Изобретение относится к медицине, точнее - к фармакологии, конкретно - к синтетическим биологически активным соединениям гетероциклического ряда, обладающим противовирусной, иммуностимулирующей (интерферониндуцирующей), антихламидийной, антиагрегационной, гепатопротекторной и антимикробной активностями.
Соединения предназначены, в основном, для использования в медицинской практике для лечения вирусных инфекций, инфекций, вызванных хламидиями, заболеваний, сопровождающихся иммунодефицитом, в частности злокачественных новообразований, а также туберкулеза. Кроме того, указанные соединения могут быть использованы для тех же целей в ветеринарии.
Уровень техники
Как известно, одну из наиболее серьезных проблем современной медицины представляют микробные и вирусные заболевания, многие из которых крайне плохо поддаются лечению, что связано как с недостаточной эффективностью существующих препаратов, так и быстрой изменчивостью микробов, приводящей к появлению устойчивых форм [1, 2].
Часто вирусные заболевания протекают на фоне снижения активности иммунной системы организма и сопровождаются вторичными инфекциями, это же относится и к онкологическим заболеваниям. Поэтому проблема разработки эффективных противовирусных и противоопухолевых препаратов тесно связана с поиском средств лечения иммунодефицитных состояний различного происхождения.
Известные противовирусные препараты можно условно разделить на 2 группы по типам механизмов их действия. Действие препаратов первой группы связано с подавлением репродукции вирусов в организме [1]. Противовирусные препараты второй группы оказывают эффект не столько за счет воздействия на сами вирусы, сколько за счет стимуляции иммунной защиты организма и усиления выработки эндогенных интерферонов [3] . Данное изобретение относится ко второй группе. Интерфероны и их индукторы используются также для лечения ряда опухолевых заболеваний [4].
Химическими аналогами заявляемых соединений являются арил-гетерилсульфамиды - этазол, диакарб, сульфадимезин и бутазоламид. Рассмотрим их подробнее.
Этазол-2-(4-Ами-обензолсульфамидо)-5-этил-1,3,4-тиадиазол.
Этазол известен антибактериальной активностью в отношении стрептококков, пневмококков, менингококков, гонококков, кишечной палочки, патогенных анаэробных микрорганизмов. Он описан в работе [5].
Диакарб-2-Ацетиламино-1,3,4-тиадиазол-5-сульфамид - имеет нижеприведенную структурную химическую формулу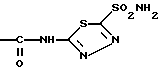
Диакарб применяется в качестве диуретического средства, а также для лечения глаукомы. Он также описан в работе [5]. Сульфадимезин - 2-(4-Аминобензолсульфамидо)-4,6-диметилпиримидин - имеет нижеприведенную структурную химическую формулу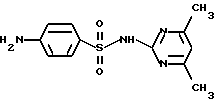
Сульфадимезин применяется при пневмококковых, стрептококковых, менингококковых инфекциях, сепсисе, гонорее, а также при инфекциях, вызванных кишечной палочкой и другими микроорганизмами. Он также описан в [5].
Наиболее близким аналогом - и по химической природе, и по назначению -является Бутазоламид-2-Бутириламино-1,3,4-тиадиазол-5-сульфамид. Он имеет нижеприведенную структурную химическую формулу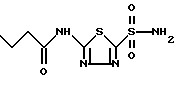
Бутазоламид применяют только в качестве антибактериального средства. Он подробно описан в работе [6].
К сожалению, спектр его биологической активности сравнительно узок, к тому же уже появились бактерии, устойчивые к нему.
Задача изобретения
Задачей изобретения является получение новых химических соединений, обладающих широкой биологической активностью, в том числе противовирусной активностью (по отношению к вирусам простого герпеса), иммуностимулирующей активностью (за счет индукции выработки эндогенных интерферонов в организме), антихламидийной, гепатопротекторной, антиагрегационной и антимикробной активностью. Другими словами, задача изобретения сводится к химическому синтезу биологически активных веществ, превосходящих прототип по противовирусной, иммуностимулирующей, противомикробной активностям, а также по широте действия.
Сущность изобретения
Поставленная задача решается путем синтеза новых соединений Арил- и гетериламидов карбоалкоксисульфаниловых кислот общей формулы (1)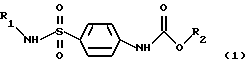
где R1 выбран из группы арилов и гетерилов, R2 выбран из группы алкилов.
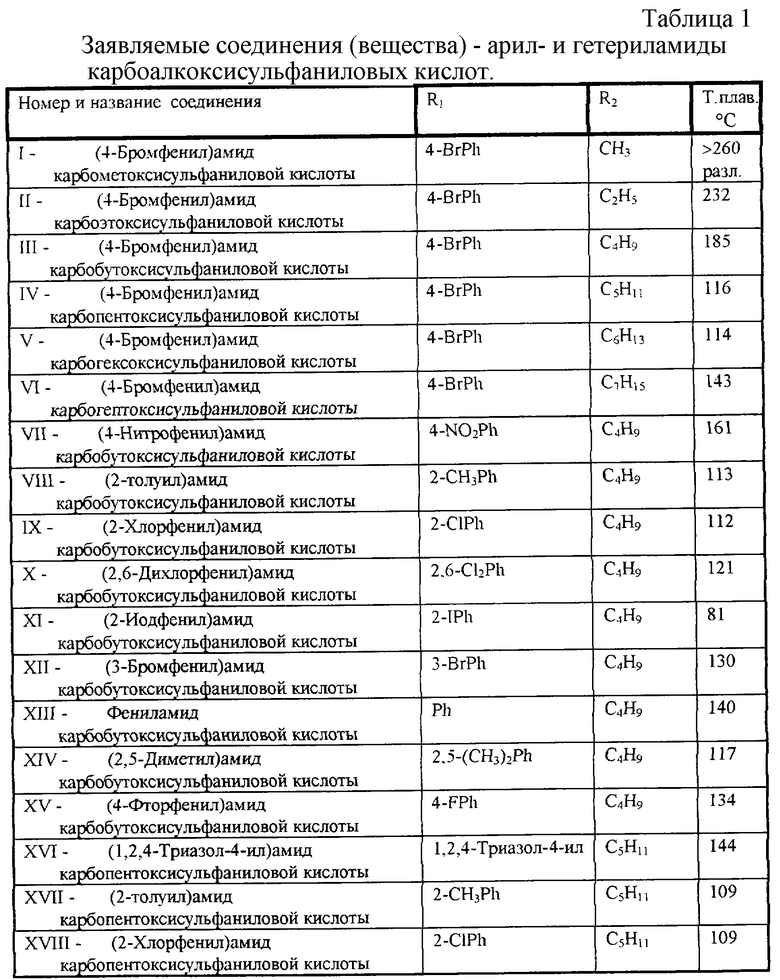
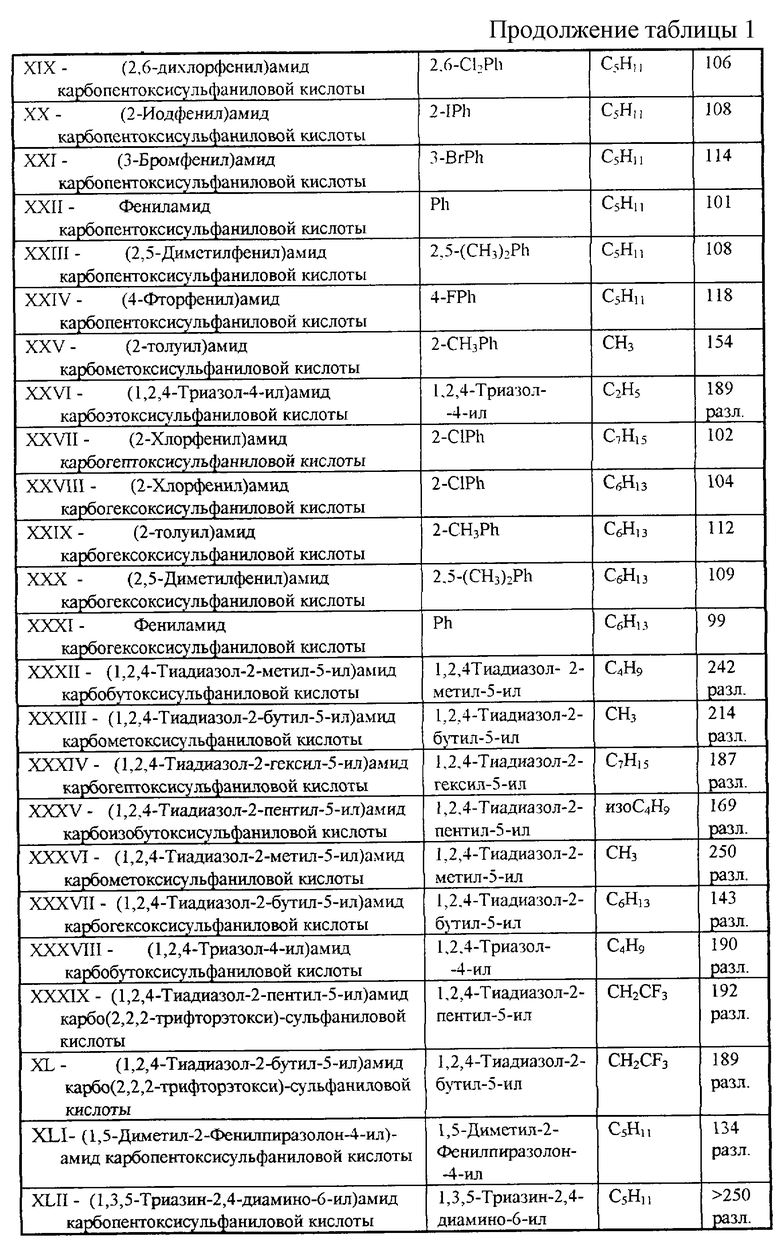
Перечень синтезированных и испытанных нами арил- и гетериламидов карбоалкоксисульфаниловых кислот приведен в Таблице 1.
Заявленные вещества новы, поскольку они неизвестны из доступных источников информации.
Наличие широкого спектра эффективной биологической активности заявленных веществ не вытекает явным образом из предшествующего уровня техники, т.е. неочевидно для специалиста.
Раскрытие изобретения.
Сущность изобретения поясняют приведенные далее:
Общий способ получения заявляемых соединений;
Пример синтеза (4-бромфенил)амида карбометоксисульфаниловой кислоты (I);
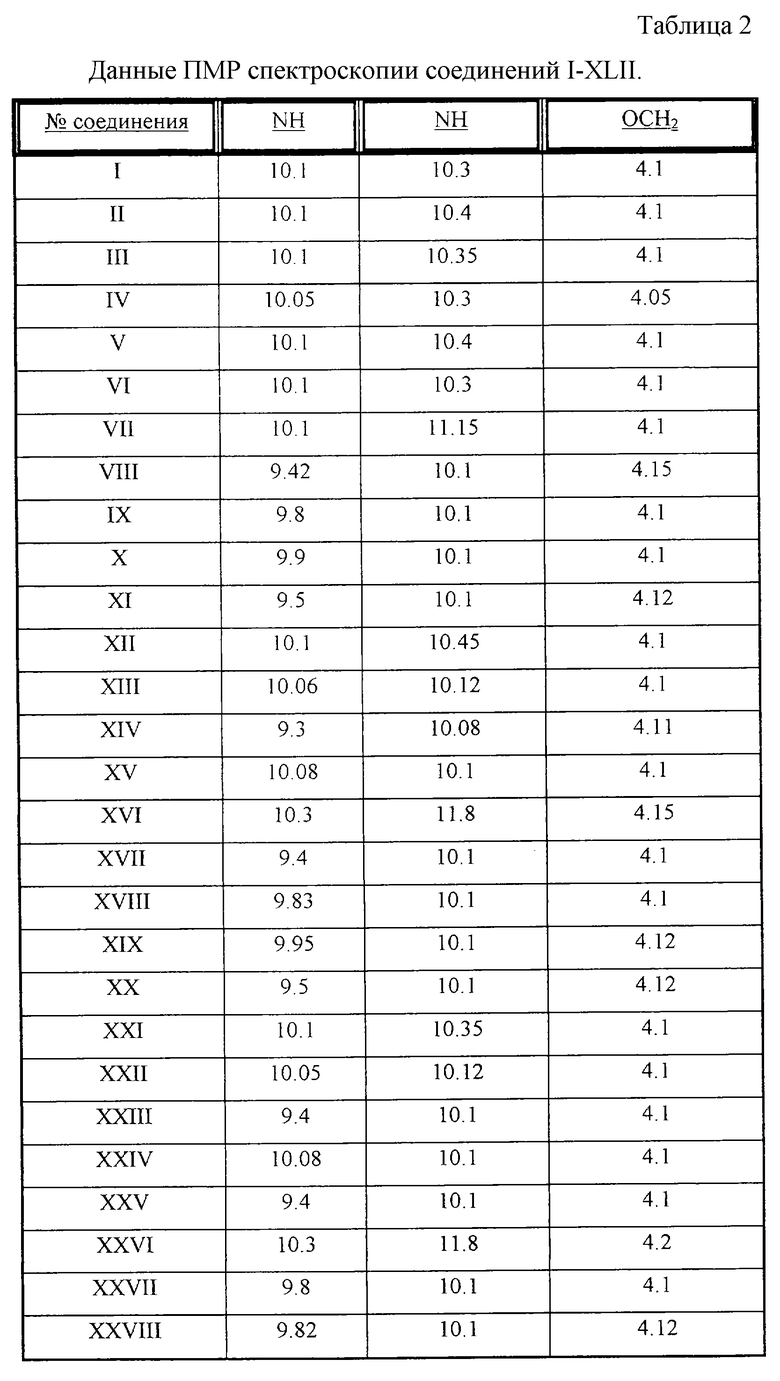
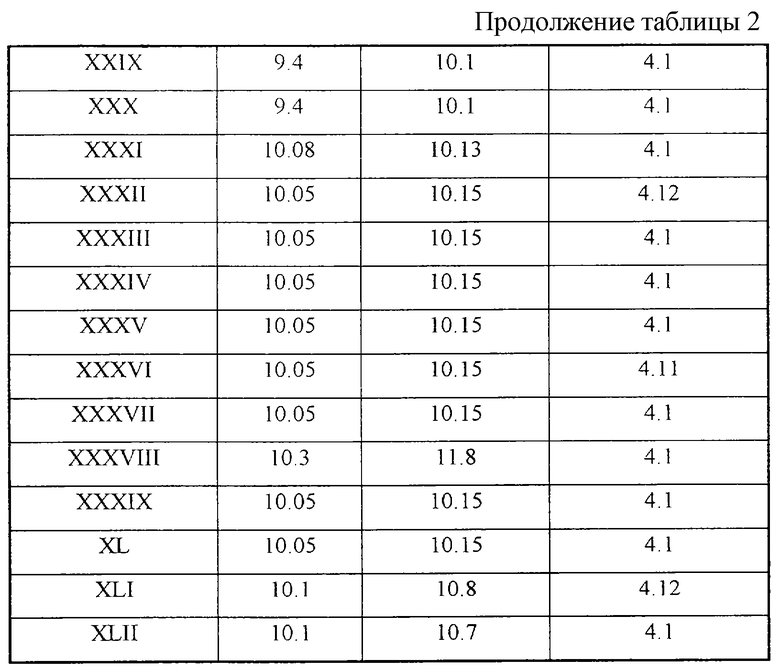
Данные ПМР-спектроскопии соединений I-XLII (с Таблицей 2);
Данные экспериментов по определению биологической активности заявляемых соединений в сопоставлении с широко распространенными средствами того же назначения, а именно:
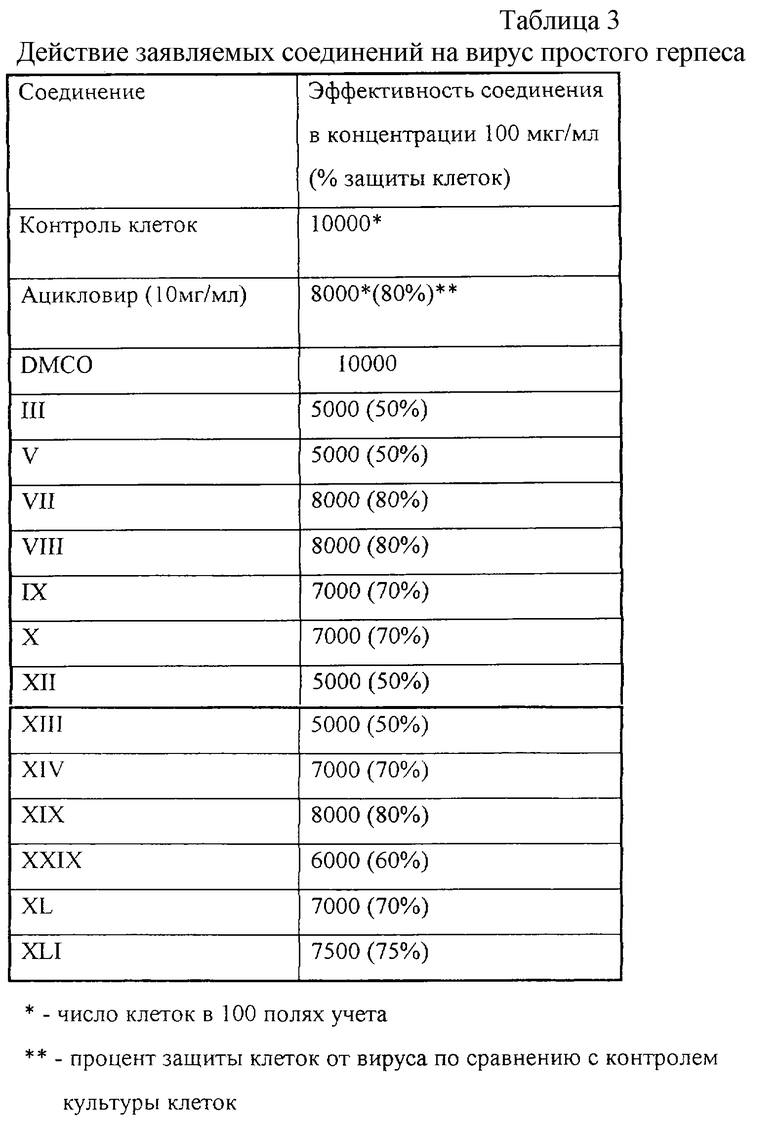
Эксперимент 1. Определение антигерпетического действия заявляемых соединений на примере вируса простого герпеса (с Таблицей 3),
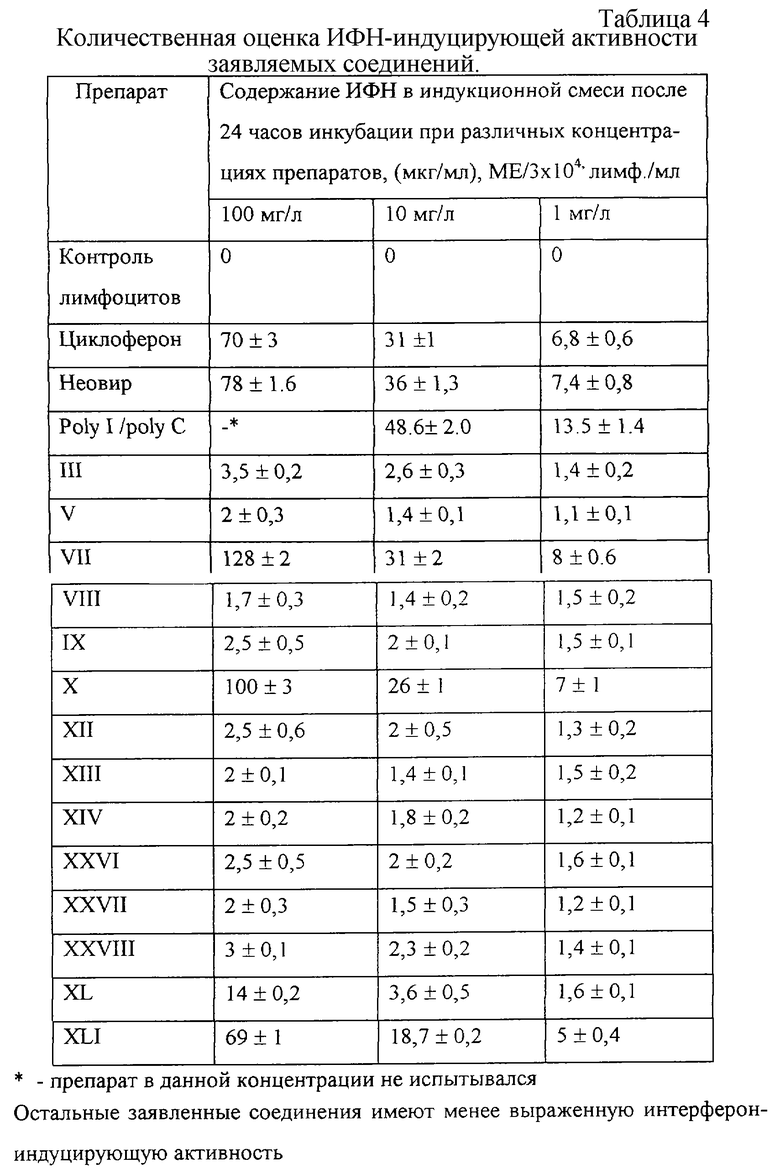
Эксперимент 2. Определение интерферониндуцирующей активности заявляемых соединений (с Таблицей 4),
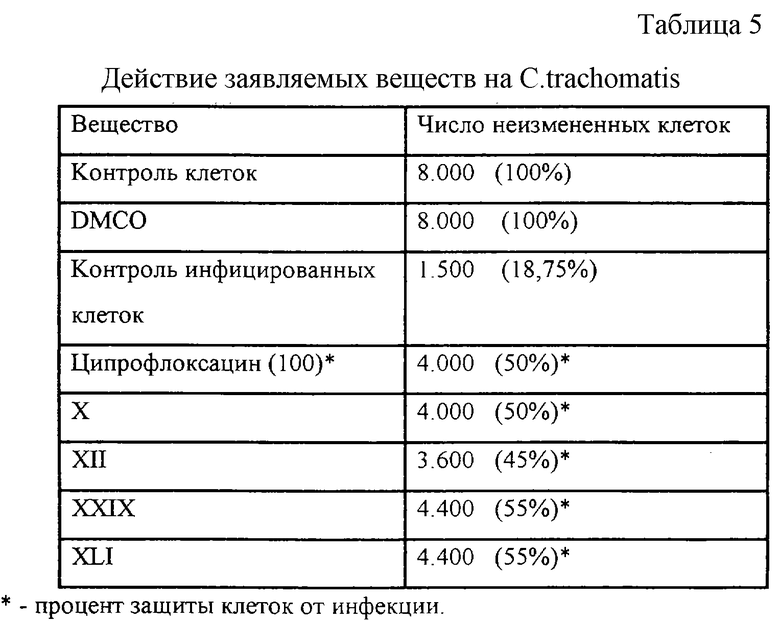
Эксперимент 3. Определение антихламидийного действия заявляемых соединений на примере Chlamydia trachomatis (с Таблицей 5),
Эксперимент 4. Определение гепатопротекторного действия заявляемых соединений (с Таблицей 6),
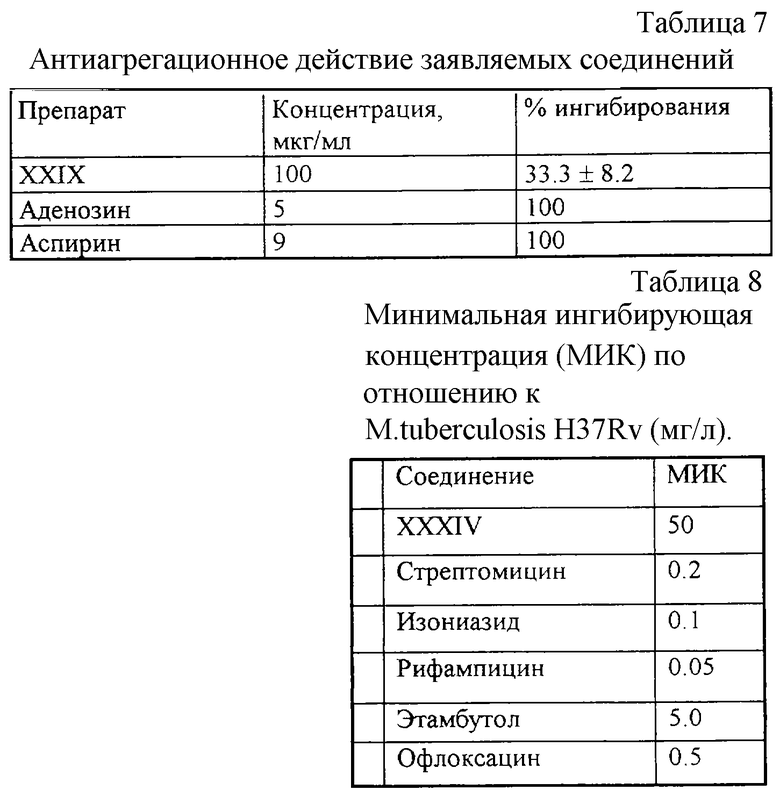
Эксперимент 5. Определение антиагрегационных свойств заявляемых соединений (с Таблицей 7),
Эксперимент 6. Определение антимикробного действия соединений (с Таблицей 8),
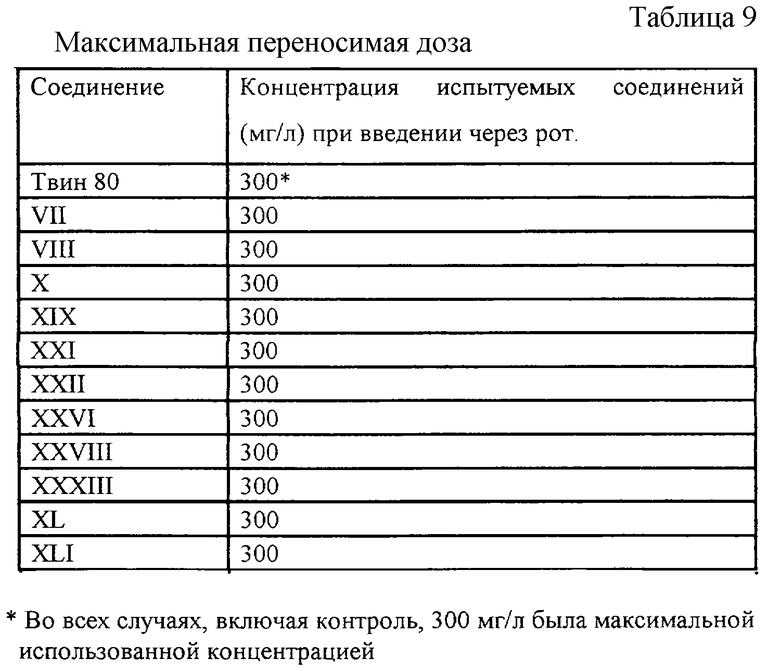
Эксперимент 7. Определение максимальной переносимой дозы (с Таблицей 9).
Общий способ получения соединений I - XLII
Способ получения целевых замещенных амидов карбоалкоксисульфаниловых кислот основан на постадийном взаимодействии фенилизоцианата, карбаматов, хлорсульфоновой кислоты и замещенных ароматических и гетероциклических аминов.
I стадия.
К фенилизоцианату по каплям при перемешивании добавляют соответствующий спирт (R2ОН) в мольном соотношении 1:1. Через 1-10 ч (в зависимости от реакционной способности спирта) реакционная смесь затвердевает в виде бесцветных кристаллов. Выход целевого продукта - количественный. Чистота полученного эфира фенилкарбаминовой кислоты достаточна для его дальнейшего использования.
II стадия.
К хлорсульфоновой кислоте, нагретой до 28-30oС, при перемешивании медленно прибавляют карбамат, поддерживая температуру реакционной массы не выше 50-55oС и выдерживают ее при указанной температуре в течение 2 ч. Полученную сульфомассу при перемешивании выливают на лед, поддерживая температуру смеси около 20oС. Образующийся мелкодисперсный осадок, как правило бесцветный, отфильтровывают и промывают ледяной водой до центральной реакции фильтрата. Осадок сушат на воздухе, а затем - в вакуум-эксикаторе над пятиокисью фосфора.
III стадия.
К смеси замещенного арил- или гетериламина (в зависимости от целевого продукта выбирают из возможных R1) и пиридина при температуре 82-88oС порциями прибавляют полученный на предыдущей стадии хлорангидрид карбоалкоксисульфаниловой кислоты. Во время внесения последних порций хлорангидрида проверяют рН среды. Если рН 1-2, то добавляют пиридин до щелочной реакции. Затем к реакционной массе добавляют горячую воду, подкисляют смесь соляной кислотой до рН 3,0-3,5 и охлаждают до 20oС. Полученный осадок фильтруют, промывают водой и сушат. Выход продуктов составляет примерно 30%.
Пример синтеза (4-бромфенил)амида карбометоксисульфаниловой кислоты (I).
I стадия. К 59,5 г фенилизоцианата, помещенного в стакан, по каплям при перемешивании прибавляют 16 г метанола. Наблюдают разогрев реакционной массы. Через 1 ч смесь затвердевает в виде бесцветных кристаллов. Выход метилового эфира фенилкарбаминовой кислоты количественный.
II стадия. К 241 г хлорсульфоновой кислоты, нагретой до 30oС, при перемешивании медленно прибавляют 76 г метилового эфира карбаминовой кислоты, поддерживая температуру массы не выше 35oС. Затем медленно нагревают смесь до 50oС и выдерживают ее при температуре 50-55oС в течение 2 ч. Полученную сульфомассу при перемешивании выливают на лед, поддерживая температуру не выше 20oС. Образовавшийся мелкодисперсный бесцветный осадок отфильтровывают, промывают ледяной водой до рН фильтрата 7. Осадок сушат на воздухе, а затем в вакуум-эксикаторе над пятиокисью фосфора. Выход хлорангидридакарбометоксисульфаниловой кислоты составит 75 г (60% от теоретического).
III стадия. К смеси 17,2 г 4-броманилина и 15,8 г пиридина при температуре 85oС порциями прибавляют 35 г хлорангидрида карбометоксисульфаниловой кислоты. рН среды к окончанию прибавления хлорангидрида составил 9. Затем к реакционной массе добавляют горячую воду, подкислляют смесь соляной кислотой до рН 3, и охлаждают до комнатной температуры. Полученный осадок отфильтровывают, промывают водой и высушили. Выход целевого продукта составляет 11,5 г (30%).
Остальные заявляемые соединения синтезируют аналогично, используя другие R1, заметная разница только в выходе целевого продукта.
Соединения общей формулы (1) представляют собой бесцветные или слегка окрашенные кристаллические вещества, растворимые в диметилсульфоксиде, хлороформе, пиридине.
Индивидуальность веществ доказана методом тонкослойной хроматографии на пластинках Silufol UV-254, элюент четыреххлористый углерод - изопропанол=9: 1.
Структура синтезированных веществ доказана методом ПМР-спектроскопии - см.Таблицу 2.
Данные экспериментов по определению биологической активности заявляемых соединений.
Эксперимент 1. Определение антивурусного действия заявляемых соединений на примере вируса простого герпеса
Антивирусная активность изучалась по отношению к вирусу простого герпеса I типа (ВПГ-1 /Ленинград/248/88) по общепринятому методу [7]. Вирусы выращивали на перевиваемой культуре клеток Vero, полученной из банка клеточных культур Института цитологии РАН.
Схема постановки опыта
К клеткам, выращенным на среде RPMI-1640 с 10% сыворотки плода коровы и помещенным в лунки 96-луночного планшета, добавляли вирус в конечной концентрации 102 Ig ТИД50/мл и заявляемые соединения, растворенные в ДМСО, в конечной концентрации 100, 10 и 1 мг/л. Для каждой испытанной концентрации препарата использовали 5 независимых лунок. Планшет инкубировали в течение 60 мин при 37oС в СО2-инкубаторе.После инкубации вирус удаляли и снова вносили свежую среду, содержащую заявляемые соединения в использованных концентрациях. Результаты оценивали по наличию цитопатогенного действия вируса на клетки через 36 ч культивирования при 37oС в СО2-инкубаторе.
В опыте были использованы следующие контроли:
1 Контроль культуры клеток (способность к нормальному росту).
2. Контроль вируса (оценка способности к репродукции).
3. Контроль антивирусной активности противовирусного препарата - ацикловира.
4. Контроль соединений (токсичность соединений).
5. Контроль растворителя (ДМСО) на токсичность.
Для оценки цитопатического действия вируса подсчитывали число неизмененных клеток в 100 полях, образованных специальной сеткой окуляр-микрометра инвертированного микроскопа. Полученные результаты представлены в Таблице 3.
Полученные результаты указывают, что приведенные в таблице 3 заявляемые соединения обладают антигерпетической активностью, сравнимой с таковой у стандартного препарата ацикловира. Остальные заявляемые соединения имели менее выраженную активность в процессе подавления репродукции вируса герпеса в выбранных условиях эксперимента.
Эксперимент 2. Определение интерферониндуцирующей активности заявляемых соединении
Индукцию синтеза интерферонов заявляемыми препаратами проводили на первичной культуре человеческих лимфоцитов (именно данные клетки в организме человека являются основными продуцентами интерферонов).
Для получения культуры лимфоцитов использовали свежую (12 ч после забора) кровь здоровых доноров (не второй группы). Для выделения лимфоцитов гепаринизированная кровь, полученная от здорового донора, подвергалась центрифугированию в градиенте плотности фиколлверографин 1,71 г/см3 для выделения фракции иммунокомпетентных клеток.
Указанная фракция отбиралась и разводилась питательной средой RPMI-1640, содержащей 5% сыворотки плода коровы, 0,3 мг/мл L-глутамина, 100 ед/мл пенициллина, 50 мг/мл стрептомицина. Концентрацию лимфоцитов учитывали после окрашивания метиленовым синим и подсчета количества клеток в камере гемоцитометра. Исходные растворы заявляемых веществ разводили питательной средой RPMI-1640 так, чтобы конечные концентрации веществ составляли ряд: 100 мг/л, 10 мг/л, 1 мг/л после внесения суспензии лимфоцитов. Конечная концентрация лимфоцитов в индукционной смеси составила 3•106 клеток/мл. Параллельно с опытными пробами проставлялись следующие контроли:
1. Контроль спонтанной продукции интерферонов (ИФН) лимфоцитами.
2. Контроль протекания процесса при воздействии стандартизированного индуктора ИФН N-метил-N-(а,D-глюкопиранозил)аммоний-10-метиленкарбоксилат акридона (циклоферон).
3. Контроль протекания процесса при воздействии стандартизированного индуктора ИФН - Неовира (натрия 10-метиленкарбоксилат-9-акридон) с соответствующим содержанием DMCO в опытных пробах.
4. Контроль спонтанной продукции интерферонов в присутствии DMCO, в количестве, соответствующем испытуемым образцам.
Контрольные и опытные образцы инкубировали 24 ч при 37oС. После инкубации пробы центрифугировались при 2000 g для осаждения клеточных элементов и из проб отбирался ИФН-содержащий супернатант, который анализировали на количественное содержание ИФН. Осадок клеток ресуспендировали в прежнем объеме питательной среды, окрашивали витальным красителем - трипановым синим и подсчитывали число клеток в камере гемоцитометра (как описано выше) для определения цитотоксического действия препаратов.
Количественное определение содержания ИФН в контрольных и опытных образцах производили с использованием иммуноферментной тест-системы на ИФН производства ТОО "Протеиновый контур" ProCon IF2 plus. Для определения количества интерферона в пробе использовали твердофазный иммуноферметный метод с использованием пероксидазы хрена в качестве индикаторного фермента. Активность связанной пероксидазы измеряли с использование автоматического фотометра для микропланшетов с микропроцессором при длине волны 450 нм.
Для подсчета результатов параллельно определяли активность ИФН у стандартных растворов ИФН, содержащих известное количество препарата. На основании полученных результатов строилась калибровочная кривая, позволяющая при использовании микропроцессора автоматического фотометра получать данные, выраженные в Международных Единицах активности (ME). Результаты анализа выражаются в ME активности ИФН на мл в данной индукционной системе, содержащей 3•106 лимфоцитов/мл.
Каждая опытная и контрольная точка исследовалась в 4 параллелях.
Контроли иммуноферментной реакции.
1. Контроль DMCO с питательной средой.
2. Контроль компонентов системы (согласно инструкции). Все результаты учитывались только при соответствии контролей паспортным данным системы.
Полученные результаты подвергались статистическому анализу по t-критерию и расчетом доверительного интервала при р 0,05. Произведен анализ сходимости результатов в параллельных опытах.
В результате проведенных исследований установлено, что среди заявляемых соединений имеются пробы, обладающие способностью индуцировать синтез ИФН (Таблица 4).
Эксперимент 3. Определение антихламидийного действия заявляемых соединений на примере Chlamydia trachomatis
Антихламидийную активность заявляемых соединеий изучали по отношению к C. trachomatis D323 - стандартному штамму из коллекции кафедры микробиологии С.Петербургского Государственного медицинского университета им.ак. И.П. Павлова. Данный штамм был выделен от больного с хламидийным уретритом, имеет морфологию и физиологическую активность, характерную для представителей данного вида, чувствителен к действию препаратов, используемых для лечения хламидийной инфекции.
В работе использованы клеточные культуры МсСоу и L929, полученные из Банка клеточных культур Института цитологии РАН.
Схема постановки опыта
Клетки выращивали во флаконах из нейтрального стекла в среде RPMI-1640 с добавлением 10% сыворотки плода коровы. Опыт ставили в стеклянных (лишенных токсичности) плоскодонных флаконах с покровными стеклами. Клетки вносили в среду в конечной концентрации 1•10 кл/мл. После получения монослоя в пробирки вносили стандартные заражающие дозы хламидий, хранящиеся в замороженном состоянии при -70oС. Одновременно к клеткам добавляли испытуемые соединения в конечной концентрации 100 мг/л. Пробу центрифугировали при 2400g в течение 60 мин при комнатной температуре и инкубировали при 37oС в течение 2 ч. После этого меняли питательную среду на новую, содержащую 5% сыворотки плода коровы и циклогексимид (2 мкг/мл), с повторным внесением заявляемых соединений в той же концентрации.
Параллельно дублировали пробы, используя среду без циклогексемида, чтобы исключить его влияние на изучаемые субстанции. Пробы инкубировали в течение 48 ч в СО2-инкубаторе.
Контроли включали:
1. Контроль культуры клеток.
2. Контроль действия растворителей.
3. Контроль действия хламидий в отсутствии каких бы то ни было препаратов.
4. Контроль чувствительности хламидий к стандартному антимикробному препарату - ципрофлоксацину.
5. Контроль испытуемых соединений на токсичность по отношению к культурам клеток.
Оценку результатов проводили путем выявления хламидийных цитоплазматических включений с помощью метода иммунофлюоресценции (MicroTrac Chlamydia trachomatis Direct Specimen Test) и хламидийных антигенов с помощью CylaMonoScreen (Russian-British Joint Venture 66 Regent's Parc Road London NW1 7SX) [8, 9]. Эффект действия препарата определяли, анализируя состояние монослоя и число клеток с ЦПВ по сравнению с контролем (культура клеток, зараженная С. trachomatis D323), при этом учитывали число неизмененных клеток в 100 полях зрения, полученных при использовании окуляр-микрометра. Результаты контрольных проб, удовлетворяющие требованиям эксперимента: контроль культуры клеток - морфология клеток и состояние монослоя соответствуют данному типу клеток, контроль роста хламидий в культуре клеток - наличие ЦПВ в монослое;
контроль действия стандартного антимикробного препарата - уменьшение числа ЦПВ в монослое по сравнению с предыдущим контролем;
контроль токсичности заявляемых соединений - токсичность отсутствует;
контроль действия растворителей - токсическое действие на клетки отсутствует.
Результаты проведенных испытаний представлены в Таблице 5.
Полученные данные свидетельствуют, что заявляемые соединения XXVIII и XLIII обладают выраженной активностью против хламидий, превосходящей таковую у стандартного препарата - ципрофлоксацина.
Остальные заявляемые соединения обладают менее выраженной активностью по защите клеток от хламидий в выбранных условиях эксперимента.
Эксперимент 4. Определение гепатопротекторного действия заявляемых соединений
Крысам подкожно вводили 50%-ный раствор CCl4 на оливковом масле (1 мл/кг). Исследуемые вещества применяли перорально в дозе 20 мг/кг за 30 мин до и через 7 ч после введения ССl4. Через сутки в крови животных определяли активность аланинаминотрансферазы (АлАТ)-маркерного фермента повреждения печеночной паренхимы. Уменьшение ферментемии более 30% по сравнению с группой контроля расценивали как наличие гепатопротекторных свойств препарата. В качестве препарата сравнения использовали витамин Е в дозе 100 мг/кг.
Остальные заявленные соединения имеют менее выраженную гепатопротекторную активность
Эксперимент 5. Определение антиагрегационных свойств заявляемых соединений
Агрегацию тромбоцитов человека исследовали в обогащенной тромбоцитами плазме здоровых доноров 50±3 лет с помощью агрегометра ФРМ-1. Исследуемую субстанцию добавляли к образцам плазмы крови (100 мкг/мл) перед индуцированием агрегации с помощью аденозиндифосфата (2,5 мкмоль/л). Амплитуду максимальной необратимой агрегации тромбоцитов в контрольных пробах (растворитель) принимали за 100%, а в опытных - рассчитывали в процентах от контроля. Ингибирование агрегации более чем на 50% при данной концентрации оценивали как значимую. В качестве препаратов сравнения использовали аденозин (5 мкг/мл) и аспирин (9 мкг/мл), ингибирующих агрегацию при этих концентрациях на 100%.
Остальные заявленные соединения специально не проверялись на наличие антиагрегационной активности, но мы имеем сведения о том, что все заявленные соединения имеют антиагрегационное действие, выраженное для разных соединений в разной степени.
Эксперимент 6. Определение антимикробного действия соединений
Для определения антимикробной активности был использован стандартный штамм Mycobacterium tuberculosis H37Rv, чувствительный ко всем антимикробным препаратам. Оценку антимикобактериального действия проводили методом серийных разведений.
Соединения растворяли в диметилсульфоксиде (ДМСО) и титровали в среде N-1 так, что данный препарат содержался в отдельных пробирках со средой в концентрациях от 200 до 0,025 мг/л. Концентрация препарата в среде соседних пробирок отличалась в два раза. В контроле использовали ДМСО, который титровали так же, как и препарат. Результат учитывали после 72- часового культивирования бактерий при 37oС. M. tuberculosis H37Rv выращивали на среде Сотона, содержащей 10% лошадиной сыворотки, и плотность микробной суспензии при засеве составляла 50•106.
В качестве контроля были использованы известные туберкулостатические препараты. Результаты, полученные для использованного штамма, приведены в Таблице 8.
Приведенные в Таблице 8 данные показывают, что соединение XXXIV обладает антимикробной активностью по отношению к использованному штамму М. tuberculosis в концентрации 50 мг/л.
У остальных заявленных соединений антимикробная активность по отношению к штамму М. tuberculosis не обнаружена.
Эксперимент 7. Определение максимальной переносимой дозы.
Испытуемые соединения вводили перорально с помощью желудочного зонда (300 мг/кг) или внутрибрюшинно (100 мг/кг) белым нелинейным мышам массой 18-20 г (по 3 самца и 3 самки в каждой из испытуемых групп), после чего наблюдали за их состоянием на протяжении 72 ч. Отсутствие симптоматики, свойственной токсическим эффектам, и отсутствие гибели животных в течение указанного времени позволяли сделать вывод о низкой токсичности изучаемого соединения [10].
Остальные заявленные соединения показали схожую токсичность.
Полученные результаты свидетельствуют, что при приеме через рот заявляемые соединения в концентрации 300 мг/кг не обладают острой токсичностью для мышей.
Промышленная применимость.
Приведенные выше примеры и практические результаты синтеза и анализа заявляемых соединений подтверждают возможность лабораторного и промышленного синтеза заявляемых соединений средствами, освоенными современной фармацевтической промышленностью, а также их строгую идентификацию общепринятыми методами контроля.
Серия экспериментов по определению биологической активности показала, что заявляемые соединения обладают биологической активностью по отношению к различным микроорганизмам - вирусу простого герпеса, микобактериям, хламидиям; выраженной интерферониндуцирующей и гепатопротекторной активностью и антиагрегационными свойствами. Высокая интерферониндуцирующая активность данного вещества также указывает на возможность их использования в лечении некоторых раковых заболеваний. Определены токсичность и максимально переносимые дозы.
Приведенные сведения доказывают достижение задач, поставленных изобретением: синтезированы новые соединения - арил- и гетериламиды карбоалкоксисульфаниловых кислот, обладающие низкой токсичностью и широким спектром ярко выраженного биологического действия, в частности, противовирусной, иммуностимулирующей, антибактериальной, антихламидийной, антиагрегационной, гепатопротекторной и антимикробной активностями.
Таким образом, заявляемые вещества удовлетворяют всем требованиям, предъявляемым к изобретению: они новы, неочевидны и промышленно применимы.
Источники информации
1. Chatis P.A., Crumpacker C.S. Resistance of herpesviruses to antiviral drugs. Antimicrob. Agents Chemother. 1992, 36: 1589-1595.
2. Pharmaceutical microbiology. Ed. by W. B. Hugo and A.D. Russel. Blackwell Scientific Publications, Oxford, 1987, 511р.
3. Esteban M. , Paez E. Antiviral and antiproliferartive properties of interferons: mechanism of action. Prog. med.virol. 1985, 32:159-173.
4. Tanneberg S., Hrelia P. Interferons in precancer and cancer prevention: where are we? J. of Interferon and cytokine res. 1996, 16,5: 339-346.
5. Машковский М.Д. Лекарственные средства. В 2 ч. - M.: Медицина, 1993.
6. Vaughan J.R.,Eichen J.A. and Anderson G.W., Heterocyclic Sulfonamides as Carbonic Anhydrase Inhibitors 2-Acylamido-and 2-Sulfonamodo-1,3,4-thiadiazole-5-Sulfonamides, The journal of Organic Chemistry, 1956. V. 21. Р. 700 - прототип.
7. Gentry G.A., Lawrency N., Lushbaugh N. Isolation and differentiation of Herpes simplex virus and Trichomonas vaginalis in cell culture, J. of Clinical Microbiology 1985, Vol. 22, No. 2, P. 199-204.
8. Wang S-P., Grayston J.T. Serotyping of Chlamydia trachomatis by inderect fluorescent-antibody staining of inclusions in cell culture with monoclonal antibodies. J. of Clinical Microbiology, 1991, Vol.29, No. 7, P. 1295-1298.
9. Judson В. A., Lambert P.P. Improved Syva Micro Trac Chlamydia trachomatis direct test method. Journal of Clinical Microbiology, 1988, Vol.26, No. 12, P.2657-2658
10. Irwin S., Psychopharmacology, 1968, 13, P. 222-257.

Изобретение относится к арил- и гетериламидам карбоалкоксисульфаниловых кислот формулы 1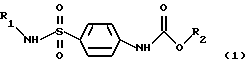
где R1 выбран из группы арилов или гетерилов, R2 выбран из группы алкилов. Задачей изобретения является получение новых химических соединений, обладающих широкой биологической активностью, в том числе противовирусной активностью (по отношению к вирусам простого герпеса), иммуностимулирующей активностью (за счет индукции выработки эндогенных интерферонов в организме), антихламидийной. Приведен общий способ их синтеза и результаты индентификации, соединения 1 обладают широким спектром биологической активности, в том числе противовирусной активностью по отношению к вирусном простого герпеса, иммуностимулирующей активностью, антихламидной, гепатопротекторной, антиагрегационной и антимикробной активностью. 42 з.п. ф-лы, 9 табл.
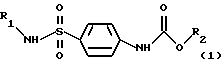
где R1 выбран из группы арилов и гетерилов, R2 выбран из группы алкилов.
| J | |||
| Org | |||
| Chemistry, 1956, v.21, p.700 | |||
| DE 3544409 A1, 16.10.1986 | |||
| Способ получения 4-фениловых эфиров 3-амино-5сульфамоилбензойных кислот | 1974 |
|
SU516347A3 |
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |

