Область изобретения
Настоящее изобретение относится к новому химическому синтезу ингибиторов рибонуклеотидредуктазы.
Предпосылки создания изобретения
Рак в настоящее время является одной из основных причин смертности, и эффективное лечение многих твердых опухолей все еще недостижимо. Предполагается, что новые противоопухолевые лекарственные средства, обладающие сильным ингибирующим действием на рибонуклеотидредуктазу, представляющую собой незаменимый фермент для репликации клеток, окажется полезным дополнением к лекарственному режиму лечения рака, существующему в настоящее время.
Хорошо известно, что восстановительное превращение рибонуклеотидов в соответствующие деоксирибонуклеотиды является ключевой стадией в биосинтезе ДНК. Так как деоксирибонуклеотиды присутствуют в клетках млекопитающих на чрезвычайно низком уровне, исследователи предположили, что ингибитор рибонуклеотидредуктазы может быть более эффективным, чем ингибитор ДНК-полимеразы, при блокировании синтеза ДНК. См. Cory and Chiba "Combination Chemotherapy Directed at the Components of Nucleoside Diphosphate Reductase", Ingibitors of Ribonucleoside diphosphate reductase Activity, ("Комбинированная химиотерапия, направленная на компоненты нуклеозиддифосфатредуктазы", Ингибиторы активности рибонуклеозиддифосфатредуктазы) Соry, J.G. and Cory A.M., Eds.: Pergamon Press; Oxford, 1989, pp. 245-264. Соответственно в данной работе предполагается, что разработка сильных ингибиторов рибонуклеотидредуктазы могла бы привести к созданию сильного оружия против рака.
В течение многих лет значительный интерес привлекало исследование новых тиосемикарбазонов α-(N)-гетероциклических карбоксальдегидов (НСТ), класса наиболее мощных ингибиторов рибонуклеозиддифосфатрелуктазы. Сообщалось о ряде НСТ, таких как тиосемикарбазон 5-гидроксипиридин-2-карбоксальдегид (5НР), тиосемикарбазон 4-метил-5-амино-1-формилизохинолин (MAIQ-1), тиосемикарбазон 5-(ацетиламино)пиридин-2-карбоксальдегид (5-ААР), тиосемикарбазон 3- и 5-аминопиридин-2-карбоксальдегид (3-АР и 5-АР) и их 4-метилэамещенные производные (3-АМР и 5-АМР), тиосемикарбазон 3- и 5-гидрокси-4-метилпиридин-2-карбоксальдегид (3-НМР и 5-НМР). См. DeConti et al., Cancer Res., 1972, 32, 1455-1462; Agrawa1 et al., J. Med. Chem., 1976, 19, 970-972; French et al., J. Med. Chem. , 1974, 17, 172-181; Lui et al., J. Med. Chem., 1992, 35, 3672-3677; Wang et al., J. Med. Chem., 1992, 35, 3667-3671.
Исследование взаимоотношения структура-активность ряда НТС показало, что как 3-АР, так и 3-АМР проявляют намного лучший терапевтический эффект против L1210 лейкемии, М-109 карциномы легкого и А2780 карциномы яичника человека, чем другие НТС, известные к настоящему времени. Liu et al., J. Med. Chem., 1992, 35, 3672-3677; Agrawal et al., "The Chemistry and Biological. Activity of the α-(N)-Heterocyclic Carboxaldehyde Thiosemicarbazones". Progress in Medicinal Chemistry; Ellis G. P.; West G.B., Eds.; Elsevier/North-Holland Biomedical Press: New York, 1978; Vol. 15, pp. 321-356. Кроме того, 3-АР и 3-АМР являются сильнодействующими агентами с существенной противоопухолевой активностью по сравнению с гидроксимочевиной (HU), одобренным ингибитором рибонуклеотидредуктазы, используемым в клинической практике. Более крупномасштабное исследование этих соединений требует масштабного получения этих агентов.
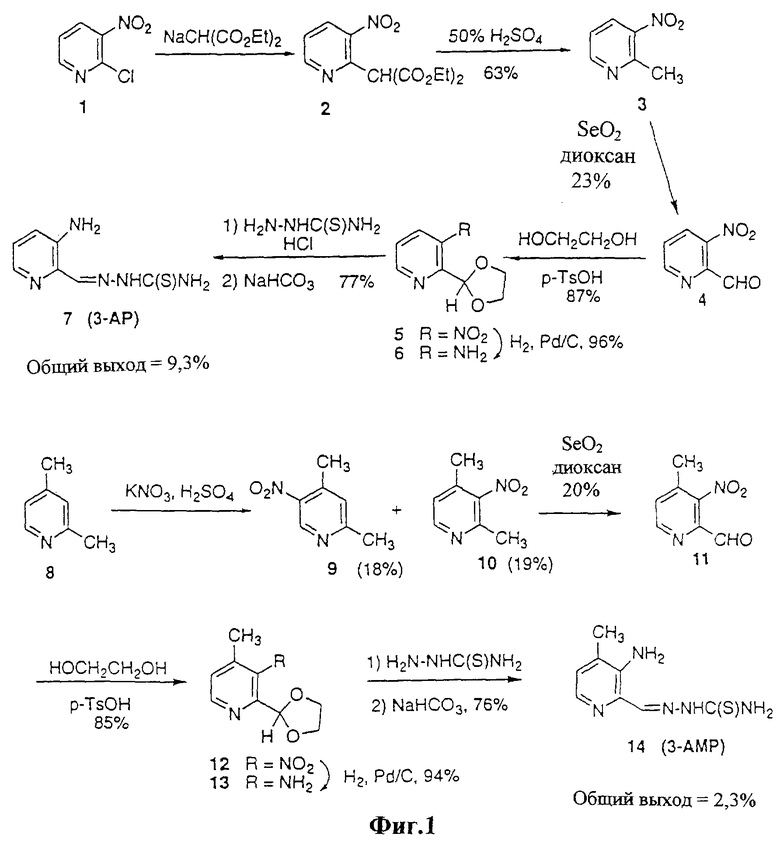
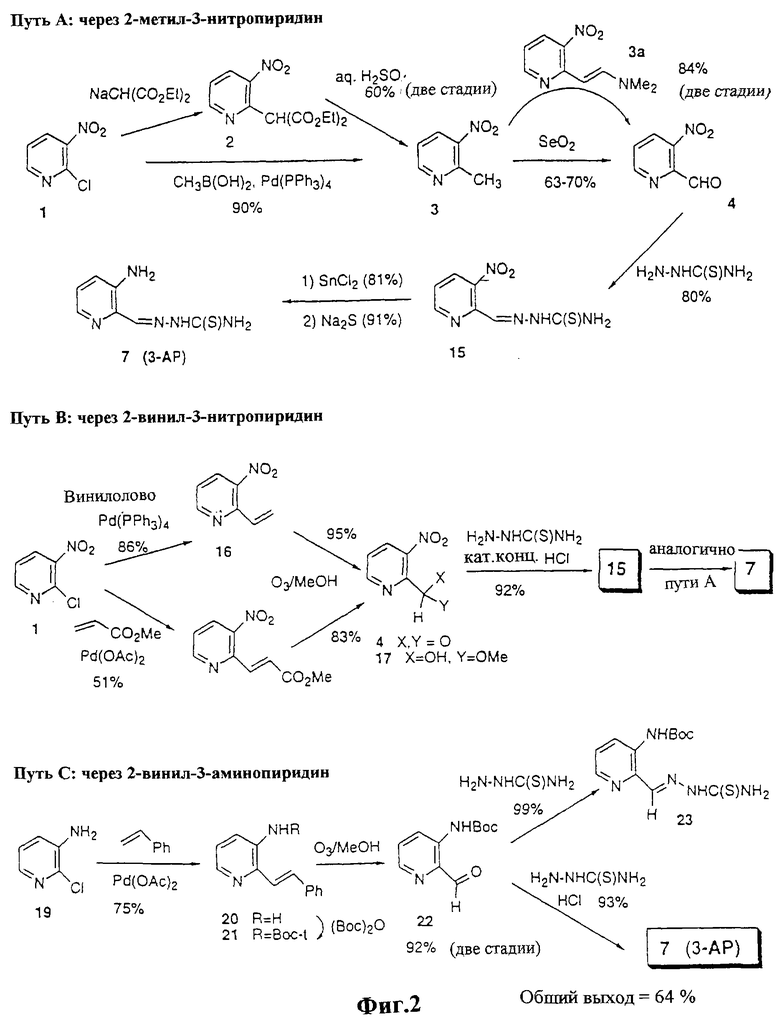
Как подчеркнуто на фиг.1, первый синтез 3-АР и 3-АМР был осуществлен в лабораториях др. Алана Сарторелли (Dr. Alan Sartorelli) Йельского университета. Хотя предшествующий синтез имел в качестве преимущества низкую стоимость материалов, используемых в способе получения, схема синтеза оказалась трудной из-за длинной последовательности реакций, низких выходов и трудностей проведения. По этой причине авторы настоящего изобретения исследовали синтез и разработали новые способы получения 3-АР и 3-АМР.
Задачи изобретения
Задачей изобретения является разработка эффективного химического синтеза тиосемикарбазона 3-аминопиридин-2-карбоксальдегида (3-АР) и тиосемикарбазона 3-амино-4-метилпиридин-2-карбоксальдегида (3-АМР).
Краткое описание изобретения
Настоящее изобретение относится к усовершенствованному эффективному химическому синтезу тиосемикарбазона 3-аминопиридин-2-карбоксальдегида (3-АР) и тиосемикарбазона 3-амино-4-метилпиридин-2-карбоксальдегида (3-АМР).
В настоящем изобретении разработан эффективный синтез 3-АР и 3-АМР. Согласно способу по настоящему изобретению целевые химиотерапевтические агенты получают из легко доступных исходных веществ с общими выходами, превышающими 30%. Для сравнения при использовании альтернативных синтетических способов по предшествующему уровню техники общий выход составляет менее 10%.
В настоящем изобретении, которое относится к новому синтезу 3-АР и 3-АМР, особенной ключевой стадией является реакция винилирования по Стилле (Stille) или Хеку (Heck), которая протекает в С-2 положении пиридинового фрагмента, приводя с высокими выходами к 2-винилпиридиновому промежуточному соединению (часто до 70%), которое легко может быть преобразовано в результате ряда синтетических стадий, протекающих с высокими выходами, в 3-АР или 3-АМР, соответственно. В этом способе пиридиновый фрагмент Р, ниже, преобразовывают в 2-винильное производное 2-VP с использованием реакции винилирования по Хеку или Стилле, как показано на схеме 2.
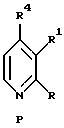
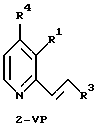
где R представляет Cl, Br, I, OMs, OTf или OTs;
R1 представляет NО2, NH2, NHP, NPP', N3 или CO2R2;
P и Р' представляют защитные группы;
R2 представляет Me, Et, Pr или i-Pr;
R3 представляет H, C1-C20алкил, арил, замещенный арил или CO2R2; и
R4 представляет Н или СН3.
Защитные группы, которые могут использоваться в качестве Р и Р', включают сложноэфирные группы, такие какС(О)OR5, где R5 представляет алкильную группу, такую как метил, этил, пропил, изопропил, бутил и трет-бутил, фенил, замещенный фенил, бензил и замещенный бензил, наряду с многими другими. Также могут использоваться другие аминозащитные группы, которые хорошо известны в данной области.
Реакция 2-винилирования предпочтительно протекает при использовании реагента, выбранного из следующих: Вu3SnСН=СНР3, (OH)2-B-CH=CHR3, ClZnCH= CHR3 и XMgCH=CHR3 (реактив Гриньяра, где Х представляет галоген, такой как I, Br, Cl, наряду с другими). Реакция винилирования протекает в присутствии трифенилфосфина (РРh3) и/или тетракис(трифенилфосфин)палладия [Pd(РРh3)4), обычно в растворителе, таком как толуол, ксилол или другом органическом растворителе, при нагревании. При введении винильной группы в С-2 положение пиридина такая группа может быть впоследствии превращена в альдегид (путем озонолиза или эквивалентного способа), который может быть затем превращен в тиосемикарбазон карбоксальдегида при получении 3-АР или 3-АМР. При альтернативной реакции винилирования с использованием стирола, 3-метилпропеноата или подобных винилирующих реагентов в сочетании с ацетатом палладия [Pd(OAc)2] и трифенилфосфином (РРh3) будут вводить винильную группу в С-2 положение пиридинового фрагмента, которая легко может быть превращена в 2-карбоксальдегид или в конечном счете в семикарбазон или гидразон карбоксальдегида.
Другие важные аспекты настоящего изобретения включают усовершенствование синтеза предшествующего уровня. По предшествующему уровню техники превращение производного 2-хлорпиримидина в 2-метилпиримидиновое производное представляло двухстадийный процесс, имеющий общий выход только 60%. По настоящему изобретению такое превращение осуществляют в одну стадию с выходом приблизительно 90%. При осуществлении такого мотивирования в условиях Сузуки (Suzuki) настоящее изобретение помимо уменьшения числа требуемых для синтеза стадий и повышения общего выхода также упрощает обработку и, следовательно, упрощает коммерческое крупномасштабное производство. Высокий выход в такой реакции является неожиданным результатом.
Кроме того, тогда как по предшествующему уровню техники требуется трудное превращение 2-карбоксальдегида в ацеталь и восстановление нитрогруппы в 3-положении перед взаимодействием с тиосемикарбазидом во 2-положении, авторы настоящего изобретения открыли более короткий и более эффективный способ, в котором 2-карбоксальдегид непосредственно взаимодействует с тиосемикарбазидом с последующим простым восстановлением нитрогруппы.
В предпочтительных аспектах настоящего изобретения указанные способы имеют то преимущество, что допускают получение больших количеств 3-АР или 3-АМР без использования дорогих исходных веществ, приводя к конечному продукту с высоким общим выходом. Способами по данному изобретению получают противораковые соединения высокой степени чистоты с выходами, подходящими для крупномасштабного и коммерческого получения. Настоящие способы адресуются к относительно низким выходам способов по предшествующему уровню техники и делают организацию серийного производства 3-АР и 3-АМР экономически жизнеспособной.
Краткое описание фигур
На фиг.1, схема 1, показан синтез 3-АР и 3-АМР по предшествующему уровню техники, каждый синтез протекает через пиридиновое 2-карбоксальдегидное промежуточное соединение (4 и 11). В случае 3-АР синтез протекает, завершаясь общим выходом примерно 9,3%. В случае 3-АМР синтез заканчивается с общим выходом примерно 2,3%.
На фиг. 2, схема 2, показаны два новых синтеза 3-АР из 2-хлор-3-нитропиридина 1 и новый синтез из 2-хлор-3-аминопиридина 19. Эти синтезы приводят к конечному результату с общими выходами в диапазоне от 31,5% до 68,4%. Действительно, химический процесс, описанный для синтеза С, был использован для получения 3-АР в количествах, превышающих 20 г, в одну стадию.
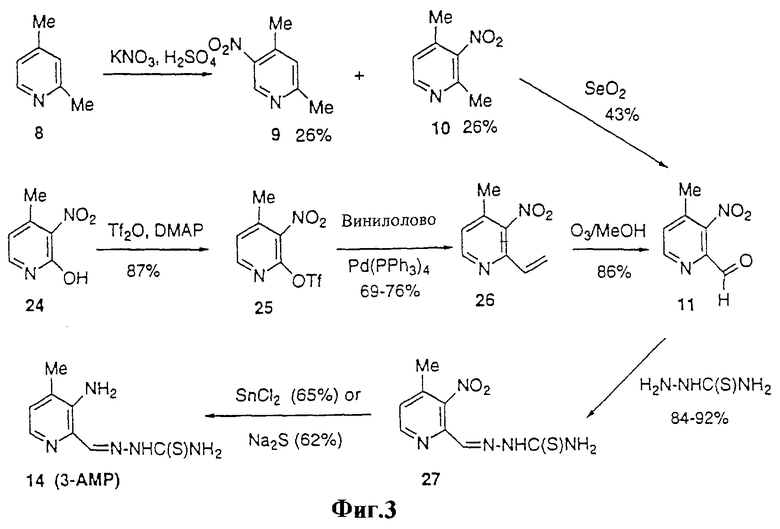
На фиг. 3, схема 3, приведены два новых синтеза 3-АМР, через промежуточный 2-карбоксальдегид 11. Эти синтезы приводят к конечному результату с общими выходами от 5,8% до 34%.
Подробное описание изобретения
Термин "неоплазия" используется в данном описании для обозначения патологического процесса, который приводит к образованию и росту раковой или злокачественной опухоли, т.е. ненормальной ткани, которая растет в результате быстрого размножения (пролиферации) клеток, часто более быстро, чем нормальная ткань, и продолжает расти после воздействия, которое инициирует прекращение нового роста. Злокачественные опухоли имеют частичный или полный дефицит структурной организации и функциональной координации с нормальной тканью и большей частью проникают в окружающие ткани, давая метастазы в нескольких местах, и, вероятно, появляются вновь после попытки удаления и вызывают смерть пациента, если не предпринять адекватное лечение. Как он используется в данном описании, термин "неоплазия" используется для описания всех раковых болезненных состояний и охватывает или включает в себя патологический процесс, связанный с злокачественными гематогенными, асцитными и плотными опухолями.
Термин "защищенный" используется для обозначения фосфатной группы или гидроксильной группы в любом одном или нескольких из промежуточных соединений, которые защищены от нежелательных взаимодействий, но защита которых легко может быть удалена при определенных условиях. Защитные группы, которые могут использоваться в этих целях, включают, например, трихлорэтил, этил, цианоэтил, триметилсилилэтил, силилэтил, трет-бутилдиметилсилил, трет-бутил, трифенилсилил и трет-бутилдифенилсилил, наряду со многими другими, включая сложноэфирные группы, такие как метильная, этильная, пропильная, изопропильная, бутильная и трет-бутильная сложноэфирные группы, наряду с другими. Защитные группы могут быть выбраны в широком диапазоне из класса силильных защитных групп, защитных групп простых эфиров и защитных групп сложных эфиров, каждая защитная группа выбирается по ее способности защищать фрагмент от нежелательного протекающего взаимодействия, легкости ее удаления и химической совместимости. Как обсуждалось ранее, аминные группы в промежуточных соединениях по настоящему изобретению также могут быть защищены.
Усовершенствованные синтезы 3-АР и 3-АМР по настоящему изобретению, имеющие более высокие выходы и более безопасно и легко осуществляемые, подчеркнуты на схемах 2 и 3 соответственно. Как доказано на фиг.2, для синтеза 3-АР были разработаны три синтетических пути. Благоприятным образом было уменьшено число стадий в способе, известном из предшествующего уровня техники (смотри фиг. 1, схема 1), за счет исключения стадий введения ацетальной защиты и снятия защиты в синтезе по предшествующему уровню в данной области. В соответствии с путями А и В, как показано на схеме 2, 2-карбоксальдегид 4 непосредственно превращают в гидразон 15 и последующие восстановления функциональной нитрогруппы в аминную группу проводят с использованием или дихлорида олова (см. Atwal K.S., et al., J. Med. Chem., 1996, 39, 305-313, для описания реакций данного типа), или сульфида натрия (восстановление Зинина, смотри Porter H. K., Org. React., 1973, 20, 455-483). По пути С N-Boc-защищенный 2-карбоксальдегид 22 непосредственно вводят в реакцию с тиосемикарбазидом и одновременно удаляют защитную группу с получением 3-АР. Эти общие подходы приводят к 3-АР с хорошими выходами.
Три различных подхода показаны на схеме 2 для синтеза 2-карбоксальдегидных пиридиновых промежуточных соединений (соединения 4 и 22). В первом из этих трех подходов, показанном как путь А, метилирование 2-хлорпиридинового соединения 1 с последующим окислением 2-метильной группы диоксидом селена приводит к 2-карбоксальдегидному соединению 4, которое легко превращается в 3-АР в две стадии с высокими выходами. В альтернативном подходе, показанном как ответвление от пути А, 2-метил-3-нитропииридин 3 взаимодействует с диметилформамид-диметилацеталем (ДМФДМА, хотя в данной реакции можно использовать любые подобные ацетальные аналоги ДМФДМА) с получением промежуточного соединения 3а, последующее окисление периодатом натрия (NaIО4) приводит к альдегиду 4 с общим выходом 84%, значительное улучшение по сравнению со способом окисления 2-метил-3-нитропиридина 3 диоксидом селена с получением альдегида 4. Полный синтез 3-АР из 2-хлор-3-нитропиридина 1 может быть осуществлен с общим выходом 55%.
В альтернативном способе, показанном как верхнее ответвление от пути В, введение 2-винильной группы с использованием палладий-опосредованной реакции винилирования Стилле в качестве ключевой стадии (см. Attwood M.R., et al., Tetrahedron Lett. , 1996, 37, 2731; Skoda-Foldes R., et al., Tetrahedron Lett. , 1996, 37, 2085; и Sabramanyam С., et al., Tetrahedron Lett., 1996, 37, 459) с последующим озонолизом приводит к 2-карбоксальдегидному соединению 4 с очень высоким выходом. В третьем способе, показанном как путь С и нижнее ответвление от пути В, используется палладий-опосредованная реакция винилирования Хека (смотри Sit S.Y., et al., Bioorg. Med. Chem. Lett., 1996, 6, 499; и Ojima I., et al., Chem. Rev., 1996, 96, 635-662, для описаний реакций этого типа) с последующим озонолизом для получения карбоксальдегидных соединений 4 или 22 (где R представляет защитную группу для 3-аминоположения, включая t-Boc группу, сложноэфирную группу или подобную защитную группу, которые хорошо известны в данной области и согласуются с химией, используемой в данной реакционной последовательности), которые легко превращаются в 3-АР с использованием тиосемикарбазида с последующим использованием условий (например, кислоты) для удаления группы, защищающей аминогруппу, или непосредственно при использовании тиосемикарбазида в НСl. Выход 3-АР для каждого из трех альтернативных синтетических путей превышает в 3-7 раз выход по способу синтеза в соответствии с предшествующим уровнем техники, неожиданно благоприятный результат. Это представляет большое преимущество для промышленного масштабного получения 3-АР для клинических целей и конечного терапевтического применения.
На фиг.3 представлены два эффективных пути синтеза 3-АМР. Как проиллюстрировано на фиг.3, синтез, начинающийся с диметилпиридина 8, уменьшает число стадий первоначального синтеза и увеличивает общий выход. Нитрозирование диметилпиридина 8 с последующим окислением 2-метильной группы приводит к 2-карбоксальдегиду 11, который непосредственно подвергают взаимодействию с тиосемикарбазидом, получая 2-тиосемикарбазон 27 с высоким выходом. Наконец, 2-тиосемикарбазон 27 превращают в 3-АМР или в условиях восстановления по Зинину, или с использованием дихлорида олова, приводя к выходу реакции, превышающему примерно в три раза выход в синтезе 3-АР по предшествующему уровню техники. Синтез, начинающийся с замещенного аналога пиридина 24, обеспечивает более эффективный синтез 2-карбоксальдегида 11 за счет использования реакции винилирования Стилле, протекающей с высокими выходами, после превращения 2-гидроксильной группы в аналоге пиридина 24 в 2-OTf группу. Озонолиз 2-винильной группы приводит к 2-карбоксальдегиду 11 с высоким выходом. 2-карбоксальдегид 11 взаимодействует с тиосемикарбазидом с образованием с высоким выходом тиосемикарбазона 27, который восстанавливают в 3-АМР для завершения синтеза с использованием или условий восстановления по Зинину, или дихлорида олова. Путь синтеза, включающий винилирование Стилле с получением ключевого промежуточного соединения 11, исключает трудные реакции нитрования и окисления 2-метильной группы и неожиданно высоко улучшает общий выход, превышающий в 15 раз выход 3-АМР в способе получения по предшествующему уровню техники.
Хотя предпочтительные синтетические химические способы были описаны выше, обычному специалисту в данной области будет очевидно, что для получения аналогичных результатов можно использовать замену стадий или эквивалентные стадии. Например, специалист может легко заменить некоторые реагенты и фактически все растворители, используемые для получения промежуточных соединений, как указано на различных схемах. Образование 2-винилпиридиновых промежуточных соединений, например, можно легко проводить с использованием любых подходящих винил-образующих реагентов или сочетания реагентов (включая трифенилфосфин или тетракистрифенилфосфин палладий в качестве подходящих) и любого подходящего растворителя. Окисляющие агенты для превращения винильных групп или бензильных (метильных) групп в альдегиды (во 2-положении пиридинового фрагмента) включают диоксид селена и озон, но также могут использоваться другие подходящие окисляющие агенты. В случае гидрогенолиза (например, 3-нитрогруппы в 3-аминогруппу) подходит применение SnCl2 или Na2S. Обычный специалист в данной области легко способен заменить применение одной защитной группы на другую защитную группу, согласующуюся с общей химией, используемой при синтезе.
В частности, ключом к эффективному синтезу 3-АР или 3-АМР является протекающее с высоким выходом введение метильной или винильной группы, предпочтительно винильной группы, предпочтительно винила, во 2-положение пиридинового фрагмента с использованием реакций винилирования Хека или Стилле или реакции метилирования в условиях Сузуки. Эти реакции протекают во 2-положении пиридинового фрагмента с высоким выходом (обычно превышающем 50% и, в большинстве случаев, превышающем 70%). Преимуществом реакции винилирования является то, что 2-винильная группа легко может быть преобразована с использованием протекающей с высоким выходом реакции озонолиза, проводимой в полярном растворителе, таком как метанол, в 2-карбоксальдегидную группу для дальнейшего превращения в 2-тиосемикарбазон. Предпочтительная реакция винилирования представляет собой реакцию винилирования Хека или Стилле, осуществляемую с использованием реагента, выбранного среди следующих: Bu3SnCH= CHR3, (OH)2-B-SnCH=CHR3, ClZnCH=CHR3 и XMgCH=CHR. Реакция винилирования протекает в присутствии трифенилфосфина (РРh3) и/или тетракис(трифенилфосфин)палладия [Pd(PPh3)4] обычно в растворителе, таком как толуол, при нагревании. Преимущество реакции метилирования в условиях Сузуки по сравнению с двухстадийным метилированием по предшествующему уровню техники заключается в значительном повышении выхода, уменьшении числа стадий и, следовательно, требуемой обработки и легкости масштабирования синтеза для коммерческого производства. Альтернативно, для введения винильной группы во 2-положение 3-нитропиридина, можно выгодным образом осуществить взаимодействие 2-метил-3-нитропиридина с диметилформамид-диметилацеталем (ДМФДМА) с получением 2-диметиламино-винил-3-ритрспиридина - соединения 3а, последующее окисление которого периодатом натрия (NaIO4) приводит к карбоксальдегиду 4 с неожиданно высоким 84%-ным выходом.
При введении метильной или винильной группы в С-2 положение пиридина эта группа может быть превращена в альдегид, который окончательно будет превращен в тиосемикарбазон карбоксальдегида. Тогда как в предшествующем уровне техники требовалось сложное превращение 2-карбоксальдегида в ацеталь и восстановление нитрогруппы в 3-положении перед взаимодействием с тиосемикарбазидом по 2-положению, настоящий способ является более коротким и более эффективным способом, в котором 2-карбоксальдегид непосредственно взаимодействует с тиосемикарбазидом с последующим простым восстановлением нитрогруппы. Восстановление 3-нитрогруппы в данном пути синтеза легко осуществляется с использованием стандартных условий восстановления (SnC12 или Nа2S, наряду с другими способами восстановления). Синтез 3-АР или 3-АМР согласно настоящему изобретению завершается с неожиданно высоким выходом по меньшей мере в 30%, исходя из легко доступных исходных веществ. В некоторых воплощениях выход может достигать 55% или более того.
Настоящее изобретение далее описано, только для иллюстрации, в следующих примерах. Специалисту в данной области следует понимать, что эти примеры не являются ограничивающими и что изменение деталей может быть сделано, не отступая от сути и в пределах объема настоящего изобретения.
Примеры
В данном разделе приведены подробные условия реакций и характеристики каждого соединения в следующих далее методиках. Все ЯМР спектры зарегистрированы при 300 МГц для 1Н и 75 МГц для 13С на ЯМР спектрометре QE Plus 300 МГц. Масс-спектры регистрировали на масс-спектрометре VG ZAB-SE и приборе VG 70-SE-4F. В данный раздел также включены некоторые существенные ссылки. Все растворители перегоняли перед использованием.
Синтез соединения 3 (2-метид-3-нитропиридина) (3)
Способ 1. В колбу, содержащую диэтилмалонат (20 г, 0,125 моль), добавляют натрий (2,0 г, 0,087 моль). Реакционную смесь перемешивают в течение 1 часа при комнатной температуре, а затем нагревают до 120oС (температура масляной бани) в течение 50 минут. К этой желтой суспензии твердого вещества добавляют толуол (120 мл) с последующим добавлением раствора 2-хлор-3-нитропиридина 1 (12,8 г, 0,08 моль) в 40 мл толуола. Реакционную смесь нагревают при температуре кипения с обратным холодильником в течение 8 часов и затем перемешивают в течение ночи при комнатной температуре. Растворитель удаляют при пониженном давлении и остаток растворяют в 30%-ной Н2SО4 (60 мл). Эту реакционную смесь нагревают при 125oС (масляная баня) в течение 7 часов, охлаждают и выливают на лед (200 г). Реакционную смесь нейтрализуют насыщенным раствором NаНСO3, фильтруют через целит, экстрагируют несколько раз диэтиловым эфиром. Объединенные экстракты сушат над безводным Na2SO4. Растворитель упаривают и остаток перегоняют при пониженном давлении, получая 6,65 г (60%) целевого продукта 3.
Способ 2. Смесь 2-хлор-3-нитропиридина 1 (793 мг, 5,0 ммоль), метилбороновой кислоты (329 мг, 5,5 ммоль), Рd(РРh3)4 (578 мг, 0,5 ммоль) и К2СО3 (2,073 г, 15,0 ммоль) в диоксане (25 мл) нагревают при кипении с обратным холодильником в течение 2 дней. Затем смесь охлаждают до комнатной температуры и фильтруют. Растворитель удаляют и остаток подвергают флэш-хроматографии (50% этилацетат в гексанах), получая 623 мг (90%) 2-метил-3-нитропиридина 3.
1Н-ЯМР (СDСl3) δ 2,88 (с, 3Н), 7,36 (дд, 1Н, J=4,8 и 8,4 Гц), 8,28 (дд, 1Н, J=1,2 и 8,1 Гц, 8,73 (дд, 1Н, J=1,2 и 4,8 Гц).
Синтез соединения 2-карбоксальдегида (4)
К раствору 2-метил-3-нитропиридина 3 (2,07 г, 0,015 моль) в 35 мл диоксана добавляют диоксид селена (1,88 г, 0,017 моль). Реакционную смесь нагревают при кипении с обратным холодильником в течение 16 часов, затем охлаждают до комнатной температуры и фильтруют. Растворитель удаляют при пониженном давлении и остаток очищают флэш-хроматографией на силикагеле (гексаны-ЕtOАс=1:1), получая 1,60 г (70%) целевого альдегида 4.
1H-ЯМР (CDCl3) δ 7,71 (д, 1Н, J=4,8 и 8,2 Гц), 8,29 (дд, 1H, J=1,1 и 8,0 Гц), 9,01 (д, 1H, J=1,1 и 4,5 Гц), 10,31 (с, 1H).
Альтернативный синтез соединения 2-карбоксальдегида (4)
Раствор 2-метил-3-нитропиридина (3) (276 мг, 2 ммоль) и диметилформамид-диметилацеталя (ДМФДМА) (477 мг, 4 ммоль) в диметилформамиде (ДМФ) (1 мл) нагревают при 140oС в атмосфере N2 в течение 7 часов, затем перемешивают при комнатной температуре в течение ночи. Растворитель удаляют при пониженном давлении и остаток высушивают в вакууме. Реакционная смесь была абсолютно чистой и содержала один продукт, соединение 3а - 2-диметиламиновинил-3-нитропиридин, который используют на следующей стадии окисления без дополнительной очистки. Раствор 3а, полученного выше, и NaIО4 (1,283 г, 6 ммоль) в 50%-ном водном ТГФ (20 мл), перемешивают при комнатной температуре в течение 2 часов, фильтруют и экстрагируют несколько раз СН2Сl2. Объединенные экстракты промывают насыщенным солевым раствором и сушат над безводным Na2SO4. Выделяют флэш-хроматографии на силикагеле (гексаны - EtOAc=1:1), получая 256 мг (84%) 2-карбоксальдегида 4.
Синтез гидразонового соединения 15 из 2-карбоксальдегида (4)
Смесь карбоксальдегида 4 (750 мг, 4,93 ммоль) и тиосемикарбазида (540 мг, 5,92 ммоль) в 70%-ном этаноле (25 мл) перемешивают при комнатной температуре в течение 6 часов, фильтруют и промывают Н2О, C2H5OH, диэтиловым эфиром и сушат в вакууме, получая 893 мг (80%) целевого гидразона 15.
1Н-ЯМР (ДМСО-d6) δ 7,09 (уш., 1Н), 7,67 (дд, 1Н, J=4,9 и 8,2 Гц), 8,27 (с, 1H), 8,38 (д, 1Н, J=7,7 Гц), 8,60 (уш., 1Н), 8,85 (д, 1Н, J=4,4 Гц), 11,97 (с, 1Н).
13С-ЯМР (ДМСО-d6) δ 125,1, 131,7, 338,3, 144,7, 145,6, 152,9, 179,3.
CIMS рассчитано для C7H7N5O2S 226 (МН+), найдено 226.
Синтез 2-винилпиридинового соединения 16
Раствор 2-хлор-3-нитропиридина 1 (417 мг, 2,63 ммоль), Pd(PPh3)4 (32 мг, 0,026 ммоль), трифенилфосфина (20 мг, 0,078 ммоль) и винилтрибутилолова (1,00 г, 3,16 ммоль) в толуоле (15 мл) нагревают при кипении с обратным холодильником в течение 2 часов. Реакционную смесь охлаждают до комнатной температуры и затем гасят водой (10 мл). Полученную смесь экстрагируют EtOAc (3•30 мл). Объединенные органические слои сушат (Na2SO4) и фильтруют. Фильтраты концентрируют в вакууме и остаток очищают хроматографией на силикагеле (20-30% EtOAc/гексаны), получая 339 мг (86%) целевого продукта 16 в виде бледно-желтого масла.
1Н-ЯМР (СDСl3) δ 8,75 (дд, 1Н, J=1,3 и 4,5 Гц), 8,16 (дд, 1Н, J=1,3 и 8,1 Гц), 7,23-7,36 (м, 2Н), 6,59 (дд, 1Н, J=1,6 и 16,7 Гц), 5,70 (дд, 1H, J= 1,6 и 10,4 Гц);
LRMS m/e 151 (МН+).
Синтез 2-полуацетального и 2-карбоксальдегидного соединений (17 и 4) из 2-винилпиридинового соединения (16)
Метанольный раствор (20 мл) 2-винилпиридина 16 (800 мг, 5,33 ммоль) подвергают озонолизу при -78oС в течение 15 минут. Реакцию гасят (при -78oС) Me2S (2,2 мл) и полученную реакционную смесь перемешивают в течение ночи при комнатной температуре. Растворитель упаривают и остаток подвергают хроматографии на короткой колонке (4'' силикагель), получая 850 мг (95%) смеси продуктов 17 и 4 (17/4=2:3).
Спектр 1Н-ЯМР соответствует опубликованным ранее литературным данным (Santorelli et al., J. Med. Chem., 1992, 35, 3672-3677).
1ЯМР полуацеталь 17 (CDCl3): δ 8,78 (м, 1Н), 8,34 (м, 1Н), 7,57 (м, 1Н), 6,10 (д, 1Н, J=8,7 Hz), 5,53 (м, 1Н), 3,47 (с, 3Н).
Синтез 2-винил-сложноэфирного соединения 18
Смесь 2-хлор-3-нитропиридина 1 (1,20 г, 7,6 ммоль), метилакрилата (1,31 г, 15,2 ммоль), триэтиламина (0,92 г, 9,1 ммоль), трифенилфосфина (0,60 г, 2,28 ммоль), Pd(OAc)2 (0,17 г, 0,76 ммоль) и 15 мл ДМФ в запаянной ампуле нагревают при 120oС в течение 24 часов. Реакционную смесь охлаждают до комнатной температуры и затем гасят реакцию водой (10 мл). Полученную смесь экстрагируют ЕtOАс (3•30 мл). Объединенные органические слои промывают насыщенным солевым раствором, сушат над Na2SO4 и фильтруют. Фильтраты концентрируют и остаток очищают хроматографией на силикагеле (гексан: ЕtOАс=4: 1), получая 0,80 г (51%) целевого продукта 18 в виде желтого твердого вещества.
1H-ЯМР (СDСl3) δ 8,85 (дд, 1Н, J=1,5 и 4,5 Гц), 8,16 (дд, 1Н, J=1,5 и 8,1 Гц), 7,49 (дд, 1Н, J=4,8 и 8,4 Гц), 7,22 (д, 1Н, J=15,3 Гц), 3,85 (c, 3Н).
LRMS m/e 201 (МН+).
HRMS, рассчитанной для С9Н8N2О4 208,0484, найдено 208,0484.
Синтез 2-полуацетального и 2-карбоксальдегидного соединений (17 и 4) из 2-винил-сложноэфирного соединения (18)
Раствор 18 (0,44 г, 5,33 ммоль) в метанол-метилхлориде (12:1; 130 мл) подвергают озонолизу при -78oС, контроль за ходом реакции осуществляют по ТСХ. После завершения реакции избыток О3 удаляют, барботируя через реакционную смесь O2 при -78oС в течение 5 минут. Затем реакцию гасят (при -78oС) с использованием Me2S (5 мл) и полученную реакционную смесь перемешивают при комнатной температуре в течение ночи. Растворитель упаривают и остаток очищают хроматографией на силикагеле (гексаны:ЕtOAc=4:1 до гексаны:ЕtOАс=1: 1), получая 0,286 г (83%) смеси продуктов 17 и 4 (17/4=1:2).
Синтез гидразонового соединения 15 из смеси полуацеталя и карбоксальдегида (17 и 4)
К водно-этанольному раствору (10 мл этанола и 5 мл воды) альдегида 4 и полуацеталя 17 (850 мг, 4,85 ммоль) добавляют при комнатной температуре конц. НСl (1 мл), а затем тиосемикарбазид (483 мг, 5,34 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 6 часов. В этот момент желтое твердое вещество собирают фильтрованием. Полученное таким образом твердое вещество промывают последовательно водой и этанолом три раза, а затем сушат в высоком вакууме в течение 1 часа, получая 1,0 г целевого продукта 15 с выходом 92%.
Синтез соединения 7 (3-АР) из гидразона (15)
Способ 1. К раствору SnCl2•2H2O (2,256 г, 10 ммоль) в 6 мл этанола добавляют нитросоединение 15 (450 мг, 2,0 ммоль). Реакционную смесь нагревают при кипении с обратным холодильником в течение ночи в атмосфере N2 и фильтруют. Твердый сырой продукт растворяют в 30 мл горячей воды и фильтруют. Фильтрат затем доводят до рН 7,5 насыщенным раствором NaHCO3 и перемешивают при комнатной температуре в течение 30 минут, фильтруют, промывают H2O, C2H5OH и диэтиловым эфиром. Полученный желтый твердый продукт дополнительно несколько раз экстрагируют ТГФ. Объединенные ТГФ-экстракты упаривают и остаток сушат в вакууме, получая 316 мг (81%) 3-АР.
Способ 2. Смесь нитросоединения 15 (450 мг, 2,0 мг) и Na2S (468 мг, 6 ммоль) в смеси H2O/С2H5OH (1:1, 20 мл) перемешивают при комнатной температуре в течение 18 часов, концентрируют. Остаток доводят до pН 7,5 1Н раствором НСl, фильтруют, промывают Н2О, C2H5OH, СН2Сl2 и сушат в вакууме, получая 355 мг (91%) 3-АР 7.
1Н-ЯМР (ДМСО-d6) δ 6,43 (уш., 2Н), 7,07 (м, 2Н), 7,80 (дд, 1Н, J=1,2 и 4,2 Гц), 7,95 (уш., 1Н), 8,15 (уш., 1Н), 8,31 (с, 1Н), 11,29 (с, 1Н).
13С-ЯМР (ДМСО-d6) δ 122,2, 124,4, 132,8, 137,1, 144,0, 149,2, 177,0.
LRMS (FAB) m/e 196 (МH+).
HRM3, рассчитано для C7H9N5S 196,0657, найдено 196,0657.
Синтез 2-аминопиридинового производного 20
Способ 1. Реакция, осуществляемая в запаянной ампуле
Суспензию 2-хлор-3-аминопиридина 19 (1,28 г, 10,0 ммоль), стирола (5,72 мл, 50,0 ммоль), бикарбоната натрия (1,68 г, 20,0 ммоль), трифенилфосфина (1,31 г, 5,0 ммоль) и Pd(OAc)2 (0,12 г, 0,50 ммоль) в ДМФ (20 мл) нагревают при 130oС в течение 24 часов в запаянной ампуле. После этого реакционную смесь охлаждают до комнатной температуры и гасят реакцию насыщенным раствором NаНСО3 (10 мл) и водой (10 мл). Реакционную смесь экстрагируют EtOAc (3х50 мл). Объединенные органические слои промывают насыщенным солевым раствором, сушат над сульфатом натрия и фильтруют. Фильтраты концентрируют в вакууме, остаток хроматографируют (25% этилацетат в гексанах), получая 1,47 г (75%) целевого продукта 20.
Способ 2. Реакция, осуществляемая при 1 атмосфере
Суспензию 2-хлор-3-аминопиридина 19 (20 г, 155,6 ммоль), стирола (89 мл, 778 ммоль), бикарбоната натрия (26 г, 311 ммоль), трифенилфосфина (2J г, 78 ммоль) и Pd(OAc)2 (1,74 г, 7,8 ммоль) нагревают при 135oС в течение 48 часов. После этого реакционную смесь охлаждают до комнатной температуры и затем добавляют 100 мл этилацетата. Эту смесь фильтруют через целит и фильтрат концентрируют в вакууме. Остаток хроматографируют (25%-ный этилацетат в гексанах), получая 22,7 г (74%) целевого продукта 20 в виде желтого твердого вещества.
Синтез альдегида 22 из производного 2-аминопиридина 20
Производное 2-аминопиридина 20 (5,00 г, 25,51 ммоль) растворяют в теплом бутаноле (100 мл). К этому теплому раствору (~40oС) добавляют (t-Boc)2O (6,68 г, 30,61 ммоль). После перемешивания этого раствора при комнатной температуре в течение нескольких часов добавляют дополнительное количество (t-Boc)2O (2,78 г, 12,76 ммоль). Реакционную смесь дополнительно перемешивают в течение 15 часов при комнатной температуре. После этого из молочнообразной суспензии упаривают растворитель. После удаления растворителя полученный остаток помещают в 100 мл смеси EtOAc/Et2O (1:1). Полученный раствор промывают насыщенным солевым раствором. Органический слой отделяют и сохраняют. Водный слой опять экстрагируют такой же смесью растворителей (3х50 мл). Объединенные органические слои сушат и концентрируют в вакууме, получая 10 г сырого продукта 21 в виде светло-коричневого твердого продукта.
Сырое N-Boc защищенное производное пиридина 21 (~25,5 ммоль) растворяют в МеОН (120 мл) и дихлорметане (30 мл). Полученный раствор охлаждают до -78oС и подвергают озонолизу в течение -45 минут. Реакцию останавливают Me2S (8 мл) и перемешивают при комнатной температуре в течение ночи. Растворители удаляют в вакууме, остаток очищают хроматографией на силикагеле (10-15% этилацетат в гексанах), получая 5,23 г (92% для двух стадий) целевого альдегида 22 в виде белого твердого вещества.
Синтез соединения 7 (3-АР) из альдегида 22
К смеси альдегида 22 (1,468 г, 6,61 ммоль) и тиосемикарбазида (622 мг, 7,27 ммоль) в смеси EtOH/Н2О (22,5 мл, содержание этанола 67%) добавляют 3 мл конц. НСl. Полученный раствор нагревают при кипении с обратным холодильником в течение 3 часов. Реакционную смесь охлаждают до комнатной температуры и фильтруют. Сырую желтоватую соль 3-АР-НСl переносят в колбу. В эту колбу добавляют 40 мл горячей воды и 8 мл 10% NAHCO3. Смесь перемешивают при комнатной температуре в течение 1 часа (при рН~7,5). Твердое вещество отфильтровывают и затем промывают водой (10 мл), EtOH (3 мл) и Et2O (10 мл). Полученное твердое вещество сушат в высоком вакууме в течение нескольких часов, получая 1,195 г (93%) целевого 3-АР 7.
Синтез соединения 10
Дымящую серную кислоту (500 г, 5,1 моль) медленно прибавляют к 2,4-лутидину 8 (55 мл, 0,48 моль) и охлаждают на ледяной бане при перемешивании. Затем медленно добавляют нитрат калия (87,5 г, 0,86 моль). Реакционную смесь перемешивают при комнатной температуре в течение 1 часа и затем нагревают при 110oС в течение дополнительных 7 часов, охлаждают до комнатной температуры и перемешивают в течение ночи. Реакционную смесь выливают на лед (1,0 кг) и нейтрализуют до рН 9 твердым NaOH, экстрагируют эфиром. Объединенные экстракты сушат над безводным Nа2SO4. Растворитель удаляют и остаток перегоняют при пониженном давлении, получая 37,83 г (53%) смеси 3-нитролутидина 10 и 5-нитролутидина 9. Дополнительное разделение с помощью перегонки дает чистый 3-нитролутидин 10.
1Н-ЯМР (CDC13) δ 2,35 (с, 3Н), 2,57 (с, 3Н), 7,13 (д, 1Н, J=4,8 Гц), 8,46 (д, 1Н, J=5,0 Гц).
Синтез соединения 11
Смесь 3-нитролутидина 10 (760 мг, 5 ммоль) и диоксида селена (555 мг, 5 ммоль) в 15 мл диоксана кипятят в течение 14 часов в атмосфере N2, добавляют дополнительно SеO2 (555 мг, 5 ммоль). Реакционную смесь кипятят с обратным холодильником еще 8 часов, фильтруют через целит. Растворитель удаляют при пониженном давлении и выделяют остаток флэш-хроматотрафией на ситикагеле (гексан:ЕtOАс=2:1), получая 357 мг (43%) альдегида 11.
1Н-ЯМР (CDC13) δ 2,42 (с, 3Н), 7,54 (д, 1Н, J=4,9 Гц), 8,76 (д, 1Н, J= 4,9Гц), 10,02 (с, 1Н).
Синтез гидразона 27 из 2-карбоксальдегида (11)
Смесь альдегида 11 (3,110 г, 18,73 ммоль) и тиосемикарбазида (2,65 г, 29,08 ммоль) в 70%-ном этаноле перемешивают при комнатной температуре в течение 8 часов, фильтруют, промывают Н2O, C2H5OH, диэтиловым эфиром и сушат в вакууме, получая 4,12 г (92%) гидразона 27.
1Н-ЯМР (ДМСО-d6) δ 2,30 (с, 3Н), 6,68 (уш., 1Н), 7,58 (д, 1Н, J=5,1 Гц), 8,04 (с, 1Н), 8,65 (д, 1Н, J=4,8 Гц), 8,72 (уш., 1Н), 12,01 (с, 1Н).
13С-ЯМР (ДМСО-d6) 615,7, 126,5, 137,7, 139,6, 142,3, 144,9, 150,5, 178,8.
LRMS m/e 240 (МН+).
HRMS, рассчитано для C8H9N5O2S 240,0555, найдено 240,0557.
Синтез 2-O-Tf соединения 25
К 2-гидрокси-3-нитро-4-метилпиридину 24 (3,08 г, 20 ммоль) и 4-диметиламинопиридину (2,44 г, 20 ммоль), растворенным в 5 мл CH2Cl2, медленно добавляют ангидрид трифторметансульфокислоты (5,7 г, 21 ммоль) при 0oС. Реакционную смесь перемешивают при 0oС в течение ночи, затем разбавляют 200 мл CH2Cl2 с последующим промыванием водой и насыщенным солевым раствором, сушат над МgSO4. Сырое соединение после удаления растворителя хроматографируют на колонке с силикагелем, используя 50%-ный этилацетат в гексане, получая 4,95 г целевого соединения 25 (87%).
1Н-ЯМР (ДМСО-d6) δ 8,38 (д, 1Н, J=5,1 Гц), 7,39 (д, 1Н, J=5,1 Гц), 2,53 (с, 3Н).
Синтез 2-винилпиридинового соединения 26
Смесь трифлата (соль трифторметансульфокислоты) 2-гидрокси-3-нитро-4-метилпиридина 25 (7,74 г, 27,06 ммоль), трибутилвинилолова (10,3 г, 32,47 ммоль) и тетракис(трифенилфосфин)палладия (0) (1,56 г, 1,35 ммоль) в 100 мл безводного толуола нагревают при кипении с обратным холодильником в течение 3 часов, затем охлаждают до комнатной температуры и гасят реакцию, добавляя 20 мл насыщенного солевого раствора. Затем смесь экстрагируют этилацетатом (3х100 мл) и объединенные органические слои сушат над MgSO4. Сырой продукт после упаривания растворителя очищают колоночной хроматографией на силикагеле (гексан: этилацетат= 5:3), получая 2,82 г целевого соединения 26 (выход 70%).
1Н-ЯМР (ДМСО-d6) δ 8,51 (д, 1Н, J=5,1 Гц), 7,13 (д, 1Н, J=5,1 Гц), 6,66 (дд, 1Н, J=10,5 и 12,3 Гц), 6,55 (д, 1Н, J=1,8 Гц), 5,63 (дд, 1Н, J=10,5 и 1,8 Гц), 2,32 (с, 3Н).
HRMS, рассчитано для C8H8N2O2 164,0586, найдено 164,0586.
Синтез 2-карбоксальдегида 11 из 2-винилпиридинового соединения 26
Раствор олефина 26 (4,03 г, 24,54 ммоль) в 100 мл метанола подвергают озонолизу при -78oС и реакцию контролируют с помощью ТСХ. После завершения реакции избыток О3 удаляют, барботируя через реакционную смесь О2 при -78oС в течение 5 минут. Реакцию гасят (при -78oС) Me2S (10 мл) и полученную реакционную смесь перемешивают при комнатной температуре в течение ночи. Растворитель упаривают и остаток очищают хроматографией на силикагеле (гексаны: ЕtOАс=4:1 до гексаны: ЕtOАс=2:1), получая 3,51 г (96%) альдегида 11.
Синтез соединения 14 (3-АМР) из гидразонового соединения 27
Способ 1. К раствору SnC12•2H2O (2,256 г, 10 ммоль) в 6 мл этанола добавляют нитросоединение 27 (478 мг, 2 ммоль). Реакционную смесь нагревают при кипении с обратным холодильником в атмосфере N2 в течение ночи, охлаждают и фильтруют. Твердый продукт растворяют в 30 мл горячей воды и фильтруют. Фильтрат затем доводят до рН 7,5 насыщенным раствором NaHCO3 и перемешивают при комнатной температуре в течение 30 минут, фильтруют и промывают Н2О, С2Н5ОН и диэтиловым эфиром. Полученный желтый твердый продукт затем несколько раз экстрагируют ТГФ. Объединенные экстракты упаривают досуха, получая 227 мг 3-АМР 14. Первый этанольный фильтрат упаривают и остаток доводят до рН 8 насыщенным раствором NаНСО3, экстрагируют дважды ТГФ. Объединенные ТГФ-экстракты упаривают и твердый остаток промывают H2O, C2H5OH и диэтиловым эфиром. Для дополнительной очистки твердый продукт несколько раз экстрагируют ТГФ. Объединенные ТГФ-экстракты упаривают, получая дополнительную порцию 3-АМР (45 мг). Общий выход: 65% (272 мг).
Способ 2. Раствор нитросоединения 27 (120 мг, 0,5 ммоль) и Na2S (117 мг, 1,5 ммоль) в 6 мл смеси 1:1 H2O/C2H5OH нагревают при кипении с обратным холодильником в течение 3 часов в атмосфере N2. Растворитель концентрируют и затем доводят до рН 7 1Н раствором НСl, фильтруют и промывают Н2О, C2H5OH, CH2Cl2 и сушат в вакууме, получая 65 мг (62%) 3-АМР 14.
1Н-ЯМР (ДМСО-d6) δ 2,16 (с, 3Н), 6,16 (уш., 2Н), 6,99 (д, 1Н, J=4,4 Гц), 7,76 (д, 1Н, J=4,4 Гц), 7,93 (уш., 1Н), 8,17 (уш., 1Н), 8,34 (с, 1Н), 11,33 (с, 1Н).
FABMS, рассчитано для C8H11N5S 210, найдено 210 (MH+).
Следует понимать, что описанные выше примеры и воплощения приведены с целью подкрепления описания настоящего изобретения примерами и не должны рассматриваться как ограничивающие изобретение каким-либо образом. Различные модификации или изменения, которые могут быть сделаны в приведенном выше описании обычным специалистом в данной области, также рассматриваются в настоящем изобретении и должны быть включены в суть и объем данной заявки и последующей формулы изобретения.
Значения используемых сокращения представлены в таблице.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТИОСЕМИКАРБАЗОНЫ ПИРИДИН-2-КАРБОКСАЛЬДЕГИДОВ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1998 |
|
RU2199531C2 |
| 3-КАРБОКСАЛЬДЕГИДНЫЕ ПРОИЗВОДНЫЕ ХИНОЛИНА | 1995 |
|
RU2097379C1 |
| БИЦИКЛИЧЕСКИЕ ПИРИДИНЫ И АНАЛОГИ В КАЧЕСТВЕ МОДУЛЯТОРОВ СИРТУИНА | 2010 |
|
RU2550821C2 |
| СОЕДИНЕНИЯ (2-КАРБОКСИАМИДО)(3-АМИНО)ТИОФЕНА | 2004 |
|
RU2339633C2 |
| СТЕРЕОИЗОМЕРНО ОБОГАЩЕННЫЕ 3-АМИНОКАРБОНИЛЬНЫЕ БИЦИКЛОГЕПТЕНОВЫЕ ПИРИМИДИНДИАМИНЫ И ИХ ПРИМЕНЕНИЯ | 2005 |
|
RU2416604C2 |
| ПРОИЗВОДНЫЕ 3-АРИЛТИОИНДОЛ-2-КАРБОКСАМИДОВ И ИХ АНАЛОГИ КАК ИНГИБИТОРЫ КАЗЕИНКИНАЗЫ Iε | 2005 |
|
RU2391098C2 |
| ХИНАЗОЛИНОВЫЕ СОЕДИНЕНИЯ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2135481C1 |
| СПОСОБЫ ПОЛУЧЕНИЯ 2-(7-ХЛОР-1,8-НАФТИРИДИН-2-ИЛ)-3-(5-МЕТИЛ-2-ОКСОГЕКСИЛ)-1-ИЗОИНДОЛИНОНА | 2003 |
|
RU2318823C2 |
| АМИДЫ 3-ЗАМЕЩЕННОЙ 5- И 6-АМИНОАЛКИЛИНДОЛ-2-КАРБОНОВОЙ КИСЛОТЫ И РОДСТВЕННЫЕ АНАЛОГИ КАК ИНГИБИТОРЫ КАЗЕИНКИНАЗЫ IΕ | 2005 |
|
RU2369599C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ПИРИДИНА, ОБЛАДАЮЩИЕ АНТИ-HELICOBACTER PYLORI АКТИВНОСТЬЮ | 2007 |
|
RU2433126C2 |
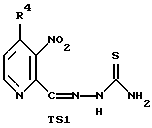
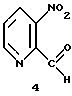
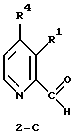
Изобретение относится к усовершенствованному способу получения соединений формулы (I), где R4 представляет Н или СН3, включающему взаимодействие соединения формулы 2-С, в котором R1 представляет группу NO2, с тиосемикарбазидом с получением тиосемикарбазона формулы ТS1, с последующим восстановлением полученного соединения. Предложен также способ получения тиосемикарбазона формулы TS2, включающий взаимодействие соединения 2-С, в котором R1 представляет группу NHP с тиосемикарбазидом. Предложены также способы получения промежуточных соединений 2-С, где R1 представляет NO2, NH2, NHP, NPP', N3 или CO2R2, P и Р' представляют защитные группы, R2 представляет C1-С3алкил, R4 представляет Н или СН3, и соединения формулы (4). Предлагаемые способы получения тиосемикарбазонов пиридин-2-карбоксальдегидов позволяют увеличить выход целевых продуктов до 30% из легко доступных исходных веществ. 4 н. и 17 з.п. ф-лы, 3 ил., 1 табл.
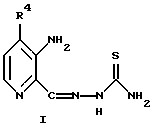
где R1 представляет NO2, NH2, NHP, NPP', N3 или CO2R2;
Р и Р' представляют защитные группы;
R2 представляет метил, этил, пропил или изопропил;
R4 представляет Н или СН3,
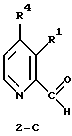
включающий реакцию винилирования соединения формулы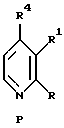
где R представляет Cl, Br, I, -O-S(О2)-СН3, -O-S(O2)-СF3 или -O-S(O2)-С6Н4-СН3-пара;
R1, R2, R4, Р и Р' определены выше, с получением соединения формулы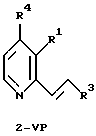
где R1, R2, R4, P и Р' определены выше;
R3 представляет H, C1-C20алкил, арил, замещенный арил или CO2R2;
с последующим озонолизом полученного соединения 2-VP с получением соединения 2-С.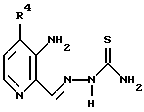
где R4 представляет Н или СН3,
включающий взаимодействие соединения формулы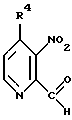
с тиосемикарбазидом с получением тиосемикарбазона формулы TS1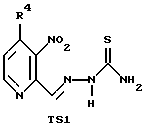
с последующим восстановлением полученного соединения TS1 для восстановления нитрогруппы до аминогруппы.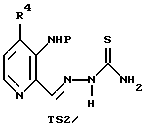
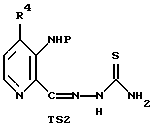
где R4 представляет Н или СН3;
Р представляет защитную группу,
включающий взаимодействие соединения формулы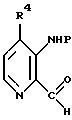
с тиосемикарбазидом с получением указанного выше тиосемикарбазона.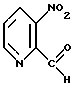
включающий взаимодействие соединения формулы 3: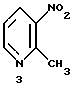
с диметилформамид-диалкилацеталем с образованием соединения формулы 3а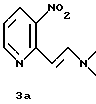
и последующее взаимодействие соединения 3а с окисляющим агентом с образованием соединения 4, при этом указанное соединение 4 выделяют по крайней мере с 75%-ным выходом из расчета на указанное соединение 3.
21. Способ по п.19, в котором указанный окисляющий агент представляет периодат натрия.
| LIU M.C | |||
| et al., Synthesis and Anti-Tumor Activity of Amino Derivatives of Pyridine-2-Carboxaldehyde Thiosemicarbazone, Journal of Medicinal Chemistry, 1992, v.35, № 20, р.3672-3677 | |||
| Способ получения производных 2-бензоил-3-амино-6-галоидпиридинов или их солей | 1974 |
|
SU597338A3 |
| ПРОИЗВОДНЫЕ ДИАМИНОТРИФТОРМЕТИЛПИРИДИНА ИЛИ ИХ СОЛИ СО ЩЕЛОЧНЫМИ ИЛИ ЩЕЛОЧНОЗЕМЕЛЬНЫМИ МЕТАЛЛАМИ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ИНГИБИРУЮЩАЯ ФОСФОЛИПАЗУ A | 1991 |
|
RU2057123C1 |

