Изобретение относится к органической химии, а именно к способам получения высокоэффективного, мощного взрывчатого вещества 2,4,6,8,10,12-гексанитро-2,4,6,8,10,12-гексаазатетрацикло[5,5,0,03,11,05,9]додекана (ГАВ).
Существует ряд способов получения 2,4,6,8,10,12-гексанитро-2,4,6,8,10,12-гексаазатетрацикло[5,5,0,03,11,05,9]додекана (ГАВ) из 4,10-диформил-2,6,8,12-тетраацетил-2,4,6,8,10,12-гексаазатетрацикло[5,5,0,03,11,05,9]додекана (ДФ).
Известен способ (Wardle и др. US Pat 6.147.209. 14.11.2000), в котором осуществляют гидрогенолиз 4,10-дибензил-2,6,8,12-тетраацетил-2,4,6,8,10,12-гексаазатетрацикло[5,5,0,03,11,05,9]додекана (ДБ) в среде муравьиной кислоты и проводят нитрование ДФ. Существенным недостатком данного способа является использование на стадии нитрования кристаллического ДФ, который получают из ДБ упариванием под вакуумом, что требует сложного аппаратурного оформления, увеличенной продолжительности нагрева и доли побочных процессов (в результате гидролиза ДФ образуются растворимые продукты, ведущие к снижению выхода ДФ).
Также известен способ по патенту № 2199540 (бюллетень №6 от 27.02.2003), принятый за прототип, в котором осуществляют гидрирование ДБ с последующим нитрованием ДФ раствором нитрата аммония в азотной кислоте. В известном способе на стадии нитрования используют кристаллический ДФ, выделяемый из муравьиной кислоты с применением дополнительных растворителей. Реализация указанного способа в промышленных условиях приводит к значительному увеличению продолжительности нагрева. Это увеличивает долю побочных процессов (гидролиз ДФ с образованием растворимых продуктов) и приводит к снижению выхода ДФ.
Известно, что гидрирование ДБ сопровождается образованием солей моноформил 2,6,8,12-тетраацетил-2,4,6,8,10,12-гексаазатетрацикло[5,5,0,03,11,05,9]додекана (ФТА) и 2,6,8,12-тетраацетил-2,4,6,8,10,12-гексаазатетрацикло[5,5,0,03,11,05,9]додекана (ТА), которые при нитровании образуют ГАВ (Sanderson A.J., Warner R., Wardle R.B., World Pat. WO 00/52011). Эти же продукты образуются из ДФ при длительной выдержке или сильном нагревании водосодержащих растворов. Побочные продукты еще менее стабильны, чем ДФ, и в условиях выделения подвергаются гидролизу с образованием смеси продуктов, не образующих при нитровании ГАВ.
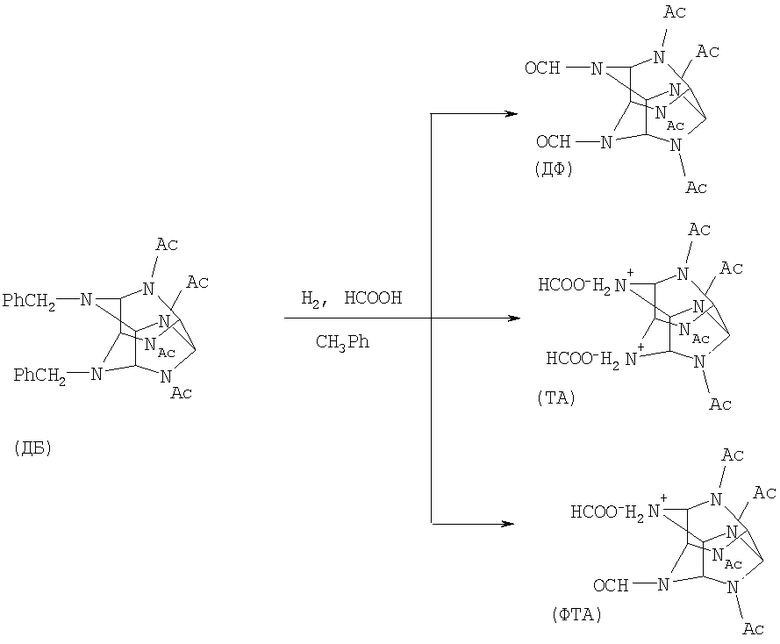
Таким образом, в силу имеющих место потерь ДФ и побочно образующихся солей известный способ предполагает увеличенные расход сырья и количество примесей в конечном продукте ГАВ. Кроме того, способ насыщен технологическими операциями, одна из которых - стадия сушки - является пожароопасной.
Задачей настоящего изобретения является создание способа получения ГАВ с высоким выходом целевого продукта (в пересчете на ДБ) при одновременном снижении насыщенности технологическими операциями и повышении безопасности процесса путем предотвращения потерь ДФ и формиатных солей ТА и ФТА на стадиях, предшествующих нитрованию, и их полного использования на стадии нитрования.

Поставленная задача решается предложенным способом получения ГАВ, при котором осуществляют гидрирование ДБ и нитрование полученной реакционной смеси раствором нитрата аммония в азотной кислоте. Согласно изобретению реакционную смесь перед нитрованием частично упаривают под вакуумом при температуре 45-50°С или частично упаривают и обрабатывают уксусным ангидридом, или частично упаривают с добавлением уксусной кислоты.
Предлагаемый способ отличается от прототипа иным агрегатным состоянием продукта, используемого на стадии нитрования. В прототипе нитрованию подвергают кристаллический ДФ (продукт дебензилирования ДБ), а в предлагаемом способе - раствор, получающийся обработкой реакционной смеси со стадии гидрирования ДБ. Нитрование растворов ДФ (обработанной реакционной смеси) изменяет условия нитрования и позволяет обеспечить достижение заявленного технического результата.
Гидрирование ДБ в среде муравьиной кислоты приводит к образованию ДФ, толуола и воды. Прямое нитрование полученной реакционной смеси приводит к получению ГАВ, сильно загрязненного продуктами нитрования толуола (нитротолуол, динитротолуол, тринитротолуол), содержание которых может достигать 30-40%. Кроме того, вследствие невысокой концентрации ДФ нитрование реакционной смеси требует большого расхода азотной кислоты.
Поэтому предлагается полученную реакционную смесь перед нитрованием подвергать обработке альтернативными приемами.
Одна из альтернатив - частичное упаривание реакционной смеси после гидрирования под вакуумом при температуре 45-50°С. Состав и содержание компонентов реакционной смеси после гидрирования и после частичного ее упаривания приведены в Таблице.
Концентрирование полученной после гидрирования смеси при указанных условиях позволяет полностью удалить толуол и получить раствор с концентрацией ДФ, близкой к насыщению.
Нитрование частично упаренной смеси раствором нитрата аммония в азотной кислоте дает ГАВ с выходом 85% (в расчете на исходный ДБ).
Другой предлагаемой альтернативой предусматривается обработка частично упаренной реакционной смеси уксусным ангидридом. В процессе обработки проходит ряд химических реакций. Вода, реагируя с уксусным ангидридом, превращается в уксусную кислоту. Муравьиная кислота с уксусным ангидридом образует смешанный ангидрид уксусной и муравьиной кислот.
(СН3СО)2O+НСООН=(СН3СО)О(ОСН)+СН3СООН
Последний, являясь малостойким соединением, при нагревании до кипения разлагается на уксусную кислоту и окись углерода.
(СН3СО)О(ОСН)=СН3СООН+СО
В результате получают раствор ДФ в безводной уксусной кислоте или с небольшим содержанием муравьиной кислоты. Нитрование смеси, обработанной уксусным ангидридом, сопровождается меньшим тепловым эффектом и меньшим выделением окислов азота.
Еще одна альтернатива предусматривает проведение частичного упаривания с добавлением уксусной кислоты. Поскольку муравьиная кислота обладает более высокой летучестью, в результате частичного упаривания реакционной смеси после гидрирования образуется раствор с концентрацией ДФ до 40%, а содержание муравьиной кислоты не превышает 10%. Такой состав раствора также обеспечивает на стадии нитрования умеренное выделение окислов азота. Выход ГАВ составил 93% (в пересчете на ДБ).
Применение на стадии нитрования обработанной реакционной смеси позволяет значительно повысить растворимость в нитросмеси как ГАВ, так и промежуточных продуктов нитрования. Это обеспечивает полноту нитрования и содержание примесей в готовом продукте не более 1%.
В известных методах для выделения ДФ в индивидуальном виде требуются дополнительные операции, а выход ДФ составляет 85-90%. Поэтому выход ГАВ в пересчете на ДБ (две стадии) не может превышать 85-90%. Разработанный способ позволяет исключить стадию выделения ДФ и связанные с ней неизбежные потери ДФ и формиатных солей ТА и ФТА и обеспечить выход ГАВ не ниже 85%. Кроме того, способ позволяет исключить стадию сушки ДФ от органических остатков пожароопасных растворителей, тем самым повысить безопасность процесса.
Сведения, подтверждающие возможность осуществления способа.
Пример 1.
В плоскодонную колбу с магнитной мешалкой загружают 9,9 г ДБ, 2,5 г катализатора (6% Pd на угле) и 37 г 88% муравьиной кислоты. Колбу герметизируют пробкой с краном и газоподводящей трубкой. С помощью вакуума откачивают воздух и подают водород. Процесс проводят при температуре 40-45°С. После завершения поглощения водорода процесс останавливают. Реакционную смесь фильтруют от катализатора. Отработанный катализатор промывают муравьиной кислотой.
Отфильтрованную смесь упаривают под вакуумом при температуре 45-50°С до веса 15 г. Состав отфильтрованной и частично упаренной смеси приведен в Таблице.
В реактор с мешалкой, термометром и обратным холодильником загружают 73,5 г 99% азотной кислоты и 21,5 г нитрата аммония. После растворения нитрата аммония в реактор при охлаждении ледяной водой дозируют частично упаренный раствор. Убирают охлаждение, нагревают смесь до кипения и выдерживают при температуре 112-118°С 10-12 часов. Реакционную смесь охлаждают до комнатной температуры и сливают в смесь льда с водой. Продукт отфильтровывают, промывают водой и сушат на воздухе. Получают 7,1 г ГАВ (85%) с содержанием основного вещества 99,0%.
Пример 2.
В плоскодонную колбу с магнитной мешалкой загружают 9,9 г ДБ, 2,5 г катализатора (6% Pd на угле) и 37 г 88% муравьиной кислоты. Колбу герметизируют пробкой с краном и газоподводящей трубкой. С помощью вакуума откачивают воздух и подают водород. Процесс проводят при температуре 40-45°С. После завершения поглощения водорода процесс останавливают. Реакционную смесь фильтруют от катализатора. Отработанный катализатор промывают муравьиной кислотой.
Отфильтрованную смесь упаривают под вакуумом до веса 15 г и переносят в реактор с мешалкой, термометром и нисходящим холодильником. Туда же при температуре не выше 45°С приливают 16 г уксусного ангидрида. Реакционную смесь кипятят в течение 2 часов, отгоняют 2,6 г уксусной кислоты и получают 23,5 г раствора ДФ в уксусной кислоте (содержание муравьиной кислоты 5%).
В реактор с мешалкой, термометром и обратным холодильником загружают 73,5 г 99% азотной кислоты и 21,5 г нитрата аммония. После растворения нитрата аммония в реактор при охлаждении ледяной водой дозируют приготовленный раствор. Убирают охлаждение, нагревают смесь до кипения и выдерживают при температуре 112-118°С 10-12 часов. Реакционную смесь охлаждают до комнатной температуры и сливают в смесь льда с водой. Продукт отфильтровывают, промывают водой и сушат на воздухе. Получают 7,1 г ГАВ (85%) с содержанием основного вещества 99,2%.
Пример 3.
В плоскодонную колбу с магнитной мешалкой загружают 9,9 г ДБ, 2,5 г катализатора (6% Pd на угле) и 37 г 88% муравьиной кислоты. Колбу герметизируют пробкой с краном и газоподводящей трубкой. С помощью вакуума откачивают воздух и подают водород. Процесс проводят при температуре 40-45°С. После завершения поглощения водорода процесс останавливают. Реакционную смесь фильтруют от катализатора. Отфильтрованную смесь упаривают под вакуумом до веса 14-16 г.
Отработанный катализатор промывают двумя порциями по 15 г ледяной уксусной кислоты. В частично упаренную смесь доливают 20 грамм промывной уксусной кислоты и повторно упаривают до веса 14-16 г. Добавляют остаток промывной уксусной кислоты и упаривают до веса 19,1 г. Получают раствор ДФ в уксусной кислоте (содержание муравьиной кислоты 4%, воды 2%).
В реактор с мешалкой, термометром и обратным холодильником загружают 73,5 г 99% азотной кислоты и 22,3 г нитрата аммония. После растворения нитрата аммония в реактор при охлаждении ледяной водой дозируют приготовленный раствор. Убирают охлаждение, нагревают смесь до кипения и выдерживают при температуре 112-118°С 10-12 часов. Реакционную смесь охлаждают до комнатной температуры и сливают в смесь льда с водой. Продукт отфильтровывают, промывают водой и сушат на воздухе. Получают 7,8 г ГАВ (93%) с содержанием основного вещества 99,4%.
Реализация данного изобретения позволит удовлетворить давно существующую потребность в безопасном и технологичном способе получения целевого продукта.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ 2,4,6,8,10,12-ГЕКСАНИТРО-2,4,6,8,10,12-ГЕКСААЗАТЕТРАЦИКЛО[5,5,0,0,0]ДОДЕКАНА | 2008 |
|
RU2360916C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2, 4, 6, 8, 10, 12-ГЕКСАНИТРО-2, 4, 6, 8, 10, 12-ГЕКСААЗАТЕТРАЦИКЛО [5, 5, 0, 0, 0]ДОДЕКАНА | 2001 |
|
RU2199540C2 |
| СПОСОБ ПОЛУЧЕНИЯ 2,4,6,8,10,12-ГЕКСАНИТРО-2,4,6,8,10,12-ГЕКСААЗАТЕТРАЦИКЛО[5,5,0,03,11,05,9]ДОДЕКАНА | 2015 |
|
RU2607517C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2,6,8,12-ТЕТРААЦЕТИЛ-2,4,6,8,10,12-ГЕКСААЗАТЕТРАЦИКЛО[5,5,0,03,11 ,05,9]ДОДЕКАНА | 2015 |
|
RU2610695C2 |
| Способ получения 4,10-диформил-2,6,8,12-тетраацетил-2,4,6,8,10,12-гексаазатетрацикло[5,5,0,0,0]додекана | 2021 |
|
RU2772755C1 |
| СПОСОБ ПОЛУЧЕНИЯ КРИСТАЛЛИЧЕСКОГО 2,4,6,8,10,12-ГЕКСАНИТРО-2,4,6,8,10,12-ГЕКСААЗАТЕТРАЦИКЛО[5,5,0,0,0]ДОДЕКАНА С ЗАДАННЫМ ПОЛИМОРФНЫМ СОСТАВОМ (ВАРИАНТЫ) | 2010 |
|
RU2452739C9 |
| СПОСОБ ПОЛУЧЕНИЯ ТЕТРААЦЕТИЛДИФОРМИЛГЕКСААЗАИЗОВЮРЦИТАНА | 2003 |
|
RU2266907C9 |
| ОДНОРЕАКТОРНЫЙ СПОСОБ ПОЛУЧЕНИЯ 4,10-ДИ(2-ЭТОКСИАЦЕТИЛ)-2,6,8,12-ТЕТРААЦЕТИЛ-2,4,6,8,10,12-ГЕКСААЗАТЕТРАЦИКЛО[5,5,0,0,0]ДОДЕКАНА | 2023 |
|
RU2834298C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГАММА-ПОЛИМОРФНОЙ МОДИФИКАЦИИ 2,4,6,8,10,12-ГЕКСАНИТРО-2,4,6,8,10,12-ГЕКСААЗАТЕТРАЦИКЛО[5,5,0,0]ДОДЕКАНА | 2011 |
|
RU2447075C1 |
| Способ получения катализатора и способ его применения для многократного использования в промышленном процессе двухстадийного гидрогенолиза при производстве 2,4,6,8,10,12-гексанитро-2,4,6,8,10,12-гексаазатетрацикло[5,5,0,0,0]додекана | 2016 |
|
RU2641694C1 |
Описывается усовершенствованный способ получения 2,4,6,8,10,12-гексанитро-2,4,6,8,10,12-гексаазатетрацикло[5,5,0,03,11,05,9]додекана, заключающийся в гидрировании 4,10-дибензил-2,6,8,12-тетраацетил-2,4,6,8,10,12-
гексаазатетрацикло[5,5,0,03,11,05,9]додекана в муравьиной кислоте с последующим частичным упариванием под вакуумом реакционной смеси при температуре 45-50°С, возможно с добавлением уксусной кислоты или обработкой частично упаренной смеси уксусным ангидридом и нитрованием полученной реакционной смеси раствором нитрата аммония в азотной кислоте. Описываемый способ позволяет упростить и повысить безопасность процесса. 1 табл.
Способ получения 2,4,6,8,10,12-гексанитро-2,4,6,8,10,12-гексаазатетрацикло
[5,5,0,03,11,05,9]додекана, включающий гидрирование 4,10-дибензил-2,6,8,12-тетраацетил-2,4,6,8,10,12-гексаазатетрацикло[5,5,0,03,11,05,9]додекана в муравьиной кислоте и нитрование полученной реакционной смеси раствором нитрата аммония в азотной кислоте, отличающийся тем, что реакционную смесь перед нитрованием частично упаривают под вакуумом при температуре 45-50°С или частично упаривают и обрабатывают уксусным ангидридом, или частично упаривают с добавлением уксусной кислоты.
| СПОСОБ ПОЛУЧЕНИЯ 2, 4, 6, 8, 10, 12-ГЕКСАНИТРО-2, 4, 6, 8, 10, 12-ГЕКСААЗАТЕТРАЦИКЛО [5, 5, 0, 0, 0]ДОДЕКАНА | 2001 |
|
RU2199540C2 |
| US 6147209 A1, 14.11.2000 | |||
| US 5973149 A1, 26.10.1999 | |||
| СПОСОБ ПОЛУЧЕНИЯ ГЕКСАНИТРОГЕКСААЗАИЗОВЮРТЦИТАНА | 1997 |
|
RU2157810C1 |
| EP 0919556 A1, 02.09.1999 | |||
| Устройство для преобразования серии импульсов | 1980 |
|
SU984015A1 |
| US 5693794 A1, 02.12.1997. | |||

