Область изобретения
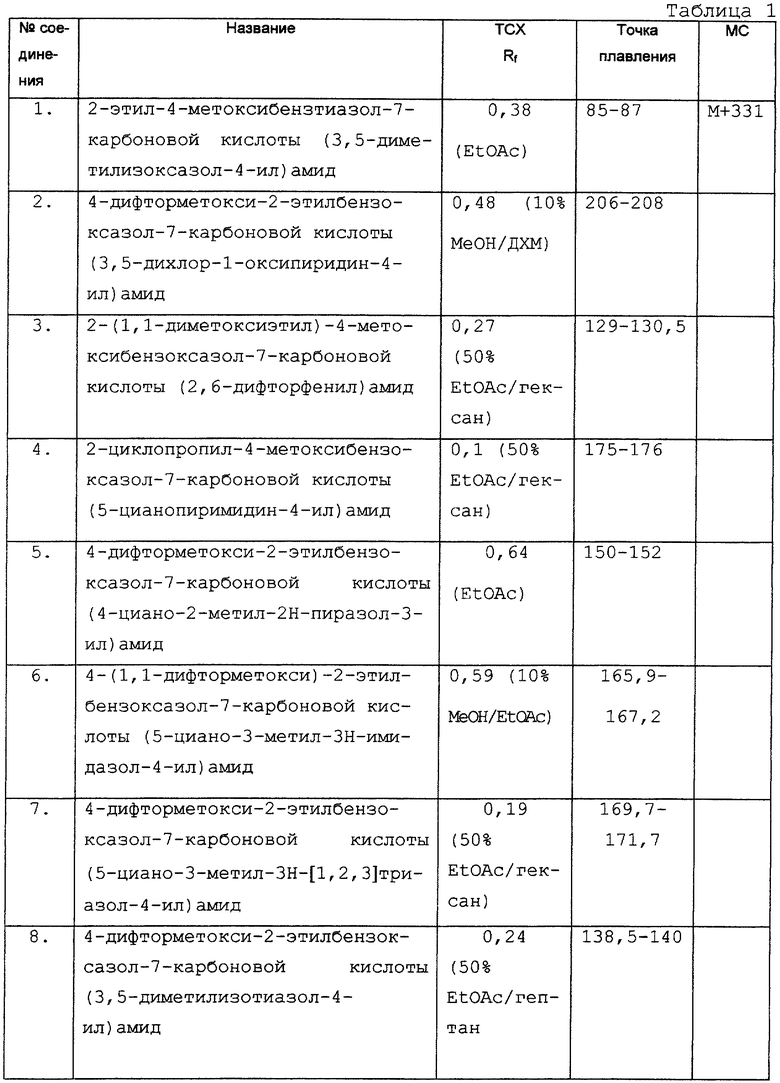
Изобретение относится к новым гетероциклическим соединениям и их фармацевтически приемлемым солям, способам их получения и составлению рецептуры на их основе и применению в качестве лекарственных средств.
Предпосылки создания изобретения
2-Тиенилбензоксазолы с антиагрегирующей активностью описаны в Eur.J.Med. Chem., (1994) 29:75.
В EP-A-0116938 и J.Med.Chem. (1987) 30, 62 описаны гетероарилоксикарбоксамиды в качестве ингибиторов липолиза, полезные для лечения ишемической болезни сердца и гипертриглицеридемии.
В WO-A-9406783 и WO-A-9406782 описаны гетероарилсульфонамиды, обладающие инсектицидной, нематоцидной, акарицидной и фунгицидной активностями.
В WO-A-9604251 описаны арилоксипроизводные гетероарильных соединений в качестве ингибиторов брадикинина.
Гетероарильные соединения описаны в качестве антагонистов фибриногена в WO-A-9408962.
В EP-A-0498722 описаны амидные производные гетероарильных соединений. В EP-A-456067 раскрываются хиноксалины в качестве усилителей продуктивности животных.
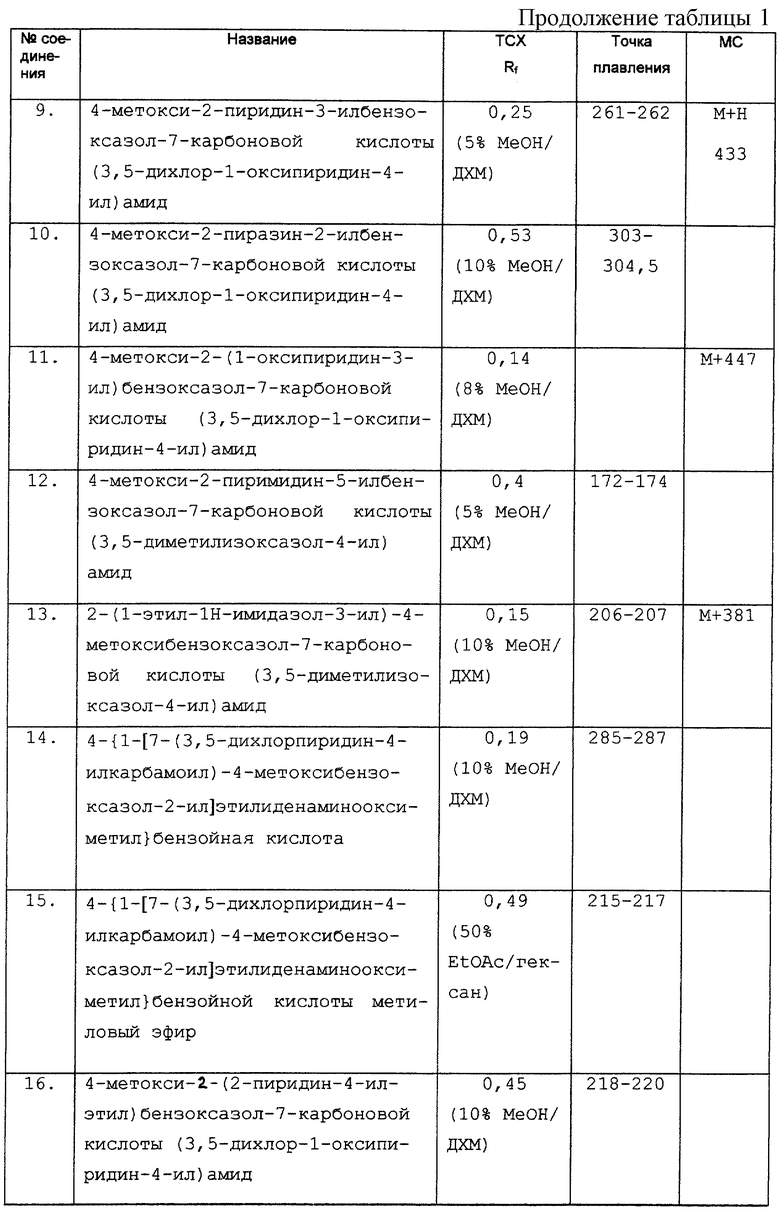
Бензимидазолы описаны в качестве антагонистов допамина в WO-A-9422839.
DE-F-4237617 раскрывает имидазолы в качестве антипаразитических агентов.
Фосфодиэстеразы (PDE) и фактор некроза опухоли (TNF), их способ действия и терапевтическая полезность их ингибиторов описаны в WO-A-9720833 и PCT/GB 97/01361, содержание которых включено в данное описание в качестве ссылки. В тех же документах описаны карбоксамиды, полезные в качестве ингибиторов PDE и TNF.
Краткое изложение сущности изобретения
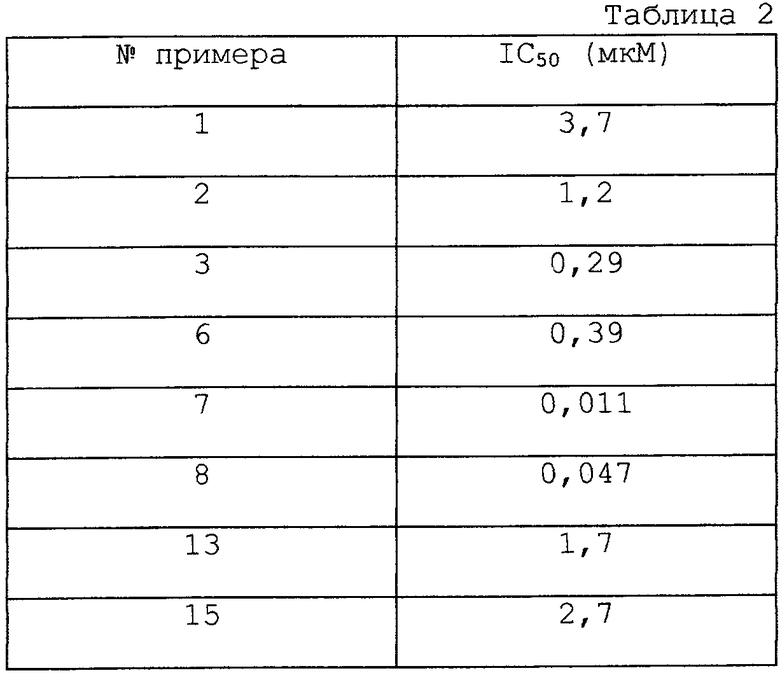
Данное изобретение относится к фармацевтическому применению соединения формулы (i), представленной ниже, для лечения болезненных состояний, например, болезненных состояний, связанных с белками, которые опосредуют клеточную активность, например, путем ингибирования фактора некроза опухоли и/или путем ингибирования фосфодиэстеразы IV. Согласно изобретению новые соединения имеют формулу (i):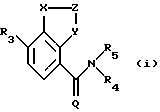
где (1) Х представляет N, и (a) Z представляет =CR1-CR2=, и Y представляет N, (b) Z представляет =CR1, и Y представляет О, S или NR4, или (с) Z представляет = CR1-N= , и Y представляет CR2, или (2) Х представляет NR4, Z представляет - CR1=, и Y представляет N;
Q представляет О или S;
R1 и R2 являются одинаковыми или разными и, каждый, представляют COR6, C(=NOR6)R13, алкил-C(=NOR6)R13, NR8R9, CON(R6)2, галоген, CF3, CN, CO2H, CO2H, R6, CO-гет, где гет представляет собой гетероциклическое кольцо (такое как морфолин или пиперидин), присоединенное через атом N в кольце и необязательно замещенное одним или несколькими R14, или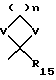
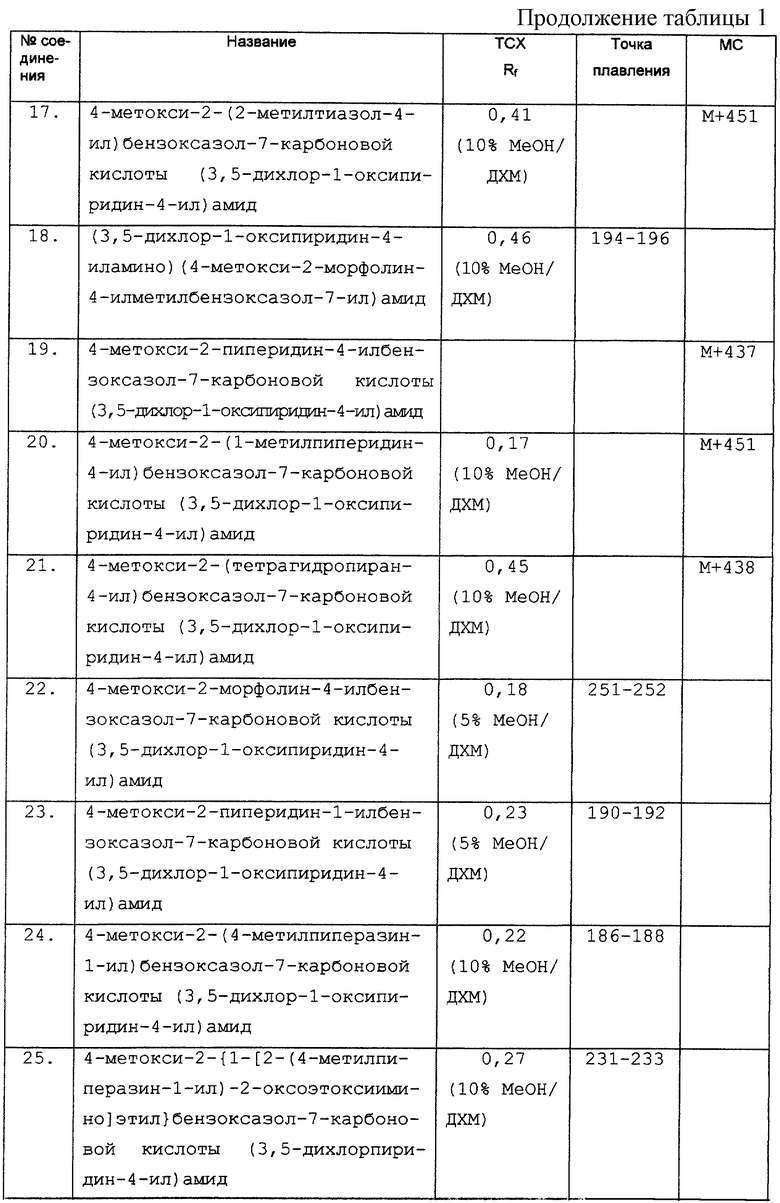
R3 представляет ОН, тиоалкил или C1-6 алкокси или циклоалкокси, каждый из которых необязательно замещен одним или несколькими галогенами;
R4 представляет Н или алкил;
R5 представляет арил или гетероарил, любой из которых может необязательно быть замещенным одним или несколькими заместителями, выбранными из галогена, необязательно галогензамещенного алкила, гидрокси, необязательно галогензамещенного алкокси, CO2H, CO2R10, CONR11R12, COR10, SO2R10, SO2NR11R12, NR9R9 и CN;
каждый R6 независимо представляет Н или группу, выбранную из алкила, циклоалкила, арила, гетероарила, гетероцикло, арилалкила, гетероарилалкила и гетероциклоалкила, и любая из этих групп необязательно замещена в любом положении заместителем R7;
R7 представляет алкил, гидрокси, OR10, NR8R9, CN, CO2H, CO2R10, CONR11R12; или COR10;
R8 представляет H, алкил, циклоалкил, арил, гетероарил, гетероцикло, арилалкил, гетероарилалкил, гетероциклоалкил, алкилкарбонил, алкоксикарбонил, арилкарбонил, гетероарилкарбонил, гетероциклокарбонил, алкилсульфонил арилсульфонил, гетероарилсульфонил или гетероциклосульфонил;
R9 представляет H, алкил, циклоалкил, арил, гетероарил гетероцикло, аралкил, гетероарилалкил или гетероциклоалкил; или NR8R9 представляет гетероциклическое кольцо (такое как морфолин или пиперидин), необязательно замещенное R14;
R10 представляет алкил, циклоалкил, арил, гетероарил, гетероцикло, арилалкил, гетероарилалкил или гетероциклоалкил;
R11 и R12 являются одинаковыми или различными и, каждый, представляют H или R10;
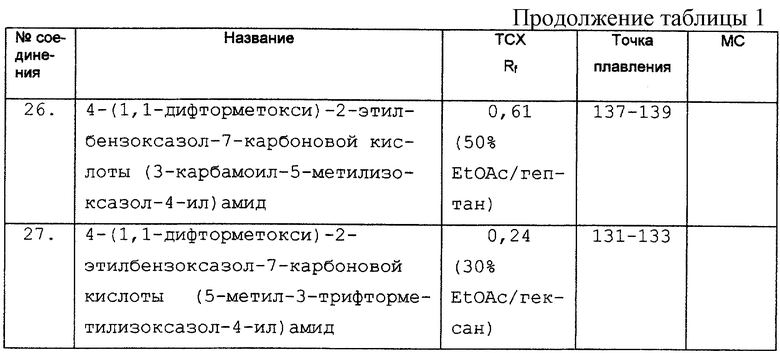
R13 представляет R10, необязательно замещенный одним или несколькими R7;
R14 представляет алкил, арилалкил или гетероарилалкил; и
R15 представляет алкил, V представляет O или S, и n составляет 2-4;
и их фармацевтически приемлемые соли.
Соединения по изобретению имеют бициклические арильные ядра. В зависимости от определений X, Y и Z они представляют (1a) хиноксалины, (1b) бензоксазолы, бензтиазолы или бензимидазолы, (1c) хиназолины или (2) бензимидазолы (замещенные иначе, чем соединения 1b). Предпочтительные соединения определены в зависимых пунктах формулы изобретения.
Описание изобретения
Подходящими фармацевтически приемлемыми солями являются соли фармацевтически приемлемых оснований и соли присоединения фармацевтически приемлемых кислот. Некоторые из соединений формулы (i), содержащие кислотную группу, образуют соли с основаниями. Подходящие фармацевтически приемлемые соли оснований включают соли металлов, такие как соли щелочных металлов, например, соли натрия или соли с органическими аминами, такие как с этилендиамином.
Некоторые из соединений формулы (i), содержащие аминогруппу, образуют соли присоединения кислот. Подходящие соли присоединения кислот включают фармацевтически приемлемые неорганические соли, такие как сульфат, нитрат, фосфат, борат, гидрохлорид и гидробромид и фармацевтически приемлемые соли присоединения органических кислот, такие как ацетат, тартрат, малеат, цитрат, сукцинат, бензоат, аскорбат, метансульфат, α-кетоглутарат, α-глицерофосфат и глюкоза-1-фосфат. Фармацевтически приемлемые соли соединений формулы (i) получают с использованием общепринятых способов.
Для специалистов в данной области очевидно, что некоторые из (соединений формулы (i) могут существовать в более чем одной таутомерной форме. Данное изобретение включает все таутомерные формы. Очевидно, что соединения согласно изобретению могут содержать один или более асимметрически замещенных атомов углерода. Присутствие одного или нескольких таких асимметрических центров в соединении формулы (i) может приводить к увеличению числа стереоизомеров, и в каждом случае следует понимать, что изобретение охватывает все такие стереоизомеры, включая энантиомеры и диастереоизомеры, и их смеси, включая рацемические смеси.
При использовании здесь термин алкил, используемый сам по себе или как часть другой группы, включает алкильные группы с линейной и разветвленной цепью, содержащей до 6 атомов. Алкокси означает алкил-О-группу, в которой алкильная группа является такой, так описано ранее. Циклоалкил включает неароматическую циклическую или мультициклическую кольцевую систему из примерно 3-10 атомов углерода. Циклический алкил может, необязательно, быть частично ненасыщенным. Циклоалкокси означает циклоалкил-О-группу, в которой циклоалкил является таким, как определено выше. Арил относится к ароматической моноциклической или мультициклической карбоциклической группе, содержащей примерно 6-10 атомов углерода. Арилалкил означает арил-алкильную группу, в которой арил и алкил, являются такими, как определено выше. Гетероарилалкил означает гетероарил-алкильную группу и гетероциклоалкил означает гетероцикло-алкильную группу. Алкилкарбонил означает алкил-CO-группу, в которой алкильная группа такая, как определено ранее. Арилкарбонил означает арил-CO-группу, в которой арильная группа является такой, как определено ранее. Гетероарилкарбонил означает гетероарил-CO-группу и гетероциклокарбонил означает гетероцикло-CO-группу. Арилсульфонил означает арил-SO2-группу, в которой арильная группа является такой, как определено ранее. Гетероарилсульфонил означает гетероарил-SO2-группу и гетероциклосульфонил означает гетероцикло-SO2-группу. Алкоксикарбонил означает алкилокси-CO-группу, в которой алкокси группа является такой, как определено ранее. Алкилсульфонил означает алкил-SO2-группу, в которой алкильная группа такая, как определено ранее. Гетероциклическое кольцо означает примерно 5-10 членную моноциклическую или мультициклическую кольцевую систему (которая может быть насыщенной или частично ненасыщенной), в которой один или несколько атомов кольцевой системы представляют собой элемент, отличный от углерода, выбранный из атомов азота, кислорода или серы. Гетероарил означает 5-10 членную ароматическую моноциклическую или мультициклическую углеводородную кольцевую систему, в которой один или несколько атомов в кольцевой системе представляют собой элемент, отличный от углерода, выбранный из азота, кислорода или серы; при желании, атом N может быть в виде N-оксида. Гетероцикло означает примерно 5-10-членную насыщенную или частично насыщенную моноциклическую или мультициклическую углеводородную кольцевую систему, в которой один или несколько атомов в кольцевой системе представляют собой элемент, отличный от углерода, выбранный из азота, кислорода или серы. Галоген означает фтор, хлор, бром или иод.
"TNF-опосредованное заболевание или болезненное состояние" означает любое или все болезненные состояния, в которых играет роль TNF или посредством продуцирования самого TNF, или посредством вызываемого TNF высвобождения другого цитокина, такого как IL-1 или IL-6, но не ограничиваясь ими. Болезненное состояние, при котором, например, IL-1 является главным компонентом, чье продуцирование или действие обостряются или секретируется в ответ на TNF, следует, таким образом, рассматривать как болезненные состояния, опосредованные TNF. Так как TNF-β (также известный как лимфотоксин) имеет близкую структурную гомологию с TNF-α (также известный как кахетин), и так как каждый из них вызывает аналогичные биологические ответы и связывается с одними и теми же клеточными рецепторами, то оба и TNF-β, и TMF-α ингибируются соединениями по настоящему изобретению, и, следовательно, оба упоминаются совместно как "TNF", если особо не оговорено иного.
Данное изобретение относится к способу опосредования или ингибирования ферментативной активности или каталитической активности PDE IV у млекопитающих, нуждающихся в этом, и к способу ингибирования продуцирования TNF у млекопитающих, нуждающихся в этом, который включает введение указанному млекопитающему эффективного количества соединения формулы (i) или его фармацевтически приемлемой соли.
Ингибиторы PDE IV полезны для лечения различных аллергических и воспалительных заболеваний, включая: астму, хронический бронхит, хроническую обструкцию дыхательных путей, хроническое воспалительное заболевание легких, идиопатический дерматит, идиопатическую) экзему, крапивницу, аллергический ринит, аллергический конъюнктивит, весенний конъюнктивит, воспаление глаза, аллергическую реакцию глаза, эозинофильную гранулему, псориаз, болезнь Бехета, эритематоз, анафилатический геморрагический нефрит, воспаление сустава, артрит, ревматоидный артрит и другие заболевания артритного характера, такие как ревматоидный спондилез и остеоартрит, септический шок, язвенный колит, болезнь Крона, реперфузионное повреждение миокарда и мозга, хронический гломерулонефрит, эндотоксический шок и синдром возрастных респираторных заболеваний. Кроме того, ингибиторы РDЕ IV полезны для лечения несахарного диабета и заболеваний, связанных с ингибированием мозгового метаболизма, таких как одряхление мозга, старческое слабоумие (болезнь Альцгеймера), нарушение памяти, вызванного болезнью Паркинсона, депрессии и мультиинфарктного слабоумия. Ингибиторы РDЕ IV также полезны для лечения заболеваний, улучшение при которых происходит за счет нейропротекторной активности, таких как остановка сердца, удар и интермиттирующая (перемежающаяся) хромота. Кроме того, ингибиторы PDE IV могут быть полезными в качестве гастропротекторов. Предпочтительное воплощение терапевтических способов по настоящему изобретению представляет собой лечение астмы.
Вирусы, предлагаемые для лечения в данном описании, представляют собой те, которые продуцируют TNF в результате инфицирования, или те, которые чувствительны к ингибированию, так, например, за счет снижения репликации, непосредственно или косвенно, при действии ингибиторов TNF формулы (i). Такие вирусы включают, но не ограничиваются ими, ВИЧ-1, ВИЧ-2 и ВИЧ-3, цитомегаловирус (ЦМВ), вирус гриппа, аденовирус и вирусы группы герпеса, как, например, но не ограничиваясь ими, Herpes zoster (Опоясывающий герпес) и Негрез simplex (Простой герпес).
Данное изобретение более конкретно относится к способу лечения млекопитающего, пораженного вирусом иммунодефицита человека (ВИЧ), который включает введение такому млекопитающему эффективного TNF-ингибирующего количества соединения формулы (i) или его фармацевтически приемлемой соли.
Соединения по данному изобретению также полезны для ветеринарного лечения животных, отличающихся от человека, нуждающихся в ингибировании продуцирования TNF. TNF-опосредованные заболевания для лечения, терапевтически или профилактически, у животных включают болезненные состояния, такие, как отмеченные выше, но, в особенности, вирусные инфекции. Примеры таких вирусов включают, но не ограничиваются ими, вирус иммунодефицита кошек (ВИК) или другие ретровирусные инфекции, такие как вирус инфекционной анемии лошадей, вирус артрита коз, Visna вирус (вызывает поражение центральной нервной системы у овец), Maedi вирус и другие лентивирусы.
Соединения по данному изобретению также полезны при лечении паразитарной, дрожжевой грибковой и грибковой инфекции, когда такие дрожжи и грибы чувствительны к позитивной регуляции посредством THF или будут вызывать продуцирование TNF in vivo. Предпочтительное болезненное состояние для лечения является грибковым менингитом.
Соединения формулы (i) находятся предпочтительно в фармацевтически приемлемой форме. Под фармацевтически приемлемой формой подразумевается, помимо прочего, фармацевтически приемлемый уровень чистоты, за исключением обычных фармацевтических добавок, таких как разбавители и носители, и не включая материалов, рассматриваемых как токсические при обычных уровнях дозировки. Фармацевтически приемлемый уровень чистоты обычно составляет по крайней мере 50%, не включая обычные фармацевтические добавки, предпочтительно 75%, более предпочтительно 90% и еще более предпочтительно 95%.
Кроме того, изобретение относится к способу получения соединения формулы (i), в которой R1-R15 и Q, X, Y и Z являются такими, как определено выше. Следует иметь в виду, что функциональные группы, такие как амино, гидроксильная или карбоксильная группа, присутствующие в различных соединениях, описываемых ниже, и которые желательно сохранить, может быть необходимо перевести в защищенную форму перед началом каких-либо реакций. В таких случаях удаление защитных групп может быть конечной стадией конкретной реакции. Подходящие защитные группы для таких функциональных групп очевидны для специалистов в данной области. Конкретные подробности см. в Protective Groups in Organic Synthesis, Wiley Interscience, TW Greens. Таким образом, способ получения соединений формулы (i), в которых R1 содержит -OH-, включает удаление защиты (например, путем гидрогенолиза или гидролиза) соединения формулы (i), в котором R3 содержит подходящую -OP, где P представляет подходящую защитную группу (например, бензил).
Способ получения соединения формулы (i) включает взаимодействие соответствующей карбоновой кислоты формулы (ii) с подходящим амином формулы (iii)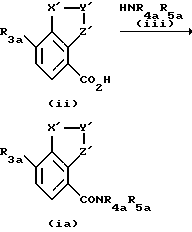
где R3a представляет R3, как он определен по отношению к формуле (i), или группу, способную превращаться в R3, и R4a и R5a аналогично представляют R4 и R5 или группы, превращаемые в R4 и R5, соответственно, и X', Y' и Z' представляют X, Y и Z или группы, превращаемые в X, Y и Z, соответственно; и, после этого, при желании, превращение любой группы R3a в R3 и/или R4a в R4, и/или R5a в R5, и/или X' в X, и/или Y' в Y, и/или Z' в Z. Взаимодействие карбоновой кислоты формулы (ii) с амином формулы (iii) можно осуществлять при любых подходящих условиях, известных специалисту. Предпочтительно, перед взаимодействием с амином формулы (iii) карбоновую кислоту превращают в хлорангидрид кислоты, смешанный ангидрид или другой активированный промежуточный продукт. Предпочтительно, взаимодействие, с амином формулы (iii) осуществляют в присутствии подходящего основания, например, аминного основания, такого как триэтиламин, предпочтительно в подходящем растворителе, таком как дихлорметан. В некоторых случаях могут потребоваться более сильное основание, такое как гидрид натрия и полярный растворитель, такой как диметилформамид.
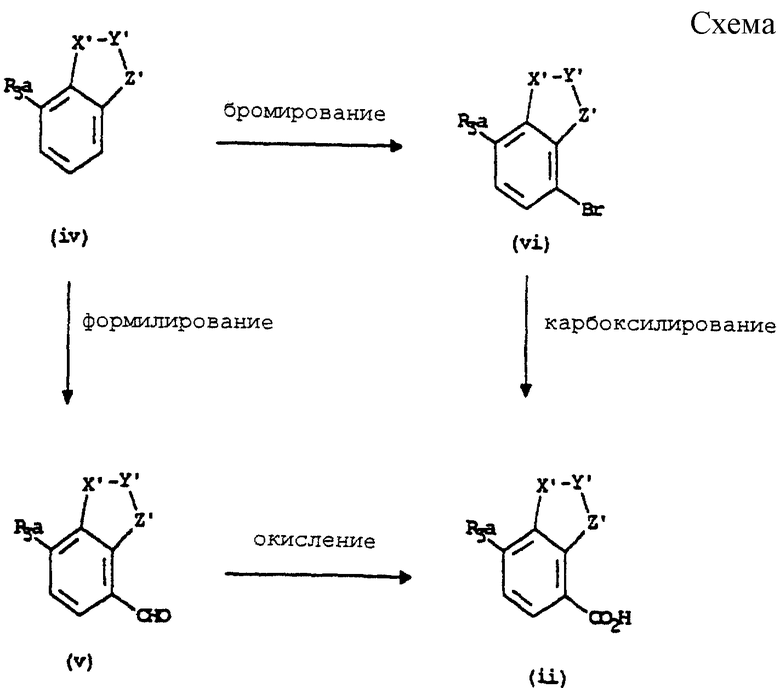
Карбоновые кислоты формулы (ii) являются или коммерчески доступными ранее описанными соединениями, или их получают в стандартных условиях, известных специалисту в данной области, Например, карбоновую кислоту формулы (ii) обычно получают из соединения формулы (iv) или формилированием, получая альдегид формулы (v), с последующим окислением, с получением кислоты формулы (ii), или бромированием, получая бромид формулы (vi), с последующим карбоксилированием с получением кислоты формулы (ii) (см. схему, представленную в конце описания).
Формилирование соединения формулы (iv) может быть проведено в стандартных условиях, известных специалистам в данной области, например, с использованием оксихлорида фосфора и диметилформамида при повышенной температуре. Окисление альдегида формулы (v) может быть осуществлено с использованием соответствующих условий, известных специалистам в данной области, например, с использованием хлорита натрия и фосфата натрия в воде/трет-бутаноле в присутствии поглотителя кислоты, такого как 2-метил-2-бутен. Бромирование соединения формулы (iv) может быть осуществлено с использованием стандартных условий, например, с использованием брома в подходящем растворителе, таком как метанол. Карбоксилирование бромида формулы (vi) обычно может быть выполнено при использовании металлоорганического катализатора, такого как палладиевый катализатор, в присутствии подходящего основания в подходящем растворителе.
Соединение формулы (iv) может быть коммерчески доступным ранее описанным соединением, или оно может быть получено с использованием стандартных условий, известных специалистам в данной области. Например, способы описаны в ЕР-A-0701907, ЕР-A-0116938, DE-A-4237417, J. Med.Chem. (1987) 30, 62, J. Chem. Soc. Perkin Trans.I (1982) 357 и J.Chem.Soc. Perkin Trans.I (1949) 3012, J. Chem.Soc. (1928) 2393 и J.Chem.Soc. (1964) 4645.
Амины формулы (iii) являются или коммерчески доступными ранее описанными соединениями, или их получают с использованием стандартных условий, известных специалистам в данной области.
Соединение формулы (i) также может быть получено путем взаимопревращения других соединений формулы (i). Например, соединение, в котором R1 содержит спиртовую функцию может быть получено восстановлением соединения формулы (i), в котором R1 содержит карбонильную функцию.
В качестве другого примера, соединения, в которых R1 и/или R2 содержит оксим, могут быть получены из соединений, в которых R1 и/или R2 содержит карбонильную группу. Такое преобразование может быть выполнено с использованием любых соответствующих стандартных условий, известных специалистам в данной области. Соединения формулы (i), в которых R1 и/или R2 содержат карбонильную группу, могут быть восстановлены с использованием стандартных условий, известных специалистам в данной области (например, боргидридом натрия в подходящем растворителе) с получением соединений, в которых R1 и/или R2 содержат спиртовую группу. Соединения, в которых R1 и/или R2 являются алкилами, могут быть получены восстановлением соединений, в которых R1 и/или R2 представляют CO-алкил, с использованием обычных условий, известных специалистам в данной области (например, гидразин гидрат в присутствии подходящего основания в соответствующем растворителе). Другие превращения могут быть осуществлены для соединений формулы (i), в которых R1 и/или R2 содержат карбонильную группу. Такие превращения включают, но не ограничиваются ими, восстановительное аминирование и алкилирование. Любое из вышеуказанных превращений может быть выполнено или в конце синтеза или для любого подходящего промежуточного продукта. Соединения формулы (i), в которых Z представляет CS, могут быть получены из соединений формулы (i), в которых Z представляет собой CO, используя любые подходящие условия, известные специалистам в данной области, например, с использованием реагента Лоусона.
Следует иметь в виду, что когда необходим индивидуальный стереоизомер формулы (i), то он может быть получен обычными способами разделения, такими как высокоэффективная жидкостная хроматография, или описанными здесь способами синтеза, выполненными с использованием соответствующего гомохирального исходного вещества.
Соединение формулы (i), или его соответствующая фармацевтически приемлемая соль и/или его фармацевтически приемлемый сольват могут вводиться сами по себе или, предпочтительно, в виде фармацевтической композиции, включающей также фармацевтически приемлемый носитель.
Соответственно, настоящее изобретение относится к фармацевтической композиции, включающей соединение формулы (i), или его соответствующую фармацевтически приемлемую соль и/или его фармацевтически приемлемый сольват и фармацевтически приемлемый носитель.
Активное соединение может быть введено в состав рецептуры для введения любым подходящим путем, предпочтительный путь введения зависит от заболевания, при котором необходимо лечение, и предпочтительно оно находится в виде единичной дозированной формы или в форме, которую человек-пациент может ввести себе как единичную дозу. Преимущественно композиция подходит для перорального, ректального, местного, парентерального введения или введения через дыхательные пути. Препараты могут быть составлены для получения медленного высвобождения активного ингредиента.
Термин парентеральный, как он использован здесь, включает подкожные инъекции, внутривенные, внутримышечные, внутригрудинные инъекции или инфузионные способы. В дополнение к лечению теплокровных животных, таких как мыши, крысы, лошади, крупный рогатый скот, овцы, собаки, кошки и т.д., соединения по изобретению эффективны при лечении людей.
Композиции по изобретению могут быть в форме таблеток, капсул, саше, ампул, порошков, гранул, пастилок, суппозиториев, восстанавливаемых порошков или жидких препаратов, как, например, пероральные или стерильные парентеральные растворы или суспензии. Препараты для местного введения используются, где это подходит.
Для достижения согласованности введения предпочтительно, чтобы композиция по изобретению находилась в виде единичной дозы.
Примером единичной дозы для перорального введения могут быть таблетки и капсулы, которые могут содержать обычные эксципиенты, такие как связывающие агенты, например, сироп, гуммиарабик, желатин, сорбит, трагакант или поливинилпирролидон; наполнители, например, микрокристаллическая целлюлоза, лактоза, сахар, кукурузный крахмал, фосфат кальция, сорбит или глицин; смазывающие агенты для таблеток, например, стеарат магния; разрыхляющие агенты, например, крахмал, поливинилпирролидон, гликоляткрахмал натрий или микрокристаллическая целлюлоза; или фармацевтически приемлемые смачивающие агенты, такие как лаурилсульфат натрия.
Твердые композиции для перорального введения могут быть получены обычными способами смешивания, наполнения, таблетирования или тому подобными. Операции повторного смешивания могут использоваться для равномерного распределения активного агента в тех композициях, где применяются большие количества наполнителей.
Безусловно, такие операции являются обычными в технике. Таблетки могут быть покрыты оболочкой способами, хорошо известными в обычной фармацевтической практике, в частности, оболочкой для растворения в кишечнике.
Пероральные жидкие препараты могут быть в форме, например, эмульсий, сиропов или эликсиров, или они могут быть представлены в виде сухого продукта, воссоздаваемого водой или другими подходящими растворителями перед употреблением. Такие жидкие препараты могут содержать обычные добавки, такие как суспендирующие агенты, например, сорбит, сироп, метилцеллюлоза, желатин, гидроксиэтилцеллюлоза, карбоксиметилцеллюлоза, гель стеарата алюминия, гидрированные пищевые жиры; эмульгирующие агенты, например, лецитин, сорбитан моноолеат или гуммиарабик, неводные растворители (которые могут включать пищевые масла), например, миндальное масло, фракционированное кокосовое масло, сложные эфиры масел, такие как сложные эфиры глицерина, пропиленгликоля или этилового спирта; консерванты, например, метил- или пропил п- гидроксибензоат или сорбиновая кислота; и, при желании, обычные вкусовые агенты или красители.
Композиции также могут подходяще быть представлены для введения в дыхательный тракт в виде нюхательного порошка или аэрозоля, или раствора для распыления, или в виде тонкоизмельченного порошка для инсуфляции, самого по себе или в сочетании с инертным носителем, таким как лактоза. В таком случае подходят частицы активного соединения, имеющие диаметр менее 50 микрон, так от 0,1 до 50 микрон, предпочтительно менее 10 микрон, например, от 1 до 10 микрон, от 1 до 5 микрон или от 2 до 5 микрон. Когда это возможно, в состав могут быть включены небольшие количества других противоастматиков и бронходилятаторов, например, амины симпатомиметики, такие как изопреналин, изоэтарин, салбутамол, фенилэфрин и эфедрин; кортикостероиды, такие как преднизолон, и стимуляторы надпочечников, такие как АСТН.
Для парентерального введения получают жидкие единичные дозированные формы с использованием соединения и стерильного растворителя и, в зависимости от используемой концентрации соединение может быть суспендировано или растворено в растворителе. При получении растворов соединение может быть растворено в воде для инъекции и простерилизовано фильтрованием перед наполнением в подходящие пузырьки или ампулы и герметически закупорено. Преимущественно, в растворителе могут быть растворены адьюванты, такие как местные анестетики, консервант и буферирующие агенты. Для увеличения стабильности композиция может быть заморожена после наполнения пузырьков и вода удалена в вакууме. Суспензии для парентерального введения получают, по существу, аналогичным образом, за исключением того, что соединение суспендируют в растворителе вместо растворения, и кроме того стерилизация не может быть выполнена путем фильтрования. Соединение может быть простерилизовано воздействием этиленоксида перед суспендированием в стерильном растворителе. Преимущественно, поверхностно-активное вещество или смачивающий агент включают в состав композиции для облегчения равномерного распределения соединения.
Композиции могут содержать от 0,1% до 99% по весу, предпочтительно от 10 до 60% по весу активного вещества в зависимости от способа введения.
Соединения формулы (i) или, если соответствует, их фармацевтически приемлемая соль и/или его фармацевтически приемлемый сольват также могут вводиться в виде рецептур для местного применения в сочетании с подходящими эксципиентами для местного применения.
Рецептуры для местного применения могут быть представлены, например, в виде мазей, кремов или лосьонов, импрегнированного перевязочного материала, гелей, гелевых карандашей, спреев и аэрозолей, и могут содержать подходящие обычные добавки, такие как консерванты, растворители для облегчения проникновения лекарства и мягчительные средства в мазях и кремах. Рецептуры могут содержать совместимые обычные носители, такие как основы кремов и мазей и этанол или олеиловый спирт в лосьонах.
Подходящие рецептуры крема, лосьона, геля, карандаша, мази, спрея или аэрозоля, которые могут быть использованы для соединений формулы (i) или, если соответствует, их фармацевтически приемлемой соли представляют собой обычные рецептуры, хорошо известные в данной области, например, как описано в стандартных справочниках, таких как Harry's Cosmeticology, опубликованной Leonard Hill Books, Remington's Pharmaceutical Sciences и British and US Pharmacopoeias (Фармакопея Британии и Соединенных Штатов).
Подходяще, соединение формулы (i) или, если соответствует, его фармацевтически приемлемая соль, составляют примерно от 0,5 до 20% по весу рецептуры, предпочтительно, от примерно 1 до 10%, например от 2 до 5%. Доза соединения по изобретению, полезного для лечения будет изменяться обычным образом в зависимости от серьезности заболеваний, веса пациента и относительной эффективности соединения. Однако в качестве общей рекомендации, единичная доза может составлять от 0,1 до 100 мг, как, например, от 0,5 до 200, 0,5 до 100 или от 0,5 до 10 мг, например, 0,5, 1, 2, 3, 4 или 5 мг; и такие единичные дозы могут вводиться, более, чем один раз в день, например, 2, 3, 4, 5 или 6 раз в день, но предпочтительно 1 или 2 раза в день, так чтобы общая дневная доза для взрослого пациента весом 70 кг составляла в диапазоне от 0,1 до 1000 мг, то есть в диапазоне примерно от 0,001 до 20 мг/кг/день, как, например, от 0,007 до 3, от 0,007 до 1,4, от 0,007 до 0,14 или от 0,01 до 0,5 мг/кг/день, например, 0,01, 0,02, 0,04, 0,05, 0,06, 0,08, 0,1 или 0,2 мг/кг/день, и такая терапия может продолжаться в течение нескольких недель или месяцев.
При использовании в данном описании термин "фармацевтически приемлемый" включает материалы, подходящие как для человека, так и для ветеринарного применения.
Следующие примеры иллюстрируют изобретение
Промежуточное соединение 1
2-Амино-3-нитроанизол
К 2-амино-3-нитрофенолу (4,0 г), растворенному в тетрагидрофуране (80 мл) при температуре окружающей среды, добавляли иодид тетрабутиламмония (0,4 г), гидроксид натрия (4,0 г) в воде (40 мл) и иодметан (3,4 мл). Эту смесь перемешивали в течение ночи, а затем концентрировали в вакууме. Остаток выливали в воду (200 мл) и экстрагировали этилацетатом (2х200 мл), затем промывали водным бикарбонатом натрия (100 мл) и насыщенным солевым раствором (100 мл). Раствор сушили над безводным сульфатом магния, фильтровали и упаривали в вакууме, получая указанное в заголовке соединение в виде темного твердого вещества (4,4 г).
ТСХ Rf 0,7 (50%-ный этилацетат в гексане)
Промежуточное соединение 2
2,3-Диаминоанизол
2-Амино-3-нитроанизол (4,3 г) гидрировали в этилацетате (200 мл) с использованием в качестве катализатора 10% палладия на угле в атмосфере водорода при температуре окружающей среды. После завершения реакционную смесь фильтровали через целит и фильтрат упаривали в вакууме, получая указанное в заголовке соединение в виде коричневой жидкости (3,60 г)
ТСХ Rf 0,65 (этилацетат)
Промежуточное соединение 3
5-Метоксихиноксалин
Гидрат бисульфита глиоксаля натрия (10,0 г) в воде (80 мл) нагревали до 60oC, затем добавляли раствор 2,3-диаминоанизола (3,40 г) в этаноле (40 мл). Затем перемешиваемую смесь нагревали при 80oC в течение 1 часа перед добавлением концентрированной соляной кислоты (6 капель). Нагревание продолжали в течение 1 часа. Смесь оставляли охлаждаться в течение ночи, концентрировали в вакууме и выливали в водный раствор карбоната калия (40 мл). Этилацетатные экстракты (3х100 мл) промывали водой (100 мл) и насыщенным солевым раствором (50 мл), затем сушили над безводным сульфатом магния, фильтровали и упаривали в вакууме, получая указанное в заголовке соединение в виде желтого твердого вещества (3,07 г).
ТСХ Rf 0,40 (этилацетат)
Промежуточное соединение 4
8-Метоксихиноксалин-5-карбоновая кислота
К 5-метоксихиноксалину (2,3 г) в метаноле (50 мл) при -20oC в атмосфере инертного газа добавляли в течение 15 минут раствор брома (0,76 мл) в метаноле (10 мл). После перемешивания в течение 4 ч реакционную смесь оставляли при -20oC на ночь. Смесь выливали в водный раствор метабисульфита натрия (100 мл), подщелоченный бикарбонатом натрия и экстрагировали этилацетатом (3х100 мл). Эти экстракты промывали водой (100 мл) и насыщенным солевым раствором (50 мл), затем сушили над безводным сульфатом магния, фильтровали и упаривали в вакууме, получая коричневое твердое вещество (2,45 г). Очистка колоночной хроматографией с использованием в качестве элюента 50%-ного этилацетата в гексане привела к не совсем белому твердому веществу (0,46 г) в виде смеси 5-бром- и 5,6-дибромзамещенного продуктов. Это твердое вещество, трифенилфосфин (0,5 г), бис (трифенилфосфин) палладий (II) хлорид (1,0 г) и триэтиламин (6 мл) растворяли в тетрагидрофуране (200 мл) и нагревали при 80oC в аппарате Парра в атмосфере газообразного монооксида углерода при 1380 кПа (200 фунтов на квадратный дюйм). Через 6 дней смесь охлаждали до температуры окружающей среды и концентрировали в вакууме. Смесь подщелачивали до pH 14 1 М раствором гидроксида натрия и экстрагировали этилацетатом (2х200 мл). Эти экстракты повторно экстрагировали 1 М раствором гидроксида натрия (2х100 мл) и объединенные водные фазы подкисляли до pH 5 ледяной уксусной кислотой. Этилацетатные экстракты (2х200 мл) кислой водной фазы промывали водой (100 мл) и насыщенным солевым раствором (50 мл), затем сушили над безводным сульфатом магния, фильтровали и упаривали в вакууме, получая указанное в заголовке соединение в виде твердого вещества (0,19 г).
ТСХ Rf 0,20 (этилацетат)
Промежуточное соединение 5
4-Метокси-2-трифторметилбензимидазол
Раствор 2,3-диаминоанизола (1,0 г) в трифторуксусной кислоте (15 мл) кипятили с обратным холодильником в течение 5 часов и затем перемешивали при комнатной температуре в течение ночи. Избыток трифторуксусной кислоты удаляли в вакууме, а остаток распределяли между этилацетатом (50 мл) и водой (50 мл). Органическую фазу промывали насыщенным раствором бикарбоната натрия (50 мл) и водой (50 мл). Сушка над безводным сульфатом натрия и удаление растворителя в вакууме привели к коричневому остатку. Очисткой колоночной хроматографией, элюируя 50%-ным этилацетатом в гексане, получали указанное в заголовке соединение в виде желтого твердого вещества (1,4 г).
ТСХ Rf 0,64 (50%-ный этилацетат в гексане)
Промежуточное соединение 6
2-(1-Гидроксиэтил)-4-метоксибензимидазол
2,3-Диаминоанизол (5,88 г) и молочную кислоту (5,6 мл) объединяли, обрабатывали концентрированной соляной кислотой (45 мл) и нагревали при 100oC в течение 18 часов. Реакционную смесь охлаждали до 0oC, нейтрализовали раствором гидроксида аммония и экстрагировали этилацетатом (3х45 мл). Объединенные органические слои сушили над сульфатом магния, фильтровали и фильтрат упаривали в вакууме, а остаток очищали флэш-хроматографией на диоксиде кремния, элюируя этилацетатом, получая требуемый продукт в виде красновато-коричневого твердого вещества (4,91 г).
ТСХ Rf 0,125 (этилацетат)
Промежуточное соединение 7
2-Ацетил-4-метоксибензимидазол
Раствор 2-(1-гидроксиэтил)-4-метоксибензимидазола (2,18 г) в уксусной кислоте (8,5 мл) нагревали при 100oC и обрабатывали раствором триоксида хрома (0,85 г) в воде (3 мл). Через 10 минут реакционную смесь выливали в воду (110 мл), осадок удаляли фильтрованием через слой целита и продукт экстрагировали дихлорметаном (3х100 мл). Объединенные органические слои сушили над сульфатом магния, фильтровали и упаривали в вакууме, получая требуемый продукт в виде светло-коричневого твердого вещества (1,4 г).
ТСХ Rf 0,6 (этилацетат)
Промежуточное соединение 8
7-Метокси-3-метил-2-трифторметилбензимидазол
К раствору 4-метокси-2-трифторметилбензимидазола (1,4 г) в тетрагидрофуране (40 мл) под азотом добавляли гидрид натрия (0,32 г; 60% дисперсия в масле). Смесь перемешивали в течение 20 минут при комнатной температуре прежде чем добавить метилиодид (1,35 г). Перемешивание продолжали в течение ночи. Реакцию останавливали добавлением воды (10 мл) и растворитель упаривали в вакууме. Добавляли этилацетат (50 мл) и органический слой промывали насыщенным раствором бикарбоната натрия (20 мл), водой (20 мл) и рассолом (20 мл). После сушки над безводным сульфатом магния с последующим удалением растворителя в вакууме получали указанное в заголовке соединение (1/6 г) в виде масла, которое при стоянии превращалось в твердое вещество.
ТСХ Ra 0,75 (50%-ный этилацетат в гексане)
Следующие соединения были получены аналогичным образом.
Промежуточное соединение 9
2-Ацетил-7-метокси-3-метилбензимидазол
Получали из 2-ацетил-7-метоксибензимидазола (0,5 г). Очистка флэш-хроматографией на диоксиде кремния при элюировании 50%-ным этилацетатом в гексане дала указанное в заголовке соединение в виде белого твердого вещества (0,24 г).
ТСХ Rf 0,38 (50%-ный этилацетат в гексане)
Промежуточное соединение 10
7-Метокси-3-пропил-2-трифторметилбензимидазол
Получали из 4-метокси-2-трифторметилбензимидазола (2,4 г) и бромистого пропила (3,02 мл). Очистка флэш-хроматографией на диоксиде кремния при элюировании 15%-ным этилацетатом в гексане давала указанное в заголовке соединение в виде белого твердого вещества (2,03 г).
ТСХ Rf 0,4 (20%-ный этилацетат в гексане)
Промежуточное соединение 11
4-Бром-7-метокси-3-метил-2-трифторметилбензимидазол
К раствору 7-метокси-3-метил-2-трифторметилбензимидазола (1,4 г) в хлороформе (50 мл) в атмосфере азота добавляли N- бромсукцинимид (1,2 г). Смесь перемешивали в течение 20 минут, а затем реакцию останавливали добавлением 5% раствора метабисульфита натрия (50 мл) и органический слой отделяли. Промывание водой (50 мл), сушка над безводным сульфатом магния и удаление растворителя в вакууме приводили к оранжевому маслу. Очистка флэш-хроматографией при элюировании 50%-ным этилацетатом в гексане давала указанное в заголовке соединение в виде оранжевого твердого вещества (1,73 г)
ТСХ Rf 0,79 (50%-ный этилацетат в гексане)
Следующие соединения были получены по аналогичной методике.
Промежуточное соединение 12
2-Ацетил-4-бром-7-метокси-3-метилбензимидазол
Получали из 2-ацетил-7-метокси-3-метилбензимидазола (0,24 г). Очистка флэш-хроматографией на диоксиде кремния при элюировании 50%-ным этилацетатом в гексане давала желаемый продукт в виде белого твердого вещества (0,23 г).
ТСХ Rf 0,5 (50%-ный этилацетат в гексане)
Промежуточное соединение 13
4-Бром-7-метокси-3-пропил-2-трифторметилбензимидазол
Получали из 7-метокси-3-пропил-2-трифторметилбензимидазола (2,03 г). Очистка флэш-хроматографией на диоксиде кремния при элюировании 15%-ным этилацетатом в гексане давала указанное в заголовке соединение в виде белого твердого вещества (0,69 г).
ТСХ Rf 0,4 (20%-ный этилацетат в гексане)
Промежуточное соединение 14
7-Meтoкcи-3-мeтил-2-тpифтopмeтилбeнзимидaзoл-4-кapбoнoвaя кислота
Смесь 4-бром-7-метокси-3-метил-2-трифторметилбензимидазола (1,7 г), бис(трифенилфосфин) палладий (II) хлорида (0,26 г), трифенилфосфина (0,48 г) и триэтиламина (7,7 мл) в тетрагидрофуране (30 мл) и воде (10 мл) нагревали при 80oC в аппарате Парра в атмосфере газообразного монооксида углерода при 1240 кПа (180 фунтов на квадратный дюйм). Через 3 дня смесь охлаждали до температуры окружающей среды и концентрировали в вакууме. Смесь подщелачивали до pH 14 1 М раствором гидроксида натрия и экстрагировали этилацетатом (2х50 мл). Водную фазу подкисляли до pH 5 ледяной уксусной кислотой. Полученный осадок отфильтровывали и промывали водой, получая указанное в заголовке соединение (0,81 г) в виде не чисто белого твердого вещества.
ТСХ Rf 0,37 (50%-ный этилацетат в гексане)
Следующие соединения получали аналогичным образом.
Промежуточное соединение 15
2-Ацетил-7-метокси-3-метилбензимидазол-4-карбоновая кислота
Получали из 2-ацетил-4-бром-7-метокси-3-метилбензимидазола. Очистка флэш-хроматографией на диоксиде кремния при элюировании 50%-ным этилацетатом в гексане давала требуемый продукт в виде белого твердого вещества (1,38 г).
ТСХ Rf 0,75 (50%-ный этилацетат в гексане)
Промежуточное соединение 16
7-Метокси-3-пропил-2-трифторметилбензимидазол-4-карбоновая кислота
Получали из 4-бром-7-метокси-3-пропил-2-трифторметилбензимидазола (0,69 г). Растирание с трет-бутилметиловым эфиром давало указанное в заголовке соединение в виде белого твердого вещества (0,29 г).
ТСХ Rf 0,4 (50%-ный этилацетат в гексане)
Промежуточное соединение 17
4-Бром-7-метокси-2-трифторметилбензимидазол
Раствор 7-метокси-2-трифторметилбензимидазола (5,0 г) в хлороформе (100 мл) охлаждали до 0oC и добавляли N-бромсукцинимид (4,5 г). Смесь перемешивали в течение 2 ч. Затем ее промывали 5%-ным водным метабисульфитом натрия (50 мл), сушили над сульфатом магния, упаривали в вакууме и очищали флэш-хроматографией, элюируя 25%-ным этилацетатом в гексане, получая указанное в заголовке соединение в виде белого твердого вещества (1,0 г).
ТСХ Rf 0,29 (20%-ный этилацетат в гексане)
Промежуточное соединение 18
4-Бром-7-метокси-3-(4-метоксибензил)-2-трифторметилбензимидазол
К раствору 4-бром-7-метокси-2-трифторметилбензимидазола (1,0 г) в N,N-диметилформамиде (20 мл) добавляли гидрид натрия (0,16 г; 60% дисперсия в масле). Смесь перемешивали при комнатной температуре в течение 10 минут, а затем добавляли 4-метоксибензилхлорид (0,56 мл) и каталитическое количество иодида тетрабутиламмония. Реакционную смесь нагревали при 90oC в течение 6 ч, затем выливали в воду (100 мл) и экстрагировали этилацетатом (2х100 мл). Объединенные
органические фазы промывали водой (100 мл) и насыщенным солевым раствором (50 мл), сушили над сульфатом магния, упаривали в вакууме и очищали флэш-хроматографией, элюируя 33%-ным этилацетатом в гексане, получая указанное в заголовке соединение в виде белого твердого вещества (0,52 г).
ТСХ Rf 0,31 (201-ный этилацетат в гексане)
Промежуточное соединение 19
7-Метокси-3-(4-метоксибензил)-2-трифторметилбензимидазол-4-карбоновая кислота
Смесь 4-бром-7-метокси-3-(4-метоксибензил)-2-трифторметилбензимидазола (520 мг), бис(трифенилфосфин)палладий (II) хлорида (90 мг), трифенилфосфина (110 мг) и триэтиламина (1,8 мл) в тетрагидрофуране (50 мл) и воде (15 мл) нагревали при 80oC в аппарате Парра в атмосфере газообразного монооксида углерода при 1240 кПа (180 фунтов на квадратный дюйм). Через 5 дней смесь охлаждали до температуры окружающей среды и концентрировали в вакууме. Смесь подщелачивали 1 М раствором гидроксида натрия и экстрагировали этилацетатом (2х50 мл). Водную фазу подкисляли до pH 6 ледяной уксусной кислотой и экстрагировали этилацетатом (2х75 мл). Объединенные экстракты сушили над сульфатом магния и упаривали в вакууме, получая указанное в заголовке соединение в виде кремового твердого вещества (310 мг).
ТСХ Rf 0,12 (50%-ный этилацетат в гексане)
Промежуточное соединение 20
2-Этил-4-гидроксибензоксазол
Смесь гидрохлорида 2-аминорезорцина (5,0 г) и триэтилортопропионата (13,7 мл) нагревали при 150oC в течение 2 ч, а затем выливали в смесь воды (140 мл) и этанола (35 мл). Смесь интенсивно перемешивали в течение 30 минут при комнатной температуре, полученный осадок удаляли фильтрованием и сушили, получая указанное в заголовке соединение в виде бежевого твердого вещества (4,11 г).
ТСХ Rf 0,40 (50%-ный этилацетат в гексане)
Промежуточное соединение 21
4-Гидроксибензоксазол
Гидрохлорид 2-аминорезорцина (2 г) и триэтилхлорформиат (4,5 мл) кипятили с обратным холодильником в атмосфере азота в течение 3 ч. После охлаждения до комнатной температуры реакционную смесь выливали в смесь воды (70 мл) и этанола (20 мл). Смесь интенсивно перемешивали в течение 30 минут, затем оставляли на ночь при комнатной температуре. Образовавшийся бежевый осадок отфильтровывали и сушили азеотропной перегонкой с толуолом, получая указанное в заголовке соединение в виде бежевого твердого вещества (1,2 г).
Промежуточное соединение 22
2-Этил-4-метоксибензоксазол
2-Этил-4-гидроксибензоксазол (4,17 г) растворяли в тетрагидрофуране (73 мл) при комнатной температуре. Добавляли иодид тетрабутиламмония (0,4 г), а затем раствор гидроксида натрия (3,89 г) в воде (40 мл). Смесь перемешивали в течение 10 минут перед добавлением иодметана (3,13 мл). Затем перемешивание продолжали в течение ночи. Сырую смесь упаривали на силикагеле и очищали флэш-хроматографией, элюируя 25%-ным, а затем 50%-ным этилацетатом в гексане, получая указанное в заголовке соединение в виде жидкости цвета соломы (3,05 г).
ТСХ Rf 0,50 (50%-ный этилацетат в гексане)
Следующее соединение получали аналогичным образом
Промежуточное соединение 23
4-Метоксибензоксазол
Получали из 4-гидроксибензоксазола, получая указанное в заголовке соединение (90 мг) в виде коричневого твердого вещества.
ТСХ Rf 0,41 (50%-ный этилацетат в гексане)
Промежуточное соединение 24
2-(1-Гидроксиэтил)-4-метоксибензоксазол
4-Метоксибензоксазол (6,0 г) растворяли в тетрагидрофуране (225 мл) и охлаждали до -78oC в атмосфере азота. Добавляли н-бутиллитий (26,5 мл 1,6 М раствора в гексанах) и смесь перемешивали при -78oC в течение 30 минут перед добавлением эфирата бромида магния (11,5 г). Полученную гетерогенную смесь перемешивали при -45oC в течение 15 минут, а затем охлаждали до -78oC. Из предварительно охлажденного (шприца добавляли раствор ацетальдегида (2,3 мл). Смесь перемешивали при -78oC в течение 3 ч, оставляли нагреваться до комнатной температуры и перемешивали в течение ночи. Реакцию останавливали водным бикарбонатом натрия (50 мл, постепенно) и тетрагидрофуран упаривали в вакууме. Остаток экстрагировали дихлорметаном (3х150 мл). Объединенные органические фазы сушили над сульфатом магния, упаривали в вакууме и очищали флэш-хроматографией, элюируя 30%-50%-ным этилацетатом в гексане, получая указанное в заголовке соединение в виде коричневого твердого вещества (6,25 г)
ТСХ Rf 0,14 (30%-ный этилацетат в гексане)
Промежуточное соединение 25
2-Ацетил-4-метоксибензоксазол
Раствор оксалилхлорида (0,25 мл) в дихлорметане (6,5 мл) охлаждали до -55oC в атмосфере азота. По каплям добавляли раствор диметилсульфоксида (0,44 мл) в дихлорметане (1,3 мл) и смесь перемешивали в течение 5 минут при -55oC прежде чем добавить раствор 2-(1-гидроксиэтил)-4-метоксибензоксазола (0,5 г) в дихлорметане (2,5 мл). Перемешивание продолжали в течение 15 минут при -55oC, затем добавляли триэтиламин (1,8 мл). Смесь перемешивали при -55oC в течение 5 минут, а затем оставляли нагреваться до комнатной температуры. Смесь выливали в воду (25 мл) и слои разделяли. Водную фазу экстрагировали дихлорметаном (20 мл). Объединенные органические фазы сушили над сульфатом магния, упаривали в вакууме и очищали флэш-хроматографией, элюируя 30%-ным этилацетатом в гексане, с получением указанного в заголовке соединения в виде белого твердого вещества (350 мг).
ТСХ Rf 0,45 (50%-ный этилацетат в гексане)
Промежуточное соединение 26
4-Метокси-2-(2-метил-[1,3]-диоксолан-2-ил)бензоксазол
2-Ацетил-4-метоксибензоксазол (200 мг), п-толуолсульфоновую кислоту (239 мг), этиленгликоль (0,29 мл) и толуол (10 мл) кипятили с обратным холодильником в условиях Дина-Старка в течение 2 ч. После охлаждения до комнатной температуры толуол упаривали и остаток распределяли между водой (20 мл) и этилацетатом (20 мл). Водную фазу экстрагировали этилацетатом (20 мл). Объединенные органические фазы промывали водой (40 мл), водным бикарбонатом натрия (2х40 мл) и водой (40 мл), сушили над сульфатом магния, упаривали в вакууме и очищали флэш-хроматографией, элюируя 30%-ным этилацетатом в гексане с получением указанного в заголовке соединения в виде белого твердого вещества (129 мг).
ТСХ Rf 0,32 (30%-ный этилацетат в гексане)
Промежуточное соединение 27
7-Бром-2-этил-4-метоксибензоксазол
2-Этил-4-метоксибензоксазол (2,81 г) растворяли в метаноле (80 мл) в атмосфере азота и раствор охлаждали до -78oC. По каплям добавляли бром (0,73 мл). Смесь постепенно нагревали до комнатной температуры и перемешивали в течение 3,5 ч. Метанол упаривали в вакууме и остаток распределяли между этилацетатом и водным бикарбонатом натрия. Объединенные органические фазы промывали 5% водным метабисульфитом натрия, упаривали на диоксиде кремния и очищали флэш-хроматографией, элюируя 25%-50%-ным этилацетатом в гексане, с получением смеси указанного в заголовке соединения и 2-этил-4-метоксибензоксазола в виде бледно-желтой жидкости (2,28 г).
TCX Rf 0,50 (50%-ный этилацетат в гексане)
Промежуточное соединение 28
2-Этил-4-метоксибензоксазол-7-карбоновая кислота
Смесь 7-бром-2-этил-4-метоксибензоксазола (0,7 г), трифенилфосфина (0,273 г), бис(трифенилфосфин)палладий (II) хлорида (0,125 г) и триэтиламина (3,9 мл) в тетрагидрофуране (19 мл) и воде (6,2 мл) нагревали при 80oC в аппарате Парра в атмосфере газообразного монооксида углерода при давлении 140 фунтов на квадратный дюйм (965,3 кПа) в течение 3 дней. Затем смесь охлаждали до температуры окружающей среды и концентрировали в вакууме. Смесь подщелачивали до pH 14 1 М раствором гидроксида натрия и экстрагировали этилацетатом (2х50 мл). Водную фазу подкисляли до pH 5 ледяной уксусной кислотой и экстрагировали дихлорметаном. Объединенные дихлорметановые экстракты сушили (MgSO4) и упаривали в вакууме, получая указанное в заголовке соединение в виде бежевого твердого вещества (0,40 г).
TCX Rf 0,30 (50%-ный этилацетат в гексане)
Промежуточное соединение 29
7-Бром-4-метокси-2-(2-метил-[1,3]-диоксолан-2-ил)бензоксазол
4-Метокси-2-(2-метил-[1,3] -диоксолан-2-ил)бензоксазол (129 мг), N-бромсукцинимид (107 мг) и ацетонитрил (5 мл) объединяли и перемешивали при комнатной температуре в атмосфере азота в течение 4 ч. Смесь распределяли между водой (20 мл) и этилацетатом (20 мл). Водную фазу экстрагировали этилацетатом (20 мл). Объединенные органические фазы промывали водой (2х50 мл), сушили над сульфатом магния, упаривали в вакууме и очищали колоночной хроматографией, элюируя 30%-ным этилацетатом в гексане, с получением указанного в заголовке соединения в виде белого твердого вещества (95 мг).
ТСХ Rf 0,41 (30%-ный этилацетат в гексане)
Следующее соединение получали аналогичным способом.
Промежуточное соединение 30
7-Бром-4-метоксибензоксазол
Получали из 4-метоксибензоксазола, выделяя указанное в заголовке соединение (635 мг).
ТСХ Rf 0,51 (30%-ный этилацетат в гексане)
Промежуточное соединение 31
4-Метокси-2-(2-метил-[1,3] -диоксолан-2-ил)бензоксазол-7-карбоновая кислота
7-Бром-4-метокси-2-(2-метил-[1,3] -диоксолан-2-ил)бензоксазол (480 мг), ацетат палладия (34 мг), 1,3-бис(дифенилфосфино)пропан (126 мг), триэтиламин (0,21 мл), воду (15 мл) и тетрагидрофуран (30 мл) объединяли и нагревали в течение 3 дней при 90oC в аппарате Парра под давлением 1035 кПа (150 фунтов на квадратный дюйм) монооксида углерода. После охлаждения до комнатной температуры тетрагидрофуран упаривали в вакууме, остаток распределяли между этилацетатом (50 мл) и водой (50 мл). Водный слой экстрагировали этилацетатом (50 мл). Объединенные органические экстракты сушили над сульфатом магния, упаривали в вакууме и очищали флэш-хроматографией, элюируя этилацетатом, с получением указанного в заголовке соединения в виде твердого белого вещества (190 мг).
ТСХ Rf 0,51 (этилацетат)
Промежуточное соединение 32
4-Метоксибензоксазол-7-карбоновая кислота
7-Бром-4-метоксибензоксазол (630 мг), триэтиламин (3,85 мл), трифенилфосфин (290 мг), бис (трифенилфосфин) палладий хлорид (88 мг), воду (20 мл) и тетрагидрофуран (40 мл) объединяли и нагревали при 90oC в аппарате Парра под давлением 1035 кПа (150 фунтов на квадратный дюйм) монооксида углерода в течение 3 дней. После охлаждения до комнатной температуры тетрагидрофуран упаривали. Остаток распределяли между этилацетатом (50 мл) и водой (50 мл). Водную фазу подкисляли до pH 4 уксусной кислотой и слои разделяли. Водную фазу экстрагировали этилацетатом (2х30 мл), а затем дихлорметаном (2х30 мл). Объединенные органические фазы сушили над сульфатом магния и упаривали в вакууме. Очистка остатка флэш-хроматографией при элюировании этилацетатом и растирание с диэтиловым эфиром приводили к указанному в заголовке соединению в виде белого твердого вещества (125 мг). При стоянии из водного слоя осаждалось белое твердое вещество, которое удаляли фильтрованием, и сушили в вакууме при 45oC в течение 1 часа, получая дополнительную порцию указанного в заголовке соединения в виде белого твердого вещества (49 мг).
ТСХ Rf 0,50 (этилацетат)
Пример 1
8-Метоксихиноксалин-5-[N-(3,5-дихлорпирид-4-ил)]карбоксамид
8-Метоксихиноксалин-5-карбоновую кислоту (0,19 г), растворенную в дихлорметане (16 мл), обрабатывали в инертной атмосфере оксалилхлоридом (0,3 мл), затем добавляли 2 капли N,N-диметилформамида и перемешивали при температуре окружающей среды в течение ночи. Реакционную смесь упаривали в вакууме и проводили азеотропную отгонку с сухим толуолом (2х15 мл), получая дигидрохлорид хлорангидрида кислоты в виде коричневого твердого вещества (0,3 г). Раствор этого твердого вещества в сухом N,N-диметилформамиде (10 мл) добавляли при 60oC к смеси 4-амино-3,5-дихлорпиридина (0,16 г), гидрида натрия (0,15 г; 60% дисперсия в масле) и N,N-диметилформамида (10 мл), которую перед этим уже перемешивали при температуре окружающей среды в течение 1 часа. Через 2 часа смесь оставляли охлаждаться в течение ночи перед упариванием в вакууме. Остаток фильтровали через слой диоксида кремния с использованием горячего этилацетата и фильтрат упаривали в вакууме, получая следующий остаток. Его очищали колоночной хроматографией с использованием в качестве элюента 10%-ного метанола в этилацетате, получая указанное в заголовке соединение в виде не совсем белого твердого вещества (0,09 г)
ТСХ Rf 0,65 (10%-ный метанол в этилацетате)
Т.пл. 205-208oC
Следующее соединение было получено по аналогичной методике.
Пример 2
2-Ацетил-7-метокси-3-метилбензимидазол-4-[N-(пиридин-4-ил)] карбоксамид
Получали из 2-ацетил-7-метокси-3-метилбензимидазол-4-карбоновой кислоты (0,50 г) и 4-аминопиридина (0,18 г). Очистка флэш-хроматографией на диоксиде кремния при элюировании 10%-ным метанолом в этилацетате дала указанное в заголовке соединение в виде не совсем белого твердого вещества (0,10 г)
ТСХ Rf 0,31 (10%-ный метанол в этилацетате)
Т.пл. 274-275oC
Пример 3
7-Метокси-3-метил-2-трифторметилбензимидазол-4- [N-(3,5-дихлорпирид-4-ил)]карбоксамид
7-Метокси-3-метил-2-трифторметилбензимидазол-4-карбоновую кислоту (0,40 г), растворенную в дихлорметане (20 мл), обрабатывали оксалилхлоридом (0,28 мл) в атмосфере азота, затем добавляли 2 капли N,N-диметилформамида и перемешивали при температуре окружающей среды в течение ночи. Реакционную смесь упаривали в вакууме, получая гидрохлорид хлорангидрида кислоты в виде коричневого твердого продукта. Раствор этого твердого продукта в сухом N, N-диметилформамиде (10 мл) добавляли при температуре окружающей среды к смеси 4-амино-3,5-дихлорпиридина (0,29 г), гидрида натрия (0,30 г, 60% дисперсия) и N, N-диметилформамида (10 мл), которую перед этим уже перемешивали в течение 1 ч при температуре окружающей среды. Через 2 ч смесь оставляли охлаждаться в течение ночи, затем упаривали в вакууме. Остаток очищали колоночной хроматографией с использованием в качестве элюента 50%-ного этилацетата в гексане, получая указанное в заголовке соединение в виде белого твердого вещества (0,16 г).
ТСХ Rf 0,34 (50%-ный этилацетат в гексане)
Т.пл. 228-229oC
Следующие соединения были получены аналогичным способом.
Пример 4
7-Метокси-3-пропил-2-трифторметилбензимидазол-4-[N-(3,5-дихлорпиридин-4-ил)]карбоксамид
Получали из 7-метокси-3-пропил-2-трифторметилбензимидазол-4-карбоновой кислоты (0,28 г) и 4-амино-3,5-дихлорпиридина (0,18 г). Очистка флэш-хроматографией на диоксиде кремния при элюировании 40%-ным этилацетатом в гексане давала указанное в заголовке соединение в виде белого твердого вещества (0,006 г).
ТСХ Rf 0,21 (40%-ный этилацетат в гексане)
Т.пл. 246-247oC
Пример 5
2-Ацетил-7-метокси-3-метилбензимидазол-4-[N-(3,5-дихлорпирид-4-ил)] карбоксамид
Получали из 2-ацетил-7-метокси-3-метилбензимидазол-4-карбоновой кислоты (1,38 г). Очистка флэш-хроматографией при элюировании 10%-ным метанолом в дихлорметане приводила к указанному в заголовке соединению (0,75 г) в виде бежевого твердого вещества.
ТСХ Rf 0,5 (10%-ный метанол в дихлорметане)
Т.пл. 290-291oC
Пример 6
2-Этил-4-метоксибензоксазол-7-[N-(4-пиридил)]карбоксамид
К раствору 2-этил-4-метоксибензоксазол-7-карбоксилата (0,40 г) в дихлорметане (20 мл) при комнатной температуре в атмосфере азота добавляли оксалилхлорид (0,32 мл). После перемешивания в течение 10 минут добавляли сухой N, N-диметилформамид (2 капли). Перемешивание продолжали в течение ночи, получая желтый раствор, который упаривали досуха в вакууме. Остаток растворяли в дихлорметане (20 мл). Добавляли триэтиламин (0,53 мл), затем 4-аминопиридин (0,20 г) и смесь перемешивали при комнатной температуре в течение ночи. Смесь выпаривали на диоксиде кремния и очищали флэш-хроматографией, элюируя дихлорметаном, а затем 10%-ным метанолом в дихлорметане, получая указанное в заголовке соединение в виде твердого вещества рыжевато-коричневого цвета (0,08 г).
ТСХ Rf 0,45 (10%-ный метанол в этилацетате)
Т.пл. 140-142oC
Пример 7
2-Этил-4-метоксибензоксазол-7-[N-(3,5-дихлорпирид-4-ил)] карбоксамид
2-Этил-4-метоксибензоксазол-7-карбоксилат (1,24 г), 4-диметиламинопиридин (каталитическое количество), 4-нитрофенол (1,17 г) и гидрохлорид 1-этил-3-(3-диметиламинопропил)карбодимида (1,6 г) в дихлорметане (60 мл) перемешивали при комнатной температуре в течение 48 ч. Смесь упаривали на диоксиде кремния и хроматографировали, элюируя 50%-ным этилацетатом в гексане, получая 4-нитрофенил 2-этил-4-метоксибензоксазол-7-карбоксилат. 4-Амино-3,5-дихлорпиридин (0,57 г) растворяли в N,N-диметилформамиде (20 мл) при комнатной температуре в атмосфере азота. Добавляли гидрид натрия (0,20 г, 60% дисперсия) и смесь перемешивали в течение 5 ч, затем добавляли 4-нитрофениловый сложный эфир (1,2 г) и перемешивали дополнительно 18 ч. Реакционную смесь упаривали на диоксиде кремния и очищали флэш-хроматографией, элюируя 50%-ным этилацетатом в гексане с выделением указанного в заголовке соединения в виде белого твердого вещества (0,44 г).
ТСХ Rf 0,20 (40%-ный этилацетат в гексане)
Масс-спектр (CI): наблюдался пик [М+Н]+
Пример 8
2-Этил-4-метоксибензоксазол-7-[N-(3,5-дихлорпиридин-4-ил-N-оксид)] карбоксамид
Раствор 2-этил-4-метоксибензоксазол-7-[N-(3,5-дихлорпиридин-4-ил)] карбоксамида (0,05 г) в хлороформе (10 мл) обрабатывали 36-40% перуксусной кислотой в уксусной кислоте (0,03 мл) и перемешивали в течение 14 дней при комнатной температуре. Реакционную смесь распределяли между дихлорметаном (20 мл) и водой (20 мл). Органический слой отделяли, сушили над сухим сульфатом магния, фильтровали и фильтрат упаривали в вакууме. Очистка флэш-хроматографией на диоксиде кремния при элюировании 10%-ным метанолом в этилацетате давала желаемый продукт в виде белого твердого вещества (0,018 г).
ТСХ Rf 0,38 (10%-ный метанол в этилацетате)
Т.пл. 201-203oC
Пример 9
4-Метоксибензоксазол-7-[N-(3,5-дихлорпиридин-4-ил)]карбоксамид
Смесь 4-метоксибензоксазол-7-карбоновой кислоты (0,16 г), диметиламинопиридина (каталитическое количество), 4-нитрофенола (0,17 г) и гидрохлорида 1-этил-3-(3-диметиламинопропил)карбодиимида (0,17 г) в дихлорметане (30 мл) перемешивали в атмосфере азота при комнатной температуре в течение ночи. Смесь разбавляли дихлорметаном (20 мл) и промывали водой (2х50 мл). Объединенные органические фазы сушили над сульфатом магния и упаривали в вакууме с получением 4-нитрофенил 4-метоксибензоксазол-7-карбоксилата. 4-Амино-3,5-дихлорпиридин (0,15 г) растворяли в N,N-диметилформамиде (5 мл) при комнатной температуре в азоте. Добавляли гидрид натрия (40 мг, 60% дисперсия в масле) и смесь перемешивали в течение 1 ч перед добавлением 4-нитрофенилового сложного эфира (0,26 г) в N,N-диметилформамиде (20 мл). После перемешивания в течение 60 ч N,N-диметилформамид упаривали в вакууме и остаток распределяли между этилацетатом (40 мл) и водой (40 мл). Объединенные органические фазы промывали водой (2х40 мл), сушили над сульфатом магния, упаривали на диоксиде кремния и очищали флэш-хроматографией при элюировании 50%-ным этилацетатом в гексане, получая указанное в заголовке соединение в виде белого твердого вещества (51 мг).
ТСХ Rf 0,19 (50%-ный этилацетат в гексане)
Т.пл. 193-194,5oC
Следующие соединения были получены аналогичным способом.
Пример 10
7-Метокси-3-(4-метоксибензил)-2-трифторметилбензимидазол-4-[N-(3,5-дихлорпиридин-4-ил)]карбоксамид
Получали из 7-метокси-3-(4-метоксибензил)-2- трифторметилбензимидазол-4-карбоновой кислоты, выделяя указанное в заголовке соединение (140 мг) в виде белого твердого вещества.
ТСХ Rf 0,35 (50%-ный этилацетат в гексане)
Т.пл. 184,5-186oC
Пример 11
2-(2-Метил-[1,3] диоксолан-2-ил)-4-метоксибензоксазол-7-[N-(3,5-дихлорпиридин-4-ил)]карбоксилат
Получали из 2-(2-метил-[1,3] диоксолан-2-ил)-4-метоксибензоксазол-7-карбоновой кислоты, выделяя указанное в заголовке соединение (58 мг) в виде белого твердого вещества.
ТСХ Rf 0,61 (этилацетат)
Т.пл. 155-157oC
Пример 12
N-(3,5-Дихлорпирил-4-ил)-7-метокси-2-трифторметилбензимидазол-4-карбоксамид
7-Метокси-3-(4-метоксибензил)-2-трифторметилбензимидазол- 4-[N-(3,5-дихлорпирид-4-ил)]карбоксамид (50 мг) перемешивали в трифторуксусной кислоте (3 мл) в течение 2 ч при комнатной температуре. Избыток трифторуксусной кислоты удаляли в вакууме и остаток распределяли между этилацетатом (25 мл) и водным бикарбонатом натрия (25 мл). Водную фазу экстрагировали этилацетатом (25 мл). Объединенные органические фазы сушили над сульфатом магния и упаривали в вакууме, получая твердое вещество, которое растирали с диэтиловым эфиром, получая указанное в заголовке соединение в виде кремового твердого вещества (19 мг).
ТСХ Rf 0,30 (50%-ный этилацетат в гексане)
Т.пл. 300-301oC
Пример 13
2-(1-Гидроксиимино)этил-7-метокси-3-метил-бензимидазол-4-[N-(3,5-дихлорпиридил)]карбоксамид
Смесь 2-ацетил-7-метокси-3-метилбензимидазол-4-[N-(3,5-дихлорпирид- 4-ил)] карбоксамида (0,60 г), гидрохлорида гидроксиламина (1,05 г) и пиридина (1,22 мл) в толуоле (40 мл) кипятили в условиях Дина-Старка в течение 21 ч. Растворитель упаривали в вакууме и остаток растирали с водой и отфильтровывали. Осадок сушили в вакууме, выделяя указанное в заголовке соединение (0,57 г) в виде бежевого твердого вещества.
Т.пл. 272-273oC
Масс-спектр: наблюдался пик [М+Н]+
Пример 14
3-Метил-2-(1-(2-метилтиазол-4-илметокси)иминоэтил)-7-метоксибензимидазол-4-[N-(3,5-дихлорпирид-4-ил-)]карбоксамид
К раствору 2-(1-гидроксиимино)этил-7-метокси-3-метилбензимидазол-4-[N-(3,5-дихлорпиридил)] карбоксамида (200 мг) в диметилформамиде (20 мл) при комнатной температуре в атмосфере инертного газа добавляли гидрид натрия (60%, 43 мг). Через 1 ч добавляли 4-хлорметил-2-метилтиазол (217 мг) и диметилформамид (5 мл) и перемешивание продолжали при комнатной температуре в течение 18 ч. Реакцию останавливали добавлением воды (25 мл) и экстрагировали этилацетатом (3х25 мл). Объединенные органические фазы сушили (сульфат магния), фильтровали и упаривали на силикагеле. Очистка флэш-хроматографией при элюировании этилацетатом дала указанное в заголовке соединение в виде белого твердого вещества (28 мг).
Т.пл. 220-221oC
Масс-спектр: наблюдался пик [М+Н]+
Следующее соединение получали аналогичным образом.
Пример 15
3-Метил-2-[1-(3-диметиламинопропилокси)иминоэтил] -7-метоксибензимидазол-4-[N-(3,5-дихлорпирид-4-ил)карбоксамид
Получали с использованием 2-(1-гидроксиимино)этил-7-метокси-3-метилбензимидазол-4-[N-(3,5-дихлорпиридил)] карбоксамида (330 мг) и гидрохлорида N, N-диметиламинопропилхлорида (383 мг). Очистка флэш-хроматографией при элюировании 20%-ным метанолом в дихлорметане приводила к указанному в заголовке соединению (96 мг) в виде не совсем белого твердого вещества.
ТСХ Rf 0,25 (20%-ный метанол в дихлорметане)
Т.пл. 186-187oC
Методы анализа
Анализы, использованные для подтверждения ингибирующей активности соединений формулы (i) в отношении фосфодиэстеразы IV представляют собой обычные методы анализа, описанные в Schilling et al. , Anal. Biochem., 216:154 (1994), Thompson and Strada, Adv. Cycl. Nuci. Res., 8:119 (1979) и Gristwood and Owen, Br.J. Pharmacol., 87:91P (1986).
Соединения формулы (i) проявили в этих анализах активность на уровне, сопоставимом с уровнем, который, как предполагается, полезен при лечении болезненных состояний, связанных с фосфодиэстеразой IV.
Способность соединений формулы (i) ингибировать продуцирование TNF в человеческих моноцитах измерена следующим образом. Моноядерные клетки периферической крови получали стандартными способами, из свежеполученных образцов крови. Клетки помещали в RPMI1640 + 1% фетальной телячьей сыворотки в присутствии и в отсутствие ингибиторов. Добавляли LPS (100 нг/мл) и культуры инкубировали в течение 22 ч при 37oC в атмосфере 95% воздуха/5% CO2. Надосадочные жидкости исследовали на ТНFα с помощью анализа ELISA (твердофазный иммуноферментный анализ) с использованием коммерчески доступных наборов для анализа.
Активность in vivo на клеточной эозинофильной модели определяли с использованием способов, описанных в Hellewell et al., Br.J. Pharmacol., 11:811 (1994) и Br.J. Pharmacol., 110:416 (1993). Активность в легочной модели измеряли с использованием способов, описанных в Kallos and Kallos, Int. Archs. Allergy Appl. lmmunol., 73:77 (1984) и Sanjar et al., Br.J. Pharmacol., 99: 679 (1990).
Дополнительная легочная модель, которая позволяет измерить ингибирование астматических ответов на ранней и на поздней фазе, а также ингибирование гиперреактивности дыхательных путей, описана в Broadley et al. Pulmonary Pharmacol., 7:311 (1994), J.Immunological Methods 190:51 (1996) и British J. Pharmacol., 116:2351 (1995). Соединения по изобретению проявили активность в этой модели.
Дополнительные экспериментальные данные представлены в табл. 1.
Данные по биологической активности представлены в табл. 2.
| название | год | авторы | номер документа |
|---|---|---|---|
| КАРБОКСАМИДЫ БЕНЗОФУРАНА И ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1997 |
|
RU2162467C2 |
| ХИНОЛИНОВЫЕ КАРБОКСАМИДЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1997 |
|
RU2170730C2 |
| N-ОКСИДЫ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, ОБЛАДАЮЩИЕ ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ TNF И PDE-IV | 1999 |
|
RU2205830C2 |
| ПРОИЗВОДНЫЕ ГИДРОКСАМОВОЙ И КАРБОНОВОЙ КИСЛОТЫ, ИМЕЮЩИЕ MMP ИНГИБИРУЮЩУЮ АКТИВНОСТЬ, И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1998 |
|
RU2210567C2 |
| ПРОИЗВОДНЫЕ БЕНЗАНИЛИДА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРОЯВЛЯЮЩАЯ АНТАГОНИЗМ К 5-HT РЕЦЕПТОРАМ | 1992 |
|
RU2077535C1 |
| АРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 1996 |
|
RU2182574C2 |
| ПИПЕРИДИНИЛЗАМЕЩЕННЫЕ МЕТАНОАНТРАЦЕНЫ И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 1994 |
|
RU2128178C1 |
| ПРОИЗВОДНЫЕ 3-(5-ТЕТРАЗОЛИЛБЕНЗИЛ)АМИНОПИПЕРИДИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1994 |
|
RU2136675C1 |
| 5-ГЕТЕРОЦИКЛО-1,5-БЕНЗОДИАЗЕПИНЫ И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1995 |
|
RU2152939C1 |
| ПРОИЗВОДНЫЕ МЕТАНАНТРАЦЕНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АНТИДОПАМИНЕРГИЧЕСКОЙ АКТИВНОСТЬЮ | 1992 |
|
RU2099328C1 |
Изобретение относится к новым бициклическим карбоксамидам формулы (i), в которой (1) X представляет N и (а) Z представляет =СR1-CR2 и Y представляет N, (в) Z представляет =СR1 и Y представляет О, S или NR4, или (с) Z представляет = СR1-N= и Y представляет СR2, или (2) X представляет NR4, Z представляет СR1= и Y представляет N, Q представляет О, R1 и R2 представляют СОR6, С(= NOR6)R13, алкил-С(=NOR6)R13, NR8R9, CF3, или R6, R3 представляет С1-6 алкоксигруппу, R4 представляет Н или алкил, R5 представляет гетероарил, необязательно замещенный галогеном, алкилом, CONR11R12, CF3 или CN, арил, замещенный галогеном; R6 представляет Н, алкил, циклоалкил, арил, гетероарил, гетероцикл, арилалкил, гетероарилалкил или гетероциклоалкил, R7 представляет алкил, гидрокси, OR10, NR8R9, CN, СO2H, СO2R10, CONR11R12, R8 и R9 представляют Н или алкил, или NR8R9 представляет гетероциклическое кольцо, необязательно замещенное R14, R10 представляет алкил, гетероцикл, R11 и R12 представляют Н или алкил, R13, R14 и R15 представляют алкил, Y представляет О и n равно 2-4, их фармацевтически приемлемым солям. Соединения формулы (i) обладают ингибирующей PDE и TNF активностью и могут найти применение в медицине в качестве агента для ингибирования фосфодиэстеразы и фактора некроза опухоли. 2 с. и 11 з.п. ф-лы, 2 табл.
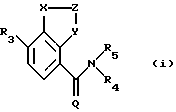
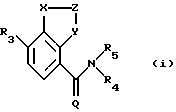
в которой (1) Х представляет N, и (a) Z представляет =CR1-CR2= и Y представляет N, (b) Z представляет =CR1, и Y представляет О, S или NR4, или (с) Z представляет =CR1-N= и Y представляет CR2, или (2) Х представляет NR4, Z представляет -СR1= и Y представляет N;
Q представляет О;
R1 и R2 являются одинаковыми или разными и, каждый, представляют COR6, С(=NOR6)R13, алкил-С(=NОR6)R13, NR8R9, СF3, R6, или циклическую группу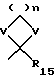
R3 представляет C1-6 алкоксигруппу, необязательно замещенную галогеном;
R4 представляет Н или алкил;
R5 представляет гетероарил, необязательно замещенный галогеном, алкилом, CONR11R12, CF3 или CN, арил, замещенный галогеном;
каждый R6 независимо представляет Н или группу, выбранную из алкила, необязательно замещенного R7, циклоалкила, арила, гетероарила, необязательно замещенного R7, гетероцикла, необязательно замещенного R7, арилалкила, замещенного R7, гетероарилалкила и гетероциклоалкила;
R7 представляет алкил, гидрокси, OR10, NR8R9, CN, CO2H, CO2R10, CONR11R12;
R8 и R9 каждый представляет Н или алкил, или NR8R9 представляет гетероциклическое кольцо, необязательно замещенное R14;
R10 представляет алкил, гетероцикл;
R11 и R12 являются одинаковыми и каждый представляет Н;
R13 представляет алкил;
R14 представляет алкил;
R15 представляет алкил;
V представляет О;
n=2-4;
и их фармацевтически приемлемые соли.
2-ацетил-7-метокси-3-метилбензимидазол-4-[N-(пиридин-4-ил)]карбоксамида,
7-метокси-3-пропил-2-трифторметилбензимидазол-4-[N-(3,5-дихлорпиридин-4-ил)]карбоксамида,
2-ацетил-7-метокси-3-метилбензимидазол-4-[N-(3,5-дихлорпирид-4-ил)]карбоксамида,
2-этил-4-метоксибензоксазол-7-[N-(3,5-дихлорпирид-4-ил)]карбоксамида,
2-этил-7-метоксибензоксазол-4-[N-(3,5-дихлорпиридин-4-ил-N-оксид)]карбоксамида,
4-метоксибензоксаэол-7-[N-(3,5-дихлорпиридин-4-ил)]карбоксамида,
7-метокси-3-(4-метоксибензил)-2-трифторметилбензимидазол-4-[N-(3,5-дихлорпиридин-4-ил)]карбоксамида,
2-(2-метил-[1,3] диоксолан-2-ил)-4-метоксибензоксазол-7-[N-(3,5-дихлорпиридин-4-ил)карбоксилата,
N-(3,5-дихлорпирид-4-ил)-7-метокси-2-трифторметилбензимидазол-4-карбоксамида,
2-(1-гидроксиимино)этил-7-метокси-3-метил-бензимидазол-4-[N-(3,5-дихлорпиридил)]карбоксамида,
3-метил-2-(1-(2-метилтиазол-4-илметокси)иминоэтил)-7-метоксибензимидазол-4-[N-(3,5-дихлорпирид-4-ил)]карбоксамида и
3-метил-2-[1-(3-диметиламинопропилокси)иминоэтил] -7-метоксибензимидазол-4-[N-(3,5-дихлорпирид-4-ил)]карбоксамида.
Приоритет по пунктам:
15.11.1996 по пп.8 и 9;
22.04.1997 по пп.7 и 10;
17.11.1997 по пп.1-6 и 11-13.
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| Приспособление в пере для письма с целью увеличения на нем запаса чернил и уменьшения скорости их высыхания | 1917 |
|
SU96A1 |
| Способ получения карбостирильных производных | 1981 |
|
SU1169535A3 |

