Изобретение относится к органической химии, а именно к способам получения 2,4,6,8,10,12-гексанитро-2,4,6,8,10,12-гексаазатетрацикло[5,5,0,03,11, 05,9]додекана (ГАВ), используемого в качестве высокоэффективного взрывчатого вещества.
Аналоги заявляемого способа характеризуются наличием стадии нитрозирования и отличаются друг от друга используемыми нитрующими агентами.
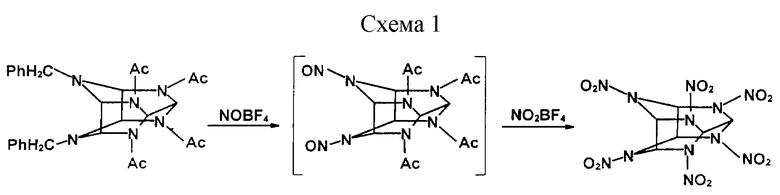
Известен способ (Nielsen А.Т. et all, Tetrahedron, 54 (1998), 11793-11812; Nielsen A. T. US Pat. 5693794, 02.12.97), в котором 4,10-дибензил-2,6,8,12-тетраацетил-2,4,6,8,10,12-гексаазатетрацикло [5,5,0,03,11, 05,9] додекан (ДБ) в растворе водного сульфолана предварительно нитрозируют тетрафторборатом нитрозония, а затем нитруют тетрафторборатом нитрония (см. схему 1 в конце описания).
На 0,03 моль ДБ берут 0,12 моль тетрафторбората нитрозония. Дозировку проводят в течение 30 мин при менее 25oС. Затем массу выдерживают 1 ч при 55-60oС. К охлажденной смеси быстро дозируют тетрафторборат нитрония (0,36 моль) и смесь выдерживают 4 ч при 55-60oС. Избыток тетрафторбората нитрозония удаляют либо фильтрованием горячего раствора, либо постепенным разбавлением реакционной массы водой. Образующийся осадок отфильтровывают, промывают водой, получая аморфный продукт (1% воды) с выходом 90,5% в пересчете на сухое вещество.
Недостатком описанного метода, препятствующим его широкому использованию в лабораторной практике, является необходимость применения тетрафторборатов нитрозония и нитрония - дорогих токсичных малостабильных веществ с высокой коррозионной активностью, требующих повышенной осторожности в обращении, специального оборудования и приемов дозировки. Использование в качестве растворителя сульфолана удорожает процесс.
Потребность в использовании тетрафторборатов отпадает при получении ГАВ нитрозированием ДБ четырехокисью азота (N2О4) с последующим нитрованием реакционной массы серно-азотной смесью ( Bescond et all, US Pat. 5973149, 26.10.99) (см. схему 2 в конце описания).
Согласно этому способу, принятому за прототип, к 3,307 моль жидкого N2O4 прибавляют 0,259 моль ДБ. Смесь выдерживают 20 ч при 15-16oС. Затем к охлажденной до 0oС реакционной массе дозируют 667 мл серно-азотной смеси (20/80 об. %), что соответствует 12,8 моль азотной кислоты. Далее массу нагревают, удаляя N2O4 , и выдерживают 4 ч при 73-75oС. После охлаждения до 40oС смесь выливают в воду со льдом, твердый осадок отфильтровывают, получая 104 г ГАВ (97%) чистотой не менее 95%.
Несмотря на достаточно высокий выход конечного продукта, существенным недостатком данного способа является использование N2O4 - малостабильного вещества, требующего специального оборудования и повышенной осторожности в обращении. Кроме того, нитрозобензильные производные, образующиеся в результате нитрозирования, являются термолабильными и при нитровании понижают стабильность реакционной массы, тем самым увеличивая опасность данного процесса. К недостаткам способа по прототипу также относятся его значительная продолжительность (24 ч) и насыщенность способа технологическими операциями.
Задачей заявляемого изобретения является получение целевого продукта (ГАВ) без использования малостабильных, высоколетучих, токсичных и дорогих реагентов при одновременном повышении технологичности процесса.
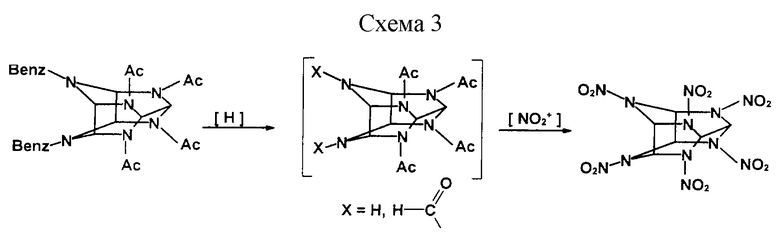
Поставленная задача решается предложенным способом получения ГАВ, включающим получение ДБ и нитрование, особенность заключается в том, что ДБ предварительно дебензилируют гидрированием, а затем полученный продукт нитруют смесью азотной кислоты с нитратом аммония в массовом соотношении 9:1 (см. схему 3 в конце описания).
Предлагаемый способ отличается от прототипа иным действием, осуществляемым над ДБ, - его дебензилируют гидрированием (в прототипе ДБ нитрозируют N2O4); использованием иного нитрующего агента - смеси азотной кислоты с нитратом аммония (в прототипе - серно-азотная смесь), что позволяет считать предложенный способ соответствующим критерию "новизна".
Сравнение заявляемого способа с прототипом и другими способами получения ГАВ, выявленными в уровне техники, показало, что неизвестно техническое решение поставленной задачи, в котором бы имело место предложенное сочетание признаков.
В отсутствие нитрата аммония нитрование не проходит до конца даже при использовании азотной кислоты с высоким модулем, и конечный продукт содержит недонитрованные фрагменты, что было установлено ИК- и ЯМР-спектроскопией. Т. е. нитрат аммония способствует полноте нитролиза, что позволяет получать целевой продукт с высоким выходом. Этот результат является неожиданным, поскольку добавка нитрата аммония, как правило, повышающая стабильность нитромасс за счет снижения кислотности, в данном случае позволяет повысить и нитрующую активность среды, что в других случаях не наблюдалось.
Только предлагаемая совокупность отличительных от прототипа признаков с остальными существенными признаками заявляемого способа позволяет не только сохранить высокий выход на стадии нитрования и качество целевого продукта, но и, в целом, существенно повысить технологичность процесса получения ГАВ, а именно это дает основание считать предлагаемый способ обладающим изобретательским уровнем.
Сведения, подтверждающие возможность осуществления способа.
Пример 1. В трехгорлой колбе вместимостью 500 см3, оснащенной мешалкой, прямым холодильником с приемником и термометром, растворяют 38 г нитрата аммония (N2O4) в 230 мл 95-98% азотной кислоты (5,5 моль). Затем вносят 23 г продукта гидрирования ДБ, полученного восстановлением ДБ в среде муравьиной кислоты, а именно 4,10-диформил-2,6,8,12-тетраацетил-2,4,6,8,10,12-гексаазатетрацикло [5,5,0,03,11,05,9]додекана (ДФ), и нагревают реакционную массу до кипения. Затем выдерживают реакционную массу в течение 5-6 ч, конденсируя отгоняющуюся азотную кислоту при помощи прямого холодильника. После накопления в приемнике 100 мл азотной кислоты и появления в колбе кристаллического осадка реакционную массу постепенно охлаждают до 40-50oС и разбавляют водой. Образующийся осадок целевого продукта отфильтровывают и промывают на фильтре водой до нейтральной реакции промывных вод. Получают 23,7 г ГАВ (98%).
Пример 2. Процесс ведут аналогично последовательности операций, приведенной в примере 1, но только с использованием в качестве продукта гидрирования ДБ 2,6,8,12-тетраацетил-2,4,6,8,10,12-гексаазатетрацикло [5,5,0,03,11, 05,9] додекана (ТА), полученного восстановлением ДБ в среде уксусной кислоты.
Нитрование ДФ и ТА проходит с образованием в качестве побочного продукта уксусной кислоты, а кроме того, при нитровании ДФ формильные фрагменты в жестких условиях реакции полностью окисляются до СО2 и Н2О, т.е. побочные продукты не являются термолабильными в отличие от прототипа.
Технологическое оформление заявляемого способа значительно проще, чем у ближайшего аналога. Операция дозировки субстрата к перемешиваемой смеси азотная кислота-нитрат аммония проводится при комнатной температуре без дополнительного охлаждения (в прототипе дозировка требует дополнительного охлаждения). Выдержку кипящей реакционной массы осуществляют с одновременной отгонкой азотной кислоты (в прототипе отсутствует отгонка нитрующего агента в процессе выдержки кипящей реакционной массы). За счет этого сокращена общая продолжительность процесса в сравнении с ближайшим аналогом и значительно повышена безопасность. Кроме того, отогнанная азотная кислота, по данным анализа, соответствует концентрации 96% и используется в процессе повторно. Это позволяет не только повысить экономичность способа, но и существенно сократить количество утилизируемой отработанной кислоты.
Реализация данного способа позволяет удовлетворить давно существующую потребность в получении целевого продукта без использования малостабильных, высоколетучих, токсичных и дорогих реагентов при одновременном повышении технологичности процесса; использовать стандартное оборудование и доступные реактивы, следовательно, предложенное техническое решение соответствует критерию промышленной применимости.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 2,4,6,8,10,12-ГЕКСАНИТРО-2,4,6,8,10,12-ГЕКСААЗАТЕТРАЦИКЛО[5,5,0,0,0]ДОДЕКАНА | 2008 |
|
RU2360916C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2,4,6,8,10,12-ГЕКСАНИТРО-2,4,6,8,10,12-ГЕКСААЗАТЕТРАЦИКЛО [5,5,0,0,0]ДОДЕКАНА | 2006 |
|
RU2355693C2 |
| СПОСОБ ПОЛУЧЕНИЯ ТЕТРААЦЕТИЛДИФОРМИЛГЕКСААЗАИЗОВЮРЦИТАНА | 2003 |
|
RU2266907C9 |
| СПОСОБ ПОЛУЧЕНИЯ КРИСТАЛЛИЧЕСКОГО 2,4,6,8,10,12-ГЕКСАНИТРО-2,4,6,8,10,12-ГЕКСААЗАТЕТРАЦИКЛО[5,5,0,0,0]ДОДЕКАНА С ЗАДАННЫМ ПОЛИМОРФНЫМ СОСТАВОМ (ВАРИАНТЫ) | 2010 |
|
RU2452739C9 |
| СПОСОБ ПОЛУЧЕНИЯ 2,4,6,8,10,12-ГЕКСАНИТРО-2,4,6,8,10,12-ГЕКСААЗАТЕТРАЦИКЛО[5,5,0,03,11,05,9]ДОДЕКАНА | 2015 |
|
RU2607517C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2,6,8,12-ТЕТРААЦЕТИЛ-2,4,6,8,10,12-ГЕКСААЗАТЕТРАЦИКЛО[5,5,0,03,11 ,05,9]ДОДЕКАНА | 2015 |
|
RU2610695C2 |
| Способ получения 4,10-диформил-2,6,8,12-тетраацетил-2,4,6,8,10,12-гексаазатетрацикло[5,5,0,0,0]додекана | 2021 |
|
RU2772755C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГАММА-ПОЛИМОРФНОЙ МОДИФИКАЦИИ 2,4,6,8,10,12-ГЕКСАНИТРО-2,4,6,8,10,12-ГЕКСААЗАТЕТРАЦИКЛО[5,5,0,0]ДОДЕКАНА | 2011 |
|
RU2447075C1 |
| СПОСОБ ДЕБЕНЗИЛИРОВАНИЯ 2,6,8,12-ТЕТРААЦЕТИЛ-2,4,6,8,10,12-ГЕКСААЗАИЗОВЮРЦИТАНА | 2010 |
|
RU2448110C1 |
| ОДНОРЕАКТОРНЫЙ СПОСОБ ПОЛУЧЕНИЯ 4,10-ДИ(2-ЭТОКСИАЦЕТИЛ)-2,6,8,12-ТЕТРААЦЕТИЛ-2,4,6,8,10,12-ГЕКСААЗАТЕТРАЦИКЛО[5,5,0,0,0]ДОДЕКАНА | 2023 |
|
RU2834298C1 |

Изобретение относится к органической химии, конкретно к способу получения 2,4,6,8,10,12-гексанитро-2,4,6,8,10,12-гексаазатетрацикло-[5,5,0,03,11, 05,9] додекана, используемого в качестве высокоэффективного взрывчатого вещества. Описывается способ получения 2,4,6,8,10,12-гексанитро-2,4,6,8,10,12-гексаазатетрацикло[5,5,0,03,11, 05,9] додекана, включающий получение 4,10-дибензил-2,6,8,12-тетраацетил-2,4,6,8,10,12-гексаазатетрацикло[5,5,0,03,9, 05,11] додекана и нитрование, при этом 4,10-дибензил-2,6,8,12-тетраацетил-2,4,6,8,10,12-гексаазатетрацикло-[5,5,0,03,11, 05,9] додекан предварительно дебензилируют гидрированием, а затем полученный продукт нитруют смесью азотной кислоты с нитратом аммония в массовом соотношении 9:1. Технический результат - получение целевого продукта без использования малостабильных высоколетучих токсичных и дорогих реагентов при одновременном повышении технологичности процесса.
Способ получения 2,4,6,8,10,12-гексанитро-2,4,6,8,10,12-гексаазатетрацикло[5,5,0,03,11, 05,9] додекана, включающий получение 4,10-дибензил-2,6,8,12-тетраацетил-2,4,6,8,10,12-гексаазатетрацикло[5,5,0,03,9, 05,11] додекана и нитрование, отличающийся тем, что 4,10-дибензил-2,6,8,12-тетраацетил-2,4,6,8,10,12-гексаазатетрацикло[5,5,0,03,11, 05,9]додекан предварительно дебензилируют гидрированием, а затем полученный продукт нитруют смесью азотной кислоты с нитратом аммония в массовом соотношении 9:1.
| US 5973149 A1, 26.10.1999 | |||
| СПОСОБ ПОЛУЧЕНИЯ ГЕКСАНИТРОГЕКСААЗАИЗОВЮРТЦИТАНА | 1997 |
|
RU2157810C1 |
| US 5693794 A1, 02.12.1997. | |||

