Область техники
Изобретение относится к способу получения кларитромицина в виде кристаллов формы II, а также к новым промежуточным соединениям, используемым в указанном способе.
Уровень техники
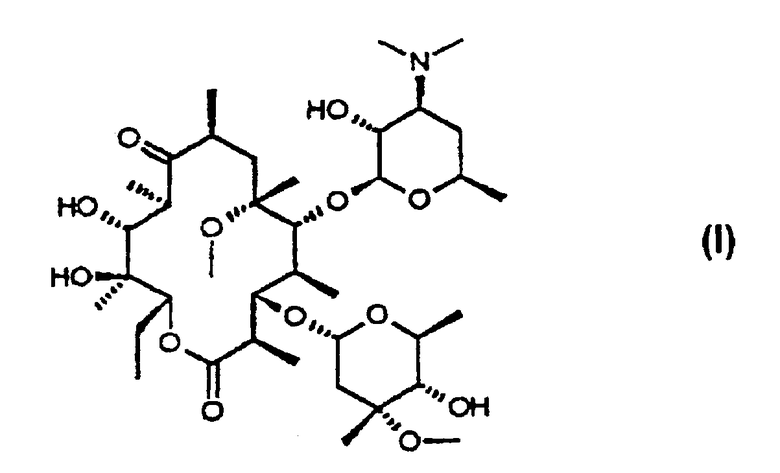
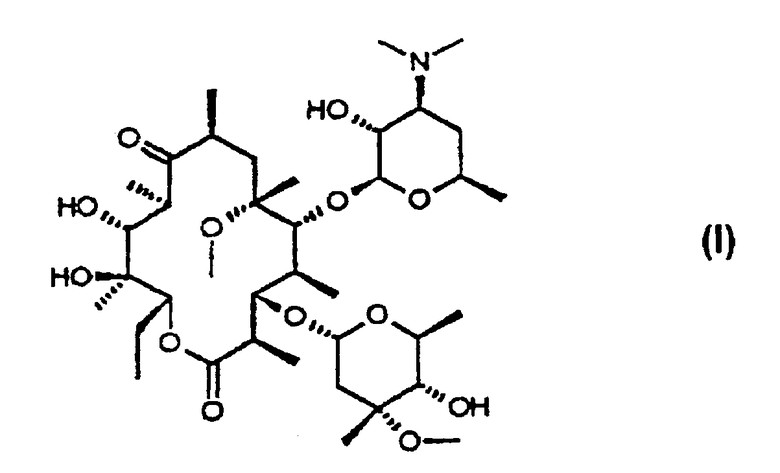
Кларитромицин, 6-О-метилэритромицин А, представляет собой полусинтетический макролидный антибиотик формулы (I), который проявляет сильную антибактериальную активность в отношении широкого спектра бактерий, включая грам-положительные бактерии, некоторые грам-отрицательные бактерии, анаэробные бактерии, Mycoplasma, Chlamidia и Helicobacter pylori; и благодаря своей высокой стабильности в кислой среде желудка он может быть введен перорально для лечения многих инфекционных заболеваний, а также для предотвращения рецидива язвенных заболеваний при использовании в сочетании с другими лекарственными средствами
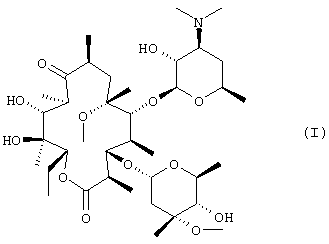
Сообщалось, что кларитромицин существует, по меньшей мере, в трех различных кристаллических формах, “форма О”, “форма I” и “форма II” (опубликованные международные заявки WO 98/04573 и WO 98/31699). Эти кристаллические формы могут быть идентифицированы с помощью инфракрасной спектроскопии, дифференциальной сканирующей калориметрии и порошковой дифракционной спектроскопии рентгеновских лучей. Форма II, которая является термодинамически более устойчивой, чем форма I, используется в лекарственных препаратах, имеющихся в настоящее время на рынке.
Сообщалось о различных способах получения кларитромицина, например, в патентах ЕР 0147062, ЕР 0158467, ЕР 195960 и ЕР 260938, и в патентах США 4990602, 5837829, 5929219, 5892008, 5864023 и 5852180. В соответствии с наиболее широко применяемыми способами, которые описаны далее, в качестве промежуточного соединения используют 9-оксимпроизводные эритромицина А.
Способ 1), описанный в патенте ЕР 0158467, включает следующие стадии: защита оксимной гидроксигруппы, а также 2’-гидроксигруппы и 3’-диметиламиногруппы 9-оксима эритромицина А бензильной группой и бензилоксикарбонильными группами, соответственно; метилирование 6-гидроксигруппы; и удаление защитных групп и оксимной группы с получением кларитромицина. Однако этот способ требует использования избыточного количества едкого и токсичного бензилоксикарбонилхлорида и непригоден для коммерческого использования ввиду необходимости проведения стадий гидрогенолиза, которые сложно осуществить в промышленных масштабах.
Способ 2), описанный в патенте ЕР 0195960, включает следующие стадии: защита оксимной гидроксигруппы, 2’-гидроксигруппы и 3’-диметиламиногруппы 9-оксима эритромицина А бензильными группами; метилирование 6-гидроксигруппы; и удаление защитных групп и оксимной группы с получением кларитромицина. Однако этот способ осложнен проблемами, возникающими при удалении защитной группы.
Способ 3), описанный в патенте ЕР 0260938, включает следующие стадии: защита оксимной гидроксигруппы 9-оксима эритромицина А бензильной или замещенной бензильной группой; защита 2’и 4’’-гидроксигрупп силильными группами, метилирование 6-гидроксигруппы; и удаление защитных групп и оксимной группы с получением кларитромицина. Однако в соответствии с этим методом защита оксимных групп также удаляется посредством проведения реакции гидрогенолиза, которая непригодна для масштабного производства.
Далее способ 4), описанный в патенте США 5837829, включает следующие стадии: защита оксимной гидроксигруппы, и 2’и 4’’-гидроксигрупп 9-оксима эритромицина А силильными группами; метилирование 6-гидроксигруппы; и удаление защитных групп и оксимной группы с получением кларитромицина. Однако этот способ требует проведения стадии метилирования в исключительно безводных условиях вследствие неустойчивости 9-оксимсилильной группы по отношению к воде, а также сопряжен с трудностями обеспечения безопасности при работе с гидридом натрия.
В дополнение, способ 5), описанный в патенте США 4990602, включает следующие стадии: защита оксимной гидроксигруппы 9-оксима эритромицина А производным кеталя; защита 2’и 4’’-гидроксигрупп силилькыми группами; метилирование 6-гидроксигруппы; и удаление защитных групп и оксимной группы с получением кларитромицина. Хотя этот метод дает относительно высокий выход, равный 45-50%, и высокую селективность, равную 90%, на стадии метилирования, для его осуществления требуется использование большого избытка (от 2,3 до 10 эквивалентов) агента для зашиты оксимных групп.
Таким образом, известным из предшествующего уровня техники способам 1)-5) присущ ряд проблем, которые следует решить для того, чтобы разработать усовершенствованный способ получения кларитромицина. Кроме того, продукт - кларитромицин, полученный указанными выше методами, не является кларитромицином фармацевтической степени чистоты, который должен представлять собой чистую кристаллическую форму II кларитромицина, а представляет собой кларитромицин, который не обладает необходимым для фармацевтического использования качеством в смысле чистоты и кристалличности. Соответственно, требуется дополнительная стадия очистки и специальная стадия кристаллизации для превращения кларитромицина, который не обладает необходимым для фармацевтического использования качеством, в чистый кларитромицин в виде кристаллов формы II, которая используется в настоящее время для получения лекарственных препаратов.
Сообщалось о нескольких способах получения кристаллов формы II из кларитромицина, который не имел необходимого для фармацевтического использования качества. Например, кристаллы формы 0 или формы I нагревают в вакууме при температуре от 70 до 110°С в течение длительного периода времени с получением при этом кристаллической формы II (см. опубликованные международные заявки WO 98/04573 и WO 98/31699), однако проблемой, присущей этому методу, является низкая производительность.
В качестве альтернативы кристаллы формы II могут быть получены посредством перекристаллизации кристаллов формы I из смеси хлороформ/простой изопропиловый эфир (см. Kerck Index 12th ed., pp.395) или посредством перекристаллизации кристаллов формы I из органического растворителя или смеси органического растворителя и воды с умеренным выходом (см. опубликованную международную заявку WO 98/04574). В соответствии с этими методами, поскольку превращение кристаллов формы I в кристаллы формы II не сопровождается повышением степени чистоты, кристаллы формы I, обладающие высокой степенью чистоты, должны быть получены заблаговременно из неочищенного кларитромицина за счет снижения выхода кларитромицина и повышения стоимости производства.
В соответствии с этим сохраняется необходимость в разработке способа получения с высоким выходом кларитромицина в виде кристаллов формы II, характеризующегося высокой чистотой.
Сущность изобретения
В соответствии с вышеизложенным первым объектом настоящего изобретения является разработка способа получения кристаллов кларитромицина в виде формы II, высокой чистоты и с высоким выходом.
Другим объектом изобретения являются новые промежуточные соединения, полученные в соответствии с указанным способом.
В соответствии с одним из аспектов изобретения предложен способ получения кларитромицина в виде кристаллов формы II (формула I), включающий следующие стадии:
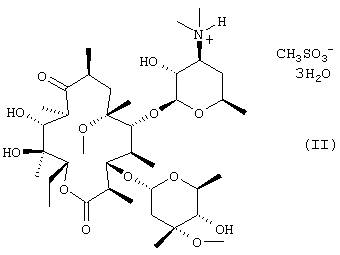
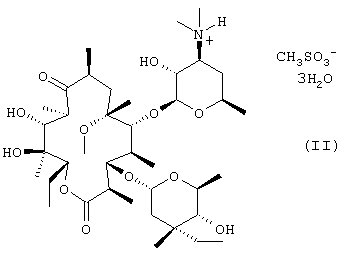
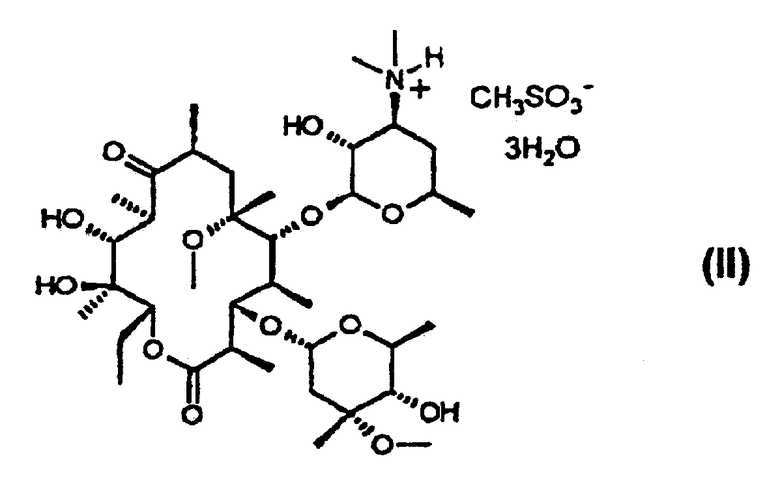
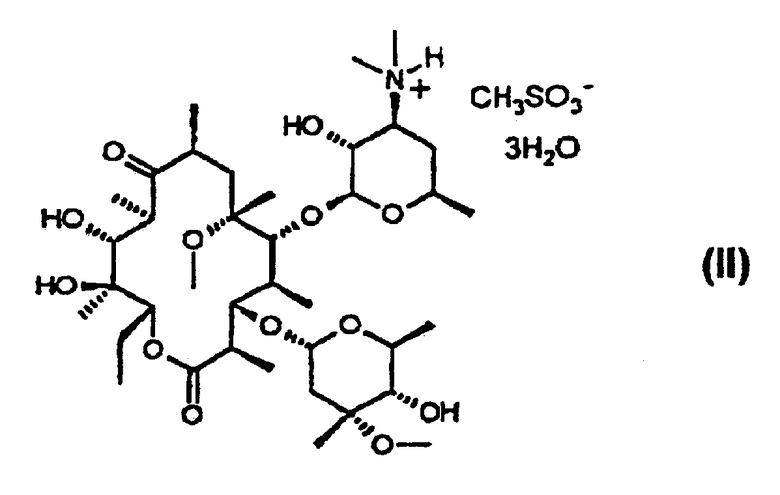
(a) обработку кларитромицина, не обладающего фармацевтической степенью чистоты, метансульфоновой кислотой в смеси смешивающегося с водой органического растворителя и воды с получением при этом кристаллического тригидрата мезилата кларитромицина формулы (II); и
(b) нейтрализацию кристаллического тригидрата мезилата кларитромицина, полученного на стадии (а), водным раствором аммиака в смеси смешивающегося с водой органического растворителя и воды, где термин “кларитромицин, не обладающий фармацевтической степенью чистоты”, относится к кларитромицину любой чистоты или любой степени кристалличности, включая неочищенный продукт, полученный согласно следующему способу получения кларитромицина:
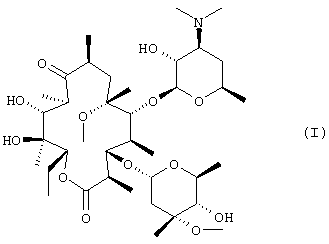
В соответствии с другим аспектом изобретения предложен усовершенствованный способ получения кларитромицина, не обладающего фармацевтической степенью чистоты, включающий следующие стадии:
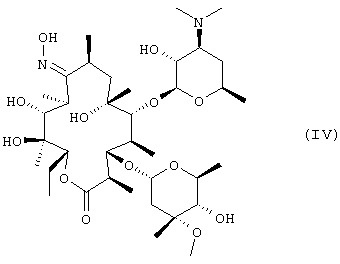
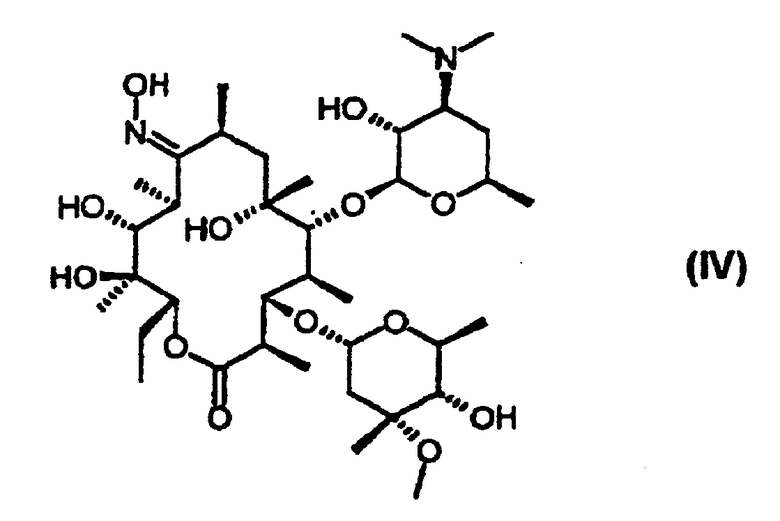
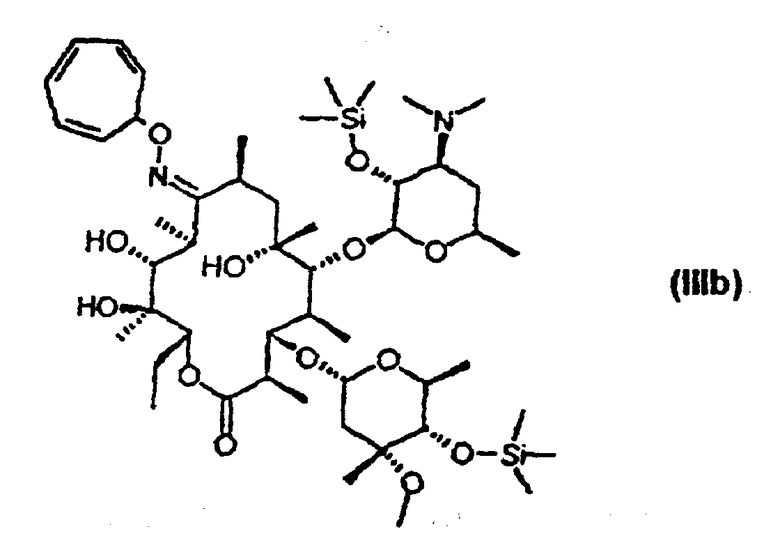
защиту 9-оксимной гидроксигруппы 9-оксима эритромицина А формулы (IV) или его соли тропильной группой и 2’- и 4’’-гидроксигрупп триметилсилильными группами с получением 9-O-тропилоксима 2’, 4’’-O-бис(триметилсилил)эритромицина А формулы (IIIb);
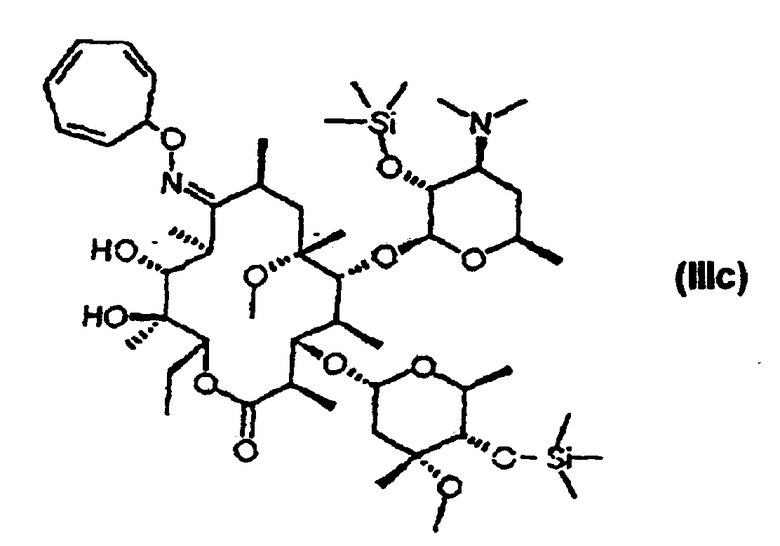
взаимодействие 9-O-тропилоксима 2’,4’’-O-бис(триметилсилил)эритромицина А с метилирующим агентом с получением 9-O-тропилоксима 2’, 4’’-O-бис(триметилсилил)-6-O-метил-эритромицина А формулы (IIIс); и удаление защитных групп и оксимной группы 9-O-тропилоксима 2’,4’’-O-бис(триметилсилил)-6-O-метилэритромицина А:
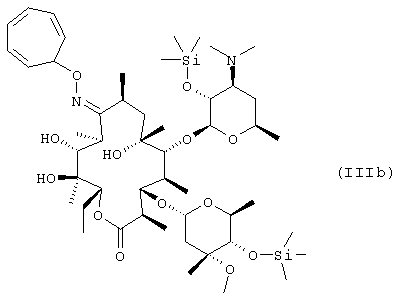
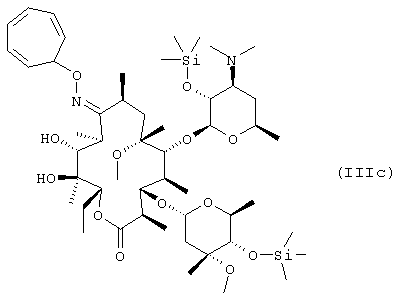
В соответствии с еще одним аспектом изобретения предложены кристаллический тригидрат мезилата кларитромицина формулы (II); и 9-O-тропилоксимное производное эритромицина А формулы (III):
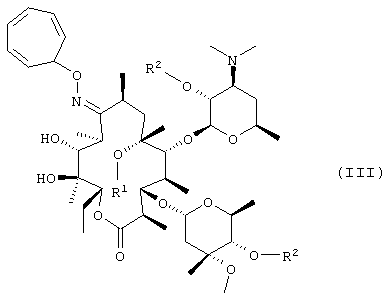
в которой
R1 представляет собой водород или метильную группу; и
R2 представляет собой водород или триметилсилильную группу (если R1 представляет собой метильную группу, то R" представляет собой триметилсилильную группу).
Краткое описание чертежей
Указанные выше и иные объекты и признаки изобретения будут ясны из приведенного далее описания изобретения, вместе со следующими сопровождающими чертежами, где:
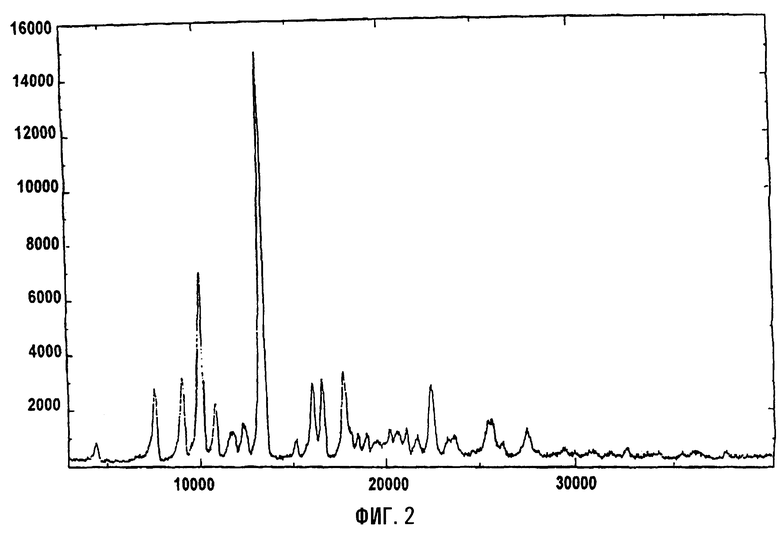
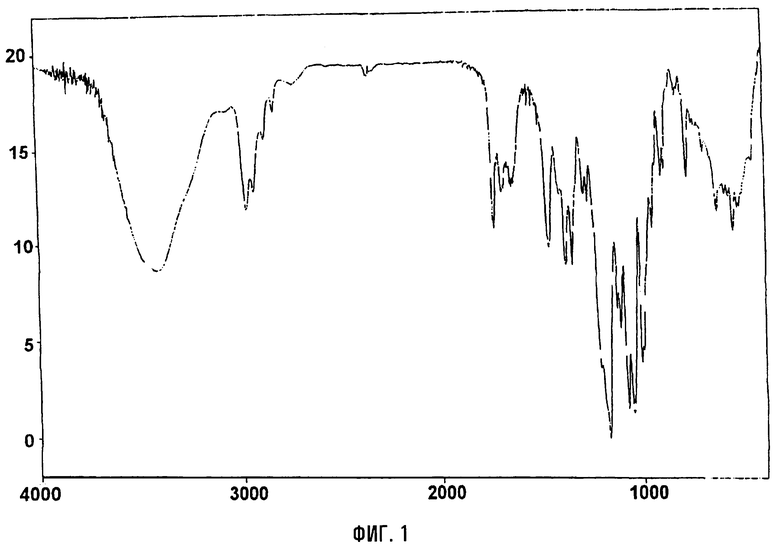
На фигурах 1 и 2 представлены инфракрасный спектр и спектр порошковой дифракционной спектроскопии рентгеновских лучей тригидрата кларитромицинмезилата, соответственно.
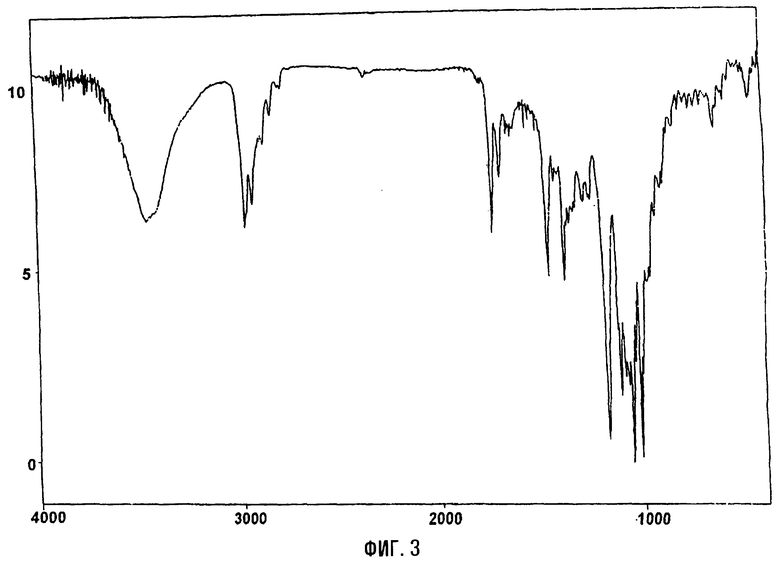
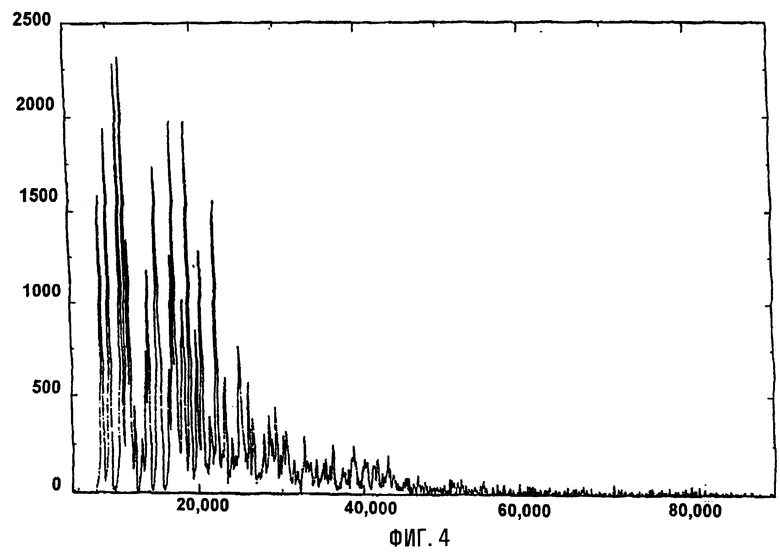
На фигурах 3 и 4 представлены инфракрасный спектр и спектр порошковой дифракционной спектроскопии рентгеновских лучей кларитромицина в виде кристаллов формы II, соответственно.
Подробное описание изобретения
Термин “кларитромицин, не обладающий фармацевтической степенью чистоты”, в том смысле, в котором он используется в описании, относится к кларитромицину любой чистоты или любой степени кристалличности, и к кларитромицину в неочищенном состоянии, полученному в соответствии со способом его получения.
Согласно изобретению кристаллический тригидрат мезилата кларитромицина формулы (II) получают посредством обработки кларитрсмицина, не обладающего фармацевтической степенью чистоты, полученного с использованием метансульфоновой кислоты в смеси смешивающегося с водой органического растворителя и воды.
Конкретно, сырье - кларитромицин растворяют или суспендируют в смеси смешивающегося с водой органического растворителя, например ацетона, этанола или изопропанола, и воды при температуре от комнатной до 45°С. Смесь растворителей содержит воду в количестве более чем 3 эквивалента, предпочтительно, от 3 до 15 эквивалентов, в расчете на используемый кларитромицин.
После этого метансульфоновую кислоту, неразбавленную или растворенную в той же самой смеси растворителей, добавляют к суспензии в количестве от 0,9 до 1,1 эквивалента, в расчете на количество кларитромицина. Смесь может быть выдержана при температуре от комнатной до 45°С, в течение от 30 минут до 3 часов. Полученную смесь охлаждают до температуры от 0°С до 5°С, и перемешивают в течение от 1 до 5 часов. В заключение, образовавшиеся кристаллы отфильтровывают, промывают той же самой смесью растворителей и высушивают при температуре от комнатной до 45°С, получая при этом кристаллический тригидрат мезилата кларитромицина.
В случае необходимости, полученные таким образом кристаллы тригидрата мезилата кларитромицина могут быть дополнительно перекристаллизованы из той же самой смеси растворителей обычным образом.
Тригидрат мезилата кларитромицина, полученный как указано выше, нейтрализуют водным раствором аммиака в смеси смешивающегося с водой органического растворителя и воды, и кларитромицин виде кристаллов формы II может быть подвергнут перекристаллизации.
В частности, тригидрат мезилата кларитромицина растворяют в смеси смешивающегося с водой органического растворителя и воды при комнатной температуре. Затем раствор отфильтровывают для того, чтобы удалить примеси, и фильтрат нейтрализуют до значения рН от 9 до 12 посредством добавления водного раствора аммиака. Полученный раствор перемешивают в течение 30 минут или более для того, чтобы вызвать осаждение кристаллов. В заключение выпавшие кристаллы отфильтровывают, промывают той же самой смесью растворителей и высушивают при температуре от комнатной до 60°С, получая кларитромицин в виде кристаллов формы II.
Смешивающийся с водой органический растворитель, который может быть использован в соответствии с указанным выше способом, представляет собой ацетон, этанол, изопропанол или их смесь.
Вода и смешивающийся с водой органический растворитель могут быть смешаны в объемном соотношении от 30:70 до 70:30.
Способ, являющийся предметом изобретения, очень прост и позволяет получить кларитромицин в виде кристаллов формы II с высоким выходом при незначительных затратах на производство.
Изобретение относится также к усовершенствованному способу получения кларитромицина, не обладающего фармацевтической степенью чистоты, который является высокоэффективным, характеризующийся высоким способом получения кларитромицина в виде кристаллов формы II, если его объединить с указанным выше способом получения кларитромицина в виде кристаллов формы II.
Первая стадия этого усовершенствованного способа получения кларитромицина, не обладающего фармацевтической степенью чистоты, может быть осуществлена таким образом, как показано на схеме 1:
Схема 1
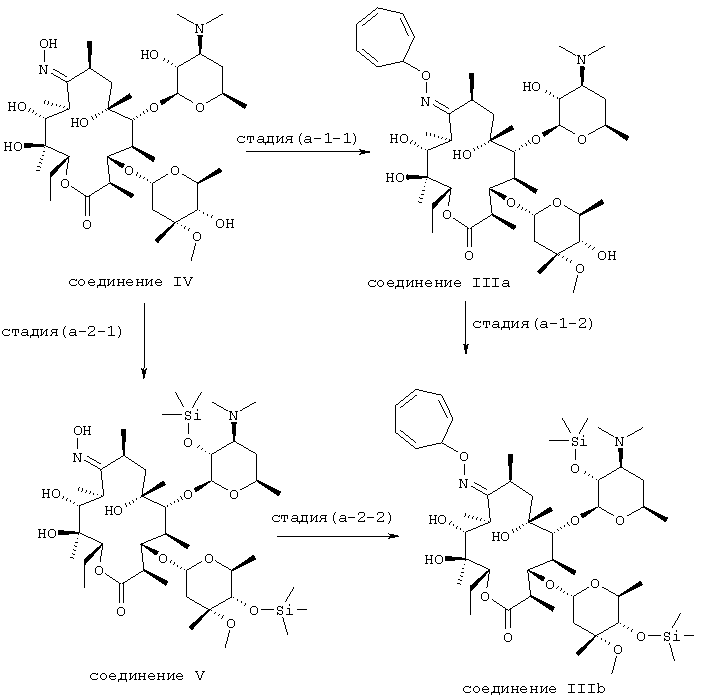
А именно, 9-O-тропилоксим эритрсмицина А формулы (IIIa) может быть получен взаимодействием 9-оксима эритромицина А формулы (IV) с тетрафторборатом тропилия в апротонном полярном растворителе в присутствии основания при температуре от 0 до 60°С.
Тетрафторборат тропилия может быть использован в количестве от 1 до 1,3 эквивалентов в расчете на 9-оксим эритромицина А формулы (IV).
Примерами апротонного полярного растворителя, который подходящим образом может быть использован для проведения вышеуказанной реакции, являются тетрагидрофуран, 1,4-диоксан, этилацетат, ацетонитрил, N,N-диметилформамид, дихлорметан или их смесь, а основание может быть выбрано из группы, включающей третичный амин, например триэтиламкн, трипропиламин, диэтилизопропиламин, трибутиламин, 1,8-диазабицикло[5.4.0]ундец-7-ен, 1,5-диазабицикло[4.3.0]нон-5-ен и 1,4-диазабицикло[2.2.2]октан; карбонат натрия; карбонат калия; гидроксид натрия; гидроксид калия, трет-бутоксид калия; и гидрид натрия. Основание может быть использовано в количестве от 1 до 1,5 эквивалентов в расчете на количество 9-оксима эритромицина А формулы (IV).
Затем, 9-O-тропилоксим 2’, 4’’-O-бис(тркметилсилил)эритромицина А формулы (IIIb) получают взаимодействием 9-O-тропилоксима эритромицина А формулы (IIIa), полученного, как указано выше, с хлоридом аммония и 1,1,1,3,3,3-гексаметилдисилазаном в органическом растворителе, например, таком как N,N-диметилформамид или ацетонитрил, при температуре от комнатной до 50°С. Количество используемых хлорида аммония и 1,1,1,3,3,3-гексаметилдисилазана находится от 0,5 до 1,5 эквивалентов и от 2 до 4 эквивалентов, соответственно, в расчете на количество 9-O-тропилоксима эритромицина А формулы (IIIa).
В качестве альтернативы соединение IIIb может быть получено следующим образом.
9-оксим 2’,4’’-O-бис(триметилсилил)эритромицина А формулы (V) получают взаимодействием 9-оксима эритромицина А формулы (IV) с хлоридом аммония и 1,1,1,3,3,3-гексаметилдисилазаном в органической кислоте, например, такой как N,N-диметилформамид, при температуре от комнатной до 50°С. Количество используемых хлорида аммония и 1,1,1,3,3,3-гексаметилдисилазана составляет от 0,5 до 1,5 эквивалентов и от 2 до 4 эквивалентов, соответственно, в расчете на 9-оксим эритромицина А формулы (IV).
Затем 9-O-тропилоксим 2’,4’’-O-бис(триметилсилил)-эритромицина А формулы (IIIb) получают взаимодействием 9-оксима 2’и 4’’-бис(триметилсилил)эритромицина А формулы (V), полученного выше, с тетрафторборатом тропилия в апротонном полярном органическом растворителе в присутствии основания при температуре от 0 до 60°С. Условия реакции, а также растворитель и основание, используемые для проведения этой реакции, могут быть точно такими же, как и при получении соединения формулы (IIIa).
На второй стадии 9-O-тропилоксим 2’,4’’-O-бис(триметилсилил) эритромицина А формулы (IIIb), полученного на первой стадии, метилируют с использованием мотивирующего агента, например, метилиодида в среде растворителя в присутствии основания с получением 9-O-тропилоксим 2’,4’’-O-бис(триметилсилил) -6-O-метилэритромицина А формулы (IIIс).
Метилиодид может быть использован в количестве от 1 до 1,5 эквивалентов, в расчете на количество 9-O-тропилоксима 2’,4’’-O-бис(триметилсилил)эритромицина А формулы (IIIb).
Примером растворителя, который подходящим образом может быть использован в вышеуказанной реакции, является смесь тетрагидрофурана и диметилсульфоксида в объемном соотношении от 2:1 до 1:2.
Примеры оснований, которые подходящим образом могут быть использованы в вышеуказанной реакции, включают гидроксид калия; гидрид калия; трет-бутоксид калия; гидрид натрия и их смесь. Основание может быть использовано в количестве от 1 до 1,3 эквивалентов в расчете на 9-O-тропилоксим 2’,4’’-O-бис(триметилсилил)эритромицина А формулы (IIIb). В том случае, когда в качестве основания используют гидроксид калия, предпочтительным является порошок гидроксида калия с размером частиц 600 мкм.
В заключение, 9-O-тропилоксим 2’,4’’-O-бис(триметилсилил)-6-O-метилэритромицина А формулы (IIIс), полученный на предыдущей стадии, обрабатывают муравьиной кислотой и бисульфитом натрия в водно-спиртовом растворителе для того, чтобы удалить защитные группы и оксимную группу 9-O-тропилоксима 2’,4’’-O-бис(триметилсилил)-6-O-метилэритромицина А формулы (IIIc), при температуре от комнатной до температуры кипения используемого растворителя, с получением при этом кларитромицина.
Количество используемых муравьиной кислоты и бисульфита натрия составляет от 1 до 2 эквивалентов и от 2 до 5 эквивалентов, соответственно, в расчете на количество 9-O-тропилоксима 2’,4’’-O-бис(триметилсилил)-6-O-метилэритромицина А формулы (IIIc).
Водно-спиртовый растворитель, который может быть подходящим образом использован в соответствии с приведенной выше реакцией, представляет собой смесь спирта, например, такого как метанол, этанол и изопропанол, и воды в объемном соотношении от 2:1 до 1:2.
Описанный выше способ получения кларитромицина, который не обладает необходимым для фармацевтического использования качеством, является значительно более простым и характеризуется высоким выходом чистого продукта, по сравнению со способами, известными из предшествующего уровня техники.
Следующие ссылочные примеры и примеры предназначены для дополнительной иллюстрации изобретения, не ограничивая его объем; и экспериментальные методы, используемые в соответствии с изобретением, могут быть осуществлены в соответствии со ссылочным примером и примерами, приведенными ниже в описании, если не указывается иное.
Кроме того, проценты, приведенные для твердого вещества в смеси твердых веществ, жидкого вещества в жидкости и для твердого вещества в жидкости приведены для соотношений мас./мас., об./об. и мас./об., соответственно, если специально не указывается иное.
Ссылочный пример: получение 9-оксима эритромицина А
31,9 г Эритромицина А растворяют в 50 мл метанола. Добавляют 15,1 г гидроксиламина-НСl и 15,1 мл триэтиламина и кипятят с обратным холодильником в течение 24 часов. Полученный раствор охлаждают до температуры ниже 5°С, и перемешивают в течение 2 часов. Образовавшиеся кристаллы отфильтровывают, промывают холодным метанолом и высушивают, получая при этом 32,8 г 9-оксима-НСl эритромицина А с выходом 96%.
Полученный, как указано выше, 9-оксим-НСl эритромицина А суспендируют в 100 мл метанола и добавляют 20 мл концентрированного водного раствора аммиака. Полученный раствор перемешивают при температуре от 0 до 5°С в течение нескольких часов. После этого образовавшееся твердое вещество отфильтровывают, промывают водой и высушивают, получая при этом 26,0 г указанного в заглавии соединения с выходом 80%.
Пример 1: получение 9-O-тропилоксима эритромицина А [стадия (A-1-1)]
11,23 г 9-оксима эритромицина А, полученного в соответствии со ссылочным примером, растворяют в 75 мл N,N-диметилформамида. Добавляют 3,20 г тетрафторбората тропилия и 3,14 мл триэтиламина, полученную смесь перемешивают при температуре от 30 до 40°С, в течение 4 часов. Полученный раствор охлаждают до комнатной температуры и добавляют к нему 200 мл воды, после чего дважды экстрагируют, с использованием 100 мл этилацетата. Органические слои объединяют, дважды промывают водой, высушивают над сульфатом магния и концентрируют при пониженном давлении, получая при этом 12,6 г 9-O-тропилоксима эритромицина А в виде вспененного вещества с выходом, равным 100%, это вещество перекристаллизовывают, используя ацетонитрил, с получением при этом 10,95 г указанного в заглавии соединения в виде белого порошка, с выходом 87%.
Т.пл. 120-122°С
1H-ЯМР (CDCl3, ч/млн): δ 6,68 (м, 2Н, тропил 4’’’-Н и 5’’’-Н), 6,32 (м, 2Н, тропил 3’’’-Н и 6’’’-Н), 5,67 (м, 2Н, тропил 2’’’-Н и 7’’’-Н), 5,12 (дд, 1Н, 13-Н), 4,93 (д, 1Н, 1’’-Н), 4,56 (т, 1Н, тропил 1’’’-Н), 4,43 (д, 1Н, 1’-Н), 4,07 (дд, 1Н, 3-Н), 4,02 (дкв., 1Н, 5’’-Н), 3,67 (д, 1Н, 11-Н), 3,57 (д, 1Н, 5-Н), 3,50 (ддкв., 1Н, 5’-Н), 3,49 (дкв., 1Н, 10-К), 3,33 (с, 3Н, cladinose 3’’-ОСН3), 3,25 (дд, 1Н, 2’-Н), 3,04 (дд, 1Н, 4’’-Н), 2,92 (ддкв., 1Н, 8-Н), 2,65 (дкв., 1H, 2-Н), 2,45 (ддд, 1Н, 3’-Н), 2,38 (дд, 1Н, 2’’-Heq), 2,30 (д, 6Н, desosamine 3’-N (СН3)2), 2,23 (ддкв., 1H, 4-H), 2,03~1,45 (м, 6Н, 4’-Heq, 7-H2, 2’’-Нах и 14-Н2), 1,43 (с, 3Н, 18-Н), 1,32 (д, 3Н, 6’’-Н), 1,26 (с, 3Н, 7’’-Н), 1,21~1,02 (м, 19Н, 4’-Hax, 6’-Н3, 16-Н3, 20-Н3, 21-Н3, 17-Н3 и 19-Н3), 0,85 (т, 3Н, 15-Н3).
13С-ЯМР (СDСl3, ч/млн): δ 175,5 (С9), 172,9 (C1), 131,4 и 131,5 (тропил С4’’’ и С5’’’), 125,2 и 125,4 (тропил С3’’’ и С6’’’), 124,4 и 125,5 (тропил С2’’’ и С7’’’), 103,4 (С1’), 96,7 (C1’’), 83,6 (С5), 80,4 (С6), 78,5 (С3), 78,1 (тропил Cl’’), 77,3 (С4’’), 75,7 (С13), 74,7 (С12), 73,1 (С3’’), 71,4 (С2’), 71,0 (С11), 69,2 (С5’), 66,0 (С5’’), 65,9 (С3’), 49,9 (С8’’), 45,1(С2), 40,7 (С7’ и С8’), 39,4 (С4), 38,2 (С7), 35,5 (С2’’), 33,4 (С8), 29,1 (С4’), 27,3 (С10), 26,8 (С19), 21,9 (С6’), 21,8 (С7’’), 21,5 (С14), 19,1 (С18), 19,1 (6’‘), 16,6 (С21), 16,5 (С16), 14,9 (С20), 11,0 (С15), 9,5 (С17).
Пример 2: получение 9-O-тропилоксима эритромицина А [стадия (а-1-1)]
Повторяют процедуру, описанную в примере 1, с тем исключением, что вместо триэтиламина используют 3,11 г карбоната калия, получая при этом 10,7 г указанного в заглавии соединения в виде белого порошка с выходом 85%.
Физико-химические характеристики и данные ЯМР-спектроскопии для этого продукта идентичны характеристикам и данным, полученным для соединения, синтезированного в соответствии с примером 1.
Пример 3: получение 9-O-тропилоксима эритромицина А [стадия (а-1-1)]
11,23 г 9-оксима эритромицина А, полученного в соответствии со ссылочным примером, растворяют в 75 мл N,N-диметилформамида и охлаждают до 0°С. Добавляют 2,19 г трет-бутоксида калия и полученную смесь перемешивают в течение 15 минут. После этого туда же добавляют 3,29 г тетрафторбората тропилия и перемешивают при температуре от 0 до 5°С в течение 2 часов, затем повторяют процедуру, описанную в примере 1, с получением 11,33 г указанного в заглавии соединения в виде белого порошка с выходом 90%.
Физико-химические характеристики и данные ЯМР-спектроскопии для этого продукта идентичны характеристикам и данным, полученным для соединения, синтезированного в соответствии с примером 1.
Пример 4: получение 9-O-тропилоксима эритромицина А [стадия (а-1-1)]
Повторяют процедуру, описанную в примере 3, с тем исключением, что вместо N,N-диметилформамида используют тетрагидрофуран, а взаимодействие осуществляют в течение 3 часов, получая при этом 11,46 г указанного в заглавии соединения в виде белого порошка с выходом 91%.
Физико-химические характеристики и данные ЯМР-спектроскопии для этого продукта идентичны характеристикам и данным, полученным для соединения, синтезированного согласно примеру 1.
Пример 5: получение 9-O-тропилоксима эритромицина А [стадия (а-1-1)]
11,23 г 9-оксима эритромицина А, полученного в соответствии со ссылочным примером, растворяют в 120 мл ацетонитрила. Добавляют 3,20 г тетрафторбората тропилия, затем добавляют по каплям 3,14 мл триэтиламина, при повышении температуры от 30 до 40°С. Полученный раствор перемешивают при той же самой температуре в течение 4 часов и охлаждают до 0°С, после чего перемешивают в течение 1 часа. Образованное твердое вещество отфильтровывают, промывают холодным ацетонитрилом и сушат с получением 11,83 г указанного в заголовке соединения в виде белого порошка с выходом 94%.
Физико-химические характеристики и данные ЯМР-спектроскопии для этого продукта идентичны характеристикам и данным, полученным для соединения, синтезированного согласно примеру 1.
Пример 6: получение 9-O-тропилоксима 2’,4’’-O-бис(триметилсилил)эритромицина А [стадия (а-1-2)]
12,59 г 9-O-тропилоксима эритромицина А, полученного в соответствии с примером 1, растворяют в 75 мл N,N-диметилформамида. Добавляют 1,20 г хлорида аммония и 6,3 мл 1,1,1,3,3,3-гексаметилдисилазана, затем перемешивают смесь при температуре, изменяющейся от 30 до 40°С, в течение 4 часов. Полученный раствор охлаждают до комнатной температуры и добавляют к нему 200 мл воды, затем дважды экстрагируют, используя 100 мл этилацетата. Органические слои объединяют и дважды промывают водой, высушивают над сульфатом магния и концентрируют при пониженном давлении, получая при этом 14,5 г 9-O-тропилоксима 2’,4’’-O-бис(триметилсилил)эритромицина А в виде вспененного вещества с выходом 98%.
1Н-ЯМР (СDСl3, ч/млн): δ 6,68 (м, 2Н, тропил 4’’’-Н и 5’’’-Н), 6,30 (м, 2Н, тропил 3’’’-Н и 6’’’-Н), 5,66 (м, 2Н, тропил 2’’’-Н и 7’’’-Н), 5,10 (дд, 1Н, 13-Н), 4,90 (д, 1Н, 1’’-Н), 4,53 (т, 1Н, тропил 1’’-Н), 4,40 (д, 1Н, 1’-Н), 4,25 (дкв., 1Н, 5’’-Н), 4,19 (дд, 1Н, 3-Н), 3,69 (д, 1Н, 11-Н), 3,66 (ддкв., 1Н, 5’-Н), 3,62 (дкв., 1Н, 10-Н), 3,58 (д, 1Н, 5-Н), 3,32 (с, 3Н, cladinose 3’’-ОСН3), 3,18 (дд, 1Н, 2’-Н), 3,16 (дд, 1Н, 4’’-Н), 2,85 (ддкв., 1Н, 8-Н), 2,70 (дкв., 1Н, 2-Н), 2,55 (ддд, 1Н, 3’-Н), 2,39 (дд, 1Н, 2’’-Heq), 2,25 (д, 6Н, desosamine 3‘-N (СН3)2), 2,00-1,40 (м, 7Н, 4-Н, 4’-Heq, 7-Н2, 2’’-Н,х и 14-Н2), 1,42 (с, 3Н, 18-Н), 1,21 (д, 3Н, 6’’-Н), 1,18 (с, 3Н, 7’’-Н), 1,25-0,99 (м, 19Н, 4’-Нах, 6’-Н3, 16-Н3, 20-Н3, 21-Н3, 17-Н3 и 19-Н3), 0,87 (т, 3Н, 15-Н3) 0,16 (с, 9Н, 4’’-OSi(СН3)3), 0,11 (с, 9Н, 2’-OSi(СН3)3).
13С-ЯМР (CDCl3, ч/млн): δ 176,1 (С9), 172,6 (Cl), 131,6 и 131,3 (тропил 04’’’ и С5’’’), 125,2 и 125,0 (тропил С3’’’ и С6’’’), 124,7 и 124,7 (тропил С2’’’ и С7’’’), 103,1 (Cl’), 97,0 (Cl’’), 81,8 (С5), 81,3 (С6), 79,8 (С3), 78,2 (и Cl’’’), 77,3 (С4’’), 75,9 (С13), 74,7 (С12), 73,7 (С3’’), 73,6 (С2’), 71,0 (С11), 68,1(С5’), 65,5 (С5’’), 65,3 (С3’), 50,1 (С8’’), 45,1 (С2), 41,4 (С7’ и С8’), 40,3 (С4), 38,9 (С7), 36,3 (С2’’), 33,5 (С8), 30,1 (С4’), 27,4 (С10), 26,8 (С19), 22,6 (С6’), 22,2 (С7’’), 21,6 (С14), 19,8 (С18), 19,0 (6’’), 16,6 (С21), 16,2 (С16), 14,8 (С20), 11,1 (С15), 10,0 (С17), 1,41 (4’’-OSi(СН3)3), 1,31 (2’-OSi(СH3)3).
Пример 7: получение 9-O-тропилсксима 2’,4’’-O-бис-(триметилсилил)эритромицина А из 9-оксима эритромицина А [стадия (а-1-1) и (а-1-2)]
11,23 г 9-оксима эритромицина А, полученного в соответствии со ссылочным примером, растворяют в 75 мл N,N-диметилформамида и охлаждают до 0°С. Добавляют 2,19 г трет-бутоксида калия и полученную смесь перемешивают в течение 15 минут. После этого туда же добавляют 3,20 г тетрафторбората тропилия и перемешивают при температуре от 0 до 5°С в течение 3 часов. После этого добавляют 1,34 г хлорида аммония и 10,0 мл 1,1,1,3,3,3-гексаметилдисилазана, перемешивают смесь при температуре, изменяющейся от 35 до 40°С в течение 4 часов. Полученный раствор охлаждают до комнатной температуры и добавляют к нему 200 мл воды, затем дважды экстрагируют, используя 100 мл этилацетата. Органические слои объединяют и дважды промывают водой, высушивают над сульфатом магния и концентрируют при пониженном давлении, получая при этом 13,5 г 9-O-тропилоксима 2’,4’’-O-бис(триметилсилил)эритромицина А в виде вспененного вещества с выходом 90%.
Данные 1Н-ЯМР-спектроскопии для этого продукта идентичны данным, полученным для соединения, синтезированного согласно примеру 6.
Пример 8: получение 9-оксима 2’,4’’-O-бис-(триметилсилил)эритромицина А [стадия (а-2-1)]
32,8 г 9-оксима-НСl эритромицина А, полученного в соответствии со ссылочным примером, растворяют в 125 мл N,N-диметилформамида. Добавляют 1,13 г хлорида аммония и 23 мл 1,1,1,3,3,3-гексаметилдисилазана, перемешивают смесь при температуре, изменяющейся от 40 до 45°С, в течение 2 часов. Постепенно добавляют 15 мл 4н раствора гидроксида натрия, 100 мл воды и 50 мл гексана, полученную смесь перемешивают в течение 2 часов. Образовавшееся твердое вещество отфильтровывают, получая при этом 31,4 г 9-оксима 2’,4’’-O-бис(триметилсилил)эритромицина А с выходом 81%.
Пример 9: получение 9-O-тропилоксима 2’,4’’-О-бис-(триметилсилил) эритромицина А [стадия (а-2-2)]
8,93 г 9-оксима 2’, 4’’-O-бис-(триметилсилил)эритромицина А, полученного в соответствии с примером 8, растворяют в 40 мл тетрагидрофурана и охлаждают до 0°С. Добавляют 0,52 г 60% гидрида натрия и полученную смесь перемешивают в течение 20 минут. Затем добавляют туда же 2,14 г тетрафторбората тропилия и перемешивают при температуре от 0 до 5°С в течение 3 часов, после чего повторяют процедуру согласно примеру 6, получая при этом 9,44 г 9-O-тропилоксима 2’,4’’-O-бис-(триметилсилил)эритромицина А в виде белого вспененного вещества с выходом 96%.
Данные 1Н-ЯМР спектроскопии для этого продукта идентичны данным, полученным для соединения, синтезированного согласно примеру 6.
Пример 10: получение 9-O-тропилоксима 2’,4’’-O-бис-(триметилсилил)эритромицина А [стадия (а-2-2)]
13,4 г 9-оксима 2’,4’’-O-бис-(триметилсилил)эритромицина А, полученного в соответствии с примером 8, и 2,67 г тетрафторбората тропилия растворяют в 50 мл дихлорметана при комнатной температуре. Затем добавляют 2,3 мл триэтиламина и перемешивают при той же самой температуре в течение 3 часов, после чего добавляют воду. Органические слои объединяют и промывают водой, высушивают над сульфатом магния и концентрируют при пониженном давлении, получая при этом 14,6 г 9-O-тропилоксима 2’,4’’-O-бис-(триметилсилил)эритромицина А в виде белого вспененного вещества с выходом 99%.
Данные 1H-ЯМР спектроскопии для этого продукта идентичны данным, полученным для соединения, синтезированного согласно примеру 6.
Пример 11: получение 9-O-тропилоксима 2’,4’’-O-бис-(триметилсилил)-6-O-метилэритромицина А
14,75 г 9-O-тропилоксима 2’,4’’-O-бис(триметилсилил)-эритромицина А, полученного согласно предыдущим примерам, растворяют в смеси 60 мл тетрагидрофурана и 60 мл диметилсульфоксида и охлаждают до 0°С. Добавляют туда же 1,21 мл метилиодида и 1,09 г 85% гидроксида калия. После этого полученную смесь перемешивают при температуре от 0 до 5°С в течение 4 часов. Добавляют 150 мл воды и дважды экстрагируют с использованием 100 мл этилаиетата. Органические слои объединяют и промывают дважды водой, высушивают над сульфатом магния, после чего концентрируют при пониженном давлении, получая при этом 14,71 г 9-O-тропилоксима 2’,4’’-O-бис(триметилсилил)-6-O-метилэритромицина А в виде белого вспененного вещества с выходом 98%.
lH-ЯMP (СDСl3, ч/млн): δ 6,65 (м, 2Н, тропил 4’’’-Н и 5’’’-Н), 6,17 (м, 2Н, тропил 3’’’-Н и 6’’’-Н), 5,61 (м, 2Н, тропил 2’’’-Н и 7’’’-Н), 5,11 (дд, 1Н, 13-Н), 4,91 (д, 1Н, 1’’-Н), 4,57 (т, 1Н, тропил 1’’’-Н), 4,43 (д, 1Н, 1’-Н), 4,23 (дкв., 1Н, 5’’-Н), 4,16 (дд, 1Н, 3-Н), 4,16 (д, 1Н, 11-Н), 3,80 (ддкв., 1Н, 5’-Н), 3,68 (дкв., 1Н, 10-Н), 3,64 (д, 1Н, 5-Н), 3,34 (с, 3Н, cladinose 3’’-ОСН3), 3,09 (с, 3Н, 6-ОСН3), 3,19 (дд, 1Н, 2’-H), 3,16 (дд, 1Н, 4’’-H), 2,87 (ддкв., 1Н, 8-Н), 2,60 (дкв., 1Н, 2-Н), 2,59 (ддд, 1Н, 3’-Н), 2,38 (дд, 1Н, 2’’-Heq), 2,25 (д, 6Н, desosamine 3’-N (СН3)2), 2,00~1,41 (м, 7Н, 4-Н, 4’-Heq, 7-H2, 2’’-Нах и 14-Н2), 1,46 (с, 3Н, 18-Н), 1,28 (д, 3H, 6''-H), 1,18 (c, 3H, 7''-H), 1,22-0,95 (м, 19H, 4'-Hax, 6'-H3, 16-Н3, 20-Н3, 21-Н3, 17-Н3 и 19-Н3), 0,85 (т, 3Н, 15-Н3), 0,16 (с, 9Н, 4''-OSi(СН3)3), 0,11 (с, 9Н, 2’-OSi(СН3)3).
13С-ЯМР (СDСl3, ч/млн): δ 176,3 (С9), 171,4 (Cl), 131,4 и 131,4 (тропил С4’’’ и С5’’’), 126,0 и 125,1 (тропил С3’’ и С6’’1), 123,4 и 123,1 (тропил С2’’’ и 07’’’), 103,0 (С1'), 96,6 (C1’’), 81,3 (С5), 79,4 (С6), 79,3 (тропил СГ’’), 78,4 (С3), 77,1 (С4’’), 77,0 (С13), 74,3 (С12), 73,7 (С3’’), 73,6 (С2’), 71,5 (С11), 67,6 (С5’), 65,5 (С5’’), 65,5 (C3’), 51,5 (6-ОСН3), 50,1 (С8’’), 45,8 (С2), 41,4 (С7’ и С8’), 40,0 (С4), 38,2 (С7), 36,2 (С2’’), 33,4 (С8), 30,0 (С4’), 26,7 (С10), 22,6 (С19), 21,6 (С6’), 22,4 (С7’’), 20,6 (С14), 19,8 (С18), 19,1 (6’’), 16,6 (С21), 16,5 (С16), 15,5 (С20), 11,1 (С15), 10,1 (С17), 1,46 (4’’-OSi(СН3)3), 1,29 (2’-OSi(СН3)3).
Пример 12: получение 9-O-тропилоксима 2’,4’’-О-бис-(триметилсилил)-6-O-метилэритромицина А из 9-оксима 2’,4’’-O-бис(триметилсилил)эритромицина А
8,93 г 9-оксима 2’,4’’-O-бис(триметилсилил)эритромицина А, полученного согласно примеру 8, растворяют в 40 мл тетрагидрофурана и охлаждают до 0°С. Добавляют туда же 1,46 г трет-бутоксида калия и полученную смесь перемешивают в течение 20 минут. Затем добавляют 2,14 г тетрафторбората тропилия и перемешивают при температуре от 0 до 5°С в течение 3 часов. Добавляют к смеси 40 мл диметилсульфоксида, 0,81 мл метилиодида и 0,73 г 85%-ного порошка гидроксида калия, перемешивают в течение 4 часов при температуре от 0 до 5°С.
После этого повторяют процедуру выделения, описанную в примере 11, получая при этом 9,27 г 9-O-тропилоксима 2’,4’’-O-бис(триметилсилил)-6-O-метилэритромицина А в виде белого вспененного вещества с выходом 93%.
Данные 1H-ЯМР спектроскопии этого продукта идентичны данным, полученным для соединения, синтезированного согласно примеру 11.
Пример: исследование свойств
Неочищенные продукты, полученные в соответствии с описанными выше стадиями метилирования 6-гидроксигрупп 9-O-тропилоксима 2’,4’’-O-бис (триметилсилил) эритромицина А, анализировали методом высокоэффективной жидкостной хроматографии (ВЭЖХ) для оценки селективности 6-O-метилирования и сравнения с данными, полученными для соответствующих стадий, проводимых согласно известному уровню техники, результаты приведены в табл. 1.
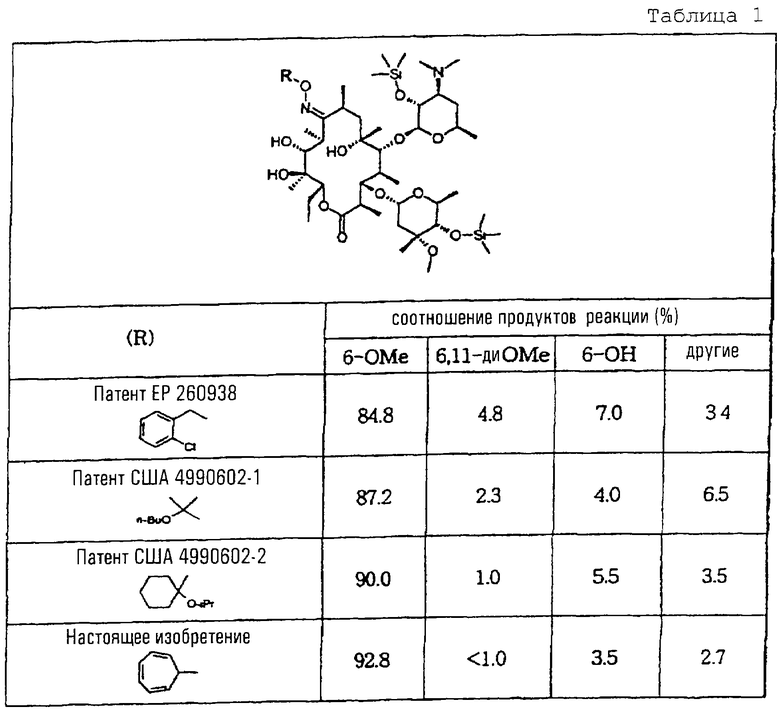
Как свидетельствуют приведенные выше результаты, 9-O-тропилоксим 2’,4’’-O-бис (триметилсилил) эритромицина А в соответствии с изобретением при метилировании 6-гидроксигрупп характеризуется наибольшей селективностью.
Пример 13. Получение кларитромицина
13,3 г 9-O-тропилоксима 2’,4’’-O-бис(триметилсилил)-6-O-метилэритромицина А, полученного в соответствии с примером 11, растворяют в 70 мл этанола. Постепенно добавляют 0,75 мл муравьиной кислоты, 70 мл воды и 4,16 г бисульфита натрия, и полученную смесь кипятят с обратным холодильником в течение 2 часов. Полученный раствор охлаждают ниже 10° С и добавляют к нему 140 мл воды и 50 мл гексана. Доводят значение рН полученного раствора до 10 добавлением 2 н. раствора гидроксида натрия. Смесь перемешивают при температуре ниже 10°С в течение 1 часа и затем отфильтровывают образовавшееся твердое вещество, промывают водой, после чего высушивают при приблизительно 50°С, получая при этом 7,46 г неочищенного кларитромицина в виде белого порошка с выходом 75%.
Т.пл.: 220~223°С (например, в литературе для кристаллов формы I т. пл. составляет 222~225°С).
1Н-ЯМР (СDСl3, ч/млн): δ 5,06 (дд, 1Н, 13-Н), 4,92 (д, 1Н, 1’’-Н), 4,44 (д, 1Н, 1’-Н), 4,02 (дкв., 1Н, 5’’-Н), 3,78 (дд, 1Н, 3-Н), (дкв., 1Н, 5’’-Н), 3,77 (д, 1Н, 11-Н), 3,67 (д, 1Н, 5-Н), 3,57 (ддкз., 1Н, 5’-Н), 3,33 (с, 3Н, 3’’-ОСН3), 3,20 (дд, 1Н, 2’-Н), 3,04 (с, 3Н, 6-ОСН3), 3,03 (дд, 1Н, 4’’-Hz), 2,99 (дкв., 1Н, 10-Н), 2,87 (дкв., 1Н, 2-Н), 2,58 (ддкв., 1H, 8-Н), 2,40 (ддд, 1Н, 3’-Н), 2,37 (д, 1Н, 2’’-Неq), 2,28 (с, 6Н, 3’-N (СН3)2), 2,00-1,80 (м, 3Н, 4-Н,7-Н2, 14-Н1), 1,75-1,40 (м, 3Н, 4’-Heq, 2’’-Нах и 14-Н1), 1,41 (с, 3Н, 18-Н), 1,13 (с, 3Н, 6-СН3), 1,31 (д, 3Н, 6’’-Н3), 1,30-1,05 (м, 22Н, 7’’-Н3, 4’-Нах 6’-Н3, 16-Н3, 20-Н3, 21-Н3, 17-Н3 и 19-Н3), 0,84 (т, 3Н, 15-Н3).
13С-ЯМР (СDСl3, ч/млн): δ 221,0 (С9), 175,9 (C1), 102,8 (С1), 96,0 (C1’’), 80,7 (С5), 78,4 (С6), 78,4 (С4’’), 77,9 (С3), 76,6 (С13), 74,2 (С12), 72,6 (C3’’), 70,9 (С2’), 69,0 (С11), 68,7 (С5’), 65,8 (С5’’), 65,5 (C3’), 50,6 (6-ОСН3), 49,4 (С8’’), 45,2 (С8), 45,0 (С2), 40,2 (С7’ и С8’), 39,4 (С7), 39,3 (С4), 37,2 (С10), 34,8 (С2’’), 28,5 (С4’), 21,4 (С6’), 21,4 (С7’’), 21,0 (С14), 19,7’ (С18), 18,6 (6’’), 17,9 (С19), 15,9 (С21), 15,9 (С16), 12,2 (С20), 10,6 (С15), 9,0 (С17).
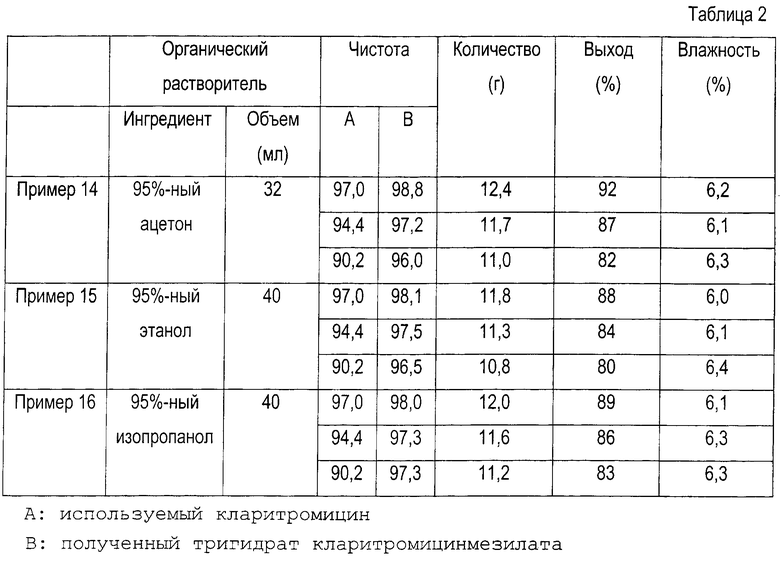
Пример 14. Получение тригидрата кларитромицинмезилата
11,2 г кларитромицина (15 ммоль), имеющего чистоту 97%, растворяют в 32 мл 95%-ного ацетона и добавляют по каплям метансульфоновую кислоту, после чего перемешивают при комнатной температуре в течение 1 часа. Затем полученную суспензию охлаждают до 0°С и перемешивают в течение 3 часов. Образовавшиеся кристаллы отфильтровывают, промывают холодным ацетоном и высушивают при 45°С, получая при этом 10,3 г кристаллического тригидрата кларитромицинмезилата (чистота: 98,8%, выход: 92%).
Т.пл.: 160~161°С
Содержание влаги: 6,2% (по Карлу Фишеру, теоретическое содержание влаги для тригидрата составляет 6,02%).
ИК (КВr, см-1): 3428, 2978, 2939, 1734, 1686, 1459, 1378, 1347, 1170, 1110, 1077, 1051, 1010, 954, 908, 892, 780.
1Н-ЯМР (CDCl3, ч/млн): δ 5,07 (дд, 1Н, 13-Н), 4,91 (д, 1Н, 1’’-Н), 4,60 (д, 1Н, 1’-Н), 3,98 (дкв., 1Н, 5’’-Н), 3,76 (ддкв., 1Н, 5’-Н), 3,76 (д, 11Н, 1-Н), 3,72 (дд, 1Н, 3-Н), 3,70 (дд, 1Н, 5-Н), 3,55 (ддд, 1Н, 3’-Н), 3,47 (дкв., 1Н, 2’-Н), 3,35 (с, 3Н, 3’’-ОСН3), 3,07 (дд, 1Н, 4’’-Н), 3,04 (с, 3Н, 6-ОСН3), 3,03 (с, 1Н, 10-Н), 2,93 (с, 6Н, 3’-N (СН3)2), 2,87 (дкв., 1Н, 8-Н), 2,78 (с, 3Н, СН3SО
13С-ЯМР (CDCl3, ч/млн): δ 221,3 (9-С), 176,4 (1-С), 102,4 (1’-С), 97,0 (1’’-С), 82,3 (5-С), 79,5 (3-С), 78,9 (6-С), 78,3 (4’’-С), 77,4 (13-С), 74,9 (12-С), 73,6 (3’’-С), 70,6 (2’-С), 69,7 (11-С), 67,7 (5’-С), 66,7 (5’’-С), 66,1 (3’-С), 51,1 (22-С), 49,9 (8’’-С), 45,7 (8-С), 45,6 (2-С), 39,8 (7’-С, 8’-С и 7-С), 39,7 (4-0, 38,0 (10-С), 35,7 (2’’-С), 31,6 (4’-С), 22,0 (6’-С), 21,6 (7’’-С и 14-C), 20,5 (18-С), 19,2 (6’’-С), 18,5 (19-C, 16,7 (21-C), 16,5 (16-C, 12,8 (20-C), 11,2 (15-C), 9,9 (17-С).
Вышеуказанную процедуру повторяют, используя партии менее чистого кларитромицина, полученные результаты приведены в таблице 2.
Инфракрасный спектр и спектр порошковой дифракционной спектроскопии рентгеновских лучей тригидрата кларитромицинмезилата приведены на фиг.1 и 2, соответственно.
Примеры 15 и 16: получение тригидрата кларитромицинмезилата
В каждом из примеров 15 и 16 повторяют процедуру, описанную в примере 14, с тем исключением, что вместо ацетона используют различные органические растворители. Полученные результаты приведены в табл.2.
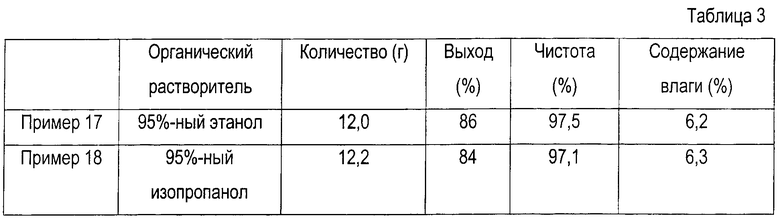
Пример 17: перекристаллизация тригидрата кларитромицинмезидата 14,0 г тригидрата кларитромицинмезилата, имеющего чистоту 92%, суспендируют в 50 мл 95%-ного этанола, кипятят с обратным холодильником до полного растворения, охлаждают до 0°С и перемешивают в течение 4 часов. После этого образовавшиеся кристаллы отфильтровывают, промывают ацетоном, охлажденным до 0°С, и высушивают при 45°С, получая при этом 12,0 г очищенного кристаллического тригидрата кларитромицинмезилата (чистота: 97,5%, выход: 86% и содержание влаги: 6,2%).
Пример 18: перекристаллизация тригидрата кларитромицинмезилата
Повторяют процедуру, описанную в примере 17, с тем исключением, что вместо этанола используют 95%-ный изопропанол. Результаты приведены в табл.3.
Пример 19. Получение кларитромицина в виде кристаллов формы II из тригидрата кларитромицинмезилата
14,0 г тригидрата кларитромицинмезилата (чистота 96°с) растворяют в смеси 42 мл этанола и 84 мл воды и отфильтровывают для того, чтобы удалить нерастворимые ингредиенты. Добавляют к фильтрату по каплям 4,8 мл концентрированного водного раствора аммиака и перемешивают в течение 3 часов. После этого образовавшиеся кристаллы отфильтровывают и высушивают в течение ночи на воздухе при 55°С, получая при этом 11,3 г кларитромицина в виде кристаллов формы II (чистота: 97,2%, выход: 97%).
Инфракрасный спектр и спектр порошковой дифракционной спектроскопии рентгеновских лучей кларитромицина в виде кристаллов формы II приведены на фигурах 3 и 4 соответственно.
Пример 20. Получение кларитромицина в виде кристаллов формы II из неочищенного кларитромицина
93,1 г неочищенного кларитромицина (0,124 моль) растворяют в 260 мл 95%-ного ацетона и фильтруют для того, чтобы удалить нерасторимые ингредиенты. Добавляют к фильтрату по каплям 10 г метансульфоновой кислоты (0,105 моль) и перемешивают в течение 1 часа. После этого полученную суспензию охлаждают до 0°С и полученный раствор перемешивают в течение 3 часов. Образовавшиеся кристаллы отфильтровывают, промывают холодным ацетоном и высушивают при 45°С, получая при этом 87,5 г кристаллического тригидрата кларитромицинмезилата (чистота: 93%, выход: 78% и содержание влаги: 6,4%).
87,5 г тригидрата кларитромицинмезилата (0,097 моль) суспендируют в 306 мл 95%-ного этанола, кипятят с обратным холодильником в течение 30 минут, для того, чтобы достичь полного растворения, и после этого медленно охлаждают до 0°С при перемешивании. Затем смесь перемешивают при 0°С в течение 4 часов, и образовавшиеся кристаллы отфильтровывают, промывают ацетоном, охлажденным до 0°С, и высушивают при 45°С, получая при этом 74,4 г очищенного кристаллического тригидрата кларитромицинмезилата (чистота: 97%, выход: 85%, и содержание влаги: 6,1%).
74,4 г тригидрата кларитромицинмезилата (0,083 моль), полученного как описано выше, суспекдируют в смеси 220 мл этанола и 440 мл воды, и фильтруют для того, чтобы удалить нерастворившиеся ингредиенты. Добавляют к фильтрату по каплям 25 мл концентрированного водного раствора аммиака и перемешивают при комнатной температуре в течение 3 часов. После этого образовавшиеся кристаллы отфильтровывают и высушивают в течение ночи при 55°С, получая при этом 60,0 г кларитромицина в виде кристаллов формы II (чистота: 98,0%, и зыход: 97%).
Сравнительный пример
93,1 г неочищенного кларитромицина (0,124 моль) перекристаллизовывают из 600 мл этанола и высушивают, получая при этом 65,2 г кларитромицина в виде кристаллов формы I (чистота: 94%, выход: 70%), в соответствии с методикой, описанной в опубликованной международной заявке WO 98/04573.
65,2 г кларитромицина в виде кристаллов формы I (0,087 моль) суспендируют в 510 мл этанола и кипятят с обратным холодильником в течение 1 часа для того, чтобы растворить большую часть кристаллов. Раствор подвергают горячему фильтрованию для удаления нерастворившихся ингредиентов, затем фильтрат охлаждают до 10°С и перемешивают в течение 2 часов. После этого образовавшиеся кристаллы отфильтровывают и высушивают при 50°С, получая при этом 54,1 г очищенного кларитромицина в виде кристаллов формы I (чистота: 97,1%, выход: 83%).
Затем из кларитромицина в виде кристаллов формы I получают кларитромицин в виде кристаллов формы II в соответствии с методом, раскрытым в опубликованной международной заявке WO 98/04574.
А именно, 54,1 г очищенного кларитромицина в виде кристаллов формы I (0,072 моль) суспендируют в 270 мл этилацетата и кипятят с обратным холодильником в течение 1 часа. Нерастворившиеся компоненты удаляют посредством горячего фильтрования, к фильтрату добавляют 40 мл этилацетата и кипятят с обратным холодильником. Раствор охлаждают до 50°С, добавляют к нему 270 мл простого изопропилового эфира и охлаждают до 5°С. Образовавшиеся кристаллы отфильтровывают и высушивают, получая при этом 41,7 г кларитромицина в виде кристаллов формы II (чистота: 97,2, выход: 77%).
Поскольку варианты осуществления изобретения описаны и проиллюстрированы примерами, является очевидным, что, не выходя за рамки предмета изобретения, могут быть осуществлены различные изменения и модификации, которые ограничены лишь объемом, определенным прилагаемой формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения кларитромицина | 2000 |
|
RU2225413C1 |
| СПОСОБЫ ПОЛУЧЕНИЯ МАКРОЛИДОВ И КЕТОЛИДОВ, И ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ ДЛЯ ИХ ПОЛУЧЕНИЯ | 2011 |
|
RU2608390C2 |
| НОВЫЕ ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И СПОСОБ ПОЛУЧЕНИЯ ИЗ НИХ МАКРОЛИДНОГО АНТИБИОТИКА | 1999 |
|
RU2208615C2 |
| НОВЫЕ КЕТОАЗОЛИДЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1999 |
|
RU2211222C2 |
| Способ получения 9-оксимных производных эритромицина А | 1985 |
|
SU1577700A3 |
| Способ получения эфиров пиперидин-4,4-дикарбоновых кислот | 2021 |
|
RU2765464C1 |
| СПОСОБ ПОЛУЧЕНИЯ АНТАГОНИСТОВ РЕЦЕПТОРОВ CGRP | 2013 |
|
RU2672056C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И СПОСОБЫ, ВКЛЮЧАЮЩИЕ КОМБИНАЦИИ 2-АЛКИЛИДЕНОВЫХ ПРОИЗВОДНЫХ 9-НОР-ВИТАМИНА D И БИСФОСФОНАТА | 2004 |
|
RU2326695C2 |
| 9-О-ОКСИМОВЫЕ ПРОИЗВОДНЫЕ ЭРИТРОМИЦИНА А И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1995 |
|
RU2152951C1 |
| МАКРОЛИДЫ И ФАРМАЦЕВТИЧЕСКИЙ СОСТАВ НА ИХ ОСНОВЕ | 2000 |
|
RU2243230C2 |
Кларитромицин в виде кристаллов формы II (формула I) получают путем обработки кларитромицина метансульфоновой кислотой в смеси смешивающегося с водой органического растворителя и воды с образованием кристаллического тригидрата мезилата кларитромицина формулы (II), который подвергают нейтрализации водным раствором аммиака в смеси смешивающегося с водой органического растворителя и воды. Кларитромицин, не обладающий фармацевтической степенью чистоты, получают путем защиты 9-оксимной гидроксигруппы 9-оксима эритромицина А или его соли тропильной группой, а 2’- и 4’’-гидроксигрупп - триметилсилильными группами с получением 9-О-тропилоксима 2’,4’’-O-бис(триметилсилил)эритромицина А, который подвергают взаимодействию с метилирующим агентом с получением 9-О-тропилоксима 2’,4’’-O-бис(триметилсилил)-6-О-метилэритромицина А формулы, а затем удаляют защитные группы и оксимную группу. Также изобретение относится к промежуточным соединениям формулы (II) и формулы (III), где R1 представляет собой водород или метил, а R2 представляет собой водород или триметилсилил, причем если R1 представляет собой метил, то R2 представляет собой триметилсилил. Технический результат - повышение выхода и улучшение чистоты целевого продукта. 4 с. и 12 з.п. ф-лы, 3 табл., 4 ил.
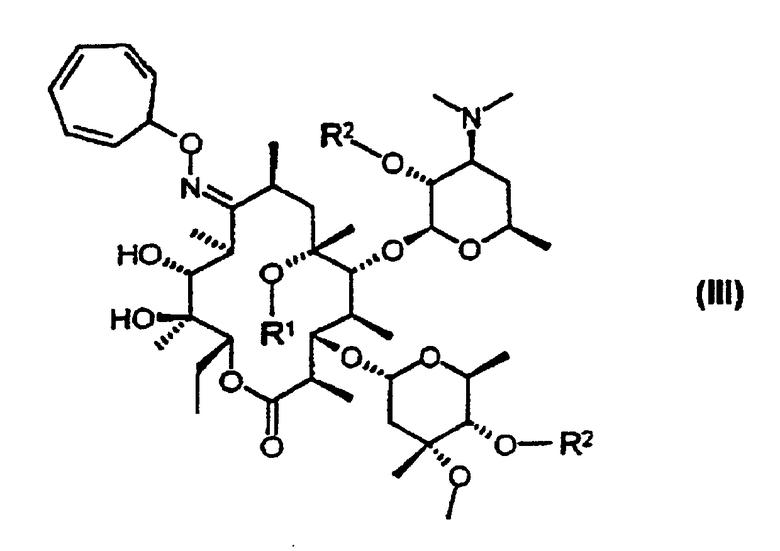
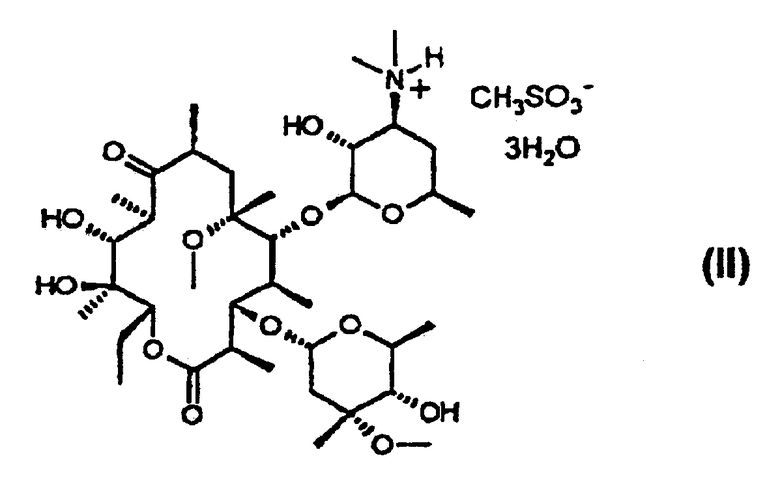
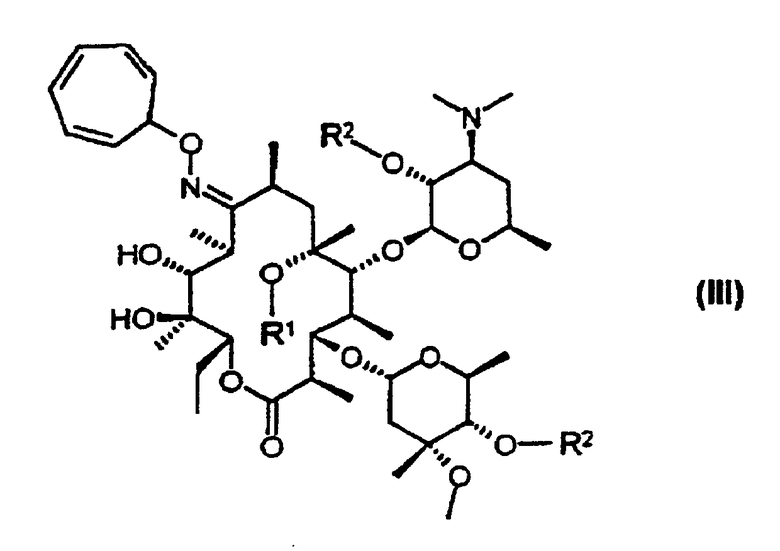
где R1 представляет собой водород или метил;
R2 представляет собой водород или триметилсилил, причем, если R1 представляет собой метил, то R2 представляет собой триметилсилил.
| СПОСОБ АВТОМАТИЧЕСКОГО УРАВНОВЕШИВАНИЯ ДЕТАЛЕЙ | 0 |
|
SU260938A1 |
| US 4990602 A, 05.02.1991 | |||
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| О-МЕТИЛЬНЫЕ ПРОИЗВОДНЫЕ АЗИТРОМИЦИНА А, ОБЛАДАЮЩИЕ АНТИБАКТЕРИАЛЬНОЙ АКТИВНОСТЬЮ, АЗИТРОМИЦИНЫ В КАЧЕСТВЕ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ ДЛЯ ПОЛУЧЕНИЯ О-МЕТИЛЬНЫХ ПРОИЗВОДНЫХ АЗИТРОМИЦИНА А И СПОСОБ ПОЛУЧЕНИЯ О-МЕТИЛЬНЫХ ПРОИЗВОДНЫХ АЗИТРОМИЦИНА А | 1991 |
|
RU2045533C1 |

