Изобретение относится к новым гетероциклическим соединениям и их препаратам, а также к применению их в качестве лекарственных средств.
Предпосылки создания изобретения
Европейская Патентная заявка ЕР-А-0498722 описывает производные хинолина как ингибиторы ангиотензина А2 и эндотелина.
Модели действия фосфодиэстераз (PDE), а также факторов некроза опухолей (TNF) и терапевтическая полезность их ингибиторов описаны в WO-A-9744036 и Патенте США 5804588, содержание которых включено здесь в качестве ссылок. Эти публикации подробно раскрывают хинолинкарбоксамиды, обладающие такой ингибирующей активностью.
Краткое содержание изобретения
Данное изобретение предлагает новые соединения, терапевтически полезные, в частности, для лечения болезненных состояний, связанных с белками, которые опосредуют клеточную активность, например, посредством ингибирования TNF и/или PDE IV. Согласно данному изобретению, эти соединения являются соединениями формулы (i):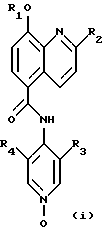
где R1 представляет собой СН3, CH2F, CHF2 или СF3;
R2 представляет собой СН3 или СF3;
R3 представляет собой F, Cl, Br, CN или СН3; и
R4 представляет собой Н, F, Cl, Br, CN или СН3;
или их фармацевтически приемлемыми солями.
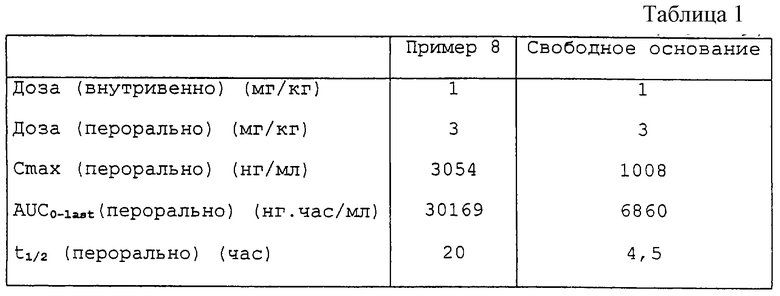
Вкратце, соединения данного изобретения являются N-окисями соответствующих оснований, которые описаны (некоторые конкретно) в WO-A-9744036. Эти новые соединения имеют превосходную растворимость, а также улучшенные метаболическую стабильность и фармакокинетический профиль. Особо предпочтительным является соединение Примера 8.
В данном изобретении предлагается также способ опосредования или ингибирования ферментативной активности, каталитической активности PDE IV у нуждающегося млекопитающего и ингибирования продуцирования TNF у нуждающегося в этом млекопитающего, который включает введение указанному млекопитающему эффективного количества соединения формулы (i) или его фармацевтически приемлемой соли.
Краткое описание чертежа
Приложенный чертеж представляет собой график, который показывает данные рК после орального введения крысам определенной дозы соединения данного изобретения и известного соединения (для сравнения).
Описание изобретения
Определенные соединения формулы (i), которые содержат основную группу, образуют соли присоединения кислот. Подходящие соли присоединения кислот включают фармацевтически приемлемые неорганические соли, такие как сульфат, нитрат, фосфат, борат, гидрохлорид и гидробромид, и фармацевтически приемлемые соли присоединения органических кислот, такие как ацетат, тартрат, малеат, цитрат, сукцинат, бензоат, аскорбат, метансульфат, α-кетоглутарат, α-глицерофосфат и глюкозо-1-фосфат. Фармацевтически приемлемые соли соединений формулы (i) получают, используя общеизвестные методики.
Соединения данного изобретения можно получить посредством N-окисления соответствующих свободных оснований. Такие свободные основания известны, или их можно легко получить способом, раскрытым в WO-A-9744036. Например, соединение формулы (i) можно получить, обрабатывая свободное основание перуксусной кислотой в уксусной кислоте в подходящем растворителе, таком как хлороформ, или перекисью водорода в уксусной кислоте.
Данное изобретение включает профилактику и лечение TNF-опосредованных заболеваний или болезненных состояний, под которыми подразумевают любые и все болезненные состояния, в которых участвует TNF, - либо путем продуцирования самого TNF, либо когда TNF вызывает высвобождение другого цитокина, такого как интерлейкин-1 (IL-1) или интерлейкин-6 (IL-6), но не ограничиваясь ими. Следовательно, болезненным состоянием, опосредованным TNF, считают болезненное состояние, при котором, например, IL-1 является основным компонентом, а его продуцирование или действие усиливается или осуществляется в ответ на TNF. Так как TNF-β (известный так же, как лимфотоксин) является близким структурным гомологом TNF-α (известным так же как, кахектин), и каждый из них вызывает аналогичные биологические реакции и связывается одним и тем же клеточным рецептором, оба, TNF-β и TNF-α, ингибируются соединениями настоящего изобретения, и поэтому имеют здесь общее обозначение "TNF", если специально не указано иначе.
Ингибиторы PDE IV полезны при лечении многих аллергических и воспалительных заболеваний, включая астму, хронический бронхит, атопический дерматит, эндогенную экзему, крапивницу, аллергический ринит, аллергический конъюнктивит, весенний конъюнктивит, воспаление глаз, аллергические реакции глаз, эозинофильную гранулему, псориаз, болезнь Бечета (Bechet's disease), эритематоз, аллергический геморрагический нефрит (anaphylactoid purpura nephritis), воспаление суставов, артрит, ревматоидный артрит и другие артритные состояния, такие как ревматоидный спондилит и остеоартрит, септический шок, неспецифический язвенный колит, болезнь Крона (Crohne's disease), поражение миокарда и мозга вследствие повторной перфузии (reperfusion injury of the myocardium and brain), хронический гломерулонефрит, эндотоксический шок и респираторный дистресс-синдром у взрослых. Кроме того, ингибиторы PDE IV полезны при лечении несахарного диабета и состояний, связанных с церебральным метаболическим ингибированием, таких как церебральная старость, старческое слабоумие (болезнь Альцгеймера), нарушение памяти, связанное с болезнью Паркинсона, депрессия и мультиинфарктное слабоумие (multi-infarct dementia). Ингибиторы PDE IV также применимы при состояниях, при которых показаны нейропротекторы, таких как остановка сердца, припадок и перемежающаяся хромота. Кроме того, ингибиторы PDE IV могут быть полезны в качестве гастрозащитных агентов. Особым вариантом терапевтических методов настоящего изобретения является лечение астмы.
Предполагаемые для лечения данным способом вирусы представляют собой такие вирусы, которые продуцируют TNF в результате инфекции, или вирусы, которые чувствительны к ингибированию (такому как снижение репликации, прямое или косвенное) посредством ингибиторов TNF формулы (i). Такие вирусы включают (но не ограничены этим) ВИЧ-1, ВИЧ-2 и ВИЧ-3, цитомегаловирус (CMV), грипп, аденовирус и вирусы группы герпеса, такие как Herpes zoster и Herpes simplex (но не ограничены только ими).
Более подробно, данное изобретение касается способа лечения млекопитающего, пораженного вирусом иммунодефицита человека (ВИЧ), который включает введение такому млекопитающему эффективного TNF-ингибирующего количества соединения формулы (i) или его фармацевтически приемлемой соли.
Соединения данного изобретения можно также использовать в комплексном ветеринарном лечении животных, отличных от человека, нуждающихся в ингибировании продуцирования TNF. Подлежащие лечению, терапевтическому или профилактическому, опосредованные TNF заболевания включают болезненные состояния, такие как перечисленные выше, но, в особенности, вирусные инфекции. Примеры таких вирусов включают (но не ограничены этим) кошачий вирус иммунодефицита (FIV) или другие ретровирусные инфекции, такие как вирус лошадиной инфекционной анемии, вирус козьего артрита, вирус visna, вирус maedi и другие лентивирусы.
Соединения данного изобретения полезны также при лечении паразитарных, дрожжевых и грибковых инфекций, когда дрожжи и грибки чувствительны к позитивной регуляции посредством TNF или вызывают продуцирование TNF in vivo. Предпочтительным для такого лечения болезненным состоянием является грибковый менингит.
Предпочтительно, чтобы соединения формулы (i) находились в фармацевтически приемлемой форме. Под фармацевтически приемлемой формой понимают, среди прочего, фармацевтически приемлемый уровень чистоты, исключая обычные фармацевтические добавки, такие как разбавители и носители, и не включая никакие материалы, считающиеся токсичными при обычной дозировке. Фармацевтически приемлемый уровень чистоты обычно составляет, по меньшей мере, 50%, исключая обычные фармацевтические добавки, предпочтительно 75%, более предпочтительно 90% и еще более предпочтительно 95%. Используемое здесь выражение "фармацевтически приемлемый" включает вещества, подходящие для применения человеком и для ветеринарного использования.
Соединение формулы (i) или в подходящих случаях его фармацевтически приемлемую соль и/или ее фармацевтически приемлемый сольват можно вводить в организм сами по себе или предпочтительно в виде фармацевтической композиции, включающей также фармацевтически приемлемый носитель.
Таким образом, настоящее изобретение предлагает фармацевтическую композицию, включающую соединение формулы (i) или в подходящих случаях его фармацевтически приемлемую соль и/или ее фармацевтически приемлемый сольват и фармацевтически приемлемый носитель.
Активное соединение можно приготовить для приема любым удобным способом, предпочтительный способ приема зависит от требующих лечения заболеваний, и предпочтительный препарат представляет собой стандартную дозированную форму или форму, которую человек может принимать сам в виде разовой дозы. Преимущественно композиция подходит для орального, ректального, локального, парентерального введения или введения через дыхательные пути. Препараты могут быть предназначены для медленного высвобождения активного компонента.
Используемый здесь термин парентеральный включает подкожные инъекции, внутривенные, внутримышечные, внутригрудинные инъекции или вливания. В дополнение к лечению теплокровных животных, таких как мыши, крысы, лошади, крупный рогатый скот, овцы, собаки, кошки и др., соединения настоящего изобретения эффективны для лечения людей.
Композиции данного изобретения могут иметь вид таблеток, капсул, саше, ампул, порошков, гранул, лепешек, суппозиториев, порошков для получения нужного состава или жидких препаратов, таких как растворы или суспензии для орального приема или стерильные растворы или суспензии для парентерального приема. Когда это удобно, препараты для локального применения также рассматриваются как приемлемые.
Для обеспечения равномерности (постоянства) приема предпочтительно, чтобы композиция данного изобретения представляла собой стандартную дозу. Стандартные дозированные формы для орального приема могут представлять собой таблетки и капсулы и могут содержать обычные наполнители (excipients), такие как связующие агенты, например, сироп, аравийскую камедь, желатин, сорбит, трагакант или поливинилпирролидон; наполнители (fillers), например, микрокристаллическую целлюлозу, лактозу, сахар, кукурузный крахмал, фосфат кальция, сорбит или глицин; смазывающие вещества для получения таблеток, например, стеарат магния; разрыхлители, например, крахмал, поливинилпирролидон, натрий крахмал гликолат или микрокристаллическую целлюлозу; или фармацевтически приемлемые увлажняющие агенты, такие как лаурилсульфат натрия.
Твердые композиции для орального приема можно получать обычными способами смешивания, заполнения, таблетирования или подобными. Повторные стадии смешивания можно применять для распределения активного ингредиента по композициям с использованием больших количеств наполнителей.
Такие методики, конечно, общеизвестны для специалистов. На таблетки можно нанести покрытие в соответствии со способами, хорошо известными в обычной фармацевтической практике, в частности, покрыть энтеросолюбильной оболочкой.
Жидкие препараты для орального применения могут быть, например, в виде эмульсий, сиропов или эликсиров или могут представлять собой сухой продукт для получения перед применением нужного состава с водой или другим подходящим растворителем. Такие жидкие препараты могут содержать обычные добавки, такие как суспендирующие агенты, например, сорбит, сироп, метилцеллюлозу, желатин, гидроксиэтилцеллюлозу, карбоксиметилцеллюлозу, гель стеарата алюминия, пищевые гидрированные жиры; эмульгаторы, например, лецитин, сорбитанмоноолеат или аравийскую камедь; неводные растворители (которые могут включать пищевые масла), например, миндальное масло, кокосовое масло, сложные эфиры масляных кислот, такие как эфиры глицерина, пропиленгликоля или этилового спирта; консерванты, например, метил- или пропил-пара-гидроксибензоат или сорбиновую кислоту; и, если требуется, обычные вкусовые или подкрашивающие агенты.
Композиции могут также быть подходящими для введения в дыхательные пути в виде порошка для вдыхания через нос, или аэрозоля, или раствора для ингалятора, или микродисперсного порошка для инсуффляции, самого по себе или в комбинации с инертным носителем, таким как лактоза. В таком случае удобно, чтобы частицы активного соединения имели диаметр менее 50 мкм, например, от 0,1 до 50 мкм, предпочтительно менее 10 мкм, например, от 1 до 10 мкм, от 1 до 5 мкм или от 2 до 5 мкм. В подходящих случаях можно включать небольшие количества других противоастматических или бронхолитических средств, например, симпатомиметические амины, такие как изопреналин, изоэтарин, салбутамол, фенилэфрин и эфедрин; кортикостероидов, таких как преднизолон, и стимуляторов надпочечников, таких как АСТН.
Для парентерального введения готовят жидкие стандартные дозированные формы, используя соединение изобретения и стерильный носитель, и в зависимости от получаемой концентрации соединение может быть суспендировано или растворено в данном носителе. При получении растворов это соединение можно растворять в воде для инъекций и стерилизовать посредством фильтрования перед заполнением подходящих флаконов или ампул и закупориванием.
Полезно растворить в носителе адъюванты, такие как анестетики локального действия, консерванты и буферные агенты. Для повышения стабильности после заполнения флаконов композицию можно заморозить и удалить воду в вакууме. Суспензии для парентерального применения получают по существу таким же способом за исключением того, что соединение суспендируют в носителе, а не растворяют, и для стерилизации нельзя применять фильтрование. Соединение можно стерилизовать, подвергая действию этиленоксида перед суспендированием в стерильном носителе. Полезно включить в композицию поверхностно-активное соединение или смачивающий агент для облегчения однородного распределения соединения.
Эти композиции могут содержать от 0,1 до 99 вес.%, предпочтительно от 10 до 60 вес.% активного соединения в зависимости от способа введения.
Соединение формулы (i) или его подходящую фармацевтически приемлемую соль и/или ее фармацевтически приемлемый сольват можно также применять в виде препарата локального действия в комбинации с обычными наполнителями для препаратов локального действия.
Препараты локального действия могут представлять собой, например, мази, кремы или лосьоны, перевязочные материалы с пропиткой, гели, гелевые палочки, спреи и аэрозоли и содержать обычные полезные добавки, такие как консерванты, растворители, способствующие прониканию лекарства, и смягчающие агенты в мазях и кремах. Эти препараты могут содержать обычные совместимые носители, такие как основы для кремов и мазей и этанол или олеиловый спирт для лосьонов.
Подходящие составы кремов, лосьонов, гелей, карандашей, мазей, спреев или аэрозолей, которые можно применять для соединений формулы (i) или в подходящих случаях для их фармацевтически приемлемых солей, представляют собой обычные хорошо известные специалистам препараты, например, как описано в стандартных трудах, таких как Harry's Cosmeticology, опубликованный Leonard Hill Books, Remington's Pharmaceutical Sciences и British and US Pharmacopoeias.
Удобно, чтобы соединение формулы (i) или в подходящих случаях его фармацевтически приемлемая соль содержали примерно от 0,5 до 20 вес.% препарата, желательно примерно от 1 до 10 вес.%, например, от 2 до 5%.
Доза используемого при лечении соединения данного изобретения обычно зависит от тяжести заболеваний, веса пациента и относительной эффективности соединения. Однако, как правило, подходящая разовая доза может составлять от 0,1 до 1000 мг, а именно, от 0,5 до 200 мг, от 0,5 до 100 мг или от 0,5 до 10 мг, например, 0,5, 1, 2, 3, 4 или 5 мг; и такие стандартные дозы можно принимать более одного раза в день, например, 2, 3, 4, 5 или 6 раз в день, но предпочтительно 1 или 2 раза в день, таким образом, общая дневная доза для взрослого человека весом 70 кг составляет примерно от 0,1 до 1000 мг, то есть примерно от 0,001 до 20 мг/кг/день, а именно, от 0,007 до 3, от 0,007 до 1,4, от 0,007 до 0,14 или от 0,01 до 0,5 мг/кг/день, например, 0,01, 0,02, 0,04, 0,05, 0,06, 0,08, 0,1 или 0,2 мг/кг/день, и такое лечение может продолжаться в течение ряда недель или месяцев.
Способы исследования
Исследования, применяемые для подтверждения ингибирующей активности соединений формулы (i) относительно фосфодиэстеразы IV, представляют собой стандартные методики исследования, описанные в работах Schilling и др., Anal. Biochem. 216: 154 (1994), Thompson and Strada, Adv. Cycl. Nucl. Res. 8:119 (1979) и Gristwood and Owen, Br. J. Pharmacol. 87:91P (1986).
В этих исследованиях соединения формулы (i) демонстрируют активность на уровне, соответствующем тому, что считается полезным действием при лечении болезненных состояний, связанных с фосфодиэстеразой IV.
Способность соединений формулы (i) ингибировать продуцирование TNF в моноядерных клетках периферической крови человека (PBMC's) измеряют следующим образом. PBMC's получают из свежеотобранной крови или "Buffy coats" (лейкоцитных пленок) стандартными методиками. Клетки высевают в RPMI1640 + 1% фетальной телячьей сыворотки в присутствии или в отсутствие ингибиторов. Добавляют LPS (липополисахарид (эндотоксин); 100 нг/мл) и культуры инкубируют в течение 22 ч при 37oС в атмосфере 95% воздуха/5% СО2. Супернатанты проверяют на TNFα посредством исследования ELISA (исследование на иммуносорбенте, связанном с ферментом), используя коммерчески доступные наборы.
Определения
Активность определяют на модели легких морских свинок, применяя методики, описанные в работах Mauser и др., Am. Rev. Respir. Dis. 148:1623 (1993) и Am. J. Respir. Crit. Care Med. 152:467 (1995).
Фармакокинетический профиль соединений данного изобретения определяют у крыс, которым в правую сонную артерию введена канюля для сбора крови. Соединение для внутривенного введения готовят в виде подходящего состава, например, в 10% (объем/объем) диметилсульфоксиде, 50% (объем/объем) PEG 400 (полиэтиленгликоль 400) в воде, и дозирование проводят посредством катетера, введенного в левую шейную вену. Образцы отбирают через 5 мин, 0,5, 1, 2, 4, 6 и 8 ч после введения дозы. Соединение для орального введения готовят в виде подходящего состава, например, в 0,4% (вес/объем) метилцеллюлозе в воде. Образцы крови отбирают через 0,5, 1, 2, 4, 6 и 8 ч после введения дозы. В некоторых случаях образцы отбирают через 12 ч после введения дозы. Посредством центрифугирования каждого образца крови получают плазму и затем определяют концентрацию лекарства, применяя стандартные методы, такие как жидкостная хроматография - масс-спектрометрия с последующим осаждением белка.
Результаты представлены ниже в табл. 1 и показаны также на приложенном чертеже. Чертеж представляет собой график данных рК после орального введения крысам дозы соединения; зависимость PC (концентрация в плазме; нг/мл) от времени t (время; часы). ▪ Представляет соединение примера 8, а • свободное основание. Очевидно преимущество нового соединения.
Растворимость соединения примера 8 в воде при рН 7 составляет 0,2 мг/мл. Растворимость соответствующего свободного основания при тех же условиях составляет 0,002 мг/мл. Другие представленные в примерах соединения также демонстрируют подходящую растворимость.
В нижепредставленной табл. 2 приведены данные испытаний как in vitro, так и in vivo.
Данные in vitro получены с использованием очищенного фермента PDE4, выделенного из человеческих клеток U937. Тест проводился в соответствии с методикой, описанной выше.
Данные in vivo получены на модели легких морских свинок, с применением методики, описанной выше, и представляют собой % подавления эозинофилии. Эта модель соответствует астме.
Кроме того, соединение по примеру 8 также оценивали на модели нейтрофилии у крыс, которая соответствует COPD - хроническому обструктивному заболеванию легких. Подавление притока нейтрофилов соединением примера 8 в этой модели составило 62% при оральном введении в дозе 3 мг/кг. Модель на крысах подробно описана в Pulmonary Pharmacology & Therapeuticals (2001) 14, 157-164, Spond et al.
Соединения по изобретению по-видимому проявляют низкую токсичность, поскольку ни в одном из тестов не была обнаружена сколько-нибудь заметная токсичность.
Следующие далее Примеры иллюстрируют данное изобретение.
Промежуточный продукт 1 - 2-Трифторметилхинолин-8-ол.
Раствор 8-метокси-2-трифторметилхинолина (10,0 г) в 48% бромистоводородной кислоте (40 мл) перемешивают при кипячении с обратным холодильником в течение ночи. Реакционную смесь выливают в воду (200 мл) и доводят рН до 12,5, используя 46-48% раствор гидроксида натрия. После экстракции дихлорметаном (2•25 мл) подкисляют водный слой до рН 5,3, добавляя 37% раствор соляной кислоты. Затем смесь экстрагируют дихлорметаном (2•100 мл) и объединенные органические экстракты промывают водой, сушат над сульфатом натрия, фильтруют и удаляют растворитель в вакууме, получая продукт (9,3 г) в виде белого твердого вещества.
Масс-спектр [М+Н] 214
Промежуточный продукт 2 - 8-(трет-Бутилдиметилсиланилокси)-2-трифторметилхинолин
Раствор 2-трифторметилхинолин-8-ола (11,5 г), трет-бутилдиметилсилилхлорида (8,9 г) и триэтиламина (6,5 г) в дихлорметане (60 мл) перемешивают в течение ночи при комнатной температуре. Реакционную смесь промывают водой (2•50 мл), сушат над сульфатом натрия, фильтруют и удаляют растворитель в вакууме, получая продукт (17,9 г) в виде белого твердого вещества.
Масс-спектр [М+Н] 328.
Следующий промежуточный продукт получают по аналогичной методике.
Промежуточный продукт 3 - 8-(трет-Бутилдиметилсиланилокси)-2-ме тилхинолин
Готовят из 8-гидроксихинолина (10 г), получая продукт (17 г) в виде оранжевого масла.
Тонкослойная хроматография (ТСХ) Rf 0,90 (10% метанол в этилацетате).
Промежуточный продукт 4 - 5-Бром-8-(трет-бутилдиметилсиланилокси)-2-трифторметилхинолин
Раствор 8-(трет-бутилдиметилсиланилокси)-2-трифторметилхинолина (17,5 г) в дихлорметане (100 мл) обрабатывают N-бромсукцинимидом (10,5 г) при 15oС. Смесь перемешивают при 20oС в течение 25 мин, промывают 1% раствором сульфата натрия (100 мл) и водой (50 мл). Органический слой отделяют, сушат над сульфатом магния, фильтруют и удаляют растворитель в вакууме, получая продукт (21,4 г) в виде темного масла.
Масс-спектр [М+Н] 406.
Следующий промежуточный продукт получают по аналогичной методике.
Промежуточный продукт 5 - 5-Бром-8-(трет-бутилдиметилсиланилокси)-2-метилхинолин
Готовят из 8-(трет-бутилдиметилсиланилокси)-2-метилхинолина (0,63 г), получая продукт (0,66 г) в виде желтого масла.
ТСХ Rf 0,90 (дихлорметан).
Промежуточный продукт 6 - 5-Бром-2-трифторметилхинолин-8-ол.
Раствор 5-бром-8-(трет-бутилдиметилсиланилокси)-2-трифторметилхинолина (21 г) в метаноле (150 мл) обрабатывают 37% раствором соляной кислоты (5 мл) и водой (5 мл). Смесь перемешивают при комнатной температуре в течение 12 ч и при 45oС в течение 2 ч. Метанол удаляют в вакууме, а остаток распределяют между 10% раствором гидроксида натрия (100 мл) и дихлорметаном (50 мл). Водный слой нейтрализуют 37% раствором соляной кислоты до рН 7,2 и экстрагируют дихлорметаном (4•50 мл). Объединенные органические экстракты сушат над сульфатом магния, фильтруют и удаляют растворитель в вакууме, получая продукт (12 г) в виде твердого вещества кремового цвета.
Масс-спектр [М] 292.
Промежуточный продукт 7 - 5-Бром-2-метилхинолин-8-ол
Раствор 5-бром-8-(трет-бутилдиметилсиланилокси)-2-метилхинолина (16,3 г) в тетрагидрофуране (500 мл) по каплям обрабатывают раствором тетрабутиламмонийфторида (1,0 М в тетрагидрофуране, 54 мл). Смесь перемешивают в течение 10 мин, разбавляют дихлорметаном (750 мл) и промывают водой (3•250 мл). Органический раствор сушат над сульфатом магния, фильтруют и удаляют растворитель в вакууме, получая оранжевое масло. Перекристаллизация из водного метанола дает продукт (7,65 г) в виде белого твердого вещества.
ТСХ Rf 0,58 (10% метанол в дихлорметане).
Промежуточный продукт 8 - 5-Бром-8-дифторметокси-2-трифторметилхинолин
К перемешиваемому раствору 5-бром-2-трифторметилхинолин-8-ола (12,0 г) в диоксане (120 мл) добавляют 47% раствор гидроксида натрия (12 мл). Смесь нагревают до 78oС и пропускают через нее хлордифторметан (7,4 г) в течение 3 ч. После охлаждения смесь разбавляют водой (80 мл) и удаляют растворитель в вакууме. Полученную суспензию фильтруют и остаток на фильтре промывают дихлорметаном (50 мл), а затем водой (50 мл). Органический слой отделяют, а водный слой экстрагируют дихлорметаном (50 мл). Объединенные органические экстракты промывают 0,5% раствором гидроксида натрия (100 мл), сушат над сульфатом магния и удаляют растворитель в вакууме. Остаток переносят в трет-бутилметиловый эфир (100 мл), мутный раствор фильтруют и удаляют растворитель в вакууме, получая продукт (11,7 г) в виде грязно-белого твердого вещества.
Масс-спектр [М+Н] 342.
Следующий промежуточный продукт получают по аналогичной методике.
Промежуточный продукт 9 - 5-Бром-8-дифторметокси-2-метилхинолин
Готовят из 5-бром-2-метилхинолин-8-ола (1,0 г), получая коричневое твердое вещество. Очистка посредством перекристаллизации из метанола дает продукт (0,96 г) в виде грязно-белого твердого вещества.
ТСХ Rf 0,86 (50% этилацетат в гексане).
Промежуточный продукт 10 - 8-Дифторметокси-2-трифторметилхинолин-5-карбоновая кислота
Смесь 5-бром-8-дифторметокси-2-трифторметилхинолина (6,0 г), трифенилфосфина (0,3 г), бис(трифенилфосфин)палладий(II) хлорида (0,15 г), 47% раствора гидроксида натрия (4,5 г) и воды (12 мл) в тетрагидрофуране (50 мл) продувают газообразным монооксидом углерода в реакторе Парра (Parr pressure reactor) при давлении 7 бар. Смесь нагревают до 100oС в течение 24 ч. После охлаждения и вентиляции реакционную смесь распределяют между раствором гидроксида натрия (1,5 г в 50 мл) и трет-бутилметиловым эфиром (100 мл). Органический раствор экстрагируют раствором гидроксида натрия (2•1,5 г в 50 мл). Объединенные водные экстракты перемешивают с активированным углем (1,5 г) в течение 15 мин и затем фильтруют. Фильтрат подкисляют до рН 4, используя 37% раствор соляной кислоты, а полученный осадок кремового цвета отделяют фильтрованием и промывают водой (20 мл). Сырой продукт очищают перекристаллизацией из толуола, получая продукт (1,8 г) в виде твердого вещества кремового цвета.
Масс-спектр [М+Н] 308.
Следующий промежуточный продукт получают по аналогичной методике.
Промежуточный продукт 11 - 8-Дифторметокси-2-метилхинолин-5-карбоновая кислота
Готовят из 5-бром-8-дифторметокси-2-метилхинолина (5,72 г), получая продукт (2,88 г) в виде коричневого твердого вещества.
ТСХ Rf 0,60 (10% метанол в дихлорметане).
Промежуточный продукт 12 - 4-Нитрофениловый эфир 8-дифторметокси-2-трифторметилхинолин-5-карбоновой кислоты
Раствор 8-дифторметокси-2-трифторметилхинолин-5-карбоновой кислоты (0,5 г) в дихлорметане (50 мл) обрабатывают 4-нитрофенолом (0,25 г), 4-диметиламинопиридином (каталитическое количество) и 1-(3-диметиламинопропил)-3-этил-карбодиимидом (0,35 г) и смесь перемешивают при комнатной температуре в течение 12 ч. Реакционную смесь промывают водой (50 мл), сушат над сульфатом натрия, фильтруют и удаляют растворитель в вакууме. Остаток очищают колоночной хроматографией на диоксиде кремния, элюируя дихлорметаном, и получают продукт (0,47 г) в виде твердого вещества кремового цвета.
ТСХ Rf 0,75 (5% этилацетат в дихлорметане).
Следующие промежуточные продукты получают по аналогичной методике.
Промежуточный продукт 13 - 4-Нитрофениловый эфир 8-дифторметокси-2-метилхинолин-5-карбоновой кислоты
Готовят из 8-дифторметокси-2-метилхинолин-5-карбоновой кислоты (0,50 г). Очистка колоночной хроматографией на диоксиде кремния при элюировании 50% этилацетатом в гексане дает продукт (0,63 г) в виде желтого твердого вещества.
ТСХ Rf 0,73 (10% метанол в дихлорметане).
Промежуточный продукт 14 - 4-Нитрофениловый эфир 8-метокси-2-трифторметилхинолин-5-карбоновой кислоты.
Готовят из 8-метокси-2-трифторметилхинолин-5-карбоновой кислоты (0,60 г), получая указанное в заголовке соединение (0,75 г) в виде желтого твердого вещества.
ТСХ Rf 0,64 (50% этилацетат в гексане).
Промежуточный продукт 15 - (3-Хлорпиридин-4-ил)амид 8-дифторметокси-2-метилхинолин-5-карбоновой кислоты.
К перемешиваемому раствору 4-амино-3-хлорпиридина (136 мг) в N,N-диметилформамиде (2 мл) в атмосфере азота добавляют гидрид натрия (60% дисперсия в масле, 42 мг). Реакционную смесь перемешивают при комнатной температуре в течение 1 ч. Затем добавляют 4-нитрофениловый эфир 8-дифторметокси-2-метилхинолин-5-карбоновой кислоты (200 мг) и продолжают перемешивание в течение 18 ч. Удаляют растворитель в вакууме, а полученный остаток очищают колоночной хроматографией на диоксиде кремния, элюируя 50% этилацетатом в гексане, и получают продукт (155 мг) в виде белого твердого вещества.
TCX Rf 0,3 (50% этилацетат в гексане).
Следующие промежуточные продукты получают по аналогичной методике.
Промежуточный продукт 16 - (3-Метилпиридин-4-ил)амид 8-дифторметокси-2-метилхинолин-5-карбоновой кислоты
Готовят из 4-нитрофенилового эфира 8-дифторметокси-2-метилхинолин-5-карбоновой кислоты (500 мг) и 4-амино-3-метилпиридина (170 мг). Очистка колоночной хроматографией на диоксиде кремния при элюировании 10% метанолом в дихлорметане дает продукт (200 мг) в виде бледно-желтого твердого вещества.
TCX Rf 0,55 (10% метанол в этилацетате).
Промежуточный продукт 17 - (3-Хлорпиридин-4-ил)амид 8-дифторметокси-2-трифторметилхинолин-5-карбоновой кислоты
Готовят из 4-нитрофенилового эфира 8-дифторметокси-2-трифторметилхинолин-5-карбоновой кислоты (466 мг) и 4-амино-3-хлорпиридина (283 мг). Очистка колоночной хроматографией на диоксиде кремния при элюировании 15% этилацетатом в дихлорметане дает продукт (297 мг) в виде белого твердого вещества.
TCX Rf 0,26 (15% этилацетат в дихлорметане).
Промежуточный продукт 18 - (3,5-Дихлорпиридин-4-ил)амид 8-дифторметокси-2-трифторметилхинолин-5-карбоновой кислоты
Готовят из 4-нитрофенилового эфира 8-дифторметокси-2-трифторметилхинолин-5-карбоновой кислоты (480 мг) и 4-амино-3,5-дихлорпиридина (360 мг). Очистка колоночной хроматографией на диоксиде кремния при элюировании 20% этилацетатом в гексане дает продукт (424 мг) в виде белого твердого вещества.
TCX Rf 0,42 (20% этилацетат в гексане).
Промежуточный продукт 19 - (3,5-Дифторпиридин-4-ил)амид 8-дифторметокси-2-трифторметилхинолин-5-карбоновой кислоты
Готовят из 4-нитрофенилового эфира 8-дифторметокси-2-трифторметилхинолин-5-карбоновой кислоты (390 мг) и 4-амино-3,5-дифторпиридина (120 мг). Очистка колоночной хроматографией на диоксиде кремния при элюировании 10% этилацетатом в дихлорметане дает продукт (180 мг) в виде белого твердого вещества.
TCX Rf 0,27 (15% этилацетат в дихлорметане).
Промежуточный продукт 20 - (3,5-Дифторпиридин-4-ил)амид 8-метокси-2-трифторметилхинолин-5-карбоновой кислоты
Готовят из 4-нитрофенилового эфира 8-метокси-2-трифторметилхинолин-5-карбоновой кислоты (425 мг) и 4-амино-3,5-дифторпиридина (282 мг). Очистка колоночной хроматографией на диоксиде кремния при элюировании 5% метанолом в дихлорметане дает продукт (162 мг) в виде белого твердого вещества.
TCX Rf 0,34 (5% метанол в дихлорметане).
Промежуточный продукт 21 - (3-Хлорпиридин-4-ил)амид 8-метокси-2-трифторметилхинолин-5-карбоновой кислоты
К перемешиваемому раствору 4-амино-3-хлорпиридина (124 мг) в N,N-диметилформамиде (5 мл) в атмосфере азота добавляют гидрид натрия (60% дисперсия в масле, 52 мг). Реакционную смесь перемешивают при комнатной температуре в течение 1 ч. Затем добавляют 8-метокси-2-трифторметилхинолин-4-карбонилхлорид (360 мг) и продолжают перемешивание в течение 18 ч. Удаляют растворитель в вакууме и полученный остаток распределяют между этилацетатом (2•50 мл) и водой (50 мл). Органический слой отделяют, сушат над сульфатом магния, фильтруют и удаляют растворитель в вакууме. Очистка колоночной хроматографией на диоксиде кремния с элюированием этилацетатом дает продукт (330 мг) в виде бледно-розового твердого вещества.
ТСХ Rf 0,41 (этилацетат).
Т.пл. 192-194oС.
Следующий промежуточный продукт получают по аналогичной методике.
Промежуточный продукт 22 - 3-(Метилпиридин-4-ил)амид 8-метокси-2-трифторметилхинолин-5-карбоновой кислоты
Готовят из 8-метокси-2-трифторметилхинолин-4-карбонилхлорида (430 мг) и 4-амино-3-метилпиридина (170 мг). Очистка колоночной хроматографией при элюировании 10% метанолом в этилацетате дает продукт (160 мг) в виде белого твердого вещества.
ТСХ Rf 0,29 (10% метанол в этилацетате).
Промежуточный продукт 23 - (3-Метилпиридин-4-ил)амид 8-дифторметокси-2-трифторметилхинолин-5-карбоновой кислоты
Раствор 8-дифторметокси-2-трифторметилхинолин-4-карбоновой кислоты (0,50 г) в дихлорметане (30 мл) перемешивают при комнатной температуре в атмосфере азота. Добавляют оксалилхлорид (0,28 мл), а затем N,N-диметилформамид (1 каплю) и продолжают перемешивание в течение ночи. Удаляют растворитель в вакууме, получая 8-дифторметокси-2-трифторметилхинолин-4-карбонилхлорид (650 мг) в виде грязно-белого твердого вещества.
К перемешиваемому раствору 8-дифторметокси-2-трифторметилхинолин-4-карбонилхлорида (650 мг) в дихлорметане (40 мл) в атмосфере азота добавляют триэтиламин (0,68 мл) и 4-амино-3-метилпиридин (352 мг). Реакционную смесь перемешивают в течение 18 ч. Удаляют растворитель в вакууме, а полученный остаток очищают колоночной хроматографией на диоксиде кремния при элюировании 5% метанолом в дихлорметане и получают продукт (563 мг) в виде не совсем белого твердого вещества.
ТСХ Rf 0,53 (10% метанол в дихлорметане).
Следующий промежуточный продукт получают по аналогичной методике.
Промежуточный продукт 24 - (3,5-Диметилпиридин-4-ил)амид 8-метокси-2-трифторметилхинолин-5-карбоновой кислоты
Готовят из 8-метокси-2-трифторметилхинолин-4-карбонилхлорида (500 мг) и 4-амино-3,5-диметилпиридина (210 мг). Очистка посредством растирания с ацетоном и эфиром дает продукт (82 мг) в виде бледно-желтого твердого вещества.
ТСХ Rf 0,42 (10% метанол в дихлорметане с 1% гидроксида аммония).
Пример 1 (3-хлор-1-оксипиридин-4-ил)амид 8-дифторметокси-2-метилхинолин-5-карбоновой кислоты
Перуксусную кислоту (36-40% в уксусной кислоте, 0,1 мл) добавляют к раствору (3-хлорпиридин-4-ил)амида 8-дифторметокси-2-метилхинолин-5-карбоновой кислоты (50 мг) в хлороформе (10 мл) при комнатной температуре. После перемешивания в течение ночи реакционную смесь разбавляют дихлорметаном (20 мл) и промывают водой (10 мл). Органическую фазу сушат над сульфатом магния и удаляют растворитель в вакууме, получая белое твердое вещество. Очистка колоночной хроматографией при элюировании 10% метанолом в этилацетате дает продукт (25 мг) в виде белого твердого вещества.
ТСХ Rf 0,2 (10% метанол в этилацетате).
Соединения следующих Примеров получают по аналогичной методике.
Пример 2 (3-Хлор-1-оксипиридин-4-ил)амид 8-дифторметокси-2-трифторметилхинолин-5-карбоновой кислоты
Готовят из (3-хлорпиридин-4-ил)амида 8-дифторметокси-2-трифторметилхинолин-5-карбоновой кислоты (261 мг), получая продукт (223 мг) в виде твердого вещества кремового цвета.
ТСХ Rf 0,4 (этилацетат).
Т. пл. 212-213oС.
Пример 3 (3-Хлор-1-оксипиридин-4-ил)амид 8-метокси-2-трифторметилхинолин-5-карбоновой кислоты
Готовят из (3-хлорпиридин-4-ил)амида 8-метокси-2-трифторметилхинолин-5-карбоновой кислоты (50 мг), получая продукт (25 мг) в виде грязно-белого твердого вещества.
ТСХ Rf 0,7 (10% метанол в этилацетате).
Т. пл. 261,5-262,5oС.
Пример 4 (3,5-Дифтор-1-оксипиридин-4-ил)амид 8-метокси-2-трифторметилхинолин-5-карбоновой кислоты
Готовят из (3,5-дифторпиридин-4-ил)амида 8-метокси-2-трифторметилхинолин-5-карбоновой кислоты (120 мг) с перемешиванием при комнатной температуре в течение 2 недель. За этот период добавляют избыток перуксусной кислоты (4•0,5 мл). Очистка колоночной хроматографией при элюировании 5-10% метанолом в дихлорметане дает продукт (28 мг) в виде желтого твердого вещества.
ТСХ Rf 0,09 (5% метанол в дихлорметане).
Т.пл. 268-269oС (разлагается).
Пример 5 (3,5-Дифтор-1-оксипиридин-4-ил)амид 8-дифторметокси-2-трифторметилхинолин-5-карбоновой кислоты
Готовят из (3,5-дифторпиридин-4-ил)амида 8-дифторметокси-2-трифторметилхинолин-5-карбоновой кислоты (160 мг) с перемешиванием при комнатной температуре в течение 2 недель. За этот период добавляют избыток перуксусной кислоты (3•0,1 мл). Очистка колоночной хроматографией при элюировании 15% этилацетатом в дихлорметане с увеличением количества метанола до 10% в дихлорметане дает продукт (120 мг) в виде желтого твердого вещества.
ТСХ Rf 0,69 (2% метанол в дихлорметане).
Т.пл. 219-220oС.
Пример 6 (3-Метил-1-оксипиридин-4-ил)амид 8-дифторметокси-2-трифторметилхинолин-5-карбоновой кислоты
Готовят из (3-метилпиридин-4-ил)амида 8-дифторметокси-2-трифторметилхинолин-5-карбоновой кислоты (316 мг), который перемешивают в присутствии перуксусной кислоты (2•0,18 мл) в течение двух дней. Очистка колоночной хроматографией при элюировании 10% метанолом в дихлорметане дает продукт (267 мг) в виде белого твердого вещества.
TCX Rf 0,25 (10% метанол в дихлорметане).
Т.пл. 210-212oС.
Пример 7 (3,5-Диметил-1-оксипиридин-4-ил)амид 8-метокси-2-трифторметилхинолин-5-карбоновой кислоты
Готовят из (3,5-диметилпиридин-4-ил)амида 8-метокси-2-трифторметилхинолин-5-карбоновой кислоты (56 мг), который перемешивают в присутствии перуксусной кислоты (2•О/05 мл) в течение двух дней. Очистка колоночной хроматографией при элюировании смесью 1% гидроксид аммония/10% метанол в дихлорметане дает продукт (37 мг) в виде белого твердого вещества.
TCX Rf 0,22 (1% гидроксид аммония/10% метанол в дихлорметане).
Т.пл. 237-239oС.
Пример 8 (3,5-Дихлор-1-оксипиридин-4-ил)амид 8-метокси-2-трифторметилхинолин-5-карбоновой кислоты
(3,5-Дихлорпиридин-4-ил)амид 8-метокси-2-трифторметил-хинолин-5-карбоновой кислоты (200 мг) перемешивают в присутствии перуксусной кислоты (36-40% в уксусной кислоте, 0,1 мл) в хлороформе при 50oС в течение 5 дней. Добавляют еще перуксусную кислоту (0,1 мл) и реакционную смесь нагревают еще 2 дня. Очистка колоночной хроматографией при элюировании 10% метанолом в этилацетате дает продукт (123 мг) в виде белого твердого вещества.
TCX Rf 0,17 (10% метанол в этилацетате).
Т.пл. 280-281oС.
Пример 9 (3,5-Дихлор-1-оксипиридин-4-ил)амид 8-дифторметокси-2-трифторметилхинолин-5-карбоновой кислоты
Получают из (3,5-дихлорпиридин-4-ил)амида 8-дифторметокси-2-трифторметилхинолин-5-карбоновой кислоты (415 мг) аналогично с получением (3,5-дихлор-1-оксипиридин-4-ил)амида 8-метокси-2-трифторметилхинолин-5-карбоновой кислоты. Очистка колоночной хроматографией при элюировании смесью 1% гидроксид аммония/10% метанол в дихлорметане дает указанное в заголовке соединение в виде твердого вещества кремового цвета (360 мг).
ТСХ Rf 0,5 (1% гидроксид аммония/10% метанол в дихлорметане).
Т.пл. 244-245oС.
Пример 10 (3-Метил-1-оксипиридин-4-ил)амид 8-дифторметокси-2-метилхинолин-5-карбоновой кислоты.
Гидрид натрия (60% дисперсия в масле, 0,11 г) добавляют к перемешиваемому раствору 3-метил-1-оксипиридин-4-иламина (0,2 г) в N,N-диметилформамиде (10 мл) в атмосфере азота при комнатной температуре в присутствии молекулярных сит. После перемешивания в течение 1 ч добавляют 4-нитрофениловый эфир 8-дифторметокси-2-метилхинолин-5-карбоновой кислоты и реакционную смесь перемешивают в течение ночи. Удаляют растворитель в вакууме и остаток распределяют между этилацетатом (50 мл) и водой (2•50 мл). Органическую фазу сушат над сульфатом магния и концентрируют в вакууме. Остаток промывают небольшим количеством этилацетата и сушат, получая продукт (50 мг) в виде бледно-желтого твердого вещества.
ТСХ Rf 0,27 (1% триэтиламин/20% метанол в дихлорметане).
Т.пл. 231,5-233,5oС.
Пример 11 (3-Метил-1-оксипиридин-4-ил)амид 8-метокси-2-трифторметилхинолин-5-карбоновой кислоты.
Добавляют триэтиламин (0,55 мл) и 4-диметиламинопиридин (каталитическое количество) к перемешиваемой суспензии 3-метил-1-оксипиридин-4-иламина (0,23 г) в дихлорметане (40 мл) в атмосфере азота при комнатной температуре. Добавляют гидрохлоридную соль 8-метокси-2-трифторметилхинолин-5-карбонилхлорида (0,6 г) и реакционную смесь перемешивают в течение ночи. Удаляют растворитель в вакууме и остаток распределяют между этилацетатом (50 мл) и водой (3•50 мл). Осадок в органической фазе отфильтровывают и сушат в вакууме при 45oС, получая продукт (0,2 г) в виде белого твердого вещества.
ТСХ Rf 0,12 (этилацетат).
Т.пл. 249,5-250,5oС.
| название | год | авторы | номер документа |
|---|---|---|---|
| ХИНОЛИНОВЫЕ КАРБОКСАМИДЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1997 |
|
RU2170730C2 |
| БИЦИКЛИЧЕСКИЕ КАРБОКСАМИДЫ И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1997 |
|
RU2176642C2 |
| КАРБОКСАМИДЫ БЕНЗОФУРАНА И ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1997 |
|
RU2162467C2 |
| ПРОИЗВОДНЫЕ ГИДРОКСАМОВОЙ И КАРБОНОВОЙ КИСЛОТЫ, ИМЕЮЩИЕ MMP ИНГИБИРУЮЩУЮ АКТИВНОСТЬ, И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1998 |
|
RU2210567C2 |
| ПРОИЗВОДНЫЕ БЕНЗАНИЛИДА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРОЯВЛЯЮЩАЯ АНТАГОНИЗМ К 5-HT РЕЦЕПТОРАМ | 1992 |
|
RU2077535C1 |
| ПРОИЗВОДНЫЕ 3-(5-ТЕТРАЗОЛИЛБЕНЗИЛ)АМИНОПИПЕРИДИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1994 |
|
RU2136675C1 |
| ПРОИЗВОДНЫЕ ЛАКТАМА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ЯВЛЯЮЩАЯСЯ АНТАГОНИСТОМ 5-ОКСИТРАПТАМИНА /5-НТ/ НА 5- НТ -РЕЦЕПТОРАХ ИЛИ ИХ ФИЗИОЛОГИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ И СОЛОВАТЫ | 1992 |
|
RU2067980C1 |
| (ДИГИДРО)ПИРРОЛО[2,1-А]ИЗОХИНОЛИНЫ | 2009 |
|
RU2495037C2 |
| ПРОИЗВОДНЫЕ АНТРАНИЛОВОЙ КИСЛОТЫ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1997 |
|
RU2195454C2 |
| ПРОИЗВОДНЫЕ ИНДОЛА | 1994 |
|
RU2144535C1 |

Изобретение относится к N-оксидам гетероциклических соединений формулы (I), где R1 представляет СН3, CH2F, CHF2, CF3; R2 представляет СН3, СF3; R3 представляет F, Cl, Br, СН3; R4 представляет Н, F, Cl, Br, СН3. Изобретение может быть использовано в медицине, в качестве терапевтического средства для лечения астмы, эозинофилии, нейтрофилии, хронического бронхита, хронического заболевания легких и хронического обструктивного заболевания дыхательных путей. 1 с. и 5 з.п. ф-лы, 1 ил., 2 табл.
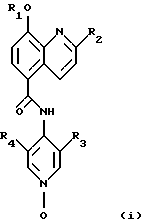
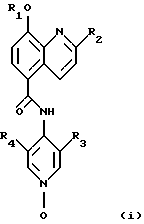
где R1 представляет собой СН3, CH2F, CHF2 или СF3;
R2 представляет собой СН3 или СF3;
R3 представляет собой F, Cl, Вr или СН3;
R4 представляет собой Н, F, Cl, Вr или СН3,
или его фармацевтически приемлемая соль.
(3-хлор-1-оксипиридин-4-ил)амида 8-дифторметокси-2-метилхинолин-5-карбоновой кислоты,
(3-хлор-1-оксипиридин-4-ил)амида 8-дифторметокси-2-трифторметилхинолин-5-карбоновой кислоты,
(3-хлор-1-оксипиридин-4-ил)амида 8-метокси-2-трифторметилхинолин-5-карбоновой кислоты,
(3,5-дифтор-1-оксипиридин-4-ил)амида 8-метокси-2-трифтор-метилхинолин-5-карбоновой кислоты,
(3,5-дифтор-1-оксипиридин-4-ил)амида 8-дифторметокси-2-трифторметилхинолин-5-карбоновой кислоты,
(3-метил-1-оксипиридин-4-ил)амида 8-дифторметокси-2-трифторметилхинолин-5-карбоновой кислоты,
(3,5-диметил-1-оксипиридин-4-ил)амида 8-метокси-2-трифторметилхинолин-5-карбоновой кислоты,
(3,5-дихлор-1-оксипиридин-4-ил)амида 8-метокси-2-трифторметилхинолин-5-карбоновой кислоты,
(3,5-дихлор-1-оксипиридин-4-ил)амида 8-дифторметокси-2-трифторметилхинолин-5-карбоновой кислоты,
(3-метил-1-оксипиридин-4-ил)амида 8-дифторметокси-2-метилхинолин-5-карбоновой кислоты,
(3-метил-1-оксипиридин-4-ил)амида 8-метокси-2-трифторметилхинолин-5-карбоновой кислоты.
| RU 95105470 A1, 10.04.1997 | |||
| WO 9748697 A1, 24.12.1997 | |||
| WO 9744036 A1, 27.11.1997. |

