Настоящее изобретение относится к способу получения хирального соединения, в частности к способу получения хирального соединения, которое можно использовать в качестве промежуточного продукта для получения фунгицидных средств, предпочтительно позаконазола.
Уровень техники
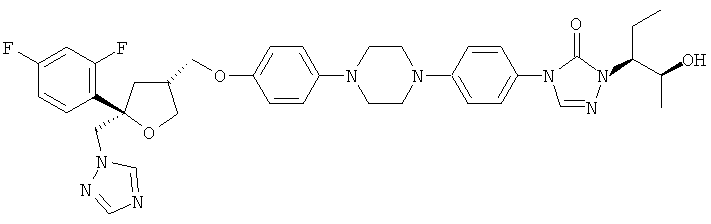
Позаконазол (регистрационный № CAS 171228-49-2; название CAS: 2,5-ангидро-1,3,4-тридезокси-2-C-(2,4-дифторфенил)-4-[[4-[4-[4-[1-[(1S,2S)-1-этил-2-гидроксипропил]-1,5-дигидро-5-оксо-4Н-1,2,4-триазол-4-ил]фенил]-1-пиперазинил]фенокси]метил]-1-(1Н-1,2,4-триазол-1-ил)-D-трео-пентит) является триазольным фунгицидным лекарственным средством, обладающим структурой:
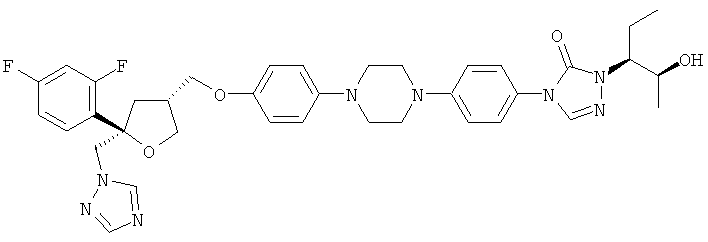
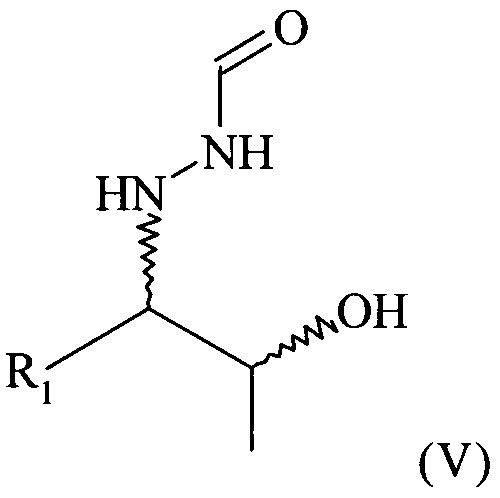
Позаконазол используется, например, для предупреждения и/или лечения инвазивных грибковых инфекций, вызванных штаммами Candida, штаммами Mucor, штаммами Aspergillus, штаммами Fusarium или штаммами Coccidioides, у пациентов с ослабленным иммунитетом и/или у пациентов, у которых заболевание устойчиво к воздействию других фунгицидных средств, таких как амфотерицин B, флуконазол или итраконазол, и/или у пациентов, которые не переносят эти фунгицидные средства. Одним из важных промежуточных продуктов для получения позаконазола является соединение формулы (V)

предпочтительно соединение формулы (V), в которой R1 = этил
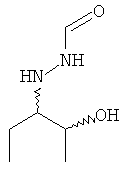 .
.
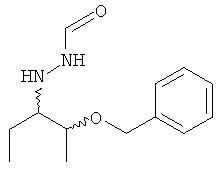
В настоящее время отсутствует способ, который дает соединение формулы (V), предпочтительно соединение формулы (V), в которой R1 = этил, в особенности с высокой энантиомерной, диастереоизомерной чистотой и выходом. До настоящего времени в литературе описано только соответствующее содержащее бензильную защитную группу соединение, представленное ниже. К сожалению, соединение формулы (V), в которой R1 = этил, затруднительно получить путем удаления бензильной защитной группы при стандартных условиях гидрогенолиза.
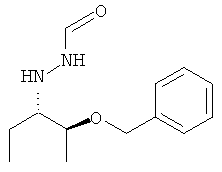
Обычным промежуточным продуктом для способа получения позаконазола является соединение формулы
 .
.
Способ получения этого промежуточного продукта раскрыт в WO 95/17407. Суммарный выход этого способа составляет примерно 25% и диастереоизомерная чистота изомера формулы
находится в диапазоне 94-99% и энантиомерная чистота зависит от качества исходного вещества, метилового эфира молочной кислоты. WO 95/17407 не описана очистка указанного промежуточного продукта.
В WO 97/22579 и публикации Saksena et al., Tetrahedron Lett. 2004, 45 (44), 8249-8251, раскрыты улучшенные способы проведения реакции Гриньяра в последовательности реакций, описанной в WO 95/17407. Описана стадия силилирования и добавление трет-BuMgCl при реакции Гриньяра с получением промежуточного продукта чистотой 95% без проведения дополнительной очистки с помощью хроматографии. Однако авторы настоящего изобретения, хотя и являющиеся высококвалифицированными специалистами в этой конкретной области химии, не смогли воспроизвести эти результаты. Несмотря на разумную модификацию методики, описанной в WO 97/22579 и публикации Saksena et al., промежуточный продукт всегда получали в виде сложной смеси, которую для обеспечения заявленной чистоты было необходимо дважды обработать с помощью хроматографии.
Другой способ, раскрытый в WO 96/33163, включает стереохимическое разделение промежуточного продукта путем образования соли с использованием хиральных кислот (например, дибензоил-L-винной кислоты, L-ДБВК) и кристаллизации полученных диастереоизомерных солей для получения указанного выше промежуточного продукта с высокой оптической чистотой. В другом способе, раскрытом в WO 97/33178, необходима защита одного гидразинового атома азота для введения хирального центра восстановлением дорогостоящими реагентами с селективностью по искомому изомеру, составляющей 0-94%. Ни в WO 96/33163, ни в WO 97/33178 не описана очистка продуктов за исключением стереохимического разделения.
Все эти способы предшествующего уровня техники обладают значительными недостатками.
Во-первых, в описанных методиках необходимо использовать защитной группы для группы ОН при синтезе промежуточного гидразида, и при последующем получении фунгицидных средств. Кроме того, жесткие условия проведения реакции синтеза могут рассматриваться, как запретительные для широко использующихся защитных групп, включая силиловые простые и сложные эфирные группы. Представляется, что достаточно стабильны только простые эфиры, и это может быть причиной того, что во всех конкретных примерах раскрыт только бензиловый эфир.
Во-вторых, ни один из этих способов предшествующего уровня техники не позволяет прямо получить незащищенные соединения формулы (V) и, например, необходима защита группы OH соединения формулы (V) желательной защитной группой, подобранной для последующих реакций.
В-третьих, как отмечено выше, трудоемкая очистка маслообразных продуктов с помощью хроматографии или стереохимического разделения необходима для компенсации недостаточной химической селективности и стереоселективности реакций предшествующего уровня техники.
В-четвертых, регулировка множества стадий окисления реакций предшествующего уровня техники, включая большой избыток дорогостоящих реагентов, увеличивает количество отдельных стадий и уменьшает суммарный выход. Эти недостатки значительно уменьшают выход искомого гидразида формулы (V) и предпочтительно формулы (V), в которой R1 = этил
и соответственно их защищенных производных. Одновременно образуется большое количество нежелательных побочных продуктов, что делает способ предшествующего уровня техники еще более неудовлетворительным.
Поэтому объектом настоящего изобретения является разработка эффективного способа получения хиральных гидразидов, предпочтительно получения соединения формулы (V), в которой R1 = этил
которое можно эффективно использовать в качестве промежуточного продукта для получения азольных фунгицидных средств, предпочтительно позаконазола.
Другим объектом настоящего изобретения является разработка эффективного способа получения кристаллического соединения формулы (V) и предпочтительно формулы (V), в которой R1 = этил
и, кроме того, соединений на основе этого кристаллического соединения, в которых группа ОН соответствующим образом защищена.
Другим объектом настоящего изобретения является получение самого кристаллического соединения, а также указанного соответствующим образом защищенного соединения.
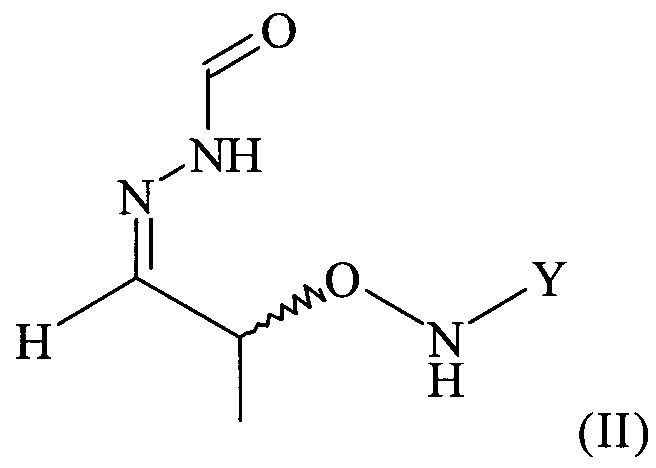
Согласно изобретению неожиданно было установлено, что указанные выше задачи решаются способом, в котором на первой стадии получают соединение формулы
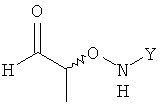
в которой Y обозначает необязательно замещенный арильный фрагмент, и его вводят в реакцию с H2N-NH-CHO в подходящем растворителе, в результате чего получают хиральное соединение формулы
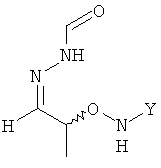 .
.
Краткое изложение сущности изобретения
Настоящее изобретение относится к способу получения хирального соединения, включающему
(1) использование хирального соединения формулы (I)
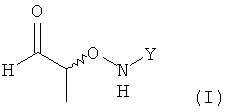 ,
,
в которой Y обозначает необязательно замещенный арильный фрагмент, предпочтительно необязательно замещенный фенильный фрагмент, более предпочтительно незамещенный фенил;
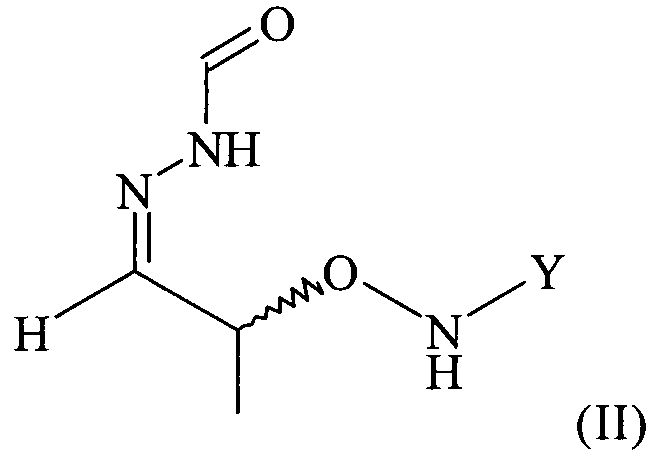
(2) реакцию соединения формулы (I) с H2N-NH-CHO в растворителе с получением соединения формулы (II)
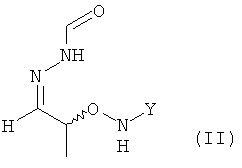 .
.
Кроме того, настоящее изобретение относится к способу получения хирального соединения, включающему
(1) использование хирального соединения формулы (I)
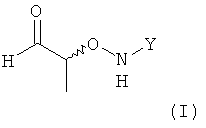 ,
,
в которой Y обозначает необязательно замещенный арильный фрагмент, предпочтительно необязательно замещенный фенильный фрагмент, более предпочтительно незамещенный фенил;
(2) реакцию соединения формулы (I) с H2N-NH-CHO в растворителе с получением соединения формулы (II)
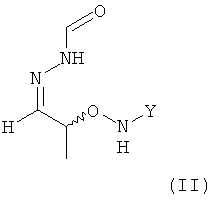 ;
;
(3) выделение соединения формулы (II) из реакционной смеси, полученной на стадии (2), с помощью жидкостной экстракции, где до стадии (3) предпочтительно проводят замену растворителя;
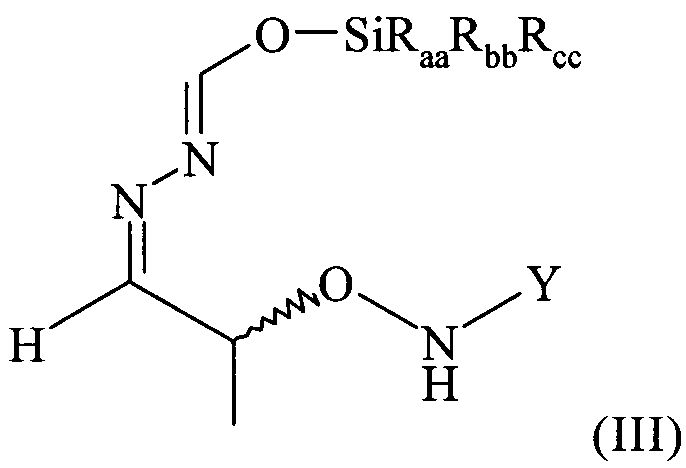
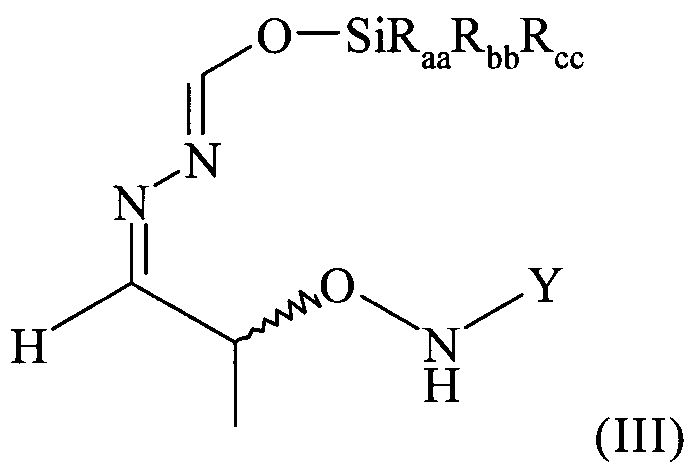
(4) реакцию соединения формулы (II) в растворителе с силилирующим реагентом, содержащим остаток -SiRaaRbbRcc, с получением соединения формулы (III)
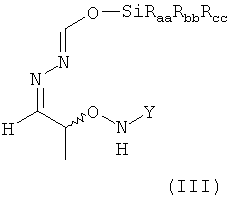
в которой остатки Raa, Rbb и Rcc могут быть одинаковыми или разными и предпочтительно представляют собой алкильные или арильные остатки, более предпочтительно алкильные остатки, содержащие от 1 до 6 атомов углерода, предпочтительно от 1 до 4 атомов углерода, более предпочтительно 1 или 2 атома углерода;
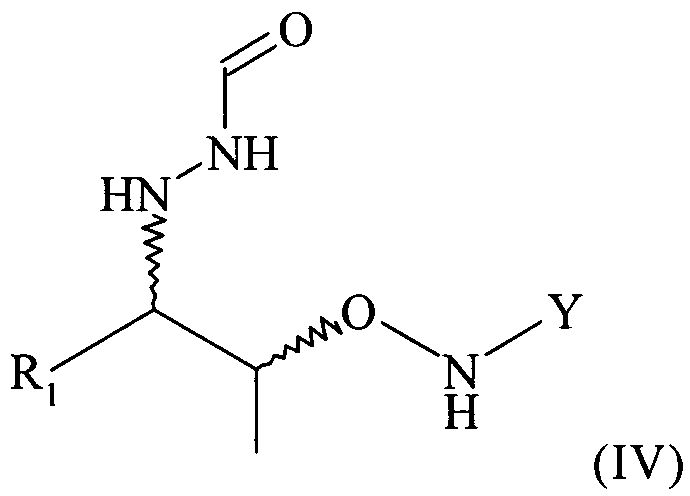
(5) реакцию соединения формулы (II) или реакцию соединения формулы (III) с нуклеофильным соединением, содержащим нуклеофильный остаток R1, в растворителе с получением соединения формулы (IV)
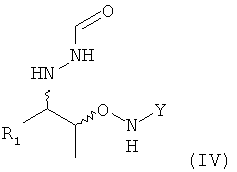 ;
;
(6) восстановление соединения формулы (IV), предпочтительно путем гидрирования, с получением соединения формулы (V)
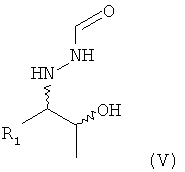 ;
;
(7) необязательно реакцию соединения формулы (V) в растворителе с силилирующим реагентом, содержащим остаток -SiRaRbRc, с получением соединения формулы (VI)
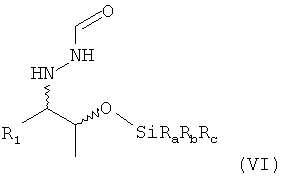 ,
,
в которой остатки Ra, Rb и Rc могут быть одинаковыми или разными и предпочтительно представляют собой алкильные или арильные остатки.
Настоящее изобретение также относится к хиральному соединению, которое можно получить или получают способом, предлагаемый в настоящем изобретении.
В частности, настоящее изобретение относится к предпочтительно кристаллическому хиральному соединению формулы (V), в которой R1 предпочтительно означает алкильный остаток, предпочтительно содержащий от 1 до 6 атомов углерода, более предпочтительно от 1 до 4 атомов углерода, еще более предпочтительно 1 или 2 атома углерода, наиболее предпочтительно 2 атома углерода, R1 предпочтительно обозначает этил, где предпочтительно не менее 95%, более предпочтительно не менее 97%, еще более предпочтительно не менее 99% молекул указанного кристаллического соединения находятся в виде изомера формулы (Va)
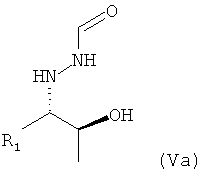 ,
,
предпочтительно в виде изомера формулы (Vb)
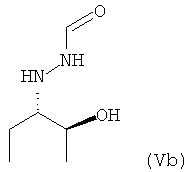 .
.
Кроме того, настоящее изобретение также относится к применению хирального соединения, предлагаемого в настоящем изобретении, предпочтительно соединения формулы (Vb), для получения фунгицидного средства, предпочтительно для получения позаконазола.
Краткое описание чертежей
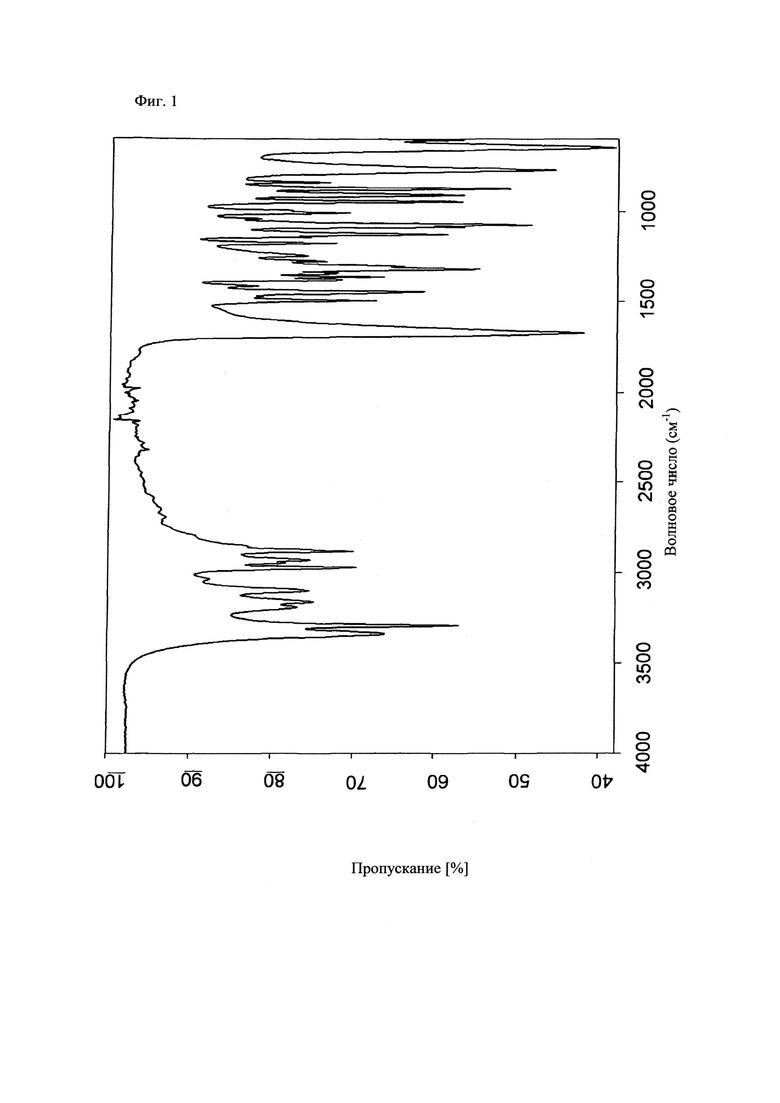
На фиг.1 приведен инфракрасный спектр (ИК) соединения формулы (V), полученного в примере 3 настоящего изобретения. На фиг.1 по оси y отложено пропускание в %, а по оси x отложено волновое число в см-1. В частности, могут обнаруживаться следующие пики в ИК-спектре: 3341, 3298, 2970, 2881, 1674, 1497, 1447, 1319, 1125, 1071, 945, 910, 876, 775 и 650 +/-2 см-1.
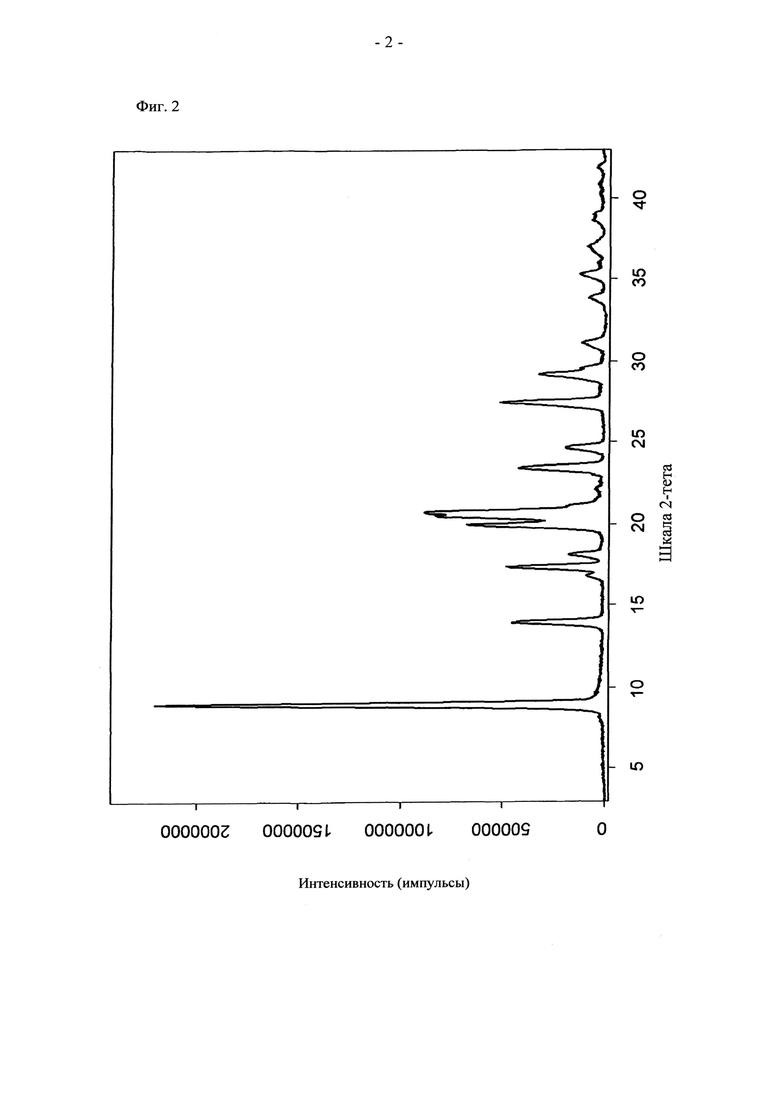
На фиг.2 приведена рентгенограмма соединения формулы (V), полученного в примере 3 настоящего изобретения. На фиг.2 по оси y отложена интенсивность, измеренная в импульсах за 300 с (линейная шкала), а по оси x отложено положение, указанное в значениях 2-тета в градусах. В частности, могут обнаруживаться следующие пики ПРГ: 9,0, 13,9, 17,4, 18,1, 20,0, 20,7, 23,4, 24,6, 27,5 и 29,2 +/-0,2°2-тета.
Подробное описание изобретения
Как отмечено выше, настоящее изобретение относится к способу получения хирального соединения, включающему
(1) использование хирального соединения формулы (I)
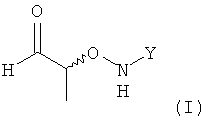 ,
,
в которой Y обозначает необязательно замещенный арильный фрагмент, предпочтительно необязательно замещенный фенильный фрагмент, более предпочтительно незамещенный фенил;
(2) реакцию соединения формулы (I) с H2N-NH-CHO в растворителе с получением соединения формулы (II)
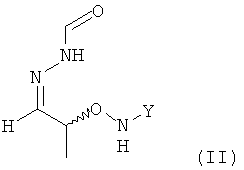 .
.
Стадия (1)
На стадии (1) настоящего изобретения используют соединение формулы (I), которое взаимодействует на стадии (2) с H2N-NH-CHO в подходящем растворителе.
Обычно на получение соединения формулы (I) не налагают специальных ограничений. В частности, можно использовать любую подходящую методику получения соединения формулы (I). В предпочтительном варианте осуществления настоящего изобретения являются предпочтительными такие методики, которые позволяют получить соединение формулы (I) с высокой энантиоселективностью. В еще более предпочтительном варианте осуществления настоящего изобретения являются предпочтительными такие методики, которые позволяют получить соединения (I) где не менее 95%, более предпочтительно не менее 97%, более предпочтительно не менее 99% молекул хирального соединения формулы (I), полученного на стадии (1), содержатся в виде изомера формулы (Ia)
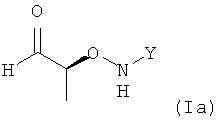 .
.
При Y = фенил получение соединения формулы (I) по реакции с использованием органического катализатора с превосходной энантиоселективностью является известным. Дается ссылка на публикации Brown et al., J. Chem. Soc. 2003, 125 (36), 10808-10809 и Cordova et al., Chem. Eur. J. 2004, 10 (15), 3673-3684; а также Hayashi et al. J. Org. Chem. 2005, 69 (18), 5966-5973.
В предпочтительном варианте осуществления соединение формулы (I), предлагаемое в настоящем изобретении, получают по реакции пропионового альдегида в подходящем растворителе с соединением формулы (i)
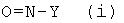 .
.
Остаток Y в соединении формулы (i) обозначает необязательно замещенный арильный фрагмент, предпочтительно необязательно замещенный фенильный фрагмент, более предпочтительно незамещенный фенил. Термин "арильный фрагмент" при использовании в контексте настоящего изобретения означает карбоциклическую ароматическую группу, такую как фенил или нафтил и т.п. Термин "замещенный арильный фрагмент" и "замещенный фенил" соответственно при использовании в контексте настоящего изобретения означает такой арильный фрагмент и фенил соответственно, которые предпочтительно содержат 1, 2 или 3 заместителя, которые предпочтительно выбраны из группы, включающей галоген, алкил и C1-алкоксигруппу или C2-алкоксигруппу, или C3-алкоксигруппу, или C4-алкоксигруппу, или C5-алкоксигруппу, или C6-алкоксигруппу. Термин "алкил" при использовании в контексте настоящего изобретения означает линейные или разветвленные алкильные фрагменты, которые предпочтительно содержат 1, 2, 3, 4, 5 или 6 атомов углерода. Термин "галоген" при использовании в контексте настоящего изобретения предпочтительно означает атом хлора, брома или йода.
Поэтому, предпочтительно, если остаток Y в соединении формулы (i) не означает бензильный фрагмент, предпочтительно не означает алкиларильный фрагмент. Термин "алкиларильный фрагмент" при использовании в контексте настоящего изобретения означает алкильный фрагмент, который замещен по меньшей мере одним арильным фрагментом. Для иллюстрации, бензильный фрагмент представляет собой алкиларильный фрагмент, где алкилом является метил, который замещен одним фенильным фрагментом.
Таким образом, в предпочтительном варианте осуществления настоящего изобретения соединением формулы (i) является нитрозобензол.
На реакцию пропионового альдегида с соединением формулы (i) не налагают специальных ограничений при условии, что получают соединение формулы (I), предпочтительно соединение (I), где не менее 95%, более предпочтительно не менее 97%, более предпочтительно не менее 99% молекул хирального соединения формулы (I), полученного на стадии (1) содержатся в виде изомера формулы (Ia)
.
Предпочтительно, если в реакции получения соединения формулы (I) используют по меньшей мере один, предпочтительно точно один органический катализатор. Более предпочтительно, если в качестве органического катализатора используют пролин (Pro), еще более предпочтительно D-пролин (D-Pro)
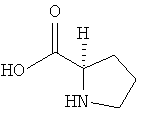 .
.
В необязательном варианте осуществления настоящего изобретения по меньшей мере один подходящий промотор можно использовать для промотирования реакции, предпочтительно реакции с использованием органического катализатора на стадии (1). Хотя можно использовать любой подходящий промотор, в контексте настоящего изобретения предпочтение отдается производному мочевины и 1-(2-диметиламиноэтил)-3-фенилмочевина является особенно предпочтительной.
Согласно изобретению было установлено, что проведение этой реакции в присутствии кислоты является еще более предпочтительным. Хотя обычно может не быть специальных ограничений, предпочтение отдается кислотам Бренстеда, в особенности органическим кислотам Бренстеда. В других предпочтительных вариантах осуществления используют уксусную кислоту или пропионовую кислоту или смесь этих кислот. а количество используемых кислот не налагают специальных ограничений. Каталитические количества являются предпочтительными. По сравнению с количествами использующегося органического катализатора является еще более предпочтительным использование кислоты в меньшем количестве. В возможных вариантах осуществления и в пересчете на 1 экв. соединения формулы (i) количество органического катализатора может находиться в диапазоне от 0,15 до 0,5, более предпочтительно в диапазоне от 0,2 до 0,4, еще более предпочтительно в диапазоне от 0,25 до 0,35 экв. и количество кислоты может находиться в диапазоне от 0,01 до 0,1, более предпочтительно в диапазоне от 0,02 до 0,09, еще более предпочтительно в диапазоне от 0,03 до 0,08 экв.
Химическая природа подходящего растворителя, в котором проводят реакцию на стадии (1), зависит главным образом от конкретных исходных веществ, использующегося катализатора или катализаторов, кислоты или кислот, если они используются, и/или содержащегося промотора или промоторов, если они используются. Можно использовать один растворитель или смесь двух или большего количества растворителей. В предпочтительном варианте осуществления настоящего изобретения, в котором пропионовый альдегид вводят в реакцию с нитрозобензолом в присутствии D-Pro и в присутствии уксусной кислоты и/или пропионовой кислоты, реакцию предпочтительно проводят в дихлорметане (ДХМ) в качестве растворителя.
Температура, при которой проводят реакцию на стадии (1), зависит главным образом от конкретных исходных веществ, использующегося катализатора или катализаторов, кислоты или кислот, если они используются, и/или содержащегося промотора или промоторов, если они используются. В предпочтительном варианте осуществления настоящего изобретения, в котором предпочтительно пропионовый альдегид вводят в реакцию с нитрозобензолом в присутствии D-Pro и в присутствии уксусной кислоты и/или пропионовой кислоты в ДХМ в качестве растворителя, реакцию предпочтительно проводят при температуре в диапазоне от -15 до +5°C, более предпочтительно от -12 до +3°C, еще более предпочтительно от -10 до 0°C.
Атмосфера, в которой проводят реакцию получения соединения формулы (I), обычно зависит от конкретных исходных веществ, использующегося растворителя или растворителей, использующегося катализатора или катализаторов, кислоты или кислот, если они используются, и/или содержащегося промотора или промоторов, если они используются. В предпочтительном варианте осуществления настоящего изобретения, в котором пропионовый альдегид вводят в реакцию с нитрозобензолом в присутствии D-Pro, реакцию проводят в инертной или в основном инертной атмосфере, такой как атмосфера N2.
Таким образом, по реакции, которую проводят на стадии (1), соединение формулы (I) предпочтительно получают по меньшей мере в одном растворителе, предпочтительно ДХМ, где не менее 95%, более предпочтительно не менее 97%, более предпочтительно не менее 99% молекул хирального соединения формулы (I), полученного на стадии (1), содержатся в виде изомера формулы (Ia)
.
Стадия (2)
Обычно можно выделить соединение формулы (I) из реакционной смеси, полученной на стадии (1). Однако в особенно предпочтительном варианте осуществления настоящего изобретения такое выделение не требуется. Поэтому настоящее изобретение также относится к описанному выше способу, в котором после стадии (1) соединение формулы (I) не выделяют из реакционной смеси, полученной на стадии (1), до реакции с H2N-NH-CHO на стадии (2).
Таким образом, в возможном варианте осуществления реакционную смесь, полученную на стадии (1), можно использовать на стадии (2) без обработки. Альтернативно, реакционную смесь можно дополнительно разбавить по меньшей мере одним подходящим растворителем, или, при желании, подходящее количество растворителя можно удалить из реакционной смеси с помощью подходящего концентрирования.
На стадии (2) настоящего изобретения соединение формулы (I) вводят в реакцию с H2N-NH-CHO в растворителе. Этот растворитель может представлять собой смесь растворителей, если, например, реакционную смесь, полученную на стадии (1), смешивают с H2N-NH-CHO (формилгидразин), содержащимся в растворителе, который отличается от растворителя, содержащегося в реакционной смеси, полученной на стадии (1). В особенно предпочтительном варианте осуществления настоящего изобретения растворителем, который содержится в реакционной смеси, полученной на стадии (1), является тот же растворитель, который используют для проведения реакции на стадии (2). Предпочтительно, если этим растворителем является ДХМ.
Как и для реакции на стадии (1), температура, при которой проводят реакцию на стадии (2), зависит главным образом от конкретных соединений, которые вводят в реакцию друг с другом, использующегося катализатора или катализаторов, кислоты или кислот, если они используются, и/или содержащегося промотора или промоторов, если они используются. В предпочтительном варианте осуществления настоящего изобретения предпочтительно, в котором пропионовый альдегид вводят в реакцию с нитрозобензолом в присутствии D-Pro и в присутствии уксусной кислоты и/или пропионовой кислоты в ДХМ в качестве растворителя на стадии (1), и, если также реакцию на стадии (2) проводят в ДХМ в качестве растворителя, реакцию на стадии (2) предпочтительно проводят при температуре в диапазоне от -10 до +20°C, предпочтительно от -5 до +5°C.
Предпочтительно, если реакцию на стадии (2) проводят в присутствии по меньшей мере одного молекулярного сита. Характеристики пор по меньшей мере одного молекулярного сита можно адаптировать в соответствии с конкретными требованиями. Однако являются предпочтительными такие сита, которые обладают диаметрами пор, определенными в соответствии со стандартом DIN 66131, находящимися в диапазоне от 0,3 до 0,5 нм (нанометров; от 3 до 5 Å), предпочтительно от 0,35 до 0,45 нм (от 3,5 до 4,5 Å), и диаметр пор, равный примерно 0,4 нм (4 Å), является особенно предпочтительным.
Атмосфера, в которой проводят реакцию на стадии (2) для получения соединения формулы (II), также, обычно зависит от конкретных соединений, которые вводят в реакцию друг с другом, использующегося растворителя или растворителей, использующегося катализатора или катализаторов, кислоты или кислот, если они используются, и/или содержащегося промотора или промоторов, если они используются. В предпочтительном варианте осуществления настоящего изобретения, в котором пропионовый альдегид вводят в реакцию с нитрозобензолом в присутствии D-Pro и в присутствии уксусной кислоты и/или пропионовой кислоты в ДХМ в качестве растворителя на стадии (1), и, если также реакцию на стадии (2) проводят в ДХМ в качестве растворителя, реакцию на стадии (2) проводят в инертной или в основном инертной атмосфере, такой как атмосфера N2.
Стадия (3)
Из реакционной смеси, полученной на стадии (2), можно выделить соединение формулы (II). Поскольку на то, как проводят такое выделение, не налагают специальных ограничений, жидкостная экстракция является предпочтительной методикой, предлагаемым в настоящем изобретении. До стадии такой экстракции предпочтительно проводят подходящую замену растворителя. При замене растворителя особенно предпочтительно заменять растворитель, в котором получают соединение формулы (II) на стадии (2), на растворитель или смесь двух или большего количества растворителей, которые являются подходящими для предпочтительной следующей стадии настоящего изобретения, такой как стадия реакции (4), подробно описанная ниже в настоящем изобретении. Конкретный тип растворителя зависит от конкретного соединения формулы (II) и конкретного типа последующей реакции. В случае, если последующей реакцией является реакция, описанная ниже в настоящем изобретении на стадии (4), и в случае, если соединение формулы (II) используют на стадии (3) предпочтительно содержащимся в ДХМ, предпочтительно заменять этот растворитель на МТБЭ (метил-трет-бутиловый эфир), этилацетат или смесь МТБЭ и этилацетата. Особенно предпочтительным является МТБЭ.
В контексте настоящего изобретения предпочтительные экстрагирующие реагенты выбраны из группы, включающей воду, предпочтительно содержащую по меньшей мере одну подходящую соль, органические простые эфиры, органические сложные эфиры и смесь двух или большего количества из них, где указанная соль предпочтительно выбрана из группы, включающей хлорид натрия, гидрокарбонат натрия и хлорид аммония. Более предпочтительным экстрагирующим реагентом является вода, содержащая по меньшей мере одну подходящую соль, предпочтительно содержащая одну подходящую соль, более предпочтительно содержащая хлорид натрия. Применительно к количеству соли, содержащейся в воде, типичными вариантами осуществления являются водные растворы, содержащие от 10 до 30 мас.% соли.
Кроме того, можно соответствующим образом кристаллизовать соединения формулы (II), полученные на стадии (3). На кристаллизацию не налагают специальных ограничений при условии, что кристаллизуется по меньшей мере часть соединения формулы (II).
Поэтому настоящее изобретение также относится к самому необязательно кристаллическому соединению формулы (II)
,
в которой Y обозначает необязательно замещенный арильный фрагмент, предпочтительно необязательно замещенный фенильный фрагмент, более предпочтительно незамещенный фенил, где предпочтительно не менее 95%, более предпочтительно не менее 97%, еще более предпочтительно не менее 99% молекул указанного необязательно кристаллического соединения находятся в виде изомера формулы (IIa)
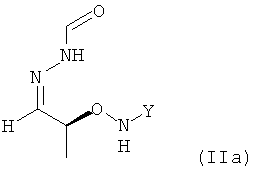 .
.
В частности, настоящее изобретение относится к необязательно кристаллическому соединению формулы (II), в которой Y = фенил, где не менее 99% молекул указанного необязательно кристаллического соединения находятся в виде изомера формулы (IIb)
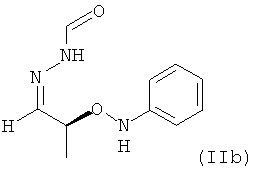 .
.
Преимуществом способа, предлагаемого в настоящем изобретении, явно является тот факт, что стадии (1), (2), и (3) можно провести в одном реакционном сосуде. Не требуется сложный перенос реакционных смесей, что, в частности, позволяет упростить крупномасштабное производство.
Стадия (4)
Как отмечено выше, в качестве экстрагирующего реагента можно использовать водный раствор соли. Поэтому до последующей стадии реакции может потребоваться удаление оставшейся воды из смеси, полученной жидкостной экстракции. Для такого удаления воды в контексте настоящего изобретения можно использовать по меньшей мере одно молекулярное сито. Характеристики пор по меньшей мере одного молекулярного сита можно адаптировать в соответствии с конкретными требованиями. Однако являются предпочтительными такие сита, которые обладают диаметрами пор, определенными в соответствии со стандартом DIN 66131, находящимися в диапазоне от 0,3 до 0,5 нм (от 3 до 5 Å), предпочтительно от 0,35 до 0,45 нм (от 3,5 до 4,5 Å), и диаметр пор, равный примерно 0,4 нм (4 Å) является особенно предпочтительным. Содержание воды в полученных смесях наиболее предпочтительно составляет не менее 0,1 мас.%, более предпочтительно не менее 0,05 мас.%.
В предпочтительном варианте осуществления настоящего изобретения соединение формулы (II), предпочтительно содержащееся в растворителе, указанном выше, еще более предпочтительно после указанной выше замены растворителя, предпочтительно содержащееся в МТБЭ и необязательно после удаления оставшихся следов воды вводят в реакцию на стадии (4) с силилирующим реагентом. Таким образом, растворителем, использующимся для этой реакции, наиболее предпочтительно является МТБЭ. По этой реакции на стадии (4) получают соединение формулы (III)
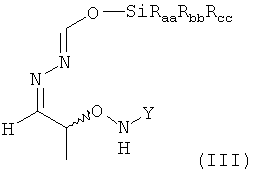
в которой остатки Raa, Rbb и Rcc могут быть одинаковыми или разными и предпочтительно представляют собой алкильные или арильные остатки, более предпочтительно алкильные остатки, содержащие от 1 до 6 атомов углерода, предпочтительно от 1 до 4 атомов углерода, более предпочтительно 1 или 2 атома углерода.
Термин "арильный остаток" при использовании в контексте настоящего изобретения означает карбоциклическую ароматическую группу, такую как фенил или нафтил и т.п. Термин "алкильный остаток" при использовании в контексте настоящего изобретения означает линейные или разветвленные алкильные фрагменты, которые предпочтительно содержат 1, 2, 3, 4, 5 или 6 атомов углерода. Предпочтительно, если остатки Raa, Rbb и Rcc могут быть одинаковыми или разными и представляют собой алкильные остатки. Более предпочтительно, если алкильные остатки содержат 1, 2, 3 или 4 атомов углерода, такие как метил, этил, пропил или бутил. Более предпочтительно, если алкильные остатки содержат 1 или 2 атома углерода. Еще более предпочтительно, если остатки Raa, Rbb и Rcc являются одинаковыми, предпочтительно представляют собой метил.
На сами силилирующие реагенты не налагают специальных ограничений при условии, что по меньшей мере часть соединения формулы (II) силилируют с получением соединения формулы (III). В наиболее предпочтительных вариантах осуществления настоящего изобретения, в которых Raa, Rbb и Rcc обозначают метил, наиболее предпочтительным силилирующим реагентом является гексаметилдисилазан, триметилхлорсилан, бис-триметилсилилацетамид или смесь двух или трех из этих соединений, предпочтительно бис-триметилсилилацетамид.
Температура, при которой проводят реакцию на стадии (4), зависит главным образом от конкретной природы соединений формулы (III), от конкретной природы силилирующего реагента и/или растворителя. Предпочтительные температуры в контексте настоящего изобретения находятся в диапазоне от 15 до 70°C.
Из полученной реакционной смеси, можно соответствующим образом кристаллизовать соединение формулы (III), полученное на стадии (4). На кристаллизацию не налагают специальных ограничений при условии, что кристаллизуется по меньшей мере часть соединения формулы (III).
Поэтому настоящее изобретение также относится к самому соединению формулы (III)
,
которое в возможном варианте осуществления может быть по меньшей мере частично кристаллическим, в которой Y обозначает необязательно замещенный арильный фрагмент, предпочтительно необязательно замещенный фенильный фрагмент, более предпочтительно незамещенный фенил, в которой остатки Raa, Rbb и Rcc могут быть одинаковыми или разными и предпочтительно представляют собой алкильные или арильные остатки, более предпочтительно алкильные остатки, содержащие от 1 до 6 атомов углерода, предпочтительно от 1 до 4 атомов углерода, более предпочтительно 1 или 2 атома углерода, предпочтительно метил, где предпочтительно не менее 95%, более предпочтительно не менее 97%, более предпочтительно не менее 99% молекул указанного хирального соединения находятся в виде изомера формулы (IIIa)
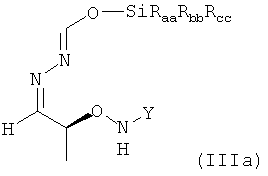 .
.
В частности, настоящее изобретение относится к соединению формулы (III), в которой Y = фенил, которое в возможном варианте осуществления может быть по меньшей мере частично кристаллическим, где не менее 99% молекул указанного необязательно кристаллического соединения находятся в виде изомера формулы (IIIb)
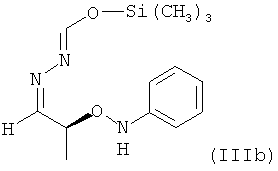 .
.
Как отмечено выше, на стадии (4) настоящего изобретения соединение формулы (III) получают содержащимся в растворителе, которым наиболее предпочтительно является МТБЭ. В этом варианте осуществления соединение формулы (III) не кристаллизуют. В особенно предпочтительном варианте осуществления настоящего изобретения эту реакционную смесь без дополнительной очистки направляют на дополнительную стадию (5), которая подробно описана ниже в настоящем изобретении.
Поэтому настоящее изобретение также относится к описанному выше способу, в котором после стадии (4) соединение формулы (III) не выделяют из реакционной смеси, полученной на стадии (4), до реакции с нуклеофильным соединением на стадии (5).
Стадия (5)
На этой предпочтительной стадии (5) настоящего изобретения соединение формулы (III) вводят в реакцию с нуклеофильным соединением, содержащим нуклеофильный остаток R1, в растворителе с получением соединения формулы (IV)
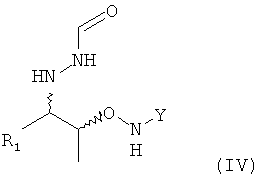 .
.
В альтернативном возможном способе, предлагаемом в настоящем изобретении, и соединение формулы (II), как это подробно описано выше в настоящем изобретении, можно ввести в реакцию с нуклеофильным соединением, содержащим нуклеофильный остаток R1, в растворителе с получением соединения формулы (IV)
.
В альтернативном возможном способе, необязательно можно проводить стадию (4) настоящего изобретения.
На нуклеофильное соединение, содержащее нуклеофильный остаток R1, не налагают специальных ограничений при условии, что реакция этого нуклеофильного соединения с соединением формулы (III) или с соединением формулы (II) позволяет получить соединение формулы (IV), этим нуклеофильным соединением предпочтительно является соединение Гриньяра R1MgX, в котором X предпочтительно выбран из группы, включающей Cl, Br и I. Предпочтительно, если R1 представляет собой линейный или разветвленный алкильный остаток, который предпочтительно содержит от 1 до 6 атомов углерода, а именно, 1, 2, 3, 4, 5 или 6 атомов углерода, еще более предпочтительно от 1 до 4 атомов углерода, а именно, 1, 2, 3, или 4 атома углерода, еще более предпочтительно 1 или 2 атома углерода, еще более предпочтительно 2 атома углерода.
Поэтому в особенно предпочтительном варианте осуществления настоящего изобретения нуклеофильное соединение, содержащее нуклеофильный остаток R1, представляет собой соединение Гриньяра CH3CH2MgX, в котором X выбран из группы, включающей Cl, Br и I. Предпочтительно, если нуклеофильным соединением, содержащим нуклеофильный остаток R1, является CH3CH2MgCl.
Растворитель или растворители, которые используют для проведения реакции на стадии (5) настоящего изобретения, можно выбрать соответствующим требованиям конкретной природы соединения формулы (III) или соединения формулы (II) и конкретной природы нуклеофильного соединения. Предпочтительно, если растворителем, использующимся в реакции на стадии (5) настоящего изобретения, является толуол или по меньшей мере один простой эфир, где простой эфир предпочтительно выбран из группы, включающей тетрагидрофуран (ТГФ), МТБЭ и смесь ТГФ и МТБЭ.
Температура, при которой проводят реакцию на стадии (5), зависит главным образом от конкретной природы соединения формулы (III) или соединения формулы (II), от конкретной природы нуклеофильного соединения и/или использующегося растворителя. Предпочтительные температуры в контексте настоящего изобретения находятся в диапазоне от -80 до 0°C, предпочтительно от -75 до -10°С, более предпочтительно от -70 до -25°C.
Преимуществом способа, предлагаемого в настоящем изобретении, явно является тот факт, что стадии (4) и (5) можно провести в одном реакционном сосуде. Не требуется сложный перенос реакционных смесей, что, в частности, позволяет упростить крупномасштабное производство.
Кроме того, настоящее изобретение относится к новому соединению формулы (II) и также к новому соединению формулы (III), которое неожиданно оказалось более реакционноспособным по отношению к реагентам Гриньяра. Таким образом, в свою очередь, реакцию Гриньяра, описанную выше, можно провести в мягких условиях с практически количественной диастереоизомерной селективностью.
Из реакционной смеси, полученной на этой стадии (5), соединение формулы (IV) можно выделить, предпочтительно после остановки реакции соответствующим реагентом, таким как вода или спирт. Для остановки предпочтительно используют спирт, более предпочтительно используют метанол. Поскольку на то, как проводят такое разделение, не налагают специальных ограничений, жидкостная экстракция является предпочтительной методикой, предлагаемым в настоящем изобретении. До стадии такой экстракции можно провести подходящую замену растворителя. В контексте настоящего изобретения предпочтительные экстрагирующие реагенты выбраны из группы, включающей воду предпочтительно содержащую по меньшей мере одну подходящую соль, органические простые эфиры, органические сложные эфиры, и смесь двух или большего количества из них, где указанная соль предпочтительно выбрана из группы, включающей хлорид натрия, гидрокарбонат натрия и хлорид аммония. Более предпочтительным экстрагирующим реагентом является вода, содержащая по меньшей мере одну подходящую соль, предпочтительно содержащая одну подходящую соль, более предпочтительно содержащая хлорид аммония или хлорид натрия. Для количества соли, содержащейся в воде типичными вариантами осуществления являются водные растворы, содержащие от 10 до 30 мас.% соли.
Из полученной реакционной смеси можно соответствующим образом кристаллизовать соединение формулы (IV), полученное на стадии (5). На кристаллизацию не налагают специальных ограничений при условии, что кристаллизуется по меньшей мере часть соединения формулы (IV).
Поэтому настоящее изобретение также относится к самому соединению формулы (IV)
,
которое в возможном варианте осуществления может быть по меньшей мере частично кристаллическим, в которой Y обозначает необязательно замещенный арильный фрагмент, предпочтительно необязательно замещенный фенильный фрагмент, более предпочтительно незамещенный фенил, в которой R1 предпочтительно означает алкильный остаток, предпочтительно содержащий от 1 до 6 атомов углерода, более предпочтительно от 1 до 4 атомов углерода, еще более предпочтительно 1 или 2 атома углерода, наиболее предпочтительно 2 атома углерода, R1 предпочтительно обозначает этил, где предпочтительно не менее 95%, более предпочтительно не менее 97%, еще более предпочтительно не менее 99% молекул указанного хирального соединения находятся в виде изомера формулы (IVa)
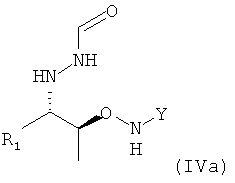 .
.
В частности, настоящее изобретение относится к соединению формулы (IV), в которой Y = фенил и R1 = этил, которое в возможном варианте осуществления может быть по меньшей мере частично кристаллическим, где не менее 99% молекул указанного необязательно кристаллического соединения находятся в виде изомера формулы (IVb)
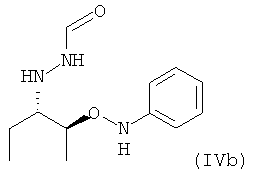 .
.
Как отмечено выше, на стадии (5) настоящего изобретения получают соединение формулы (IV) необязательно после жидкостной экстракции, содержащимся в растворителе или смеси растворителей, использовавшихся для реакции соединения формулы (III) с нуклеофильным соединением. В этом варианте осуществления, соединение формулы (IV) не кристаллизуют. В особенно предпочтительном варианте осуществления настоящего изобретения эту реакционную смесь без дополнительной очистки направляют на дополнительную стадию (6), которая подробно описана ниже в настоящем изобретении. Как уже указано для реакционной смеси, полученной на стадии (4) и направленной на стадию (5), в этом случае также отмечается, что не требуется дополнительная и трудоемкая очистка, такая как очистка с помощью хроматографии, что делает способ, предлагаемый в настоящем изобретении, совершенно прямым и особенно подходящим крупномасштабного производства.
Стадия (6)
В предпочтительном варианте осуществления настоящего изобретения способ, предлагаемый в настоящем изобретении, дополнительно включает (6) восстановление соединения формулы (IV), предпочтительно путем гидрирования, с получением соединения формулы (V)
.
Для восстановления соединения формулы (V) гидрирование является предпочтительной методикой. Хотя и зависящее от конкретной химической природы соединения (V) и использующегося растворителя, гидрирование, предлагаемое в настоящем изобретении, предпочтительно проводят при температуре в диапазоне от 15 до 35°C, более предпочтительно от 20 до 30°С. Типичная температура может находиться в диапазоне от 20 до 25°С. Давление водорода, при котором восстанавливают соединение формулы (IV), предпочтительно находится в диапазоне от 0,5 до 50 бар, предпочтительно от 1 до 20 бар, более предпочтительно от 1 до 10 бар. Типичные давления водорода находятся, например, в диапазоне от 1 до 5 бар или от 1 до 2 бар.
Обычно используют подходящий катализатор гидрирования. Хотя обычно могут использоваться гомогенные катализаторы, гетерогенные катализаторы являются предпочтительными. В качестве гетерогенных катализаторов применимы элементы подгрупп 8, 9, 10, 11 и 12 Периодической системы элементов. В качестве примеров можно отметить Pt, Rh, Ru, Co, Fe, Cu (например, в виде хромита меди), Zn (например, в виде хромита цинка), Pd или Ni. В контексте настоящего изобретения Pd является особенно предпочтительным. Эти элементы могут быть нанесены на соответствующую подложку. Например, можно отметить такие подложки, которые обеспечивают лучшее распределение элементов и более значительную площадь поверхности. В частности, для Pd предпочтительной подложкой является уголь. Таким образом, палладий на угле, обозначаемый в настоящем изобретении, как Pd/C, в контексте настоящего изобретения является особенно предпочтительным катализатором.
Поэтому настоящее изобретение также относится к описанному выше способу, в котором на стадии (6) гидрирование проводят при температуре в диапазоне от 15 до 35°C, предпочтительно от 20 до 30°C, при давлении водорода, находящемся в диапазоне от 0,5 до 50 бар, предпочтительно от 1 до 20 бар, более предпочтительно от 1 до 10 бар, в присутствии катализатора, содержащего благородный металл, предпочтительно катализатора, содержащего палладий, наиболее предпочтительно катализатора Pd/C.
На растворитель, в котором проводят восстановление, предпочтительно гидрирование, не налагают специальных ограничений при условии, что по меньшей мере часть соединения формулы (IV) восстанавливают. В способе, предлагаемом в настоящем изобретении, предпочтительно используют смесь растворителей, которая содержит по меньшей мере один спирт, предпочтительно по меньшей мере один спирт, содержащий от 1 до 4 атомов углерода, а именно, 1, 2, 3 или 4 атома углерода. Более предпочтительно, если смесь растворителей содержит по меньшей мере один спирт, содержащий от 1 до 3 атомов углерода, а именно, 1, 2 или 3 атома углерода. Более предпочтительно, если смесь растворителей содержит по меньшей мере один спирт, выбранный из группы, включающей метанол, этанол и изопропанол. Наиболее предпочтительным спиртом является метанол.
Кроме по меньшей мере одного спирта, наиболее предпочтительно метанола, смесь растворителей может дополнительно содержать по меньшей мере один дополнительный растворитель, который не является спиртом, предпочтительно дополнительный растворитель, который выбран из группы, включающей ТГФ, толуол, МТБЭ и смесь двух или большего количества из них.
Таким образом, предпочтительные смеси растворителей, предлагаемые в настоящем изобретении, использующиеся для восстановления, предпочтительно гидрирования на стадии (6) настоящего изобретения, содержат, более предпочтительно в основном содержат по меньшей мере один спирт, содержащий от 1 до 4 атомов углерода, предпочтительно по меньшей мере один спирт, выбранный из группы, включающей метанол, этанол и изопропанол, и по меньшей мере один растворитель, выбранный из группы, включающей ТГФ, толуол, МТБЭ и смесь двух или большего количества из них. Особенно предпочтительные смеси растворителей, предлагаемые в настоящем изобретении, использующиеся для восстановления, предпочтительно гидрирования на стадии (6) настоящего изобретения, содержат, более предпочтительно в основном содержат метанол и по меньшей мере один растворитель, выбранный из группы, включающей ТГФ, толуол и МТБЭ, предпочтительно МТБЭ.
Из реакционной смеси, полученной указанным выше восстановлением, катализатор восстановления предпочтительно выделяют по меньшей мере по одной подходящей методике, такой как фильтрование. Выделенный таким образом катализатор затем можно промыть по меньшей мере один раз, предпочтительно таким же растворителем или смесью растворителей, которая используется для реакции восстановления, более предпочтительно спиртом, использующимся на стадии (6).
Обычно в возможном варианте осуществления настоящего изобретения соединение формулы (V), полученное на стадии (6), наиболее предпочтительно после выделения катализатора, можно использовать для необязательной дополнительной реакции содержащимся в растворителе или смеси растворителей, использующейся для реакции восстановления и необязательно с промывкой катализатора.
Согласно изобретению неожиданно было установлено, что восстановительное расщепление на стадии (6) настоящего изобретения дает соединение формулы (V) в виде кристаллического соединения. Поэтому в предпочтительном варианте осуществления настоящего изобретения соединение формулы (V) кристаллизуют. Поэтому настоящее изобретение относится к способу, определенному выше, который дополнительно включает кристаллизацию соединения формулы (V). В этом контексте следует отметить, что в предшествующем уровне техники, в частности в WO 96/33163, где раскрыто некристаллическое содержащее бензильную защитную группу соединение формулы (V), в которой R1 = этил, полагают, что соединение формулы (V) недоступно в виде кристаллического соединения. Таким образом, настоящее изобретение впервые относится к способу, который позволяет получить соединение формулы (V) в кристаллическом виде, в особенно предпочтительном варианте осуществления настоящего изобретения обладающее стереохимической чистотой, соответствующей равному не менее 99% содержанию изомера формулы (Va), где указанную стереохимическую чистоту определяют, как это описано в примере 3 настоящего изобретения. Кроме того, новый способ, включающий новые соединения, описанные выше, приводит к очень высокому суммарному выходу предпочтительно кристаллического соединения формулы (V), который составляет примерно от 30 до 40% в пересчете на исходное вещество, нитрозобензол.
Поэтому настоящее изобретение также относится к предпочтительно кристаллическому хиральному соединению формулы (V)
,
в которой R1 предпочтительно означает алкильный остаток, предпочтительно содержащий от 1 до 6 атомов углерода, более предпочтительно от 1 до 4 атомов углерода, более предпочтительно 1 или 2 атома углерода, наиболее предпочтительно 2 атома углерода, R1 предпочтительно обозначает этил, где предпочтительно не менее 95%, предпочтительно не менее 97%, более предпочтительно не менее 99% молекул указанного кристаллического соединения находятся в виде изомера формулы (Va)
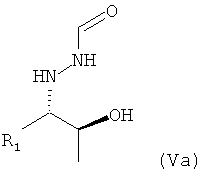 ,
,
предпочтительно в виде изомера формулы (Vb)
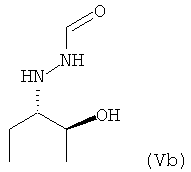 .
.
В частности, настоящее изобретение относится к кристаллическому соединению формулы (V), в которой R1 = этил,

где предпочтительно не менее 95%, предпочтительно не менее 97%, более предпочтительно не менее 99% молекул указанного кристаллического соединения находятся в виде изомера формулы (Vb), обладающего рентгенограммой, содержащей по меньшей мере следующие отражения:
где 100% относится к интенсивности наиболее интенсивного пика на рентгенограмме.
В предпочтительном варианте осуществления настоящего изобретения кристаллизацию соединения формулы (V) проводят с использованием смеси циклогексан (СНХ) и МТБЭ. Температура кристаллизации предпочтительно находится в диапазоне от -10 до +10°C, более предпочтительно от -5 до +5°C. Для улучшения условий кристаллизации можно дополнительно проводить соответствующее внесение затравочных кристаллов в реакционную смесь, полученную на стадии (6).
После отделения кристаллов, например, путем подходящего фильтрования, кристаллы можно промыть подходящим промывочным реагентом, предпочтительно смесью МТБЭ и СНХ.
В необязательном варианте осуществления настоящего изобретения полученное таким образом кристаллическое соединение формулы (V) можно перекристаллизовать по меньшей мере один раз. Из возможных вариантов осуществления перекристаллизации, перекристаллизацию соединения формулы (V) можно предпочтительно проводить из изопропилацетата или из МИБК (метилизобутилкетон) или из смеси изопропилацетата и МИБК. Такую перекристаллизацию необязательно можно провести при дополнительном присутствии по меньшей мере одного реагента, подходящего для связывания некоторых примесей, все еще содержащихся в соответствующей смеси. В частности, в качестве подходящего дополнительного реагента можно отметить древесный уголь. Поэтому настоящее изобретение относится к описанному выше способу, в котором после кристаллизации соединения формулы (V) это соединение перекристаллизовывают по меньшей мере один раз из изопропилацетата или из МИБК, или из смеси изопропилацетата и МИБК, необязательно в присутствии древесного угля.
Полученные таким образом кристаллы после кристаллизации и/или после перекристаллизации можно соответствующим образом высушить. Предпочтительно, если температуры сушки находятся в диапазоне от 10 до 40°C, более предпочтительно от 15 до 35°C, еще более предпочтительно от 20 до 30°C при давлении, равном менее 1 бар, предпочтительно менее 500 мбар, более предпочтительно менее 100 мбар.
Как уже указано выше, в предпочтительном варианте осуществления соединение формулы (V), предпочтительно формулы (V), в которой R1 = этил, получают предпочтительно в виде кристаллического соединения, обладающего энантиомерной чистотой, соответствующей равному не менее 99% содержанию изомера формулы (Va), без необходимости использования трудоемких методик очистки, таких как очистка с помощью хроматографии. Поэтому настоящее изобретение также относится к описанному выше способу, в котором соединение (V) не подвергают очистке с помощью хроматографии. Таким образом, ясно, что способ, предлагаемый в настоящем изобретении, намного превосходит способы предшествующего уровня техники, в частности, для среднемасштабного или крупномасштабного получения соединения формулы (V). Следовательно, способ, предлагаемый в настоящем изобретении, обладает значительными преимуществами, в особенности в качестве способов, в которых соединение формулы (V) используют в качестве исходного вещества для получения широко использующихся соединений, таких как, например, фунгицидные средства.
Таким образом, настоящее изобретение, в частности, относится к применению описанного выше способа получения исходного вещества для получения фунгицидного средства, предпочтительно позаконазола.
В первом варианте осуществления настоящего изобретения предпочтительно кристаллическое соединение формулы (V), предпочтительно соединение формулы (V), в которой R1 = этил, можно использовать без обработки по меньшей мере для одной дополнительной подходящей реакции, например, в качестве исходного вещества для получения фунгицидного средства, предпочтительно позаконазола. Для полноты следует отметить, что предпочтительно кристаллическое соединение также можно использовать для любой возможной реакции и использование не ограничивается использованием в качестве исходного вещества для получения фунгицидного средства.
Во втором варианте осуществления настоящего изобретения соединение формулы (V), предпочтительно соединение формулы (V), в которой R1 = этил, до использования в качестве исходного вещества по меньшей мере для одной другой подходящей реакции, например, в качестве исходного вещества для получения фунгицидного средства, предпочтительно позаконазола, необязательно можно ввести по меньшей мере в одну реакцию, где группы ОН соответствующим образом по меньшей мере частично защищены. Обычно не налагают специальных ограничений при условии, что группы ОН соответствующим образом защищены. Обычные приемлемые группы описаны, например, в публикации Greene et al., "Protective Groups in Organic Synthesis", 2nd ed., John Wiley & Sons, New York 1991 10-142.
Стадия (7)
В одном варианте осуществления настоящего изобретения группа OH соединения формулы (V) соответствующим образом силилирована. Предпочтительно, если силилирование проводят для получения соединения формулы (VI)
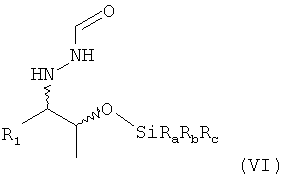 ,
,
в которой остатки Ra, Rb и Rc могут быть одинаковыми или разными и предпочтительно представляют собой алкильные или арильные остатки. Поэтому настоящее изобретение относится к способу, определенному выше, который дополнительно включает
(7) реакцию соединения формулы (V) в растворителе с силилирующим реагентом, содержащим остаток -SiRaRbRc, с получением соединения формулы (VI)
,
в которой остатки Ra, Rb и Rc могут быть одинаковыми или разными и предпочтительно представляют собой алкильные или арильные остатки.
Термин "арильный остаток" при использовании в контексте настоящего изобретения означает карбоциклическую ароматическую группу, такую как фенил или нафтил и т.п. Термин "алкильный остаток" при использовании в контексте настоящего изобретения означает линейные или разветвленные алкильные фрагменты, которые предпочтительно содержат 1, 2, 3, 4, 5 или 6 атомов углерода.
В возможном варианте осуществления настоящего изобретения соединение формулы (VI) можно соответствующим образом кристаллизовать.
Поэтому настоящее изобретение также относится к самому необязательно кристаллическому хиральному соединению формулы (VI),
,
в которой R1 предпочтительно означает алкильный остаток, предпочтительно содержащий от 1 до 6 атомов углерода, более предпочтительно от 1 до 4 атомов углерода, более предпочтительно 1 или 2 атома углерода, наиболее предпочтительно 2 атома углерода, R1 предпочтительно обозначает этил, в которой остатки Ra, Rb и Rc могут быть одинаковыми или разными и предпочтительно представляют собой алкильные или арильные остатки, где предпочтительно не менее 95%, более предпочтительно не менее 97%, еще более предпочтительно не менее 99% молекул указанного необязательно кристаллического соединения находятся в виде изомера формулы (VIa)
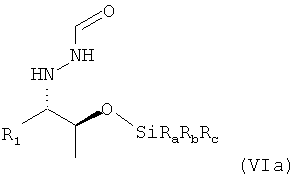 ,
,
предпочтительно в виде изомера формулы (VIb)
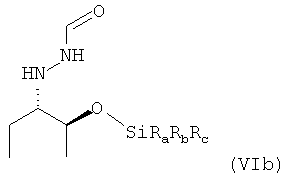 .
.
Чистоту соединения формулы (VI) можно определить по методикам, известным в данной области техники.
Обычно настоящее изобретение также относится к хиральному соединению, которое можно получить или получают способом, включающим любую из стадий, указанных выше. В частности, настоящее изобретение относится к хиральному соединению, которое можно получить или получают способом, включающим стадии (1), (2) и (3), или включающим стадии (1), (2), (3) и (4), или включающим стадии (1), (2), (3), (4) и (5), или включающим стадии (1), (2), (3), и (5) без стадии (4), или включающим стадии (1), (2), (3), (4), (5) и (6), или включающим стадии (1), (2), (3), (5) и (6) без стадии (4), или включающим стадии (1), (2), (3), (4), (5), (6) и (7), или включающим стадии (1), (2), (3), (5), (6) и (7) без стадии (4).
Кроме того, в возможном варианте осуществления настоящее изобретение также относится по меньшей мере к одной подходящей соли соединения формулы (II) и/или по меньшей мере к одной подходящей соли соединения формулы (III), и/или по меньшей мере к одной подходящей соли соединения формулы (IV), и/или по меньшей мере к одной подходящей соли соединения формулы (V), и/или по меньшей мере к одной подходящей соли соединения формулы (VI).
Применение
Как уже указано выше, предпочтительно кристаллическое соединение формулы (V) и/или необязательно кристаллическое соединение формулы (VI), которые оба необязательно получают или можно получить способом, предлагаемым в настоящем изобретении, предпочтительно кристаллическое соединение формулы (V), в которой R1 = этил
,
где предпочтительно не менее 95%, более предпочтительно не менее 97% и особенно предпочтительно не менее 99% молекул указанного кристаллического соединения находятся в виде изомера формулы (Vb)
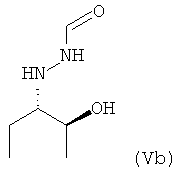
используют в качестве исходного вещества для дополнительной реакции, предпочтительно в качестве исходного вещества для получения фунгицидного средства.
Таким образом, настоящее изобретение также относится к способу получения фунгицидного средства, где предпочтительно кристаллическое соединение формулы (V), в частности предпочтительно кристаллическое соединение формулы (V), в которой R1 = этил, и/или необязательно кристаллическое соединение формулы (VI), в частности необязательно кристаллическое соединение формулы (VI), в которой R1 = этил, оба необязательно получают или можно получить способом, предлагаемым в настоящем изобретении, предпочтительно кристаллическое соединение формулы (V), в которой R1 = этил
,
где предпочтительно не менее 95%, более предпочтительно не менее 97%, и особенно предпочтительно не менее 99% молекул указанного кристаллического соединения находятся в виде изомера формулы (Vb)
используют в качестве исходного вещества для последующей реакции, предпочтительно в качестве исходного вещества для получения фунгицидного средства.
В предпочтительном применении или способе, предлагаемом в настоящем изобретении, предпочтительно кристаллическое соединение формулы (V), в которой R1 = этил, и/или необязательно кристаллическое соединение формулы (VI), в которой R1 = этил, необязательно получают или можно получить способом, предлагаемым в настоящем изобретении, предпочтительно кристаллическое соединение формулы (V), в которой R1 = этил, вводят в реакцию с анилином или его производным и фосгеном или его производным или изоцианатом, предпочтительно с производным фосгена.
Поскольку на химическую природу производного анилина не налагают специальных ограничений при условии, что в конечном счете получают фунгицидное средство, производным анилина предпочтительно является соединение формулы:
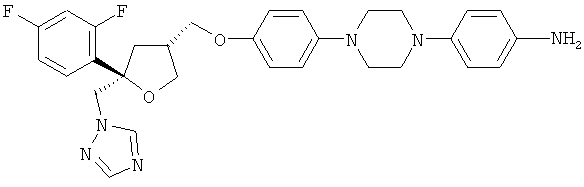
Поскольку на химическую природу производного фосгена не налагают специальных ограничений при условии, что в конечном счете получают фунгицидное средство, производным фосгена предпочтительно является соединение формулы:

В особенно предпочтительном применении или способе, предлагаемом в настоящем изобретении, в конечном счете получаемым фунгицидным средством является позаконазол формулы:
Настоящее изобретение дополнительно иллюстрируется приведенными ниже примерами.
Примеры
Пример 1: Синтез соединения формулы (II), в которой Y = фенил
a) 1,70 кг Нитрозобензола (ММ (молекулярная масса): 107,11; 15,9 моля; 1,0 экв.) растворяли в 6,16 л ДХМ путем перемешивания в атмосфере N2 при температуре, равной 20-25°C.
b) В отдельном сосуде 2,76 кг пропионового альдегида (ММ: 58,08; плотность: 0,798 г/мл; 47,5 моля; 3,0 экв.) и 5,28 л ДХМ охлаждали в атмосфере N2 до температуры, равной от -6 до -4°C. К полученной смеси добавляли 0,057 кг ледяной уксусной кислоты (MM: 60,05; плотность: 1,049 г/мл; 0,95 моля; 0,06 экв.) и 0,55 кг D-пролина (MM: 115,13; 4,75 моля; 0,3 экв.) и получали мелкодисперсную суспензию.
c) К этой суспензии, полученной на стадии b), добавляли 0,4 л раствора нитрозобензола, полученного на стадии a). О начале реакции свидетельствовало обесцвечивание суспензии и повышение температуры до +3°C в течение 1 мин. Затем оставшийся раствор нитрозобензола (примерно 7 л), раствора нитрозобензола, полученного на стадии a), добавляли со скоростью, обеспечивающей поддержание температуры реакционной смеси, равной от -5 до -3°C. Смесь постепенно темнела и после завершения добавления превращалась в прозрачный раствор. Затем перемешивание продолжали в течение 10 мин и ВЭЖХ указывала на полное превращение. Соединение формулы (i), в которой Y = фенил, получали в качестве промежуточного продукта с энантиомерной чистотой, выраженной в виде энантиомерного избытка (ЭИ)>98%, т.е. более 99% молекул хирального соединения формулы (I), в которой Y = фенил, содержались в виде изомера формулы (Ia), в которой Y = фенил. Энантиомерную чистоту определяли по методике, аналогичной описанной в публикации Brown et al., J. Chem. Soc. 2003, 125 (36), 10808-10809 и с использованием Chiralcel OD-H и смеси н-гептан/изопропанол/диэтиламин в качестве элюента.
d) Затем при 0-5°C добавляли 8,8 л ДХМ, 3,0 кг молекулярного сита, обладающего порами диаметром 0,4 нм (4 Å; выпускается фирмой Aldrich) и 3,14 кг формилгидразина (ММ: 60,06; 52 моля; 3,3 экв.), что приводило к повышению температуры. Перемешивание продолжали при 0-5°C в атмосфере N2. Через 3 ч ВЭЖХ указывала на полное превращение.
e) Полученное твердое вещество отфильтровывали и промывали с помощью 4,4 л ДХМ. Раствор концентрировали до 1/4 его исходного объема при температуре бани, равной <10°C. Затем добавляли 28 л МТБЭ и полученный раствор концентрировали до 1/4 его исходного объема путем отгонки при температуре бани, равной <10°C. Затем полученный органический слой разбавляли с помощью 7 л МТБЭ и 5 раз экстрагировали с помощью 28 л 20% водного раствора хлорида натрия. Затем добавляли 8,8 л ДХМ для азеотропного удаления воды. Полученный раствор концентрировали и получали 12 кг раствора соединения формулы (II), в которой Y = фенил, в МТБЭ, который содержал 2,86 кг указанного соединения (MM 207,23; выход 80%).
f) Этот раствор, содержащий соединение формулы (II), в которой Y = фенил, использовали на следующей стадии (пример 2) без дополнительной очистки.
Пример 2: Синтез соединений формулы (III) и (IV), в которой Y = фенил и Raa, Rbb и Rcc = метил и R1 = этил
a) 0,887 кг Соединения формулы (II), полученного в примере 1 (ММ: 207,23; 4,28 моля; 1,0 экв.), используемого в виде раствора в МТБЭ (полная масса 290 г), который содержал примерно 2 мас.% H2O, сушили над 1,54 кг молекулярного сита, обладающего порами диаметром 0,4 нм (4 Å; выпускается фирмой Aldrich) при температуре, равной 20-25°C в течение 30 мин. Таким образом содержание воды уменьшали до значения, равного менее 0,1 мас.%. Затем молекулярное сито удаляли фильтрованием и промывали с помощью 1,7 л МТБЭ. Полученный раствор разбавляли с помощью 16 л МТБЭ. Содержание воды в этом растворе составляло примерно 0,05% (0,5 моля; 0,12 экв.).
b) Затем к фильтрату добавляли 2,8 л БСА (бис-триметилсилилацетамид) (ММ: 203,43; плотность: 0,832 г/мл; 11,5 моля; 2,7 экв.). Полученный раствор перемешивали при 20-25°C. Через 1 ч по данным 1H-ЯМР силилирование завершалось. Соединение формулы (III), в которой Y = фенил и Raa, Rbb и Rcc = метил, получали в качестве промежуточного продукта.
c) Затем раствор охлаждали до температуры, равной от -70 до -60°C, и 8,65 л раствора этилхлорида магния в ТГФ (2 моль/л; ММ: 88,82; плотность: 0,978 г/мл; 17,3 моля; 4,0 экв.) добавляли со скоростью, обеспечивающей поддержание температуры реакционной смеси, равной от -70 до -60°C. Эту смесь перемешивали в течение 1 ч при температуре, равной -60°C. Затем, температуру повышали до -25°C, и перемешивание продолжали. Через 3 ч ВЭЖХ указывала на полное превращение. Затем реакцию останавливали путем проводимого по каплям добавления 5,5 кг MeOH при температуре, равной -25°C. При остановке смеси давали нагреться до 0-15°C.
d) Полученный органический слой после этого дважды экстрагировали при температуре, равной 20-25°C, с использованием по 30 л 10% водного раствора хлорида аммония и один раз с помощью 30 л 20% водного раствора хлорида натрия. Органический слой (20 кг) содержал примерно 0,89 кг соединения формулы (IV), в которой Y = фенил и R1 = этил (ММ: 237,30; выход 80%), и его использовали на следующей стадии (пример 3.3) без дополнительной очистки.
Пример 3: Синтез соединений формулы (V), в которой R1 = этил
a) 0,89 кг Соединения формулы (IV) (ММ: 237,30; 3,76 моля; 1,0 экв.), полученного в соответствии с примером 2 и использующегося в виде раствора в МТБЭ (полная масса равна 20 кг), разбавляли с помощью 1 л MeOH при температуре, равной 20-25°C.
b) Затем добавляли 0,9 кг палладия на угле (Pd/C; 5% Pd; 50% воды) и полученный раствор энергично перемешивали при 20-25°C. Реакцию восстановления проводили при давлении H2, равном 1 атм. Сосуд трижды откачивали и заполняли с помощью H2 при давлении, равном 1 атм. После перемешивания в течение 1,5 ч в атмосфере H2 ВЭЖХ указывала на полное превращение. Затем суспензию фильтровали и катализатор промывали с помощью 1,8 л МТБЭ/МеОН (1/1 об./об.). Объединенные фильтраты концентрировали с образованием желтого масла и получали неочищенное соединение формулы (V), в которой R1 = этил, содержащее примерно 40% (примерно 0,55 кг) соединения формулы (V), в которой R1 = этил.
c) Это масло разбавляли с помощью 13,9 л МТБЭ при температуре, равной 20-25°C, и в полученный раствор вводили затравочные кристаллы. После перемешивания в течение 1 ч при температуре, равной 20-25°C, получали мелкодисперсную суспензию. Затем добавляли 17 л СНХ (циклогексан), смесь охлаждали до 0°C и перемешивали при 0°C в течение 3 ч и получали вязкую суспензию соединения формулы (V), в которой R1 = этил. Полученные таким образом кристаллы собирали и промывали с помощью 2,2 л холодной (0°С) смеси МТБЭ и СНХ (1/1 об./об.) и после сушки получали 0,44 кг соединения формулы (V), в которой R1 = этил (ММ 146,19; выход 80%; выход 64% в пересчете на соединение формулы (II)). Более 99% молекул хирального соединения формулы (V), в которой R1 = этил, получали в виде изомера формулы (Vb).
10,4 г Этого желтого вещества добавляли к 50 мл изопропилацетата и полученную смесь нагревали при температуре, равной от 85 до 89°C, до образования раствора. К желтому раствору добавляли 0,5 г активированного угля и после перемешивания в течение нескольких минут горячую смесь фильтровали и ей при перемешивании давали охладиться примерно до 5°C. После перемешивания в течение от 2 до 3 ч, осадившийся продукт отфильтровывали, промывали с помощью 5 мл изопропилацетата и сушили при комнатной температуре в вакууме в течение ночи и получали 8,54 г продукта в виде почти белого твердого вещества (температура плавления от 78 до 80°C), т.е. соединение формулы (V), в которой R1 = этил, где более 99% молекул указанного соединение получали в виде изомера формулы (Vb).
Получение затравочных кристаллов, использующихся на этой стадии c):
100 г Неочищенного масла, полученного выше на стадии b), очищали с помощью колоночной хроматографии с использованием 800 г силикагеля 60 (0,063-0,200 мм, Merck) в качестве стационарной фазы и смеси ДХМ/метанол = 20/1 в качестве подвижной фазы. Собирали фракции, содержащие искомый продукт в чистом виде, что устанавливали с помощью ТСХ (тонкослойная хроматография) (Merck, силикагель 60 F254, подвижная фаза СНХ/этилацетат = 1/1). После выпаривания растворителей полученное твердое вещество перекристаллизовали из диэтилового эфира. Полученные кристаллы собирали и после сушки использовали в качестве затравочных кристаллов (20°C, <100 мбар).
d) ИК-спектр и рентгенограмма приведены на фиг.1 и 2.
Экспериментальные данные получали следующим образом:
Энантиомерную чистоту соединения формулы (V), в которой R1 = этил, полученного на стадии c), определяли с помощью ВЭЖХ следующим образом:
Хроматография
Прибор для ВЭЖХ: Agilent 1200
Колонка: Waters XBridge C18, 2,5 мкм, 50×4,6 мм (order no. 186003090)
Система: градиентный режим
Элюент A: буферный раствор pH 7,0
Элюент B: буферный раствор pH 7,0/ацетонитрил = 2/8 (об./об.)
Скорость потока: 1,8 мл/мин
Температура печи: 40°C
Инжектируемый объем: 10 мкл (микролитров)
Время остановки: 20 мин
Детектирование: λ (лямбда) = 260 нм
Градиентный режим:
Буферный раствор pH 7,0 готовили по следующей методике: растворяли 7,0 мл триэтиламина в 900 мл воды, значение pH устанавливали равным 7,0 с помощью H3PO4 и разбавляли до 1000 мл водой.
Раствор реагента готовили по следующей методике: растворяли от 80 до 90 мг (S)-(-)-α-метилбензилизоцианата в ацетонитриле и разбавляли до 1,0 мл ацетонитрилом.
Приготовление образца:
a) Исходный исследуемый раствор готовили по следующей методике: Растворяли от 38 до 42 мг исследуемого вещества, отвешенного с точностью 0,01 мг, в 1,0 мл ацетонитрила.
b) Исследуемый раствор готовили по следующей методике:
В сосуде для ВЭЖХ смешивали 100 мкл (микролитров) исходного исследуемого раствора и 100 мкл (микролитров) раствора реагента. Выдерживали при комнатной температуре (от 20 до 25°C) в течение 30 мин, добавляли 800 мкл (микролитров) буферного раствора с pH 7,0 и энергично встряхивали. Затем раствор охлаждали в бане со льдом (0°C) в течение еще 30 мин (происходило осаждение реагента) и образец фильтровали через фильтр с отверстиями 0,2 мкм (микрометров) непосредственно в другой сосуд для ВЭЖХ.
Данные инфракрасных спектров (ИК) получали в кювете МКИ Golden Gate™ Single Reflection Diamond ATR (ослабленное полное отражение) в спектрометре Bruker Tensor 27 FTIR с разрешением 4 см-1 при условиях окружающей среды. Для получения спектра количество порошкообразного образца, помещающееся на кончике шпателя, помещали на поверхность алмаза. Затем образец припрессовывали к алмазу сапфировым стержнем и регистрировали спектр. Спектр алмаза без образца использовали в качестве спектра фона. Типичная точность значений волновых чисел находится в диапазоне примерно ±2 см-1. Таким образом, для большинства инфракрасных спектрометров при стандартных условиях пик в инфракрасном спектре, который появляется при 1716 см-1, может появиться в диапазоне от 1714 от 1718 см-1.
Рентгенограммы (порошковые рентгенограммы, ПРРГ, рентгенограммы ПРГ) получали на порошковом дифрактометре Unisantis XMD 300 с детектором положения с использованием оптики с параллельными пучками при следующих условиях: анод трубки: Cu, 40 кВ, 0,8 мА; 3-43° тета/2-тета; одновременное детектирование областей по 10° за шаг с разрешением детектора, равным 1024, длительность подсчета 300 с за шаг. Образцы исследовали при комнатной температуре в стандартном держателе образца на вращающемся устройстве для образца. Типичная точность значений 2-тета находится в диапазоне примерно ±0,2°2-тета. Таким образом, для большинства дифрактометров спектрометров при стандартных условиях дифракционный пик, который появляется при 5,0°2-тета, может появиться в диапазоне от 4,8 до 5,2°2-тета.
ВЭЖХ для определения завершения превращения, указанного в примере 1, c) и d), примере 2), c) и примере 3, b) проводили следующим образом:
Колонка: Zorbax Eclipse XDB-C18, 150*4,6 мм, 5 мкм (микрометров).
Система: градиентный режим
Буфер: 2,10 г KH2PO4+4,28 г K2HPO4/2,0 л H2O, с помощью 85% H3PO4 устанавливали значение pH, равное 6,5
Подвижная фаза A: 20 мМ фосфатный буфер pH 6,5/ацетонитрил, 85/15, об./об.
Подвижная фаза B: 20 мМ фосфатный буфер pH 6,5/ацетонитрил, 50/50, об./об.
Растворитель: H2O/ацетонитрил = 50/50 об./об.
Скорость потока: 1,5 мл/мин
Температура печи: 60°C
Инжектируемый объем: 5-20 мкл (микролитров)
Время остановки: 30 мин
Детектирование: λ (лямбда) = 210 нм (детектор Agilent 1200)
Автоматический пробоотборник: 5°C
Градиентный режим:
Приготовление образца:
Раствор образца для ВЭЖХ в примере 1), с) готовили по следующей методике: растворяли примерно 100 мкл (микролитров) реакционной смеси в 0,2 мл изопропанола, добавляли примерно 50 мг NaBH4 и перемешивали в течение 10 мин при 25°C. Экстрагировали с помощью 0,2 мл этилацетата и 0,5 мл буфера 5% KH2PO4 (pH 7,0). Разбавляли 50 мкл (микролитров) полученного органического слоя в мерной колбе объемом 10 мл и доводили до метки растворителем. Массы образцов подбирали в соответствии с требованиями прибора.
Раствор образца для ВЭЖХ в примере 1), d), примере 2), c) и примере 3), b) готовили по следующей методике: растворяли примерно 100 мкл реакционной смеси в 2 мл ацетонитрила в мерной колбе объемом 20 мл и f доводили до метки растворителем. Массы образцов подбирали в соответствии с требованиями прибора.
Перечень цитированных документов
1. WO 95/17407.
2. WO 97/22579.
3. Saksena et al., Tetrahedron Lett. 2004, 45 (44), 8249-8251.
4. WO 96/33163.
5. WO 97/33178.
6. Brown et al., J. Chem. Soc. 2003, 125 (36), 10808-10809.
7. Cordova et al., Chem. Eur. J. 2004, 10 (15), 3673-3684.
8. Hayashi et al., J. Org. Chem. 2005, 69 (18), 5966-5973.
9. Greene et al., "Protective Groups in Organic Synthesis", 2nd ed., John Wiley & Sons, New York 1991 10-142.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ХИРАЛЬНЫХ ТРИАЗОЛОНОВ | 2011 |
|
RU2585760C2 |
| ПОЛУЧЕНИЕ ПРОМЕЖУТОЧНЫХ ПРОДУКТОВ ДЛЯ СИНТЕЗА ПОЗАКОНАЗОЛА | 2011 |
|
RU2580318C2 |
| ЦИКЛИЧЕСКИЕ КАРБОНИЛЬНЫЕ СОЕДИНЕНИЯ С БОКОВЫМИ ПЕНТАФТОРФЕНИЛКАРБОНАТНЫМИ ГРУППАМИ, ИХ ПОЛУЧЕНИЕ И ПОЛУЧАЕМЫЕ ИЗ НИХ ПОЛИМЕРЫ | 2011 |
|
RU2572246C2 |
| ОЧИСТКА ПОЗАКОНАЗОЛА И ПРОМЕЖУТОЧНЫХ ПРОДУКТОВ ДЛЯ СИНТЕЗА ПОЗАКОНАЗОЛА | 2011 |
|
RU2585683C2 |
| СПОСОБ ОБРАЩЕНИЯ (S)-(+)- И (R)-(-)-10,11-ДИГИДРО-10-ГИДРОКСИ-5Н-ДИБЕНЗ/b, f/АЗЕПИН-5-КАРБОКСАМИДА И ИХ ОПТИЧЕСКИ ОБОГАЩЕННЫХ СМЕСЕЙ | 2005 |
|
RU2382772C2 |
| СПОСОБ СИНТЕЗА НУКЛЕОЗИДНЫХ АНАЛОГОВ | 2000 |
|
RU2200738C2 |
| ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ В СИНТЕЗЕ АНАЛОГОВ ЗЕАРАЛЕНОНОВЫХ МАКРОЛИДОВ | 2008 |
|
RU2497803C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ДИБЕНЗОТИАЗЕПИНА | 1999 |
|
RU2233275C2 |
| СПОСОБ СТЕРЕОСЕЛЕКТИВНОГО ПОЛУЧЕНИЯ ХИРАЛЬНЫХ 2-[(ГЕТЕРО)АРИЛАЛКИЛСУЛЬФАНИЛ]ПИРИМИДИНОВ И ПРОДУКТОВ, ПОЛУЧЕННЫХ ИЗ НИХ | 2019 |
|
RU2813355C2 |
| ПРОИЗВОДНЫЕ ПИРИМИДИНОВЫХ 2,3-ДИДЕЗОКСИНУКЛЕОЗИДОВ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2163909C2 |
Изобретение относится к способам получения хиральных соединений, в частности к способу получения хирального соединения формулы (II). Способ включает реакцию хирального соединения формулы (I) с H2N-NH-CHO в растворителе с получением соединения формулы (II). В формулах (I) и (II) Y обозначает замещенный или незамещенный арильный фрагмент, где замещенный арильный фрагмент содержит 1, 2 или 3 заместителя, которые выбраны из группы, включающей галоген, алкил и C1-C6алкоксигруппу. Изобретение относится также к способам получения хиральных соединений формул (III)-(V) с использованием способа получения хирального соединения формулы (II), хиральным соединениям формул (II)-(V) и их применению для получения фунгицидного средства, предпочтительно позаконазола. В формулах (III)-(V) Y имеет значение, указанное для соединений формул (I) и (II); Raa, Rbb и Rcc могут быть одинаковыми или разными и представляют собой алкильные или арильные остатки; R1 означает алкильный остаток. 9 н. и 22 з.п. ф-лы, 2 ил., 3 пр.

1. Способ получения хирального соединения формулы (II)
,
включающий
(1) реакцию хирального соединения формулы (I)
,
в которой Y обозначает замещенный или незамещенный арильный фрагмент, предпочтительно замещенный или незамещенный фенильный фрагмент, более предпочтительно незамещенный фенил, где замещенный арильный фрагмент и замещенный фенильный фрагмент содержат 1, 2 или 3 заместителя, которые выбраны из группы, включающей галоген, алкил и C1-алкоксигруппу или C2-алкоксигруппу, или C3-алкоксигруппу, или C4-алкоксигруппу, или C5-алкоксигруппу, или C6-алкоксигруппу;
с H2N-NH-CHO в растворителе с получением соединения формулы (II).
2. Способ по п.1, в котором соединение формулы (I) получают по реакции пропионового альдегида в растворителе с соединением формулы (i)
O=N-Y (i),
предпочтительно с нитрозобензолом, в присутствии каталитической системы, предпочтительно содержащей по меньшей мере один органический катализатор, более предпочтительно пролин (Pro), более предпочтительно D-Pro, указанная каталитическая система необязательно дополнительно содержит промотор, предпочтительно производное мочевины, более предпочтительно 1-(2-диметиламиноэтил)-3-фенилмочевину.
3. Способ по п.2, в котором реакцию пропионового альдегида с соединением формулы (i) проводят при температуре в диапазоне от -15 до +5°С, предпочтительно от -12 до +3°С, более предпочтительно от -10 до 0°С, предпочтительно в дихлорметане (ДХМ) в качестве растворителя.
4. Способ по п.2 или 3, в котором реакцию пропионового альдегида с соединением формулы (i) проводят в присутствии каталитического количества кислоты, предпочтительно уксусной кислоты или пропионовой кислоты.
5. Способ по п.1, в котором соединение формулы (I) вводят в реакцию с H2N-NH-CHO в присутствии молекулярного сита, предпочтительно обладающего диаметрами пор, определенными в соответствии со стандартом DIN 66131, находящимися в диапазоне от 0,3 до 0,5 нм (от 3 до 5  ).
).
6. Способ по п.1, в котором соединение формулы (I) вводят в реакцию с H2N-NH-CHO при температуре в диапазоне от -10 до +20°С, предпочтительно от -5 до +5°С, предпочтительно в дихлорметане (ДХМ) в качестве растворителя.
7. Способ по п.1, в котором соединение формулы (I) не выделяют из реакционной смеси до реакции с H2N-NH-CHO.
8. Способ по п.1, дополнительно включающий
(2) выделение соединения формулы (II) из реакционной смеси с помощью жидкостной экстракции, где до стадии (2) предпочтительно проводят замену растворителя.
9. Способ получения хирального соединения формулы (III) из соединения формулы (II)
 ,
,
включающий
(3) реакцию соединения формулы (II) в растворителе с силилирующим реагентом, содержащим остаток -SiRaaRbbRcc, с получением соединения формулы (III)
,
в которой остатки Raa, Rbb и Rcc могут быть одинаковыми или разными и представляют собой алкильные или арильные остатки, предпочтительно алкильные остатки, содержащие от 1 до 6 атомов углерода, предпочтительно от 1 до 4 атомов углерода, предпочтительно 1 или 2 атома углерода, и
где Y обозначает замещенный или незамещенный арильный фрагмент, предпочтительно замещенный или незамещенный фенильный фрагмент, более предпочтительно незамещенный фенил, где замещенный арильный фрагмент и замещенный фенильный фрагмент содержат 1, 2 или 3 заместителя, которые выбраны из группы, включающей галоген, алкил и C1-алкоксигруппу, или C2-алкоксигруппу, или C3-алкоксигруппу, или C4-алкоксигруппу, или C5-алкоксигруппу, или C6-алкоксигруппу.
10. Способ по п.9, в котором силилирующим реагентом является гексаметилдисилазан, триметилхлорсилан, бис-триметилсилилацетамид или смесь двух или трех из этих соединений, предпочтительно бис-триметилсилилацетамид.
11. Способ по п.9, в котором реакцию на стадии (3) проводят при температуре в диапазоне от 15 до 70°С.
12. Способ получения хирального соединения формулы (IV) из соединения формулы (III)
 ,
,
включающий
(4) реакцию соединения формулы (III) с нуклеофильным соединением, содержащим нуклеофильный остаток R1, в растворителе с получением соединения формулы (IV)
,
в котором нуклеофильное соединение представляет собой соединение Гриньяра R1MgX, в котором X выбран из группы, включающей Cl, Br и I, и в котором R1 означает алкильный остаток, предпочтительно содержащий от 1 до 6 атомов углерода, более предпочтительно от 1 до 4 атомов углерода, более предпочтительно 1 или 2 атома углерода, наиболее предпочтительно 2 атома углерода, и
где Y обозначает замещенный или незамещенный арильный фрагмент, предпочтительно замещенный или незамещенный фенильный фрагмент, более предпочтительно незамещенный фенил, где замещенный арильный фрагмент и замещенный фенильный фрагмент содержат 1, 2 или 3 заместителя, которые выбраны из группы, включающей галоген, алкил и C1-алкоксигруппу, или C2-алкоксигруппу, или C3-алкоксигруппу, или C4-алкоксигруппу, или C5-алкоксигруппу, или C6-алкоксигруппу.
13. Способ по п.12, в котором нуклеофильным соединением является CH3CH2MgCl.
14. Способ по п.12, в котором на стадии (4) соединение формулы (III) вводят в реакцию с нуклеофильным соединением при температуре в диапазоне от -80 до 0°С, предпочтительно от -75 до -10°С, более предпочтительно от -70 до -25°С.
15. Способ по п.12, в котором в реакции соединения формулы (III) с нуклеофильным соединением на стадии (4) в качестве растворителя используют толуол или по меньшей мере один простой эфир, простой эфир предпочтительно выбран из группы, включающей тетрагидрофуран (ТГФ), метил-трет-бутиловый эфир (МТБЭ) и смесь ТГФ и МТБЭ.
16. Способ по п.12, в котором после
(3) реакции соединения формулы (II) в растворителе с силилирующим реагентом, содержащим остаток -SiRaaRbbRcc, с получением соединения формулы (III)
 ,
,
в которой остатки Raa, Rbb и Rcc могут быть одинаковыми или разными и представляют собой алкильные или арильные остатки, предпочтительно алкильные остатки, содержащие от 1 до 6 атомов углерода, предпочтительно от 1 до 4 атомов углерода, предпочтительно 1 или 2 атома углерода;
соединение формулы (III) не выделяют из реакционной смеси, полученной на стадии (3), до реакции с нуклеофильным соединением на стадии (4).
17. Способ получения хирального соединения формулы (V) из соединения формулы (IV)
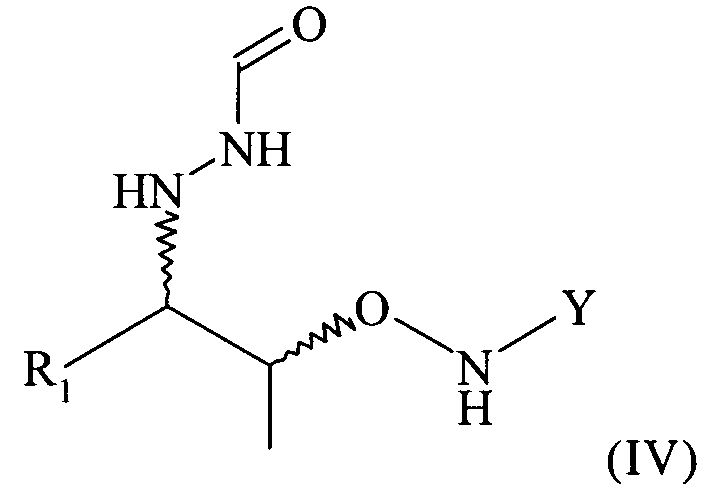 ,
,
включающий
(5) восстановление соединения формулы (IV), предпочтительно путем гидрирования, с получением соединения формулы (V)
,
в котором R1 означает алкильный остаток, предпочтительно содержащий от 1 до 6 атомов углерода, более предпочтительно от 1 до 4 атомов углерода, более предпочтительно 1 или 2 атома углерода, наиболее предпочтительно 2 атома углерода, и
где Y обозначает замещенный или незамещенный арильный фрагмент, предпочтительно замещенный или незамещенный фенильный фрагмент, более предпочтительно незамещенный фенил, где замещенный арильный фрагмент и замещенный фенильный фрагмент содержат 1, 2 или 3 заместителя, которые выбраны из группы, включающей галоген, алкил и C1-алкоксигруппу, или C2-алкоксигруппу, или C3-алкоксигруппу, или C4-алкоксигруппу, или C5-алкоксигруппу, или C6-алкоксигруппу.
18. Способ по п.17, в котором на стадии (5) гидрирование проводят при температуре в диапазоне от 15 до 35°С, предпочтительно от 20 до 30°С, при давлении водорода, находящемся в диапазоне от 0,5 до 50 бар, предпочтительно от 1 до 20 бар, более предпочтительно от 1 до 10 бар, в присутствии катализатора, содержащего благородный металл, предпочтительно катализатора, содержащего палладий, наиболее предпочтительно катализатора Pd/C.
19. Способ по п.17, в котором для восстановления соединения формулы (IV) на стадии (5) используют смесь растворителей, включающую по меньшей мере один спирт, содержащий от 1 до 4 атомов углерода, предпочтительно метанол, этанол, изопропанол, наиболее предпочтительно метанол.
20. Способ по п.19, дополнительно включающий кристаллизацию соединения формулы (V).
21. Способ по п.20, в котором соединение формулы (V) кристаллизуют из смеси МТБЭ и циклогексана (СНХ).
22. Способ по п.17, в котором соединение формулы (V) не подвергают очистке с помощью хроматографии.
23. Хиральное соединение формулы (II)
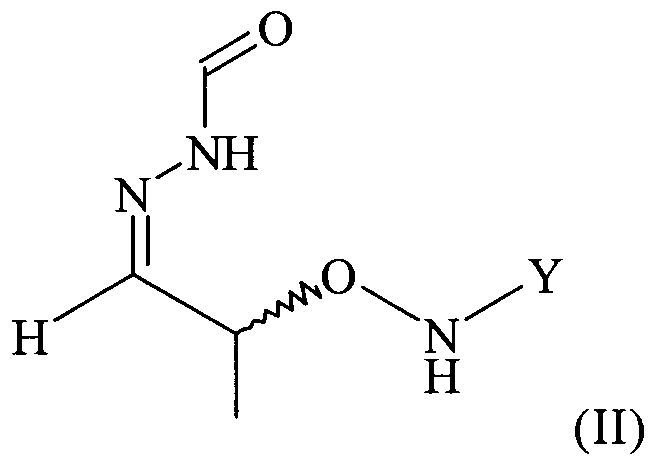 ,
,
в которой Y обозначает незамещенный арильный фрагмент, где предпочтительно не менее 95%, более предпочтительно не менее 97%, более предпочтительно не менее 99% молекул указанного соединения находятся в виде изомера формулы (IIa)
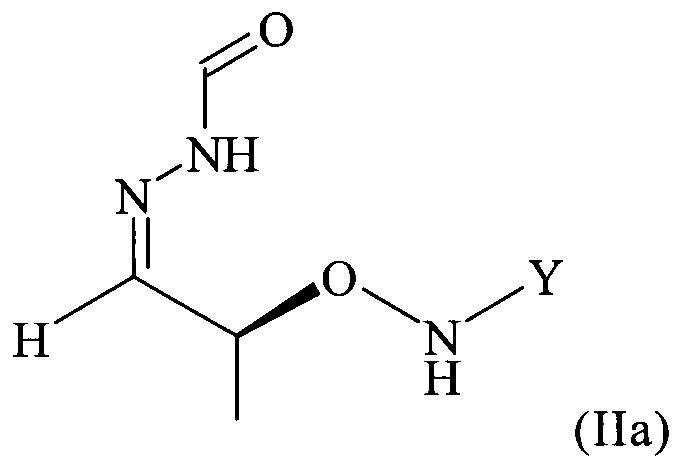 .
.
24. Хиральное соединение формулы (III)
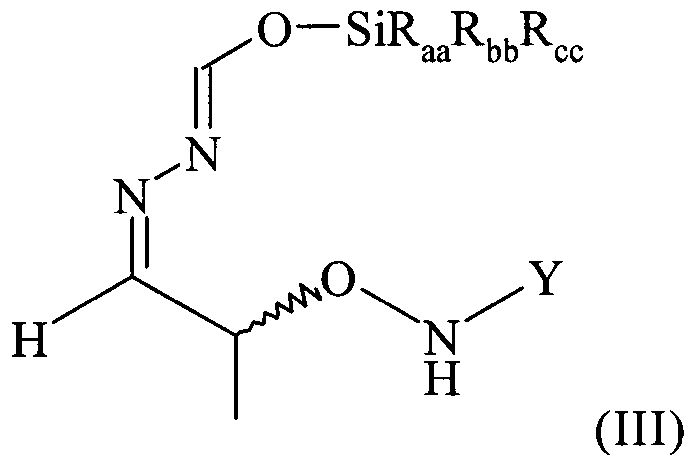 ,
,
в которой Y обозначает незамещенный арильный фрагмент, в которой остатки Raa, Rbb и Rcc могут быть одинаковыми или разными и представляют собой алкильные остатки, содержащие от 1 до 6 атомов углерода, предпочтительно от 1 до 4 атомов углерода, более предпочтительно 1 или 2 атома углерода, предпочтительно метил, где предпочтительно не менее 95%, более предпочтительно не менее 97%, более предпочтительно не менее 99% молекул указанного хирального соединения находятся в виде изомера формулы (IIIa)
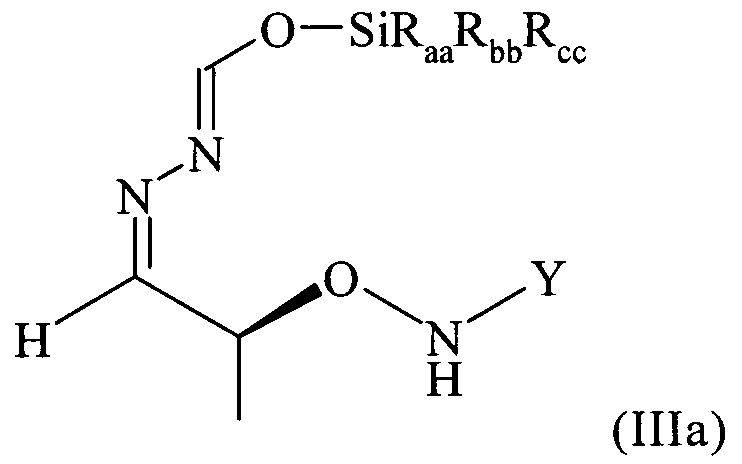 .
.
25. Хиральное соединение формулы (IV)
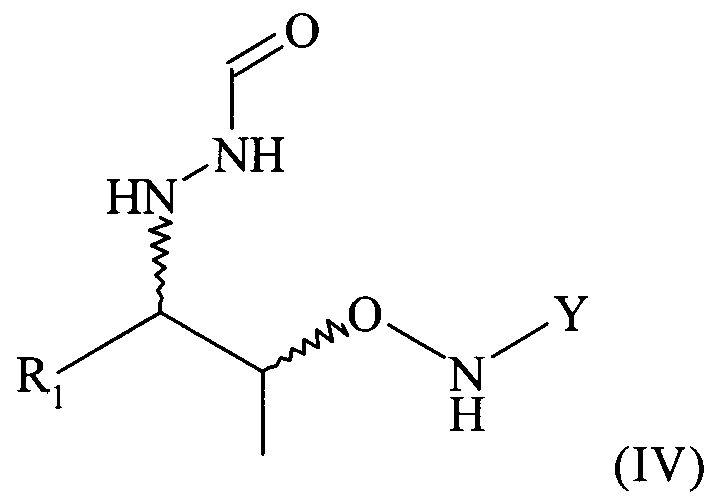 ,
,
в которой Y обозначает незамещенный арильный фрагмент, в которой R1 означает алкильный остаток, содержащий от 1 до 6 атомов углерода, более предпочтительно от 1 до 4 атомов углерода, более предпочтительно 1 или 2 атома углерода, наиболее предпочтительно 2 атома углерода, R1 предпочтительно обозначает этил, где предпочтительно не менее 95%, более предпочтительно не менее 97%, более предпочтительно не менее 99% молекул указанного хирального соединения находятся в виде изомера формулы (IVa)
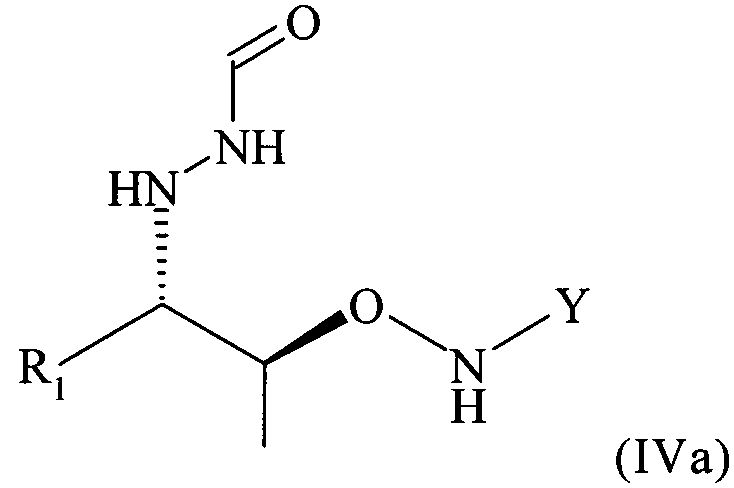 .
.
26. Хиральное соединение формулы (V)
 ,
,
в которой R1 означает алкильный остаток, содержащий от 1 до 6 атомов углерода, более предпочтительно от 1 до 4 атомов углерода, более предпочтительно 1 или 2 атома углерода, наиболее предпочтительно 2 атома углерода, R1 предпочтительно обозначает этил, где предпочтительно не менее 95%, предпочтительно не менее 97%, более предпочтительно не менее 99% молекул указанного соединения находятся в виде изомера формулы (Va)
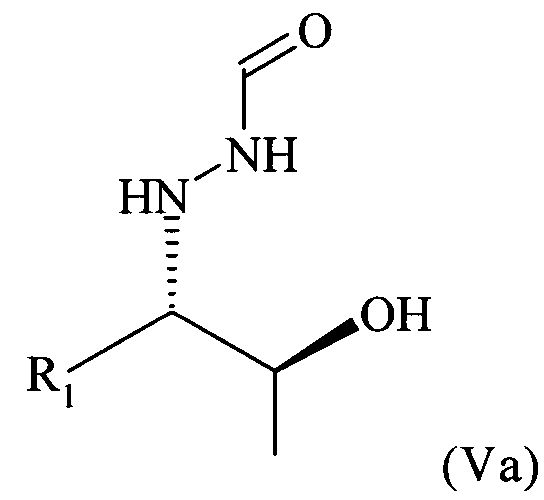 ,
,
предпочтительно в виде изомера формулы (Vb)
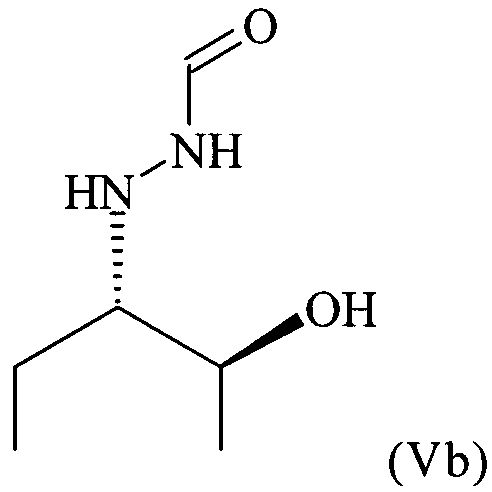 .
.
27. Хиральное соединение по п.26 формулы (V), в которой R1 = этил, в кристаллической форме
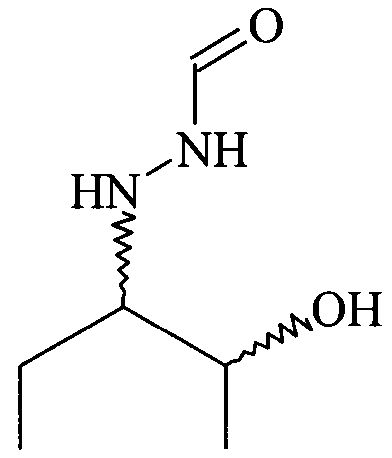 ,
,
где предпочтительно не менее 95%, предпочтительно не менее 97%, более предпочтительно не менее 99% молекул указанного кристаллического соединения находятся в виде изомера формулы (Vb), обладающего рентгенограммой, содержащей по меньшей мере следующие отражения:
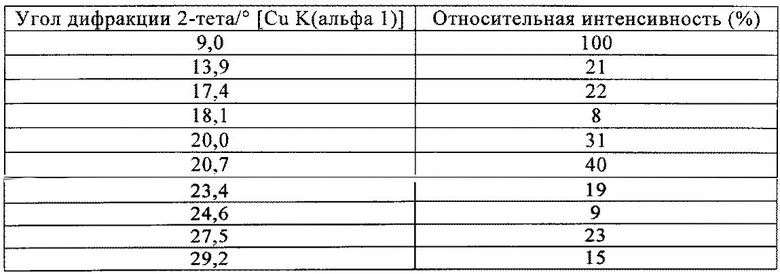
,
где 100% относится к интенсивности наиболее интенсивного пика на рентгенограмме.
28. Применение хирального соединения по п.26 или 27 или хирального соединения, которое получают способом по любому из пп.17-22, для получения фунгицидного средства.
29. Применение по п.28, в котором для получения фунгицидного средства хиральное соединение вводят в реакцию с производным анилина
и производным фосгена
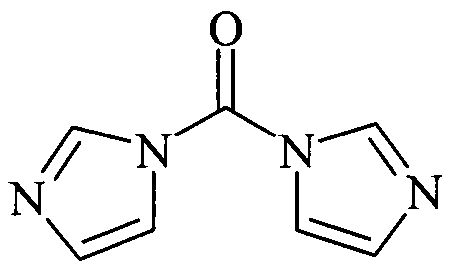 .
.
30. Применение по п.28 или 29, в котором хиральным соединением является хиральное соединение по п.26 или 27 или хиральное соединение, которое получают способом по любому из пп.17-22, хиральным соединением предпочтительно является кристаллическое хиральное соединение формулы (V), в которой R1=этил
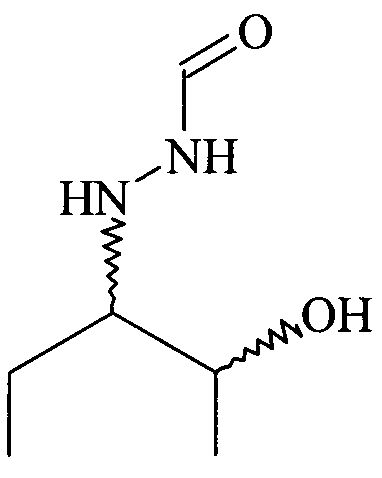 ,
,
где не менее 99% молекул указанного кристаллического соединения находятся в виде изомера формулы (Vb)
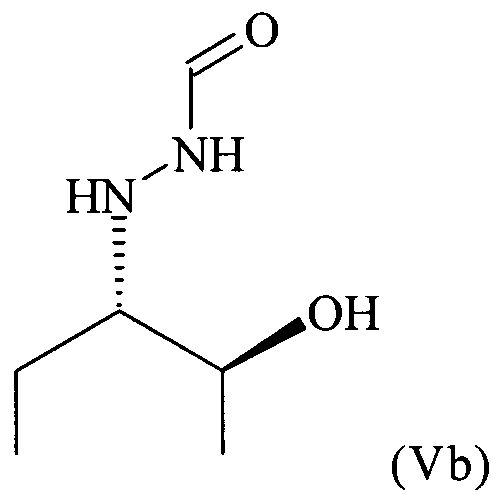 .
.
31. Применение по п.30, в котором фунгицидным средством является
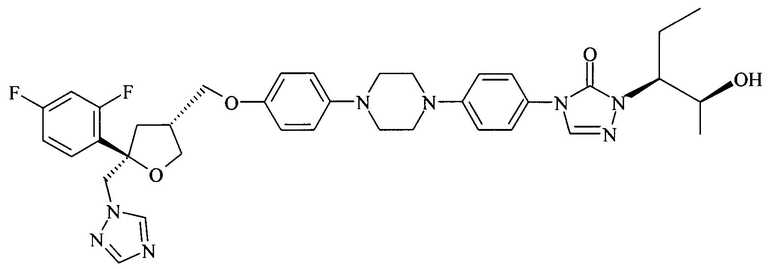 .
.
| A | |||
| K | |||
| SAKSENA et al., Stereoselective Grignard additions to N-formyl hydrazone: a concise synthesis of Noxafil side chain and a synthesis of Noxafil, TETRAHEDRON LETTERS, 2004, Vol | |||
| Железобетонный фасонный камень для кладки стен | 1920 |
|
SU45A1 |
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| ОПТИЧЕСКИ АКТИВНЫЕ ПРОИЗВОДНЫЕ 2-ИМИДАЗОЛИН-5-ОНОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФУНГИЦИДНАЯ КОМПОЗИЦИЯ, СПОСОБ ОБРАБОТКИ КУЛЬТУР | 1994 |
|
RU2126390C1 |

