Область изобретения
Настоящее изобретение относится к способу получения кларитромицина с использованием N-оксида эритромицина А и к новому промежуточному соединению, полученному этим способом.
Предпосылки создания изобретения
Кларитромицин, 6-O-метилэритромицин А, представляет собой полусинтетический макролидный антибиотик, который обладает широким диапазоном антибактериальной активности против грамположительных бактерий, некоторых грамотрицательных бактерий, анаэробных бактерий, Mycoplasma, Chlamidia и Helicobacter pylori.
Кларитромицин может быть получен посредством метилирования 6-гидроксигруппы эритромицина А. Однако осуществление селективного метилирования 6-гидроксигруппы является трудным, поскольку эритромицин А, помимо 6-гидроксигруппы, имеет четыре реакционноспособные гидроксигруппы, а также диметиламиногруппу, которая может подвергаться кватернизации в процессе реакции метилирования.
Для решения этой проблемы были разработаны различные способы получения кларитромицина.
Так, например, был описан общий способ получения кларитромицина с использованием 9-оксим производного эритромицина А в качестве промежуточного соединения (см. ЕР патенты №№0158467, 0195960, 0269938 и 0272110 и публикации международных заявок №№ WO 97/36912 и WO 97/36913). Хотя этот способ дает относительно высокий выход, однако он является неэффективным из-за низкой производительности, обусловленной тем фактом, что данный способ требует осуществления ряда реакционных стадий, включая стадии оксимирования, защиты оксимной группы, удаления оксимзащитной группы и деоксимирования.
Другой общий способ получения кларитромицина описан в ЕР патентах №№0147062 и 0177696. Способ, описанный в ЕР патенте №0147062, включает стадии: защиты 2-гидрокси- и аминогрупп эритромицина А бензилоксикарбонильными группами; метилирования 6-гидроксигруппы; удаления двух бензилоксикарбонильных защитных групп; и метилирования аминогруппы с получением кларитромицина. Однако при массовом производстве этот способ, помимо трудности использования разделения продукта с помощью хроматографии на колонках, имеет еще тот недостаток, что он дает низкий выход от 13,1 до 18,5% и требует использования избыточного количества бензилоксикарбонилхлорида.
В соответствии с этим существует необходимость в разработке усовершенствованного способа получения кларитромицина.
Краткое описание изобретения
Следовательно, целью настоящего изобретения является обеспечение способа получения кларитромицина.
Другой целью настоящего изобретения является получение нового промежуточного соединения, полученного указанным способом.
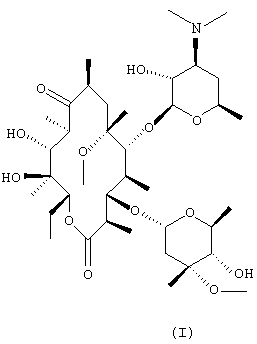
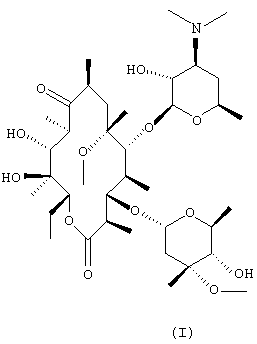
В соответствии с одним из аспектов настоящее изобретение предлагает способ получения кларитромицина формулы (I), включающий стадии:
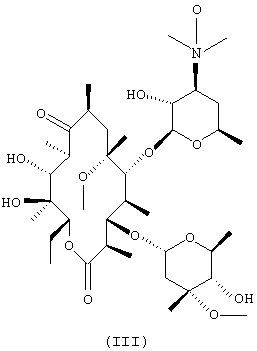
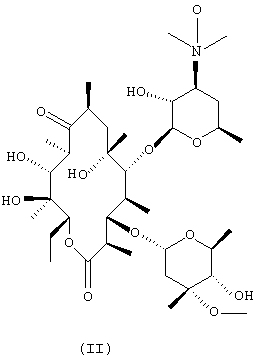
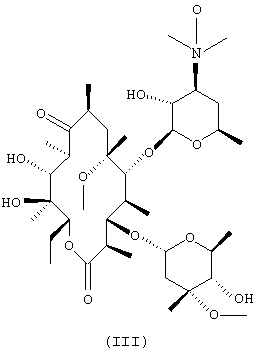
(a) взаимодействия N-оксида эритромицина А формулы (II) с метилирующим агентом с получением N-оксида 6-O-метилэритромицина А формулы (III);
(b) обработки N-оксида 6-O-метилэритромицина А, полученного на стадии (а), восстановителем с получением кларитромицина:

В соответствии с другим аспектом настоящее изобретение предлагает N-оксид 6-O-метилэритромицина А формулы (III).
Подробное описание изобретения
Соединение формулы (I) может быть получено из N-оксида эритромицина А, как описано ниже.
Стадия (а)
N-оксид 6-O-метилэритромицина А формулы (III) получают посредством реакции N-оксида эритромицина А формулы (II), полученного из эритромицина А с выходом более чем 98%, согласно хорошо известному методу [Е.Н. Flynn et al., J. Am. Chem. Soc., 76, 3121 (1954) & P.H. Jones & Е.К. Rowley., J.Org.Chem., 33, 665(1968)] с метилирующим агентом в органическом растворителе в присутствии основания.
Примерами метилирующих агентов, которые могут быть подходящим образом использованы в настоящем изобретении, являются метилбромид, метилиодид, диметилсульфат, метил-п-толуолсульфонат, метилметансульфонат и их смеси. Метилирующий агент может быть использован в количестве от 1 до 3 эквивалентов относительно количества N-оксида эритромицина А формулы (II).
Примеры растворителей, которые могут быть использованы в вышеуказанной реакции метилирования, включают N,N-диметилформамид, N,N-диметилацетамид, N-метил-2-пирролидон, диметилсульфоксид, сульфолан, N,N,N’,N’,N”,N”-гексаметилфосфорамид, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, 1,2-диэтоксиэтан, 2-метоксиэтиловый эфир, 2-этоксиэтиловый эфир, 1,2-бис(2-метоксиэтокси)этан, диметиловый эфир тетраэтиленгликоля, ацетон, ацетонитрил и их смеси.
Кроме того, в рассматриваемом способе может быть использовано основание, выбранное из группы, состоящей из гидридов, гидроксидов и алкоксидов щелочных металлов, например гидрида натрия, гидрида калия, гидроксида натрия, гидроксида калия и трет-бутоксида калия. Основание может быть использовано в количестве от 0,9 до 2 эквивалентов относительно количества N-оксида эритромицина А формулы (II). Реакция метилирования может быть проведена при температуре от -15 до 40°С, предпочтительно от 0°С до комнатной температуры.
После завершения реакции метилирования к полученной смеси добавляют воду и смесь экстрагируют хлороформом. Экстракт концентрируют, и к остатку добавляют ацетон, и затем перемешивают для осаждения побочных продуктов. Полученную смесь фильтруют для удаления побочных продуктов и фильтрат концентрируют с получением неочищенного N-оксида 6-O-метилэритромицина А (выход: 67-73% и чистота: 50-57%).
Неочищенный N-оксид 6-O-метилэритромицина А, полученный, как описано выше, может быть использован как в стадии (b), или он может быть очищен перекристаллизацией из этилацетата с получением очищенного N-оксида 6-O-метилэритромицина А (чистота: 83-88% и выход: 40-44%), который может быть дополнительно очищен перекристаллизацией из хлороформа с получением кристаллов N-оксида 6-0-метилэритромицина А, имеющих чистоту более 95%.
Стадия (b)
Кларитромицин формулы (I) получают посредством реакции N-оксида 6-O-метилэритромицина А, полученного на стадии (а), с восстановителем в органическом растворителе для удаления N-оксида.
Примерами подходящих восстановителей, которые могут быть использованы в настоящем изобретении, являются водород в присутствии катализатора гидрирования, такого как палладий, никелевый катализатор Ренея или окись платины (PtO2); сплав никеля и алюминия (сплав Ni-Al), объединенный с гидроксидом калия; металлический цинк в присутствии муравьиной кислоты или уксусной кислоты; гидротеллурид натрия (NaTeH); иодид самария (SmI2); дихлорид олова (SnCl2); гексабутилдиолово (Bu3SnSnBu3); циклогексен и тетраксид осмия (OsO4); и сульфат железа (2), и предпочтительными восстановителями для осуществления настоящего изобретения являются дихлорид олова (SnCl2); гексабутилдиолово (Вu3SnSnВu3); сплав никеля и алюминия, объединенный с гидроксидом калия; и водород в присутствии никелевого катализатора Ренея или катализатора окиси платины (PtO2).
В случае использования дихлорида олова в качестве восстановителя могут быть использованы растворители, такие как метанол, этанол, изопропанол, этилацетат, ацетонитрил, ацетон, тетрагидрофуран, 1,2-диметоксиэтан, дихлорметан, хлороформ и их смеси. Количество используемого дихлорида олова составляет от 1 до 3 молярных эквивалентов относительно количества N-оксида 6-O-метилэритромицина А формулы (III), и реакция может быть осуществлена при температуре от 0°С до температуры кипения используемого растворителя, предпочтительно от 10 до 45°С. После завершения реакции кларитромицин может быть выделен (1) нейтрализацией реакционной смеси основанием, таким как триэтиламин; добавлением к этой смеси воды; и экстракцией этой смеси органическим растворителем; или (2) добавлением к реакционной смеси воды; нейтрализацией полученной смеси основанием, таким как бикарбонат натрия, карбонат натрия, раствор гидроксида натрия и водный раствор аммиака; и экстракцией смеси органическим растворителем.
В случае использования гексабутилдиолова в качестве восстановителя могут быть использованы растворители, такие как этилацетат, ацетонитрил, ацетон, тетрагидрофуран, 1,2-диметоксиэтан и их смеси. Гексабутилдиолово используется в количестве от 1 до 3 молярных эквивалентов относительно количества N-оксида 6-O-метилэритромицина А формулы (III), и реакция может быть осуществлена при температуре в пределах от комнатной температуры до температуры кипения используемого растворителя.
Если в качестве восстановителя используется сплав Ni-Al вместе с гидроксидом калия, то в качестве растворителя используют смесь воды и низшего спирта, например метанола и этанола. Количества используемого сплава Ni-Al и гидроксида калия составляют от 0,5 до 3 г и от 2 до 20 моль соответственно в расчете на один моль N-оксида 6-O-метилэритромицина А формулы (III). Реакция может быть осуществлена при температуре в пределах от комнатной температуры до температуры кипения используемого растворителя.
Если способом проведения реакции стадии (b) является каталитическое гидрирование, то в качестве растворителя может быть использован низший спирт или смесь воды и низшего спирта, а в качестве катализатора может быть использован катализатор гидрирования, такой как никелевый катализатор Ренея, при температуре в пределах от комнатной температуры до температуры кипения используемого растворителя в атмосфере водорода.
Реакция стадии (b) может быть также осуществлена с использованием неорганического восстановителя, такого как бисульфит натрия (NаНSО3), сульфит натрия (Nа2SO3), тиосульфат натрия (Nа2S2О3), гидросульфит натрия (Na2S2O4), пиросульфат натрия (Na2S2O5), тионат натрия (Na2S2O6), бисульфит калия (КНSО3), тиосульфат калия (К2S2О3) и пиросульфат калия (К2S2O5). Эта реакция может быть осуществлена в смеси воды и низшего спирта, такого как метанол, этанол и изопропанол, при температуре от 0°С до температуры кипения используемого растворителя. Восстановитель может быть использован в количестве от 1 до 20 эквивалентов, предпочтительно от 1 до 4 эквивалентов относительно количества N-оксида 6-O-метилэритромицина А формулы (III).
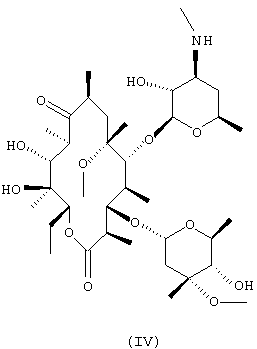
При осуществлении реакции стадии (b) с использованием оксидов серы в качестве восстановителя, помимо кларитромицина формулы (I), может образовываться 3’-N-диметил-6-О-метилэритромицин А формулы (IV) в качестве побочного продукта. В этом случае, побочные продукты могут быть превращены в кларитромицин путем метилирования вторичной аминогруппы указанного побочного продукта с использованием муравьиной кислоты и формальдегида в соответствии с известным методом [см. Eschweiler & Clarke, Org.React., 5, 290(1945)]
Нижеследующий сравнительный пример и примеры приводятся только в целях иллюстрации настоящего изобретения и никоим образом не ограничивают его объема; экспериментальные методы, используемые в настоящем изобретении, могут быть осуществлены в соответствии с указанными сравнительным примером и примерами, приведенными ниже, если не оговорено иначе.
Кроме того, проценты, указанные ниже для твердого вещества в твердой смеси, жидкого вещества в жидкости и твердого вещества в жидкости даны в мас./мас., об./об. и мас./об. соответственно, если не оговорено иначе.
Сравнительный пример
Получение N-оксида эритромицина А
220,2 г (0,3 моль) эритромицина А растворяют в смеси 1500 мл метанола и 1000 мл воды. К раствору по каплям добавляют 79 мл (0,9 моль) 35% перекиси водорода при температуре от 15 до 20°С и перемешивают при комнатной температуре в течение 20 часов методом, аналогичным описанному в Е.Н. Flynn et al., J. Am. Chem. Soc., 76, 3121 (1954) & P.H. Jones et al., J. Org.Chem., 33, 665 (1968). Полученную смесь концентрируют до половины объема при пониженном давлении и последовательно экстрагируют 1000-мл и 500-мл порциями хлороформа. Объединенный хлороформный экстракт промывают рассолом, сушат над безводным сульфатом магния, а затем концентрируют при пониженном давлении с получением пенистого остатка. К остатку добавляют 1000 мл ацетона и перемешивают при комнатной температуре в течение 3 часов. Образованный кристаллический продукт фильтруют и сушат в течение ночи при 45°С с получением 220 г N-оксида эритромицина А с выходом 98%.
Т.пл. 218~221°С (т.пл. в литературе: 222~224°С).
1H-ЯМР (CDCl3, м.д.): δ 5,00(дд, 1Н, 13-Н), 4,86(д, 1Н, 1"-Н), 4,50(д, 1Н, 1'-Н), 3,34 (с, 3Н, кладиноза -ОСН3), 3,17 (д, 6Н, дезозамин -N(СН3)2), 1,43(с, 3Н, 18-Н), 0,82(т, 3Н, 15-Н).
МС (м/z): ESI 750[M+1]+.
Пример 1
Получение N-оксида 6-O-метилэритромицина А
75,0 г (0,1 моль) N-оксида эритромицина А, полученного в сравнительном примере, суспендируют в смеси 450 мл диметилсульфоксида и 450 мл тетрагидрофурана и охлаждают до 5°С. Затем добавляют 8,1 мл иодметана и 7,26 г порошкообразного 85% гидроксида калия и смесь перемешивают в течение 1 часа. К полученной смеси добавляют 1000 мл холодной воды и последовательно экстрагируют 1000-мл и 500-мл порциями хлороформа. Объединенный экстракт дважды промывают 500 мл воды, сушат над безводным сульфатом магния, а затем концентрируют при пониженном давлении. К пенистому остатку добавляют 500 мл ацетона и перемешивают при комнатной температуре в течение 5 часов. Полученную смесь фильтруют для удаления осажденных побочных продуктов. Фильтрат концентрируют при пониженном давлении с получением 52,7 г неочищенного N-оксида 6-O-метилэритромицина А, имеющего чистоту 57%.
К полученному неочищенному продукту добавляют 300 мл этилацетата и смесь перемешивают с получением кристаллов. Кристаллы фильтруют и сушат в течение ночи при 45°С с получением 30,3 г N-оксида 6-O-метилэритромицина А в виде кристаллического порошка, имеющего чистоту 88%.
Полученный кристаллический порошок перекристаллизовывают из хлороформа с получением 20,1 г целевого соединения, имеющего чистоту выше, чем 95%, и выход 27%.
Т.пл.: 204~207°С.
ИК(КВr, см-1): 3446, 2972, 2938, 1733, 1693, 1462, 1379, 1169, 1111, 1079. 1H-ЯМР (CDCl3, м.д.): δ 5,06(дд, 1Н, 13-H), 4,93(д, 1Н, 1”-Н), 4,55(д, 1Н, 1’-Н), 4,02(дкв, 1Н, 5''-Н), 3,76(д, 1Н, 11-Н), 3,73(дд, 1Н, 3-Н), 3,70(д, 1Н, 5-Н), 3,65 (ддкв, 1Н, 5’-Н), 3,38 (с, 3Н, 3"-ОСН3), 3,19(д, 6Н, 3’-N(CH3)2), 3,05 (с, 3Н, 6-ОСН3), 2,91(дкв, 1Н, 2-Н), 2,59(ддкв, 1Н, 8-Н), 1,41(с, 3Н, 18-Н), 1,12(с, 3Н, 6-СН3), 0,83 (т, 3Н, 14-СН3).
13С-ЯМР (СDСl3, м.д.): δ 221,4(С1=O), 176,2(С9=О), 102,9(С1’), 96,5(С1”), 81,6(С5), 79,0(С6), 78,7(С3), 78,2(С4”), 77,6(С3’), 77,0(С13), 76,7(С2’), 74,7(С12), 73,1(С3”), 69,5 (С11), 67,4(С5’), 66,3(C5”), 59,1(С8’), 52,8(С7’), 51,0(С22), 50,1(С8”), 45,7(С8), 45,4(С2), 39,7(С7), 39,6(С4), 37,7(С10), 35,3(С2”), 32,0(С4’), 22,0(С6’), 21,7(C7”), 21,4 (С14), 20,2(C18), 19,1(C6”), 18,4(С19), 16,4(С21), 16,3(С16), 12,7(С20), 11,0(С15), 9,4(С17), МС (м/z): ESI 764[М+1]+.
Пример 2
Получение N-оксида 6-O-метилэритромицина А
75,0 г (0,1 моль) N-оксида эритромицина А, полученного в сравнительном примере, суспендируют в смеси 450 мл диметилсульфоксида и 450 мл тетрагидрофурана и охлаждают до 5°С. Затем добавляют 8,1 мл иодметана и 7,26 г порошкообразного 85% гидроксида калия и смесь перемешивают в течение 1,5 часов. К полученной смеси добавляют 1000 мл холодной воды и последовательно экстрагируют 1000-мл и 500-мл порциями хлороформа. Объединенный экстракт дважды промывают 500 мл воды, сушат над безводным сульфатом магния, а затем концентрируют при пониженном давлении с получением пенистого остатка. К этому остатку добавляют 500 мл ацетона и смесь перемешивают при комнатной температуре в течение 5 часов. Полученную смесь фильтруют для удаления осажденных побочных продуктов. Фильтрат концентрируют при пониженном давлении с получением 50,4 г неочищенного N-оксида 6-O-метилэритромицина А, имеющего чистоту 55%.
Пример 3
Получение кларитромицина с использованием дигидрата дихлорида олова в качестве восстановителя
3,82 г (5 ммоль) N-оксида 6-O-метилэритромицина А, полученного в примере 1, суспендируют в 30 мл изопропанола. Затем добавляют 2,26 г (2,0 ммоль) дигидрата дихлорида олова и смесь перемешивают при температуре от 30 до 40°С в течение 2 часов. К полученной смеси добавляют насыщенный раствор бикарбоната натрия и смесь дважды экстрагируют этилацетатом. Объединенный органический слой промывают водой, сушат над безводным сульфатом магния, а затем концентрируют при пониженном давлении с получением 3,60 г кларитромицина в виде белого порошка с выходом 96%.
Т.пл. 220~223°С (т.пл. в литературе: 222~225°С).
1H-ЯМР (CDCl3, м.д.): δ 5,06(дд, 1Н, 13-Н), 4,92(д, 1Н, 1''-Н), 4,44(д, 1Н, 1'-Н), 4,02(дкв, 1Н, 5”-Н), 3,78(дд, 1Н, 3-Н), 3,77(д, 1Н, 11-Н), 3,67(д, 1Н, 5-Н), 3,57(ддкв, 1Н, 5’-Н), 3,33(с, 3Н, 3”-ОСН3), 3,20(дд, 1Н, 2’-Н), 3,07~2,95 (м, 2Н, 10-Н и 4”-Н), 3,03 (с, 3Н, 6-ОСН3), 2,87(дкв, 1Н, 2-Н), 2,58(ддкв, 1Н, 8-Н), 2,40(ддд, 1Н, 3’-Н), 2,37(д, 1Н, 2”-Н), 2,28 (с, 6Н, 3’-N(СН3)2), 2,00~1,80 (м, 3Н, 4-Н и 7-Н2), 1,41 (с, 3Н, 18-Н), 1,13(с, 3Н, 6-СН3), 0,85(т, 3Н, 14-СН3).
Пример 4
Получение кларитромицина с использованием гексабутилдиолова в качестве восстановителя
3,82 г (5 ммоль) N-оксида 6-O-метилэритромицина А, полученного в примере 1, суспендируют в 50 мл тетрагидрофурана. Затем добавляют 5,6 мл (2,2 ммоль) гексабутилдиолова и смесь кипятят с обратным холодильником в течение 24 часов. Растворитель концентрируют при пониженном давлении и к остатку добавляют изопропиловый эфир и гексан. Образовавшиеся кристаллы фильтруют и промывают гексаном, в результате чего получают 3,55 г кларитромицина в виде белого порошка с выходом 95%.
Пример 5
Получение кларитромицина с использованием сплава никеля и алюминия в качестве восстановителя
3,82 г (5 ммоль) N-оксида 6-O-метилэритромицина А, полученного в примере 1, суспендируют в смеси 100 мл метанола и 50 мл 1н гидроксида калия. Затем в течение 30 минут добавляют 5 г сплава никеля-алюминия, поддерживая при этом температуру 35-40°С, и перемешивают в течение 3 часов. Полученную смесь разбавляют метанолом и фильтруют через слой целита для удаления твердых веществ. Фильтрат концентрируют при пониженном давлении для удаления метанола и остаток разбавляют водой. Образовавшиеся кристаллы фильтруют, промывают водой, а затем сушат с получением 3,44 г кларитромицина в виде белого порошка с выходом 92%.
Пример 6
Получение кларитромицина путем гидрирования с использованием никелевого катализатора Ренея
3,82 г (5 ммоль) N-оксида 6-O-метилэритромицина А, полученного в примере 1, суспендируют в 100 мл этанола. Затем добавляют 0,25 г никелевого катализатора Ренея W4 и смесь перемешивают при температуре от 40 до 50°С в атмосфере водорода в течение 3 часов. Полученную смесь фильтруют через слой целита при 50°С или выше для удаления израсходованного катализатора. Фильтрат концентрируют при пониженном давлении до 30 мл и охлаждают до комнатной температуры. Образовавшиеся кристаллы фильтруют и сушат с получением 3,44 г кларитромицина в виде белого порошка с выходом 92%.
Пример 7
Получение кларитромицина с использованием бисульфита натрия в качестве восстановителя
3,82 г (5 ммоль) N-оксида 6-O-метилэритромицина А, полученного в примере 1, суспендируют в смеси 15 мл этанола и 15 мл воды. Затем добавляют 1,05 г (10 ммоль) бисульфита натрия и смесь перемешивают при комнатной температуре в течение 1 часа. Полученную смесь концентрируют, добавляют воду, а затем эту смесь доводят до рН 10 посредством добавления 10% карбоната натрия. Полученную смесь три раза экстрагируют этилацетатом, органические слои объединяют, промывают водой и рассолом, а затем сушат над безводным сульфатом магния. Растворитель удаляют при пониженном давлении и полученный остаток растворяют в 20 мл этанола. После добавления 0,55 мл муравьиной кислоты и 0,8 мл 35% формальдегида смесь кипятят с обратным холодильником в течение 2 часов. Полученную смесь концентрируют до половины объема и добавляют 20 мл воды, после чего полученную смесь доводят до рН 11 добавлением 10% карбоната натрия и получают 3,44 г кларитромицина в виде белого порошка с выходом 92%.
Пример 8
Получение кларитромицина
Процедуру примера 1 повторяют с использованием 75,0 г (0,1 моль) N-оксида эритромицина А, полученного в сравнительном примере, в результате чего получают 54,0 г неочищенного N-оксида 6-O-метилэритромицина А, имеющего чистоту примерно 54%.
54,0 г полученного таким образом N-оксида 6-O-метилэритромицина А растворяют в 400 мл дихлорметана и к этому раствору добавляют 32,6 г (144 ммоль) дигидрата дихлорида олова, а затем перемешивают при комнатной температуре в течение 1,5 часов. К полученной смеси добавляют 300 мл 10% карбоната натрия и дихлорметан удаляют при пониженном давлении. Остаток последовательно экстрагируют 400-мл и 200-мл порциями этилацетата, органические слои объединяют, дважды промывают 300 мл воды, а затем сушат над безводным сульфатом магния. Растворитель удаляют при пониженном давлении, к остатку добавляют 100 мл этанола, кипятят с обратным холодильником в течение 30 минут и перемешивают при комнатной температуре в течение 3 часов. Образовавшиеся твердые вещества собирают и сушат с получением 24,0 г кларитромицина в виде белого кристалла с выходом 32%.
Пример 9
Получение кларитромицина
Процедуру примера 1 повторяют с использованием 75,0 г (0,1 моль) N-оксида эритромицина А, полученного в сравнительном примере, в результате чего получают 33,2 г N-оксида 6-O-метилэритромицина А в виде кристаллического порошка, имеющего чистоту примерно 85%.
33,2 г чистого 85% N-оксида 6-O-метилэритромицина А растворяют в смеси 70 мл этанола и 70 мл воды, к полученному раствору добавляют 6,52 г (62,6 ммоль) бисульфита натрия, а затем смесь перемешивают при комнатной температуре в течение 1 часа. Полученную смесь концентрируют до небольшого объема, к концентрату добавляют воду, а затем доводят до рН 10 добавлением 10% карбоната натрия. Полученную смесь три раза экстрагируют этилацетатом, органические слои объединяют, промывают водой и рассолом, а затем сушат над безводным сульфатом магния. Растворитель удаляют при пониженном давлении и остаток растворяют в 80 мл этанола. К раствору этанола добавляют 5,0 мл муравьиной кислоты и 7,1 мл 35% формальдегида, а затем его кипятят с обратным холодильником в течение 2 часов. К полученной смеси добавляют 200 мл воды, рН доводят до 11 путем добавления концентрированного водного раствора аммиака, а затем охлаждают до комнатной температуры. Образовавшиеся твердые вещества фильтруют и сушат, в результате чего получают 22,5 г кларитромицина в виде белого порошка с выходом 30%.
Хотя настоящее изобретение проиллюстрировано и описано выше на конкретных вариантах его осуществления, очевидно, что в настоящее изобретение могут быть внесены различные изменения и модификации без отклонения от сущности настоящего изобретения, которое определяется объемом формулы изобретения.
Изобретение относится к способу получения кларитромицина формулы (I), включающему взаимодействие N-оксида эритромицина А формулы (II) с метилирующим агентом с получением N-оксида 6-О-метил-эритромицина А формулы (III) и обработку N-оксида 6-О-метилэритромицина А восстановителем. Технический результат - повышение выхода целевого продукта. 8 з.п. ф-лы.
(a) взаимодействия N-оксида эритромицина А формулы (II) с метилирующим агентом с получением N-оксида 6-O-метил-эритромицина А формулы (III);
(b) обработки N-оксида 6-O-метилэритромицина А, полученного на стадии (а), восстановителем с получением кларитромицина:
| US 5864023 A, 26.01.1999 | |||
| US 5919916 A, 06.07.1999 | |||
| О-МЕТИЛЬНЫЕ ПРОИЗВОДНЫЕ АЗИТРОМИЦИНА А, ОБЛАДАЮЩИЕ АНТИБАКТЕРИАЛЬНОЙ АКТИВНОСТЬЮ, АЗИТРОМИЦИНЫ В КАЧЕСТВЕ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ ДЛЯ ПОЛУЧЕНИЯ О-МЕТИЛЬНЫХ ПРОИЗВОДНЫХ АЗИТРОМИЦИНА А И СПОСОБ ПОЛУЧЕНИЯ О-МЕТИЛЬНЫХ ПРОИЗВОДНЫХ АЗИТРОМИЦИНА А | 1991 |
|
RU2045533C1 |
| D.R | |||
| HILL et al | |||
| Tetrahedron Lett., 1996, v | |||
| Пишущая машина | 1922 |
|
SU37A1 |
| Устройство для многократного телефонирования | 1919 |
|
SU787A1 |

