Область техники, к которой относится изобретение
Настоящее изобретение относится к способу получения производных эритромицина и кристаллам фумарата производных эритромицина, полученных указанным способом.
Предпосылки создания изобретения
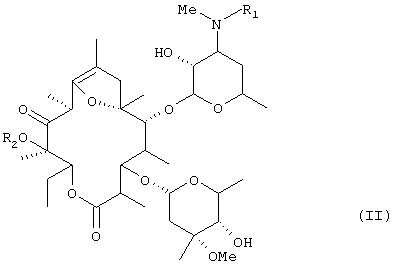
Соединения, представленные общей формулой (II)
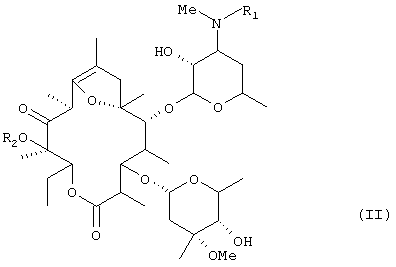
(где R1 представляет низшую алкильную группу и R2 представляет низшую алкильную группу) описаны в публикации №56873/1994 патента Японии и т.д. и известны как обладающие способностью усиливать движение пищеварительных трактов.
Способ получения этих соединений описан в публикации №56873/1994 патента Японии, Bioorg. & Med. Chem. Lett., т.4, №11, стр. 1347, 1994 и т.д.
Однако способы, описанные в этих ссылочных материалах, имеют некоторые недостатки, такие как большое число стадий, чрезмерное использование колоночной хроматографии для очистки и применение реагентов (например, йода), не пригодные для крупносерийного производства, что делает эти способы непригодными для коммерческих процессов. Кроме того, необходимо, чтобы соединения были высококачественными с точки зрения устойчивости, однородности и соответствия стандартам в случае их использования в качестве фармацевтических препаратов или исходных материалов для них такого типа, который может быть обеспечен способом по настоящему изобретению.
Раскрытие сущности изобретения
Были проведены интенсивные исследования в поисках выхода из этой ситуации и был найден эффективный способ получения и очистки фумаратов соединений, представленных общей формулой (II):
(где R1 представляет низшую алкильную группу и R2 представляет низшую алкильную группу) и, кроме того, было обнаружено, что кристаллы фумарата, очищенные предлагаемым способом, имеют более высокое качество как фармацевтические препараты или исходные материалы для них, чем кристаллы, которые получали до сих пор. На основе этих находок и было создано настоящее изобретение.
Таким образом, настоящее изобретение относится к способу получения фумарата соединения общей формулы (II):
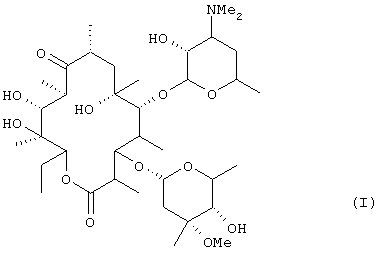
(где R1 представляет низшую алкильную группу и R2 представляет низшую алкильную группу) из эритромицина А [формула (I)]:
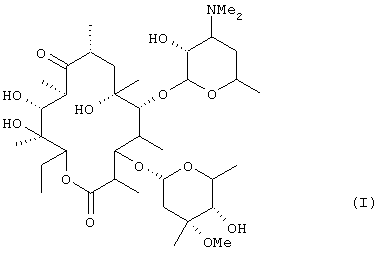
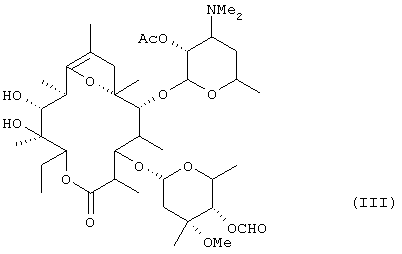
включающему стадии ацетилирования гидроксильной группы в положении 2' эритромицина А, формилирования гидроксильной группы в положении 4" с последующим осуществлением реакции образования полукеталя и получением в результате этого соединения формулы (III):
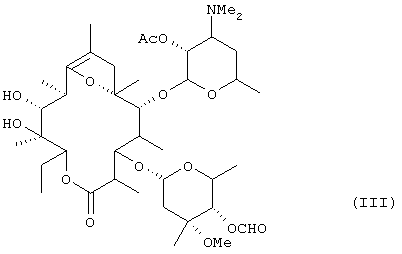
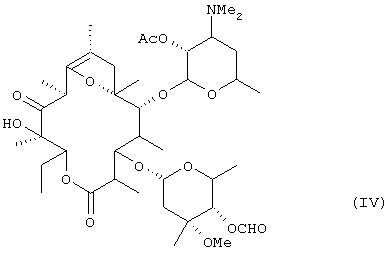
окисления гидроксильной группы в положении 11 соединения (III) с получением соединения формулы (IV):
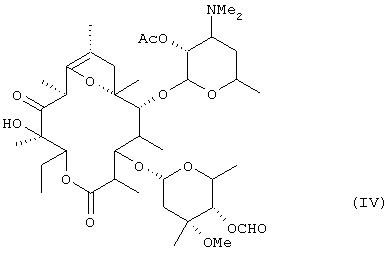
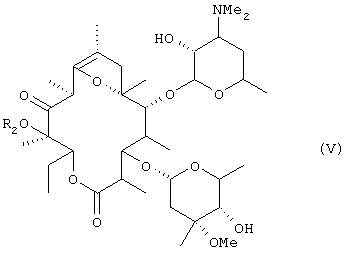
алкилирования гидроксильной группы в положении 12 соединения (IV), удаления ацетильной группы в положении 2' и формильной группы в положении 4" с получением соединения общей формулы (V):
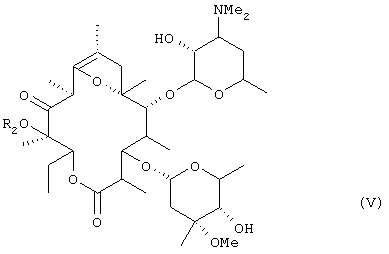
(где R2 представляет низшую алкильную группу), осуществления взаимодействия соединения (V) с бензилоксикарбонилхлоридом в оснóвных условиях, затем удаления введенной бензилоксикарбонильной группы, алкилирования атома азота в положении 3', преобразования соединения в фумарат в неочищенной кристаллической форме, перекристаллизации неочищенного кристалла из спиртового растворителя и осуществления другой перекристаллизации из водного этилацетата.
Изобретение также относится к способу получения фумарата соединения общей формулы (II):
(где R1 представляет низшую алкилиьную группу и R2 представляет низшую алкильную группу) из эритромицина А [формула (I)]:
включающему стадии ацетилирования гидроксильной группы в положении 2' эритромицина А, формилирования гидроксильной группы в положении 4" с последующим осуществлением реакции образования полукеталя и получением в результате этого соединения формулы (III):
окисления гидроксильной группы в положениим 11 соединения (III) с получением соединения формулы (IV):
алкилирования гидроксильной группы в положении 12 соединения (IV), удаления ацетильной группы в положении 2' и формильной группы в положении 4" с получением соединения общей формулы (V):
(где R2 представляет низшую алкильную группу), осуществления взаимодействия соединения (V) с бензилоксикарбонилхлоридом в оснóвных условиях, затем удаления введенной бензилоксикарбонильной группы, алкилирования атома азота в положении 3' и преобразования соединения в фумарат.
Из описанных выше реакций ацетилирование гидроксильной группы в положении 2' эритромицина А, формилирование гидроксильной группы в положении 4" и реакцию образования полукеталя предпочтительно осуществляют в одном резервуаре. Термин "один резервуар" при использовании в настоящем изобретении означает, что представляющие интерес реакции осуществляют в одну стадию без выделения и очистки продукта реакции на каждой стадии.
Реакцию алкилирования гидроксильной группы в положении 12 и реакцию удаления ацетильной группы в положении 2' и формильной группы в положении 4" предпочтительно также осуществляют в одном резервуаре.
В конкретном предпочтительном случае ацетилирование гидроксильной группы в положении 2' эритромицина А и формилирование гидроксильной группы в положении 4" и реакцию образования полукеталя осуществляют в одном резервуаре и, кроме того, реакцию алкилирования гидроксильной группы в положении 12 и реакцию удаления ацетильной группы в положении 2' и формильной группы в положении 4" также осуществляют в одном резервуаре.
Настоящее изобретение касается также способа получения соединения формулы (III):
из эритромицина А формулы (I):
путем осуществления в одном резервуаре ацетилирования гидроксильной группы в положении 2' эритромицина А, формилирования гидроксильной группы в положении 4" и реакции образования полукеталя.
Настоящее изобретение касается также способа получения соединения общей формулы (VI):
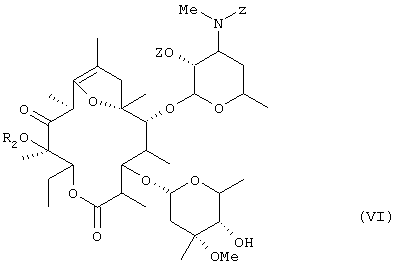
(где R2 представляет низшую алкильную группу и Z представляет бензилоксикарбонильную группу) путем осуществления взаимодействия соединения общей формулы (V):
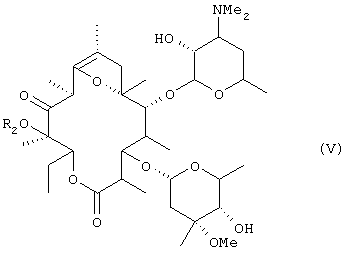
(где R2 представляет низшую алкильную группу) с бензилоксикарбонилхлоридом в оснóвных условиях.
Настоящее изобретение относится также к способу очистки фумарата соединения общей формулы (II):
(где R1 представляет низшую алкильную группу и R2 представляет низшую алкильную группу) путем перекристаллизации неочищенного кристалла фумарата соединения общей формулы (II):
(где R1 представляет низшую алкильную группу и R2 представляет низшую алкильную группу) из спиртового растворителя и путем осуществления другой перекристаллизации из водного этилацетата.
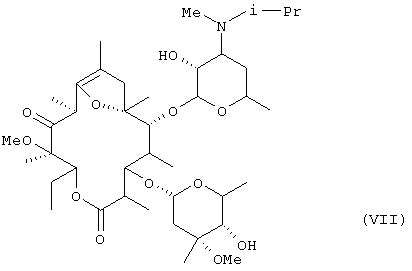
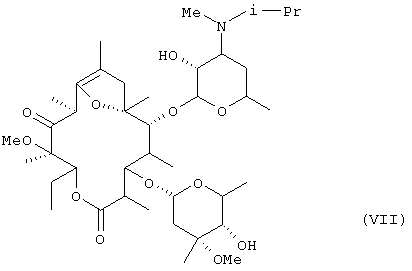
В дополнительном аспекте настоящее изобретение относится к кристаллу фумарата соединения формулы (VII):
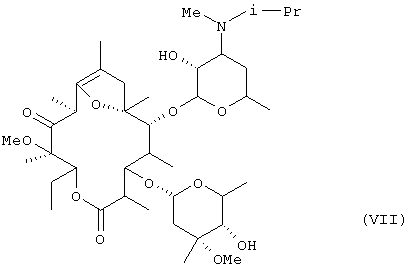
в котором молярное отношение соединения (VII) к фумаровой кислоте составляет 2:1 и который получают путем перикристаллизации из водного этилацетата.
Краткое описание чертежей.
Фиг.1 показывает порошковую рентгенограмму для кристаллической формы А соединения VII.
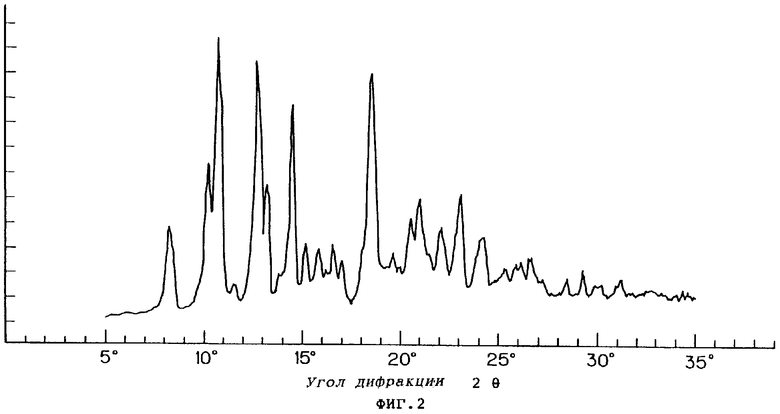
Фиг.2 показывает порошковую рентгенограмму для кристаллической формы C соединения VII.
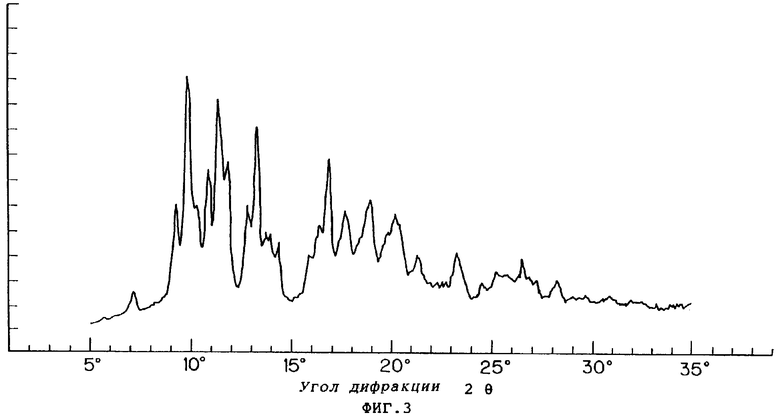
Фиг.3 показывает порошковую рентгенограмму для кристаллической формы D соединения VII.
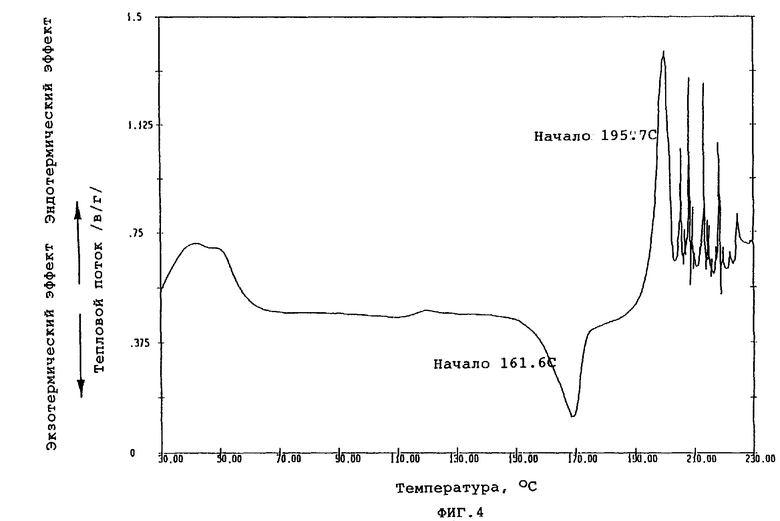
Фиг.4 показывает кривую DSC (ДСК - дифференциальная сканирующая калориметрия), полученную путем термического анализа кристаллической формы А.
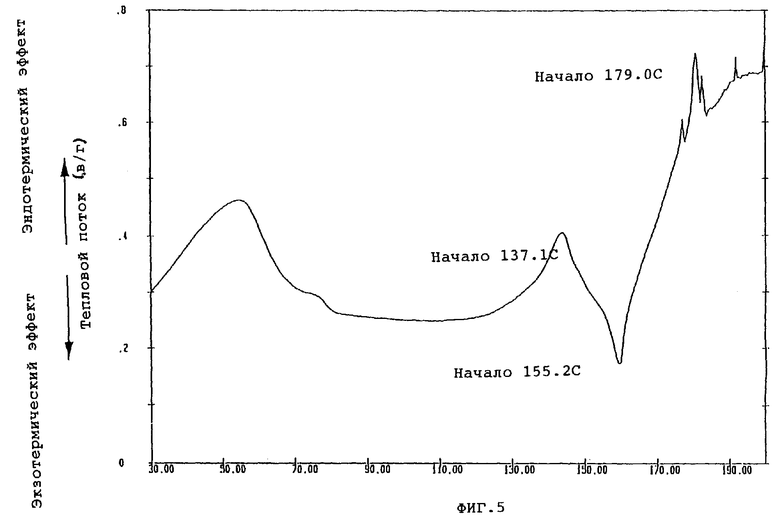
Фиг.5 показывает кривую DSC, полученную путем термического анализа кристаллической формы С.
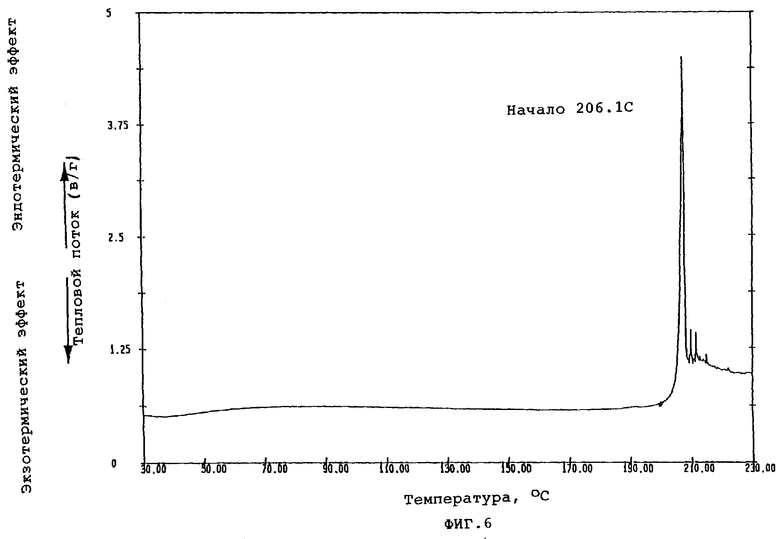
Фиг.6 показывает кривую DSC, полученную путем термического анализа кристаллической формы D.
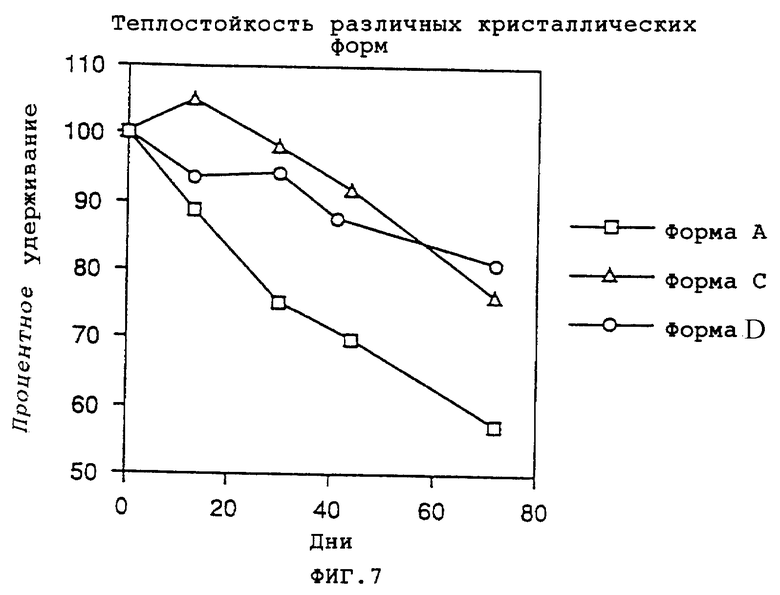
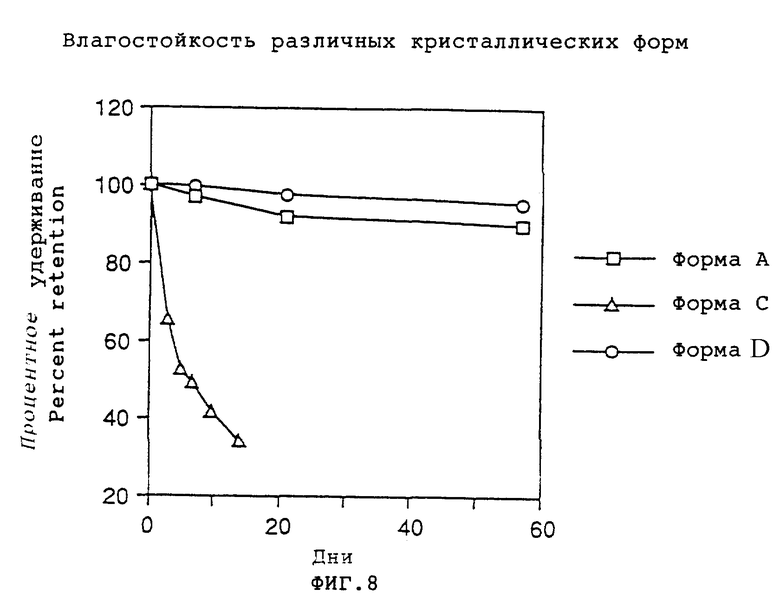
Фиг.7 представляет график, показывающий процентное удерживание кристаллических форм А, С и D при испытании на теплостойкость.
Фиг.8 представляет график, показывающий процентное удерживание кристаллических форм А, С и D при испытании на влагостойкость.
Наилучший способ осуществления изобретения.
Используемый в данном описании термин "низшая алкильная группа" охватывает неразветвленные или разветвленные алкильные группы, имеющие 1-6 углеродных атомов, и их конкретные примеры включают метильную, этильную, н-пропильную, изопропильную, н-бутильную, изобутильную, втор-бутильную, трет-бутидьную, пентильную или гексильную группы, из которых предпочтительными являются метильная, этильная, н-пропильная и изопропильная группы. Особенно предпочтительным примером группы R1 является изопропильная группа и группы R2 - метильная группа.
Ниже схематически показаны примеры способа получения (маршрут 1 реакции) по настоящему изобретению.
Маршрут 1-1 реакции
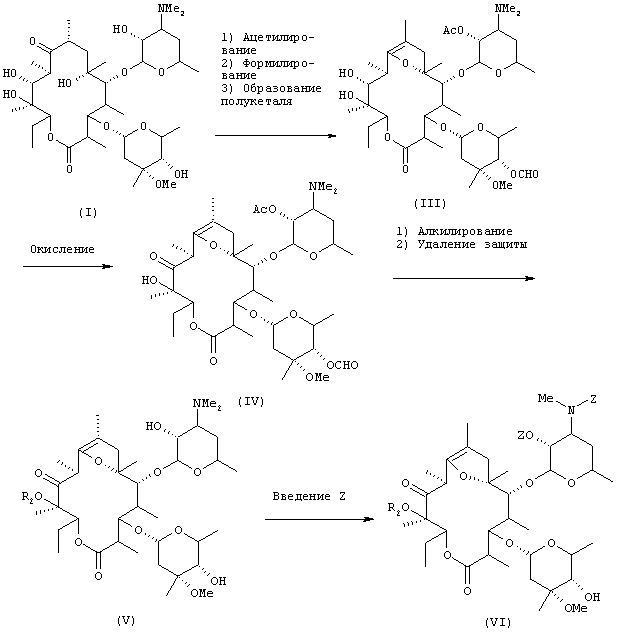
(где R2 представляет низшую алкильную группу и Z представляет бензилоксикарбонильную группу).
Маршрут 1-2 реакции
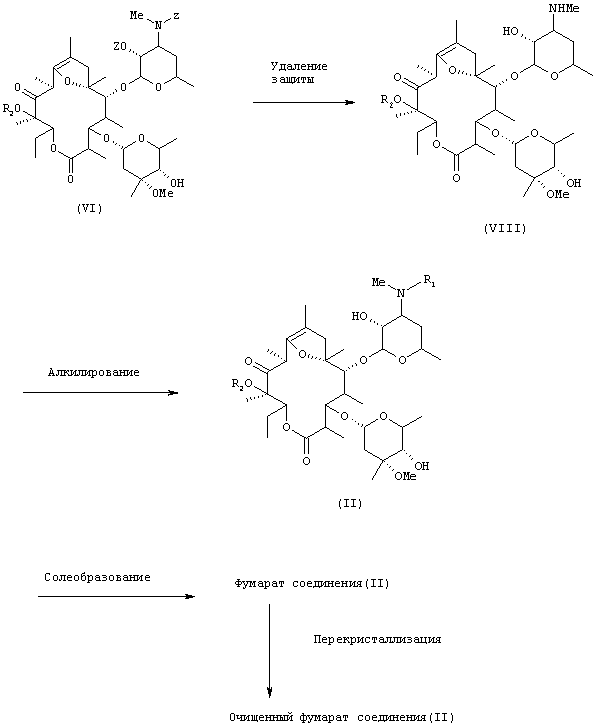
(где R1 представляет низшую алкильную группу, R2 представляет низшую алкильную группу и Z представляет бензилоксикарбонильную группу).
Таким образом, гидроксильную группу в положении 2' эритромицина [соединение формулы (I)] ацетилируют в присутствии основания, после чего гидроксильную группу в положении 4" формилируют и затем подвергают реакции образования полукеталя, в результате чего получают соединение формулы (III). Три стадии реакции, т.е. ацетилирование, формилирование и образование полукеталя, предпочтительно осуществляют в одном резервуаре.
Примеры основания для применения в ацетилировании, которое является реакцией первой стадии, включают неорганические основания и органические основания, такие как амины; предпочтительными примерами являются органические основания, такие как пиридин, триэтиламин, диизопропилэтиламин, пирролидин, пиперидин, морфолин, диэтиламин и диизопропиламин, из которых более предпочтительным является пиридин. Предпочтительными для использования растворителями являются растворители, инертные по отношению к трем стадиям реакции, т.е. ацетилированию, формилированию и образованию полукеталя, и в качестве их примеров могут служить этилацетат, ацетон, дихлорметан и хлороформ, из которых более предпочтительно являются этилацетат и ацетон, а из них наиболее предпочтительным является этилацетат. Примеры ацетилирующего агента включают уксусный ангидрид, ацетилхлорид и ацетат натрия, причем предпочтительными являются уксусный ангидрид и ацетилхлорид, из которых уксусный ангидрид является наиболее предпочтительным. Температура реакции находится предпочтительно в пределах от примерно 0°С до примерно 50°С, причем более предпочтительными являются комнатная температура и температуры вблизи нее. Время реакции находится в пределах обычно от примерно 30 минут до 3 часов, предпочтительно от 1 часа до 2 часов.
Предпочтительные примеры формилирующего агента, применяемого при формилировании, которое является реакцией второй стадии, включают смеси муравьиная кислота - уксусный ангидрид и формиат натрия - ацетиллохлорид, из которых более предпочтительной является смесь муравьиная кислота - уксусный ангидрид. Примеры основания, которое можно использовать, включают неорганические основания и органические основания, такие как амины, предпочтительными примерами которых являются пиридин, триэтиламин, диизопропилэтиламин, пирролидин, пиперидин, морфолин, диэтиламин и диизопропиламин, из которых более предпочтительным является пиридин. Следует, однако, отметить, что при последовательном осуществлении реакций ацетилирования и формилирования основание, используемое в первой реакции, может служить и для второй реакции, что исключает необходимость использования дополнительного основания. Температура реакции формилирования находится предпочтительно в пределах от примерно -40°С до примерно 5°С, более предпочтительно в пределах от -20°С до 0°С. Время реакции находится в пределах обычно от примерно 1 часа до примерно 1 дня, предпочтительно от примерно 5 часов до примерно 12 часов.
Образование полукеталя, которое является реакцией третьей стадии, осуществляют в кислых условиях. Термин "кислые условия" означает присутствие кислоты в реакционной системе. Кислота может быть органической, которой предпочтительно является карбоновая кислота, такая как уксусная или муравьиная кислота, предпочтительной является уксусная кислота. При осуществлении реакции первой - третьей стадии в одном резервуаре уксусную или муравьиную кислоту вводят в систему с предшествующей стадии реакции и, следовательно, предполагаемая реакция будет протекать без добавления кислоты на третьей стадии. Температура реакции третьей стадии находится предпочтительно в пределах от примерно комнатной температуры до примерно 60°С, более предпочтительно в пределах от 40°С до 50°С. Время реакции находится в пределах обычно от примерно 1 часа до примерно 1 дня, предпочтительно от примерно 2 часов до примерно 12 часов.
Полученное соединение формулы (III) подвергают реакции окисления, чтобы окислить гидроксильную группу в положении 11. Примеры окислителей включают органические окислители, такие как диметилсульфоксид и реактив Периодинан Десс-Мартина (Dess - Martin Periodinane reagent), и оксиды металлов, такие как тетроксид рутения, и предпочтительные примеры включают смеси диметилсульфоксид - дициклогексилкарбодиимид и диметилсульфоксид - трифторуксусный ангидрид, из которых особенно предпочтительной является смесь диметилсульфоксид - трифторуксусный ангидрид. Могут быть использованы любые растворители, инертные по отношению к реакции; при использовании в качестве окислителя смеси диметилсульфоксид - трифторуксусный ангидрид предпочтительно используют в качестве растворителей галогенсодержащие растворители, такие как хлороформ и дихлорметан, из которых более предпочтительным является дихлорметан. Температура реакции окисления находится предпочтительно в пределах от примерно -60°С до примерно 0°С, более предпочтительно в пределах от примерно -20°С до примерно -10°С. Время реакции находится в пределах обычно от примерно 30 минут до примерно 5 часов, предпочтительно от 1 часа до 2 часов.
Полученное соединение формулы (IV) подвергают взаимодействию с алкилирующим агентом в оснóвных условиях, чтобы алкилировать гидроксильную группу в положении 12. Затем удаляют защитные группы в положениях 2' и 4". В этом случае алкилирование и реакцию удаления защитных групп предпочтительно осуществляют в одном резервуаре.
Примерами алкилирующего агента, используемого при алкилировании, которое является реакцией первой стадии, являются алкилгалогениды, алкилтозилаты и алкилимидаты, из которых предпочтительными являются алкилтозилаты и алкилгалогениды. Особенно предпочтительной в качестве алкильной части этих алкилирующих агентов является метильная группа. Конкретные примеры метилирующего агента включают метилиодид и метилтозилат, причем предпочтительным является метилтозилат. Примеры оснований, которые можно использовать, включают гидриды металлов, гидроксиды металлов и алкоксиды металлов, причем предпочтительными являются гидриды металлов, из которых особенно предпочтительным является гидрид натрия. Можно использовать любые растворители, инертные по отношению к реакции, но предпочтительными являются апротонные полярные растворители, причем более предпочтительными являются диметилимидазолидинон, диметилформамид, диметилацетамид, тетрагидрофуран и ацетонитрил, из которых особенно предпочтительными являются диметилимидазолидинон и диметилформамид. Температура реакции находится предпочтительно в пределах от примерно 0°С до примерно 60°С, более предпочтительно от 0°С до 30°С. Время реакции находится в пределах обычно от примерно 1 часа до примерно 12 часов, предпочтительно от 2 часов до 8 часов.
Удаление защитных групп, которое является реакцией второй стадии, осуществляют обычными методами осуществления реакции удаления ацетильных и формильных групп, предпочтительно в основных условиях. Примеры оснований, которые можно использовать, включают неорганические основания, такие как бикарбонат натрия и карбонат калия, из которых более предпочтительным является бикарбонат натрия. Можно использовать любые растворители, инертные по отношению к реакции, но предпочтительными являются спиртовые растворители, причем более предпочтительными являются метанол и этанол. Температура реакции находится предпочтительно в пределах от примерно 40°С до примерно 80°С, более предпочтительно от 50°С до 60°С. Время реакции находится в пределах обычно от примерно 1 часа до примерно 12 часов, предпочтительно от 3 часов до 8 часов.
Если реакцию алкилирования и удаления защитных групп нужно осуществить в одном резервуаре, то алкилирование, которое является реакцией первой стадии, может быть выполнено с использованием основания в избыточном количестве, скажем в количестве 2 эквивалентов или более, предпочтительно около 2 эквивалентов; полученная в результате этого основность основания устраняет необходимость использования дополнительного основания в реакции второй стадии. При необходимости, растворители на каждой стадии могут быть необязательно обменены путем, например, пополнения растворителя для реакции первой стадии растворителем для реакции второй стадии.
Полученное таким образом соединение общей формулы (V) подвергают взаимодействию с избыточным количеством бензилоксикарбонилхлорида в оснóвных условиях, в результате чего указанное соединение преобразуется в соединение общей формулы (VI), и затем введенную бензилоксикарбонильную группу обычным способом удаляют, преобразуя тем самым соединение общей формулы (VI) в соединение общей формулы (VIII), которое в свою очередь подвергают взаимодействию с алкилирующим агентом в оснóвных условиях, в результате чего оно превращается в соединение общей формулы (II), которое обрабатывают обычным способом для преобразования в фумарат. Серия реакций, состоящая из бензилоксикарбонилирования, дебензилоксикарбонилирования, алкилирования и преобразования в фумарат, может быть проведена до конечной стадии, т.е. образования фумарата соединения общей формулы (II), без очистки продуктов, полученных на соответственных стадиях.
Предпочтительным примером бензилоксикарбонилирующего агента для применения на первой стадии является бензилоксикарбонилхлорид. Примеры оснований, которые можно использовать, включают неорганические основания, такие как бикарбонат натрия и карбонат калия, из которых предпочтительным является бикарбонат натрия. Можно использовать любые растворители, инертные по отношению к реакции, но предпочтительными являются растворители на основе ароматических углеводородов, причем более предпочтительными являются толуол и т.д. Температура реакции находится предпочтительно в пределах от примерно 30°С до примерно 80°С, более предпочтительно от 45°С до 70°С и наиболее предпочтительно находится на уровне 60°С и около этого. Время реакции находится в пределах обычно от примерно 2 часов до примерно 12 часов, предпочтительно от 4 часов до 8 часов. Следует отметить, что бензилоксикарбонилирующий агент должен использоваться в избыточном количестве по отношению к соединению общей формулы (V), предпочтительно в количестве 9-15 эквивалентов, более предпочтительно в количестве 10-12 эквивалентов.
Удаление бензилоксикарбонильной группы, которое является реакцией второй стадии, осуществляют обычным методом удаления защиты. Примерным методом удаления защиты является каталитическое гидрирование, предпочтительно с использованием катализатора палладий на углероде. Источником водорода обычно является водород, но можно также использовать формиат аммония. Если в качестве источника водорода используют водород, то каталитическое гидрирование можно осуществлять при давлении выше атмосферного, находящемся в пределах предпочтительно от примерно 2 до 5 атмосфер, более предпочтительно от 3 до 4 атмосфер. Можно использовать любые растворители, инертные по отношению к реакции, но предпочтительными являются спиртовые растворители и подобные, причем более предпочтительным является метанол, этанол и т.д. Температура реакции находится в пределах от примерно 0°С до примерно 50°С, предпочтительно от примерно 10°С до 30°С, более предпочтительно находится на уровне комнатной температуры и около нее. Время реакции находится в пределах от примерно 30 минут до примерно 3 часов, предпочтительно от 1 часа до 2 часов. Если в качестве источника водорода используют формиат аммония, то можно использовать любые растворители, инертные по отношению к реакции, но предпочтительными являются спиртовые растворители и подобные, причем более предпочтительными являются метанол, этанол и т.д. Температура реакции находится предпочтительно в пределах от примерно 50°С до примерно 100°С, более предпочтительно от 60°С до 90°С. Время реакции находится в пределах обычно от примерно 30 минут до примерно 3 часов, предпочтительно от 1 часа до 2 часов.
Примеры алкилирующего агента для использования при алкилировании, которое является реакцией третьей стадии, включают алкилгалогениды и алкилтозилаты, причем предпочтительными являются алкилгалогениды. В качестве алкильной части этих алкилирующих агентов особенно предпочтительной является изопропильная группа. Предпочтительные примеры изопропилирующего агента включают изопропилиодид. Примеры оснований, которые можно использовать, включают органические основания, такие как амины, и неорганические основания; причем предпочтительные примеры включают диизопропилэтиламин, триэтиламин, морфолин, пиперидин, пирролидин и пиридин, из которых особенно предпочтительным является триэтиламин. Можно использовать любые растворители, инертные по отношению к реакции, но предпочтительными являются апротонные полярные растворители, спиртовые растворители и подобные, причем более предпочтительными являются диметилмидазолидинон, диметилформамид, диметилацетамид, ацетонитрил, тетрагидрофуран, метанол, этанол и т.д., из которых особенно предпочтительными являются диметилимидазолидинон, диметилформамид и ацетонитрил. Температура реакции находится предпочтительно в пределах от примерно 50°С до примерно 100°С, более предпочтительно от 60°С до 80°С. Время реакции находится в пределах обычно от примерно 3 часов до примерно 10 дней, предпочтительно от 5 часов до 10 часов.
Преобразование в фумарат, которое является реакцией четвертой стадии, осуществляют обычным методом солеобразования. Предпочтительными для использования растворителями являются спиртовые растворители, растворители на основе простых эфиров, такие как тетрагидрофуран, ацетон и т.д., причем более предпочтительными являются метанол, этанол, изопропанол и т.д. Температура реакции находится предпочтительно в пределах от примерно -20°С до примерно 50°С, более предпочтительно от примерно -15°С до примерно комнатной температуры. Время реакции находится в пределах обычно от примерно 1 часа до примерно 6 часов, предпочтительно от 3 часов до 4 часов.
При необходимости полученный таким образом фумарат соединения общей формулы (II) очищают. Предпочтительным методом очистки является перекристаллизация. Примеры растворителей для перекристаллизации включают необязательно водосодержащие растворители на основе сложных эфиров, спиртовые растворители, растворители на основе простых эфиров и их смеси, предпочтительными примерами являются этанол, смесь метанола и изопропанола и смесь этилацетата и воды, из этих растворителей более предпочтительными являются смесь метанола и изопропанола, смесь этилацетата и воды и т.д. В этих смешанных системах растворителей отношение метанола к изопропанолу может находиться в пределах от примерно 10:90 до примерно 50:50, предпочтительно от примерно 20:80 до примерно 30:70, отношение этилацетата к воде может находиться в пределах от 99,5:0,5 до 97:3, предпочтительно от 99:1 до 98:2, более предпочтительное значение составляет примерно 98,5:1,5.
При осуществлении перекристаллизации с использованием в качестве растворителя этилацетата, одного или в смеси с водой, были получены кристаллы (кристаллические формы С и D), которые отличались от продукта (кристаллическая форма А) перекристаллизации из смеси метанола и изопропанола, описанного в публикации №56873/1994 патента Японии. Результаты порошковой рентгенографии и термического анализа (DSK, ДСК - дифференциальная сканирующая калорометрия) этих кристаллов показаны на фиг.1-6.
В кристалле (кристаллическая форма А), который был получен путем перекристаллизации из смеси метанола и изопропанола, молярное отношение соединения формулы (VII) к фумаровой кислоте составляло 2:1. Кристалл, полученный путем перекристаллизации с использованием в качестве растворителя этилацетата, одного или в смеси с водой, был один из формы С, в котором молярное отношение соединения формулы (VII) к фумаровой кислоте составляло 1:1, или из формы D, в котором указанное молярное отношение составляло 2:1.
Было установлено, что из этих кристаллических форм, кристалл формы D, который был получен перекристаллизацией из смеси этилацетата и воды фумарата (например, кристаллической формы, А) соединения формулы (VII), который был частично очищен путем, например, перекристаллизации с использованием смеси метанола и изопропанола, был лучшего качества, такого как более высокая стойкость, чем другие формы кристалла, с точки зрения применения в качестве фармацевтического препарата или исходного материала для него.
Чтобы получить кристаллическую форму D, перекристаллизацию с использованием смеси этилацетата и воды предпочтительно осуществляют следующим образом: сначала частично очищенный фумарат соединения формулы (VII) суспендируют или растворяют в этлацетате при температуре, близкой к комнатной, и после добавления воды, охлаждают суспензию или раствор до температуры в пределах от примерно -10°С до примерно -20°С.
Следующие далее примеры даны для дополнительной иллюстрации настоящего изобретения и ни в коей мере не должны считаться как ограничивающие. В последующем описании в данных спектрального анализа методом 1H-ЯМР указаны лишь характеристические пики.
Пример 1: Синтез полукетальной формы [соединение формулы (III)]
Эритромицин А (20,0 г, 0,027 моль) растворяют в уксусном ангидриде (3,34 г, 0,033 моль), пиридине (3,45 г, 0,044 моль) и этилацетате (80 мл) и раствор перемешивают при комнатной температуре в течение 1 часа. Затем к раствору добавляют по каплям при охлаждении льдом (0°С) муравьиную кислоту (11,29 г, 0,245 моль) и уксусный ангидрид (12,52 г, 0,123 моль) и смесь перемешивают 3 часа при охлаждении льдом. Затем смесь постепенно возвращают к комнатной температуре и оставляют стоять в течение ночи.
Смесь нагревают при 40-50°С в течение примерно 2 часов. Затем смесь возвращают к комнатной температуре, растворяют в этилацетате (120 мл) и раствор промывают ледяной водой (60 мл × 2). Этилацетатный слой нейтрализуют насыщенным водным раствором бикарбоната натрия (120 мл) и твердым бикарбонатом натрия (8 г или более). Этилацетатный слой отделяют, промывают водой (40 мл × 3) и насыщенным рассолом (40 мл) и сушат безводным сульфатом натрия в течение ночи.
После фильтрования осуществляют концентрирование в вакууме. Остаток нагревают с обратным холодильником с гексаном (136 мл) в течение примерно 30 минут и затем охлаждают. После добавления этилацетата (24 мл) смесь охлаждают до 0°С при перемешивании. Полученный кристалл отделяют и промывают гексаном (20 мл), получив в результате указанное в заголовке соединение (15,8 т, 74%) в виде белого кристалла.
Т.пл.: 200-208°С (этилацетат-гексан).
1H-ЯМР (CDCl3): 0,89 (3Н, т, 13-СН2СН3), 2,05 (3Н, с, 2'-ОСОСН3), 2,27 (6Н, с, 3'-N(СН3)2), 3,36 (3Н, с, 3"-ОСН3), 3,83 (1Н, с, 11-СН(ОН)), 8,20 (1Н, с, 4"-OCHO).
Пример 2: Синтез оксоформы [соединение формулы (IV)]
Часть (10,0 г, 0,013 моль) соединения, полученного в примере 1, и диметилсульфоксид (2,64 г, 0,032 моль) растворяют в дихлорметане (50 мл). Охлаждают реакционную систему до -20°С смесью льда с хлоридом натрия и к реакционной смеси добавляют по каплям при -10°С или ниже трифторуксусный ангидрид (3,36 г, 0,016 моль) и перемешивают смесь в течение 20 минут. Затем, поддерживая температуру реакционной системы на уровне -20°С, добавляют по каплям триэтиламин (3,49 г, 0,034 моль) при -10°С или ниже и продолжают перемешивание в течение 20 минут. После добавления насыщенного водного раствора бикарбоната натрия (50 мл) перемешивают смесь в течение 20 минут. После промывки водой (50 мл × 3) смесь сушат безводным сульфатом натрия в течение ночи.
Затем дихлорметан концентрируют в вакууме, к сиропообразному остатку добавляют гексан (100 мл) и перемешивают смесь, пока она была горячей, для образования раствора. Охладив раствор, добавляют дихлорметан (2,5 мл) и смесь перемешивают при комнатной температуре в течение 2,5 часов. Кристалл извлекают путем фильтрования и промывают 2,5% дихлорметангексан (30 мл) с получением указанного соединения (6,49 г, 65%) в виде белого кристалла.
Т. пл.: 186-188°С (дихлорметан - гексан).
1H-ЯМР (CDCl3): 0,90 (3Н, т, 13-СН2СН3), 2,04 (3Н, с, 2'-ОСОСН3), 2,26 (6Н, с, 3'-N(CH3)2), 3,33 (3Н, с, 3"-ОСН3), 4,53 (1Н, д, 1'-Н), 4,84 (1Н, д, 1"-Н), 4,97 (1Н, дд, 13-Н), 8,21 (1Н, с, 4"-ОСНО).
13C-ЯМР (CDCl3): 208,4 (11-СО).
Пример 3: Синтез формы с удаленной защитой [соединение формулы (V) (R2: метил)].
К диметилформамиду (30 мл) добавляют 60% гидрида натрия (1,02 г, 0,026 моль); к смеси добавляют соединение (10,0 г, 0,013 моль), полученное в примере 2, и полученную смесь перемешивают в течение 30 минут. Затем добавляют по каплям метилтозилат (2,38 г, 0,013 моль) и смесь перемешивают сначала при 0-5°С в течение 1 часа, затем при 15-20°С в течение 1,5 часов. После добавления метанола (60 мл) смесь нагревают при 60°С в течение 5 часов. Нагретую смесь оставляют стоять в течение ночи.
Всю смесь концентрируют в вакууме и концентрат добавляют по каплям к теплой воде (150 мл) при 40°С при перемешивании для выделения осадка. В течение дополнительных 30 минут продолжают перемешивание в теплой воде (150 мл) при 40°С для выделения осадка. Высушив осадок при 50°С в течение 4 часов, получают неочищенную форму с удаленной защитой (7,9 г, 85%).
Неочищенную форму с удаленной защитой растворяют в ацетоне (12,6 мл) и добавляют 10% водный аммиак (5,9 мл) для кристаллизации. Для разделения фаз и промывки смесь перемешивают сначала при 15-25°С в течение 1 часа, а затем при температуре от -5 до -10°С в течение 1 часа. После сушки при 50°С в течение 3 часов получают указанное в заголовке соединение (5,5 г, 59%) в виде бледно-желтого кристалла.
Т. пл.: 168-174°С (водный аммиак - ацетон).
1H-ЯМР (CDCl3): 0,85 (3Н, т, 13-СН2СН3), 1,68 (3Н, с, 8-СН3), 2,28 (6Н, с, 3'-N(СН3)2), 3,06 (3Н, с, 12-ОСН3), 3,34 (3Н, с, 3"-ОСН3), 4,37 (1Н, д, 1'-Н), 4,97 (1Н, д, 1"-Н), 5,63 (1Н, дд, 13-Н).
Пример 4: Синтез бензилоксикарбонильной формы [соединение формулы (VI) (R2: метил)].
К толуолу (55 мл) добавляют соединение (5,5 г, 0,0076 моль), полученное в примере 3, и твердый бикарбонат натрия (9,5 г, 0,113 моль). Затем добавляют по каплям бензилоксикарбонилхлорид (18,0 г, 0,106 моль) при 70-80°С при перемешивании и смесь нагревают при той же температуре в течение 4 часов. Затем жидкую реакционную смесь оставляют стоять в течение ночи при комнатной температуре.
К жидкой реакционной смеси добавляют пиридин (4,02 г, 0,05 моль) при перемешивании в течение 30 минут. Затем добавляют насыщенный водный раствор бикарбоната натрия (38,5 мл) и смесь перемешивают 10 минут с последующим добавлением этилацетата (38,5 мл). Смесь перемешивают для разделения фаз и полученный органический слой промывают водой. Затем органический слой промывают насыщенным рассолом (38,5 мл) и сушат безводным сульфатом натрия.
Всю смесь концентрируют в вакууме. К остатку добавляют ацетонитрил (27,5 мл) и полученный раствор промывают гексаном (187 мл × 5) для разделения фаз. Ацетонитриловый слой концентрируют в вакууме. К остатку добавляют метанол (13,5 мл) и смесь перемешивают сначала при 15-25°С в течение 1 часа, а затем при 0°С или ниже в течение 1 часа. Выпавший кристалл извлекают путем фильтрования и высушивают при 50°С в течение 3 часов, получая в результате указанное в заголовке соединение (4,0 г, 54%) в виде белого кристалла.
Т. пл.: 122-126°С (метанол).
1H-ЯМР (CDCl3): 0,96 (3Н, т, 13-СН2СН3), 1,68 (3Н, с, 8-СН3), 3,03-3,37 (3Н, д, 3"-ОСН3), 3,06 (3Н, с, 12-ОСН3), 5,03-5,21 (4Н, м, CH2C6H5×2), 5,63 (1Н, дд, 13-Н), 7,28-7,34 (10Н, м, 2'-ОСОСН2С6Н5, 3'-NOCOCH2C6H5).
Пример 5: Синтез дебензилоксикарбонилированной формы [соединение формулы (VIII) R2:метил)].
К метанолу (36,8 мл) добавляют соединение (4,0 г, 0,004 моль), полученное в примере 4, 10% палладий на углероде (0,4 г) и формиат аммония (1,03 г) и смесь нагревают с обратным холодильником в течение 1 часа. Отфильтровывают палладий на углероде и затем отгоняют в вакууме метанол. Остаток растворяют в этилацетате (40 мл) и раствор промывают насыщенным водным раствором бикарбоната натрия (16 мл) для разделения фаз. Полученный органический слой промывают сначала водой (16 мл × 2), а затем насыщенным рассолом (16 мл) и сушат безводным сульфатом натрия. Отфильтровав безводный сульфат натрия, органический слой концентрируют в вакууме с получением неочищенной дебензилоксикарбонилированной формы (2,0 г, 69%) в виде белого кристалла.
Т. пл.: 187-190°С (как суспендировано в гексане для очистки).
1H-ЯМР (CDCl3): 0,95 (3Н, т, 13-СН2СН3), 1,68 (3Н, с, 8-СН3), 2,47 (3Н, с, 3'-NHCH3), 3,06 (3Н, с, 12-ОСН3), 3,21 (1Н, дд, 2"-Н), 3,33 (3Н, с, 3"-ОСН3), 4,37 (1Н, д, 1'-Н), 4,96 (1Н, д, 1"-Н), 5,61-5,65 (1Н, дд, 13-Н).
Пример 6: Синтез фумаратной формы [фумарат соединения формулы (VII)].
К диметилимидазолидинону (35 мл) добавляют соединение (10 г, 0,014 моль), полученное в примере 5, изопроилиодид (23,8 г, 0,14 моль) и триэтиламин (16,95 г, 0,17 моль) для образования раствора, который нагревают при 70-75°С в течение 7-8 часов и оставляют стоять в течение ночи. После экстрагирования и промывки этилацетатом (200 мл) и 2,5% водным аммиаком (75 мл) раствор промывают водой (150 мл × 2), а затем насыщенным рассолом (100 мл) и сушат безводным сульфатом натрия. Затем этилацетат концентрируют в вакууме, остаток и фумаровую кислоту (0,84 г, 0,0073 моль) растворяют в метаноле (25 мл). При перемешивании к раствору медленно добавляют по каплям изопропанол (75 мл) с получением фумаратной формы. Жидкую реакционную смесь, содержащую фумаратную форму, перемешивают сначала при комнатной температуре в течение 1 часа, затем при 0°С в течение 1 часа и наконец при -15°С в течение 1 часа, с последующим фильтрованием в вакууме с получением указанного в заголовке соединения (7,9 г, 69%) в виде белого кристалла.
Т. пл.: 194-197°С (метанол - изопропанол).
1H-ЯМР (CDCl3+ДМСО-d6): 0,94 (3Н, т, 13-СН2СН3), 1,73 (3Н, с, 8-СН3), 3,05 (3Н, с, 12-ОСН3), 3,08 (1Н, дд, 4"-H), 3,35 (3Н, с, 8"-ОСН3), 4,43 (1Н, д, 1'-Н), 4,96 (1Н, д, 1"-Н), 5,60-5,63 (1Н, дд, 13-Н), 6,78 (1Н, с, 1/2 (=СН-СООН)2).
Пример 7: Частичная очистка фумаратной формы [фумарат соединения формулы (VII)].
Соединение (10,0 г, 0,0123 моль), полученное в примере 6, растворяют в метаноле (25 мл) и к раствору медленно добавляют по каплям изопропанол (75 мл). Жидкий реакционный раствор перемешивают сначала при комнатной температуре в течение 1 часа, затем при 0°С в течение 1 часа и наконец при -15°С в течение 1 часа, чтобы осуществить кристаллизацию. Фильтрование в вакууме дало частично очищенный продукт указанного в заголовке соединения (9,25 г, 92,5%) в виде белого кристалла.
Т. пл.: 194-197°С (метанол - изопропанол).
1H-ЯМР (CDCl3+ДМСО-d6): 0,94 (3Н, т, 13-СН2СН3), 1,73 (3Н, с, 8-СН3), 3,05 (3Н, с, 12-ОСН3), 3,08 (1Н, дд, 4"-Н), 3,35 (3Н, с, 8"-ОСН3), 4,43 (1Н, д, 1'-Н), 4,96 (1Н, д, 1"-Н), 5,60-5,63 (1Н, дд, 13-Н), 6,78 (1Н, с, 1/2 (=СН-СООН)2).
Пример 8: Полная очистка фумаратной формы [фумарат соединения формулы (VII)].
Частично очищенный фумарат (10 г, 0,0123 моль), полученный в примере 7, растворяют в этилацетате (100 мл) при комнатной температуре. Затем добавляют по каплям воду (1,5 мл) и раствор перемешивают сначала при комнатной температуре в течение 1 часа, затем при 0°С в течение 1 часа и наконец при -10°С в течение 4 часов. Фильтрование в вакууме дало полностью очищенный продукт указанного в заголовке соединения (9,04 г, 90,4%) в виде белого кристалла.
Т. пл.: 199-200°С (1,5% вода - этилацетат).
1H-ЯМР (CDCl3+ДМСО-d6): 0,94 (3Н, т, 13-СН2СН3), 1,73 (3Н, с, 8-СН3), 3,05 (3Н, с, 12-ОСН3), 3,08 (1Н, дд, 4"-H), 3,35 (3Н, с, 8"-ОСН3), 4,43 (1Н, д, 1'-Н), 4,96 (1Н, д, 1"-Н), 5,60-5,63 (1Н, дд, 13-Н), 6,78 (1Н, с, 1/2 (=СН-СООН)2).
Пример 9: Синтез фумаратной формы [фумарат соединения формулы (VII)].
К толуолу (55 мл) добавляют соединение (9,5 г, 0,013 моль), полученное в примере 3, и твердый бикарбонат натрия (16,4 г, 0,195 моль). Затем добавляют по каплям бензилоксикарбонилхлорид (31,3 г, 0,183 моль) при 70°С или выше при перемешивании и смесь нагревают при той же самой температуре в течение 4 часов и затем оставляют стоять в течение ночи при комнатной температуре.
К жидкой реакционной смеси добавляют пиридин (6,94 г, 0,086 моль) с последующим перемешиванием в течение 30 минут. Затем добавляют насыщенный водный раствор бикарбоната натрия (66,5 мл), перемешивают смесь в течение 10 минут и добавляют этилацетат (66,5 мл). Полученную жидкую смесь перемешивают для отделения органического слоя, последовательно промывают водой и насыщенным рассолом (66,5 мл) и сушат безводным сульфатом натрия.
Всю смесь концентрируют в вакууме и остаток растворяют в метаноле (128 мл). К раствору добавляют 10% палладий на углероде (1,28 г) и смесь перемешивают при комнатной температуре в течение 1 часа в атмосфере водорода при давлении выше атмосферного (3-4 атмосферы). Затем отфильтровывают палладий на углероде и отгоняют в вакууме метанол. Остаток растворяют в этилацетате (120 мл) и промывают насыщенным водным раствором бикарбоната натрия (50 мл) для разделения фаз. Полученный органический слой промывают водой (50 мл × 2), а затем насыщенным рассолом (50 мл) и сушат безводным сульфатом натрия. Отфильтровав безводный сульфат натрия, концентрируют в вакууме этилацетат с получением дебензилоксикарбонилированной формы [соединение формулы (VIII) (R2: метил)] в виде масла.
Не очищая, растворяют дебензилоксикарбонилированную форму в диметилимидазолидиноне вместе с изопропилиодидом (20,0 г, 0,118 моль) и триэтиламином (13,2 г, 0,131 моль) и раствор нагревают при 70-75°С в течение 7-8 часов, после чего оставляют стоять в течение ночи. После экстрагирования и промывки этилацетатом (100 мл) и 2,5% водным аммиаком (50 мл) раствор промывают водой (50 мл × 2) и насыщенным рассолом (50 мл) и затем сушат безводным сульфатом натрия. Затем концентрируют в вакууме этилацетат, остаток и фумаровую кислоту (0,76 г, 0,0066 моль) растворяют в метаноле (25,0 мл). При перемешивании к раствору медленно добавляют по каплям изопропанол (75,0 мл) для кристаллизации. Раствор перемешивают сначала при комнатной температуре в течение 1 часа, затем при 0°С в течение 1 часа, и, наконец, при -15°С в течение 1 часа. Последующие фильтрование в вакууме и сушка дали указанное в заголовке соединение (6,5 г, 60,0%) в виде белого кристалла.
Т. пл.: 194-197°С (метанол - изопропанол).
1H-ЯМР (CDCl3+ДМСО-d6): 0,94 (3Н, т, 13-СН2СН3), 1,73 (3Н, с, 8-СН3), 3,05 (3Н, с, 12-ОСН3), 3,08 (1Н, дд, 4"-Н), 3,35 (3Н, с, 8"-OCH3), 4,43 (1Н, д, 1'-Н), 4,96 (1Н, д, 1"-Н), 5,60-5,63 (1Н, дд, 13-Н), 6,78 (1Н, с, 1/2 (=СН-СООН)2).
Пример 10: Испытание на стойкость кристаллов фумарата соединения формулы (VII).
Кристаллы фумарата соединения формулы (VII) испытывают на их стойкость, которая зависит от кристаллической формы.
Испытуемыми кристаллами были кристаллическая форма А, которую получали путем перекристаллизации фумарата соединения формулы (VII) из смеси метанол - изопропанол, и кристаллические формы С и D, которые получали путем перекристаллизации из этилацетата, используемого в качестве растворителя или отдельно или в смеси с водой.
Каждый из кристаллов точно взвешивают и подвергают ускоренному испытанию в термостатической камере, заполненной нагретым воздухом при 80°С. Пробы каждого кристалла извлекают из камеры с заданными интервалами времени и всю порцию растворяют в 50% ацетонитриле с получением концентрации приблизительно 1 мг/мл. К 2 мл раствора добавляют 2 мл раствора внутреннего стандарта (состоящего из 100 мкг циклогексилпарабензоата, растворенного в 2 мл 50% ацетонитрила), после чего общее количество смеси доводят до 10 мл и 100 мкл смеси подвергают ВЭЖХ (высокоэффективная жидкостная хроматография) при условиях, указанных ниже, и определяют процентное удерживание каждого кристалла из отношения площадей пиков пробы и внутреннего стандарта.
Условия для измерения путем ВЭЖХ
Результаты показаны на фиг.7. В условиях испытания удерживание кристаллической формы А падало до примерно 60% на 70-й день, тогда как удержание кристаллических форм С и D составляло примерно 80% даже на 70-й день.
Пример 11: Испытание на стойкость кристаллов фумарата соединения формулы (VII) в условиях влажности.
Кристаллы фумарата соединения формулы (VII) испытывают на их стойкость в условиях влажности, которая зависит от кристаллической формы. Метод и условия испытания были такими же, как в примере 10, за исключением того, что ускоренное испытание проводят в эксикаторе при 80°С, который конденсировали до относительной влажности 75% насыщенным водным раствором хлорида натрия.
Результаты показаны на фиг.8, где можно видеть, что кристаллические формы А и D были намного более стойкими в условиях влажности, чем кристаллическая форма С.
Результаты двух описанных выше испытаний показывают, что кристаллическая форма D является более стойкой, чем другие формы.
Пример 12: Получение кристаллической формы С фумарата.
Кристаллическую форму А фумарата общей формулы (VII) (200г) растворяли в этилацетате (1.5 л) и метаноле (150 мл) и смесь концентрировали досуха. После этого добавляли этилацетат (1.6 л) и смесь перемешивали при 25°С в течение 1 часа. Из части (820 мл) полученной суспензии собирали твердое вещество фильтрацией и сушили при 50°С в течение 4 часов с получением кристаллической формы С фумарата общей формулы (VII) (58,9 г).
Пример 13: Получение кристаллической формы С фумарата.
К кристаллической форме А (15,0 г, 18,4 мол) в 500-мл круглодонной колбе добавляли AcOEt (125 мл) при 25°С. После перемешивания в течение 2 часов при той же температуре кристаллическое вещество собирали фильтрованием с получением кристаллов С-формы в виде белых кристаллов (8,2 г, 54,7%).
1H-ЯМР (DMSO-d6): 0,84 (3Н, т, 13-СН2СН3), 1,68 (3Н, с, 8-СН3), 2,89 (1Н, дд, 4"-Н), 2,99 (3Н, с, 12-ОСН3), 3,22 (3Н, с, 8"-ОСН3), 4,41 (1Н, д, 1'-Н), 4,65 (1Н, д, 1"-Н), 5,39-5,42 (1Н, дд, 13-Н), 6,54 (2Н, с, (=СН-СООН)2).
Промышленная применимость изобретения.
Способ по настоящему изобретению имеет следующие преимущества: (1) очистку на каждой из стадий реакции, необходимую для получения очищенной формы конечного продукта, можно производить просто путем перекристаллизации и (2) ацетилирование гидроксильной группы в положении 2' эритромицина А, формилирование гидроксильной группы в положении 4" и реакцию образования полукеталя можно осуществлять в одном резервуаре и, кроме того, реакцию алкилирования гидроксильной группы в положении 12, а также реакцию удаления ацетильной группы в положении 2' и формильной группы в положении 4" также можно осуществлять в одном резервуаре. Таким образом, способ по настоящему изобретению может дать целевой продукт при меньшем числе стадий, чем известные способы, и, следовательно, даст значительные выгоды в коммерческом производстве.
Кроме того, способ по настоящему изобретению может дать кристаллическую форму фумарата соединения формулы (VII), которая имеет лучшее качество, такое как более высокая стойкость, чем кристалл, получаемый до сих пор, с точки зрения применения в качестве фармацевтического препарата или исходного материала для него.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения кларитромицина | 2000 |
|
RU2225413C1 |
| СПОСОБ ПОЛУЧЕНИЯ И ОЧИСТКИ ПРОИЗВОДНЫХ ЭРИТРОМИЦИНА | 2001 |
|
RU2248981C2 |
| СПОСОБ ПОЛУЧЕНИЯ КЛАРИТРОМИЦИНА В ВИДЕ КРИСТАЛЛОВ ФОРМЫ II | 2001 |
|
RU2230748C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФОРМОТЕРОЛА ИЛИ ЕГО ПРОИЗВОДНЫХ | 1991 |
|
RU2079486C1 |
| ПРОИЗВОДНОЕ ХИНОНА ИЛИ ЕГО ФАРМАКОЛОГИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1992 |
|
RU2049776C1 |
| СПОСОБ ПОЛУЧЕНИЯ 1-(3, 4-ДИХЛОРБЕНЗИЛ)-5-ОКТИЛБИГУАНИДА ИЛИ ЕГО СОЛИ | 2007 |
|
RU2440978C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ НА ИХ ОСНОВЕ | 1991 |
|
RU2057126C1 |
| РОДСТВЕННОЕ ВИТАМИНУ A СОЕДИНЕНИЕ И СПОСОБЫ ЕГО ПОЛУЧЕНИЯ | 1998 |
|
RU2188193C2 |
| СПОСОБ ПОЛУЧЕНИЯ СУЛЬФОНИЛАМИНОПИРИМИДИНОВЫХ СОЕДИНЕНИЙ (ВАРИАНТЫ), ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2002 |
|
RU2301801C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНОГО БЕНЗОКСАЗИНА, СПОСОБЫ ПОЛУЧЕНИЯ ЕГО ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ | 2000 |
|
RU2258069C2 |
Описываются кристаллические формы А, С и D фумарата производного эритромицина формулы
которые получают путем перекристаллизации неочищенного кристалла фумарата из спиртового растворителя (форма А) и дополнительно из этилацетата (форма С) или дополнительно из водного этилацетата (форма D), способы получения промежуточных соединений. Полученные кристаллические формы имеют лучшее качество, в частности высокую стойкость, что важно при получении фармацевтических препаратов. 11 н. и 5 з.п. ф-лы, 8 ил.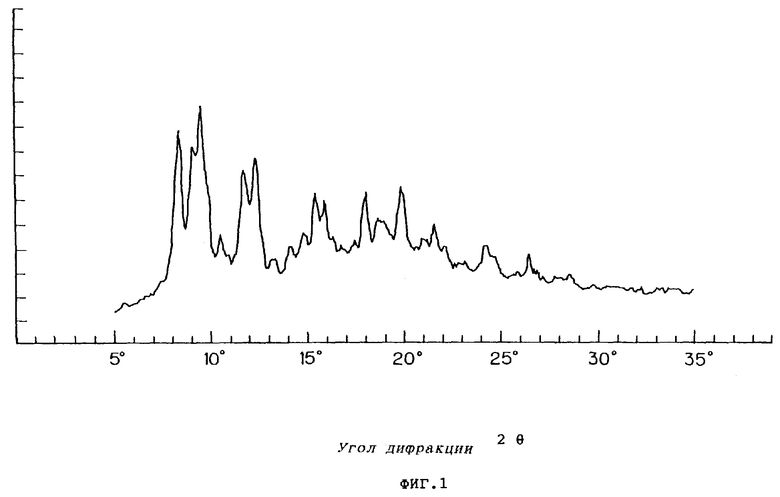
в котором молярное отношение соединения общей формулы (VII) к фумаровой кислоте составляет 2:1, включающий стадию перекристаллизации неочищенного кристалла фумарата соединения общей формулы (VII) из спиртового растворителя с последующей перекристаллизацией из водного этилацетата.
где R1 представляет низшую алкильную группу и R2 представляет низшую алкильную группу, из эритромицина А формулы (I)
включающий стадии ацетилирования гидроксильной группы в положении 2' эритромицина А,
формилирования гидроксильной группы в положении 4" и осуществления реакции образования полукеталя с получением соединения формулы (III):
окисления гидроксильной группы в положении 11 соединения (III) с получением соединения формулы (IV):
алкилирования гидроксильной группы в положении 12 соединения (IV), удаления ацетильной группы в положении 2' и формильной группы в положении 4" с получением соединения общей формулы (V):
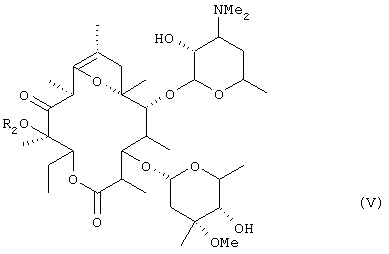
где R2 представляет низшую алкильную группу,
взаимодействия соединения (V) с бензилоксикарбонилхлоридом при основных условиях, затем удаления введенной бензилоксикарбонильной группы и алкилирования атома азота в положении 3' при условии, что когда R1 представляет метил, указанный способ не включает стадии взаимодействия соединения (V) с бензилоксикарбонилхлоридом при основных условиях, удаления введенной бензилоксикарбонильной группы и алкилирования атома азота в положении 3'.
где R1 представляет низшую алкильную группу и R2 представляет низшую алкильную группу,
из эритромицина А, включающий все стадии способа по п.2, и стадию превращения соединения, полученного способом по п.2, в фумарат.
из эритромицина А, включающий все стадии способа по п.2, и стадию превращения соединения, полученного способом по п.2, в кристаллическую форму А фумарата путем перекристаллизации из спиртового растворителя.
из эритромицина А формулы (I)
включающий следующие реакции:
a) ацетилирование гидроксильной группы в положении 2',
b) формилирование гидроксильной группы в положении 4", и
c) образование полукеталя,
где ацетилирование гидроксильной группы в положении 2', формилирование гидроксильной группы в положении 4" и реакцию образования полукеталя осуществляют в одном резервуаре.
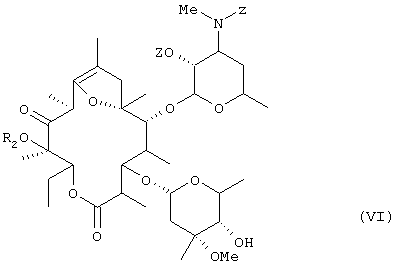
где R2 представляет низшую алкильную группу и Z представляет бензилоксикарбонильную группу, отличающийся тем, что соединение общей формулы (V)
где R2 имеет значение, определенное выше, подвергают взаимодействию с бензилоксикарбонилхлоридом в основных условиях.
в котором молярное отношение соединения общей формулы (VII) к фумаровой кислоте составляет 1:1, и которая может быть получена перекристаллизацией неочищенного кристалла фумарата соединения общей формулы (VII) из спиртового растворителя с последующей перекристаллизацией из этилацетата.
в котором молярное отношение соединения общей формулы (VII) к фумаровой кислоте составляет 2:1 и которая может быть получена путем перекристаллизации неочищенного кристалла фумарата общей формулы (VII) из спиртового растворителя с последующей перекристаллизацией из водного этилацетата.
где R1 представляет низшую алкильную группу и R2 представляет низшую алкильную группу,
путем перекристаллизации неочищенного кристалла фумарата соединения общей формулы (II) из спиртового растворителя с последующей перекристаллизацией из водного этилацетата.
характеризующаяся 2θ пиками порошковой рентгенограммы при 8,3, 12,8 и 18,5°.
характеризующаяся 2θ пиками порошковой рентгенограммы при 7,1 и 14,2°.
| СПОСОБ ПОЛУЧЕНИЯ КРИСТАЛЛИЧЕСКОГО 9-ДЕОКСО-11-ДЕОКСИ- 9,11-{ИМИНО[2- (2-МЕТОКСИЭТОКСИ)- ЭТИЛИДЕН]ОКСИ} -(9S)-ЭРИТРОМИЦИНА | 1992 |
|
RU2045534C1 |
| Способ борьбы с насекомыми и клещами | 1975 |
|
SU643068A3 |
| Способ получения эритромицина аскорбината | 1963 |
|
SU249562A1 |
| 0 |
|
SU158467A1 | |
| WO 9805674 А1, 12.02.1998 | |||
| Jones P.H., Perum TJ | |||
| J | |||
| Med | |||
| Chem | |||
| Контрольный висячий замок в разъемном футляре | 1922 |
|
SU1972A1 |

