Техническая область
Изобретение относится к новым солям оптически активного сульфоксидного производного, обладающим отличной антагонистической активностью в отношении рецепторов субстанции Р и рецепторов нейрокинина А.
Известный уровень техники
Несмотря на то, что имеется немного сообщений о низкомолекулярном соединении непептидного типа, обладающем антагонистической активностью в отношении рецепторов субстанции Р (рецепторы NK1) и рецепторов нейрокинина А (рецепторы NK2), например, описанные ниже соединения А, В и С являются именно такими соединениями. Согласно описанию публикации РСТ WO 94/17045 соединение В обладает антагонистической активностью как в отношении рецепторов NK1, так и в отношении рецепторов NK2. Фармакологические исследования, проведенные авторами настоящего изобретения, показали, однако, что антагонистическая активность соединения B в отношении рецепторов NK1 in vitro является весьма слабой. Помимо этого, при пероральном приеме всех этих соединений возникает ряд проблем, таких как недостаточная активность как в отношении рецепторов NK1, так и в отношении рецепторов NK2.
В течение продолжительного периода времени авторы настоящего изобретения проводили широкомасштабное исследование по синтезу производных, обладающих антагонистической активностью в отношении тахикинина (в частности, антагонизм в отношении субстанции Р, антагонистическая активность в отношении нейрокининов А и В), и их фармакологической активности. В результате было установлено, что по сравнению с описанными выше известными соединениями некоторые новые соли оптически активного вещества спиро[бензо[с]тиофен-1(3Н), 4'-пиперидин] -2-оксида, имеющего абсолютную S-конфигурацию, демонстрируют более хорошее всасывание при пероральном приеме, а также отличную антагонистическую активность как в отношении рецепторов NK1, так и в отношении рецепторов NK2, что завершило настоящее изобретение.
Целью настоящего изобретения является создание вышеописанного соединения. Другой целью настоящего изобретения является создание лекарственного средства, включающего вышеописанное соединение в качестве эффективного ингредиента, в частности в качестве профилактического агента или средства (композиции для профилактики или лечения) для лечения заболеваний, опосредованных тахикинином. Еще одной целью настоящего изобретения является обеспечение применения вышеописанного соединения для изготовления лекарственного средства, в частности профилактического агента или средства (композиции для профилактики или лечения) для лечения заболеваний, опосредованных тахикинином, или обеспечение способа профилактики или лечения заболеваний, опосредованных тахикинином, который включает введение фармакологически эффективного количества этого соединения теплокровному животному.
Примеры такого профилактического агента или средства включают ингибиторы рецепторов NK1. и/или рецепторов NK2. Примеры заболеваний включают заболевания центральной нервной системы, такие как беспокойство, депрессия, психоз и шизофрения; нейродегенеративные заболевания, такие как деменция при СПИДе, сенильная деменция Альцгеймера, болезнь Альцгеймера, синдром Дауна, демиелинизирующая болезнь, амиотрофический латеральный склероз, нейропатия, периферическая нейропатия и невралгия; респираторные заболевания, такие как хроническое обструктивное заболевание легких, бронхит, пневмония, бронхоконстрикция, астма и кашель; воспалительные заболевания, такие как воспалительное заболевание кишечника (ВЗК), псориаз, фиброз, эпифизарный остеомиелит, дегенеративный артрит и ревматоидный артрит; экзема; аллергии, такие как ринит; заболевания гиперчувствительности, такие как гиперчувствительность к винам; офтальмологические заболевания, такие как конъюнктивит, весенний конъюнктивит, весенний катар, деструкция гематоофтальмического барьера, вызванная различными воспалительными заболеваниями глаз, повышенное внутриглазное давление и миоз; кожные заболевания, такие как контактный дерматит, атопический дерматит, крапивница и другие экземоподобные дерматиты; злоупотребления некоторыми веществами, такие как алкогольная зависимость; соматические заболевания, вызванные стрессом; симпатическая рефлекторная дистрофия, такая как синдром руки и плеча; дистимия; нежелательные иммунные реакции, такие как отторжение трансплантатов; заболевания, относящиеся к иммуностимуляции, такие как системная красная волчанка или иммуносупрессия; расстройства пищеварения, такие как заболевания, вызванные нарушениями нервной регуляции органов, колит, язвенный колит и болезнь Крона; рвота, такая как рвота, обусловленная побочными эффектами лучевой и химиотерапии, ядами, токсинами, беременностью, вестибулярными расстройствами, послеоперационными состояниями, непроходимостью желудочно-кишечного тракта, пониженной перистальтикой желудочно-кишечного тракта, висцеральной болью, мигренью, повышенным внутричерепным давлением, пониженным внутричерепным давлением или побочными реакциями, индуцированными введением различных лекарственных средств; функциональные болезни мочевого пузыря, такие как цистит и недержание мочи; эозинофилия, вызванная коллагенозами, склеродермией или инвазией Fasciola hepatica; заболевания, обусловленные нарушениями кровотока вследствие вазодилатации или вазоконстрикции, такие как стенокардия, мигрень и болезнь Рейнольдса; боли, обусловленные ноцицептивным восприятием, такие как головная боль при мигрени, головная боль и зубная боль.
Новыми солями оптически активного сульфоксидного производного по настоящему изобретению являются гидрохлорид или фумарат 1-{2-[(2R)-(3,4-дихлорфенил)-4-(3,4,5-триметоксибензоил)морфолин-2-ил] этил}спиро[бензо(с)тиофен-1(3Н),4'-пиперидин]-(2S)-оксида.
Новое лекарственное средство по настоящему изобретению включает соединение, выбранное из описанных выше соединений, в качестве активного ингредиента;
новый профилактический агент или средство для лечения заболеваний, опосредованных тахикинином, по настоящему изобретению включает соединение, выбранное из описанных выше соединений, в качестве активного ингредиента;
новый ингибитор рецепторов NK1 и/или рецепторов NK2 по настоящему изобретению включает соединение, выбранное из описанных выше соединений, в качестве активного ингредиента;
новый профилактический агент или средство для лечения астмы и/или бронхита по настоящему изобретению включает соединение, выбранное из описанных выше соединений, в качестве активного ингредиента;
новый профилактический агент или средство для лечения ринита по настоящему изобретению включает соединение, выбранное из описанных выше соединений, в качестве активного ингредиента;
новый профилактический агент или средство для лечения аллергии по настоящему изобретению включает соединение, выбранное из описанных выше соединений, в качестве активного ингредиента;
новый профилактический агент или средство для лечения недержания мочи по настоящему изобретению включает соединение, выбранное из описанных выше соединений, в качестве активного ингредиента.
Применение для получения лекарственного средства по настоящему изобретению включает применение соединения, выбранного из описанных выше соединений;
применение для получения профилактического агента или средства для лечения заболеваний, опосредованных тахикинином, по настоящему изобретению включает использование соединения, выбранного из описанных выше соединений;
применение для получения ингибитора рецепторов NK1 и/или рецепторов NK2 по настоящему изобретению включает использование соединения, выбранного из описанных выше соединений;
применение для получения профилактического агента или средства для лечения астмы и/или бронхита по настоящему изобретению включает использование соединения, выбранного из описанных выше соединений;
применение для получения профилактического агента или средства для лечения ринита по настоящему изобретению включает использование соединения, выбранного из описанных выше соединений;
применение для получения профилактического агента или средства для лечения аллергии по настоящему изобретению включает использование соединения, выбранного из описанных выше соединений;
применение для получения профилактического агента или средства для лечения недержания мочи по настоящему изобретению включает использование соединения, выбранного из описанных выше соединений.
В солях оптически активного сульфоксидного производного по настоящему изобретению 1-{2-[(2R)-(3,4-дихлорфенил)-4-(3,4,5-триметоксибензоил)морфолин-2-ил] этил} спиро[бензо(с)тиофен-1(3Н), 4'-пиперидин]-(2S)-оксид представляет собой соединение следующей структурной формулы (I):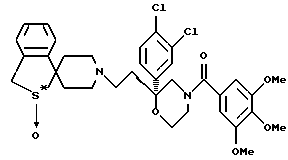
(в которой >S*-->O представляет собой сульфоксидную группу, в которой атом кислорода соединен с атомом серы в абсолютной конфигурации S).
Из гидрохлорида и фумарата 1-{2-[(2R)-(3,4-дихлорфенил)-4-(3,4,5-триметоксибензоил)морфолин-2-ил] этил} спиро[бензо(с)тиофен-1(3Н),4'-пиперидин] -(2S)-оксида согласно настоящему изобретению более предпочтительным является гидрохлорид.
Гидрохлорид и фумарат 1-{2-[(2R)-(3,4-дихлорфенил)-4-(3,4,5-триметоксибензоил)морфолин-2-ил] этил} спиро[бензо(с)тиофен-1(3Н), 4'-пиперидин] -(2S)-оксида согласно настоящему изобретению могут являться сольватами, абсорбирующими воду или растворитель для перекристаллизации, если их оставляют на воздухе или перекристаллизовывают. Такие соли также входят в объем настоящего изобретения.
Соли оптически активного сульфоксидного производного по настоящему изобретению можно получить путем превращения 1-{2-[(2R)-(3,4-дихлорфенил)-4-(3,4,5-триметоксибензоил)морфолин-2-ил] этил}спиро[бензо(с)тиофен-1(3Н),4'-пиперидин] -(2S)-оксида, полученного согласно ссылочным примерам, описанным ниже, в гидрохлорид или фумарат с помощью известных способов.
Новые соли оптически активного сульфоксидного производного по настоящему изобретению обладают отличным антагонистическим действием как в отношении рецепторов субстанции Р, так и в отношении рецепторов нейрокинина А, и, помимо этого, они имеют низкую токсичность и поэтому пригодны в качестве профилактического агента или средства для лечения заболеваний, опосредованных тахикинином. Примерами таких заболеваний являются заболевания центральной нервной системы, такие как беспокойство, депрессия, психоз и шизофрения; нейродегенеративные заболевания, такие как деменция при СПИДе, сенильная деменция Альцгеймера, болезнь Альцгеймера, синдром Дауна, демиелинизирующая болезнь, амиотрофический латеральный склероз, нейропатия, периферическая нейропатия и невралгия; респираторные заболевания, такие как хроническое обструктивное заболевание легких, бронхит, пневмония, бронхоконстрикция, астма и кашель; воспалительные заболевания, такие как воспалительное заболевание кишечника (ВЗК), псориаз, фиброз, эпифизарный остеомиелит, дегенеративный артрит и ревматоидный артрит; экзема; аллергии, такие как ринит; заболевания гиперчувствительности, такие как гиперчувствительность к винам; офтальмологические заболевания, такие как конъюнктивит, весенний конъюнктивит, весенний катар, деструкция гематоофтальмического барьера, вызванная различными воспалительными заболеваниями глаз, повышенное внутриглазное давление и миоз; кожные заболевания, такие как контактный дерматит, атопический дерматит, крапивница и другие экэемоподобные дерматиты; злоупотребления некоторыми веществами, такие как алкогольная зависимость; соматические заболевания, вызванные стрессом; симпатическая рефлекторная дистрофия, такая как синдром руки и плеча; дистимия; нежелательные иммунные реакции, такие как отторжение трансплантатов; заболевания, относящиеся к иммуностимуляции, такие как системная красная волчанка или иммуносупрессия; расстройства пищеварения, такие как заболевания, вызванные нарушениями нервной регуляции органов, колит, язвенный колит и болезнь Крона; рвота, такая как рвота, обусловленная побочными эффектами лучевой и химиотерапии, ядами, токсинами, беременностью, вестибулярными расстройствами, послеоперационными состояниями, непроходимостью желудочно-кишечного тракта, пониженной перистальтикой желудочно-кишечного тракта, висцеральной болью, мигренью, повышенным внутричерепным давлением, пониженным внутричерепным давлением или побочными реакциями, индуцированными введением различных лекарственных средств; функциональные болезни мочевого пузыря, такие как цистит и недержание мочи; эозинофилия, вызванная коллагенозами, склеродермией или инвазией Fasciola hepatica; заболевания, обусловленные нарушениями кровотока вследствие вазодилатации или вазоконстрикции, такие как стенокардия, мигрень и болезнь Рейнольдса; боли, обусловленные ноцицептивным восприятием, такие как головная боль при мигрени, головная боль и зубная боль.
Примеры путей введения солей оптически активного сульфоксидного производного по настоящему изобретению включают пероральное введение посредством, например, таблеток, капсул, гранул, порошков или сиропов, и парентеральное введение посредством инъекции, суппозитория и т.п. Такие фармацевтические препараты можно изготовить хорошо известными способами с использованием вспомогательных веществ, таких как наполнители (примеры включают органические наполнители, такие как производные сахаров, например лактоза, сахароза, декстроза, маннит и сорбит; производные крахмала, например кукурузный крахмал, картофельный крахмал, α-крахмал, декстрин и карбоксиметилкрахмал; производные целлюлозы, например кристаллическая целлюлоза, малозамещенная гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза, карбоксиметилцеллюлоза, карбоксиметилцеллюлозакальций и карбоксиметилцеллюлозанатрий с внутренними поперечными сшивками; аравийская камедь; декстран и пуллулан; и неорганические наполнители, такие как производные кремния, например легкий кремневый ангидрид, синтетический силикат алюминия и алюмометасиликат магния; фосфаты, например фосфат кальция; карбонаты, например карбонат кальция; и сульфаты, например сульфат кальция), смазывающие агенты (примеры включают стеариновую кислоту; соли стеариновой кислоты с металлами, такие как стеарат кальция и стеарат магния; тальк; коллоидный диоксид кремния; воски, такие как пчелиный воск и спермацет; борная кислота; адипиновая кислота; сульфаты, такие как сульфат натрия; гликоль; фумаровая кислота; бензоат натрия; DL-лейцин; натриевые соли алифатических кислот; лаурилсульфаты, такие как лаурилсульфат натрия и лаурилсульфат магния; кремневые кислоты, такие как кремневый ангидрид и силикат гидрат; и вышеописанные производные крахмала), связывающие агенты (примеры включают поливинилпирролидон, макрогол и соединения, сходные с приведенными выше примерами наполнителей), разрыхлители (примеры включают соединения, сходные с приведенными выше примерами наполнителей, и химически модифицированные крахмалы и целлюлозы, такие как кроскармеллоза-натрий, карбоксиметилкрахмал-натрий и поперечно сшитый поливинилпирролидон), стабилизаторы (примеры включают параоксибензоаты, такие как метилпарабен и пропилпарабен; спирты, такие как хлорбутанол, бензиловый спирт и фенилэтиловый спирт; хлорид бензалкония; фенол и производные фенола, такие как крезол; тимерозал; дегидроуксусную кислоту и сорбиновую кислоту), корригенты (примеры включают обычно применяемые в таких случаях подсластители, подкислители и отдушки) и/или разбавители.
Доза соединения по настоящему изобретению варьируется в зависимости от состояния и возраста пациента, пути введения и т.п. Соединение вводится перорально в количестве от 0,01 мг/кг веса (предпочтительно 0,1 мг/кг веса, нижний лимит) до 100 мг/кг веса (предпочтительно 50 мг/кг веса, верхний лимит) в виде однократной дозы; с другой стороны, соединение вводится внутривенно в количестве от 0,01 мг/кг веса (предпочтительно 0,05 мг/кг веса, нижний лимит) до 100 мг/кг веса (предпочтительно 50 мг/кг веса, верхний лимит) в виде однократной дозы. Желательно вводить соединение от одного до нескольких раз в день, в зависимости от состояния пациента.
Наилучшие варианты осуществления настоящего изобретения
Настоящее изобретение далее описывается более подробно с помощью примеров, ссылочных примеров, тестов и примеров композиций.
Пример 1
Гидрохлорид 1-{ 2-[(2R)-(3,4-дихлорфенил)-4-(3,4,5-триметоксибензоил)морфолин-2-ил]этил}спиро[бензо(с)тиофен-1(3Н),4'-пиперидин]-(2S)-оксида
В 220 мл 2-пропанола растворяют 21,4 г (31,8 ммоль) 1-{2-[(2R)-(3,4-дихлорфенил)-4-(3,4,5-триметоксибензоил)морфолин-2-ил]этил}спиро[бензо(с)тиофен-1(3Н),4'-пиперидин]-(2S)-оксида. К полученному раствору по каплям добавляют 39,8 мл (159 ммоль) 4 н. раствора хлористого водорода в диоксане при 0oС в течение 20 минут, после чего перемешивают в течение 30 минут. Реакционную смесь концентрируют до сухости путем отгонки растворителя при пониженном давлении. К остатку добавляют 220 мл диэтилового эфира, после чего дистиллируют при пониженном давлении. После двукратного повторения этой процедуры к остатку добавляют 110 мл диэтилового эфира с получением кристаллов. Эти кристаллы собирают фильтрованием и промывают диэтиловым эфиром, в результате чего получают 20,99 г целевого соединения.
[α]
Температура плавления: от 162 до 166oС
Спектр ядерного магнитного резонанса (400 МГц, CDCl3) δ миллионные доли: 13,2 (1Н, ш), 7,25-7,70 (7Н, м), 6,74 (2Н, с), 2,93-4,60 (14Н, М), 4,49 (1Н, д, J= 16 Гц), 4,10 (1Н, д, J=16 Гц), 3,87 и 3,94 (всего 9Н, с каждый), 2,63 (1Н, д, J=15 Гц), 2,47 (1Н, м), 2,20 (1Н, м), 1,91 (1Н, д, J=15 Гц)
Инфракрасный спектр поглощения: νмакс см-1 (KBr): 3429, 2963, 2937, 2482, 2404, 1635, 1584
Масс-спектрометрия (FAB) m/z: 673 (свободная форма, (М+Н)+)
Элементный анализ (для С34Н39N2O6SCl3•0,6Н2O (%))
Рассчитано: С 56,65, Н 5,62, N 3,89, S 4,45, Cl 14,75
Найдено: С 56,40, Н 5,91, N 3,75, S 4,16, Cl 14,82
Анализ посредством высокоэффективной жидкостной хроматографии:
Колонка: TSK-гель ODS-80TS (250•4,6 мм диаметр) (продукт компании TOSOH CORPORATION)
Растворитель: смесь 45: 55 0,1%-ного раствора ацетата аммония в ацетонитриле и 0,1%-ного водного раствора ацетата аммония
Скорость потока: 1,0 мл/мин
Время задержки: 21,0 мин
Пример 2
Фумарат 1-{2-[(2R)-(3,4-дихлорфенил)-4-(3,4,5-триметоксибензоил)морфолин-2-ил]этил}спиро[бензо(с)тиофен-1(3Н),4'-пиперидин]-(2S)-оксида
В 230 мл этилацетата растворяют 400 мг (3,45 ммоль) фумаровой кислоты, после чего добавляют 2,32 г (3,44 ммоль) 1-{2-[(2R)(3,4-дихлорфенил)-4-(3,4,5-триметоксибензоил)морфолин-2-ил] этил}спиро[бензо(с)тиофен-1(3Н),4'-пиперидин] -(2S)-оксида к полученному раствору для его растворения в этом растворе. Этот раствор оставляют стоять в течение ночи. Растворитель реакционной смеси отгоняют при пониженном давлении с получением остатка. Этот остаток растворяют в 5 мл метанола, а затем добавляют 200 мл диизопропилового эфира с получением кристаллов. Эти кристаллы собирают фильтрованием, в результате чего получают 2,52 г целевого соединения.
[α]
Температура плавления: от 151 до 155oС
Спектр ядерного магнитного резонанса (400 МГц, CDCl3) δ миллионные доли: 7,2-7,7 (7Н, м), 6,62 (2Н, с), 6,57 и 6,55 (всего 2Н, с каждый), 4,54 (1Н, д, J=17 Гц), 3,94 (1Н, д, J=17 Гц), 1,8-4,5 (18Н, м), 3,77 и 3,69 (всего 9Н, с каждый)
Инфракрасный спектр поглощения: νмакс см-1 (KBr): 3422, 2839, 1711, 1637, 1584, 1465, 1239, 1127
Масс-спектрометрия (FAB) m/z: 673 (свободная форма, (М+Н)+)
Элементный анализ (для C38H42N2O10SCl2•H2O (%))
Рассчитано: С 56,50, Н 5,49, N 3,47, S 3,97, Cl 8,78
Найдено: С 56,77, Н 5,39, N 3,34, S 3,55, Cl 8,33
Ссылочный пример 1
Гидрохлорид спиро[бензо[с]тиофен-1(3Н),4'-пиперидин]-(2S)-оксида
Ссылочный пример 1(а)
1'-Трет-бутоксикарбонил-спиро[бензо[с]тиофен-1(3Н),4'-пиперидин]
В 800 мл тетрагидрофурана растворяют 81,0 г (0,40 моль) 2-бромбензилтиола, после чего по каплям добавляют 516 мл (0,84 моль) н-бутиллития (1,6 моль, раствор в гексане) при -78oС в течение 6 часов. После перемешивания при той же температуре в течение 1,5 часов к реакционной смеси по каплям добавляют раствор 79,5 г (0,40 моль) N-трет-бутоксикарбонил-4-пиперидона в 800 мл тетрагидрофурана в течение 3 часов, а затем смесь перемешивают еще 1 час. К реакционной смеси добавляют насыщенный водный раствор хлорида аммония и смесь экстрагируют этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия и высушивают над безводным сульфатом натрия, а растворитель отгоняют при пониженном давлении с получением остатка. К этому остатку добавляют 2 литра 4 н. серной кислоты и смесь нагревают с обратным холодильником в течение 14 часов. При охлаждении льдом реакционную смесь подщелачивают 350 г (8,75 моль) гидроксида натрия, после чего добавляют 102 г (0,47 моль) ди-трет-бутилбикарбоната. Полученную смесь перемешивают в течение 1 часа. Реакционную смесь экстрагируют метиленхлоридом. Органический слой промывают насыщенным водным раствором хлорида натрия, а затем высушивают над безводным сульфатом натрия. Остаток, полученный после отгонки растворителя из экстракта при пониженном давлении, очищают посредством хроматографии на колонке с силикагелем (элюирующий растворитель; н-гексан : этилацетат 97: 3), в результате чего получают 56 г целевого соединения в виде белых кристаллов.
Температура плавления: от 131,0 до 132,5oС (н-гексан-этилацетат)
Спектр ядерного магнитного резонанса (400 МГц, CDCl3) δ миллионные доли: 7,28-7,24 (3Н, м), 7,17-7,15 (1Н, м), 4,23 (2Н, ш, с), 4,19 (2Н, с), 3,02 (2Н, ш, с), 2,07 (2Н, дт, J=4,4, 13 Гц), 1,88 (2Н, м), 1,49 (9Н, с)
Инфракрасный спектр поглощения: νмакс см-1 (KBr): 2970, 1680, 1428, 1234, 1163
Масс-спектрометрия (FAB) m/z: 306 (М+Н)+
Ссылочный пример 1(b)
1'-Трет-бутоксикарбонил-спиро[бензо[с]тиофен-1(3Н),4'-пиперидин]-2-оксид
В 420 мл хлороформа растворяют 42,0 г (0,14 моль) 1'-трет-бутоксикарбонил-спиро[бензо[с] тиофен-1(3Н), 4'-пиперидин] а, полученного в ссылочном примере 1(а), после чего добавляют 12,7 г (0,15 моль) бикарбоната натрия. К полученной смеси малыми порциями добавляют 28,0 г (содержание 85%, 0,14 моль) м-хлорпербензойной кислоты при охлаждении льдом. После перемешивания смеси в течение 30 минут при охлаждении льдом к ней добавляют 10 г иодида калия и смесь перемешивают при комнатной температуре в течение 30 минут. Реакционную смесь промывают водой и насыщенным водным раствором хлорида натрия и высушивают над безводным сульфатом натрия, а растворитель отгоняют при пониженном давлении с получением остатка. Остаток очищают посредством хроматографии на колонке с силикагелем (элюирующий растворитель н-гексан : этилацетат 1:1), в результате чего получают 42 г указанного в заглавии соединения в виде белых кристаллов.
Температура плавления: от 103o до 107oС (диизопропиловый эфир)
Спектр ядерного магнитного резонанса (400 МГц, CDCl3) δ миллионных долей: 7,37-7,32 (3Н, м), 7,25-7,23 (1Н, м), 4,37 (1H, д, J=16,7 Гц), 4,13 (2Н, ш, с), 4,05 (2Н, д, J=16,7 Гц), 3,21 (2Н, ш, с), 2,43 (1H, м), 2,21 (1H, м), 1,70 (1H, м), 1,61 (1H, м), 1,50 (9Н, с)
Инфракрасный спектр поглощения: νмакс см-1 (KBr): 2985, 1686, 1429, 1368, 1286, 1167
Масс-спектрометрия (FAB) m/z: 322 (М+Н)+
Ссылочный пример 1(с)
Спиро[бензо[с]тиофен-1(3Н),4'-пиперидин]-2-оксид
В 420 мл 2-пропанола растворяют 42,0 г (0,13 моль) 1'-трет-бутоксикарбонил-спиро[бензо[с]тиофен-1(3Н),4'-пиперидин]-2-оксида, полученного в ссылочном примере 1(b), после чего добавляют 150 мл 4 н. раствора хлористого водорода в диоксане при охлаждении льдом и смесь перемешивают в течение 4 часов. К реакционной смеси добавляют 200 мл диэтилового эфира. После этого смесь оставляют стоять в течение 1 часа при охлаждении льдом, в результате чего получают кристаллы. Эти кристаллы собирают фильтрованием. Полученные кристаллы растворяют в 200 мл 5%-ного водного раствора гидроксида натрия. Этот раствор экстрагируют метиленхлоридом. Органический слой высушивают над безводным сульфатом натрия и растворитель отгоняют при пониженном давлении, в результате чего получают 21,7 г целевого соединения в виде белого аморфного продукта.
Ссылочный пример 1(d)
(S)-(+)-манделат спиро[бензо[с]тиофен-1(3Н),4'-пиперидин]-(2S)-оксида
В 3350 мл ацетонитрила растворяют 33,51 г (0,15 моль) спиро[бензо[с]тиофен-1(3Н),4'-пиперидин]-2-оксида, полученного в ссылочном примере 1(с), после чего добавляют 11,52 г (75,7 ммоль) (S)-(+)-миндальной кислоты. Полученный раствор оставляют стоять при комнатной температуре в течение ночи. Кристаллы, выпавшие в осадок в реакционной смеси, собирают фильтрованием, в результате чего получают 19,62 г целевого соединения в виде белых кристаллов. Фильтрат концентрируют при пониженном давлении с получением остатка. Остаток растворяют в 5%-ном водном растворе гидроксида натрия, после чего экстрагируют метиленхлоридом. Органический слой высушивают над безводным сульфатом натрия и растворитель отгоняют при пониженном давлении с получением 22,01 г (99,5 ммоль) остатка. Этот остаток растворяют в 2200 мл ацетонитрила при нагревании и в полученном растворе растворяют 7,22 г (47,5 ммоль) (R)-(-)-миндальной кислоты. Полученный раствор оставляют стоять при комнатной температуре в течение ночи, в результате чего получают кристаллы. Эти кристаллы собирают фильтрованием, в результате чего получают 15,91 г (R)-(-)-манделата спиро[бензо[с] тиофен-1(3Н), 4'-пиперидин]-(2R)-оксида в виде белых кристаллов. Фильтрат снова концентрируют при пониженном давлении и остаток растворяют в 5%-ном водном растворе гидроксида натрия. Полученный раствор экстрагируют метиленхлоридом. Органический слой высушивают над безводным сульфатом натрия и растворитель отгоняют при пониженном давлении с получением 11,51 г (52,0 ммоль) остатка. Этот остаток растворяют в 1100 мл ацетонитрила при нагревании, а затем добавляют 3,95 г (26,0 ммоль) (S)-(+)-миндальной кислоты. Полученный раствор оставляют стоять при комнатной температуре в течение ночи с получением кристаллов. Эти кристаллы собирают фильтрованием с получением 4,73 г целевого соединения в виде белых кристаллов. Порции целевого соединения, полученные описанным способом, объединяют, 24,00 г растворяют в 9,6 литрах ацетонитрила при нагревании и раствор оставляют стоять при комнатной температуре в течение ночи с получением 20,13 г кристаллов. Оптическая чистота этих кристаллов составляет 99,8% по результатам анализа ВЭЖХ 1'-трет-бутоксикарбонил-спиро[бензо[с]тиофен-1(3Н),4'-пиперидин]-(2S)-оксида, приготовленного из этих кристаллов.
Температура плавления: от 197 до 200oС
[α]
Инфракрасный спектр поглощения: νмакс см-1 (KBr): 3388, 3029, 1629, 1332, 1017
Масс-спектрометрия (EI) m/z: 221 (свободная форма М+)
Ссылочный пример 1(е)
1'-Трет-бутоксикарбонил-спиро[бензо[с] тиофен-1(3Н),4'-пиперидин]-(2S)-оксид
В 200 мл 5%-ного водного раствора гидроксида натрия растворяют 19,88 г (53,2 ммоль) (S)-(+)-манделата, синтезированного в ссылочном примере 1(d), после чего экстрагируют метиленхлоридом (200 мл, трижды). Органический слой высушивают над безводным сульфатом магния, а затем растворитель отгоняют при пониженном давлении, в результате чего получают остаток. В 300 мл метиленхлорида растворяют 11,80 г этого остатка, после чего последовательно добавляют 11,2 мл (79,8 ммоль) триэтиламина и 17,4 г (79,8 ммоль) ди-трет-бутилбикарбоната, при охлаждении льдом. После перемешивания при комнатной температуре в течение ночи реакционную смесь разбавляют 200 мл метиленхлорида, промывают 10%-ным водным раствором лимонной кислоты и насыщенным водным раствором бикарбоната натрия, а затем высушивают над безводным сульфатом магния. Растворитель отгоняют при пониженном давлении, в результате чего получают остаток. Этот остаток очищают посредством хроматографии на колонке с силикагелем (элюирующий растворитель н-гексан : этилацетат от 4:6 до 3:7), после чего перекристаллизовывают из диизопропилового эфира, в результате чего получают 13,1 г целевого соединения в виде белых кристаллов.
Температура плавления: от 129,0 до 130,5oС (диизопропиловый эфир)
[α]
Анализ ВЭЖХ:
Колонка: Chiral Cel OD (250•4,6 мм диаметр)
Элюирующий растворитель: н-гексан : 2-пропанол 80:20
Скорость потока: 0,8 мл/мин
Время задержки: 18,1 мин
Спектр ядерно-магнитного резонанса, инфракрасный спектр поглощения и масс-спектральный анализ кристаллов были идентичны результатам для рацемической формы, полученной в ссылочном примере 1(b).
Ссылочный пример 1(f)
Гидрохлорид спиро[бензо[с]тиофен-1(3Н),4'-пиперидин]-(2S)-оксида
В 130 мл 2-пропанола растворяют 13,0 г (40,4 ммоль) 1'-трет-бутоксикарбонил-спиро[бензо[с]тиофен-1(3Н),4'-пиперидин]-(2S)-оксида, полученного в ссылочном примере 1(е), после чего добавляют 50 мл 4 н. раствора хлористого водорода в диоксане при охлаждении льдом. После перемешивания в течение одного часа при охлаждении льдом реакционную смесь перемешивают в течение еще 6 часов при комнатной температуре. Реакционную смесь концентрируют при пониженном давлении с получением остатка. К этому остатку добавляют 200 мл диэтилового эфира и растворитель отгоняют из полученной смеси при пониженном давлении (трижды). Остаток перекристаллизовывают из 300 мл смеси метанола и диэтилового эфира 1:2, в результате чего получают 9,10 г целевого соединения в виде белых кристаллов.
Температура плавления: от 209,5 до 210,5oС
[α]
Ссылочный пример 2
1'-Трет-бутоксикарбонил-спиро[бензо[с] тиофен-1(3Н),4'-пиперидин]-(2S)-оксид
В 5 мл метиленхлорида растворяют 250 мг (0,82 ммоль) 1'-трет-бутоксикарбонил-спиро[бензо[с] тиофен-1(3Н),4'-пиперидин]а, полученного в ссылочном примере 1(а). К полученному раствору добавляют 308 мг (0,82 ммоль) (3'S, 2R)-(-)-N-(фенилсульфонил)(3,3-дихлоркамфорил)оксазолидина, полученного в соответствии со способом F. A. Davis et al. (J.Am.Chem.Soc., 114, 1428 (1992)), и полученную смесь перемешивают при комнатной температуре в течение ночи. К реакционной смеси добавляют 500 мг иодида калия, после чего перемешивают при комнатной температуре в течение 30 минут. Реакционную смесь промывают последовательно водой и насыщенным водным раствором хлорида натрия и высушивают над безводным сульфатом натрия, а затем растворитель отгоняют при пониженном давлении. Остаток очищают посредством хроматографии на колонке с силикагелем (элюирующий растворитель н-гексан : этилацетат от 1:2), в результате чего получают 245 г целевого соединения.
Оптическая чистота: 94% ее
Ссылочный пример 3
3-(3,4-Дихлорфенил)-3-бутен-1-ол-трет-бутилдиметилсилиловый эфир
Ссылочный пример 3(а)
Метил 3-(3,4-дихлорфенил)-3-бутеноат
К 300 мл диэтилового эфира добавляют 11,31 г (0,47 моль) металлического магния куском, после чего добавляют небольшое количество иода. Смесь оставляют стоять 1 час, после чего по каплям медленно добавляют раствор 102,87 г (0,46 моль) 1-бром-3,4-дихлорбензола в диэтиловом эфире (150 мл). К реакционной смеси добавляют 150 мл диэтилового эфира, а затем медленно добавляют 60,33 г (44,3 ммоль) безводного хлорида цинка и полученную смесь перемешивают течение 1 часа. После добавления к реакционной смеси 3,10 г (4,42 ммоль) хлорида бис (трифенилфосфин) палладия к полученной смеси по каплям добавляют раствор 34,15 мл (42,8 ммоль) дикетона в диэтиловом эфире (600 мл). Реакционную смесь перемешивают при комнатной температуре в течение 30 минут.
Реакционную смесь выливают в 1 литр 1 н. хлористоводородной кислоты, которую перед этим охлаждали в смеси воды и льда, и смесь экстрагируют диэтиловым эфиром (500 мл, трижды). Органические слои объединяют и экстрагируют 1 н. водным раствором гидроксида натрия (700 мл, трижды). Водные слои объединяют и подкисляют концентрированной хлористоводородной кислотой при охлаждении льдом. Полученный раствор экстрагируют диэтиловым эфиром (500 мл, трижды) и органические слои высушивают над безводным сульфатом магния, а растворитель отгоняют при пониженном давлении с получением остатка, который растворяют в 350 мл метанола. К этому раствору добавляют 10 мл концентрированной серной кислоты, после чего нагревают с обратным холодильником в течение 30 минут. После охлаждения реакционной смеси до комнатной температуры ее нейтрализуют насыщенным водным раствором бикарбоната натрия, а метанол отгоняют при пониженном давлении с получением остатка, который экстрагируют метиленхлоридом (200 мл, трижды). Органические слои объединяют, высушивают над безводным сульфатом магния, а затем концентрируют при пониженном давлении с получением остатка. Этот остаток дистиллируют при пониженном давлении, в результате чего получают 69,13 г (62%) целевого соединения в виде светло-желтого масла.
Температура кипения: от 144 до 146oС (5 мм рт.ст.)
Спектр ядерного магнитного резонанса (400 МГц, CDCl3) δ миллионных долей: 7,51 (1Н, д, J=2,2 Гц), 7,40 (1Н, д, J=8,2 Гц), 7,25 (1Н, дд, J=8,2, 2,2 Гц), 5,55 (1Н, с), 5,30 (1H, с), 3,67 (3Н, с), 3,49 (2Н, с)
Ссылочный пример 3(b)
3-(3,4-Дихлорфенил)-3-бутен-1-ол-трет-бутилдиметилсилиловый эфир
В 500 мл безводного тетрагидрофурана суспендируют 11,76 г (0,28 моль) гидрида литийалюминия. К суспензии по каплям добавляют раствор 69,06 г (0,28 моль) метил 3-(3,4-дихлорфенил)-3-бутеноата, полученного в ссылочном примере 3(а), в безводном тетрагидрофуране (500 мл), при 0oС в течение 15 минут, в атмосфере азота. После перемешивания при той же температуре в течение 30 минут к реакционной смеси постепенно добавляют 500 мл воды и 500 мл 10%-ного водного раствора гидроксида натрия. Полученную смесь перемешивают в течение еще 1 часа при комнатной температуре.
Реакционную смесь фильтруют через целитовый фильтр. Фильтрат экстрагируют этилацетатом (500 мл, трижды). Органические слои объединяют и высушивают над безводным сульфатом магния, а затем концентрируют при пониженном давлении с получением остатка. Этот остаток растворяют в 250 мл безводного диметилформамида. К полученному раствору последовательно добавляют 47,12 мл (0,34 моль) триэтиламина, 6,88 г (0,06 моль) 4-диметиламинопиридина и 50,96 г (0,34 моль) трет-бутилдиметилсилилхлорида при охлаждении льдом и
смесь перемешивают в течение 2 часов при охлаждении льдом.
К реакционной смеси добавляют литр этилацетата. Полученную смесь промывают последовательно 10%-ной хлористоводородной кислотой, которую предварительно охлаждали льдом, и насыщенным водным раствором хлорида натрия, а затем высушивают над безводным сульфатом магния. Остаток, полученный после отгонки растворителя из смеси при пониженном давлении, очищают посредством флэшхроматографии на колонке с силикагелем (элюирующий растворитель н-гексан : этилацетат от 50: 1 до 20:1), в результате чего получают 43,52 г (47%) целевого соединения в виде бесцветного масла.
Спектр ядерного магнитного резонанса (400 МГц, CDCl3) δ миллионные доли: 7,50 (1Н, д, J= 2,1 Гц), 7,38 (1Н, д, J=8,1 Гц), 7,24 (1Н, дд, J=8,1, 2,1 Гц), 5,35 (1Н, с), 5,16 (1H, с), 3,70 (2Н, т, J=6,9 Гц), 2,67 (2Н, т, J=6,9 Гц), 0,89 (9Н, с), 00,0 (6Н, с)
Ссылочный пример 4
3-(3,4-Дихлорфенил)-3-бутен-1-ол-трет-бутилдиметилсилиловый эфир
Ссылочный пример 4(а)
3-(3,4-Дихлорфенил)-3-оксо-1-пропанол
В 2,4 литрах этанола растворяют 119 г (0,46 моль) этил 3-(3,4-дихлорфенил)-3-оксопропионата. К полученному раствору добавляют 115 мл (0,68 моль) этилортоформиата и 4,4 г (2,28 ммоль) п-толуолсульфоновой кислоты, после чего нагревают с обратным холодильником в течение 8 часов. Реакционную смесь выливают в 1 литр насыщенного водного раствора бикарбоната натрия и смесь экстрагируют этилацетатом (700 мл, трижды). Органические слои объединяют, промывают насыщенным водным раствором хлорида натрия, а затем высушивают над безводным сульфатом натрия. После отгонки растворителя при пониженном давлении остаток растворяют в 800 мл тетрагидрофурана. Полученный раствор по каплям добавляют к суспензии 25,9 г (0,68 моль) гидрида литийалюминия в 4 литрах тетрагидрофурана в течение 1 часа при охлаждении льдом. После перемешивания при 0oС в течение 2 часов добавляют 250 мл воды и 125 мл 10%-ного водного раствора гидроксида натрия и смесь перемешивают при комнатной температуре в течение еще 1 часа. Реакционную смесь фильтруют через целитовый фильтр. Фильтрат выливают в 1 литр насыщенного водного раствора хлорида натрия, после чего экстрагируют этилацетатом. Органический слой высушивают над безводным сульфатом натрия и концентрируют при пониженном давлении. Остаток растворяют в 500 мл хлороформа. При охлаждении льдом к полученному раствору по каплям добавляют 500 мл 50%-ной трифторуксусной кислоты в течение 30 минут и смесь перемешивают в течение 30 минут. Реакционную смесь разбавляют 300 мл метиленхлорида. Органический слой промывают водой и насыщенным водным раствором бикарбоната натрия и высушивают над безводным сульфатом натрия, а затем растворитель отгоняют при пониженном давлении. Остаток очищают посредством хроматографии на колонке с силикагелем (элюирующий растворитель н-гексан : этилацетат 9: 1), в результате чего получают 46 г соединения в виде белых кристаллов.
Спектр ядерного магнитного резонанса (270 МГц, CDCl3) δ миллионные доли: 8,05 (1Н, д, J= 2,0 Гц), 7,79 (1Н, дд, J=2,0, 8,1 Гц), 7,57 (1Н, д, J=8,1 Гц), 4,04 (2Н, м), 3,19 (2Н, т, J=5,3 Гц), 2,44 (1Н, т, J=6,6 Гц, D2O исчезает)
Ссылочный пример 4(b)
3-(3,4-Дихлорфенил)-3-оксо-1-пропанол-трет-бутилдиметилсилиловый эфир
В 460 мл диметилформамида растворяют 46,0 г (0,21 моль) 3-(3,4-дихлорфенил)-3-оксо-1-пропанола, полученного в ссылочном примере 4(а), после чего добавляют 35 мл (0,25 моль) триэтиламина и 38,0 г (0,25 моль) трет-бутилдиметилхлорсилана при охлаждении льдом. Полученную смесь перемешивают при 0oС в течение 2 часов. Реакционную смесь выливают в воду и смесь экстрагируют этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия и высушивают над безводным сульфатом натрия, растворитель отгоняют при пониженном давлении, в результате чего получают остаток, который очищают посредством хроматографии на колонке с силикагелем (элюирующий растворитель н-гексан : этилацетат 96:4), в результате чего получают 66,1 г целевого соединения в виде белых кристаллов.
Спектр ядерного магнитного резонанса (270 МГц, CDCl3) δ миллионные доли: 8,06 (1Н, д, J= 2,0 Гц), 7,80 (1Н, дд, J=2,0, 8,3 Гц), 7,55 (1Н, д, J=8,3 Гц), 4,04 (2Н, т, J=6,3 Гц), 3,13 (2Н, т, J=6,3 Гц), 0,85 (9Н, с), 0,04 (6Н, с)
Ссылочный пример 4(с)
3-(3,4-Дихлорфенил)-3-бутен-1-ол-трет-бутилдиметилсилиловый эфир
К 2 литрам высушенного бензола добавляют 215 г (0,60 моль) бромида метилтрифенилфосфония и 54 г (0,48 моль) т-бутоксида калия и смесь перемешивают при комнатной температуре в течение 9 часов. В 800 мл бензола растворяют 40 г (0,12 моль) 3-(3,4-дихлорфенил)-3-оксо-1-пропанол-трет-бутилдиметилсилилового эфира, полученного в ссылочном примере 4(b), и полученный раствор по каплям добавляют к реакционной смеси в течение 2,5 часов. К реакционной смеси добавляют 1 литр воды и перемешивают при охлаждении льдом в течение 30 минут. Органический слой промывают водой и насыщенным водным раствором хлорида натрия и высушивают над безводным сульфатом натрия, после чего растворитель отгоняют при пониженном давлении с получением остатка, который очищают посредством хроматографии на колонке с силикагелем (элюирующий растворитель н-гексан), в результате чего получают 23,5 г целевого соединения. Данные различных спектральных анализов идентичны данным, полученным в ссылочном примере 3(b).
Ссылочный пример 5
3-(3,4-Дихлорфенил)-3-бутен-1-ол-трет-бутилдиметилсилиловый эфир
К 2 мл диэтилового эфира добавляют 129 мг (5,31 ммоль) металлического магния куском, после чего добавляют малое количество иода. К полученной смеси по каплям добавляют раствор 1,01 г (4,47 ммоль) 3,4-дихлорбромбензола в диэтиловом эфире (1 мл) и смесь перемешивают при комнатной температуре в течение 1 часа в атмосфере азота с получением реагента Гриньяра. В 5 мл высушенного тетрагидрофурана растворяют 500 мл (1,60 ммоль) 3-иод-3-бутен-1-ол-трет-бутилдиметилсилилового эфира и 34 мг (0,048 ммоль) бистрифенилфосфин палладия (II) хлорида, после чего по каплям добавляют реагент Гриньяра при комнатной температуре в атмосфере азота. По мере нагревания растворитель отгоняют из реакционной смеси с получением остатка. Остаток хранят при 60oС в течение 1 часа, а затем его выливают в водный раствор хлорида аммония и смесь экстрагируют этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия и высушивают над безводным сульфатом натрия, а растворитель отгоняют при пониженном давлении. Остаток очищают посредством хроматографии на колонке с силикагелем (элюирующий растворитель н-гексан), в результате чего получают 422 мг целевого соединения. Данные различных спектральных анализов этого продукта идентичны данным, полученным в ссылочном примере 3(b).
Ссылочный пример 6
1-{ 2-[(2R)-(3,4-дихлорфенил)-4-(3,4,5-триметоксибензоил)морфолин-2-ил] этил}спиро[бензо[с]тиофен-1(3Н),4'-пиперидин]-(2S)-оксид
Ссылочный пример 6(а)
4-Трет-бутилдиметилсилилокси-(2R)-(3,4-дихлорфенил)бутан-1,2-диол
В смеси 500 мл 2-метил-2-пропанола и 500 мл воды растворяют 790 мг (1,01 ммоль) (DHQD)2-PHAL (гидрохинидин 1,4-фталазиндииловый диэфир), 100,19 г (0,30 моль) K3Fe(CN)6 (феррицианид калия), 42,06 г (0,30 моль) карбоната калия и 0,516 мл (0,20 моль) тетраоксида осмия (0,393 М раствор в толуоле), после чего добавляют 33,61 г (0,10 моль) 3-(3,4-дихлорфенил)-3-бутен-1-ол-трет-бутилдиметилсилиловый эфир при 0oС. Полученную смесь перемешивают в течение 5 часов при 0oС. К реакционной смеси добавляют 150 г сульфата натрия и смесь перемешивают при комнатной температуре в течение 1 часа. Реакционную смесь экстрагируют этилацетатом (800 мл, трижды). Органические слои объединяют и высушивают над безводным сульфатом магния. После отгонки растворителя при пониженном давлении остаток очищают посредством флэшхроматографии на колонке с силикагелем (элюирующий растворитель н-гексан : этилацетат от 5: 1 до 1:1), в результате чего получают 32,3 г (87%) целевого соединения в виде бесцветного масла.
Оптическая чистота: 97% ее
[α]
Спектр ядерного магнитного резонанса (400 МГц, CDCl3) δ миллионные доли: 7,57 (1Н, д, J= 2,1 Гц), 7,43 (1Н, д, J=8,1 Гц), 7,24 (1Н, дд, J=8,1, 2,1 Гц), 5,00 (1Н, с), 3,80 (1H, ддд, J=10,4, 3,8, 3,8 Гц), 3,5-3,7 (3Н, м), 2,51 (1H, дд, J= 8,0, 5,2 Гц), 2,37 (1H, ддд, J=15,0, 11,1, 4,0 Гц), 1,86 (1H, ддд, J=15,0, 2,9, 2,9 Гц), 0,89 (9Н, с), 0,04 (3Н, с), -0,01 (3Н, с)
Ссылочный пример 6(b)
4-(Трет-бутилдиметилсилилокси)-(2R)-(3,4-дихлорфенил)-1-[N-(трет-бутоксикарбонил)-N-(2-гидроксиэтил)амино]-2-бутанол
В 80 мл пиридина растворяют 39,9 г (109 ммоль) 4-трет-бутилдиметилсилилокси-(2R)-(3,4-дихлорфенил)бутан-1,2-диола, полученного в ссылочном примере 6(а), после чего добавляют 31,3 г (164 ммоль) п-толуолсульфонилхлорида. Смесь перемешивают при комнатной температуре в течение 2 дней в атмосфере азота. Реакционную смесь разбавляют водой и экстрагируют этилацетатом. Органический слой промывают водой и насыщенным водным раствором хлорида натрия и высушивают над безводным сульфатом натрия. Затем растворитель отгоняют при пониженном давлении с получением остатка, который растворяют в 600 мл ацетонитрила. К раствору добавляют 35,0 г (329 ммоль) перхлората лития и 33,4 г (547 ммоль) 2-аминоэтанола. Эту смесь нагревают с обратным холодильником в течение 16 часов. После охлаждения до комнатной температуры реакционную смесь разбавляют этилацетатом, промывают насыщенным водным раствором хлорида натрия и высушивают над безводным сульфатом натрия, после чего растворитель отгоняют при пониженном давлении с получением остатка, который растворяют в 700 мл метиленхлорида. К этому раствору добавляют 22,8 мл (164 ммоль) триэтиламина и 26,3 г (120 ммоль) ди-трет-бутилбикарбоната. Смесь перемешивают при комнатной температуре в течение 12 часов. Реакционную смесь выливают в воду и полученную смесь экстрагируют метиленхлоридом. Органический слой промывают насыщенным водным раствором хлорида натрия и высушивают над безводным сульфатом натрия, а растворитель отгоняют при пониженном давлении с получением остатка, который очищают посредством хроматографии на колонке с силикагелем (элюирующий растворитель н-гексан : этилацетат от 4:1 до 7:3), в результате чего получают 49,9 г целевого соединения.
[α]
Спектр ядерного магнитного резонанса (400 МГц, CDCl3) δ миллионные доли: 7,30-7,75 (3Н, м), 5,30 и 5,57 (всего 1Н, каждый из ш.с), 3,05-4,00 (9Н, м), 2,00-2,40 (2Н, м), 1,53 (9Н, с), 0,94 (9Н, с), 0,09 (3Н, с), 0,07 (3Н, с)
Инфракрасный спектр поглощения: νмакс см-1 (KBr): 3420, 2957, 2933, 2885, 2861, 1687
Масс-спектрометрия (FAB) m/z: 508 (М+Н)+
Ссылочный пример 6(с)
2-[4-(трет-бутоксикарбонил)-(2R)-3,4-дихлорфенил)морфолин-2-ил] этанол-трет-бутилдиметилсилиловый эфир
В 600 мл высушенного толуола растворяют 49,9 г (98,1 ммоль) 4-(трет-бутилдиметилсилилокси)-(2R)-(3,4-дихлорфенил)-1-[N-(трет-бутоксикарбонил)-N-(2-гидроксиэтил)амино]-2-бутанола, полученного в ссылочном примере 6(b), и 30,9 г (118 ммоль) трифенилфосфина. К этому раствору по каплям добавляют 51,3 г (118 ммоль) 40%-ного раствора диэтилазодикарбоксилата в толуоле при комнатной температуре в атмосфере азота, после чего перемешивают в течение 2 часов. Растворитель отгоняют из реакционной смеси при пониженном давлении с получением остатка, который очищают посредством хроматографии на колонке с силикагелем (элюирующий растворитель н-гексан : этилацетат от 47:3 до 23:2), в результате чего получают 43,2 г целевого соединения.
[α]
Спектр ядерного магнитного резонанса (400 МГц, CDCl3) δ миллионные доли: 7,56 (1Н, ш. с), 7,43 (1Н, д, J=9 Гц), 7,28 (1Н, дд, J=2,9 Гц), 3,00-4,55 (8Н, м), 1,80-2,10 (2Н, м), 1,35-1,60 (9Н, ш.с), 0,85 (9Н, с), -0,01 (6Н, с)
Инфракрасный спектр поглощения: νмакс см-1 (СНСl3): 2957, 2931, 2859, 1687
Масс-спектрометрия (FAB) m/z: 490 (M+H)+
Ссылочный пример 6(d)
Гидрохлорид (2R)-(3,4-дихлорфенил)-2-(2-гидроксиэтил)морфолина
В 600 мл 4 н. раствора хлористого водорода в диоксане растворяют 43,1 г (87,9 ммоль) 2-[4-(трет-бутоксикарбонил)-(2R)-(3,4-дихлорфенил)морфолин-2-ил]этанол-трет-бутилдиметилсилилового эфира, полученного в ссылочном примере 6(с).
Полученный раствор перемешивают при 60oС в течение 4 часов. После отгонки растворителя из реакционной смеси при пониженном давлении к остатку добавляют диэтиловый эфир. Растворитель из этой смеси отгоняют при пониженном давлении, в результате чего получают остаток, который перекристаллизовывают из этанола/этилацетата, с получением 24,1 г целевого соединения.
[α]
Спектр ядерного магнитного резонанса (400 МГц, DMSO-D6) δ миллионные доли: 8,60-9,80 (2Н, ш.с), 7,72 (1Н, с), 7,70 (1Н, д, J=9 Гц), 7,44 (1Н, дд, J= 2, 9 Гц), 4,53 (1H, ш.с), 3,89 (1H, дт, J=4, 13 Гц), 3,75 (1H, д, J=14 Гц), 3,68 (1H, м), 3,30-3,45 (2Н, м), 2,93-3,13 (3Н, м), 2,09 (1H, м), 1,90 (1H, м)
Инфракрасный спектр поглощения: νмакс см-1 (KBr): 3378, 2966, 2893, 2812, 2783, 2724, 2656, 2530, 1598
Масс-спектрометрия (FAB) m/z: 276 ((M+H)+ свободная форма)
Ссылочный пример 6(е)
2-[(2R)-(3,4-дихлорфенил)-4-(3,4,5-триметоксибензоил)морфолин-2-ил] этанол
В 500 мл метиленхлорида суспендируют 22,9 г (82,9 ммоль) гидрохлорида (2R)-(3,4-дихлорфенил)-2-(2-гидроксиэтил)морфолина, полученного в ссылочном примере 6(d). К этой суспензии добавляют 27,6 мл (1,99 ммоль) триэтиламина, 21,0 г (91,0 ммоль) 3,4,5-триметоксибензоилхлорида и 100 мг 4-диметиламинопиридина, после чего перемешивают при комнатной температуре в течение 12 часов. Реакционную смесь выливают в воду и смесь экстрагируют метиленхлоридом. Органический слой промывают насыщенным водным раствором хлорида натрия и высушивают над безводным сульфатом натрия, а растворитель отгоняют при пониженном давлении с получением остатка, который очищают посредством хроматографии на колонке с силикагелем (элюирующий растворитель метиленхлорид : ацетон от 4:1 до 7:3), в результате чего получают 30,0 г целевого соединения.
[α]
Спектр ядерного магнитного резонанса (400 МГц, CDCl3) δ миллионные доли: 6,80-7,80 (3Н, м), 6,47 (2Н, с), 3,40-4,80 (8Н, м), 3,84 и 3,86 (всего 9Н, с каждый), 1,75-2,25 (2Н, м)
Инфракрасный спектр поглощения: νмакс см-1 (KBr): 3429, 2940, 2838, 1630, 1585
Масс-спектрометрия (EI) m/z: 469 (М+)
Ссылочный пример 6(f)
Метансульфонат 2-[(2R)-(3,4-дихлорфенил)-4-(3,4,5-триметоксибензоил)морфолин-2-ил]этанола
В 500 мл метиленхлорида растворяют 30,0 г (63,8 ммоль) 2-[(2R)-(3,4-дихлорфенил)-4-(3,4,5-триметоксибензоил)морфолин-2-ил] этанола, полученного в ссылочном примере 6(е), после чего последовательно добавляют 11,5 мл (83,0 ммоль) триэтиламина и 5,93 мл (76,6 ммоль) метансульфонилхлорида при охлаждении льдом. В атмосфере азота смесь перемешивают при комнатной температуре в течение 2 часов. Реакционную смесь разбавляют метиленхлоридом, промывают 1 н. хлористоводородной кислотой и насыщенным водным раствором хлорида натрия, а затем высушивают над безводным сульфатом натрия. Растворитель отгоняют при пониженном давлении с получением остатка. Остаток очищают посредством хроматографии на колонке с силикагелем (элюирующий растворитель н-гексан : этилацетат от 1:4 до 1:9), в результате чего получают 34,8 г целевого соединения.
[α]
Спектр ядерного магнитного резонанса (400 МГц, CDCl3) δ миллионные доли: 6,90-7,80 (3Н, м), 6,52 (2Н, с), 3,40-4,35 (8Н, м), 3,86 и 3,87 (всего 9Н, с каждый), 2,93 (3Н, с), 2,10-2,55 (2Н, м)
Инфракрасный спектр поглощения: νмакс см-1 (KBr): 2999, 2966, 2939, 2875, 1634, 1585
Масс-спектрометрия (FAB) m/z: 548 (М+Н)+
Ссылочный пример 6(g)
1-{ 2-[(2R)-(3,4-дихлорфенил)-4-(3,4,5-триметоксибензоил)морфолин-2-ил] этил}спиро[бензо[с]тиофен-1(3Н),4'-пиперидин]-(2S)-оксид
В 150 мл безводного диметилформамида суспендируют 15,00 г (27,4 ммоль) мезилированного соединения, полученного в ссылочном примере 6(f), 7,76 г (30,1 ммоль) гидрохлорида спиро[бензо[с]тиофен-1(3Н), 4'-пиперидин]-(2S)-оксида, 6,89 г (82,0 ммоль) бикарбоната натрия и 6,81 г (41,0 ммоль) иодида калия, после чего нагревают до 80oС в течение 8 часов в атмосфере азота. Реакционную смесь выливают в 400 мл насыщенного водного раствора хлорида натрия и экстрагируют этилацетатом. Органический слой высушивают над безводным сульфатом магния, а затем растворитель отгоняют при пониженном давлении с получением остатка. Этот остаток очищают посредством хроматографии на колонке с силикагелем (элюирующий растворитель метиленхлорид : метанол от 40:1 до 20:1), кристаллизуют из н-гексана, в результате чего получают 15,5 г целевого соединения в виде белых кристаллов.
[α]
Анализ ВЭЖХ:
Колонка: YMC-Pack ODS-A (250•4,6 мм диаметр)
Элюирующий растворитель: СН3СN:H2О = 40:60, 0,1% ацетат аммония
Скорость потока: 1,0 мл/мин
Время задержки: 23,7 мин
Спектр ядерного магнитного резонанса (400 МГц, CDCl3) δ миллионные доли: 7,1-7,8 (7Н, м), 6,49 (2Н, ш.с), 4,31 (1Н, д, J=16,8 Гц), 3,99 (1Н, д, J= 16,8 Гц), 3,86 и 3,84 (всего 9Н, с каждый), 3,3-4,0 (6Н, м), 1,5-3,1 (12Н, м)
Инфракрасный спектр поглощения: νмакс см-1 (KBr): 2939, 1636, 1584, 1464, 1426, 1329, 1237, 1128
Масс-спектрометрия (FAB) m/z: 673 (М+Н)+
Элементный анализ (для C34H38N2O6SCl2•0,5Н2О (%))
Рассчитано: С 59,82, Н 5,76, N 4,10, S 4,70, Cl 10,39
Найдено: С 60,20, Н 6,14, N 4,04, S 4,54, Cl 10,38
Ссылочный пример 7
1-{ 2-[(2R)-(3,4-дихлорфенил)-4-(3,4,5-триметоксибензоил)морфолин-2-ил] этил}спиро[бензо[с]тиофен-1(3Н),4'-пиперидин]-(2S)-оксид
Ссылочный пример 7(а)
2-[(2R)-(3,4-дихлорфенил)-4-(3,4,5-триметоксибензоил)морфолин-2-ил] этанал
В 10 мл метиленхлорида растворяют 0,88 мл (10,1 ммоль) оксалилхлорида. К полученному раствору по каплям добавляют раствор 0,79 мл (11,1 ммоль) диметилсульфоксида в метиленхлориде (5 мл) при -78oС в атмосфере азота, после чего перемешивают в течение 30 минут. К реакционной смеси по каплям добавляют раствор 950 мг (2,02 ммоль) 2-[(2R)-(3,4-дихлорфенил)-4-(3,4,5-триметоксибензоил)морфолин-2-ил] этанола, полученного в ссылочном примере 6(е), в метиленхлориде (10 мл) и смесь перемешивают в течение 4 часов. К реакционной смеси добавляют 2,24 мл (16,2 ммоль) триэтиламина, после чего перемешивают при комнатной температуре в течение 2 часов. Реакционную смесь выливают в воду и экстрагируют метиленхлоридом. Органический слой промывают водой и насыщенным водным раствором хлорида натрия и высушивают над безводным сульфатом натрия, а растворитель отгоняют при пониженном давлении с получением остатка. Этот остаток очищают посредством хроматографии на колонке с силикагелем (элюирующий растворитель метиленхлорид : ацетон от 23:2 до 21:4), в результате чего получали 878 мг целевого соединения.
[α]
Спектр ядерного магнитного резонанса (400 МГц, СDСl3) δ миллионные доли: 9,56 (1Н, с), 6,90-7,80 (3Н, м), 6,50 (2Н, с), 3,40-4,60 (6Н, м), 3,85-3,87 (всего 9Н, с каждый), 2,70-3,05 (2Н, м)
Инфракрасный спектр поглощения: νмакс см-1 (KBr): 2962, 2930, 2838, 1723, 1636, 1585
Масс-спектрометрия (FAB) m/z: 468 (М+Н+)
Ссылочный пример 7(b)
1-{ 2-[(2R)-(3,4-дихлорфенил)-4-(3,4,5-триметоксибензоил)морфолин-2-ил] этил}спиро[бензо[с]тиофен-1(3Н),4'-пиперидин]-(2S)-оксид
В 1 мл метанола растворяют 150 мг (0,32 ммоль) 2-[(2R)-(3,4-дихлорфенил)-4-(3,4,5-триметоксибензоил)морфолин-2-ил] этанала, полученного в ссылочном примере 7(а), и 99 мг (0,38 ммоль) гидрохлорида спиро[бензо[с]тиофен-1(3Н),4'-пиперидин]-(2S)-оксида. К полученному раствору добавляют 100 мг молекулярных сит 3А (порошок) и 209 мг (3,33 ммоль) цианборгидрида натрия, после чего нагревают с обратным холодильником в течение 8 часов в атмосфере азота. Реакционную смесь фильтруют через целитовый фильтр. Фильтрат выливают в воду и экстрагируют этилацетатом. Органический слой промывают насыщенным водным раствором хлорида натрия и высушивают над безводным сульфатом натрия, а растворитель отгоняют при пониженном давлении с получением остатка. Этот остаток очищают посредством хроматографии на колонке с силикагелем (элюирующий растворитель метиленхлорид : метанол от 97:3 до 19:1), в результате чего получают 184 мг целевого соединения. Данные различных спектральных анализов этого продукта идентичны данным, полученным в ссылочном примере 6.
Тест 1
Ингибирующий эффект в отношении повышенной проницаемости сосудов.
Ингибирующий эффект в отношении повышенной проницаемости сосудов, вызванной субстанцией Р (SP), агонистом рецепторов NK1, оценивают на основании утечки пигмента в качестве показателя, с использованием морских свинок (вес тела приблизительно 400 г, самцы морских свинок Хартли).
Пигмент (синий Эванса: 40 мг/кг) вводят в бедренную вену морской свинки под пентобарбиталовым наркозом (30 мг/кг, и/п) и немедленно после этого внутривенно вводят SP (1 мкг/кг) для того, чтобы вызвать усиление проницаемости кровеносных сосудов. Спустя пятнадцать минут после инъекции морских свинок под хлороформным наркозом умерщвляют и оценивают количество пигмента, просочившегося в главные бронхи, согласно методике Harada (J.Pharm. Pharmacol. 23, 218 (1971)). Испытуемое вещество суспендируют в 0,5%-ной суспензии трагаканта и вводят морским свинкам перорально за 1 час перед индукцией SP. Его ингибирующее действие оценивают по соотношению просочившегося пигмента у животных из группы испытуемого вещества и у животных из группы, не получавшей испытуемого вещества. В таблице 1 показана 50%-ная ингибирующая доза (ID50) и степень ингибирования при пероральном введении дозы 3,3 мг/кг.
Соединения настоящего изобретения демонстрируют активность, эквивалентную активности ранее известного соединения С при испытании на антагонистическую активность in vivo в отношении рецепторов NK1.
Тест 2
Ингибирующий эффект в отношении бронхоконстрикции
Ингибирующий эффект в отношении бронхоконстрикции, вызванной [Nle10]-NKA[4-10] , агонистом рецепторов NK2, оценивают на основании давления в дыхательных путях в качестве показателя согласно модифицированной методике Konzett-Roessler [Naunyn-Schmiedebergs Arch. Exp. Pathol. Pharmakol. 195, 71 (1940)] с использованием морских свинок (вес тела приблизительно 500 г, самцы морских свинок Хартли).
Немедленно после канюляции трахеи морских свинок под фенобарбиталовым наркозом (30 мг/кг, п/к) и введения галламина (20 мг/кг, в/в) животных искусственно вентилируют с помощью респираторного насоса с постоянным объемом (Ugo-Basile, 7025) с частотой 60 в минуту и дыхательным объемом 8 мл/кг. Давление в дыхательных путях во время искусственной вентиляции амплифицируют с помощью преобразователя давления (Nihon Koden, TP-200T), установленного в ответвлении трахеостомической канюли, определяют (Nihon Koden, AP-610G) и записывают с помощью записывающего устройства (Nihon Koden, WT-685G). Через пять минут после предварительного воздействия атропином (1 мг/кг, в/в) и пропранололом (1 мг/кг, в/в) внутривенно вводят 4 мкг/кг [Nle10]-NKA[4-10], чтобы вызвать бронхоконстрикцию, а затем измеряют давление в дыхательных путях в течение 10 минут. Испытуемое вещество приготавливают так же, как описано в тесте 1 и вводят перорально за один час до индукции [Nle10]-NKA[4-10] . Ингибирующий эффект определяли по площади под кривой внутреннего давления в дыхательных путях в группе, получившей испытуемое вещество, и в группе, не получившей испытуемого вещества. В таблице 2 показана 50%-ная (ID50) ингибирующая доза.
(Когда соединение А подвергают испытанию при внутривенном введении до описанного выше теста с пероральным введением, оно показывает величину ID50 более 10 мг/кг. Таким образом, тест с пероральным введением для этого соединения не проводился).
Соединения настоящего изобретения демонстрируют активность, значительно превосходящую активность ранее известного соединения при испытании на антагонистическую активность in vivo в отношении рецепторов NK2.
Как следует из таблиц 1 и 2, соединения по настоящему изобретению демонстрируют отличное антагонистическое действие как в отношении рецепторов NK1, так и в отношении рецепторов NK2. Более конкретно эти соединения демонстрируют антагонистическое действие в отношении рецепторов NK1 на том же уровне, что и ранее известные соединения, и антагонистическое действие в отношении рецепторов NK2, превосходящее действие ранее известных соединений.
Пример композиции 1
Порошки
Порошки можно изготовить путем смешивания 5 г соединения примера 1, 895 г лактозы и 10 г кукурузного крахмала в мешалке. Эти порошки содержат соединение примера 1 в количестве 5 мг/г.
Пример композиции 2
Гранулы
После смешивания 5 г соединения примера 1, 865 г лактозы и 100 г малозамещенной гидроксипропилцеллюлозы к полученной смеси добавляют 300 г 10%-ного водного раствора гидроксипропилцеллюлозы, после чего перемешивают. Гранулы можно получить путем гранулирования замешенной массы в экструзионном грануляторе и высушивания полученных гранул. Полученные гранулы содержат соединение примера 1 в количестве 5 мг/г.
Пример композиции 3
Капсулы
Таблетки можно изготовить путем смешивания 5 г соединения примера 1, 115 г лактозы, 58 г кукурузного крахмала и 2 г стеарата магния в мешалке V-образной формы, а затем наполнением капсул 3 по 180 мг полученной смеси. Каждая капсула содержит соединение примера 1 в количестве 5 мг.
Пример композиции 4
Таблетки
Таблетки можно изготовить путем смешивания 5 г соединения примера 1, 90 г лактозы, 34 г кукурузного крахмала, 20 г кристаллической целлюлозы и 1 г стеарата магния в мешалке, а затем таблетированием полученной смеси в машине для таблетирования.
Возможности промышленного применения
Новые соли оптически активного сульфоксидного производного по настоящему изобретению демонстрируют отличную антагонистическую активность как в отношении рецепторов субстанции Р, так и в отношении рецепторов нейрокинина А, обладают малой токсичностью и поэтому пригодны в качестве профилактического агента или средства для лечения заболеваний, опосредованных тахикинином.
Методы испытаний
1. Испытание связывания NK1 рецептора
(a) Получение неочищенных образцов легочных мембран
Неочищенные образцы легочных мембран получали из легких мужских особей морских свинок Hartley. А именно, животных умерщвляли путем exaanguination из брюшной аорты под анестезией хлороформом, после чего сразу вырезали легкое и ткани дыхательных путей.
После перфузии вырезанного легкого в буфере (1) (50 мМ Трис-HC1, рН 7,4) легкие разрезали, получая тонкие срезы, и затем гомогенизировали в буфере (2) (буфер (1), содержащий 120 мМ хлорида натрия и 5 мМ хлорида калия), используя Политрон.
Тканевые конгломераты удаляли из гомогената путем пропускания через найлоновое сито (50 мкм) и отделения центрифугированием (30000•g, 30 минут, 4oС).
Осадок снова суспендировали в охлажденном льдом буфере (3) (буфер (1), содержащий 10 мМ ЭДТУ и 300 мМ хлорида калия), затем давали выстояться на льду в спокойном состоянии в течение 60 минут, после чего полученную суспензию центрифугировали и промывали дважды (30000•g, 15 минут, 4oС).
Неочищенные образцы мембран хранили при -80oС до использования.
(b) Испытание связывания рецептора
250 мкл раствора образца неочищенных легочных мембран добавляли к 250 мкл смешанного раствора испытываемого лекарственного средства и [3Н]-вещества Р (конечная концентрация: 1 нМ) (50 мМ Трис-HC1, рН 7,4, 6 мМ хлорида марганца, 800 мкг/мл альбумина бычьей сыворотки, 8 мкг/мл химостатина, 8 мкг/мл лейпептина, 80 мкг/мл бацитрацина и 20 мкг/мл фосфорамидона) и затем инкубировали в течение 30 минут при комнатной температуре.
По завершении реакции мембранный компонент извлекали на GF/B стекловолоконный фильтр (Whatman) с использованием автоматической фильтровальной системы (Brandel).
В связи с этим стеклянный фильтр предварительно обрабатывали в течение примерно 4 часов 0,1%-ным раствором полиэтиленимина, чтобы свести к минимуму неспецифическое связывание.
Фильтр, содержащий мембранный компонент, переносили в пластиковую минипробирку, содержащую 4 мл Pico Fluor®, и измеряли радиоактивность при помощи жидкостного сцинтилляционного счетчика (Beckman, LCS3500).
2. Испытание связывания NK2 рецептора
(а) Получение неочищенных образцов мембран подвздошной кишки
Неочищенные образцы мембран получали из подвздошной кишки мужских особей морских свинок Hartley. А именно, животных умерщвляли путем exaanguination из брюшной аорты под анестезией хлороформом, после чего сразу вырезали подвздошную кишку.
Вырезанную кишку разделяли на ее содержимое, секреты и эпителий путем соскабливания слайдовым стеклом. После получения тонких срезов в буфере (1) (50 мМ Трис-HC1, рН 7,4) образцы гомогенизировали в буфере (2) (буфер (1), содержащий 120 мМ хлорида натрия и 5 мМ хлорида калия), используя Политрон.
Тканевые конгломераты удаляли из гомогената путем пропускания через найлоновое сито (50 мкм) и отделения центрифугированием (30000•g, 30 минут, 4oС).
Осадок снова суспендировали в охлажденном льдом буфере (3) (буфер (1), содержащий 10 мМ ЭДТУ и 300 мМ хлорида калия), затем давали выстояться на льду в спокойном состоянии в течение 60 минут, после чего полученную суспензию центрифугировали и промывали дважды (30000•g, 15 минут, 4oС).
Неочищенные образцы мембран хранили при -80oС до использования.
(b) Испытание связывания рецептора
250 мкл раствора неочищенного образца подвздошной кишки добавляли к 250 мкл смешанного раствора испытываемого лекарственного средства и [3Н]-SR-48968 (Amersham, конечная концентрация: 1 нМ) (50 мМ Трис-HC1, рН 7,4, 6 мМ хлорида марганца, 800 мкг/мл альбумина бычьей сыворотки, 8 мкг/мл химостатина, 8 мкг/мл лейпептина, 80 мкг/мл бацитрацина и 20 мкг/мл фосфорамидона) и затем инкубировали в течение 30 минут при комнатной температуре.
По завершении реакции мембранный компонент извлекали на GF/B стекловолоконный фильтр (Whatman) с использованием автоматической фильтровальной системы (Brandel).
В связи с этим стеклянный фильтр предварительно обрабатывали в течение примерно 4 часов 0,1%-ным раствором полиэтиленимина, чтобы свести к минимуму неспецифическое связывание.
Фильтр, содержащий мембранный компонент, переносили в пластиковую минипробирку, содержащую 4 мл Pico Fluor®, и измеряли радиоактивность при помощи жидкостного сцинтилляционного счетчика (Beckman, LCS3500).
3. Испытание связывания NK3 рецептора
(а) Получение неочищенных образцов церебральных мембран
Неочищенные образцы церебральных мембран получали из мозга мужских особей морских свинок Hartley. А именно, животных умерщвляли путем exaanguination из брюшной аорты под анестезией хлороформом. После перфузии буфером (1) (50 мМ Трис-HC1, рН 7,4) от правого желудочка мозг сразу же энуклеировали. Вырезанный мозг гомогенизировали в буфере (2) (буфер (1), содержащий 120 мМ хлорида натрия и 5 мМ хлорида калия), используя Политрон. Тканевые конгломераты удаляли из гомогената путем пропускания через найлоновое сито (50 мкм) и отделения центрифугированием (30000•g, 30 минут, 4oС). Осадок снова суспендировали в охлажденном льдом буфере (3) (буфер (1), содержащий 10 мМ ЭДТУ и 300 мМ хлорида калия), затем давали выстояться на льду в спокойном состоянии в течение 60 минут, после чего полученную суспензию центрифугировали и промывали дважды (30000•g, 15 минут, 4oС). Полученное вещество суспендировали в буфере (1) с получением неочищенных образцов мембран. Их хранили при -80oС до использования в испытании связывания рецептора.
(b) Испытание связывания рецептора
Пробирку, которую использовали для проведения реакции, заранее обрабатывали буфером (1), содержащим 5 мг/мл альбумина бычьей сыворотки (BSA). К 100 мкл буфера (1), содержащего [3Н]-сенктид, 6 мМ хлорида марганца, 800 мкг/мл BSA, 8 мкг/мл химостатина, 8 мкг лейпептина, 80 мкг/мл бацитрацина и 20 мкг/мл фосфорамидона, добавляли 150 мкл буфера (1), содержащего 400 мкг/мл BSA и испытываемое соединение. К полученной смеси добавляли 250 мкл раствора образца неочищенных церебральных мембран (доведенного до концентрации белка 1 мг/мл) для того, чтобы начать реакцию (в это время конечная концентрация [3H]-сенктида в реакционной фазе составляла 2,5 нМ).
После инкубации при комнатной температуре в течение 60 минут мембранный компонент извлекали на GF/B стекловолоконный фильтр (Whatman) с использованием автоматической фильтровальной системы (Brandel), который был предварительно обработан 0,1%-ным полиэтиленимином в течение примерно 4 часов, после чего его три раза промывали 5 мл охлажденного льдом буфера (4) (5 мМ трис-хлористоводородной кислоты, содержащей 400 мкг/мл BSA и 0,01% додецилсульфата натрия, рН 7,4).
Фильтр, содержащий мембранный компонент, переносили в пластиковую минипробирку, содержащую 4 мл Pico Fluor®, и измеряли радиоактивность при помощи жидкостного сцинтилляционного счетчика (Aloka, LCS3500).
Для определения радиоактивности из-за неспецифического связывания [3Н] -сенктида (связывание на других участках, а не на рецепторе, например на фильтре) эксперимент осуществляли с добавлением избыточного количества сенктида (конечная концентрация 10 мкМ) и измеряли радиоактивность.
Степень ингибирования связывания сенктид-рецептор, благодаря испытываемому соединению, вычисляли с использованием следующего уравнения:
Степень ингибирования (%) [1-(С-А)/(В-А)]•100
А: радиоактивность из-за неспецифического связывания
В: радиоактивность в растворе образца без добавления испытываемого соединения
С: радиоактивность в растворе образца при добавлении испытываемого соединения
Результаты испытаний приведены в таблице 3.
Соединениями примеров 1 и 2 являются соответственно гидрохлорид и фумарат. Они оба преобразуются в свободное основание при введении в организм. Свободное основание является активным действующим началом, которое связывается с рецепторами в проведенных испытаниях. Соединение примера 2 демонстрирует по существу такую же активность, что и соединение примера 1.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ СПИРОПИПЕРИДИНА, ЛЕКАРСТВЕННОЕ СРЕДСТВО, СПОСОБ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ | 1999 |
|
RU2184735C2 |
| АЦИЛИРОВАННЫЕ ГЕТЕРОАЛИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ, ЛЕКАРСТВЕННОЕ СРЕДСТВО, ОБЛАДАЮЩЕЕ СЕЛЕКТИВНОЙ АНТАГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ ПРОТИВ NK-РЕЦЕПТОРОВ, СПОСОБ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЯ | 1998 |
|
RU2174122C1 |
| ПРОИЗВОДНЫЕ ИНДАНОЛА | 2005 |
|
RU2323937C1 |
| ПРОИЗВОДНЫЕ АМИНОСПИРТОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И СПОСОБ ПОЛУЧЕНИЯ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 2001 |
|
RU2233839C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ N-АКРИЛОИЛПИПЕРАЗИНА | 1990 |
|
RU2024513C1 |
| НОВЫЕ КАРБОКСИЗАМЕЩЕННЫЕ ЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ КАРБОКСАМИДА | 1997 |
|
RU2199535C2 |
| α,ω ДИАРИЛАЛКАНЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1993 |
|
RU2105752C1 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, ПРОЯВЛЯЮЩАЯ АНТАГОНИСТИЧЕСКОЕ ДЕЙСТВИЕ В ОТНОШЕНИИ РЕЦЕПТОРОВ ТАХИКИНИНА | 1996 |
|
RU2135494C1 |
| ПРОИЗВОДНЫЕ БЕНЗОПИРАНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1991 |
|
RU2038354C1 |
| ФУНГИЦИДНЫЕ ПРОИЗВОДНЫЕ ОКСЕТАНА И ИХ СОЛИ | 1992 |
|
RU2044736C1 |



Изобретение относится к солям сульфоксидным производным, именно к гидрохлориду или фумарату 1-{ 2-[(2R)-(3,4-дихлорфенил)-4-(3,4,5-триметоксибензоил)морфолин-2-ил] этил} спиро[бензо(с)тиофен-1(3Н),4'-пиперидин]-(2S)-оксида, которые обладают хорошей всасываемостью при приеме внутрь и демонстрируют отличную антагонистическую активность как в отношении рецепторов NK1, так и в отношении рецепторов NK2. 5 с. и 18 з.п. ф-лы, 3 табл.
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| Оправка для крепления хонинговальной головки | 1978 |
|
SU776893A1 |
| ТРИНУС Ф.П | |||
| Фармакотерапевтический справочник | |||
| - Киев: Здоровья, 1989, с | |||
| Способ закалки пил | 1915 |
|
SU140A1 |

