Настоящее изобретение относится к соединениям, которые связываются с Н3-рецепторами гистамина, и к способам получения таких соединений.
Гистамин хорошо известен как медиатор некоторых реакций гиперчувствительности организма, таких как, аллергическая сыпь, сенная лихорадка и астма. В настоящее время для лечения этих состояний широко применяются сильные антагонисты гистамина, так называемые "антигистамины".
В 1940 году было замечено, что некоторые физиологические эффекты гистамина, такие как, повышенная секреция кислоты в желудке и стимуляция сердечной мышцы, не блокируются антигистаминами, существующими в то время. Это обстоятельство позволило предположить, что существует, по крайней мере, два различных типа рецепторов гистамина, называемых H1- и Н2-рецепторами. Позже были идентифицированы антагонисты Н2 (такие как, циметидин, ранитидин и фамотидин), которые стали важными средствами для лечения язв желудка.
В начале 1980 годов было установлено, что гистамин также играет определенную роль как нейромедиатор центральной нервной системы. Arrang et al., Nature, 302, 832-837 (1983) высказали предположение о существовании третьего подтипа гистаминового рецептора (Н3), расположенного пресинаптически на гистаминергических нервных окончаниях. Arrang и др. предположили, что Н3-рецептор участвует в ингибировании синтеза и высвобождения гистамина по типу обратной связи. Существование Н3-рецептора было затем подтверждено при разработке селективных Н3-агонистов и антагонистов (Arrang et al., Nature, 327, 117-123 (1987)). Впоследствии было показано, что Н3-рецептор регулирует высвобождение других нейромедиаторов как в центральной нервной системе, так и в периферических органах, в частности, в легких и в желудочно-кишечном тракте. Кроме того, сообщалось, что Н3-рецепторы регулируют высвобождение гистамина из тучных клеток и энтерохромаффин-подобных клеток.
В настоящее время существует потребность в сильных и селективных Н3-лигандах (как агонистах, так и антагонистах), которые могут быть использованы в качестве инструментов при исследовании роли гистамина как нейромедиатора, а также его роли как нейро-, эндо- и паракринного гормона. Было также предположено, что Н3-лиганды могут быть использованы в качестве терапевтических средств при ряде показаний, включая их использование в качестве седативных средств, регуляторов сна, противосудорожных средств, регуляторов гипоталамино-гипофизарной секреции, антидепрессантов, и модуляторов мозгового кровообращения, а также для лечения астмы и синдрома раздражения кишечника.
В патентной литературе был описан ряд имидазольных производных, которые предлагалось использовать в качестве Н3-лигандов. Представительными являются описания в ЕР-А-0197840, ЕР-А-0214058, ЕР-А-0458661, ЕР-А-0494010, ЕР-А-0531219, WO 91/17146, WO 92/15567, WO 93/01812, WO 93/12093, WO 93/12107, WO 93/12108, WO 93/14070, WO 93/20061, WO 94/17058, WO 95/06037, WO 95/11894, WO 95/14007, US-A-4988689 и US-А-5217986.
Настоящее изобретение относится к новому классу лигандов Н3-рецептора, имеющих сульфонамидную или сульфамидную группу, расположенную на имидазоловом кольце.
Настоящее изобретение относится к соединению формулы: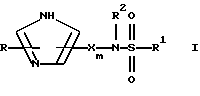
или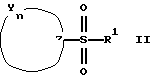
где R представляет от 0 до 2 заместителей, выбранных из группы, включающей C1-C6 алкил, C1-C6 алкокси, C1-C6 алкилтио, карбокси, C1-C6 карбоалкокси, нитро, тригалогенметил, гидрокси, амино, C1-C6 алкиламино, ди(С1-С6 алкил)амино, арил, C1-C6 алкиларил, галоген, сульфамоил и циано;
R1 представляет C4-С20 гидрокарбил (в котором один или несколько атомов водорода могут быть замещены галогеном, и вплоть до четырех атомов углерода [а в частности, от 0 до 3 атомов углерода] могут быть замещены атомами кислорода, азота или серы, при условии, что R1 не содержит группу -0-0-);
R2 представляет Н или C1-C15 гидрокарбил (в котором один или несколько атомов водорода могут быть замещены галогеном, и вплоть до трех атомов углерода могут быть замещены атомами кислорода, азота или серы, при условии, что R2 не содержит группу -0-0-);
m=3-15 (предпочтительно 3-10, например, 4-9);
n=2-6;
каждая группа Х независимо представляет 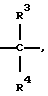 либо одна группа Х представляет -N(R4)-, -0- или -S- (при условии, что эта группа Х не является смежной с группой -NR2-), а остальные группы Х независимо представляют
либо одна группа Х представляет -N(R4)-, -0- или -S- (при условии, что эта группа Х не является смежной с группой -NR2-), а остальные группы Х независимо представляют 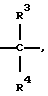 , где R3 представляет Н, C1-C6 алкил, С2-С6 алкенил, -CO2R5, -CONR5 2, -CR5 2OR6 или -OR5 (где R5 и R6 представляют Н или C1-С3 алкил), а R4 представляет Н или C1-C6 алкил;
, где R3 представляет Н, C1-C6 алкил, С2-С6 алкенил, -CO2R5, -CONR5 2, -CR5 2OR6 или -OR5 (где R5 и R6 представляют Н или C1-С3 алкил), а R4 представляет Н или C1-C6 алкил;
каждая группа Y независимо представляет -C(R3)R4-, или вплоть до двух групп Y представляют -N(R4)-, -0- или -S-, а остальные группы Y независимо представляют -С(R3)R4-, где R3 определен выше, а одна группа R4 по своей структуре представляет имидазоил, имидазоилалкил, замещенный имидазолил или замещенный имидазоилалкил, а остальные группы R4 представляют Н или C1-C6 алкил, и
Z представляет >C(R7)NR2- или >N-, где R7 представляет любую из групп, указанных выше для R3, и их фармацевтически приемлемым солям.
Настоящее изобретение также относится к производным ("пролекарственным" соединениям), которые разлагаются in vivo с образованием соединений формулы (I) или (II). Пролекарственные соединения обычно (но не всегда) имеют более низкую активность по отношению к рецептору-мишени, чем соединения, на которые они разлагаются. Пролекарственные соединения могут быть, в частности, использованы в случае, когда нужные соединения имеют химические или физические свойства, которые затрудняют их введение или делают его неэффективным. Так, например, нужное соединение может быть лишь плохо растворимым, либо оно может плохо проходить через эпителий слизистой оболочки, либо оно может иметь слишком короткий период полужизни в плазме. Дополнительное обсуждение пролекарственных соединений можно найти в работах Stella, V.J. et al., "Prodrugs", Drug Delivery Systems, pp. 112-176 (1985), и Drugs, 29, pp. 455-473 (1985).
Пролекарственные формы фармакологически активных соединений настоящего изобретения обычно представляют собой соединения формул (I) или (II), имеющих кислотную группу, которая является этерифицированной или амидированной. Такими этерифицированными кислотными группами являются группы -COOR8, где R8 представляет C1-C5 алкил, фенил, замещенный фенил, бензил, замещенный бензил или одну из следующих групп: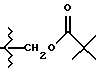
или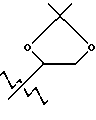
Амидированными кислотными группами являются группы формулы -CONR9R10, где R9 представляет Н, C1-C5 алкил, фенил, замещенный фенил, бензил, или замещенный бензил, а R10 представляет -ОН или одну из групп, указанных выше для R9.
Соединения формул (I) или (II), имеющих аминогруппу, могут быть дериватизированы кетоном или альдегидом, таким как, формальдегид, с образованием основания Манниха. Это может быть осуществлено путем гидролиза с кинетикой первого порядка в водном растворе.
Фармацевтически приемлемыми солями кислотных соединений настоящего изобретения являются соли, образуемые щелочными металлами и щелочноземельными металлами, такими как, натрий, калий, кальций и магний, a также соли, образуемые органическими основаниями. Подходящими органическими основаниями являются амины, такие как, N-метил-D-глюкамин.
Фармацевтически приемлемыми солями основных соединений настоящего изобретения являются соли, полученные из органических или неорганических кислот. Подходящими кислотами являются хлористоводородная кислота, бромистоводородная кислота, трифторуксусная кислота, фосфорная кислота, щавелевая кислота, малеиновая кислота, янтарная кислота и лимонная кислота.
Соединения настоящего изобретения могут существовать в различных энантиомерных, диастереомерных и таутомерных формах. При этом следует отметить, что настоящее изобретение включает различные энантиомеры, диастереомеры и таутомеры, как в чистом виде, так и в виде смеси энантиомеров, диастереомеров и таутомеров.
Используемый в настоящем описании термин "гидрокарбил" относится к моновалентным группам, состоящим из углерода и водорода. Так, например, гидрокарбильными группами являются алкильная, алкенильная, и алкинильная группы (имеющие как прямые, так и разветвленные цепи), циклоалкильная (включая полициклоалкил), циклоалкенильная и арильная группы, а также их комбинации, такие как, алкиларильная, алкениларильная, алкиниларильная, циклоалкиларильная и циклоалкениларильная группы.
Используемый в настоящем описании термин "карбоциклическая" группа означает одну или несколько замкнутых цепей или колец, которые целиком состоят из атомов углерода. Такими группами являются алициклические группы (такие как, циклопропил, циклобутил, циклопентил, циклогексил и адамантил), группы, содержащие алкильные и циклоалкильные части (такие как, адамантанметил), и ароматические группы (такие как, фенил, нафтил, инданил, флуоренил, (1,2,3,4)-тетрагидронафтил, инденил и изоинденил).
Используемый в настоящем описании термин "арил" относится к ароматическим карбоциклическим группам, включая вышеупомянутые группы.
"Гетероциклическая" группа включает одну или несколько замкнутых цепей или колец, которые имеют, по крайней мере, один атом, не являющийся атомом углерода, в замкнутой цепи или кольце. В качестве примеров могут служить бензимидазолил, тиенил, фуранил, пирролил, имидазолил, пиразолил, тиазолил, изотиазолил, оксазолил, пирролидинил, пирролинил, имидазолидинил, имидазолинил, пиразолидинил, тетрагидрофуранил, пиранил, пиронил, пиридил, пиразинил, пиридазинил, пиперидил, пиперазинил, морфолинил, "тионафтил, бензофуранил, изобензофурил, индолил, оксииндолил, изоиндолил, индазолил, индолинил, 7-азаиндолил, изоиндазолил, бензопиранил, кумаринил, изокумаринил, хинолил, изохинолил, нафтридинил, циннолинил, хиназолинил, пиридопиридил, бензоксазинил, хиноксадинил, хроменил, хроманил, изохроманил и карболинил.
Если в настоящем описании упоминается замещенная карбоциклическая группа (такая как, замещенный фенил) или замещенная гетероциклическая группа, то подразумевается, что число их заместителей составляет от 1 до 3, и эти заместители выбирают из группы, включающей C1-C6 алкил, C1-C6 алкокси, C1-C6 алкилтио, карбокси, C1-C6 карбоалкокси, нитро, тригалогенметил, гидрокси, амино, C1-C6 алкиламино, ди(C1-C6 алкил)амино, арил, C1-C6 алкиларил, галоген, сульфамоил и циано. Радикал R, если он присутствует, представляет одну или две из вышеуказанных групп, а предпочтительно C1-С3 алкил или галоген.
Используемый в настоящем описании термин "галоген" означает фтор, хлор, бром и йод.
R2 предпочтительно выбирают из группы, включающей Н, C1-С6 алкил, C1-C6 циклоалкил, C1-C6 гидроксиалкил, C1-C6 алкил-гидроксиалкил, арил C1-C6 алкил и замещенный арил C1-C6 алкил. Например, R2 может быть Н или C1-С3 алкилом.
В некоторых вариантах настоящего изобретения -Хm- представляет C1-С8 алкиленовую группу, например, бутиленовую группу.
Примерами R1 являются арил-содержащие группы (такие как, фенил, замещенный фенил, нафтил и замещенный нафтил) и (циклоалкил)алкильные группы (такие как, циклогексилпропил и адамантилпропил).
Предпочтительно, R1 представляет группу формулы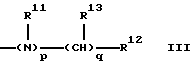
где р=0 или 1;
R11 представляет Н или C1-С3 алкил,
q=0-4,
R12 представляет карбоциклическую, замещенную карбоциклическую, гетероциклическую или замещенную гетероциклическую группу, и
R13 независимо выбирают из группы, включающей Н, C1-C6 алкил, C1-C6 циклоалкил, C1-C6 гидроксиалкил, C1-C6 алкилгидроксиалкил, арил C1-C6 алкил, и замещенный арил C1-C6 алкил.
Предпочтительно, R13 представляет водород.
Имидазоилалкильные группы в соединениях настоящего изобретения обычно имеют от 1 до 8 атомов углерода в алкильной цепи.
Обнаружено, что ряд соединений в предшествующих работах показывают значительные отклонения в своей активности, как было определено в двух анализах с использованием подвздошной кишки, описанных ниже. Анализ данных, полученных в указанных функциональном анализе и в анализе на связывание с радиоактивным лигандом, а также в других подобных биоанализах, дает основание предположить, что такое отклонение может быть связано, по крайней мере, частично, с остаточной эффективностью, свойственной этим структурам. Фактически, это означает, что указанные конкретные соединения могут действовать как агонисты, по крайней мере, в некоторых тканях.
Неожиданно обнаружено, что когда m=3 или более, предпочтительно от 3 до 9, а особенно от 4 до 8, то соединения, описанные в настоящей заявке, не показывают значительного расхождения в этих двух анализах. Таким образом, эти соединения могут считаться истинными антагонистами по отношению к действию нативного гормона, то есть, они потенциально не являются ни частичными, ни полными агонистами. В одном из своих аспектов настоящее изобретение относится к использованию этих соединений в качестве антагонистов гистамина, и к изготовлению лекарственных препаратов для этих целей.
Соединения настоящего изобретения, которые представляют собой сульфонамиды, могут быть получены посредством реакции соответствующим образом защищенного производного соединения формулы: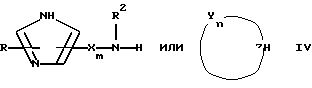
с сульфонилхлоридом формулы R1SO2Cl. Подходящими защитными группами для имидазольной части являются тритильная и N,N-диметилсульфамоильная группы. Любые другие функциональные группы в этих реакциях могут быть защищены с использованием реагентов, хорошо известных специалистам.
Реакцию с сульфонилхлоридом осуществляют в подходящем безводном растворителе, таком как, осушенный N,N-диметилформамид или осушенный дихлорметан, в присутствии основания, такого как, триэтиламин. Обычно, реакцию осуществляют при комнатной температуре в течение нескольких часов.
Соединения настоящего изобретения, которые представляют собой сульфамиды, могут быть, в основном, получены путем:
a) реакции хлорсульфонилизоцианата с соответствующим спиртом формулы R14ОH; где является трет-бутилом или бензилом;
b) реакции продукте, стадии а) соответствующим образом защищенным производным соединения формулы IV, указанной выше;
c) реакции продукта стадии b) с основанием, таким как, гидрид натрия, а затем с соединением формулы R1-Br, где атом брома связан с атомом углерода группы R1; и
d) обработки продукта стадии с) кислотой для удаления группы R14OCO и других защитных групп.
В предпочтительных вариантах осуществления настоящего изобретения, реагент, используемый в стадии с) имеет формулу: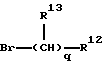
где q, R12 и R13 являются такими, как они были определены выше.
Особенно предпочтительными спиртами для использования в стадии а) являются трет-бутанол и бензиловый спирт.
На фиг. 1 проиллюстрирован синтез N-[5-(4(5)-имидазоил)пентил]-N'-(4-хлорфенил)метилсульфамида, осуществленный этим способом.
В альтернативном способе сульфамиды настоящего изобретения могут быть получены путем:
a) реакции хлорсульфонилизоцианата с соответствующим спиртом формулы R14ОН, где R14 является трет-бутилом или бензилом;
b) реакции продукта стадии а) с соответствующим образом защищенным производным соединения формулы R1-H, где атом водорода связан с атомом азота группы R1;
c) реакции продукта стадии b) с соответствующим образом защищенным производным соединения формулы: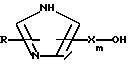
и d) обработки продукта стадии с) кислотой для удаления группы R14OCO- и других защитных групп.
В предпочтительных вариантах осуществления изобретения реагент, используемый в стадии b) имеет формулу: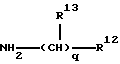
где q, R12 и R13 являются такими, как они были определены выше. На фиг.2 проиллюстрирован синтез N-[4-(4(5)-имидазоил) бутил]-N'-циклогексилметилсульфамида, осуществленный указанным способом.
Фармацевтически приемлемые соли кислотных или основных соединений настоящего изобретения могут быть, разумеется, получены стандартными способами, такими как, проведение реакции свободного основания или кислоты с, по крайней мере, стехио-метрическим количеством нужной солеобразующей кислоты или нужного солеобразующего основания.
Подразумевается, что соединения настоящего изобретения могут быть введены перорально или парентерально, включая внутривенное, внутримышечное, внутрибрюшинное, подкожное и ректальное введение, а также путем местного применения.
Для перорального введения соединения настоящего изобретения получают, в основном, в виде таблеток или капсул, либо в виде водного раствора или суспензии.
Таблетки для перорального введения могут включать активный ингредиент, смешанный с фармацевтически приемлемыми носителями, таким как, инертные разбавители, дезинтегрирующие агенты, связующие агенты, смазывающие агенты, подслащивающие агенты, ароматизирующие аренты, красители и консерванты. Подходящими инертными разбавителями являются карбонат натрия и кальция, фосфат натрия и кальция, и лактоза; а подходящими дезинтегрирующими агентами являются кукурузный крахмал и альгиновая кислота. Связующими агентами могут быть крахмал и желатин, а замасливающим агентом, если он присутствует, является, в основном, стеарат магния, стериновая кислота или тальк. Если необходимо, то для замедления абсорбции лекарственного средства в желудочно-кишечном тракте, таблетки могут быть покрыты таким материалом, как глицерилмоностеарат или глицерилдистеарат.
Капсулы для перорального введения могут быть жесткими желатиновыми капсулами, в которых активный ингредиент смешан с твердым разбавителем, и мягкими желатиновыми капсулами, в которых активный ингредиент смешан с водой или маслом, таким как, арахисовое масло, жидкое парафиновое масло или оливковое масло.
Для внутримышечного, внутрибрюшинного, подкожного и внутривенного введения, соединения настоящего изобретения, в основном, готовят в виде стерильных водных растворов или суспензий, забуференных для получения соответствующих рН и изотоничности. Подходящими водными наполнителями являются раствор Рингера и изотонический раствор хлорида натрия. Водные суспензии настоящего изобретения могут включать суспендирующие агенты, такие как, как производные целлюлозы, альгинат натрия, поливинил-пирролидон и трагакантовая камедь, а также смачивающий агент, такой как, лецитин. Подходящими консервантами для водных суспензий являются этил- и н-пропил-п-гидроксибензоат.
Эффективные дозы соединений настоящего изобретения могут быть определены стандартными методами. Точный уровень дозы, требуемый для каждого конкретного пациента, зависит от ряда факторов, включая, тяжесть заболевания, подвергаемого лечению, способ введения лекарственного средства, и вес пациента. Однако, в основном, предусматривается, что суточная доза (вводимая в виде разовой дозы или в виде дробных доз) составляет в пределах от 0,001 до 5000 мг в день, а обычно, от 1 до 1000 мг в день, а более конкретно от 10 до 200 мг в день. В пересчете на единицу веса тела, типичная доза будет, в основном, составлять от 0,01 мкг/кг до 50 мг/кг, а в частности, от 10 мкг/кг до 10 мг/кг, например, от 100 мкг/кг до 2 мг/кг.
Кроме того, настоящее изобретение проиллюстрировано нижеследующими примерами.
ПРИМЕР 1
N-[2-(4(5)-имидазоил)этил]-2-нафталинсульфонамид
К суспензии гистамина (824 мг, 7,41 ммоль) в сухом N,N-диметилформамиде (10 мл) добавляли триэтиламин (2,07 мл, 14,8 ммоль) и 2-нафталинсульфонилхлорид (1,68 г, 7,41 ммоль). Смесь перемешивали при комнатной температуре в течение 48 часов, выливали в воду (50 мл), и экстрагировали этилацетатом (3х20 мл). Объединенные органические экстракты промывали насыщенным солевым раствором (3х20 мл), растворитель выпаривали при пониженном давлении, и остаток очищали с помощью колоночной флэш-хроматографии (двуокись кремния, 1% NН3 вод. (880)/10% MeOH/CH2Cl2). Полученное масло (Rf 0,17) кристаллизовали из тетрагидрофурана и получали целевое соединение в виде белого твердого вещества (440 мг, 20%): 1H-ЯМР (300 Мгц, ДМСО-d6) 11,73 (1Н, с), 8,41 (1Н, с), 8,13 (2Н, м), 8,02 (1Н, д), 7,80 (1Н, дд), 7,67 (2Н, м), 7,44 (1Н, д), 6,74 (1Н, с), 2,99 (2Н, т), 2,58 (2Н, т). Соль малеиновой кислоты была получена путем лиофилизации эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане. Анализ для C19H19N3O6S:
Найдено: С 54,67; Н 4,69; N 10,07%.
Вычислено: С 54,67; Н 4,59; N 10,07%.
ПРИМЕР 2
N-[2-(4(5)-имидазоил)этил]бензолсульфонамид
Стадия а: N-[2-(1-бензолсульфонил-имидазоил-4-ил)этил]бензолсульфонамид. К суспензии гистамина (337 мг, 3,04 ммоль) в сухом дихлорметане (60 мл) добавляли триэтиламин (1,27 мл, 9,10 ммоль) и бензолсульфонилхлорид (775 мкл, 6,08 ммоль). Смесь перемешивали при комнатной температуре в течение 6 часов, растворитель выпаривали при пониженном давлении, и остаток растворяли в этилацетате (50 мл). Гидрохлорид триэтиламина отфильтровывали, и фильтрат упаривали при пониженном давлении. Остаток кристаллизовали из этилацетата/гексана и получали продукт в виде белого твердого вещества (1,04 г, 93%).
Стадия b. К суспензии продукта стадии а) (1,04 г, 2,82 ммоль) в этаноле (115 мл) добавляли раствор карбоната натрия (1,20 г, 11,3 ммоль) в воде (85 мл). Смесь перемешивали в течение 24 часов и этанол удаляли при пониженном давлении и при температуре окружающей среды. Водную смесь экстрагировали хлороформом (6х50 мл), и объединенные экстракты промывали насыщенным солевым раствором и сушили безводным сульфатом натрия. После фильтрации и упаривания при пониженном давлении получали целевое соединение в виде белого твердого вещества (567 мг, 80%): 1H-ЯМР (300 Мгц, ДМСО-d6) 7,77 (2Н, м), 7,60 (3Н, м), 7,46 (1Н, с), 6,74 (1Н, с), 2,94 (2Н, т), 2,57 (2Н, т). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане. Анализ для C15H17N3O6S 1,0 Н2О:
Найдено: С 46,65; Н 4,74; N 10,95%.
Вычислено: С 46,77; Н 4,97; N 10,91%.
ПРИМЕР 3
N-[2-(4(5)-имидазоил)этил]-1-нафталинсульфонамид
Cтадия а: N-[2-(1-(1-нафталинсульфонил)-4-имидазоил)этил]-нафталинсульфонамид. К суспензии гистамина (393 мг, 3,54 ммоль) в сухом дихлорметане (60 мл) добавляли триэтиламин (1,48 мл, 10,6 ммоль) и 1-нафталинсульфонилхлорид (2,01 г, 8,85 ммоль). Смесь перемешивали при комнатной температуре в течение 18 часов, растворитель выпаривали при пониженном давлении, и остаток растворяли в этилацетате (100 мл). Гидрохлорид триэтиламина удаляли путем фильтрации, и фильтрат упаривали при пониженном давлении. После колоночной флэш-хроматографии (двуокись кремния; 10% этилацетат/гексан) получали продукт (Rf 0,35) в виде бесцветной пены (1,32 г, 76%).
Стадия b. К раствору продукта стадии а (1,18 г, 2,41 ммоль) в этаноле (100 мл) добавляли раствор карбоната натрия (1,02 г, 9,64 ммоль) в воде (35 мл). Смесь перемешивали в течение 18 часов, и нерастворившееся вещество удаляли путем фильтрации. Этанол выпаривали при пониженном давлении при температуре окружающей среда. Образовавшийся осадок собирали путем фильтрации и сушили в вакууме при 50oС. После колоночной флэш-хроматографии (двуокись кремния; 1% NН3 вод. (880)/10% MeOH/CH2Cl2) было получено целевое соединение (Rf 0,29) в виде белого твердого вещества (366 мг, 50%): 1Н-ЯМР (300 Мгц, ДМСО-d6): 11,70 (1Н, шир.с), 8,64 (1Н, дд), 8,21 (1Н, д), 8,09 (2Н, м), 8,03 (1Н, т) 7,68 (3Н, м), 7,42 (1Н, д), 6,66 (1Н, с), 2,97 (2Н, м), 2,55 (2Н, м). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане. Анализ для C19H19N3O6S•0,6 Н2О:
Найдено: С 53,42; Н 4,73; N 9,69%.
Вычислено: С 53,31; Н 4,75; N 9,82%.
ПРИМЕР 4
N-[2-(4(5)-имидазоил)этил]-3-циклогексилпропансульфонамид
Стадия а: 3-Циклогексилпропансульфонилхлорид. Смесь 1-хлор-3-циклогексилпропана (8,00 г, 50 ммоль), тиомочевины (3,80 г, 50 ммоль) и иодида натрия (100 мг) в этаноле (40 мл) нагревали с обратным холодильником в течение 3 часов. Растворитель выпаривали при пониженном давлении, и остаток растирали с диэтиловым эфиром. Продукт собирали путем фильтрации, промывали диэтиловым эфиром и сушили воздухом, в результате чего получали белое твердое вещество (6,64 г), которое суспендировали в воде (50 мл)/дихлорметане (50 мл). Газообразный хлор при интенсивном перемешивании барботировали в смесь в течение 30 минут, поддерживая при этом температуру ниже 20oС. Органический слой отделяли, а затем промывали охлажденным на льду 10%-ным водным бисульфитом натрия (2х50 мл), насыщенным водным раствором бикарбоната натрия (2х50 мл) и водой (50 мл), после чего сушили сульфатом магния. После фильтрации и упаривания был получен продукт в виде бесцветного масла (3,60 г, 34%).
Стадия b. К суспензии гистамина (222 мг, 2,00 ммоль) в дихлорметане (10 мл) по каплям в течение 5 минут добавляли триэтиламин (278 мкл, 2,00 ммоль), и раствор продукта стадии а (224 мг, 1,00 ммоль) в дихлорметане (2 мл). Смесь перемешивали в течение 18 часов, и растворитель выпаривали при пониженном давлении с получением белого твердого вещества. После колоночной флэш-хроматографии (двуокись кремния; 1% NН3 вод. (880)/10% MeOH/CH2Cl2) получали целевое соединение (Rf 0,25) в виде бесцветного масла (100 мг, 33%): 1H-ЯМР (300 Мгц, CDCl3) 8,80 (1Н, шир.с), 7,52 (1H, дд), 6,83 (1Н, с), 5,80 (1H, шир. с), 3,38 (2Н, т), 2,93 (2Н, т), 2,84 (2Н, т), 1,78 (2Н, м), 1,65 (5Н, м), 1,25 (6Н, м), 0,88 (2Н, м).
ПРИМЕР 5
N-[2-(4(5)-имидазоил)этил]-(3-(1-адамантил)пропан)сульфонамид
3-(1-адамантил) пропансульфонилхлорид (700 мг, 2,50 ммоль), полученный способом, в стадии (а) примера 4, подвергали реакции с гистамином (555 мг, 5,00 ммоль) способом, описанным в стадии b примера 4. Таким образом было получено целевое соединение в виде бесцветной пены (340 мг, 39%): 1H-ЯМР (300 Мгц, CDCl3) 7,55 (1H, с), 6,86 (1H, с), 5,50 (1H, шир.с.), 3,41 (2Н, т), 2,93 (2Н, т), 2,86 (2Н, т), 1,94 (3Н, с), 1,80-1,59 (8Н, м), 1,46 (6Н, с), 1,12 (2Н, м). Соль малеиновой кислоты получали лиофилизацией эквимолярного растворa продукта и малеиновой кислоты в воде/диоксане. Анализ для С22Н33N3О6S•0,5 Н2О:
Найдено: С 55,55; H 7,20; N 8,83%.
Вычислено: С 55,44; Н 7,19; N 8,82%.
ПРИМЕР 6
N-[5-(4(5)-имидазоил)пентил]-2-нафталинсульфонамид
Стадия а: N-[5-(1-(N', N'-диметилсульфамоил)имидазол-4-ил)пентил]-2-нафталинсульфонамид. К раствору 5-(5-аминопентил)-1-(N,N-диметилсульфамоил)имидазола1 (166 мг, 0,64 ммоль) и триэтиламина (107 мкл, 0,77 ммоль) в сухом дихлорметане (2 мл), охлажденному на льду, в атмосфере аргона добавляли 2-нафталинсульфонилхлорид (175 мг, 0,77 ммоль). Раствор перемешивали в течение 45 минут при 0oС, а растворитель выпаривали. Остаток подвергали колоночной флэш-хроматографии (двуокись кремния; аммиак (880)/метанол/дихлорметан, 0,5: 5: 95) и получали продукт (Rf 0,76) в виде бесцветной пены (224 мг, 79%).
Стадия b: раствор продукта стадии а (224 мг, 0,50 ммоль) в смеси этанола (4 мл) и 1М хлористоводородной кислоты (4 мл) нагревали с обратным холодильником в течение 6 часов, а растворитель удаляли в вакууме. Остаток подвергали колоночной флэш-хроматографии (двуокись кремния; аммиак (880)/метанол/дихлорметан, 0,5: 5: 95-1:10:90), получали целевое соединение (Rf 0,26; 1: 10:90 ) и аммиак (880)/метанол/дихлорметан) в виде бесцветной пены (138 мг, 81%): 1H-ЯМР (300 Мгц, ДМСО-d6) 11,74 (1Н, шир.с), 8,41 (1H, д), 8,13 (2Н, т), 8,02 (1Н, д), 7,80 (1Н, дд), 7,67 (3Н, м), 7,44 (1Н, с), 6,61 (1Н, с), 2,79 (2Н, дд), 2,37 (2Н, т), 1,40 (4Н, м), 1,22 (2Н, дд). Анализ для C18H21N3O2S:
Найдено: С 63,04; Н 6,22; N 12,19%.
Вычислено: С 62,95; H 6,16; N 12,23%.
ПРИМЕР 7
N-[4-(4(5)-имидазоил)бутил]-2-нафталинсульфонамид
Стадия а: 4-(4-хлорбутил)-1-(трифенилметил)имидазол. Раствор 5-(4-хлорбутил)-2-(трет-бутилдиметилсилил)-1-(N, N-ди-метилсульфамоил)имидазола1 (8,80 г, 23,2 ммоль) в смеси этанола (100 мл) и 2М хлористоводородной кислоты (100 мл) нагревали с обратным холодильником в течение 2 часов. Этанол выпаривали и водный раствор экстрагировали диэтиловым эфиром (2х50 мл). Водный слой выпаривали, и остаток растворяли в сухом дихлорметане (100 мл). Затем добавляли триэтиламин (6,50 мл, 46,6 ммоль) и трифенилметилхлорид (7,10 г, 25,5 ммоль), и раствор перемешивали в течение 18 часов, промывали водой, и сушили сульфатом магния. После фильтрации и упаривания получали коричневое масло. Остаток подвергали колоночной флэш-хроматографии (двуокись кремния; 5% метанол/дихлорметан) и получали продукт в виде желтого масла (7,20 г, 77%).
Стадия b: 4-(4-фталимидобутил)-1-(трифенилметил)имидазол. К раствору продукта стадии b (7,20 г, 18,0 ммоль) в сухом N,N-диметилформамиде (150 мл) в атмосфере аргона добавляли фталимид калия (1,67 г, 9,00 ммоль). Смесь перемешивали и нагревали при 100oС в течение 5 часов, а затем охлаждали до комнатной температуры, и выливали в воду со льдом (150 мл). Полученный белый осадок собирали путем фильтрации. Остаток растворяли в дихлорметане (100 мл), промывали насыщенным солевым раствором, и сушили сульфатом магния. После выпаривания растворителя, остаток очищали с помощью колоночной флэш-хроматографии (двуокись кремния; 5% метанол/дихлорметан), и выделяли продукт в виде желтого масла (4,50 г, 98%).
Стадия с: 4-(4-аминобутил)-1-(трифенилметил)имидазол. К суспензии продукта стадии b (3,00 г, 5,86 ммоль) в этаноле (30 мл) добавляли гидрат гидразина (1,5 мл, 25,8 ммоль). Смесь нагревали с обратным холодильником в течение 2 часов и оставляли для охлаждении до комнатной температуры. Осадок удаляли путем фильтрации. Фильтрат выпаривали, а остаток растирали с хлороформом. Твердое вещество снова удаляли путем фильтрации. Затем фильтрат выпаривали, и процесс растирания повторяли, в результате чего получали продукт в виде желтого масла (2,05 г, 92%).
Стадия d: N-[4-(1-(трифенилметил)имидазол-4-ил)бутил]-2-нафталинсульфонамид. К раствору продукта стадии (с) (465 мг, 1,22 ммоль) и триэтиламина (185 мкл, 1,33 ммоль) в сухом дихлорметане (15 мл) добавляли 2-нафталинсульфонилхлорид (227 мг, 1,22 ммоль). Раствор перемешивали в течение 2 часов и растворитель выпаривали. Остаток подвергали колоночной флэш-хроматографии (двуокись кремния; 5% метанол/дихлорметан) и получали продукт в виде бесцветной пены (588 мг, 84%).
Стадия е: раствор продукта стадии d (588 мг, 1,02 ммоль) в трифторуксусной кислоте (5 мл) перемешивали в течение 18 часов и растворитель выпаривали. Остаток подвергали колоночной флэш-хроматографии (двуокись кремния; 1: 10: 90 аммиак (880)/метанол/дихлорметан) и получали целевое соединение (Rf 0,24) в виде белого твердого вещества (96 мг, 63%): 1H-ЯМР (300 Мгц, ДМСО-d6) 11,70 (1Н, шир.с), 8,41 (1Н, с), 8,13 (2Н, т), 8,03 (1H, д), 7,80 (1Н, дд), 7,67 (3Н, м), 7,45 (1Н, с), 6,62 (1H, с), 2,76 (2Н, дд), 2,38 (2Н, т), 1,48 (2Н, м), 1,41 (2Н, дд). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане. Анализ для С21Н23N3О6S•0,5 Н2О:
Найдено: С 56,55; Н 5,31; N 9,31%.
Вычислено: С 56,62; Н 5,20; N 9,43%.
ПРИМЕР 8
N-[6-(4(5)-имидазоил)гексил]-2-нафталинсульфонамид
Целевое соединение получали способом, описанным в примере 6, с использованием 5-(6-аминогексил)-1-(N, N-диметилсульфамоил)имидазола1 в качестве субстрата в стадии а. Продукт от двух стадий получали в виде белого твердого вещества: 1H-ЯМР (300 Мгц, ДМСО-d6) 11,72 (1H, шир.с), 8,41 (1H, дд), 8,13 (2Н, дд), 8,03 (1H, д), 7,80 (1H, дд), 7,67 (3Н, м), 7,44 (1H, с), 6,62 (1H, с), 2,74 (2Н, дд), 2,37 (2Н, т), 1,42 (2Н, м), 1,33 (2Н, м), 1,17 (4Н, м). Анализ для C19H23N3O2S•0,9 H2O:
Найдено: С 60,99; H 6,59; N 11,17%.
Вычислено: С 61,07; H 6,69; N 11,24%.
ПРИМЕР 9
N-[5-(4(5)-имидазоил)пентил]-(4-хлорфенил)метансульфонамид
Стадия а: N-[5-(1-(N',N'-диметилсульфамоил)имидазол-4-ил)пентил]-(4-хлорфенил)метансульфонамид. К раствору 5-(5-аминопентил)-1-(N,N-диметилсульфамоил)имидазола1 (412 мг, 1,58 ммоль) и триэтиламина (264 мкл, 1,90 ммоль) в сухом дихлорметане (5 мл), охлажденному в атмосфере аргона до -78oС, по каплям добавляли раствор (4-хлорфенил)метансульфонилхлорида (533 мг, 2,37 ммоль) в сухом дихлорметане (5 мл). Полученный раствор перемешивали в течение 18 часов, нагревали до комнатной температуры, и растворитель выпаривали. После колоночной флэш-хроматографии (двуокись кремния; 0,5:5:95, аммиак (880)/метанол/дихлорметан) остатка получали продукт (Rf 0,66; 1:10:90, аммиак (880)/метанол/дихлорметан) в виде бесцветного масла (307 мг, 43%).
Стадия b: раствор продукта стадии а (275 мг, 0,61 ммоль) в смеси этанола (4 мл) и 1М хлористоводородной кислоты (4 мл) нагревали с обратным холодильником в течение 18 часов, и растворитель удаляли в вакууме. Остаток подвергали колоночной флэш-хроматографии (двуокись кремния; 1:10:90 аммиак (880)/метанол/дихлорметан) и получали продукт (Rf 0,34; 1:10:90, аммиак (880)/метанол/дихлорметан) в виде белого кристаллического твердого вещества (181 мг, 87%): 1Н-ЯМР (300 Мгц, ДМСО-d6) 11,75 (1Н, шир.с), 7,46 (1Н, с), 7,43 (2Н, дд), 7,37 (2Н, д), 7,04 (1Н, т), 6,68 (1Н, с), 4,31 (2Н, с), 2,87 (2Н, д), 2,45 (2Н, т), 1,50 (2Н, м), 1,40 (2Н, м), 1,27 (2Н, м). Анализ для C15H20ClN3O2S:
Найдено: С 52,88; H 6,13; N 12,28%.
Вычислено: С 52,70; Н 5,90; N 12,29%.
ПРИМЕР 10
N-[4-(4(5)-имидазоил)бутил]-(4-хлорфенил)метансульфонамид
Целевое соединение получали способом, описанным в примере 9, с использованием 4-(4-аминобутил)-1-(трифенилметил)ими- дазола (пример 7, стадия с) в качестве субстрата в стадии (а). Продукт от двух стадий получали в виде белого твердого вещества: 1H-ЯМР (300 Мгц, ДМСО-d6) 7,47 (1Н, с), 7,43 (2Н, дд), 7,37 (2Н, д), 7,05 (1Н, т), 6,69 (1Н, с), 4,31 (2Н, с), 2,89 (2Н, дд), 2,46 (2Н, т), 1,55 (2Н, м), 1,45 (2Н, м). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане. Анализ для С18Н22СINзО6S:
Найдено: С 48,68; Н 5,14; N 9,32%.
Вычислено: С 48,70; Н 5,00; N 9,47%.
ПРИМЕР 11
N-[6-(4(5)-имидазоил)гексил]-(4-хлорфенил)метансульфонамид
Целевое соединение получали способом, описанным в примере 9, с использованием 5-(6-аминогексил)-1-(N, N-диметилсульфамоил)имидазола1 в качестве субстрата в стадии а. Продукт от двух стадий получали в виде белого кристаллического твердого вещества: 1H-ЯМР (300 Мгц, ДМСО-D6) 11,71 (1Н, шир.с), 7,46 (1H, с), 7,43 (2Н, ддд), 7,37 (2Н, ддд), 7,05 (1Н, т), 6,68 (1Н, с), 4,31 (2Н, с), 2,86 (2Н, дд), 2,46 (2Н, т), 1,52 (2Н, м), 1,37 (2Н, м), 1,27 (4Н, м). Анализ для С16Н22СIN3О2S:
Найдено: С 51,51; Н 6,28; N 15,05%.
Вычислено: С 51,81; Н 6,25; N 15,11%.
ПРИМЕР 12
N-[5-(4(5)-имидазоил)пентил]-N'-(4-хлорфенил)метилсульфамид
Стадия а: N-[5-(1-(N",N"-диметилсульфамоил)имидазол-4-ил)пентил]-N'-трет-бутоксикарбонилсульфамид. К раствору хлорсульфонилизоцианата (211 мкл, 2,42 ммоль) в сухом дихлорметане (3 мл), охлажденному на льду, в атмосфере аргона по каплям добавляли раствор сухого т-бутанола (346 мкл, 3,63 ммоль) в сухом дихлорметане (3 мл). Раствор нагревали до комнатной температуры, перемешивали в течение 10 минут, и в атмосфере аргона по каплям добавляли к охлажденному льдом раствору 5-(5-аминопентил)-1-(N, N-диметилсульфамоил)имидазола1 (484 мг, 1,86 ммоль) и триэтиламина (388 мкл, 2,79 ммоль) в сухом дихлорметане (6 мл). Смесь перемешивали в течение 18 часов, нагревали до комнатной температуры, и растворитель выпаривали. После колоночной флэш-хроматографии (двуокись кремния; 0,5:5:95, аммиак (880)/метанол/дихлорметан) остатка получали продукт (Rf 0,29; 1:10:90 аммиак(880)/метанол/дихлорметан) в виде бесцветного масла (400 мг, 49%).
Стадия b. N'-(4-хлорфенил)метил-N-[5-(1-(N", N"-диметилсульфамоил)имидазол-4-ил)пентил]-N'-трет-бутоксикарбонилсульфамид. К раствору продукта стадии а) (390 мл, 0,89 ммоль) и 4-хлорбензилбромида (183 мг, 0,89 ммоль) в сухом N,N-диметилформамиде (3 мл), охлажденном в атмосфере аргона до -15oС, добавляли гидрид натрия-(36 мг, 0,89 ммоль, 60% дисперсия в масле). Полученную смесь перемешивали в течение 18 часов и оставляли для медленного нагревания до температуры окружающей среды. Затем добавляли воду (15 мл) и смесь экстрагировали этилацетатом (4х10 мл). Объединенные органические слои промывали четыре раза водой, сушили сульфатом натрия и упаривали. После колоночной флэш-хроматографии (двуокись кремния; аммиак (880)/метанол/дихлорметан= 0,2: 2:98) остатка получали продукт (Rf 0,29; аммиак (880)/метанол/дихлорметан= 1: 10: 90) в виде бесцветного масла (378 мг 75%).
Стадия с. Суспензию продукта стадии b) (374 мг, 0,66 ммоль) в смеси этанола (5 мл) и 1М соляной кислоты (5 мл) нагревали с обратным холодильником в течение 18 часов и растворитель удаляли в вакууме. Остаток подвергали флэш-хроматографии (двуокись кремния; аммиак (880)/метанол/дихлорме-тан=1: 10:90) и получали продукт (Rf 0,24) в виде белого твердого вещества (132 мг, 5%): 1H-ЯМР (300 Мгц, ДМСО-d6) 7,46 (1Н, с), 7,43 (5Н, м), 6,87 (1Н, т), 6,68 (1Н, с), 4,03 (2Н, д), 2,78 (2Н, дд), 2,45 (2Н, т), 1,58 (2Н, м), 1,47 (2Н, м), 1,27 (2Н, м). Анализ для G15H21ClN4O2S:
Найдено: С 50,33; H 5,88; N 15,55%.
Вычислено: С 50,48; Н 5,93; N 15,70%.
ПРИМЕР 13
N-[4-(4(5)-имидазоил)бутил]-N'-(4-хлорфенил)метилсульфамид
Целевое соединение получали способом, описанным в примере 12, с использованием 4-(4-аминобутил)-1-(трифенилметил)имидазола (пример 7, стадия с) в качестве субстрата в стадии а. Продукт от трех стадий получали в виде белого твердого вещества: 1H-ЯМР (300 Мгц, ДМСО-d6) 11,68 (1Н, шир.с), 7,47 (1H, д), 7,36 (4Н, м), 6,86 (1H, т), 6,68 (1H, с), 3,98 (2Н, д), 2,79 (2Н, дд), 2,45 (2Н, т), 1,53 (2Н, м), 1,43 (2Н, м). Анализ для C14H19ClN4O2S:
Найдено: С 49,08; H 5,61; N 16,23%.
Вычислено: С 49,05; Н 5,59; N 16,34%.
ПРИМЕР 14
N-[3-(4(5)-имидазоил)пропил]-N'-(4-хлорфенил)метилсульфамид
Стадия а: 4-(3-фталимидопропил)-1-(трифенилметил)имидазол. К раствору 3-[1-(трифенилметил)имидазол-4-ил] пропан-1-ола (2,24 г, 6,07 ммоль) в сухом тетрагидрофуране (10 мл) в атмосфере аргона добавляли фталимид (1,16 г, 7,89 ммоль) и трифенилфосфин (2,07 г, 7,89 ммоль). Суспензию охлаждали на льду и по каплям добавляли раствор диэтилазодикарбоксилата (1,24 мл, 7,89 ммоль) в сухом тетрагидрофуране (10 мл). Смесь нагревали до комнатной температуры и перемешивали в течение 2 часов. Затем добавляли диэтиловый эфир (20 мл). Осадок собирали путем фильтрации и сушили в вакууме с получением белого твердого продукта (2,08 г, 68%).
Стадия b: 4-(3-аминопропил)-1-(трифенилметил)имидазол. К суспензии продукта стадии а (4,09 г, 8,22 ммоль) в этаноле (82 мл) добавляли гидрат гидразина (2,32 мл, 41,1 ммоль). Смесь нагревали с обратным холодильником в течение 3 часов и оставляли для охлаждения до комнатной температуры. Осадок удаляли путем фильтрации. Фильтрат выпаривали и остаток растирали с хлороформом. Твердое вещество снова удаляли путем фильтрации. Фильтрат упаривали, и процесс растирания повторяли, в результате чего получали продукт в виде желтого масла с количественным выходом.
Стадия с: N-[3-(1-(трифенилметил)имидазол-4-ил)пропил]-N'-трет-бутоксикарбонил-N'(4-хлорфенил) метил-сульфамид. Продукт стадии b преобразовывали в нужный продукт способом, описанным в примере 12, стадии а и b.
Стадия d: раствор продукта стадии с (308 мг, 0,46 ммоль) в трифторуксусной кислоте (3 мл) перемешивали в течение 18 часов, а растворитель выпаривали. После колоночной флэш-хроматографии (двуокись кремния; 1:10:90, аммиак (880)/метанол/дихлорметан) остатка получали целевое соединение (Rf 0,18) в виде белого твердого продукта (96 мг, 63%): 1Н-ЯМР (300 Мгц, ДМСО-d6) 11,77 (1Н, шир. с), 7,37 (4Н, м), 6,99 (1Н, т), 6,72 (1Н, с), 3,98 (2Н, д), 2,81 (2Н, дд), 2,48 (2Н, м), 1,72 (2Н, м). Анализ для C13H17ClN4O2S:
Найдено: С 47,30; Н 5,26; N 16,95%.
Вычислено: С 47,49; Н 5,21; N 17,04%.
ПРИМЕР 15
N-[6-(4(5)-имидазоил)гексил]-N'-(4-хлорфенил)метилсульфамид
Целевое соединение получали способом, описанным в примере 12, с использованием 5-(6-аминогексил)-1-(N, N-диметилсульфамоил)имидазола1 в качестве субстрата в стадии а. Продукт от трех стадий получили в виде белого твердого вещества: 1H-ЯМР (300 Мгц, ДМСО-d6) 7,46 (1Н, д), 7,35 (5Н, м), 6,86 (1Н, т), 6,67 (1Н, с), 3,98 (2Н, д), 2,75 (2Н, дд), 2,45 (2Н, т), 1,52 (2Н, м), 1,38 (2Н, м), 1,25 (4Н, м). Анализ для C16H23ClN4O2S:
Найдено: С 51,51; Н 6,28; N 15,05%.
Вычислено: С 51,81; Н 6,25; N 15,11%.
ПРИМЕР 16
N-[2-(4(5)-имидазоил)этил]-3,4-дихлорбензолсульфонамид
Целевое соединение получали способом, описанным в примере 2, с использованием гистамина и 3,4-дихлорбензолсульфо1 нилхлорида (полученного, в основном, как описано в примере 4 в стадии а) в качестве субстратов в стадии (а). Продукт от двух стадий получали в виде белого кристаллического твердого вещества: 1Н-ЯМР (300 Мгц, ДМСО-d6) 11,80 (1Н, шир.с), 7,94 (1Н, д), 7,90 (1Н, м), 7,8 (1Н, м), 7,72 (1Н, дд), 7,47 (1Н, с), 6,76 (1Н, с), 3,00 (2Н, т), 2,59 (2Н, т).
ПРИМЕР 17
N-[2-(4(5)-имидазоил)этил]-2-фенилэтансульфонамид
Стадия а: 4-(2-фталимидоэтил)-1-(трифенилметил)имидазол. К суспензии 4(5)-(2-фталимидоэтил)имидазола2 (838 мг, 3,48 ммоль) в сухом дихлорметане (10 мл) в атмосфере аргона добавляли триэтиламин (728 мкл, 5,22 ммоль) и трифенилметилхлорид (1,16 г, 4,18 ммоль). После колоночной флэш-хроматографии (двуокись кремния; 0,2:2:98-1:10:90 аммиак (880)/метанол/дихлорметан) остатка получали продукт в виде пены (1,20 г, 71%).
Стадия b: 4-(2-аминоэтил)-1-(трифенилметил)имидазол. К суспензии продукта стадий а (1,20 г, 2,48 ммоль) в этаноле (25 мл) добавляли гидрат гидразина (702 мл, 12,4 ммоль). Смесь нагревали с обратным холодильником в течение 90 минут и
оставляли для охлаждения до комнатной температуры. Осадок удаляли путем фильтрации. Фильтрат упаривали, и остаток растирали с хлороформом. Твердое вещество снова удаляли путем фильтрации. Фильтрат упаривали, и процесс растирания повторяли с получением продукта и виде желтого масла (818 мг, 93%).
Стадия с: N-[2-(1-трифенилметил)имидазоил-4-ил)этил]-2-фенилэтансульфонамид. К раствору продукта стадии b (353 мг, 1,00 ммоль) и триэтиламин (154 мкл, 1,10 ммоль) в сухом дихлорметане (5 мл) добавляли раствор 2-фенилэтансульфонилхлорида (полученного, в основном, как описано в стадии а примера 4) (205 мг, 1,00 ммоль) в сухом дихлорметане (2 мл). Раствор перемешивали в течение 30 минут, промывали водой и сушили сульфатом магния. После фильтрации и упаривания получали продукт (Rf 0,68; аммиак (880)/метанол/дихлорметан= 1:10:90) в виде белого твердого вещества (450 мг, 86%).
Стадия d: раствор продукта стадии с (440 мг, 0,84 ммоль) в трифторуксусной кислоте (4 мл) перемешивали в течение 18 часов и растворитель выпаривали. После колоночной флэш-хроматографии (двуокись кремния; аммиак (880)/метанол/дихлорметан= 1: 10:90) остатка получали целевое соединение (Rf 0,25) в виде масла (61 мг, 26%): 1H-ЯМР (300 Мгц, CDCl3) 7,83 (1Н, с), 7,20 (5Н, м), 6,93 (1H, с), 3,39 (2Н, м), 3,27 (2Н, м), 3,08 (2Н, м), 2,88 (2Н, м). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане. Анализ для C17H21N3O6S:
Найдено: С 51,35; Н 5,41; N 10,62%.
Вычислено: С 51,64; Н 5,35; N 10,63%.
ПРИМЕР 18
N-[2-(4(5)-имидазоил)этил]-3-фенилпропансульфонамид
Целевое соединение получали способом, описанным в примере 17, с использованием 3-фенилпропансульфонилхлорида (полученного, в основном, как описано в стадии а примера 4) в качестве субстрата в стадии с. Продукт (Rf 0,26); аммиак (880)/метанол/дихлорметан=1:10:90) от четырех стадий получали в виде бесцветного масла 1Н-ЯМР (300 Мгц, СDСl3) 9,80 (1Н, шир.с), 7,50 (1Н, с), 7,42 (2Н, т), 7,20 (1Н, м), 7,13 (2Н, м), 6,80 (1Н, с), 3,30 (2Н, т), 2,95 (2Н, т), 2,79 (2Н, т), 2,70 (2Н, т), 2,07 (2Н, квинтет). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане. Анализ для C18H23N3O6S:
Найдено: С 52,69; Н 5,71; N 10,23%.
Вычислено: С 52,80; Н 5,66; N 10,27%.
ПРИМЕР 19
N-[2-(4(5)-имидазоил)этил]-2-нафтилметансульфонамид
Целевое соединение получали способом, описанным в примере 17, с использованием 2-нафтилметансульфонилхлорида (полученного, в основном, как описано в примере 4 в стадии а) в качестве субстрата в стадии с. Продукт (Rf 0,27); аммиак (880)/метанол/дихлорметан=1:10:90) от четырех стадий получали в виде белого твердого вещества: 1Н-ЯМР (300 Мгц, ДМСО-d6) 11,75 (1Н, шир. с), 7,89 (4Н, м), 7,51 (4Н, м), 7,17 (1Н, т), 6,79 (1Н, с), 4,46 (2Н, с), 3,17 (2Н, дд), 2,66 (2Н, м). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане. Анализ для C20H21N3O6S•0,5 Н2О:
Найдено: С 54,30; Н 5,09; N 9,55%.
Вычислено: С 54,33; Н 5,03; N 9,53%.
ПРИМЕР 20
(Е)-N-[2-(4(5)-имидазоил)этил]-2-фенилэтенсульфонамид
Целевое соединение получали способом, описанным в примере 17, с использованием транс-β-стиролсульфонилхлорида в качестве субстрата в стадии с. Продукт (Rf 0,13); аммиак (880)/метанол/дихлорметан=1:10:90) от четырех стадий получали в виде бесцветной пены: 1H-ЯМР (300 Мгц, CDCl3) 7,56 (1Н, д), 7,42 (6Н, м), 6,84 (1Н, с), 6,73 (1H, д), 3,36 (2Н, т), 2,86 (2Н, т). Анализ для С13Н15N3O2S•0,5 Н2O:
Найдено: C 54,41; H 5,49; N 14,69%.
Вычислено: С 54,53; H 5,63; N 14,67%.
ПРИМЕР 21
N-[2-(4(5)-имидазоил)этил]фенилметансульфонамид
Целевое соединение получали способом, описанным в примере 9, с использованием фенилметансульфонилхлорида в качестве субстрата в стадии с. Продукт (Rf 0,27), аммиак (880)/метанол/дихлорметан=1:10:90) от 4 стадий получали в виде белого твердого вещества: 1Н-ЯМР (300 Мгц, ДМСО-d6) 7,50 (1H, с), 7,31 (5Н, м), 7,20 (1H, т), 6,80 (1H, с), 4,27 (2Н, с), 3,13 (2H, м), 2,63 (2H, т). Анализ для C12H15N3О2S:
Найдено: С 54,39; H 5,73; N 15,60%.
Вычислено: С 54,32; Н 5,70; N 15,84%.
ПРИМЕР 22
N-[2-(4(5)-имидазоил)этил]-8-хинолинсульфонамид
Целевое соединение получали способом, описанным в примере 17, с использованием 8-хинолинсульфонилхлорида в качестве субстрата в стадии с. Продукт (Rf 0,34; аммиак (880)/метанол/дихлорметан=1:10:90) от четырех стадий был получен в виде бесцветного кристаллического твердого вещества: 1H-ЯМР (300 Мгц, CDCl3) 8,82 (1Н, дд), 8,42 (1Н, дд), 8,25 (1H, дд), 8,05 (1Н, д), 7,64 (1H, дд), 7,51 (1H, дд), 7,38 (1H, д), 6,66 (1H, д), 3,17 (2Н, т), 2,74 (2H, т). Анализ для C14H14N4О2S:
Найдено: С 55,33; Н 4,81; N 18,47%.
Вычислено: С 55,61; Н 4,67; N 18,53%.
ПРИМЕР 23
N-[2-(4(5)-имидазоил)этил]-2-циклогексилэтансульфонамид
Целевое соединение получали способом, описанным в примере 17, с использованием 2-циклогексилэтансульфонилхлорида (полученного, в основном, как описано в стадии а примера 4) в качестве субстрата в стадии с. Продукт (Rf 0,27); аммиак (880)/метанол/дихлорметан=1:10:90) от четырех стадий был получен в виде белого твердого вещества: 1H-ЯМР (300 Мгц, ДМСО-d6) 11,80 (1H, шир. с), 7,51 (1H, с), 7,05 (1H, т), 6,81 (1H, с), 3,13 (2H, дд), 2,90 (2H, м), 2,65 (2H, т), 1,62 (5Н, м), 1,47 (2Н, м), 1,16 (4Н, м), 0,86 (2Н, м). Анализ для С13Н23N3О2S:
Найдено: С 54,68;, Н 8,20; N 14,71%.
Вычислено: С 54,70; Н 8,12; N 14,72%.
ПРИМЕР 24
N-[2-(4(5)-имидазоил)этил]-(3,4-дихлорфенил)метансульфонамид
Целевое соединение получали способом, описанным в примере 9, с использованием (3,4-дихлорфенил)метансульфонилхлорида (полученного, в основном, как описано в стадии а примера 4) в качестве субстрата в стадии а. Продукт (Rf 0,31; аммиак (880)/метанол/дихлорметан=1:10:90) от четырех стадий был получен в виде белого твердого вещества: 1H-ЯМР (300 Мгц, ДМСО-d6) 11,85 (1Н, шир. с), 7,60 (3Н, м), 7,32 (1Н, дд), 7,23 (1H, т), 6,82 (1H, с), 4,36 (2Н, с), 3,16 (2Н, дд), 2,65 (2Н, т). Анализ для C12H13Cl2N3О2S:
Найдено: С 42,79; Н 3,98; N 12,49%.
Вычислено: С 43,12; Н 3,92; N 12,57%.
ПРИМЕР 25
N-[2-(4(5)-имидазоил)этил]-(4-хлорфенил)метансульфонамид
Целевое соединение было получено способом, описанным в примере 9, с использованием (4-хлорфенил)метансульфонилхлорида (полученного, в основном, как описано в стадии а примера 4) в качестве субстрата в стадии а. Продукт (Rf 0,19; аммиак (880)/метанол/дихлорметан=1:10: 90) от четырех стадий был получен в виде белого твердого вещества: 1Н-ЯМР (300 Мгц, ДМСО-d6) 7,54 (1H, д), 7,43 (2H, д), 7,34 (2Н, д), 7,16 (1H, т), 6,82 (1Н, с), 4,31 (2Н, с), 3,14 (2Н, дд), 2,65 (2Н, т). Анализ для C12H14ClN3О2S:
Найдено: С 48,01; Н 4,80; N 14,14%.
Вычислено: С 48,08; Н 4,71; N 14,02%.
ПРИМЕР 26
N-[3-(4(5)-имидазоил)пропил]-(4-хлорфенил)метансульфонамид
Стадия а: N-[3-(1-(трифенилметил)имидазоил-4-ил)пропил] -(4-хлорфенил)метансульфонамид. 4-(3-аминопропил)-1-(трифенилметил)имидазол (стадия b примера 14) (593 мг, 1,61 ммоль) подвергали реакции с (4-хлорфенил)метансульфонилхлоридом (полученным, в основном, как описано в стадии (а) примера 4) (545 мг, 2,42 ммоль) в присутствии триэтиламина (270 мкл, 1,94 ммоль) способом, описанным в стадии (а) примера 9. Таким образом, был выделен продукт в виде бесцветной пены (489 мг, 62%).
Стадия b: продукт стадии а деблокировали способом, описанным в стадии d примера 17, и выделяли целевое соединение (Rf 0,17; аммиак (880)/метанол/дихлорметан=1:10:90) в виде белого твердого вещества с количественным выходом: 1Н-ЯМР (300 Мгц, ДМСО-d6) 11,77 (1Н, шир.с), 7,50 (1Н, с), 7,43 (2Н, д), 7,37 (2Н, д), 7,14 (1H, т), 6,72 (1Н, с), 4,32 (2Н, с), 2,92 (2Н, дд), 2,46 (2Н, т), 1,70 (2Н, м). Анализ для C13H16ClN3O2S:
Найдено: С 49,54; Н 5,38; N 13,12%.
Вычислено: С 49,76; Н 5,14; N 13,39%.
ПРИМЕР 27
N-[3-(4(5)-имидазоил)пропил]бензолсульфонамид
Целевое соединение получали методом, описанным в примере 26, с использованием бензолсульфонилхлорида в качестве субстрата в стадии а. Продукт (Rf 0,16; аммиак (880)/метанол/дихлорметан=1:10:90); от четырех стадий получали в виде белого твердого вещества 1Н-ЯМР (300 Мгц, CDCl3) 7,86 (2Н, д), 7,54 (4Н, м), 6,76 (1H, с), 3,04 (2Н, т), 2,67 (2Н, т), 1,81 (2Н, квинт.). FAB-MC; [М++Н] 266; точный анализ масс-спектра для C12H16N3O2S:
Найдено: 266,0936.
Вычислено: 266,0963.
Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане.
ПРИМЕР 28
N-[3-(4(5)-имидазоил)пропил]-2-нафталинсульфонамид
Целевое соединение получали методом, описанным в примере 26, с использованием 2-нафталинсульфонилхлорида в качестве субстрата в стадии а. Продукт (Rf 0,17; аммиак (880)/метанол/дихлорметан=1:10:90) от четырех стадий получали в виде белого твердого вещества: 1Н-ЯМР (300 Мгц, ДМСО-d6) 8,40 (1Н, с), 8,10 (2Н, т), 8,02 (1Н, д), 7,74 (4Н, м), 7,44 (1Н, с), 6,62 (1Н, с), 2,78 (2Н, дд), 2,43 (2Н, т), 1,62 (2Н, квинт.). Анализ для соединения C16H17N3O2S:
Найдено: С 60,69; H 5,51; N 13,18%.
Вычислено: С 60,93; Н 5,43; N 13,32%.
ПРИМЕР 29
N-[7-(4(5)-имидазоил)гептил]-2-нафталинсульфонамид
Стадия а: 2-(трет-бутилдиметилсилил) -1-(N,N-диметилсульфамоил)имидазол. Раствор 1-(N, N-диметилсульфамоил)имидазола1 (4,48 г, 25,6 ммоль) в сухом тетрагидрофуране (100 мл) охлаждали в атмосфере аргона до -78oС. Затем в течение 30 минут добавляли н-бутиллитий (1,5М в гексане)(18,0 мл, 27,0 ммоль), и раствор перемещивали еще 30 минут. К полученному коричневому раствору в течение 15 минут добавляли раствор трет-бутилдиметилсилилхлорида в сухом тетрагидрофуране (20 мл). Раствор нагревали до комнатной температуры и перемешивали в течение 24 часов. После добавления насыщенного раствора хлорида аммония (100 мл) и диэтилового эфира (100 мл), эфирный экстракт промывали насыщенным солевым раствором, и сушили сульфатом магния. После фильтрации фильтрат упаривали, и получали маслянистый остаток, который очищали с помощью колоночной флэш-хроматографии (двуокись кремния; элюент: этилацетат) и получали продукт виде твердого вещества янтарного цвета (6,97 г, 94%).
Стадия b: 5-(7-бромгептил)-2-(трет-бутилдиметилсилил)-1-(N, N-диметилсульфамоил)имидазол. Раствор продукта стадии (а) (2,50 г, 8,64 ммоль) в сухом тетрагидрофуране (30 мл) охлаждали в атмосфере аргона до -78oС. Затем добавляли н-бутиллитий (1,5М в гексане) (8,50 мл, 12,7 ммоль) в течение 15 минут, и раствор перемешивали еще 30 минут. Затем добавляли в течение 10 минут раствор 1,7-дибромгептана (4,60 г, 17,3 ммоль) в сухом тетрагидрофуране (6 мл). Раствор перемешивали в течение 30 минут, нагревали до комнатной температуры и перемешивали в течение еще 18 часов. После добавления насыщенного раствора хлорида аммония (50 мл) и этилацетата (50 мл), органический экстракт промывали насыщенным солевым раствором и сушили сульфатом натрия. После фильтрации фильтрат упаривали и очищали с помощью колоночной флэш-хроматографии (двуокись кремния; 20% этилацетат/гексан) с получением продукта (Rf 0,55) в виде белого твердого вещества (2,48 г, 62%).
Стадия с: 1-(N, N-диметилсульфамоил)-5-(7-фталимидогептил) имидазол. К раствору продукта стадии b (2,10 г, 4,50 ммоль) в сухом N,N-диметилформамиде (10 мл) в атмосфере аргона добавляли фталимид калия (1,67 г, 9,00 ммоль). Смесь перемешивали и нагревали при 100oС в течение 18 часов, а затем охлаждали до комнатной температуры. После добавления воды (75 мл) смесь экстрагировали дихлорметаном (3х40 мл). Объединенные экстракты упаривали, а остаток растворяли в этилацетате (75 мл), и раствор пять раз промывали насыщенным солевым раствором. После выпаривания растворителя остаток очищали с помощью колоночной флэш-хроматографии (двуокись кремния; этилацетат), и выделяли продукт в виде желтого масла (1/75 г, 93%).
Стадия d: 5-(7-аминогептил)-1-(N,N-диметилсульфамоил)имидазол. Продукт стадии (с) деблокировали способом, описанным в стадии (b) примера 17.
Стадия е: целевое соединение получали методом, описанным в примере 6, с использованием 5-(7-аминогептил)-1-(N,N-диметилсульфамоил)имидазола в качестве субстрата в стадии а. Продукт (Rf 0,38; аммиак (880)/метанол/дихлорметан= 1: 10: 90) от двух стадий получали в виде белого твердого вещества: 1H-ЯМР (300 Мгц, ДМСО-d6) 8,41 (1Н, с), 8,13 (2Н, дд), 8,02 (1Н, дд), 7,80 (1Н, дд), 7,67 (3Н, м), 7,45 (1Н, д), 6,64 (1Н, с), 2,75 (2Н, дд), 2,38 (2Н, т), 1,43 (2Н, м), 1,32 (2Н, м), 1,13 (6Н, м).
ПРИМЕР 30
N-[8-(4(5)-имидазоил)октил]-2-нафталинсульфонамид
Целевое соединение получали способом, описанным в примере 6, с использованием 5-(8-аминооктил)-1-(N,N-диметилсульфамоил)имидазола1 в качестве субстрата в стадии а. Продукт (Rf 0,39; аммиак (880)/метанол/дихлорметан=1:10: 90) от двух стадий получали в виде белого твердого вещества: 1Н-ЯМР (300 Мгц, ДМСО-d6) 11,72 (1Н, шир.с), 8,40 (1Н, д), 8,13 (2Н, дд), 8,02 (1Н, дд), 7,80 (1Н, дд), 7,65 (3Н, м), 7,45 (1Н, д), 6,65 (1Н, д), 2,75 (2Н, дд), 2,41 (2Н, т), 1,45 (2Н, квинт.), 1,32 (2Н, квинт.), 1,12 (8Н, м).
ПРИМЕР 31
N-[10-(4(5)-имидазоил)децил]-3-нафталинсульфонамид
Целевое соединение получали способом, описанным в примере 6, с использованием 5(10-аминодецил)-1-(N,N-диметилсульфамоил)имидазола1 в качестве субстрата в стадии а. Продукт (Rf 0,33; аммиак (880)/метанол/дихлорметан=1:10: 90) от двух стадий получали в виде белого твердого вещества: 1H-ЯМР (300 Мгц, ДМСО-d6) 8,40 (1H, д), 8,12 (2Н, дд), 8,02 (1Н, дд), 7,80 (1H, дд), 7,66 (3Н, м), 7,46 (1Н, д), 6,66 (1H, с), 2,75 (2Н, дд), 2,44 (2Н, т), 1,47 (2Н, м), 1,31 (2Н, м), 1,10 (12Н, м).
ПРИМЕР 32
N-[7-(4(5)-имидазоил)гептил]-(4-хлорфенил)метансульфонамид
Целевое соединение получали способом, описанным в примере 9, с использованием 5-(7-аминогептил)-1-(N,N-диметилсульфамоил)имидазола (стадия d примера 29) в качестве субстрата в стадии а. Продукт (Rf 0,30; аммиак (880)/метанол/дихлорметан= 1: 10: 90) от двух стадий получали в виде белого твердого вещества: 1H-ЯМР (300 Мгц, ДМСО-d6) 7,56 (1H, с), 7,42 (2Н, д), 7,37 (2Н, д), 7,04 (1H, т), 6,72 (1H, с), 4,30 (2Н, с), 2,85 (2Н, дд), 2,48 (2Н, м), 1,53 (2Н, м), 1,37 (2Н, м), 1,24 (6Н, м).
ПРИМЕР 33
N-[8-(4(5)-имидазоил)октил]-(4-хлорфенил)метансульфонамид
Стадия а: 5-(8-бромоктил)-2-(трет-бутилдиметилсилил)-1-(N, N-диметилсульфамоил)имидазол. Раствор 2-(трет-бутилдиметилсилил)-1-(N, N-диметилcульфамоил)имидазола (стадия а примеpa 29) (2,62 г, 9,05 ммоль) в сухом тетрагидрофуране (30 мл) охлаждали в атмосфере аргона до -78oС. Затем добавляли н-бутиллитий (1,5М в гексане) (7,25 мл, 10,9 ммоль) в течение 15 минут, и раствор перемешивали еще 30 минут. После этого добавляли 1,8-дибромоктан (2,55 мл, 13,6 ммоль) в сухом тетрагидрофуране (5 мл) в течение 10 минут. Раствор перемешивали в течение 2 часов, нагревали до комнатной температуры, и перемешивали в течение 18 часов. После добавления насыщенного раствора хлорида аммония (30 мл) и диэтилового эфира (30 мл), эфирный экстракт промывали насыщенным солевым раствором и сушили сульфатом натрия. После фильтрации фильтрат упаривали, и очищали с помощью колоночной флэш-хроматографии (двуокись кремния; 20% этилацетат/гексан) с получением продукта (Rf 0,43) в виде белого твердого вещества (2,36 г, 54%).
Стадия b: 2-(трет-бутилдиметилсилил)-1-(N,N-диметилсульфамоил)-5-(8-фталимидооктил)имидазол (А) и 1-(N,N-диметилсульфамоил)-5-(8-фталимидооктил)имидазол (В).
К раствору продукта стадии (а) (2,39 г, 4,97 ммоль) в сухом диметилформамиде (16 мл) в атмосфере аргона добавляли фталимид калия (1,84 г, 9,95 ммоль). Смесь перемешивали и нагревали при 100oС в течение 18 часов, а затем охлаждали до комнатной температуры. После добавления воды (80 мл), смесь экстрагировали дихлорметаном (3х40 мл). Объединенные экстракты упаривали, остаток растворяли в этилацетате (80 мл), и раствор четыре раза промывали насыщенным солевым раствором. После выпаривания растворителя остаток очищали с помощью колоночной флэш-хроматографии (двуокись кремния; этилацетат), в результате чего получали соединение (A) (Rf 0,72) в виде масла янтарного цвета (639 мг, 23%) и соединение (В) (Rf 0,27) в виде маслянистого твердого вещества (1,56 г, 73%).
Стадия с: 5-(8-аминооктил)-2-(трет-бутилдиметилсилил)-1-(N,N-диметилсульфамоил)имидазол. Соединение А стадии b деблокировали методом, описанным в стадии b примера 17.
Стадия d: целевое соединение получали способом, описанным в примере 9, с испозованием 5-(8-аминооктил)-2-(трет-бутилдиметилсилил)-1-(N, N-диметилсульфамоил)имидазола, продукта предыдущей реакции, в качестве субстрата в стадии а. Продукт (Rf 0,42; аммиак (880)/метанол/дихлорметан=1:10:90) был получен в виде белого твердого вещества: 1H-ЯМР (300 Мгц, ДМСО-d6) 7,47 (1Н, д), 7,43 (2Н, д), 7,36 (2Н, д), 7,03 (1Н, т), 6,68 (1Н, с), 4,30 (2H, с), 2,85 (2Н, дд), 2,46 (2Н, т), 1,53 (2Н, м), 1,37 (2Н, м), 1,23 (8Н, м). Анализ для С18Н26СIN3O2S:
Найдено: С 55,99, Н 7,04; N 10,67%.
Вычислено: С 56,31, H 6,83; N 10,94%.
ПРИМЕР 34
N-[10-(4(5)-имидазоил)децил]-(4-хлорфенил)метансульфонамид
Целевое соединение получали способом, описанным в примере 9, с использованием 5-(10-аминодецил)-1-(N, N-диметилсульфамоил)имидазола1 в качестве субстрата в стадии а. Продукт (Rf 0,37; аммиак (880)/метанол/дихлорметан=1: 10: 90) двух стадий получали в виде белого твердого вещества: 1Н-ЯМР (300 Мгц, ДМСО-d6), 7,46 (1Н, дд), 7,43 (2Н, дд), 7,37 (2Н, д), 7,04 (1Н, т), 6,67 (1Н, c), 4,31 (2Н, с), 2,85 (2Н, дд), 2,45 (2Н, т), 1,53 (2Н, м), 1,37 (2H, м), 1,21 (12Н, м).
ПРИМЕР 35
N-[2-(4(5)-имидазоил]этил]-N'-фенилметилсульфамид
4-(2-аминоэтил)-1-(тpифeнилмeтил)имидaзoл (стадия b примера 17) преобразовывали N-2-[1-(трифенилметил)имидазол-4-ил]этил-N'-трет-бутоксикарбонил-сульфамид способом, описанным в стадии а примера 12. Доследующую реакцию алкилирования с бензилбромидом проводили, в основном, как описано в стадии b примера 12, и получали N-[2-(1-(трифенилметил)имидазол-4-ил)этил]-N'-трет-бутоксикарбонил-N'-фенилметилсульфамид. Конечную реакцию снятия защиты проводили способом, описанным в стадии b примера 17, и получали целевое соединение (Rf 0,21; аммиак (880)/метанол/дихлорметан=1:10:90) в виде бесцветного масла: 1H-ЯМР (300 Мгц, ДMСО-d6) 7,50 (1Н, с), 7,30 (6Н, м), 6,95 (1Н, т), 6,78 (1Н, с), 3,96 (2Н, с), 3,03 (2Н, дд), 2,65 (2Н, т). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане. Анализ для C16H20N4O6S:
Найдено: С 48,05; Н 5,10; N 13,87%.
Вычислено: С 48,48; Н 5,09; N 14,13%.
ПРИМЕР 36
N-[3-(4(5)-имидазоил)пропил]-N'-фенилметилсульфамид
Целевое соединение получали способом, описанным в примере 35, с использованием 4-(3-аминопропил)-1-(трифенилметил)имидазола (пример 14, стадия b) в качестве исходного субстрата, и это соединение представляло собой белое твердое вещество (Rf 0,10; аммиак (880) /метанол/дихлорметан=1:10:90): 1Н-ЯМР (300 Мгц, ДМСО-d6) 11,75 (1Н, шир.с), 7,49 (1Н, с), 7,30 (5Н, м), 7,25 (1Н, м), 6,96 (1Н, т), 6,71 (1H, с), 3,98 (2Н, с), 2,82 (2Н, м), 2,48 (2Н, м), 1,72 (2Н, м). Анализ для С13Н18N4O2S:
Найдено: С 53,03; H 6,19; N 18,89%.
Вычислено: С 53,04; H 6,16; N 19,03%.
ПРИМЕР 37
N-[2-(4(5)-имидазоил)этил]-N'-(4-хлорфенил) метилсульфамид
Целевое соединение получали способом, описанным в примере 35, с использованием 4-хлорбензилбромида в стадии алкилирования, и это соединение представляло белое твердое вещество (Rf 0,20; аммиак (880)/метанол/дихлорметан= 1: 10: 90): 1H-ЯМР (300 Мгц, ДМСО-d6) 11,75 (1H, шир. с. ), 7,51 (1Н, с), 7,36 (5Н, м), 6,99 (1Н, т), 6,75 (1Н, с), 3,96 (2Н, с), 3,01 (2Н, дд), 2,65 (2Н, т). Анализ для C12H15ClN4O2S:
Найдено: С 45,47; H 4,83; N 17,93%.
Вычислено; С 45,79; H 4,80; N 17,80%.
ПРИМЕР 38
N-[7-(4(5)-имидазоил)гептил]-N'-(4-хлорфенил)метилсульфамид
Целевое соединение получали способом, описанным в примере 12, с использованием в качестве субстрата в стадии (а) 5-(7-аминогептил)-1-(N,N-диметилсульфамоил)имидазола (стадия d примера 29). Продукт (Rf 0,28; аммиак (880) /метанол/дихлорметан=1:10:90) от трех стадий был получен в виде белого твердого вещества: 1Н-ЯМР (300 Мгц, ДМСО-d6), 7,45 (1Н, с), 7,36 (5Н, м), 6,84 (1Н, т), 6,67 (1Н, с), 3,98 (2Н, с), 2,79 (2Н, дд), 2,46 (2Н, т), 1,53 (2Н, м), 1,37 (2Н, м), 1,23 (6Н, м). Анализ для C17H25ClN4O2S:
Найдено: С 53,17; Н 6,62; N 14,53%.
Вычислено: С 53,05; Н 6,55; N 14,56%.
ПРИМЕР 39
N-[8-(4(5)-имидазоил)октил]-N'-(4-хлорфенил)метилсульфамид
Целевое соединение получали способом, описанным в примере 12, с использованием 5-(8-аминооктил)-1-(N,N-диметилсульфамоил)имидазола1 в качестве субстрата в стадии а. Продукт (Rf 0,30; аммиак (880)/метанол/дихлорметан=1:10: 90) от трех стадий получали в виде белого твердого вещества: 1Н-ЯМР (300 Мгц, ДМСО-d6) 7,45 (1Н, с), 7,36 (5Н, м), 6,82 (1Н, т), 6,67 (1Н, с), 3,98 (2Н, д), 2,74 (2Н, дд), 2,45 (2Н, т), 1,55 (2Н, т), 1,37 (2Н, м), 1,23 (8H, м).
ПРИМЕР 40
N-[10-(4(5)-имидазоил)децил]-N'-(4-хлорфенил)метилсульфамид
5-(10-аминодецил)-2-(трет-бутилдиметилсилил)-1-(N, N-диметилсульфамоил)имидазол получали способом, описанным в примере 33, стадии a, b и с, с использованием 1,10-дибромдекана в качестве алкилирующего реактива в стадии а. Этот амин превращали в целевое соединение способом, описанным в примере 12. Продукт (Rf 0,41; аммиак (880)/метанол/дихлорметан=1:10: 90) был выделен в виде белого твердого вещества: 1Н-ЯМР (300 Мгц, ДМСО-d6) 7,45 (1Н, с), 7,35 (5Н, м), 6,82 (1Н, т), 6,66 (1Н, с), 3,98 (2Н, д), 2,74 (1Н, дд), 2,45 (2Н, т), 1,53 (2Н, м), 1,37 (2Н, м), 1,21 (12Н, м). Анализ для C20H31ClN4O2S:
Найдено: С 55,97; Н 7,55; N 12,88%.
Вычислено: С 56,26; Н 7,32; N 13,12%.
ПРИМЕР 41
N-(4-(4(5)-имидазоил)бутил]-N'-(3,4-дихлорфенил)метилсульфамид
Целевое соединение получали способом, описанным в примере 12, с использованием 5-(4-аминобутил)-1-(N,N-диметилсульфамоил)имидазола1 в качестве субстрата в стадии а и 3,4-дихлорбензилбромида в стадии b. Продукт (Rf 0,26; аммиак (880)/метанол/дихлорметан= 1: 10:90) получали в виде белого твердого вещества: 1Н-ЯМР (300 Мгц, СDСl3) 7,51 (1Н, д), 7,42 (2Н, м), 7,19 (1Н, дд), 6,76 (1Н, д), 4,17 (2Н, с), 3,06 (2Н, т), 2,62 (2Н, т), 1,68 (2Н, квинт.), 1,59 (2Н, квинт. ). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане.
ПРИМЕР 42
N-[4-(4(5)-имидазоил)бутил]-N'-(3-хлорфенил)метилсульфамид
Целевое соединение получали способом, описанным в примере 12, с использованием 5-(4-аминобутил)-1-(N,N-диметилсульфамоил)имидазола1 в качестве субстрата в стадии (а) и 3-хлорбензилбромида в стадии b. Продукт (Rf 0,26; аммиак (880)/метанол/дихлорметан= 1: 10: 90) получали в виде белого твердого вещества: 1Н-ЯМР (300 Мгц, ДМСО-d6) 11,73 (1Н, шир.с), 7,47 (1Н, д), 7,35 (5Н, м), 6,88 (1Н, т), 6,69 (1Н, с), 4,00 (2Н, д), 2,79 (2Н, дд), 2,45 (2Н т), 1,55 (2Н, м), 1,42 (2Н, м). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане.
ПРИМЕР 43
N-[4-(4(5)-имидазоил)бутил]-N'-(2-хлорфенил)метилсульфамид
Целевое соединение получали способом, описанным в примере 12, с использованием 5-(4-аминобутил)-1-(N,N-диметилсульфамоил)имидазола1 в качестве субстрата в стадии (а) и 2-хлорбензилбромида в стадии b. Продукт (Rf 0,26; аммиак (880)/метанол/дихлорметан= 1:10:90) был получен в виде белого твердого вещества: 1H-ЯМР (300 Мгц, ДМСО-d6) 11,75 (1Н, шир.с), 7,53 (1Н, д), 7,47 (1Н, с), 7,35 (4Н, м), 6,95 (1Н, т), 6,68 (1Н, с), 4,09 (2Н, д), 2,83 (2Н, дд), 2,45 (2Н, т), 1,55 (2Н, м), 1,46 (2Н, м). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане.
ПРИМЕР 44
N-[4-(4(5)-имидазоил)бутил]-N'-(4-иодфенил)метилсульфамид
Целевое соединение получали способом, описанным в примере 12, с использованием 5-(4-аминобутил)-1-(N,N-диметилсульфамоил)имидазола1 в качестве субстрата в стадии (а) и 4-иодбензилбромида в стадии (b). Продукт (Rf 0,24; аммиак (880)/метанол/дихлорметан= 1: 10:90) получали в виде белого твердого вещества: 1H-ЯМР (300 Мгц, ДМСО-d6) 11,75 (1Н, шир.с), 7,67 (2Н, д), 7,49 (1Н, с), 7,33 (1Н, т), 7,14 (2Н, д), 6,85 (1Н, т), 6,70 (1Н, с), 3,94 (2Н, д), 2,79 (2Н, дд), 2,46 (2Н, т), 1,55 (2Н, м), 1,46 (2Н, м). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане.
ПРИМЕР 45
N-[4-(4(5)-имидазоил)бутил]-N'-(4-бромфенил)метилсульфамид
Стадия а: N-[4-(1-(N",N"-диметилсульфамоил)имидазол-4-ил)бутил]-N'-трет-бутоксикарбонилсульфамид. 5-(4-аминобутил)-1-(N, N-диметилсульфамоил)имидазол1 превращали в нужный продукт способом, описанным в стадии а примера 12.
Стадия b: N-[4-(1-(N",N"-диметилсульфамоил)имидазол-4-ил)бутил]-N'-(4-бромфенил)метил-N'-трет-бутоксикарбонилсульфамид (А). Продукт стадии а (500 мг, 1,27 ммоль) подвергали реакции с 4-бромбензилбромидом, как описано в стадии b примера 12. Неочищенную продуктовую смесь очищали с помощью колоночной флэш-хроматографии (двуокись кремния; 50% этилацетат/дихлорметан) и получали продукт (A) (Rf 0,37) в виде желтого масла (267 мг, 35%) и N,N'-ди-[(4-бромфенил)метил] -N-[4-(1-(N", N"-диметилсульфамоил)имидазол-4-ил)бутил]-N'-трет-бутоксикарбонилсульфамид (В) (Rf 0,56) (220 мг, 23%).
Стадия с: продукт (А) от стадии b деблокировали способом, описанным в примере 12 (стадия с), и получали целевое соединение (Rf 0,28; аммиак (880)/метанол/дихлорметан= 1: 10:90) в виде белого твердого вещества: 1Н-ЯМР (300 Мгц, ДМСО-d6) 7,50 (3Н, м), 7,35 (1Н, т), 7,28 (2Н, д), 6,87 (1Н, т), 6,71 (1Н, с), 3,96 (2Н, д), 2,78 (2Н, дд), 2,46 (2Н, т), 1,55 (2Н, м), 1,42 (2Н, м). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане. Анализ для С18Н23ВrN4O6S•1,5 Н2O:
Найдено: С 40,66; Н 4,90; N 10,67%.
Вычислено: С 40,76; Н 4,94; N 10,56%.
ПРИМЕР 46
N-[4-(4(5)-имидазоил)бутил]-N'-(4-фторфенил)метилсульфамид
Целевое соединение получали способом, описанным в примере 12, с использованием 5-(4-аминобутил)-1-(N,N-диметилсульфамоил)имидазола1 в качестве субстрата в стадии а и 4-фторбензилбромида в стадии b. Продукт (Rf 0,26; аммиак (880)/метанол/дихлорметан= 1: 10:90) получали в виде белого твердого вещества: 1H-ЯМР (300 Мгц, ДМСО-d6) 11,67 (1Н, шир.с), 7,46 (1Н, с), 7,35 (3Н, м), 7,14 (2Н, м), 6,85 (1Н, т), 6,68 (1Н, с), 3,97 (2Н, д), 2,79 (2Н, дд), 2,46 (2Н, м), 1,54 (2Н, м), 1,45 (2Н, м). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане. Анализ для C18H23FN4O2S•2,0 Н2О:
Найдено: С 44,92; Н 5,61; N 11,42%.
Вычислено: С 45,18; Н 5,69; N 11,71%.
ПРИМЕР 47
N-[4-(4(5)-имидазоил)бутил]-N'-(4-трифторметил)фенил)-метилсульфамид
Целевое соединение получали способом, описанным в примере 12, с использованием 5-(4-аминобутил)-1-(N,N-диметилсульфамоил)имидазола1 в качестве субстрата в стадии а и 4-(трифторметил)бензилбромида в стадии b. Продукт (Rf 0,26; аммиак (880)/метанол/дихлорметан= 1:10:90) от трех стадий получали в виде белого твердого вещества: 1Н-ЯМР (300 Мгц, ДМСО-d6) 11,70 (1Н, шир.с), 7,68 (2Н, д), 7,54 (2Н, д), 7,44 (2Н, м), 6,90 (1Н, т), 6,68 (1Н, с), 4,09 (2Н, д), 2,80 (2Н, дд), 2,45 (2Н, т), 1,55 (2Н, м), 1,45 (2Н, м). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане. Анализ для С19Н23F3N4O6S•2,0 Н2О:
Найдено: С 43,17; Н 4,99; N 10,88%.
Вычислено: С 43,18; H 5,15; N 10,60%.
ПРИМЕР 48
N-[4-(4(5)-имидазоил)бутил]-N'-(4-метоксифенил)метилсульфамид
Целевое соединение получали способом, описанным в примере 12, с использованием [5-(4-аминобутил)-1-(N, N-диметилсульфамоил)имидазола1 в качестве субстрата в стадии а и 4-метоксибензилбромида в стадии b. Продукт (Rf 0,26; аммиак (880)/метанол/дихлорметан= 1: 10: 90) получали в виде белого твердого вещества: 1H-ЯМР (300 Мгц, ДМСО-d6) 11,70 (1Н, шир.с), 7,47 (1Н, с), 7,22 (2Н, д), 7,18 (1Н, т), 6,87 (2Н, д), 6,79 (1Н, т), 6,68 (1Н, с), 3,90 (2Н, д), 3,72 (3Н, с), 2,79 (2Н, дд), 2,45 (2Н, т), 1,55 (2Н, м) 1,44 (2Н, м). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане. Анализ для C19H26N4O7S:
Найдено: С 50,19; Н 5,80; N 12,36%.
Вычислено: С 50,21; H 5,77; N 12,33%.
ПРИМЕР 49
N-[4-(4(5)-имидазоил)бутил]-N'-(4-бифенил)метилсульфамид
5-(4-аминобутил)-2-(трет-бутилдиметилсилил)-1-(N, N-диметилсульфамоил)имидазол выделяли в виде побочного продукта во время получения 5-(4-аминобутил)-1-(N,N-диметилсульфамоил)имидазола1 способом, аналогичным описанному в примере 33. Его превращали в целевое соединение способом, описанным в примере 12, с использованием (4-хлорметил)бифенила в качестве субстрата в стадии b. В стадию b, кроме того, вносили изменения, заключающиеся в нагревании реакционой смеси при 50oС в течение 2 часов с последующей обработкой. Продукт (Rf 0,28; аммиак (880)/метанол/дихлорметан=1:10:90) получали в виде белого твердого вещества: 1H-ЯМР (300 Мгц, ДМСО-d6) 11,75 (1Н, шир.с), 7,63 (4Н, м), 7,42 (5Н, м), 7,33 (2Н, м), 6,86 (1Н, т), 6,69 (1Н, с), 4,03 (2Н, д), 2,82 (2Н, дд), 2,46 (2Н, т), 1,54 (2Н, м), 1,45 (2Н, м). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане. Анализ для С24Н28N4O6S•1,5 Н2О:
Найдено: С 54,71; Н 5,87; N 10,80%.
Вычислено: С 54,63; H 5,92; N 10,61%.
ПРИМЕР 50
N-[4-(4(5)-имидазоил)бутил]-N'-2-нафтилметилсульфамид
Целевое соединение получали способом, описанным в примере 12, с использованием (5-(4-аминобутил)-2-(трет-бутилдиметилсилил)-1-(N, N-диметилсульфамоил)имидазола (пример 49) в качестве субстрата в стадии (а) и 2-бромметилнафталина в стадии b. Продукт (Rf 0,24; аммиак (880)/метанол/дихлорметан=1:10:90) получали в виде белого твердого вещества: 1H-ЯMP (300 Мгц, ДМСО-d6) 11,70 (1Н, шир.с), 7,85 (4Н, м), 7,48 (4Н, м), 7,40 (1Н, т), 6,88 (1Н, т), 6,71 и 6,58 (1Н, 2 х шир.с), 4,16 (2Н, д), 2,83 (2Н, дд), 2,43 (2Н, м), 1,53 (2Н, м), 1,45 (2Н, м). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане.
Найдено: С 55,49; Н 5,56; N 11,59%.
Вычислено: С 55,68; Н 5,52; N 11,81%.
Анализ для C22H26N4O6S.
ПРИМЕР 51
N-[4-(4(5)-имидазоил)бутил]-N'-циклогексилметилсульфамид
Стадия a: (Z)-4-[4-(1,3-диоксолан-2-ил)бут-2-енил]-1-(трифенилметил)имидазол. Суспензию бромида [2-(1,3-диоксолан-2-ил)этил] трифенилфосфонил (48,54 г, 109 ммоль) в сухом тетрагидрофуране (500 мл) охлаждали в атмосфере аргона до -20oС. Затем добавляли по каплям н-бутиллитий (1,6М в гексане) (68,3 мл, 109 ммоль), и раствор перемешивали в течение 1 часа. После медленного добавления по каплям раствора [1-(трифенилметил)имидазол-4-ил] карбальдегида4 (36,80 г, 109 ммоль) в сухом тетрагидрофуране (500 мл), реакционную смесь перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь концентрировали в вакууме, добавляли воду, и смесь фильтровали через слой целита. Фильтрат экстрагировали дихлорметаном (2х500 мл) и объединенные экстракты сушили сульфатом магния. После фильтрации и упаривания получали желтое масло. В результате колоночной флэш-хроматографии (двуокись кремния; 10-20% этилацетат/гексан) был получен продукт в виде желтого масла (19,73 г, 42%).
Стадия b: 4-[4-(1,3-диоксолан-2-ил)бутил] -1-(трифенилметил)имидазол. Раствор продукта стадии а в этаноле гидрировали в присутствии каталитического количества 10% палладия-на-угле при атмосферном давлении и температуре в течение 18 часов. Продукт был получен в виде бесцветного масла с количественным выходом.
Стадия с: 4-[1-(трифенилметил)имидазол-4-ил]бутан-1аль. Суспензию продукта стадии b (19,8 г, 46,6 ммоль) в смеси ацетона (300 мл) и 2М хлористоводородной кислоты (50 мл) перемешивали при комнатной температуре в течение 20 часов. Смесь нейтрализовали бикарбонатом натрия, фильтровали, и фильтрат экстрагировали дихлорметаном (3х100 мл). Объединенные экстракты сушили сульфатом магния, фильтровали, и упаривали с получением продукта в виде бесцветного масла (16,1, 91%).
Стадия d: 4-[1-(трифенилметил)имидазол-4-ил]бутан-1-ол. Раствор продукта стадии (16,1 г, 42,4 ммоль) в этаноле (300 мл) охлаждали в атмосфере аргона до 0oС. После добавления боргидрида натрия (1,57 г, 42,4 ммоль) смесь перемешивали в течение 4 часов, и осторожно гасили путем добавления насыщенного хлорида аммония. Смесь экстрагировали дихлорметаном (3х100 мл). Объединенные экстракты сушили сульфатом магния, фильтровали, и упаривали с образованием белого твердого вещества, которое растворяли в 5% метаноле/дихлорметане, и осаждали диэтиловым эфиром. Таким образом был получен продукт в виде бесцветного кристаллического твердого вещества (9,34 г, 58%).
Стадия е: N-трет-бутоксикарбонил-N'-циклогексилметилсульфамид. Циклогексилметиламин преобразовывали в нужный продукт, используя, в основном, тот же метод, описанный в стадии (а) примера 12.
Стадия f: N-трет-бутоксикарбонил-N-[4-[1-(трифенилметил)имидазол-4-ил] бутил] -N'-циклогексилметилсульфамид. К раствору продукта стадии d (764 мг, 2,00 ммоль), продукта стадии е (642 мг, 2,20 ммоль) и трифенилфосфина (576 мг, 2,20 ммоль) в сухом тетрагидрофуране (20 мл) в атмосфере аргона в течение 10 минут добавляли раствор диэтилазодикарбоксилата (383 мг, 2,20 ммоль) в сухом тетрагидрофуране (5 мл). Смесь перемешивали в течение 2 часов, а затем растворитель выпаривали, и остаток очищали с помощью колоночной флэш-хроматографии (двуокись кремния; 30% этилацетат/дихлорметан). Таким образом был выделен продукт в виде бесцветной пены (800 мг, 60%).
Стадия g: раствор продукта стадии f (800 мг, 1,20 ммоль) в смеси этанола (15 мл) и 2М хлористоводородной кислоты (5 мл) нагревали с обратным холодильником в течение 2 часов. Растворитель выпаривали и остаток очищали с помощью колоночной флэш-хроматографии (двуокись кремния; аммиак (880)/метанол/дихлорметан= 1: 10: 90). Таким образом было выделено целевое соединение (Rf 0,28) в виде белого твердого вещества (184 мг, 49%. Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане: 1H-ЯМР (300 Мгц, ДМСО-d6) 8,87 (1H, с), 7,36 (1H, с), 6,70 (1Н, т), 6,02 (1H, с), 2,80 (2Н, дд), 2,61 (4Н, м), 1,63 (7Н, м), 1,44 (3Н, м), 1,13 (3Н, м), 0,83 (2Н, м).
ПРИМЕР 52
N-[4-(4(5)-имидазоил)бутил]-N'-адамант-1-илметилсульфамид
Целевое соединение получали способом, описанным в примере 51, с использованием 1-адамант-1-илметиламина в качестве субстрата в стадии е. Продукт (Rf 0,32; аммиак (880)/метанол/дихлорметан=1:10:90) получали в виде белого твердого вещества: 1H-ЯМР (300 Мгц, ДМСО-d6) 11,66 (1H, шир.с), 7,45 (1H, с), 6,70 (1H, шир.с), 6,66 (1H, т), 6,57 (1H, т), 2,78 (2Н, м), 2,48 (4Н, м), 1,91 (3Н, с), 1,56 (8Н, м), 1,44 (8Н, м). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане.
ПРИМЕР 53
N-[3-(4(5)-имидазоил)пропил]-2-(4-хлорфенил)этансульфонамид
Стадия а: N-[3-(1-(трифенилметил)имидазоил-4-ил)пропил] -2-(4-хлорфенил)этансульфонамид. 4-(3-аминопропил)-1-(трифенилметил)имидазол (пример 14, стадия b) (500 мг, 1,01 ммоль) и 2-(4-хлорфенил)этансульфонилхлорид (полученный, в основном, как описано в стадии а примера 4) (266 мг, 1,01 ммоль) вместе подвергали реакции в присутствии триэтиламина (155 мкл, 1,94 ммоль) способом, описанным в стадии а примера 9. Продукт был выделен в виде бесцветной пены (494 мг, 86%).
Стадия b: продукт стадии а (494 мг, 0,87 ммоль) деблокировали способом, описанным в стадии d примера 17, и целевое соединение получали в виде белого твердого вещества (154 мг, 54%): 1H-ЯМР (300 Мгц, МеОН-d4) 7,66 (1Н, с), 7,38 (2Н, д), 7,63 (2Н, д), 6,90 (1Н, с), 3,37 (2Н, м), 3,14 (4Н, м), 2,74 (2Н, т), 1,94 (2Н, квинтет). Анализ для C14H18ClN3O2S:
Найдено: С 51,25; H 5,61: N 12,72%.
Вычислено: С 51,29; Н 5,53; N 12,82%.
ПРИМЕР 54
N-[5-(4(5)-имидазоил)пентил]-2-(4-хлорфенил)этансульфонамид
Стадия а: N-[5-(2-(трет-бутилдиметилсилил)-1-(N', N'-диметилсульфамоил)имидазол-4-ил)пентил] -2-(4-хлорфенил)этансульфонамид. 5-(5-аминопентил)-2-(трет-бутилдиметилсилил)-1-(N, N-диметилсульфамоил)имидазол был выделен в виде побочного продукта в процессе получения 5-(5-аминопентил)-1-(N, N-диметилсульфамоил)имидазола1, как описано в примере 33. Этот продукт подвергали реакции 2-(4-хлорфенил)этансульфонилхлоридом (полученным, в основном, как описано в стадии а примера 4) способом, описанным в стадии а примера 9. Продукт был получен в виде желтого масла.
Стадия b: продукт стадии (а) (494 мг, 0,87 ммоль) деблокировали способом, описанным в стадии (с) примера 12, и получали целевое соединение виде белого твердого вещества (227 мг, 92%): 1H-ЯМР (300 Мгц MеОН-d4) 7,58 (1Н, с), 7,34 (2Н, д), 7,29 (2Н, д), 6,80 (1H, с), 3,31 (2Н, м), 3,09 (4Н, м), 2,63 (2Н, м), 1,70 (2Н, м), 1,60 (2Н, м), 1,44 (2Н, м). Анализ для С16Н22СIN3O2S:
Найдено: С 54,04; H 6,27; N 11,55%.
Вычислено: С 54,00; Н 6,23; N 11,83%.
ПРИМЕР 55
N-[4-(4(5)-имидазоил)бутил]-2-(4-хлорфенил)этансульфонамид
Целевое соединение получали способом, описанным в примере 54, с использованием 5-(4-аминобутил)-1-(N,N-диметилсульфамоил)имидазола1 в качестве аминового компонента в стадии а. Продукт был получен в виде белого твердого вещества: 1H-ЯMP (300 Мгц, СDСl3) 7,56 (1Н, с), 7,30 (2Н, дд), 7,16 (2Н, д), 6,78 (1Н, с), 4,55 (1Н, шир.с), 3,25 (2Н, м), 3,09 (4Н, м), 2,64 (2Н, т), 1,72 (2Н, квинт.), 1,58 (2Н, квинт.). Анализ для C15H20ClN3O2S:
Найдено: С 52,52; Н 5,92; N 12,11%.
Вычислено: С 51,70; Н 5,90; N 12,29%.
ПРИМЕР 56
N-[3-(4(5)-имидазоил)пропил]-N'-(4-хлорфенил)этилсульфамид
Стадия а: N-трет-бутоксикарбонил-N'-2-(4-хлорфенил)этилсульфамид. 2-(4-хлорфенил)этиламин преобразовывали в продукт с использованием, в основном, той же методики, описанной в стадии а примера 12.
Стадия b: N-трет-бутоксикарбонил-N-[3-[1-трифенилметил)имидазол-4-ил] пропил]-N'-2-(4-хлорфенил)этилсульфамид. Продукт стадии а и 3-[1-(трифенилметил)имидазол-4-ил] пропан-1-ол3 вместе подвергали реакции в условиях, описанных в стадии (f) примера 51. Продукт был выделен в виде белой пены.
Стадия с: продукт стадии b деблокировали способом, описанным в стадии (с) примера 12, и получали целевое соединение в виде белого твердого вещества: 1H-ЯМР (300 Мгц, МеОН-d4) 7,58 (1Н, с), 7,28 (4Н, м), 6,82 (1Н, с), 3,20 (2Н, т), 2,86 (4Н, м), 2,64 (2Н, т), 1,83 (2Н, квинт). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане. Анализ для С18Н23СIN4О2S:
Найдено: С 46,87; Н 5,14; N 12,36;
Вычислено: С 47,11; Н 5,05; N 12,21%.
ПРИМЕР 57
N-[4-(4(5)-имидазоил)бутил]-N'-2-(4-хлорфенил)этилсульфамид
Целевое соединение получали способом, описанным в примере 56, с использованием 4-[1-(трифенилметил)имидазол-4-ил]бутан-1-ола (стадия d примера 51) в стадии b. Продукт был получен в виде бесцветного масла: 1Н-ЯМР (300 Мгц, MeOH-d4) 7,53 (1Н, с), 7,24 (4Н, м), 6,75 (1Н, с), 3,15 (2Н, т), 2,83 (4Н, м), 2,57 (2Н, т), 1,57 (4H, м). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане. Анализ для C19H25ClN4O6S:
Найдено: С 48,28; Н 5,44; N 11,62%.
Вычислено: С 48,25; Н 5,33; N 11,85%.
ПРИМЕР 58
N-[6-(4(5)-имидазоил)гексил]-N'-(4-бромфенил)метилсульфамид
Целевое соединение получали способом, описанным в примере 12, с использованием 5-(6-аминогексил)-1-(N,N-диметилсульфамоил)имидазола1 в качестве субстрата в стадии а и 4-бромбензилбромида в стадии b. Продукт был получен в виде белого твердого вещества: 1H-ЯМР (300 Мгц, ДМСО-d6) 7,52 (3Н, м), 7,37 (1Н, т), 7,31 (2Н, д), 6,86 (1Н, т), 6,70 (1Н, с), 3,99 (2Н, д), 2,77 (2Н, дд), 2,48 (2Н, т), 1,55 (2Н, м), 1,41 (2Н, м), 1,28 (4Н, м). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане. Анализ для С20Н27ВrN4О6S:
Найдено: С 44,92; H 5,17; N 10,58%.
Вычислено: С 45,20; H 5,12; N 10,54%.
ПРИМЕР 59
N-[6-(4(5)-имидазоил)гексил]-N'-(4-фторфенил)метилсульфамид
Целевое соединение получали способом, описанным в примере 12, с использованием 5-(6-аминогексил)-1-(N, N-диметилсульфамоил)имидазола1 в качестве субстрата в стадии а и 4-фторбензилбромида в стадии b. Продукт был получен в виде белого твердого вещества: 1Н-ЯМР (300 Мгц, ДМСО-d6) 11,50 (1Н, шир. с), 7,48 (1Н, с), 7,35 (3Н, м), 7,16 (2Н, м), 6,86 (1Н, т), 6,70 (1Н, с), 4,00 (2Н, д), 2,78 (2Н, дд), 2,48 (2Н, т), 1,55 (2Н, м), 1,42 (2Н, м), 1,28 (4Н, м). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане. Анализ для C20H27FN4O6S:
Найдено: С 51,25; Н 5,79; N 11,72%.
Вычислено: С 51,05; Н 5,78; N 11,91%.
ПРИМЕР 60
N-[6-(4(5)-имидазоил)гексил]-N'-(4-трифторметил)фенил)-метилсульфамид
Целевое соединение получали способом, описанным в примере 12, с использованием 5-(6-аминогексил)-1-(N, N-диметилсульфамоил)имидазола1 в качестве субстрата в стадии а и 4-(трифторметил)бензилбромида в стадии b. Продукт был получен в виде белого твердого вещества: 1Н-ЯМР (300 Мгц, ДМСО-d6) 11,70 (1H, шир. с), 7,70 (2Н, д), 7,58 (2Н, д), 7,49 (1Н, с), 7,47 (1Н, с), 6,91 (1Н, т), 6,70 (1Н, с), 4,12 (2Н, д), 2,78 (2Н, дд), 2,48 (2Н, т), 1,54 (2Н, м), 1,41 (2Н, м), 1,27 (4Н, м). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане.
ПРИМЕР 61
N-[6-(4(5)-имидазоил)гексил]-N'-(4-иодфенил)-сульфамид
Целевое соединение получали способом, описанным в примере 12, с использованием 5-(6-аминогексил)-1-(N,N-диметилсульфамоил)имидазола1 в качестве субстрата в стадии а и 4-иодбензилбромида в стадии b. Продукт был получен в виде белого твердого вещества: 1Н-ЯМР (300 Мгц, ДМСО-d6) 11,75 (1Н, шир.с), 7,67 (2Н, д), 7,45 (1H, с), 7,33 (1Н, т), 7,14 (1H, д), 6,82 (1H, т), 6,67 (1Н, с), 3,94 (2Н, д), 2,74 (2Н, дд), 2,46 (2Н, т), 1,52 (2Н, м), 1,37 (2Н, м), 1,25 (4Н, м). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане. Анализ для C20H27IN4O6S:
Найдено: С 41,62; H 4,75; N 9,59%.
Вычислено: С 41,53; H 4,71; N 9,69%.
ПРИМЕР 62
N-[6-(4(5)-имидазоил)гексил]-N'-(4-бифенил)-метилсульфамид
Целевое соединение получали способом, описанным в примере 49, с использованием 5-(6-аминогексил)-1-(N,N-диметилсульфамоил)имидазола1. Продукт был получен в виде белого твердого вещества: 1H-ЯМР (300 Мгц, ДМСО-d6) 11,75 (1Н, шир. с), 7,62 (4Н, м), 7,42 (5Н, м), 7,33 (2Н, м), 6,83 (1Н, т), 6,66 (1Н, т), 4,03 (2Н, д), 2,78 (2Н, дд), 2,44 (2Н, т), 1,51 (2Н, м), 1,38 (2Н, м), 1,26 (4Н, м). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане.
ПРИМЕР 63
N-[5-(4(5)-имидазоил)пентил]-N'-(4-(трифторметил)фенил)метилсульфамид
Целевое соединение получали способом, описанным в примере 12, с использованием 5-(5-аминопентил)-2-(трет-бутилдиметилсилил)-1-(N, N-диметилсульфамоил)имидазола (стадия а примера 54) в качестве субстрата в стадии (а) и 4-(трифтор-метил)бензилбромида в стадии b. Продукт был получен в виде белого кристаллического твердого вещества: 1Н-ЯМР (300 Мгц, MeOH-d4) 7,62 (2Н, д), 7,56 (3Н, м), 6,75 (1Н, с), 4,40 (2Н, с), 2,92 (2Н, т), 2,57 (2Н, т), 1,63 (2Н, квинт.), 1,55 (2Н, квинт.), 1,38 (2Н,м).
ПРИМЕР 64
N-[5-(4(5)-имидазоил)пентил]-N'-(4-бромфенил)метилсульфамид
Целевое соединение получали способом, описанным в примере 12, с использованием 5-(5-аминопентил)-2-(трет-бутилдиметилсилил)-1-(N, N-диметилсульфамоил)имидазола (стадия а примера 54) в качестве субстрата в стадии (а) и 4-бромбензилбромида в стадии b. Продукт был получен в виде белого кристаллического твердого вещества: 1H-ЯМР (300 Мгц, MeOH-d4) 7,53 (1Н, с), 7,47 (2Н, д), 7,28 (2Н, д), 6,75 (1Н, с), 4,08 (2Н, с), 2,90 (2Н, т), 2,57 (2Н, т), 1,63 (2Н, квинт.), 1,50 (2Н, квинт.), 1,38 (2Н, м).
ПРИМЕР 65
N-[5-(4(5)-имидазоил)пентил]-N'-(4-фторфенил)-метилсульфамид
Целевое соединение получали способом, описанным в примере 12, с использованием 5-(5-аминопентил)-1-(N,N-диметилсульфамоил)имидазола1 в качестве субстрата в стадии а и 4-фторбензилбромида в стадии b. Продукт был получен в виде белого твердого вещества: 1H-ЯМР (300 Мгц, MeOH-d4) 7,54 (1Н, с), 7,37 (2Н, дд), 7,04 (2Н, т), 6,75 (1Н, с), 4,09 (2Н, с), 2,90 (2Н, т), 2,50 (2Н, т), 1,64 (2Н, квинт.), 1,52 (2Н, квинт.), 1,37 (2Н, м).
ПРИМЕР 66
N-[5-(4(5)-имидазоил)пентил]-N'-(4-иодфенил)-метилсульфамид
Целевое соединение получали способом, описанным в примере 12, с использованием 5-(5-аминопентил)-1-(N,N-диметилсульфамоил)имидазола1 в качестве субстрата в стадии а и 4-иодбензилбромида в стадии b. Продукт был получен в виде белого твердого вещества: 1H-ЯМР (300 Мгц, MeOH-d4) 7,67 (2Н, д), 7,54 (1Н, с), 7,16 (2Н, а), 6,76 (1Н, с), 4,06 (2Н, с), 2,89 (2Н, т), 2,58 (2Н, т), 1,63 (2Н, квинт.), 1,50 (2Н, квинт.), 1,36 (2Н, м).
ПРИМЕР 67
N,N'-ди-[(4-бромфенил)метил]-N-[4-(4(5)-имидазоил)бутил]-сульфамид
N,N'-ди-[(4-бромфенил)метил]-N-[4-(1-(N",N"-диметилсульфамоил)имидазол-4-ил)бутил] -N'-трет-бутоксикарбонилсульфамид (пример 45, стадия b, продукт (В)) деблокировали способом, описанным в стадии с примера 12, и получали целевое соединение (Rf 0,45; аммиак (880)/метанол/дихлорметан=1:10: 90) в виде белого твердого вещества: 1Н-ЯМР (300 Мгц, СDСl3) 7,43 (5Н, м), 7,16 (4Н, д), 6,68 (1Н, с), 4,27 (2Н, с), 4,12 (2Н, с), 3,13 (2Н, т), 2,53 (2H, м), 1,53 (4Н, м). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислотой в воде/диоксане. Анализ для С20Н28Вr2N4O6S:
Найдено: С 44,62; Н 4,29; N 8,35%.
Вычислено: С 44,66; Н 4,20; N 8,33%.
ПРИМЕР 68
N'-(4-хлорфенил)метил-N,N'-диметил-N-[4-(4(5)-имидазоил)-бутилсульфамид
Стадия а: N, N'-диметил-N-[4-(1-(N",N"-диметилсульфамоил)имидазол-4-ил)бутил]-N'-трет-бутоксикарбонил-сульфамид. Раствор N-[4-(1-(N",N"-диметилсульфамоил)имидазол-4-ил)бутил] -N'-трет-бутоксикарбонилсульфамида (стадия а примера 45) (686 мг, 1,67 ммоль) и иодметана (218 мкл, 3,50 ммоль) в безводном N,N-диметилформамиде (4 мл) охлаждали в атмосфере аргона до 0oС. После добавления гидрида натрия (147 мг, 3,67 ммоль), реакционную смесь перемешивали и нагревали до комнатной температуры в течение ночи. Реакцию гасили насыщенным солевым раствором и экстрагировали этилацетатом (2х50 мл). Объединенные экстракты три раза промывали водой, сушили сульфатом магния, фильтровали и упаривали с получением желтого масла. После колоночной флэш-хроматографии (двуокись кремния; аммиак (880)/метанол/дихлорметан=0,5:5:95) получали продукт в виде бесцветного масла (356 мг, 48%).
Стадия b: N, N'-диметил-N-[4-(1-(N",N"-диметилсульфамоил)имидазол-4-ил)бутил] сульфамид. Раствор продукта стадии а (352 мг, 0,80 ммоль) в трифторуксусной кислоте (1 мл) перемешивали в течение 30 минут, и кислоту выпаривали. Остаток растворяли в дихлорметане, нейтрализовали насыщенным раствором бикарбоната натрия, и экстрагировали дихлорметаном (3х10 мл). Объединенные экстракты сушили сульфатом магния, фильтровали, и упаривали с получением продукта (271 мг) в виде бесцветного масла с количественным выходом.
Стадия с: N'-(4-хлоpфeнил)мeтил-N, N'-димeтил-N-[4-(1-(N",N"-диметилсульфамоил)имидазол-4-ил)бутил] сульфамид. Продукт стадии b (315 мг, 0,93 ммоль) подвергали реакции алкилирования с 4-хлорбензилбромидом способом, описанным в стадии b примера 12. После колоночной флэш-хроматографии (двуокись кремния; аммиак (880)/метанол/дихлорметан=0,5:5:95) получали продукт в виде светло-желтого масла (325 мг, 75%).
Стадия d: продукт стадии с (325 мг, 0,70 ммоль) деблокировали способом, описанным в стадии с примера 12, и получали целевое соединение в виде белого твердого вещества (81 мг, 32%): 1H-ЯМР (300 Мгц, СDС13) 7,55 (1Н, д), 7,33 (2Н, дд), 7,26 (2Н, дд), 6,78 (1Н, c), 4,26 (2Н, с), 3,24 (2Н, т), 2,81 (3Н, с), 2,65 (5Н, м), 1,68 (4H, м). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане. Анализ для C20H27ClN4O6S:
Найдено: С 49,04; Н 5,73; N 11,39%.
Вычислено: С 49,33; Н 5,59; N 11,51%.
ПРИМЕР 69
N'-(4-хлорфенил)метил-N-[4-(4(5)-имидазоил)бутил)-N-метилсульфамид
Стадия а: N'-(4-хлорфенил)метил-N-[4-(1-(N", N"-диметилсульфамоил)имидазол-4-ил)бутил] -N'-трет-бутоксикарбонилсульфамид. 5-(4-аминобутил)-1-(N, N-диметилсульфамоил)имидазол1 преобразовывали в продукт способом, аналогичным описанному в стадиях а и b примера 12.
Стадия b: N'-(4-хлорфенил)метил-N-[4-(1-(N", N"-диметилсульфамоил)имидазол-4-ил)бутил] -N-метил-N'-трет-бутоксикарбонилсульфамид. Продукт стадии а (310 мг, 0,58 ммоль) подвергали реакции метилирования, как описано в стадии а примера 68. Продукт был получен в виде светло-желтого масла (309 мг, 97%).
Стадия с: продукт стадии b (325 мг, 0,70 ммоль) деблокировали способом, описанным в стадии с примера 12, и получали целевое соединение в виде белого твердого вещества (80 мг, 42%): 1H-ЯМР (300 Мгц, CDCl3) 7,50 (1H, c), 7,32 (4Н, м), 6,77 (1Н, с), 4,17 (2Н, с), 3,18 (2Н, т), 2,77 (3Н, с), 2,64 (2Н, т), 1,66 (4Н, м). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане. Анализ для C19H25ClN4O6S:
Найдено: С 48,33; Н 5,41; N 11,56%.
Вычислено: С 48,25; Н 5,33; N 11,85%.
ПРИМЕР 70
N'-(4-хлорфенил)метил-N-[4-(4(5)-имидазоил)-бутил)-N-метилсульфамид
Стадия а: N'-(4-хлорфенил)метил-N-[4-(1-(N", N"-диметилсульфамоил)имидазол-4-ил)бутил] сульфамид. Трет-бутоксикарбонильную группу N'-(4-хлорфенил)метил-N-[4-(1-(N", N"-диметилсульфамоил)имидазол-4-ил)бутил] -N'-трет-бутоксикарбонилсульфамида (примера 10) (414 мг, 0,77 ммоль) удаляли способом, описанным в стадии b примера 68. Продукт был получен в виде белого твердого вещества (290 мг, 86%).
Стадия b: N'-(4-хлорфенил)метил-N-[4-(1-(N", N"-диметилсульфамоил)имидазол-4-ил)бутил] -N'-метилсульфамид. Продукт стадии а (290 мг, 0,66 ммоль) подвергали реакции метилирования способом, описанным в стадии а примера 68, с использованием иодметана (43 мкл, 0,69 ммоль) и гидрида натрия (28 мг, 0,70 ммоль). Продукт был получен в виде бесцветного масла (49 мг, 17%).
Стадия с: продукт стадии b (49 мг, 0,11 ммоль) деблокировали способом, описанным в стадии с примера 12, и получали целевое соединение в виде бесцветного масла (22 мг, 58%): lH-ЯМР (300 Мгц, CDCl3) 7,56 (1Н, с), 7,32 (2Н, дд), 7,26 (2Н, дд), 6,76 (1Н, с), 4,26 (2H, с), 3,09 (2Н, т), 2,68 (3Н, с), 2,64 (2Н, т), 1,71 (2Н, м), 1,61 (2Н, м). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта малеиновой кислоты в воде/диоксане. Анализ для C19H25ClN4O6S:
Найдено: С 48,36; Н 5,52; N 11,61%.
Вычислено: С 48,25; Н 5,33; N 11,85%.
ПРИМЕР 71
(R)-(+)-N-[2-(4(5)-имидазоил)-1-метилэтил]-2-нафталинсульфонамид
Целевое соединение получали способом, описанным в примере 3, с использованием (R)-α-метилгистамина в качестве аминового компонента. Продукт (Rf 0,18; аммиак (880)/метанол/дихлорметан=1:10:90) получали в виде белой пены: 1H-ЯМР (300 Мгц, СDС13), 8,42 (1H, шир.с), 7,93 (3Н, м), 7,80 (1Н, м), 7,57 (2Н, м), 7,52 (1H, с), 6,72 (1Н, с), 3,61 (1H, м), 2,72 (1H, дд), 1,57 (1H, дд), 1,11 (3Н, д). [α]D = + 41,8o(с=0,91; метанол/дихлорметан=1:9). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане. Анализ для С20Н21N3О6S:
Найдено: С 55,36; Н 5,08; N 9,40%.
Вычислено: С 55,67, Н 4,90; N 9,37%.
ПРИМЕР 72
N-[2-(4(5)-имидазоил)транс-циклопропил]-2-нафталинсульфонамид
Стадия а: 2-(1-трифенилметил)имидазоил-4-ил)транс-циклопропиламин. 1R-(2-нафтил)этиловый эфир[2-(1-(трифенилметил)имидазоил-4-ил)-транс-циклопропил] карбоновой кислоты получали путем синтеза Бюргера соответствующего этилового эфира с использованием R-(+)-2-нафтил-этанола5. Затем его растворяли в метаноле/тетрагидрофуране (1:1) и гидрировали в присутствии каталитического количества 10% палладия-на-угле при атмосферном давлении и температуре в течение 18 часов. После фильтрации и упаривания реакционной смеси получали аминовый продукт.
Стадия b: N-[2-(1-(трифенилметил)имидазоил-4-ил)транс-циклопропил]-2-нафталинсульфонамид получали в результате реакции продукта стадии а с 2-нафталинсульфонилхлоридом, как описанно в стадии (а) примера 6.
Стадия с: продукт стадии b деблокировали способом, описанным в стадии d примера 17, и выделяли целевое соединение (Rf 0,26; аммиак (880)/метанол/дихлорметан= 1: 10: 90) в виде белой пены: 1H-ЯМР (300 Мгц, CDCl3) 8,44 (1Н, с), 7,92 (3Н, м), 7,84 (2Н, дд), 7,62 (2H, м), 7,47 (1Н, с), 6,72 (1H, с), 2,48 (1H, м), 2,15 (1H, м), 1,27 (1H, м), 1,21 (1H, м).
ПРИМЕР 73
3-(4(5)-имидазоил-2S-(2-нафталин)сульфонаминопропионовой кислоты метиловый эфир
Стадия а: 3-(1-(трифенилметил)имидазоил-4-ил)-2S-(2-нафталин)сульфонаминопропионовой кислоты метиловый эфир. К раствору гидрохлорида метилового эфира трифенилметилгистидина (488 мг, 1,00 ммоль) и триэтиламина (304 мкл, 2,20 ммоль) в сухом дихлорметане (20 мл) в атмосфере аргона добавляли 2-нафталинсульфонилхлорид (238 мг, 1,05 ммоль). Раствор перемешивали в течение одного часа, промывали водой (2х20 мл), сушили сульфатом магния, фильтровали, и упаривали. Остаток перекристаллизовывали из этилацетата/гексана (1:2) и получали продукт в виде белого твердого вещества (438 мг, 73%).
Стадия b: продукт стадии а (550 мг, 0,91 ммоль) деблокировали способом, описанным в стадии b примера 17, и получали целевое соединение (Rf 0,32; аммиак (880)/метанол/дихлорметан= 1: 10:90) в виде белого твердого вещества (114 мг, 35%): 1H-ЯМР (300 Мгц, ДМСО-d6) 11,75 (1H, шир.с), 8,45 (1H, д), 8,28 (1H, д), 8,09 (1H, дд), 8,02 (2Н, т), 7,68 (3Н, м), 7,39 (1Н, д), 6,72 (1H, с), 4,08 (1H, дд), 3,21 (3Н, с), 2,76 (2Н, с). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане.
ПРИМЕР 74
N-[1S-гидроксиметил-2-(4(5)-имидазоил)этил]-2-нафталинсульфонамид
Стадия а: N-[1S-гидроксиметил-2-(1-(трифенилметил)имидазоил-4-ил)этил] -2-нафталинсульфонамид. К раствору продукта от стадии (а) примера 73 (438 мг, 0,73 ммоль) в метаноле (6 мл) и тетрагидрофуране (12 мл) небольшими порциями в течение нескольких часов добавляли, перемешивая при этом, смесь борогидрида натрия (378 мг, 10,0 ммоль) и хлорида лития (420 мг, 10,0 ммоль). Смесь концентрировали в вакууме и распределяли между этилацетатом и водой. Органический слой промывали насыщенным солевым раствором, сушили сульфатом магния, и упаривали с получением продукта в виде белой пены (305 мг, 73%).
Стадия b: продукт стадии а (300 мг, 0,52 ммоль) деблокировали способом, описанным в стадии d примера 17, и получали целевое соединение (Rf 0,32; аммиак (880)/метанол/дихлорметан= 1: 10:90) в виде белого твердого вещества (56 мг, 33%): 1H-ЯМР (300 Мгц, ДМСО-d6) 8,34 (1Н, с), 8,09 (1Н, д), 8,00 (2Н, м), 7,67 (3Н, м), 7,41 (1Н, д), 6,69 (1Н, с), 3,25 (3Н, м), 2,64 (1Н, дд), 2,46 (1H, м). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане.
ПРИМЕР 75
3-(4(5)-имидазоил)-2S-(2-нафталин)сульфонаминопропионовая кислота
Стадия а: 3-(1-(трифенилметил)имидазоил-4-ил)-2S-(2-нафталин)сульфонаминопропионовая кислота. К раствору трифенилметилгистидина (398 мг, 1,00 ммоль) и триэтиламина (304 мкл, 2,20 ммоль) в сухом дихлорметане (20 мл) в атмосфере аргона добавляли 2-нафталинсульфонилхлорид (238 мг, 1,05 ммоль). Раствор перемешивали в течение одного часа и добавляли воду (20 мл). Затем добавляли разбавленную уксусную кислоту до тех пор, пока рН не становился равным 5. Полученную смесь встряхивали. Органический слой промывали насыщенным солевым раствором, сушили сульфатом магния, и упаривали с получением продукта в виде бледно-желтого твердого вещества (486 мг, 83%).
Стадия b: раствор продукта стадии а (486 мг, 0,83 ммоль) в трифторуксусной кислоте (5 мл) перемешивали в течение 18 часов, и растворитель выпаривали. Остаток растирали с диэтиловым эфиром и полученное белое твердое вещество перекристаллизовывали из пропан-2-ола/диэтилового эфира. Соль трифторуксусной кислоты целевого соединения (Rf 0,07; 20% метанол/дихлорметан) выделяли в виде белого твердого вещества (115 мг, 30%); 1H-ЯМР (300 Мгц, ДМСО-d6) 8,30 (1Н, д), 8,00 (4Н, м), 7,68 (3Н, м), 6,98 (1H, c) 4,06 (1H, дд), 2,93 (1Н, дд), 2,81 (2Н, дд). Анализ для С18Н16F3N3О6S:
Найдено: С 47,08; H 3,59; N 9,20%.
Вычислено: С 47,06; Н 3,51; N 9,15%.
ПРИМЕР 76
N-[5-(4(5)-имидазоил)пентил]-N'-(2-гидроксиэтил)сульфамид
Стадия а: N-[5-(2-(трет-бутилдиметилсилил)-1-(N", N"-диметилсульфамоил)имидазол-4-ил)пентил] -N'-(2-гидроксиэтил)сульфамид. 5-(5-аминопентил)-2-(трет-бутилдиметилсилил)-1-(N, N-диметилсульфамоил)имидазол (стадия а примера 54) и 2-бромэтанол оставляли для реакции с хлорсульфонилизоцианатом, как описано в стадии а примера 12. Раствор полученного соединения в этаноле обрабатывали 2М раствором гидроксида натрия (2 эквивалента), и нагревали обратным холодильником в течение 2 минут, после чего, растворитель выпаривали. К остатку добавляли воду и смесь экстрагировали дихлорметаном. Органический слой сушили сульфатом магния, фильтровали, и упаривали. Остаток очищали с помощью колоночной флэш-хроматографии (двуокись кремния; 2-4% метанол/дихлорметан) и получали продукт в виде бесцветного масла.
Стадия b: продукт стадии b деблокировали способом, описанным в стадии с примера 12. Целевое соединение (Rf 0,35; аммиак (880)/метанол/дихлорметан=1: 10: 90) получали в виде бесцветного масла: 1H-ЯМР (300 Мгц, ДМСО-d6) 11,70 (1Н, шир. с), 7,46 (1Н, с), 7,10 (1Н, т), 6,87 (1Н, т), 6,68 (1Н, с), 3,60 (2Н, т), 3,12 (25, дд), 2,79 (2Н, дд), 2,46 (2Н, т), 1,61 (2Н, квинт.), 1,46 (2Н, квинт.), 1,37 (2Н, квинт.).
ПРИМЕР 77
N-[4-(4(5)-имидазоил)бутил]-(4-бромфенил)метансульфонамид
Целевое соединение получали способом, описанным в примере 9, с использованием 5-(4-аминобутил)-1-(N, N-диметилсульфамоил)имидазола1 и (4-бромфенил)метансульфонилхлорида (полученного, в основном, как описано в стадии а примера 4) в качестве субстратов в стадии а. Продукт от двух стадий был получен в виде белого твердого вещества: 1Н-ЯМР (300 Мгц, ДМСО-d6) 7,56 (1Н, с), 7,47 (2Н, д), 7,30 (2Н, д), 7,04 (1Н, т), 6,69 (1Н, с), 4,28 (2Н, c), 2,87 (2Н, дд), 2,46 (2Н, м), 1,53 (2Н, квинт.), 1,41 (2Н, квинт.). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане.
ПРИМЕР 78
N-[4-(4(5)-имидазоил)бутил]-(4-трифторметилфенил)метансульфонамид
Целевое соединение получали способом, описанным в примере 9, с использованием 5-(4-аминобутил)-1-(N, N-диметилсульфамоил)имидазола1 и (4-тpифтopмeтилфeнил)мeтaнcyльфoнилxлoрида (полученного в основном, как описано в стадии а примера 4) в качестве субстратов в стадии а. Продукт был получен в виде белого твердого вещества: 1Н-ЯМР (300 Мгц, МеОН-d4) 7,67 (2Н, д), 7,60 (2Н, д), 7,55 (1Н, с), 6,76 (1Н, с), 4,39 (2Н, с), 3,00 (2Н, т), 2,57 (2H, т), 1,64 (2Н, квинт.), 1,48 (2Н, квинт.). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане.
ПРИМЕР 79
N-[4-(4(5)-имидазоил)бутил]-(4-фторметилфенил)метансульфонамид
Целевое соединение получали способом, описанным в примере 9, с использованием 5-(4-аминобутил)-1-(N, N-диметилсульфамоил) имидазола1 и (4-фтopмeтилфeнил)мeтaнcyльфoнилxлoрида (полученного в основном, как описано в стадии а примера 4) в качестве субстратов в стадии а. Продукт был получен в виде белого твердого вещества: 1Н-ЯМР (300 Мгц, ДМСО-d6) 7,47 (1Н, с), 7,39 (2Н, дд), 7,18 (2Н, т), 7,01 (1Н, т), 6,69 (1Н, с), 4,28 (2Н, с), 2,86 (2Н, дд), 2,48 (2Н, квинт. ), 1,52 (2Н, квинт.), 1,42 (2Н, квинт.). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане.
ПРИМЕР 80
N-[4-(4(5)-имидазоил)бутил]-N'-(1R)-фенилэтил)-сульфамид
Целевое соединение получали способом, описанным в примере 51, с использованием (R)-(+)-α-метилбензиламина в качестве субстрата в стадии е. Продукт (Rf 0,24; аммиак (880)/метанол/дихлорметан=1:10:90) получали в виде белого твердого вещества: 1H-ЯМР (300 Мгц, ДМСО-d6) 11,80 (1Н, шир.с), 7,47 (1H, с), 7,28 (5Н, м), 7,21 (1H, дд), 6,66 (1Н, с), 6,65 (1H, т), 4,29 (1Н, м), 2,65 (2Н, м), 2,40 (2Н, т), 1,47 (2Н, м), 1,41 (3Н, д), 1,36 (2Н, м). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане.
ПРИМЕР 81
N-[4-(4(5)-имидазоил)бутил]-N'-(1S)-фенилэтил)-сульфамид
Целевое соединение получали способом, описанным в примере 51, с использованием, (S)-(+)-α-метилбензиламина в качестве субстрата в стадии е. Продукт (Rf 0,24; аммиак (880)/метанол/дихлорметан=1:10:90): получали в виде белого твердого вещества: 1H-ЯМР (300 Мгц, ДМСO-d6) 11,80 (1Н, шир.с), 7,47 (1Н, с), 7,28 (5Н, м), 7,21 (1Н, дд), 6,66 (1Н, с), 6,65 (1Н, т), 4,29 (1Н, м), 2,65 (2H, м), 2,40 (2Н, т), 1,47 (2Н, м), 1,41 (3Н, д), 1,36 (2Н, м). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане.
ПРИМЕР 82
N-[5-(4(5)-имидазоил)пентил]-N'-(4-бифенил)метилсульфамид
Целевое соединение получали способом, описанным в примере 49, с использованием и 5-(5-аминопентил)-1-(N,N-диметилсульфамоил)имидазола1. Продукт получали в виде белого твердого вещества: 1H-ЯМР (300 Мгц, MeOH-d4) 7,60 (4Н, м), 7,52 (1Н, с), 7,43 (4Н, м), 7,32 (1H, дд), 6,72 (1Н, с), 4,16 (2Н, с), 2,91 (2Н, т), 2,55 (2Н, т), 1,59 (2Н, квинт.), 1,51 (2Н, квинт), 1,38 (2Н, квинт.).
ПРИМЕР 83
N-[5-(4(5)-имидазоил)бутил]-N'-(1,1-дифенил)метилсульфамид
Целевое соединение получали способом, описанным в примере 51, с использованием С,С-дифенилметиламина в качестве субстрата в стадии е. Продукт (Rf 0,28; аммиак (880)/метанол/дихлорметан=1:10:90) получали в виде белого твердого вещества: 1Н-ЯМР (300 Мгц, ДМСО-d6) 11,70 (1Н, шир.с), 8,03 (1Н, д), 7,46 (1Н, с), 7,27 (10Н, м), 6,76 (1Н, т), 6,63 (1Н, с), 5,42 (1Н, д), 2,52 (2Н, т), 2,32 (4Н, т), 1,36 (2Н, квинт.), 1,19 (2Н, квинт.). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане.
ПРИМЕР 84
N-[4-(4(5)-имидазоил)бутил]-(4-бифенил)-метансульфонамид
Целевое соединение получали способом, описанным в примере 9, с использованием 5-(4-аминобутил)-1-(N, N-диметилсульфамоил)имидазола1 и (4-бифенил)метансульфонилхлорида (полученного, как описано в стадии а примера 4) в качестве субстратов в стадии а. Продукт был получен в виде белого твердого вещества: 1Н-ЯМР (300 Мгц, МеОН-d4) 7,63 (4Н, м), 7,56 (1Н, с), 7,48 (4Н, м), 7,32 (1Н, м), 6,76 (1Н, с), 4,34 (2Н, д), 3,00 (2Н, т), 2,58 (2Н, т), 1,65 (2Н, квинт.), 1,52 (2Н, квинт.). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане.
ПРИМЕР 85
N-[4-(4(5)-имидазоил)бутил]-N-метил-2-(4-хлорфенил)этансульфонамид
N-[4(1-(N', N'-диметилсульфамоил)имидазол-4-ил)бутил] -2-(4-хлорфенил)этансульфонамид (промежуточное соединение синтеза примера 55) подвергали реакции метилирования с иодметаном, в основном, как описано в стадии а пример 68. Реакция снятия защиты, описанная в стадии b примера 9, давала целевое соединение в виде бесцветного масла: 1Н-ЯМР (300 Мгц, MeOH-d4), 7,55 (1H, д), 7,27 (4Н, м), 6,78 (1H, с), 3,26 (2Н, м), 3,21 (2Н, т), 3,06 (2Н, м), 2,83 (3Н, с), 2,63 (2Н, т), 1,64 (4Н, м). Соль малеиновой кислоты получали лиофилизацией эквимолярного раствора продукта и малеиновой кислоты в воде/диоксане.
Библиография
1. R. C.Vollinga, W.M.P.B.Mange, E.Leurs, H.Timmerman J.Med.Chem. 1995, 38, 266.
2. J.C.Emmett, F.H.flolloway, Turner J.C.S.Perkin I 1979, 1341.
3. H. Stark, K.Purand, A.Huls, X.Ligneau, M.Garbarg, J.-C.Schwartz, W. Schunack J.Мed.Chem. 1996, 39, 1220.
4. J.L.Kelley, C.A.Miller, E.W.McLean J.Med.Chem. 1977, 20, 721.
5. A.Burger, M.Bernabe, P.W.Collins J.Med.Chem. 1970, 13,33. Н3 receptor bioassay.
Биологическую активность соединений, описанных в примерах, измеряли с помощью анализа с использованием продольной подвздошной мышцы, мышечно-кишечного сплетения, который был описан Paton&Aboo Zar, (J.Physiol. 194, 13-33 (1968)). В этом анализе были использованы самцы морских свинок Dunkin-Hartley (250-300 г). 50 см - часть подвздошной кишки в области, примыкающей к слепой кишке, удаляли, после отбрасывания концевой части в 20 см. Сегменты подвздошной кишки (3 см) очищали путем пропускания через подвздошную кишку буфера Кребса-Хенселейта, содержащего 3 мкМ мепирамина, с помощью пипетки Пастера (размер: 13,8 см длиной и 0,65 см в диаметре). Во избежание нежелательного повреждения ткани, буфер Кребса-Хенселейта пропускали через сегмент подвздошной кишки, когда он находился в горизонтальном положении на чашке Петри. Поэтому, подвздошная кишка не растягивалась, и буфер свободно протекал. Затем каждый сегмент пропускали через пипетку Пастера, и слой продольной мышцы и прилипшее мышечно-кишечное сплетение отделяли с использованием влажной хлопковой ваты энергичными движениями, направленными тангенциально от мезентериального прикрепления. Ткани суспендировали в 20 мл бане для органов, содержащей буфер Кребса-Хенселейта при 37oС±1oС в атмосфере газа 95% CO2/5%O2. Ткани лигировали двумя параллельными проволоками из нержавеющей стали, расположенными между двумя платиновыми электродами (0,76 см длиной и 0,06 см в диаметре). Все измерения регистрировали изометрически (датчик Grass FTО3). После первоначальной нагрузки в 1 г ткани подвергали стимулированию электрическими импульсами с частотой 0,1 Гц, и продолжительностью 0,5 мсек, как описано Kosterlitz & Watt (1968). Сначала ткани стимулировали при сверхмаксимальном напряжении (в 1,3 раза превышающем максимальное) в течение 30 минут, после чего ткани промывали и снова стимулировали. Затем вводили "пробную дозу" селективного агониста Н3-рецептора гистамина, R-(α)-метилгистамина (0,3 мкМ) (Arrang et al., 1987). После продуцирования ответа, "пробную дозу" удаляли и ткани путем "отмывки" (6 промывок за 60 минут), причем в течение указанного периода времени электрическую стимуляцию отключали. Затем перед добавлением исследуемого лекарственного средства ткани снова стимулировали и оставляли для стабилизации, помещая их на произвольно расположенное блок-основание в бани для органов. По истечении периода инкубирования устроили одну интегральную кривую Е/[А]. Данные экспериментальной кривой Е/[А] выражали как процент ингибирования высоты пика электрически стимулированного сокращения. Величины аффинности антагониста вычисляли по степени правого сдвига кривых Е/[А] для R-(α)-метилгистамина с использованием методов Шильдера (Arunlatcsharna & Schild, 1949).
Анализ на связывание радиолиганда Н3-гистамина
Получение мембран
Были использованы самцы морских свинок Dunkin-Hartley (200-300 г). Тонкую кишку быстро удаляли (вырезали ~ 5 см из слепой кишки и 5 см из желудка), и помещали в охлажденный льдом 20 мМ буфер Hepes-NaOH (рН 7,4 при 21±3oС). Ткань нарезали на ~ 10 см сегменты промывали охлажденным на льду 20 мМ буфером Hepes-NaOH, и помещали в сосуд, содержащий свежий буфер при 4oС. 10 см сегменты подвздошной кишки растягивали на стеклянной пипетке, и мышечно-кишечное сплетение с продольной мышцей отделяли от кругового слоя мышечной оболочки с использованием влажной хлопковой ваты. Мышечно-кишечное сплетение с продольной мышцей сразу помещали в охлажденный льдом раствор Viaspan® (~100 мл для ткани от 3 морских свинок) и помещали в холодильник на 24 часа.
Предварительно смоченную ткань взвешивали и измельчали ножницами. Затем ткань гомогенизировали в Viaspan® с использованием политрона (Kinematica AG; PT-DA 3020/2TS, 3х~ 1-2 с). Затем к ткани добавляли 50 мл 500 мМ Триса-НС1 (рН 6,9 при 21±3oС), и смесь центрифугировали при 39800хg в течение 12 минут при 4oС. Супернатант отбрасывали и снова гомогенизировали в 100 мл охлажденного льдом 20 мМ буфера Hepes-NaOH (рН 7,4 при 21±3oС) с использованием гомогенизатора из "тефлона в стекле" (положение 10; 3 х). Гомогенат снова центрифугировали при 39,800хg, и осадок ресуспендировали в 20 мМ буфера Hepes-NaOH (рН 7,4 при 21±3oС) до получения концентрации ткани 50 мг/мл с использованием политрона (Brinkman, PT 10,3х~1с).
Условия инкубирования
Мембраны мышечно-кишечного сплетения с продольной подвздошной мышцей морской свинки (400 мкл) инкубировали в течение 165 минут при 21±3oС в конечном объеме 500 мкл с 20 мМ буфером Hepes-NaOH, содержащем [3Н]-R-(α)-метилгистамина (50 мкл; 3 нМ) и конкуретное соединение. Полное и неспецифическое связывание [3H] -R-(α)-метилгистамина определяли с использованием 50 мкл буфера и 50 мкл 10 мкМ тиоперамида, соответственно. Анализ завершали путем быстрой фильтрации через фильтры Whatman GF/B, предварительно вымоченные (2 час) в 0,1% полиэтиленимине, с использованием сборщика клеток (Brandel Cell Harvester). Фильтры промывали (3х3 мл) охлажденным на льду 50 мМ Трис-НС1 (рН 6,9 при 21±3oС), переносили в сцинтилляционные сосуды, и добавляли 5 мл жидкой сцинтилляционной смеси, а через 4 часа определяли уровень связанной радиоактивности путем посчета (4 мин) в жидкостном сцинтилляционном счетчике Весkman.
Анализ данных
Данные анализировали с использованием призмы GraphPad и общего уравнения для кривой конкурентного связывания с переменной крутизной Хилла (nН).
где Х - log концентрации конкурирующего соединения.
Y - связывание, полученное при каждой концентрации X.
рIC50 - концентрация конкурента, требуемая для конкурентного ингибирования половины специфического связывания.
IC50 преобразовывали в K1 с использованием уравнения Cheng Prussoff:
K1=IC50/(1+(L/KD)),
где IC50 - концентрация конкурента, требуемая для конкурентного ингибирования половины специфического связывания,
L - концентрация используемого радиолиганда.
КD - константа равновесия диссоциации для радиолиганда, определенная с помощью эксперимента путем насыщения.
В описанных двух анализах были получены результаты, которые указаны в таблице, приведенной в конце описания.
| название | год | авторы | номер документа |
|---|---|---|---|
| БЕНЗОТРИАЗЕПИНЫ В КАЧЕСТВЕ ЛИГАНДОВ РЕЦЕПТОРОВ ГАСТРИНА И ХОЛЕЦИСТОКИНИНА | 2002 |
|
RU2304438C2 |
| СПОСОБ ПОЛУЧЕНИЯ АМИДОВ НЕНАСЫЩЕННЫХ КИСЛОТ | 1989 |
|
RU2010025C1 |
| ПРОИЗВОДНЫЕ 4-МЕРКАПТОПИРРОЛИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ ФАРНЕЗИЛТРАНСФЕРАЗЫ | 1996 |
|
RU2191773C2 |
| ПРОИЗВОДНЫЕ 3-ОКСО-1-ЦИКЛОБУТЕНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2002 |
|
RU2296753C2 |
| АРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 1996 |
|
RU2198878C2 |
| ПИРРОЛОИНДОЛЫ, ПИРИДИНОИНДОЛЫ И АЗЕПИНОИНДОЛЫ В КАЧЕСТВЕ АГОНИСТОВ 5-НТ2С | 1999 |
|
RU2232162C2 |
| АРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 1996 |
|
RU2182574C2 |
| ИНГИБИТОРЫ АКТИВНОСТИ АКТ | 2010 |
|
RU2579513C2 |
| ПРОИЗВОДНЫЕ ИНДОЛА, ИХ ОПТИЧЕСКИЕ ИЗОМЕРЫ И ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1991 |
|
RU2095360C1 |
| АМИДИНПРОИЗВОДНЫЕ | 1992 |
|
RU2130014C1 |




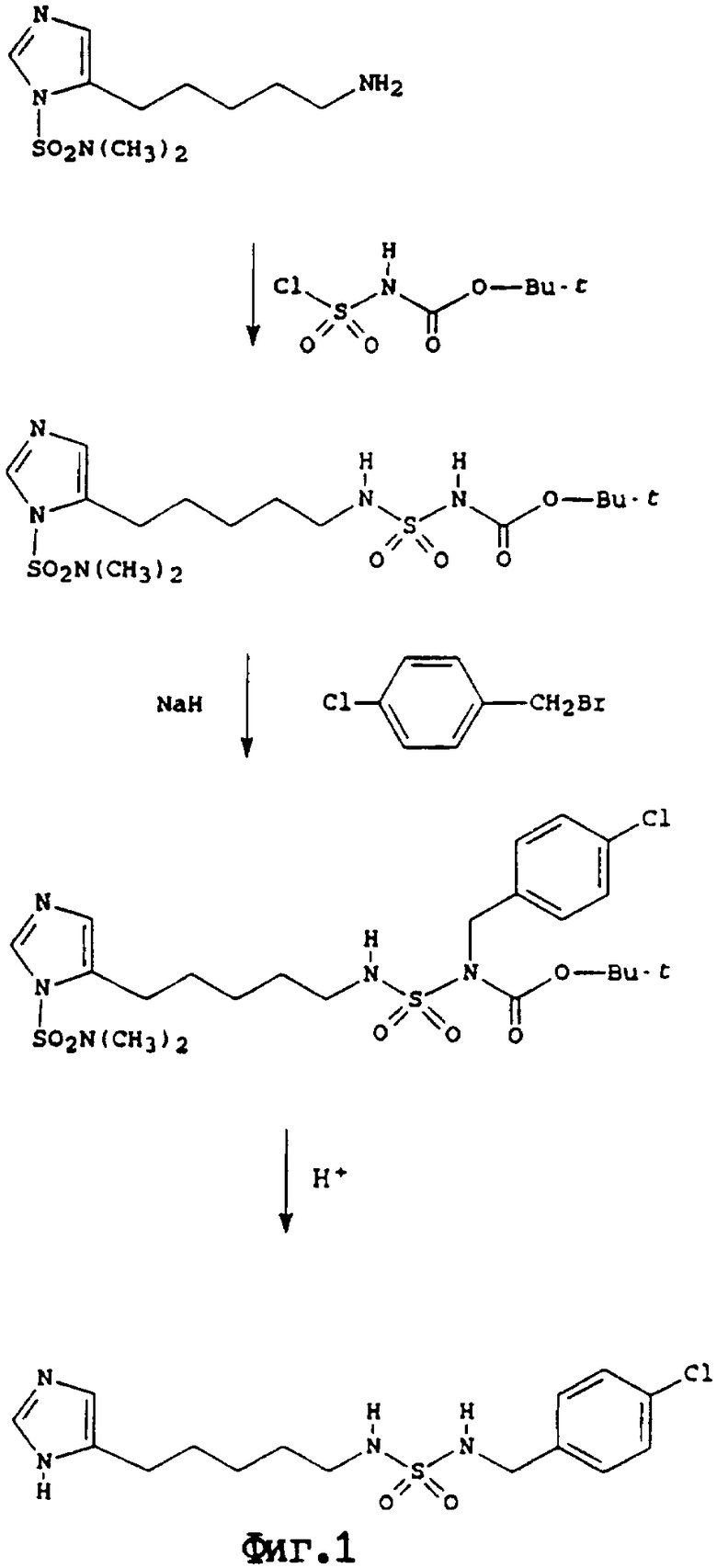
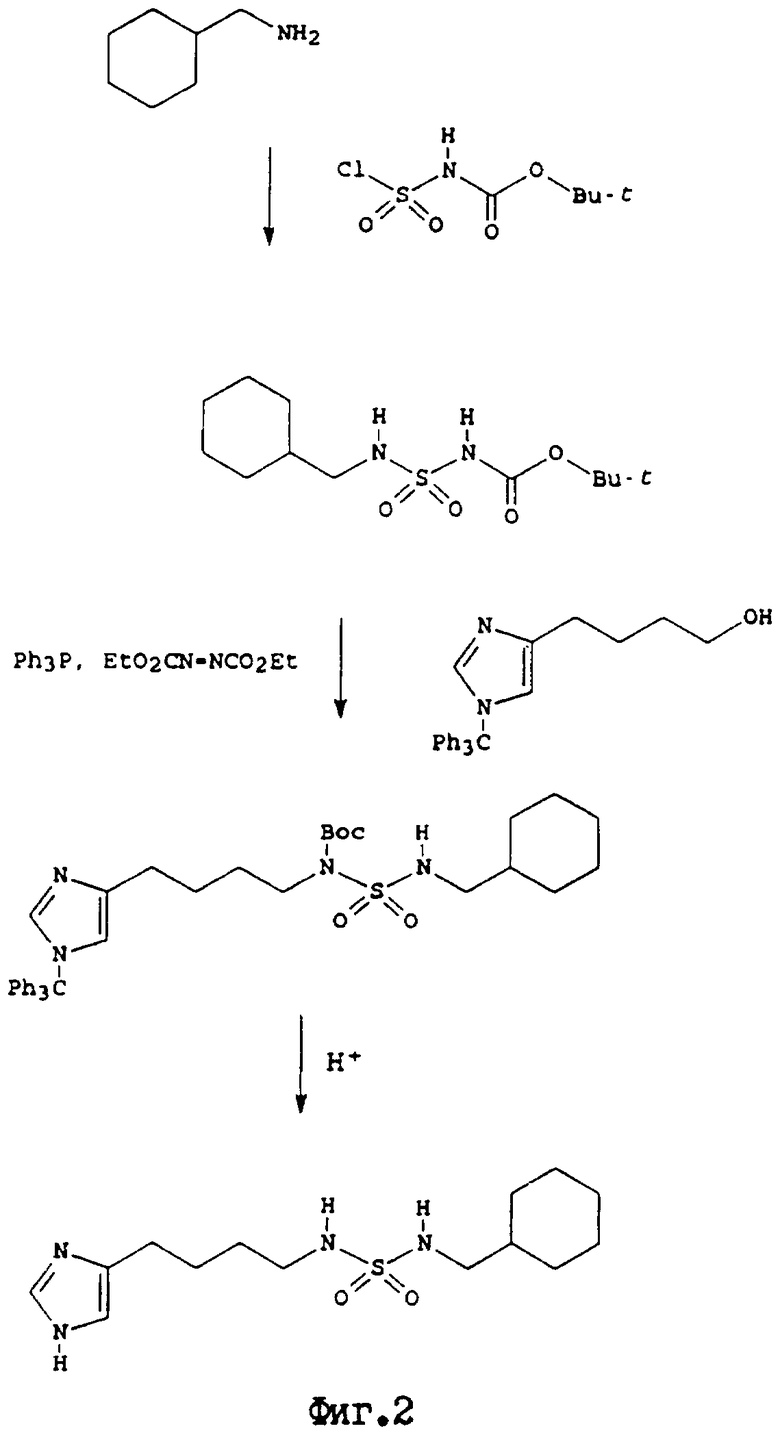
Описываются сульфонамиды общей формулы I, где Х представляет С3-С9 алкиленовую группу, R2 представляет Н или C1-С3 алкил, R11 представляет Н или C1-С3 алкил, p представляет 0 или 1, q представляет 0-3, R12 представляет фенил или нафтил, необязательно замещенный галогеном, метокси- или трифторметилом, 4-бифенил, циклогексил или адамантил и R13 независимо выбран из Н, метила и фенила, и способы их получения. Соединения I являются лигандами Н3-рецепторов гистамина и могут быть использованы в качестве седативных средств, регуляторов сна, противосудорожных средств, антидепрессантов и модуляторов мозгового кровообращения, а также для лечения астмы и синдрома раздражения кишечника. 4 с. и 6 з.п. ф-лы, 1 табл., 2 ил.
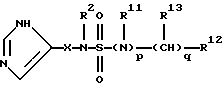
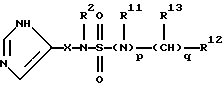
где X представляет С3-С9 алкиленовую группу;
R2 представляет Н или С1-С6алкил;
R11 представляет Н или C1-С3 алкил;
p представляет 0 или 1;
q представляет 0-3;
R12 представляет фенил или нафтил, необязательно замещенный галогеном, метокси- или трифторметилом, 4-бифенил, циклогексил или адамантил;
R13 независимо выбран из Н, метила и фенила.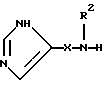
с сульфонилхлоридом формулы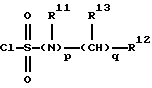
в которой значения R2, R11, R12, R13, р, q определены в пп. 1-6.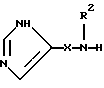
с) взаимодействие продукта, полученного на стадии б), с основанием, таким, как гидрид натрия, и затем с соединением формулы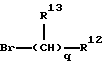
и д) взаимодействие продукта, полученного на стадии с), с кислотой, причем значения R2, R12, R13, X и q определены в пп. 1-6.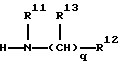
с) взаимодействие продукта, полученного на стадии б), с соответствующим образом защищенным производным соединения формулы
д) обработка продукта, полученного на стадии с), кислотой, причем значения R12, R11, R13, q определены в любом из пп. 1-5.
Приоритет по признакам и пунктам:
09.02.1996 по п. 1, значение X-C1-5 алкилен и пп. 5 и 6 формулы;
21.11.1996 по п. 1, значение X-C6-10 алкилен;
все оставшиеся пункты имеют приоритет по дате подачи заявки РСТ.
| ЕР, 082648 А, 29.06.1983 | |||
| DE, 3219113 А, 24.11.1983 | |||
| WO, 93/14070 А, 22.07.1993 | |||
| US, 3497591 А, 24.02.1970 | |||
| US, 5447728 А, 05.09.1995 | |||
| SU, 1802811 A3, 15.03.1993. |
