаз включает в себя диспергированные кристаллические частицы или кристаллизацию из раствора, это будет иметь место в трех основных стадиях: индуцирование перенасыщения, образование центров кристаллизации и рост кристаллов. При данной температуре и концентрации раствор может быть насыщен или охлаждением, или удалением растворителя. Также возможным является добавление третьего компонента, который уменьшает растворимость растворенного вещества, или провести химическую реакцию в растворителе, в котором полученный в результате продукт имеет низкую растворимость. При дальнейшем охлаждении или концентрировании возникает перенасыщенная метастабильная область. Маловероятно, что низкие уровни перенасыщения приводят к самопроизвольному образованию центров кристаллов, но рост кристаллов может быть инициирован посредством добавления зародышей кристаллизации. При более низких температурах или более высоких концентрациях, которые попадают на кривую ограничивающую метастабильную область, самопроизвольное зародышеобразование является фактически несомненным и рост кристаллов также происходит при этих условиях.
Когда граница метастабильной области превышена, скорость зародышеобразования быстро возрастает и процесс кристаллизации становится неконтролируемым. Следовательно, желательно поддерживать состояние раствора внутри метастабильной области. На ширину области под кривой метастабильной зоны наиболее сильно влияет перемешивание, скорость охлаждения, присутствие растворимых добавок, растворитель и тепловая история конкретного раствора.
Зародышеобразование вызывает образование маленьких центров, вокруг которых растут кристаллы. Таким образом, без зародышеобразования рост кристаллов не может произойти. Когда вещество кристаллизуется из раствора, зародышеобразование и рост кристаллов происходят одновременно в течение широкого промежуточного температурного диапазона. Зародышеобразование зависит от степени переохлаждения, причем низкие степени переохлаждения приводят к маленькому зародышеобразованию или к его отсутствию. Однако скорость зародышеобразования возрастает до максимума и затем падает так, что избыточное охлаждение может подавить скорость кристаллизации посредством ограничения числа образовавшихся центров. Самопроизвольное зародышеобразование имеет место, когда достаточное число молекул с низкой кинетической энергией сталкиваются друг с другом в среде, где их совместное притяжение перекрывает их индивидуальную инерцию. Как только достигнут определенный размер, центры становятся стабильными в существующих условиях, и с падением температуры присутствуют молекулы с более низкой энергией и скорость зародышеобразования возрастает. Эти обстоятельства частично характеризуют образование обсужденной выше димерной примеси, которая является особенно причиняющей затруднения в растворах промежуточного продукта, представляющего собой сложный эфир формулы (I), как описано выше.
Образование центров кристаллизации или зародышеобразование также представляет собой процесс, который определяет размер кристаллов продукта и далее играет существенную роль в определении ряда физических свойств упомянутых кристаллов и, что более важно в данном случае, их чистоту.
Что касается роста кристаллов, при более высоких температурах молекулы обладают слишком большой энергией, чтобы оставаться захваченными в кристаллической решетке, в то время как при более низких температурах больше молекул удерживается и скорость роста возрастает. В конечном счете, диффузия к поверхности кристалла и ориентация на поверхности кристалла становится подавленной при еще более низких температурах. Осаждение на поверхности кристалла вызывает истощение молекул в непосредственной близости. Таким образом, движущая сила роста кристаллов обеспечивается структурой градиента концентрации от перенасыщения в растворе до более низких концентраций на поверхности кристалла. Соответственно высокий уровень перенасыщения способствует высокой скорости роста кристаллов.
Правильное расположение и подходящая ориентация по отношению к кристаллической решетке приводит к потере кинетической энергии вовлеченными молекулами. Эта совокупность, имеющая отношение к теплу кристаллизации, должна отводиться прочь, т. е. передаваться какой-то поверхности от всего раствора и, таким образом, на скорость роста кристаллов влияют как скорость теплопереноса, так и изменения, которые имеют место на упомянутой поверхности. Например, хорошо известно, что перемешивание системы увеличивает теплоперенос, уменьшая тепловое сопротивление соседних к кристаллу слоев жидкости до тех пор, пока изменения на поверхности кристалла не станут контролирующим эффектом. Вначале перемешивание быстро увеличивает скорость роста, уменьшая толщину этого граничного слоя и диффузионное сопротивление. Однако когда перемешивание интенсифицируют, достигается предельное значение, которое определяется кинетикой реакции на поверхности.
Несколько стадий, через которые проходит растущее звено или предшественник в течение роста кристалла, обнаруживают критические факторы, например, транспорт через объем раствора к участку соударения, не обязательно к участку роста кристалла, адсорбцию при соударении, где предшественники теряют молекулы растворителя и растворитель переносится обратно в раствор, диффузию предшественников от участка соударения к участку роста и внедрение в кристаллическую решетку после десольватации, в течение которого также возможно, что растворитель адсорбируется прежде, чем уйдет в раствор. Все эти процессы зависят от морфологии области раздела фаз.
Используются различные модели, чтобы идентифицировать механизмы роста поверхности кристалла и, следовательно, также процессы на границе раздела фаз. Например, используют модели объемной диффузии и поверхностной диффузии, так же как модели двухразмерного зародышеобразования и спирального роста. Кроме того, общие скорости роста измеряют в соответствии с различными известными методами, но с точки зрения теории роста кристалла наиболее часто используют линейную скорость плоскости кристалла. Далее, измерение скоростей зародышеобразования и кинетики зародышеобразования достигают посредством различных подходов. Один из них представляет собой измерение периода индукции, который представляет собой время, протекающее между достижением перенасыщения и появления твердой фазы в изучаемой системе. Считается, что индукционный период обратно пропорционален скорости зародышеобразования. В кристаллизаторе как зародышеобразование, так и рост кристаллов завершаются в течение перенасыщения, и оба эти процесса дают вклад в распределение размеров конечного продукта.
Для того чтобы получить кристаллы с высокой однородностью по составу и, следовательно, с высокой чистотой, важным является, чтобы линейная скорость роста поддерживалась бы постоянной по всему растущему разделу фаз, т.е. чтобы форма кристалла оставалась неизменной в течение роста.
Растворимые примеси, от которых отделяют осадок конечного продукта, могут или увеличивать, или уменьшать скорость зародышеобразования. Например, нерастворимые вещества могут действовать в качестве ядер и посредством этого содействовать кристаллизации. Примеси могут также влиять на форму кристалла. Из-за присутствия этих примесей состав твердого осадка отличается от состава сосуществующей жидкости в течение кристаллизации. Это явление относят к сегрегации и это важно для роста кристаллов по разнообразию причин, и центральный вопрос в каждом случае представляет собой следующее - в какой степени состав кристаллов отражает состав питательной среды, из которой он растет.
В зависимости от их вклада в свободную энергию Гиббса кристалла примеси являются или частично отвергнуты, или избирательно захвачены растущей поверхностью раздела фаз. Таким образом, коэффициент сегрегации определяют, основываясь на переносе примеси через поверхность раздела фаз. Далее, известно, что взаимодействие примеси с растворителем и комплексообразование приводит к сложной концентрационной зависимости коэффициента сегрегации. Сегрегация также важна по отношению к самой кинетике роста кристаллов, поскольку примеси могут сильно влиять на кинетику роста. Когда кристалл растет из раствора, содержащего примеси, он будет, как правило, отвергать примесь, если она менее растворима в кристалле, чем в растворе. В то время как поверхность раздела движется, примесь может быть отвергнута в раствор более быстро, чем она может быть унесена диффузией. Следовательно, концентрация примеси в твердом веществе будет определяться концентрацией примеси в обогащенном диффузионном слое, а не значением концентрации в растворе. Соответственно сегрегация, исполненная контролируемым способом, может быть с выгодой применена для очистки материалов.
Хорошо известно, что огромные различия в максимуме достижимого перенасыщения и скоростей зародышеобразования кристаллов могут происходить из правильного выбора системы растворитель - растворенное вещество. Далее, есть существенные различия в максимуме достижимого перенасыщения, ΔCмакс., когда растворитель изменяется от полярного к неполярному и существует очевидная корреляция между ΔCмакс. и растворимостью. Чем выше растворимость, тем ниже перенасыщение, при котором происходит зародышеобразование; таким образом, зародышеобразование происходит легче, когда раствор более концентрированный. Выбор растворителя также имеет значительное влияние на рост кристаллов. Кинетика роста кристаллов, растущих из раствора, определяется двумя факторами, связанными с природой растущей поверхности раздела фаз: степенью молекулярной неоднородности и природой адсорбции растворителя на поверхности.
Когда желаемые параметры для процедур разделения фаз выбраны в соответствии с выше обсужденными принципами и как описано здесь, и затем применены к способу по настоящему изобретению, полученные в результате конкретные реализации способа очистки будут затем выполнены в подходящей аппаратуре для получения желаемого результата. Цель разделения фаз или самого способа кристаллизации состоит в получении оптимальной основы аморфных или кристаллических веществ требуемой формы, распределения размеров, чистоты и выхода. Где подразумевается кристаллизация, это достигается поддержанием степени перенасыщения, при котором зародышеобразование и рост кристаллов происходит при соответствующих скоростях. Кроме растворимости растворенного вещества и температуры другие важные факторы включают термическую стабильность растворенного вещества, природу присутствующих примесей и степень требуемой гидратации.
Растворенное вещество промежуточного продукта, представляющего собой сложный эфир, в способе по настоящему изобретению является в значительной степени нерастворимым в горячем растворителе, являющемся уксусной кислотой, с самого начала процесса. Однако промежуточный продукт, представляющий собой сложный эфир, который растворен на этой стадии, будет существенно увеличивать перенасыщение с увеличением температуры и осаждение огромной части растворенного вещества обычно проводят в подходящей аппаратуре кристаллизатора посредством охлаждения горячего концентрированного раствора. Таким образом, маточные растворы, которые подвергают кристаллизации посредством выпаривания, могут быть охлаждены, чтобы дать дополнительную массу кристаллов. И наоборот, может быть использована аппаратура кристаллизатора, которая использует однократное испарение.
В такой аппаратуре горячий раствор пропускают в вакуумную камеру, в которой имеет место как выпаривание, так и охлаждение. Оптимально, когда кристаллизатор, который используют, производил бы кристаллы одинакового размера, которые облегчают удаление маточного раствора и промывку. Если большие количества жидкости захвачены в массе кристаллов, сушка будет приводить к загрязненному продукту, неприемлемому с точки зрения настоящего изобретения. Дальнейшей выгодой является то, что кристаллы одинакового размера с меньшей вероятностью слеживаются при хранении.
Производство порции больших однородных кристаллов может быть достигнуто с использованием реакционных сосудов с мешалкой, в которых имеет место медленно контролируемое или полностью естественное охлаждение. Когда имеет место кристаллизация, степень перенасыщения и концентрация растворенного вещества падает, в конечном счете достигая насыщения, где рост прекращается. Более плотный контроль за этим процессом может быть получен посредством внесения искусственных зародышей кристаллизации в перенасыщенный раствор в отсутствие естественного зародышеобразования. Непрерывное производство больших одинаковых кристаллов можно достичь, используя кристаллизаторы Осло или Кристалл, в которых метастабильный, перенасыщенный раствор поступает на дно массы растущих кристаллов, на которых осаждается растворенное вещество. Кристаллы псевдоожижают с помощью циркуляции раствора и классификации, т.е. расслаивание в этой зоне позволяет удалять достаточно большие кристаллы из донной части кристаллизатора.
Кристаллизаторы обычно классифицируют по способу, которым раствор перенасыщается, например охлаждающий кристаллизатор или выпаривающий кристаллизатор. Вакуумный кристаллизатор использует оба процесса. Дозированную кристаллизацию в охлаждающем кристаллизаторе проводят в закрытых резервуарах, оборудованных мешалкой, в которых удельная теплоемкость раствора и теплота кристаллизации отводятся посредством рубашек или змеевиков, через которые пропускают рециркулирующую охлаждающую воду. Перемешивание является важным для предотвращения температурного градиента в таких резервуарах, противодействуя осаждению и неправильному росту кристаллов в донной части резервуара и облегчая рост кристаллов.
Там, где желательно проводить процесс кристаллизации на непрерывной основе, аппаратура кристаллизатора может принять форму желоба, охлаждаемого таким же образом, как описано выше для резервуара. Раствор входит на одном конце и кристаллы и жидкость разгружают с другого конца. Перемешивание в такой аппаратуре может быть достигнуто, используя медленно движущийся шнек, который работает в растворе и поднимает кристаллы от охлаждаемой поверхности, чтобы распределить их по раствору и медленно транспортировать их через желоб. Может быть также использовано качание всего желоба в комбинации с отражательными перегородками, которые увеличивают время пребывания раствора в желобе. Оба этих типа кристаллизаторов отличаются низкими коэффициентами теплопереноса, а более быстрый теплообмен может быть достигнут при использовании устройства труба в трубе, в котором кристаллизующаяся жидкость проходит по центральной трубе с противотоком охладителя в кольцеобразном зазоре между трубами. Перемешивание в этом типе аппаратуры часто достигается при использовании вала, который вращается в центральной трубе и несет лопасти, которые слегка задевают поверхность теплопереноса, позволяя получить высокие коэффициенты теплопереноса.
Выпаривающие кристаллизаторы могут быть простыми чашеобразными устройствами или перемешиваемыми реакционными сосудами. Для больших уровней производительности применяют нагревательные элементы для нагревания, а циркуляционную трубу, которая должна быть достаточно огромной, чтобы накапливать поток суспензии, обычно оборудуют крыльчаткой, с усиленной циркуляцией, увеличивающей теплоперенос к кипящей жидкости. Непрерывный процесс, в котором является важным точный контроль размера кристаллического продукта, может быть выполнен с использованием кристаллизатора Осло, который насыщает раствор посредством выпаривания. В вакуумном кристаллизаторе, как правило, горячий концентрированный раствор подают в кристаллизационную камеру с перемешиванием, которую поддерживают при низком давлении. Раствор кипит и адиабатически охлаждается до температуры кипения, соответствующей рабочему давлению кристаллизатора. За концентрированием следует кристаллизация и продукт удаляют из донной части сосуда.
Ниже дается начало рабочего примера реализации настоящего изобретения для цели его дальнейшей иллюстрации, но без какого-либо намерения посредством этого ограничить настоящее изобретение, на которое здесь направлены пункты формулы изобретения.
ПРИМЕР
Очистка промежуточного продукта, представляющего собой сложный эфир карбазола
В реакционный сосуд было добавлено 30,0 г конкретной производственной партии промежуточного продукта, представляющего собой сложный эфир карбазола, диэтиловый сложный эфир (6-хлор-2-карбазолил)метилмалоновой кислоты, который, как было предварительно определено, имеет 0,6% по весу примеси димера спирооксиндола, имеющего следующую структуру: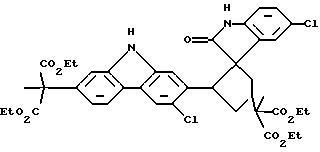
Промежуточный продукт, представляющий собой сложный эфир карбазола, объединили с 90 мл ледяной уксусной кислоты и нагрели до 50-55oС при перемешивании. Образовалась тонкая суспензия, которую перемешивали в течение примерно 2,5 ч при этой температуре. Суспензию затем медленно охладили до 20-25oС, перемешивали дополнительные 2 ч, затем отфильтровали и высушили. Выход полученного конечного продукта сложного эфира карбазола был 23,14 г (77%), который содержал 0,028% по весу примеси димера спирооксиндола.
Изобретение относится к способу очистки ди(С1-С6) алкилового сложного эфира (6-хлор-2-карбазолил) метилмалоновой кислоты формулы I, где Ra и Rb должны быть одинаковыми и выбраны из группы (C1-С6) алкила, включающему фазовое отделение димера спирооксиндола формулы IV от указанного сложного эфира ледяной уксусной кислотой при температуре 30-110oС. Способ обеспечивает получение очищенного сложного эфира с чистотой 99,80% по весу, так что количество присутствующих в нем примесей равно 0,2% или менее по весу, что позволяет использовать его в качестве лекарственного препарата для животных. 3 з.п.ф-лы.
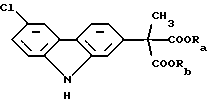
где Ra и Rb должны быть одинаковыми и выбраны из группы, состоящей из C1-C6 алкила;
включающий фазовое отделение димера спирооксиндола формулы IV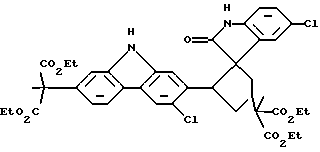
от указанного сложного эфира карбазола, где растворитель, используемый для проведения указанного фазового отделения, представляет собой ледяную уксусную кислоту, температуру которой поддерживают от 30 до 110oС.
| US 4264500, 28.04.1981 | |||
| GB 1385620, 26.02.1975 | |||
| US 4226774, 07.10.1980 | |||
| Способ выделения карбазола | 1984 |
|
SU1225838A1 |

