Область изобретения
Изобретение относится к новым способам получения 3(S)-[(5-хлор-1Н-индол-2-карбонил)-амино] -2(R)-гидрокси-4-фенил-масляной кислоты, которая дополнительно может быть подвергнута реакции, протекающей с образованием соединений, которые обладают ингибиторной в отношении гликогенфосфорилазы активностью. Предложены также новые промежуточные соединения, используемые в этих способах. Эти ингибиторы гликогенфосфорилазы пригодны для использования в лечении млекопитающих, особенно людей, с гликогенфосфорилаза-зависимыми заболеваниями или состояниями, в том числе с гиперхолестеринемией, гипергликемией, гиперинсулинемией, гиперлипидемией, гипертензией, атеросклерозом, диабетом и миокардиальной ишемией.
Предпосылки изобретения
В Международных патентных заявках PCT/IB95/00443, опубликованной как WO 96/39385, и PCT/IB95/00442, опубликованной как WO 96/39384, раскрыты новые замещенные N-(индол-2-карбонил)-амиды, производные и промежуточные соединения, в том числе 3(S)-[(5-хлор-1Н-индол-2-карбонил)-амино]-2(R)-гидрокси-4-фенил-масляная кислота, способы получения таких соединений, фармацевтические композиции, содержащие такие соединения или производные, и способы лечения гликогенфосфорилаза-зависимых заболеваний или состояний путем введения таких соединений или производных.
Как описано в этой связи, 3(S)-[(5-хлор-1Н-индол-2-карбонил)-амино]-2(R)-гидрокси-4-фенил-масляная кислота может быть получена путем сочетания хлорангидрида кислоты с аминокислотой с получением эфира 3(S)-[(5-хлор-1Н-индол-2-карбонил)-амино] -2(R)-гидрокси-4-фенил-масляной кислоты, с которого защита может быть затем удалена путем водно-щелочного гидролиза с получением соответствующей кислоты.
Согласно настоящему изобретению предложены новые способы получения 3(S)-[(5-хлор-1Н-индол-2-карбонил)-амино] -2(R)-гидрокси-4-фенил-масляной кислоты, при которых используют на две стадии меньше, чем при известных из уровня техники способах, указанных выше, что в свою очередь обеспечивают более быстрый, более легкий и менее дорогой способ производства. Раскрытые в данном описании способы генерируют всего два промежуточных продукта, которые нет необходимости выделять, и в результате единственного выделения обеспечивают получение 3(R)-[(5-хлор-1Н-индол-2-карбонил)-амино]-2(R)-гидрокси-4-фенил-масляной кислоты. Кроме того, этот продукт также нет необходимости выделять, то есть способ можно продолжить так, как описано, например, в вышеупомянутых WO 96/39385 и/или WO 96/39384, с образованием описанных в них ингибиторов гликогенфосфорилазы.
Все цитированные здесь документы, включая вышеупомянутые, во всей их полноте включены в данное описание ссылкой.
Краткое описание изобретения
Настоящее изобретение относится к новым способам получения 3(S)-[(5-хлор-1Н-индол-2-карбонил)-амино]-2(R)-гидрокси-4-фенил-масляной кислоты.
Настоящее изобретение также относится к новым промежуточным соединениям, образующимся в ходе способов по этому изобретению.
В первом аспекте этого изобретения предложены способы получения 3(S)-[(5-хлор-1Н-индол-2-карбонил)-амино]-2(R)-гидрокси-4-фенил-масляной кислоты, соединения формулы 6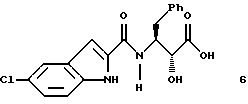
при которых проводят стадии:
получения раствора, содержащего соединение формулы 2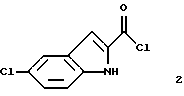
включающего в себя, последовательно, добавление соединения формулы 1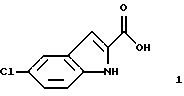
к апротонному растворителю А, добавление некоторого количества каталитического апротонного растворителя при перемешивании в атмосфере инертного газа и добавление активирующего агента;
получения соединения формулы 4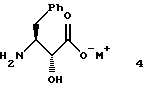
где M+ представляет собой любой одновалентный катион, из соединения формулы 3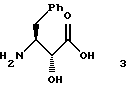
включающего в себя, последовательно, добавление соединения формулы 3 и основания к смеси апротонного растворителя В и протонного растворителя при температуре от приблизительно -20oС до приблизительно температуры дефлегмации указанной смеси и поддерживание рН указанной смеси от приблизительно рН 8 до приблизительно рН 13;
получения соединения формулы 5 путем сочетания указанных соединений формул 2 и 4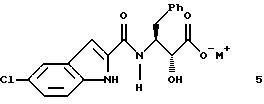
включающего в себя, последовательно, добавление указанного раствора к указанной смеси в атмосфере инертного газа при поддерживании указанной температуры и после завершения указанного добавления выдерживание указанной смеси до достижения комнатной температуры;
добавления некоторого количества органического растворителя к указанной смеси; и
экстрагирования указанного соединения формулы 6 в указанном органическом растворителе, включающего в себя, последовательно, разделение водного слоя А и органического слоя А, обработку указанного органического слоя А водным раствором кислоты или водным раствором кислоты и Н2О, отделение водного слоя Б от органического слоя Б и сохранение указанного органического слоя Б;
при условии, что когда M+ представляет собой N(C1-C6алкил)4 +, после указанного основания к указанной смеси добавляют тетра-С1-С6алкиламмонийгалогенид.
В предпочтительном воплощении указанного первого аспекта указанный апротонный растворитель А и указанный апротонный растворитель Б каждый независимо представляет собой ТГФ, толуол или CH2Cl2. Апротонный растворитель А и апротонный растворитель Б каждый предпочтительно представляет собой толуол.
В другом предпочтительном воплощении указанного первого аспекта указанный каталитический апротонный растворитель представляет собой ДМФ.
В другом предпочтительном воплощении указанного первого аспекта указанный инертный газ представляет собой N2.
В другом предпочтительном воплощении указанного первого аспекта указанный активирующий агент представляет собой оксалилхлорид или тионилхлорид. Тионилхлорид является особенно предпочтительным активирующим агентом. В предпочтительном воплощении, там где указанным активирующим агентом является тионилхлорид, указанное добавление завершают через приблизительно 16 ч.
В другом предпочтительном воплощении указанного первого аспекта указанный M+ представляет собой Li+, Na+, K+, Cs+ или тетра-С1-С6алкиламмоний. Особенно предпочтительный M+ представляет собой Na+, K+ или NBu4 +. Особенно предпочтительный тетра-С1-С6алкиламмонийгалогенид представляет собой ТБАБ (тетрабутиламмонийбромид).
В другом предпочтительном воплощении указанного первого аспекта указанное основание представляет собой бикарбонат натрия, гидроксид натрия, фосфат натрия, карбонат калия, двухосновный фосфат калия или трехосновный фосфат калия. Особенно предпочтительные основания включают в себя бикарбонат натрия, бикарбонат калия и трехосновный фосфат калия. Трехосновный фосфат калия является особенно предпочтительным основанием. В предпочтительном воплощении, там где указанное основание является карбонатом калия, указанный M+ представляет собой NBu4 +, а указанный тетра-С1-С6алкиламмонийгалогенид представляет собой ТБАБ. В предпочтительном воплощении, там где указанным основанием является бикарбонат натрия, указанный M+ представляет собой Na+. В указанном воплощении, там где указанное основание является бикарбонатом натрия, а указанный М+ представляет собой Na+, указанный апротонный растворитель Б представляет собой ТГФ, указанный протонный растворитель представляет собой H2O и указанная температура составляет приблизительно 65oС. В предпочтительном воплощении, там где указанное основание является трехосновным фосфатом калия, указанный M+ представляет собой K+. В предпочтительном воплощении, там где указанное основание является трехосновным фосфатом калия, а указанный М+ представляет собой K+, указанный апротонный растворитель Б представляет собой ТГФ, указанный протонный растворитель представляет собой Н2О и указанная температура составляет приблизительно -5oС.
В другом предпочтительном воплощении указанного первого аспекта указанный протонный растворитель представляет собой Н2О или ROH, где R представляет собой С1-C4алкил. Особенно предпочтительные протонные растворители включают в себя Н2О и МеОН. Особенно предпочтительным протонным растворителем является Н2О.
В другом предпочтительном воплощении указанного первого аспекта указанный рН поддерживают указанным основанием.
В другом предпочтительном воплощении указанного первого аспекта указанный рН составляет от приблизительно рН 11 до приблизительно рН 13.
В другом предпочтительном воплощении указанного первого аспекта указанный органический растворитель представляет собой EtOAc или СН2Сl2. Особенно предпочтительным органическим растворителем является СН2Cl2.
В другом предпочтительном воплощении указанного первого аспекта указанный водный раствор кислоты представляет собой водную HCl или водную H2SO4. Особенно предпочтительным водным раствором кислоты является водная HCl.
В другом предпочтительном воплощении указанного первого аспекта указанный сохраненный органический слой Б концентрируют, переносят в гексаны или гептаны, гранулируют в указанных гексанах или указанных гептанах в течение некоторого периода времени в атмосфере инертного газа, полученную суспензию фильтруют и остаток (содержащий соединение формулы 6) сушат. Предпочтительным периодом времени является время в течение ночи. Предпочтительным инертным газом является N2.
В другом предпочтительном воплощении указанного первого аспекта перед добавлением к указанной смеси указанное соединение формулы 2 выделяют, растворяют в апротонном растворителе В и добавляют к указанной смеси путем добавления указанного апротонного растворителя В, содержащего указанное соединение формулы 2, к указанной смеси. Предпочтительный способ выделения включает в себя, последовательно, добавление гексанов или гептанов к указанному раствору, фильтрование полученной суспензии и сушку остатка. Апротонный растворитель В предпочтительно представляет собой ТГФ, толуол или CH2Cl2 и особенно предпочтительно ТГФ.
В особенно предпочтительном воплощении указанного первого аспекта указанный способ получения указанного соединения формулы 6 включает в себя стадии: получения раствора, содержащего соединение формулы 2, включающего в себя, последовательно, добавление соединения формулы 1 к толуолу, добавление некоторого количества ДМФ при перемешивании в атмосфере N2 и добавление SOCl2; получения соединения формулы 4, где M+ представляет собой Na+, из соединения формулы 3, включающего в себя, последовательно, добавление указанного соединения формулы 3 и NaHCO3 к смеси ТГФ и Н2О при приблизительно 65oС; получения соединения формулы 5, где M+ представляет собой Na+, путем сочетания указанных соединений формул 2 и 4, включающего в себя, последовательно, добавление указанного раствора к указанной смеси в атмосфере N2 при поддерживании температуры приблизительно 65oС и после завершения указанного добавления выдерживание указанной смеси до достижения комнатной температуры; добавления некоторого количества EtOAc к указанной смеси; экстрагирования указанного соединения формулы 6 в указанный EtOAc, включающего в себя, последовательно, разделение указанного водного слоя А и указанного органического слоя А, обработку указанного органического слоя А водным раствором кислоты, отделение указанного водного слоя Б от указанного органического слоя Б и сохранение указанного органического слоя Б; и выделения указанного соединения формулы 6, включающего в себя, последовательно, концентрирование указанного сохраненного органического слоя Б, перенос указанного концентрированного слоя Б в гексаны или гептаны, гранулирование указанного замещенного органического слоя Б в указанных гексанах или гептанах в течение ночи в атмосфере N2, фильтрование полученной суспензии и сушку остатка.
В особенно предпочтительном воплощении указанного первого аспекта указанный способ получения указанного соединения формулы 6 включает в себя стадии: получения раствора, содержащего соединение формулы 2, включающего в себя, последовательно, добавление соединения формулы 1 к толуолу, добавление некоторого количества ДМФ при перемешивании в атмосфере N2 и добавление SOCl2; получения соединения формулы 4, где М+ представляет собой К+, из соединения формулы 3, включающего в себя, последовательно, добавление указанного соединения формулы 3 и K2PO4 к смеси ТГФ и Н2О при приблизительно -5oС; получения соединения формулы 5, где М+ представляет собой K+, путем сочетания указанных соединений формул 2 и 4, включающего в себя, последовательно, добавление указанного раствора к указанной смеси в атмосфере N2 при поддерживании температуры приблизительно -5oС и после завершения указанного добавления выдерживание указанной смеси до достижении комнатной температуры; добавления некоторого количества CH2Cl2 к указанной смеси; экстрагирования указанного соединения формулы 6 в указанном CH2Cl2, включающего в себя, последовательно, разделение указанного водного слоя А и указанного органического слоя А, обработку указанного органического слоя А 1М HCl, отделение указанного водного слоя Б от указанного органического слоя Б и сохранение указанного органического слоя Б; и выделения указанного соединения формулы 6, включающего в себя, последовательно, концентрирование указанного сохраненного органического слоя Б путем дистилляции, перенос указанного концентрированного слоя в гептаны или гексаны до тех пор, пока температура паров не достигнет приблизительно 95oС, охлаждение до температуры окружающей среды, фильтрование полученной суспензии и сушку остатка.
В дополнительном особенно предпочтительном воплощении указанного первого аспекта указанное соединение формулы 6 дополнительно подвергают реакции, протекающей с образованием замещенных N-(индол-2-карбонил)-амидов и производных, раскрытых в вышеупомянутых WO 96/39385 и/или WO 96/39384, как описано в них, например, используя Методику А (пептидное сочетание с использованием ДЭК).
Например, как описано в вышеупомянутом WO 96/39385, соединение формулы 6, полученное новыми способами по этому изобретению, может быть дополнительно подвергнуто реакции с получением, например:
[(1S)-((R)-гидрокси-диметил-карбамоил-метил)-2-фенил-этил] -амида 5-хлор-1 Н-индол-2-карбоновой кислоты,
{(1S)-[(R)-гидрокси-(метокси-метил-карбамоил)-метил]-2-фенил-этил}-амида 5,6-дихлор-1Н-индол-2-карбоновой кислоты,
{(1S)-[(R)-гидрокси-(метокси-метил-карбамоил)-метил]-2-фенил-этил}-амида 5-хлор-1Н-индол-2-карбоновой кислоты,
((1S)-{ (R)-гидрокси-[2-гидрокси-этил)-метил-карбамоил]-метил}-2-фенил-этил)-амида 5-хлор-1Н-индол-2-карбоновой кислоты,
{ (1S)-[(R)-гидрокси-(метил-пиридин-2-ил-карбамоил)-метил]-2-фенил-этил} -амида 5-хлор-1Н-индол-2-карбоновой кислоты,
((1S)-{(R)-гидрокси-[метил-(2-пиридин-2-ил-этил)-карбамоил]-метил}-2-фенил-этил)-амида 5-хлор-1Н-индол-2-карбоновой кислоты,
гидрохлорида [(1S)-бензил-(2R)-гидрокси-3-(4-метил-пиперазин-1-ил)-3-оксо-пропил]-амида 5-хлор-1Н-индол-2-карбоновой кислоты,
[(1S)-бензил-(2R)-гидрокси-3-(3-гидрокси-азетидин-1-ил)-3-оксо-пропил] -амида 5-хлор-1Н-индол-2-карбоновой кислоты,
((1S)-бензил-(2R)-гидрокси-3-изоксазолидин-2-ил-3-оксо-пропил)-амида 5-хлор-1Н-индол-2-карбоновой кислоты,
((1S)-бензил-(2R)-гидрокси-3-[1,2] оксазинан-2-ил-3-оксо-пропил)-амида 5-хлор-1Н-индол-2-карбоновой кислоты,
[(1S)-бензил-(2R)-гидрокси-3-((3S)-гидрокси-пирролидин-1-ил)-3-оксо-пропил]-амида 5-хлор-1Н-индол-2-карбоновой кислоты,
[(1S)-бензил-3-((3S, 4S)-дигидрокси-пирролидин-1-ил)-(2R)-гидрокси-3-оксо-пропил]-амида 5-хлор-1Н-индол-2-карбоновой кислоты,
[(1S)-бензил-3-((ЗR, 4S)-дигидрокси-пирролидин-1-ил)-(2R)-гидрокси-3-оксо-пропил]-амида 5-хлор-1Н-индол-2-карбоновой кислоты или
((1S)-бензил-(2R)-гидрокси-3-морфолин-4-ил-3-оксо-пропил] -амида 5-хлор-1Н-индол-2-карбоновой кислоты.
Как описано в вышеупомянутом WO 96/39384, соединение формулы 6, полученное новыми способами по этому изобретению, может быть дополнительно подвергнуто реакции с получением, например:
[(1S)-бензил-2-(3-гидроксиимино-пирролидин-1-ил)-2-оксо-этил] -амида 5-хлор-1Н-индол-2-карбоновой кислоты,
[2-(цис-3,4-дигидрокси-пирролидин-1-ил)-2-оксо-этил] -амида 5-хлор-1Н-индол-2-карбоновой кислоты,
[2-((3S, 4S)-дигидрокси-пирролидин-1-ил)-2-оксо-этил] -амида 5-хлор-1Н-индол-2-карбоновой кислоты,
[(1S)-бензил-2-(цис-3,4-дигидрокси-пирролидин-1-ил)-2-оксо-этил] -амида 5-хлор-1Н-индол-2-карбоновой кислоты,
[2-(1,1-диоксо-тиазолидин-3-ил)-2-оксо-этил] -амида 5-хлор-1Н-индол-2-карбоновой кислоты,
(2-оксо-2-тиазолидин-3-ил-этил)-амида 5-хлор-1Н-индол-2-карбоновой кислоты,
[(1S)-(4-фтор-бензил)-2-(4-гидрокси-пиперидин-1-ил)-2-оксо-этил] -амида 5-хлор-1Н-индол-2-карбоновой кислоты,
[(1S)-бензил-2-((3RS)-гидрокси-пиперидин-1-ил)-2-оксо-этил] -амида 5-хлор-1Н-индол-2-карбоновой кислоты,
[2-оксо-2-((1RS)-оксо-1-тиазолидин-3-ил)-этил] -амида 5-хлор-1Н-индол-2-карбоновой кислоты,
[(1S)-бензил-(2-фтор-бензил)-2-(4-гидрокси-пиперидин-1-ил)-2-оксо-этил] -амида 5-хлор-1Н-индол-2-карбоновой кислоты,
[(1S)-бензил-2-((3S, 4S)-дигидрокси-пирролидин-1-ил)-2-оксо-этил] -амида 5-хлор-1Н-индол-2-карбоновой кислоты,
[(1S)-бензил-2-(3-гидрокси-азетидин-1-ил)-2-оксо-этил] -амида 5-хлор-1Н-индол-2-карбоновой кислоты,
[(1S)-бензил-2-(3-гидроксиимино-азетидин-1-ил)-2-оксо-этил] -амида 5-хлор-1Н-индол-2-карбоновой кислоты или
[(1S)-бензил-2-(4-гидроксиимино-пиперидин-1-ил)-2-оксо-этил] -амида 5-хлор-1Н-индол-2-карбоновой кислоты.
Во втором аспекте этого изобретения предложены новые соединения формулы 4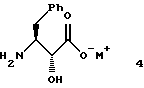
где М+ представляет собой тетра-С1-С6алкиламмоний. Предпочтительным воплощением указанного второго аспекта является новое соединение формулы 4а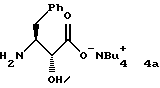
В третьем аспекте этого изобретения предложены новые соединения формулы 5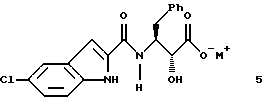
где M+ представляет собой тетра-C1-С6алкиламмоний. Предпочтительным воплощением указанного второго аспекта является новое соединение формулы 5а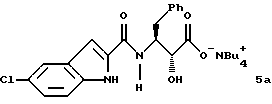
Это изобретение также включает в себя в еще одном аспекте масштабирование новых способов для обеспечения коммерческих количеств любого из соединений, используемых и образуемых в ходе этих способов, включая исходные вещества, промежуточные соединения и конечный продукт. Например, добавление по каплям (предпочтительно с использованием капельной воронки) раствора, содержащего соединение формулы 2, к смеси, содержащей соединение формулы 4.
Специалистам будут полностью понятны термины, используемые здесь в описании и в прилагаемой формуле изобретения для описания настоящего изобретения; тем не менее, если здесь не указано иначе, следующие термины являются такими, как представлено непосредственно ниже.
"Алкил" означает радикал с прямой или разветвленной углеводородной цепью, причем "C1-C6-алкил" включает в себя метил, этил, пропил, изопропил, бутил, втор-бутил, трет-бутил, пентил, изопентил, гексил и изогексил, a "C1-С4алкил" включает в себя метил, этил, пропил, изопропил, бутил, вторичный бутил и третичный бутил.
"Апротонный" означает растворитель без доступного протона.
"Протонный" означает растворитель с доступным протоном.
Если не указано иначе, по всему этому документу и прилагаемой формуле изобретения: % - процент, АЦН - ацетонитрил, oС - градус Цельсия, CH2Cl2 - метиленхлорид, см - сантиметр или сантиметры, ДЭК - 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид, ДМФ - диметилформамид, экв. или эквив. - эквивалент или эквиваленты, EtOAc - этилацетат, г - грамм или граммы, ч - час или часы, HCl - соляная кислота, HClO4 - перхлорная кислота, Н2O - вода, H2SO4 - серная кислота, ЖХВД - жидкостная хроматография высокого давления, K2СО3 - карбонат калия, K3РO4 - трехосновный фосфат калия, М - молярная (концентрация), МеОН - метанол, MgSO4 - сульфат магния, мин - минута или минуты, мл - миллилитр или миллилитры, мм - миллиметр или миллиметры, ммоль - миллимоль или миллимоли, МС - масс-спектр, н. - нормальная (концентрация), NaHCO3 - бикарбонат натрия, NaOH - гидроксид натрия, NBu4 + - тетрабутиламмоний или ТБА, нм - нанометр или нанометры, ЯМР - спектр протонного ядерного магнитного резонанса, ОФ ЖХВД - обратнофазная ЖВХД, КТ - комнатная температура, ТБАБ - тетрабутиламмонийбромид, ТГФ - тетрагидрофуран, мкл - микролитр или микролитры, мкм - микромоль или микромоли, УФ -ультрафиолетовое излучение, и об./об. - объем/объем.
Подробное описание изобретения
Согласно этому изобретению предложены новые способы получения соединения формулы 6 из соединений формул 1 и 3. Эти исходные вещества являются известными соединениями, которые могут быть получены специалистами. Кроме того, оба эти соединения коммерчески доступны, как описано более подробно ниже в Примерах.
Соединение формулы 2 может быть получено путем помещения соединения формулы 1 в любой подходящий апротонный растворитель, предпочтительно толуол, добавления (предпочтительно по каплям) любого подходящего активирующего агента для получения хлорангидрида кислоты, предпочтительно SOCl2, а затем добавления любого подходящего каталитического апротонного растворителя, предпочтительно ДМФ. Этот раствор, содержащий соединение формулы 2, затем может быть добавлен прямо к раствору, содержащему соединение формулы 4. Альтернативно и предпочтительно, соединение формулы 2 сначала может быть выделено путем добавления любого подходящего понижающего растворимость растворителя, предпочтительно гексанов, к раствору, содержащему соединение формулы 2, фильтрования раствора, а затем сушки остатка. Выделенное соединение формулы 2 затем может быть растворено в любом подходящем апротонным растворителе, предпочтительно ТГФ, и добавлено к раствору, содержащему соединение формулы 4.
Соединение формулы 4 может быть получено путем добавления соединения формулы 3 и любого подходящего основания, предпочтительно K3РO4, к смеси любого подходящего апротонного растворителя, предпочтительно ТГФ, и любого подходящего протонного растворителя, предпочтительно Н2О, при КТ в атмосфере любого подходящего инертного газа, предпочтительно N2, и полученная суспензия может быть охлаждена, предпочтительно в смеси льда и ацетона. Сразу после этого для поддерживания рН от приблизительно рН 8 до приблизительно рН 13, предпочтительно от приблизительно рН 11 до приблизительно рН 13, подают вышеупомянутое основание.
Затем раствор, содержащий соединение формулы 2, может быть добавлен в атмосфере любого подходящего инертного газа, предпочтительно N2, в течение любого подходящего периода времени, предпочтительно от приблизительно 1 часа до приблизительно 2 часов, при охлаждении, поддерживаемом в процессе добавления, с последующим продолжением охлаждения в течение любого подходящего периода времени, предпочтительно от приблизительно 1 ч до приблизительно 2 ч, а затем нагреванием до КТ с выделением соединения формулы 5.
Соединение формулы 6 затем может быть получено из соединения формулы 5 путем добавления любого подходящего апротонного растворителя, предпочтительно CH2Cl2, разделения водного и органического слоев, промывания органического слоя водной кислотой, предпочтительно HCl, предпочтительно 1М HCl, концентрирования промытого органического слоя, предпочтительно путем дистилляции, переноса концентрированного органического слоя в любую подходящую органическую жидкость, предпочтительно гептаны, охлаждения до КТ в течение любого подходящего периода времени, например в течение ночи, фильтрования полученной суспензии и сушки полученного твердого вещества или остатка, т.е. соединения формулы 6, под вакуумом в течение любого подходящего периода времени, например в течение ночи.
Проверка того, что новые способы, предложенные согласно этому изобретению, обеспечивают получение соединения формулы 6, может быть легко проведена специалистами. Присутствие соединения формулы 6, а также относительная и абсолютная чистота его в полученном высушенном твердом веществе могут быть определены ЖВХД, а именно по ЖВХД УФ% площади и по сравнению с внешним стандартом. Химическая структура полученного в результате соединения формулы 6 может быть проверена путем масс- и ЯМР-спектроскопии.
Как это очевидно специалистам, могут быть использованы любые подходящие количества любых веществ, используемых в новых способах по этому изобретению, в зависимости от, например, желаемого количества 3(S)-[(5-хлор-1Н-индол-2-карбонил)-амино]-2(R)-гидрокси-4-фенил-масляной кислоты.
Однако более конкретно на стадии получения соединения формулы 2 количества апротонного растворителя А, каталитического апротонного растворителя и активирующего агента берут в расчете на количество соединения формулы 1:
для апротонного растворителя А - предпочтительно от приблизительно 4 мл/г соединения формулы 1 до приблизительно 30 мл/г соединения формулы 1, более предпочтительно от приблизительно 7 мл/г до приблизительно 20 мл/г и наиболее предпочтительно приблизительно 10 мл/г;
для каталитического апротонного растворителя - предпочтительно от приблизительно 0,0001 мл/г соединения формулы 1 до приблизительно 0,25 мл/г соединения формулы 1, более предпочтительно от приблизительно 0,002 мл/г до приблизительно 0,1 мл/г и наиболее предпочтительно приблизительно 0,0004 мл/г;
для активирующего агента - предпочтительно от приблизительно 0,95 моль/моль соединения формулы 1 до приблизительно 2,0 моль/моль соединения формулы 1, более предпочтительно от приблизительно 1,1 моль/моль до приблизительно 1,5 моль/моль и наиболее предпочтительно 1,3 моль/моль.
Точно так же на стадии получения соединения формулы 4 количества основания, апротонного растворителя Б и протонного растворителя берут в расчете на количество соединения формулы 3:
для основания - предпочтительно от приблизительно 1,9 моль/моль соединения формулы 3 до приблизительно 3,0 мол/моль соединения формулы 3, более предпочтительно от приблизительно 2,0 моль/моль до приблизительно 2,3 моль/моль и наиболее предпочтительно приблизительно 2,1 моль/моль;
для апротонного растворителя Б - предпочтительно от приблизительно 3 мл/г соединения формулы 3 до приблизительно 30 мл/г соединения формулы 3, более предпочтительно от 4 мл/г до приблизительно 10 мл/г и наиболее предпочтительно приблизительно 5 мл/г;
для протонного растворителя - предпочтительно от приблизительно 3 мл/г соединения формулы 3 до приблизительно 30 мл/г соединения формулы 3, более предпочтительно от приблизительно 4 мл/г до приблизительно 10 мл/г и наиболее предпочтительно приблизительно 5 мл/г.
Далее, на стадии добавления органического растворителя количество органического растворителя берут в расчете на количество соединения формулы 3, т. е. предпочтительно от приблизительно 3 мл/г соединения формулы 3 до приблизительно 30 мл/г соединения формулы 3, более предпочтительно от приблизительно 4 мл/г до приблизительно 10 мл/г и наиболее предпочтительно приблизительно 5 мл/г.
Как обсуждалось выше, в ходе новых способов образуются новые промежуточные соединения, предложенные согласно этому изобретению. Первыми по времени являются новые промежуточные соединения формулы 4, где M+ представляет собой тетра-С1-С6алкиламмоний, полученные из соединения формулы 3, затем в результате сочетания соединения формулы 4 и соединения формулы 2 образуются новые промежуточные соединения с получением новых промежуточных соединений формулы 5. Предпочтительные промежуточные соединения включают в себя соединения вышеупомянутых формул 4а и 5а, где M+ представляет собой NBu4 + (например, из ТБАБ).
Основываясь на данном описании, специалистам будет понятно, как легко получить новые соединения формул 4, 4а, 5 и 5а, используя стандартные методы синтеза. Например, соединение формулы 4а может быть получено путем добавления соединения формулы 3 и любого подходящего основания, предпочтительно K3РO4, к смеси любого предпочтительного апротонного растворителя, предпочтительно ТГФ, и любого подходящего протонного растворителя, предпочтительно Н2О, и тетрабутиламмонийбромида при КТ в атмосфере любого подходящего инертного газа, предпочтительно N2, и охлаждения полученной суспензии, предпочтительно в смеси льда в ацетоне. Соединение формулы 2, полученное как обсуждалось выше, затем может быть добавлено, как также обсуждалось выше, к этому раствору, содержащему соединение формулы 4а, с получением соединения формулы 5а. Как описано выше для соединений формул 4 и 5, ни одно из соединений формул 4а и 5а нет необходимости выделять, и предпочтительно не нужно, в ходе способа по этому изобретению перед соответствующими следующими стадиями их обработки.
Специалистам должно быть ясно из настоящего описания, что в зависимости от, например, требуемого масштаба при использовании в новых способах нагрев может быть обеспечен любым подходящим способом, например обычно удобно использовать нагревательную рубашку. Точно так же любой подходящий способ охлаждения может быть использован при осуществлении способов по данному изобретению, например выдерживание при КТ, ледяная баня, холодная комната или просто удаление источника тепла.
Данное изобретение включает в себя также любые геометрические и оптические изомеры этих новых промежуточных соединений. Включены также изотопно-меченые соединения формул 4а и 5а, которые идентичны соединениям, приведенным в формулах 4а и 5а, но при условии, что один или более чем один атом заменен на атом, имеющий атомную массу или массовое число, отличные от атомной массы или массового числа естественно существующего в природе. Примеры изотопов, которые могут быть введены в такие новые промежуточные соединения по этому изобретению, включают в себя изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, такие как 2H, 3H, 13С, 14С, 15N, 18О, 17О, 31Р, 32P, 35S, 18F и 36Cl соответственно.
Не будучи связанными какой-либо конкретной теорией, специалисты поймут, что карбоксилатная соль соединения формулы 3 делает соединение формулы 3 более растворимым в апротонном растворителе, в котором происходит сочетание соединения формулы 2 с соединением формулы 3. Как также поймут специалисты, такая повышенная растворимость соединения формулы 3 по существу исключает нежелательный гидролиз соединения формулы 2 обратно в соединение формулы 1, который в противном случае негативно влияет на получение соединения формулы 4 и, следовательно, на выход 3(S)-[(5-хлор-1Н-индол-2-карбонил)-амино]-2(R)-гидрокси-4-фенил-масляной кислоты.
Несмотря на то что соединение формулы 6, получение которого обеспечено способами по этому изобретению, дополнительно может быть подвергнуто реакции с получением соединений и производных, описанных в вышеупомянутых WO 96/39385 и/или WO 96/39384, способы по этому изобретению благодаря образованию карбоксилатной соли соединения формулы 3 исключают стадии защиты и удаления защиты карбоксильной группы, описанные в вышеупомянутых WO 96/39385 и/или WO 96/39385. Как должно быть понятно специалистам, меньшее общее количество стадий и использование промежуточных соединений, которые сохраняют свою растворимость, не подвергаются заметной самодимеризации или гидролизу до сочетания и не требуют выделения перед их последующим сочетанием, обеспечивают преимущества, которые реализуются, в частности, в промышленных масштабах.
Настоящее изобретение включает в себя любые подходящие способы получения соединений формул 2 и 4, например, такие, которые специалисты, основываясь на данном описании, могут выбрать для защиты конкретной функциональной группы, например для предотвращения вмешательства такой функциональной группы в реакции на других центрах в пределах молекулы или для сохранения цельности такой функциональной группы, в любом данном случае. Необходимость и тип такой защиты легко определят специалисты, и они будут зависеть от, например, природы функциональности и условий выбранного способа получения [T.W. Green, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1991]. Подходящие защитные группы для любой конкретной функциональной группы могли бы включать группы, которые не являются существенно химически активными в реакционных условиях, описанных для новых способов, и которые могут быть удалены без существенного химического изменения других функциональных групп любого данного промежуточного соединения. Как должно быть понято специалистам, защитные группы могут быть удалены так, как это требуется при любом данном способе получения, например на более поздней стадии.
Специалистам будет понятно на основании этого описания и описаний, которые даны в вышеупомянутых WO 96/39385 и WO 96/39384, как легко получить соединения, раскрытые в WO 96/39385 и WO 96/39384, из соединения формулы 6, используя стандартные методы синтеза, такие как, например, вышеупомянутые Методика А, описанная в них и приведенная непосредственно ниже.
Методика А (Пептидное сочетание с использованием ДЭК)
Раствор первичного амина (0,1-0,7М, 1,0 эквив., или гидрохлорида первичного амина и 1,0-1,3 эквив. триэтиламина на эквив. HCl) в дихлорметане (или любом подходящем растворителе) обрабатывают последовательно при 25oС конкретной карбоновой кислотой (0,95-1,2 эквив.), гидратом гидроксибензотриазола (1,2-1,8 эквив., обычно 1,5 эквив. по отношению к карбоновой кислоте) и ДЭК (0,95-1,2 эквив., соответственно в молярном отношении к карбоновой кислоте) и смесь перемешивают в течение 14-20 ч. При реакциях сочетания в больших масштабах, например >50 мл растворителя, смесь концентрируют в этой точке и остаток растворяют в EtOAc). Смесь разбавляют EtOAc, промывают 2-3 раза 1н. или 2н. NaOH, 2-3 раза 1н. или 2н. HCl (если продукт содержит ионизуемую аминную функциональную группу, кислоту не используют), органический слой сушат над MgSO4 и концентрируют, получая неочищенный продукт, который очищают хроматографией на силикагеле, тритурацией или перекристаллизацией с использованием подходящих растворителей. Очищенные продукты анализируют посредством ОФ-ЖХВД. Реакции проводят при температуре от 0 до 25oС. Эти реакции проводят с предварительным охлаждением реакционного сосуда в изолированной ледяной бане, которой дают нагреться до КТ в течение нескольких часов. Специалистам будет ясно (например, из описания в вышеупомянутых WO 96/39385 и WO 96/39384), как модифицировать Методику А в зависимости от, например, окончательного соединения, которое стремятся получить из соединения формулы 6, и такие модификации считаются частью этого изобретения.
Настоящее изобретение иллюстрируется следующими ПРИМЕРАМИ, которые приведены только в целях иллюстрации и не ограничивают изобретение. Более того, должно быть понято, что другие изменения и модификации, которые могут быть применены на практике, также являются частью изобретения и, как таковые, также находятся в объеме прилагаемой формулы изобретения: специалисты признают или будут способны установить без лишнего экспериментирования эквиваленты конкретных воплощений этого изобретения, описанных здесь, и подразумевается, что такие эквиваленты охвачены прилагаемой формулой изобретения.
В ПРИМЕРАХ оценку идентичности и чистоты продукта новых способов по этому изобретению обеспечивали анализом ОФ-ЖХВД. Более конкретно, анализ ОФ-ЖХВД проводили, используя прибор Waters (34 Marple Street, Milford, Massachusetts 01757) Symmetry C8, 5 мкм, 15 см длиной х 3,9 мм внутренний диаметр, температура в колонке 40oС. Подвижную фазу А [800:200:4, деионизированная Н2O:МеОН:НСlO4, об./об.] готовили, на 1 л, смешиванием 800 мл деионизованной Н2О, 200 мл МеОН и 4 мл Н2О и дегазацией под вакуумом [где объемы компонентов могут быть доведены до получения соответствующих объемов в любом данном случае]. Подвижную Фазу Б [800:200 АЦН:МеОН, об./об.] готовили, на 1 л, смешиванием 800 мл АЦН и 200 мл МеОН и дегазацией под вакуумом [где объемы компонентов могут быть доведены до получения соответствующих объемов для любого данного случая] . Растворители непрерывно дегазируют путем продувки гелием или использованием встроенного дегазатора, детектирование при УФ 220 нм, скорость потока 2,0 мл/мин, объем впрыска 20 мкл, время прохода 65 мин [включая переуравновешивание между впрысками]. Время удерживания 3(S)-[(5-хлор-1Н-индол-2-карбонил)-амино] -2(R)-гидрокси-4-фенил-масляной кислоты составляет приблизительно 15 минут в этих условиях хроматографии.
Соединение формулы 1, используемое в ПРИМЕРАХ, было приобретено у Merck SA (Zone Inindustrielle 45300, Tithivier, France).
Соединение формулы 3, используемое в ПРИМЕРАХ, было приобретено у Nippon Kayaku (Fujimi Building, 11-12 Fujimi 1-Chone, Chiyoda-Ka 102, Japan).
ПРИМЕРЫ
ПРИМЕР 1
Получение Соединения формулы 2
А. Если активирующий агент является тионилхлоридом
Соединение формулы 1 [50,00 г, 255,6 ммоль] добавляют к толуолу [500 мл] . Добавляют ДМФ [0,02 мл, 0,2556 ммоль, (0,001 экв.)]. Суспензию перемешивают в атмосфере N2. По каплям добавляют тионилхлорид [24,24 мл, 332,3 ммоль, (1,30 экв.)], а затем смесь нагревают до 85oС и реакцию продолжают в течение 18 ч. Раствор охлаждают до 40oС и концентрируют вакуумной дистилляцией до объема приблизительно 100-150 мл, после чего вакуум нарушают и по каплям добавляют гексаны [250 мл] в течение приблизительно 1 ч при 40oС. Суспензию затем оставляют охлаждаться до КТ и перемешивают, гранулируют, фильтруют и сушат. Выход соединения формулы 2 составляет 51,98 г [твердое вещество, 95%] .
Б. Если активирующий агент является оксалилхлоридом
Оксалил хлорид [4,6 мл, 53,8 ммоль, (1,05 экв.)] добавляют к суспензии соединения формулы 1 [10,0 г, 51,3 ммоль, (1,0 экв.)] в CH2Cl2 [200 мл]. Затем медленно добавляют пару капель ДМФ. После прекращения выделения газа добавляют дополнительно ДМФ [10 мл] и раствор перемешивают до гомогенного состояния.
ПРИМЕР 2
Получение соединения формулы 4
А. Если М+ представляет собой К+
Соединение формулы 3 [5,00 г, 25,6 ммоль] и K3РO4 [11,42 г, 53,8 ммоль, (2,10 экв.)] перемешивают в ТГФ (25 мл) и H2O (25 мл) при КТ. Раствор затем охлаждают до приблизительно -5oС.
Б. Если М+ представляет собой Na+
Соединение формулы 3 [75,00 г, 384,2 ммоль] перемешивают в ТГФ [375 мл] и H2O [375 мл]. Добавляют NaHCO3 [80,68 г, 960,5 ммоль, (2,50 экв.)]. Суспензию затем перемешивают и нагревают до приблизительно 65oС до образования прозрачного раствора.
В. Если М+ представляет собой NBu4 +
1. Тетрабутиламмоний-3(S)-амино-2(R)-гидрокси-4-фенил-бутират
К соединению формулы 3 [10,0 г, 51,3 ммоль, (1,0 экв.)] добавляют метанол [200 мл], затем добавляют K2СО3 [7 г, 51,3 ммоль, (1,0 экв.), тонкоизмельченный] , после чего добавляют ТБАБ [17,0 г, 51,3 ммоль, (1,0 экв.)]. Суспензию перемешивают до тех пор, пока она не станет легкотекучей. Затем добавляют метиленхлорид [100 мл] с последующим добавлением КНСО3 [20,0 г, 20 ммоль, (4 экв.), тонкоизмельченный].
2. Тетрабутиламмоний-3(S)-амино-2(R)-гидрокси-4-фенил-бутират
Воду [20 мл] и СН2Сl2 [200 мл] добавляют к соединению формулы 3 [10,0 г, 51,3 ммоль, (1,0 экв.)], затем добавляют K2СО3 [35 г, 256 ммоль, (5,0 экв.)] , после чего добавляют ТБАБ [17,0 г, 51,3 ммоль (1,0 экв.)]. Суспензию перемешивают до тех пор, пока она не станет лекготекучей.
ПРИМЕР 3
Сочетание, завершение и выделение, а также анализ
А. Соединение ПРИМЕРА 1.А. с соединением ПРИМЕРА 2.А.
1. Сочетание. 3(S)-[(5-хлор-1Н-индол-2-карбонил)-амино]-2(R)-гидрокси-4-фенил-бутират калия
Определенное количество соединения ПРИМЕРА 1.А. [5,47 г, 25,6 ммоль] растворяют в ТГФ [25 мл]. Этот раствор затем добавляют к раствору, полученному в ПРИМЕРЕ 2.А. в течение периода времени 80 мин при -5oС. Полученную суспензию перемешивают в течение 2 ч при -5oС и оставляют нагреваться до КТ.
2. Завершение и выделение
Метиленхлорид [25 мл] добавляют к реакционной смеси после окончания реакции сочетания и эту смесь перемешивают в течение 15 мин. Водный и органический слои затем разделяют. Органический слой обрабатывают 1М HCl [2 х 25 мл]. Обработанный кислотой органический слой затем концентрируют дистилляцией и переносят в гептаны, постепенно добавляя гептаны до тех пор, пока температура паров не достигнет 95oС, а затем охлаждают до температуры окружающей среды. Полученную суспензию фильтруют и твердое вещество сушат в вакуумной печи. Выход 3(S)-[(5-хлор-1Н-индол-2-карбонил)-амино]-2(R)-гидрокси-4-фенил-масляной кислоты составляет 9,16 г (твердое вещество, 96%).
3. Анализ
а. Твердое вещество имеет чистоту более 99% по данным ЖХВД УФ% площади.
б. 1НЯМР (400 МГц, CDCl3/ДМСО-d6) δ 10,99 (s, 1Н, NH), 7,61 (а, 1Н, NH), 6,82-7,34 (m, 9Н), 4,63-4,69 (m, 1H, СН3), 3,99-4,00 (m, 1H, С2Н), 2,84-2,95 (m, 2H, С4Н).
в. МС (М-1) = 371 для C19H17ClN2O4.
Б. Соединение ПРИМЕРА 1.А. с соединением ПРИМЕРА 2.Б.
1. Сочетание. 3(S)[(5-хлор-1Н-индол-2-карбонил)-амино]-2(R)-гидрокси-4-фенил-бутират натрия
Соединение формулы 2, полученное таким же общим способом, как описано в ПРИМЕРЕ 1.А. [82,24 г, 384,2 ммоль], растворяют в ТГФ [190 мл]. Этот раствор затем добавляют к раствору, полученному в ПРИМЕРЕ 2.Б. в течение периода 50 мин при 65oС. Полученную суспензию перемешивают в течение приблизительно 3,5 ч при 65oС и оставляют охлаждаться до КТ.
2. Завершение и выделение
Этилацетат [375 мл] добавляют к реакционной смеси после окончания реакции сочетания. Полученную смесь перемешивают, а затем дают ей отстояться. Водный и органический слои затем разделяют. Органический слой обрабатывают 1М HCl [375 мл] и H2О [375 мл]. Водный и органический слои затем разделяют. Органический слой затем концентрируют дистилляцией. Смесь EtOAc/ТГФ концентрируют и удаляют, в то же время заменяя гептанами [375 мл]. Полученную суспензию гранулируют и сушат в вакуумной печи. Выход 3(S)-[(5-хлор-1Н-индол-2-карбонил)-амино] -2(R)-гидрокси-4-фенил-масляной кислоты составляет 132,34 г (твердое вещество, 92,4%).
3. Анализ
По данным ЖХВД УФ% площади твердое вещество оказалось чистым и показало такие же значения ЯМР и МС, как и в ПРИМЕРЕ 3.А.
В. Соединение ПРИМЕРА 1.Б. с соединением ПРИМЕРА 2.В.1.
1. Сочетание. Тетрабутиламмоний-3(S)-[(5-хлор-1Н-индол-2-карбонил)-амино]-2(R)-гидрокси-4-фенил-бутират
Раствор ПРИМЕРА 1.Б. по каплям добавляют к раствору ПРИМЕРА 2.В.1., используя капельную воронку, в течение периода времени 2 ч. После добавления одной четверти раствора соединения формулы 2 добавляют K2СО3 [7,0 г, 51,3 ммоль, (1,0 экв.), тонкоизмельченный]. После добавления трех четвертей раствора соединения формулы 2 добавляют K2СО3 [7,0 г, 51,3 ммоль, (1,0 экв.). тонкоизмельченный]. рН реакционной смеси измеряют перед добавлением соединения формулы 2 [рН 11] и в конце добавления [рН 9] путем опускания полоски бумаги для определения рН прямо в реакционную смесь.
2. Завершение и выделение
Реакционную смесь после окончания реакции сочетания фильтруют для удаления неорганических солей [осадок на фильтре промывают МеОН до тех пор, пока ЖХВД не покажет отсутствие продукта], разбавляют EtOAc и летучие вещества удаляют под вакуумом. EtOAc-слой [примерно 500 мл] затем промывают 3 раза 1М H2SO4 [100 мл, охлажденная во льду]. Чтобы убедиться, что соли ТБА удалены, отбирают аликвоту и анализируют ее ЯМР. После этого следуют 2 промывки рассолом [100 мл] , сушка над сульфатом натрия, фильтрация и концентрирование под вакуумом. Продукт концентрируют до приблизительно 100 мл EtOAc и осаждают при добавлении гексана [около 100 мл]. Суспензию перемешивают в течение ночи, а затем фильтруют. Выход 3(S)-[(5-хлор-1Н-индол-2-карбонил)-амино]-2(R)-гидрокси-4-фенил-масляной кислоты составляет 16,62 г [твердое вещество, 87%].
3. Анализ
Твердое вещество имеет чистоту более 99,5% по данным ЖХВД УФ% площади и показывает такие же данные ЯМР и МС, как и данные, которые приведены в ПРИМЕРЕ 3.А.
Г. Соединение ПРИМЕРА 1.Б. с соединением ПРИМЕРА 2.В.2.
1. Сочетание. Тетрабутиламмоний-3(S)-[(5-хлор-1Н-индол-2-карбонил)-амино]-2(R)-гидрокси-4-фенил-бутират
Раствор ПРИМЕРА 1.Б. по каплям добавляют к смеси ПРИМЕРА 2.В.2., используя капельную воронку, в течение 2 ч. рН суспензии все время составляет приблизительно рН 11 [определяют, опуская полоску бумаги для определения рН прямо в реакционную смесь] . Реакционную смесь разбавляют МеОН, a CH2Cl2 удаляют под вакуумом. рН доводят до рН 13-14 1н. NaOH и перемешивают реакционную смесь в течение ночи.
2. Завершение и выделение
Реакционную смесь разбавляют EtOAc и летучие вещества удаляют под вакуумом. Смесь [содержащую приблизительно 500 мл EtOAc] доводят до рН 1 6М HCl. EtOAc-слой отделяют, а затем дважды промывают 1М HCl. За этим следуют 2 промывки рассолом [100 мл], сушка над сульфатом натрия, фильтрование и концентрирование под вакуумом. Продукт концентрируют до приблизительно 100 мл EtOAc и осаждают при добавлении гексанов [приблизительно 100 мл]. Суспензию перемешивают в течение ночи, а затем фильтруют. Выход 3(S)-[(5-хлор-1Н-индол-2-карбонил)-амино] -2(R)-гидрокси-4-фенил-масляной кислоты составляет 16,59 г [твердое вещество, 87%].
3. Анализ
Твердое вещество имеет чистоту более 98% чистоты по данным ЖХВД УФ% площади и показывает такие же данные ЯМР и МС, как в ПРИМЕРЕ 3.А.
Изобретение относится к новому способу получения соединения формулы 6, который включает получение раствора соединения формулы 2 путем последовательного добавления соединения формулы 1 к апротонному растворителю А, взятому в количестве 4-30 мг/г, апротонного растворителя, взятого в количестве 0,0001-0,25 мл/г, при перемешивании в атмосфере инертного газа и добавление активирующего агента, взятого в количестве 0,95-2,0 моль/моль от указанного соединения 1; получение соединения формулы 4 путем последовательного добавления соединения формулы 3 и основания, взятого в количестве 1,9-3,0 моль/моль к смеси апротонного растворителя Б, взятого в количестве 3-30 мг/г и протонного растворителя, взятого в количестве 3-30 мл/г от указанного соединения 3, при температуре от -20oС до температуры дефлегмации указанной смеси и поддержания рН указанной смеси 8-13; получение соединения формулы 5, включающего в себя, последовательно, добавление указанного раствора к указанной смеси в атмосфере инертного газа при поддерживании указанной температуры, с последующим выдерживанием полученной смеси до достижения комнатной температуры и добавления к ней органического растворителя в количестве 3-30 мл/г и экстрагирования соединения формулы 6 в указанный органический растворитель, включающий в себя, последовательно, разделение водного слоя А и органического слоя А, обработку указанного органического слоя А водным раствором кислоты или водным раствором кислоты и воды, отделение водного слоя Б от органического слоя Б, где М+ представляет одновалентный катион при условии, что, если М+ представляет N(C1-C6алкил)4 +, после указанного основания к указанной смеси добавляют тетра - C1-С6алкиламмония галогенид. Способ обеспечивает получение соединения 6 за меньшее число стадий за счет образования соединений формулы 4 и 5. 3 с. и 23 з.п. ф-лы.
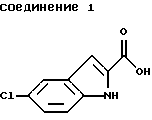
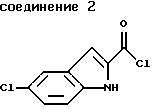
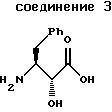
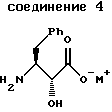
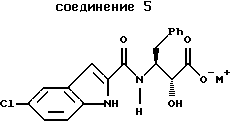
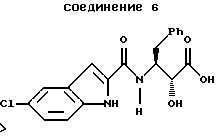
указанное соединение формулы 1 представляет собой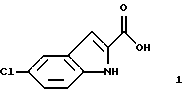
указанное соединение формулы 2 представляет собой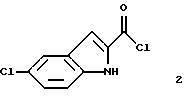
указанное соединение формулы 3 представляет собой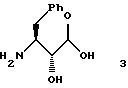
указанное соединение формулы 4 представляет собой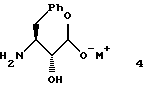
указанное соединение формулы 5 представляет собой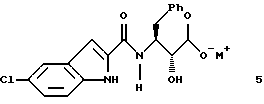
и указанное соединение формулы 6 представляет собой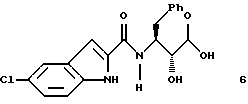
2. Способ по п. 1, где указанный апротонный растворитель А и указанный апротонный растворитель Б каждый независимо представляет собой ТГФ, толуол или CH2Cl2.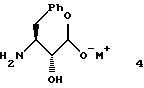
где M+ представляет собой тетра-С1-С6алкиламмоний.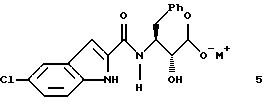
где М+ представляет собой тетра-С1-С6алкиламмоний.
| WO 9639384 А1, 12.12.1996 | |||
| WO 9639385 А1, 12.12.1996 | |||
| WO 9734864 А, 25.09.1997 | |||
| Способ выделения солей аспарагиновойКиСлОТы | 1978 |
|
SU797572A3 |

