Изобретение относится к области органической химии, а именно к способу получения пептидов, обладающих биологической активностью, в частности противоопухолевой активностью, что позволяет использовать их в медицинской практике.
Известен способ получения производных пептидов в растворе в присутствии конденсирующего агента (патент РФ №2047621, С 07 К 7/02, 1995 г.). В известном способе производные аминокислот со свободной карбоксильной группой активируют путем растворения в диметилформамиде (ДМФА) и добавлением раствора диизопропилкарбодиимида в ДМФА. Затем эфиры других аминокислот растворяют в N-метилпиридине и осуществляют сочетание с предварительно деблокированной 20%-ным раствором пиперидина в диметилформамиде смолой. После чего проводят отщепление пептида от смолы при одновременном удалении защитных групп боковых цепей.
Таким образом, в известном способе необходимо провести присоединение к смоле первой аминокислоты, к которой далее присоединяется после активации следующая аминокислота или фрагмент со свободной аминогруппой, что усложняет способ, увеличивая его стадийность. Известный способ позволяет получить производные пептидов, обладающих способностью тормозить действие ретровирусных протеаз. При этом речь идет об индивидуальных стереоизомерах пептидов, и в случае получения смеси стереоизомеров, в частности смеси диастереомеров, они разделяются известными способами путем фракционной кристаллизации или с помощью хроматографии. Но в медицинской практике часто необходимо использование дипептидов, обогащенных DL- или D,L+L,D-диастереомерами.
Известен способ получения дипептидов в растворе в присутствии конденсирующего агента и триэтиламина (ТЭА) ("Химико-фармацевтический журнал", "Медицина", 1970, номер 10, стр.25-27). В известном способе дипептиды 4-ди(2-хлорэтил)амино-DL-фенилаланина (сарколизина) получены путем двухстадийного синтеза, на первой стадии при взаимодействии N-ацетилсарколизина с хлорангидридом изовалериановой кислоты в присутствии ТЭА в среде хлороформа получают смешанный ангидрид N-ацетилсарколизина и изовалериановой кислоты, на второй стадии без выделения ангидрида его вводят в реакцию с эфиром DL- или L-аминокислот. Наряду с другими дипептидами получен этиловый эфир N-ацетилсарколизина-DL-валина (асалин), который успешно применяется в медицинской практике при лечении некоторых форм злокачественных новообразований.
Известным способом могут быть получены дипептиды N-ацетилсарколизина. Для поиска новых лекарственных средств и проведения научных исследований необходимы пептиды различных производных аминокислот, обогащенные нужным диастереомером.
Таким образом, перед авторами стояла задача разработать способ, позволяющий расширить ассортимент получаемых дипептидов, в частности не только дипептиды N-ацетил-сарколизина, но и дипептиды N-ацетил-L-фенилаланина, при этом обогащенные DL- или D,L+L,D-диастереомерами.
Поставленная задача решена в предлагаемом способе получения дипептидов взаимодействием N-защищенной аминокислоты с этерифицированным производным аминокислоты со свободной аминогруппой в органическом растворителе в присутствии конденсирующего агента и триэтиламина, в котором в качестве N-защищенной аминокислоты используют N-ацетил-L-фенилаланин или N-ацетил-4[ди-(2-хлорэтил)амино]-DL-фенилаланин, в качестве этерифицированного производного аминокислоты - метиловый, или этиловый, или бензиловый, или трет-бутиловый эфир L-аланина, или L-фенилаланина, или L- и DL-валина, а в качестве конденсирующего агента - изобутилхлоркарбонат, при этом триэтиламин присутствует с 20%-ным избытком по отношению к стехиометрии.
В настоящее время не известен способ получения дипептидов, обогащенных DL- или DL+LD-диастереомерами, который может быть охарактеризован предлагаемой совокупностью отличительных признаков, в частности предлагаемыми исходными, взаимодействие которых осуществляется в присутствии изобутилхлоркарбоната и ТЭА, взятого с 20%-ным избытком к стехиометрии.
Предлагаемый способ позволяет получить дипептиды, обогащенные DL- или DL+LD-диастереомерами, путем взаимодействия N-защищенной аминокислоты в среде органического растворителя методом смешанных ангидридов, который сопровождается образованием энантиомерных оксазолонов. Эти оксазолоны, присутствующие в рацемических смесях, обнаруживают выраженные различия в скоростях реакций с хиральными эфирами аминокислот. Осуществление способа путем использования свойства смешанных ангидридов образовывать рацемические оксазолоны, которые не только с разными скоростями реагируют с аминокислотой, но и превращаются друг в друга, позволяет получать широкий ассортимент дипептидов, обогащенных DL- или DL+LD-диастереомерами. Применение в качестве конденсирующего агента изобутилхлоркарбоната позволяет на стадии образования смешанных ангидридов использовать в качестве исходной N-защищенной аминокислоты как N-ацетил-L-фенилаланин, так и N-ацетил-4[ди-(2-хлорэтил)-амино]-DL-фенилаланин, расширяя тем самым ассортимент получаемых пептидов. Проведение процесса в присутствии 20%-ного избытка к стехиометрии ТЭА позволяет получить уже после первичной обработки реакционной массы соотношение диастереомеров значительно более чем 50:50. При этом при меньшем или большем содержании триэтиламина не удается достигнуть желаемого соотношения.
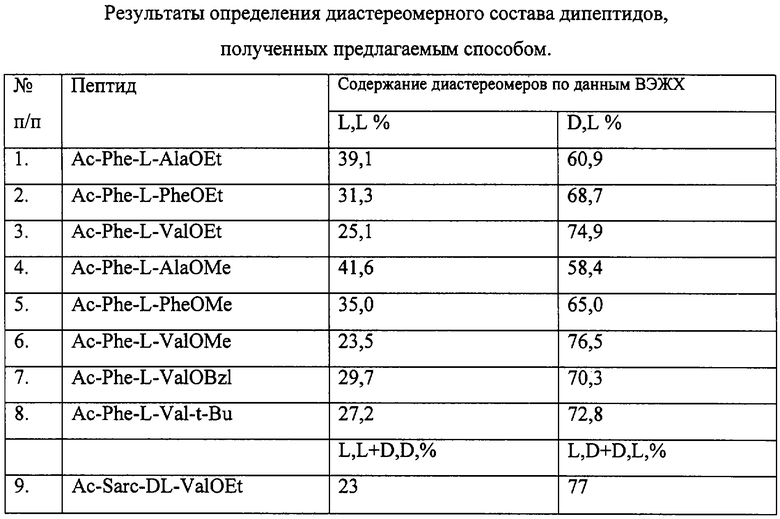
Предлагаемый способ осуществляют следующим образом. N-ацетил-L-фенилаланин или его производное N-ацетил-4[ди-(2-хлорэтил)амино]-DL-фенилаланин растворяют в тетрагидрофуране (ТГФ), полученный раствор охлаждают до -13°С или до 0°С (в случае производного фенилаланина) и добавляют по каплям ТЭА с избытком 20% к стехиометрии; затем добавляют изобутилхлоркарбонат при той же температуре. Реакционную массу выдерживают 30 мин при перемешивании и температуре -13°С или 0°С (в случае производного фенилаланина) и добавляют в растворе ТГФ гидрохлорид эфира L-аланина, или L-фенилаланина, или L-и DL-валина, нейтрализованный эквимолярным количеством ТЭА и предварительно охлажденный в холодильнике. Реакционную массу перемешивают 1 час при температуре -13°С, 1 час при температуре 0°С и затем 3 часа при комнатной температуре. После чего оставляют на ночь без перемешивания. В случае производного фенилаланина реакционную массу перемешивают 2 часа при температуре 0°С, затем 3 часа при комнатной температуре и оставляют на ночь без перемешивания. Осадок отфильтровывают, фильтрат упаривают в вакууме досуха. Остаток растворяют в хлороформе и промывают 5%-ным раствором NаНСО3, водой, 5%-ным раствором соляной кислоты и снова водой, затем сушат над Na2SO4. После отгонки растворителя и сушки остатка в вакууме получают смесь D,L- и L,L-диастереомеров, в которой значительно превалирует D,L-диастереомер (см. таблицу).
Предлагаемый способ иллюстрируется следующими примерами
Пример 1. Метиловые эфиры N-ацетил-L- и N-ацетил-О-фенилаланил-L-валина
К охлажденному до -13°С раствору 0,75 г (3,62 ммоль) N-ацетил-L-фенилаланина в 17 мл ТГФ при перемешивании по каплям добавляют 0,61 мл (4,34 ммоль) ТЭА, что составляет 20% избытка к стехиометрии, и 0,48 мл (3,62 ммоль) изобутилхлоркарбоната. Реакционную массу перемешивают 30 мин при температуре -13°С, добавляют охлажденную до температуры -10°С смесь, полученную перемешиванием 0,61 г (3,62 ммоль) гидрохлорида метилового эфира L-валина и 0,51 мл (3,62 ммоль) ТЭА, 3 мл ДМФА в 9 мл ТГФ. Реакционную массу выдерживают при перемешивании и температуре -13°С в течение 1 часа, затем при температуре 0°С в течение 1 часа, далее при комнатной температуре в течение 3 часов, после чего оставляют при комнатной температуре на 17 часов. Осадок отфильтровывают, ТГФ отгоняют в вакууме. Фильтрат растворяют в хлороформе, промывают 5%-ным NаНСО3, водой, 5%-ным раствором НСl, затем водой до рН 7 и сушат над Na2SO4. Хлороформный раствор упаривают в вакууме досуха. После высушивания в вакууме над Р2О5 получают соединение в виде бесцветного кристаллического порошка.
Получают 0,88 г (75,9%) смеси диастереомеров. По данным ВЭЖХ - 23,5% L,L-диастереоизомера и 76,5% D,L-диастереоизомера. Rf 0,62 (бензол-уксусная кислота-метанол 11:1:2). Найдено, %: С 63,83; Н 7,60; N 8,73. C7H24N2O4. Вычислено, %: С 63,73; Н 7,55; N 8,75. УФ-спектр (λ, нм): 261,2 (бензольное кольцо). ИК-спектр (ν, см-1): 3280 (вал. кол. NH); 3070 (вал. кол. СН аромат.); 1723 (С=0 слож. эфира); 1625 (амид. полоса I); 1525 (деф. кол. NH, амид. полоса II).
1Н ЯМР-спектр (CDCl3, δ, м.д.): 0,73д (СН3-5’ D,L-изомера, Iд=6,75 Гц); 0,75д (СН3-4’ D,L, Iд=6,5 Гц); 0,83д и 0,87д (2×СН3-4’,5’,L,L-изомера, Iд=6,8Гц); 1,98 с (СН3-СО, D,L+L,L); 2,01-2,14 м (СН3’, D,L+L,L); 3,03дд и 3,11дд (СН2-3, АВ-система D,L,IАВ=13,7 Гц; I3А-2=6,2 Гц; I3В-2=8,2 Гц); 3,05д (СН2-3, L,L изом., I3-2=7,2 Гц); 4,40дд (СН-2’, D,L-изомера; I2’-1=8,4 Гц, I2’-3’=5,0 Гц); 4,41дд (СН-2’, LL-изом., I2’-1’=8,4 Гц, I2’-3’=5,0 Гц), 4,71дт (СН-2, L,L-изомера, I2-1=7,8 Гц; I2-3=7,2 Гц); 4,76 д.д.д. (СН-2, L,L-изомера, I2-1=8,1 Гц; I2-3B-8,2 Гц; I2-3A=6,3 Гц); 6,23д (NH-1, D,L-изомера; I1-2=8,1 Гц); 6,28 д (NH-1, L,L-изомера, I1-2=7,8 Гц); 6,37 д (NH-1’, D,L+L,L, I1’-2’=8,4 Гц); 7,12-7,32 м (Ph от D,L- и L,L-изомеров).
Пример 2. Этиловые эфиры N-ацетил-DL-сарколизил-DL-валина
1,5 г (4,3 ммоль) N-ацетилсарколизина растворяют в 36 мл ТГФ. Раствор охлаждают до температуры -2 - 0°С и добавляют по каплям 0,73 мл (5,2 ммоль) ТЭА, что составляет 20% избытка к стехиометрии, поддерживая температуру около 0°С, затем добавляют по каплям 0,57 мл (4,3 ммоль) изобутилхлоркарбоната. Реакционную массу выдерживают 30 мин при температуре 0°С и перемешивании, затем добавляют 0,79 г (4,3 ммоль) гидрохлорида этилового эфира D,L-валина, предварительно охлажденного до температуры 0°С и нейтрализованного 0,61 мл (4,3 ммоль) ТЭА. Реакционную массу перемешивают 2 часа при температуре 0°С, затем 3 часа при комнатной температуре и оставляют на ночь без перемешивания. Осадок отфильтровывают, фильтрат упаривают в вакууме досуха. Остаток растворяют в хлороформе и промывают 5%-ным раствором NaHCO3, водой 5%-ным раствором НСl, затем водой до рН 7 и сушат над Na2SO4. Хлороформный раствор упаривают в вакууме досуха. После высушивания в вакууме над P2O5 получают соединение в виде бесцветного кристаллического порошка.
Получают 1,67 г (81,5%) дипептида с соотношением диастереомеров (L,D+D,L):(L,L+D,D)=77:23 по данньм ВЭЖХ. После высушивания в вакууме над P2O5 получают вещество желтоватого цвета. Rf 0,5 (бензол-уксусная кислота:метанол 11:1:2). Найдено, %: С 55,26; Н 6,88; N 8,49; Cl 15,26; C22H33N3O4Cl2. Вычислено, %: С 55,69; Н 7,01; N 8,86; Cl 14,95. УФ-спектр (λ, нм): 260. ИК-спектр (ν, см-1): 3280 (NH), 3070 (вал.кол.аром. кольца), 1735 (вал. кол. С=0 в сложном эфире); 1635 (амид.полоса I); 1540 (амид.полоса II).
1Н ЯМР (СОСl3, δ, м.д.): 0,76д и 0,78д (6Н, 2×СН3 Val; Iд=6,7 Гц); 1,25т (3Н, СН3 эф, IT=7,2 Гц); 1,99с (3Н, СН3СО); 2,03сп.д (1Н, СН(3’), Icп=6,7 Гц, Iд=4,7 Гц); 2,92д.д. и 3,02д.д. (АВ-система, СН2(3); IАВ=13,9 Гц; IВ-2=8,1 Гц; IA-2=6,0 Гц); 3,60м и 3,7м (8Н, N(CH2CH2Cl2)); 4,14д.кв. и 4,18д.кв. (система АВ, ОCH2эф, IAB=10,8 Гц, Iкв=7,2 Гц); 4,39д.д. (1Н, СН(2’), I2’-1’=8,6 Гц; I2’-3’=4,7 Гц); 4,69д.д.д. (1Н, СН(2), I2-в=8,1 Гц, I2-1=7,9 Гц, I2-A=6,0 Гц); 6,22д. (1Н, NH, I1-2=7,9 Гц); 6,35д. (1Н, NH, I1’-2’=8,6 Гц); 6,61д. (2Н, 2×СНаром., Iд=8,8 Гц); 7,11д. (2Н, 2×СНаром., Iд.=8,8 Гц).
Таким образом, предлагаемый способ позволяет получить дипептиды, обогащенные DL- или DL+LD-диастереомерами, а следовательно, позволяет расширить ассортимент получаемых пептидов, используемых как в научно-исследовательских целях, так и в медицинской практике.
| название | год | авторы | номер документа |
|---|---|---|---|
| ДИПЕПТИДЫ В КАЧЕСТВЕ КОРМОВЫХ ДОБАВОК | 2010 |
|
RU2536467C2 |
| СПОСОБ ПОЛУЧЕНИЯ ТРИТЕРПЕНОВЫХ ГЛИКОПЕПТИДОВ | 1994 |
|
RU2083587C1 |
| СПОСОБ ПОЛУЧЕНИЯ N-КАРБОКСИАНГИДРИДОВ ИЛИ N-ТИОКАРБОКСИАНГИДРИДОВ УРЕТАНЗАЩИЩЕННЫХ АМИНОКИСЛОТ | 1989 |
|
RU2007396C1 |
| ПРОИЗВОДНЫЕ ГИДРАЗИНА ИЛИ ИХ СОЛИ, ФАРМАЦЕВТИЧЕСКОЕ СРЕДСТВО, АМИНОАЛКИЛГИДРАЗИНЫ ИЛИ ИХ СОЛИ | 1992 |
|
RU2092492C1 |
| 4-Диметиламинопиридиниевые соли @ -защищенных аминокислот в качестве промежуточных продуктов для синтеза пептидов | 1984 |
|
SU1253975A1 |
| СПОСОБ ПОЛУЧЕНИЯ L-КАРНОЗИНА И ЕГО ГОМОЛОГОВ | 1992 |
|
RU2084457C1 |
| ПРОИЗВОДНЫЕ 5-АМИНО-4-ОКСИГЕКСАНОВОЙ КИСЛОТЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1992 |
|
RU2067585C1 |
| ПРОИЗВОДНЫЕ N-АЦИЛПРОЛИЛДИПЕПТИДОВ | 1993 |
|
RU2119496C1 |
| СПОСОБ ПОЛУЧЕНИЯ ОПТИЧЕСКИ АКТИВНЫХ АЛЬФА-АМИНОАЦЕТАЛЕЙ | 2009 |
|
RU2507194C2 |
| СПОСОБ ПОЛУЧЕНИЯ КАРБОКСИЗАЩИЩЕННЫХ ГЛИКОПЕПТИДОВ ГЛИЦИРРИЗИНОВОЙ КИСЛОТЫ | 1992 |
|
RU2057139C1 |
Изобретение относится к области органической химии, а именно к способу получения пептидов, обладающих биологической активностью, в частности противоопухолевой активностью, что позволяет использовать их в медицинской практике. Предлагается способ получения дипептидов взаимодействием N-защищенной аминокислоты с этерифицированным производным аминокислоты со свободной аминогруппой в органическом растворителе в присутствии конденсирующего агента и триэтиламина, в котором в качестве N-защищенной аминокислоты используют N-ацетил-L-фенилаланин или N-ацетил-4[ди-(2-хлорэтил)амино]-DL-фенилаланин, в качестве этерифицированного производного аминокислоты - метиловый, или этиловый, или бензиловый, или трет-бутиловый эфир L-аланина или L-фенилаланина или L- и DL-валина, а в качестве конденсирующего агента - изобутилхлоркарбонат, при этом триэтиламин присутствует с 20%-ным избытком по отношению к стехиометрии. Предлагаемый способ позволяет получить дипептиды, обогащенные DL- или DL + LD-диастереомерами, а следовательно, позволяет расширить ассортимент получаемых пептидов, используемых как в научно-исследовательских целях, так и в медицинской практике. 1 табл.
Способ получения дипептидов взаимодействием N-защищенной аминокислоты с этерифицированным производным аминокислоты в органическом растворителе в присутствии конденсирующего агента и триэтиламина, отличающийся тем, что в качестве N-защищенной аминокислоты используют N-ацетил-L-фенилаланин или N-ацетил-4[ди-(2-хлорэтил)амино]-DL-фенилаланин, в качестве этерифицированного производного аминокислоты - метиловый, или этиловый, или бензиловый, или трет-бутиловый эфир L-аланина, или L-фенилаланина, или L- и DL-валина, а в качестве конденсирующего агента - изобутилхлоркарбонат, при этом триэтиламин присутствует с 20%-ным избытком по отношению к стехиометрии.
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПЕПТИДОВ ИЛИ ИХ ФИЗИОЛОГИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ | 1990 |
|
RU2047621C1 |

