Изобретение относится к сульфонамидсодержащему соединению индола и его антиангиогенному действию. Более подробно, изобретение относится к противоопухолевому средству, супрессору метастазирования злокачественной опухоли, лекарственному средству против диабетической ретинопатии, лекарственному средству против ревматоидного артрита и лекарственному средству против гематомы, основанным на антиангиогенном эффекте.
Было выяснено, что существует тесная связь между пролиферацией злокачественной опухоли и ангиогенезом. Таким образом, когда ангиогенез не происходит в очаге злокачественной опухоли, злокачественная опухоль остается в состоянии покоящейся опухоли. Однако выяснилось, что при развитии ангиогенеза кислород и питательные вещества из крови поступают к ткани опухоли, что способствует пролиферации и метастазированию злокачественной опухоли, что приводит к клинически злокачественному состоянию. Соответственно, можно ожидать, что при супрессии ангиогенеза злокачественной опухоли могут подавляться пролиферация и метастазирование злокачественной опухоли. Поскольку ангиогенные сосуды состоят из эндотелиальных клеток и интерстициальных клеток хозяина, мишенью антиангиогенного препарата являются не злокачественные клетки, а подобные нормальные клетки хозяина. Поскольку злокачественные клетки не являются непосредственной мишенью, можно ожидать наличие эффективного воздействия на злокачественную опухоль, невосприимчивую к известным противоопухолевым средствам, и, кроме того, предполагается, что вероятность формирования толерантной злокачественной опухоли, что является большой проблемой в терапии злокачественных опухолей, мала. Кроме того, ангиогенез является опухолево-специфическим явлением, и у взрослых лиц ограничен образованием эндометрии и прочими процессами, сопровождающими менструальный цикл. Соответственно, его нежелательный эффект предположительно мал по сравнению с известными противоопухолевыми средствами. Недавно было экспериментально доказано в доклинических испытаниях, что антиангиогенные средства способны супрессировать и далее уменьшить пролиферацию злокачественной опухоли в моделях с трансплантированной опухолью и препятствовать образованию толерантной злокачественной опухоли, и в клинических испытаниях показана корреляция между ангиогенезом и малигнизацией для многих злокачественных опухолей, таких как злокачественная опухоль молочной железы, злокачественная опухоль предстательной железы, злокачественная опухоль легких и злокачественная опухоль ободочной кишки.
В тканях злокачественной опухоли непрерывно происходят процессы апоптоза и пролиферации злокачественных клеток, и известно, что в зависимости от баланса между ними образуется прогрессирующая злокачественная опухоль или покоящаяся опухоль. Антиангиогенное средство непосредственно не уничтожает злокачественные клетки, а прекращает поступление питательных веществ, таким образом, смещая указанный баланс в сторону апоптоза, что приводит опухоль в состояние покоя или к уменьшению злокачественной опухоли, вследствие чего оно является средством, которое, возможно, проявит прекрасный эффект (продление жизни, ингибирование рецидива и супрессия метастазирования) при длительной терапии.
На доклинической стадии существуют антиангиогенные средства с различными механизмами действия, но поскольку их противоопухолевый эффект на доклинической стадии оказался недостаточным, их приемлемость на клинической стадии еще является сомнительной, и поэтому имеется большой спрос на антиангиогенные препараты с достоверным эффектом.
Также известно, что ангиогенез участвует в развитии ретинопатии и ретинита. При пролиферации кровеносных сосудов в сетчатке происходит ухудшение зрения и в случае прогрессии наступает слепота. В настоящее время не существует эффективного лекарственного средства против этого заболевания, и имеется спрос на эффективные лекарственные средства.
WO 9301182 относится к противоопухолевым средствам со специфической тирозинкиназной ингибиторной активностью соединений, имеющих индоловый скелет, но они представляют собой соединения индолилметилен-2-индолинона и отличны от соединений по настоящему изобретению. Аналогично, WO 964016 относится к противоопухолевым средствам со специфической тирозинкиназной ингибиторной активностью соединений, имеющих индольный скелет, но они представляют собой 2-индолинон-3-метиленовые соединения и отличны от соединений по настоящему изобретению. JP-A 7-165708 и JP-A 8-231505 относятся к сульфонамидным соединениям, содержащим индольную структуру. Однако соединения, конкретно раскрытые в JP-A 7-165708 и содержащие в индольном кольце два заместителя, отличных от арил (гетероарил) сульфониламиногруппы, ограничены и имеют только шесть комбинаций данных заместителей, т.е. (1) 3-Сl и 4-Сl; (2) 3-Сl и 4-ОСН3; (3) 3-Сl и 4-ОН; (4) 3-Сl и 4-СН3; (5) 3-Сl и 4-CN; и (6) 3-CN и 5-Вr. Комбинаций (a) 3-CN и 4-СН3; (b) 3-Cl и 5-Вr; (с) 3-Сl и 4-Вr; и (d) 3-Br и 4-СН3 не имеется. Относительно 4-галогенмонозамещенных соединений, имеется описание для 4-Вr-замещенных соединений, но их сульфонильная составляющая представляет собой только п-нитрофенольное соединение. Кроме того, соединения индола, раскрытые в JP-A 8-231505 представляют собой только 3-галоген- или 3-цианомонозамещенные соединения. В данных выложенных публикациях совсем не имеется описания антиангиогенного эффекта, а также не содержится его предсказания.
Целью настоящего изобретения является создание нового антиангиогенного препарата и обеспечение противоопухолевого препарата с высокой безопасностью и надежным эффектом по сравнению с традиционными противоопухолевыми препаратами, который было бы возможно принимать длительное время.
Авторами настоящего изобретения проведено интенсивное исследование, показавшее, что сульфонамидсодержащее соединение индола, представленное следующей формулой, достигает поставленной цели, и тем самым осуществили настоящее изобретение. То есть настоящее изобретение относится к сульфонамидсодержащему соединению индола, представленному следующей формулой (I), его фармацевтически приемлемой соли или ее гидратам.

В указанной формуле R1 представляет собой атом водорода, атом галогена или цианогруппу; R2 и R3 являются одинаковыми или отличными, и каждый представляет собой атом водорода, C1-C4 низшую алкильную группу или атом галогена; R4 представляет собой атом водорода или C1-C4 низшую алкильную группу; и кольцо А представляет собой цианофенильную группу, аминосульфонилфенильную группу, аминопиридильную группу, аминопиримидильную группу, галогенпиридильную группу или цианотиофенильную группу, при условии, что случаи, когда и R1, и R2, и R3 представляют собой атомы водорода, когда оба R2 и R3 представляют собой атомы водорода, или когда кольцо А представляет собой аминосульфонильную группу, и оба R1 и R2 представляют собой атомы галогена, исключены. Кроме того, когда кольцо А представляет собой цианофенильную группу, 2-амино-5-пиридильную группу или 2-галоген-5-пиридильную группу, и R1 представляет собой цианогруппу или галогеновую группу и по крайней мере один из R2 и R3 не должен быть атомом водорода.
Настоящее изобретение относится к способу профилактики или лечения заболевания, при котором ингибирование ангиогенеза в очаге опухоли, ревматоидного артрита или диабетической ретинопатии является эффективным для профилактики или лечения путем введения фармакологически эффективной дозы вышеуказанного соединения индола, его фармацевтически приемлемой соли или ее гидратов пациенту.
Кроме того, настоящее изобретение относится к применению вышеуказанного соединения индола, его фармацевтически приемлемой соли или ее гидратов для производства профилактического или лекарственного средства против заболевания, при котором антиангиогенный препарат является эффективным для профилактики или лечения.
Кроме того, настоящее изобретение относится к антиангиогенному средству, противоопухолевому средству, терапевтическому средству против злокачественной опухоли поджелудочной железы, терапевтическому средству против рака ободочной кишки, терапевтическому средству против рака желудка, терапевтическому средству против злокачественной опухоли молочной железы, терапевтическому средству против злокачественной опухоли предстательной железы, терапевтическому средству против рака легких, терапевтическому средству против злокачественной опухоли яичников, супрессору метастазирования злокачественной опухоли, терапевтическому средству против диабетической ретинопатии, терапевтическому средству против ревматоидного артрита и терапевтическому средству против гематомы, содержащему в качестве эффективного ингредиента вышеуказанное соединение индола, их фармацевтически приемлемую соль или ее гидраты. Изобретение относится к способу профилактики, лечения и усовершенствования применения любого из данных фармацевтических средств. Кроме того, изобретение относится к применению вышеуказанного соединения для получения любого из данных фармацевтических средств.
В вышеуказанной формуле (I) атом галогена означает атом фтора, атом хлора, атом брома или атом йода. C1-C4 низшая алкильная группа означает линейную или разветвленную алкильную группу, такую как метильная группа, этильная группа, н-пропильная группа, н-бутильная группа, изопропильная группа, изобутильная группа и трет-бутильная группа.
Соединение индола, представленное выше формулой (I), может образовывать соль с кислотой или с основанием. Настоящее изобретение также включает соль соединения индола (I). Примерами соли, образованной с кислотой, является соль неорганической кислоты, такая как гидрохлорид, гидробромид или сульфат, и соль органической кислоты, такой как уксусная кислота, молочная кислота, янтарная кислота, фумаровая кислота, малеиновая кислота, лимонная кислота, бензойная кислота, метансульфоновая кислота или п-толуолсульфоновая кислота. Примерами соли, образованной с основанием являются неорганические соли, такие как соль натрия, соль калия или соль кальция и соли органических оснований, таких как триэтиламин, аргинин или лизин.
Само собой разумеется, что включены все гидраты подобного соединения и его фармацевтически приемлемой соли. Хотя соединения настоящего изобретения обладают сильным антиангиогенным эффектом, также включены соединения, подвергаемые метаболизму in vivo, такому как окисление, восстановление, гидролиз и сопряжение. Настоящее изобретение, кроме того, включает соединения, в результате метаболизма которых in vivo, такого как окисление, восстановление и гидролиз, получается соединение по настоящему изобретению.
Соединение по настоящему изобретению (I) может быть получено различными способами, и представительным из них будет являться следующий.
Соединение может быть получено взаимодействием сульфоновой кислоты, представленной формулой (II)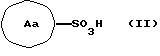
(в формуле кольцо Аа представляет собой цианофенильную группу, аминосульфонилфенильную группу, аминопиридильную группу, аминопиримидильную группу, галогенпиридильную группу или цианотиофенильную группу) или их реакционноспособное производное, с соединением, представленным формулой (III)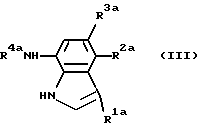
(в формуле R1a представляет собой атом водорода, атом галогена или цианогруппу; и R2a и R3a являются одинаковыми или отличаются и каждый представляет собой атом водорода, C1-C4 низшую алкильную группу или атом галогена, при условии, что исключаются случаи, когда все R1a, R2a и R3a представляют собой атомы водорода или когда оба R2a и R3a представляют собой атомы водорода).
Примерами реакционноспособного производного сульфоновой кислоты (II) являются обычно и хорошо утилизируемые реакционноспособные производные, такие как сульфонилгалогенид, ангидрид сульфоновой кислоты и N-сульфонилимидазолид и особенно подходящим примером является сульфонилгалогенид. Хотя не налагается особенного ограничения на природу растворителя, применяемого в реакции, предпочтительными являются такие, которые растворяют вещества и не вступают с ними во взаимодействие. Например, могут применяться пиридин, тетрагидрофуран, диоксан, бензол, этиловый эфир, дихлорметан, диметилформамид и смешанный растворитель, состоящий из двух или нескольких выбранных из них. Кроме того, при высвобождении кислоты в ходе реакции, как в случае применения в реакции сульфонилгалогенида, предпочтительным является проведение реакции в присутствии подходящего удаляющего кислоту вещества, и поэтому особенно подходящим является применение основного растворителя, такого как пиридин. В случае применения нейтрального растворителя может быть добавлено основное вещество, такое как карбонат щелочного металла или органический третичный амин. Конечно, растворитель, который может быть использован, не ограничивается данными здесь примерами. Обычно настоящую реакцию проводят при комнатной температуре, но, если необходимо, может быть проведено охлаждение или нагревание. Время реакции обычно составляет от 10 минут до 20 часов и необязательно выбирается в зависимости от природы соединений и температуры реакции.
Когда аминогруппа в полученном продукте защищена, при необходимости может быть проведен обычный способ удаления защитной группы, такой как обработка кислотой, обработка щелочью и каталитическое восстановление, вследствие чего возможно получение соединения индола (I), содержащего свободную аминогруппу.
Далее будут проиллюстрированы способы получения исходных соединений (II), их реакционноспособных производных и (III), применяемых в данном изобретении.
Исходное соединение (II) и его реакционноспособное производное включают как известные соединения, так и новые соединения. В случае новых соединений они могут быть получены применением уже опубликованного способа синтеза известных соединений или с помощью его сочетания. Например, новый сульфонилхлорид можно получить с помощью способа, использующего методы синтеза, указанные в Chem. Ber. , 90, 841 (1957); J. Med. Chem., 6, 307 (1963); J. Chem. Soc. (c), 1968, 1265; Chem. Lett., 1992, 1483; J. Am. Chem. Soc., 59, 1837 (1937); J. Med. Chem. , 23, 1376 (1980); J. Am. Chem. Soc., 70, 375 (1948); J. Am. Chem. Soc., 78, 2171 (1956) и прочие.
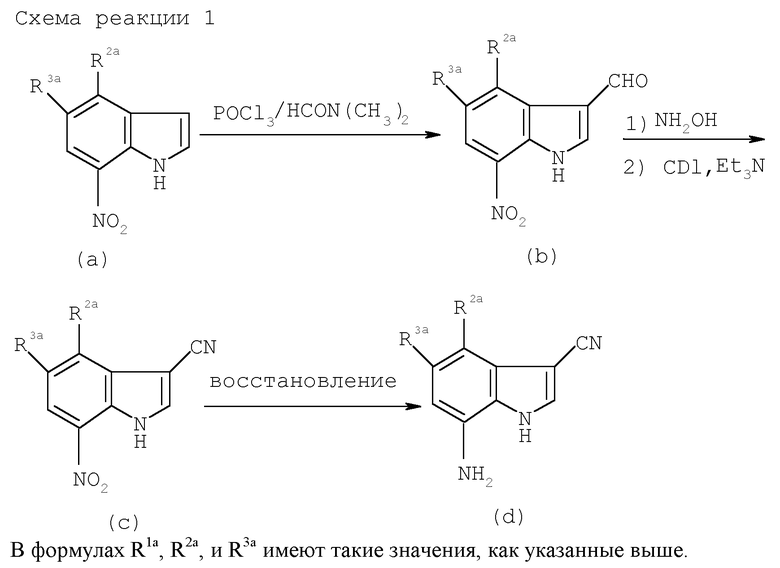
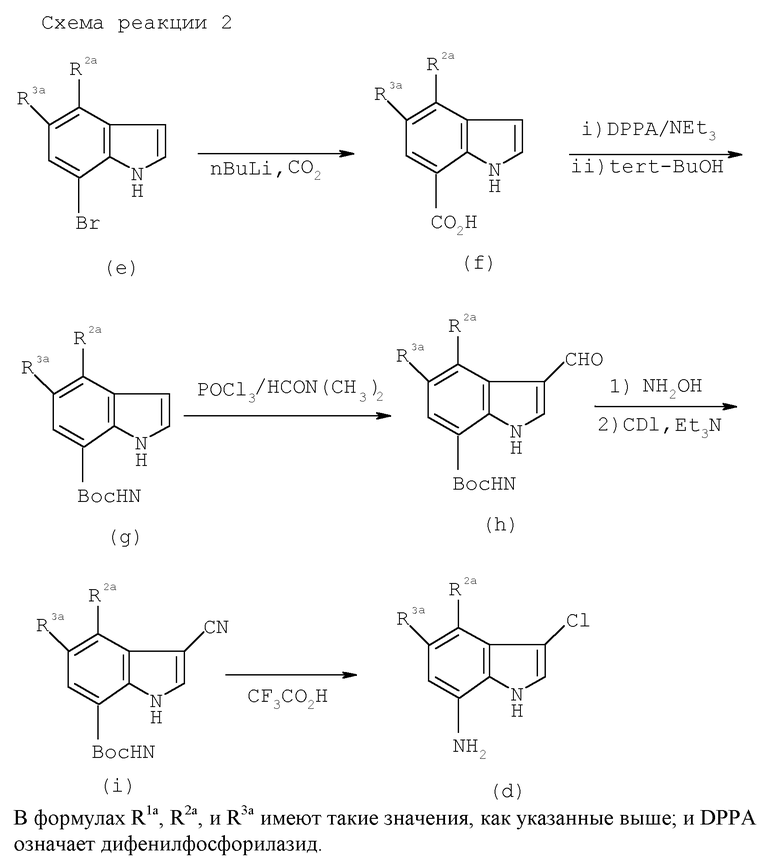
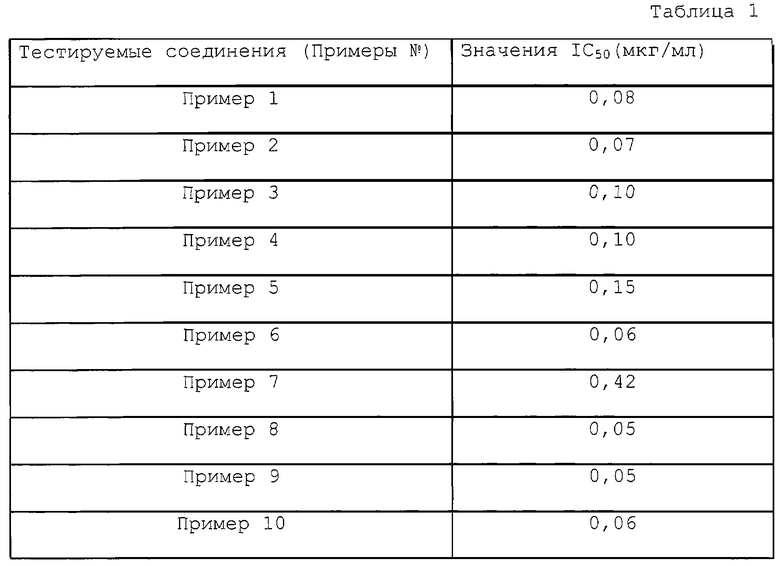
Когда R1a и R3a представляют собой атомы водорода и R2a является атомом галогена в исходном соединении (III), его можно получить известным способом синтеза. Когда R2a и R3a являются одинаковыми или отличаются, и каждый представляет собой атом водорода, C1-C4 низшую алкильную группу или атом галогена (случай, когда оба являются атомами водорода исключается) и R1a представляет собой цианогруппу, его можно получить следующим способом (см. схемы реакции в конце описания).
Когда R1a представляет собой атом галогена, соединение можно получить таким образом, когда соединение формулы (а) или формулы (d) в вышеуказанной схеме реакции (1) и (2) галогенируют обычным способом и восстанавливают нитрогруппу или удаляют защитную группу аминогруппы.
Когда соединение по настоящему изобретению применяют в качестве лекарственного средства, его можно вводить как орально, так и парентерально. Доза варьирует в зависимости от степени выраженности симпотома, возраста, пола, массы тела и индивидуальной чувствительности пациента, способа введения, периода введения, интервала введения, свойства фармацевтического препарата, типа препарата, типа эффективного ингредиента и прочего и особенно не ограничена. В случае внутривенного введения доза составляет 1-2000 мг, предпочтительно 1-500 мг и более предпочтительно 5-1000 мг, хотя в случае перорального введения она обычно составляет 10-6000 мг, предпочтительно около 50-4000 мг и более предпочтительно 100-3000 мг в сутки для взрослых и обычно вводится один раз в сутки или разделяется на три раза в сутки.
Когда готовят твердый препарат для перорального применения, к основному ингредиенту добавляют наполнитель и, если необходимо, связующее вещество, дезинтегрирующее вещество, смазывающее вещество, краситель, корригент и прочее с последующей обработкой обычным способом для получения таблеток, таблеток в оболочке, гранул, мелких гранул, порошков, капсул и прочего.
Примерами наполнителя являются лактоза, кукурузный крахмал, сахароза, глюкоза, сорбит, кристаллическая целлюлоза и диоксид кремния; примерами связующего вещества являются поливиниловый спирт, этилцеллюлоза, метилцеллюлоза, аравийская камедь, гидроксипропилцеллюлоза и гидроксипропилметилцеллюлоза; примерами смазывающего вещества являются стеарат магния, тальк и кремнезем; примерами красителя являются вещества, которые приемлемы для добавления к фармацевтическим препаратам; и примерами ароматизаторов являются порошок какао, ментол, ароматические масла, масло перечной мяты, борнеол и порошок корицы. Конечно, если необходимо, не составляет проблемы покрыть соответствующим образом подобные таблетки и гранулы сахарной оболочкой, желатиновой оболочкой или другими.
При приготовлении инъекции, если необходимо, к основному ингредиенту добавляют вещество, регулирующее рН, буфер, суспендирующее вещество, солюбилизатор, стабилизатор, изотонизирующее вещество, консервант и прочее с последующей обработкой в соответствии с традиционной методикой для получения инъекций для внутривенного, подкожного и внутримышечного введения. В то же время, если необходимо, оно может быть преобразовано в соответствии с традиционной методикой в лиофильно высушенный продукт.
Примерами суспендирующего вещества являются метилцеллюлоза, полисорбат 80, гидроксиэтилцеллюлоза, аравийская камедь, трагакантовый порошок, натрийкарбоксиметилцеллюлоза и полиоксиэтиленсорбитанмонолаурат.
Примерами солюбилизатора являются полиоксиэтиленгидрогенизированное касторовое масло, полисорбат 80, никотинамид, полиоксиэтиленсорбитанмонолаурат, макрогол и этиловый эфир жирных кислот касторового масла.
Примерами стабилизатора являются сульфит натрия и метасульфит натрия. Примерами консерванта являются метилпарагидроксибензоат, этилпарагидроксибензоат, сорбиновая кислота, фенол, крезол и хлоркрезол.
Действие соединений по настоящему изобретению будет показано с помощью следующих фармакологических экспериментальных примеров.
Фармакологический экспериментальный пример 1.
Антиангиогенный эффект.
Степень ингибирования ангиогенеза, который наблюдали при инкубировании частей аорты крысы с коллагеном, определяли как антиангиогенный эффект. Таким образом, аорту, выделенную из организма самца крысы линии Sprague-Dawley (возраста 10-12 недель), промывали раствором Хэнкса так, что окружающие жировые ткани были тщательно удалены.
Аорту разрезали для получения частей площадью 2 мм и их оставляли в 24-луночном планшете эндотелиальными клетками наружу. Затем вносили 500 мкл нейтрализованного коллагена типа I (Cell Matrix Type I-A; производства Nitta Gelatin) в каждую лунку и оставляли при комнатной температуре приблизительно 20 минут в ламинаре для затвердевания геля. После затвердевания геля туда добавляли 500 мкл среды 131 (производства Chlorella Kogyo) с последующей инкубацией в СO2-инкубаторе (5% СО2) при 37oС. На следующий день культуральную среду заменяли 500 мкл среды MCDB 131, содержащей тестируемые соединения, и продолжали инкубацию. После трех дней среду опять заменяли 500 мкл среды MCDB 131, содержащей тестируемые соединения и на стадии 7-го дня от начала добавления тестируемого соединения подсчитывали число капилляров, образовавшихся вокруг аорты, под микроскопом. Раствор, содержащий тестируемые соединения, получали в трехкратной разводящей системе, где наибольшая концентрация составляла 10 мкг/мл.
Степень ингибирования вычисляли по следующей формуле и определяли 50% концентрацию ингибирования (IC50) для каждого тестируемого соединения (см. табл.1).
Степень ингибирования (%) = (С - Т)/С•100,
С: количество капилляров в отсутствие соединения,
Т: количество капилляров при добавлении соединения.
Фармакологический экспериментальный пример 2.
Эффект ингибирования роста эндотедиальных клеток.
Эндотелиальные клетки, взятые из пупочной вены человека (HUVEC; производства Sanko Junyaku), инкубированные в среде EGM (производство Sanko Junyaku), содержащей 100 единиц пенициллина и 100 мкг/мл стрептомицина, доводили до 0,8-1•104 клеток на мл и каждые 100 мкл отдельно вносили в 96-луночную плашку. После инкубирования в CO2 инкубаторе (5% СО2) при 37oС в течение ночи, туда добавляли 100 мкл среды EGM, содержащей тестируемое соединение, разведенное трехкратно, с последующей инкубацией в течение трех дней. Число клеток на данный момент измеряли с помощью метода МТТ. Таким образом, туда добавляли 50 мкл фосфатного буфера, содержащего 0,33% бромида 3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолия (МТТ), и продолжали инкубацию в течение 3-4 часов. Затем, после удаления супернатантной жидкости культуры, добавляли 100 мкл диметилсульфоксида (ДМСО), для растворения формазана, который образовался в клетках, и измеряли поглощение при длине волны 540 нм с помощью планшетного считывающего устройства (производство Corona Denki).
Степень ингибирования вычисляли по следующей формуле и определяли 50% концентрацию ингибирования (IC50) для каждого соединения (см. табл.2).
Степень ингибирования (%) = (С - Т)/С•100,
С: поглощение в отсутствие соединения,
Т: поглощение при добавлении соединения.
Фармакологический экспериментальный пример 3.
Эффект ингибирования пролиферации меланомы мыши В16
Клетки меланомы мыши В16, инкубированные в модифицированной среде Dulbecco-Eagle (DMED; производство Nissui Seiyaku), содержащей 10% эмбриональной бычьей сыворотки, 100 единиц/мл пенициллина и 100 мкг/мл стрептомицина, доводили до 2•104 клеток/мл и каждые 100 мкл отдельно помещали в 96-луночную плашку. После проводили инкубацию в СО2 инкубаторе (5% СО2) при 37oС в течение одной ночи, затем добавляли 100 мкл вышеуказанной культуры, содержащей тестируемое соединение, разведенное в трехкратной серии, с последующей инкубацией в течение трех дней и измеряли число клеток на тот момент с помощью метода МТТ. Кроме того, проводили обработку 0,33% раствором МТТ в течение 1-2 часов.
Степень ингибирования вычисляли по следующей формуле и определяли 50% концентрацию ингибирования (IC50) для каждого соединения (см. табл.3).
Степень ингибирования (%) = (С - Т)/С•100,
С: поглощение в отсутствие соединения,
Т: поглощение при добавлении соединения.
Из фармакологического экспериментального примера 1 ясно, что соединения по настоящему изобретению проявляют четкое антиангиогенное действие. Из фармакологических экспериментальных примеров 2 и 3 ясно, что эффект ингибирования пролиферации соединениями по настоящему изобретению клеток меланомы В16 был в 5-100 раз слабее, чем эндотелиальных клеток, и что они специфически воздействуют на эндотелиальные клетки в кровеносном сосуде.
Между тем, оценку противоопухолевого эффекта проводили в соответствии с методом Koyanagi и др. (Cancer Res., 54, 1702-1706, 1994), применяя клеточную линию КР-1, происходящую из злокачественной опухоли поджелудочной железы человека, и клеточную линию НСТ 116, происходящую из злокачественной опухоли ободочной кишки человека. Вышеупомянутые малигнизированные клетки человека (5•106 клеток) подкожно трансплантировали голым мышам (KSN) 6-7-недельного возраста и начали введение соединения по настоящему изобретению со стадии, когда они достигли размера приблизительно 100 мм3. В экспериментах применяли группу из десяти мышей, которым не вводили лекарство, а в группе, которой вводили лекарство, для каждой дозы использовали пять мышей. Непрерывно, два раза в день вводили дозу 50 мг/кг, 100 мг/кг или 200 мг/кг per os и сравнивали размер опухоли на 22-й день от начала введения с группой, которой не вводили лекарство. Результат составил 37%, 30% и 11% соответственно в клеточной линии КР-1, происходящей из злокачественной опухоли поджелудочной железы человека, и 0,2%, 0,3% и 0,0% соответственно в клеточной линии НСТ 116, происходящей из злокачественной опухоли ободочной кишки человека, в случае группы, которой для примера вводили соединение по примеру 1. Таким образом, соединение по примеру 1 показало существенный противоопухолевый эффект.
Из вышеприведенных результатов можно предположить, что соединение по настоящему изобретению оказывает превосходный эффект с точки зрения эффективности и безопасности по сравнению с известными бактерицидными противоопухолевыми препаратами, которые непосредственно воздействуют на малигнизированные клетки.
Как указано выше в экспериментальных примерах, соединения по настоящему изобретению обладают превосходным антиангиогенным действием и являются пригодными в качестве противоопухолевых средств против злокачественной опухоли поджелудочной железы, злокачественной опухоли ободочной кишки, рака желудка, злокачественной опухоли молочной железы, злокачественной опухоли предстательной железы, рака легких и злокачественной опухоли яичников и в качестве лекарственных средств против диабетической ретинопатии, ревматоидного артрита и гематомы.
Примеры
Здесь и далее будут проиллюстрированы примеры получения, демонстрирующие производство исходных соединений для соединений по настоящему изобретению, и примеры, демонстрирующие соединения по настоящему изобретению, хотя разумеется, что настоящее изобретение этим не ограничивается.
Пример получения 1. Этилпируват-N-(5-метил-2-нитрофенил)гидразон
К смешанному раствору из 160 мл воды и 170 мл концентрированной хлористо-водородной кислоты добавляли 75,0 г (493 ммоль) 5-метил-2-нитроанилина с последующим перемешиванием. Водный раствор 80 мл 36,0 г (517 ммоль) нитрита натрия по каплям добавляли туда при -20oС. Реакционный раствор добавляли к раствору, полученному растворением этил-2-метилацетата в 100 мл этанола, с последующим добавлением 200 мл 12 н. водного раствора гидроксида калия при -20oС с перемешиванием в течение 30 минут. После перемешивания смеси при той же температуре в течение 30 минут добавляли 100 мл концентрированной хлористо-водородной кислоты и полученные преципитаты собирали с помощью фильтрации, промывали водой и сушили в вакууме в течение ночи. Туда добавляли смешанный раствор из диэтилового эфира и гексана, полученные кристаллы собирали фильтрацией с получением 130 г соединения, указанного в заголовке.
1Н-ЯМР (ДМСО-d6) δ (м.д.); 1,29 (3Н, t, J=7,2 Гц), 2,16 (3Н, с), 2,40 (3Н, с), 4,25 (2Н, кв, J=7,2 Гц), 6,91 (1Н, дд, J=8,8, 2,0 Гц), 7,63 (1Н, с), 8,07 (1Н, д, J=8,8 Гц), 10,69 (1Н, с).
Пример получения 2. Этил-4-метил-7-нитро-1Н-индол-2-карбоксилат
К 250 мл суспензии 25,0 г (94,2 ммоль) соединения по примеру получения 1 в ксилоле добавляли 100 г полифосфорной кислоты с последующим нагреванием с обратным холодильником в течение 3 часов. К реакционному раствору добавляли 80 мл воды и 300 мл этилацетата при охлаждении льдом. Полученные нерастворимые вещества отфильтровывали с последующим промыванием 1,5 литрами этилацетата, и полученный фильтрат экстрагировали этилацетатом. Органическую фазу последовательно промывали насыщенным раствором бикарбоната натрия, водой и солевым раствором, сушили над сульфатом магния и концентрировали до высушивания. К полученному остатку добавляли смешанный раствор трет-бутилметилового эфира и гексана, и полученные кристаллы собирали фильтрацией с получением 11,1 г соединения, указанного в заголовке.
1Н-ЯМР (ДМСО-d6) δ (м.д.); 1,35 (3Н, т, J=7,2 Гц), 2,65 (3Н, с), 4,38 (2Н, кв, J=7,2 Гц), 7,16 (1H, д, J=8,4 Гц), 7,51 (1Н, с), 8,19 (1Н, д, J=8,4 Гц), 11,29 (1Н, ушир.с).
Пример получения 3. 4-Метил-7-нитро-1Н-индол-2-карбоновая кислота
К 150 мл раствора 11,0 г (44,3 ммоль) соединения по примеру получения 2 в тетрагидрофуране добавляли 150 мл 1 н. водного раствора гидроксида натрия с последующим нагреванием при перемешивании по 80oС в течение 30 минут. Реакционный раствор концентрировали, к полученному остатку при охлаждении льдом добавляли 40 мл 5 н. хлористо-водородной кислоты для доведения до рН 1, и полученные преципитаты фильтровали и промывали водой. Преципитаты растворяли в 300 мл тетрагидрофурана и экстрагировали этилацетатом. Органическую фазу промывали насыщенным солевым раствором, сушили над сульфатом магния и концентрировали до высушивания с получением 9,60 г соединения, указанного в заголовке.
1Н-ЯМР (ДМСО-d6) δ (м.д.); 2,62 (3Н, с), 7,13 (1Н, д, J=8,0 Гц), 7,42 (1Н, с), 8,15 (1Н, д, J=8,0 Гц), 11,00 (1Н, ушир.с).
Пример получения 4. 4-Метил-7-нитро-1Н-индол
В 60 мл 1,3-диметил-2-имидазолидинона растворяли 9,58 г (43,5 ммоль) соединения по примеру получения 3, туда добавляли 1,04 г (4,35 ммоль) основного карбоната меди и смесь нагревали при перемешивании до 180oС в течение 4 часов. К реакционному раствору добавляли 120 мл этилацетата при охлаждении льдом, полученные нерастворимые вещества отфильтровывали и полученный фильтрат экстрагировали этилацетатом. Органическую фазу последовательно промывали водой и насыщенным солевым раствором и сушили над сульфатом магния. После концентрирования полученный остаток очищали на хроматографической колонке с силикагелем с получением 4,87 г соединения, указанного в заголовке.
1H-ЯМР (ДМСО-d6) δ (м.д.); 2,59 (3Н, с), 6,74 (1Н, с), 7,03 (1Н, д, J= 8,4 Гц), 7,48 (1H, с), 8,00 (1Н, д, J=8,4 Гц), 11,86 (1Н, ушир.с).
Пример получения 5. 3-Формил-4-метил-7-нитро-1Н-индол
К 12 мл (154 ммоль) диметилформамида добавляли 1,5 мл (16,1 ммоль) оксихлорида фосфора при 0oС в атмосфере азота с последующим перемешиванием при комнатной температуре в атмосфере азота в течение 20,5 часов. Раствор (20 мл) 2,0 г (11,4 ммоль) соединения по примеру получения 4 в диметилформамиде добавляли туда при 0oС с последующим нагреванием до 90oС в течение 21 часа при перемешивании. К реакционному раствору добавляли 100 мл 1 н. водного раствора гидроксида натрия при охлаждении льдом с последующим экстрагированием этилацетатом. Органическую фазу последовательно промывали водой и насыщенным солевым раствором, сушили над сульфатом магния и концентрировали до высушивания. К полученному остатку добавляли смешанный раствор трет-бутилметилового эфира и гексана, и полученные кристаллы собирали фильтрованием с получением 2,23 г соединения, указанного в заголовке.
1H-ЯМР (ДМСО-d6) δ (м.д.); 2,90 (3Н, с), 7,21 (1Н, д, J=8,4 Гц), 8,11 (1H, д, J=8,4 Гц), 8,39 (1Н, с), 10,01 (1H, с), 12,71 (1H, ушир.с).
Пример получения 6. 3-циано-4-метил-7-нитро-1Н-индол
В 100 мл диметилформамида растворяли 2,21 г (10,8 ммоль) соединения по примеру получения 5 с последующим добавлением туда 900 мг (13,0 ммоль) гидроксиламингидрохлорида и 1,05 мл (13,0 ммоль) пиридина. После нагревания до 60oС при перемешивании в течение 40 минут к реакционному раствору добавляли 53,9 ммоль 1,1'-карбонилдиимидазола (53,9 ммоль) при охлаждении льдом. После нагревания при 60oС в течение дальнейших 30 минут при перемешивании к реакционному раствору добавляли 3,0 мл (21,5 ммоль) триэтиламина с последующим нагреванием при той же температуре дополнительно в течение 1 часа при перемешивании. К реакционной смеси добавляли 50 мл ледяной воды при охлаждении льдом с последующей экстракцией этилацетатом. Органическую фазу последовательно промывали водой и насыщенным солевым раствором, сушили над сульфатом магния и концентрировали до высушивания. К полученному остатку добавляли смешанный раствор трет-бутилметилового эфира и гексана, и полученные кристаллы собирали фильтрацией с получением 1,95 г соединения, указанного в заголовке.
1Н-ЯМР (ДМСО-d6) δ (м.д.); 2,78 (3Н, с), 7,22 (1Н, д, J=8,0 Гц), 8,14 (1Н, д, J=8,0 Гц), 8,41 (1Н, с), 12,76 (1Н, ушир.с).
Пример получения 7. 7-Бром-4-метил-1Н-индол
К 300 мл раствора 65,0 г (301 ммоль) 2-бром-5-метилнитробензола в тетрагидрофуране добавляли 1 литр 1,0 М раствора винилбромида магния (1 моль) в тетрагидрофуране при -60oС в атмосфере азота при перемешивании в течение 1 часа. К реакционному смешанному раствору добавляли насыщенный водный раствор хлорида аммония и этилацетата, и полученные нерастворимые вещества отфильтровывали. Полученный фильтрат сушили над сульфатом магния, концентрировали и затем полученный остаток очищали с помощью хроматографии на колонке с силикагелем с получением 35,5 г соединения, указанного в заголовке.
1Н-ЯМР (ДМСО-d6) δ (м.д.); 2,42 (3Н, с), 6,55 (1Н, с), 6,73 (1Н, д, J= 7,6 Гц), 7,16 (1Н, д, J=7,6 Гц), 7,35 (1Н, с), 11,24 (1Н, ушир.с).
Пример получения 8. 4-Метил-1Н-индол-7-карбоновая кислота
К раствору (200 мл) 35,5 г (169 ммоль) соединения по примеру получения 7 в тетрагидрофуране добавляли 1,6 М раствор (350 мл) бутиллития (384 ммоль) в гексане, в атмосфере азота при -78oС, при перемешивании. После перемешивания в течение 40 минут при охлаждении льдом в реакционный раствор вводили диоксид углерода при -50oС и перемешивали в течение 15 минут. К реакционной смеси добавляли воду при той же температуре, растворитель выпаривали и полученный преципитат собирали с помощью фильтрации и промывали водой. Преципитат растворяли в 300 мл тетрагидрофурана, сушили над сульфатом магния и далее концентрировали до высушивания с получением 25,9 г соединения, указанного в заголовке.
1Н-ЯМР (ДМСО-d6) δ (м.д.); 2,51 (3Н, с), 6,53 (1Н, с), 6,88 (1Н, д, J= 7,6 Гц), 7,31 (1Н, с), 7,62 (1Н, д, J=7,6 Гц), 10,99 (1Н, ушир.с), 12,79 (1H, ушир.с).
Пример получения 9. 7-(N-трет-Бутоксикарбонил)амино-4-метил-1H-индол
В 80 мл толуола суспендировали 7,0 г (40,0 ммоль) соединения по примеру получения 8, далее туда добавляли 22 мл (160 ммоль) и 11,2 мл (52 ммоль) дифенилфосфорилазида в атмосфере азота и смесь перемешивали при комнатной температуре в течение 30 минут. К реакционному раствору добавляли 8 мл (84 ммоль) трет-бутанола, смесь нагревали при перемешивании при 100oС в течение 2,5 часов и далее реакционный раствор концентрировали. Полученный остаток растворяли в этилацетате, последовательно промывали 0,1 н. хлористо-водородной кислотой, водой и насыщенным солевым раствором, сушили над сульфатом магния и концентрировали до высушивания. К полученному остатку добавляли смешанный раствор из диэтилового эфира и гексана, и полученные кристаллы собирали фильтрацией с получением 7,87 г соединения, указанного в заголовке.
1Н-ЯМР (ДМСО-d6) δ (м.д.); 1,48 (9Н, с), 2,38 (3Н, с), 6,37-6,44 (1H, м), 6,68 (1Н, д, J=8,4 Гц), 7,22-7,31 (2Н, м), 8,86 (1Н, ушир.с), 10,73 (1Н, ушир.с).
Пример получения 10. 7-(N-трет-Бутоксикарбонил)амино-3-формил-4-метил-1H-индол
К 400 мл (5,2 моль) диметилформамида добавляли 40 мл (429 ммоль) оксихлорида фосфора при 0oС в атмосфере азота с последующим перемешиванием при той же температуре в течение 25 минут. При 0oС туда добавляли 74,0 г (300 ммоль) соединения по примеру получения 9 с последующим перемешиванием при комнатной температуре в течение 1,5 часов. К реакционной смеси добавляли 250 мл 5 н. водного раствора гидроксида натрия при охлаждении льдом до установления значения рН 8. Для отделения органической фазы туда добавляли тетрагидрофуран, этилацетат и воду с последующим промыванием водой и солевым раствором последовательно. После высушивания сульфата магния выпаривали растворитель. К полученному остатку добавляли смешанный раствор диэтилового эфира и гексана, и полученные кристаллы собирали фильтрацией с получением 53,7 г соединения, указанного в заголовке.
1H-ЯМР (ДМСО-d6) δ (м.д.); 1,50 (9Н, с), 2,71 (3Н, с), 6,90 (1H, д, J= 7,6 Гц), 7,32-7,41 (1H, м), 8,21 (1H, д, J=1,6 Гц), 8,99 (1H, ушир.с), 9,93 (1Н, с), 11,88 (1H, ушир.с).
Пример получения 11. 7-(N-трет-Бутоксикабонил)амино-3-циано-4-метил-1Н-индол
В 50 мл диметилформамида растворяли 4,43 г (16,2 ммоль) соединения по примеру получения 10 с последующим добавлением туда же 1,35 г (19,4 ммоль) гидроксиламингидрохлорид и 1,6 мл (19,8 ммоль) пиридина. После нагревания при перемешивании при 60oС в течение 45 минут, к реакционному раствору добавляли 1,1-карбоилдиимидазол (80,8 ммоль) при охлаждении льдом. После нагревания при перемешивании при 60oС в течение дальнейших 30 минут к реакционному раствору добавляли 4,5 мл (32,3 ммоль) триэтиламина с последующим нагреванием при перемешивании при той же температуре в течение следующих 30 минут. К реакционной смеси при охлаждении льдом добавляли воду с последующей экстракцией этилацетатом. Органическую фазу последовательно промывали водой и солевым раствором, сушили над сульфатом магния и затем концентрировали с получением 4,27 г соединения, указанного в заголовке.
1Н-ЯМР (ДМСО-d6) δ (м.д.); 1,49 (9Н, с), 2,60 (3Н, с), 6,89 (1Н, д, J= 8,0 Гц), 7,34-7,42 (1Н, м), 8,20 (1Н, д, J=2,8 Гц), 9,04 (1Н, ушир.с), 11,80 (1Н, ушир.с).
Пример получения 12. 7-Амино-3-циано-4-метил-1Н-индол
В смешанном растворе из 100 мл тетрагидрофурана и 100 мл метанола суспендировали 12,6 г (62,6 ммоль) соединения по примеру получения 6, и проводили гидрирование при 3-кратном атмосферном давлении и при температуре окружающей среды в присутствии 430 мг (1,87 ммоль) оксида платины. После отфильтровывания фильтрат концентрировали до высушивания, к остатку добавляли смешанный раствор трет-бутилметилового эфира и гексана и собирали кристаллы с помощью фильтрации с получением 10,7 г соединения, указанного в заголовке. В 400 мл дихлорметана растворяли 50,5 г (186 ммоль) соединения по примеру получения 11 и туда же добавляли 210 мл (2,76 ммоль) трифторуксусной кислоты при 0oС в атмосфере азота с последующим перемешиванием при комнатной температуре в течение 40 минут. К реакционному раствору добавляли 5 н. водный раствор гидроксида натрия при -20oС до значения рН 7. Растворитель удаляли, и остаток экстрагировали этилацетатом. Органическую фазу последовательно промывали водой и солевым раствором, сушили над сульфатом магния и концентрировали до высушивания. К полученному остатку добавляли смешанный раствор диэтилового эфира и гексана, и кристаллы собирали фильтрацией с получением 24,5 г соединения, указанного в заголовке.
1H-ЯМР (ДMCO-d6) δ (м.д.); 2,47 (3Н, с), 5,07 (2Н, с), 6,34 (1Н, д, J= 7,6 Гц), 6,64 (1Н, д, J=7,6 Гц), 8,10 (1Н, с), 11,70 (1H, ушир.с).
Пример получения 13. 3-Цианобензолсульфонилхлорид
К смешанному раствору 200 мл воды и 250 мл концентрированной хлористо-водородной кислоты добавляли 25,0 г (212 ммоль) 3-цианоанилина с последующим перемешиванием. Водный раствор (80 мл) 15,5 г (223 ммоль) нитрита натрия добавляли по каплям туда же при -10oС. Реакционный раствор добавляли к уксусной кислоте, насыщенной диоксидом серы (полученной растворением диоксида серы в 250 мл уксусной кислоты с последующим добавлением 2,1 г одновалентного хлорида меди) при охлаждении льдом и перемешивании. После 1 часа реакционный раствор вносили в 500 мл ледяной воды и экстрагировали диэтиловым эфиром. Экстракт последовательно промывали насыщенным водным раствором бикарбоната натрия, водой и солевым раствором и сушили над сульфатом магния. Растворитель выпаривали, к остатку добавляли смешанный раствор диэтилового эфира и гексана, и кристаллы собирали фильтрацией с получением 16,0 г соединения, указанного в заголовке.
1Н-ЯМР (ДМСО-d6) δ (м.д.); 7,55 (1Н, т, J=8,0 Гц), 7,78 (1Н, дд, J=8,0, 1,2 Гц), 7,86-7,92 (2Н, м).
Пример получения 14. 4-Сульфамоилбензолсульфонилхлорид
К смешанному раствору 80 мл воды и 50 мл концентрированной хлористо-водородной кислоты добавляли 25,0 г (145 ммоль) 4-аминобензолсульфонамида с последующим перемешиванием. Водный раствор (20 мл) 10,5 г (152 ммоль) нитрита натрия добавляли туда по каплям при температуре от -13oС до -10oС в течение 15 минут. После 10 минут реакционный раствор добавляли к смешанному раствору, насыщенному диоксидом серы (полученному насыщением диоксидом серы смешанного раствора из 150 мл уксусной кислоты и 12,5 мл концентрированной хлористо-водородной кислоты с последующим добавлением 3,7 г одновалентного хлорида меди) при -30oС при перемешивании. После 1 часа к реакционному раствору добавляли ледяную воду (500 мл), и полученный преципитат собирали фильтрацией. Преципитат растворяли в смешанном растворе из 450 мл толуола и 150 мл этилацетата. После полученные нерастворимые вещества отфильтровывали, фильтрат экстрагировали этилацетатом. Органическую фазу последовательно промывали насыщенным раствором бикарбоната натрия и солевым раствором и сушили над сульфатом магния. Растворитель выпаривали, к полученному остатку добавляли 100 мл толуола, и кристаллы собирали фильтрацией с получением 20,9 г соединения, указанного в заголовке.
1H-ЯМР (ДМСО-d6) δ (м.д.); 7,65-7,69 (2Н, м), 7,71-7,78 (4Н, м).
Пример получения 15. 5-Бром-3-хлор-7-нитро-1Н-индол
К раствору 12,00 г (49,8 ммоль) 5-бром-7-нитро-1Н-индола в 140 мл тетрагидрофурана добавляли 1,4 мл диметилформамида и 6,98 г (52,3 ммоль) N-хлорсукцинимида с последующим перемешиванием при комнатной температуре в течение ночи. Туда же добавляли 10% водный раствор тиосульфата натрия с последующей экстракцией этилацетатом. Органическую фазу последовательно промывали водой и солевым раствором, сушили над сульфатом магния и концентрировали до высушивания с получением 14,84 г соединения, указанного в заголовке.
1Н-ЯМР (ДМСО-d6) δ (м. д.); 7,79 (1Н, с), 8,15 (1Н, с), 8,23 (1Н, с), 12,32 (1Н, ушир.с).
Пример получения 16. 7-Амино-5-бром-3-хлор-1H-индолгидрохлорид
К раствору (250 мл) 14,84 г (53,9 ммоль) соединения по примеру получения 15 в метаноле добавляли 70 мл концентрированной хлористо-водородной кислоты и 31,97 г (269 ммоль) порошкообразного олова, с последующим перемешиванием при комнатной температуре в течение 80 минут. После туда же добавляли 5 н. водный раствор гидроксида натрия при охлаждении льдом для доведения до значения рН 10, полученный преципитат отфильтровывали и фильтрат экстрагировали этилацетатом. Органическую фазу последовательно промывали насыщенным раствором бикарбоната натрия и солевым раствором, сушили над сульфатом магния и концентрировали. Затем полученный остаток очищали с помощью хроматографии на колонке с силикагелем с получением 14,35 г 7-амино-5-бром-3-хлор-1Н-индола. Его растворяли в этилацетате, и туда же добавляли смешанный раствор (17 мл) 4 н. хлороводорода и этилацетата. Полученный преципитат собирали фильтрацией и промывали гексаном с получением 13,23 г соединения, указанного в заголовке.
1H-ЯМР (ДМСО-d6) δ (м.д.); 5,11 (3Н, ушир.с), 6,64 (1Н, с), 6,93 (1H, с), 7,50 (1Н, д, J=2,0 Гц), 11,38 (1H, ушир.с).
Пример получения 17. Этилпируват-2-(4-метил-2-нитрофенил)гидразон
В 110 мл воды суспендировали 30,00 г (0,197 моль) 4-метил-2-нитроанилина с последующим добавлением туда же 66 мл концентрированной хлористо-водородной кислоты. Водный раствор (35 мл) 16,33 г (0,231 моль) нитрита натрия добавляли по каплям туда же при 10oС или ниже с последующим перемешиванием в течение 40 минут при охлаждении льдом для получения раствора соли диазония. В смешанном растворе 150 мл этанола и 300 мл воды растворяли 28,43 г (0,197 моль) этил-2-метилацетоацетата с последующим добавлением туда же 120 мл водного раствора 53,36 г (0,808 моль) гидроксида калия при охлаждении льдом. Затем туда же добавляли по каплям предварительно приготовленный раствор соли диазония при той же температуре и перемешивали при охлаждении льдом в течение 20 минут. После добавляли туда же концентрированную хлористо-водородную кислоту для доведения до значения рН 1, полученный преципитат собирали фильтрацией, промывали водой и сушили в вакууме над пентаоксидом фосфора с получением 46,42 г соединения, указанного в заголовке.
1H-ЯМР (ДМСО-d6) δ (м.д.); 1,40 (3Н, т, J=7,2 Гц), 2,23 (3Н, с), 2,36 (3Н, с), 4,35 (2Н, кв, J=7,2 Гц), 7,44 (1Н, дд, J=8,8, 1,6 Гц), 7,93 (1Н, д, J=8,8 Гц), 8,00 (1H, с), 10,87 (1Н, ушир.с).
Пример получения 18. Этил-5-метил-7-нитро-1Н-индол-2-карбоксилат
К раствору (320 мл) 15,92 г (60,0 ммоль) соединения по примеру получения 17 в ксилоле добавляли полифосфорную кислоту с последующим нагреванием при перемешивании в течение ночи. Туда же добавляли воду и этилацетат, полученные нерастворимые вещества отфильтровывали и органическую фазу отделяли. Органическую фазу последовательно промывали водой и солевым раствором, сушили над сульфатом магния и концентрировали. Затем полученный остаток очищали с помощью хроматографии на колонке с силикагелем с получением 7,32 г соединения, указанного в заголовке.
1H-ЯМР (ДМСО-d6) δ (м.д.); 1,34 (3Н, т, J=7,0 Гц), 2,47 (3Н, с), 4,36 (2Н, кв, J= 7,0 Гц), 7,35 (1H, с), 7,99 (1Н, с), 8,11 (1Н, с), 11,25 (1Н, ушир.с).
Пример получения 19. 5-Метил-7-нитро-1Н-индол
К раствору (80 мл) 7,86 г (31,7 ммоль) соединения по примеру получения 18 в тетрагидрофуране добавляли 150 мл 1 н. водного раствора гидроксида натрия при охлаждении льдом с последующим перемешиванием при комнатной температуре в течение 3,5 часов. При охлаждении льдом добавляли туда же 2 н. хлористо-водородную кислоту для доведения до значения рН 1 с последующей экстракцией этилацетатом. Органическую фазу последовательно промывали водой и солевым раствором, сушили над сульфатом магния и затем концентрировали до высушивания с получением 7,13 г 5-метил-7-нитро-1Н-индол-2-карбоновой кислоты. Полученное соединение растворяли в 160 мл 1,3-диметил-2-имидазолидинoна с последующим добавлением 716 мг (3,24 ммоль) основного карбоната меди и перемешиванием при 185oС в течение 2 часов. Реакционный раствор вносили в воду, полученные нерастворимые вещества отфильтровывали, и полученный фильтрат экстрагировали этилацетатом. Органическую фазу последовательно промывали водой и солевым раствором, сушили над сульфатом магния и концентрировали. Затем полученный остаток очищали с помощью хроматографии на колонке с силикагелем с получением 4,50 г соединения, указанного в заголовке.
1Н-ЯМР (ДМСО-d6) δ (м.д.); 2,46 (3Н, с), 6,62 (1Н, д, J=2,8 Гц), 7,47 (1Н, д, J=2,8 Гц), 7,87 (1H, с), 7,92 (1Н, с), 11,77 (1Н, ушир.с).
Пример получения 20. 3-Бром-5-метил-7-нитро-1Н-индол
К раствору (70 мл) 4,50 г (25,5 ммоль) соединения по примеру получения 19 в тетрагидрофуране добавляли 0,7 мл диметилформамида и 4,78 г (26,9 ммоль) N-бромсукцинимида с последующим перемешиванием при комнатной температуре в течение 70 минут. Туда же добавляли 10% водный раствор тиосульфата натрия с последующей экстракцией этилацетатом. Органическую фазу последовательно промывали водой и солевым раствором, сушили над сульфатом магния и затем концентрировали до высушивания с получением 6,53 г соединения, указанного в заголовке.
1H-ЯМР (ДМСО-d6) δ (м. д.); 2,50 (3Н, с), 7,67 (1Н, с), 7,73 (1Н, с), 8,02 (1Н, с), 12,10 (1Н, ушир.с).
Пример получения 21. 7-Амино-3-бром-5-метил-1Н-индол
В смешанном растворе 150 мл метанола и 75 мл воды суспендировали 6,76 г (26,5 ммоль) соединения по примеру получения 20, и затем добавляли туда же 11,34 г (212 ммоль) хлорида аммония и 5,92 г (106 ммоль) порошкообразного железа. После перемешивания при 80oC в течение 1 часа полученные нерастворимые вещества отфильтровывали. К фильтрату добавляли насыщенный раствор бикарбоната натрия для доведения до значения рН 8 с последующей экстракцией этилацетатом. Органическую фазу последовательно промывали насыщенным раствором бикарбоната натрия, водой и солевым раствором, сушили над сульфатом магния и концентрировали. Затем полученный остаток очищали с помощью хроматографии на колонке с силикагелем с получением 3,30 г соединения, указанного в заголовке.
1Н-ЯМР (ДМСО-d6) δ (м.д.); 2,24 (3Н, с), 5,08 (2Н, ушир.с), 6,20 (1Н, с), 6,41 (1Н, с), 7,35 (1Н, с), 10,86 (1Н, ушир.с).
Пример получения 22. 6-Амино-3-пиридинсульфонилхлорид
К 123,8 г (1,06 моль) хлорсульфоновой кислоты добавляли порциями 10,00 г (0,106 моль) 2-аминопиридина при охлаждении льдом. Туда же добавляли тионилхлорид (50,56 г, 0,425 моль) с последующим нагреванием с обратным холодильником в течение 2,5 часов и дальнейшим перемешиванием при 150oС в течение 7 часов. Реакционный раствор вносили в ледяную воду, нейтрализовали добавлением туда раствора бикарбоната натрия и экстрагировали этилацетатом. Органическую фазу последовательно промывали насыщенным раствором бикарбоната натрия, водой и солевым раствором, сушили над сульфатом магния и затем концентрировали до высушивания. Полученный остаток суспендировали в этиловом эфире и нерастворимые вещества отфильтровывали. Фильтрат концентрировали до высушивания и полученный остаток перекристаллизовывали из смеси этиловый эфир-гексан с получением 6,58 г соединения, указанного в заголовке.
Пример получения 23. 4,7-Дибром-1Н-индол
Из 62,0 г (0,224 моль) 2,5-дибромнитробензола получали 27,2 г соединения, указанного в заголовке, таким же способом, описанным в примере получения 1 JP-A 7-165708.
1H-ЯМР (ДМСО-d6) δ (м.д.); 6,52 (1Н, д, J=3,2 Гц), 7,18 (1Н, д, J=8,0 Гц), 7,26 (1Н, д, J=8,0 Гц), 7,53 (1Н, д, J=3,2 Гц), 11,75 (1Н, ушир.с).
Пример получения 24. 7-Амино-4-бром-1Н-индолгидрохлорид
К раствору (300 мл) 27,2 г (98,9 ммоль) соединения по примеру получения 23 в тетрагидрофуране по каплям добавляли 186 мл (116,3 ммоль) 1,6 М раствор н-бутиллития в гексане в атмосфере азота при -78oС с последующим перемешиванием в течение 1 часа при охлаждении льдом. После повторного охлаждения до -78oС туда же по каплям добавляли 28 мл (0,13 ммоль) дифенилфосфорилазида и смесь перемешивали при -78oС в течение 1 часа и затем при -40oС в течение 1 часа. После добавления туда же при -40oС 150 г 3,4 М раствора бис (2-метоксиэтокси)алюмогидрида натрия в толуоле, его перемешивали при комнатной температуре в течение 1 часа. Туда же добавляли воду (120 мл), полученные нерастворимые вещества собирали фильтрованием и фильтрат экстрагировали этиловым эфиром. Органическую фазу последовательно промывали насыщенным раствором бикарбоната натрия и солевым раствором и сушили над сульфатом магния. После его концентрировали, полученный остаток растворяли в этиловом эфире, туда же добавляли 50 мл смешанного раствора 4 н. хлористо-водородной кислоты и этилацетата, и полученный преципитат собирали фильтрацией с получением 14,5 г соединения, указанного в заголовке.
1H-ЯМР (ДМСО-d6) δ (м.д.); 6,41-6,43 (1Н, м), 6,80 (1H, д, J=8,0 Гц), 7,16 (1H, д, J=8,0 Гц), 7,54 (1H, т, J=2,8 Гц), 11,57 (1H, ушир.с).
Пример получения 25. 7-Бром-4-хлор-1Н-индол
Соединение, указанное в заголовке, получали способом, подобным описанному в примере получения 23.
1H-ЯМР (ДМСО-d6) δ (м.д.); 6,60-6,61 (1H, м), 7,04 (1H, д, J=8,1 Гц), 7,2 (1Н, д, J=8,1 Гц), 7,53 (1H, т, J=2,7 Гц), 11,74 (1H, ушир.с).
Пример получения 26. 7-Амино-4-хлор-1Н-индолгидрохлорид
Соединение, указанное в заголовке, получали способом, подобным описанному в примере получения 24.
1Н-ЯМР (ДМСО-d6) δ (м.д.); 6,54-6,55 (1H, м), 7,05 (1H, д, J=8,1 Гц), 7,11 (1Н, д, J=8,1 Гц), 7,60 (1H, т, J=2,7 Гц), 11,82 (1H, ушир.с).
Пример получения 27. 5-Бром-2-тиофенкарбоксиальдегид
К раствору (80 мл) 10,0 г (41,3 ммоль) 5-дибромтиофена в тетрагидрофуране добавляли по каплям 27,0 мл (43,4 ммоль) 1,6 М раствора н-бутиллития в гексане в атмосфере азота при -78oС с последующим перемешиванием при той же температуре в течение 10 минут. Затем добавляли туда же 3,5 мл (45,5 ммоль) диметилформамида при той же температуре с последующим перемешиванием в течение 20 минут. Туда же добавляли воду с последующей экстракцией этилацетатом. Органическую фазу последовательно промывали 0,1 н. водным раствором хлористо-водородной кислоты, водой и солевым раствором и сушили над сульфатом магния. Проводили концентрирование до высушивания с получением 6,4 г соединения, указанного в заголовке.
1H-ЯМР (ДМСО-d6) δ (м.д.); 7,49 (1Н, д, J=4,0 Гц), 7,87 (1Н, д, J=3,9 Гц), 9,81 (1Н, с).
Пример получения 28. 5-Бром-2-тиофенкарбонитрил
К раствору 8,2 г (43,1 ммоль) соединения по примеру получения 27 в 40 мл диметилформамида добавляли 3,3 г (51,7 ммоль) гидроксиламингидрохлорида и 4,1 г (51,7 ммоль) пиридина с последующим перемешиванием при комнатной температуре в течение 30 минут. Затем добавляли туда же 34,9 г (215,5 ммоль) 1,1'-карбонилдиимидазола при охлаждении льдом с последующим перемешиванием при комнатной температуре в течение 1 часа. К реакционному раствору добавляли ледяную воду с последующей экстракцией этилацетатом. Органическую фазу последовательно промывали 0,1 н. водным раствором хлористо-водородной кислоты, водой и солевым раствором, и сушили над сульфатом магния. После проводили концентрирование, полученный остаток очищали с помощью хроматографии на колонке с силикагелем с получением 6,7 г соединения, указанного в заголовке.
1Н-ЯМР (ДМСО-d6) δ (м.д.); 7,45 (1Н, д, J=4,0 Гц), 7,84 (1Н, д, J=4,0 Гц).
Пример получения 29. 5-Бензилтио-2-тиофенкарбонитрил
В 10 мл диметилсульфоксида суспендировали 585 мг (13,4 ммоль; 55% в масле) гидрида натрия, и затем добавляли туда же 1,4 г (11,2 ммоль) бензилмеркаптана при охлаждении льдом с последующим перемешиванием в течение 10 минут. Затем туда же добавляли 2,1 г (11,2 ммоль) соединения по примеру получения 14 с последующим перемешиванием при комнатной температуре в течение 1 часа. К реакционному раствору добавляли воду с последующей экстракцией этилацетатом. Органическую фазу последовательно промывали водой и солевым раствором и сушили над сульфатом магния. После проводили концентрирование, полученный остаток очищали с помощью хроматографии на колонке с силикагелем с получением 1,51 г соединения, указанного в заголовке.
1Н-ЯМР (ДМСО-d6) δ (м.д.); 4,26 (2Н, с), 7,18 (1Н, д, J=4,0 Гц), 7,27-7,30 (5Н, м), 7,83 (1Н, д, J=4,0 Гц).
Пример получения 30. 4-Бром-1Н-индолкарбоновая кислота
Из 51 г соединения по примеру получения 23 получали 34 г соединения, указанного в заголовке, способом, подобным описанному в примере получения 8.
1Н-ЯМР (ДMCO-d6) δ (м.д.); 6,51-6,52 (1Н, м), 7,35 (1H, д, J=8,0 Гц), 7,48 (1H, т, J=2,8 Гц), 7,66 (1H, д, J=8 Гц), 11,4 (1H, ушир.с), 13,2 (1H, ушир.с).
Пример получения 31. 7-(N-трет-Бутоксикарбонил)амино-4-бром-1Н-индол
Из 34 г соединения по примеру получения 30 получали 32 г соединения, указанного в заголовке, способом, подобным описанному в примере получения 9.
1Н-ЯМР (ДМСО-d6) δ (м.д.); 1,51 (9Н, с), 6,38-6,39 (1H, м), 7,13 (1H, д, J=8,0 Гц), 7,44-7,46 (2Н, м), 9,11 (1Н, ушир.с), 11,2 (1H, ушир.с).
Пример получения 32. 7-(N-трет-Бутоксикарбонил)амино-4-бром-3-хлор-1H-индол
Соединение, указанное в заголовке, получали обработкой N-хлорсукцинимидом раствора соединения по примеру получения 31 в смеси тетрагидрофуран/диметилформамид.
1Н-ЯМР (ДМСО-d6) δ (м.д.); 1,50 (9Н, с), 7,19 (1Н, д, J=8,4 Гц), 7,45 (1H, д, J= 8,4 Гц), 7,62 (1H, д, J=2,8 Гц), 9,08 (1H, ушир.с), 11,41 (1H, ушир.с).
Пример получения 33. 7-Амино-4-бром-3-хлор-1Н-индолгидрохлорид
Соединение (10,87 г, 31,5 ммоль) по примеру получения 32 растворяли в 120 мл метанола, и добавляли туда же 20 мл концентрированной хлористо-водородной кислоты с последующим перемешиванием при 60oС в течение 40 минут. После завершения реакции удаляли растворитель и полученный остаток трехкратно подвергали азеотропной дистилляции этанолом. Полученный остаток промывали эфиром с получением 8,5 г соединения, указанного в заголовке.
1H-ЯМР (ДМСО-d6) δ (м.д.); 6,67 (1Н, д, J=8,0 Гц), 7,13 (1Н, д, J=8,0 Гц), 7,65 (1Н, д, J=2,8 Гц), 11,74 (1Н, ушир.с).
Пример получения 34. 2-Амино-5-пиримидинсульфонилхлорид
В воде со льдом охлаждали 21 мл (0,316 моль) хлорсульфоновой кислоты и добавляли туда же порциями 3 г (0,032 моль) при перемешивании. Далее добавляли туда же 9,2 мл (0,126 моль) тионилхлорида с последующим перемешиванием при 150oС в течение 70 часов. Реакционный раствор возвращали к комнатной температуре и добавляли воду и смесь экстрагировали этилацетатом. Экстракт сушили над сульфатом натрия и концентрировали до высушивания с получением 1,7 г соединения, указанного в заголовке.
1H-ЯМР (ДМСО-d6) δ (м.д.); 5,97 (2Н, ушир.), 8,83 (2Н,с).
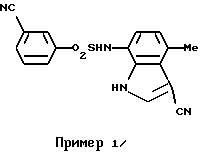
Пример 1. 3-Циано-N-(3-циано-4-метил-1Н-индол-7-ил)бензолсульфонамид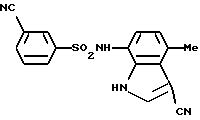
Соединение (2,00 г, 11,7 ммоль) по примеру получения 12 растворяли в 60 мл тетрагидрофурана, и затем добавляли туда же 4,0 мл (49,5 ммоль) пиридина и 2,60 г (12,9 ммоль) соединения по примеру получения 13. После перемешивания при комнатной температуре в течение 16 часов, туда же добавляли 2 н. хлористо-водородную кислоту для доведения до значения рН 1-2, и смесь экстрагировали этилацетатом. Органическую фазу последовательно промывали водой и солевым раствором, сушили над сульфатом магния и концентрировали. Затем полученный остаток очищали с помощью хроматографии на колонке с силикагелем с получением 3,90 г соединения, указанного в заголовке. Точка плавления: 220-221oС (перекристаллизация из смеси этанол/н-гексан).
1H-ЯМР (ДМСО-d6) δ (м.д.); 2,55 (3Н, с), 6,50 (1Н, д, J=8,0 Гц), 6,77 (1Н, д, J=8,0 Гц), 7,71 (1Н, т, J=8,0 Гц), 7,90 (1Н, д, J=8,0 Гц), 8,05-8,13 (2Н, м), 8,16 (1Н, с), 10,11 (1Н, ушир.с), 12,01 (1Н, ушир.с).
Пример 2. 6-Хлор-N-(3-циано-4-метил-1Н-индол-7-ил)-3-пиридинсульфонамид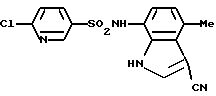
Соединение (700 мг, 4,09 ммоль) по примеру получения 12 растворяли в 20 мл тетрагидрофурана, и затем добавляли туда же 1,3 мл (16,1 ммоль) пиридина и 950 мг (4,48 ммоль) 6-хлор-3-пиридинсульфонилхлорида. После перемешивания при комнатной температуре в течение 2 часов туда же добавляли 1 н. хлористо-водородную кислоту для доведения до значения рН 1-2 и смесь экстрагировали этилацетатом. Органическую фазу последовательно промывали водой и солевым раствором, сушили над сульфатом магния и концентрировали. Затем полученный остаток очищали с помощью хроматографии на колонке с силикагелем с получением 1,16 г соединения, указанного в заголовке.
Точка плавления: 262-2630oС (перекристаллизация из смеси этанол/гексан).
1H-ЯМР (ДМСО-d6) δ (м.д.); 2,57 (3Н, с), 6,55 (1Н, д, J=7,6 Гц), 6,82 (1Н, д, J= 7,6 Гц), 7,69 (1Н, д, J=8,4 Гц), 8,01 (1Н, дд, J=8,4, 2,4 Гц), 8,17 (1Н, д, J=2,8 Гц), 8,60 (1Н, д, J=2,4 Гц), 10,21 (1Н, ушир.с), 12,03 (1Н, ушир.с).
Пример 3. N-(3-Бром-5-метил-1Н-индол-7-ил)-4-сульфамоилбензолсульфонамид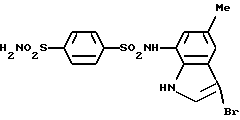
Соединение (200 мг, 0,89 ммоль) по примеру получения 22 растворяли в 6 мл тетрагидрофурана, и затем добавляли туда же 0,3 мл (3,71 ммоль) пиридин и 300 мг (1,17 ммоль) соединения по примеру получения 14. После перемешивания при комнатной температуре в течение 48 часов, добавляли туда же 1 н. хлористо-водородную кислоту для доведения до значения рН 1-2 и смесь экстрагировали этилацетатом. Органическую фазу последовательно промывали водой и солевым раствором, сушили над сульфатом магния и концентрировали. Затем смешанный раствор диэтилового эфира и гексана добавляли к полученному остатку и кристаллы собирали фильтрацией с получением 387 мг соединения, указанного в заголовке.
Точка плавления: 196-197oС (перекристаллизация из смеси этанол/н-гексан).
1H-ЯMP (ДMCO-d6) δ (м. д.); 2,24 (3Н, с), 6,60 (1Н, с), 6,98 (1Н, с), 7,44 (1Н, с), 7,55 (2Н, ушир.с), 7,85-7,95 (4Н, м), 10,13 (1H, ушир.с), 11,01 (1H, ушир.с).
Пример 4. 6-Амино-N-(5-бром-3-хлор-1Н-индол-7-ил)-3-пиридинсульфонамид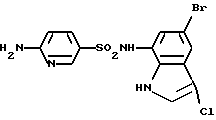
Соединение (1,00 г, 3,55 ммоль) по примеру получения 16 суспендировали в 25 мл тетрагидрофурана, и затем добавляли туда же 0,86 мл (10,6 ммоль) пиридина и 718 мг (3,73 ммоль) соединения по примеру получения 8 при охлаждении льдом. После перемешивания при комнатной температуре в течение 3 часов добавляли туда же воду и смесь экстрагировали этилацетатом. Органическую фазу последовательно промывали водой и солевым раствором, сушили над сульфатом магния и концентрировали. Затем полученный остаток очищали с помощью хроматографии на колонке с силикагелем с получением 1,27 г соединения, указанного в заголовке. Точка плавления: начало окрашивания около 237oС и разложение при 240-242oС (перекристаллизация из смеси этанол/вода).
1H-ЯМР (ДМСО-d6) δ (м.д.); 6,37 (1H, д, J=8,8 Гц), 6,94 (2Н, ушир.с), 6,97 (1Н, с), 7,36 (1Н, с), 7,54-7,57 (2Н, м), 8,16 (1H, д, J=2,8 Гц), 9,94 (1H, ушир.с), 11,17 (1Н, ушир.с).
Гидрохлорид: 1H-ЯMP (ДМСО-d6) 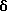 (м.д.); 6,59 (1Н, д, J=9,2 Гц), 7,00 (1H, с), 7,40 (1Н, с), 7,56 (1Н, д, J=2,4 Гц), 7,70 (1Н, дд, J=9,2, 2,0 Гц), 8,20 (1Н, д, J=2,0 Гц), 10,20 (1H, ушир.с), 11,37 (1H, ушир.с).
(м.д.); 6,59 (1Н, д, J=9,2 Гц), 7,00 (1H, с), 7,40 (1Н, с), 7,56 (1Н, д, J=2,4 Гц), 7,70 (1Н, дд, J=9,2, 2,0 Гц), 8,20 (1Н, д, J=2,0 Гц), 10,20 (1H, ушир.с), 11,37 (1H, ушир.с).
Пример 5. N-(3-Бром-5-метил-1Н-индол-7-ил)-3-цианобензолсульфонамид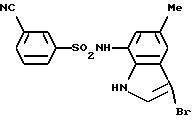
К раствору (6 мл) 260 мг (1,16 ммоль) соединения по примеру получения 21 в тетрагидрофуране добавляли 0,19 мл (2,35 ммоль) пиридина и 280 мг (1,39 ммоль) 3-цианобензолсульфонилхлорида при охлаждении льдом, и смесь перемешивали при комнатной температуре в течение ночи. Туда же добавляли 0,2 н. хлористо-водородную кислоту, и смесь экстрагировали этилацетатом. Органическую фазу последовательно промывали водой и солевым раствором, сушили над сульфатом магния и концентрировали. Затем полученный остаток очищали с помощью хроматографии на колонке с силикагелем с получением 360 мг соединения, указанного в заголовке.
Точка плавления: начало разложения около 148oС и быстрое разложение при 163-164oС (перекристаллизация из смеси этанол/н-гексан)
1Н-ЯМР (ДМСО-d6) δ (м. д.); 2,25 (3Н, с), 6,54 (1Н, с), 7,01 (1Н, с), 7,42 (1Н, д, J= 2,8 Гц), 7,71 (1Н, т, J=7,6 Гц), 7,93 (1Н, д, J=7,6 Гц), 8,07-8,11 (2Н, м), 10,09 (1Н, ушир.с), 11,04 (1Н, ушир.с).
Пример 6. N-(4-Бром-1Н-индол-7-ил)-4-цианобензолсульфонамид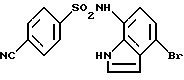
Соединение, указанное в заголовке, (686 мг) получали обработкой 700 мг (2,8 ммоль) соединения по примеру получения 24 и 685 мг (3,4 ммоль) 4-цианобензолсульфонилхлорида способом, подобным описанному в примере 1, точка плавления: 214-216oС.
1Н-ЯМР (ДМСО-d6) δ (м.д.); 6,35 (1Н, д, J=2,6 Гц), 6,53 (1Н, д, J=8,0 Гц), 7,04 (1Н, д, J=8,0 Гц), 7,41 (1H, т, J=2,8 Гц), 7,85 (2Н, д, J=8,0 Гц), 8,00 (2Н, д, J=8,0 Гц), 10,24 (1Н, ушир.с), 11,19 (1Н, ушир.с).
Пример 7. 6-Амино-N-(4-хлор-1Н-индол-7-ил)-3-пиридинсульфонамид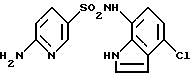
Соединение, указанное в заголовке, (961 мг) получали обработкой 1330 мг (6,4 ммоль) соединения по примеру получения 22 и 1000 мг (4,9 ммоль) соединения по примеру получения 12 способом, подобным описанному в примере 1. Точка плавления: 204-206oС.
1Н-ЯМР (ДМСО-d6) δ (м.д.); 6,38 (1Н, д, J=9,0 Гц), 6,43 (1Н, т, J=2,2 Гц), 6,77 (1Н, д, J=7,7 Гц), 6,86 (2Н, ушир.с), 7,42 (1Н, т, J=2,6 Гц), 7,56 (1Н, дд, J= 2,6, 9,0 Гц), 8,14 (1Н, д, J=2,6 Гц), 9,70 (1Н, ушир.с), 11,07 (1Н, ушир.с).
Пример 8. 6-Aмино-N-(3-бром-4-хлор-1Н-индол-7-ил)-3-пипиридинсульфонамид и его гидрохлорид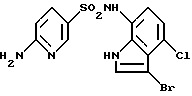
К раствору (10 мл) 650 мг (2,0 ммоль) соединения по примеру 7 в тетрагидрофуране добавляли 1 мл диметилформамида и 359 мг (2,0 ммоль) N-бромсукцинимида с последующим перемешиванием при комнатной температуре в течение ночи. Туда же добавляли 0,2 н. водный раствор хлористо-водородной кислоты с последующей экстракцией этилацетатом. Органическую фазу последовательно промывали водным раствором тиосульфата натрия, водой и солевым раствором, сушили над сульфатом магния и концентрировали. Затем полученный остаток очищали с помощью хроматографии на колонке с силикагелем с получением 662 мг соединения, указанного в заголовке.
1H-ЯМР (ДMCO-d6) δ (м.д.); 6,38 (1Н, д, J=8,8 Гц), 6,76 (1Н, д, J=8,4 Гц), 6,88 (2Н, ушир.с), 6,97 (1Н, д, J=8,4 Гц), 7,52-7,56 (2Н, м), 8,12 (1Н, д, J=2,4 Гц), 9,68 (1Н, ушир.с), 11,44 (1H, ушир.с).
В 3 мл ацетона растворяли 660 мг полученного соединения, указанного в заголовке, добавляли туда же 0,62 мл 4 н. хлористо-водородной кислоты в этилацетате и полученный преципитат собирали фильтрацией с получением 590 мг гидрохлорида соединения, указанного в заголовке.
Точка плавления: начало постепенного разложения около 267oС.
1Н-ЯМР (ДМСО-d6) δ (м.д.); 6,65 (1Н, д, J=9,2 Гц), 6,78 (1Н, д, J=8,1 Гц), 6,98 (1Н, д, J=8,2 Гц), 7,57 (1Н, д, J=2,6 Гц), 7,73 (1Н, дд, J=2,0, 9,0 Гц), 8,15 (1Н, д, J=2,4 Гц), 10,00 (1Н, ушир.с), 11,67 (1H, ушир.с).
Пример 9. N-(3-Бром-5-метил-1Н-индол-7-ил)-5-циано-2-тиофенсульфонамид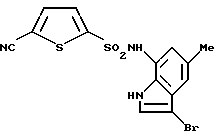
В раствор 1,3 г (5,6 ммоль) соединения по примеру получения 29 в 15 мл концентрированной хлористо-водородной кислоты (15 мл) вводили газообразный хлор при охлаждении льдом. После перемешивания в течение 30 минут реакционный раствор добавляли к смеси воды со льдом с последующей экстракцией этилацетатом. Органическую фазу последовательно промывали водой и солевым раствором, сушили над сульфатом магния и концентрировали. Полученный остаток добавляли к раствору 1,2 г (5,35 ммоль) соединения по примеру получения 22 в 6 мл пиридина с последующим перемешиванием при комнатной температуре в течение ночи. Туда же добавляли воду с последующей экстракцией этилацетатом. Органическую фазу последовательно промывали 1 н. водным раствором хлористо-водородной кислоты, водой и солевым раствором, сушили над сульфатом магния и концентрировали. Затем остаток очищали с помощью хроматографии на колонке с силикагелем с получением 1227 мг соединения, указанного в заголовке.
Точка плавления: 166-169oС (разложение).
1Н-ЯМР (ДМСО-d6) δ (м. д.); 2,30 (3Н, с), 6,65 (1Н, с), 7,07 (1Н, с), 7,44 (1Н, с), 7,54 (1Н, д, J=4,0 Гц), 7,94 (1Н, д, J=4,0 Гц), 10,47 (1H, ушир.с), 11,04 (1Н, ушир.с).
Пример 10. 2-амино-N-(4-бром-3-хлор-1Н-индол-7-ил)-5-пиримидинсульфонамид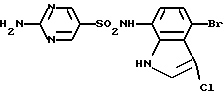
К 5 мл раствора 712 мг (2,52 ммоль) соединения по примеру получения 33 в пиридине добавляли 513 мг (2,65 ммоль) соединения по примеру получения 34 и смесь перемешивали в течение 15 часов. К реакционному раствору добавляли воду, и затем проводили экстракцию смешанным раствором этилацетата и тетрагидрофурана (10:1). Органическую фазу сушили над сульфатом магния, и затем концентрировали и очищали с помощью хроматографии на колонке с силикагелем с получением 950 мг соединения, указанного в заголовке.
Точка плавления: 285-289oС.
1Н-ЯМР (ДМСО-d6) δ (м.д.); 6,75 (1H, д, J=8,0 Гц), 7,19 (1Н, д, J=8,0 Гц), 7,59 (1Н, д, J=3,0 Гц), 7,65 (2Н, с), 8,37 (2Н, с), 9,82 (1Н, с), 11,43 (1Н, с).
1) Антипролиферативная активность в отношении меланомы мышей В16 (in vitro)
Экспериментальный метод, табл.4
См. фармакологический экспериментальный пример 3.
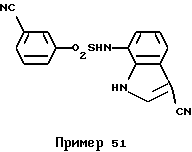
Ингибирующую активность соединения по примеру 51 ЕР 0673937 (пример 51) в отношении пролиферации клеток меланомы мышей В16 (IC50) составляет 1,6 мкг/мл, тогда как активность соединения по примеру 1 настоящей заявки (пример 1) составляет 10 мкг/мл. В сравнении с примером 51 цитотоксичность (антипролиферативная активность) соединения по примеру 1 в отношении клеток опухоли составляет одну шестую от этого показателя для соединения по примеру 51. В то же время, как показано в таблице фармакологического экспериментального примера 1 в описании настоящего изобретения, ингибирующая активность (IC50) соединения по примеру 1 в модели ангиогенеза (in vitro) составляет 0,8 мкг/мл и является в 125 раз большей, чем цитотоксичность. Поэтому предполагают, что терапевтический эффект соединения по примеру 1 на клетки опухоли (in vivo), описанный ниже, является результатом антиангиогенного действия, и соединение по примеру 1, как это ожидалось, имеет меньше побочных действий в сравнении с соединением по примеру 51.
2) Ингибирующая активность в модели ангиогенеза мышей (in vivo)
Экспериментальный метод
Ингибирующую активность соединений оценивали методом дорсального воздушного мешочка (Funahashi et al., Oncol.Res. 11, 319-329, 1999). Кольцо Millipore (от фирмы Millipore) уплотняли 0,22 мкм мембранным фильтром (HAWPO: Millipore), получая таким образом камеру. Линию клеток опухоли, полученную из раковых клеток толстой кишки человека (WiDr) (1 или 1,5 х 107 клеток), суспендированую в коллагеновом геле Типа I (type I-A: Nitta Gelatin), инъецировали в камеру и герметично закрывали. Затем камеры, содержащие опухолевые клетки или не содержащие их, имплантировали в дорсальный воздушный мешочек, образованный у самок мышей C57BL/6N в возрасте 6-8 недель путем инъецирования 10 мл воздуха через иглу размера 25. Примерно через 6 часов после имплантации перорально вводили испытываемые соединения один раз в день в течение 4 дней. На 4 день после начала эксперимента мышам вводили через хвостовую вену меченные 51Сr эритроциты мыши. Через 1 час под анестезией вырезали часть кожи вокруг камеры. После замораживания точно отделяли только ту ткань, которая соприкасалась с камерой. Меченые клетки подсчитывали при помощи γ-счетчика, a затем определяли объем крови. Величину ангиогенеза определяли путем вычитания объема крови камеры с или без опухолевых клеток. В этом эксперименте использовали 8 мышей для контрольной группы, 5 мышей использовали для испытываемого соединения при каждой дозе, табл.5.
Ингибирующее действие на ангиогенез, индуцированный опухолевыми клетками (in vivo) - см. табл.5.
При дозе 100 мг/кг соединения по примеру 51 ангиогенез ингибировался до 70,6% в модели дорсального воздушного мешочка мыши. Однако при 200 мг/кг наблюдали уменьшение веса тела, что указывало на то, что эта доза является токсичной. Таким образом, соединение по примеру 51 показало лишь незначительный эффект при максимально переносимой дозе (100 мг/кг).
Тогда как соединение по примеру 1, в котором один атом водорода в положении 4 (R2) индольного кольца соединения по примеру 51 замещен метильной группой, ингибировало ангиогенез до 53,1% при дозе 50 мг/кг, до 57,2% при дозе 100 мг/кг, до 49,6% при дозе 200 мг/кг и до 12,7% при дозе 400 мг/кг соответственно.
При сравнении соединения по примеру 51 и соединения по примеру 1, первое из указанных соединений показывает слабое ингибирующее действие на ангиогенез, тогда как соединение по примеру 1 показывает замечательное ингибирующее действие на ангиогенез.
3) Противоопухолевый эффект на модели ксенотрансплантата мыши (in vivo)
Экспериментальный метод
Противоопухолевый эффект определяли в соответствии с методом Koyanagi et al. (cancer Res., 54, 1702-1706, 1994). Опухолевые клетки НСТ116, полученные из клеток рака толстой кишки человека (5 х 106 клеток), подкожно имплантировали "голым" мышам (KSN) в возрасте от 6 до 7 недель. Через 1 неделю перорально вводили испытываемые соединения, суспендированные в 0,5% метилцеллюлозе, два раза в день по 5 дней в неделю в течение 2 недель. В этом эксперименте использовали по 5 мышей для контрольной группы и группы, которую обрабатывали соединением. На 12 день подсчитывали число мышей, не имеющих опухоли.
Терапевтический эффект испытываемых соединений на опухолевых клетках толстой кишки человека НСТ116, подкожно имплантированных модели мыши
Безопухолевый коэффициент на день 12 (два раза в день, 5 дней/неделю в течение 2 недель).
Терапевтический эффект соединения по примеру 1 и соединения по примеру 51 в отношении опухоли толстой кишки человека НСТ 116 (in vivo), табл.6.
У мышей, обработанных соединением по примеру 51, 4 из 5 обработанных мышей погибли при введении дозы 100 мг/кг в течение 12 дней, и не наблюдали ни одной не имеющей опухоли мыши при дозе 50 мг/кг. Тогда как у мышей, обработанных соединением по примеру 1, не возникало никаких побочных эффектов, и терапевтическая эффективность была определенно подтверждена. Это значит, что соединение по примеру 1 является значительно улучшенным для использования.
Из показанных выше результатов испытаний необходимо отметить то, что соединение по примеру 1 настоящей заявки является неожиданно превосходящим соединение по примеру 51 ЕР 0673937 в свете наблюдаемых in vivo отсутствия побочных эффектов, антиангиогенного действия и противоопухолевого действия.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОР ЭКСПРЕССИИ ИНТЕГРИНА | 2001 |
|
RU2240826C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ СУЛЬФОНАМИДНЫЕ ГРУППЫ, ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ И АНТИАНГИОГЕННЫЙ АГЕНТ НА ИХ ОСНОВЕ | 2000 |
|
RU2239631C2 |
| АЗОТСОДЕРЖАЩИЕ АРОМАТИЧЕСКИЕ ПРОИЗВОДНЫЕ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ИХ СОДЕРЖАЩАЯ, СПОСОБ ЛЕЧЕНИЯ И ПРИМЕНЕНИЕ | 2003 |
|
RU2310651C2 |
| ПРОИЗВОДНЫЕ СУЛЬФОНАМИДА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ | 1994 |
|
RU2128648C1 |
| ПРОИЗВОДНЫЕ КОНДЕНСИРОВАННЫХ ПОЛИЦИКЛИЧЕСКИХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1996 |
|
RU2167877C2 |
| ПРОИЗВОДНЫЕ 7-АМИНО-1Н-ИНДОЛА | 1994 |
|
RU2121997C1 |
| ПРОИЗВОДНЫЕ АНТРАНИЛОВОЙ КИСЛОТЫ ИЛИ ИХ ФАРМАКОЛОГИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ ДЛЯ ИХ ПОЛУЧЕНИЯ И ЛЕКАРСТВЕННЫЙ ПРЕПАРАТ НА ИХ ОСНОВЕ | 1994 |
|
RU2128644C1 |
| ПРОИЗВОДНЫЕ 2-АМИНОПИРИДИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ | 2001 |
|
RU2250898C2 |
| ПРОИЗВОДНОЕ ИНДОЛА, СОДЕРЖАЩЕЕ ПИПЕРИДИНОВЫЙ ЦИКЛ | 2005 |
|
RU2332413C1 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И ИХ ПРИМЕНЕНИЯ | 2002 |
|
RU2358733C2 |



Изобретение относится к новым производным индола формулы I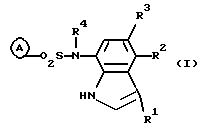
где R1 - H, галоген, CN; R2 и R3 одинаковые или отличные - Н, C1-C4 алкил, галоген; R4 - Н, С1-С4 алкил; А означает цианофенильную, аминосульфонилфенильную, аминопиридильную, аминопиримидильную, галогенпиридильную или циантиофенильную группы, при условии, что случаи, когда все R1, R2 и R3 - Н, когда оба R2 и R3 - Н или когда кольцо А - аминосульфонилфенильная группа и оба R1 и R2 - атомы галогена, исключены; и, кроме того, когда кольцо А представляет цианофенильную группу, 2-амино-5-пиридильную группу или 2-галоген-5-пиридильную группу и R1 представляет собой цианогруппу или галогеновую группу, по крайней мере один из R2 и R3 не должен быть атомом водорода. Соединения I проявляют антиангиогенное действие и могут быть использованы в качестве противоопухолевых средств, особенно против злокачественных опухолей поджелудочной железы, ободочной кишки, желудка, молочной железы, опухоли предстательной железы, легких, яичников и др. 15 с. и 12 з.п.ф-лы, 6 табл.
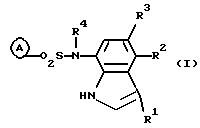
где R1 - атом водорода, атом галогена или цианогруппа;
R2 и R3 являются одинаковыми или отличньми, и каждый представляет собой атом водорода, C1-С4 алкильную группу или атом галогена;
R4 представляет собой атом водорода или C1-C4 алкильную группу;
кольцо А представляет собой цианофенильную группу, аминосульфонилфенильную группу, аминопиридильную группу, аминопиримидильную группу, галогенпиридильную группу или цианотиофенильную группу,
при условии, что случаи, когда все R1, R2 и R3 представляют собой атомы водорода, когда оба R и R3 представляют собой атомы водорода или когда кольцо А представляет собой аминосульфонилфенильную группу и оба R1 и R2 представляют собой атомы галогена, исключены; и, кроме того, когда кольцо А представляет собой цианофенильную группу, 2-амино-5-пиридильную группу или 2-галоген-5-пиридильную группу и R1 представляет собой цианогруппу или галогеновую группу, по крайней мере один из R2 и R3 не должен быть атомом водорода.
1) 3-циано-N-(3-циано-4-метил-1Н-индол-7-ил)бензолсульфонамид,
2) 6-хлор-N-(3-циано-4-метил-1Н-индол-7-ил)-3-пиридинсульфонамид,
3) N-(3-бром-5-метил-1Н-индол-7-ил)-4-сульфамоилбензолсульфонамид,
4) 6-амино-N-(5-бром-3-хлор-1Н-индол-7-ил)-3-пиридинсульфонамид,
5) N-(3-бром-5-метил-1 Н-индол-7-ил)-3-цианобензолсульфонамид,
6) N-(4-бpом-1Н-индол-7-ил)-4-цианобензолсульфонамид,
7) 6-амино-М-(4-хлор-1Н-индол-7-ил)-3-пиридинсульфонамид,
8) 6-aмино-N-(3-бром-4-хлор-1Н-индол-7-ил)-3-пиридинсульфонамид,
9) N-(3-бром-5-метил-1Н-индол-7-ил)-5-циано-2-тиофенсульфонамид и
10) 2-амино-N-(4-бром-3-хлор-1Н-индол-7-ил)-5-пиримидинсульфонамид.
1) 3-циано-N-(3-циано-4-метил-1Н-индол-7-ил)бензолсульфонамид,
2) 6-хлор-N-(3-циано-4-метил-1Н-индол-7-ил)-3-пиридинсульфонамид,
3) N-(3-бром-5-метил-1Н-индол-7-ил)-4-сульфамоилбензолсульфонамид,
4) 6-амино-N-(5-бром-3-хлор-1Н-индол-7-ил)-3-пиридинсульфонамид,
5) N-(3-бром-5-метил-1Н-индол-7-ил)-3-цианобензолсульфонамид,
6) 6-амино-N-(3-бром-4-хлор-1Н-индол-7-ил)-3-пиридинсульфонамид,
7) N-(3-бpoм-5-мeтил-1H-индoл-7-ил)-5-циaнo-2-тиoфeнcyльфoнaмид и
8) 2-амино-N-(4-бром-3-хлор-1Н-индол-7-ил)-5-пиримидинсульфонамид.
| Устройство для калибровки широкоапертурных излучателей шума | 1976 |
|
SU673937A1 |
| RU 95112848 A1, 20.03.1997 | |||
| JP 08129447, 09.12.1997 | |||
| Шпиндель хлопкоуборочной машины | 1975 |
|
SU560554A1 |
| Электромагнитное дозирующее устройство | 1975 |
|
SU614887A1 |
| RU 95122639 A1, 09.27.1997. | |||

