Настоящее изобретение относится к периодическому способу получения полиамидов из аминонитрилов и воды при повышенной температуре и повышенном давлении в присутствии катализатора.
В патенте США US 4629776 описан каталитический способ получения полиамидов из ω-аминонитрилов, таких как ω-аминокапронитрил (АКН). АКН подвергают взаимодействию с водой в присутствии каталитического количества окисленного соединения серы в качестве катализатора. Например, в качестве катализатора используют серную кислоту.
В патенте США US 4568736 описан аналогичный способ получения полиамидов. В качестве катализатора в нем используется кислородсодержащее соединение фосфора, фосфорная кислота или фосфоновая кислота.
Полное отделение катализаторов в обоих способах практически невозможно. Наличие катализаторов в полимере может препятствовать синтезу высокомолекулярных полимеров и затруднять дальнейшие стадии переработки, например прядение. Кроме того, полученные полимеры содержат большое количество летучих компонентов, что делает затруднительной переработку полиамидов.
В европейской заявке на патент ЕР-А-0479306 описан способ получения полиамидов из ω-аминонитрилов. В этом способе ω-аминонитрилы подвергаются взаимодействию с водой в присутствии кислородсодержащего соединения фосфора в качестве катализатора. Этот способ предусматривает, что по достижении температуры реакции в пределах от 200 до 260°С производят непрерывное удаление аммиака и воды путем сброса давления при одновременном непрерывном добавлении воды, причем давление выбирают в пределах от 14 до 24·106 Па (14-24 бар).
В немецкой заявке на патент DE-A-4339648 описан способ получения капролактама путем взаимодействия нитрилов карбоновых аминокислот с водой, причем способ осуществляется в жидкой фазе с применением гетерогенных катализаторов. В качестве гетерогенных катализаторов могут быть использованы кислые, основные или амфотерные оксиды элементов второй, третьей или четвертой главных групп Периодической системы элементов. Катализатор применяется, например, в форме прутков.
Задача настоящего изобретения состоит в том, чтобы предложить способ получения полиамидов из аминонитрилов, обеспечивающий улучшенный гидролиз реагентов, в частности гидролиз кислотных амидных групп, и улучшенную молекулярно-массовую структуру. Способ должен обеспечивать практически полное удаление применяемого катализатора из реакционной смеси и обладать высокой эффективностью. Кроме того, полученные продукты должны обладать улучшенной способностью к термообработке.
Согласно изобретению поставленная задача решается с помощью периодического способа получения полиамида путем взаимодействия смеси, содержащей, по меньшей мере, один аминонитрил и необязательно мономеры, применяемые для получения полиамида, с водой, причем способ включает следующие стадии:
(1) взаимодействие смеси с водой при температуре от 90 до 400°С и давлении от 0,1 до 35·106 Па с получением реакционной смеси,
(2) последующее взаимодействие реакционной смеси при температуре от 150 до 400°С и давлении, более низком, чем давление на стадии 1, причем температуру и давление выбирают так, что получаются первая газовая фаза и первая жидкая или первая твердая фаза, или смесь первой твердой и первой жидкой фаз, и отделяют первую газовую фазу от первой жидкой или первой твердой фазы или от смеси первой жидкой и первой твердой фаз, и
(3) смешение первой жидкой или первой твердой фазы или смеси первой жидкой и первой твердой фаз с газообразной или жидкой фазой, содержащей воду, при температуре от 150 до 370°С и давлении от 0,1 до 30·106 Па с получением продуктсодержащей смеси,
причем стадию (1) проводят в присутствии катализатора на основе кислоты Бренстеда, выбранного из бета-цеолитного катализатора, слоистого силикатного катализатора или катализатора неподвижного слоя и состоящего, в основном, из диоксида титана (ТiO2) с 70-100 мас.% анатаза и 0-30 мас.% рутила, причем в катализаторе до 40 мас.% диоксида титана могут быть заменены оксидом вольфрама, и стадии (2) и (3) могут проводиться в присутствии этого катализатора.
Далее изобретение относится к периодическому способу получения полиамида путем взаимодействия смеси, содержащей, по меньшей мере, один аминонитрил и необязательно мономеры, применяемые для получения полиамида, с водой, причем способ включает следующие стадии:
(1) взаимодействие смеси с водой при температуре от 90 до 400°С и давлении от 0,1 до 35·106 Па с получением реакционной смеси,
(2) последующее взаимодействие реакционной смеси при температуре от 150 до 400°С и давлении, более низком, чем давление на стадии 1, причем температуру и давление выбирают так, что получаются первая газовая фаза и первая жидкая или первая твердая фаза или смесь первой твердой и первой жидкой фаз, и отделяют первую газовую фазу от первой жидкой или первой твердой фазы или от смеси первой жидкой и первой твердой фазы,
(3) смешение первой жидкой или первой твердой фазы или смеси первой жидкой и первой твердой фаз с газообразной или жидкой фазой, содержащей воду, при температуре от 150 до 370°С и давлении от 0,1 до 30·106 Па с получением продуктсодержащей смеси,
(4) дополнительная конденсация продуктсодержащей смеси при температуре от 200 до 350°С и давлении, более низком, чем давление на стадии 3, причем температуру и давление выбирают так, что получаются вторая, содержащая воду и аммиак, газовая фаза и вторая жидкая или вторая твердая фаза или смесь второй жидкой и второй твердой фазы, соответственно содержащие полиамид,
причем стадию (1) проводят в присутствии катализатора на основе кислоты Бренстеда, выбранного из бета-цеолитного катализатора, слоистого силикатного катализатора или катализатора неподвижного слоя и состоящего, в основном, из диоксида титана (TiO2) с 70-100 мас.% анатаза и 0-30 мас.% рутила, причем в катализаторе до 40 мас.% диоксида титана могут быть заменены оксидом вольфрама, и стадии (2) и (3) могут проводиться в присутствии этого катализатора.
В предлагаемом способе на стадии (3) газообразную или жидкую фазу, содержащую воду, предпочтительно добавляют в количестве от 50 до 1500 мл воды на 1 кг первой жидкой или первой твердой фазы или смеси первой жидкой и первой твердой фаз.
Далее изобретение относится к периодическому способу получения полиамида путем взаимодействия смеси, содержащей, по меньшей мере, один аминонитрил и необязательно мономеры, применяемые для получения полиамида, с водой, причем способ включает следующие стадии:
(1) взаимодействие смеси с водой при температуре от 90 до 400°С и давлении от 0,1 до 35·106 Па с получением реакционной смеси,
(2) последующее взаимодействие реакционной смеси при температуре от 150 до 400°С и давлении, более низком, чем давление на стадии 1, причем температуру и давление выбирают так, что получаются первая газовая фаза и первая жидкая или первая твердая фаза или смесь первой твердой и первой жидкой фаз, и отделяют первую газовую фазу от первой жидкой или первой твердой фазы или от смеси первой жидкой и первой твердой фаз, и
(4) дополнительная конденсация первой жидкой или первой твердой фазы или смеси первой жидкой и первой твердой фаз при температуре от 200 до 350°С и давлении, более низком, чем давление на стадии 3, причем температуру и давление выбирают так, что получаются вторая, содержащая воду и аммиак, газовая фаза и вторая жидкая или вторая твердая фаза или смесь второй жидкой и второй твердой фаз, соответственно содержащие полиамид,
причем стадию (1) проводят в присутствии катализатора на основе кислоты Бренстеда, выбранного из бета-цеолитного катализатора, слоистого силикатного катализатора или катализатора неподвижного слоя и состоящего, в основном, из диоксида титана (TiO2) с 70-100 мас.% анатаза и 0-30 мас.% рутила, причем в катализаторе до 40 мас.% диоксида титана могут быть заменены оксидом вольфрама, и стадия (2) может проводиться в присутствии этого катализатора.
Принципиальная идея предлагаемого согласно изобретению способа описана в неопубликованной заявке DE-A-19709390 с более ранним приоритетом.
В качестве аминонитрила в смеси могут применяться принципиально все аминонитрилы, то есть соединения, которые имеют как, по меньшей мере, одну амино-, так и, по меньшей мере, одну нитрильную группу. Среди них предпочтительны ω-аминонитрилы, причем среди последних особенно предпочтительны ω-аминоалкилнитрилы с 4-12 атомами углерода, в частности с 4-9 атомами углерода в алкиленовом остатке, или аминоалкиларилнитрил с 8-13 атомами углерода, причем в качестве этих последних предпочтительны такие аминоалкиларилнитрилы, которые содержат между ароматическим звеном и амино- и нитрильной группой алкиленовую группу с, по меньшей мере, одним атомом углерода. Среди аминоалкиларилнитрилов предпочтительны, в частности, такие, которые содержат амино- и нитрильную группу в 1,4-положении относительно друг друга. В качестве ω-аминоалкилнитрила особенно предпочтительно применяют линейные ω-аминоалкилнитрилы, в которых алкиленовый остаток (-СН2-) содержит, как правило, от 4 до 12 атомов углерода, в частности от 4 до 9 атомов углерода, такие, как, например, 6-амино-1-цианопентан (6-аминокапронитрил), 7-амино-1-цианогексан, 8-амино-1-цианогептан, 9-амино-1-цианооктан, 10-амино-1-цианононан, особенно предпочтителен 6-аминокапронитрил.
6-Аминокапронитрил обычно получают гидрированием адипонитрила с помощью известных способов, например как описано в немецких заявках на патент DE-A 836938, DE-A 848654 или в патенте CШA US 5151543.
Могут использоваться также смеси нескольких аминонитрилов или смеси аминонитрила с другими сомономерами, например капролактамом, или смесь, подробнее описываемая ниже.
В особом варианте изобретения, в частности, при использовании сополиамидов или полиамидов с разветвленной, соответственно удлиненной цепью предусматривается, что вместо чистого 6-аминокапронитрила применяется следующая смесь, содержащая:
от 50 до 99,99, предпочтительно от 80 до 90 мас.% 6-аминокапронитрила,
от 0,01 до 50, предпочтительно от 1 до 30 мас.%, по меньшей мере, одной дикарбоновой кислоты, выбранной из группы, состоящей из алифатических α,ω-дикарбоновых кислот с 4-10 атомами углерода, ароматических дикарбоновых кислот с 8-12 атомами углерода и циклоалкандикарбоновых кислот с 5-8 атомами углерода,
от 0 до 50, предпочтительно от 0,1 до 30 мас.% α,ω-диамина с 4-10 атомами углерода,
от 0 до 50, предпочтительно от 0 до 30 мас.% α,ω-динитрила с 2-12 атомами углерода, а также
от 0 до 50, предпочтительно от 0 до 30 мас.% α,ω-аминокислоты, содержащей 5-12 атомов углерода, или соответствующего лактама,
от 0 до 10 мас.%, по меньшей мере, одной неорганической кислоты или ее солей, причем сумма всех указанных в процентах отдельных компонентов составляет 100%.
В качестве дикарбоновых кислот можно использовать алифатические дикарбоновые α,ω-кислоты, содержащие от 4 до 10 атомов углерода, например янтарную, глутаровую, адипиновую, пимелиновую, пробковую, азелаиновую, себациновую кислоты, предпочтительно адипиновую и себациновую кислоты, особенно предпочтительно адипиновую кислоту, и ароматические дикарбоновые кислоты, содержащие от 8 до 12 атомов углерода, например терефталевую кислоту, а также циклоалкандикарбоновые кислоты, содержащие от 5 до 8 атомов углерода, например циклогександикарбоновую кислоту.
В качестве α,ω-диамина с 4-10 атомами углерода можно использовать тетра-метилендиамин, пентаметилендиамин, гексаметилендиамин, гептаметилен-диамин, октаметилендиамин, нонаметилендиамин и декаметилендиамин, предпочтительно гексаметилендиамин.
Кроме того, можно применять также соли названных дикарбоновых кислот и диаминов, в частности соль адипиновой кислоты и гексаметилендиамина, так называемую соль АГ.
В качестве α,ω-динитрила с 2-12 атомами углерода предпочтительно применяются алифатические динитрилы, такие как 1,4-дицианбутан (адиподинитрил), 1,5-дицианпентан, 1,6-дициангексан, 1,7-дициангептан, 1,8-дицианоктан, 1,9-дицианнонан, 1,10-дициандекан, особенно предпочтительным является адиподинитрил.
При желании можно применять также диамины, динитрилы и аминонитрилы, являющиеся производными от разветвленных алкиленовых или ариленовых, или алкилариленовых соединений.
В качестве α,ω-аминокислоты с 5-12 атомами углерода можно применять 5-аминопентановую, 6-аминогексановую, 7-аминогептановую, 8-аминооктановую, 9-аминононановую, 10-аминодекановую, 11-аминоундекановую и 12-аминододекановую кислоты, предпочтительно 6-аминогексановую кислоту.
В соответствии с изобретением на первой стадии (стадия 1) аминонитрил нагревают с водой при температуре от 90 до 400°С, предпочтительно приблизительно от 180 до 310°С и, в частности, от 220 до 270°С, причем давление устанавливают на уровне от 0,1 до 15·106 Па, предпочтительно от 1 до 10·106 Па и, в частности, от 4 до 9·106 Па. При этом давление и температуру на этой стадии согласовывают друг с другом так, что получаются одна жидкая или одна твердая фаза и смесь жидкой и твердой фаз и газообразная фаза.
В соответствии с изобретением воду применяют в молярном отношении аминоалкилнитрила к воде в пределах от 1:1 до 1:10, предпочтительно от 1:2 до 1:8, особенно предпочтительно от 1:2 до 1:6, причем применение воды в избытке, в пересчете на используемый аминоалкилнитрил, предпочтительно.
В рассматриваемом варианте изобретения жидкая или твердая фаза или смесь жидкой и твердой фаз соответствует реакционной смеси, в то время как газообразную фазу отделяют. При этом на данной стадии газообразная фаза может быть сразу отделена от жидкой или твердой фазы или от смеси жидкой и твердой фаз, или же реакционная смесь, образующаяся на данной стадии, может присутствовать в виде двухфазной смеси, то есть в виде жидкой и газообразной, твердой и газообразной или жидкой/твердой и газообразной фаз. Давление и температура могут быть согласованы друг с другом так, чтобы реакционная смесь была однофазной, то есть представляла собой одну твердую или одну жидкую фазу.
Отделение газовой фазы может производиться в сепараторах с мешалкой или без мешалки или каскадах сепараторов, а также в выпарных аппаратах, например в циркуляционных испарителях или пленочных испарителях, таких как пленочный экструдер, или в дисковых реакторах, позволяющих получать увеличенную поверхность раздела фаз. В ряде случаев необходимо перекачивание реакционной смеси, соответственно применение петлевого реактора, для увеличения поверхности раздела фаз. Кроме того, отделению газовой фазы способствует добавление водяного пара или инертного газа в жидкую фазу.
При заранее выбранной температуре предпочтительно устанавливать давление так, чтобы оно было ниже равновесного давления паров аммиака, но выше равновесного давления паров остальных компонентов в реакционной смеси при заданной температуре. Такой режим благоприятствует отделению аммиака и тем самым ускоряет гидролиз кислотных амидных групп.
На стадии 1 могут применяться реакторы с мешалкой или каскады реакторов. При двухфазном режиме предпочтительно применять котлы или колонные реакторы.
Относительно времени пребывания реакционной смеси на первой стадии никаких ограничений не существует; тем не менее, как правило, оно выбирается в пределах от приблизительно 10 минут до приблизительно 10 часов, предпочтительно от приблизительно 30 минут до приблизительно 6 часов.
Хотя относительно степени превращения нитрильных групп на первой стадии также не существует никаких ограничений, тем не менее, в основном, из экономических соображений, конверсия нитрильных групп на первой стадии в общем случае составляет не менее приблизительно 70 мол.%, предпочтительно не менее приблизительно 95 мол.% и, в частности, не менее приблизительно от 97 до 99 мол.%, в пересчете на число молей использованного аминонитрила.
Степень превращения нитрильных групп определяется посредством ИК-спектроскопии (CN-валентные колебания при волновом числе 2247), ЯМР или жидкостной хроматографии высокого давления, предпочтительно посредством ИК-спектроскопии.
В другом предпочтительном варианте изобретения смесь аминонитрила с водой непрерывно нагревают в теплообменнике и нагретую таким образом смесь вводят в поддерживаемый при той же температуре реактор. Само собой разумеется, можно нагревать аминонитрил и воду также и раздельно.
Далее, в соответствии с изобретением не исключается проведение взаимодействия на стадии 1 также в присутствии кислородсодержащих соединений фосфора, в частности фосфорной кислоты, фосфористой кислоты и фосфорноватистой кислоты, а также их солей с щелочными и щелочно-земельными металлами и солей аммония, таких как Na3PO4, NaH2PO4, Na2HPO4, NaH2PO3, Na2HPO3, NaH2PO2, К3РO4, КН2РO4, К2НРO4, КН2РО3, К2НРО3, КН2РO2, причем молярное отношение ω-аминонитрила к соединениям фосфора находится в пределах от 0,01:1 до 1:1, предпочтительно от 0,01:1 до 0,1:1.
В соответствии с изобретением стадия (1) и необязательно стадия (2) и/или (3) проводятся в присутствии катализатора на основе кислоты Бренстеда, выбранного из бета-цеолитного катализатора, слоистого силикатного катализатора или катализатора неподвижного слоя и состоящего, в основном, из диоксида титана (TiO2) с 70-100 мас.% анатаза и 0-30 мас.% рутила, причем в катализаторе до 40 мас.% диоксида титана могут быть заменены оксидом вольфрама. На стадии (4) катализатор неподвижного слоя, как правило, не применяется. Перед дополнительной конденсацией на стадии (4) гетерогенный катализатор может быть легко отделен от реакционной смеси, так как вязкость реакционной смеси еще относительно невелика. Это позволяет, по меньшей мере, практически полностью, предпочтительно полностью, отделить катализатор от продуктсодержащей смеси.
Катализатор с высоким содержанием анатаза особенно пригоден, если применяется чистый аминокапронитрил (АКН) с очень малым содержанием примесей или вообще без примесей. Как правило, такой катализатор содержит от 80 до 100 мас.% анатаза и от 0 до 20 мас.% рутила, предпочтительно он состоит в основном или полностью из анатаза. При использовании загрязненного аминокапронитрила, содержащего приблизительно от 1 до 3 мас.% примесей, в основном, применяют катализатор с более высокой долей рутила. Предпочтительный катализатор содержит от 70 до 80 мас.% анатаза и от 20 до 30 мас.% рутила, предпочтительно около 30 мас.% рутила.
Объем пор катализатора составляет, как правило, от 0,1 до 5 мл/г, особенно предпочтительно от 0,2 до 0,5 мл/г. Средний диаметр пор преимущественно равен от 0,005 до 0,1 мкм, особенно предпочтительно от 0,01 до 0,06 мкм. При работе с высоковязкими продуктами средний диаметр пор следует выбирать большим. Твердость при резании составляет, как правило, более 20 Н, предпочтительно более 25 Н. БЭТ-поверхность составляет, как правило, более 40 м2/г, особенно предпочтительно более 100 м2/г. При меньшей БЭТ-поверхности следует выбирать соответственно больший насыпной объем, чтобы обеспечить достаточную активность катализатора. Особенно предпочтительные катализаторы имеют следующие свойства: содержание анатаза 100%; объем пор 0,3 мл/г; средний диаметр пор 0,02 мкм; твердость при резании 32 Н; БЭТ-поверхность 116 м2/г или содержание анатаза 84 мас.%; содержание рутила 16 мас.%; объем пор 0,3 мл/г; средний диаметр пор 0,03 мкм; твердость при резании 26 Н; БЭТ-поверхность 46 м2/г. При этом катализаторы могут быть изготовлены из имеющихся в продаже порошков, например, предлагаемых фирмами Degussa, Finti или Kemira. Возможна замена диоксида титана оксидом вольфрама, при этом оксидом вольфрама заменяется до 40 мас.%, предпочтительно до 30 мас.%, особенно предпочтительно до 15-25 мас.% диоксида титана. Приготовление готовых форм катализатора может осуществляться в соответствии с технологией, описанной в работе: Ertl, Knozinger, Weitkamp: "Handbook of heterogenous catalysis", VCH Weinheim, 1997, стр.98 и следующие.
Предлагаемый периодический способ осуществляется преимущественно в замкнутой реакционной системе в 3 или 4 стадии. Ниже подробнее описываются отдельные стадии реакции.
Можно работать с концентрированными реакционными смесями с массовым отношением аминокапронитрила к воде от 1:1 до 1:10.
В соответствии с изобретением полученная на первой стадии реакционная смесь подвергается дальнейшему взаимодействию на второй стадии при температуре приблизительно от 150 (200) до 400 (350)°С, предпочтительно при температуре приблизительно от 100 до 330 (300)°С и, в частности, в пределах приблизительно от 230 до 290 (270)°С и при давлении, более низком, чем давление на первой стадии. Как правило, давление на второй стадии приблизительно, по меньшей мере, на 0,5·106 Па ниже, чем давление на первой стадии, причем в общем случае давление находится в пределах приблизительно от 0,01 до 45·106 Па, предпочтительно от 0,5 до 15·106 Па и, в частности, от 2 до 6·106 Па. (Значения в скобках: с катализатором).
При этом температуру и давление на второй стадии выбирают так, что получаются первая газовая фаза и первая жидкая или первая твердая фаза или смесь первой жидкой или первой твердой фаз, и первую газовую фазу отделяют от первой жидкой или первой твердой фазы или от смеси первой жидкой и первой твердой фаз.
Первую газообразную фазу, состоящую, в основном, из аммиака и водяного пара, удаляют в основном непрерывно с помощью дистилляционного устройства, такого как дистилляционная колонна. Органические компоненты дистиллята, необязательно отделенные при этой дистилляции, - в подавляющем большинстве случаев речь идет о не прореагировавшем аминонитриле - могут быть возвращены полностью или частично в процесс на первой и/или на второй стадии. Время пребывания реакционной смеси на второй стадии не подлежит никаким ограничениям, однако в общем случае оно составляет приблизительно от 10 минут до 5 часов, предпочтительно приблизительно от 30 минут до 3 часов.
Продуктовый трубопровод между первой и второй стадией, при необходимости, содержит насадку, например кольца Рашига или смесительные элементы Зульцера, которые обеспечивают сброс давления реакционной смеси в газовую фазу. На стадии 3 первую жидкую или первую твердую фазу или смесь первой жидкой или первой твердой фаз с газообразной или жидкой фазой, содержащей воду, смешивают с водой или водяным паром. Количество добавленной воды (в виде жидкости), как правило, составляет приблизительно от 50 до 1500 мл, более предпочтительно от 100 до 500 мл, в каждом случае в пересчете на 1 кг первой жидкой или первой твердой фаз или смеси первой жидкой или первой твердой фаз. Указанная добавка воды позволяет компенсировать ее потери, происходящие в первую очередь на стадии 2, и способствует гидролизу кислотных амидных групп в реакционной смеси. Преимуществом изобретения, обусловленным такой добавкой воды, является то, что для применения смеси исходных продуктов, такой как смесь, используемая на первой стадии, требуется лишь небольшой избыток воды.
Содержащая воду газообразная или жидкая фаза, как правило, перед ее вводом на стадию 3 предварительно подогревается в теплообменнике и затем смешивается с первой жидкой или первой твердой фазой или смесью первой жидкой или первой твердой фаз. При этом в реакторе, при необходимости, могут быть предусмотрены смесительные элементы, способствующие перемешиванию компонентов.
Стадия 3 проводится при температуре от 150 до 370°С, преимущественно от 180 до 300°С, особенно предпочтительно при температуре от 200 до 280°С, и давлении от 0,1 до 30·106 Па, преимущественно, от 1 до 10·106 Па, особенно предпочтительно при давлении от 2 до 7·106 Па.
При этом давление и температура могут быть подобраны в соответствии друг с другом так, что реакционная смесь будет представлять собой однофазную жидкость или однофазное твердое тело. В другом варианте изобретения давление и температура выбираются так, что получаются одна жидкая или одна твердая фаза или смесь твердой или жидкой фаз и одна газообразная фаза. В этом варианте жидкая или твердая фаза или смесь жидкой и твердой фаз соответствует продуктсодержащей смеси, в то время как газообразную фазу отделяют. При этом в рамках данной стадии газообразная фаза либо может быть сразу отделена от жидкой или твердой фазы или от смеси твердой и жидкой фаз, либо образующаяся во время этой стадии реакционная смесь будет представлять собой двухфазную жидко-газообразную, твердо-газообразную или жидко/твердо-газообразную смесь.
При заранее выбранной температуре давление может быть выбрано так, что оно будет меньше равновесного давления паров аммиака, однако больше равновесного давления паров остальных компонентов в реакционной смеси при заданной температуре. Это, в частности, способствует отделению аммиака и тем самым ускоряет гидролиз кислотных амидных групп.
Применяемые на этой стадии аппараты/реакторы идентичны аппаратам и реакторам, применяемым на рассмотренной выше стадии 1.
Время пребывания на третьей стадии также не подлежит никаким ограничениям, однако из экономических соображений его в общем случае выбирают в пределах приблизительно от 10 минут до 10 часов, как правило, в пределах от 6 до 8 часов, особенно предпочтительно в пределах приблизительно от 60 минут до 6 часов.
Получаемая на стадии 3 продуктсодержащая смесь затем может быть подвергнута дальнейшей переработке, как описано ниже.
В предпочтительном варианте изобретения продуктсодержащая смесь со стадии 3 подвергается дополнительной конденсации на четвертой стадии при температуре приблизительно от 200 до 350°С, как правило, приблизительно от 220 до 300°С и, в частности, приблизительно от 240 до 270°С. Стадия 4 проводится при давлении, которое ниже давления на стадии 3 и предпочтительно находится в пределах приблизительно от 5 до 1000·103 Па, более предпочтительно приблизительно в пределах от 10 до 300·103 Па. В рамках этой стадии температуру и давление выбирают так, что получаются одна вторая газовая фаза и одна вторая жидкая или твердая фаза или смесь второй жидкой и второй твердой фаз, которые содержат полиамид.
Дополнительная конденсация, согласно стадии 4, проводится преимущественно так, что относительная вязкость полиамида (измеренная при температуре 25°С и концентрации 1 г полимера на 100 мл в 96%-ной (масс.) серной кислоте) имеет значение в пределах приблизительно от 1,6 до 3,5.
В предпочтительном варианте изобретения можно с помощью инертного газа, такого как, например, азот, отогнать из жидкой фазы необязательно присутствующую в ней воду.
Время пребывания реакционной смеси на стадии 4 определяется, в частности, заданной относительной вязкостью, температурой, давлением и количеством добавленной на стадии 3 воды.
Если стадия 3 проводится как однофазная, то в продуктовом трубопроводе между стадией 3 и стадией 4 могут быть необязательно использованы насадочные тела, состоящие, например, из колец Рашига или смесительных элементов Зульцера, которые обеспечивают контролируемый сброс давления реакционной смеси в газовой фазе.
Еще один вариант изобретения предусматривает отказ от стадии 3 и получение полиамида с помощью стадий (1), (2) и (4).
Предпочтительно этот вариант проводится следующим образом.
На стадии 1, по меньшей мере, один аминоалкилнитрил нагревают с избытком воды до температуры в пределах приблизительно от 220 до 270°С и при давлении приблизительно от 4 до 9·106 Па, причем давление и температуру согласовывают друг с другом так, что реакционная смесь представляет собой однофазную жидкость, и причем степень конверсии нитрильных групп составляет не меньше 95 мол.%, в пересчете на количество молей использованного аминоалкилнитрила, и получают реакционную смесь.
Реакционную смесь обрабатывают на стадии 2 так же, как описано выше, или при температуре в пределах приблизительно от 220 до 300°С и при давлении приблизительно от 1 до 7·106 Па, причем давление на второй стадии по меньшей мере на 0,5·106 Па ниже, чем давление на стадии 1. Одновременно отделяют образовавшуюся первую газовую фазу от первой жидкой фазы.
Полученную на стадии 2 первую жидкую фазу обрабатывают на стадии 4 так же, как на стадии 1, или при температуре в пределах приблизительно от 220 до 300°С и при давлении приблизительно в пределах от 10 до 300·103 Па, причем образовавшуюся при этом вторую, содержащую воду и аммиак, газовую фазу отделяют от второй жидкой фазы. На этой стадии относительную вязкость полученного полиамида (измеренную, как указано выше) устанавливают подбором температуры и времени пребывания на заданное значение в пределах приблизительно от 1,6 до 3,5.
Затем полученную таким образом вторую жидкую фазу выгружают обычными методами и, если это необходимо, подвергают переработке.
Вышеописанные способы, то есть последовательность стадий (1)-(3) или (1), (2) и (4), или (1)-(4), предпочтительно осуществляются периодически, то есть в одном реакторе последовательно во времени.
Еще один предпочтительный вариант настоящего изобретения предусматривает возможность возврата, по меньшей мере, одной из газовых фаз, полученных на соответствующих стадиях, в, по меньшей мере, одну из предшествующих стадий.
Предпочтительно, далее, выбирать на стадии 1 или на стадии 3 или как на стадии 1, так и на стадии 3 температуру и давление так, чтобы получались одна жидкая или одна твердая фаза или смесь жидкой и твердой фаз и газообразная фаза, и отделять газообразную фазу.
Далее, а рамках предлагаемого способа можно также осуществлять удлинение цепи или разветвление цепи, или комбинацию того и другого. С этой целью на отдельных стадиях процесса производится добавление известных специалисту веществ, используемых для разветвления, соответственно удлинения цепи полимеров. Предпочтительно эти вещества добавляют на стадиях 3 или 4.
В качестве используемых для этого соединений следует назвать:
Трифункциональные амины или карбоновые кислоты в качестве разветвителей цепи, соответственно сшивающих агентов. Примеры подходящих, по меньшей мере, трифункциональных аминов или карбоновых кислот описаны в европейский заявке на патент ЕР-А-0345648. По меньшей мере, трифункциональные амины имеют, по меньшей мере, три аминогруппы, способные к взаимодействию с карбоксильными группами. Они, как правило, не содержат карбоксильных групп. По меньшей мере, трифункциональные карбоновые кислоты имеют, по меньшей мере, три способные к взаимодействию с аминами карбоксильные группы, которые могут присутствовать, например, в форме их производных, таких как сложные эфиры. Карбоновые кислоты, как правило, не содержат аминогрупп, способных к реакции с карбоксильными группами. Примеры подходящих карбоновых кислот включают тримезиновую кислоту, тримеризованные жирные кислоты, которые могут быть получены, например, из масляной кислоты и могут содержать от 50 до 60 атомов углерода, нафталинполикарбоновые кислоты, такие как нафталин-1,3.5,7-тетракарбоновая кислота. Как правило, карбоновые кислоты представляют собой определенные органические соединения и не являются полимерными соединениями.
Примеры аминов с, по меньшей мере, 3 аминогруппами включают, например, нитрилотриалкиламин, в частности нитрилотриэтанамин, диалкилентриамины, в частности диэтилентриамин, триалкилентетрамины и тетраалкиленпентамины, причем алкиленовые остатки предпочтительно представляют собой этиленовые остатки. Далее, в качестве аминов могут использоваться дендримеры. Желательно, чтобы дендримеры имели общую формулу
(R2N-(CH2)n)2N-(CH2)x-N((CH2)n-NR2)2, (I)
в которой
R обозначает водород или -(CH2)n-NR
где R1 обозначает водород или -(CH2)n-NR
где R2 обозначает водород или -(CH2)n-NR
где R3 обозначает водород или -(CH2)n-NH2,
n имеет целочисленное значение от 2 до 6 и
х имеет целочисленное значение от 2 до 14.
Предпочтительно n имеет целочисленное значение 3 или 4, а х имеет целочисленное значение от 2 до 6, предпочтительно от 2 до 4, в частности 2. Остатки R независимо друг от друга могут также иметь указанное значение. Остаток R предпочтительно является водородом или остатком -(CH2)n-NH2.
Подходящими карбоновыми кислотами являются кислоты с 3-10 карбоксильными группами, предпочтительно с 3 или 4 карбоксильными группами. Предпочтительными карбоновыми кислотами являются кислоты с ароматическими и/или гетероциклическими ядрами. Примерами являются бензильный, нафтильный, антраценовый, бифенильный, трифенильный остатки или гетероциклы, такие как пиридин, бипиридин, пиррол, индол, фуран, тиофен, пурин, хинолин, фенантрен, порфирин, фталоцианин, нафталоцианин. Предпочтительны 3,5,3',5'-бифенилтетракарбоновая кислота-фталоцианин, нафталоцианин, 3,5,5',5'-бифенилтетракарбоновая кислота, 1,3,5,7-нафталинтет-ракарбоновая кислота, 2,4,6-пиридинтрикарбоновая кислота, 3,5,3',5'-бипи-ридинтетракарбоновая кислота, 3,5,3',5'-бензофенонтетракарбоновая кислота, 1,3,6,8-акридинтетракарбоновая кислота, особенно предпочтительны 1,3,5-бензолтрикарбоновая (тримезиновая) кислота и 1,2,4,5-бензолтетракарбоновая кислота. Соединения такого типа получаются технически или могут быть получены по способу, описанному в немецкой заявке на патент DE-A-4312182. При использовании орто-замещенных ароматических соединений образование имида предпочтительно предотвращается путем выбора подходящих температур реакции.
Эти вещества являются, по меньшей мере, трифункциональными, предпочтительно, по меньшей мере, тетрафункциональными. При этом количество функциональных групп может составлять от 3 до 16, предпочтительно от 4 до 10, особенно предпочтительно от 4 до 8. В предлагаемых способах применяются либо, по меньшей мере, трифункциональные амины, либо, по меньшей мере, трифункциональные карбоновые кислоты, но не смеси соответствующих аминов или соответствующих карбоновых кислот. Однако небольшие количества, по меньшей мере, трифункциональных аминов могут содержаться в трифункциональных карбоновых кислотах, и наоборот.
Указанные вещества присутствуют в количестве от 1 до 50 мкмоль/г полиамида, предпочтительно от 1 до 35, особенно предпочтительно от 1 до 20 мкмоль/г полиамида. Предпочтительно эти вещества содержатся в количестве от 3 до 150, особенно предпочтительно от 5 до 100, в частности от 10 до 70 мкмоль/г полиамида в пересчете на эквиваленты. Эквиваленты относятся при этом к количеству функциональных аминогрупп или карбоксильных групп.
Дифункциональные карбоновые кислоты или дифункциональные амины служат в качестве средств удлинения цепи. Они имеют 2 карбоксильные группы, которые могут взаимодействовать с аминогруппами, или соответственно 2 аминогруппы, которые могут взаимодействовать с карбоновыми кислотами. Кроме карбоксильных групп или аминогрупп, дифункциональные карбоновые кислоты или амины не содержат никаких других функциональных групп, которые могут реагировать с аминогруппами или соответственно с карбоксильными группами. Предпочтительно они вообще не содержат никаких других функциональных групп. Примеры пригодных дифункциональных аминов включают амины, которые образуют с дифункциональными карбоновыми кислотами соли. Они могут быть линейно-алифатическими, такими как алкилендиамин, содержащий от 1 до 14 атомов углерода, предпочтительно алкилендиамин, содержащий от 2 до 6 атомов углерода, например гексилендиамин. Они также могут быть циклоалифатическими. Примерами являются изофорондиамин, дицицикан, ларомин. Могут также применяться разветвленные алифатические диамины, примером которых является триме-тилгексаметилендиамин (ТМД) (который производится фирмой Huls AG). Кроме того, диамины могут быть ароматически-алифатическими, например может применяться п-ксилилендиамин. Все амины могут быть замещены в углеродном остове алкильными остатками, содержащими от 1 до 12, предпочтительно от 1 до 14 атомов углерода.
Дифункциональными карбоновыми кислотами являются, например, кислоты, которые образуют с дифункциональными диаминами соли. Это могут быть линейные алифатические дикарбоновые кислоты, предпочтительно дикарбоновые кислоты с 4-20 атомами углерода. Примеры таких алифатических кислот включают адипиновую, азелаиновую, себациновую, субериновую кислоты. Они могут быть также ароматическими кислотами. Примеры таких ароматических кислот включают изофталевую, терефталевую, нафталинди-карбоновую кислоты, а также димеризованные фумаровые кислоты.
Дифункциональные основные структурные элементы (с) применяются, как правило, в количествах от 1 до 55, особенно предпочтительно от 1 до 30, в частности от 1 до 15 мкмоль/г полиамида.
Полученную на стадии 3 продуктсодержащую смесь или вторую жидкую или вторую твердую фазу или смесь второй жидкой и второй твердой фаз (со стадии 4), содержащие полиамид, предпочтительно расплав полимера, согласно изобретению, выгружают из реактора обычными методами, например с помощью насоса. Затем полученный полиамид может быть подвергнут переработке известными методами, например методами, подробно описанными в немецкой заявке на патент DE-A 4321683.
Согласно предпочтительному варианту изобретения, дальнейшее снижение содержания циклического димера в полученном по изобретению полиамиде-6 возможно путем экстракции полиамида сначала водным раствором капролактама и затем водой и/или путем газофазной экстрации (например, как описано в европейской заявке на патент ЕР-А-0284968). Образующиеся во время этой дополнительной обработки низкомолекулярные компоненты, такие как капролактам и его линейные, а также циклические олигомеры, можно возвращать на первую и/или вторую, и/или третью стадию.
На всех стадиях процесса в исходную смесь и реакционную смесь могут быть добавлены регуляторы цепи, например алифатические и ароматические карбоновые и дикарбоновые кислоты, и катализаторы, например кислородсодержащие фосфорные соединения, в количестве от 0,01 до 5 мас.%, предпочтительно от 0,2 до 3 мас.%, в пересчете на количество использованных полиамидобразующих мономеров и аминонитрилов. Подходящими регуляторами цепи являются, например, пропионовая, уксусная, бензойная, терефталевая кислоты, а также триацетондиамин.
Добавки и наполнители, такие как пигменты, красители и стабилизаторы, вводятся в реакционную смесь, как правило, перед грануляцией, предпочтительно на второй, третьей и четвертой стадиях процесса. Особенно предпочтительно вводить добавки и наполнители в том случае, если реакционная, соответственно полимерная смесь в ходе дальнейшего процесса уже не подвергается превращению в присутствии катализаторов неподвижного слоя. В качестве добавок составы могут содержать от 0 до 40 мас.%, предпочтительно от 1 до 30 мас.%, в пересчете на весь состав, одного или несколько каучуков, модифицирующих ударную вязкость.
Например, могут применяться обычные модификаторы ударной вязкости, пригодные для полиамидов и/или полиариленэфиров.
Каучуки, повышающие вязкость полиамидов, обладают в общем случае двумя признаками: они содержат эластомерную часть, которая имеет температуру стеклования ниже -10°С, предпочтительно ниже -30°С, и они содержат, по меньшей мере, одну функциональную группу, которая может взаимодействовать с полиамидом. Подходящими функциональными группами являются, например, карбоксильная группа, кислотные ангидридная, эфирная, амидная, имидная группы, аминная, гидроксильная, эпоксидная, уретановая и оксазолиновая группы.
В качестве каучуков, повышающих вязкость смесей, необходимо назвать, например, следующие: ЕР-, соответственно EPDM-каучуки, привитые с вышеназванными функциональными группами. Подходящими прививочными агентами являются, например, малеиновый ангидрид, итаконовая кислота, акриловая кислота, глицидилакрилат и глицидилметакрилат.
Эти мономеры могут быть привиты на полимер в расплаве или в растворе, при необходимости, в присутствии радикального инициатора, такого как кумолгидроксипероксид.
Описанные под рубрикой “полимеры А” сополимеры α-олефинов, в том числе, в частности, этиленовые сополимеры, вместо их применения в качестве полимера А могут применяться в качестве каучуков и в качестве таковых могут быть подмешаны к составам по изобретению.
В качестве другой группы подходящих эластомеров следует назвать привитые каучуки со структурой ядро-оболочка. При этом речь идет о полученных в эмульсии привитых каучуках, состоящих из, по меньшей мере, одного твердого и одного мягкого компонента. Под твердым компонентом обычно понимается полимер с температурой стеклования, по меньшей мере, 25°С, под мягким компонентом - полимер с температурой стеклования максимально 0°С. Эти продукты имеют структуру, состоящую из ядра и, по меньшей мере, одной оболочки, причем эта структура получается в результате последовательных прибавлений мономеров. Мягкие компоненты являются производными в общем случае бутадиена, изопрена, алкилакрилатов, алкилметакрилатов или силоксанов и необязательно других сомономеров. Подходящие силоксановые ядра могут быть получены, например, из циклических олигомерных октаметилтетрасилоксана или тетравинилтетраметилтетрасилоксана. Последние могут быть, например, подвергнуты взаимодействию с γ-меркаптопропилметилдиметоксисиланом в присутствии сульфоновых кислот с получением мягких силоксановых ядер. Силоксаны могут быть также сшиты путем проведения реакции полимеризации в присутствии силанов с гидролизуемыми группами, такими как галоген, или алкоксигруппами, такими как тетраэтоксисилан, метилтриметоксисилан или фенилтриметоксисилан. В качестве пригодных сомономеров следует назвать, например, стирол, акрилонитрил и сшивающие или графт-активные мономеры с более чем одной способной к полимеризации двойной связью, такие как диаллилфталат, дивинилбензол, бутандиолдиакрилат или триаллил(изо)цианурат. Твердые компоненты являются в общем случае производными стирола, α-метилстирола и их сополимеров, причем в этом случае сомономерами являются предпочтительно акрилонитрил, метакрилонитрил и метилметакрилат.
Предпочтительные привитые каучуки со структурой ядро-оболочка содержат мягкое ядро и твердую оболочку или твердое ядро, первую мягкую оболочку и, по меньшей мере, еще одну твердую оболочку. Встройка функциональных групп, таких как карбонильная, карбоксильная группа, кислотные ангидридная, амидная, имидная группа, карбонокислотная эфирная группа, аминная, гидроксильная, эпоксидная, оксазолиновая, уретановая, мочевинная, лактамная или галогенбензильная группы, осуществляется в этом случае предпочтительно путем добавления соответствующим образом функционализованных мономеров при полимеризации последней оболочки. Подходящими функционализованными мономерами являются, например, малеиновая кислота, малеиновый ангидрид, моно- и диэфиры малеиновой кислоты, трет-бутил-(мет-)акрилат, акриловая кислота, глицидил(мет-)акрилат и винилоксазолин. Доля мономеров с функциональными группами составляет в общем случае от 0,1 до 25 мас.%, предпочтительно от 0,25 до 15 мас.%, в пересчете на общую массу привитого каучука со структурой ядро-оболочка. Массовое отношение мягкого компонента к твердому составляет в общем случае от 1:9 до 9:1, предпочтительно от 3:7 до 8:2.
Такие каучуки, которые повышают вязкость полиамидов, сами по себе известны и описаны, например, в европейской заявке на патент ЕР-А-0208187.
Другой группой подходящих модификаторов ударной вязкости являются термопластические сложные полиэфирные эластомеры. Под сложными полиэфирными эластомерами понимаются при этом сегментированные сополи-эфирэстеры, содержащие длинноцепные сегменты, производные, как правило, от поли(алкилен)эфиргликолей, и короткоцепные сегменты, производные от низкомолекулярных диолов и дикарбоновых кислот. Такие продукты известны и описаны в литературе, например в патенте США US 3651014. Соответствующие продукты также имеются в продаже под названиями Hytrel®(фирма Du Pont), Arnitel® (фирма Akzo), Pelprene® (фирма Toyobo Co. Ltd).
Само собой разумеется, могут применяться также смеси различных каучуков.
В качестве других добавок следует назвать, например, технологические добавки, стабилизаторы и замедлители окисления, средства против разложения под действием тепла и разложения под действием ультрафиолетового излучения, внутренние смазки и смазки для извлечения изделия из формы, огнезащитные средства, красители и пигменты и пластификаторы. Доля этих добавок составляет в общем случае до 40, предпочтительно до 15 мас.%, в пересчете на общую массу состава.
Пигменты и красители содержатся, как правило, в количествах до 4, предпочтительно от 0,5 до 3,5 и, в частности, от 0,5 до 3 мас.%.
Пигменты для окрашивания термопластов общеизвестны, см., например, R. Gachter und H. Muller, Taschenbuch der Kunststoffadditive, Carl Hanser Verlag, 1983, стр.494-510. Из двух наиболее распространенных кристаллических модификаций диоксида титана (рутил или анатаз) для окрашивания в белый цвет формовочных масс по изобретению, в основном, применяется модификация рутила.
Черными пигментами, которые могут быть использованы согласно изобретению, являются черный железоокисный пигмент (Fе2O3), черный шпинелевый пигмент (Cu(Cr,Fe)2O4), марганцевая черная (смесь из диоксида марганца, диоксида кремния и оксида железа), кобальтовая чернь и сурьмяный черный пигмент и особенно предпочтительно сажа, которая применяется большей частью в форме печной или газовой сажи (см. об этом G. Benzing, Pigmente fur Anstrichmittel, Expert-Verlag (1988), стр.78 и следующие).
Само собой разумеется, что в соответствии с настоящим изобретением для настройки определенных цветовых тонов могут применяться неорганические цветные пигменты, такие как хромоксидный зеленый, или органические цветные пигменты, такие как азопигменты и фталоцианины. Эти пигменты широко представлены в торговле.
Далее, может быть целесообразным применять указанные пигменты, соответственно красители в смеси, например сажу с фталоцианинами меди, так как в этом случае, как правило, облегчается диспергирование красителя в термопластах.
Замедлители окисления и стабилизаторы теплового разложения, которые могут добавляться к термопластическим массам, согласно изобретению, представляют собой, например, галогениды металлов группы 1 Периодической системы элементов, например галогениды натрия, калия, лития, необязательно в сочетании с галогенидами меди (I), например хлоридами, бромидами или иодидами. Галогениды, особенно галогениды меди, могут также содержать богатые электронами п-лиганды. В качестве примера таких медных комплексов следует назвать комплексы галогенида меди, например, с трифенилфосфином. Кроме того, могут быть использованы фторид и хлорид цинка. Далее, можно использовать стерически затрудненные фенолы, гидрохиноны, замещенные представители этой группы, вторичные ароматические амины, необязательно в комбинации с фосфорсодержащими кислотами, соответственно солями этих кислот, а также смеси этих соединений, предпочтительно в концентрации до 1 мас.% в пересчете на массу смеси.
Примеры УФ-стабилизаторов включат различные замещенные резорцины, салицилаты, бензотриазолы и бензофеноны, которые в общем случае применяются в количестве до 2 мас.%.
Внутренними смазками и смазками для извлечения изделия из формы, добавляемыми, как правило, в количестве до 1 мас.% от термопластической массы, являются стеариновая кислота, стеариновый спирт, алкильные эфиры и амиды стеариновой кислоты, а также эфиры пентаэритрита с длинноцепными жирными кислотами. Пригодны также кальциевые, цинковые и алюминиевые соли стеариновой кислоты, а также диалкилкетоны, например дистеарилкетон.
Далее настоящее изобретение относится к полиамиду, получаемому одним из вышеописанных способов.
Ниже изобретение подробнее поясняется на примерах.
Примеры:
Получение катализатора:
Общая методика
Катализатор 1: Бета-цеолитный порошок
В качестве катализатора применялся бета-цеолит фирмы Uetikon (Zeokat-Beta) следующего состава: SiO2 91%, Аl2О3 7,8%, Nа2О 0,5%, K2O 0,7%, БЭТ-поверхность 700 м2/г, размер пор в ангстремах  =7,6×6,7; 5,5×5,5, величина частиц 0,2-0,5 мм.
=7,6×6,7; 5,5×5,5, величина частиц 0,2-0,5 мм.
Катализатор 2: Бета-цеолитные прутки
220 г бета-цеолита из примера 1 уплотняли в смесителе с 5% Walocel® и 230 г воды в течение 45 минут. Затем массу подвергали прессованию в 2-миллиметровые прутки при давлении 70 бар. Прутки сушили при 110°С и в течение 16 часов кальцинировали при 500°С.
195 г этих прутков подвергали обмену с 3 л 20-процентного раствора NH4Cl при 80°С в течение 2 часов и затем промывали 10 литрами воды. После этого проводили второй обмен также с 3 л 20-процентного раствора NH4Cl при 80°С в течение 2 часов и продукт полностью отмывали от хлора. После сушки при 110°С проводили кальцинацию в течение 5 часов при 500°С.
Катализатор 3: слоистый силикат типа К10®
К10® представляет собой обработанный кислотой монтмориллонит фирмы Sad-Chemie. Он имеет БЭТ-поверхность 180-220 м2/г и ионообменный эквивалент, равный 40-50 мг-эквивалент/100 г.
Катализаторы 4 и 5: ТiO2-прутки из 100-процентного, соответственно 84-процентного анатаза
Катализаторы получали в соответствии с описанием, приведенным в работе: Erti, Knozinger, Weitkamp: "Handbook of heterogenous catalysis", VCH Weinheim, 1997; стр.98 и следующие. Указанные в этом описании в качестве особенно предпочтительных модификации смешивали с водой, силиказолем и глицерином, экструдировали и кальцинировали при 500°С.
Катализатор 6: Диоксиднотитано-оксидновольфрамовый катализатор
Применяемый катализатор получали путем тщательного перемешивания стандартного диоксида титана VKR 611 (фирма Schachtleben) с оксидом вольфрама и последующего формования полученной массы в прутки согласно примеру 2 или 4.
Катализатор имеет следующую спецификацию: 20 мас.% WO3, 80 мас.% TiO2; БЭТ-поверхность 73 м2/г, общая кислотность (pKs 6,8) 0,56 ммоль/г; общая кислотность (pKs 3) 0,035 ммоль/г.
Реакционные смеси, состоящие из ε-аминокапронитрила (АКН) и воды при молярном отношении компонентов от 1:1 до 1:10, после полного их перемешивания вводили выборочно с кислотой или без кислоты в 1-литровый автоклав.
В случае применения катализатора автоклав засыпали либо только гранулированным катализатором, либо слоем, состоящим из гранулированного катализатора и колец Рашига в качестве наполнителя. Применялись прутково-образные ТiO2-катализаторы с диаметром от 0,5 до 6 мм, длиной от 1 до 10 мм и удельной поверхностью приблизительно 50 м2/г.
Автоклав закрывали и вводили инертный газ - азот путем многократного повышения давления с последующей декомпрессией.
Чистота применяемого аминокапронитрила составляет 99%. Массовая доля кислот дана в пересчете на применяемое количество аминокапронитрила.
Приготовление образца и метод анализа:
Определяли так называемую относительную вязкость (RV) в качестве меры молекулярно-массовой структуры и степени полимеризации в 96%-ной серной кислоте в качестве растворителя при 25°С с помощью вискозиметра Уб-белоде. Концентрация полиамида составляет 1 г/100 мл для экстрагированного материала и 1,1 г/100 мл для неэкстрагированного материала. Неэкстрагированные полимеры перед анализом сушили в течение 20 часов в вакууме. Для экстракции гранулят перемешивали с водой с возвратом жидкого потока в реакционное пространство.
Примеры 1.1-1.8
Реакционную смесь (180 г) с отношением аминокапронитрила к воде (АКН:Н2O), равным 1:2 или 1:4, нагревали в реакторе в течение 75 минут до температуры 270°С, поддерживая давление 18 бар путем регулирования с помощью расширительного клапана. Гранулированный катализатор (400 г) полностью покрывал реакционную смесь. По истечении одного часа времени реакции, проводившейся при давлении 18 бар, давление в автоклаве в течение одного часа снижали до окружающего (1 бар) и автоклав опорожняли с непрерывным получением прутка. В качестве эталонного опыта (сравнительные примеры V 1.1 и V 1.6, см табл.1) служил вышеописанный способ, проводившийся аналогично без катализатора.

Пример 2.1
Реакционную смесь (180 г) с отношением аминокапронитрила к воде (АКН:Н2O), равным 1:1, нагревали в реакторе в течение 75 минут до температуры 270°С, поддерживая давление 18 бар путем регулирования с помощью расширительного клапана. Гранулированный катализатор (400 г) полностью покрывал реакционную смесь. По истечении трех часов времени реакции, проводившейся при давлении 18 бар, давление в автоклаве в течение одного часа снижали до окружающего (1 бар), реакционную смесь дополнительно конденсировали в течение трех часов при 270°С в потоке азота и затем выгружали в виде непрерывного прутка в водяную баню. В качестве эталонного опыта (сравнительный пример V 2.1, см. табл.2) служил вышеописанный способ, проводившийся аналогично без катализатора.

Примеры 3.1-3.2
Реакционную смесь (180 г) с отношением аминокапронитрила к воде (АКН:Н2O), равным 1:2,5, нагревали в реакторе в течение 75 минут до температуры 270°С, поддерживая давление 18 бар путем регулирования с помощью расширительного клапана. Гранулированный катализатор (400 г) полностью покрывал реакционную смесь. По истечении одного часа времени реакции, проводившейся при давлении 18 бар, давление в автоклаве в течение одного часа снижали до окружающего (1 бар), реакционную смесь дополнительно конденсировали в течение одного часа при 270°С в потоке азота и затем выгружали в водяную баню. В качестве эталонного опыта (сравнительный пример V 3.1, см. табл.3) служил вышеописанный способ, проводившийся аналогично без катализатора.

Пример 4.1
Реакционную смесь (180 г) с отношением аминокапронитрила к воде (АКН:H2O), равным 1:4, нагревали в реакторе в течение 75 минут до температуры 270°С, поддерживая давление 18 бар путем регулирования с помощью расширительного клапана. Гранулированный катализатор (400 г) полностью покрывал реакционную смесь. По истечении 45 минут времени реакции, проводившейся при давлении 18 бар, давление в автоклаве в течение 10 минут снижали до окружающего (1 бар) и автоклав затем опорожняли с непрерывным получением прутка. Относительная вязкость продукта обозначается как предконденсат (RV). Продукт затем на следующей стадии процесса подвергали термообработке в потоке азота при 160°С в течение 48 часов. Вязкость термообработанного продукта обозначается как RV (термообработанный продукт).
Пример 5.1
Реакционную смесь (180 г) с отношением аминокапронитрила к воде (АКН:Н2О), равным 1:4, нагревали в реакторе в течение 75 минут до температуры 270°С, поддерживая при этом давление 18 бар путем регулирования с помощью расширительного клапана. Гранулированный катализатор (400 г) полностью покрывал реакционную смесь. По достижении конечной температуры 270°С давление в автоклаве в течение 10 минут снижали до окружающего (1 бар) и автоклав затем опорожняли с непрерывным получением прутка. Относительная вязкость продукта обозначается как RV (предконденсат). Полученный продукт затем на следующей стадии процесса подвергали термообработке в потоке азота при 160°С в течение 48 часов. Вязкость термообработанного продукта обозначается как RV (термообработанный продукт).
Пример 6.1
Автоклав содержал насыпную смесь, состоящую из 100 г гранулированного катализатора и 340 г колец Рашига (диаметр и длина: 6 мм). 250 г реакционной смеси с отношением аминокапронитрила к воде (АКН:Н2O), равным 1:4, нагревали в реакторе в течение одного часа до температуры 270°С, поддерживая при этом давление 18 бар путем регулирования с помощью расширительного клапана. Гранулированный катализатор (400 г) полностью покрывал реакционную смесь. По достижении конечной температуры 270°С давление в автоклаве в течение 10 минут снижали до окружающего (1 бар) и автоклав затем опорожняли с непрерывным получением прутка. Относительная вязкость продукта обозначается как RV (предконденсат). Полученный продукт затем снова нагревали с водой (30 г воды на 100 г реакционной смеси) в автоклаве без катализатора до 260°С и дополнительно конденсировали в потоке азота при давлении окружающей среды в течение 3 часов. Вязкость дополнительно конденсированного продукта обозначается как RV (вторичный конденсат).
Для сравнительных примеров (V 4.1, V 5.1 и V 6.1, см. табл.4) вышеописанные способы 4.1, 5,1 и 6.1 проводились без катализатора. Так как взаимодействие реакционной смеси в отсутствие катализатора не приводит к получению твердого гранулированного продукта, то от последующей конденсации, соответственно температурной обработки отказались.

Пример 7
180 г указанных в таблице А исходных веществ в присутствии 400 г указанного в таблице А катализатора нагревают в автоклаве в течение 75 минут до температуры T1, указанной в таблице А. При этом устанавливается давление p1, указанное в таблице А. Реакцию проводят в течение указанного в таблице А времени Z1. Затем в течение 30 минут автоклав доводят до указанного в таблице А давления р2 с установлением температуры Т2. Параметры Т2 и p2 поддерживают в течение указанного в таблице А времени t2. Затем добавляют 30 г воды на 100 г продукта реакции и получаемую реакционную смесь поддерживают при указанных в таблице А температуре Т3 и давлении р3 в течение указанного в таблице А времени t3. Затем автоклав доводят до атмосферного давления и содержимое автоклава выпускают через сито (для отделения катализатора). Определяют относительную вязкость (RV) не экстрагировавшегося полиамида, которая также указана в таблице А.
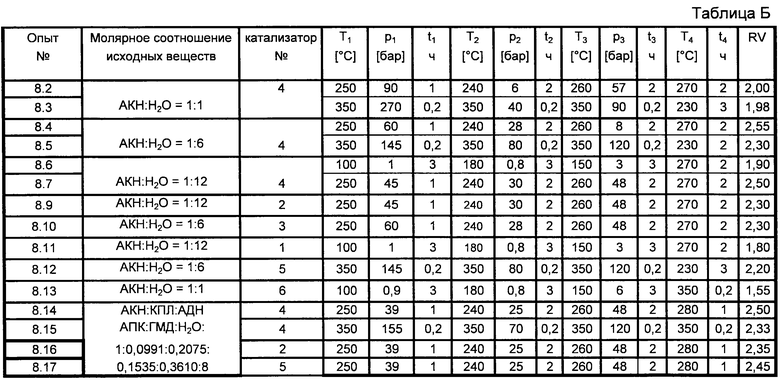
Пример 8
180 г указанных в таблице Б исходных веществ в присутствии 400 г указанного в таблице Б катализатора нагревают в автоклаве в течение 75 минут до температуры T1, указанной в таблице Б. При этом устанавливается давление p1, указанное в таблице Б. Реакцию проводят в течение указанного в таблице Б времени t1. Затем в течение 30 минут автоклав доводят до указанного в таблице Б давления р2 с установлением температуры T2. Параметры T2 и р2 поддерживают в течение указанного в таблице Б времени t2. Затем автоклав доводят до атмосферного давления и содержимое автоклава выпускают через сито (для отделения катализатора). Продукт реакции и 30 г воды на 100 г продукта реакции подают в автоклав и смесь нагревают до температуры Т3, указанной в таблице Б, в течение указанного в таблице Б времени t3. При этом устанавливается давление р3. Затем автоклав доводят до атмосферного давления. После дополнительной конденсации при указанной в таблице Б температуре Т4 в течение указанного в таблице Б времени t4 в атмосфере азота выпускают продукт реакции. Определяют относительную вязкость (RV) не экстрагировавшегося полиамида, которая также указана в таблице Б.
Пример 9
180 г указанных в таблице В исходных веществ в присутствии 400 г указанного в таблице В катализатора нагревают в автоклаве в течение 75 минут до температуры T1, указанной в таблице В. При этом устанавливается давление p1, указанное в таблице В. Реакцию проводят в течение указанного в таблице В времени t1. Затем в течение 30 минут автоклав доводят до указанного в таблице В давления р2 с установлением температуры Т2. Параметры Т2 и р2 поддерживают в течение указанного в таблице В времени t2. Затем автоклав доводят до атмосферного давления. После дополнительной конденсации при указанной в таблице В температуре Т3 в течение указанного в таблице В времени t3 содержимое автоклава выпускают через сито (для отделения катализатора). Определяют относительную вязкость (RV) не экстрагировавшегося полиамида, которая также указана в таблице В.
| название | год | авторы | номер документа |
|---|---|---|---|
| НЕПРЕРЫВНЫЙ СПОСОБ ПОЛУЧЕНИЯ ПОЛИАМИДА (ЕГО ВАРИАНТЫ) | 1999 |
|
RU2235737C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИАМИДОВ ИЗ СОЕДИНЕНИЙ АМИНОКАРБОНОВОЙ КИСЛОТЫ | 1999 |
|
RU2215754C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИАМИДА ИЗ АМИНОНИТРИЛА И ПОЛИАМИД, ПОЛУЧАЕМЫЙ ИМ | 1997 |
|
RU2214425C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИАМИДОВ | 1998 |
|
RU2221819C2 |
| СПОСОБ ПОЛУЧЕНИЯ АМИНОНИТРИЛА И ДИАМИНА | 1999 |
|
RU2210564C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИМЕРНЫХ СМЕСЕЙ ИЗ АМИНОНИТРИЛА И ТЕРМОПЛАСТИЧНЫХ ПОЛИМЕРОВ | 1999 |
|
RU2219194C2 |
| СПОСОБ ЦИКЛИЗИРУЮЩЕГО ГИДРОЛИЗА АМИНОНИТРИЛА В ЛАКТАМ | 1999 |
|
RU2199528C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЛАКТАМА | 1996 |
|
RU2167860C2 |
| СПОСОБ ЭКСТРАКЦИИ ПОЛИАМИДНЫХ ЧАСТИЦ И СПОСОБ ПОЛУЧЕНИЯ ПОЛИАМИДНЫХ ЧАСТИЦ | 1999 |
|
RU2224575C2 |
| ПОЛУЧЕНИЕ АМИНОНИТРИЛОВ (ВАРИАНТЫ) | 1999 |
|
RU2220132C2 |


Изобретение относится к периодическому способу получения полиамидов из аминонитрилов, которые обладают улучшенной способностью к термообработке. Способ включает три стадии. На первой стадии происходит взаимодействие аминонитрила или его смеси с мономерами и водой при температуре 90-400°С и давлении 1-350 бар с получением реакционной смеси. На второй стадии эта смесь взаимодействует при температуре от 150 до 400°С и давлении, более низком, чем давление на первой стадии, причем температуру и давление выбирают так, что получаются газовая и жидкая фазы, причем газовую фазу отделяют от жидкой фазы. На третьей стадии смешивают указанную жидкую фазу с газообразной или жидкой фазой, содержащей воду, при температуре от 150 до 370°С и давлении от 1 до 300 бар с получением продукта, причем первую стадию проводят в присутствии катализатора на основе кислоты Бренстеда, выбранного из группы, включающей бета-цеолитный катализатор, слоистый силикатный катализатор и неподвижный слой катализатора, состоящего в основном из диоксида титана, включающего 70-100 мас.% анатаза и 0-30 мас.% рутила, причем до 40 мас.% диоксида титана могут быть заменены оксидом вольфрама и вторая и третья стадии могут проводиться в присутствии этого катализатора. Изобретение позволяет полностью удалить применяемый катализатор из реакционной смеси. Способ получения полиамидов является эффективным и обеспечивает улучшенный гидролиз реагентов, в частности гидролиз кислотных амидных групп. 3 с. и 21 з.п. ф-лы, 7 табл.
(а)-(в) проводят удлинение цепи, или разветвление цепи, или комбинацию того и другого.
| DE 3534817 A1, 02.04.1987 | |||
| EP 0227866 A1, 08.07.1987 | |||
| US 4568736 A, 04.02.1986 | |||
| US 4629776 A, 16.12.1986 | |||
| US 5109104 A, 28.04.1992 | |||
| Способ заделки биологически вредных отходов в битум | 1972 |
|
SU479306A3 |
| Непрерывный способ получения полиамидов | 1974 |
|
SU620494A1 |

