Изобретение относится к имидазопиридинам, в частности к некоторым производным 4-замещенным-1-/2-метилимидазо [4,5-с]пирид-1-ил/-бензола и алкилбензола. Соединения обладают антагонистической активностью как в отношении гистамина (H1), так и тромбоцитактивирующего фактора и свойствами, которые позволяют их использовать в лечении аллергических воспалительных заболеваний как дыхательных путей, таких как аллергический ринит, синусит и астма, так и кожи, таких как атопический дерматит и крапивница.
Острые симптомы аллергического ринита, например чихание, выделения из носа и глаз, а также зуд, как правило, хорошо контролируются H1-антагонистами. Однако эти лекарственные средства оказывают слабое или не оказывают успокаивающего действия на застойные явления, причину как сосудорасширения, так и эдемы, приводящие к увеличению проницаемости сосудов. Кроме того, H1-антагонисты не оказывают влияния на аккумуляцию воспалившихся клеток, которые в свою очередь влияют как на замедленное проявление болезни, так и на гиперчувствительность к аллергену в случае хронического заболевания. Сильная эдемогенная активность ТАФ с его известным высвобождением и активацией многих типов воспалительных клеток свидетельствует о том, что антагонист ТАФ должен быть эффективен в отношении гистаминнезависимого застоя и воспалительных признаков аллергического ринита. Соединения предлагаемого изобретения являются так ТАФ-, так и H1-антагонистами и поэтому обладают способностью снижать интенсивность симптомов, практически большинства из симптомов, характерных для аллергического ринита.
Дополнительно следует отметить, что хотя гистамин способствует развитию бронхоспазма на аллерген при астме, он незначительно влияет на замедленный ответ на бронхоспазм или на неспецифическую гиперраздражительность бронхов, связанную с аккумуляцией воспалившихся клеток в нижних дыхательных путях. Включение ТАФ в этот ответ на воспаление наряду с его бронхоспазматической активностью поддерживает активность двойного ТАФ /H1-антагониста при лечении астмы. Аналогично следует ожидать, что двойной ТАФ /H1-антагонист по своему действию будет превосходить антигистаминные препараты, предназначенные для лечения аллергических кожных заболеваний, таких как атопический дерматит и крапивница, поскольку хотя антигистаминные препараты и снижают зуд и покраснение, они менее эффективны в отношении таких реакций как волдыри, связанных с наплывом воспалительных клеток. Соединения настоящего изобретения, по-видимому, будут иметь также значение в лечении других заболеваний: патофизиологические изменения, которые вызываются ими, связаны как с гистаминными, так и с независимыми от гистамина случаями воспалений.
В нашей Европейской патентной заявке N 0310386 мы раскрываем серию ТАФ-антагонистов на основе дигидропиридина, в которых заместитель в положении 2 включает, в частности, (2-метилимидазо[4,5-с]пирид-1-ил)фенильную группу. Патент США N 3326924 раскрывает некоторые аза-дибензоциклогептены в качестве антигистаминных препаратов, включающих 3-аза-5-(4-пиперидилиден)-10,11-дигидро-5H-дибензо[a, d]-циклогептен и его 4-аза-аналог (5,6-дигидро-11/4-пипередилиден)-11H- бензо[5,6] -циклогепта[1,2-b]пиридин, а также 7- и 8-хлоро и 7,8- и 9-метилпроизводные на его основе.
Согласно настоящему изобретению обеспечиваются соединения общей формулы: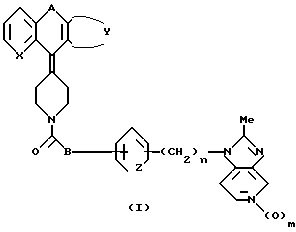
и фармацевтически приемлемые соли на их основе:
в формуле 1 - X это - CH или N; Z это CH=CH или S; А это - CH2CH2, CH= CH, CH/CH/CH2, или COCH2; B это - направленная связь или -CH2-, -CH(CH3)2- или CH(CH3)2-, или когда Z - это - CH=CH, то B может образовывать циклопентановое кольцо, сконденсированное с присоединенным бензольным кольцом; Y завершает сконденсированное кольцо, которое представлено следующими формулами:
где
R - H, галоид или C1 - C4 алкил; n равно 0, 1 или 2; m равно 0 или 1.
В приведенном выше определении термин галоид означает фтор, хлор, бром или иод; алкильные и алкоксильные группы, содержащие 3 или более атомов углерода могут быть как неразветвленными, так и содержать разветвленную углеродную цепочку, и связывающая группа A может быть прикреплена в любом плане, когда она асимметрична. Если соединения содержат центры асимметрии, то соединение может существовать в виде энантиометров и диастереомеров. Подобного рода изомеры могут быть разделены физическими методами, например, путем фракционной кристаллизации или хроматографическим путем как самих родственных соединений, так и их солей или производных на их основе. Изобретение включает все энантиомеры вне зависимости: разделены они или нет.
Фармацевтически приемлемыми солями присоединения соединений формулы 1, которые образуют эти соли, являются те соли, которые образованы из кислот, дающих нетоксичные кислые соли присоединения, например, такими солями могут быть гидрохлориды, гидробромиды, сульфаты или бисульфаты, фосфаты или кислые фосфаты, ацетаты, цитраты, фумараты, глюконаты, лактаты, малеаты, сукцинаты и соли винной кислоты - тартраты.
В предпочтительных вариантах настоящего изобретения X - N, Z это -CH=CH и Y завершает бензосконденсированное кольцо формулы: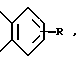
в которой,
в частности, R - H или Cl, B - предпочтительно направленная связь или CH2, n равно 0 и m равно 0. В частности, предпочтительны соединения, в которых R - Cl, B - направленная связь, A - CH2CH2, n равно 0 и m равно 0.
Особенно предпочтительно соединение 4-(8-хлор-5,6-дигидро-11H-бензо[5,6] -циклогепта[1,2-b] -пирид-11-илиден)- 1-[4-/2-метилимидазо-[4,5-c]-пирид-1-ил/-бензоил]пиперидин.
Соединения изобретения, в которых m равно 0, могут быть получены следующим путем, который включает реакцию производного пиперидина формулы II с кислотой (или активированным производным на основе этой кислоты) формулы III:
Реакция соединения формулы II и соединения формулы III осуществляется с использованием техники традиционного амидирования. Таким образом в одном процессе реакции осуществляется при взаимодействии реагентов, растворимых в органическом растворителе, например дихлорметане, с использованием диимидсочетающего средства, например 3-(диметиламинопропил)-1-этилкарбодиимида или N, N'-дициклогексилкарбодиимида, преимущественно, в присутствии 1-гидроксибензотриазоло и органического основания такого, как N-метилморфолин. Реакция обычно завершается за 2-24 ч и проводится при комнатной температуре, после чего продукт выделяют стандартными способами, а именно используя промывание водой или фильтрацию, для удаления побочного продукта - мочевины, а затем упаривают (отгоняют) растворитель. Далее продукт может быть очищен путем перекристаллизации или методом хроматографии, если в этом есть необходимость.
Возможно использование ряда последовательных реакций превращений. Так восстановление продукта, в котором A - COCH2, с использованием в качестве восстановителя, например, борогидрида натрия, дает соответствующее соединение, в котором A - CH/OH/CH2, N - оксиды (в которых m равно 1) получают окислением соответствующего имидазопиридина (m равно 0), используя, например, 3-хлорпербензойную кислоту.
Исходные материалы формулы II получают согласно литературным методам, описанным в работах Enhelhardt et al., J.Med.Chem. 1965, 8, 829; Waldvogel et al. , Helvetsca Chimica Acta, 1976, 59, 866 и в патенте США N 3326924. Исходные материалы формулы III получают согласно процедурам, описанным в Европейском патенте 03100386 и так, как это описано здесь ниже.
Активность соединений настоящего изобретения как антагонистом H1 продемонстрирована на их способности ингибировать гистамининдуцированный спазм трахеи морской свинки in vitro. Тестирование проводят следующим образом.
Вырезанные в виде спирали полоски трахеи морской свинки суспендировали при растяжении в ванночках для ткани, содержащих 15 мл непрерывно продуваемого газом (95% O2, 5% CO2) буфера Кребса (118 милимоль NaCl, 4,62 милимоль KCl, 1,16 милимоль MgSO4, 1,18 милимоль KH2PO4, 25 милимоль NaHCO3, 11 милимоль глюкозы, 2,5 милимоль CaCl2) и индометацин (2-микромоль). Вслед за период установления равновесия (45 мин) в ванночки добавляют дигидрохлорид гистамина в таком количестве, чтобы его конечная концентрация составляла бы 10-5M, и изометрически измеряют возникший спазм (сокращение). Через 30 мин после наступившего спазма (сокращения) ткань промывают буферным раствором до полного релаксирования в исходном состояние. Затем вновь вызывают сокращение добавлением гистамина (30 мин) и вновь промывают. Затем в третий раз проводят с помощью гистамина сокращение ткани, не без последующей промывки. Через 30 мин выдержанной концентрации в ванночки с тканью добавляют первую дозу тестируемого соединения и определяют его действие в течение 20 мин. Возрастающие кумулятивные дозы соединения добавляют каждые 20 мин и определяют количественно действие добавок тестируемого соединения. Значение ИК50 рассчитывают как концентрацию соединения, необходимую для того, чтобы вызвать релаксацию ткани на 50% после третьей контракции, вызванной введением гистамина.
Активность соединений настоящего изобретения как ТАФ-антагониста демонстрируется путем определения способности ингибировать тромбоцит, агрегирующего активность ТАФ in vitro. Тестирование осуществляют следующим образом.
Образцы крови кролика, помещенные в 0,1 объем (77 милимоль) динатрий этилендиамин тетрауксусной кислоты центрифугировали при 150 г в течение 15 мин для получения плазмы, обогащенной тромбоцитами. Затем эту плазму центрифугировали при 2000 г в течение 10 мин и получили сгусток тромбоцитов, который промыли буферным раствором (4 мМ KH2PO4, 6 мМ Na2HPO4, 100 мМ NaCl, 55 мМ глюкозы и 0,1% бычьего сывороточного альбумина, pH 7,25) и вновь ресуспендировали в буферном растворе до концентрации 2 • 108 тромбоцит/мл. Промытые тромбоциты предварительно инкубировали при перемешивании в течение 2 мин при 25oC в агрегометре в присутствии АДФ ловушки 1 мМ креатин фосфата (27 U/мл креатин фосфокинаты и 10 мМ MgCl2), хлорида кальция 1 мМ и либо одного наполнителя (диметилсульфоксид), либо наполнителя, содержащего одного из тестируемых соединений. C18-ТАФ добавляют в концентрации, достаточной для получения максимального агрегационного ответа в отсутствии тестируемого соединения (10-8 - 10-9 М) и затем измеряют агрегацию тромбоцитов по увеличению светопропускания раствора. Эксперимент повторяют в присутствии нескольких концентраций тестируемого соединения и концентрацию тестируемого соединения, которая потребовалась для снижения реакции на 50% от максимального значения записывают как значение ИК50.
Результаты представлены в табл.1.
Активность соединений формулы I как в отношении ТАФ, так и в отношении H1-антагониста демонстрируется in vivo по их способности ингибировать увеличение проницаемости кожных сосудов, индуцированной подкожными инъекциями ТАФ или гистамина мышам. Животные получали перорально или были инъецированы внутривенно тестируемым соединением. Через 45 мин (для перорального приема соединения) и через 10 мин (при инъекции) каждое животное однократно инъецировали подкожно либо гистамином (13,5 нанамоль), либо ТАФ (30 пикомоль) в спину непосредственно за головой. Сразу после инъецирования внутривенно ввели краситель голубой Эванса (250 мкл, 6,25 мг/мл). Через 30 мин животные умерщвлялись введением летальной дозы пентобарбитона, кожа со спины удалялась и области подкожного окрашивания вырезались (отштамповывались). Степень окрашивания вырезанных штампов определяли путем экстрагирования красителя из штампов формамидом в течение 24 ч при 70oC с последующим определением поглощения на длине волны 620 нм в спектрофотометре. ИД50 рассчитывали как концентрацию соединения, которое снижает поглощение (окрашивание) на 50% в сравнении с контрольным необработанным вариантом (т.е. вариантом, когда животное не получало тестируемое соединение). Ряд соединений также испытывались орально на мышах на проницаемость кожных сосудов против PAF и гистамина. Результаты представлены в табл.2.
В случае терапевтического применения соединения формулы I, как правило вводят в смеси с фармацевтически приемлемым носителем, выбранным в зависимости от способа введения препарата и стандартно используемым в фармацевтической практике. Например, соединения настоящего изобретения могут вводиться в виде таблеток, содержащих в качестве наполнителя крахмал или лактозу, или в капсулах или шариках либо без наполнителя, либо вместе с наполнителем, или в виде эликсиров или суспензий, содержащих отдушки или окрашивающие компоненты. Они также могут инъецироваться, например, внутривенно, внутримышечно или подкожно. В случае парентерального введения препараты, содержащие соединения настоящего изобретения, лучше всего готовить в виде стерильного водного раствора, который может содержать другие вещества, например, достаточное количество соли или глюкозы для того, чтобы вводимый раствор был изотоничен по отношению к крови.
Для введения препарата больному в период лечения или на этапе профилактики аллергических и воспалительных болезней, дозы, вводимые перорально, обычно находятся в диапазоне 2 - 1000 мг ежедневно для пациента, средняя масса которого составляет 70 кг. Таким образом в случае типичного взрослого больного каждая таблетка или капсула будет содержать 1 - 500 мг активного соединения в подходящем фармацевтически приемлемом носителе или наполнителе. Характерные дозировки активного соединения при внутривенном введении составляет 1 - 10 мг на одноразовую дозу. При лечении аллергической астмы и ринита предпочтительным способом введения препарата может быть введение в нос или ингаляции с помощью ингалятора или аэрозоля. При таком способе введения доза может варьироваться в пределах 0,1 - 50 мг для одноразового приема. Фактически необходимую дозу врач устанавливает на практике в зависимости от индивидуальных особенностей больного, его возраста, веса и реакции на препарат. Вышеприведенные дозы являются усредненными примерами и, конечно, они могут варьироваться в каждом конкретном случае, т.е. быть и выше, и ниже указанных, и все они попадают в рамки настоящего изобретения.
Следовательно в качестве еще одного аспекта настоящее изобретение обеспечивает фармацевтический состав, содержащий соединение формулы I и фармацевтически приемлемый носитель или разбавитель.
Изобретение также охватывает соединение формулы I или фармацевтически приемлемую соль на основе этого соединения, для использования в медицине, в частности, при лечении аллергических и воспалительных заболеваний человека.
Далее изобретение будет проиллюстрировано примерами получения соединений формулы 1. Чистоту соединений обычно проверяли методами тонкослойной хроматографии, используя Кизельгель 60 фирмы Merck, F254 пластины. 1H-ЯМР спектры были записаны на спектрометрах Nicolet QE-300 или Brucker AC-300 и во всех случаях они совпали с предложенными структурами.
Значения химических сдвигов приведены в миллионных долях от тетраметилсилана, для обозначения основных пиков использованы стандартные скрещения: с - синглет, д - дублет, т - триплет, м - мультиплет и уш. - уширение.
Пример 1. 4-(8-Хлоро-5,6-дигидро-11H-бензо[5,6]циклогепта[1,2-b]- пирид-11-илиден/-1-[4-/2-метилимидазо[4,5-c]пирид-1-ил/- бензоил]пиперидин гемигидрат.
Раствор 4-(8-хлоро-5,6-дигидро-11H-бензо[5,6]-циклогепта [1,2-b]пирид-11-илиден)пиперидина (224 мг, 0,75 ммоль) и 4-(2-метилимидазо[4,5-c]пирид-1-ил)бензойной кислоты (0,75 ммоль, 190 мг) в дихлорметане (12 мл) последовательно обработали 1-гидроксибензотриазолгидратом (102 мг, 0,75 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорида (290 мг, 1,5 ммоль) и 4-метилморфолином (165 микролитр, 1,5 ммоль). Смесь перемешивали при комнатной температуре 16 ч, промыли водой, высушили над сульфатом магния и упарили. Остаток очистили, использовав хроматографирование на силикагеле, и в качестве элюента применяли смесь дихлорметана плюс 3 - 4% метанола. Соответствующие фракции объединили и отогнали растворитель, в результате получили озаглавленное соединение (113 мг, 27%) в виде бесцветной пены, которое было охарактеризовано как гемигидрат.
Найдено, %: C 71,1; H 5,2; N 12,3; C33H28ClN5•0,5 H2O.
Рассчитано, %: C 71,4; H 5,3; N 12,6.
Примеры 2 - 6. Примеры следующих соединений были получены при взаимодействии 4-(8-хлоро-5,6-дигидро-11H-бензо[5,6]циклогепта- [1,2-b]пирид-11-илиден)-пиперидина с соответствующей кислотой, используя метод, описанный в примере 1, и были охарактеризованы как показано в табл. 3.
Пример 7. 4-(5H-Дибензо[a,d]циклогептен-5-илиден)-1-[4- (2-метилимидазо[4,5-c]пирид-1-ил)бензоил]пиперидин гемигидрат.
Это соединение было получено так, как описано в примере 1, но и с использованием в качестве исходного реагента 4-(5H-дибензо[a,d]циклогептен-5-илиден)пиперидин гидрохлорида (J. Med. Chem. 1965, 8, 829) вместо 4-(8-хлоро-5,6-дигидро-11H- бензо[5,6]циклогепта[1,2-b]-пиридин-11-илиден)пиперидин. Озаглавленное соединение получили в виде пены бесцветной и охарактеризовали как гемигидрат.
Найдено, %: C 78,9; H 5,6; N 11,0. C33H28N4O • 0,5H2O.
Рассчитано, %: C 78,9; H 5,8; N 10,8.
Пример 8. 1-[4-(2-Метилимидазо[4,5-c]пирид-1-ил/бензоил]-4-) 10-оксо-9,10-дигидро-4H-бензо[4,5]циклогепта[1,2-b]тиофен- 4-илиден)пиперидин.
Это соединение было получено так, как описано в примере 1, с использованием в качестве исходного материала 4-(10-оксо-9,10- дигидро-4H-бензо[4,5] циклогепта[1,2-b] тиофен-4-илиден)-пиперидина) (Helvetica Acta, 1976, 59, 866) вместо 4-(8-хлоро-5,6-дигидро-11H- бензо[5,6]циклогепта[1,2-b]-пирид-11-илиден)пиперидина. Озаглавленное соединение получено в виде пены и охарактеризовано как моногидрат.
Найдено, %: C 70,1; H 5,1; N 10,1. C32H26N4O2 • H2O.
Рассчитано, %: C 70,0; H 5,1; N 10,2.
Пример 9. 4-(8-Хлоро-5,6-дигидро-11H- бензо[5,6]циклогепта[1,2-b]-пирид-11-илиден)-1- [4-(2-метилимидазо[4,5-c] пирид-1-ил-5-оксид)-бензол] пиперидин.
Раствор 3-хлоропербензойной кислоты (50%: 126 мг, 0,37 ммоль) в дихлорметане (3 мл) по каплям добавляли в течение 10 мин к перемешиваемому, охлажденному льдом раствору 4-(8-хлоро-5,6-дигидро- 11H-бензо[5,6]циклогепта[1,2-b] пиридин-11-илиден)-1-[4- (2-метилимидазо[4,5-c]пирид-1-ил)бензоил] пиперидин гемигидрата (200 мг, 0,37 ммоль)) (пример 1). Смесь перемешивали при 0oC в течение 19 ч, обработали еще одной порцией 3-хлоропербензойной кислоты (25 мг), перемешивали при охлаждении льдом еще 25 ч, промыли насыщенным водным раствором гидрокарбоната натрия и водой, высушили над сульфатом магния и упарили. Остаток прохроматографировали на силикагеле, использовав смесь дихлорметан плюс 5% метанола плюс 0,1 - 0,5% насыщенного водного раствора аммиака в качестве элюента. Соответствующие фракции собирали и отогнали растворитель, получив в результате озаглавленное соединение (44 мг, 21%) в виде бесцветного стекла, т. пл. 176 - 180oC, которое охарактеризовано ЯМР/1H-ЯМР спектром.
1H-ЯМР (CDCl3/ δ = 9,03/1H, с/; 8,39 /1Н, д, J = 8 Гц/, 8,04 /1H, с/, 7,64 /2H, д, J = 8 Гц/, 7,37 /2H, д, J = 8 Гц/, 6,95 - 7,25 /6H, м/, 3,25 - 4,15 /4H, м/, 1,7 - 3,0 /8H, м/.
Пример 10. 4-(10-Гидрокси-9,10-4H-бензо[4,5] циклогепта[1,2-b] тиофен- 4-илиден)-1-[4-/2-метилимидазо[4,5-c]-пирид-1-ил)бензоил]-пиперидин.
Раствор 1-[4-(2-метилимидазо[4,5-c] пирид-1-ил)бензоил] -4- (10-оксо-9,10-дигидро-4H-бензо[4,5] циклогепта[1,2-b]тиофен- 4-илиден)пиперидина (200 мг, 0,38 моль) (пример 8) и борогидрида натрия (200 мг) в метаноле (20 мл) перемешивали при комнатной температуре и упарили. Остаток разделили между дихлорметаном и водой и органический слой промыли водой, высушили над безводным сульфатом магния и упарили. Остаток растерли с гексаном, и в результате получили озаглавленное соединение (70 мг, 35%) в виде бесцветной пены, которое охарактеризовано как дигидрат.
Найдено,%: C 67,9; H 5,4; N 9,6. C32H28N4O2S • 2H2O.
Рассчитано, %: C 67,5; H 5,6; N 9,9.
Пример 11. 4-(8-Хлоро-5,6-дигидро-6-оксо-11H- бензо[5,6] циклогепта[1,2-b] -пирид-11-илиден)-1-[4- (2-метилимидазо[4,5-c]пирид-1-ил)бензоил] пиперидин.
Это соединение получили так, как описано в примере 1, использовав в качестве исходного регента 4-(8-хлоро-5,6-дигидро-6-оксо- 11H-бензо[5,6]-циклогепта[1,2-b]пирид-11-илиден)пиперидин (J. Org. Chem., 1990, 55, 3341) вместо 4-(8-хлоро-5,6-дигидро-11H-бензо[5,6] - циклогепта[1,2-b] пирид-11-илиден)пиперидина. Озаглавленное соединение было получено в виде бесцветной смолы и охарактеризовано, как содержащее 1,5 эквивалента воды.
Найдено, %: C 67,2; H 5,2; N 11,4. C33H26ClN5O2 • 1,5 H2O.
Рассчитано, %; C 67,0; H 4,9; N 11,9.
Пример 12. 4-(10-11-Дигидро-5H-дибензо[a, d]циклогептен- 5-илиден)-1-[4-(2-метилимидазо[4,5-c]пирид-1-ил)бензоил]пиперидин.
Это соединение было получено так, как описано в примере 1, с использованием в качестве исходного реагента 4-(10,11-дигидро-5H-дибензо[a,d]циклогептен-5-илиден)пиперидина (Европейский патент ЕР-А-034723, 1989) вместо 4-(8-хлоро-5,6-дигидро-11H-бензо[5,6] циклогепта[1,2-b] пиридин- 11-илиден)пиперидина. Озаглавленное соединение получили в виде бесцветной пены (248 мг, 48%) и охарактеризовали как гемигидрат.
Найдено, %: C 78,4; H 5,9; N 10,7. C34H30N4 • 0,5 H2O.
Рассчитано, %: C 78,5; H 6,0; N 10,8.
Пример 13. 4-(8-Хлоро-11H-бензо[5,6] циклогепта[1,2-пирид-11-илиден)- 1-[4-(2-метилимидазо[4,5-c]пирид-1-ил)бензоил]-пиперидин.
Это соединение было получено так, как описано в примере 1, с использованием в качестве исходного реагента 4-(8-хлоро-11H-бензо [5,6]циклогепта[1,2-b] пирид-11-илиден)пиперидина (WO 88/03138), 1988, вместо 4-(8-хлоро-5,6-дигидро-11H-бензо[5,6] циклогепта[1,2-b] пирид- 11-илиден)пиперидина. Озаглавленное соединение получили в виде бесцветной смолы, Rf 0,25 (силикагель, растворитель: CH2Cl2, CH3OH, NH4OH - 98:7:1). Соединение охарактеризовано полученным масс-спектром: М/е. М+ = 543 (C33H26ClN5O).
Пример 14. 4-(5,6-Дигидро-11H-бензо[5,6] циклогепта[1,2-b]пирид- 11-илиден)-1-[4-(2-метилимидазо[4,5-c]пирид-1-ил)бензоил]пиперидин.
Это соединение получили так, как описано в примере 1, используя в качестве исходного реагента 4-(5,6-дигидро-11H- бензо[5,6]циклогепта[1,2-b]пирид-11-илиден)пиперидин (J. Med. Chem. , 1972, 15, 750) вместо 4-(8-хлоро-5,6-дигидро-11H- бензо[5,6] циклогепта[1,2-b] пирид-11-илиден)пиперидина. Озаглавленное соединение получили в виде светло-коричневого твердого продукта с т. пл. 243-245oC, которое было охарактеризовано, как содержащее 0,67 эквивалента воды.
Найдено, %: C 75,5; H 5,9; N 13,4. C33H29N5O.
Рассчитано, %: C 75,7; H 5,8; N 13,4.
Пример 15. 4-(8-Хлоро-5,6-дигидро-6-гидрокси- 11H-бензо[5,6]циклогепта[1,2-b] пирид-11-илиден)-1-[4- (2-метилимидазо[4,5-c] пирид-1-ил)бензоил] пиперидин.
Это соединение было получено так, как описано в примере 10, путем восстановления борогидридом натрия 4-(8-хлоро-5,6-дигидро-6-оксо-11H-бензо[5,6] циклогепта[1,2-b] пирид- 11-илиден)-1-[4-(2-метилимидазо[4,5-c] пирид-1-ил)бензоил]пиперидина (пример 11). Озаглавленное соединение получали в виде бесцветной смолы, Rf 0,30 (силикагель, растворитель: CH2Cl2, CH3OH, NH4OH = 93 : 7 : 1). Соединение охарактеризовано его масс-спектром: M/e, M+ = 561 (C33H28ClN5O2).
Пример 16. 4-(8-Хлоро-5,6-дигидро-11H- бензо[5,6]циклогепта[1,2,-b]пирид-11-илиден)-1- [4-(2-метилимидазо[4,5-c]пирид-1-ил)-тиен-2-оил]пиперидин.
Это соединение получено так, как описано в примере 1, с использованием в качестве исходного реагента 5-(2-метилимидазо- [4,5-c]пирид-1-ил)тиофен-2-карбоновой кислоты (препаративный пример 8, 185 мг) вместо 4-(2-метилимидазо-[4,5-c] пирид-1-ил)бензойной кислоты. Озаглавленное соединение получили в виде желтовато-коричневой пены (121 мг, 38,5%), которое охарактеризовали как полученный гидрат.
Найдено, %: C 64,3; H 4,8; N 12,0. C31H26ClN5O • 1,5H2O.
Рассчитано, %: C 64,3; H 5,0; N 12,1.
Пример 17. 4-(8-Метил-5,6-дигидро-11H- бензо[5,6]циклогепта[1,2-b]пирид-11-илиден)-1-[4- (2-метилимидазо[4,5-c]пирид-1-ил)бензоил]пиперидин.
Это соединение получено так, как описано в примере 1, с использованием в качестве исходного реагента 4-(8-метил-5,6- дигидро-11H-бензо[5,6]циклогепта[1,2-b] пирид-11-илиден)пиперидина (J. Med. Chem., 1991, 34, 457-461 (300 мг) вместо 4-(8-хлоро-5,6-дигидро-11H-бензо[5,6] циклогепта[1,2-b] пирид- 11-илиден)пиперидина. Озаглавленное соединение получили в виде стекла (270 мг. 49,6%), которое охарактеризовали, как содержащее 0,25 моль дихлорметана.
Найдено, %: C 74,8; H 6,0; N 12,7. N34H31N5O • 0,25CH2Cl2.
Рассчитано, %: C 75,2; H 5,8; N 12,8.
Пример 18. 4-(8-Хлоро-5,6-дигидро-5-оксо-11H-бензо[5,6] - циклогепта[1,2-b] пирид-11-илиден)-1-[4-(2-метилимидазо[4,5-c] пирид- 1-ил)бензоил] пиперидин.
Это соединение было получено так, как описано в примере 1, с использованием в качестве исходного реагента 4-(8-хлоро-5,6-дигидро- 5-оксо-11H-бензо[5,6] циклогепта[1,2-b] пирид-11-илиден)пиперидина (J. Org. Chem., 1990, 55, 3341; 80 мг) вместо 4-(8-хлоро-5,6-дигидро-11H- бензо[5,6]циклогепта[1,2-b] пирид-11-илиден)пиперидина. Озаглавленное соединение получили в виде желтовато-коричневой пены (60 мг, 43,5%), Rf 0,08 (силикагель, растворитель: CH3CO2C2H5, CH3OH, NH4OH = 80 : 20 : 1), м/е, M+ = 559 (C33H26ClN5O2).
Пример 19. 4-(8-Хлоро-5,6-дигидро-5-гидрокси-11H- бензо[5,6]циклогепта[1,2-b] пирид-11-илиден)-1-[4- (2-метилимидазо[4,5-c] пирид-1-ил)бензоил] пиперидин.
Это соединение получено так, как описано в примере 10, путем восстановления боргидридом натрия 4-(8-хлоро-5,6-дигидро-5- оксо-11H-бензо[5,6] циклогепта[1,2-b] пирид-11-илиден)-1-[4- (2-метилимидазо[4,5-c] пирид-1-ил)бензоил] пиперидина (из примера 18, 40 мг). Озаглавленное соединение было получено в виде пены желтого цвета (35 мг, 87%). Оно было охарактеризовано как гидрат, содержащий 0,33 эквивалента этилацетата.
Найдено, %: C 67,7; H 5,2; N 11,4. C33H28ClN5O2 • H2O • 0,33 C4H8O2.
Рассчитано, %: C 67,7; H 5,1; N 11,5.
Препаративный пример 1. 4-(2-Метилимидазо[4,5,-c]пирид-1-ил)бензойная кислота.
Смесь 4-(2-метилимидазо[4,5-c]пирид-1-ил)бензонитрил (EP-0310386) (12,0 мг, 51,3 ммоль) и гидроксида натрия (22,0 г, 0,55 ммоль) в смеси этанола (55 мл) и воды (55 мл) нагревали в токе азота при кипении 1,5 ч, охладили и сконцентрировали при пониженном давлении. Остаток коричневого цвета растворили в ледяной воде и добавили 33 мл ледяной уксусной кислоты. Выпавший осадок собрали, промыли водой и высушили в вакууме при 70oC. В результате получили озаглавленное соединение (9,1 г, 70%) в виде твердого продукта светло-желтого цвета, которое охарактеризовали 1H-ЯМР спектром.
ЯМР-спектр /DMCO-d6/ δ : 2,50 /3H, с/, 7,25 /1H, д, J = 5 Гц/, 7,72 /2H, д, J = 8 Гц/ 8,16 /2H, д, J = 8 Гц/, 8,92 /1H, с/.
Препаративный пример 2. Этил 4-/2-метилимидазо[4,5-c] пирид- 1-ил/фенилацетат.
a) Этил 4-аминофенилацетат (17,7 г, 0,1 моль) и гидрокарбонат натрия (8,4 г, 0,1 моль) перемешивали в этаноле (200 мл). 4-Хлоро-3-нитропиридин (15,9 г, 0,1 моль) в виде раствора в этаноле (50 мл) добавили в первому раствору и перемешивали при комнатной температуре в течение 3 ч. Затем смесь упаривали до небольшого объема и вылили в этилацетат (500 мл) и раствор промыли водой (200 мл). Органическую фазу проэкстрагировтали 0,5 M хлористоводородной кислотой и объединенные водные экстракты подщелочили добавлением 2 M гидроксида натрия, а затем проэкстрагирвровали дихлорметаном. Объединенные органические экстракты высушивали над сульфатом натрия, отфильтровали и упарили до сухости. Остаток перекристаллизовали из водного раствора этанола. В результате получили этил 4-(3-нитропирид-4-иламино)-фенилацетат (7,32 мг) с т. пл. 124 - 126oC. Из маточного раствора дополнительно выделили 8,56 г этого вещества.
b) 15,7 г Вышеуказанного продукта прогидрировали под давлением 60 фунт/дюйм2 (4,1 бар или 4,1•105 н/м2/ над катализатором - 5%-ный палладий, нанесенный на углеродную подложку, в течение 3 ч при комнатной температуре. После фильтрации и упаривания растворителя получили этил 4-(3-аминопирид-4-аламино)фенилацетат (14,1 г).
c) Этил 4-(3-аминопирид-4-иламино)фенилацетат (14,1 г, 52 ммоль), уксусную кислоту (100 мл) и уксусный ангидрид (100 мл) перемешали и нагревали при кипении в атмосфере азота 1,5 ч. Охлажденный раствор упарили до сухости и подщелочили добавлением 10%-ного водного раствора гидрокарбоната натрия, затем проэкстрагировали дихлорметаном. Объединенные органические слои упарили до сухости и очистили хроматографированием на силикагеле, использовав в качестве элюента смесь дихлорметана и этанола. В результате получили этил 4-(2-метилимидазо [4,5-c]пирид-1-ил)фенилацетата (13,6 г).
Препаративный пример 3. 4-(2-Метилимидазо[4,5-c]пирид-1- ил)фенилуксусная кислота.
Раствор этил 4-(2-метилимидазо[4,5-c] пирид-1-ил)фенилацетата (750 мг, 2,54 ммоль) и гидроксида натрия (160 мг, 4,0 ммоль) в смеси этанола (5 мл) и воды (5 мл) перемешивали при комнатной температуре 16 ч и после этого упарили. Остаток растворили в воде, подкислили 2 M хлористоводородной кислотой до pH 4 - 5 и проэкстрагировали 1-бутанолом. Объединенные 1-бутанольные экстракты упарили и остаток растерли с диэтиловыми эфиром. Полученный твердый продукт собрали, промыли гексаном и высушили, получив в результате озаглавленное соединение (267 мг, 39%) в виде желтовато-коричневого твердого продукта с т. пл. 226 - 230oC. Соединение охарактеризовали 1H-ЯМР спектром.
ЯМР-спектр (DMCO -d6/ δ : 8,86 /1H, с/, 8,23 /1H, д, J = 8 Гц/, 7,48 /4H, с/, 7,15 /1H, д, J = 8 Гц/, 3,70 /2H, с/, 2,42 /3H, с).
Препаративный пример 4. 4-[(2-Метилимидазо[4,5,-с]пирид-1-ил)метил]бензойная кислота.
Этил 4-[(2-метилимидазо[4,5, -с] пирид-1-ил)метил]бензоат гидролизовали согласно процедуре, описанной в препаративном примере 3, приведенном выше, в результате получили озаглавленное соединение в виде бесцветного твердого продукта, которое использовали непосредственно в примере 3.
Препаративный пример 5. 5-(2-Метилимидазо[4,5-с]пирид-1-ил)индан-2-карбоновая кислота.
a) Дымящую азотную кислоту (40 мл, 1,5 г/мл) по каплям добавляли к перемешиваемому уксусному ангидриду (80 мл), поддерживая температуру равную 0oC. После завершения прикапывания к полученному раствору при перемешивании в течение 30 мин по каплям добавляли 2-цианоиндан, поддерживая температуру в интервале -5 - 0oC. Смесь перемешивали еще 15 мин, затем вылили в лед. Смесь проэкстрагировали дихлорметаном (4•150 мл) и промыли экстракты насыщенным водным раствором бикарбоната натрия (3•150 мл), высушили над сульфатом магния и растворитель отогнали при пониженном давлении. Остаток перекристаллизовали из этанола и получили 2-циано-5-нитроиндан (12,09 г, 81%).
b) Раствор 2-циано-5-нитроиндана (11,90 г, 63,3 ммоль) в смеси метанол/дихлорметан - 1: 1 (200 мл) прогидрировали над катализатором - 10%-ным палладием на углеродной подложке (1,2 г) при давлении 30 фунтов на кв. дюйм (2,05•105 н/м3) и 20oC в течение 5 ч. Катализатор отфильтровывали и фильтрат сконцентрировали при пониженном давлении, получив в результате 5-амино-2-цианоиндан (10,4 г), который непосредственно использовали в следующей реакции. Часть перекристаллизовали из этанола и получили иглы розового цвета. Т. пл. 73 - 76oC.
c) К суспензии 5-амино-2-цианоиндана (10,4 г, 65,7 ммоль) в этаноле (150 мл) добавили при комнатной температуре 4-хлоро-3-нитропиридин (11,46 г, 72,3 ммоль). Смесь перемешивали в течение ночи при комнатной температуре и затем вылили в переохлажденный льдом водный раствор аммиака. Желтый осадок отфильтровали, частично переработали в горячем этаноле (150 мл), охладили, вновь отфильтровали и получили 2-циано-5-(3-нитропирид-4-иламино)индан (13,61 г, 74%).
d) 2-Циано-5-(3-нитропирид-4-иламино)индан (12,46 г, 44,5 ммоль) суспендировали в смеси метанол/дихлорметан = 1 : 1 (750 мл) и прогидрировали при 20oC и давлении 30 фунтов/дюйм2 (2,05 н/м2) над катализатором - 10%-ным палладием, нанесенным на углеродную подложку. Гидрирование осуществляли в течение 2 ч. Катализатор отфильтровали и фильтрат сконцентрировали при пониженном давлении, получив в результате 5-(3-амино-4-иламино)-2-цианоиндан (12,28 г, примерно количественный выход) в виде твердого продукта желтого цвета. Т.пл. 98 - 100oC.
e) Смесь 5-(3-аминопирид-4-иламино)-2-цианоиндана (12,28 г, примерно 44,5 ммоль) из стадии (d), уксусную кислоту (70 мл) и уксусный ангидрид (70 мл) нагревали с обратным холодильником при кипении в атмосфере азота 1,75 ч, охладили и сконцентрировали при пониженном давлении. Остаток в виде коричневого цвета смолы растворили в 2 М хлористоводородной кислоты (40 мл) и промыли этилацетатом (50 мл). Водный слой подщелочили добавлением 2 М водного раствора гидроксида натрия и проэкстрагировали дихлорметаном. Объединенные экстракты промыли водой (50 мл), высушили над сульфатом магния и растворитель отогнали. Остаток очистили методом колоночной хроматографии на силикагеле, проэлюировали смесью этилацетат/метанол = 7 : 1 и получили смолу коричневого цвета, которую перекристаллизовали из смеси этилацетат/метанол и получили 2-циано-5-(2-метилимидазо[4,5-c]пирид-1-ил)индан в виде беловатого порошка (9,67 г, 79%) с т.пл. 174 - 176oC.
Найдено,%: C 74,4; H 5,2; N 20,7. C17H14N4.
Рассчитано,%: C 74,4; H 5,1; N 20,4.
f) Смесь 2-циано-5-(2-метилимидазо[4,5-c]пирид-1-ил)индана (739 мг, 2,70 ммоль) 50%-ного водного раствора гидроксида натрия (1 мл) и метанола (6 мл) нагревали при кипении в атмосфере азота 9 ч, затем охладили, вылили в лед и довели pH раствора до pH 5 добавлением 2 М хлористоводородной кислоты. Выпавший осадок отфильтровали и высушили в вакууме, получив в результате озаглавленное соединение (426 мг, 54%) в виде бесцветного твердого продукта с т.пл. 264 - 267oC.
Найдено,%: C 68,9; H 5,1; N 14,1. C17H15N3O2•0,2H2O.
Рассчитано,%: C 68,8; H 5,2; N 14,1.
Препаративный пример 6. 3-(2-Метилимидазо[4,5-c]пирид-1-ил)бензойная кислота.
a) Раствор этил 3-аминобезонзоата (3,3 г, 20 ммоль) и 4-хлоро-3-нитропиридина (3,17 г, 20 ммоль) в этаноле (150 мл) перемешивали при комнатной температуре 16 ч и затем охладили, промыли этиловым спиртом, охлажденным льдом, и высушили, получив в результате 4,17 г 3-(3-нитро-4-пиридиламино)бензоат гидрохлорида в виде кристаллического продукта желтого цвета с т. пл. 201 - 204oC.
Найдено,%: C 5,2; H 4,4; N 12,9. C14H13N3O4•HCl.
Рассчитано,%: C 51,9; H 4,3; N 13,0.
Маточные растворы упарили и остаток перекристаллизовали из этанола и получили в результате еще 1,2 г названного продукта.
b) Полученный в стадии (a) продукт (5,2 г) разделили между этилацетатом и 10%-ным водным раствором карбоната натрия, органический слой промыли водой, высушили над сульфатом магния и упарили. Остаток растворили в этаноле (300 мл) и раствор перемешивали в атмосфере водорода при комнатной температуре при давлении 40 фунтов/дюйм2 (2,76 н/м2) в присутствии катализатора - 5%-ного палладия на углеродной подложке в течение 16 ч. Смесь профильтровали и фильтр упарили. В результате получили этил 3-(3-амино-4-пиридиламино)бензоат (4,8 г) в виде бесцветного твердого продукта с т.пл. 89 - 91oC.
c) Раствор продукта, полученного в стадии (b) (4,8 г) в уксусной кислоте (25 мл) и уксусном ангидриде (25 мл) нагревали при кипении в течение 2 ч, после чего упарили. Оставшееся масло вылили в воду, подщелочили, добавив твердый карбонат натрия и проэкстрагировали этилацетатом. Объединенные органические экстракты промыли водой, насыщенной хлоридом натрия, высушили над сульфатом магния и упарили. Остаток перекристаллизовали из этилацетата и получили 3-(2-метилимидазо[4,5-c]пирид-1-ил)бензоат (2,9 г) в виде бесцветного твердого продукта с т.пл. 120 - 121oC.
Найдено,%: C 69,1; H 5,4; N 14,8. C15H15N3O2.
Рассчитано,%: C 68,3; H 5,3; N 14,9.
d) Смесь продукта, полученного в стадии (c) (1,0 г) и 2 М водного раствора гидроксида натрия (2,2 мл) в этаноле (10 мл) перемешивали при комнатной температуре в течение 18 ч. Этанол отогнали в вакууме и оставшийся водный раствор нейтрализовали до pH 6 добавлением 2 М хлористоводородной кислоты. Полученный осадок собрали, промыли охлаждаемой льдом водой и диэтиловым эфиром и высушили. В результате получили 3-(2-метилимидазо[4,5-c]пиридил-1-ил)бензойную кислоту (830 мг) в виде бесцветного порошка, которое охарактеризовано, как продукт, содержащий 0,7% эквивалента воды.
C13H11N3O2.
Найдено %: C 62,8; H 4,6; N 15,5.
Рассчитано %: 0,75 H2O; C 63,0; H 4,7; N 15,7.
Препаративный пример 7. 2-(2-Метилимидазо [4,5-c] пирид-1-ил) бензойная кислота
а) Раствор этил 2-аминобензоата (3,17 г, 20 ммоль) и 4-хлор-3-нитропиридина (2,8 мл) в этаноле (150 мл) перемешивали при комнатной температуре в течение 60 ч. Полученный осадок собрали, промывали этиловым спиртом и высушили. В результате получили этил 2-(3-нитро-4-пиридиламино)бензоат гидрохлорида (3,9) в виде кристаллического продукта желтого цвета с т.пл. 192 - 205oC.
Маточные растворы упарили и остаток перекристаллизовали из этилового спирта, получив в результате еще 1,5 г продукта
b) Полученный в стадии (a) продукт (5,3 г) распределили между этилацетатом и 10%-ным водным раствором карбоната натрия и промыли органический слой водой, высушили затем над сульфатом магния и упарили. Остаток растворили в этаноле (800 мл) и раствор перемешивали в атмосфере водорода (40 фунт/дюйм2 или 2,76 н/м2) при комнатной температуре в присутствии катализатора - 5%-ного палладия, нанесенного на углеродную подложку, в течение 16 ч. Смесь отфильтровали и фильтрат упарили, получив в результате этил-2-(3-амино-4-пиридиламино)бензоат (5,1 г) в виде бесцветного масла.
c) Раствор продукта, полученного в стадии (b) (5,0 г) в уксусной кислоте (25 мл) и уксусном ангидриде (25 мл) нагревали при кипении в течение 4 ч, после чего упарили. Оставшееся масло вылили в воду, подщелочили твердым карбонатом натрия и проэкстрагировали этилацетатом. Объединенные органические слои промыли насыщенным водным раствором хлорида натрия, высушили над сульфатом магния и упарили. Остаток перекристаллизовали из этилацетата и получили этил 2-(2-метилимидазо[4,5-c]пирид-1-ил)бензоат (3,20) в виде бесцветных кристаллов с т.пл. 125 - 127oC.
d) Смесь продукта, полученного в стадии (c) (1,0 г) и 2 М водного раствора гидроксида натрия (2,2 мл) в этаноле (10 мл), перемешивали при комнатной температуре 18 ч. Этанол отогнали при пониженном давлении (в вакууме), а оставшийся водный раствор нейтрализовали до pH 6 добавлением 2 М хлористоводородной кислоты. Полученный осадок собрали, промыли охлаждаемой льдом водой и диэтиловым эфиром и высушили. В результате получили 2-(2-метилимидазо[4,5-c] пирид-1-ил)бензойную кислоту (760 мг), которая охарактеризована как продукт, содержащий 0,75 эквивалента воды.
C13H11N3O2.
Найдено, %: C 63,4; H 4,8; N 15,8.
Рассчитано,%: 0,75 H2O; C 63,O; H 4,7; N 15,7.
Препаративный пример 8. 5-(2-Метилимидазо[4,5-c]пирид-1-ил)тиофен-2-карбоновая кислота.
a) Раствор 2-циано-5-нитротиофена (Berichte, 1943, 76B, 419) (7,3 г, 47 ммоль) в этиловом спирте (150 мл) прогидрировали над катализатором - 30%-ным палладием на углеродном подложке в течение 4,5 ч при комнатной температуре и давлении 20 фунт/дюйм2 (1,4•105н/м2). После фильтрации получили раствор 2-амино-5-цианотиофена в этиловом спирте, который использовали сразу же в следующей стадии.
b) Раствор из стадии (a) (5,8 и продукта, 47 ммоль) перемешивали с 4-хлоро-3-нитропиридином (8,89 г, 56 ммоль) в атмосфере азота в темное время в течение 18 ч. Добавили 100 мл дихлорметана и триэтиламин (13 мл, 94 ммоль), затем силикагель (50 г) и растворитель отогнали в вакууме. Остаток пропустили через хроматографическую колонку, проэлюировали сначала дихлорометаном и затем смесью дихлорометан плюс 10% этилацетата. Продукт, содержащий фракции, упарили и твердый продукт темно-красного цвета суспендировали в этиловой спирте (100 мл), полученную смесь нагревали при кипении 30 мин. После охлаждения твердый продукт отфильтровали и высушили в вакууме, в результате получили 4-(5-цианотиен-2-ил)амино-3-нитропиридин (6,1 г, 52%).
C10H6N4O2.
Найдено,%: C 48,77; H 2,42; N 23,37;
Рассчитано,%: C 48,77; H 2,46; N 22,75.
c) Полученный в стадии (b) продукт прогидрировали при 20 фунт/дюйм2 (1,4•105 н/м2) и комнатной температуре над катализатором - 30%-ным палладием на углеродной подложке в течение 4 ч. После фильтрования и упаривания получили 3-амино-4-(5-цианотиен-2-ил)аминопиридин (5,35 г 100%), который сразу же использовали в стадии циклизации, следуя процедуре, описанной в препаративном примере 2 c, в результате получили 2-циано-5-(2-метилимидазо[4,5-c] пирид-1-ил)тиофен (2,25 г, 46%).
1H-ЯМР (CDCl3) δ : 9,10 /1H, c/, 8,53 /1H, д, J=6 Гц/ 7,75 /1H, д, J=4 Гц/, 7,26 /1H, д, J=6 Гц/, 7,20 /1H, д, J=4 Гц/, 2,62 /3H, c/.
d) Раствор 2-циано-5-(2-метилимидазо[4,5-c]пирид-1-ил)тиофена (2,7 г, 11 ммоль) и гидроксида натрия (1,76 г, 44 ммоль) в смеси этанола (23 мл) и воды (5 мл) нагревали при кипячении два часа, pH раствора довели до pH 6, добавляя 2М хлористоводородной кислоты и разбавив реакционную смесь водой до 500 мл. Раствор разделили на две равные порции и каждую порцию пропустили через колонку с ионообменной смолой (350 г), проэлюировав колонки сначала водой, а затем метанолом и водой, взятых в соотношении 1:1. Фракции, содержащие продукт, собрали и объединили и растворитель отогнали при пониженном давлении. Остаток растворили в кипящем изопропиловом спирте, отфильтровали и растворитель отогнали при пониженном давлении, после азеотропной перегонки с толуолом получили озаглавленное соединение (1,8 г, 63%).
1H-ЯМР (DMCO d6/, δ : 8,91 /1H, c/, 8,36 /1H, д, J=5,5 Гц/, 7,79 /1H, д, J=4,0 Гц/, 7,47 /1H, д, J=6 Гц/, 7,40 /1H, д, J=6 Гц, J=6 гц/, 2,55 /3H, c/.
Пример 20.
Фармацевтический состав.
Ингредиенты - мг/доза
1. Соединение примера 1 - 197,531 (a)
2. Микрокристаллическая целлюлоза - 49,383 (b)
3. Фосфат кальция диабазовый - 26,086 (c)
4. Натрий кроскармеллоза - 12,000 (d)
5. Повидин - 12,000 (e)
6. Стеарат магния - 3,000
Примечание.
a) Эквивалент 160 мг в расчете на теоретическую активность 81%.
b) В виде Avicel PH 101.
c) В виде безводного препарата.
d) В виде Ac-D-Sol.
e) Пиролидон поливинил в виде Kollidon 30.
Ингредиенты гранулируют с небольшим количеством воды, сушат, скринируют и заполняют готовой смесью желатиновые капсулы для получения композиции, указанной выше.
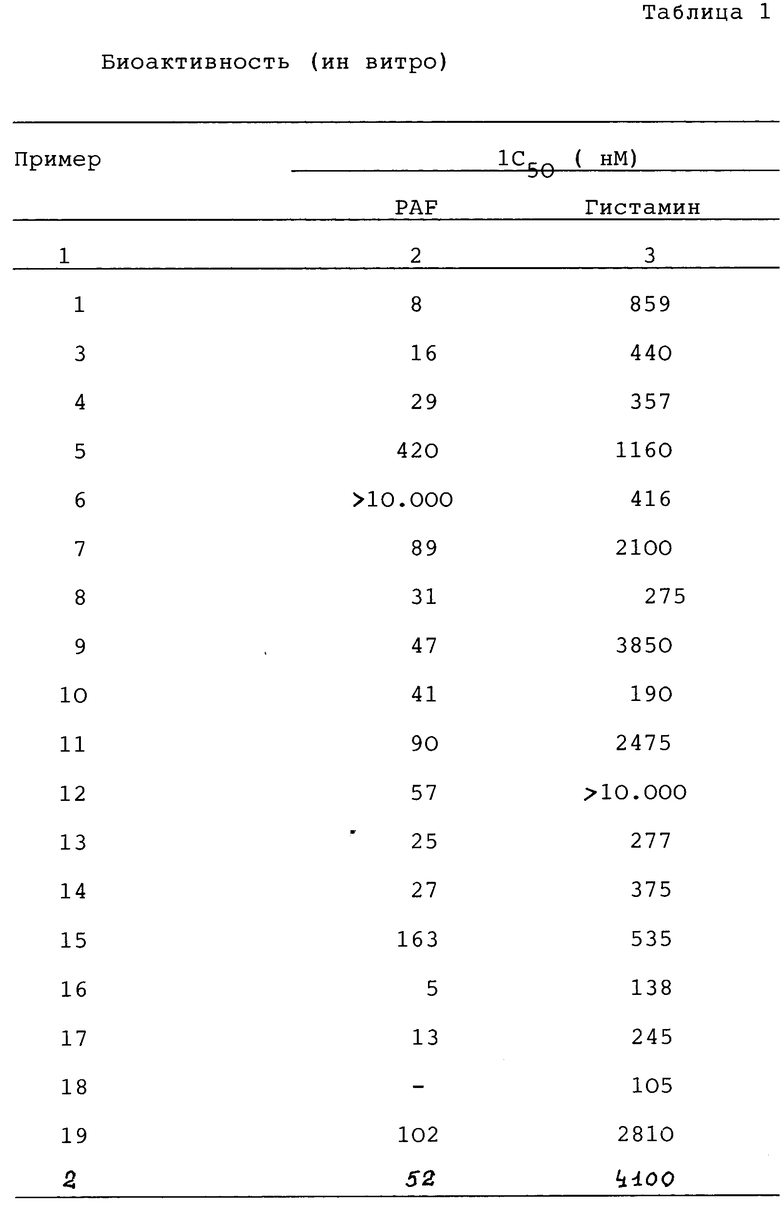

Описываются производные имидазопиридина общей формулы 1: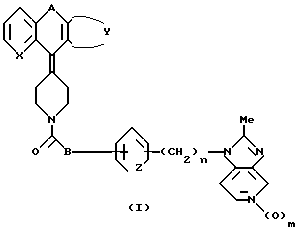
в которой X - CH или N; Z представляет -CH=CH или S; A представляет -CH2CH2, CH= CH, CH/OH/CH2 или COCH2; B означает направленную связь или -CH2-, -CH/CH3/2-, или когда Z представляет CH=CH. В может образовывать циклопентановое кольцо, слитое с прикрепленным бензольным кольцом, Y завершает сконденсированное бензо- или тиенильное кольцо, которое необязательно замещено гало- или C1-C4 алкилом; n равно 0, 1 или 2, а m равно 0 или 1, являющиеся антагонистами как ТАФ, как и гистамина H1, обладают свойствами, которые позволяют использовать их в лечении аллергических воспалительных заболеваний, таких как аллергический ренит. Описан также способ получения производных имидазопиридина формулы 1 и фармацевтическая композиция на их основе. 3 с. и 12 з.п. ф-лы, 3 табл.

где X - CH или азот;
Z - -CH=CH- или сера;
A - CH2CH2, CH=CH, CH(OH)CH2 или COCH2;
B - прямая связь или -CH2-, CH(CH3)- или -C(CH3)2-, или в том случае, когда Z - CH=CH, B может представлять циклопентановое кольцо, сконденсированное с присоединенным бензольным кольцом;
Y завершает сконденсированное кольцо, которое представляет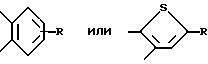
в которых R - водород, галоид или C1 - C4-алкил;
n = 0, 1 или 2;
m = 0 или 1,
или их фармацевтически приемлемые соли.
3. Соединение по п.2, отличающееся тем, что B - прямая связь или -CH2-.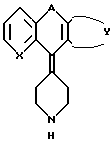
с кислотой формулы III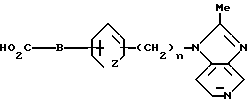
или ее активированным производным,
в которых X, Y, A, B и n имеют значения по п.1,
и при желании окисляют полученный продукт с образованием соединения, где m = 1, и, необязательно, получают его фармацевтически приемлемую соль.
9. Способ по п.8, отличающийся тем, что B - прямая связь или CH2.
| US, патент, 3326924, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
