Настоящее изобретение относится к синтезу промежуточных соединений цефалоспорина для получения цефовецина.
Цефовецин является сильнодействующим антибиотиком, предназначенным для домашних животных. Цефовецин отличается наличием хирального заместителя на тетрагидрофурановом кольце в положении С3, который ответственен за уникальную активность и стабильность.
Общая схема синтеза цефовецина из пенициллина G включает 15 трансформаций, многие из которых представляют собой последовательные стадии. Промежуточные соединения часто представляют собой различные смеси диастереоизомеров. Единые кристаллические диастереоизомеры образуются только тогда, когда происходит образование промежуточных соединений цефалоспорина. Поэтому целью являлись промежуточные соединения цефалоспорина в качестве ключевого управляемого средства при синтезе цефовецина, и их синтез является определяющим для промышленного способа получения цефовецина.
J.H. Bateson et al., The Journal of Antibiotics, 47, 253-256 (1994) предложили способ получения промежуточных соединений цефалоспорина путем превращения β-лактама в хлорсодержащее соединение с использованием тионилхлорида с последующим осуществлением реакции хлорсодержащего соединения с триалкилфосфином с образованием фосфониевой соли. Однако этот способ предусматривает использование стандартных фосфиновых реагентов, таких как триэтилфосфит, трибутилфосфин и трифенилфосфины, которые дают низкие выходы промежуточных соединений цефалоспорина.
Патенты США № 6077952 и № 6001997, а также публикация патентной заявки США № 2002/0099205 предполагают, что использование триметилфосфина (ТМФ) обеспечивает лучший выход, и он был успешно использован в промышленном масштабе. Существует ряд недостатков при использовании ТМФ в этом процессе, таких как высокие затраты, высокий разброс выходов и относительно нестабильные промежуточные соединения.
Патентная заявка Великобритании № 2300856 предлагает альтернативные способы синтеза промежуточных соединений цефалоспорина. Однако эти способы имеют относительно низкие выходы. Поэтому существует необходимость в разработке новых способов синтеза промежуточных соединений цефалоспорина.
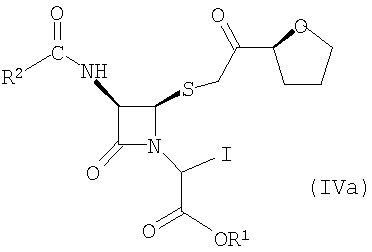
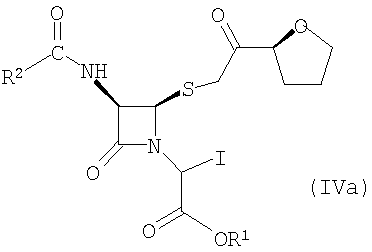
Настоящее изобретение относится к способу получения соединения формулы (IVa)

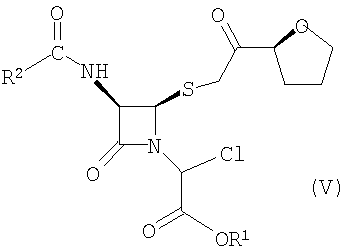
где R1 представляет собой пара-нитробензил или аллил и R2 представляет собой бензил или замещенный бензил, включающему стадию взаимодействия соединения формулы (V)
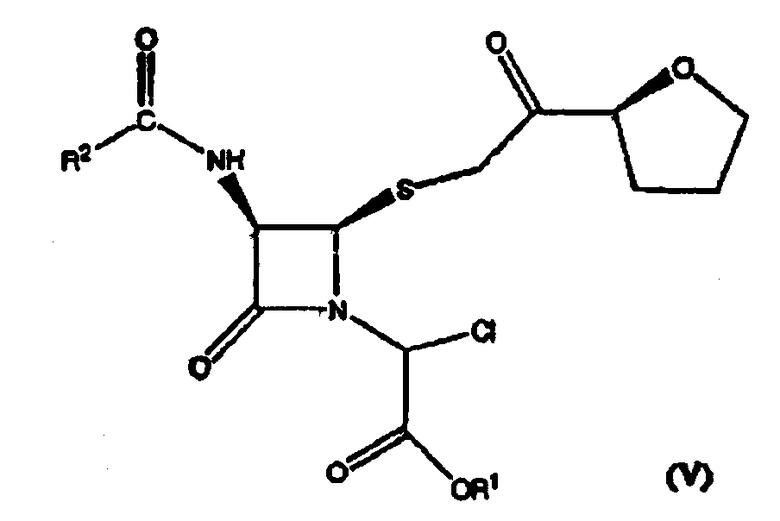
где значения радикалов R1 и R2 те же, что определены выше с солью йодистоводородной кислоты с получением соединения формулы (IVa).
Подходящие соли йодистоводородной кислоты включают, но не ограничивают объема притязаний изобретения, натриевую соль йодистоводородной кислоты, калиевую соль йодистоводородной кислоты, литиевую соль йодистоводородной кислоты, кальциевую соль йодистоводородной кислоты и аммониевую соль йодистоводородной кислоты. Предпочтительной солью йодистоводородной кислоты является натриевая соль йодистоводородной кислоты.
В предпочтительных вариантах осуществления изобретения R1 представляет пара-нитробензил, R2 представляет бензил, замещенный 1-3 заместителями, каждый из которых независимо выбран из группы, состоящей из С1-6 алкилов или атомов галогенов.
Подходящие хлорирующие агенты для превращения соединения формулы (VI) в соединение формулы (V) включают тионилхлорид и оксихлорид фосфора. Предпочтительно хлорирующим агентом является тионилхлорид.
Настоящее изобретение также относится к способу получения соединения формулы (III)

где R1 представляет пара-нитробензил или аллил; R2 представляет бензил или замещенный бензил; и R3 представляет С1-6 алкил, включающему стадию взаимодействия соединения формулы (IVa)
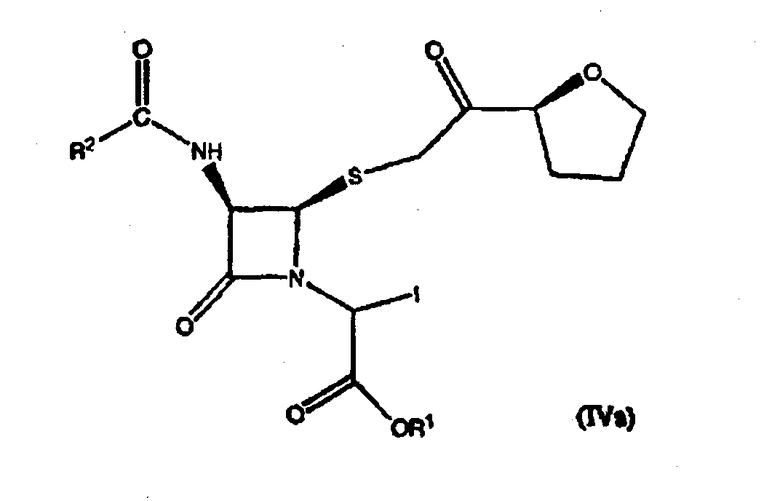
с P(OR3)3 в среде растворителя, где R1, R2 и R3 имеют значения, определенные выше.
В предпочтительном варианте осуществления изобретения R1 представляет собой пара-нитробензил в способе получения соединений формулы (III).
В другом варианте осуществления изобретения R2 представляет собой бензил в способе получения соединений формулы (III).
В другом варианте осуществления изобретения R3 представляет метил и Х представляет атом хлора в способе получения соединений формулы (III).
В другом варианте осуществления изобретения в способе получения соединений формулы (III) R1 представляет пара-нитробензил, R2 представляет бензил, R3 представляет метил и Х представляет атом хлора.
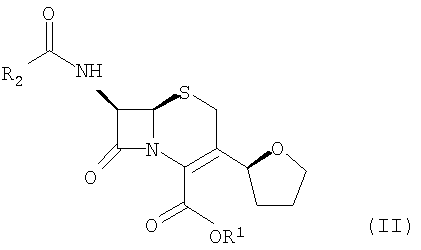
В другом предпочтительном варианте осуществления изобретения соединение формулы (III) нагревают в растворителе в присутствии LiCl и органического растворимого основания с образованием соединения формулы (II):

где R1 представляет пара-нитробензил или аллил; R2 представляет бензил или замещенный бензил; и соединение формулы (II) далее взаимодействует с R4-OH и РХ5 с образованием соединений формулы (I):
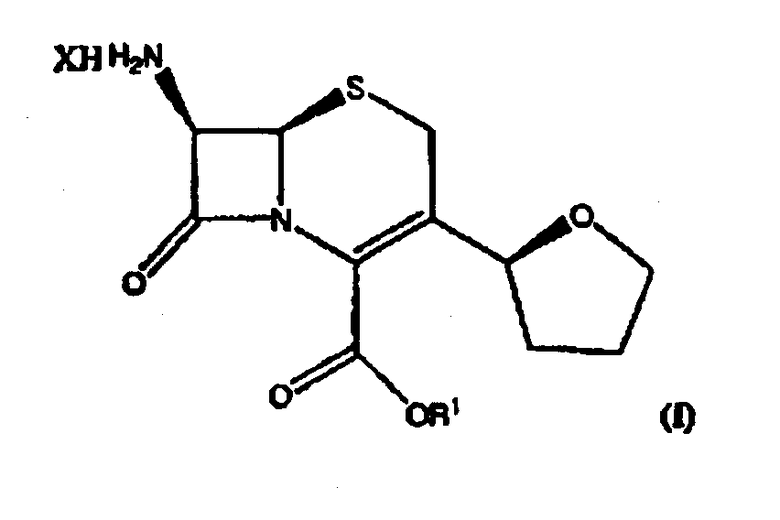
где R1 имеет значения, определенные выше, и R4 представляет собой С1-6 алкил и Х представляет атом галогена.
В другом предпочтительном варианте осуществления изобретения R1 представляет собой пара-нитробензил при превращении соединения формулы (III) в соединение формулы (I).
В другом предпочтительном варианте осуществления изобретения R2 представляет собой бензил, замещенный 1-3 заместителями, каждый из которых независимо выбран из группы, содержащей С1-6 алкил или атом галогена при превращении соединения формулы (III) в соединение формулы (I).
В другом предпочтительном варианте осуществления изобретения R3 представляет собой метил при превращении соединения формулы (III) в соединение формулы (I).
В другом предпочтительном варианте осуществления изобретения R1 представляет собой пара-нитробензил, R2 представляет собой бензил, R3 представляет метил, Х означает атом хлора; и R4 представляет изобутил при превращении соединения формулы (III) в соединение формулы (I).
В другом предпочтительном варианте осуществления изобретения органическим растворимым основанием является диизопропилэтиламин и растворителем является дихлорметан при превращении соединения формулы (III) в соединение формулы (II).
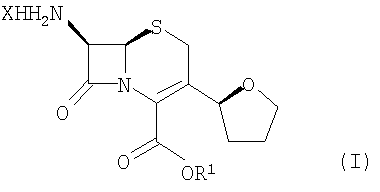
Настоящее изобретение дополнительно относится к способу получения соединения формулы (I)
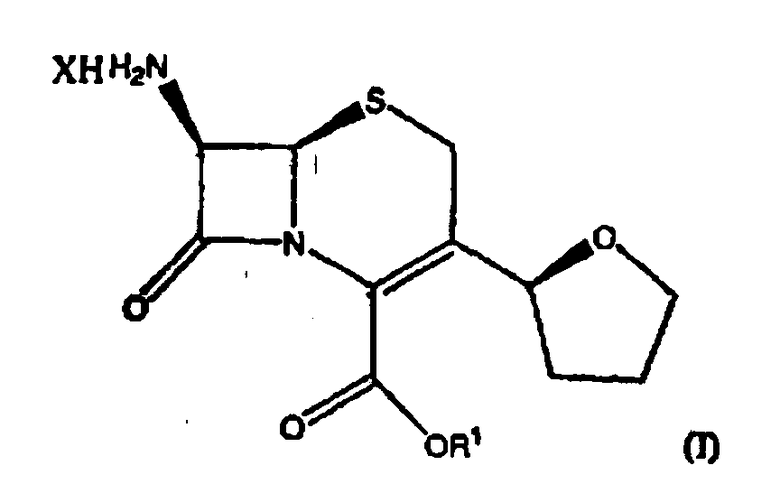
где R1 представляет собой пара-нитробензил или аллил и Х означает атом галогена, включающему следующие стадии:
(1) взаимодействие соединения формулы (V)

где R1 представляет собой пара-нитробензил или аллил и R2 представляет собой бензил или замещенный бензил с солью йодистоводородной кислоты с образованием соединения формулы (IVa)
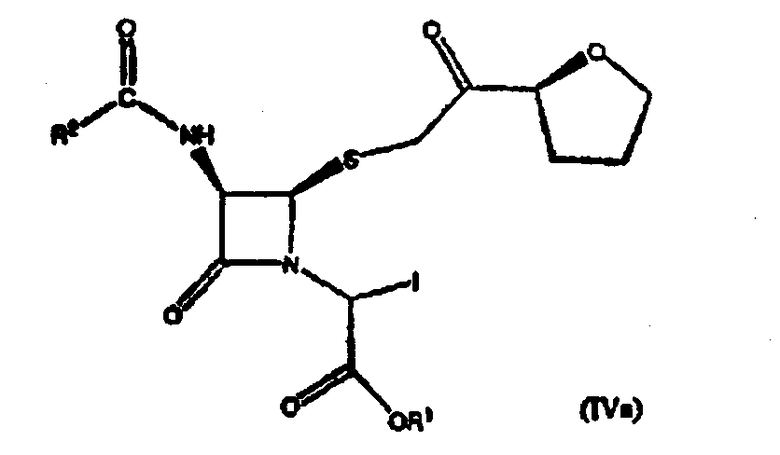
(2) взаимодействие соединения формулы (IVa) с P(OR3)3 в среде растворителя с получением соединения формулы (III)

где R1 и R2 имеют значения, определенные выше, и R3 представляет С1-6 алкил;
(3) нагревание соединения формулы (III) со стадии (2) в указанном растворителе в присутствии LiCl и органического растворимого основания с образованием соединения формулы (II);
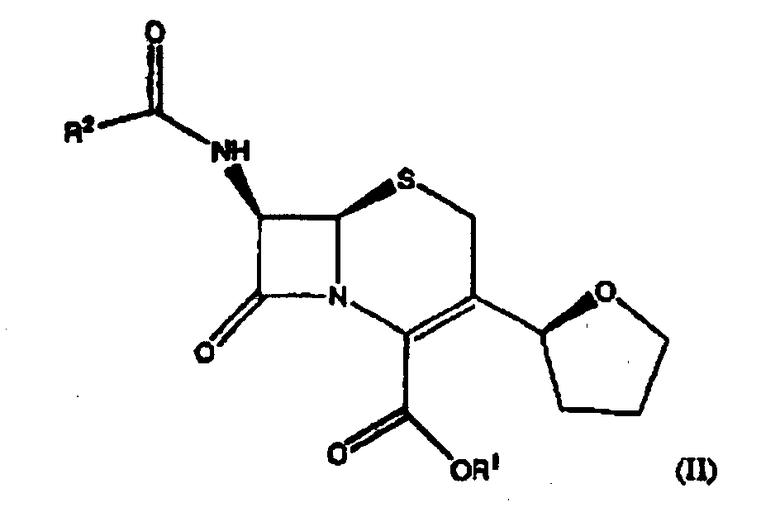
где R1 представляет пара-нитробензил или аллил и R2 представляет бензил или замещенный бензил; и
(4) взаимодействие соединения формулы (II) с R4-OHи РХ5 с образованием соединений формулы I, где R4 представляет С1-6 алкил и Х означает атом галогена.
В предпочтительном варианте R1 представляет пара-нитробензил при превращении соединения формулы (V) в соединение формулы (I).
В другом предпочтительном варианте осуществления изобретения R2 представляет бензил, замещенный 1-3 заместителями, каждый из которых независимо выбран из группы, включающей С1-6 алкил или атом галогена, при превращении соединения формулы (V) в соединение формулы (I).
В другом предпочтительном варианте осуществления изобретения R3 представляет метил при превращении соединения формулы (V) в соединение формулы (I).
В другом предпочтительном варианте осуществления изобретения Х означает атом хлора при превращении соединения формулы (V) в соединение формулы (I).
В другом предпочтительном варианте осуществления изобретения R1 представляет пара-нитробензил, R2 представляет бензил, R3 представляет метил и Х означает атом хлора при превращении соединения формулы (V) в соединение формулы (I).
Подходящие растворители, используемые в процессе превращения соединения формулы (VI) в соединение формулы (III), включают, но не ограничивают объема притязаний, толуол, ксилол, тетрагидрофуран, дихлорметан или ацетонитрил. Предпочтительно растворителем является дихлорметан.
Подходящие органические растворимые основания в процессе превращения соединения формулы (III) в соединение формулы (II) включают, но не ограничивают объема притязаний, диизопропилэтиламин («ДИПЭА»), дибутилэтиламин, метилпирролидин, этилпирролидин, метилпиперидин, этилпиперидин, этилморфолин и метилморфолин, дициклогексанметиламин, дициклогексанэтиламин и N,N′-дибутилмочевину («ДБМ»).
Предпочтительно органическое растворимое основание присутствует в процессе превращения соединения формулы (III) в соединение формулы (II) в количестве от примерно 1 до примерно 2 эквивалентов на каждый моль соединения формулы (III), предпочтительно в интервале от примерно 1,2 до примерно 1,5 эквивалентов.
Превращение соединения формулы (III) в соединение формулы (II) может быть осуществлено при температуре от примерно 0°С до примерно 60°С, предпочтительно от примерно 5°С до примерно 50°С, более предпочтительно от примерно 5°С до примерно 30°С. Вышеуказанное превращение можно проводить в течение от примерно 1 часа до примерно 16 часов, предпочтительно от примерно 4 часов до примерно 10 часов.
Использованный в настоящем описании термин «атом галогена» включат атом хлора, брома, йода и фтора.
Примеры замещенного бензила включают, но не ограничивают объема притязаний, бензил, замещенный 1-3 заместителями, каждый из которых независимо выбран из группы, включающей С1-6 алкил или атом галогена.
Настоящее изобретение дополнительно относится к соединению формулы (IV)

где R1 представляет пара-нитробензил; R2 представляет бензил, где * означает хиральный центр, который представляет абсолютную конфигурацию (R) или (S), где указанное соединение содержит (R) и (S) - изомеры в отношении 0:1 - 1:0.
Настоящее изобретение дополнительно относится к соединению формулы (IVa) или (IVb):
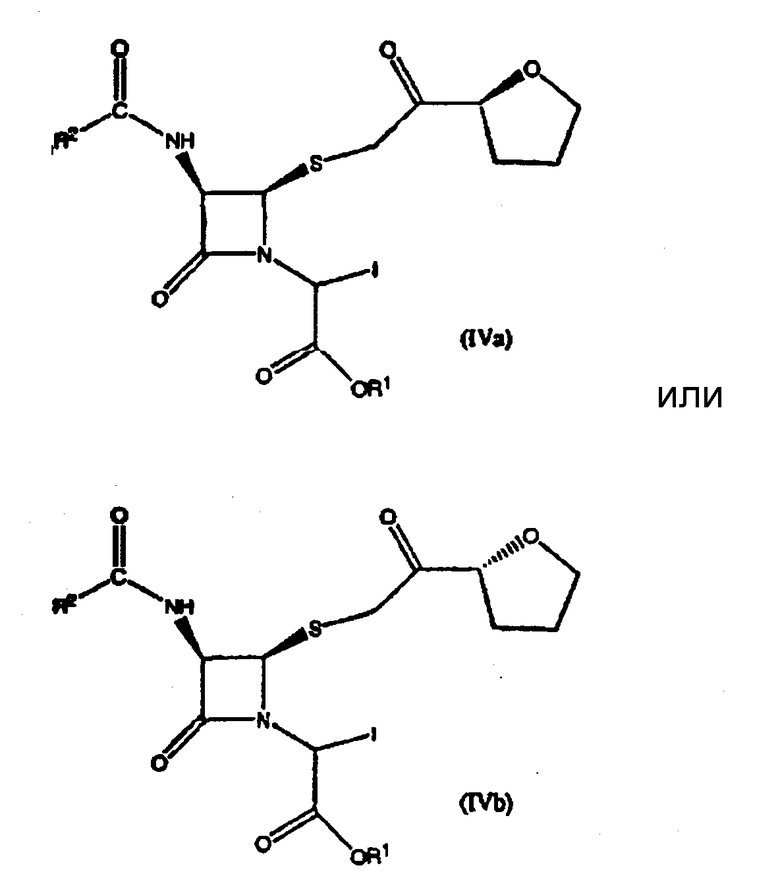
где R1 представляет пара-нитробензил, R2 представляет бензил.
Различные патенты и публикации цитированы в тексте настоящего описания. Содержание данных патентов и публикаций и содержание документов, цитированных в данных патентах и публикациях, включено в настоящее описание в качестве ссылок.
Способ настоящего изобретения и получение соединений настоящего изобретения пояснены на следующей схеме реакций. Если не указано иначе, в реакционной схеме и следующем ниже обсуждении заместители, в которых содержатся R1, R2, R3, R4 и Х, имеют значения, определенные выше.
Соединения формулы I синтезированы по следующей схеме:

где R1 представляет пара-нитробензил или аллил, R2 представляет бензил или замещенный бензил, R3 представляет С1-6алкил; Х означает атом хлора и R4 представляет изобутил.
Получение соединений формулы (VI) описано в публикации патентной заявки США № 2002/0099205, содержание которой включено в настоящее описание в качестве ссылки.
Получение хлорида
Превращение соединений формулы (VI) в соединения формулы (V) обычно осуществляют хлорированием вышеупомянутых соединений формулы (VI) с использованием агента хлорирования, такого как тионилхлорид, в среде органического растворителя, такого как толуол, ксилол, тетрагидрофуран, дихлорметан и ацетонитрил с 2-пиколином. В результате этой конверсии образуются соединения формулы V почти с количественным выходом. Установлено, что оптимальные условия для загрузки агента хлорирования соответствуют 1,1 эквивалентам, в расчете на исходную загрузку соединений формулы (VI). При более низких загрузках агента хлорирования конверсия соединений формулы V неполная.
Чтобы избежать образования побочных продуктов, эта реакция должна протекать при низкой температуре. Однако в растворе соединений формулы (VI) и 2-пиколина в дихлорметане образуется некоторое количество осадка, когда этот раствор охлаждают от комнатной температуры до -20°С. Добавление тионилхлорида к этому раствору при -20°С дает более высокое количество непрореагировавшего исходного материала, который не может быть хлорирован при добавлении избытка тионилхлорида. Поэтому часть общей загрузки тионилхлорида (10%) добавляют до начала выпадения осадка при -15°С. Затем раствор охлаждают до -20°С и оставшийся тионилхлорид медленно добавляют при этой температуре или ниже этой температуры. Продукт значительно более растворим в дихлорметане, и образования осадка при использовании этой методики не наблюдается.
Соединения формулы (VI) и формулы (V) являются смесями диастереомеров, гидрокси- и хлор-эпимеров, соответственно. Тонкослойная хроматография («ТСХ») реакционной смеси хлорирования показала чистую конверсию соединений формулы (VI) в соединения формулы (V) с небольшим количеством непрореагировавших соединений формулы (VI) и материала базовой линии. Ни один из диастереомеров не был разрешен.
Четыре возможных диастереомера были разрешены с использованием ЖХВР с обращением фаз. Однако результат ЖХВР-ОФ не соответствовал данным ТСХ. Он указывал, что реакционная смесь содержала приблизительно 50% соединений формулы (V), которые представляют главным образом один эпимер, и 50% соединений формулы (VI), также представляющих главным образом один эпимер. Данные ЖХВР с нормальной фазой соответствовали данным ТСХ и показали, что в использованных условиях реакции конверсия до соединений формулы (V) протекала более чем на 90%, а 3-10% соединений формулы (VI) оставались. Эти наблюдения позволили предположить, что один эпимер продукта быстро гидролизуется в процессе ЖХВР-ОФ, тогда как другой является относительно стабильным.
К счастью, хотя реакцию обрывали насыщенным раствором соли и сушили над сульфатом магния перед переходом к получению фосфоната, методика обработки не вызывала никакого существенного гидролиза соединений формулы (V).
Получение фосфоната
Конверсию соединений формулы V в соединения формулы III обычно проводят путем взаимодействия алкилгалогенида с триалкилфосфитом (реакция Арбузова) или производным щелочного металла диалкилфосфата (реакция Михаэлиса). Реакция Арбузова предлагает более простые реакционные условия (J. Boutagy & R. Thomas, Chem. Rev. 1, 87-99 (1974) и была разработана для получения соединений формулы (III).
Триметилфосфит, триэтилфосфит и трибутилфосфит не взаимодействуют с хлорсодержащими соединениями формулы (IVa), и поэтому хлорид заменили йодидом реакцией с солью йодистоводородной кислоты, такой как иодид натрия (реакция Финкельштейна). Процесс проводили посредством добавления йодида натрия в реакционный раствор, содержащий соединения формулы (V), и последующей обработки водой и сушки.
Вследствие низкой растворимости йодида натрия в дихлорметане эта методика дала противоречивые результаты по выходам и чистоте соединений формулы (IVa). Введение следовых количеств воды в реакционную смесь повышает растворимость йодида натрия. Однако, когда дихлорметан содержал достаточное количество воды для растворения количества йодида натрия, достаточного для протекания реакции, происходил заметный гидролиз.
Были испытаны альтернативные растворители для реакции Финкельштейна. Использования ацетона (других содержащих кетон растворителей, например, метилэтилкетона) избегали из-за его потенциальной конкуренции с внутренним кетоном в процессе кристаллизации соединений формулы (III). Ацетонитрил оказался хорошим растворителем для обмена галогенида как с точки зрения выхода, так и качества получаемого продукта. Происходила незначительная деструкция, когда реакционный раствор, содержащий соединения формулы (V), выпаривали досуха и остаток растворяли в ацетонитриле. Однако реакция обмена галогенида может быть осуществлена высушиванием и последующим концентрированием реакционного раствора, содержащего соединения формулы (V), после обработки и высушивания, а затем разбавлением ацетонитрилом с последующим добавлением йодида натрия.
Загрузка соли йодистоводородной кислотой является критическим фактором для выхода соединений формулы (IVa). Недостаточное количество соли йодистоводородной кислоты приводит к снижению выхода за счет неполного взаимодействия соединений формулы (V). Избыток соли йодистоводородной кислоты вызывает разложение соединений формулы (IVa) за счет взаимодействия с данными соединениями. Примерно 1,05 молярных эквивалентов соли йодистоводородной кислоты, в расчете на исходную загрузку соединений формулы (VI), использовали при конверсии соединений формулы (V) в соединения формулы (VI).
Соединения формулы (V) превращаются в соединения формулы (IVa) в течение нескольких минут после добавления соли йодистоводородной кислоты. Опыт авторов изобретения по синтезу Виттига предполагает, что использование, по меньшей мере, стерически затрудненного триалкилфосфита для реакции Арбузова с соединениями формулы (IVa) будет предпочтительным. Триметилфосфит («ТМФТ») дает хорошую конверсию соединений формулы (IVa) в соответствующие соединения формулы (III).
Соединения формулы (III) были получены добавлением ТМФТ к раствору, содержащему соединения формулы (IVa). Реакция ТМФТ c соединениями формулы (III) является экзотермической и требует осторожного регулирования температуры, так как при более высоких температурах повышается образование фосфатной примеси. Выделение тепла регулировали охлаждением раствора, содержащего соединения формулы IV, до температуры ниже 5°С перед медленным введением ТМФТ из раствора в дихлорметане.
Оптимальная загрузка ТМФТ составила примерно 1,45 молярных эквивалентов в расчете на исходную загрузку соединений формулы (VI). Более низкие загрузки ТМФТ давали неполную конверсию соединений формулы (V) в соединения формулы (IVa), а более высокие загрузки сопровождались возникновением проблем позднее при синтезе (в удалении защитных групп PX5) вследствие последовательной схемы процесса.
Соединения формулы (III) полностью образуются после протекания реакции в течение полутора часов при комнатной температуре. Был разработан метод испытания раствора ЖХВР для соединений формулы (III), который показал выход 75% от соединений формулы (VI). Было важно определить содержание соединений формулы (III) в реакционной смеси, чтобы последующие загрузки реагентов были основаны на этом результате.
Циклизация фосфоната
Циклизацию цефема в 6-ти членное кольцо осуществляют добавлением литиевой соли, такой хлорид лития, фторид лития и бромид лития, и органического растворимого основания, такого как ДИПЭА, в реакционный раствор, содержащий соединения формулы (III). Реакция протекает через образование (стабилизированного) фосфонатного аниона, который участвует во внутренней циклизации с образованием продукта, соединений формулы (III), который содержит полностью сформированные бициклические цефалоспориновые ядра. Для успешной циклизации требуется, по меньшей мере, два молярных эквивалента литиевой соли. Избыток литиевой соли не оказывает отрицательного действия.
Исследован ряд оснований, и диизопропилэтиламин, ДИПЭА, оказался очень эффективным в реакции циклизации. Другие растворимые основания, такие как дибутилэтиламин, метилпирролидин, этилпирролидин, метилпиперидин, этилпиперидин, этилморфолин и метилморфолин, дициклогексанметиламин, дициклогексанэтиламин и ДБМ, также могут быть использованы. Однако использование оснований, которые слабее ДИПЭА, оказалось неудачным, вероятно, вследствие того, что они неспособны депротонировать фосфонат.
Не предполагая связываться ни с какой конкретной теорией процесса, авторы изобретения полагают, что основное различие между фосфитным и фосфиновым механизмом заключается в потенциальной возможности для изомеризации двойной связи на стадии циклизации по фосфитному методу. Изомеризация двойной связи в цефалоспориновом кольце ускоряется в присутствии основания. В синтезе Виттига дает выход обработки фосфониевой соли в дихлорметане водным раствором бикарбоната натрия. Органическую фазу отделяют и позволяют протекать реакции циклизации при температуре окружающей среды, что занимает до 16 часов. Поскольку ДИПЭА является более сильным основанием, чем бикарбонат, и его трудно удалить из реакции ранее, чем до завершения циклизации, количество ДИПЭА является критическим параметром. Степень изомеризации прямо связана с количеством ДИПЭА. Оптимальное количество ДИПЭА лежит в интервале от 1,20 до 1,50 эквивалентов в расчете на молярное количество фосфоната.
Это обеспечивает полноту реакции и сводит к минимуму образование изомера с двойной связью. Количество фосфоната в реакционном растворе определяли методом ЖХВР, и расчет количеств ДИПЭА и хлорида лития проводили по этому результату.
После добавления ДИПЭА и хлорида лития раствор перемешивали при температуре окружающей среды для протекания циклизации, что требовало более 16 часов до завершения реакции. Использование более высокой температуры реакции и/или значительно более продолжительные времена реакции приводили к увеличению образования побочных продуктов и более низкому выходу.
Установлено, что остаточная вода в реакционной смеси циклизации приводит к образованию примесей и более низким выходам. Поэтому раствор фосфоната сушили над сульфатом магния перед добавлением йодида натрия, хлорида лития и ДИПЭА.
Удаление защитных групп из соединений формулы II
Конверсия соединений формулы (II) в соединения формулы (I) включает удаление защитных групп из аминогрупп в соединениях формулы (II). Удаление защитных групп предусматривает использование стандартных условий в химии цефалоспоринов, пентагалогенида фосфора, пиколина, а затем изобутанола. Соединения формул (VI) и (III) требуют присутствия ацетонитрила в реакционном растворе. Однако необходимо было удалить ацетонитрил перед проведением конечной реакции удаления защитных групп из соединений формулы (II), потому что ацетонитрил взаимодействует с пентагалогенидом фосфора. Он увеличивает растворимость соединений формулы (I) в реакционной смеси и приводит к более низкому выходу.
Существует два возможных момента для удаления ацетонитрила: после образования соединений формулы (III) или после образования соединений формулы (II). Есть два метода для удаления ацетонитрила - отгонка и фазовая экстракция. Было установлено, что удаление ацетонитрила после образования соединений формулы (VI) отгонкой влияет на содержание примесей в продукте и дает соединения формулы (II). Аналогично этому, удаление ацетонитрила экстракцией реакционной смеси, содержащей соединения формулы (IVa), приводит к образованию эмульсий, низкому выходу и извлечению, способствует вступлению в реакцию содержащейся воды на последующей стадии циклизации. Таким образом, единственной стадией, приемлемой для удаления ацетонитрила, явилась стадия сразу же перед удалением защитных групп из соединений формулы (II). Реакционную смесь экстрагировали кислым раствором для удаления солей ДИПЭА, затем насыщенным раствором соли, в результате чего удаляли некоторое количество ацетонитрила. Реакционную смесь затем дважды перегоняли для полного удаления ацетонитрила.
При осуществлении данной конверсии существуют два момента. Один из них заключается в присутствии остаточной воды, а другой - в регулировании температуры реакции/экзотермы. Эти моменты являются общими для обеих схем - фосфитной и фосфиновой. Необходимо, чтобы содержание воды было низким, и это достигается посредством перегонки с удалением ацетонитрила. Кроме того, установлено, что реакция удаления защитных групп протекает устойчиво хорошо для соединений формулы (II), которые были выделены и очищены, но меняется при использовании полученных соединений формулы (II) по таким последовательным сериям реакций из соединений формулы (VI). Это предполагает, что некоторый другой компонент(ы) реакционного раствора оказывает отрицательное влияние на протекание реакции удаления защитных групп.
Диметилфосфат («ДМФ») является побочным продуктом реакции циклизации. Было показано, что ДМФ и избыток ТМПТ отрицательно влияют на удаление защитных групп и не удаляются водной обработкой соединений формулы (II). Основываясь на этом наблюдении, избыточную загрузку ТМПТ, использованную при получении соединений формулы (III) из соединений формулы (IVa), сохраняли минимальной, что, как оказалось, составляет 1,45. Существуют некоторые данные, которые позволяют предположить, что молекулярные сита 10А удаляют фосфорные соединения из реакционной смеси.
Следующие примеры поясняют способ получения настоящего изобретения. Результаты ЯМР представлены в миллионных долях (м.д.) и получены при сравнении с опорным сигналом дейтерия из образца растворителя (дейтериохлороформа, если не указано иначе).
Далее, любой интервал цифр, цитированный в описании или параграфах далее по тексту, описывающий или заявляющий в формуле изобретения различные аспекты изобретения, как те, что представляют конкретный набор свойств, единицы измерения, условия, физическое состояние или процентное содержание, предназначен для дословного включения в описание ссылкой или иначе любого числа, попадающего в такой интервал, включая любое подмножество цифр или интервалов, относящихся к любому интервалу, указанному таким образом. Термин «примерно», использованный как указатель, или в сочетании с переменной, предназначен выражать, что числа или интервалы, указанные в описании, являются гибкими и что осуществление настоящего изобретения специалистами в данной области с использованием температур, концентраций, количеств, содержаний, числа атомов углерода и свойств, которые лежат вне интервала или отличаются от однозначного числа, приведет к достижению желательного результата.
Пример 1. Получение (3R,4R)-(4-нитрофенил)метиловый сложный эфир-α-йод-2-оксо-4-[[2-оксо-2-[(1S)-(тетрагидро-2-фуранил]этил]тио]-3-[(фенилацетил)амино]-1-азетидин уксусной кислоты
Четырехчленное кольцевое соединение (3R,4R)-(4-нитрофенил)метиловый сложный эфир-α-гидрокси-2-оксо-4-[[2-оксо-2-[(1S)-(тетрагидро-2-фуранил]этил]тио]-3-[(фенилацетил)амино]-1-азетидин уксусная кислота представляет собой смесь диастереомерных спиртов в отношении 8:2. Абсолютная стереохимия пары неизвестна на спиртовом атоме углерода. 51,19 г соединения (80% чистота, 73,4 ммоль) растворяли в 750 мл дихлорметана. Добавляли 2-пиколин (11,8 мл) (119,5 ммоль, 1,63 эквивалент) и раствор охлаждали до -15°С. Тионихлорид (7,6 мл) (104,19 ммоль, 1,42 эквивалент) добавляли за один прием (приблизительно за 3 минуты). Реакционную смесь перемешивали в течение 1 часа при температуре ниже -20°С. Ее промывали 2 х 250 мл 20%-ным раствором соли и сушили над 40 г сульфата магния в течение 10 минут при температуре окружающей среды. Осушитель отфильтровывали и промывали 100 мл дихлорметана. Фильтрат концентрировали до 150 мл на ротационном испарителе при температуре ниже 35°С. Добавляли ацетонитрил (150 мл) и раствор дополнительно концентрировали до 200 мл при температуре ниже 35°С.
Раствор охлаждали до температуры ниже 5°С. Йодид натрия (11,59 г) (119,5 ммоль, 1,05 эквивалентов от исходного соединения) загружали в раствор с получением (3R,4R)-(4-нитрофенил)метиловый сложный эфир-α-йод-2-оксо-4-[[2-оксо-2-[(1S)-(тетрагидро-2-фуранил]этил]тио]-3-[(фенилацетил)амино]-1-азетидин уксусной кислоты, которая может существовать в форме (S)-ТГФ изомера или (R)-ТГФ изомера или их смеси. Кроме того, оба (S)-ТГФ изомер и (R)-ТГФ изомера могут существовать в форме смеси йодостереомеров, которые состоят из (S)-йод изомера и (R)-йод изомера. (S)-ТГФ изомер используют для получения промежуточного соединения цефалоспорина и цефовецина. (R)-ТГФ изомер присутствует в качестве примеси во всех промежуточных соединениях процесса получения цефовецина и в конечном продукте. Однако в начальных предварительных испытаниях показано, что (R)-ТГФ изомер цефовецина натрия обладает сильным антимикробным действием сам по себе.
ЯМР данные, полученные для смеси йодсодержащего соединения, преимущественно S-серии со следами R серии, следующие:δ (400 МГц, CDCl3): 8,43 (м, 2Н, PNB-Н2,6), 7,54 (м, 2Н, PNB-Н3,5), 7,20-7,40 (м, 5Н, Bnz-H), 6,5-6,7 (м, 1Н, NH), 5,2-5,45 (м, 4Н, PNB-СН2, СН-NH&CH-NH), 5,07 (д, 1Н, J=4,8 Гц, CH-S1), 4,2-4,5 (м, 1Н, ТГФ-H2), 3,83 (м, 2Н, ТГФ-H5), 3,3-3,7 (м, 4Н, S-CH2 & Bnz-CH2), 2,1-2,2 (м, 1Н, ТГФ-H3), 1,7-1,95 (м, 3H, ТГФ-H3 & H4).
МС данные: 690,0382 (М+Na)+
Данные ЖХВР: 42,2% двух эпимеров вышеуказанных йодсодержащих соединений (Rt 12,6 & 14,5 мин), 8,4% двух эпимеров хлорных аналогов йодсодержащих соединений (Rt 12,2 & 14,1 мин), 11,4% двух эпимеров (3R,4R)-(4-нитрофенил)метиловый сложный эфир-α-гидрокси-2-оксо-4-[[2-оксо-2-[(1S)-(тетрагидро-2-фуранил]этил]тио]-3-[(фенилацетил)амино]-1-азетидин уксусной кислоты (Rt 19,6 & 20,5 мин)
Пример 2. Получение промежуточного соединения цефалоспорина
После добавления йодида натрия в примере 1 по каплям в течение 10 минут добавляли триметилфосфит (ТМФТ) (12,6 мл, 106,8 ммоль, 1,45 эквивалентов относительно исходного соединения), растворенные в дихлорметане (10 мл). В процессе добавления температуру поддерживали при или ниже 5°С. Никакого выделения тепла в таких условиях не наблюдали. Раствору позволяли нагреться до комнатной температуры в течение 1,5 часов. Содержание фосфоната определяли ЖХВР (36,49 г, 56,2 ммоль). Это соответствовало выходу 76,5% для двух стадий. Добавляли дихлорметан (500 мл) (общий объем приблизительно 700 мл). Добавляли активированный уголь (17 г) и сульфат магния (20,1 г) и смесь перемешивали в течение 10 минут. Смесь осветляли фильтрованием через слой целита, и целит промывали дихлорметаном (150 мл). Содержание фосфоната определяли ЖХВР (36,5 г, 56,1 ммоль). Добавляли хлорид лития (5,11 г) (120,5 ммоль, 2,15 эквивалентов фосфоната) и ДИПЭА (12,6 мл) (72,3 ммоль, 1,29 эквивалентов фосфоната). Раствор перемешивали при температуре окружающей среды в течение 16 часов. Реакционный раствор последовательно промывали 400 мл 1%-ного водного раствора соляной кислоты и 2 х 400 мл 20%-ного раствора соли. Органическую фазу сушили порошкообразным молекулярным ситом 4А (22,3 г) и целитом (20,3 г). Осушитель отделяли через слой диоксида кремния G (43 г) и промывали 200 мл дихлорметана. Раствор концентрировали до образования густого масла на ротационном испарителе при температуре менее 35°С и добавляли дихлорметан (350 мл). Этот раствор затем повторно концентрировали до образования густого масла на ротационном испарителе при температуре менее 35°С и добавляли дихлорметан. Определяли количество воды, которое составило 140 млн ч. Содержание продукта циклизации определяли методом ЖХВР, которое составило 25,76 г (49,2 ммоль, 67,0% выход из 3; 87,6% для циклизации).
Раствор охлаждали до -55°С и загружали пентахлорид фосфора (30,4 г) (147,4 ммоль, 3,0 эквивалентов продукта циклизации). Через 5 минут добавляли 2-пиколин (29 мл) (293,6 ммоль, 6,0 эквивалентов продукта циклизации), поддерживая температуру ниже -40°С. Наблюдали выделение тепла. Раствор перемешивали в течение 1 часа при температуре ниже -20°С. На этой стадии реакционная смесь представляла собой густую суспензию. Ее охлаждали до температуры ниже -50°С и загружали изобутанол (205 мл) (2,02 моль). Это сопровождалось нагреванием реакционной смеси до -30°С. Раствору позволяли нагреться до температуры окружающей среды, и после перемешивания в течение 1 часа добавляли зародышевый кристалл кристаллообразования промежуточного соединения цефалоспорина. Раствор перемешивали в течение 16 часов в закрытой системе во избежание испарения дихлорметана. Твердое вещество собирали фильтрованием. Твердое вещество промывали 2 х 100 мл дихлорметана. Твердое вещество высушивали до постоянной массы при 40°С под высоким вакуумом с получением промежуточного соединения цефалоспорина (18,4 г) (41,64 ммоль, 56,7% выход из (3R,4R)-(4-нитрофенил)метиловый сложный эфир-α-гидрокси-2-оксо-4-[[2-оксо-2-[(1S)-(тетрагидро-2-фуранил]этил]тио]-3-[(фенилацетил)амино]-1-азетидин уксусной кислоты, 84,6% выход из продукта циклизации).
Получали три дополнительные партии промежуточного соединения цефалоспорина с очень близкими выходами (50-55%) на 50 г навеске. Суммарный выход сравним с наилучшим выходом, достигаемым в фосфиновом способе.
Установили, что партии промежуточных соединений цефалоспорина, полученные с использованием этого способа, имеют характеристики в отношении примесей, аналогичные для продуктов, полученных оригинальным фосфиновым способом (Виттига). Их использовали для получения цефовецина, который отвечает всем существующим в настоящее время спецификациям к испытаниям на выделение лекарственного вещества.
Пример 3. Получение и идентификация фосфоната
51,8 г (3R,4R)-(4-нитрофенил)метиловый сложный эфир-α-гидрокси-2-оксо-4-[[2-оксо-2-[(1S)-(тетрагидро-2-фуранил]этил]тио]-3-[(фенилацетил)амино]-1-азетидин уксусной кислоты (RD2424, 80%, 73,4 ммоль) растворяли в 750 мл дихлорметана. Добавляли 12 мл 2-пиколина (121,5 ммоль, 1,63 эквивалентов к АЛАТ) и раствор охлаждали до -15°С. Добавляли 7,5 мл тионилхлорида (102,82 ммоль, 1,38 эквивалентов к АЛАТ). Реакционную смесь перемешивали в течение 1 часа при температуре ниже -20°С. Ее промывали 2 х 250 мл 20%-ным раствором соли и сушили над 40 г сульфата магния в течение 10 минут при температуре окружающей среды. Осушитель отфильтровывали и промывали 100 мл дихлорметана. Фильтрат концентрировали до 100 мл на ротационном испарителе при температуре менее 35°С. Добавляли 150 мл ацетонитрила и раствор дополнительно концентрировали до 200 мл на ротационном испарителе при температуре менее 35°С. Раствор охлаждали до температуры менее 4°С. Загружали 11,6 г (77,4 ммоль, 1,04 эквивалентов к (3R,4R)-(4-нитрофенил)метиловый сложный эфир-α-гидрокси-2-оксо-4-[[2-оксо-2-[(1S)-(тетрагидро-2-фуранил]этил]тио]-3-[(фенилацетил)амино]-1-азетидин уксусной кислоте) йодида натрия с последующим добавлением триметилфосфита (110,22 ммоль, 1,48 эквивалентов к (3R,4R)-(4-нитрофенил)метиловый сложный эфир-α-гидрокси-2-оксо-4-[[2-оксо-2-[(1S)-(тетрагидро-2-фуранил]этил]тио]-3-[(фенилацетил)амино]-1-азетидин уксусной кислоте), растворенного в дихлорметане (10 мл), по каплям, в течение 15 минут. Температуру поддерживали ниже 4°С в процессе добавления реагента, и выделения тепла не наблюдали. Раствор перемешивали в течение 1,5 часов. Дохлорметан (500 мл) добавляли так, что общий объем составлял ˜ 700 мл. Добавляли активированный уголь (17 г), 13х молекулярные сита (40,00 г) и сульфат магния (20,1 г) и раствор перемешивали в течение 10 минут. Его фильтровали через слой целита и промывали дихлорметаном (100 мл). Фильтрат концентрировали на ротационном испарителе при температуре менее 35°С до густого масла. Его растирали с простым диэтиловым эфиром (2×500 мл, второй промывной раствор хранили при 4°С в течение 16 часов перед декантированием), и полукристаллическое вещество сушили под вакуумом с получением твердого вещества желтого цвета (51,89 г, ЖХВР 60,9%, 65,4 выход).
ИК (диск из KBr): 3300sh, 3281s, 2958s, 1779s, 1678s, 1607m, 1524s, 1454m, 1349s, 1261s, 1035s, 850m, 739m, 697m см-1.
ЯМР (1H 400 МГц, CDCl3): 1,88 (м, 3Н), 2,12 (м, 1Н), 3,37-3,54 (2×дд, 2Н), 3,64 (с, 2Н), 3,75-3,80 (м, 6Н), 3,87 (м, 2Н), 3,90 (м, 1Н), 4,95 (дд, 1Н (J1 HP=24,8 Гц)), 5,15-5,30 (дд, 0,5H (J=4,7, 1 Гц)), 5,30 (м, 2,5 Н), 5,46 (м(2×ддд)1Н), 6,36&6,46 (2×д, 1Н), 7,27-7,28 (м, 5Н), 7,55 (д, 2Н), 8,21 (м, 2Н) м.д.
Пример 4. Циклизация фосфоната
11,31 г Фосфоната из примера 3 растворяли в смеси дихлорметана (140 мл) и ацетонитрила (30 мл). К полученному раствору добавляли 1,33 г LiCl (31,38 ммоль) и 3,30 мл ДИПЭА (18,95 ммоль). Раствор перемешивали при температуре окружающей среды в течение 16 часов. Реакционный раствор последовательно промывали 80 мл 1%-ного раствора HCl и 80 мл 20%-ного раствора соли. Органическую фазу осушали порошкообразными молекулярными ситами 4А (4,20 г), молекулярными ситами 13х (4,26 г) и целитом (4,11 г). Осушитель отделяли через слой диоксида кремния (30 г) и промывали 150 мл дихлорметана. Раствор концентрировали на ротационном испарителе при температуре менее 35°С с получением густого масла. Это масло растирали с простым диэтиловым эфиром (2 х 100 мл) и полутвердое вещество высушивали под вакуумом с получением твердого вещества золотисто-желтого цвета (2,78 г, ЖХВР 87,9%, выход 44%).
ИК: (диск из KBr): 3276s, 3029m, 2949s, 2872m, 1783s, 1725s, 1666s, 1630s, 1610s, 1520m, 1454m, 1345s, 1219s, 1103s, 1053s, 926m, 852s, 768m, 737s, 700m см-1.
ЯМР (1Н 400 МГц): 1,55 (м, 1Н), 1,9 (м, 2Н), 2,35 (м, 1Н), 3,25 (д, 1Н SCH2), 3,65 (д, 1Н, SCH2), 3,6 (д, 2Н PhCH2CO), 3,8-3,9 (м, 2Н), 4,9 (м, 1Н), 4,95 (д, 1Н), 5,25 (дд, 2Н NO2PhCH2O), 5,8 (дд, 1Н), 6,1 (д, 1H, NH), 7,23-7,35 (м, 5Н), 7,55 (д, 2Н), 8,2 (д, 2Н).
Пример 5. Получение соединения формулы (IVb)
Соединение формулы (Vb):
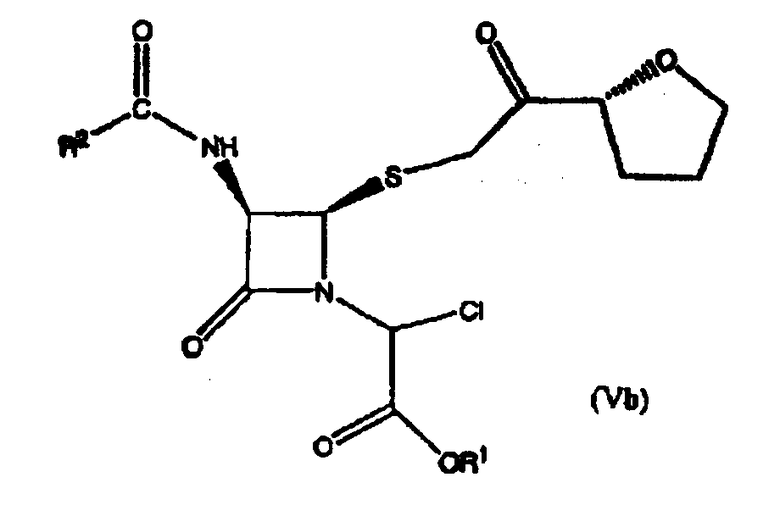
превращали в соединение формулы (IVb) посредством добавления соли йодистоводородной кислоты, где R1 представляет пара-нитробензил; R2 представляет бензил.
Хотя изобретение описано и пояснено с помощью некоторых конкретных вариантов его осуществления, специалисты в данной области поймут, что могут быть сделаны различные адаптации, изменения, модификации, замещения, исключения или добавления к методикам и протоколам без отклонения от существа и объема притязаний изобретения. Поэтому предполагается, что изобретение определяется объемом формулы изобретения, которая представлена ниже, и что данная формула изобретения может быть интерпретирована так широко, как допускает здравый смысл.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ И СЛОЖНОЭФИРНЫЕ ПРОИЗВОДНЫЕ, ИСПОЛЬЗУЕМЫЕ ДЛЯ ПОЛУЧЕНИЯ ЦЕФАЛОСПОРИНОВ | 2001 |
|
RU2237671C1 |
| СПОСОБ ВЗАИМОДЕЙСТВИЯ И ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ, ПРИГОДНЫЕ ДЛЯ ПОЛУЧЕНИЯ ЦЕФАЛОСПОРИНОВ | 2001 |
|
RU2237670C1 |
| СПОСОБ ПОЛУЧЕНИЯ N-ЗАМЕЩЕННЫХ САЛИЦИЛАМИДОВ | 2005 |
|
RU2404158C2 |
| RGD-СОДЕРЖАЩИЕ ПЕПТИДЫ | 2001 |
|
RU2214416C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 3-ПИРРОЛИН-2-КАРБОНОВОЙ КИСЛОТЫ | 1997 |
|
RU2199529C2 |
| ПОЛУЧЕНИЕ ПЕПТИДОВ СОМАТОСТАТИНА | 2004 |
|
RU2360921C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗО[1,2-C]ПИРИМИДИНИЛУКСУСНОЙ КИСЛОТЫ | 2005 |
|
RU2373208C2 |
| СПОСОБ СИНТЕЗА ПЕПТИДОВ, СОДЕРЖАЩИХ ПОДСТРУКТУРУ 4-ГИДРОКСИПРОЛИНА | 2003 |
|
RU2392265C2 |
| СПОСОБ ПОЛУЧЕНИЯ 4-(БЕНЗИМИДАЗОЛИЛМЕТИЛАМИНО)БЕНЗАМИДИНОВ | 2005 |
|
RU2401264C2 |
| СПОСОБ ПОЛУЧЕНИЯ СЛОЖНОГО ЭФИРА СУЛЬФОКСИДА 3-ЭКЗОМЕТИЛЕНЦЕФАМА | 1992 |
|
RU2010795C1 |
Настоящее изобретение относится к способу получения соединения формулы 1
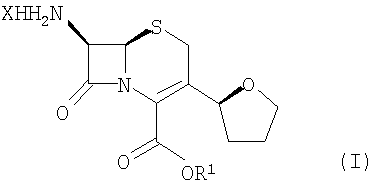
где R1 представляет пара-нитробензил или аллил; и Х представляет атом галогена, включающий следующие стадии: (1) взаимодействие соединения формулы (IVa)
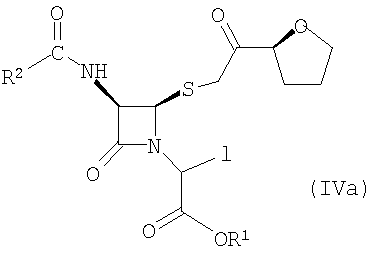
где R1 представляет собой пара-нитробензил или аллил и R2 представляет собой бензил или замещенный бензил; с Р(OR3)3 в среде растворителя с получением соединения формулы (III)
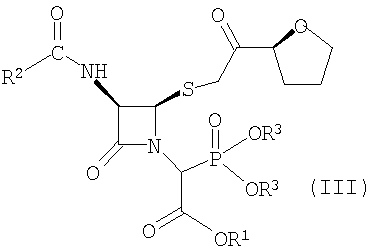
где R1 представляет пара-нитробензил или аллил; R2 представляет бензил или замещенный бензил; и R3 представляет C1-6 алкил; и последующее (2) нагревание указанного соединения формулы (III) в растворителе в присутствии LiCl и органического растворимого основания с образованием соединения формулы (II):
где R1 представляет пара-нитробензил или аллил; R2 представляет бензил или замещенный бензил; и (3) взаимодействие соединения формулы (II) с R4-OH и PX5 с образованием соединений формулы (I), которое является промежуточным в синтезе цефалоспорина. 6 н. и 8 з.п. ф-лы.
где R1 представляет собой пара-нитробензил или аллил и R2 представляет собой бензил или замещенный бензил;
включающий стадию взаимодействия соединения формулы V
где значения радикалов R1 и R2 те же, что определены выше;
с солью йодистоводородной кислоты с получением соединения формулы (IVa).

где R1 представляет пара-нитробензил или аллил; R2 представляет бензил или замещенный бензил; и R3 представляет C1-6 алкил,
включающий стадию взаимодействия соединения формулы (IVa), определенному по п.1, с Р(OR3)3 в среде растворителя.
где R1 представляет пара-нитробензил или аллил; и Х представляет атом галогена, включающий следующие стадии:
(1) взаимодействие соединения формулы (IVa)
где R1 представляет собой пара-нитробензил или аллил и R2 представляет собой бензил или замещенный бензил;
с Р(OR3)3 в среде растворителя с получением соединения формулы (III)
где R1 представляет пара-нитробензил или аллил; R2 представляет бензил или замещенный бензил; и R3 представляет C1-6 алкил, и последующее
(2) нагревание указанного соединения формулы (III) в растворителе в присутствии LiCl и органического растворимого основания с образованием соединения формулы (II)
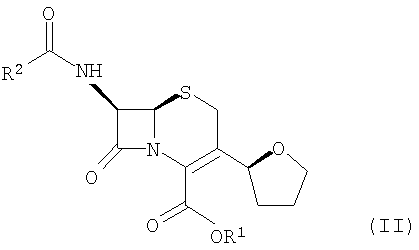
где R1 представляет пара-нитробензил или аллил; R2 представляет бензил или замещенный бензил; и
(3) взаимодействие соединения формулы (II) с R4-OH и РХ5 с образованием соединений формулы (I).
где R1 представляет собой пара-нитробензил или аллил; и Х означает атом галогена; включающий следующие стадии:
(1) взаимодействие соединения формулы (V)
где R1 представляет собой пара-нитробензил или аллил и R2 представляет собой бензил или замещенный бензил;
с солью йодистоводородной кислоты с образованием соединения формулы (IVa), как описано в п.1;
(2) взаимодействие соединения формулы (IVa) с Р(OR3)3 в среде растворителя с получением соединения формулы (III), как описано в п.2, где R3 представляет C1-6 алкил;
(3) нагревание соединения формулы (III) со стадии (2) в указанном растворителе в присутствии LiCl и органического растворимого основания с образованием соединения формулы (II), как описано в п.4; и
(4) взаимодействие соединения формулы (II) с R4-OH и PX5 с образованием соединений формулы I; где R4 представляет C1-6 алкил и Х означает атом галогена.

где R1 представляет пара-нитробензил; R2 представляет бензил; где * означает хиральный центр, который представляет абсолютную конфигурацию (R) или (S);
причем указанное соединение содержит (R) и (S) изомеры в отношении 0:1-1:0.
или

где R1 представляет пара-нитробензил; R2 представляет бензил.
| РЕГУЛЯТОР УРОВНЯ ВОДЫ | 0 |
|
SU246199A1 |
| УСТРОЙСТВО ОХЛАЖДЕНИЯ МИКРОСХЕМ ГРАФИЧЕСКОГО ВИДЕОАДАПТЕРА | 2003 |
|
RU2300856C2 |
| Топчак-трактор для канатной вспашки | 1923 |
|
SU2002A1 |
| US 60001997 А, 14.12.1999 | |||
| RU 21504771 С1, 10.06.2000. | |||

