Настоящее изобретение относится к способу очистки гетерополикислоты, в частности к способу очистки кремневольфрамовой кислоты.
Хорошо известно, что гетерополикислоты, практически свободные от посторонних катионов (в дальнейшем упоминаются как "свободные гетерополикислоты"), являются ценными химическими соединениями, которые могут быть использованы в качестве кислотных катализаторов для проведения реакций органических веществ, таких, как реакция присоединения низших алифатических карбоновых кислот к олефинам с образованием соответствующих сложных эфиров. Один такой способ представлен в заявке ЕР А-0757027, поданной авторами настоящего изобретения, в которой описано применение свободных гетерополикислот в качестве катализаторов получения алифатических сложных эфиров, таких, как этилацетат, в ходе проведения вышеупомянутой реакции присоединения. Способы синтеза гетерополикислот также хорошо известны. Так, в частности, такие способы описаны, например, North, E.O. в "Organic Synthesis", 1, с.129 (1978), редактор H.S. Booth, Robert E. Krelger publishing company, Huntington, New York, и Tatsuhiko H. и др. в Kogyo Kagaku Zasshi, 72 (9), 1945-48 (1969).
Известные способы получения гетерополикислот обычно включают следующие три стадии:
а. Реакционная стадия, на которой добавлением соляной кислоты в кипящий водный раствор вольфрамата натрия и силиката натрия получают вольфрамокремниевую кислоту. При нейтрализации и гидролизе в результате реакции образуется концентрированный соляной раствор, включающий структурное звено вольфрамокремниевой кислоты Кеггина. В общем эту реакцию суммарно можно представить следующим образом:
12Na2WO4+Na2SiO3+НСl→H4SiW12O40·nH2O+NaCl+H2O
Перед очисткой продукт фильтруют, подкисляют и охлаждают.
б. Стадия выделения и очистки продукта, на которой охлажденный раствор продукта со стадии (а) очищают осуществлением по меньшей мере одной стадии жидкостно-жидкостной экстракции с использованием органического растворителя. Применяют такой растворитель, который способен образовывать комплекс растворитель/гетерополикислота, который в значительной степени нерастворим в подкисленном водном соляном растворе. Образовавшаяся фаза растворитель/комплекс способна освобождать концентрированный соляной раствор от загрязняющих неорганических солей.
в. Стадия регенерации продукта, на которой из фазы растворитель/комплекс, полученной на стадии б), регенерируют водный раствор гетерополикислоты. Этой регенерации достигают добавлением воды для вытеснения растворителя, участвующего в образовании фазы органический растворитель/комплекс. Органический растворитель удаляют из смеси отгонкой. Органический растворитель удобно удалять в виде азеотропа растворитель/вода. При охлаждении этот азеотроп может образовывать двухфазную смесь. Более плотной из этих двух фаз является водная фаза, которую можно возвращать в испаритель. Менее плотной фазой является органическая фаза, которую можно выделять, например, для повторного использования. Количество добавляемой и удаляемой воды можно регулировать для приготовления водного раствора гетерополикислоты концентрацией от 20 до 80 мас.%.
Далее образовавшийся концентрированный водный раствор полученной гетерополикислоты можно применять для пропитки неорганической подложки.
При осуществлении этих известных способов синтеза в качестве возможных растворителей для очистки продукта применяют диэтиловый эфир, метилэтилкетон и этилацетат. При создании настоящего изобретения было установлено, что этим материалам присущи недостатки. Так, например, диэтиловый эфир обладает высокой летучестью и низкой температурой самовоспламенения; метилэтилкетон обуславливает возникновение окраски вследствие того, что целевая кислота катализирует реакцию альдольного типа с этим кетоновым растворителем; а сложные эфиры, такие, как этилацетат, обладают способностью подвергаться гидролизу в присутствии синтезируемой гетерополикислоты и всех содержащихся в ней нейтрализующих кислот.
Было установлено, что эти недостатки можно уменьшить, если при осуществлении стадий очистки применять приемлемый растворитель.
В соответствии с первым объектом настоящего изобретения предлагается способ очистки гетерополикислоты, который включает:
обработку водного раствора, включающего (I) гетерополикислоту и (II) примеси солей, осуществлением по меньшей мере одной стадии жидкостно-жидкостной экстракции органическим растворителем,
характеризующийся тем, что органический растворитель включает дигидрокарбиловый простой эфир, содержащий по крайней мере 5 углеродных атомов.
Другими словами, в качестве дигидрокарбилового эфира используют такой продукт, у которого общее число углеродных атомов в двух гидрокарбильных звеньях, по одному с каждой стороны эфирного кислородного атома, равно по крайней мере 5.
Осуществление стадии жидкостно-жидкостной экстракции позволяет гетерополикислоте in situ образовывать с органическим растворителем комплекс. Это дает возможность разделить экстракт на две или большее число фаз, включая помимо прочего плотную органическую фазу, которая обычно опускается в основание или в направлении днища сепараторного сосуда. Эта фаза включает растворенный комплекс гетерополикислоты.
В предпочтительном варианте плотную органическую фазу выделяют. Ее можно смешивать с водой с получением отдельного слоя разбавленной водной смеси, из которой можно выделить концентрированный раствор (от 20 до 80 мас.%) гетерополикислоты, свободной от органических примесей.
Исходный водный раствор гетерополикислоты можно приготовить реакцией (I) водного раствора одной или нескольких солей щелочных или щелочно-земельных металлов, выбранных из вольфрамата и молибдата, с (II) силикатом щелочного или щелочно-земельного металла или фосфатом щелочного или щелочно-земельного металла. В предпочтительном варианте эту реакцию проводят кипячением с обратным холодильником. В результате этой реакции получают неочищенный водный раствор, включающий гетерополикислоту выбранной соли и все другие соли, образовавшиеся in situ или используемые в качестве реагента. Далее этот раствор можно фильтровать, охлаждать и обрабатывать на стадии жидкостно-жидкостной экстракции согласно первому объекту настоящего изобретения.
В предпочтительном варианте гетерополикислота в водном растворе представляет собой кремневольфрамовую кислоту.
В соответствии со вторым объектом настоящего изобретения предлагается способ синтеза гетерополикислоты, практически свободной от посторонних катионов, причем этот способ включает:
а) реакцию (I) водного раствора одной или нескольких солей щелочных или щелочно-земельных металлов, выбранных из вольфрамата и молибдата, при кипячении с обратным холодильником с (II) силикатом щелочного или щелочно-земельного металла или фосфатом щелочного или щелочно-земельного металла в присутствии кислоты с получением неочищенного водного раствора, включающего гетерополикислоту выбранной соли и все другие соли, образовавшиеся in situ или используемые в качестве реагента;
б) фильтрование и охлаждение этого неочищенного водного раствора, включающего гетерополикислоту;
в) очистку охлажденного неочищенного водного раствора со стадии (б) по меньшей мере одной жидкостно-жидкостной экстракцией органическим растворителем для предоставления гетерополикислоте возможности образовывать in situ комплекс с органическим растворителем и предоставление экстракту возможности разделиться на две или большее число фаз, включая, в частности, плотную органическую фазу, которая содержит растворенный комплекс гетерополикислоты, причем эта плотная органическая фаза находится ниже водной фазы;
г) выделение этой плотной органической фазы со стадии (в) и ее тщательное смешение с водой с получением отдельного слоя разбавленной водой смеси и
д) выделение из разбавленной водной смеси концентрированного (от 20 до 80 мас.%) водного раствора гетерополикислоты, практически свободной от органических примесей,
где органический растворитель, используемый на стадии жидкостно-жидкостной экстракции (в), включает дигидрокарбиловый простой эфир, содержащий по меньшей мере 5 углеродных атомов.
В предпочтительном варианте органический растворитель, используемый на стадии жидкостно-жидкостной экстракции (в), представляет собой дигидрокарбиловый простой эфир, содержащий по меньшей мере 5 углеродных атомов.
Дигидрокарбиловый простой эфир может содержать 5-20 углеродных атомов, а предпочтительный содержит 5-10 углеродных атомов. В качестве дигидрокарбилового простого эфира можно использовать такой, у которого по меньшей мере одна из гидрокарбильных групп представляет собой алкильную группу с разветвленной цепью. Предпочтительной разветвленной алкильной группой является трет-бутил или - амил. По меньшей мере одной из гидрокарбильных групп в простом эфире может быть метальная, этильная или пропильная группа. В предпочтительном варианте эта гидрокарбильная группа представляет собой метальную или этильную группу, а наиболее предпочтительна метальная группа. Может оказаться эффективным применение простых эфиров, которые по разные стороны от эфирного кислородного атома содержат разные гидрокарбильные группы. Так, например, простой эфир может включать метальную группу с одной стороны относительно эфирного кислородного атома и бутильную или амильную группу с другой.
Используемый простой эфир должен быть по существу не смешивающимся с водой. Другими словами, простой эфир должен быть по существу нерастворимым в воде, а вода должна быть по существу нерастворимой в этом простом эфире. Для того чтобы содействовать удалению из экстракта любой соли, простой эфир, когда он образует с ГПК комплекс, должен быть не смешивающимся с водой.
Температура кипения приемлемых простых эфиров обычно превышает 40°С, предпочтительно находится в пределах между 40 и 60°С, более предпочтительно в пределах между 50 и 60°С, например в пределах между 55 и 56°С. Для некоторых целей применения могут оказаться также приемлемыми простые эфиры, температура кипения которых превышает 90°С.
Подходящие простые эфиры включают пропилбутиловый эфир (например, пропилтрет-бутиловый эфир), этилпропиловый эфир, дипропиловый эфир [например, ди-н-пропиловый эфир (с tкип 88-90°С) и диизопропиловый эфир], бутилгликолевый эфир, дибутиловый эфир (например, диизобутиловый эфир, дитрет-бутиловый эфир, ди-н-бутиловый эфир), метилбутиловый эфир, [например, метил-н-бутиловый эфир (с tкип 70-71°С), метилвторбутиловый эфир (с tкип 75°С), метил-трет-бутиловый эфир (с tкип 55-56°С)], метиламиловый эфир [например, 4-трет-амилметиловый эфир (с tкип 85-86°С], бутилэтиловый эфир [например, н-бутилэтиловый эфир (с tкип 91-92°С), трет-бутилэтиловый эфир (с tкип 72-73°С] и пропилбутиловый эфир. Среди них предпочтительны метилтрет-амиловый эфир, а особенно метил-трет-бутиловый эфир (в дальнейшем "МТБЭ"). Преимущество применения таких простых эфиров в качестве растворителей состоит в том, что вследствие их низкой растворимости в воде они, по-видимому, снижают общее содержание органического компонента в любом отходящем потоке.
Стадию а) можно осуществлять приготовлением водного раствора по меньшей мере одной вольфраматной или молибдатной соли щелочного или щелочно-земельного металла. Для сведения к минимальной степени загрязненности целевой готовой гетерополикислоты посторонними ионами такие растворы могут быть приготовлены с использованием деминерализованной воды.
В предпочтительном варианте готовят раствор молибдата натрия или вольфрамата натрия.
Далее водный раствор по меньшей мере одной вольфраматной или молибдатной соли щелочного или щелочно-земельного металла кипятят с обратным холодильником совместно с силикатом или фосфатом щелочного или щелочно-земельного металла (стадия а). Силикат или фосфат можно использовать в виде твердого вещества или водного раствора. Предпочтительнее применять силикат.
В результате реакции образуется смесь, представляющая собой неочищенный раствор смешанных гетерополикислот выбранной соли, которую вначале можно получать в виде соли частично подкисленной гетерополикислоты (например, Н2Na2-гетерополикислоты) или полностью подкисленной гетерополикислоты (например, H4-гетерополикислоты). После этого конечный раствор фильтруют, охлаждают и, что предпочтительно, подкисляют.
Реакцию на стадии а) проводят в присутствии кислоты, которая нейтрализует реакционную смесь. Полагают, что во время осуществления этой стадии нейтрализации собирают гетерополикислотный компонент. Приемлемые для стадии нейтрализации кислоты включают минеральные кислоты, такие, как соляная кислота.
В предпочтительном варианте кислоту вводят в реакционную смесь на стадии а) посредством потока флегмы. Добавление кислоты регулируют таким образом, чтобы предотвратить осаждение таких соединений, как, например, вольфрамовая кислота.
В более предпочтительном варианте введение соляной кислоты в реакционную смесь осуществляют на двух стадиях: во-первых, как сказано выше, во время стадии а) с тем, чтобы нейтрализовать реакционную смесь, а во-вторых, перед тем, как обработанную кипячением реакционную смесь подвергают жидкостно-жидкостной экстракции на стадии в). Это второе подкисление можно проводить в процессе охлаждения и фильтрации смеси со стадии а), но в предпочтительном варианте проводят после стадии охлаждения и фильтрования. Цель второй стадии добавления кислоты заключается в том, чтобы (I) гарантировать соответствующий гидролиз солевых реагентов, (II) перевести гетерополикислоту, содержащуюся в реакционной смеси, в Н4-форму и (III) предотвратить ионизацию водного раствора. Поскольку ионизация гетерополикислоты в растворе способствует ее растворению в воде, подкисление раствора добавлением кислоты, в особенности сильной минеральной кислоты, подавляет такую тенденцию гетерополикислоты к ионизации. Это не только снижает ее водорастворимость, но также дает возможность упростить ее экстрагирование путем экстракции растворителем.
Далее подкисленный, охлажденный и профильтрованный раствор подвергают по меньшей мере одной обработке на стадии жидкостно-жидкостной экстракции (стадия в). В предпочтительном варианте осуществляют множество таких стадий экстракции. Жидкостно-жидкостную экстракцию проводят с использованием органического растворителя, который in situ образует с гетерополикислотой комплекс. Необходимо отметить, что образовывать такой комплекс способны не все растворители. Так, например, циклогексан образовывать с гетерополикислотой такой комплекс неспособен.
После этого жидкостно-жидкостному экстракту дают возможность разделиться на две или большее число фаз. Комплекс гетерополикислоты в экстракционном органическом растворителе растворим и образует раствор, который выделяется в виде плотной органической фазы.
В некоторых вариантах выполнения изобретения экстракт разделяется на три фазы: верхняя, органическая фаза, которая включает главным образом растворитель со следами гетерополикислоты, средняя, водная фаза, которая включает преимущественно воду и некоторое количество кислот и солей, используемых или образующихся во время реакции синтеза, и наконец, нижняя, плотная органическая фаза, которая включает представляющий интерес комплекс целевой гетерополикислоты. Поведение такой системы во время жидкостно-жидкостной экстракции необычно в том отношении, что комплекс гетерополикислоты и экстракционного растворителя образуется, когда эти компоненты смешивают между собой, в нижней, плотной органической фазе.
Эффективность экстракции обычно зависит от степени освобождения этих фаз в экстракте. Так, в частности, органическая фаза, включающая комплекс органического растворителя и гетерополикислоты, из-за наличия незахваченных солей/водной фазы неорганических соединений как реагентов и всех солей, образующихся in situ во время реакции, вначале может казаться мутной. От такого помутнения можно избавиться, например, методами сепарации. Подходящие методы сепарации обработки включают центрифугирование и фильтрование.
Раствор, содержащий комплекс гетерополикислоты, отделяют от другой фазы (фаз). В предпочтительном варианте этот раствор смешивают с водой, которую перед стадией смешения целесообразно деминерализовать и/или деионизировать. Органический растворитель можно соответствующим образом удалить из конечной смеси азеотропной перегонкой в колонне. Предпочтительные органические растворители способны образовывать смеси для декантации, которые позволяют упростить отделение нижнего водного слоя от верхней органической фазы. Водный слой может быть возвращен в дистилляционную колонну или удален из колонны в виде головной фракции. Количество воды, используемой для приготовления смеси, можно регулировать перед дистилляцией. Точно так же можно регулировать количество воды, остающейся после перегонки, для приготовления раствора гетерополикислоты с концентрацией гетерополикислоты 20-80 мас.%.
Другой метод приготовления водного раствора свободной гетерополикислоты состоит в отгонке органической фазы под вакуумом с целью удаления растворителя и последующего растворения остатка водой. Однако при сушке под вакуумом следует соблюдать осторожность, поскольку длительная выдержка при повышенной температуре под вакуумом даже в умеренных условиях (например, 100°С, 16 ч, 0,01 мм рт.ст.) может привести к потере некоторого количества гетерополикислоты из-за образования побочных продуктов.
Понятие "гетерополикислота" в данном случае и во всем тексте описания использовано как охватывающее свободные кислоты. Гетерополикислоты можно применять при приготовлении катализаторов этерификации и катализаторов присоединения олефинов. Что касается этих целей применения, то гетерополикислоты могут быть использованы в виде свободных кислот и/или неполных солей. Синтез гетерополикислот в соответствии с настоящим изобретением дает возможность регулировать чистоту таких солей.
Гетерополикислота или анионный компонент ее соответствующей соли как правило включает от 2 до 18 связанных кислородными атомами атомов поливалентных металлов, которые называют периферийными атомами. Эти периферийные атомы симметрично окружают один или несколько центральных атомов. Обычно функцию периферийных атомов выполняет один или несколько атомов молибдена, вольфрама, ванадия, ниобия, тантала и других металлов. Как правило, центральным является атом кремния или фосфора, но им может служить любой из широкого разнообразия атомов элементов групп с I по VIII Периодической таблицы элементов. К ним относятся, в частности, ионы двухвалентной меди; ионы двухвалентных бериллия, цинка, кобальта и никеля; ионы трехвалентных бора, алюминия, галлия, железа, церия, мышьяка, сурьмы, фосфора, висмута, хрома и родия; ионы четырехвалентных кремния, германия, олова, титана, циркония, ванадия, серы, теллура, марганца, никеля, платины, тория, гафния, церия и ионы других редкоземельных металлов; ионы пятивалентных фосфора, мышьяка, ванадия, сурьмы; ионы шестивалентного теллура и ионы семивалентного йода. Такие гетерополикислоты известны также как "полиоксоанионы", "полиоксометаллаты" или "металлоксидные кластеры". Структуры некоторых хорошо известных анионов названы по фамилиям первых исследователей в данной области техники, например структуры Кеггина, Уэллса-Доусона и Андерсона-Эванса-Перлова.
Обычно гетерополикислоты обладают высокой молекулярной массой, например в интервале от 700 до 8500, и включают димерные комплексы. Им свойственна относительно высокая растворимость в полярных растворителях, таких, как вода и другие кислородсодержащие растворители, в особенности если они являются свободными кислотами и в случае некоторых солей, причем их растворимость можно регулировать выбором соответствующих противоионов. Конкретные примеры гетерополикислот, которые могут быть синтезированы по настоящему изобретению, включают:
12-вольфрамофосфорную кислоту H3[PW12O40]·H2O
12-молибдофосфорную кислоту Н3[РМо12O40]·Н2О
12-вольфрамокремниевую кислоту H4[SiW12O40]·H2O
12-молибдокремниевую кислоту H4[SiMo12O40]·H2O
Настоящее изобретение особенно эффективно при синтезе и очистке кремневольфрамовой кислоты.
Очищенными или синтезированными по настоящему изобретению гетерополикислотами можно пропитывать носитель. Этого можно добиться растворением гетерополикислоты в дистиллированной воде, а затем введением носителя в приготовленный таким образом водный раствор. Носитель целесообразно оставлять для пропитки в кислотном растворе в течение нескольких часов с периодическим перемешиванием вручную, а по прошествии этого времени ее рекомендуется отфильтровывать с применением воронки Бюхнера с целью удалить избыток кислоты.
Далее приготовленный таким образом влажный катализатор целесообразно поместить в сушильный шкаф, в котором его выдерживают при повышенной температуре в течение нескольких часов для сушки, и по прошествии этого времени ему дают остыть до комнатной температуры в эксикаторе. Катализатор можно также эффективно сушить с использованием тока нагретого газа, такого, как, например, азот или воздух. Массу введенного катализатора в граммах на литр определяют вычитанием массы использованного носителя из массы подвергаемого сушке катализатора.
По другому варианту носитель можно пропитывать катализатором с применением метода начальной влажности, сушить с использованием тока нагретого газа, такого, как, например, азот или воздух.
После этого нанесенный на носитель катализатор (содержание которого выражают в пересчете на массу) можно применять в таких процессах, как, например, гидратация олефинов с получением спиртов или присоединение к олефинам алифатических монокарбоновых кислот с получением соответствующих сложных эфиров. Приемлемое содержание гетерополикислоты, осажденной/введенной пропиткой в носитель, предназначенной для использования в ходе проведения реакции присоединения при получении сложных эфиров, находится в интервале от 10 до 60 мас.%, предпочтительно от 20 до 50 мас.%, более предпочтительно от 20 до 35 мас.% (что соответствует введенному количеству в интервале примерно от 100 до 215 г/л) в пересчете на общую массу гетерополикислоты и носителя.
Далее сущность настоящего изобретения проиллюстрирована со ссылкой на приведенные ниже примеры.
Примеры
Для иллюстрации ключевых технологических особенностей, имеющих решающее значение факторов и связанных с ними временных характеристик, ниже подробно описан синтез, проводимый в лабораторном масштабе. Общий выход продукта в лабораторных экспериментах в пересчете на вольфрам составлял >95%. Следует отметить, что молекулярная масса вольфрамокремниевой кислоты была равна 2878 или, если ее получали в виде гидрата, 2986.
Все используемые реактивы приобретали на фирме Aldrich:
раствор силиката натрия -
жидкое стекло каталог №33, 844-3
гидрат вольфрамата натрия каталог №22, 333-6
соляная кислота, 37 мас.% каталог №25, 814-8
вода - сорта для ВЭЖХ каталог №27, 073-3
метил-трет-бутиловый эфир (МТБЭ) каталог №17, 978-7
Вместо раствора силиката натрия может быть использован твердый силикат натрия. Вместо воды сорта для ВЭЖХ можно использовать высококачественную деионизированную воду. Для контактирования с реагентами и реакционными продуктами допускали только ПТФЭ и стекло. Было установлено, что контактирование вольфрамокремниевой кислоты с металлами, такими, как нержавеющая сталь, приводило к образованию темно-голубого продукта. Перед применением аппаратуру очищали соляной кислотой.
А. Собирали следующий аппарат: 3-литровая снабженная фланцем колба с электронагревателем, оборудованная установленной наверху эффективной мешалкой лопастного типа из политетрафторэтилена (ПТФЭ), холодильником Либиха и капельной воронкой. В процессе такой сборки никаких попыток исключить воздух не предпринимали.
В колбу загружали 500 г вольфрамата натрия, 37,5 г раствора силиката натрия, 1000 мл воды.
Реакционную смесь нагревали до кипения и во время такого нагрева вольфрамат натрия полностью растворялся с образованием прозрачного раствора. Нагревание до температуры кипения имело, как было установлено, существенное значение для успешного синтеза, поскольку, если этого не делать, во время добавления соляной кислоты (300 мл в течение 20 мин) образуется светло-желтый осадок вольфрамовой кислоты. Даже при кипячении реакционной смеси с обратным холодильником (примерно 105°С) скорость добавления соляной кислоты следует регулировать с целью предотвратить осаждение вольфрамовой кислоты по месту добавления. Во время этого добавления вследствие образования осадка диоксида кремния реакционная смесь становилась слегка мутной и бледно-желтой. Затем смесь охлаждали и фильтровали через стеклянный фильтр типа фильтра Бюхрнера с пористостью №3 и возвращали в колбу. Количество бледно-желтого осадка составляло примерно 10 г. После этого почти бесцветный прозрачный раствор вновь нагревали до кипения, а затем в смесь в течение 10 мин добавляли дополнительные 200 мл соляной кислоты. Далее бледно-желтой смеси давали остыть до комнатной температуры с получением водного раствора (2131,5 г). Никакого последующего осаждения не отмечали.
Б. В однолитровую делительную воронку загружали 321,7 г (255 мл) водного раствора с вышеописанной стадии (А), 143 мл МТБЭ. Смесь встряхивали в течение 2 мин и давали отстояться с разделением на три слоя. Начальное отстаивание с расслоением происходило быстро, но было установлено, что на границе раздела между верхним слоем и водным слоем в течение 3 мин образовывались капельки комплекса, и эти капельки опускались в нижний слой. По прошествии 10 мин получали следующие слои:
органический верхний слой, 50 мл, 58,4 г (содержание вольфрамокремниевой кислоты: <1 г)
средний слой, 320 мл, 321,7 г
плотный нижний слой, 60 мл, 89,2 г (содержание вольфрамокремниевой кислоты: 54,2 г).
Выход продукта в результате этих экстракций показывал, что возможно достижение выхода выделенной вольфрамокремниевой кислоты (в пересчете на вольфрам) >95%. В ходе проведения других экстракций первой реакционной смеси вместо МТБЭ может быть использован верхний органический слой.
По внешнему виду бесцветный плотный нижний слой по прошествии 10 мин был слегка мутным, обладал вязкостью, аналогичной вязкости этиленгликоля, а содержание натрия составляло >300 част./млн. При длительном стоянии (16 ч) помутнение исчезало, а содержание натрия в этом прозрачном продукте было равным <10 част./млн. Эффективность такого освобождения рассола от плотного нижнего слоя определяла чистоту конечного продукта, а так же то, была ли потребность в одной или нескольких обратных экстракциях.
Когда существовала необходимость в обратной экстракции, ее можно было проводить следующим образом. Для подавления ионизации и, следовательно, растворимости вольфрамокремниевой кислоты в воде ко всей свежей воде следовало, как было установлено, добавить дополнительное количество соляной кислоты.
В однолитровую делительную воронку загружали:
185 мл, 299,3 г продукта (комплекса вольфрамокремниевой кислоты/МТБЭ из прямой экстракции - оценивали как 171,9 г вольфрамокремниевой кислоты)
210 мл, 245,5 г 3 н. соляной кислоты.
При встряхивании (в течение 2 мин) смесь становилась мутноватой и при стоянии на четкие фазы не разделялась (водный слой оставался мутноватым).
Было установлено, что с целью упростить разделение необходимо было добавлять дополнительно 25 мл МТБЭ, предположительно для компенсации потери МТБЭ в водной фазе.
Верхний слой: 45 мл.
Средний слой: 235 мл (верхний + средний слои - 303,8 г).
Плотный нижний слой: 140 мл, 256,5 г.
Плотный нижний слой сушили и выход выделенной вольфрамокремниевой кислоты оценивали как равный 98%. Примечательно то, что вязкость и плотность продукта обратной экстракции оказывались более высокими: так, например, начальная плотность образца составляла 1,39, а плотность материала после обратной экстракции - 1,83. Эта тенденция к повышению вязкости и плотности продолжалась и на последующих обратных экстракциях, что приводило к получению продукта с плотностью 2,5 и такой же вязкостью, как у глицерина.
Было установлено, что одной или двух обратных экстракций в течение двадцатиминутного времени отстаивания при таком масштабе было достаточно для удаления всех следов рассола, хотя дополнительными экстракциями можно было добиться дальнейшего улучшения.
Это проиллюстрировано данными таблицы, полученными с 20-минутным периодом отстаивания.
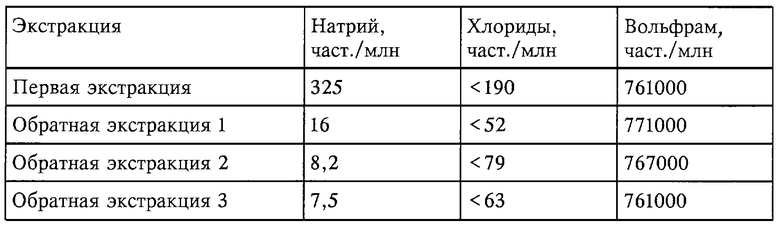
На эффективность этой экстракции влияет осветление комплексного слоя из водного слоя. Этим осветлению/demisting можно содействовать осуществлением такого технического приема, как применение демистеров или центрифуги.
Г1. Данное испытание показывает, каким образом метод добавления соляной кислоты влияет на синтез гетерополикислот.
Метод синтеза гетерополикислотного катализатора, описанный North E.O. в работе "Organic Synthesis", 1, с.129 (редактор H.S. Booth, Robert E. Krieger publishing company Huntington, New York 1978), повторяли в половине от указанного масштаба. Аппарат собирали так, как изложено в статье. Из материалов для контактирования с реактивами допускали только ПТФЭ и стекло Ругех®. Реакцию проводили на воздухе. По одному варианту осуществления метода Норта (North) реакцию проводили без доведения реакционной смеси до кипения перед добавлением соляной кислоты. Процесс описан ниже, а влияние добавления соляной кислоты при температуре кипения описано в разделе Г2.
3-литровую трехгорлую круглодонную колбу снабжали стеклянным карманом для термопары, капельной воронкой с уравновешенным давлением, ПТФЭ валом мешалки и лопастью и холодильником. В колбу загружали 500 г дигидрата вольфрамата натрия, 37,5 г силиката натрия и 1 л воды. Смесь перемешивали и в капельную воронку загружали 300 мл водной соляной кислоты концентрацией 37 мас.%. Включали нагреватель и по каплям в течение 90 мин добавляли кислоту, причем за это время смесь доводили до слабого кипения. Сразу же при добавлении кислоты наблюдали образование белого, а затем желтого осадка, в дальнейшем смесь становилась более вязкой вплоть до достижения 80° С, после чего вязкость снижалась и осадок становился известково-зеленым. Было установлено, что твердое вещество представляло собой вольфрамовую кислоту. Данные этих наблюдений не противоречили изложенному в статье Норта, в которой сообщается о небольшом количестве осадка кремниевой кислоты. В результате для повторного растворения осадка принимали решение добавить дополнительно 200 мл водной 37%-ной (по массе) соляной кислоты, определив, что значение рН смеси перед дополнительным добавлением кислоты соответствовало кислой среде. Дальнейшим кипячением смеси повторно растворить осадок не удавалось.
Г2. Полагали, что ответственным за образование осадка был метод добавления соляной кислоты (водная НС1) согласно разделу Г1. В статье Tatsuhiko H. и др., опубликованной в Kogyo Kagaku Zasshi, 72 (9), 1945-48 (1969), описана операция добавления как проводимая при повышенной температуре. Экспериментально температуру смеси определяли как равную 100-106°С, причем когда добивались устойчивого кипения, температура содержимого реактора составляла 106°С. Кроме того, как фактор, возможно относящийся к описываемым явлениям, рассматривают также предварительное уравновешивание реагентов. Таким образом, эксперимент, описанный в разделе Г1, повторяли со следующими изменениями в экспериментальном процессе (в соответствии с изобретением). В данном случае перед добавлением соляной кислоты при повышенной температуре смесь вольфрамата натрия, силиката натрия и воды доводили до кипения и выдерживали при кипении в течение 2 ч.
Раствор медленно становился мутным. После завершения операции добавления кислоты, которая продолжалась в течение 30 мин, до отключения нагревателя реакционную смесь выдерживали при кипячении в течение дополнительного часа. Смеси давали остыть без перемешивания в течение ночи, причем было отмечено, что за это время происходило выпадение некоторого дополнительного количества осадка, после чего смесь декантировали и фильтровали. Этот осадок легко удаляли промывкой водой. Получали примерно 10 г бледно-желтого воскоподобного твердого вещества, а фильтрат был прозрачным и почти бесцветным. Этот профильтрованный раствор возвращали в аппарат и с перемешиванием добавляли дополнительные 200 мл 37%-ной (по массе) водной соляной кислоты, и за это время температура повышалась примерно на 5°С. Прозрачной смеси давали остыть и ее использовали для последующих экспериментов с прямой жидкостно-жидкостной экстракцией.
Выход водного раствора: 2131,5 г.
Д. Воспроизводимость синтеза гетерополикислоты
После предыдущего успешного эксперимента, описанного в разделе Г2, метод этого раздела Г2 осуществляли в полном описанном масштабе с получением материала для изучения очистки и проверки воспроизводимости синтеза. Другими словами, использовали двойные количества в сравнении с количествами в предыдущих экспериментах. Так, например, в общем загружали 1000 г вольфрамата натрия, 75 г силиката натрия и 1000 мл 37%-ной (по массе) водной НСl. Было отмечено, что в точке вхождения кислоты в контакт с раствором во время начала добавления водной НСl появлялся желтый осадок, а при продолжении добавления кислоты этот осадок вновь растворялся. Полагали, что это было возможной причиной слабой окраски, которую замечали в конечном продукте. Было отмечено, что фильтрование для устранения осадка диоксида кремния в наибольшей степени устраняло также окраску раствора. Второе добавление соляной кислоты аналогичной концентрации обуславливало появление дополнительной бледно-желтой окраски.
Расчетный выход после экстракции МТБЭ
Общий выход гетерополикислоты в эксперименте вышеприведенного раздела Д составлял 54,2/321,7×4111,9=692,77 г.
Формульная масса (Fw) гетерополикислоты составляла 2878,94 г/моль без учета гидратной воды (или 2986 г/моль с гидратной водой, 6Н2О).
Формульная масса (Fw) вольфрамата натрия была равной 329,86 г/моль.
В расчетах гетерополикислоту Г оценивали как содержавшую 12 атомов вольфрама на молекулу, вследствие чего выход продукта составлял:
692,77/2878×12=2,88 моля (или 2,78 моля с учетом гидратной воды)
1000/329,87=3,03 моля
Выход=2,88/3,03=95% (или 92% с учетом гидратной воды)
Е. Усовершенствованный синтез гетерополикислоты
Предыдущие синтезы, описанные в разделах Г2 и Д, давали возможность узнать, как улучшить применяемые оборудование и метод синтеза, например, осуществлением предварительного уравновешивания или введением соляной кислоты с потоком флегмы в качестве средства снижения концентрации соляной кислоты по месту вхождения в контакт с реакционной смесью и, следовательно, ослабления окраски конечного раствора гетерополикислоты.
5-литровую круглодонную колбу снабжали ПТФЭ механической мешалкой, двойным холодильником, оборудованным 1-литровой делительной воронкой для введения в конденсатный поток, стеклянным карманом для термопары, термопарой, регулятором температуры и электронагревателем.
В колбу загружали 998,9 г вольфрамата натрия и дистиллированную воду. Затем добавляли 75,5 г силиката натрия. Смесь перемешивали и кипятили с обратным холодильником в течение 2 ч с целью обеспечить возможность образования структурных звеньев гетерополикислоты. В делительную воронку загружали приблизительно 600 мл водной 37%-ной (по массе) НСl, после чего кислоту по каплям добавляли совместно с конденсатным потоком с целью его разбавить и уменьшить количество появлявшегося желтого осадка. Скорость добавления кислоты задавали на уровне 1 капля/2 с вначале, а после добавления 200 мл кислоты ее повышали до 2 капель/с (приблизительно 20 мл/мин). В процессе такого добавления вследствие образования диоксида кремния смесь становилась белой и мутной. Если скорость введения оказывалась чрезмерно высокой, конденсатный поток становился слишком кислым, и по месту вхождения в контакт образовывался желтый осадок.
Далее кипячение смеси с обратным холодильником поддерживали в течение дополнительных 45 мин и ей давали остыть в течение ночи без перемешивания. Смесь фильтровали под вакуумом для удаления избытка диоксида кремния, колбу очищали и аппарат разбирали. Фильтрат возвращали в колбу. Фильтрованием собирали 13,3 г твердых частиц.
Прозрачный неокрашенный фильтратный раствор вновь доводили до кипения и в конденсатный поток со скоростью 30 мл/мин добавляли дополнительно 400 мл водной 37%-ной (по массе) НСl. По месту вхождения в контакт с раствором желтый осадок не появлялся, но с течением времени в массе реакционной смеси появлялась желтая окраска и возникало слабое помутнение.
Кипячение смеси с обратным холодильником поддерживали в течение еще одного часа и ей давали остыть в течение ночи. Фильтрование конечного раствора позволяло устранить окраску и помутнение смеси. В конечном итоге собирали 4089,7 г раствора.
Расчетный выход после экстракции МТБЭ
Общий выход гетерополикислоты в эксперименте вышеприведенного раздела Е составлял 23,1/142×4089,7=665,3 г.
Fw гетерополикислоты составляла 2878,94 г/моль (или 2986 г/моль с гидратной водой, 6Н2О).
Fw вольфрамата натрия была равной 329,86 г/моль.
В расчетах гетерополикислоту оценивали как содержавшую 12 атомов W на молекулу, вследствие чего выход продукта составлял:
665,3/2878×12=2,77 моля (или 2,67 моля с учетом гидратной воды)
1000/329,87=3,03 моля
Выход=2,77/3,03=91% (или 88% с учетом гидратной воды)
Ж. Исследование очистки продукта, полученного реакцией, описанной в разделе Д, без жидкостной экстракции. Вышеупомянутый метод очистки гетерополикислоты, описанный Нортом, применяли для жидкостно-жидкостной экстракции диэтиловым эфиром (ДЭЭ) с целью выделить из реакционной смеси гетерополикислоту в виде ее комплекса. Выделенный материал в дальнейшем очищали либо кристаллизацией и/либо обратной экстракцией. Сообщалось также о применении для таких процессов разделения других растворителей, например этилацетата и метилэтилкетона (МЭК). Оба эти материала обладают более высокой температурой самовоспламенения, чем ДЭЭ, что позволяет повысить безопасность при масштабировании.
В соответствии с настоящим изобретением в качестве безопасной альтернативы ДЭЭ предлагается новый экстрагент, метил-трет-бутиловый эфир (МТБЭ). Целью программы очистки является сравнительная оценка этих растворителей.
Выбор экстрагента/растворителя
Данную работу вели с целью определить оптимальный растворитель для экстракции. Операции экстракции проводили добавлением органического растворителя в раствор гетерополикислоты, полученной в примере Д, декантированием нижней плотной органической фазы, включавшей гетерополикислоту, и затем удалением растворителя из этой фазы. Этого добивались с помощью роторного испарителя. Гетерополикислоту сушили досуха и подвергали анализу для определения чистоты продукта. Эффективность экстракции оценивали определением массы выделенной гетерополикислоты и содержания примесей в продукте. Результаты каждого испытания экстрагента/растворителя представлены ниже.
Циклогексан (не в соответствии с изобретением)
Было установлено, что гетерополикислота нерастворима в циклогексане, вследствие чего этот растворитель оказывается непригодным.
Метилэтилкетон (2-бутанон) (не в соответствии с изобретением)
При добавлении метилэтилкетонового (МЭК) растворителя появлялись две фазы: нижняя, органическая фаза и верхняя, водная фаза. Твердую гетерополикислоту выделяли из органической фазы в виде желтого вещества. Это, по-видимому, объясняется тем, что МЭК растворитель вступал в катализируемую кислотой реакцию альдольной конденсации, которая сопровождалась возникновением бледно-желтой окраски (реакционные побочные продукты) с повышением ее интенсивности с течением времени, а также ее появлением как во время стадий нагрева (в роторном испарителе), так и в выделенном кетоне. Из этого можно было заключить, что МЭК как экстрагент/растворитель был непригодным.
Диэтиловый эфир (ДЭЭ) (не в соответствии с изобретением)
Из-за высокой горючести и низкой температуры самовоспламенения ДЭЭ предпринимали меры предосторожности. Загружаемые материалы: образец из примера Д: 250 мл (316,1 г) диэтиловый эфир: 250 мл (177 г благодаря плотности).
ДЭЭ вначале вводили медленно, с ополаскиванием, и отмечали отделение от верхней, водной фазы нижней, плотной фазы гетерополикислота/ДЭЭ. Последующее добавлением ДЭЭ вызывало образование над водной фазой третьей, верхней фазы ДЭЭ. Верхняя фаза ДЭЭ не растворяла плотную фазу гетерополикислота/ДЭЭ, как это показано ниже.
Смесь встряхивали в течение 5 мин и оставляли отстаиваться в течение 30 с. Появлялись три следующие фазы:
верхняя: преимущественно диэтиловый эфир - 200 мл (149 г)
средняя: водная - 140 мл (258,2 г)
нижняя: комплекс гетерополикислоты - 50 мл (85 г).
Нижняя фаза имела молочную окраску. Эту нижнюю фазу собирали и выпаривали на роторном испарителе. Выделяли 75,1 г белого микрокристаллического твердого продукта. Верхнюю фазу выпаривали досуха. Выделяли 0,7 г белого твердого продукта (демонстрировавшего низкую растворимость комплекса гетерополикислота/диэтиловый эфир в избытке диэтилового эфира).
Если исходить из этих результатов, то очевидно, по-видимому, что ДЭЭ может выполнять функции растворителя для экстракции гетерополикислоты из полученной реакционной смеси. Однако средний, водный слой содержал, как было установлено, значительное количество диэтилового эфира, и еще один недостаток состоит в том, что ДЭЭ характеризуется низкой температурой самовоспламенения и высокой горючестью.
Метил-трет-бутиловый эфир (МТБЭ) (в соответствии с настоящим изобретением)
В 1-литровую делительную воронку загружали: раствор гетерополикислоты из эксперимента раздела Д - 255 мл (321,7 г), МТБЭ - 63 мл (45,1 г).
Эту смесь встряхивали в течение 2 мин и давали ей отстояться в течение 5 мин. Она обладала молочной окраской, расслоение протекало очень медленно. Появлялись только две фазы. Добавляли дополнительно 30 мл (21,7 г) МТБЭ и экстракцию повторяли. Вновь можно было видеть только две фазы. Вводили последнюю дополнительную добавку 50 мл (37,2 г) МТБЭ. Экстракцию повторяли и давали отстояться в течение 10 мин. Три фазы обладали менее мутным внешним видом, чем тот, который отмечали до этого.
Верхний слой: 50 мл (58,4 г)
Средний слой: 320 мл (268,6 г)
Нижний слой: 60 мл (89,2 г)
Нижний слой выпаривали досуха, получая 54,2 г белого твердого вещества.
Выход=2,88/3,03=95% (или 92% с гидратной водой).
Судя по этим результатам, МТБЭ в качестве простого эфира оказывается эффективным при выделении гетерополикислоты. Он предпочтительнее диэтилового эфира благодаря своим более высокой температуре самовоспламенения и более низкой растворимости в водном слое.
Этилацетат
В 1-литровую делительную воронку загружали: раствор гетерополикислоты - 150 мл (178,6 г), этилацетат - 50 мл (43,6 г).
После введения этилацетата появлялась молочная окраска, которая во время 2-минутного встряхивания постепенно исчезала. Далее смеси давали отстояться в течение 5 мин. Внешне все три фазы выглядели прозрачными, на каждой из межфазных границ было видно всего по несколько пузырьков. Разделение протекало намного быстрее, чем в случае МББЭ.
Верхний слой: от 10 до 15 мл (13,1 г)
Средний слой: 120 мл (146,4 г)
Нижний слой: 40 мл (61,1 г)
Нижний слой выпаривали досуха, получая 34,0 г белого твердого вещества.
Выход=3,26/3,03>100%.
При определении выхода в этом случае не принимали во внимание возможное присутствие в твердом продукте захваченной им воды.
Обратная экстракция
Целью этих экспериментов являлось еще большее снижение содержания примесей в гетерополикислоте в сравнении с тем, которое было достижимым в результате одной прямой экстракции. Основными примесями являются неорганические соли, в особенности галогениды щелочных металлов, такие, как, например, хлорид натрия. Было установлено, что прямой жидкостно-жидкостной экстракцией удаляли значительную часть примесей, содержавшихся в реакционной смеси, включавшей полученную гетерополикислоту. Для дополнительной очистки продукта Норт предлагает два следующих метода: (I) перекристаллизация (которая в промышленном масштабе оказывается, что очевидно, дорогостоящей) и (II) жидкостно-жидкостная обратная экстракция. В данном случае исследовали этот последний метод.
Этилацетат (не в соответствии с изобретением)
Загружаемые материалы:
продукт из предыдущего эксперимента с прямой экстракцией этилацетатом:
293,0 г (170 мл, плотность - 1,72, гетерополикислота по приближенному расчету - 179,2 г)
водная 3 н. НСl: 202,6 г (180 мл)
После добавления кислоты смесь приобретала молочную окраску. После встряхивания в течение 2 мин и стояния в течение 15 мин появлялась только одна мутная фаза с очень медленным осаждением белого твердого вещества.
В смесь добавляли дополнительно 41 г (45 мл) этилацетата и ее вновь встряхивали. Никакого улучшения не отмечали.
Вводили семь дополнительных добавок этилацетата при общем количестве добавленного этилацетата 509 г. Отдельная фаза не образовывалась, но при введении каждой дополнительной добавки усиливалась флокуляция твердого вещества. В результате этих наблюдений приходили к заключению, что, вероятно, происходил гидролиз этилацетата, который препятствовал разделению фаз. Таким образом, в качестве растворителя для обратной экстракции этилацетат является неприемлемым.
Метил-трет-бутиловый эфир (МТБЭ) (в соответствии с настоящим изобретением)
Для оценки приемлемости растворителя проводили ряд последовательных обратных экстракций с использованием МТБЭ.
Обратная экстракция 1
Загружаемые материалы:
комплекс гетерополикислоты/МТБЭ как продукт прямой экстракции - 185 мл (299,3 г, по приближенному расчету - 171,89 г гетерополикислоты) смешивали с водной НСl: (в данном случае использовали 37%-ную [по массе] и в дальнейшем было установлено, что можно также применять 3 н.) - 210 мл (245,5 г).
При встряхивании смесь приобретала молочную окраску и наблюдали медленное расслоение без верхнего слоя. Дополнительно добавляли 18,4 г МТБЭ ('25 мл) и смесь встряхивали. Смесь оставалась окрашенной в молочный цвет, но по прошествии 10 мин образовывалось три слоя, которые медленно осветлялись, становились менее мутными.
Верхний слой: 45 мл
Средний слой: 235 мл (верхний + средний слои - 303,8 г)
Нижний слой: 140 мл (256,5 г)
Нижний слой был более вязким, чем первый базовый экстракционный образец (плотность образца - 1,39, плотность продукта обратной экстракции - 1,83).
Образец продукта обратной экстракции после первой обратной экстракции сушили и взвешивали, установив, что выход гетерополикислоты составлял 167,93/171,89×100=97,7%.
Обратная экстракция 2
Загружаемые материалы: продукт обратной экстракции 1-120 мл (234,2 г, 153,33 г гетерополикислоты) смешивали с водной НСl: (37%-ная [по массе]) - 120 мл (140,2 г)
В результате встряхивания вышеупомянутой смеси получали две фазы, но нижняя фаза начинала налипать на поверхности и обладала густой молочной окраской.
В эту смесь добавляли 11,5 г (11,5 г, '16 мл) МТБЭ и смесь встряхивали повторно. На этот раз расслоение проходило быстрее и в течение примерно 5 мин образовывалась нижняя фаза, которая легче поддавалась обработке.
Верхний слой: 40 мл
Средний слой: 135 мл (масса верхнего + среднего слоев - 194,6 г)
Нижний слой: 75 мл (масса: 187,2 г, плотность: 2,496)
Образец нижнего слоя сушили и взвешивали, в результате чего устанавливали, что выход гетерополикислоты составлял 146,7/153,33×100=95,7%.
Обратная экстракция 3
Загружаемые материалы:
продукт обратной экстракции 2 - 68 мл (159,9 г, 125,34 г гетерополикислоты) смешивали с водной 37%-ной (по массе) НСl - 70 мл (81,8 г),
МТБЭ - 9 мл (6,8 г).
Образовавшуюся смесь молочной окраски отстаивали в течение 2-3 мин с получением следующих трех фаз:
Верхний слой: 13 мл
Средний слой: 80 мл (масса верхнего + среднего слоев - 117 г)
Нижний слой: 50 мл (масса: 129 г, плотность: 2,58, несколько больше, чем у вышеприведенного).
Продукт этой экстракции сушили досуха и взвешивали. Масса продукта: 102 г Выход гетерополикислоты: 102/125,34×100=81,4%.
Эти результаты показывают, что МТБЭ является превосходным растворителем для обратной экстракции и позволяет получать гетерополикислоту, в существенной степени свободную от посторонних катионов.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ХИРАЛЬНЫХ ТРИАЗОЛОНОВ | 2011 |
|
RU2585760C2 |
| РЕАКЦИОННАЯ РЕКТИФИКАЦИЯ ДЛЯ ДЕГИДРАТАЦИИ СМЕШАННЫХ СПИРТОВ | 2006 |
|
RU2419595C2 |
| СПОСОБ ИЗВЛЕЧЕНИЯ КАТАЛИЗАТОРА | 2008 |
|
RU2484900C2 |
| СПОСОБ ПОЛУЧЕНИЯ ХИРАЛЬНЫХ ГИДРАЗИДОВ | 2011 |
|
RU2588570C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГИПОАЛЛЕРГЕННЫХ МОХОВЫХ МАСЕЛ | 1994 |
|
RU2095400C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПЛЕВРОМУТИЛИНОВ | 2007 |
|
RU2512591C2 |
| СПОСОБ СИНТЕЗА МАНДИПРОПАМИДА И ЕГО ПРОИЗВОДНЫХ | 2006 |
|
RU2470914C9 |
| СПОСОБ ПОЛУЧЕНИЯ СЛОЖНЫХ ЭФИРОВ ЦИКЛОПРОПАНА ИЗ 3-МЕТИЛБУТ-2-ЕН-1-АЛЯ | 1994 |
|
RU2120936C1 |
| СПОСОБ ПОЛУЧЕНИЯ НИЗШИХ АЛИФАТИЧЕСКИХ СЛОЖНЫХ ЭФИРОВ | 1999 |
|
RU2225386C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ КАРБОНОВОЙ КИСЛОТЫ И ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ | 1994 |
|
RU2145959C1 |
Изобретение относится к получению и очистке гетерополикислоты. Способ включает реакцию водного раствора солей щелочных или щелочно-земельных вольфраматов или модибдатов с силикатом или фосфатом щелочного или щелочно-земельного металла. Реакцию проводят при кипячении с обратным холодильником. Полученный раствор очищают от примесей. Обработку водного раствора, включающего гетерополикислоту и примеси солей, осуществляют по меньшей мере одной стадией жидкостно-жидкостной экстракции органическим растворителем. В качестве растворителя используют дигидрокарбиловый простой эфир, содержащий по крайней мере 5 углеродных атомов. Результат изобретения: повышение степени очистки гетерополикислоты. 2 с. и 12 з.п. ф-лы.
а) реакцию (I) водного раствора одной или нескольких солей щелочных или щелочноземельных металлов, выбранных из вольфрамата и молибдата, при кипячении с обратным холодильником с (II) силикатом щелочного или щелочноземельного металла или фосфатом щелочного или щелочноземельного металла с получением неочищенного водного раствора, включающего гетерополикислоту выбранной соли и все другие соли, образовавшиеся in situ или используемые в качестве реагента; б) фильтрование и охлаждение этого неочищенного водного раствора, включающего гетерополикислоту; в) очистку охлажденного неочищенного водного раствора со стадии (б) по меньшей мере одной жидкостно-жидкостной экстракцией органическим растворителем для предоставления гетерополикислоте возможности образовывать in situ комплекс с органическим растворителем и предоставление экстракту возможности разделиться на две или большее число фаз, включая, в частности, плотную органическую фазу, которая содержит растворенный комплекс гетерополикислоты, причем эта плотная органическая фаза находится ниже водной фазы; г) выделение этой плотной органической фазы со стадии (в) и ее тщательное смешение с водой с получением отдельного слоя разбавленной водой смеси и д) выделение из разбавленной водной смеси концентрированного (от 20 до 80 мас.%) водного раствора гетерополикислоты, практически свободной от органических примесей, где органический растворитель, используемый на стадии жидкостно-жидкостной экстракции (в), включает дигидрокарбиловый простой эфир, содержащий по меньшей мере 5 углеродных атомов.
| НИКИТИНА Е.А | |||
| Гетерополисоединения | |||
| - М.: Госхимиздат, 1962, с.110-111, 134-136,158-161, 200-203 | |||
| СПОСОБ ВЫДЕЛЕНИЯ КАТАЛИЗАТОРА СИНТЕЗА ФЕНОЛА И АЦЕТОНА | 1979 |
|
SU1019711A1 |
| СПОСОБ ПОЛУЧЕНИЯ ГЕТЕРОПОЛИКИСЛОТ | 1992 |
|
RU2076071C1 |
| US 3243258 A, 29.03.1966. | |||

