Изобретение относится к способу получения алкансульфохлоридов жидких н-алканов преимущественно состава C12-C18 (RH) под воздействием смеси газов - сернистого ангидрида (SO2) и хлора (Cl2) - при мольном соотношении 1,1:1 в присутствии органических перекисей как инициаторов процесса.
Свободнорадикальное сульфохлорирование алканов нормального строения реализуется в технике с целью получения сульфонатов - важных ионогенных ПАВ (эмульгирующих, моющих и смачивающих веществ):
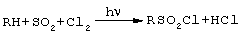
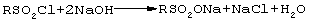
Сульфохлориды лабильны при нагревании (выделяется SO2), отсюда температура как инициатор процесса не применима. Промышленным активатором процесса при температуре 30-35°С является ультрафиолетовый (УФ) свет [Н.Н Лебедев. Химия и технология основного органического нефтехимического синтеза. М., "Химия", 1988, с.323-326].
Известно применение органических перекисей для инициирования свободнорадикальной реакции. Перекиси ацилов, например бензоила, лаурила и др., в процессе сульфохлорирования не эффективны. Хорошо зарекомендовали себя ацетилциклогексилсульфонилперекись, тримерная перекись ацетона, диоксиперекись пропаналя [Ф.Азингер. Химия и технология парафиновых углеводородов. М., Гостоптехиздат, 1959, с.369]. В их присутствии достигается высокая скорость замещения в ряду н-алканов состава С10-C18, однако указанные инициаторы требуют особых условий хранения или работы с ними в связи с наблюдаемой потерей активного кислорода (Оакт) и высокой чувствительностью к удару и трению.
Известно применение совершенно безопасной в обращении диоксиперекиси хлораля для переработки углеводородов состава C12-C18 темных тонов (и так называемых “неомыляемых”) до сульфохлоридов [Авт. свид. №186459, кл. 120 23/03, 1966]. Однако детальная проверка ее инициирующей способности показала, что результаты сульфохлорирования чистых парафинов по глубине замещения (по накоплению Сlомыл во времени), по достигнутой конверсии RH и газов, по выходу алкансульфохлоридов в целом хуже показателей процесса при освещении.
Известна высокая эффективность инициирования процесса сульфохлорирования алканов нормального строения в присутствии оксиметил-трет-бутилперекиси (СН3)3С-ОО-СН2OН. Оксиперекись обеспечивает высокую скорость замещения и позволяет получать практически чистые моносульфохлориды при сульфохлорировании, например, пентана или гексана при подаче газов на 20% и более (даже 100% в среде ССl4), конверсию RH при температуре 5-30°С и расходе инициатора 0,01-0,2% от RH [Авт. свид. №516683 С 07 С 143/70, 1976].
Реакционная способность н-алканов состава C10-C18 и выше и, как результат, образование побочных продуктов, особенно нежелательных для производства ПАВ дисульфохлоридов, возрастает. Так, при УФ-свете практически невозможно вести процесс при конверсии RH 25% и выше, так как при подаче газов на 22-24% превращение RH в конечном продукте содержится более 25% дисульфохлоридов.
Наиболее близким к заявленному является способ получения алкансульфохлоридов реакцией сульфохлорирования н-алканов состава C12-C18 под воздействием смеси газов SO2 и Cl2 в соотношении 1,1:1, поданных в расчете на 30% превращение RH в присутствии указанной оксиметил-трет-бутилперекиси 0,1% от RH [А.И.Рахимов. Химия и технология органических перекисных соединений. М., "Химия", 1979, с.340]. Глубина сульфохлорирования (по величине гидролизуемого хлора Сlомыл) додекана за одно и то же время взаимодействия превосходит результаты, полученные при освещении лампой ПРК-2, при этом доля дисульфохлоридов меньше.
Однако диапазон эффективной работы инициатора при использовании синтина оказался ограниченным. При больших расходах газов, например около 4×10-3 г Cl2/г RH в мин оксиперикись уступает УФ-свету. Т.е. использовать ее для цели интенсификации производства алкансульфохлоридов проблематично. В противоточной колонне при времени пребывания реагентов около 20-25 мин ее воздействие на систему незначительно.
Применение перекисей в качестве инициаторов процесса сульфохлорирования алканов требует затрат на их производство и обязательную очистку, которая в ряде случаев довольно специфична. Упростить стадию инициирования за счет использования реакционной смеси производства инициатора (без его выделения) не представляется возможным. Например, указанные перекиси на основе карбонильных соединений (в том числе окси- и диоксиперекиси) получают, используя водную перекись водорода (30% Н2O2) или формалин (40% Н2СО). Наличие воды в сульфохлораторе недопустимо.
Отсутствие простого и доступного и в то же время эффективного, превосходящего УФ-свет химического инициатора является важной задачей поиска. Изложенное выше, вероятно, ограничивает внедрение органических перекисей в практику процесса сульфохлорирования н-алканов состава C12-C18. В технике отдано предпочтение УФ-свету [Пат. ГДР №220583, С 07 С 139/02, 143/70, 1985], в том числе в каскаде реакторов [Пат. ГДР №147844, С 07 С 139/02, 143/70, 1981].
При всех достоинствах фотохимический процесс имеет ряд серьезных недостатков. Он ограничен в возможностях создания аппаратов большой единичной мощности с равномерным фотоэффектом по всему объему, требует применения сырья только светлых тонов (загрязнение увеличивает оптическую плотность раствора и снижает фотоэффект), увеличивается пожароопасность. Даже при относительно большом времени контакта реагентов в противоточном непрерывном реакторе (около 75 минут) степень превращения хлора около 90%, около 30% исходного углеводорода не участвует в процессе получения сульфохлоридов. Качество эмульгатора крайне низкое (из-за большого содержания дисульфохлоридов).
Известен способ получения алкансульфохлоридов из н-алканов C12-C18 в присутствии УФ-света с добавкой 0,5×10-3% иода [Авт. свид. №1685929, С 07 С 309/80, 1991]. Такое совершенствование фотохимического процесса приводит к определенному росту конверсии RH до моносульфохлоридов, но принципиально никак не решает проблему химического инициирования реакции.
Неприемлем для получения алкансульфохлоридов способ [Авт. свид. №1558900, С 07 С, 1990] из-за соотношения Сl2:SO2:RH, равного 2,2-6,5:1:1, и высокой температуры 50-90°С в присутствии или в отсутствие диметиформамида (ДМФА) с использованием УФ-света или ДАК. Следует иметь в виду, что известны также способы получения алкансульфохлоридов близкого состава с использованием одного из признаков, например газообразного Cl2 [Авт. свид. №1563591, С 07 С 309/00, 309/70, 1990], но хлорированию подвергают дитиокарбонаты 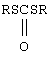 в водной среде, т.е. используют реакцию и исходные вещества, отличные от реакции сульфохлорирования алканов.
в водной среде, т.е. используют реакцию и исходные вещества, отличные от реакции сульфохлорирования алканов.
Предлагаемый способ получения алкансульфохлоридов позволяет решить проблемы, сопровождающие процесс сульфохлорирования н-алканов типа синтина и парафина усредненного состава С12-С18.
Задачей предлагаемого изобретения является расширение ассортимента эффективных инициаторов процесса перекисной природы, обеспечивающих интенсификацию производства, высокое качество сульфонатов и одновременно существенно упрощающих стадию инициирования.
Технический результат достигается тем, что получение алкансульфохлоридов путем сульфохлорирования н-алканов состава C12-C18 под воздействием смеси газов SO2 и Cl2 при мольном соотношении 1,1:1 в присутствии инициатора - органической перекиси - ведут в присутствии моно- и дипероксиацеталей структуры 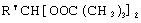 или
или 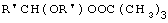 или
или 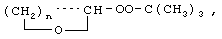
где R′=-СН3, -С2Н5, n=3,4,
которые вносят в алкан в количестве 0,05-0,15% до начала процесса или получают непосредственно внутри сульфохлоратора (in situ) при температуре 25-35°С, при удельном расходе (1,54-4,33)х10-3 г Cl2/г RH в мин периодически или непрерывно при времени пребывания реакционной массы от 75 до 25 мин при подаче смеси газов SO2 и Cl2 на 22-30% конверсию алкана.
Если алкансульфохлориды получают в присутствии пероксиацеталей, образующихся внутри сульфохлоратора (in situ), то процесс сульфохлорирования ведут с использованием растворов гидроперекиси трет-бутила в простом алифатическом или циклическом эфире, например тетрагидрофуране, в количестве 0,5% от алкана.
Моно- и дипероксиацетали устойчивы при хранении, перегоняются в вакууме при умеренных температурах без каких-либо осложнений. Инициируют свободнорадикальное сульфохлорирование алканов в широком диапазоне по времени контакта реагентов и удельному расходу газов.
Могут получаться различными способами, но наиболее доступный и быстрый способ синтеза пероксиацеталей базируется на реакции простых эфиров с гидроперекисями под воздействием хлористого сульфурила по схеме, например, для ди-трет-бутилпероксиацеталя ацетальдегида (1,1-ди-трет-бутилпероксиэтан) с выходом 90% при пониженной температуре:
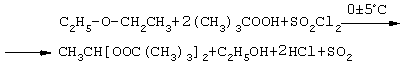
При пропускании испаренного хлора через раствор гидроперекиси трет-бутила в эфире преимущественно образуется монопероксиацеталь (наряду с диперекисью) согласно схеме, например, для 1-этокси-1-трет-бутилпероксиэтана:
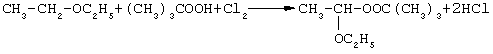
В расчете на монопероксиацеталь выход продукта около 70-80% в диапазоне температур от 0 до 25°С [Авт. свид. №1796621, С 07 С 407/00, 409/20, 1993]. В течение часа взаимодействия определенная часть гидроперекиси не конвертируется.
Без раскрытия кольца тетрагидрофуран и другие циклические эфиры реагируют с гидроперекисью и хлористым сульфурилом или хлором.
Однако более целесообразным оказался способ получения пероксиацеталей с использованием смеси Сl2 и SO2 в соотношении 1:1. В мягких условиях, при умеренной температуре, с высокой конверсией гидроперекиси быстро с высоким выходом получают из простых эфиров дипероксиацетали, а из простых циклических эфиров - пероксиацетали [Авт. свид. №1668361, С 07 Д 307/20, 1991], например 2-трет-бутилперокситетрагидрофуран, согласно схеме:
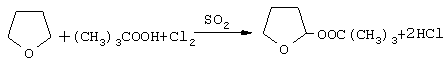
Изложенный путь синтеза пероксиацеталей удачно сочетается с процессом сульфохлорирования алканов по реагентам и температуре. За счет большей скорости взаимодействия эфира с гидроперекисью в смеси с SO2Cl2 (или смесью газов SO2 и Сl2) инициатор образуется внутри сульфохлоратора (in situ) с последующим воздействием на свободнорадикальное замещение по конечным результатам, не уступающим очищенным пероксиацеталям.
Пероксиацетали обеспечивают интенсивное ведение процесса сульфохлорирования н-алканов с высокой конверсией всех реагентов, достаточно близкой теоретически возможной, при этом алкансульфохлориды получают с высокими показателями качества.
Изобретение иллюстрируется следующими примерами по сульфохлорированию синтина (синтетического парафина со средней молекулярной массой 198 состава C12-C18) в периодических и непрерывных условиях в присутствии предлагаемых пероксиацеталей в сравнении с УФ-светом, оксиметил-трет-бутилперекисью и др. добавками.
Пример 1. В затемненный реактор из стекла, снабженный мешалкой, термометром и обратным холодильником, загружают 97,50 г (127,3 мл) синтина с добавкой свежеприготовленного (97,8%, по Оакт иодометрией) ди-трет-бутилпероксиацеталя ацетальдегида в количестве 0,1286 г, что соответствует 0,13% от RH и [Jnc2]=0,0048 моль/л.
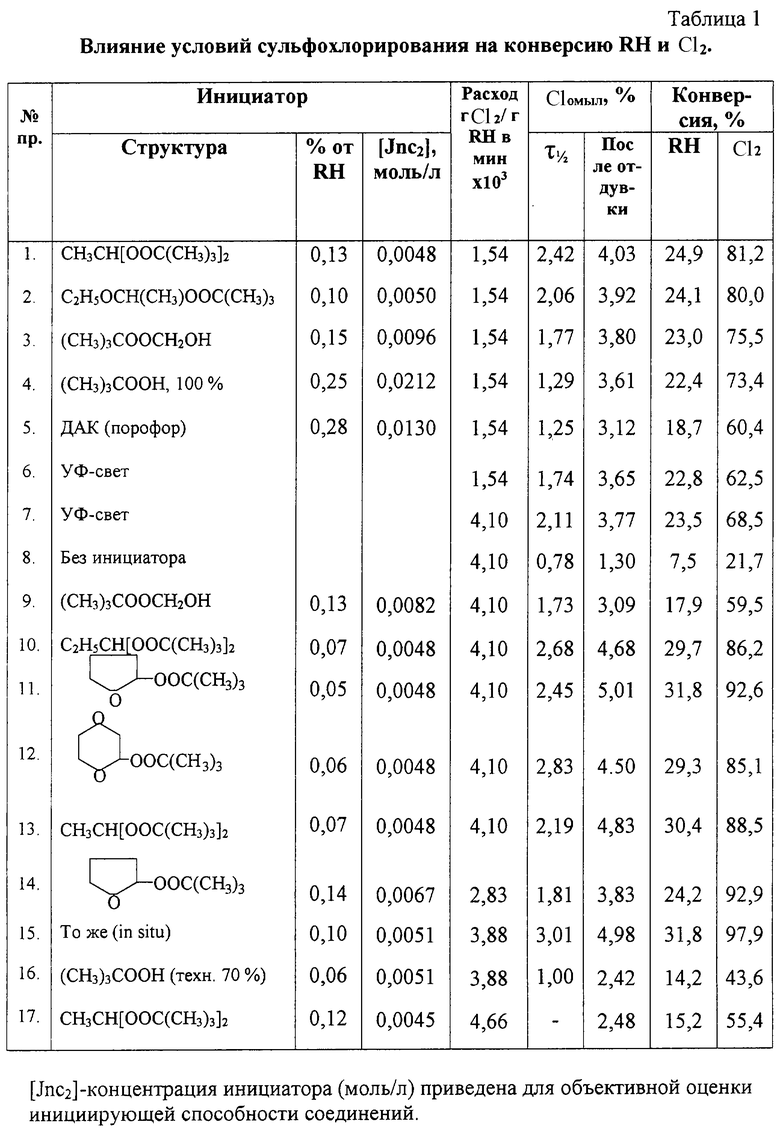
В раствор синтина с инициатором, поддерживая температуру реакции 30±2°С, подают по барботеру смесь газов SO2 и Cl2, испаренных из баллонов, в количестве 0,15 г/мин каждого по реометру в течение 71 мин до мольного соотношения RH:SO2:Cl2, равного 3,28:1,1:1 (1:0,33:0,30). Удельный расход Cl2 при этом составлял 1,54×10-3 г/г RH в минуту. Глубина сульфохлорирования оценивалась по величине Сlомыл (по Фольгарду) за время полупревращения (τ1/2) и конца процесса (см. таблицу 1).
После отдувки азотом реакционная масса (без расслоения) имела показатели: Сlомыл 4,03%, n
Примеры 2-6. Сульфохлорирование синтина вели аналогично примеру 1 в присутствии монопероксиацеталя 1-этокси-1-трет-бутилпероксиэтана (пр.2) и для сравнения добавок: оксиметил-трет-бутилперекиси (пр.3), гидроперекиси трет-бутила (пр.4), ДАК (пр.5), а также в присутствии УФ-света лампы сверхвысокого давления ОКН-11 на расстоянии 10 см от стенки сульфохлоратора (пр.6).
Глубина сульфохлорирования во всех примерах (см. таблицу 1) приведена для образцов инициатора чистотой 98-100%.
Пример 7. Сульфохлорирование синтина в количестве 73 г (95,5 мл) вели при подаче газов SO2 и Cl2 по 0,30 г/мин каждого при температуре 30±2°С в течение 30 мин при освещении лампой ОКН-11 (10 см от стенки) до мольного соотношения RH:SO2:Сl2, равного 2,9:1,1:1 (1:0,38:0,34). При этом удельный расход каждого из газов составлял 4,10×10-3 г/г RH в мин.
После отдувки азотом реакционная масса имела показатели: Сlомыл 3,77%, n
При хранении в течение суток масса расслоилась, декантацией выделено 1,47 г (1,8% от общей массы с Сlомыл 14,5%) дисульфохлоридов, плохо растворимых в углеводороде.
Примеры 8-13. При аналогичном расходе сырья, как в примере 7, в затемненном реакторе, как в примере 1, оценена глубина (см. таблицу 1) сульфохлорирования без инициатора (пр.8), в присутствии оксиметил-трет-бутилперекиси (пр.9), а также в присутствии пероксиацеталей структуры 1,1-ди-трет-бутилпероксипропана (пр.10), 2-трет-бутилперокситетрагидрофурана (пр.11), трет-бутил-пероксидиоксана-1,4 (пр.12), 1,1-ди-трет-бутилпероксиэтана (пр.13).
В условиях проведения процесса, аналогичных примеру 9 (по [Jnc2]), конверсия RH и Cl2 в присутствии гидроперекиси трет-бутила или ДАК была хуже, чем при использовании оксиметил-трет-бутилперекиси.
Пример 14. Сульфохлорирование синтина в количестве 106 г (138,4 мл) вели аналогично примеру 1, в присутствии пероксиацеталя структуры 2-трет-бутилперокситетрагидрофурана чистотой 98,9% (по Оакт иодометрией) в количестве 0,1485 г (что соответствует 0,14% от RH и [Jnc2]=0,0067 моль/л), подавая смесь газов SO2 и Cl2 по 0,30 г/мин каждого в течении 33 мин до мольного соотношения RH:SO2:Cl2, равного 3,85:1,1:1 (1:0,30:0,26). Удельный расход каждого из газов составлял 2,83×10-3 г/г RH в мин.
После отдувки азотом реакционная масса (без расслоения) имела показатели: Сlомыл. 3,83%, n
Пример 15. Сульфохлорирование синтина в количестве 72,0 г (94 мл) в присутствии пероксиацеталя структуры 2-трет-бутилперокситетрагидрофурана, получаемого внутри (in situ) сульфохлоратора, вели аналогично примеру 1 (см. также пр.11) с добавкой 0,062 г гидроперекиси трет-бутила (70% чистоты, иодометрией) в 0,5 мл тетрагидрофурана при подаче газов SO2 и Сl2 по 0,28 г/мин каждого до мольного соотношения RH:SO2:Cl2, равного 3,07:1,1:1 (1:0,36:0,32). Время реакции 30 мин.
Удельный расход каждого из газов составлял 3,88×10-3 г/г RH в мин. Концентрация инициатора (расчетная) 0,0051 моль/л или 0,1% от RH, считая на 100% чистоту.
После отдувки азотом реакционная масса имела показатели: Сlомыл 4,98%, n
При хранении в течение двух суток из реакционной массы декантацией выделено 0,94 г плохо растворимых в синтине продуктов.
Пример 16. Сульфохлорирование синтина вели аналогично примеру 15 с использованием 0,062 г технической гидроперекиси трет-бутила с содержанием основного вещества 70% (без ТГФ). Концентрация гидроперекиси в углеводороде 0,06% от RH или 0,00513 моль/л, считая на 100% чистоту.
Получено после отдувки азотом 75,86 г массы с Сlомыл 2,42%, n
Пример 17. Сульфохлорирование с запредельным удельным расходом газов вели аналогично примеру 1, подавая в 84 мл (64,36 г) синтина с добавкой 0,0772 г 1,1-ди-трет-бутилпероксиэтана смесь газов SO2 и Сl2 по 0,30 г/мин до мольного соотношения RH:SО2:Сl2, равного 3,6:1,1:1 (1:0,30:0,27). Время реакции 21 мин. Удельный расход каждого из газов составлял 4,67×10-3 г на 1 г RH в мин. Концентрация инициатора 0,12% от RH или 0,00446 моль/л.
Получено после отдувки азотом 70,44 г массы с Сlомыл 2,48%, n
Пример 18. Непрерывное сульфохлорирование осуществляли в противоточной колонне с рубашкой высотой 300 мм, внутренним диаметром 20 мм. Коэффициент заполнения не превышал 0,7. Подача углеводорода осуществлялась непрерывно дозатором через верх колонны со скоростью (4,6 мл/мин), зависимой от времени пребывания и соотношения реагентов. Вниз колонны подавалась смесь газов. Отбор продуктов производился с низа колонны по перетоку, установленному на уровне реакционной массы в сульфохлораторе. Температура реакции 30°С поддерживалась ультратермостатом УТ-15.
“Затравка” процесса производилась путем подачи SO2 и Cl2 по 0,38 г/мин каждого из газов через 114 мл синтина, содержащего 0,15% от RH чистого дипероксиацеталя структуры 1,1-ди-трет-бутилпероксиэтана, в течение 25 мин до мольного соотношения RH:SO2:Cl2, равного 3,29:1,1:1 (1:0,33:0,30). Удельный расход каждого из газов составлял 4,33×10-3 г/г RH в мин. Показатели массы при “затравке” Сlомыл 4,10%, n
Поддерживая указанное соотношение в течение нескольких часов, со временем пребывания углеводорода в сульфохлораторе 25 мин (при контроле приведенных показателей в течении каждого часа) получена реакционная масса с общими показателями Сlомыл 4,28%, Сlобщ 6,67%, n
Для сравнения в аналогичных условиях процесса (без добавки перекиси) только на рассеянном свету показатели реакционной массы: n
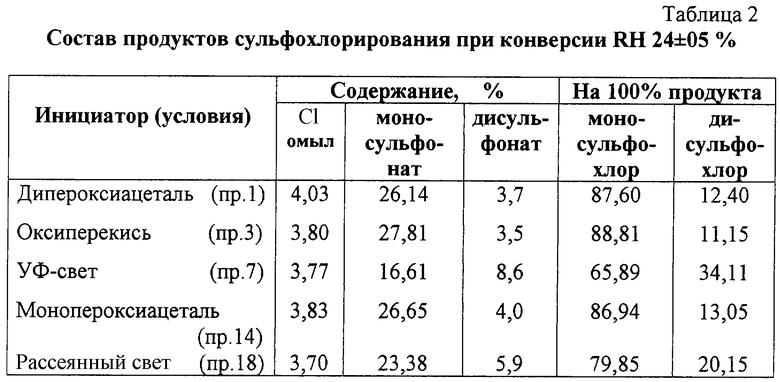
Калориметрическое определение состава продуктов сульфохлорирования при конверсии RH, близкой к величине 24±05%, достигнутой в ходе эксперимента, приведено в таблице 2.
Результаты эксперимента свидетельствуют о том, что пероксиацетали при расходе 0,1±0,05% от RH являются эффективными инициаторами сульфохлорирования синтина в широком диапазоне изменения условий реакции, при удельном расходе каждого из газов от 1,54×10-3 до 4,33×10-3 г/г RH в мин или времени контакта реагентов от 75 до 25 мин включительно.
Обсуждать воздействие инициатора при меньшем удельном расходе газов с позиции интенсификации процесса не имеет смысла. Возрастание [SO2] и [Сl2] способствует возрастанию скорости замещения. Но при расходе SO2, большем чем 4,33×10-3 г/г RH в мин, в присутствии пероксиацеталя наблюдается заметное снижение эффективности инициирования. Результаты (см. пр.17, время 21 мин), по-видимому, обусловлены непроизводительным ионным распадом перекиси. Сернистый ангидрид, как известно, является восстановителем O-O-связи. Следует иметь в виду, что интенсификация процесса в направлении уменьшения времени контакта реагентов (меньше 25 мин) вряд ли целесообразна из-за высокой экзотермичности процесса (возникают проблемы с теплосъемом.)
Эффект воздействия пероксиацеталя на систему подтверждается и при проведении процесса в противоточной колонне (см. пр.18, в незатемненном реакторе). Все другие добавки (оксиперекись, ДАК, перекись лаурила, гидроперекись) заметного влияния на глубину сульфохлорирования в колонне, в том числе при времени пребывания 40 мин, не оказывают (результаты близки влиянию рассеянного света).
Особую ценность имеет возможность получения инициатора внутри сульфохлоратора (in situ) из доступных технических продуктов, например, продемонстрированном в пр. 15 из гидроперекиси трет-бутила и тетрагидрофурана (ТГФ). В данном случае не прореагировавший ТГФ, взятый в небольшом избытке к гидроперекиси, никак не будет способствовать загрязнению конечного продукта - сульфоната ввиду хорошей растворимости ТГФ в воде в процессе переработки алкансульфохлоридов.
Определяющее влияние на состав продуктов свободнорадикального замещения имеет соотношение реагирующих веществ и достигнутая степень превращения алкана. При близкой конверсии RH (см. таблицу 2) органические перекиси указанной структуры, несомненно, более предпочтительны как инициаторы процесса, чем УФ-свет. В их присутствии реакционная масса содержит заметно меньше нежелательных продуктов дизамещения (как и параллельно образующихся продуктов хлорирования, на что указывает характер изменения хлора цепи по Сlобщ, пр.18). Визуально расслоение реакционной массы (выделение дисульфохлоридов) наблюдается при конверсии RH более 28%, тогда как при освещении (особенно в противоточной колонне) даже при конверсии 22%.
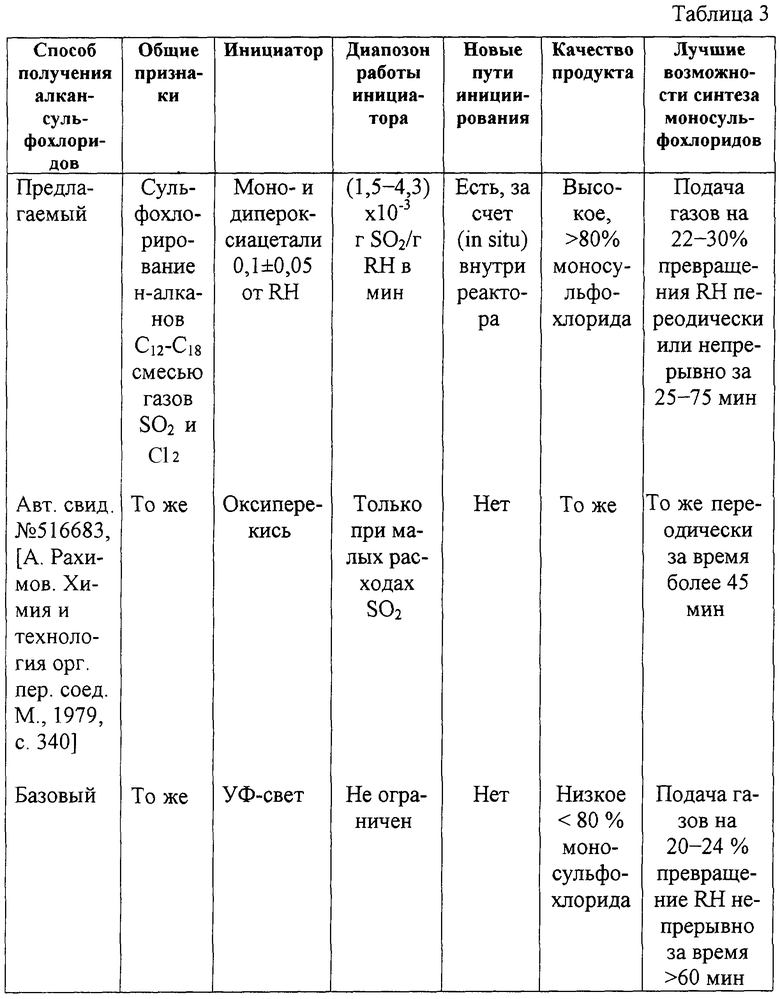
Сравнительные данные по сульфохлорированию предельных углеводородов нормального строения состава C12-C18, заявленного и известного способов, наиболее близких по целям и задачам, приведены в таблице 3.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения пероксиацеталей | 1989 |
|
SU1796621A1 |
| СПОСОБ ПОЛУЧЕНИЯ БЕНЗИЛХЛОРИДА | 2005 |
|
RU2291144C1 |
| СПОСОБ ПОЛУЧЕНИЯ ФТОРАЛКАНСУЛЬФОХЛОРИДОВ | 2010 |
|
RU2440979C1 |
| СПОСОБ ПОЛУЧЕНИЯ ВОДНЫХ РАСТВОРОВ N,N-ДИМЕТИЛДОДЕЦИЛАМИНОКСИДА | 1989 |
|
SU1839434A1 |
| СПОСОБ ПОЛУЧЕНИЯ ЭТИЛОВОГО ЭФИРА α - БРОМИЗОВАЛЕРИАНОВОЙ КИСЛОТЫ | 1993 |
|
RU2080318C1 |
| СПОСОБ ПОЛУЧЕНИЯ ОКСИДОВ АМИЛЕНОВ | 1994 |
|
RU2072995C1 |
| Способ получения 2-трет-бутилперокситетрагидрофурана | 1989 |
|
SU1668361A1 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКАНСУЛЬФОХЛОРИДОВ | 1966 |
|
SU186459A1 |
| СПОСОБ ПОЛУЧЕНИЯ АЛКАНСУЛЬФОХЛОРИДОВ | 1967 |
|
SU225181A1 |
| Способ получения сульфохлорирован-НОгО пОлиэТилЕНА | 1979 |
|
SU804642A1 |
Изобретение относится к способу получения алкансульфохлоридов реакцией сульфохлорирования н-алканов состава С12-С18 под воздействием смеси газов SO2 и Cl2 при мольном соотношении 1,1:1 в присутствии органических перекисей как инициаторов процесса с широким диапазоном эффективного действия, обеспечивающих интенсификацию процесса, высокое качество продуктов реакции и упрощающих стадию инициирования. Технический результат достигается использованием в процессе сульфохлорирования моно- и дипероксиацеталей структуры 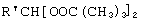 или
или 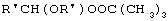 или
или 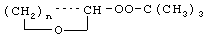 , где R′=-СН3 -С2Н5; n=3,4, которые вносят в алканы в количестве 0,05-0,15% до начала процесса или получают непосредственно внутри сульфохлоратора (in situ) при температуре 25-35°С, при удельном расходе (1,54-4,33)х10-3 г Cl2/г алканов в мин периодически или непрерывно при времени пребывания реакционной массы от 75 до 25 мин при подаче смеси газов SO2 и Cl2 на 22-30% конверсию алкана. При получении алкансульфохлоридов в присутствии пероксиацеталей, образующихся внутри сульфохлоратора (in situ), процесс сульфохлорирования ведут с использованием растворов гидроперекиси трет-бутила в простом алифатическом или циклическом эфире, например тетрагидрофуране, в количестве 0,5% от алканов. 1 з.п. ф-лы, 3 табл.
, где R′=-СН3 -С2Н5; n=3,4, которые вносят в алканы в количестве 0,05-0,15% до начала процесса или получают непосредственно внутри сульфохлоратора (in situ) при температуре 25-35°С, при удельном расходе (1,54-4,33)х10-3 г Cl2/г алканов в мин периодически или непрерывно при времени пребывания реакционной массы от 75 до 25 мин при подаче смеси газов SO2 и Cl2 на 22-30% конверсию алкана. При получении алкансульфохлоридов в присутствии пероксиацеталей, образующихся внутри сульфохлоратора (in situ), процесс сульфохлорирования ведут с использованием растворов гидроперекиси трет-бутила в простом алифатическом или циклическом эфире, например тетрагидрофуране, в количестве 0,5% от алканов. 1 з.п. ф-лы, 3 табл.
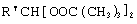
или
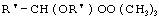
или
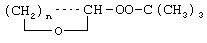
где R′= -СН3 -С2Н5;
n=3,4,
которые вносят в алканы в количестве 0,05-0,15% до начала процесса или получают непосредственно внутри сульфохлоратора (in situ) при температуре 25-35°С, при удельном расходе (1,54-4,33)х10-3 г Cl2/г алканов в 1 мин периодически или непрерывно при времени пребывания реакционной массы от 75 до 25 мин при подаче смеси газов SO2 и Sl2 на 22-30%-ную конверсию алкана.
| Способ получения алкилсульфохлоридов | 1989 |
|
SU1685929A1 |

