Область техники, к которой относится изобретение
Данное изобретение относится к регулируемому высвобождению терапевтических соединений из систем доставки лекарственных средств. Более конкретно, данное изобретение относится к эпидуральному введению терапевтических соединений с поддерживаемой скоростью высвобождения из липосомной лекарственной формы. Кроме того, изобретение относится к способу помещения эпидурального катетера в живое позвоночное.
Предпосылки создания изобретения
Устранение послеоперационной боли является серьезной проблемой для пациентов и врачей, особенно в послеоперационной палате, когда пациент просыпается от наркоза. Слишком большая доза системного опиоида, введенная с попыткой подавить боль, может потенциально вызвать угрожающее жизни угнетение дыхания. С другой стороны, либо слишком малая, либо слишком поздняя доза медикамента против послеоперационной боли может привести к тому, что пациент просыпается с невыносимой сильной болью. Кроме того, было показано, что слабо подавленная послеоперационная боль, следующая за абдоминальной или торакальной операциями, угнетает дыхательное движение грудной перегородки, живота и диафрагмы (P. R. Bromage, Textbook of Pain, P.D. Wall et al. (Eds), Churchill Livingstone, 1989, стр. 744-753), что приводит к легочному ателектазу.
Существование опиоидных рецепторов в спинном мозге было обнаружено в 1970-х годах. Вслед за первыми сообщениями о клинической эффективности в 1979 г. (М. Behar et al., Lancet 1: 527-529, 1979) эпидуральное введение опиоидов стало очень популярным для снятия послеоперационной боли (T.I. lonescu et al., Act. Anaesth. Belg. 40: 65-77, 1989; С. Jayr et al., Anesthesiology, 78: 666-676, 1993; Lurie et al., European Journal of Obstetrics and Gynecology and Reproductive Biology 49: 147-153, 1993). Эпидуральные опиоиды имеют преимущество в том, что достигается хороший местный наркоз на спинальном уровне без потери двигательного или сосудодвигательного контроля или без сниженного уровня сознания.
Инъецируемые опиоиды широко используются эпидурально в послеоперационной и послеродовой обстановке. Послеоперационные и послеродовые боли обычно продолжаются несколько дней, однако инъецируемые опиоиды имеют сравнительно короткую продолжительность действия (W. G. Brose et al., Pain 45: 11-15, 1991; R.H. Drost et al., Arzneim-Forsch/Druq Res., 38: 1632-1634, 1988; G.K. Gourlay et al. , Pain, 31: 297-305, 1987). Таким образом, для того, чтобы поддерживать надлежащее подавление боли, требуется либо непрерывное вливание, либо повторные инъекции (J.W. Kwan, Am. J. Hosp. Pharm. 47(Suppl. 1): S18-23, 1990; J.S. Anulty, International Anesthesiology Clinics 28: 17-24, 1990; R. S. Sinatra, The Vale Journal of Biology and Medicine, 64: 351-374, 1991). Непрерывное вливание или повторные инъекции дополнительно вызывают необходимость размещения катетерных систем с присоединенными инфузионными насосами или без них, причем все они требуют дорогостоящее время врача и медсестры для ухода и обслуживания. Кроме того, повторные инъекции ударной дозы вещества или непрерывные вливания могут привести к угнетению дыхания.
Позднее угнетение дыхания и апнеические эпизоды являются побочными эффектами, которые являлись самыми большими проблемами в ранних исследованиях (P.R. Bromage, Anesthesia and Analgesia 60: 461-463, 1981; E.M. Camporesi et al. , Anesthesia and. Analgesia 62: 633-640, 1983; T.L. Yaksh, Pain 11: 293-346, 1981). В недавнем нерандомизированном исследовании эпидурального морфина на 1085 пациентах, перенесших торакальную, абдоминальную или ортопедическую операции, установлено, что частота "угнетения дыхания" после эпидурального морфина составляет 0,9% (R. Stenseth et al., Acta Anaesthesiol. Scand. 29: 148-156, 1985). В качестве сравнения, частота "угрожающего жизни угнетения дыхания" у 860 пациентов, которым давали системный морфин (п. о., в.в., в.м., п.к.), составляла 0,9% (R.R. Miller et al., Drug Effects in Hospitalized Patients. John Wiley & Sons, New York, 1976). Рандомизированные исследования по сравнению эпидуральных опиоидов с системными опиоидами (в.м. или в.в.) у пациентов высокого риска показали, что подавление послеоперационной боли с помощью эпидурального опиоида приводит к превосходной анальгезии с пониженной частотой послеоперационных осложнений (N. Rawal et al. , Anesth. Analg. , 63: 583-592, 1984; М.Р. Yeager et al., Anesth. 60: 729-736, 1987).
Длительное высвобождение различных терапевтических средств после введения в липосомы, такие как мультивезикулярные липосомы, хорошо отражено в документах как для систем in vitro, так и для животных, для подоболочечного, подкожного и внутрибрюшинного пути введения, а также для пациентов-людей для подоболочечного пути введения (S. Kim, et al., J. Clin. Oncol., 11: 2186-2193, 1993; V. Russack et al. , Ann. Neurol., 34: 108-112, 1993 и М.С. Chamberlain et al., Arch. Neurol., 50: 261-264, 1993). Однако до сих пор на уровне техники не было известно длительное высвобождение эпидурально введенных соединений.
Поэтому существует потребность в новых и лучших способах введения опиоидов и других терапевтических соединений эпидурально в виде разовой дозы с тем, чтобы достигнуть поддерживаемой скорости высвобождения на терапевтически эффективных уровнях. Настоящее изобретение решает проблему ограничений, известных из предшествующего уровня, тем, что предлагается лекарственная форма с замедленным высвобождением терапевтического средства, такого как опиоид, что приводит к максимальной анальгезии сразу после разовой эпидуральной дозы и обеспечивает постепенно уменьшающуюся анальгезию в течение нескольких последующих дней.
Описание чертежей
Фиг. 1 представляет собой ряд из четырех графиков с записью анальгезирующего эффекта у крыс в течение времени после разовой эпидуральной дозы инкапсулированного в липосомы морфинсульфата (DTC401) (светлые кружки) или свободного морфинсульфата (темные кружки) для дозировок (от верхней части до нижней части) 10, 50, 175 или 250 мкг. Интенсивность анальгезии выражена в "процентах от максимально возможной анальгезии (% МВА)". Каждая точка данных представляет среднее и стандартную ошибку (СО) для 5 или 6 животных.
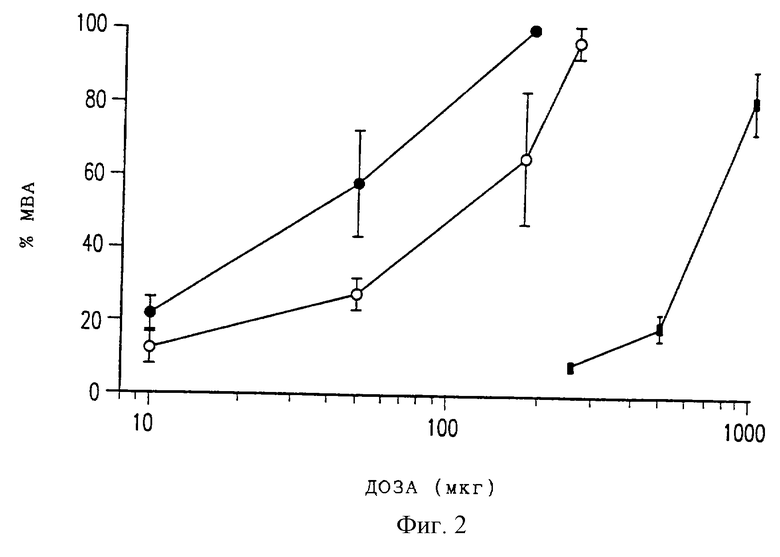
Фиг. 2 представляет собой график, показывающий кривые доза-ответ для максимальной анальгезии, измеренные для крыс после разовой эпидуральной дозы DTC401 (светлые кружки), свободного морфинсульфата (темные кружки), или же после разовой подкожной дозы свободного морфинсульфата (темные квадраты). Средний пиковый % МВА±СО получали для 5 или 6 животных.
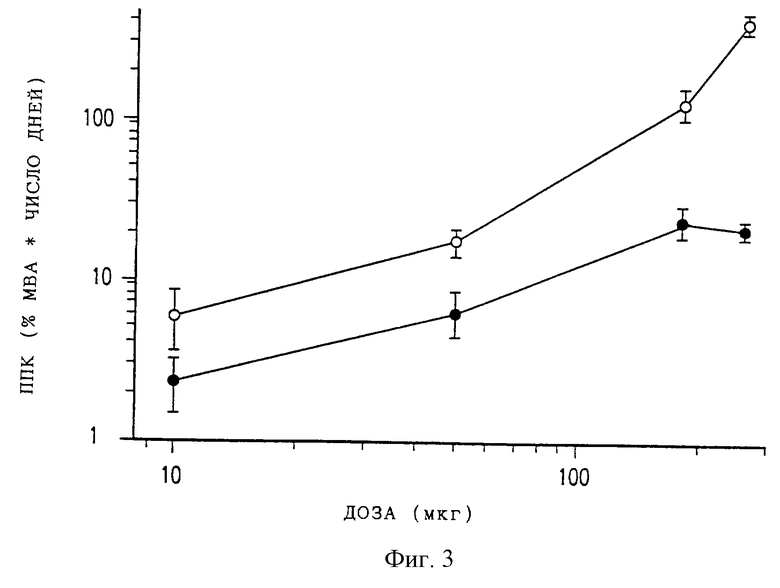
Фиг. 3 представляет собой график, на котором производится сравнение общего анальгезирующего эффекта у крыс [измеренного по площади под кривыми анальгезия-время (ППК)] для разовых доз эпидурального DTC401 (светлые кружки) или свободного морфинсульфата (темные кружки). Каждая точка данных представляет среднее и стандартную ошибку (СО) для 5 или 6 животных.
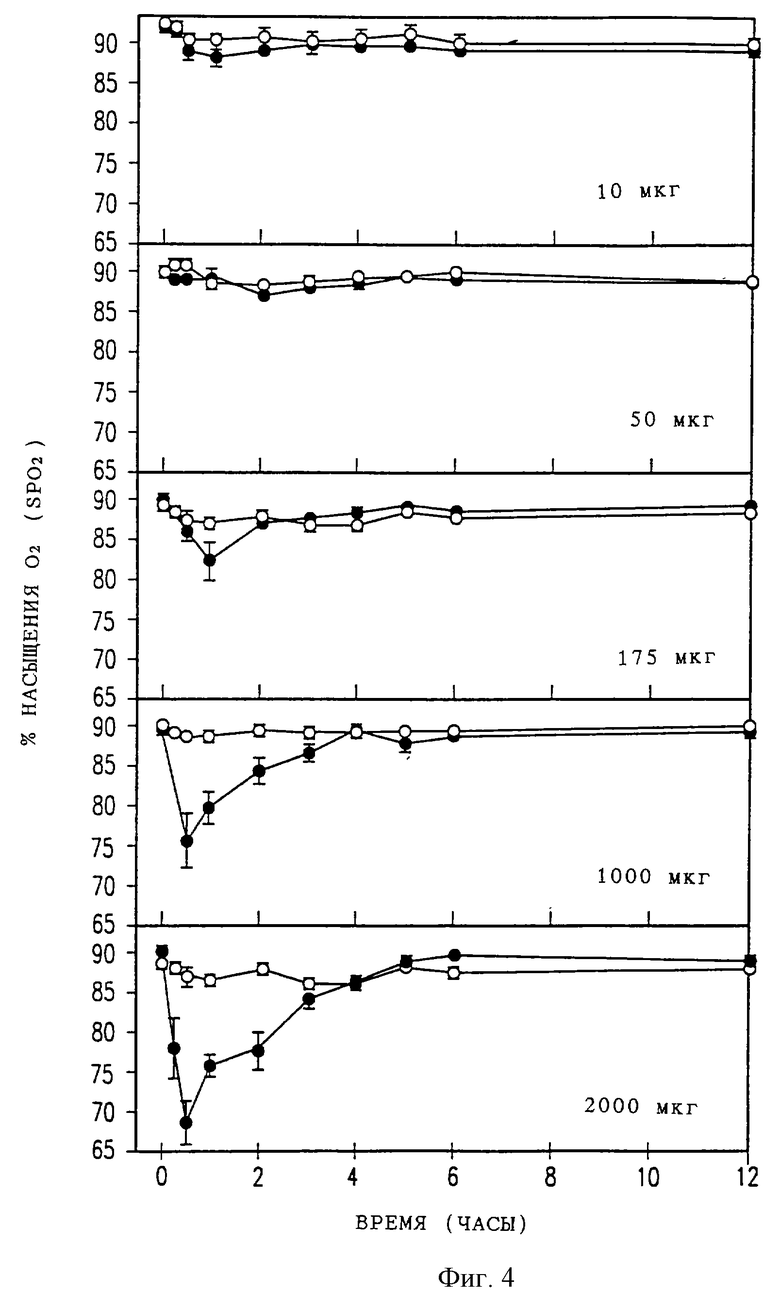
Фиг. 4 представляет собой ряд из пяти графиков, где приводится сравнение процента насыщения гемоглобина кислородом (SpO2) у крыс в течение времени после разовых эпидуральных доз (от верхней части до нижней части) 10, 50, 175, 1000 или 2000 мкг эпидурального DTC401 (светлые кружки) или свободного морфинсульфата (темные кружки). Каждая точка данных представляет среднее и стандартную ошибку (СО) для 5 животных за исключением группы с дозой 50 мкг, где n=3.
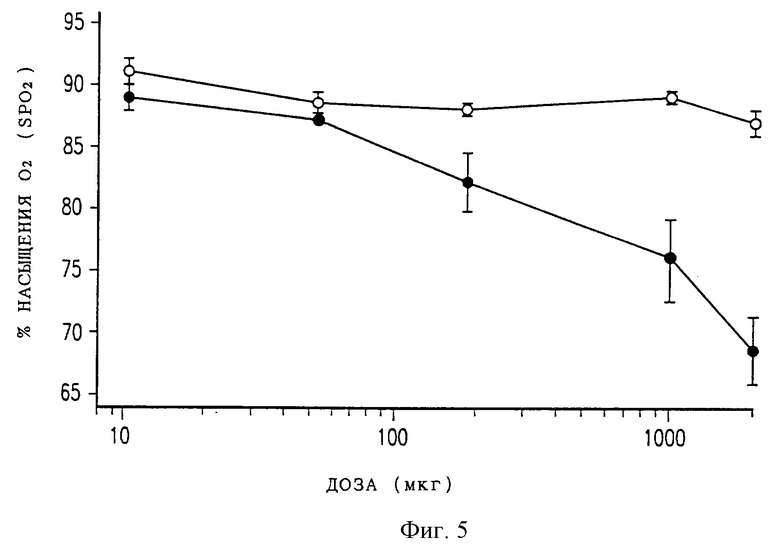
Фиг. 5 представляет собой график, где изображена кривая доза-ответ для максимального угнетения дыхания у крыс после разовой эпидуральной дозы DTC401 (светлые кружки) или свободного морфинсульфата (темные кружки). Наименьшее достигнутое значение SpO2 откладывали как функцию эпидуральной дозы морфина. Каждая точка данных представляет среднее и стандартную ошибку (СО) для 5 животных за исключением группы с дозой 50 мкг, где n=3.
На фиг. 6 изображены два графика, где сравнивается фармакокинетика в цереброспинальной жидкости (верхняя часть) и сыворотке (нижняя часть) крыс после эпидурального введения 250 мкг DTC401 (светлые кружки) или свободного морфинсульфата (темные кружки). Каждая точка данных представляет среднее и стандартную ошибку (СО) для 3 или 4 животных.
Краткое изложение сущности изобретения
Эпидуральное введение терапевтического соединения в системе доставки лекарственного средства обеспечивало удивительно более длительное высвобождение и большую продолжительность терапевтического эффекта по сравнению с применением свободного терапевтического соединения.
Следовательно, по одному из аспектов изобретения предлагается способ для длительного высвобождения терапевтического соединения путем использования системы доставки лекарственного средства, которую эпидурально вводят позвоночным, нуждающимся в такой терапии.
Позвоночное предпочтительно является млекопитающим, таким как человек. В различных предпочтительных вариантах система доставки лекарственного средства основана на липидах, особенно когда она реализуется в виде мультивезикулярной липосомы.
Признаком изобретения является возможность продолжительной доставки различных терапевтических соединений, которые в предпочтительных вариантах охватывают опиоиды или опиатные антагонисты, чтобы допустить модуляцию анальгезии. Альтернативные варианты позволяют доставлять такие терапевтические соединения, как нейротропные факторы.
Кроме того, применение лекарственной формы с длительным высвобождением в соответствии со способом изобретения облегчает эпидуральную анальгезию и снижает суммарные затраты на нее путем устранения потребности в непрерывном вливании, многократных инъекциях ударной дозы вещества или установке катетеров, а также снижает вероятность инфекции. Уменьшенная частота инъекций имеет преимущество даже в присутствии эпидуральных катетеров.
Подробное описание изобретения
В настоящем изобретении представлена система на липидной основе для доставки длительно высвобождаемого лекарственного средства, предназначенная для эпидуральной доставки терапевтического соединения с эпидуральной эффективностью, такого как опиоид. С помощью эпидурального введения соединения высвобождаются в центральную нервную систему и в цереброспинальную жидкость без пункции твердой мозговой оболочки и при поддерживаемой скорости высвобождения.
Термин "длительно высвобождаемое" означает, что терапевтическое соединение при введении в виде дозы ударного действия, инкапсулированной в лекарственной форме на липидной основе, высвобождается в течение большего периода времени по сравнению с эпидуральным введением того же лекарственного средства в свободной форме в виде инъекции ударной дозы вещества. Это необязательно означает, что концентрация терапевтического соединения остается постоянной в течение продолжительного периода времени. Как правило, после операции или после родов пациент день за днем испытывает все меньшую боль. Поэтому потребность пациента в анальгезии также уменьшается с течением времени. Используя способ эпидуральной доставки лекарственного средства согласно данному изобретению, в цереброспинальной жидкости и/или сыворотке можно поддерживать терапевтически эффективную концентрацию терапевтического соединения в течение нескольких дней, предпочтительно от примерно 2 до примерно 7 дней.
Используемый здесь термин "терапевтическое соединение" означает химическое соединение, которое пригодно для модуляции биологических процессов так, чтобы достигнуть желаемого эффекта при модуляции или лечении нежелательных состояний в живом организме. Термин "терапевтическое соединение" охватывает химические небелковые лекарственные средства, такие как антибиотики и анальгетики, а также белковые лекарственные средства, такие как цитокины, интерфероны, факторы роста и т.п.
Системы доставки лекарственных средств хорошо известны из уровня техники. Настоящее изобретение относится к любым готовым лекарственным формам с длительным высвобождением, таким как синтетические или природные полимеры в форме макромолекулярных комплексов, нанокапсул, микросфер или гранул, а также системам на липидной основе, включая эмульсии типа "масло-в-воде", мицеллы, смешанные мицеллы, синтетические мембранные везикулы и (resealed) эритроциты. Эти системы известны в общем как дисперсные системы. Дисперсные системы являются двухфазными системами, в которых одна фаза распределена во второй фазе в виде частиц или капель. Как правило, образующие систему частицы имеют диаметр 20 нм - 50 мкм. Размер частиц позволяет им суспендироваться в фармацевтическом растворе и быть введенными в эпидуральное пространство с использованием иглы или катетера и щприца.
Материалы, используемые при получении дисперсных систем, обычно являются нетоксичными и биоразлагаемыми. Таким образом, можно использовать, например, коллаген, альбумин, этилцеллюлозу, казеин, желатин, лецитин, фосфолипиды и соевое масло. Полимерные дисперсные системы можно получить способом, подобным коацервации или микрокапсулированию. Если требуется, плотность дисперсной системы можно модифицировать путем изменения удельного веса, чтобы сделать дисперсию гипербарной или гипобарной. Например, материал дисперсии можно сделать более гипербарным путем добавления иогексола, иодиксанола, метризамида, сахарозы, трегалозы, глюкозы или других биосовместимых молекул с высоким удельным весом.
Один из типов дисперсной системы, который можно использовать в соответствии с изобретением, состоит из дисперсии терапевтического средства в полимерной матрице. Терапевтическое средство высвобождается, когда полимерная матрица разлагается или подвергается биоразложению на растворимые продукты, выводящиеся из тела. Для этой цели исследовали несколько классов синтетических полимеров, включая сложные полиэфиры (Pitt et al., Controlled Release of Bioactive Materials, R. Baker, Ed., Academic Press, New York, 1980); полиамиды (Sidman et al., Journal of Membrane Science, 7:277, 1979); полиуретаны (Master, et al. , Journal of Polymer Science, Polymer Symposium, 66: 259, 1979); полиортоэфиры (Heller, et al., Polymer Engineering Science, 21: 72 7, 1981) и полиангидриды (Leong. et al., Biomaterials, 7: 364, 1986). Значительные исследования проводились на сложных полиэфирах ПМК и ПМК/ПГК. Несомненно, это является следствием соображения по удобству и безопасности. Данные полимеры легко доступны, так как их используют в качестве биоразлагаемых швов и они разлагаются на нетоксичные молочную и гликолевую кислоты (см. патент США 4,578,384, патент США 4,785,973, включенные как ссылки).
Твердые полимерные дисперсные системы могут быть синтезированы с применением таких способов полимеризации, как полимеризация в массе, межфазная полимеризация, полимеризация в растворе и полимеризация с раскрытием кольца (Odian, G. , Principles of Polymerization, 2-nd ed., John Viley & Sons, New York, 1981). С применением любого из этих способов получают разнообразие различных синтетических полимеров, имеющих широкий интервал механических, химических и биодеградационных свойств; различия в свойствах и характеристиках регулируют путем варьирования параметров реакции: температуры, концентраций реагентов, типов растворителя и времени реакции. При необходимости твердые полимерные дисперсные системы можно вначале получить в виде большей массы, которую затем размалывают или иным образом перерабатывают в частицы, достаточно малые для того, чтобы поддерживать дисперсию в подходящем физиологическом буфере (см. , например, патенты США 4,452,025; 4,389,330 и 4,696,258, включенные как ссылки).
При необходимости терапевтическое соединение можно ввести в недисперсную структуру, которую эпидурально имплантируют с помощью хирургического или механического средства. Недисперсная структура является структурой, имеющей определенную общую форму, такую как пластина, цилиндр или сфера. Механизм выделения терапевтического средства из биоразлагаемых пластин, цилиндров или сфер описан Hopfenberg (Conrolled Release Polymeric Formulations, стр. 26-32, Paul, D.R. and Harris, F.W., Eds., American Chemical Society, Washington, D.C., 1976). Простое выражение, описывающее аддитивное высвобождение из этих устройств, где высвобождение в основном контролируется разложением матрицы, выглядит следующим образом:
Mt/M∞ = 1-[1-k0t/C0α]n,
где n= 3 для сферы, n= 2 для цилиндров и n=1 для пластины. Символ α представляет радиус сферы или цилиндра или полутолщину пластины. Mt и M∞ представляют собой массы лекарства, высвободившиеся в момент времени t и на бесконечности соответственно.
В практике изобретения можно использовать любые системы доставки лекарственных средств на липидной основе. Например, можно использовать мультивезикулярные липосомы (МВЛ), многопластинчатые липосомы (также известные как многопластинчатые везикулы или "МПВ"), однопластинчатые липосомы, включая малые однопластинчатые липосомы (также известные как однопластинчатые везикулы или "МОВ") и большие однопластинчатые липосомы (также известные как большие однопластинчатые везикулы или "БОВ"), при условии, что может быть установлена поддерживаемая скорость высвобождения инкапсулированного терапевтического соединения. Однако в предпочтительном варианте осуществления изобретения система доставки лекарственного средства на липидной основе является системой мультивезикулярных липосом. Способ изготовления систем доставки лекарственных средств с контролируемым высвобождением на основе мультивезикулярных липосом описан полностью в патентных заявках США 08/352,352, поданной 7 декабря 1994 г., и 08/393,724, поданной 23 февраля 1995 г., а также в РСТ-эаявках US94/12957 и US94/04490, которые все включены как ссылки.
Композиция везикулы с синтетической мембраной обычно является комбинацией фосфолипидов, обычно в сочетании со стероидами, особенно с холестерином. Также можно использовать другие фосфолипиды или же другие липиды.
К примерам липидов, пригодных при получении везикул с синтетической мембраной, относятся фосфатидилглицерины, фосфатидилхолины, фосфатидилсерины, фосфатидилэтаноламины, сфинголипиды, цереброзиды и ганглиозиды. Иллюстративные фосфолипиды включают фосфатидилхолин яйца, дипальмитоилфосфатидилхолин, дистеароилфосфатидилхолин, диолеоилфосфатидилхолин, дипальмитоилфосфатидилглицерин и диолеоилфосфатидилглицерин.
При получении везикул, содержащих терапевтическое средство, следует принимать во внимание такие переменные, как эффективность инкапсулирования лекарственного средства, лабильность лекарственного средства, гомогенность и размер полученной совокупности везикул, отношение лекарственное средство/липид, проницаемость, неустойчивость препарата, а также фармацевтическая приемлемость лекарственной формы (Szoka et аl., Annual Reviews of Biophysics and Bioengineering, 9: 467, 1980; Deamer, et al., Liposomes, Marcel Dekker, New York, 1983, 27: Hope, et al., Chem. Phys. Lipids, 40: 89, 1986).
Применение лекарственных форм опиоидов на липидной основе исследовалось другими авторами с ограниченным успехом и ни один из них не изучал их через эпидуральный путь. Например, исследовали получение инкапсулированных в липосомы опиоидов и их активность in vitro (F. Reig., et al./ J. Microencapsulation 6: 277-283, 1989) без какого-либо эпидурального изучения in vivo. Кроме того, исследовали антиноцицепцию и побочные эффекты алфентанила, инкапсулированного в липосомы лекарственной формы и введенного с помощью спинальной доставки крысам (M.S. Wallace et al., Anestch. Analg. 79: 778-786, 1994; C. M. Bernards et al., Anesthesiology 77: 529-535, 1992). Однако, ни фармакокинетика, ни фармакодинамика данных соединений не отличались существенно от таковых для стандартных опиоидов так, чтобы гарантировать их применение в клинической практике. В данных исследованиях не изучались лекарственные формы с длительным высвобождением опиоидов, вводимых эпидурально.
Систему доставки лекарственного средства на липидной основе, включающую терапевтическое соединение, можно вводить в виде разовой дозы, например, через эпидуральный катетер. Однако в предпочтительном варианте систему доставки лекарственного средства на липидной основе инъецируют в виде разовой дозы в эпидуральное пространство вокруг спинного мозга с использованием небольшой калиброванной иглы, так что избегается помещение катетера. Предпочтительно используют иглу 18-25 калибра.
Представительный список терапевтических соединений, пригодных для эпидуральной доставки, включает опиаты: морфин, гидроморфин, кодеин, гидрокодон, леворфанол, оксикодон, оксиморфон, диацетилморфин, бупренорфин, налбупин, буторфанол, пентазоцин, метадон, фентанил, суфентанил и алфентанил. Кроме того, эпидурально можно вводить опиатные антагонисты, такие как налоксон и налтрексон, с использованием способа по изобретению, чтобы обратить или антагонизировать опиатный эффект.
Полагают, что опиоидами являются пептиды и пептидомиметики, которые связываются с одним или несколькими нейрорецепторами, такими как дельта-опиоидные, мю-опиоидные, каппа-опиоидные и эпсилон-опиоидные рецепторы, и их можно вводить для терапевтического действия в соответствии со способом по изобретению. К таким соединениям относятся энкефалины, эндорфины, казоморфин, киоторфин и их биологически активные фрагменты. Термин "биологически активный фрагмент" означает любую часть терапевтического соединения, которая в основном сохраняет биологическую активность всей терапевтической молекулы. Специалист в данной области будет знать или сможет легко определить, сохраняет ли в основном фрагмент биологическую активность всей молекулы.
Кроме опиоидов, в практике способа по изобретению также можно использовать ряд соединений, полезных терапевтически при эпидуральном введении с поддерживаемой скоростью. К этим соединениям относятся нейротропные факторы, такие как инсулиноподобный фактор роста, реснитчатый нейротропный фактор и факторы роста нервов; нейротрансмиттеры и их антагонисты, такие как допамин, эпинефрин, норепинефрин и гамма-аминомасляная кислота; локальные анестетики, такие как тетракаин, лидокаин, бупивакаин и мепивакаин; субстанция Р и родственные пептиды; а также антагонисты альфа-2-рецепторов, такие как клонидин и дексмедетомидин. Кроме того, эффективность эпидуральных опиоидов может быть повышена совместным введением локальных анестетиков, таких как лидокаин, бупиракаин и тетракаин.
В настоящем изобретении показано, что система доставки лекарственного средства на липидной основе, содержащая опиоид, такой как морфинсульфат, обладает минимальной возможностью угнетения дыхания, измеряемого как уменьшение в процентах насыщения гемоглобина кислородом (SpO2) от максимального насыщения крови кислородом или базовой величины перед введением лекарственного средства по сравнению с эпидуральным введением свободного лекарственного средства. Специалист в данной области поймет, что содержание кислорода в крови можно легко измерить с помощью таких доступных в продаже устройств, как импульсный оксигемометр.
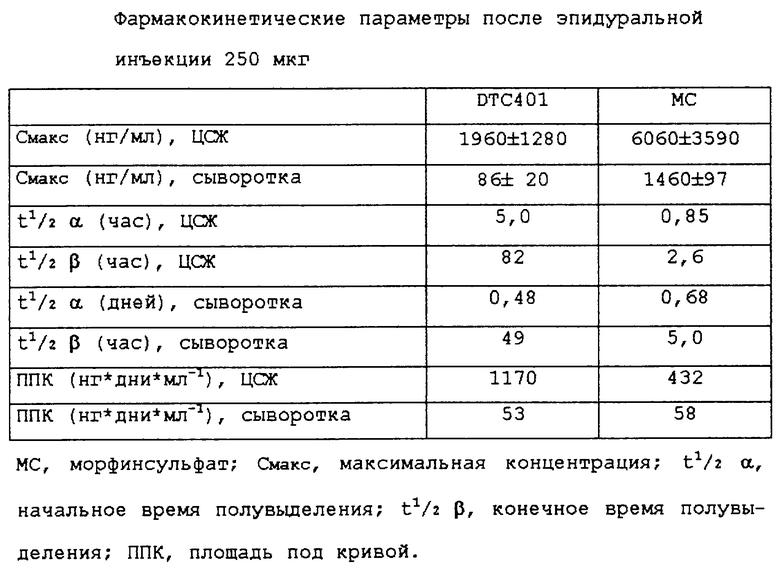
Также показано, что разовая доза длительно выделяемого опиоида, заключенного в композицию мультивезикулярных липосом и введенного эпидурально, приводит к увеличению продолжительности действия анальгезии, причем максимальная концентрация терапевтического лекарственного средства в цистернальной ЦСЖ (цереброспинальной жидкости) наблюдается в течение 60 минут после [введения] разовой эпидуральной дозы, а затем постепенно снижается в течение нескольких следующих дней, например до восьми дней. Хотя максимальная концентрация ЦСЖ (цереброспинальной жидкости) была снижена по сравнению с концентрацией после эпидурального введения свободного морфинсульфата, общая анальгезия (как показано, например, площадью под кривой (ППК) на фиг.1, 3 и в таблице 1), была во много раз увеличена по сравнению со свободным морфинсульфатом, доставленным эпидурально. Например, для крыс наблюдалось 17- и 3,1-кратное снижение максимальных концентраций морфина в сыворотке и ЦСЖ соответственно, однако ППК для ЦСЖ была в 2,8 раза увеличена после эпидурального введения 250 мкг морфинсульфата, инкапсулированного в мультивезикулярных липосомах (DTC401), по сравнению с идентичной дозой некапсулированного морфинсульфата.
Вследствие снижения максимальных концентраций морфина в сыворотке и ЦСЖ при регулируемом высвобождении эпидурально введенного морфина не было угнетения дыхания; в то время как эпидурально введенный свободный морфин при высоких дозах вызывал угнетение дыхания.
Есть три главных преимущества настоящего изобретения. Во-первых, способ эпидуральной доставки разовой дозы длительно высвобождаемого соединения дает то преимущество, что для пациента снижается риск связанных с дозой отрицательных эффектов, таких как угнетение дыхания, которое обычно связано с эпидуральными инъекциями ударного действия или с вливаниями терапевтического соединения. Во-вторых, путем введения терапевтического соединения эпидурально, а не непосредственно в цереброспинальную жидкость терапевтическое соединение не мигрирует по всему мозгу и спинному мозгу и терапевтически эффективная дозировка терапевтического соединения высвобождается локально в эпидуральное пространство в течение увеличенного периода времени, например до восьми дней. И наконец, достигается пролонгированная анальгезия без многократных инъекций или непрерывных вливаний.
Специалист в данной области поймет, что период времени, в течение которого в практике изобретения поддерживается терапевтическая скорость высвобождения, будет изменяться в зависимости от болезненного состояния, которое нужно лечить, характеристик терапевтического соединения и системы доставки лекарственного средства с длительным высвобождением, а также от общего количества соединения, инкапсулированного и введенного пациенту.
Термин "терапевтически эффективный" в том виде, в котором он относится к композициям по изобретению, означает, что терапевтическое соединение высвобождается из системы доставки лекарственного средства при концентрации, достаточной для достижения конкретного медицинского действия, для которого предназначено терапевтическое средство. Например, если терапевтическое соединение является опиоидом, то требуемым медицинским действием является анальгезия без угнетения дыхания. Точные дозировки будут изменяться в зависимости от таких факторов, как конкретное терапевтическое соединение и требуемое медицинское действие, а также таких факторов, как возраст, пол, общее состояние пациента и т. д. Специалисты в данной области могут легко принять во внимание эти факторы и использовать их для установления эффективных терапевтических концентраций, не прибегая к излишним экспериментам.
Например, интервал дозировок, подходящих для эпидурального введения морфинсульфата людям, составляет 1 - 60 мг. Для более действенных соединений могут потребоваться даже такие низкие дозировки, как 0,01 мг, а для менее действенных может потребоваться 5000 мг. Хотя можно давать дозы вне вышеуказанного интервала доз, этот интервал охватывает ширину применения практически для всех терапевтических соединений, предполагаемых для введения эпидуральным путем.
Ранее опубликованные способы эпидурального помещения крысам включают просверливание отверстия через кость поясничного позвонка и введение катетера на 1 см вверх в эпидуральное пространство. Настоящее изобретение позволяет помещать катетер сверху (то есть из области шеи) без травмы из-за хирургической процедуры. Кроме того, кончик катетера может быть помещен в любом месте вдоль позвоночного столба, а не ограничен областью поясницы, как описано в предшествующем уровне техники. Данный способ помещения катетера сверху также применим к животным, отличающимся от крыс, таким как кролики, собаки и люди.
Следующие примеры иллюстрируют, как изобретение может быть воплощено в практику. Однако понятно, что примеры приведены с целью иллюстрации, и не следует полагать, что изобретение ограничено какими-либо конкретными материалами или условиями, приведенными здесь.
ПРИМЕР 1
А. Получение мультивезикулярных липосом, инкапсулирующих морфинсульфат (DTC401) в присутствии гидрохлорида.
Стадия 1) В чистый однодрахмовый стеклянный пузырек (внутренний диаметр 1,3 см • высота 4,5 см) помещали 1 мл раствора в хлороформе (Spectrum Corp., Gardena, CA), содержащего 9,3 мкмоль диолеоиллецитина (Avanti Polar Lipids, Alabaster, AL), 2,1 мкмоль дипальмитоилфосфатидилглицерина (Avanti Polar Lipids), 15 мкмоль холестерина (Avanti Polar Lipids) и 1,8 мкмоль триолеина (Sigma). Этот раствор назван липидным компонентом.
Стадия 2) Один мл водного раствора, содержащего 20 мг/мл морфинсульфата (Sigma Chemical Co. , St. Louis, МО) и 0,1 н. соляную кислоту, добавляли в вышеуказанный однодрахмовый пузырек, содержащий липидный компонент.
Стадия 3) Для приготовления эмульсии типа "вода-в-масле" стеклянный пузырек, содержащий смесь со "стадии 2", герметизировали, прикрепляли горизонтально к головке прибора для встряхивания и перемешивания (# по каталогу S8223-1, American Scientific Products, McGaw Park, IL) и встряхивали при максимальной скорости в течение 6 минут.
Стадия 4) Для получения сферул хлороформа, суспендированных в воде, полученную на "стадии 3" эмульсию типа "вода-в-масле" разделяли на равные объемы и быстро вытесняли через пипетку Пастера с узким кончиком в каждый из двух однодрахмовых стеклянных пузырьков (внутренний диаметр 1,3 см • высота 4,5 см), каждый из которых содержал 2,5 мл воды, глюкозу (32 мг/мл) и лизин в виде свободного основания (40 мМ) (Sigma). Каждый пузырек затем герметизировали, прикрепляли к головке того же самого устройства для встряхивания и перемешивания, которое использовали на "стадии 3", и встряхивали в течение 3 секунд при максимальной скорости для образования сферул хлороформа.
Стадия 5) Для того чтобы получить мультивезикулярные липосомы, суспензии сферул хлороформа, полученные в двух пузырьках на "стадии 4", выливали на дно колбы Эрленмейера объемом 250 мл, содержащей 5 мл воды, глюкозу (32 мг/мл) и свободное основание лизина (40 мМ). Колбу держали при 37oС на встряхиваемой водяной бане, через колбу пропускали поток газообразного азота с расходом 7 л/мин, чтобы медленно испарить хлороформ в течение 10-15 минут. После этого липосомы выделяли путем центрифугирования при 600 • g в течение 5 минут; затем промывали три раза 0,9% раствором NaCl.
Б. Получение готовых лекарственных форм
Перед эпидуральной инъекцией препараты DTC401 и контрольного некапсулированного ("свободного") морфинсульфата доводили до такого состояния, чтобы в 50 мкл содержалась доза в 10, 50, 175, 250 или 1000 мкг. Кроме того, в 75 мкл объема для инъекции составляли лекарственную форму МВЛ, содержащую дозу морфинсульфата 2000 мкг для использования в исследовании угнетения дыхания. Концентрацию морфина в различных липосомных лекарственных формах определяли путем растворения 50 мкл каждого препарата в 1 мл иэопропилового спирта с последующим разбавлением в воде. Концентрацию морфина определяли с помощью ВЭЖХ с использованием опубликованной методики (S.P. Joel et al., Journal of Chromatography, 430: 394-399, 1988). Для контроля в виде плацебо была приготовлена холостая композиция мультивезикулярных липосом путем замены морфинсульфата глюкозой.
ПРИМЕР 2
А. Подготовка животных
Шести-8-месячных самцов крыс Спраг-Доли весом 205-254 г (Harlan Sprague-Dawley, San Diego, CA) содержали по 1-2 особи в клетке в термостатируемой среде при цикле чередования света и темноты через 12 часов и давали им неограниченный доступ к пище и воде. Перед каждым исследованием животных приучали к среде. Каждое животное исследовали только один раз. Всех животных содержали в соответствии с руководствами Комитета по использованию лабораторных животных и уходу за ними Института ресурсов лабораторных животных. Национальный совет по исследованиям.
Б. Эпидуральная катетеризация
Каудальную эпидуральную катетеризацию крыс осуществляли следующим образом: проводили наркоз галотаном и животных помещали в стереотаксическое устройство для горизонтального положения высотой 7 см. Голову отгибали, обращая внимание на то, чтобы животные сохраняли нормальное дыхание. Иглу 19 калибра с коротким срезом вводили под углом примерно 170o в позвоночник каудально к затылочному выступу по центральной линии, причем срез иглы был обращен вниз. Иглу продвигали каудально по направлению к C1-позвонку до тех пор, пока конец иглы не касался позвоночного отростка или posterior lamina C1. Кончик иглы осторожно продвигали в вентральный край posterior lamina. В данной точке чувствовалось небольшое ослабление и иглу продвигали еще на 1-2 мм. Старались не позволить игле проникнуть в твердую мозговую оболочку. Случайное нарушение твердой мозговой оболочки можно определить по выплеску цереброспинальной жидкости (ЦСЖ) через раструб иглы или через помещенный последовательно катетер. В заднее эпидуральное пространство через иглу вводили полиэтиленовый катетер (РЕ-10; длина 12 см; внутр. диаметр: 0,28 мм; объем: 7,4 мкл (Becton Dickinson, Sparks, MD). Катетер медленно продвигали через иглу и останавливали на уровне L примерно 1,8 см от С1. Наружную часть катетера подкожно туннелировали под скальп и закрепляли кисетным шелковым швом 3-0. Наконец, катетер промывали 10 мкл нормального солевого раствора и закупоривали проволокой из нержавеющей стали. Процедура от начала анестезии до наложения швов длилась примерно 10-15 минут. Животных оставляли до возвращения к норме и наблюдали за ними в течение 60 минув. В последующих исследованиях использовали только тех животных, которые полностью оправились от процедуры.
В. Антиноцицепция
Базовые значения ноцицепции после помещения эпидурального катетера определяли, подвергая животных стандартному тесту горячей пластинки (52,5±0,5oС), как описано в M.S. Wallace et al. (Anesth. Analg. 79: 778-786, 1994). Задержку ответа на ноцицепцию (в секундах) измеряли от момента времени, когда животные были помещены на горячую пластину, до момента времени, когда они либо лизали заднюю лапу, либо прыгали. Базовую (после предварительной обработки) величину задержки ответа определяли как 0% максимально возможной анальгезии (МВА) для каждого экспериментального животного. После этого каждому животному делали эпидуральную инъекцию 50 мкл либо DTC401, содержащего дозы эпидурального морфина в интервале от 10 до 250 мкг, раствора некапсулированного морфинсульфата либо контрольных холостых МВЛ. Кроме того, определяли антиноцицепторный эффект введенного подкожно морфинсульфата в интервале доз от 250 мкг до 1 мг. После эпидурального введения исследуемых растворов через катетер, вставленный так, как описано выше, эпидуральный катетер промывали 10 мкл 0,9% хлорида натрия.
Затем животных снова подвергали тесту горячей пластинки для измерения антиноцицепторного эффекта в заданные моменты времени: 0,5, 1, 2, 3, 4, 6, 12 и 24 часа после введения некапсулированного морфинсульфата и 0,5, 1,6 часов и 1, 2, 3, 4, 5, 6, 7 и 8 дней после введения как DTC401, так и холостых МВЛ. Антиноцицепцию определяли для 5 или 6 животных для каждой дозы и каждого лекарственного средства. Чтобы предотвратить тканевое повреждение подушечек на лапах, использовали предельное время 60 секунд. В соответствии с этим 100% МВА определялась как антиноцицепция, длящаяся ≥60 секунд. Интервал задержки от 10±2 до 60 секунд, отвечающий МВА от 0 до 100% соответственно, был чувствителен для демонстрации [зависимости] доза-ответ в исследуемом интервале доз.
Строили кривые для эффективности и угнетения дыхания как функций времени для каждой введенной дозы. Ответ на горячую пластинку рассчитывали как процент максимально возможной анальгезии (% МВА) согласно описанию Wallace и др. (см. выше):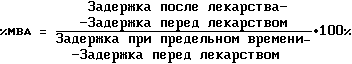
Все площади под кривыми рассчитывали по правилу трапеций до последней точки данных с использованием компьютерной программы RSTRIP [Micromath, Salt Lake City, UT].
Использовали однофакторный дисперсионный анализ (ДА) для того, чтобы раздельно определить дозовую зависимость для различных лекарственных форм и путей введения; в то время как двухфакторный ДА использовали для проведения сравнения лекарственных форм при равной дозе. При всех ДА осуществляли контроль по Ньюмену-Койлу (Newman-Keuls) для того, чтобы определить статистическую значимость; во всех испытаниях р<0,05 полагали статистически значимым. Все данные приводили как среднее±стандартная ошибка (СО).
Как показывают данные из фиг.1, эпидуральное введение DTC401 приводило к эквивалентному началу анальгезии, но продолжительность анальгезии была существенно увеличена по сравнению с введенным эпидурально свободным морфинсульфатом. При эпидуральной инъекции контрольных холостых МВЛ антиноцицепторный эффект не проявлялся (данные не показаны). Максимальные анальгезирующие эффекты эпидурального DTC401 и эпидурального и подкожного морфинсульфата зависели от дозы, как показано на фиг.2, причем максимальная возможная анальгезия эпидурального свободного морфинсульфата была больше, чем таковая для эпидурального DTC401, которая была значительно больше, чем в случае подкожно введенного свободного морфинсульфата (р<0,05 для каждого сравнения).
Существенное пролонгирование анальгезирующих эффектов у животных, которым ввели эпидуральный DTC401, легко видеть на фиг.1, а также из больших значений площадей под кривыми (ППК) для DTC401 на фиг.3. При дозе 250 мкг, приводящей к максимальным эффектам, близким к 100% МВА как для DTC401, так и для свободного морфинсульфата, время снижения до 50% МВА составляло 3, 4 дня для DTC401 по сравнению с 0,17 дня для морфинсульфата.
Г. Угнетение дыхания
Угнетение дыхания количественно оценивали с помощью импульсной оксигемометрии. Животных извлекали из их клеток, помещали в полистирольные средства механического удерживания для крыс (Plas Labs, Lansing, MI) и оставляли адаптироваться на 5 минут. Насыщение кислородом определяли по базовой линии и после разовой эпидуральной ударной дозы морфинсульфата или DTC401, в заданные моменты времени, помещая зонд импульсного оксигемометра на правую заднюю лапу (Ohmeta Medical Systems, модель 3740, Madison, WI). Дозы DTC401 и свободного морфинсульфата изменялись в интервале от 10 до 2000 мкг. Импульсную оксигемометрию осуществляли для 5 или 6 животных для каждой точки данных за исключением дозы 50 мкг, где использовали 3-х животных. Значения насыщения гемоглобина кислородом в процентах (SpO2) согласно импульсной оксигемометрии непрерывно отслеживали в реальном времени, максимальную величину, достигаемую в течение 3-минутного периода записи, определяли как насыщение кислородом.
На фиг. 4 изображено изменение во времени насыщения гемоглобина кислородом в процентах (SpO2), измеренное импульсным оксигемометром при различных дозах DTC401 и морфинсульфата. Имело место зависящее от дозы увеличение угнетения дыхания при повышении доз морфинсульфата, как показано на фиг.5; в то время как угнетение дыхания при тех же дозах DTC401 было минимальным. С другой стороны, максимальные снижения SpO2 наблюдались в течение 1 часа после эпидурального введения свободного морфинсульфата или DTC401 и не было видно никакого задержанного угнетения дыхания при любой лекарственной форме. Различие между морфинсульфатом и DTC401 по максимальному угнетению дыхания было статистически значимым (р<0,01).
Д. Фармакокинетика
Фармакокинетические исследования проводили путем измерения концентраций морфина в периферийной крови и в ЦСЖ в надлежащие моменты времени после разовой эпидуральной дозы 250 мкг DTC401 или свободного морфинсульфата. Пробы отбирали через 0,5, 1 час и 1, 3, 5, 8 дней после эпидурального введения DTC401, как описано выше, и через 0,5, 1, 3, 6, 12, 24 часа после эпидурального введения свободного морфинсульфата. Совокупность из 3 или 4 животных анестезировали с использованием галотана, и пробы ЦСЖ и крови отбирали путем цистернальной пункции и кардиальной пункции соответственно. Затем животных умерщвляли посредством передозировки галотана. Сыворотку отделяли от крови путем центрифугирования и хранили вместе с пробами ЦСЖ при -80oС до дальнейшего исследования методом радиоиммуноанализа (РИА).
Концентрации морфина в сыворотке и ЦСЖ определяли с использованием доступного в продаже набора для РИА, высокоспецифичного на морфин [Coat-A-CountTM Serum Morphine, Diagnostic Products Corp., Los Angeles, CA], как предложено изготовителем. Все измерения были проведены дважды.
На фиг. 6 изображены концентрации морфина в цистернальной ЦСЖ и в сыворотке животных, инъецированных 250 мкг свободного морфинсульфата или DTC401. В таблице 1 обобщены фармакокинетические параметры. Максимальные концентрации морфина в ЦСЖ и сыворотке после эпидурального введения DTC401 составляли соответственно 32 и 5,9% от таковых после введения морфинсульфата. Конечный период полувыведения из ЦСЖ (β) для DTC401 составлял 82 часа по сравнению с 2,6 часами для морфинсульфата. В случае DTC401 площадь под кривой (ППК) для ЦСЖ была увеличена в 2,7 раза по сравнению с морфинсульфатом, однако ППК для плазмы была очень похожей. Времена полувыведения рассчитывали путем подгонки фармакокинетических кривых к биэкспоненциальной функции. Для подгонки кривой путем итерационной нелинейной регрессии использовали программу RSTRIP.
ПРИМЕР 3
Получение DTC401 в большем масштабе
Стадия 1) В чистую пробирку для центрифуги из нержавеющей стали объемом 50 мл помещали 5 мл раствора в хлороформе, содержащего 46,5 мкмоль диолеоилфосфатидилхолина (Avanti Polar Lipids), 10,5 мкмоль дипальмитоилфосфатидилглицерина (Avanti Polar Lipids), 75 мкмоль холестерина (Sigma Chemical Co. ), 9,0 мкмоль триолеина (Avanti Polar Lipids). Этот раствор назван липидным компонентом.
Стадия 2) Пять мл водного раствора, содержащего 20 мг/мл пентагидрата морфинсульфата (Mallinckrodt Chemical Inc.) и 0,1 н. соляную кислоту, добавляли в вышеуказанную пробирку для центрифуги из нержавеющей стали, содержащую липидный компонент.
Стадия 3) Для приготовления эмульсии типа "вода-в-масле" смесь со стадии 2 перемешивали с помощью мешалки ТК (AutoHomoMixer, модель М, Tokushu Kika, Osaka, Japan) со скоростью 9000 оборотов в минуту (об/мин) в течение 9 минут.
Стадия 4) Для получения сферул хлороформа, суспендированных в воде, к полученной на стадии 3 эмульсии типа "вода-в-масле" добавляли 25 мл раствора, содержащего 4 процента глюкозы и 40 мМ лизина в воде, а затем перемешивали со скоростью 3500 об/мин в течение 120 секунд.
Стадия 5) Чтобы получить мультивезикулярные липосомы, суспензию сферул хлороформа в пробирке для центрифуги выливали на дно колбы Эрленмейера объемом 1000 мл, содержащей 25 мл 4% глюкозы и 40 мМ лизина в воде. Колбу держали при 37oС на встряхиваемой водяной бане, через колбу пропускали поток газообразного азота с расходом 7 л/мин, чтобы медленно испарить хлороформ в течение 20 минут. После этого липосомы выделяли путем 4-кратного разбавления суспензии нормальным солевым раствором и центрифугирования суспензии при 600 • g в течение 5 минут; супернатант декантировали и липосомную гранулу повторно суспендировали в 50 мл нормального солевого раствора. Липосомы снова выделяли путем центрифугирования при 600 • g в течение 5 минут. Супернатант вновь декантировали и гранулу повторно суспендировали в нормальном солевом растворе.
Предшествующее описание изобретения является примерным с целью иллюстрации и пояснения. Следует понять, что могут быть осуществлены различные модификации без отклонения от объема и сущности изобретения. Соответственно, предполагается, что последующую формулу изобретения следует интерпретировать так, что она охватывает все такие модификации.

Изобретение относится к области медицины, а именно к регулируемому высвобождению терапевтических соединений из систем доставки лекарственных средств. Соединение инкапсулируют в систему доставки лекарственного средства, имеющую замедленную скорость высвобождения. Вводят указанную систему эпидурально в разовой дозе. Система доставки лекарственного средства включает мультивезикулярные липосомы, полученные из группы, состоящей из фосфатидилхолинов яйца, дипальмитоилфосфатидилхолинов, диолеоилфосфатидилхолинов, дистеароилфосфатидилхолинов, дипальмитоилфосфатидилглицеринов, диолеоилфосфатидилглицеринов и их приемлемых комбинаций. Для уменьшения угнетения дыхания эпидурально вводят инкапсулированное анальгезирующее средство в мультивезикулярной липосомной системе доставки. Способ позволяет повысить эффективность лечения. 2 с. и 22 з.п.ф-лы, 1 табл., 6 ил.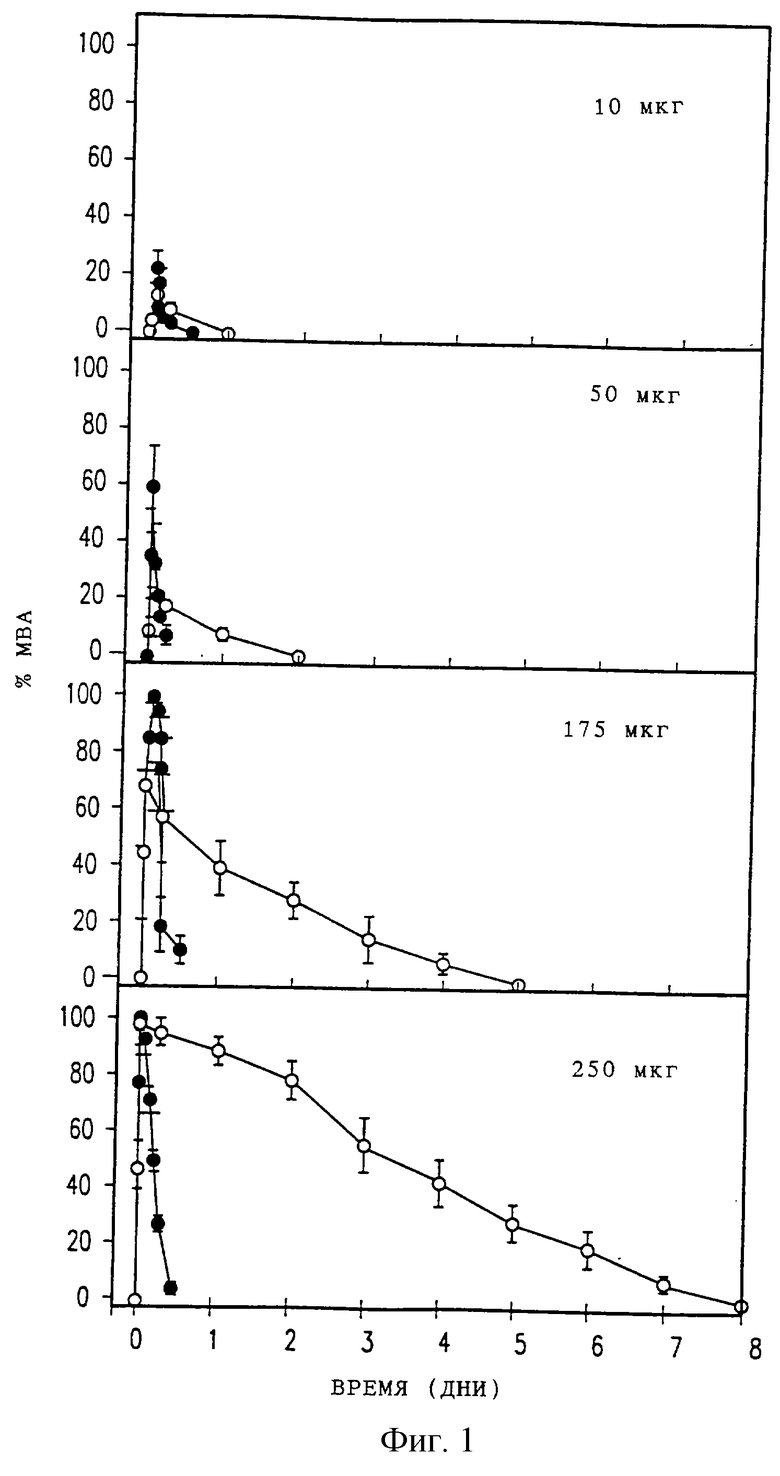
| ПРОИЗВОДНОЕ N-ЗАМЕЩЕННОГО 1-ГЕКСИЛ-4-ФЕНИЛ-4-ПИПЕРИДИНКАРБОКСАМИДА ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМАЯ СОЛЬ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1992 |
|
RU2039043C1 |
| RU 93004902 А, 10.05.1995 | |||
| RU 92016352 А, 10.02.1995 | |||
| KIM S., SCHEERER S., GEYER M.A., Howell SB | |||
| Direct cerebrospinal fluid delivery of an antiretroviral agent using multivesicular liposomes | |||
| Способ приготовления консистентных мазей | 1919 |
|
SU1990A1 |

