Изобретение относится к производньм 1,7,7-триметилбицикло[2,2,1]гептана, способу их получения и применению указанных соединений в качестве фармацевтически активного ингредиента фармацевтических композиций.
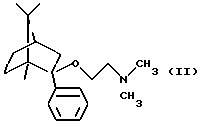
Известно, что (1R,2S,4R)-(-)-2-фенил-2-диметиламиноэтокси-1,7,7-триметилбицикло[2,2,1] гептан является соединением небензодиазепинового типа, усиливающим анксиолитическое действие. Международное непатентованное название (1R, 2S, 4R)-(-)-2-фенил-2-диметиламиноэтокси-1,7,7-триметилбицикло[2,2,1]гептана-дерамциклан (Венгерский патент 179164).
Целью настоящего изобретения являлось получение производных 1,7,7-триметилбицикло[2,2,1] гептана, имеющих структуру, подобную структуре (1R,2S, 4R)-(-)-2-фенил-2-диметиламиноэтокси-1,7,7-триметилбицикло-[2,2,1] гептана, но отличающихся от них способом взаимодействия с рецептором, механизмом действия и моделью анксиолитического теста на животных.
Указанная цель была достигнута в результате получения соединений настоящего изобретения.
Согласно одному аспекту настоящего изобретения был разработан способ получения соединения формулы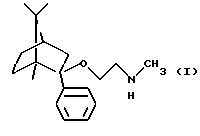
и его фармацевтически приемлемых солей присоединения кислот.
Согласно еще одному аспекту настоящего изобретения были получены фармацевтические композиции, включающие в качестве активного ингредиента соединение формулы I и его фармацевтически приемлемые соли присоединения кислот.
Согласно еще одному аспекту настоящего изобретения был разработан способ получения указанных фармацевтических композиций.
Согласно еще одному аспекту настоящего изобретения был разработан способ применения соединения формулы I и его фармацевтически приемлемых солей присоединения кислот в качестве активного ингредиента фармацевтических композиций, обладающих анксиолитическим действием.
Соединение формулы I и его фармацевтически приемлемые соли присоединения кислот обладают ценньми анксиолитическими свойствами.
Соединение формулы I может существовать в рацемической или оптически активной форме. Настоящее изобретение включает как рацемическую, так и оптически активную форму.
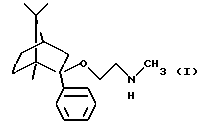
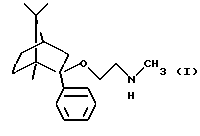
Согласно особо предпочтительной реализации настоящего изобретения был получен (1R, 2S, 4R)-(-)-2-фенил-2-метиламиноэтокси-1,7,7-триметилбицикло[2,2,1]-гептан и его фармацевтически приемлемые соли присоединения кислот.
Соединение формулы I и его фармацевтически приемлемые соли присоединения кислот могут применяться для анксиолитического лечения путем введения пациенту, нуждающемуся в таком лечении, фармацевтически активного количества соединения формулы I или его фармацевтически приемлемых солей присоединения кислот. В качестве активного ингредиента для такого лечения предпочтительно применять (1R, 2S, 4R)-(-)-2-фенил-2-метиламиноэтокси-1,7,7-триметилбицикло[2,2,1]гептан и его соли.
Фармацевтически приемлемые соли присоединения кислот соединения формулы I получают реакцией с неорганическими или органическими кислотами. Для получения, например, галогенидов водорода используют хлористоводородную или бромистоводородную кислоты; также можно использовать серную, азотную, фосфорную, уксусную, пропионовую, яблочную, молочную, малеиновую. фумаровую. винную, янтарную, метансульфокислоту, пара-толуолсульфокислоту и т.д. Особо предпочтительными солями являются соли фумаровой кислоты.
Согласно способу по настоящему изобретению соединение формулы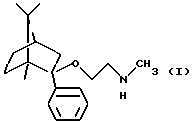
и его фармацевтически приемлемые соли присоединения кислот могут быть получены
а) деметилированием соединения формулы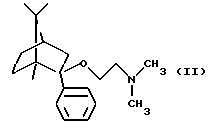
или
b) удалением защитной группы из соединения общей формулы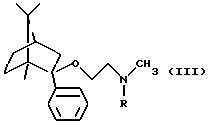
(где R является защитной группой);
и, если необходимо, разделением полученного таким образом рацемического соединения формулы I на оптически активные изомеры, и, если необходимо, превращением соединения формулы I в фармацевтически приемлемые соли присоединения кислот или выделением свободного основания из соли.
Согласно способу а) деметилирование можно предпочтительно проводить путем реакции соединения формулы II с соединением общей формулы
Hlg-COOR1 (IV)
(где R1 означает алкил или арил и Hlg означает галоген) и обработки полученного таким образом соединения общей формулы
(где R1 имеет значения, указанные выше) основанием.
В исходных соединениях общей формулы IV R1 является предпочтительно низшим алкилом с прямой или развлетвленной цепью с 1-4 атомами углерода или необязательно замещенным фенилом. Предпочтительно использовать соединения общей формулы IV, где R1 является метилом, этилом или фенилом, особенно предпочтительным является этилхлорформиат.
Реакцию соединения формулы II и хлорформиата общей формулы IV можно проводить в органическом растворителе. В качестве реакционной среды можно использовать предпочтительно ароматические углеводороды (например, бензол, толуол, ксилол). Реакцию можно проводить при нагревании, предпочтительно при температуре от 80 до 110oС, особенно предпочтительно при 80-85oС. Реакцию можно проводить, используя предпочтительно галогенформиат общей формулы IV, предпочтительно хлорформиат в 2-4 молярном избытке. Время реакции - несколько часов, предпочтительно 4-8 часов.
Реакция между соединениями формул II и IV приводит к образованию соединения общей формулы V (где R1 имеет значения, указанные выше). После окончания реакции реакционную смесь можно предпочтительно выпаривать и превращать соединение общей формулы V в соединение формулы I без выделения.
Соединение общей формулы V обрабатывают основанием. Для этого используют предпочтительно гидроксиды щелочных металлов (например, гидроксид натрия или гидроксид калия). Реакцию проводят в растворителе. В качестве реакционной среды можно использовать алифатические спирты (например, метанол, этанол и т.д.). Предпочтительно использовать этанол. Реакцию проводят при нагревании, предпочтительно с обратным холодильником. Время реакции 10-20 часов.
Реакцию можно проводить известным способом. Предпочтительно можно удалять осажденные неорганические соли путем фильтрации. выпаривать растворитель, растворять осадок в органическом растворителе (например, в галогенпроизводных углеводородов, например, дихлорэтане), экстрагировать органический раствор водой, выпаривать водный экстракт и фракционировать остаток в вакууме.
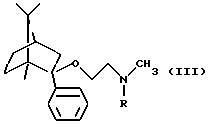
Согласно способу b) настоящего изобретения защитная группа удаляется из соединения общей формулы III. Защитной группой R может быть предпочтительно необязательно замещенный бензил, особенно предпочтительно бензил.
Бензильную группу можно удалять известным способом путем каталитического гидрирования. В качестве катализатора можно использовать предпочтительно палладий или платину, предпочтительно палладий на угле. Гидрирование можно проводить при нагревании, предпочтительно при 40-80oС. Восстановление можно проводить под давлением 1-50 бар, предпочтительно 5-10 бар. Гидрирование можно проводить в растворителе, предпочтительно в низшем спирте, особенно в этаноле.
Реакционную смесь можно обрабатывать известным способом. Например, можно отфильтровывать катализатор и выпаривать фильтрат.
Рацемическое соединение формулы I можно разделять на оптически активные изомеры. Разделение можно проводить известным способом. Таким образом, можно проводить реакцию рацемата формулы I с оптически активной кислотой (например, оптически активной винной кислотой, дитолуолвинной кислотой, камфарсульфокислотой и т.д.), разделяя полученные диастереометрические соли путем фракционной кристаллизации и выделяя из соли оптически активное свободное основание формулы I с помощью обработки основанием (например, гидроксидом щелочного металла). Также можно использовать физическое разделение (например, на хиральной колонке).
Соединение формулы I можно превращать в фармацевтически приемлемые соли присоединения кислот известными способами. Можно проводить реакцию соединения формулы I с соответствующей кислотой в подходящем растворителе в качестве среды, в результате чего осаждается полученная соль.
Согласно предпочтительной реализации настоящего изобретения (1R,2S, 4R)-(-)-2-фенил-2-метиламиноэтокси-1,7,7-триметилбицикло[2,2,1] гептан и его фармацевтически приемлемые соли присоединения кислот можно получить
a) деметилированием (1R,2S,4R)-(-)-2-фенил-2-диметиламиноэтокси-1,7,7-триметилбицикло[2,2,1]гептана, или
b) удалением бензильной группы из (1R,2S,4R)-(-)-2-бензилметиламиноэтокси-1,7,7-триметилбицикло[2,2,1]гептана, или
c) разделением рацемического (1R,2S,4R)-(-)-2-фенил-2- метиламиноэтокси-1,7,7-триметилбицикло[2,2,1] гептана формулы I на оптически активные изомеры;
и, если необходимо, превращением (1R,2S,4R)-(-)-2-фенил-2-метиламиноэтокси-1,7,7-триметилбицикло[2,2,1] гептана в его фармацевтически приемлемую соль присоединения кислоты или выделением свободного основания из соли.
Исходное соединение формулы II можно получить, как описано в Венгерском патенте 179164 или выложенной Венгерской заявке 5997/90 (Т/60996).
Исходные соединения формулы III можно получить реакцией фенилборнеола с амином общей формулы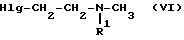
(где R1 имеет значения, указанные выше, и Hlg является галогеном).
Таким образом, например, исходное соединение формулы III, где R1 означает бензил, можно получить реакцией фенилборнеола с бензилметиламиноэтилхлоридом.
Соединения общей формулы IV имеются в продаже или могут быть получены известными способами.
Соединение формулы I и его фармацевтически приемлемые соли присоединения кислот обладают ценными анксиолитическими свойствами. Их терапевтический эффект может быть продемонстрирован следующими тестами.
В тестовых системах использовались следующие соединения:
Фумарат (1R, 2S,4R)-(-)-2-фенил-2-метиламиноэтокси-1,7,7-триметилбицикло[2,2,1]гептана (Соединение А);
Фумарат (1R, 2S, 4R)-(-)-2-фенил-2-диметиламиноэтокси-1,7,7-триметилбицикло[2,2,1]гептана (Соединение В).
Механизмы взаимодействия этих двух соединений с рецепторами значительно отличаются (Таблица 2).
Среди 5-НТ рецепторов Соединение А связывается только с 2С, а Соединение В не селективно к 2С. Более того, эти два соединения обладают различным сродством к другим группам рецепторов, например, Соединение В показывает значительное связывание с сигма рецепторами, а Соединение А - нет.
Также имеют место явные различия в механизме анксилитического действия этих двух соединений. Соединение А оказалось эффективным в тесте на поднятом плюс-лабиринте, а соединение В, что удивительно, не проявило никакого эффекта. Более того, Соединение В значительно противодействовало индуцированному mСРР чувству страха в тесте на крысах, а соединение А было совершенно не эффективно в дозе 3,0 мг/кг при внутрибрюшинном введении (Таблица 3). Эти различия были не предсказуемыми.
Другие различия были выявлены в тестах по проверке седативно-снотворного побочного действия этих соединений. Соединение А ингибировало самопроизвольную двигательную активность только в высоких дозах и усиливало наркоз, индуцированный гексабарбиталом, в меньшей степени по сравнению с Соединением В (Таблица 4).
Методы
Испытания по связыванию рецепторов
Для испытаний по связыванию рецепторов использовали различные области головного мозга самцов крыс Wistar весом 120-200 г, за исключением 5-НТ2С рецепторов, испытания которых проводили на хориоидальных сплетениях свиней. Содержание белка в мембранной фракции определяли, как описано Лоури (Lowry) [Lowry, O. H. , Rosenbrough, M.J., Farr, A.L. and Randall, R.Y.: J. Biol. Chem., 193: 265-275, 1951]. Результаты приведены в Таблице 1.
Поднятый плюс-лабиринт
Поднятый плюс-лабиринт состоял из двух открытых и двух закрытых каналов со стенкой 40 см одинакового размера (50х15 см), собранных в виде креста. Перегородки одного типа располагались напротив друг друга. Сочленение четырех каналов образовывало квадратную область в центре (15х15 см). Эта конструкция была изготовлена из дерева, поднята над полом на высоту 50 см и освещена слабым светом сверху.
За 60 минут до испытания самцам крыс Sprague-Dawley весом 220-260 г вводили тестируемые и контрольные вещества. Затем крыс помещали в квадратную область лабиринта и проводили испытание в течение 5 минут. Определяли следующие параметры:
- время, проведенное в открытых перегородках;
- время, проведенное в закрытых перегородках;
- количество входов в открытые перегородки;
- количество входов в закрытые перегородки.
Вещество считали эффективным, если наблюдалось существенное увеличение либо времени, проведенного в открытых каналах, либо количества входов в открытые каналы по сравнению с контрольной группой животных. Определение минимальных эффективных доз (МЭД) было основано на времени, проведенном в открытых каналах, для каждого исследуемого вещества (Таблица 2) [Pelow et.al., J. Neurosci. Methods, 14: 149-169, 1985].
Индуцированное тСРР чувство страха
Тесты проводили на самцах крыс Wistar весом 160-220 г по Кеннетту (Kennett) [Kennett, G.A., Whitton, P., Shan, К. аnd Curzon, G. Eur. J. Pharmacol. , 164: 445-454, 1989.]. Животным вводили либо тестируемое вещество, либо "наполнитель" (0,4% раствор метилцеллюлозы). Через 20 минут вводили подкожно либо mCCP (мета-хлорфенилпиперазин), либо физиологический раствор. Животных выдерживали в темноте еще 20 минут, затем помещали в аппарат, имеющий темное и светлое отделения (Omnitech, Digiscan, Model RXYZCM 16), и регистрировали их двигательную активность в течение 5 минут. Аппарат для тестирования состоял из одного темного и одного светлого отделений одинакового размера (39х20х29 см) с отверстием 8х8 для свободного прохода животных из одного отделения в другое. Светлое отделение освещалось красной лампой 40 Вт, расположенной на расстоянии 30 см над полом. Двигательную активность регистрировали по количеству пересечений животными инфракрасных лучей (16 лучей на расстоянии 2 см над полом и 16 лучей - 8 см над полом). Количество пересечений в светлом отделении считали мерой противодействия на индуцированное тСРР чувство страха. Данные статистически обрабатывали методом ANOVA, после чего проводили t-тест Даннета (Dunnet). Значения МЭД для двух соединений приведены в Таблице 3.
Ингибирование самопроизвольной двигательной активности
Самопроизвольную двигательную активность измеряли, как описано ранее [Borsy et. al., Arch. Int. Pharmacodyn. 124:1 - 1960.], в аппарате с 10 отделениями (Dews) по 3-3 мыши в каждом отделении. Мышам вводили либо тестируемое вещество, либо "наполнитель" за 60 минут перед тестом. Подсчитывали количество пересечений инфракрасных лучей. Значения ID50 рассчитывали с помощью анализа линейной регрессии (Таблица 4).
Усиление наркоза, индуцированного гексабарбиталом
Самцам мышей NMRI весом 20-25 г вводили перорально либо тестируемое вещество, либо "наполнитель" в количестве 20 мл/кг за 60 минут до внутривенного введения 40 мг/кг (10 мл/кг) гексабарбитала. Спящих мышей помещали на ровную поверхность на левый бок и регистрировали точное время сна и пробуждения. Животные считались проснувшимися, если они переворачивались с левого бока.
Время сна было в 2,5 раза больше, чем среднее в контрольной группе, что являлось критерием усиления действия наркоза (метод ограничения), результаты выражали в процентах увеличения по сравнению с контрольными значениями. Значения ED50 рассчитывали из кривых ответа на дозу по методу Litchfield-Wilcoxon. Значения ED50 показаны в Таблице 4.
Представленные результаты ясно показывают, что Соединение А значительно отличается от Соединения В, используемого в качестве контрольного вещества, как способом взаимодействия с рецептором (механизмом действия), так и по моделям чувства страха на животных. Описанное здесь действие соединения А существенно отличается от действия соединения В, что является удивительным, если принять во внимание подобие этих соединений.
Согласно настоящему изобретению были получены фармацевтические композиции, включающие в качестве активного ингредиента соединение формулы I или его фармацевтически приемлемые соли присоединения кислот. Предпочтительно использовать в качестве активного ингредиента (1R,2S,4R)-(-)-2-фенил-2-метиламиноэтокси-1,7,7-триметилбицикло[2,2,1] гептан или его фармацевтически приемлемую соль присоединения кислоты - особенно его фумарат.
Фармацевтические композиции по настоящему изобретению подходят для орального (например, в виде таблеток, таблеток в оболочке, твердых или мягких желатиновых капсул, растворов, суспензий, сиропов); парэнтерального (например, в виде подкожных, внутримышечных или внутривенных инъекций); ректального (например, в виде суппозиториев) или назального (например, в виде спрея, аэрозоля) введения. Активный ингредиент может мгновенно растворяться, поэтому период действия фармацевтической композиции будет определяться только периодом действия самого активного ингредиента. Также могут быть получены длительно растворяющиеся композиции, в этом случае период действия также будет зависеть от формы и компонентов фармацевтической композиции.
Фармацевтические композиции по настоящему изобретению могут быть приготовлены традиционными способами фармацевтической промышленности.
Таблетки или капсулы могут содержать в качестве наполнителя различные виды лактозы (моногидрат, безводную, высушенную), маннита или целлюлозы (высушенной, микрокристаллической). В качестве связующего агента можно использовать, например, желатин, поливинилпирролидон (молекулярный вес может быть различным), различные виды эфиров целлюлозы (например, гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, этилцеллюлозу и т.д.), гидролизованный крахмал, различные растительные смолы (например, гуммиарабик, смолу хьюара ит.д.) в водных растворах алифатических спиртов с 1-4 атомами углерода или их смесях. В качестве расщепляющего агента можно использовать, например, различные виды крахмала (картофельного, кукурузного, пшеничного) и так называемые супер-дезинтегранты, например карбоксиметилцеллюлозу (торговое название Ац-дисол (Ac-disol)), натриевый карбоксиметилкрахмал (Примойел) (Primojel), Ультраамилопектин, Экспло-Таб (Explo-Tab)), поливинилпирролидон (торговое название Полипласдон (Poliplasdone)) и т.д. В качестве вспомогательного вещества для улучшения текучести можно использовать стеараты щелочноземельных металлов (например, стеарат магния, стеарат кальция), жирные кислоты (например, стеариновую кислоту), глицериды (например, торговые названия - Прецирол (Precirol), Кутина Н (Cutina H)) парафиновое масло, силиконовые масла, эмульсии силиконовых масел, тальк или кремниевую кислоту.
Таблетирование и капсулирование можно проводить методом сухого или влажного гранулирования либо простой гомогенизацией порошка.
Длительно растворяющиеся твердые фармацевтические композиции могут быть получены любым подходящим способом. Таким образом, можно приготовить твердые таблетки, используя в качестве замедляющего растворение агента гидрофильные полимеры (например, гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу, карбоксиметилцеллюлозу, производные полиакриловой кислоты), полисахарозу (смолу хьюара, ксантановую смолу) или их смеси, либо гидрофобные полимеры (например, этилцеллюлозу, сополимеры эфиров метакриловой кислоты, поливинилацетат, поливинилбутират, и т.д. или их смеси). Также растворение активного ингредиента можно замедлить использованием смесей гидрофильных и гидрофобных полимеров или смесей полимера и жирсодержащего вещества. Твердые таблетки также можно получить в форме многослойных таблеток, в которых активные ингредиенты введены в различные слои, с помощью этого метода можно наилучшим образом сочетать механизм растворения с индивидуальными фармакокинетическими характеристиками активных ингредиентов.
Соединение формулы I и его фармацевтически приемлемые соли присоединения кислот также можно использовать в форме длительно растворяющихся драже в оболочке. Такие драже можно приготовить отдельно из каждого активного ингредиента или из смеси активных ингредиентов. Драже можно приготовить с помощью методов экструзионной грануляции, роторной грануляции или с помощью нанесения на драже плацебо. Драже можно покрыть оболочкой, используя роторную аппаратуру или оборудование с псевдоожиженным слоем. Для получения оболочки можно использовать растворы водонерастворимых полимеров в органических растворителях (предпочтительно, в C1-3 алифатических спиртах и/или C1-2 полихлорированных углеводородах, и/или ацетоне, и/или этилацетате) или водные дисперсии.
Активные ингредиенты по настоящему изобретению можно использовать в форме осмотических или диффузионно-осмотических композиций. Такие композиции можно получить путем приготовления таблеток, содержащих активный ингредиент и гидрофильные полимеры (например, гидроксипропилметилцеллюлозу), покрытия этих таблеток оболочкой, являющейся полупроницаемой (например, из ацетата целлюлозы) или проницаемой (например, из сополимера аминометакрилата) по отношению к активному ингредиенту, с помощью известных методов и создания в этой оболочке отверстий, через которые активный ингредиент может проникать в водную среду под действием осмотического давления.
Путем соответствующих способов приготовления длительно растворяющихся композиций можно регулировать скорость растворения активного ингредиента таким образом, чтобы in vitro по крайней мере 80% активного ингредиента растворялось за 2-24 часа (измерения проводят в соответствии с методами, описанными в Фармакопее).
Доза соединения формулы I может варьироваться в широких пределах, ее величина определяется в каждом конкретном случае, принимая во внимание состояние и вес тела пациента, серьезность заболевания, схему введения и т.д. В общем случае оральная дневная доза составляет около 0,01-1,0 мг/кг, предпочтительно 0,05-0,5 мг/кг.
Дополнительные детали настоящего изобретения описаны в Примерах, причем эти Примеры не ограничивают объем охраны изобретения.
Пример 1
Получение (1R, 2S, 4R)-(-)-2-фенил-2-метиламиноэтокси-1,7,7-триметилбицикло[2,2,1]гептана
К раствору 57,14 г (0,19 моль) (1R,2S,4R)-(-)-2-фенил-2-диметиламиноэтокси-1,7,7-триметилбицикло[2,2,1] гептана в 150 мл безводного толуола добавляли по каплям 61,8 г (0,57 моль) этилхлорфомиата при 80-85oС в течение 1,5 часов. Реакционную смесь нагревали при 80-85oС в течение 6 часов, затем охлаждали до 20oС, промывали водой, сушили и выпаривали.
Полученный маслянистый продукт (58,5 г) растворяли в 60 мл этанола, затем этот раствор добавляли по каплям при 50oС к раствору 72,5 г (1,29 моль) гидроксида калия в 270 мл 96% этанола. Реакционную смесь кипятили с обратным холодильником в течение 20 часов. Осажденный продукт отфильтровывали при 20oС, фильтрат выпаривали. Кубовый остаток растворяли в 100 мл дихлорэтана, раствор экстрагировали водой, сушили и выпаривали. Осадок фракционировали в вакууме. Таким образом было получено 27,09 г нужного соединения в виде желтого масла с выходом 49,6% и температурой кипения 130oС/25 Па.
Н-ЯМР: ЯМР (CDCl3)
δ 0,60-0,80 [m, 1H, С(6)-Н(ах)]; 0,88-0,90 [ss, 6H, 2хСН3],
1,17 [s, 3H, -СН3], 1,00-1,30 [m, 2H, C(5)-H(ax), C(6)-H(eq)];
1,53 [s, 1H, -NH];
1,6-1,70 [m, 1H, C(5)-H(eq)]; 1,86 [t, J=4,3, C(4)-H], 2,00 [d, 1H, J= 13,8, C(3)-H(ax)]; 2,25 [dt, 1H, J=13,3 J=3,9, C(3)-H(eq)];
2,42 [s, 3H, -N-СН3]; 2,50-2,75 [m, 2H, -N-CH2-]; 2,80-2,90 [m, 1H -O-CH2(1)] ; 3,25-3,35 [m, 1H, -O-СН2(2)-1]; 7,20-7,40 [m, 4H, Ph-H]; 7,55 [d, 1H, J=7,5, Ph-H].
Соль фумаровой кислоты получали путем добавления 11,5 г (0,04 моль) (1R, 2S, 4R)-(-)-2-фенил-2-метиламиноэтокси-1,7,7-триметилбицикло[2,2,1]гептана к кипящему раствору 4,64 г (0,04 моль) фумаровой кислоты в 50 мл безводного этанола. Осажденный кристаллический продукт отфильтровывали, промывали этанолом и сушили. Полученный (1R,2S,4R)-(-)-2-фенил-2-метиламиноэтокси-1,7,7-триметилбицикло[2,2,1] гептан-(Е)-бутендиоат (1/1) имел температуру плавления 178-180oС.
Анализ соединения формулы: C23H33NO5 (403,52)
Расчетные значения: C 68,46%; H 8,24%; N 3,47%.
Определенные значения: C 68,15%; H-8,08%; N 3,52%.
[α]
Пример 2
Получение (1R, 2S,4R)-(-)-2-фенил-2-бензилметиламиноэтокси-1,7,7-триметилбицикло[2,2,1]гептана
2,0 г (8,68 моль) (-)-фенилборнеола кипятили в толуоле с 0,53 г (11 милимоль) 50%-ного гидрида натрия, затем добавляли 46,16% раствор 3,9 г (9,8 милимоль) бензилметиламиноэтилхлорида в толуоле при температуре кипения и кипятили с обратным холодильником в течение 3 часов. Реакционную смесь промывали водой, сушили и выпаривали. Кубовый остаток очищали колоночной хроматографией (элюент: гексан: этилацетат 10:1). Таким образом было получено 2,6 г нужного соединения с выходом 79,5%. Оксалат (1/1) имел температуру плавления 194-196oС (из этанола). Анализ соединения формулы: C28H37NO5 (467,59)
Расчетные значения: C 71,92%; H 7,98%; N 2,99%.
Определенные значения: C 72,08%; H 7,83%; N 3,08%.
Получение (1R, 2S, 4R)-(-)-2-фенил-2-метиламиноэтокси-1,7,7-триметилбицикло[2,2,1]гептана
0,4 г (1,06 милимоль) (1R,2S,4R)-(-)-2-фенил-2-бензилметиламиноэтокси-1,7,7-триметилбицикло[2,2,1] гептана гидрировали в этаноле в присутствии 5%-ного катализатора палладия на угле при 60oС и давлении 10 бар в течение 6 часов. Реакционную смесь отфильтровывали, фильтрат выпаривали. Таким образом было получено 0,26 г нужного соединения в виде бесцветного масла с выходом 85,2%. Соль фумаровой кислоты получали, как описано в Примере 1. Температура плавления 2-(Е)-бутендиоата (1/1) 179-180oС (из этанола).
Анализ соединения формулы: С23Н33NO5 (403,52)
Расчетные значения: C 68,46%; H 8,24%; N 3,47%.
Определенные значения: C 68,50%; H 8,18%; N 3,42%.
Пример 3
Получение таблеток
6 весовых частей фумарата (1R,2S,4R)-(-)-2-фенил-2-метиламиноэтокси-1,7,7-триметилбицикло[2,2,1]гептана смешивали с 9 весовыми частями лактозы и 3 весовыми частями микрокристаллической целлюлозы. Полученную порошкообразную смесь гранулировали с раствором из 0,5 весовых частей поливинилпирролидона и 4 весовых частей воды, очищенной на ионообменнике, в аппаратуре для гранулирования с распылением в псевдоожиженном слое. К высушенным гранулам добавляли 1,3 весовых части карбоксиметилцеллюлозы и 0,2 весовых части стеарата магния, гранулы просеивали через 1,00 мм сито. Полученные таким образом гранулы прессовали на роторной машине для таблетирования, используя форму с диаметром 8 мм, в таблетки, имеющие среднюю массу 200 мг. Полученные таким образом таблетки массой 200 мг имели содержание активного ингредиента 60 мг.
Пример 4
Получение капсул, покрытых оболочкой
Гранулы, полученные, как описано в примере 3, помещали в желатиновые капсулы размером 2.
Пример 5
Получение длительно растворяющихся таблеток
10 весовых частей фумарата (1R,2S,4R)-(-)-2-фенил-2-метиламиноэтокси-1,7,7-триметилбицикло[2,2,1] гептана смешивали с 9 весовыми частями гидроксипропилметилцеллюлозы (Methocel К 4М, производства Clorcon Ltd.) и 10 весовыми частями лактозы. Полученную порошкообразную смесь гранулировали с раствором из 0,4 весовых частей поливинилпирролидона и 4 весовых частей изопропанола в аппаратуре для турбулентного гранулирования. К высушенным гранулам добавляли 0,3 весовых части талька и 0,3 весовых части стеарата магния, гранулы просеивали через 1,00 мм сито. Полученные таким образом гранулы прессовали в таблетки массой 300 мг, имеющие содержание активного ингредиента 100 мг, на роторной машине для таблетирования, используя форму в виде чечевицы с диаметром 10 мм.
Пример 6
Получение суппозиториев
7 весовых частей фумарата (1R,2S,4R)-(-)-2-фенил-2-метиламиноэтокси-1,7,7-триметилбицикло[2,2,1] гептана диспергировали с 53 весовыми частями Witерsol S 58 основы для суппозиториев с температурой плавления 50oС. Еще жидкую суспензию помещали в формы для суппозиториев и отверждали охлаждением до 25oС, затем суппозитории вынимали из форм. Таким образом были получены суппозитории массой 6 г и содержанием активного ингредиента 20 мг.
Соединение формулы I подпадает под общую структурную формулу соединений, описанных в ЕР 694299. Однако ни в указанной публикации, ни в других источниках информации не были представлены его физические константы, пригодные для идентификации. При этом предлагаемые согласно изобретению соединения обладают новыми и неочевидными свойствами - селективно связываться с 5-НТ2C рецепторами с меньшими побочньми эффектами.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНИЛАЛКИЛТИОПИРИМИДИНА, СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И СПОСОБ ПОЛУЧЕНИЯ | 2000 |
|
RU2245334C2 |
| ПРОИЗВОДНЫЕ 1,3-ДИОКСОЛ/4,5-h//2,3/БЕНЗОДИАЗЕПИНА, ЯВЛЯЮЩИЕСЯ ИНГИБИТОРАМИ AMPA/КАИНАТНОГО РЕЦЕПТОРА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, СПОСОБ ЛЕЧЕНИЯ | 1998 |
|
RU2208014C2 |
| ПРОИЗВОДНЫЕ 8-ЗАМЕЩЕННОГО-9H-1,3-ДИОКСОЛ/4,5-h//2,3/БЕНЗОДИАЗЕПИНА, ЯВЛЯЮЩИЕСЯ ИНГИБИТОРАМИ AMPA/КАИНАТНОГО РЕЦЕПТОРА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ | 1998 |
|
RU2208013C2 |
| ПРОИЗВОДНЫЕ 1-[2-(ЗАМЕЩЕННЫЙ ВИНИЛ)]-5Н-2,3-БЕНЗОДИАЗЕПИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ, ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ И СПОСОБ ЕГО ПОЛУЧЕНИЯ, СПОСОБ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ ЦЕНТРАЛЬНОЙ НЕРВНОЙ СИСТЕМЫ | 1996 |
|
RU2161607C2 |
| ПРОИЗВОДНОЕ 2-(1,2,4-ТРИАЗОЛ-1-ИЛ)-1,3,4-ТИАДИАЗОЛА, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ | 1998 |
|
RU2180903C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ 2,3-БЕНЗОДИАЗЕПИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ | 2000 |
|
RU2243228C2 |
| ИНГИБИТОРЫ ФЕРМЕНТА ДИАЦИЛГЛИЦЕРИН-О-АЦИЛТРАНСФЕРАЗЫ ТИПА 1 | 2008 |
|
RU2486186C2 |
| СОЛИ АРИПИПРАЗОЛА | 2006 |
|
RU2384572C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФЛУОКСЕТИНА | 1997 |
|
RU2173679C2 |
| ПРОИЗВОДНЫЕ 3(2Н)-ПИРИДАЗИНОНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЛЕЧЕНИЯ | 1992 |
|
RU2130019C1 |
Изобретение относится к применению 2-фенил-2-метиламиноэтокси-1,7,7-триметилбицикло[2,2,1] гептана формулы I, его оптически активных изомеров и фармацевтически приемлемых солей, способных селективно связываться с 5-HT2c-рецепторами, в фармацевтических композициях, обладающих анксиолитическим действием. Указанные соединения получают деметилированием соединения формулы II реакцией с Hlg-COOR1, где Hlg - галоген, R1 - алкил или арил, с получением соединения формулы V, которое обрабатывают основанием либо удалением защитной группы R из соединения формулы III и, если необходимо, разделяют полученный рацемат на оптические изомеры, и/или превращают в фармацевтически приемлемую соль, или из соли выделяют основание. Фармацевтическая композиция содержит указанные выше соединения с подходящими инертными твердыми или жидкими наполнителями или вспомогательными агентами. Композицию получают смешением указанных компонентов и, если необходимо, переводят полученную композицию в форму, пригодную для применения. 5 с. и 10 з.п. ф-лы, 4 табл.

его оптически активных изомеров или его фармацевтически приемлемых солей присоединения кислот в фармацевтических композициях, обладающих анксиолитическим действием, в качестве активного ингредиента, способного селективно связываться с 5-НТ2C-рецепторами.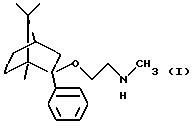
его оптически активных изомеров или его фармацевтически приемлемых солей присоединения кислот деметилированием соединения формулы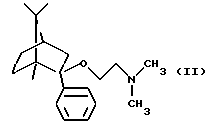
включающим реакцию соединения формулы II с соединением общей формулы
Hlg-COOR1,
где R1 - алкил или арил;
Hlg - галоген,
и обработку полученного таким образом соединения общей формулы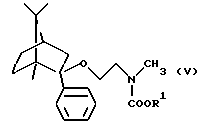
где R1 имеет значения, указанные выше,
основанием, и, если необходимо, разделением полученного таким образом рацемического соединения формулы I на оптически активные изомеры и, если необходимо, превращением соединения формулы I в его фармацевтически приемлемую соль присоединения кислоты или выделением свободного основания из соли.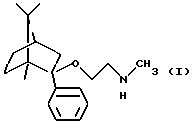
его оптически активных изомеров или его фармацевтически приемлемых солей присоединения кислот, включающий удаление защитной группы из соединения общей формулы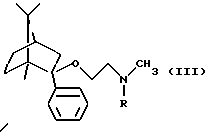
где R является защитной группой,
и, если необходимо, разделение полученного таким образом рацемического соединения формулы I на оптически активные изомеры, и, если необходимо, превращение соединения формулы I в его фармацевтически приемлемую соль присоединения кислоты или выделение свободного основания из соли.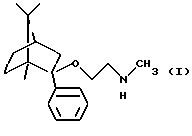
его оптически активный изомер или его фармацевтически приемлемую соль присоединения кислоты, способные селективно связываться с 5-НТ2С-рецепторами, в смеси с подходящими инертными твердыми или жидкими наполнителями или вспомогательными агентами.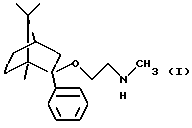
или его оптически активного изомера, или его фармацевтически приемлемой соли присоединения кислоты с подходящими инертными твердыми, или жидкими наполнителями, или вспомогательными веществами и при необходимости, превращение этой смеси в форму, пригодную для применения.
| Устройство для разделения профильного материала | 1977 |
|
SU694299A1 |
| US 4342762 A.03.08.1982 | |||
| УСТРОЙСТВО ДЛЯ ЗАБОРА ВЫСОКОТЕМПЕРАТУРНЫХ ТОПОЧНЫХ ГАЗОВ | 1994 |
|
RU2065122C1 |
| РАДИАЦИОННЫЙ БАЛАНСОМЕР | 0 |
|
SU212574A1 |
| Способ получения 2-алкил(фенил) аминометилнорборнана | 1976 |
|
SU598873A1 |

