Изобретение относится к новым замещенным фенильным производным, которые являются сильными блокаторами хлор-ионных каналов и как таковые полезны при лечении серповидно-клеточной анемии, отека головного мозга, сопровождающего ишемию или опухоли, диареи, гипертензии (в качестве диуретиков), остеопороза и для снижения внутриглазного давления для лечения таких расстройств, как глаукома.
Предпосылки изобретения
Хлор-ионные каналы выполняют огромное разнообразие специфических клеточных функций. Так, хлор-ионные каналы вносят вклад в нормальное функционирование клеток скелетных и гладких мышц. Известно, что блокаторы хлор-ионных каналов применяют при лечении отека головного мозга, сопровождающего ишемию или опухоли, диареи, гипертензии (в качестве диуретиков), остеопороза и для снижения внутриглазного давления при таких расстройствах, как глаукома. Соединения по изобретению могут также применяться при лечении аллергических и воспалительных состояний и для стимулирования заживления ран.
Использование блокаторов хлор-ионных каналов для лечения серповидно-клеточной анемии составляет новый терапевтический подход.
Серповидно-клеточная анемия и наличие гемоглобина серповидных эритроцитов было первым генетическим заболеванием, которое сумели объяснить на молекулярном уровне. Генетический дефект, лежащий в основе серповидно-клеточной анемии, вызывает замещение одной аминокислоты, приводящее к мутантному гемоглобину, гемоглобину серповидных эритроцитов.
Физические проявления серповидно-клеточной анемии представляют собой анемию и болезненные ишемические кризы вследствие окклюзии микроциркуляции деформированными эритроцитами (серповидными эритроцитами). Основная причина серповидной деформации эритроцитов и искривления (или выработки серповидных эритроцитов) представляет собой обратимую полимеризацию и застудневание гемоглобина серповидных эритроцитов, индуцируемые при низких давлениях кислорода, преобладающих в метаболически активных тканях. Серповидные эритроциты отличаются также повышенной катионной проницаемостью, приводящей к катионному истощению и клеточному обезвоживанию. Поскольку время задержки полимеризации было описано как чрезвычайно крутая функция концентрации гемоглобина серповидных эритроцитов, любое снижение клеточного объема должно значительно увеличивать вероятность выработки серповидно-клеточных эритроцитов и, таким образом, окклюзии сосудов. Соединения, которые блокируют дезоксигенирование, вызывающее потерю соли и объема (воды), могут задерживать процесс выработки серповидно-клеточных эритроцитов в достаточной степени, чтобы избежать окклюзии при переносе серповидного эритроцита через метаболически активную ткань. Подсчитано, что достаточным может быть время задержки, составляющее лишь 10 секунд.
Было выдвинуто предположение, что некоторые мембранные ионные каналы и переносчики, присутствующие в нормальных эритроцитах, участвуют в измененной мембранной проницаемости серповидных эритроцитов. Получила признание гипотеза о стимулировании Са2+-активируемого К+-канала, и несколько блокаторов этого канала были предложены в качестве терапевтических агентов для лечения серповидно-клеточной анемии [Effects of Cetiedil on Monovalent Cation Permeability in the Erythrocyte: An explanation for the Efficacy of Cetiedil in the treatment of Sickle Cell Anaemia, Berkowitz, L.R. Orringer, E.P, Blood cells, 283-288 (1982) и патент США 5273992].
Так как после эманации К+ через К-канал для поддержания электронейтральности должна следовать равная эманация Cl-, нужно, чтобы блокада хлор-ионных каналов эритроцитов была такой же эффективной, как и блокада самих К-каналов. Преимущество использования блокаторов хлор-ионных каналов заключается в том, что потеря соли, которая может происходить вследствие активации неизвестных типов К-каналов, косвенно также будет блокироваться.
Соединения по настоящему изобретению представляют собой сильнодействующие блокаторы хлор-ионных каналов, что определено сопутствующими измерениями общих проводящих потоков хлор-ионов и мембранных потенциалов в суспензиях эритроцитов, и по этой причине заявлено, что эти соединения пригодны для лечения серповидно-клеточной анемии.
Некоторые блокаторы хлор-ионных каналов и их применение уже были описаны.
Описано [ (1986), 407 (suppl. 2), pages 128-141] несколько соединений, обладающих блокирующей хлор-ионные каналы активностью. Весьма сильное соединение, описанное в этой публикации, представляет собой 5-нитро-2-(3-фенилпропиламино)бензойную кислоту. Использование блокаторов хлор-ионных каналов для лечения серповидно-клеточной анемии в этой публикации не раскрыто.
(1986), 407 (suppl. 2), pages 128-141] несколько соединений, обладающих блокирующей хлор-ионные каналы активностью. Весьма сильное соединение, описанное в этой публикации, представляет собой 5-нитро-2-(3-фенилпропиламино)бензойную кислоту. Использование блокаторов хлор-ионных каналов для лечения серповидно-клеточной анемии в этой публикации не раскрыто.
В патенте США 4889612 описаны производные Каликсарена и их применение в качестве блокаторов хлор-ионных каналов.
В патенте США 4994493 описаны некоторые производные 5-нитробензойной кислоты и их применение при лечении отека головного мозга.
В WO 96/16647 описано применение блокаторов хлор-ионных каналов для снижения внутриглазного давления, в частности применение блокаторов хлор-ионных каналов для лечения глаукомы.
Настоящее изобретение относится к ряду замещенных фенильных производных, которые представляют собой сильнодействующие блокаторы хлор-ионных каналов и к их применению при лечении, например, серповидно-клеточной анемии.
Задачи изобретения
Задача настоящего изобретения заключается в том, чтобы создать новые замещенные фенильные производные и их фармацевтически приемлемые соли, которые полезны в лечении расстройств или заболеваний, которые являются чувствительными к блокаде хлор-ионных каналов.
Еще одна задача настоящего изобретения заключается в том, чтобы разработать способ лечения расстройств или заболеваний, которые являются чувствительными к блокаде хлор-ионных каналов, таких как, например, отек головного мозга, сопровождающий ишемию или опухоли, диарея, гипертензия.
Сущность изобретения
Далее, данное изобретение составляют, среди прочего, одно или в сочетании
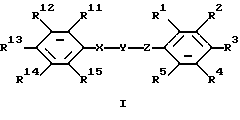
соединение, имеющее формулу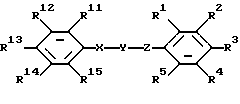
или его фармацевтически приемлемая соль,
где один из R1, R2 и R3 представляет собой 4-гидрокси-1,2,4-триазолил, 3-оксо-1,2-дигидро-1,2,4-триазолил, 2-оксо-3Н-1,3,4-оксадиазолил или тетразолил;
R4 и R5 представляют собой водород;
два других заместителя R1, R2 и R3 каждый независимо выбран из водорода, галогена, нитро, амино, ациламино, бензоиламино, фенила, нафтила, дифенила или 5- либо 6-членной гетероциклической моноциклической группы, содержащей один гетероатом, выбранный из N, S и О, где фенильная группа может быть замещена один или более чем один раз заместителями, выбранными из трифторметила, нитро, фенила, карбокси, алкоксикарбонила, аминокарбонила, диалкиламинокарбонила и анилинокарбонила;
Y представляет собой -СО- или -CS-;
Х представляет собой -NH-;
Z представляет собой NH;
один из R11, R12, R13, R14 и R15 выбран из водорода, галогена, трифторметила, -COOR7, фенила или 5- либо 6-членной гетероциклической моноциклической группы, содержащей один гетероатом, выбранный из N, S и О, а четыре других из R11, R12, R13, R14 и R15 представляют собой водород либо одни из R11 и R12, R12 и R13, R13 и R14, R14 и R15 вместе образуют циклическую структуру, а другие заместители R11, R12, R13, R14 и R15 представляют собой водород; R7 представляет собой алкил;
соединение, как оно указано выше, где один из R1, R2 и R3 представляет собой 4-гидрокси-1,2,4-триазолил, тетразолил, 3-оксо-1,2-дигидро-1,2,4-триазолил, 2-оксо-3Н-1,3,4-оксадиазолил, Z представляет собой NH и Y представляет собой СО;
соединение, как оно указано выше, причем указанное соединение представляет собой:
3-трифторметилфенил-4-нитро-2-(5-тетразолил)фенилмочевину,
3-трифторметилфенил-4-(2-нафтил)-2-(5-тетразолил)фенилмочевину,
3-трифторметилфенил-4-(3-пиридил)-2-(5-тетразолил)фенилмочевину,
3-трифторметилфенил-4-(1-нафтил)-2-(5-тетразолил)фенилмочевину,
3-трифторметилфенил-4-(4-трифторметилфенил)-2-(5-тетразолил)фенилмочевину,
3-трифторметилфенил-4-(3-фурил)-2-(5-тетразолил)фенилмочевину,
3-трифторметилфенил-4-(3-тиенил)-2-(5-тетразолил)фенилмочевину,
3-трифторметилфенил-4-(3-нитрофенил)-2-(5-тетразолил)фенилмочевину,
3-трифторметилфенил-4-(4-этоксикарбонилфенил)-2-(5-тетразолил)фенилмочевину,
3-трифторметилфенил-4-(4-диэтиламинокарбонилфенил)-2-(5-тетразолил)фенилмочевину,
3-трифторметилфенил-4-(4-аминокарбонилфенил)-2-(5-тетразолил) фенилмочевину,
3-трифторметилфенил-2-(4-гидрокси-1,2,4-триазол-3-ил)фенилмочевину,
3-трифторметилфенил-2-(3-оксо-1,2-дигидро-1,2,4-триазол-1-ил)фенилмочевину,
3-трифторметилфенил-2-(2-оксо-3Н-1,3,4-оксадиазол-5-ил) фенилмочевину,
3-трифторметилфенил-4-дифенилил-2-(3-оксо-1,2-дигидро-1,2,4-триазол-1-ил)фенилмочевину,
3-трифторметилфенил-4-амино-2-(5-тетразолил)фенилмочевину,
3-трифторметилфенил-4-ацетиламино-2-(5-тетразолил)фенилмочевину,
3-трифторметилфенил-4-бензоиламино-2-(5-тетразолил)фенилмочевину,
3-трифторметилфенил-4-(4-карбоксифенил)-2-(5-тетразолил)енилмочевину,
3-трифторметилфенил-4-(4-анилинокарбонилфенил)-2-(5-тетразолил)фенилмочевину,
4-дифенилил-2-(5-тетразолил)фенилмочевину,
3-дифенилил-2-(5-тетразолил)фенилмочевину,
5-инданил-2-(5-тетразолил)фенилмочевину,
3-бромфенил-4-бром-2-(5-тетразолил)фенилмочевину,
3-ацетилфенил-2-(5-тетразолил)фенилмочевину,
3-дифенилил-4-бром-2-(5-тетразолил)фенилмочевину,
3-(3-пиридил)фенил-4-бром-2-(5-тетразолил)фенилмочевину,
3-трифторметилфенил-4-бром-2-(5-тетразолил)фенилмочевину,
3-трифторметилфенил-2-(5-тетразолил)фенилмочевину,
3-трифторметилфенил-4-фенил-2-(5-тетразолил)фенилмочевину,
4-трифторметилфенил-2-(5-тетразолил)фенилмочевину,
3-хлорфенил-2-(5-тетразолил)фенилмочевину,
фенил-2-(5-тетразолил)фенилмочевину или
3-трифторметилфенил-4-амино-2-(5-тетразолил)фенилмочевину;
фармацевтическая композиция, имеющая активность блокатора хлор-ионных каналов нормальных и серповидных эритроцитов, содержащая терапевтически эффективное количество соединения, такого как любое из указанных выше, или его фармацевтически приемлемой соли вместе с по меньшей мере одним фармацевтически приемлемым носителем или разбавителем;
соединение, такое как указано выше, для применения в качестве активного ингредиента для приготовления лекарства для лечения расстройства или заболевания животного или человека, в лечении которого полезна блокада хлор-ионных каналов;
соединение, такое как указано выше, для применения в качестве активного ингредиента для приготовления лекарства для лечения серповидно-клеточной анемии;
способ лечения расстройства или заболевания животного или человека, в лечении которого полезна блокада хлор-ионных каналов, при котором такому животному или человеку, нуждающемуся в этом, вводят терапевтически эффективное количество соединения, такого как указано выше;
способ лечения расстройства или заболевания животного или человека, которое представляет собой серповидно-клеточную анемию, при котором такому животному или человеку, нуждающемуся в этом, вводят терапевтически эффективное количество соединения, такого как любое из указанных выше;
способ получения соединения, такого как указано выше, при котором
соединение, имеющее формулу
где W представляет собой О или S, a R11, R12, R13, R14 и R15 являются такими, как определено выше,
подвергают взаимодействию с соединением, имеющим формулу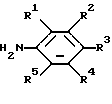
где R1, R2, R3, R4 R5 являются такими, как определено выше,
после чего полученное соединение возможно превращают в другое соединение по изобретению и/или образуют его фармацевтически приемлемую соль, используя общепринятые способы.
Примеры фармацевтически приемлемых солей присоединения соединений по изобретению включают в себя неорганические и органические соли присоединения кислоты, такие как гидрохлорид, гидробромид, фосфат, нитрат, перхлорат, сульфат, цитрат, лактат, тартрат, малеат, фумарат, манделат, бензоат, аскорбат, циннамат, бензолсульфонат, метансульфонат, стеарат, сукцинат, глутамат, гликолят, толуол-п-сульфонат, формиат, малонат, нафталин-2-сульфонат, салицилат и ацетат. Такие соли получают способами, хорошо известными из уровня техники.
Другие кислоты, такие как щавелевая кислота, хотя и не являются фармацевтически приемлемыми, могут быть использованы при получении солей, пригодных в качестве промежуточных соединений при получении соединений по изобретению и их фармацевтически приемлемых солей присоединения кислоты.
Галоген представляет собой фтор, хлор, бром или йод.
Алкил означает прямую цепь или разветвленную цепь, состоящую из от одного до шести атомов углерода, включая, но не ограничивая ими, метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, пентил и гексил. Метил, этил, пропил и изопропил являются предпочтительными группами.
Алкокси представляет собой O-алкил, где алкил является таким, как определено выше.
Амино представляет собой NН2 или NH-алкил, или N-(алкил)2, где алкил является таким, как определено выше.
Гетероарил представляет собой 5- или 6-членную моноциклическую гетероциклическую группу. Такая моноциклическая гетероарильная группа включает в себя, например, оксазол-2-ил, оксазол-4-ил, оксазол-5-ил, изоксазол-3-ил, изоксазол-4-ил, изоксазол-5-ил, тиазол-2-ил, тиазол-4-ил, тиазол-5-ил, изотиазол-3-ил, изотиазол-4-ил, изотиазол-5-ил, 1,2,4-оксадиазол-3-ил, 1,2,4-оксадиазол-5-ил, 1,2,4-тиадиазол-3-ил, 1,2,4-тиадиазол-5-ил, 1,2,5-оксадиазол-3-ил, 1,2,5-оксадиазол-4-ил, 1,2,5-тиадиазол-3-ил, 1,2,5-тиадиазол-4-ил, 1-имидазолил, 2-имидазолил, 4-имидазолил, 1-пирролил, 2-пирролил, 3-пирролил, 2-фуранил, 3-фуранил, 2-тиенил, 3-тиенил, 2-пиридил, 3-пиридил, 4-пиридил, 2-пиримидинил, 4-пиримидинил, 5-пиримидинил, 3-пиридазинил, 4-пиридазинил, 2-пиразинил, 1-пиразолил, 3-пиразолил и 4-пиразолил, 2-фурил, 3-фурил, 4-фурил, 5-фурил.
Соединения по данному изобретению могут существовать как в несольватированной, так и в сольватированной форме с фармацевтически приемлемыми растворителями, такими как вода, этанол и т.п. В общем, сольватированные формы считаются эквивалентными несольватированным формам для назначения данного изобретения.
Для специалиста в данной области техники очевидно, что соединения по настоящему изобретению содержат несколько хиральных центров и что такие соединения существуют в форме изомеров (т.е. энантиомеров). Данное изобретение включает в себя все такие изомеры и любые их смеси, в том числе рацемические смеси.
Некоторые соединения по настоящему изобретению существуют в (+) и (-) формах, а также в рацемических формах. Рацемические формы могут быть разделены на оптические антиподы известными методами, например разделением их диастереоизомерных солей с использованием оптически активной кислоты и высвобождением оптически активного аминосоединения обработкой основанием. Другой метод разделения рацематов на оптические антиподы основан на хроматографии на оптически активной матрице. Рацемические соединения по настоящему изобретению, таким образом, могут быть разделены на их оптические антиподы, например, фракционной кристаллизацией d- или l- солей (тартратов, манделатов или камфорсульфонатов). Соединения по настоящему изобретению могут быть также разделены путем образования диастереоизомерных амидов взаимодействием соединений по настоящему изобретению с оптически активной активированной карбоновой кислотой, такой как кислота, производная от (+) или (-) фенилаланина, (+) или (-) фенилглицина, (+) или (-) камфановая кислота, или путем образования диастереоизомерных карбаматов взаимодействием соединений по настоящему изобретению с оптически активным хлорформиатом или ему подобным.
Для разделения оптических изомеров могут быть использованы дополнительные способы, известные специалистам в данной области техники, и эти способы очевидны для среднего специалиста. Такие способы включают в себя способы, которые описаны в J.Jaques, A.Collet и S.Wilen "Enantiomers, Racemates, and Resolutions", John Wiley and Sons, New York (1981).
Соединения по изобретению могут быть получены различными способами. Соединения по изобретению и их фармацевтически приемлемые производные, таким образом, могут быть получены любым известным из уровня техники способом получения соединений аналогичной структуры и способами, которые продемонстрированы в типичных примерах, которые следуют далее.
Биология
Соединения по настоящему изобретению представляют собой сильнодействующие блокаторы хлор-ионных каналов в нормальных, а также серповидных эритроцитах. Способность соединений блокировать хлор-ионные каналы эритроцитов не может быть продемонстрирована классическими электрофизиологическими измерениями, такими как пэтч-клэмпинг, поскольку проводимость единицы канала находится ниже предела детекции этих методов.
Поэтому все эксперименты по определению зависимости доза-ответ проводили путем сопутствующих измерений общих проводящих потоков Сl- (JCl) и мембранных потенциалов (Vm) в суспензиях эритроцитов [Bennekou P. and Christophersen P. (1986), Flux ratio of Valinomycin-Mediated K+ Fluxes across the Human Red Cell Membrane in the presence of the Protonophore СССР. J.Membrane Biol. 93, 221-227.].
Мембранные Сl--проводимости (GCl) рассчитывали согласно следующему уравнению [Hodgkin A. L. and Huxley A.F. (1952) The components of membrane conductance in the giant axon of Loligo. J. Physiol. Lond. 116, 449-472]: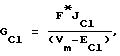
где F - константа Фарадея,
ECl - потенциал Нернста для Cl-иона.
Введение 3-трифторметилфенил-2-карбоксифенилмочевины в суспензию нормальных эритроцитов приводило к блокированию GCl более чем на 95% при КD-значении 1,3 мкМ. Это соединение с равной силой блокировало GCl из оксигенированных, так же как и из дезоксигенированных гомозиготных серповидно-клеточных эритроцитов.
Значение КD для 3-трифторметил-4-бром-2-(5-тетразолил)фенилмочевины в этом тесте составляло 1,9 мкМ.
Экспериментально индуцированные потери клеточного объема измеряли как изменения относительного объема эритроцитарной массы. Индуцирование потери массы воды и соли (KCl) добавлением к суспензии K+-ионофора валиномицина в течение 5 минут уменьшало клеточный объем на 26%. 3-Трифторметилфенил-2-карбоксифенилмочевина дозозависимо (значение IC50 1,2 мкМ) уменьшала потерю объема до 7%.
Увеличение проводимости серповидных клеток, индуцированное дезоксигенированием, оценивали по измерению внеклеточной концентрации К+ в зависимости от времени. Нормальные эритроциты показывали очень маленькие K+-потоки, которые не реагировали на дезоксигенирование и не реагировали на 10 мкМ 3-трифторметилфенил-2-карбоксифенилмочевину. К+-поток из оксигенированных серповидных эритроцитов был в 2-3 раза выше, чем из нормальных эритроцитов, и эти потоки увеличивались в 4-8 раз после дезоксигенирования. В присутствии 3-трифторметилфенил-2-карбоксифенилмочевины (10 мкМ) основной поток К+ из серповидных эритроцитов нормализовался и компонента потока, индуцированная дезоксигенированием, практически устранялась.
3-Трифторметилфенил-2-карбоксифенилмочевина нетоксична для мышей и крыс при концентрациях вплоть до 250 мг/кг внутрибрюшинно и внутривенно.
Фармацевтические композиции
Хотя возможно для применения в терапии вводить соединение по изобретению в виде химической субстанции, предпочтительно представлять активный ингредиент в виде фармацевтического препарата.
Согласно данному изобретению, таким образом, предложены также фармацевтические препараты, содержащие соединение по изобретению или его фармацевтически приемлемую соль или производное вместе с одним или более чем одним фармацевтически приемлемым носителем для него и, возможно, другими терапевтическими и/или профилактическими ингредиентами. Носитель(ли) должен быть "приемлемым" в том смысле, что он должен быть совместим с другими ингредиентами препарата и не должен быть вредным для его реципиента.
Фармацевтические препараты включают в себя такие препараты, которые пригодны для перорального, ректального, назального, местного (включая в себя буккальное и подъязычное), вагинального или парентерального (включая в себя внутримышечное, подкожное и внутривенное) введения или в форме, пригодной для введения ингаляцией или инсуффляцией.
Соединения по изобретению вместе с традиционным адъювантом, носителем или разбавителем, таким образом, могут быть представлены в форме фармацевтических композиций и их стандартных доз, и в такой форме их можно использовать как твердые вещества, такие как таблетки или заполненные капсулы, или в виде жидкостей, таких как растворы, суспензии, эмульсии, эликсиры, или в виде капсул, заполненных этими жидкостями, все они для перорального употребления; в форме суппозиториев для ректального введения либо в форме стерильных инъекционных растворов для парентерального (включая подкожное) применения. Такие фармацевтические композиции и их стандартные лекарственные формы могут включать в себя традиционные ингредиенты в подходящих соотношениях вместе с дополнительными активными соединениями или действующими началами или без них, и такие стандартные лекарственные формы могут содержать любое соответствующее эффективное количество активного ингредиента, соразмерное назначенному для употребления диапазону суточной дозы. Препараты, содержащие десять (10) миллиграммов активного ингредиента или, более широко, от 0,1 до ста (100) миллиграммов на таблетку, представляют собой соответственно пригодные типичные стандартные лекарственные формы.
Соединения по настоящему изобретению можно вводить в составе широкого разнообразия пероральных и парентеральных лекарственных форм. Специалистам в данной области техники ясно, что лекарственные формы могут включать в себя в качестве активного компонента либо соединение по изобретению, либо фармацевтически приемлемую соль соединения по изобретению.
Для приготовления фармацевтических композиций из соединений по настоящему изобретению фармацевтически приемлемые носители могут быть твердыми или жидкими. Препараты в твердой форме включают в себя порошки, таблетки, пилюли, капсулы, облатки, суппозитории и диспергируемые гранулы. Твердым носителем может быть одно или более чем одно вещество, которое может также действовать в качестве разбавителя, корригента, растворителя, смазывающего вещества, суспендирующего агента, связывающего вещества, консерванта, разрыхлителя таблеток или в качестве вещества оболочки.
В порошках носителем является тонко измельченное твердое вещество, которое находится в смеси с тонко измельченным активным компонентом.
В таблетках активный компонент смешан с носителем, имеющим необходимую связывающую способность, в подходящих соотношениях и спрессован в таблетки требуемой формы и размера.
Порошки и таблетки предпочтительно содержат от пяти или десяти до примерно семидесяти процентов активного соединения. Подходящими носителями являются карбонат магния, стеарат магния, тальк, сахар, лактоза, пектин, декстрин, желатин, трагакант, метилцеллюлоза, натриевая карбоксиметилцеллюлоза, легкоплавкий воск, масло какао и т.д. Предполагается, что термин "препарат" включает в себя препарат активного соединения с инкапсулирующим веществом в качестве носителя, обеспечивающим получение капсулы, в которой активный компонент вместе с носителями или без них окружен носителем, который, таким образом, находится в связи с этим активным компонентом. Соответственно, в число таких препаратов включены облатки и лепешки. Таблетки, порошки, капсулы, пилюли, облатки и лепешки можно использовать как твердые формы, пригодные для перорального введения.
Для приготовления суппозиториев легкоплавкий воск, такой как смесь глицеридов жирных кислот или масло какао, сначала расплавляют, и активный компонент гомогенно диспергируют в нем путем перемешивания. Расплавленную гомогенную смесь затем заливают в формы нужного размера, дают возможность остыть и в результате этого затвердеть.
Препараты, пригодные для вагинального введения могут быть представлены в виде пессариев, тампонов, кремов, гелей, паст, пен или аэрозолей, содержащих в дополнение к активному ингредиенту такие носители, которые известны в данной области техники как подходящие.
Препараты в жидкой форме включают в себя растворы, суспензии и эмульсии, например водные или водно-пропиленгликолевые растворы. Например, жидкие препараты для парентеральной инъекции могут быть приготовлены в виде растворов в водном полиэтиленгликоле.
Соединения по настоящему изобретению, таким образом, могут быть приготовлены для парентерального введения (например, инъекцией, например болюсной инъекцией или непрерывной инфузией) и могут быть представлены в стандартной лекарственной форме в ампулах, предварительно заполненных шприцах, в контейнерах для инфузии небольшого объема или многодозовых контейнерах с добавлением консервантов. Композиции могут принимать такие формы, как суспензии, растворы или эмульсии в масляных или водных носителях, и могут содержать вспомогательные агенты, такие как суспендирующие, стабилизирующие и/или диспергирующие агенты. Альтернативно активный ингредиент может быть в форме порошка, полученного асептическим выделением стерильного твердого вещества или лиофилизацией из раствора, для разведения перед использованием с соответствующим носителем, например стерильной, апирогенной водой.
Водные растворы, пригодные для перорального применения, могут быть приготовлены растворением активного компонента в воде и, при желании, добавлением подходящих пигментов, корригентов, стабилизаторов и загустителей.
Водные суспензии, пригодные для перорального применения, могут быть приготовлены диспергированием тонко измельченного активного компонента в воде с вязким веществом, таким как природные или синтетические камеди, смолы, метилцеллюлоза, натриевая карбоксиметилцеллюлоза или другие хорошо известные суспендирующие агенты.
В число препаратов по настоящему изобретению включены также препараты в твердой форме, которые предполагается непосредственно перед использованием превращать в препараты в жидкой форме для перорального введения. Такие жидкие формы включают в себя растворы, суспензии и эмульсии. В добавление к активному компоненту эти препараты могут содержать красители, корригенты, стабилизаторы, буферы, искусственные и натуральные подсластители, диспергирующие, загущающие, солюбилизирующие агенты и т.д.
Для местного введения на эпидермис соединения по изобретению могут быть приготовлены в виде мазей, кремов или лосьонов, или в виде трансдермальных пластырей. Мази и кремы могут быть приготовлены, например, с использованием водной или масляной основы с добавлением подходящих загустителей или гелеобразующих агентов. Лосьоны могут быть приготовлены с использованием водной или масляной основы, и, как правило, они также будут содержать один или более чем один эмульгирующий, стабилизирующий, диспергирующий, суспендирующий агенты, загуститель или краситель.
Препараты, пригодные для местного введения во рту, включают в себя лепешки, содержащие активный агент в корригентной основе, обычно сахарозе и аравийской камеди или трагаканте; пастилки, содержащие активный ингредиент в инертной основе, такой как желатин и глицерин или сахароза и аравийская камедь; полоскания, содержащие активный ингредиент в подходящем жидком носителе.
Растворы или суспензии наносят непосредственно в полость носа при помощи традиционных средств, например капельницей, пипеткой или аэрозолем. Препараты могут быть представлены в форме разовой дозы или в форме разделенной дозы. В последнем случае при использовании капельницы или пипетки этого можно достичь введением пациенту соответствующего предопределенного объема раствора или суспензии. В случае аэрозоля этого можно достичь, например, посредством дозирующего распылительного аэрозольного насоса.
Введение в дыхательные пути также может быть достигнуто посредством аэрозольных препаратов, в которых активный ингредиент находится в упаковке под давлением вместе с подходящим пропеллентом, таким как хлорфторуглерод (ХФУ), например дихлордифторметан, трихлорфторметан или дихлортетрафторэтан, диоксид углерода или другой подходящий газ. Аэрозоль может также содержать поверхностно-активное вещество, такое как лецитин. Дозу лекарственного средства можно контролировать при помощи дозирующего клапана.
Альтернативно активные ингредиенты могут быть представлены в форме сухого порошка, например порошковой смеси соединения в подходящей порошковой основе, такой как лактоза, крахмал, производные крахмала, такие как гидроксипропилметилцеллюлоза и поливинилпирролидон (ПВП). Удобно, если порошковый носитель будет образовывать гель в носовой полости. Порошковая композиция может быть представлена в стандартной лекарственной форме, например в капсулах или картриджах, например, из желатина, или в блистерных упаковках, из которых порошок можно вводить посредством ингалятора.
В препаратах, предназначенных для введения в дыхательные пути, в том числе препаратах для интраназального введения, соединение, как правило, будет находиться в форме частиц небольшого размера, например порядка 5 микрон или меньше. Такой размер частиц можно получить способами, известными из уровня техники, например микронизацией. При желании можно использовать препараты, приспособленные для пролонгированного высвобождения активного ингредиента.
Фармацевтические препараты предпочтительно находятся в стандартной лекарственной форме. В такой форме препараты разделены на стандартные дозы, содержащие соответствующие количества активного компонента. Стандартной лекарственной формой может быть препарат в упаковке, причем упаковка содержит обособленные доли препарата, такие как упакованные таблетки, капсулы и порошки в пузырьках или ампулах. Стандартной лекарственной формой может быть также сама капсула, таблетка, облатка или лепешка или это может быть соответствующее число любого из вышеперечисленного в упакованной форме.
Таблетки или капсулы для перорального введения и жидкости для внутривенного введения представляют собой предпочтительные композиции.
Способы лечения
Соединения по настоящему изобретению очень полезны при лечении серповидно-клеточной анемии, отека головного мозга, сопровождающего ишемию или опухоли, диареи, гипертензии (в качестве диуретиков), остеопороза и глаукомы благодаря их сильной блокирующей хлор-ионные каналы активности. Эти свойства делают соединения по данному изобретению чрезвычайно полезными при лечении серповидно-клеточной анемии, отека головного мозга, сопровождающего ишемию или опухоли, диареи, гипертензии (в качестве диуретиков), остеопороза и глаукомы, а также других расстройств, в лечении которых полезна блокирующая периферические хлор-ионные каналы активность соединений по настоящему изобретению. Соответственно, соединения по данному изобретению можно вводить животному или человеку, нуждающемуся в лечении, облегчении или устранении показания, ассоциированного с блокирующей хлор-ионные каналы активностью или чувствительного к ней. Это показание включает в себя, в частности, серповидно-клеточную анемию, отек головного мозга, сопровождающий ишемию или опухоли, диарею, гипертензию (в качестве диуретиков), остеопороз и глаукому.
Подходящий диапазон дозировки составляет от 0,1 до 500 миллиграммов в сутки, и главным образом от 10 до 70 миллиграммов в сутки, вводимых один или два раза в день в зависимости, как обычно, от конкретного способа введения, формы, в которой вводят, показания, в отношении которого назначено введение, предполагаемого субъекта и веса тела предполагаемого субъекта и, кроме того, от выбора и опыта лечащего врача или ветеринара.
Следующие далее примеры иллюстрируют изобретение, однако они не должны расцениваться как ограничивающие данное изобретение.
Пример 1
3-Трифторметилфенил-4-бром-2-(5-тетразолил)фенилмочевина
3-Трифторметилфенилизоиианат (0,41 мл, 3,0 ммоль) и 5-(2-амино-5-бромфенил)тетразол (0,6 г, 2,5 ммоль) добавляли к толуолу (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Осадок отфильтровывали и промывали толуолом, а затем петролейным эфиром с получением 0,53 г требуемого соединения, т.пл. 269-270oС.
Следующие соединения были получены по аналогии:
3-трифторметилфенил-2-(5-тетразолил)фенилмочевина, т.пл.257oС;
3-трифторметилфенил-2-(5-тетразолил)фенилтиомочевина, т. пл. >200oС (разл);
3-трифторметилфенил-4-фенил-2-(5-тетразолил)фенилмочевина, т.пл. 260oС;
4-трифторметилфенил-2-(5-тетразолил)фенилмочевина, т.пл.240oС (разл.);
3-хлорфенил-2-(5-тетразолил)фенилмочевина, т.пл.243oС (разл.);
фенил-2-(5-тетразолил)фенилмочевина, т.пл.239oС (разл.);
3-трифторметилфенил-4-нитро-2-(5-тетразолил)фенилмочевина, т. пл.204-205oС;
3-трифторметилфенил-4-(2-нафтил)-2-(5-тетразолил)фенилмочевина, т. пл. 257-258oС;
3-трифторметилфенил-4-(3-пиридил)-2-(5-тетразолил)фенилмочевина, т.пл. 148-152oС;
3-трифторметилфенил-4-(1-нафтил)-2-(5-тетразолил)фенилмочевина, т. пл. 207-208oС;
3-трифторметилфенил-4-(4-трифторметилфенил)-2-(5-тетразолил)-фенилмочевина, т.пл.135-140oС;
3-трифторметилфенил-4-(3-фурил)-2-(5-тетразолил)фенилмочевина, т. пл. 260-261oС;
3-трифторметилфенил-4-(3-тиенил)-2-(5-тетразолил)фенилмочевина, т. пл. 259-260oС;
3-трифторметилфенил-4-(3-нитрофенил)-2-(5-тетразолил)фенилмочевина, т. пл.135-140oС;
3-трифторметилфенил-4-(4-этоксикарбонилфенил)-2-(5-тетразолил)фенилмочевина, т.пл.262-263oС;
3-трифторметилфенил-4-(4-диэтиламинокарбонилфенил)-2-(5-тетразолил)фенилмочевина, т.пл. 264oС;
3-трифторметилфенил-4-(4-аминокарбонилфенил)-2-(5-тетразолил)фенилмочевина, т.пл.252-253oС;
3-трифторметилфенил-2-(4-гидрокси-1,2,4-триазол-3-ил)фенилмочевина, т. пл.220-221oС;
3-трифторметилфенил-2-(3-оксо-1,2-дигидро-1,2,4-триазол-1-ил)фенилмочевина, т.пл.>300oС;
3-трифторметилфенил-2-(2-оксо-3Н-1,3,4-оксадиазол-5-ил)фенилмочевина, т. пл.>300oС;
3-трифторметилфенил-4-дифенилил-2-(3-оксо-1,2-дигидро-1,2,4-триазол-1-ил)фенилмочевина, т.пл.166oС;
3-бромфенил-4-бром-2-(5-тетразолил)фенилмочевина, т.пл.142oС.
Пример 2
5-(2-Аминофенил)тетразол
2-Аминобензонитрил (9,44 г, 80 ммоль), азид натрия (6,24 г, 0,1 моль), хлорид аммония (5,12 г, 0,1 моль) и диметилформамид (50 мл) смешивали и нагревали при 120oС в течение ночи. Растворитель выпаривали, а остаток переносили в воду. Неочищенный продукт выделяли фильтрацией и перекристаллизовывали из воды. Выход чистого составил 8,4 г.
Аналогично были получены:
5-(2-амино-5-бромфенил)тетразол,
5-(4-амино-3-дифенил)тетразол,
5-(2-амино-5-нитрофенил)тетразол,
5-(2-амино-4-(2-нафтил)фенил)тетразол,
5-(2-амино-4-(3-пиридил)фенил)тетразол,
5-(2-амино-4-(1-нафтил)фенил)тетразол,
5-(2-aминo-4-(4-3трифтopмeтилфeнил)фeнил)тeтpaзoл,
5-(2-амино-4-(3-фурил)фенил)тетразол,
5-(2-амино-4-(3-тиенил)фенил)тетразол,
5-(2-амино-4-(4-трифторметилфенил)фенил)тетразол,
5-(2-амино-4-(3-нитрофенил)фенил)тетразол,
5-(2-амино-4-(4-этоксикарбонилфенил)фенил)тетразол,
5-(2-амино-4-(4-диэтиламинокарбонилфенил)фенил)тетразол,
5-(2-амино-4-(4-аминокарбонилфенил)фенил)тетразол.
Пример 3
2-Амино-4-фенилбензонитрил
Смесь 2-амино-5-бромбензонитрила (1,0 г, 5 ммоль), фенилбороновой кислоты (0,92 г, 7,5 ммоль), тетракис(трифенилфосфин)палладия (50 мг) и карбоната калия (3,5 г, 25 ммоль) в смеси диметоксиэтан/вода 2:1 (60 мл) нагревали при дефлегмации в течение 4 часов. После охлаждения до комнатной температуры реакционную смесь разбавляли водой и экстрагировали этилацетатом. Органическую фазу высушивали и растворитель выпаривали. Растирание в порошок с петролейным эфиром привело к получению 0,89 г требуемого соединения.
Подобным образом были получены:
2-амино-4-(2-нафтил)бензонитрил,
2-амино-4-(3-пиридил)бензонитрил,
2-aминo-4-(1-нафтил)бeнзoнитpил,
2-амино-4-(4-трифторметилфенил)бензонитрил,
2-амино-4-(3-фурил)бензонитрил,
2-амино-4-(3-тиенил)бензонитрил,
2-амино-4-(3-нитрофенил)бензонитрил,
2-амино-4-(4-этоксикарбонилфенил)бензонитрил,
2-амино-4-(4-диэтиламинокарбонилфенил)бензонитрил,
2-амино-4-(4-аминокарбонилфенил)бензонитрил,
1-(3-нитро-4-дифенилил)-1,2-дигидро-1,2,4-триазол-3-он.
Пример 4
3-Трифторметилфенил-4-амино-2-(5-тетразолил)фенилмочевина
Раствор 3-трифторметилфенил-4-нитро-2-(5-тетразолил)фенилмочевины (0,8 г, 2,0 ммоль) в 96% этаноле гидрировали над 5% палладием на угле в течение 3 часов при комнатной температуре. Реакционную смесь фильтровали через слой целита и растворитель выпаривали с получением 0,75 г требуемого продукта, т. пл. 175-180oС.
Пример 5
3-Трифторметилфенил-4-ацетиламино-2-(5-тетразолил)фенилмочевина
К раствору 3-трифторметилфенил-4-амино-2-(5-тетразолил)фенилмочевины (0,22 г, 0,6 ммоль) в 17% водном ацетате натрия (5 мл), охлажденному на ледяной бане, добавляли уксусный ангидрид (1 мл). Реакционную смесь перемешивали при 0oС в течение еще одного часа. Осадок отфильтровали и перекристаллизовывали из 96% этанола с получением 0,12 г требуемого вещества, т. пл. 280-282oС.
Пример 6
3-Трифторметилфенил-4-бензоиламино-2-(5-тетразолил)фенилмочевина
К раствору 3-трифторметилфенил-4-амино-2-(5-тетразолил)фенилмочевины (0,36 г, 1,0 ммоль) в тетрагидрофуране (40 мл) добавляли триэтиламин (0,17 мл, 1,2 ммоль). Раствор охлаждали на ледяной бане и добавляли бензоилхлорид (0,14 мл, 1,2 ммоль). Реакционную смесь перемешивали при 0oС в течение следующих 30 мин. Реакционную смесь вливали в воду. Осадок отфильтровывали и перекристаллизовывали из 96% этанола с получением 0,28 г требуемого вещества, т.пл.271-272oС.
Пример 7
4-Метилфенилбороновая кислота
К раствору 4-йодтолуола (35 г, 160,5 ммоль) в диэтиловом эфире (400 мл) добавляли н-бутиллитий (2М в пентане, 88,3 мл, 176,6 ммоль) при 0oС. После перемешивания при 0oС в течение следующих 15 минут раствор охлаждали до -60oС и добавляли трибутилборат (60,6 мл, 224,7 ммоль). Охлаждающую баню удаляли и реакционной смеси давали возможность нагреться до комнатной температуры. Раствор подкисляли соляной кислотой (2 н., 280 мл) и органическую фазу отделяли. Водную фазу экстрагировали диэтиловым эфиром (2х125 мл). Объединенные органические фазы экстрагировали гидроксидом натрия (1 н., 5х50 мл). Объединенные водные экстракты подкисляли с получением 18,6 г требуемого вещества.
Пример 8
4-Карбоксифенилбороновая кислота
К раствору 4-метилфенилбороновой кислоты (34 г, 0,25 моль) в водном гидроксиде натрия (0,5 н. , 1000 мл) добавляли перманганат калия (83 г, 0,53 моль), поддерживая температуру 35-40oС. После добавления реакционную смесь фильтровали и фильтрат подкисляли концентрированной соляной кислотой (65 мл). Продукт отфильтровывали. Получили выход 29,6 г, т.пл.228oС.
Пример 9
4-Этоксикарбонилфенилбороновая кислота
Раствор 4-карбоксифенилбороновой кислоты (15 г, 0,09 моль), 99% этанола (150 мл) и концентрированной серной кислоты (0,5 мл) нагревали до образования флегмы в течение двух дней. Объем уменьшался до приблизительно 20 мл. Остаток растирали в порошок с петролейным эфиром с получением 13,4 г требуемого вещества.
Пример 10
4-Аминокарбонилфенилбороновая кислота
Раствор 4-карбоксифенилбороновой кислоты (10 г, 0,06 моль) и тионилхлорида (875 мл) нагревали до 50-60oС в течение ночи. Тионилхлорид выпаривали. Половину остатка добавляли к концентрированному аммиаку (30 мл). Реакционную смесь нагревали до образования флегмы. Горячая фильтрация и последующее подкисление фильтрата позволили получить неочищенное вещество. Это неочищенное вещество очищали путем его суспендирования в разбавленном гидрокарбонате натрия с получением 1,09 г требуемого вещества.
Подобным образом была получена 4-диметиламинокарбонилфенилбороновая кислота.
Пример 11
3-Трифторметилфенил-4-(4-карбоксифенил)-2-(5-тетразолил)фенилмочевина
К суспензии 3-трифторметилфенил-4-(4-этоксикарбонилфенил)-2-(5-тетразолил)фенилмочевины (4,5 г, 9 ммоль) в 96% этаноле добавляли гидроксид натрия (4 н., 25 мл). Реакционную смесь нагревали до образования флегмы в течение 30 мин, затем охлаждали до комнатной температуры и подкисляли соляной кислотой. Осадок отфильтровывали с получением 3,8 г требуемого вещества, т. пл. 300oС (разл.).
Пример 12
3-Трифторметилфенил-4-(4-анилинокарбонилфенил)-2-(5-тетразолил)фенилмочевина
Смесь 3-трифторметилфенил-4-(4-карбоксифенил)-2-(5-тетразолил)фенилмочевины (1,9 г, 4 ммоль) и тионилхлорида (10 мл) нагревали до 50oС в течение 6 часов. Избыток тионилхлорида выпаривали. К остатку добавляли диэтиловый эфир с получением 2,2 г твердого вещества. Половину этого вещества суспендировали в тетрагидрофуране. Добавляли анилин (0,2 мл, 2,2 ммоль) и триэтиламин (0,5 мл, 3,6 ммоль). После перемешивания в течение 30 мин растворитель выпаривали. Остаток суспендировали в воде и добавляли небольшое количество разбавленной соляной кислоты. Твердое вещество отфильтровывали и перекристаллизовывали из 96% этанола с получением 0,2 г требуемого вещества, т.пл.>300oС.
Пример 13
4-Дифенилил-2-(5-тетразолил)фенилмочевина
К раствору N,N-карбонилдиимидазола (0,96 г, 5,0 ммоль) и имидазола (0,68 г, 10 ммоль) в тетрагидрофуране (10 мл) при 0oС добавляли 4-аминодифенил (1,0 г, 5,9 ммоль) в тетрагидрофуране (10 мл). После перемешивания при 0oС в течение 10 мин добавляли 5-(2-аминофенил)тетразол (1,14 г, 7,1 ммоль). Реакционную смесь перемешивали в течение следующих 4 часов и фильтровали. Фильтрат выпаривали до сухого состояния и неочищенный продукт очищали колоночной хроматографией. Получен выход 0,28 г, т.пл.224-226oС.
Подобным образом были получены:
3-дифенилил-2-(5-тетразолил)фенилмочевина, т.пл. 189-191oС,
5-инданил-2-(5-тетразолил)фенилмочевина, т.пл. 154-157oС,
3-ацетилфенил-2-(5-тетразолил)фенилмочевина, т.пл. 115oС,
3-дифенилил-4-бром-2-(5-тетразолил)фенилмочевина,
3-(3-пиридил)фенил-4-бром-2-(5-тетразолил)фенилмочевина.
Пример 14
4-Гидрокси-3-(2-аминофенил)-[1,2,4]триазол
Раствор 4-гидрокси-3-(2-нитрофенил)-1,2,4-триазола (0,38 г, 1,8 ммоль), полученного по H.G.O. Becker (J. Prakt. Chem., 1970, 312, 610), в 96% этаноле гидрировали над 5% палладием на угле при комнатной температуре в течение 1 часа. Реакционную смесь фильтровали через слой целита с получением, после выпаривания растворителя, требуемого вещества.
Пример 15
1-(4-Бромфенил)-1,2-дигидро-[1,2,4]триазол-3-он
Смесь гидрохлорида 4-бромфенилгидразина (5,0 г, 22,4 ммоль) и мочевины (8,1 г, 134 ммоль) нагревали до 80oС в течение ночи в 1-метил-2-пирролидиноне (25 мл). Реакционную смесь вливали в воду (250 мл) и добавляли концентрированный аммиак до основного рН. Раствор охлаждали на ледяной бане и семикарбазон отфильтровывали. Семикарбазон (1,0 г, 4,4 ммоль) перемешивали в триэтилортоформиате (8 мл) при 90oС в течение трех дней. Реакционную смесь охлаждали до комнатной температуры и неочищенное вещество отфильтровывали. Перекристаллизация из метанола позволила получить требуемый продукт.
Пример 16
1-(4-Бром-2-аминофенил)-1,2-дигидро-1,2,4-триазол-3-он
К раствору 1-(4-бромфенил)-1,2-дигидро-[1,2,4]триазол-3-она (0,25 г, 1,0 ммоль) в концентрированной серной кислоте (10 мл) при 0oС добавляли нитрат калия (0,13 г, 1,2 ммоль). Реакционную смесь перемешивали в течение следующего часа при 0oС и при комнатной температуре в течение ночи. Реакционную смесь вливали в воду и осадок отфильтровывали с получением 0,28 г требуемого нитросоединения. Нитросоединение (0,28 г, 1,0 ммоль), суспендированное в 96% этаноле, гидрировали над 5% палладием на угле с получением требуемого вещества.
Пример 17
5-(2-Аминофенил)-3Н-1,3,4-оксадиазол-2-он
Раствор 2-нитробензоилгидразина (9,05 г, 0,05 моль) в диоксане (30 мл) медленно добавляли к раствору трихлорметилхлорформиата (4,6 мл, 0,04 моль) в диоксане (30 мл) при комнатной температуре. По завершении добавления реакционную смесь нагревали до образования флегмы в течение 4 часов. Растворитель выпаривали и остаток перекристаллизовывали из 96% этанола (50 мл) с получением 7,15 г 5-(2-нитрофенил)-3Н-[1,3,4]оксадиазол-2-она, т.пл. 157-158oС. Раствор нитросоединения (2 г, 9,7 ммоль) в 96% этаноле (25 мл) гидрировали над 5% палладием на угле с получением 1,6 г требуемого амина.
Пример 18
1-(2-Амино-4-дифенилил)-1,2-дигидро-1,2,4-триазол-3-он
Раствор 1-(2-нитро-4-дифенилил)-1,2-дигидро-1,2,4-триазол-3-она в 96% этаноле (25 мл) гидрировали над 5% палладием на угле с получением требуемого амина.
Пример 19
Фармацевтическая композиция
Химическое соединение по данному изобретению может быть включено в состав фармацевтической композиции любой желаемой формы и может быть введено в любом желаемом количестве. Этот пример иллюстрирует приготовление стандартных препаратов в форме капсулы, таблетки и инъекционного раствора соответственно. В качестве активного ингредиента использовано соединение примера 1 - [3-трифторметилфенил-4-бром-2-(5-тетразолил)фенилмочевина].
Стандартный препарат в форме капсулы
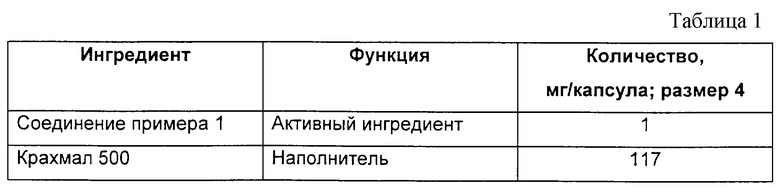
Капсулы, содержащие 1 мг активного фармацевтического ингредиента на капсулу, могут быть получены с использованием композиции, приведенной в табл.1.
Взвешивают рассчитанные количества лекарственного вещества и наполнителя, соответствующие 1 мг активного лекарственного вещества, и 117 мг наполнителя на одну капсулу и смешивают в сухом состоянии. Затем полученной смесью заполняют рассчитанное количество капсул (размер 4).
Стандартный препарат в форме таблетки
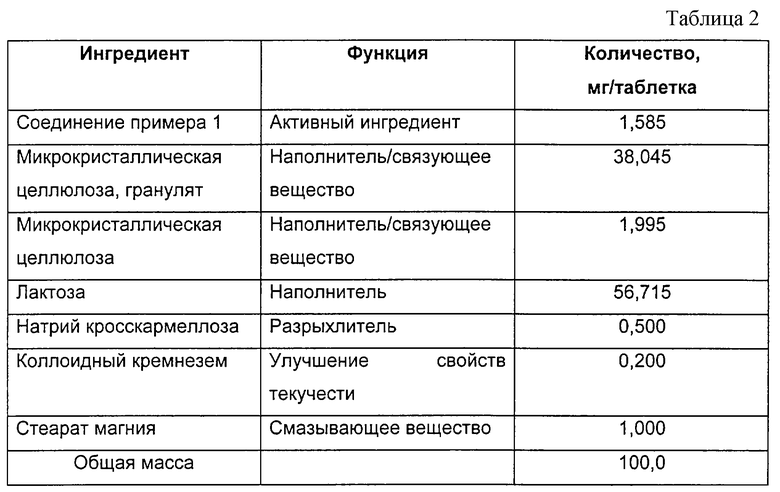
Таблетки, содержащие 1,585 мг активного ингредиента на одну таблетку, были получены с использованием композиции, приведенной в табл.2.
Активный ингредиент растворяют в гранулирующем растворе, который состоит из микрокристаллической целлюлозы и воды и который затем используют для гранулирования микрокристаллической целлюлозы. Гранулят оставляют сушиться на поддоне.
Вышеуказанный гранулят, содержащий активный фармацевтический ингредиент, микрокристаллическую целлюлозу, лактозу и натрия кросскармеллозу взвешивают, просеивают в смеситель и перемешивают.
Взвешивают стеарат магния и просеивают в смеситель вместе с вышеуказанной смесью, а затем перемешивают. Полученную в результате смесь затем прессуют в таблетки.
Стандартный препарат в форме инъекционного раствора
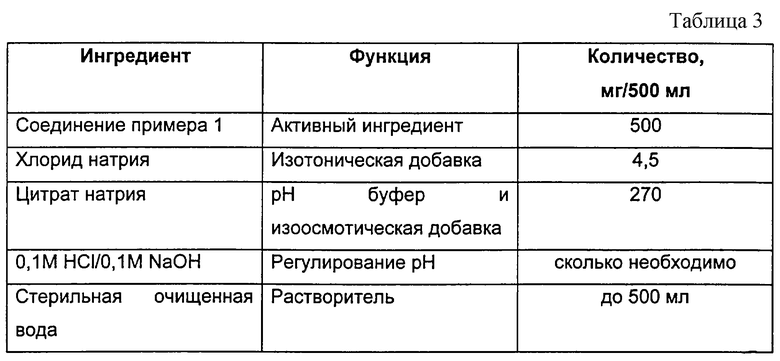
Инъекционный раствор, содержащий 1 мг/мл активного ингредиента, был получен с использованием композиции, приведенной в табл.3.
Рассчитанное количество активного ингредиента взвешивают, растворяют в стерильной очищенной воде, добавляют предписанное количество хлорида натрия и цитрата натрия и затем рН раствора доводят до желаемого значения, обычно в диапазоне от приблизительно рН 6,5.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ ФЕНИЛЬНЫЕ ПРОИЗВОДНЫЕ, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 1999 |
|
RU2218328C2 |
| ПРОИЗВОДНЫЕ [1,2,4] ТРИАЗОЛО [4,3-А]ХИНОКСАЛИНОНА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ РЕЦЕПТОРОВ АМРА | 1995 |
|
RU2149162C1 |
| ПРОИЗВОДНЫЕ ДИФЕНИЛМОЧЕВИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ БЛОКАТОРОВ ХЛОРИДНЫХ КАНАЛОВ | 2004 |
|
RU2350607C2 |
| НОВЫЕ ДИАЗАБИЦИКЛИЧЕСКИЕ АРИЛЬНЫЕ ПРОИЗВОДНЫЕ, СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И ИХ ПРИМЕНЕНИЕ | 2004 |
|
RU2338746C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА И СОДЕРЖАЩИЕ ЭТИ СОЕДИНЕНИЯ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2000 |
|
RU2243226C2 |
| ПЕСТИЦИДНЫЕ КОМПОЗИЦИИ И СВЯЗАННЫЕ С НИМИ СПОСОБЫ | 2012 |
|
RU2614976C2 |
| АРИЛКАРБОНИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ТЕРАПЕВТИЧЕСКИХ СРЕДСТВ | 2003 |
|
RU2340605C2 |
| ДИФЕНИЛЬНЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ | 1997 |
|
RU2175319C2 |
| Производные тетрагидротриазолопиримидина в качестве ингибиторов нейтрофильной эластазы человека | 2012 |
|
RU2622643C2 |
| ПРОИЗВОДНЫЕ ХИНОКСАЛИНДИОНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКОЕ СРЕДСТВО НА ИХ ОСНОВЕ | 1994 |
|
RU2140420C1 |
Описываются новые замещенные фенильные производные общей формулы I, где один из R1, R2 и R3 представляет собой 4-гидрокси-1,2,4-триазолил, 3-оксо-1,2-дигидро-1,2,4-триазолил, 2-оксо-3Н-1,3,4-оксадиазолил или тетразолил; R4 и R5 представляют собой водород; два других заместителя R1, R2 и R3 каждый независимо выбран из водорода, галогена, нитро, амино, ациламино, бензоиламино, фенила, нафтила, дифенила или 5- либо 6-членной гетероциклической моноциклической группы, содержащей 1 гетероатом, выбранный из N, S и О, где фенильная группа может быть замещена один или более чем один раз заместителями, выбранными из трифторметила, нитро, фенила, карбокси, алкоксикарбонила, аминокарбонила, диалкиламинокарбонила и анилинокарбонила; Y представляет собой -СО- или -CS-; Х представляет собой -NH-; Z представляет собой NH; один из R11, R12, R13, R14 и R15 выбран из водорода, галогена, трифторметила, -СООR7, фенила или 5- либо 6-членной гетероциклической моноциклической группы, содержащей от 1 до 3 гетероатомов, выбранных из N, S и О, а четыре других из R11, R12, R13, R14 и R15 представляют собой водород либо одни из R11 и R12, R12 и R13, R13 и R14, R14 и R15 вместе образуют циклическую структуру, а другие заместители R11, R12, R13, R14 и R15 представляют собой водород; R7 представляет собой алкил, способ их получения, фармацевтическая композиция и способы лечения, в частности серповидно-клеточной анемии. 5 с. и 4 з.п.ф-лы, 3 табл.
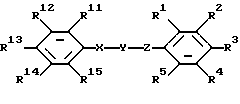
или их фармацевтически приемлемая соль,
где один из R1, R2 и R3 представляет собой 4-гидрокси-1,2,4-триазолил, 3-оксо-1,2-дигидро-1,2,4-триазолил, 2-оксо-3Н-1,3,4-оксадиазолил или тетразолил;
R4 и R5 представляют собой водород;
два других заместителя R1, R2 и R3 каждый независимо выбран из водорода, галогена, нитро, амино, ациламино, бензоиламино, фенила, нафтила, дифенила или 5- либо 6-членной гетероциклической моноциклической группы, содержащей один гетероатом, выбранный из N, S и О, где фенильная группа может быть замещена один или более чем один раз заместителями, выбранными из трифторметила, нитро, фенила, карбокси, алкоксикарбонила, аминокарбонила, диалкиламинокарбонила и анилинокарбонила;
Y представляет собой -СО- или -CS-;
Х представляет собой -NH-;
Z представляет собой NH;
один из R11, R12, R13, R14 и R15 выбран из водорода, галогена, трифторметила, -COOR7, фенила или 5- либо 6-членной гетероциклической моноциклической группы, содержащей один гетероатом, выбранный из N, S и О, а четыре других из R11, R12, R13, R14 и R15 представляют собой водород; либо одни из R11 и R12, R12 и R13, R13 и R14, R14 и R15 вместе образуют циклическую структуру, а другие заместители R11, R12, R13, R14 и R15 представляют собой водород;
R7 представляет собой алкил.
3-трифторметилфенил-4-нитро-2-(5-тетразолил)фенилмочевину,
3-трифторметилфенил-4-(2-нафтил)-2-(5-тетразолил)фенилмочевину,
3-трифторметилфенил-4-(3-пиридил)-2-(5-тетразолил)фенилмочевину,
3-трифторметилфенил-4-(1-нафтил)-2-(5-тетразолил)фенилмочевину,
3-трифторметилфенил-4-(4-трифторметилфенил)-2-(5-тетразолил)фенилмочевину,
3-трифторметилфенил-4-(3-фурил)-2-(5-тетразолил)фенилмочевину,
3-трифторметилфенил-4-(3-тиенил)-2-(5-тетразолил)фенилмочевину,
3-трифторметилфенил-4-(3-нитрофенил)-2-(5-тетразолил)фенилмочевину,
3-трифторметилфенил-4-(4-этоксикарбонилфенил)-2-(5-тетразолил)фенилмочевину,
3-трифторметилфенил-4-(4-диэтиламинокарбонилфенил)-2-(5-тетразолил)фенилмочевину,
3-трифторметилфенил-4-(4-аминокарбонилфенил)-2-(5-тетразолил)фенилмочевину,
3-трифторметилфенил-2-(4-гидрокси-1,2,4-триазол-3-ил)фенилмочевину,
3-трифторметилфенил-2-(3-оксо-1,2-дигидро-1,2,4-триазол-1-ил)-фенилмочевину,
3-трифторметилфенил-2-(2-оксо-3Н-1,3,4-оксадиазол-5-ил)фенилмочевину,
3-трифторметилфенил-4-дифенилил-2-(3-оксо-1,2-дигидро-1,2,4-триазол-1-ил)фенилмочевину;
3-трифторметилфенил-4-амино-2-(5-тетразолил)фенилмочевину,
3-трифторметилфенил-4-ацетиламино-2-(5-тетразолил)фенилмочевину,
3-трифторметилфенил-4-бензоиламино-2-(5-тетразолил)фенилмочевину,
3-трифторметилфенил-4-(4-карбоксифенил)-2-(5-тетразолил)-фенилмочевину,
3-трифторметилфенил-4-(4-анилинокарбонилфенил)-2-(5-тетразолил)-фенилмочевину,
4-дифенилил-2-(5-тетразолил)фенилмочевику,
3-дифенилил-2-(5-тетразолил)фенилмочевину,
5-инданил-2-(5-тетразолил)фенилмочевину,
3-бромфенил-4-бром-2-(5-тетразолил)фенилмочевину,
3-ацетилфенил-2-(5-тетразолил)фенилмочевину,
3-дифенилил-4-бром-2-(5-тетразолил)фенилмочевину,
3-(3-пиридил)фенил-4-бром-2-(5-тетразолил)фенилмочевину,
3-трифторметилфенил-4-бром-2-(5-тетразолил)фенилмочевину,
3-трифторметилфенил-2-(5-тетразолил)фенилмочевину,
3-трифторметилфенил-4-фенил-2-(5-тетразолил)фенилмочевину,
4-трифторметилфенил-2-(5-тетразолил)фенилмочевину,
3-хлорфенил-2-(5-тетразолил)фенилмочевину,
фенил-2-(5-тетразолил)фенилмочевину или
3-трифторметилфенил-4-амино-2-(5-тетразолил)фенилмочевину.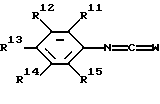
где W представляет собой О или S;
R11, R12, R13, R14 и R15 являются такими, как определено выше,
подвергают взаимодействию с соединением, имеющим формулу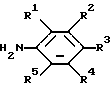
где R1, R2, R3, R4 и R5 являются такими, как определено выше,
после чего полученное соединение возможно превращают в другое соединение по изобретению и/или образуют его фармацевтически приемлемую соль, используя общепринятые способы.
| WO 9422807 А, 13.10.1994 | |||
| US 5362744 А, 08.11.1994 | |||
| RU 94016198 А1, 27.12.1995. |

