Изобретение относится к области органической химии, конкретно к способу получения 2-метил-4(5)-нитроимидазола, являющегося промежуточным продуктом в синтезе лекарственных препаратов, в частности метронидазола.
Известным методом получения 2-метил-4(5)-нитроимидазола является нитрование 2-метилимидазола смесью концентрированных азотной и серной кислот при повышенных температурах.
Подобный метод синтеза затруднительно использовать в промышленном производстве из-за высокой коррозионной активности реакционной среды, невысокого выхода продукта и его низкого качества.
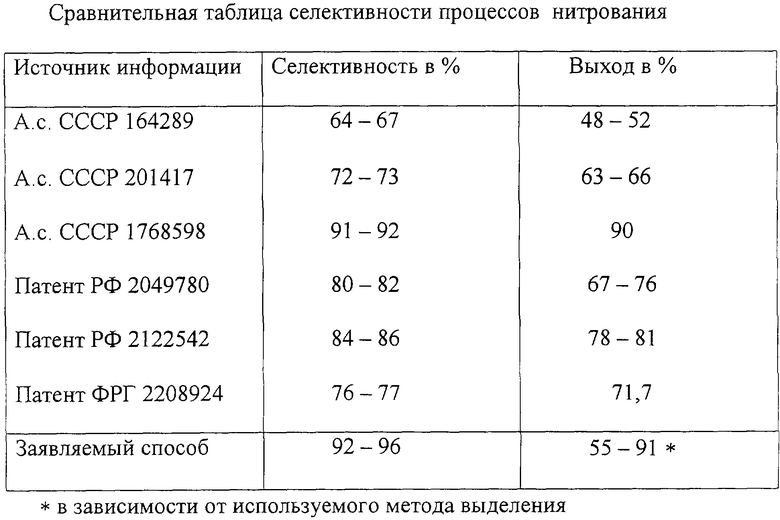
С целью повышения выхода и качества продукта в а.с. СССР 164289 (кл. 12р 9; 1964 г.) нитрование осуществляют азотнокислыми солями натрия или калия в 94-96%-ной серной кислоте при температуре 130-140oС в течение 4,5-5 часов. Получают продукт с выходом 48-52%.
Недостатками метода являются низкий выход продукта, повышенная температура синтеза, использование высококонцентрированной серной кислоты.
Для повышения выхода продукта и упрощения технологии его получения в а. с. СССР 201417 (C 07 D 49/36, 1967 г.) в смесь воды, 93%-ной серной кислоты, безводного сульфата натрия и 2-метилимидазола добавляют 99%-ную азотную кислоту, реакционную массу кипятят при 130-132oС в течение 4 часов. Получают продукт с выходом 63-66%.
Основные недостатки метода остаются прежними: использование в процессе высококонцентрированных кислот, высокая температура синтеза, низкий выход продукта.
В а. с. СССР 1768598 (C 07 D 233/92, 1990 г.) для снижения температуры нитрования авторы предлагают следующий вариант. В суспензию 2-метилимидазола в уксусной кислоте при 10oС добавляют 96%-ную серную кислоту, уксусный ангидрид, смесь выдерживают 30 мин при 60-65oС, охлаждают, добавляют 98%-ную азотную кислоту. Выдерживают 3-3,5 часа при 18-20oС, охлаждают, нейтрализуют 25%-ным водным раствором аммиака до рН 6,5-7, отфильтровывают, промывают водой, сушат. Получают продукт с выходом 90%.
Использование в процессе высококонцентрированных кислот, многокомпонентный состав реакционной среды, необходимость неоднократного охлаждения реакционной массы сильно усложняет аппаратурное оформление технологического процесса и утилизацию отходов.
В патенте РФ 2049780 (C 07 D 233/92, 1995 г.) нитрование проводят раствором нитрата аммония в 98%-ной азотной кислоте при 65-97oС в течение 1,5-6 ч при массовом соотношении реагентов на 1 мас.ч. имидазола: 1,5-5,4 нитрата аммония, 5,9-16,0 азотной кислоты, 0,10-0,35 воды. Выход продукта не превышает 76%.
Недостатками метода являются низкий выход продукта, повышенная температура синтеза, большой расход высококонцентрированной азотной кислоты.
По патенту РФ 2122542 (C 07 D 233/92, 1998 г.) нитрованию подвергают предварительно приготовленный при 25-30oС буферный раствор 2-алкилимидазола в концентрированной азотной кислоте, содержащей нитрат аммония. Для приготовления буферного раствора расходуется 50-60% расчетного количества азотной кислоты. Готовят буферный раствор, подавая составляющие компоненты дозирующими порциями. Стадию нитрования проводят в реакторе при 75-90oС концентрированной азотной кислотой в количестве 40-50% от расчетного в течение 3,5-4,5 часов. Продукт нейтрализуют щелочным агентом. Выход достигает 79-81%.
Повышенная температура синтеза, необходимость предварительного приготовления буферного раствора, порционное дозирование реагентов усложняют технологический процесс.
Наиболее близким по технической сущности является метод, заключающийся в нитровании 2-алкилзамещенного имидазола 64-72%-ной азотной кислотой в количестве 2-5 молей на 1 моль имидазола при 200-260oС в присутствии 0,5-5 молей 86-92%-ной серной кислоты на 1 моль азотной кислоты и 10-55% вес. мочевины (в расчете на имидазол). Выход 2-метил-4-нитроимидазола составляет 71,7%. Приводится описание специального реактора для нитрования (заявка ФРГ 2208924, C 07 D 233/92, 1977 г.).
Недостатками метода являются низкий выход продукта, неудовлетворительное качество продукта, высокая температура синтеза, высокая коррозионная активность реакционной среды, использование специфического оборудования.
Как видно из приведенных источников информации, для повышения выхода продукта, снижения температуры нитрования, упрощения технологического и аппаратурного оформления процесса исследователи идут по пути замены или нитрующего агента (азотной кислоты на ее соли: нитрат натрия, нитрат калия, нитрат аммония), или на изменение реакционной среды (нитрование азотной кислотой в присутствии водного бисульфата натрия, в присутствии уксусной кислоты и уксусного ангидрида), или полным исключением из процесса серной кислоты. Низкий выход продукта по указанным методам свидетельствует о протекании побочных окислительных реакций, т.е. низкой селективности процесса. Это заключение подтверждают исследования составов реакционных масс с помощью метода ЯМР на ядрах 13С. Анализы проводились на спектрометре СХР-100 и WP-80 с рабочей частотой на ядрах 1H= 90 МГц, 13C=22,63 МГц и 1H=80 МГц соответственно. Количество целевого продукта в реакционных массах определялось методом измерения интегральной интенсивности сигналов 13С ЯМР. Результаты анализов представлены в таблице.
Таким образом, указанные процессы протекают с недостаточно высокой селективностью. Различия показателей выхода объясняются различными методами выделения целевого продукта.
Целью изобретения является повышение селективности процесса, аппаратурное и технологическое упрощение процесса, снижение тепло- и энергозатрат.
Поставленная цель достигается нитрованием 2-метилимидазола нитрующей смесью, состоящей из 52-60% мас. серной кислоты, 25-30% мас. азотной кислоты, 15-18% мас. воды, при температуре 20-50oС. В ходе экспериментов авторы неожиданно установили, что процесс нитрования довольно гладко протекает при низких температурах с использовании серной и азотной кислот значительно меньших концентраций. Отличительными признаками изобретения являются: низкая температура нитрования (по прототипу 200-260oС), использование низкоконцентрированных серной и азотной кислот (по прототипу 86-92% серная и 64-72% азотная кислоты). Изобретение иллюстрируется следующими примерами.
Пример 1. В колбу емкостью 100 мл, снабженную мешалкой, термометром и водяной баней, загружают 62,0 г нитрующей смеси, имеющей следующий состав: H2SO4 - 56% мас., HNO3 - 28% мас., Н2O - 16% мас. При перемешивании к нитрующей смеси порциями добавляют 20,6 г (0,25 моля) 2-метилимидазола так, чтобы температура смеси не поднималась выше 25-30oС. После перемешивания в течение 3 часов температуру поднимают до 40oС и выдерживают еще 1 час. Реакционную массу охлаждают до комнатной температуры и оставляют стоять на 5-6 часов. Получают реакционную смесь, содержащую по данным анализа 30,1 г 2-метил-4(5)-нитроимидазола, что соответствует 96% селективности.
Пример 2. К 300 г нитрующей смеси состава: H2SO4 - 52% мас., HNO3 - 30% мас. , Н2O - 18% мас., при перемешивании и температуре не выше 30oС порциями добавляют 100 г (1,21 моль) 2-метилимидазола. Смесь перемешивают при этой температуре в течение 3 часов, затем температуру поднимают до 50oС и выдерживают в течение 1-1,5 час. Реакционную смесь охлаждают, выдерживают 2-3 часа. По данным анализа полученная реакционная смесь содержит в своем составе 144 г 2-метил-4(5)-нитроимидазола, что соответствует 93% селективности.
Пример 3. К 2,5 кг нитрующей смеси состава: H2SO4 - 60% мас., HNO3 - 25% мас. , Н2О - 15% мас., при перемешивании и температуре не выше 20-25oС порциями добавляют 410,6 г (5 моль) 2-метилимидазола. Смесь перемешивают при этой температуре в течение 3 часов, затем температуру поднимают до 50oС и выдерживают в течение 1-1,5 час. Реакционную смесь охлаждают, выдерживают 2-3 часа. По данным анализа полученная реакционная смесь содержит в своем составе 590 г 2-метил-4(5)-нитроимидазола, что соответствует 92% селективности.
Выход на выделенный продукт в зависимости от метода выделения составляет 55-91%. Температура плавления 252-258oС (разл.).
Техническим результатом изобретения является упрощение технологического и аппаратурного оформления процесса и снижение энерго- и теплозатрат за счет проведения синтеза при низких температурах, улучшение экологических условий и повышение техники безопасности труда за счет отказа от использования в процессе высококонцентрированных кислотных агентов, значительное увеличение селективности процесса.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ НИТРОВАНИЯ 2-МЕТИЛИМИДАЗОЛА | 2013 |
|
RU2523125C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2 -МЕТИЛ-4(5)-НИТРОИМИДАЗОЛА | 2012 |
|
RU2486177C1 |
| СПОСОБ ПОЛУЧЕНИЯ 4(5)-НИТРОИМИДАЗОЛА | 2016 |
|
RU2610267C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2',4,4'-ТРИНИТРОБЕНЗАНИЛИДА ИЗ АНИЛИНА И 4-НИТРОБЕНЗОЙНОЙ КИСЛОТЫ | 2014 |
|
RU2560881C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 2-R-4(5)-НИТРОИМИДАЗОЛОВ | 1992 |
|
RU2049780C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-R-4(5)-НИТРОИМИДАЗОЛОВ | 1997 |
|
RU2122542C1 |
| Способ получения цетаноповышающей присадки н-бутилнитрат | 2022 |
|
RU2780865C1 |
| Способ получения 2-метил-4/5/-нитроимидазола | 1990 |
|
SU1768598A1 |
| СПОСОБ ПОЛУЧЕНИЯ ДИХЛОР- ИЛИ ДИБРОМПИНАКОЛИНА | 2001 |
|
RU2206561C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИЗОПРЕНА | 2001 |
|
RU2184107C1 |
Изобретение относится к способу получения 2-метил-4(5)-нитроимидазола нитрованием 2-метилимидазола азотной кислотой в присутствии серной кислоты. Нитрование осуществляют при 20-50oС нитрующей смесью, состоящей из 52-60 мас. % серной кислоты, 25-30 мас.% азотной кислоты, 15-18 мас.% воды. Технический результат - повышение селективности процесса, аппаратурное и технологическое упрощение процесса, снижение тепло- и энергозатрат. Изобретение может быть использовано в фармацевтической промышленности с целью получения промежуточного продукта для синтеза лекарственного препарата метронидазола. 1 табл.
Способ получения 2-метил-4(5)-нитроимидазола нитрованием 2-метилимидазола азотной кислотой в присутствии серной кислоты, отличающийся тем, что нитрование осуществляют при 20-50oС нитрующей смесью, состоящей из 52-60 мас.% серной кислоты, 25-30 мас.% азотной кислоты, 15-18 мас.% воды.
| ВЫСАЖИВАЮЩИЙ АППАРАТ КАРТОФЕЛЕСАЖАЛКИ | 2001 |
|
RU2208924C2 |
| Демпфирующее устройство | 1986 |
|
SU1418538A1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-R-4(5)-НИТРОИМИДАЗОЛОВ | 1997 |
|
RU2122542C1 |

