Настоящее изобретение относится к способу отделения растворенной L-дигидрооротовой кислоты из раствора (далее в тексте “L-DHO”) методом хроматографии на анионообменном материале. Этот способ может использоваться при диагностике активности ингибиторов дигидрооротовой кислоты, а также при мониторинге этой активности как in vivo, так и in vitro.
L-DHO можно определять хроматографией на силикагеле с последующей химической дериватизацией и использованием колориметрических методов (Kesner, L., Aronson, F.L., Silverman, М., Chan, Р.С., Clin. Chem. 21/3 (1975) 353). В соответствие с другим способом L-DHO ферментативно превращают в оротовую кислоту под действием дегидрогеназы L-дигидрооротовой кислоты (далее в тексте "DHODH") печени крыс и, после химической дериватизации, оротат детектируют по колориметрическим изменениям (Rogers, L.E., Nicolaisen, К., Experientia 28/10 (1972) 1259). Недостатком этих способов является вмешательство посторонних материалов в комплексные физиологические растворы. Кроме этого, упомянутые методы требуют больших временных затрат в связи с трудоемкостью приготовления образцов и в связи с этим они неприменимы для рутинных анализов в крупномасштабных клинических исследованиях.
При проведении исследований, направленных на разработку усовершенствованных процессов разделения и выделения, предназначенных для получения L-дигидрооротовой кислоты, было установлено, что конечный результат может быть достигнут методом хроматографии L-DHO в смеси воды с основанием на анионообменном материале и при давлении от около 1,1 до около 40 МПа. Такой способ может применяться для количественного определения L-DHO в клеточных лизатах, сыворотке млекопитающих и людей. Указанный способ характеризуется высокой воспроизводимостью, чувствительностью и надежностью.
Как разъясняется в формуле изобретения, задача изобретения достигается способом отделения растворенной L-дигидрооротовой кислоты хроматографией из раствора, содержащего указанную кислоту и вещества-примеси, предусматривающим:
a) загрузку указанного раствора в хроматографическую колонну, содержащую устойчивый к действию давления аниоонобменный материал для связывания указанной кислоты,
b) элюирование большей части веществ-примесей из колонны водным раствором и
c) элюирование указанной кислоты из указанного анионообменного материала водным раствором, содержащим основание, под давлением от около 1,1 до около 40 МПа.
Термин "анионообменный материал, устойчивый к действию давления" относится, например, к таким материалам, как макропористый (2000) дивинилбензол/этилвинилбензольный полимер или микропористый поливинилбензиламмониевый полимер, сшитый с помощью дивинилбензола, или к их смесям, модифицированным четвертичным аммониевым соединением алканола, или к винилбензилхлорид/дивинилбензольному макропористому полимеру, или сшитому полиэтилиминовому полимеру, или к диоксиду кремния, модифицированному пропилтриметиламмонием, или поли(стиролдивинилбензол)триметиламмонию.
Особенно предпочтительными являются следующие продукты:
анионообменные колонны Ion Pas As 11, CarboPac PA 1 или CarboPac MA 1, поставляемые Dionex Corporation, Idstein, Germany,
сильно анионный GROM-SIL или слабоанионный GROM-SIL, поставляемые Grom, P 1000 SAX, Ionospher SA или Chpompack PA, поставляемые Chpompack,
PRP-X100 или RCX-10, поставляемые Hamilton.
Элюирующий раствор содержит смесь воды с основанием. Подходящие основания являются такими производными щелочных или щелочноземельных металлов, как гидроксид натрия, гидроксид калия, гидроксид магния или гидроксид кальция. Концентрация основания составляет 1-200 ммоль/л в расчете на воду или растворитель, предпочтительно 2-120 ммоль/л, особенно предпочтительно 100 ммоль/л.
Температуры в процессе хроматографической обработки составляют 0-50°С, предпочтительно 15-30°С, особенно предпочтительно 19-25°С. Рабочее давление в ходе хроматографического процесса имеет практически постоянное значение. Хроматографию можно осуществлять с использованием различных давлений, например, процесс можно проводить при давлении 1,1×106-40×106 Па (1,1-40 МПа), особенно при 4,1-5,5 МПа. Расход элюента составляет 0,2-3 мл/мин, предпочтительно 1 мл/мин.
Загрузка материала в колонну, сам способ хроматографии и элюирование L-DHO осуществляют традиционными техническими методами.
Подходящий метод элюирования представляет собой элюирование с временным градиентом по концентрации основания, причем предпочтительно, чтобы изменение времени происходило почти линейно. Может использоваться концентрационный градиент, например в начале процесса элюирования может применяться низкая концентрация основания (нулевая концентрация, как крайний случай), после чего в ходе элюирования концентрацию основания повышают. Таким способом удается достичь особенно эффективного разделения L-DHO в образцах на основе сыворотки или клеточных лизатов. Предпочтительный градиент основания может изменяться от величины, близкой к 1% NaOH (100 ммоль/л) в 99% воды (в начальный период элюирования) до примерно 60% NaOH в 40% воды (к концу элюирования), причем особенно предпочтительный интервал составляет от 1% NaOH в 99% воды (в начальный период элюирования) до 15% NaOH в 75% воды (к концу элюирования). Градиент смеси воды с основанием изменяется линейно в периоды времени от 2,5 до 14 минут и от 14 до 25 минут, причем на этих двух временных периодах глубина изменения градиента различна.
Особенно успешное элюирование может достигаться с использованием низкой концентрации основания в начале процесса разделения, например 1% концентрации за период в 2,5 минуты. В результате элюируется большая часть вещества-примеси из биологической матрицы, загруженной в колонну. Разделение аналита достигается медленным повышением градиента до 23% основания за 14 минут общего времени анализа. Затем концентрацию основания в течение 4 минут увеличивают примерно до 60% с целью осуществления элюирования прочно связанного материала. 60% концентрацию основания следует применять в течение времени не более 6 минут, после чего повторно устанавливается равновесие с помощью 1% смеси основания с водой. Следующий анализ начинают через 45 минут общего времени анализа.
Воду в градиенте смеси вода-основание следует деионизировать и дегазировать. Согласно изобретению процесс разделения осуществляют на колонне. Температура, которую в ходе анионообменной хроматографии предпочитают поддерживать постоянной, может изменяться в широких пределах. Предпочтительный температурный интервал составляет 10-50°С, особенно предпочтительно 15-25°С.
Элюирование L-DHO происходит в период с 10 по 12 минуту после начала применения градиента. Время действия процесса элюирования составляет от 13 до 25 минут. L-DHO детектируют с помощью такого детектора по проводимости, как, например, модель CD20 от Dionex Cooperation. С целью минимизации дрейфа базовой линии и уменьшения фоновой проводимости можно использовать такой анионный саморегенерирующий подавитель, как модель ASRS-1, размером 4 мм, выпускаемый Dionex Cooperation.
Способ по изобретению, главным образом, применим для аналитической хроматографии, однако он может использоваться и в препаративной хроматографии, особенно в том случае, когда процесс настоящего изобретения осуществляют с использованием системы с колонной для препаративной жидкостной хроматографии высокого давления (HPLC). Термин “препаративная хроматография” относится к способу очистки, предназначенному для получения чистых продуктов, а не только для их анализа. Количество чистых продуктов может изменяться в широких пределах, например от 1 до 1000 г, предпочтительно от 50 до 500 мг.
Способ по изобретению может использоваться для определения изменений концентраций внутриклеточной и межклеточной L-DHO за счет ингибирования дегидрогеназы дигидрооротовой кислоты (DHO-DH). Фермент DHO-DH ответственен за конверсию L-дигидрооротовой кислоты в оротовую кислоту в ходе de novo пиримидинового синтеза. Ингибирование DHO-DH приводит к накоплению L-DHO. Способ настоящего изобретения может использоваться для приготовления образца для диагностического анализа. Способ настоящего изобретения может использоваться для определения активности DHO-DH ингибиторов. DHO-DH ингибиторы представляют собой, например, 5-метил-N-[4-(трифторметил)фенил)-4-изоксазол-карбоксамид, 6-фтор-2-(2’-фтор-[1,1’-дифенил]-4-ил)-3-метил-4-хинолинкарбоновую кислоту (также известную под названием Brequinar), 2-циано-3-гидрокси-N-[4-(трифторметил)фенил]-2-гептен-6-инамид, 2-циано-N-(4-цианофенил)-3-циклопропил-3-гидрокси-2-пропенамид и 2-циано-3-гидрокси-N-[4-(трифторметил)фенил]-2-бутенамид. Способ согласно настоящему изобретению может применяться для определения концентраций L-DHO в растениях, линиях клеток, животных и людях. Определение L-DHO может использоваться для мониторинга активности DHO-DH ингибиторов в организмах растений, млекопитающих и людей.
Способ настоящего изобретения подробно описывается в следующих ниже примерах. Если не указано особо, то процентные соотношения даны в весовом выражении.
Пример 1
1.1. Химикаты и реагенты
Химикаты и реагенты приобретены в следующих фирмах:
NaOH и КОН, не содержащие карбоната (Backer, Holland)
L-Дигидрооротовая кислота L-DHO(Sigma, Munich)
HClO4 Riedel de Haen, Seelze
Eosin, хлороформ Riedel de Haen, Seelze
Среда RPMI 1640 Gibco, Eggenstein
Сыворотка эмбриона теленка Bio Whitaker, Verviers, Belguim
(FCS)
Деионизированную воду дегазируют гелием перед применением.
1.2. Хроматографическое оборудование
HPLC система состоит из следующих элементов (см. таблицу А).
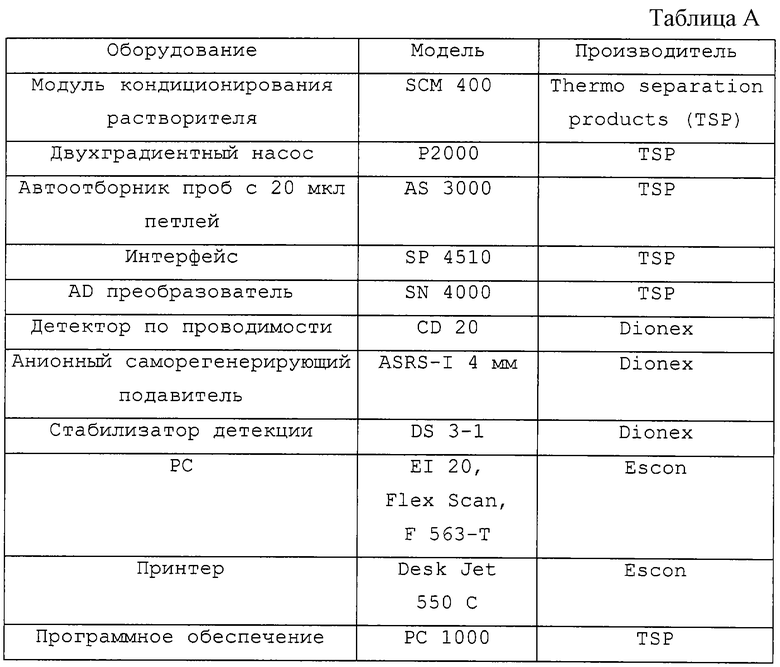
Во всех экспериментах использовали указанные материалы.
1.3. Условия HPLC
Хроматографическое разделение осуществляли с использованием колонки с анионообменной смолой IonPac AS 11 длиной 250 мм и внутренним диаметром 4 мм (размер частиц 13 мкм; P/N 044076, Dionex), снабженной предколонной an IonPac AG 11 длиной 50 мм и с внутренним диаметром 4 мм (размер частиц 13 мкм; P/N 044078, Dionex). Кроме этого, между градиентным насосом и инжекционным клапаном устанавливали анионную колонку-сепаратор АТС-1 (P/N 037151, Dionex). Для минимизации дрейфа базовой линии и уменьшения фоновой проводимости инсталлировали ASRS-I подавитель, работающий при силе тока 300 мА. Диапазон работы детектора устанавливали на 10 μS. Автоотборник проб охлаждали до 14°С, но сам анализ проводили при комнатной температуре. Подвижная фаза состояла из 100 мМ NaOH (А) и деионизированной и дегазированной воды (В). С помощью указанной системы получали следующие градиенты (см. таблицу В).
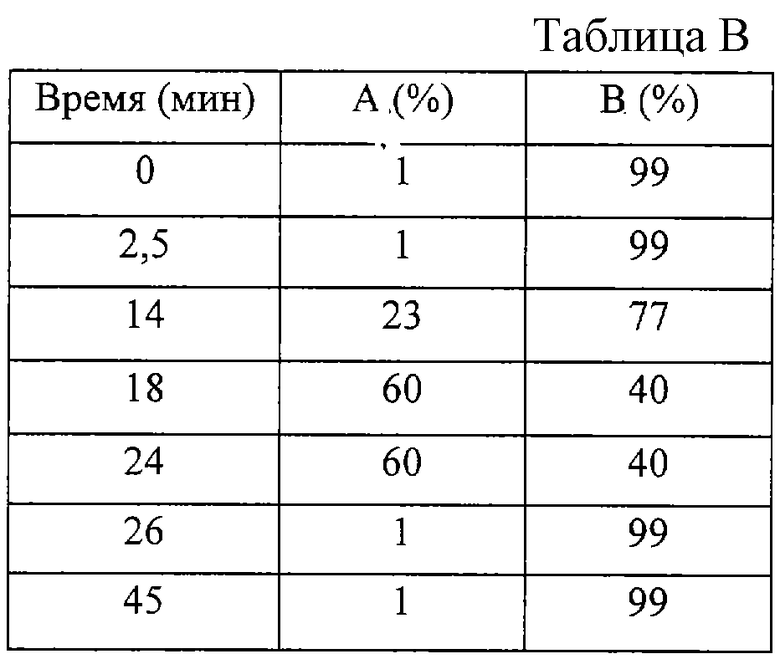
Объемная скорость составляла 1 мл/мин; время опыта 45 минут.
1.4. Стандарты и образцы для контроля качества
Стандартный исходный раствор готовили растворением 1 мг L-DHO в 1 мл воды. Аликвоты объемом в 400 мкл замораживали до -20°С. Стабильность таких растворов гарантирована в течение, по меньшей мере, 4 недель. Определенные количества исходного раствора добавляли к клеточным лизатам и к сыворотке человека или крысы и проводили проверку линейности между увеличением сигнала и концентрацией L-DHO. Точность и достоверность метода оценивали с использованием образцов для контроля качества (QS) с содержанием L-DHO в области интервала низких, средних и высоких концентраций линейной кривой зависимости сигнал/концентрация.
1.5. Приготовление образцов
1.5.1. Клетки
Клетки Jurkat получали из коллекции АТСС (TIB 182) и культивировали, как описано в разделе 1.5.1.1.
1.5.1.1. Культивирование тканевых культур
Клетки Jurkat высевали в количестве 5×105 /мл и выращивали в течение 24 часов в среде RPMI 1640, содержащей 10% сыворотки эмбриона теленка (FCS). Клетки собирали в свежей среде для лизиса, после чего общее количество клеток и процентное количество погибших клеток рассчитывали по жизнеспособности при микроскопическом исследовании с окрашиванием эозином. Через 24 часа численность клеток возрастала в 1,6-1,8 раз, причем число погибших клеток составляло менее 7%. Клетки Jurkat, используемые для определения L-DHO, имели коэффициент пролиферации 1,6-1,8 за 24 часа и при этом погибало менее 7% клеток.
1.5.1.2. Приготовление клеточных лизатов
Клетки суспендировали в определенном объеме среды и плотность клеток в образцах определяли методом витальной микроскопии с окрашиванием эозином. Примерно 10×106 клеток удаляли, осаждали в результате центрифугирования в течение 5 минут при 350 g и супернатант отбрасывали. Лизис клеток осуществляли ресуспендированием клеточного осадка в 500 мкл 1,2 М НСlO4. Полученную смесь переносили в пробирки Эппендорфа с закрывающимися крышками емкостью 2,0 мл и высокоскоростным центрифугированием в течение 2 минут осаждали белок. Надосадочную жидкость полностью удаляли, переносили в стеклянные пробирки и, после добавления 500 мкл хлороформа, тщательно перемешивали в течение 2 минут. Клеточные липиды экстрагировали хлороформом, после чего центрифугировали в течение 10 минут (при 1502 g) при 10°С. Очищенную надосадочную жидкость собирали в 2 мл пробирки Эппендорфа и хранили при -20°С до использования. Для HPLC анализа 100 мкл такой надосадочной жидкости нейтрализовали 30 мкл 6 М раствора КОН. После встряхивания в течение 5 секунд полученные образцы хранили на льду в течение 30 минут. После этого их центрифугировали в течение 5 минут со скоростью 15 000 об/мин. 10 мкл порцию прозрачной надосадочной жидкости использовали для HPLC анализа.
1.5.2. Сыворотка
С целью снижения количества белка, образец сыворотки объемом 20 мкл помещали на фильтр Microcon (10000 D, модель 10, код 42407, Amicon) и центрифугировали в течение 30 минут со скоростью 13000 об/мин. Через фильтр проходило примерно 150 мкл вещества, представляющего собой аналитический образец L-DHO. 20 мкл полученной жидкости использовали для HPLC анализа.
1.6. Количественное определение
С помощью интегратора определяли высоту пика анализируемого вещества. Строили калибровочные кривые зависимости измеренных высот пиков (y) от концентрации анализируемого вещества в различных биологических матрицах. Значение взвешенной линейной регрессии (1/y) использовали для обратного расчета концентрации L-DHO в стандартных образцах, а также для качественного контроля. Общий коэффициент корреляции R определяли с помощью PROC GLM на основе анализа ковариантной модели с использованием фактора взвешивания.
1.7. Стабильность
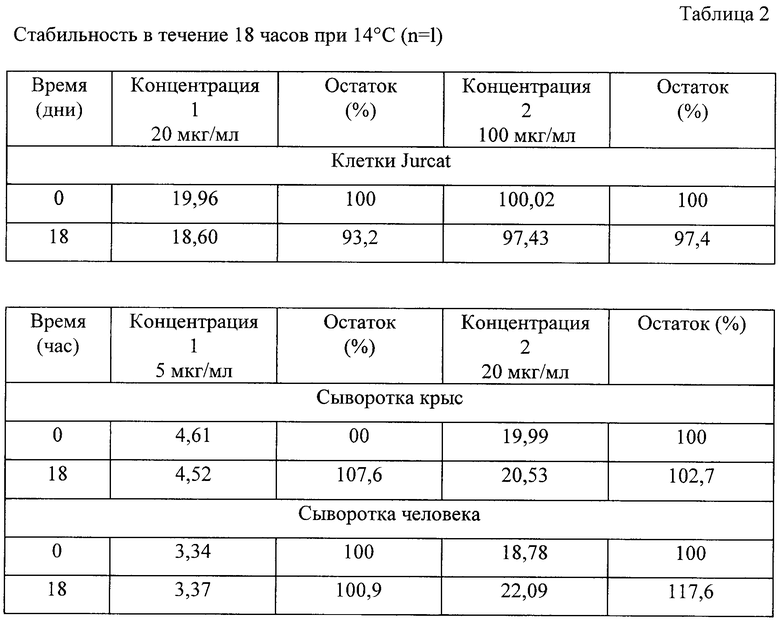
В таблице 1 представлены данные по стабильности аналита при -20°С в клеточных лизатах и образцах сыворотки после 2-3 циклов замораживания/оттаивания одного образца. В указанных выше условиях L-DHO обладала устойчивостью в клетках Jurcat в течение, по крайней мере, 4 недель. Детектируемое увеличение концентрации на величину более 10% в некоторых образцах сыворотки крыс и одном образце человеческой сыворотки, после нескольких циклов оттаивания, не находит объяснения и полученный результат свидетельствует о том, что в этих случаях точность может снижаться до 15% и в худшем случае до 29%. По этим причинам можно считать, что L-DHO в образцах сыворотки человека и крысы, в действительности, устойчива лишь в течение, по крайней мере, 1 недели.
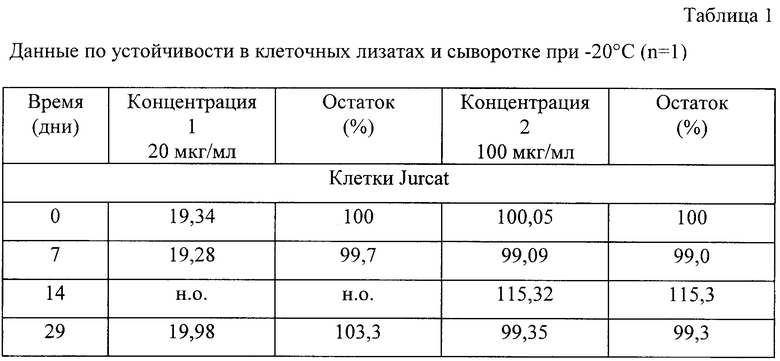
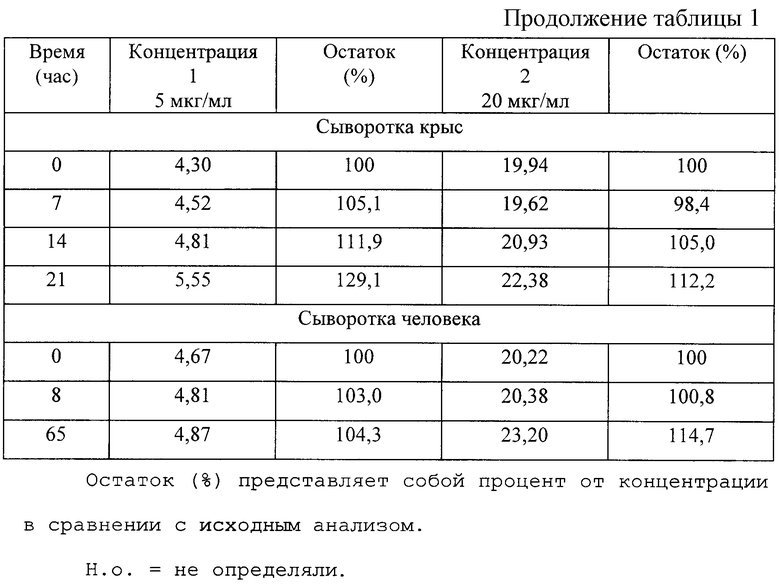
Для имитации условий пребывания образцов в автоотборнике проб перед реальным анализом, определяли стабильность в течение 18 часов при 14°С. Для этой цели перед началом анализа клеточные лизаты активизировали и обрабатывали системой 30 мкл 6 М КОН/100 мкл лизата. Соответствующим образом образцы сыворотки активизировали и затем извлекали белки, как описано в 1.5.2.
Как можно видеть из данных, представленных в таблице 1, в таких условиях количество L-DHO в клетках несколько уменьшалось до максимального значения около 7%. В образцах сыворотки, при таких условиях, аналит был стабильным. В указанных целях готовили такое количество образцов для HPLC, чтобы максимальная продолжительность пребывания в аутосэмлере была менее 18 часов.
1.8. Селективность
Сравнение не активизированных лизатов клеток Jurcat с соответствующими хроматограммами клеточных лизатов, активированных 50 мкг L-DHO/мл, позволило обнаружить наличие небольшого пика 11,983 мин, что является временем удерживания L-DHO. Весьма вероятно, что этот пик отражает естественное содержание аналита в таких клетках. Кроме этого, можно видеть, что в результате обработки клеточных лизатов КОН пик, соответствующий L-DHO, расщепляется на два пика. Вместе с тем обработка КОН является важной операцией для нейтрализации кислых клеточных лизатов перед HPLC анализом. Может быть показано, что в описанных условиях оценка высоты такого второго пика (время удерживания (КЕ)=11,954 мин) может использоваться для получения улучшенных результатов в плане линейности и воспроизводимости.
В случае опытов с образцами сыворотки крыс и людей, в контрольных опытах был обнаружен пик с тем же временем удерживания, что время удерживания L-DHO. Было предположено, что полученный результат отражает природное содержание L-DHO в организме. Исследование, по меньшей мере, 10 различных образцов обеих разновидностей показало, что естественное содержание L-DHO составляет величину ниже уровня определения порядка 1 мкг/мл.
1.9. Линейность
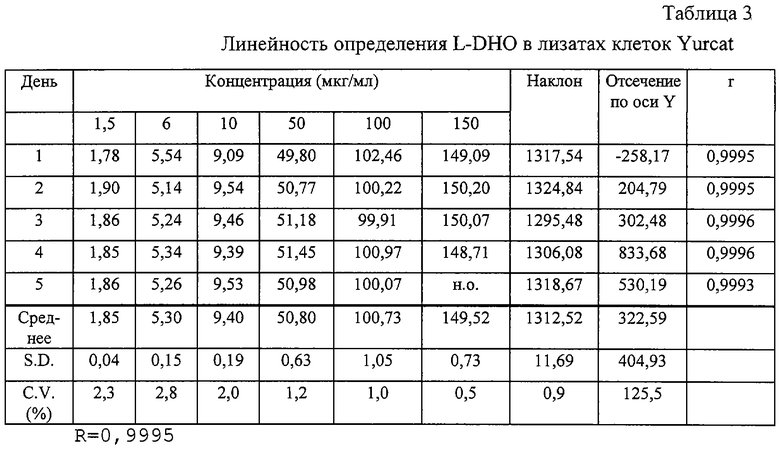
Линейность определения оценивали по пяти калибровочным кривым для линии клеток и различных образцов сыворотки. Образцы готовили и испытывали в различные пятые дни, используя концентрации L-DHO в интервале 1,5-150 мкг/мл (клеточные лизаты) и 1-30 мкг/мл (образцы сыворотки). Полученные результаты представлены в таблицах 3-5. Для определения линии регрессии использовали максимальные значения высот. На основании полученных результатов соответствующие концентрации различных стандартов получали обратным расчетом, как указано в различных таблицах.
После активизации образцы обрабатывали 30 мкл 6 М раствора КОН и после этого однократно анализировали, как описано в разделе 1.3.
После активизации сыворотку очищали от протеинов, как описано в 1.5.2.
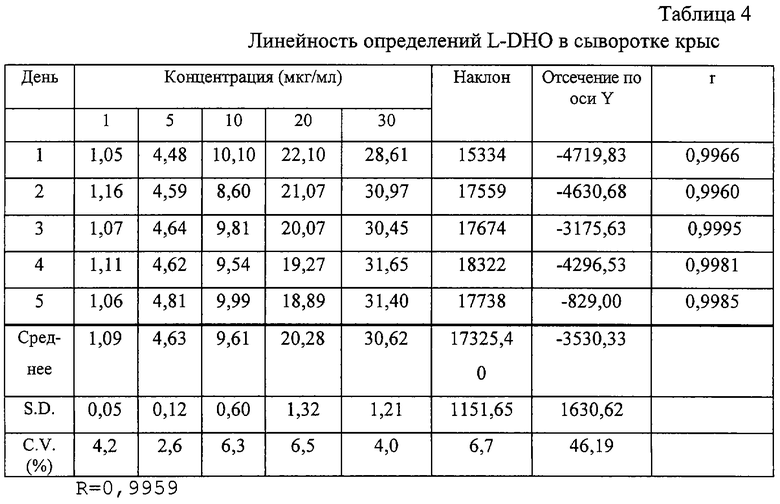
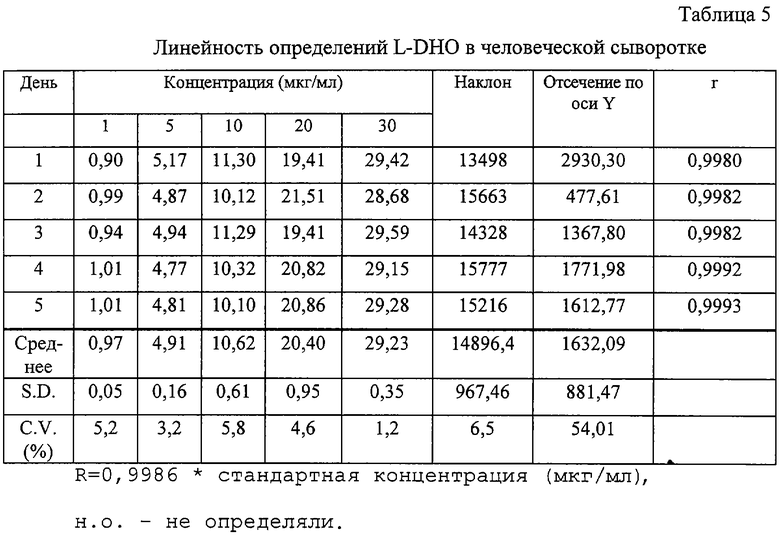
Как следует из результатов, представленных в приведенных выше таблицах, в каждом из рассматриваемых случаев доказана линейность. Этот факт отражен индивидуальными коэффициентами корреляции "r", которые во всех случаях >0,99. В каждой из приведенных таблиц представлены линии средней линейной регрессии для концентрационных кривых, полученных на различные пятые дни и они показывают, что значения наклона очень хорошо воспроизводятся при максимальной вариации 6,7%.
Полученные обратным расчетом значения стандартных концентраций в среднем дают значения C.V. менее 6,5%, что свидетельствует о высокой точности полученных значений. Общее корреляционное значение коэффициента R выше 0,99 свидетельствует об очень высокой точности и воспроизводимости метода.
1.10. Пределы количественного определения
На основании полученных результатов, предел количественного определения в клетках Jurcat составляет 1,5 мкг/мл. В образцах крысиной и человеческой сыворотки могут детектироваться количества L-DHO порядка 1 мкг/мл. Активизированные образцы в таких концентрациях имеют отношение сигнал/шум, по меньшей мере, 1:3.
1.12. Точность и прецизионность
Точность воспроизводимых определений L-DHO при трех различных концентрациях на различные пятые дни отражена в таблицах 6-8.
Точность выражали как % различий в соотношениях найденного к добавленному количеству L-DHO (регенерация). Точность определений в течение одного дня, выраженную значением C.V. (%), рассчитывали по двум значениям, полученным при двукратном измерении на одном образце в течение одного дня. Точность определения в различные дни также выражали значениями C.V. (%) и рассчитывали с использованием средних экспериментальных значений для каждого контрольного образца, полученных в различные пятые дни.
Точность определений в лизатах клеток Jurcat после активизации и нейтрализации 30 мкл 6 М раствора КОН. n=2 означает, что один образец клеточного лизата активизировали с помощью соответствующей концентрации и измерение проводили два раза.
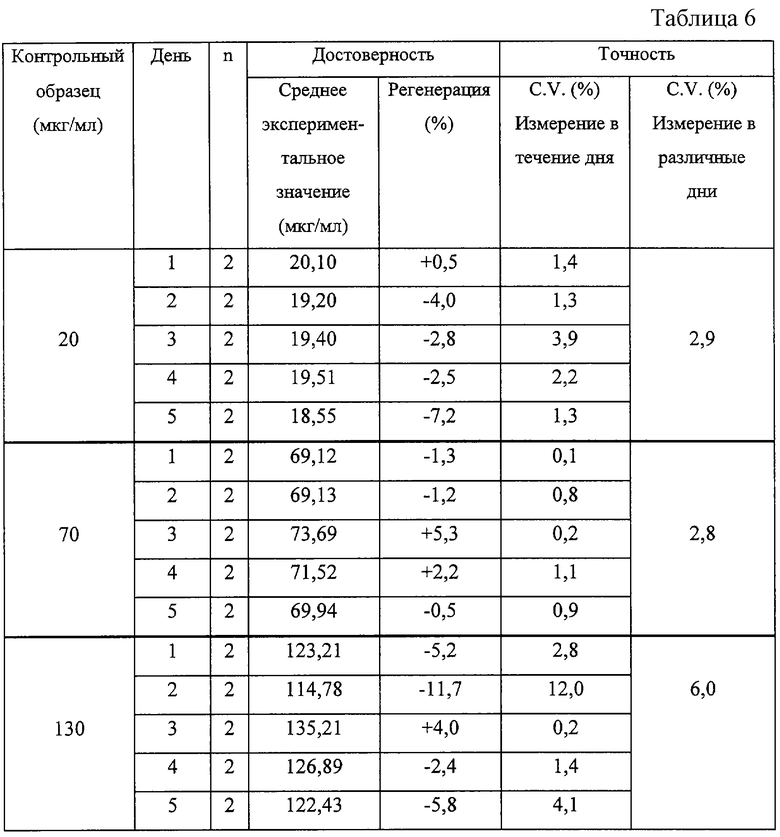
Точность определений на образцах крысиной сыворотки после активизации и последовательной депротенизации. n=2 означает, что активизации подвергали один образец сыворотки с помощью соответствующей концентрации и проводили двукратное измерение.
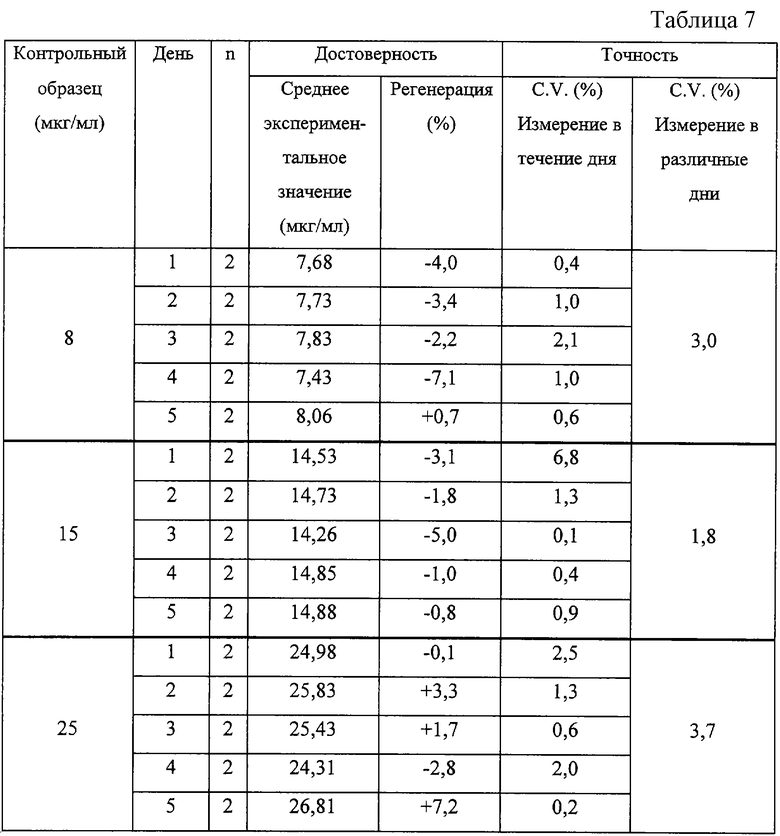
Точность измерений на образцах человеческой сыворотки после активизации и последующей депротенизации. n=2 обозначает, что активизации подвергали один образец сыворотки с помощью соответствующей концентрации и проводили двукратное измерение.
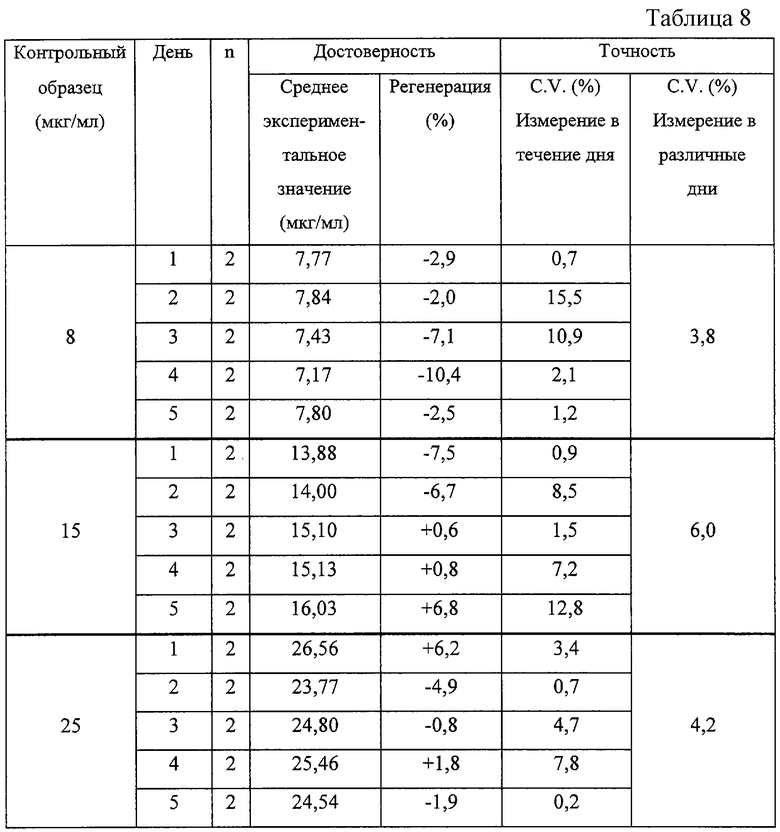
Представленные выше результаты показывают, что в большинстве случаев контрольные значения в точке максимума определяют с точностью +/-10%, в связи с чем такой метод может считаться весьма точным. Лишь в одном случае выход при определениях в клетках Jurcat несколько отличается от указанного (-11,7%). Этот результат может найти объяснение в рамках, указанных выше, дня испытаний, достигающего 12%. Внутридневная точность определения в образцах клеток Jurcat крысиной и человеческой сыворотки составляет величину ниже 5%, 7% и 10% соответственно. Точность определения в различные дни на всех исследованных матрицах была очень высокой со значением C.V. ниже 6,0%. Этот факт показывает, что полученные результаты могут рассматриваться как высоковоспроизводимые и точные.
Пример 2
2.1. Тканевые культуры
Приготовление бессывороточной среды. Порошкообразную среду Iscove (Biochrom) растворяли в 10 л бидистиллированной воды, дополненной 18,95 г NaCl, 11,43 г NаНСО3, 700 мг КСl, 10 мл 35% раствора NaOH и 0,5 мл 1 М раствора меркаптоэтанола (Riedel de Haen), и полученную смесь фильтровали в стерильных условиях. К одному литру приготовленной среды Iscove 32 перед применением добавляли 32 мг человеческого голо-трансферина, 1 г коровьего альбумина и 1,5 мл липидов (Sigma).
- Клеточная культура. Клетки A20.2.J культивировали в бессывороточной среде (37°С, 5% CO2) в виде расширенной культуры при логарифмическом клеточном росте. Клетки, отбираемые для анализа, имели коэффициент пролиферации 2,2 за 24 часа. Процент погибших клеток составлял <8% (3).
Обработка клеток N-(4-трифторметил)-2-циано-3-гидроксикротонамидом, полученным в соответствии с ЕР-0529500, далее в тексте А77 1726. А77 1726 растворяли в воде (10 мМ) и дополнительно разбавляли бессывороточной средой.
Затем в клетки добавляли соответствующее количество А77 1726 и смесь инкубировали при 37°С в атмосфере содержащей 5% СO2.
2.2. Приготовление клеточных лизатов для определения DHO
Приготовленные клетки ресуспендировали в указанной среде при подходящей плотности клеток. В зависимости от ожидаемого содержания DHO 1-50 миллионов клеток удаляли, подвергали центрифугированию (5 мин, 350 g) и надосадочную жидкость выбрасывали. Клетки лизировали путем добавления 500 мкл 1,2 М раствора НСlO4. Лизаты переносили в пробирки Эппендорфа объемом 2 мл и осаждали белок высокоскоростным центрифугированием в течение 2 минут. Подкисленные лизаты полностью удаляли, переносили в стеклянные пробирки и, после добавления 500 мкл хлороформа, тщательно перемешивали в течение 2 минут с образованием воронки. Клеточные липиды экстрагировали, после чего материал подвергали холодному центрифугированию (1502×g; 10°С). Очищенную надосадочную жидкость собирали в 2 мл пробирки Эппендорфа и хранили при -20°С до проведения анализа методом жидкостной хроматографии высокого давления (HPLC).
2.3. Определение DHO методом HPLC
Хроматографическое разделение осуществляли по методике, описанной в примере 1. Диапазон детектора по проводимости устанавливали равным 10 μS. Анализ проводили при комнатной температуре. Подвижная фаза содержала 100 мМ NaOH (А) и воду (В). С помощью такой системы получали следующий градиент (см. таблицу С).
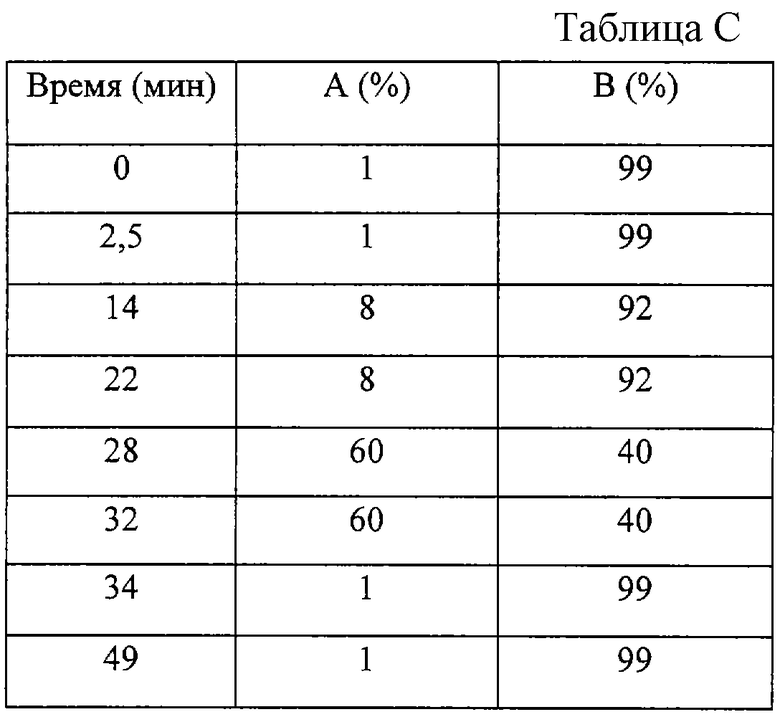
Объемная скорость составляла 1 мл/мин; продолжительность опыта 49 минут.
2.4. Результаты
Клетки A20.2.J, инкубированные с АРУ 1726, имели повышенные содержания внутриклеточной DHO (таблицы 9-11). Результаты представленные в таблице 9 демонстрируют тот факт, что уровни содержания DHO непосредственно коррелируют с количеством экстрагированных клеток.
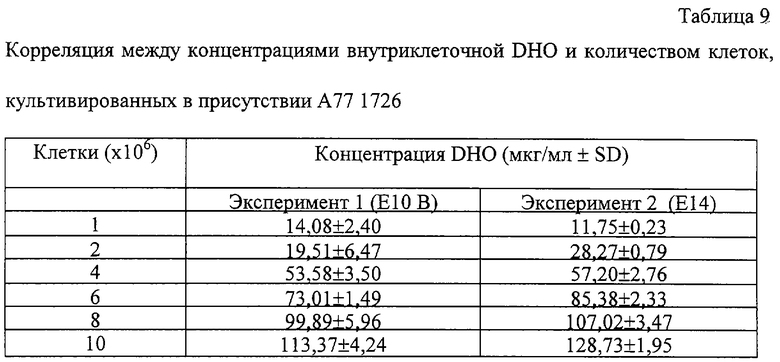
Клетки A20.2.J обрабатывали 5 мкМ А77 1726 и культивировали в течение 24 часов (37°С, 5% CO2), после чего готовили для экстракции DHO (n=3).
С целью оптимизации методов культивирования клеток и определения наилучшего молярного соотношения клетка/А77 1726, различные плотности клеток A20.2.J инкубировали совместно с А77 1726 (5 мкМ). Образцы отбирали в различные периоды времени и определяли концентрации DHO (таблица 10). В связи с тем, что концентрации DHO непосредственно коррелируют с количеством экстрагированных клеток (см. таблицу 9), в следующих ниже экспериментах концентрации DHO экстраполировали к величине 10х106 клеток в мкг/мл. Лучшее линейное увеличение уровня содержания DHO было установлено при плотности 1×106 клеток/мл. Используя указанную плотность клеток, исследовали временную зависимость увеличения уровней содержания внутриклеточной DHO в клетках, инкубированных в присутствии А77 1726. Детектируемые количества DHO могут быть определены через 1 час инкубации, независимо от концентрации лекарственного препарата (таблица 11). Линейное увеличение регистрировали при наивысшем количестве DHO, определенном через 6 часов (таблица 11). После этого наблюдалась кривая с насыщением и дальнейшего увеличения DHO не происходило.
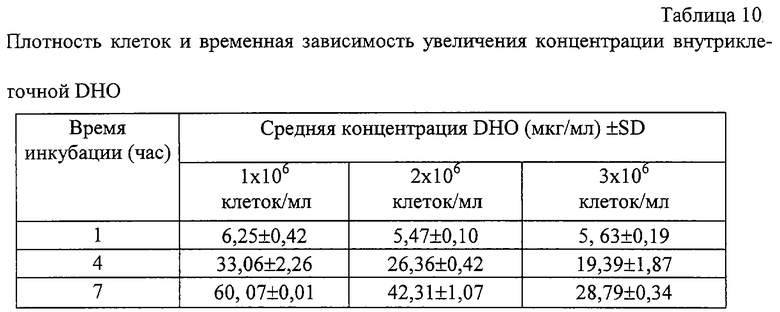
Различные количества клеток A20.2.J инкубировали совместно с А77 1726 (5 мкМ), в течение указанных периодов времени, и их концентрации внутриклеточной DHO определены для каждого образца. (* все значения экстраполированы к 10×106 клеток) (n=2).
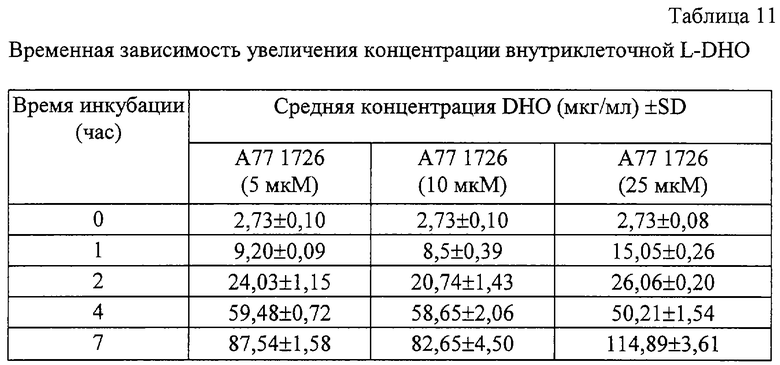
Один миллион А20.2.J-клеток/мл инкубировали совместно с различными концентрациями А77 1726 и их концентрации внутриклеточной DHO определяли в различные моменты времени. Полученные данные представлены как мкг/мл DHO±SD, экстраполированное к десяти миллионам клеток (0-4 час = n=2, 7 час = n=4).
В результате совместной инкубации опухолевых клеток A20.2.J и А77 1726 происходило быстрое накопление L-DHO за счет ингибирования DHO-DH. Концентрации внутриклеточной L-DHO коррелировали с числом клеток и зависели от времени. Мониторинг L-DHO представляет собой суррогатный маркер иммуномодулирующей активности А77 1726 у пациентов.
Пример 3
Животные: самцы крыс вида Wistar-Lewis (Mollegaard Breading Center Ltd. Ejby, DK) с весом тела 160-200 г.
Адъювантный артрит. Заболевание индуцировали инъекцией адъюванта Freund (6 мг Mycobacterium smegmatis суспендировали в 1 мл тяжелого белого парафинового масла (Merck, Darmstadt) в хвостовой корешок крыс Wistar-Lewis. Обычно патологические симптомы проявлялись на 10-14 день после индуцирования болезни.
Лекарственное лечение. Лекарственные средства суспендировали в 1% карбоксиметилцеллюлозы (СОМС). Здоровым животным (n=18) и адьювантным больным (n=18) крысам (9 день нарушения здоровья) давали перорально по 10 мг/кг N-4-трифторметилфенил)-5-метилизоксазол-4-карбоксамида, далее в тексте лефлуноамид, причем лекарство применяли дважды в день (7:30 час и 13:30 час) в течение 5 дней, т.е. в следующие моменты времени (0 часов, 6 часов, 24 часа, 30 часов, 48 часов, 54 часа, 72 часа, 78 часов, 96 часов) (см. таблицу 12). В период времени 0 часов, 3 животных из здоровой и больной групп умерщвляли с целью определения уровней содержания основного материала. Еще по 3 здоровых и 3 больных животных обрабатывали плацебо (только СОМС) в течение 5 дней.
Отбор образцов. По три животных из каждой группы умерщвляли в каждый из моментов отбора образцов. Образцы сыворотки и спленоцитов отбирали через 3, 7, 27, 51, 75 и 99 часов (см. таблицу 12). За исключением образца, отбираемого через 7 часов, все другие образцы отбирали через 3 часа после последнего применения лекарственного средства. Образец, отбираемый через 7 часов, брался после второго приема лекарства. Образцы, взятые у животных, получивших плацебо, отбирали в моменты времени 0 часов (n=3) и 99 часов (n=3).
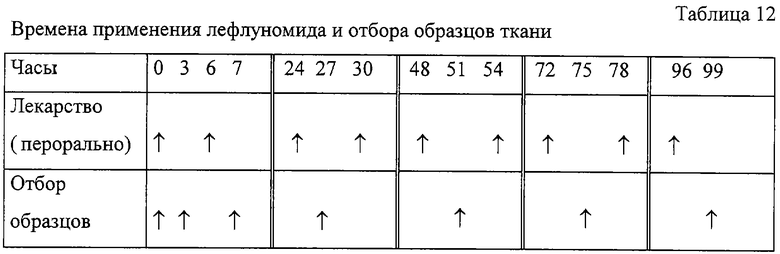
Приготовление образцов
- Образцы крови, собранные в результате пункции сердца, хранили в течение 30 минут при 4°С и затем центрифугировали в течение 10 минут при скорости вращения 3000 об/мин. Сыворотку крови отделяли и хранили в пробирках Эппендорфа при -20°С (3). Перед HPLC-анализом замороженную сыворотку оттаивали и с целью удаления протеинов 200 мкл сыворотки помещали на фильтр MICROCON (модель 10, код 42407, Amicon) и центрифугировали в течение 30 минут со скоростью вращения 13000 об/мин.
- Собирали селезенки (n=3), которые объединяли для L-DHO анализа. Отпрепарированные клетки (пропусканием через сетчатый фильтр из нержавеющей стали) обрабатывали 0,17 М раствором NH4Cl для лизирования эритроцитов. Готовили аликвоты из 50 миллионов клеток селезенки в расчете на группу, их помещали в пробирки для центрифугирования и супернатант отбрасывали. При постоянном перемешивании к клеточному осадку добавляли 500 мкл 1,2 М раствора НСlO4 для лизиса клеток и проведения центрифугирования в течение 2 минут. Кислый клеточный лизат полностью переносили в стеклянные пробирки 500 мкл хлороформа и содержимое перемешивали в течение 2 минут в мешалке Vortex. Клеточные липиды осаждали центрифугированием (10 минут, 1502 g при 10°С). Надосадочную жидкость помещали в пробирки объемом 2 мл и хранили при -20°С.
Определение концентраций А77 1726 в сыворотке осуществляли следующим образом.
Образцы сыворотки приводили к комнатной температуре и тщательно перемешивали с использованием вихревой мешалки. Образцы сыворотки с помощью пипетки (200 мкл) переносили в пробирки Эппендорфа и добавляли внутренний стандарт (А77 1726, 2 мкг в 400 мкл ацетонитрила). Затем содержимое пробирок перемешивали в мешалке Vortex и центрифугировали со скоростью 2500 об/мин (при комнатной температуре в течение 10 минут). Для проведения HPLC анализа надосадочную жидкость (400 мкл) переносили в пробирку, добавляли воду (400 мкл) и содержимое перемешивали. Использовали следующие условия HPLC анализа. Оборудование включало насос TSP P2000, автоотборник проб TSP AS1000, интегратор TSP SP4270 и УФ-детектор TSP UV100. Для детектирования использовали длину волны 292 нм. Подвижная фаза содержала 650 мл метанола (CHROMASOLV), 2,42 г бромистого тетрабутиламмония и 350 мл 0,05 М раствора ацетата аммония. Расход через колонку A CHROMPACK Sperisorb OD-2 длиной 10 см составлял 0,5 мл/мин, причем использовали предохранительную колонку (R2) с обращенной фазой длиной 1 см. В колонку вводили пробу объемом 100 мкл и время анализа составляло 7 мин.
HPLC определение концентрация L-DHO. Хроматографическое разделение осуществляли по методике, описанной в примере 1. Диапазон детектора по проводимости устанавливали на 10 μS; анализ проводили при комнатной температуре. Подвижная фаза состояла из 100 мМ NaOH (А) и воды (В). С помощью такой системы готовили следующие градиенты (см. таблицу D).
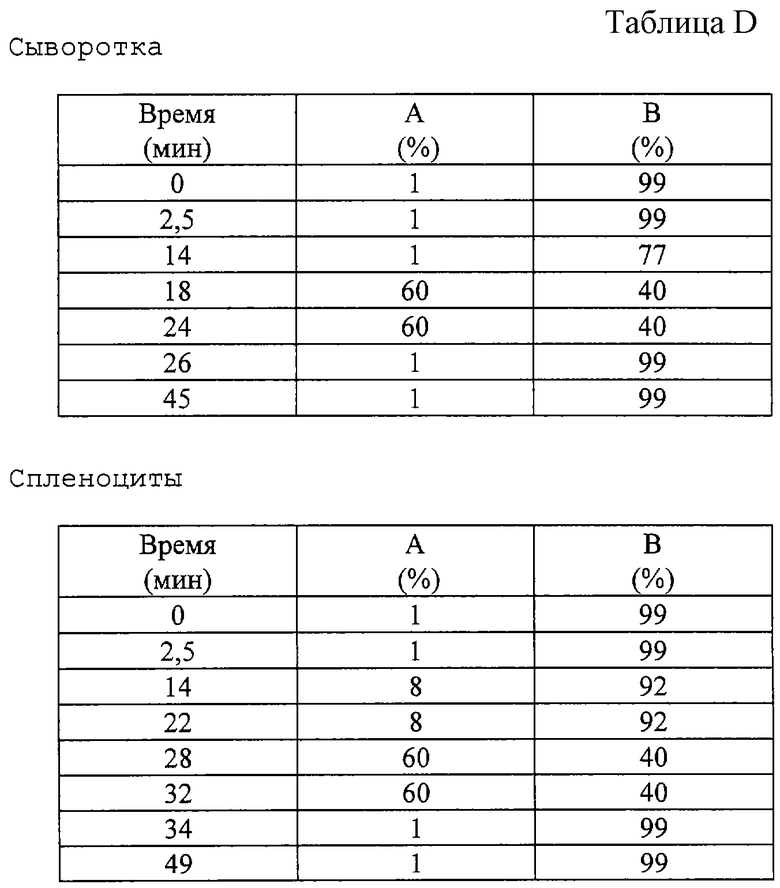
Объемная скорость 1 мл/мин.
В обеих группах испытуемых крыс, как здоровых, так и больных, наблюдались повышенные концентрации клеточной (таблица 13) и сывороточной (таблица 14) L-DHO после перорального применения лефлуномида. Такое увеличение коррелирует с сывороточными концентрациями А77 1727, определенными у этих животных (таблица 15). Вначале, через 3 часа после перорального применения лекарственного препарата, у адъювантных больных крыс достигается концентрация А77 1726 26 мкг/мл, соответствующее значение для здоровых крыс составляло 31 мкг/мл. Эти значения достигали максимального значения через час после второй дозировки (7 часов), но снижались до значений 7-12 мкг/мл по ходу эксперимента как у больных, так и у здоровых крыс. Больным крысам требовался 51 час для достижения таких концентраций, тогда как у здоровых крыс эти концентрации достигались уже через 27 часов (таблица 15). Сывороточные концентрации А77 1726 коррелируют с концентрациями L-DHO в сыворотке адъювантных больных и здоровых грызунов. В отличие от сывороточных концентраций L-DHO, которые уравновешиваются на значении примерно 5 мкг/мл в ходе эксперимента, количество L-DHO, обнаруженное в спленоцитах, падает до значения ниже уровня детекции (1,5 мг/мл) через 99 часов.
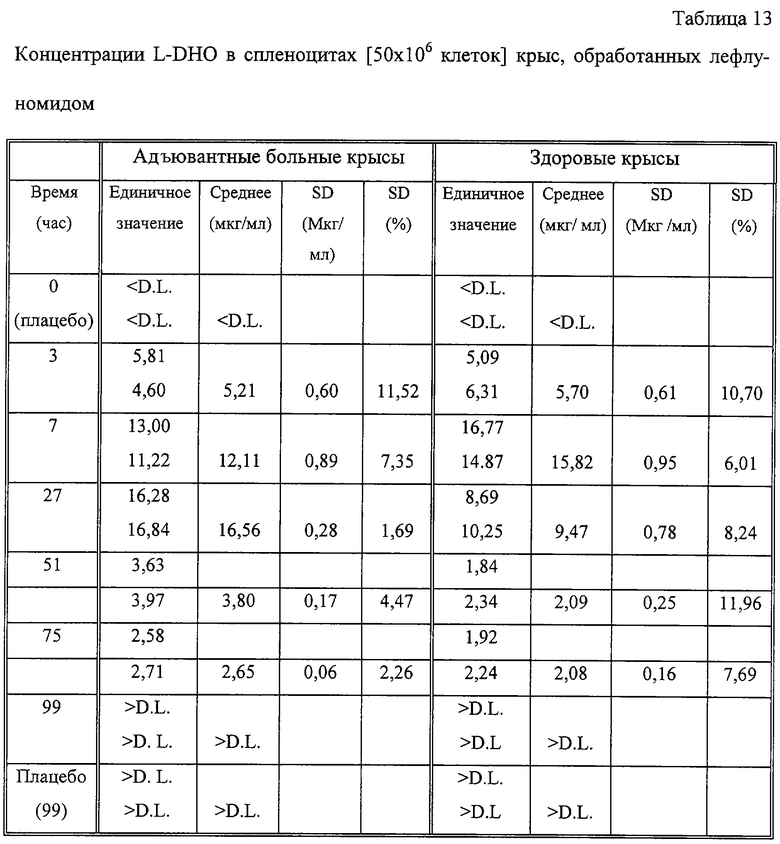
Животных обрабатывали лефлуномидом или плацебо, селезенки удаляли (n=3) и объединяли друг с другом, как описано. Объединенные спленоциты анализировали дважды. DL=предел детекции (1,5 мкг/мл).
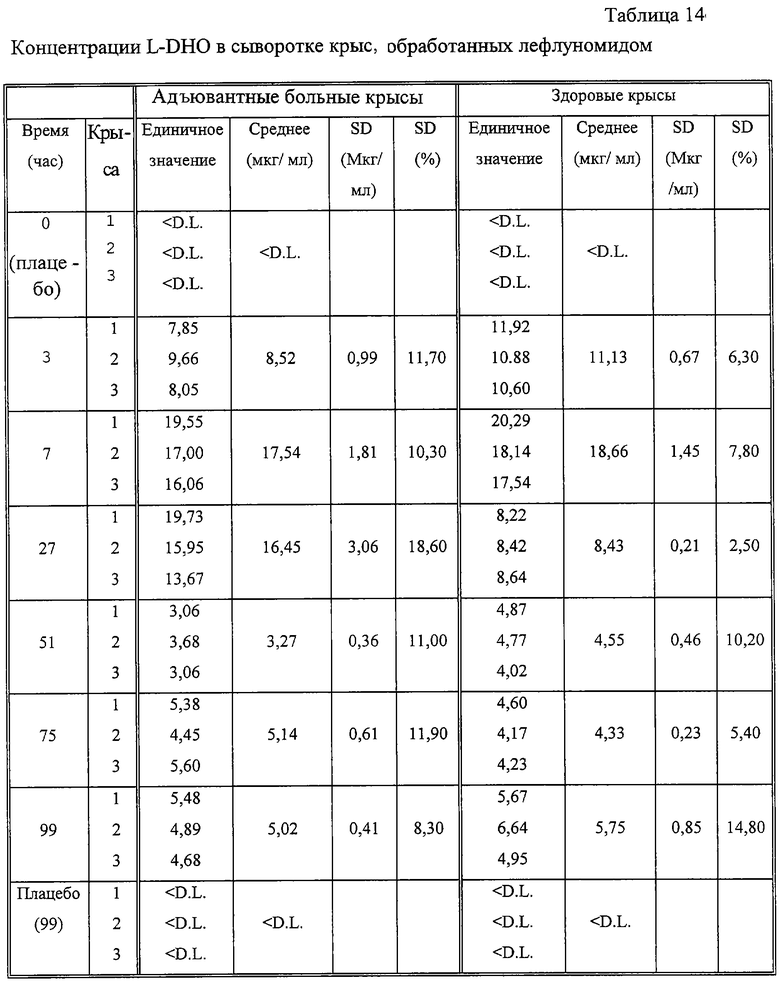
Животных обрабатывали лефлуномидом или плацебо, отбирали образцы крови, после чего их подготавливали и анализировали, как описано выше. Концентрации сывороточной L-DHO определяли для каждого животного индивидуально. DL=предел детекции (0,5 мкг/мл).
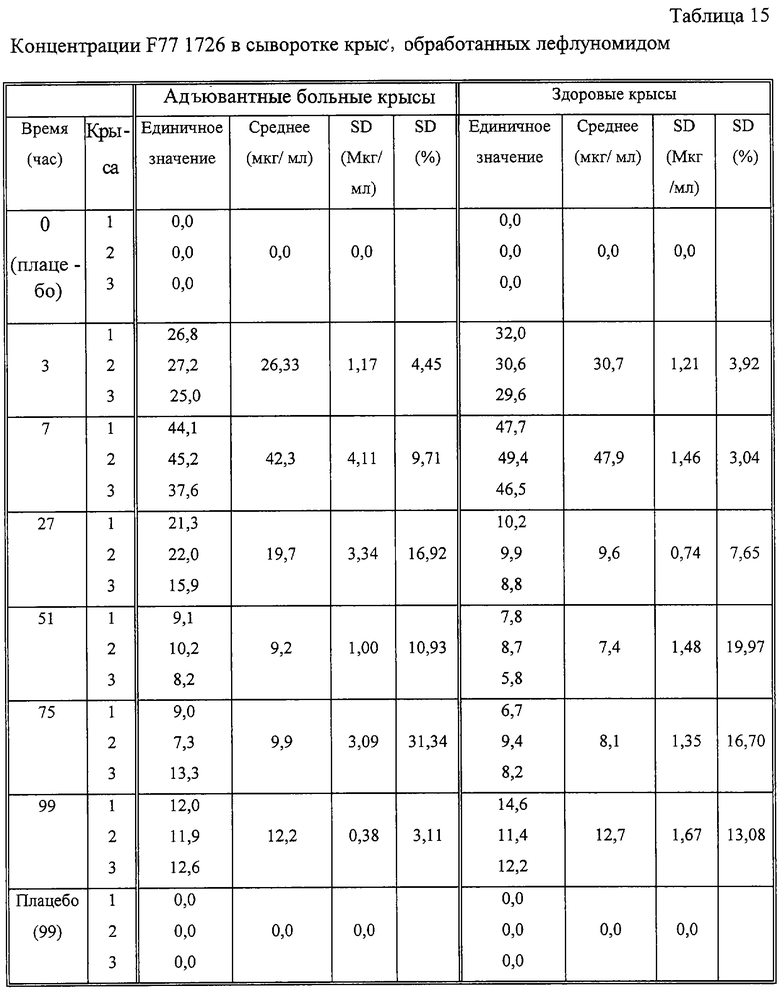
Животных обрабатывали лефлуномидом или плацебо, отбирали образцы крови, подготавливали и анализировали их по описанной выше методике. Концентрации А77 1726 в сыворотке определяли индивидуально для каждого животного.
Применяемый перорально лефлуномид очень быстро превращается in vivo в А77 1726. А77 1726 представляет собой активный метаболит лефлуномида (US 5679709). Инкубирование адъювантных больных или здоровых крыс в присутствии лефлуномида в результате приводит к быстрому накоплению L-DHO в их сыворотке и спленоцитах. Концентрация L-DHO коррелирует с концентрациями А77 1726 в сыворотке крови, что демонстрирует факт активного подавления DHO-DH под действием такой молекулы in vivo. В клинических испытаниях на людях L-DHO мониторинг может служить суррогатным маркером иммуномодулирующей активности лефлуномида на пациентах.
Настоящее изобретение относится к способу отделения растворенной L-дигидрооротовой кислоты хроматографией из раствора, содержащего указанную кислоту и вещества-примеси. Способ предусматривает: a) загрузку указанного раствора в хроматографическую колонну, содержащую устойчивый к действию давления анионообменный материал для связывания указанной кислоты, b) элюирование большей части веществ-примесей из колонны водным раствором и c) элюирование указанной кислоты из указанного анионообменного материала водным раствором, содержащим основание, например гидроксид натрия, под давлением от около 1,1 до около 40 МПа, преимущественно от около 4,1 до около 5,5 МПа. Обычно анионообменный материал выбирается, например, из дивинилбензол/этилвинил-бензольного полимера, поливинилбензиламмониевого полимера, сшитого дивинилбензолом, и др. При этом, как правило, концентрацию основания в элюирующем растворе повышают во время элюирования колонны, а L-дигидрооротовую кислоту детектируют в элюате с помощью детектора проводимости. Предлагаемый способ может использоваться для исследования in vitro и in vivo активности N-(4-трифторметилфенил)-5-метилизоксазол-4-карбоксамида, N-(4-трифторметилфенил)-2-циано-3-гидроксикротонамида и аналогичных соединений. Изобретение также относится к способу приготовления диагностического аналитического средства для определения активности ингибиторов дегидрогеназы дигидрооротовой кислоты, выбранных, например, из вышеуказанных соединений, и к способу мониторинга активности ингибиторов дегидрогеназы дигидрооротовой кислоты с использованием указанного способа отделения L-дигидрооротовой кислоты, где по концентрации L-дигидрооротовой кислоты определяют активность ингибиторов. 3 с. и 6 з.п.ф-лы, 19 табл.
a) загрузку указанного раствора в хроматографическую колонну, содержащую устойчивый к действию давления аниоонобменный материал для связывания указанной кислоты,
b) элюирования большей части веществ-примесей из колонны водным раствором, и
c) элюирования указанной кислоты из указанного анионообменного материала водным раствором, содержащим основание, под давлением от около 1,1 МПа до около 40 МПа.
5-метил-N-[4-(трифторметил)фенил)-4-изоксазол-карбоксамида,
6-фтор-2-(2’-фтор-[1,1’-дифенил]-4-ил)-3-метил-4-хинолинкарбоновой кислоты,
2-циано-3-гидрокси-N-[4-(трифторметил)фенил]-2-гептен-6-инамида,
2-циано-1N-(4-цианофенил)-3-циклопропил-3-гидрокси-2-пропенамида, и
2-циано-3-гидрокси-N-[4-(трифторметил)фенил]-2-бутенамида.
| R.DANIEL ET AL | |||
| Anal | |||
| Biochem., 1996, v.239, №2, р.130-135. |

