Изобретение касается способа получения волокон (вытягивания нити) из дисперсии политетрафторэтилена или родственных ему полимеров либо формования из такой дисперсии изделий определенной формы, при котором в конечной подвергшейся спеканию структуре фторированного полимера, а также в его промежуточной структуре практически не содержится применяемых в процессе солей, кислот и других примесей.
Исключительная устойчивость политетрафторэтилена и родственных ему полимеров к воздействию света, нагревания, растворителей, агрессивных химических реагентов и электростатическим напряжениям делает желательным применение этих полимеров и изготовленных из таких полимеров изделий для целого ряда случаев. Однако из-за сложностей, связанных с обработкой расплава и раствора этих полимеров, является очень сложным формовать волокна этих полимеров или формовать из них определенные изделия при помощи стандартных способов.
Один из способов, который применяется для того, чтобы формовать изделия или вытягивать нити из политетрафторэтилена и родственных полимеров, состоит в том, чтобы формовать изделия из полимера или вытягивать его нити из смеси водной дисперсии частиц полимера и вискозы, причем растворимой формой полимерной матрицы является ксантогенат целлюлозы, как изложено в патентах США 3655853, 3114672 и 2772444.
Хотя вискоза обычно применяется при формовании волокон из политетрафторэтилена и родственных полимеров, в использовании вискозы имеются некоторые серьезные недостатки. Вискозу получают посредством сложного и требующего много времени процесса, в котором древесную массу обрабатывают гидроксидом щелочного металла и дисульфидом углерода. Дисульфид углерода представляет собой опасный химический реагент. В связи со взрывоопасностью смесей дисульфида с воздухом при работе с ним требуются чрезвычайные тщательность и меры предосторожности. Является как непрактичным, так и небезопасным улавливать дисульфид углерода, который испаряется из коагуляционной ванны в процессе регенерации целлюлозы из вискозы (ксантогената целлюлозы) при помощи химической реакции. Таким образом, этот опасный химический реагент обычно выпускается в атмосферу, что создает проблемы для окружающей среды, а также повышает стоимость производства вискозы.
Известны альтернативы формованию с использованием вискозы, однако применение таких полимерных матриц обычно также сопряжено с использованием органического растворителя, поверхностно-активного вещества или их обоих, как это изложено в патентах США 3147323, 3118846 и 2951047.
В патенте США 3242120 (автор Steuber) описываются самоподдерживающаяся гелеобразная структура и способ вытягивания нитей или формования изделий определенной формы из водных дисперсий частиц нерастворимого в воде полимера, смешанных с водным раствором полимерной матрицы, такой как альгинат натрия или поливиниловый спирт. Эта смесь образует гелеподобную структуру при контакте с коагулирующей средой, желатинирующую полимерную матрицу. Хотя автор указанного патента перечислил соединения, которые могут служить в качестве полимерных матриц, и описал промывание волокон, формуемых из гелеподобной структуры после коалесценции частиц полимера, он не указал и не предположил, каким образом можно было бы получить промежуточное волокно, которое не содержало бы солей и других примесей.
В процессе вытягивания нити или формования из дисперсии ионы из коагуляционной ванны включаются в состав промежуточной структуры. Эти ионы, например ионы водорода, натрия и сульфат-ионы, могут вызывать серьезные проблемы при преобразовании промежуточной волоконной структуры в конечное волокно из спеченного (подвергшегося коалесценции) полимера фторированного олефина.
Типичная коагуляционная ванна, используемая при формовании из дисперсии, представляет собой кислотную ванну, содержащую серную кислоту и сульфат натрия. Кислотный остаток серной кислоты вызывает деструкцию промежуточной волоконной структуры под воздействием температуры, необходимой для коалесценции фторированного полимера. Присутствие соли, которая может иногда накапливаться до содержания, составляющего до 25% от массы волоконной структуры, является вероятной причиной получения волокна с неприемлемой механической прочностью. В большинстве случаев высокая концентрация соли в промежуточной волоконной структуре может даже препятствовать образованию спеченного волокна, поскольку очень сложно, если вообще возможно, спекать промежуточную волоконную структуру, содержащую остаточную соль.
Целью настоящего изобретения является способ, при помощи которого политетрафторэтилен и родственные ему полимеры могут быть сформованы в промежуточные изделия определенной формы или быть сформованы в волокна, которые могут быть легко промыты до состояния отсутствия накапливающихся при обработке ионов и других примесей, а затем обработаны с получением конечных спеченных продуктов.
Другой целью настоящего изобретения является способ изготовления изделий определенной формы из водных дисперсий политетрафторэтилена и родственных полимеров, который обладает преимуществами способа на основе вискозы, но свободен от недостатков, связанных с использованием ксантогената целлюлозы в качестве растворимой полимерной матрицы.
В настоящем изобретении предлагается способ вытягивания нити (формования) полностью промытого водой промежуточного волокна фторированного олефинового полимера из смеси водной дисперсии частиц фторированного олефинового полимера и раствора полимерной матрицы, включающий стадии:
(а) образования смеси водной дисперсии частиц фторированного олефинового полимера и раствора полимерной матрицы, причем полимерная матрица представляет собой простой эфир целлюлозы, имеющий степень замещения, которая составляет не более около 0,5 и не менее около 0,02;
(б) экструдирования смеси в коагуляционный раствор, содержащий соли, кислоты или их смеси, с коагуляцией полимерной матрицы и образованием промежуточной волоконной структуры; и
(в) промывания промежуточной волоконной структуры в достаточном количестве воды с близким к нейтральному значением рН, чтобы в существенной степени удалить из волоконной структуры соли, кислоты и их смеси,
причем промываемая волоконная структура имеет самоподдерживающуюся длину, составляющую по меньшей мере 30 см, и практически не содержит ионов.
Промежуточная волоконная структура согласно настоящему изобретению может быть преобразована в волокно из подвергшегося коалесценции фторированного олефинового полимера посредством воздействия на промежуточную волоконную структуру на следующих за стадией (в) дополнительных стадиях сушки и спекания волоконной структуры для осуществления окисления полимерной матрицы и коалесценции частиц фторированного олефинового полимера.
В настоящем изобретении предлагается также улучшенная промежуточная волоконная структура, состоящая в существенной степени из смеси частиц фторированного олефинового полимера, коагулировавшей полимерной матрицы и воды, отличающаяся тем, что соотношение массы частиц полимера и массы полимерной матрицы в промежуточной волоконной структуре составляет от около 3:1 до около 20:1, отличающаяся тем, что полимерная матрица представляет собой простой эфир целлюлозы, имеющий степень замещения, которая составляет не более около 0,5 и не менее около 0,02, и отличающаяся тем, что полимерная матрица образует совместно с частицами фторированного полимера промытую волоконную структуру, имеющую самоподдерживающуюся длину, составляющую по меньшей мере 30 см, и практически не содержащую ионов.
Приведенный чертеж иллюстрирует шприцевое формование волокна для тестирования целостности промежуточных волоконных структур.
Термин "политетрафторэтилен и родственные ему полимеры", как он используется в настоящем изобретении, обозначает политетрафторэтилен и полимеры, в целом известные как фторированные олефиновые полимеры, например сополимеры тетрафторэтилена и гексафторпропилена (FEP), сополимеры тетрафторэтилена и перфторалкилвиниловых простых эфиров, таких как перфторпропилвиниловый эфир (PFA) и перфторэтилвиниловый эфир, тройные сополимеры фторированных олефинов, включая тройные сополимеры вышеперечисленных мономеров, и другие сополимеры на основе тетрафторэтилена.
Термин "ПТФЭ", как он используется в настоящем изобретении, обозначает политетрафторэтилен.
Термин "водная дисперсия", как он используется в настоящем изобретении, обозначает приготовленную в воде дисперсию частиц, которая может содержать различные поверхностно-активные добавки и добавки для регулирования рН и сохранения состояния (стабилизации) дисперсии.
Термин "промежуточная волоконная структура", как он используется ниже, обозначает экструдированную и подвергшуюся коагуляции смесь раствора полимерной матрицы и дисперсии частиц полимера. Промежуточная волоконная структура согласно настоящему изобретению обладает самоподдерживающейся длиной, составляющей по меньшей мере 30 см, после того как она промыта до состояния, практически не содержащего ионов и примесей. У промежуточной волоконной структуры согласно настоящему изобретению после ее промывания в воде с близким к нейтральному рН, чтобы практически полностью удалить ионы и примеси, не проявляется существенной потери прочности или целостности, и она может быть обработана, например вытянута с умеренным коэффициентом вытяжки, и подвергнута спеканию с получением конечного волокна или изделия определенной формы из скоалесцированного полимера. Промежуточная волоконная структура согласно настоящему изобретению может быть выделена, обработана в последующих процессах обработки или же использована для производства тканей либо материала типа ватной прокладки или фетра, как это известно в данной области техники.
Как будет понятно квалифицированному специалисту в данной области, промежуточная волоконная структура включает, наряду с типичными волокнистыми структурами в виде однониточных структур и структуры типа жгута, ленты, тесьмы, пленки и т.п.
Под термином "формование из дисперсии" понимают процесс, посредством которого дисперсию частиц нерастворимого полимера смешивают с раствором растворимой полимерной матрицы и эту смесь подвергают коагуляции путем приведения смеси в контакт с коагуляционным раствором, в котором полимерная матрица становится нерастворимой.
Формование из дисперсии, в целом известное для изделий из волокон как вытягивание нитей из дисперсии, является пригодным для получения изделий определенной формы из фторированных полимеров. Эти полимеры, которые сложно формовать посредством экструзии расплава или вытягивания нитей из раствора, могут быть успешно сформованы в волокна из смеси водной дисперсии частиц фторированного полимера, смешанной с раствором подходящей полимерной матрицы. Промежуточная структура формуется при приведении этой смеси в контакт с подходящей коагуляционной ванной. Хотя промежуточная структура является механически прочной, обычно формуют конечную спеченную структуру посредством нагревания промежуточной структуры до температуры, достаточной для того, чтобы произошла коалесценция фторированных частиц полимера. При спекании полимерная матрица разлагается с образованием летучих газов и углеродного остатка.
Для того чтобы получить пригодные к употреблению коалесцированные волокна фторированного олефинового полимера, является существенным промывание промежуточной волоконной структуры до состояния, не содержащего ионов, абсорбированных из коагуляционной ванны, а также удалить другие примеси, такие как добавки и/или диспергаторы, которые присутствовали в исходной дисперсии фторполимера, и удалить вещества, которые являются вредными для процесса спекания волокон и/или для свойств конечного коалесцированного волокна фторированного полимера. Известно, что выбор полимерной матрицы не вытекает непосредственно из свойств волокон, сформованных из типичных формовочных растворов на основе рассматриваемой полимерной матрицы, и даже рабочие характеристики такой полимерной матрицы не являются предсказуемыми, исходя из свойств волокон.
В настоящем изобретении простой эфир целлюлозы присутствует в составе промежуточной волоконной структуры лишь как второстепенный составляющий компонент твердой фазы волокон, тогда как главным составляющим компонентом являются частицы фторированного полимера, на долю которых в промежуточной волоконной структуре может приходиться масса, превышающая массу полимерной матрицы в 3-20 раз. Тот факт, что в состав вытягиваемой нити может войти конкретное соединение целлюлозы, не гарантирует степень необходимой когезионной способности, которой должна обладать полимерная матрица, для того чтобы она могла обеспечить необходимую основу и структуру при изготовлении работоспособной промежуточной структуры фторполимерного волокна. Примеры 3 и 4 ниже иллюстрируют это утверждение.
Для того чтобы промежуточное волокно можно было промывать водой, полимерная матрица должна иметь строго определенное свойство быть нерастворимой в воде при близком к нейтральному значении рН и при температурах процесса. При отсутствии возможности промывания промежуточной структуры волокна в воде, которая практически не содержит ионов, как, например, вода с близким к нейтральному рН, промежуточное волокно не может быть получено в виде, практически не содержащем опасных примесей, которые могут препятствовать образованию пригодного к употреблению фторированного волокна при спекании.
Кроме того, является предпочтительным, чтобы полимерная матрица не размягчалась и не плавилась при температуре, которая существенно ниже температуры спекания, поскольку в противном случае промежуточная волоконная структура может растягиваться, ослабляться или разрушаться под воздействием собственного веса по мере того, как она нагревается до температуры спекания.
Процесс формования матрицы из ксантогената целлюлозы имеет некоторые серьезные недостатки, состоящие в том, что для формования ксантогената целлюлозы требуется использование дисульфида углерода, токсичного и крайне легко воспламенимого вещества. Кроме того, вискоза не образует стабильный раствор. Раствор вискозы самопроизвольно желатинируется по мере его старения. В промышленных процессах формования на основе вискозы самопроизвольное желатинирование вискозы представляет собой вполне реальную проблему этих процессов, приводящую к потерям и требующую обширного промывания технологической линии и очистки резервуара.
Авторы настоящего изобретения хотели найти такую замену для процесса формования матрицы из ксантогената целлюлозы, которая обладает преимуществами процесса формования вискозы, но лишена серьезных недостатков. Найдено, что простые эфиры целлюлозы, имеющие однородную степень замещения и растворимые только в крепких водных растворах щелочи, но не растворимые в воде с близким к нейтральному значением рН, дают полимерные матрицы, которые удовлетворяют требованиям настоящего изобретения.
Термин "вода с близким к нейтральному значением рН" обозначает воду, имеющую рН 6-8.
Структурными характеристиками, которые в сильной степени связаны с растворимостью простых эфиров целлюлозы, являются функциональность химических заместителей в эфирах целлюлозы и степень замещения. Под степенью замещения (СЗ) понимается та степень, в которой гидроксильные группы молекулы целлюлозы были замещены функциональными группами, образующими простой эфир.
В молекуле глюкозы имеется три гидроксильные группы на каждый цикл ангидроглюкозы. Если все три этих гидроксильные группы были замещены, степень замещения составляет 3, что является максимальным значением степени замещения.
Простые эфиры целлюлозы, используемые в процессе согласно настоящему изобретению, представляют собой такие эфиры целлюлозы, которые растворимы только при высокой концентрации гидроксида натрия в воде и не растворимы в воде с близким к нейтральному значением рН в диапазоне температур 10-90oС. Из простых эфиров целлюлозы, обладающих такими характеристиками растворимости, предпочтительными полимерными матрицами являются неионогенные эфиры целлюлозы. В дополнение к этому полимерные матрицы согласно настоящему изобретению не имеют температуры размягчения или плавления. Эти полимеры разлагаются при температуре, близкой к температуре спекания волокна, обеспечивая структуру вплоть до коалесценции фторполимера.
Авторы изобретения установили, что для обеспечения промежуточной структуры, которую можно было бы промыть до состояния, практически не содержащего солей и других примесей, необходимо использовать только те простые эфиры целлюлозы, которые являются нерастворимыми в воде с близкой к нейтральной реакцией, и которые обеспечивают после промывания промежуточную волоконную структуру, имеющую самоподдерживающуюся длину по меньшей мере 30 см. Хотя многие материалы могут образовывать гелеподобные структуры, как это иллюстрируется в списке, представленном в вышеуказанном патенте США 3242120 (столбец 13), только сочетание растворимости в растворе, имеющем концентрацию гидроксида натрия более чем около 1,3-молярная (более около 5% по массе, рассчитанное значение рН для которой составляет свыше 14), и нерастворимости коагулировавшей полимерной матрицы в воде с близкой к нейтральной реакцией обеспечивает существенные свойства полимерной матрицы согласно настоящему изобретению. Без такого сочетания свойств промежуточная волоконная структура не будет обладать свойством полностью промываться водой и не будут обеспечены приемлемые прочностные характеристики спеченного волокна.
Неионогенные простые эфиры целлюлозы, такие как гидроксипропилцеллюлоза и гидроксиэтилцеллюлоза, обеспечивают особенно хорошие формовочные композиции для вытягивания нитей фторированных полимеров из дисперсии. Значения степени замещения, которые являются характерными для полимерных матриц, согласно настоящему изобретению находятся в интервале от около 0,02 до 0,5. Однородность замещения полимерных матриц согласно настоящему изобретению является предпочтительной, и ее признаком служит прозрачность раствора, образуемого в приблизительно 10% (по массе) водном растворе гидроксида натрия.
Раствор матрицы для любой из полимерных матриц согласно настоящему изобретению или их смесей может быть получен посредством растворения конкретного простого эфира целлюлозы в растворе с содержанием гидроксида натрия от около 5 до 10% по массе. Низкое значение степени замещения, требуемое для настоящего изобретения, делает необходимым использование значительно более высоких значений рН, чем это известно из предшествующего уровня техники.
При использовании полимерной матрицы из гидроксипропилцеллюлозы является предпочтительным материал, характеризуемый тем, что он имеет вязкость по меньшей мере 90 мПа•с при растворении его в количестве 2% по массе в 10% растворе гидроксида натрия и проведении измерения при 25oС, хотя вытягиванию нити или формованию в изделия могут быть успешно подвергнуты растворы с более низкой вязкостью материала.
Является предпочтительным формовать изделия согласно настоящему изобретению посредством экструдирования смеси раствора полимерной матрицы и дисперсии частиц фторированного полимера в коагуляционную жидкость, которая обеспечивает быстрое желатинирование изделия. Сформованное изделие может быть затем промыто и подвергнуто дальнейшей обработке. Состав коагуляционных жидкостей зависит, до некоторой степени, от конкретной используемой полимерной матрицы. Пригодные к употреблению коагуляционные жидкости включают множество водных растворов, типичными представителями которых являются, без ограничения указанными растворами, 40% ацетат аммония - 5% уксусная кислота, 30% уксусная кислота или 30% раствор хлористого кальция. Особенно ценным для эфиров целлюлозы согласно настоящему изобретению является раствор, содержащий 5% серной кислоты и 18% сульфата натрия. Температура коагуляционной ванны может быть доведена до температуры, которая обеспечивает наилучшие свойства для промежуточной волоконной структуры, и в типичном случае находится в диапазоне 25-90oС. Для материалов согласно настоящему изобретению предпочтительной является температура коагуляционной ванны 40-60oС.
Является предпочтительным так регулировать температуру воды для промывки, чтобы максимизировать прочность промежуточной волоконной структуры. Полимерные матрицы согласно настоящему изобретению в общем случае являются нерастворимыми в воде при температуре приблизительно в 20oС или выше. Однако с целью обеспечить условия для повышения нерастворимости полимера и ускорения процесса промывки для промышленного процесса рекомендуется температура промывки около 50oС.
Промежуточное волокно согласно настоящему изобретению промывали до состояния, практически не содержащего ионов и примесей, без существенной потери прочности. Термин "практически не содержащий ионов и примесей" обозначает, что рН и проводимость деионизированной воды для промывки сохранялись неизменными после погружения промежуточного волокна в эту воду. Самоподдерживающаяся длина промытого промежуточного волокна составляла по меньшей мере 30 см. Хотя ниже приводится прочность на разрыв для нескольких промежуточных волоконных структур, действительная прочность на разрыв, требующаяся для того, чтобы обеспечить самоподдерживающуюся длину в 30 см, изменяется с содержанием воды в волокне. Таким образом, самоподдерживающая длина промежуточного волокна является более удобным на практике определением достаточной прочности волокна, чем конкретный диапазон значений прочности на разрыв.
Композиции для вытягивания получения нитей или формования изделий, используемые в способе согласно настоящему изобретению, готовят путем смешивания водной дисперсии частиц фторированного полимера с раствором полимерной матрицы согласно настоящему изобретению. Водные дисперсии частиц фторированного олефинового полимера, как, например, известные в технике дисперсии, могут использоваться в настоящем способе. Растворы полимерной матрицы должны быть прозрачными и должны иметь вязкость, которая гарантирует хорошее смешивание с дисперсией. Предпочтительно, концентрация полимерной матрицы в растворе составляет 3-10% по массе. Эти компоненты затем смешивают таким образом, что соотношение массы частиц полимера и массы полимерной матрицы в промежуточной волоконной структуре составляет от около 3:1 до около 20:1, предпочтительно, около 9:1.
Растворы полимерных матриц согласно настоящему изобретению являются стабильными и не желатинируются со временем. Отсутствует необходимость в постоянном взбалтывании или точном регулировании температуры растворов. Хотя композиция согласно настоящему изобретению также является стабильной при хранении, является предпочтительным, чтобы раствор полимерной матрицы и дисперсия фторированного полимера смешивались, согласно общепринятой практике в этой области техники, непосредственно перед использованием, чтобы гарантировать, что эта смесь является однородной и что частицы дисперсии фторированного полимера не будут осаждаться.
В настоящем изобретении предлагается также способ формования промежуточных и конечных изделий из фторированных полимеров, таких как пленки, ленты, тесьма и волокна различной формы, из водной дисперсии частиц фторированного полимера, включающий стадии:
(а) образования смеси водной дисперсии частиц фторированного олефинового полимера и раствора полимерной матрицы, причем полимерная матрица представляет собой простой эфир целлюлозы, имеющий степень замещения, которая составляет не более около 0,5 и не менее около 0,02, при этом полимерная матрица образует совместно с частицами фторированного полимера промытое промежуточное изделие, которое практически не содержит ионов;
(б) экструдирования смеси в коагуляционный раствор, содержащий соли, кислоты или их смеси, с коагуляцией полимерной матрицы и формованием промежуточного изделия; и
(в) промывания промежуточного изделия в достаточном количестве воды с близким к нейтральному рН, чтобы в существенной степени удалить из структуры волокна соли, кислоты или их смеси и другие примеси.
Промежуточное изделие затем может быть подвергнуто конечной обработке путем применения к нему следующих за стадией (в) дополнительных стадий сушки и спекания, чтобы окислить полимерную матрицу и вызвать коалесценцию частиц фторированного олефинового полимера.
Вязкость полимера
Вязкость раствора полимера измеряли следующим образом.
Образец раствора, для которого следовало измерить вязкость, фильтровали, помещали в вакуумную камеру и выдерживали под вакуумом до тех пор, пока следы пузырьков воздуха уже не были видны. В лабораторный стакан объемом 600 мл переносили образец в количестве, достаточном для заполнения им стакана на 10 см по высоте. Затем образец помещали в термостат при 25oС до становления температуры, постоянной по всему образцу.
Вязкость измеряли с использованием вискозиметра Брукфилда модели НВ-Т. Лабораторный стакан объемом 600 мл, содержащий образец, помещали под вискозиметром, а к вискозиметру прикрепляли шпиндель #2. Высоту вискозиметра регулировали до достижения поверхностью жидкости отметки на валу шпинделя, а положение лабораторного стакана регулировали до тех пор, пока шпиндель не был отцентрирован по отношению к образцу. Вискозиметр вращали таким образом, чтобы начал вращаться шпиндель, и регистрировали получаемые в результате значения вязкости и температуры.
По снятому показанию вискозиметра Брукфилда вычисляли вязкость посредством применения соответствующего стандарту ISO 9002 усовершенствованного переводного коэффициента Брукфилда, определяемого из номера шпинделя, числа оборотов в минуту и показания вискозиметра Брукфилда.
Прочность промежуточного волокна
Прочность структуры промежуточного волокна определяли следующим образом.
Раствор полимерной матрицы готовили с такой концентрацией, что приводимая вязкость по Брукфилду (измеренная, как описано выше) составляла 3000-7000 МПа•с при 25oС. Затем этот раствор деаэировали либо посредством помещения его под вакуум до тех пор, пока из него не выходили все пузырьки, либо посредством выдерживания раствора в течение приблизительно 24 ч или до тех пор, пока не выходили все пузырьки. Затем раствор смешивали с дисперсией ПТФЭ таким образом, что отношение по массе твердой фазы полимера ПТФЭ к 1 части (по массе) полимерной матрицы составляло 3-20. В типичной дисперсии общее содержание твердой фазы полимера составляло 60%, а отношение ПТФЭ к полимерной матрице составляло 9-1. Предпочтительный размер частиц для частиц ПТФЭ составлял от около 0,1 до около 0,17 мкм, как, например, это имеет место для полимера типа 3311 фирмы DuPont.
Эту свежеприготовленную смесь затем инжектировали при помощи шприца 1, как показано на чертеже, в объем (кончик иглы под поверхностью жидкости) соответствующей коагуляционной жидкости со скоростью около 1 мл в минуту. Состав коагуляционных жидкостей изменялся в соответствии со свойствами конкретной промежуточной волоконной структуры. Оптимальные составы и температуры коагуляционных жидкостей определяли отдельно для каждой испытывавшейся полимерной матрицы.
Как показано на чертеже, шприц 1, имеющий объем 3 см3 и снабженный иглой калибра 20, был присоединен к обслуживающему шприц насосу 2. Вращающийся с постоянной скоростью цилиндр 4, приводимый в движение мотором 3 (скорость вращения поверхности около 2 м/мин), использовался для того, чтобы протягивать промежуточную волоконную структуру через коагуляционную жидкость в контейнере 5, обеспечивая однородный диаметр волокна. Промежуточной волоконной структуре давали возможность спускаться обратно в коагуляционную жидкость после прохождения над вращающимся цилиндром.
Затем промежуточную волоконную структуру промывали в воде с близкой к нейтральной реакцией, чтобы освободить ее от солей и удалить остаточные ионы. Волокно промывали путем погружения его в деионизированную воду, которая находилась в контейнере. До и после каждого погружения волокна в воду проверяли рН и проводимость воды. После каждого погружения воду удаляли и заменяли свежей деионизированной водой. Волокно промывали до тех пор, пока рН и проводимость воды не начинали соответствовать значениям для свежей деионизированной воды.
Линейную плотность (измеряемая в денье = масса в граммах/9000 м длины) промежуточного волокна измеряли посредством определения массы высушенного волокна определенной длины. Обычно использовали нить волокна длиной приблизительно 0,3 м, так что сухая масса волокна длиной 0,3 м, умноженная на 30000, давала плотность в денье. Для каждой нити выполняли по три измерения, результаты которых усредняли.
Разрушающую нагрузку для промытой влажной промежуточной волоконной структуры определяли путем закрепления пробы волокна на образце бумаги и тестирования прочности волокна на подходящем устройстве для тестирования механических свойств (например, приборе Instron) при скорости ползуна 100 мм/мин. Значения, приведенные в таблице 1, представляют собой средние значения по пяти измерениям и приведены к удельной плотности (например, выражены в мг/денье).
Прочность на разрыв спеченного волокна
Прочность на разрыв спеченного волокна определяли в соответствии со стандартом ASTM, метод D 2256-90.
ПРИМЕРЫ
Для следующих примеров 1-15 были проведены испытания для определения прочности промежуточного волокна с использованием описанной выше методики испытания.
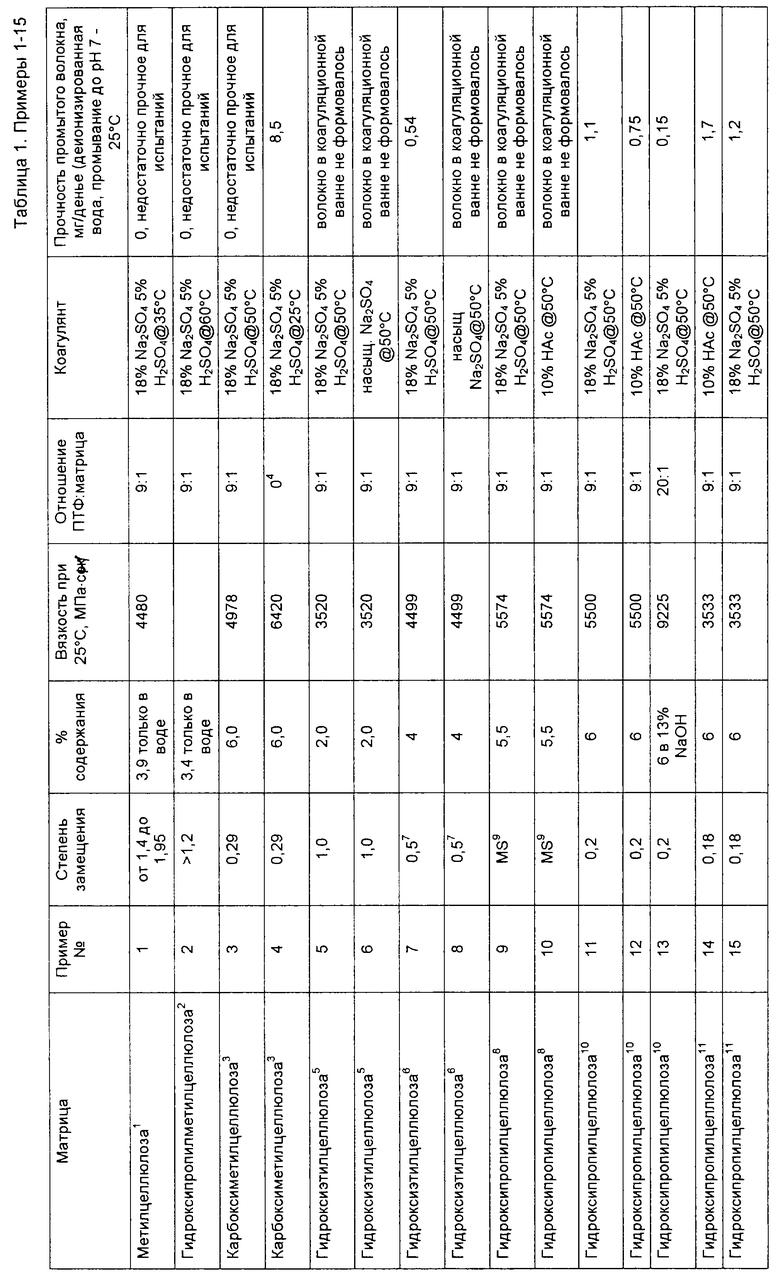
В таблице 1 приведены список испытывавшихся полимерных матриц, их степень замещения, концентрация (в процентах по массе) полимерной матрицы в растворе полимера, вязкость раствора полимерной матрицы при 25oС, массовое отношение ПТФЭ к твердой фазе полимерной матрицы в промежуточном волокне, состав коагуляционной жидкости и данные испытания на прочность промежуточной волоконной структуры.
Отношение массы ПТФЭ к массе полимерной матрицы определяли путем деления массы твердой фазы частиц полимера на массу твердой фазы полимерной матрицы в смеси для вытягивания нити. Поскольку при экструдировании волокна в коагуляционную ванну все твердые фазы полимеров превращаются в твердые фазы волокна, то же самое соотношение характеризует и состав промежуточной волоконной структуры.
НАс - уксусная кислота
Если не указано иначе, все растворы полимерной матрицы приготовлены в 10% NaOH.
1. Метилцеллюлоза в виде продукта Methocel A4C фирмы Dow.
2. Гироксипропилметилцеллюлоза в виде продукта Primaflo фирмы Hercules.
3. Получена от фирмы Akzo Nobel в виде экспериментального образца 40-С LDS.
4. 100% по массе раствора матрицы; отсутствуют частицы фторированного полимера.
5. Гидроксиэтилцеллюлоза в виде продукта Cellosize QP4400H фирмы Union Carbide.
6. Гидроксиэтилцеллюлоза, экспериментальный образец low EO MS Cellosize HEC 19636-37 фирмы Union Carbide. MS обозначает максимальное замещение этиленоксидом.
7. Сообщается мольное замещение этиленоксидом. При данном конкретном уровне замещения эта величина практически совпадает со степенью замещения.
8. Гидроксипропилцеллюлоза в виде продукта Klucel G фирмы Hercules. MS= 4,6.
9. Значение максимального мольного замещения MS, равное 4,6, обеспечено производителем; соответствующее значение степени замещения было неизвестно.
10. Гидроксипропилцеллюлоза в виде продукта HT-A фирмы Shin-Etsu.
11. Гидроксипропилцеллюлоза в виде продукта LH-22 фирмы Shin-Etsu.
Эти примеры иллюстрируют важность значения степени замещения простого эфира целлюлозы, используемого в качестве полимерной матрицы. Приводимые значения прочности промытого волокна показывают измеренную прочность на растяжение сформованных волокон. "Недостаточно прочное для испытаний" обозначает, что волокно формовалось в коагуляционной ванне, однако это волокно разрушалось при обработке. В тех случаях, когда значение степени замещения находилось вне пределов значений согласно настоящему изобретению, волокно в коагуляционной ванне либо не формовалось, либо сформованное волокно было невозможно выделить.
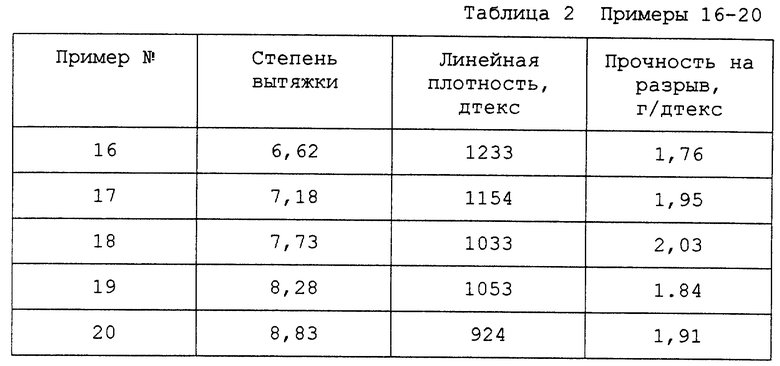
Примеры 16-20
Раствор готовили путем суспендирования 1,9 кг гидроксипропилцеллюлозы из вышеприведенных примеров 11, 12 и 13 в 15,8 л мягкой воды при температуре около 25oC. После того, как гидроксипропилцеллюлоза размокала, к смеси воды и гидроксипропилцеллюлозы добавляли 12,3 кг 23%-ного раствора гидроксида натрия. Получившуюся смесь перемешивали под вакуумом (около 3,87 кПа) в продолжение 1 ч, а затем фильтровали через 50-мкм рукавный фильтр из полипропиленового войлока в тонкопленочный деаэратор, работающий при разрежении около 3,87 кПа. Получившийся раствор имел вязкость в 4800 МПа•сек при 25oC. Поток вышеуказанного раствора соединяли с потоком дисперсии политетрафторэтилена (ПТФЭ) марки TEF 3311 (выпускаемого фирмой DuPont de Nemours and Company, Wilmington, DE) при таком соотношении объемных скоростей, что отношение по массе твердой фазы ПТФЭ к твердой фазе гидроксипропилцеллюлозы составляло 8,2, и смешивали в совмещенном (с трубой) статическом смесителе. Получившуюся смесь затем прокачивали через фильеру, содержащую 180 отверстий (диаметр 0,152 мм), погружунную под поверхность коагуляционной ванны. В состав коагуляционной ванны входили 5%-ная серная кислота и 18%-ный сульфат натрия. Температура ванны поддерживалась на уровне (55±3)oC. Получившиеся волокна затем пропускали через промывную ванну с мягкой водой, температура которой поддерживалась на уровне (58±5)oC, а затем подавали на ряд вращающихся горячих вальцов. Температура поверхности этих вальцов поддерживалась на уровне (130±5)oC, чтобы высушить волокно. Пряжу подавали на другой ряд вращающихся горячих вальцов. Температура поверхности этих вальцов поддерживалась на уровне (363±5)oC для спекания волокна. Пряжу подавали на ряде ненагретых "тянущих вальцов", на которых размещалось множество витков. Различие скоростей второго ряда горячих вальцов и "тянущих вальцов" было таково, что пряжа вытягивалась в 6,62 раза. Эта величина известна как степень вытяжки. Пряжу, выходящую из тянущих вальцов, наматывали на бумажную трубку. Получившаяся пряжа имела линейную плотность 1233 дтекс. Ее прочность на разрыв составляла 1,76 г/дтекс.
Данные для примеров 16-20 представлены в таблице 2. В примерах 17-20 волокна получали, как в примере 16, за исключением того, что степень вытяжки была такой, как указано в таблице 2.
Сравнительный пример 21
5,4%-ный Раствор ксантогената целлюлозы в 5% растворе гидроксида натрия (вискозу) получали путем взаимодействия древесной массы с гидроксидом натрия и дисульфидом углерода. Получившийся раствор имел вязкость 5400 МПа•с при 25oC. Поток вышеуказанного раствора соединяли с потоком дисперсии политетрафторэтилена (ПТФЭ) марки TEF 3311 (выпускаемого фирмой DuPont de Nemours and Company, Wilmington, DE) при таком соотношении объемных скоростей, что отношение массы твердой фазы ПТФЭ к массе твердой фазы вискозы составляло 8,2, и смешивали в совмещенном (с трубой) статическом смесителе. Получившуюся смесь затем прокачивали через фильеру, содержащую 180 отверстий (диаметр 0,152 мм), погруженную под поверхность коагуляционной ванны. В состав коагуляционной ванны входили 5%-ная серная кислота и 18%-ный сульфат натрия. Ее температура поддерживалась на уровне (59±3)oC. Получившиеся волокна затем пропускали через промывную ванну с мягкой водой, температура которой поддерживалась на уровне (46±5)oC, а затем подавали на ряд вращающихся горячих вальцов. Температура поверхности этих вальцов поддерживалась на уровне (210±5)oC, чтобы высушить волокно. Пряжу подавали на другой ряд вращающихся горячих вальцов. Температура поверхности этих вальцов поддерживалась на уровне (360±5)oC для спекания волокна. Пряжу подавали на ряд не нагретых "тянущих вальцов", на которых размещалось множество витков. Различие скоростей второго ряда горячих вальцов и "тянущих вальцов" было таково, что пряжа вытягивалась в 6,1 раза. Эта величина известна как степень вытяжки. Пряжу, выходящую из тянующих вальцов, наматывали на бумажную трубку. Получившаяся пряжа имела линейную плотность 750 дтекс. Сопротивление разрыву составляло 1,40 г/дтекс.
Изобретение относится к технологии получения волокнистых изделий, в частности к промежуточным волоконным структурам из дисперсий политетрафторэтилена и родственных ему полимеров. Способ включает образование смеси водной дисперсии фторированного олефинового полимера и раствора полимерной матрицы из простого эфира целлюлозы со степенью замещения не более около 0,5 и не менее около 0,02. Смесь экструдируют в коагуляционный раствор, который содержит соли, кислоты или их смеси. Полученную промежуточную волоконную структуру промывают в достаточном количестве воды с близким к нейтральному значением рН. Промываемая структура имеет самоподдерживающую длину по меньшей мере 30 см и практически не содержит ионов. Отношение массы частиц полимера к массе полимерной матрицы в структуре составляет от около 3 до около 20. Предусматривается дополнительное спекание изделия для осуществления окисления полимерной матрицы и коалесценции частиц фторированного олефинового полимера. Способ позволяет получать изделия с улучшенными прочностными показателями, но при этом не имеет технологических недостатков, присущих способу с использованием вискозы. 3 с. и 9 з.п.ф-лы, 2 табл., 1 ил.
| US 3118846 А, 21.01.1964 | |||
| JP 60065107 А, 13.04.1985 | |||
| US 2951047 А, 30.08.1960 | |||
| US 5762846 А, 09.06.1998 | |||
| DE 3718949 A1, 10.12.1987 | |||
| Способ получения волокна из водных суспензий политетрафторэтилена | 1960 |
|
SU132800A1 |
