Настоящее изобретение относится к производным имидазопиридина, используемым для лечения патологий, связанных с метаболическим синдромом, в частности, для лечения или предотвращения сахарного диабета.
Сахарный диабет представляет собой весьма гетерогенную группу заболеваний, которые характеризуются рядом общих свойств: гипергликемией, функциональными и количественными отклонениями показателей бета-клеток поджелудочной железы от нормы, резистентностью тканей к инсулину, а также увеличением риска развития в долгосрочной перспективе осложнений, в частности, сердечно-сосудистых осложнений.
Сахарный диабет II типа представляет собой существенную проблему здравоохранения. Распространение данного заболевания резко увеличивается в большинстве промышленно развитых стран, и еще более - в странах с развивающейся экономикой. На сегодняшний день можно говорить об эпидемии данного заболевания, вызывающего серьезные осложнения, которые вполне могут приводить к инвалидности или даже к смертельному исходу, в том числе вследствие почечной недостаточности, инфаркта миокарда или сердечно-сосудистого инсульта.
Недавно установленные количественные показатели сахарного диабета являются следующими (данные ВОЗ (Всемирной организации здравоохранения)):
- Во всем мире от сахарного диабета страдают более 220 миллионов человек.
- Сахарный диабет в 3 раза увеличивает риск возникновения инсульта.
- В западном мире сахарный диабет является первой причиной слепоты и почечной недостаточности.
- Согласно оценкам, в 2005 году сахарный диабет стал причиной гибели 1,1 миллиона человек.
- Согласно прогнозам ВОЗ, в период с 2005 по 2030 год количество смертей, вызванных сахарным диабетом, увеличится в два раза.
Во Франции уход за пациентами, страдающими от диабета, а также лечение таких пациентов оказывают существенную нагрузку на бюджет государственной системы медицинского страхования. Принимая во внимание тревожные прогнозы количества пациентов в мире, у которых будет выявлен диабет в период с сегодняшнего дня до 2030 года, многие фармацевтические и биотехнологические компании интенсивно производят инвестиции в исследование и разработку в области метаболизма и, более конкретно, в области сахарного диабета II типа с целью выведения на рынок новых альтернативных лекарственных препаратов.
В настоящее время лечение сахарного диабета II типа, способное восстановить нормальное гликемическое равновесие в течение 24 часов и не обладающее побочными действиями, отсутствует. Ни один из существующих вариантов лечения не принимает во внимание всеобъемлющую патологию данного заболевания; кроме того, существующие варианты лечения нацелены исключительно на исправление того или иного негативного явления. Средства для лечения сахарного диабета, которые недавно были выведены на рынок, не продемонстрировали какого-либо существенного улучшения гликемического контроля по сравнению с контролем, наблюдавшимся для ранее существующих вариантов лечения, а также оказывали нежелательные побочные действия, что оставляет пространство для новых потенциальных способов лечения.
Вследствие этого существует потребность в новых молекулах, используемых для лечения или предотвращения сахарного диабета или осложнений и/или сопутствующих патологий данного заболевания, предпочтительно сахарного диабета II типа.
Авторы настоящего изобретения неожиданно обнаружили, что определенные производные имидазопиридина обладают активностью ингибирования продукции глюкозы печенью, а также активностью стимулирования секреции инсулина в ответ на поступление глюкозы, и, в частности, что указанные производные имидазопиридина могут использоваться в качестве продуктов для фармацевтического применения пациентами, которые нуждаются в таких продуктах, в особенности для предотвращения и/или лечения сахарного диабета и осложнений и/или сопутствующих патологий данного заболевания (ожирение, гипертензия и т.д.), предпочтительно сахарного диабета II типа.
Подробное описание изобретения
Вследствие этого настоящее изобретение относится к производным имидазопиридина следующей общей формулы I:
 , где:
, где:
Y представляет собой атом кислорода или серы, группу SO, SO2 или -NR19, где R19 представляет собой атом водорода или C1-C6 алкильную группу, предпочтительно метильную (Me); предпочтительно Y представляет собой атом кислорода, серы или группу -NR19, в частности, атом кислорода или группу -NR19.
R1, Ra, Rb и Rc представляют собой независимо друг от друга атом водорода; атом галогена, предпочтительно Cl; C1-C6 алкильную группу, предпочтительно метильную, необязательно замещенную группой -OH; группу -OH; -O(C1-C6 алкильную) группу, такую как -OMe, -O(C1-C6алкил)O(C1-C6 алкильную) группу; группу -CN; группу -NR4R5, где R4 и R5 представляют собой независимо друг от друга атом водорода или C1-C6 алкильную группу; или группу -(C1-C6алкил)NR6R7, где R6 и R7 представляют собой независимо друг от друга атом водорода или C1-C6 алкильную группу;
R2 представляет собой
- атом водорода;
- C1-C6 алкильную группу, замещенную группой -OH, в частности, -(CH2)2OH;
- группу -(C1-C6алкил)COOR8, предпочтительно -CH2COOR8, где R8 представляет собой атом водорода или C1-C6 алкильную группу, в частности, метильную (Me), этильную (Et) или изопропильную (iPr), причем указанная алкильная группа может быть замещена группой -NH2 или -OH (предпочтительно, указанная алкильная группа не замещена), примерами групп -(C1-C6алкил)COOR8 являются –CH2COOH, -CH2COOMe, -CH2COOEt, -CH2CH(NH2)COOEt и -CH2COOiPr;
- группу -(C1-C6алкил)CONHR9, предпочтительно -CH2CONHR9, где R9 представляет собой группу -OH; C1-C6 алкильную группу, предпочтительно этильную; -O(C1-C6 алкильную) группу, предпочтительно -OEt; арильную группу, предпочтительно фенильную; гетероарильную группу, предпочтительно пиридильную; -(C=NH)NHCOO(C1-C6 алкильную) группу, предпочтительно -(C=NH)NHCOOt-бутильную; группу -(C=NH)NH2; или группу -(C1-C6алкил)NR10R11, предпочтительно -(CH2)2NR10R11, где R10 и R11 представляют собой независимо друг от друга C1-C6 алкильную группу, предпочтительно метильную; примерами групп -(C1-C6алкил)CONHR9 являются -CH2CONHOH, -CH2CONHOEt и CH2CONH(CH2)2NMe2;
- -(C1-C6алкил)COморфолиновую группу, предпочтительно -CH2COморфолиновую;
- группу -C(=O)R12, где R12 представляет собой –О(C1-C6 алкильную) группу, предпочтительно -Oизобутильную (OiBu); C1-C6 алкильную группу, предпочтительно этильную или изопропильную, необязательно замещенную группой -OH, такую как -(CH2)2OH; морфолиновую группу; NH-арильную группу, предпочтительно -NHфенильную, причем указанная арильная группа необязательно замещена группой -COOH или -COO(C1-C6алкилом), предпочтительно -COOMe; или группу -NR13R14, где R13 и R14 представляют собой независимо друг от друга C1-C6 алкильную группу, предпочтительно этильную; примерами групп -C(=O)R12 являются -C(=O)OiBu, -C(=O)(CH2)2OH, -C(=O)NEt2, -C(=O)Морфолин, -C(=O)iPr, -C(=O)NH-3-HO2C-Ph и -C(=O)NH-3-MeO2C-Ph;
- (C1-C6алкил)арильную группу, предпочтительно бензильную, причем указанная арильная группа необязательно замещена группой -CN, -COOH или -COO(C1-C6алкилом); примером замещенной (C1-C6алкил)арильной группы является -CH2-3-NC-Ph;
- (C1-C6алкил)гетероарильную группу, предпочтительно (C1-C6алкил)тиофеновую, в частности, метилтиофеновую группу;
- арильную группу, предпочтительно фенильную, причем указанная арильная группа необязательно замещена одной или несколькими группами, которые выбирают из -COOH, -COO(C1-C6алкила), предпочтительно -COOMe, C1-C6алкила, замещенного группой -OH, предпочтительно -CH2OH, -CN, -CONHOH, -NHSO2(C1-C6алкила), предпочтительно -NHSO2Me, или -CONH-(C1-C6алкил)R15R16, предпочтительно -CONH-(этил)NR15R16, где R15 и R16 представляют собой независимо друг от друга C1-C6 алкильную группу, предпочтительно метильную; примерами необязательно замещенных арильных групп являются -Ph; -2-MeO2C-Ph, -2-HO2C-Ph, -3-MeO2C-Ph, -3-HO2C-Ph, -4-MeO2C-Ph, -4-HO2C-Ph; -2-HO2HC-Ph, -3-MeSO2HN-Ph, -3-NC-Ph, -2-HONHOC-Ph, и -2-Me2N(CH2)2HNOC-Ph;
- гетероарильную группу, предпочтительно пиридильную;
- гетероциклическую группу, предпочтительно тетрагидрофурановую, в частности, содержащую один или два гетероатома, которые предпочтительно выбирают из N, S и О, причем указанная гетероциклическая группа может необязательно включать ненасыщенную связь, такая как тиазолиновая;
- лактоновую группу, содержащую от 3 до 6 членов, в частности, тетрагидрофураноновую, необязательно замещенную одной или несколькими C1-C6 алкильными группами, в частности, двумя группами, такими как две метильные группы;
- или -SO2(С1-C6 алкильную) группу, предпочтительно -SO2(изопропильную);
R3 представляет собой арильную группу, предпочтительно фенильную, или гетероарильную группу, предпочтительно пиридильную или тиенильную, причем указанная арильная группа необязательно замещена одной или несколькими группами, которые выбирают из -(C1-C6 алкильной), предпочтительно метильной, причем указанная алкильная группа необязательно замещена одним или несколькими атомами галогена, предпочтительно F, такая как, например, -CF3, или группой -CN, такая как, например, -CH2CN; атома галогена, предпочтительно F, -O(C1-C6алкила), предпочтительно -OMe, причем указанная алкильная группа необязательно замещена одним или несколькими атомами галогена, предпочтительно F, такая как, например, -OCF3 или -OCH2F; -CN; -OH; -NO2; -COOH; -NR17R18, где R17 и R18 представляют собой независимо друг от друга C1-C6 алкильную группу, предпочтительно метильную; или -NHCO-(C1-C6алкила), предпочтительно -NHCOMe; примерами необязательно замещенных арильных групп являются -Ph, -2-F3C-Ph, -3-F3C-Ph, -4-F3C-Ph, -2-F-Ph, -3-F-Ph, -4-F-Ph, -3,4-(F)2-Ph, -3-F-4-F3CO-Ph, -3-F3CO-Ph, -4-F3CO-Ph, -3-F2HCO-Ph, -3-NC-Ph, -3-HO-Ph, -2-MeO-Ph, -3-MeO-Ph, -4-MeO-Ph, -3,4-(MeO)2-Ph, -4-Me-Ph, -4-Me2N-Ph, -4-O2N-Ph, -3-HO2C-Ph, -3-MeCOHN-Ph и -4-NCH2C-Ph;
или энантиомер, диастереоизомер, гидрат, сольват, таутомер, рацемическая смесь или фармацевтически приемлемая соль указанного соединения.
Согласно конкретному варианту реализации настоящего изобретения Y представляет собой атом кислорода или группу -NH или -NMe, предпочтительно атом кислорода.
Согласно другому конкретному варианту реализации настоящего изобретения R1, Ra, Rb и Rc представляют собой независимо друг от друга атом водорода, атом галогена, в частности, Cl, или –О(C1-C6 алкильную) группу, такую как OMe, в частности, атом водорода или атом галогена. Предпочтительно три из групп R1, Ra, Rb и Rc представляют собой атом водорода, а четвертая из указанных групп представляет собой атом водорода, атом галогена, в частности, Cl, или -O(C1-C6 алкильную) группу, такую как OMe, предпочтительно атом водорода или атом галогена. Более предпочтительно, R1, Ra, Rb и Rc представляют собой атом водорода.
Согласно еще одному конкретному варианту реализации настоящего изобретения R2 представляет собой
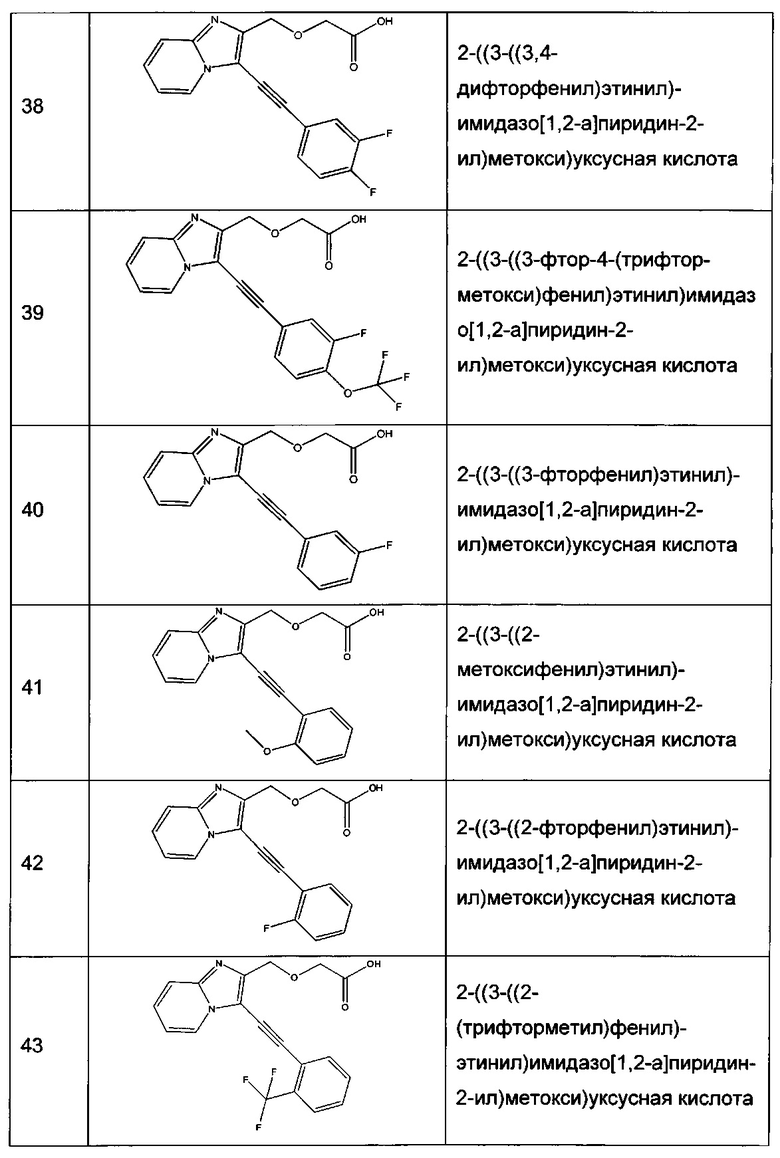
- группу (C1-C6алкил)COOR8, предпочтительно -CH2COOR8, где R8 представляет собой атом водорода или C1-C6 алкильную группу, в частности, метильную (Me), этильную (Et) или изопропильную (iPr), примерами групп -(C1-C6алкил)COOR8 являются -CH2COOH, -CH2COOMe, -CH2COOEt и -CH2COOiPr;
- группу (C1-C6алкил)CONHR9, предпочтительно -CH2CONHR9, где R9 представляет собой группу -OH; -O(C1-C6 алкильную) группу, предпочтительно -OEt, или группу -(C1-C6алкил)NR10R11, где R10 и R11 представляют собой независимо друг от друга C1-C6 алкильную группу, предпочтительно метильную; примерами групп -(С1-C6алкил)CONHR9 являются -CH2CONHOH, -CH2CONHOEt и -CH2CONH(CH2)2NMe2;
- арильную группу, предпочтительно фенильную, причем указанная арильная группа необязательно замещена одной или несколькими группами, которые выбирают из -COOH, -COO(C1-C6алкила), предпочтительно -COOMe, -CONHOH, или -CONH-(C1-C6алкил)NR15R16, предпочтительно -CONH-(этил)NR15R16, где R15 и R16 представляют собой независимо друг от друга C1-C6 алкильную группу, предпочтительно метильную; примерами необязательно замещенных арильных групп являются -Ph; -2-MeO2C-Ph, -2-HO2C-Ph, -3-MeO2C-Ph, -3-HO2C-Ph, -4-MeO2C-Ph, -4-HO2C-Ph; -2-HONHOC-Ph, и -2-Me2N(CH2)2HNOC-Ph; или
- -CONHарильную группу, предпочтительно -CONHфенильную, необязательно замещенную группой -COOH или -COO(C1-C6алкилом), предпочтительно -COOMe.
Предпочтительно, R2 представляет собой группу -(C1-C6алкил)COOR8, где R8 представляет собой атом водорода или C1-C6 алкильную группу, в частности, метильную (Me), этильную (Et) или изопропильную (iPr), или арильную группу, предпочтительно фенильную, причем указанная арильная группа необязательно замещена одной или несколькими группами, в частности, группами, которые выбирают из -COOH, -COO(C1-C6алкила), предпочтительно -COOMe. Более предпочтительно, R2 представляет собой группу -(C1-C6алкил)COOH, предпочтительно -CH2COOH.
Согласно еще одному конкретному варианту реализации настоящего изобретения R3 представляет собой арильную группу, предпочтительно фенильную, необязательно замещенную одной или несколькими группами, которые выбирают из -(C1-C6 алкильной), предпочтительно метильной, причем указанная алкильная группа необязательно замещена одним или несколькими атомами галогена, предпочтительно F, такая как, например, -CF3; атома галогена, предпочтительно F, -O(C1-C6алкила), предпочтительно -OMe, причем указанная алкильная группа необязательно замещена одним или несколькими атомами галогена, предпочтительно F, такая как, например, -OCF3 или -OCH2F; -CN; или -NO2; примерами необязательно замещенных арильных групп являются -Ph, -2-F3C-Ph, -3-F3C-Ph, -4-F3C-Ph, -2-F-Ph, -3-F-Ph, -4-F-Ph, -3,4-(F)2-Ph, -3-F-4-F3CO-Ph, -3-F3CO-Ph, -4-F3CO-Ph, -3-F2HCO-Ph, -3-NC-Ph, -2-MeO-Ph, -3-MeO-Ph, -4-MeO-Ph, -3,4-(MeO)2-Ph, -4-Me-Ph и -4-O2N-Ph.
Предпочтительно, R3 представляет собой фенильную группу, замещенную одной или несколькими группами, которые выбирают из -(C1-C6 алкильной), предпочтительно метильной, замещенной одним или несколькими атомами галогена, предпочтительно F, такой как, например, -CF3; атома галогена, предпочтительно F, -O(C1-C6алкила), предпочтительно -OMe, причем указанная алкильная группа замещена одним или несколькими атомами галогена, предпочтительно F, такая как, например, -OCF3; или -CN.
В рамках настоящего изобретения под «арильной группой» понимают ароматическое кольцо, содержащее от 5 до 8 атомов углерода, или несколько сочлененных ароматических колец, содержащих от 5 до 14 атомов углерода. В частности, арильные группы могут представлять собой моноциклические или бициклические группы, предпочтительно фенильные или нафтильные группы. Предпочтительно, арильная группа представляет собой фенильную группу (Ph).
В рамках настоящего изобретения под «гетероарильной группой» понимают любую углеводородную ароматическую группу, содержащую от 3 до 9 атомов и содержащую один или несколько гетероатомов, таких как, например, атомы серы, азота или кислорода. Гетероарильная группа согласно настоящему изобретению может быть образована одним или несколькими сочлененными кольцами. Примерами гетероарильных групп являются фурильная, изоксазильная, пиридильная, тиазолильная, пиримидильная, бензимидазольная, бензоксазольная, тиенильная, тиофенильная, бензотиазольная группы. Предпочтительно, гетероарильную группу выбирают из фурильной, пиридильной, тиазолильной, тиофенильной и тиенильной групп, предпочтительно гетероарильная группа представляет собой пиридильную, тиофенильную и тиенильную группу.
В рамках настоящего изобретения под «гетероциклической группой» понимают любую насыщенную циклическую углеводородную группу, содержащую от 3 до 9 атомов, и содержащую один или несколько гетероатомов, таких как, например, атомы серы, азота или кислорода. Гетероциклическая группа согласно настоящему изобретению может быть образована одним или несколькими сочлененными кольцами. Примерами гетероциклических групп являются тетрагидрофурановая, пирролидильная, пиперидильная, тиолановая, оксирановая, оксановая, тиановая, тиазолидиновая, морфолиновая группы. Предпочтительно, гетероциклическую группу выбирают из тетрагидрофурановой, тиазолидиновой и морфолиновой групп.
В рамках настоящего изобретения под «атомом галогена» понимают любой атом галогена, который предпочтительно выбирают из Cl, Br, I или F, в частности, которые выбирают из F, Cl или Br, в частности, из Cl или F.
В рамках настоящего изобретения под «C1-C6 алкильной группой» понимают любую алкильную группу, содержащую от 1 до 6 атомов углерода, линейную или разветвленную, в частности, метильную, этильную, н-пропильную, изо-пропильную, н-бутильную, изо-бутильную, втор-бутильную, т-бутильную, н-пентильную, н-гексильную группы. Предпочтительно, C1-C6 алкильная группа представляет собой метильную, этильную, изо-бутильную или изо-пропильную группу, в частности, метильную или этильную группу, более конкретно, метильную группу.
В рамках настоящего изобретения под «(C1-C6алкил)арильной группой» понимают любую арильную группу, как определено выше, связанную с C1-C6 алкильной группой, как определено выше. В частности, примером (C1-C6алкил)арильной группы является бензильная группа.
В рамках настоящего изобретения под «(C1-C6алкил)гетероарильной группой» понимают любую гетероарильную группу, как определено выше, связанную с C1-C6 алкильной группой, как определено выше. В частности, примером (C1-C6алкил)гетероарильной группы является метилтиофеновая группа.
В рамках настоящего изобретения под «фармацевтически приемлемым» понимают, что такое вещество можно использовать для приготовления фармацевтической композиции, которая является в общем виде безопасной, нетоксичной и использование которой не является нежелательным ни с биологической, ни с какой-либо другой точки зрения, и которая является приемлемой для ветеринарного применения, а также для приготовления фармацевтических препаратов, применяемых у человека.
В рамках настоящего изобретения под «фармацевтически приемлемыми солями соединения» понимают соли, которые являются фармацевтически приемлемыми, как определено в настоящей заявке, и которые обладают желаемой фармакологической активностью родительского соединения. Такие соли включают:
(1) соли присоединения кислоты, образованные из минеральных кислот, таких как хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и тому подобное; или образованные из органических кислот, таких как уксусная кислота, бензолсульфоновая кислота, бензойная кислота, камфорсульфоновая кислота, лимонная кислота, этансульфоновая кислота, фумаровая кислота, глюкогептоновая кислота, глюконовая кислота, глутаминовая кислота, гликолевая кислота, гидроксинафтоевая кислота, 2-гидроксиэтансульфоновая кислота, молочная кислота, малеиновая кислота, яблочная кислота, миндальная кислота, метансульфоновая кислота, муконовая кислота, 2-нафталинсульфоновая кислота, пропионовая кислота, салициловая кислота, янтарная кислота, дибензоил-L-винная кислота, винная кислота, п-толуолсульфоновая кислота, триметилуксусная кислота, трифторуксусная кислота и тому подобное; или
(2) соли, образованные в случае замещения протона кислоты, присутствующего в родительском соединении, ионом металла, например, ионом щелочного металла, ионом щелочноземельного металла или ионом алюминия; либо в результате координирования указанного протона кислоты с органическим или неорганическим основанием. Приемлемые органические основания включают диэтаноламин, этаноламин, N-метилглюкамин, триэтаноламин, трометамин и тому подобное. Приемлемые неорганические основания включают гидроксид алюминия, гидроксид кальция, гидроксид калия, карбонат натрия и гидроксид натрия.
В рамках настоящего изобретения под «сольватом соединения» понимают любое соединение, полученное в результате добавления молекулы инертного растворителя к соединению согласно настоящему изобретению; при этом сольват образуется в результате взаимной силы притяжения. Сольваты представляют собой, например, алкоголяты соединения. Гидрат представляет собой сольват, в котором используемым инертным растворителем является вода. Гидрат может представлять собой моно-, ди- или тригидрат.
В рамках настоящего изобретения под «таутомером» понимают любой изомер с получением соединений согласно настоящему изобретению, причем указанные соединения являются взаимно превращаемыми в результате обратимой химической реакции, которую называют реакцией таутомеризации. В большинстве случаев данная реакция происходит в результате миграции атома водорода, сопровождающейся изменением локализации двойной связи. В растворе соединения, способного к таутомеризации, устанавливается равновесие между 2 таутомерами. Соотношение между таутомерами также зависит от растворителя, от температуры и от значения pH. Вследствие этого таутомерия представляет собой трансформацию функциональной группы в другую группу, наиболее часто в результате сопутствующего смещения атома водорода и π-связи (двойной или тройной связи). Общеизвестные таутомеры представляют собой, например, альдегиды/кетоны - спирты или более конкретные пары енолов; амиды - имидокислоты; лактамы - лактимы; имины - енамины; енамины - енамины. В частности, таутомерия может включать таутомерию «цикл-цепь», возникающую в случае, если движение протона сопровождается трансформацией открытой структуры в одно кольцо.
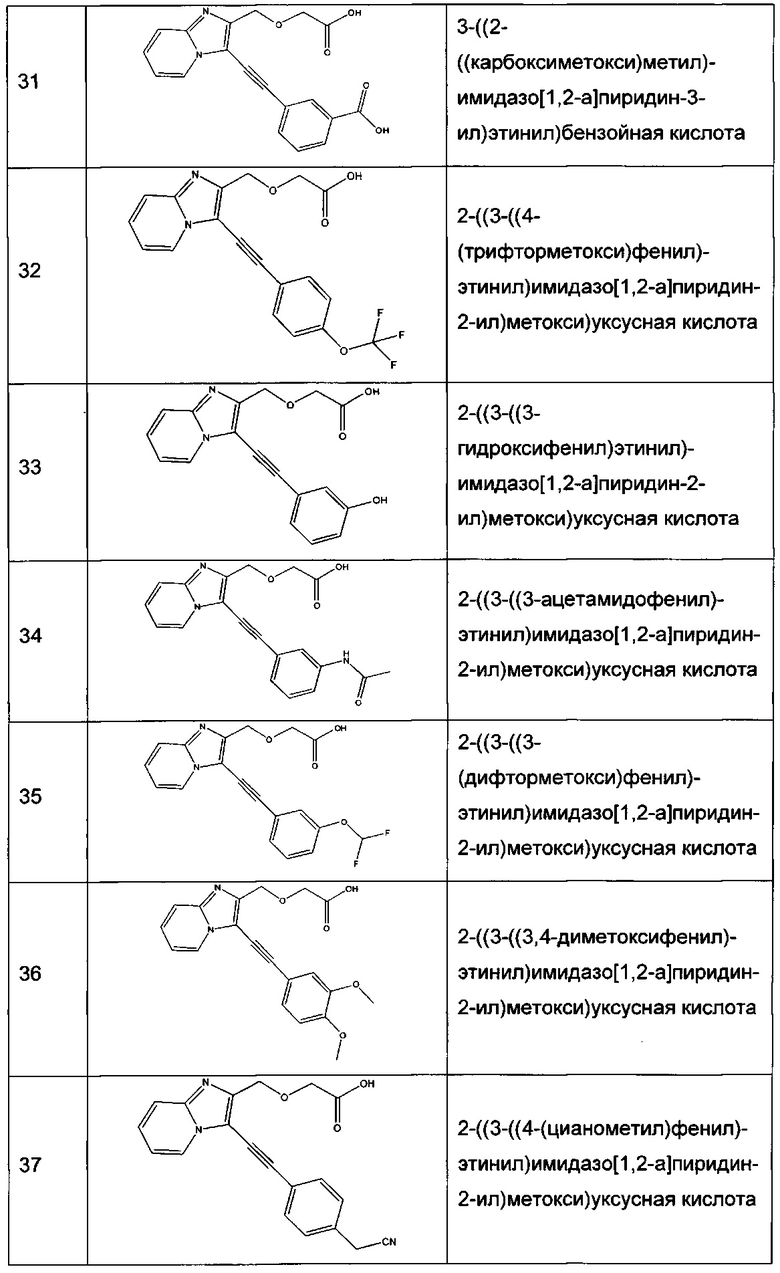
Согласно варианту реализации настоящего изобретения, вызывающему особенный интерес, производные имидазопиридина выбирают из соединений формул с 1 по 138, приведенных в таблице 1 ниже.
Согласно другому, вызывающему еще больший интерес варианту реализации настоящего изобретения производные имидазопиридина выбирают из 84 соединений, имеющих номера 1-3, 5, 7, 10-12, 14, 17, 18, 20, 23, 25-27, 29, 30, 32, 35, 38-40, 42, 44, 45, 46, 47, 48, 51, 54, 55, 57-59, 68, 72-79, 81, 83, 87-90, 92, 93, 95, 96, 101-126, 129-131 и 135 в таблице 1 ниже.
Еще более предпочтительно, что указанные производные имидазопиридина представляют собой соединения 3, 7, 14, 25, 26, 29, 44, 51, 54, 58, 59, 72, 76-79, 81, 87-90, 92, 93, 101, 102, 104, 110-112, 114-120 и 135, приведенные в таблице 1 ниже.
Настоящее изобретение также относится к фармацевтической композиции, содержащей производное имидазопиридина согласно настоящему изобретению и фармацевтически приемлемый эксципиент.
Данные композиции могут быть приготовлены в состав для введения млекопитающим, включая людей. Доза варьирует в зависимости от лечения и в зависимости от соответствующего заболевания. Данные фармацевтические композиции приспособлены для введения посредством любого приемлемого пути, например, перорального (включая буккальный и сублингвальный пути), посредством ректального, назального, местного (включая трансдермальный), вагинального, интраокулярного или парентерального (включая подкожный, внутримышечный или внутривенный) пути. Предпочтительно фармацевтические композиции приспособлены для перорального введения. Данные составы могут быть приготовлены с применением всех способов объединения активных компонентов с подходящими фармацевтически приемлемыми эксципиентами, известных специалисту в данной области техники.
Подходящие единичные лекарственные формы для перорального введения включают таблетки, желатиновые капсулы, порошки, гранулы и растворы для перорального применения или суспензии в водных или неводных жидкостях, съедобные или пищевые пены либо жидкие эмульсии типа «вода в масле» или «масло в воде». В случае если твердая композиция приготовлена в форме таблетки, главный активный компонент предпочтительно смешивают в форме порошка с приемлемым фармацевтическим эксципиентом, таким как желатин, крахмал, лактоза, стеарат магния, тальк, аравийская камедь или тому подобное. Представляется возможным покрыть таблетки сахарозой или другими подходящими материалами, либо таблетки могут быть дополнительно обработаны таким образом, чтобы данные таблетки обладали пролонгированной или отсроченной активностью и непрерывно высвобождали заранее определенное количество активного компонента.
Препарат в желатиновых капсулах получают в результате смешивания активных компонентов, предпочтительно в виде порошка, с разбавителем и в результате розлива полученной смеси в мягкие или твердые желатиновые капсулы, в частности, в желатиновые капсулы. Перед помещением в желатиновые капсулы к композиции могут быть добавлены смягчающие вещества, такие как, например, тальк, стеарат магния, стеарат кальция или полиэтиленгликоль в твердой форме. Также для улучшения доступности лекарственного препарата после приема желатиновой капсулы в состав может быть добавлено разрыхляющее или солюбилизирующее вещество, такое как, например, карбонат кальция или карбонат натрия.
Дополнительно, в случае необходимости, в смесь могут быть добавлены связывающие вещества, смягчающие вещества и подходящие разрыхлители, а также окрашивающие вещества. Подходящие связывающие вещества могут представлять собой, например, крахмал, желатин, естественные сахара, такие как, например, глюкоза или бета-лактоза, подслащивающие вещества, полученные из кукурузы, синтетические или растительные каучуки, такие как, например, камедь или альгинат натрия, карбоксиметилцеллюлозу, полиэтиленгликоль, воски и тому подобное. Смягчающие вещества, которые могут применяться в данных лекарственных формах, включают олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и тому подобное. Разрыхляющие вещества включают крахмал, метил целлюлозу, агар, бентонит, ксантановую камедь и тому подобное. Таблетки могут быть изготовлены, например, путем приготовления смеси порошка, гранулирования или сухого прессования смеси, добавления смягчающего вещества и разрыхляющего вещества и прессования смеси с получением таблеток. Смесь порошка приготавливают в результате смешивания активного компонента, к которому добавляют соответствующий разбавитель или основу и, в некоторых случаях, связывающее вещество, такое как, например, карбоксиметилцеллюлоза, альгинат, желатин или поливинилпирролидон, вещество, замедляющее растворение, такое как, например, парафин, вещество, ускоряющее всасывание, такое как, например, четвертичная соль, и/или поглощающее вещество, такое как, например, бентонит, каолин или дикальций фосфат. Смеси порошков могут быть гранулированы путем их смачивания связывающим веществом, таким как, например, сироп, крахмальная паста, мазь на основе камеди или растворы целлюлозы либо полимерные материалы, и продавливания через сито. Гранулы могут быть смазаны в результате добавления стеариновой кислоты, соли стеариновой кислоты, талька или минерального масла для того, чтобы избежать прилипания к формам, в которых осуществляется производство таблеток. Смазанную смесь затем прессуют с получением таблеток. В некоторых случаях на таблетки наносят непрозрачный или прозрачный защитный слой, состоящий из слоя шеллака, слоя сахара или полимерных материалов. К таким покрытиям можно добавить окрашивающие вещества для того, чтобы отличить данные таблетки от других.
Препарат в форме сиропа или эликсира может содержать активный компонент совместно с подслащивающим веществом, антисептическим средством, веществом, придающим вкус, а также подходящим окрашивающим веществом. Как правило, препараты в форме сиропа получают в результате растворения соединения в водном растворе с подходящим веществом, придающим вкус, тогда как эликсиры приготавливают с применением нетоксичного спиртового носителя.
Порошки или гранулы, которые могут быть диспергированы в воде, могут содержать активные компоненты, смешанные с диспергирующим веществом или со смачивающими веществами, или с суспендирующими веществами, такими как, например, этоксилированные изостеариловые спирты и полиоксиэтиленовые эфиры сорбитола, а также с усилителями вкуса и аромата или подслащивающими веществами.
Для ректального введения прибегают к применению суппозиториев, которые приготавливают со связывающими веществами, плавящимися при ректальной температуре, например, с маслом какао или полиэтиленгликолями.
Для парентерального, интраназального или интраокулярного введения применяют водные суспензии, изотонические солевые растворы или стерильные и инъецируемые растворы, которые содержат фармакологически совместимые диспергирующие вещества и/или смачивающие вещества.
Активный компонент может быть также приготовлен в состав в форме микрокапсул, в некоторых случаях с одним или несколькими дополнительными носителями.
Фармацевтические композиции, приспособленные для введения местным путем, могут быть приготовлены в состав в форме крема, мази, суспензии, лосьона, порошка, раствора, пасты, геля, спрея, аэрозолей или масел.
Фармацевтические композиции, приспособленные для введения интраназальным путем, вспомогательный эксципиент в которых находится в твердом состоянии, включают порошки, размер частиц которых находится в диапазоне, например, от 20 до 500 микрон, и которые вводят посредством ингаляции из контейнера, содержащего порошок, причем указанный контейнер располагают напротив носа.
Фармацевтические составы, приспособленные для введения вагинальным путем, могут быть введены в форме буфера, крема, геля, пасты, пены или спрея.
Согласно предпочтительному варианту реализации настоящего изобретения фармацевтическая композиция согласно настоящему изобретению дополнительно содержит другой активный компонент, предпочтительно обладающий комплементарным или синергетическим действием. В частности, данный активный агент представляет собой другой противодиабетический агент, который предпочтительно выбирают из инсулина, сульфонилмочевины, глинидов, бигуанидов, тиазолидиндионов, агонистов GLP-1R (рецептора глюкагоноподобного пептида-1), ингибиторов DPP-IV (дипептидилпептидазы-4), ингибиторов SGLT-2 (натрий-глюкозного котранспортера 2), предпочтительно выбирают из инсулина, глибенкламида, гликлазида, глипизида, глимепирида, репаглинида, натеглинида, метформина, троглитазона, розиглитазона, пиоглитазона, экзенатида, лираглутида, ситаглиптина, вилдаглиптина, саксаглиптина, алоглиптина, дапаглифлозина. Более конкретно, данный активный агент представляет собой метформин. Данный второй активный агент может быть введен в той же фармацевтической композиции, что и производное тиофена согласно настоящему изобретению. Данный второй активный агент может быть также введен отдельно, например, в то же время или способом, протяженным во времени. Предпочтительно, данный второй активный агент вводят пероральным путем.
Настоящее изобретение также относится к производному имидазопиридина согласно настоящему изобретению для применения в качестве лекарственного средства.
Настоящее изобретение также относится к применению производного имидазопиридина согласно настоящему изобретению для приготовления лекарственного препарата.
Согласно настоящему изобретению соединения формулы (I) обладают гипогликемической активностью. Указанные соединения могут вызывать уменьшение гипергликемии, более конкретно, гипергликемии сахарного диабета II типа. В частности, соединения согласно настоящему изобретению обладают гипогликемической активностью и являются вследствие этого пригодными для лечения и/или предотвращения сахарного диабета, осложнений и/или сопутствующих патологий данного заболевания, таких как, например, патологии, связанные с метаболическим синдромом, предпочтительно сахарного диабета II типа или гипергликемии. Данные лекарственные препараты являются особенно активными у лиц пожилого возраста. Под «лицами пожилого возраста» понимают лиц, мужчин или женщин, возраст которых составляет 65 лет или более.
Термин «резистентность к инсулину» в рамках настоящего изобретения относится к состоянию, при котором нормальное количество инсулина не может вызвать физиологический или нормальный молекулярный ответ.
Вследствие этого, настоящее изобретение относится к производному имидазопиридина согласно настоящему изобретению для применения в качестве лекарственного препарата, предназначенного для лечения и/или предотвращения сахарного диабета, осложнений и/или сопутствующих патологий данного заболевания, предпочтительно сахарного диабета II типа и гипергликемии.
Авторы настоящего изобретения обнаружили, что производные согласно настоящему изобретению дают возможность стимулировать секрецию инсулина клетками INS1 и ингибировать продукцию глюкозы печенью на изолированных гепатоцитах крысы.
Предпочтительно сахарный диабет выбирают из раннего, позднего, детского сахарного диабета, диабета лиц пожилого возраста и беременных женщин, в частности, лиц пожилого возраста. Предпочтительно негативные явления сахарного диабета и осложнения и/или патологии, связанные с сахарным диабетом, выбирают из гипергликемии, функциональных и количественных отклонений показателей эндокринных клеток поджелудочной железы от нормы, резистентности к инсулину, диабетической нейропатии, диабетической нефропатии, диабетической ретинопатии, воспаления, ожирения, гипертензии, сердечно-сосудистых, микрососудистых, нейрологических проблем, а также проблем с заживлением ран. Предпочтительно, данные негативные явления представляют собой гипергликемию, функциональные и количественные отклонения показателей эндокринных клеток поджелудочной железы от нормы, резистентность к инсулину и воспаление.
Предпочтительно пациент, который получает лечение, имеет факторы риска, связанные с сахарным диабетом, т.е. заболевания, возникновение которых прямо или опосредованно связано с появлением сахарного диабета. В частности, такие факторы риска включают семейный анамнез, гестационный сахарный диабет, избыточную массу тела, ожирение, недостаточную физическую нагрузку, гипертензию, высокое содержание триглицеридов, воспаление и гиперлипидемию.
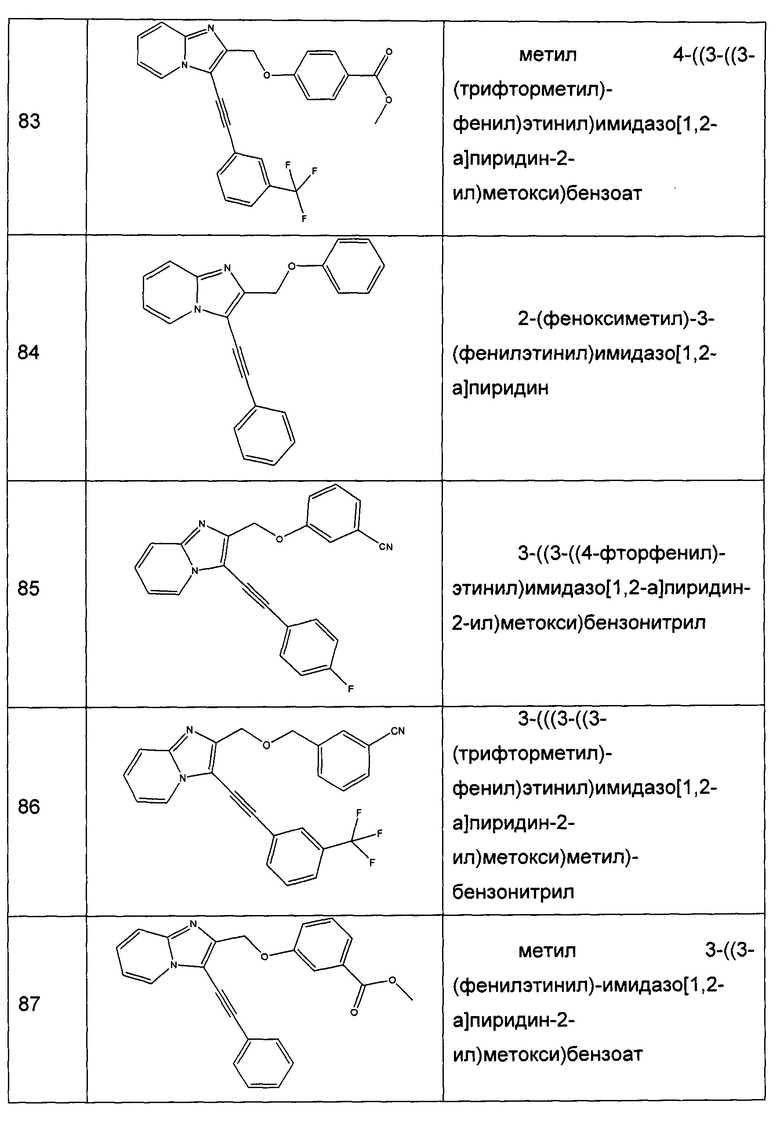
Настоящее изобретение также относится к применению производного имидазопиридина согласно настоящему изобретению для приготовления лекарственного препарата, предназначенного для лечения и/или предотвращения сахарного диабета, осложнений и/или сопутствующих патологий данного заболевания, в особенности сахарного диабета II типа и гипергликемии.
Наконец, настоящее изобретение относится к способу лечения и/или превентивного и/или профилактического лечения и/или лечения, направленного на замедление возникновения сахарного диабета, осложнений и/или сопутствующих патологий данного заболевания, предпочтительно сахарного диабета II типа и гипергликемии, причем указанный способ включает введение эффективного количества производного имидазопиридина согласно настоящему изобретению пациенту, нуждающемуся в указанном соединении.
Эффективное количество можно корректировать в зависимости от природы и тяжести патологии, которую подвергают лечению, от пути введения, а также от массы тела и возраста пациента. Как правило, единица дозирования находится в диапазоне от 0,5 мг до 2000 мг в сутки, которые принимают за один или несколько приемов, предпочтительно от 1 до 1000 мг.
Производные имидазопиридина согласно настоящему изобретению получены способами, хорошо известными специалисту в данной области техники, и частично способами, описанными ниже.
Настоящее изобретение станет более понятным после прочтения описания и следующих примеров, которые приведены в качестве неограничивающего пояснения.
Описание синтеза и общие схемы
Соединения общей формулы (I) могут быть приготовлены в результате применения или адаптирования любого способа, известного в качестве такового специалисту в данной области техники и/или достижимого специалистом в данной области техники, в особенности способов, описанных автором Larock в руководстве Comprehensive Organic Transformations, VCH Pub., 1989, или в результате применения или адаптирования способов, описанных в нижеследующих процедурах.
Синтез молекул общей формулы (I) близок, иногда идентичен синтезу, который был описан в документах с [1] по [8], при этом приведенный перечень ссылок не следует рассматривать как исчерпывающий.
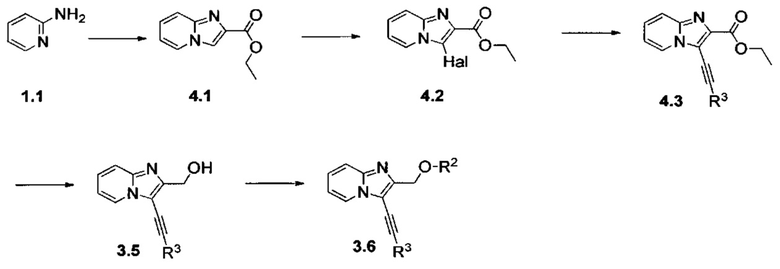
Различные группы с R1 по R3 и Y на схемах с 1 по 4 соответствуют определениям, данным ранее; «GP» означает защитную группу; «Hal» означает атом галогена.
Схема 1: Производные формулы I могут быть приготовлены согласно известному способу, начиная с 2-аминопиридина, который вступает в одноэтапную реакцию с 1,3-дихлорпропан-2-оном с образованием гетероцикла 1.2. Хлорид производных типа 1.2 является особенно подходящим для вовлечения в различные реакции О-, S- или N-алкилирования, которые проводят с алкоголятами, фенолятами и вторичными аминами в щелочной среде. После добавления цепи реакция селективной галогенизации (предпочтительно, реакция иодирования) позволяет получить производные типа 1.4, которые вовлекают в реакции Соногаширы с получением производных типа 1.5.

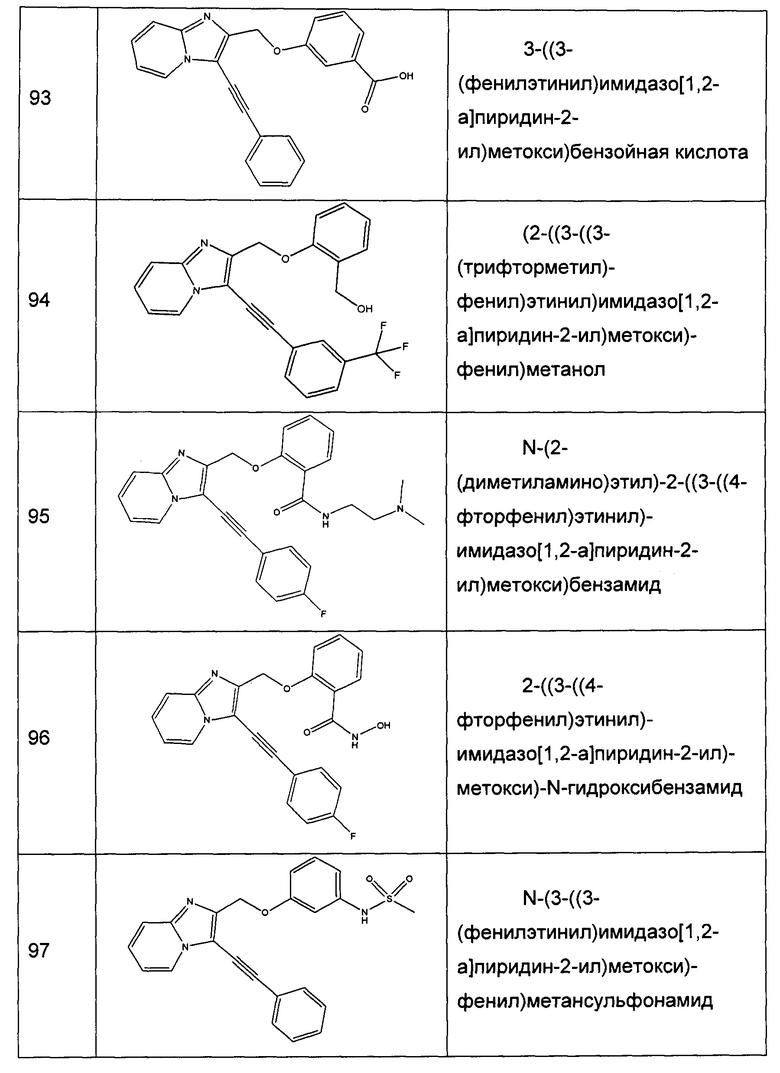
Схема 2: В конкретном случае, когда Y=NH, предпочтительно применение другого подхода для синтеза производных формулы I.

Начиная с продукта хлорирования вещества 1.2, проводят синтез аминов методом Габриэля. Производное фталимида 2.1 галогенизируют (предпочтительно иодируют), после чего вовлекают в реакции Соногаширы с получением производных типа 2.3; первичный амин выделяется в щелочных условиях. Затем производные типа 2.4 могут быть предпочтительно включены в реакции восстановительного аминирования, реакции ацилирования или дополнительно могут быть добавлены к изоцианатам с получением вторичных аминов, амидов, карбаматов или, дополнительно, карбамидов типа 2.5.
Схема 3: Альтернативный способ синтеза производных формулы I, в которых Y=O, проводили, начиная с имидазо[1,2-а]пиридин-2-илметанола 2.1, который может быть защищен подходящей защитной группой (например, триизопропилсилильной группой); указанное соединение вовлекали в реакцию галогенизации (предпочтительно, реакцию иодирования), а затем - в реакцию Соногаширы. Впоследствии защитную группу отщепляли с применением подходящих условий (фторида тетрабутиламмония в случае защиты триизопропилсилильной группой), после чего первичный спирт депротонировали щелочью и вовлекали в реакции алкилирования либо реакции ацилирования с получением производных типа 3.6.

Схема 4: Применяли второй альтернативный способ синтеза производных формулы I, у которых Y=O. В данном подходе прибегали к отличной исходной стратегии конструирования гетероцикла: к этил бромпирувату добавляли 2-аминопиридин с получением сложного эфира производного 4.1, который затем вовлекали в реакцию галогенизирования (предпочтительно, реакцию иодирования), а затем - в реакцию Соногаширы. Сложноэфирную функциональную группу восстанавливали до спирта стандартными методами (реакция с гидридом алюминия) для вовлечения производных типа 3.5 в реакции алкилирования либо реакции ацилирования.
Примеры
Оборудование и метод
Спектр протонного (1H) ядерного магнитного резонанса (ЯМР) получали на приборе Bruker Avance DPX300 (300,16 МГц). Химический сдвиг (δ) измеряли в миллионных долях (м.д.). Спектры калибровали по химическому сдвигу используемого дейтерированного растворителя. Константы взаимодействия (J) выражены в Герцах (Гц); мультиплетность представлена следующим образом: синглет (с), дублет (д), дублет дублетов (дд), триплет (т), триплет дублетов (тд), квадруплет (к), мультиплет (м). Масс-спектры (МС) получали на спектрометре Agilent Technologies MSD типа G1946A, образцы ионизировали с помощью источника «Химической ионизации при атмосферном давлении» (APCI, Atmospheric pressure chemical ionization).
Сокращения
Ряд примеров, приведенных ниже, используется для иллюстрации объема настоящего изобретения, но не для ограничения области применения настоящего изобретения.
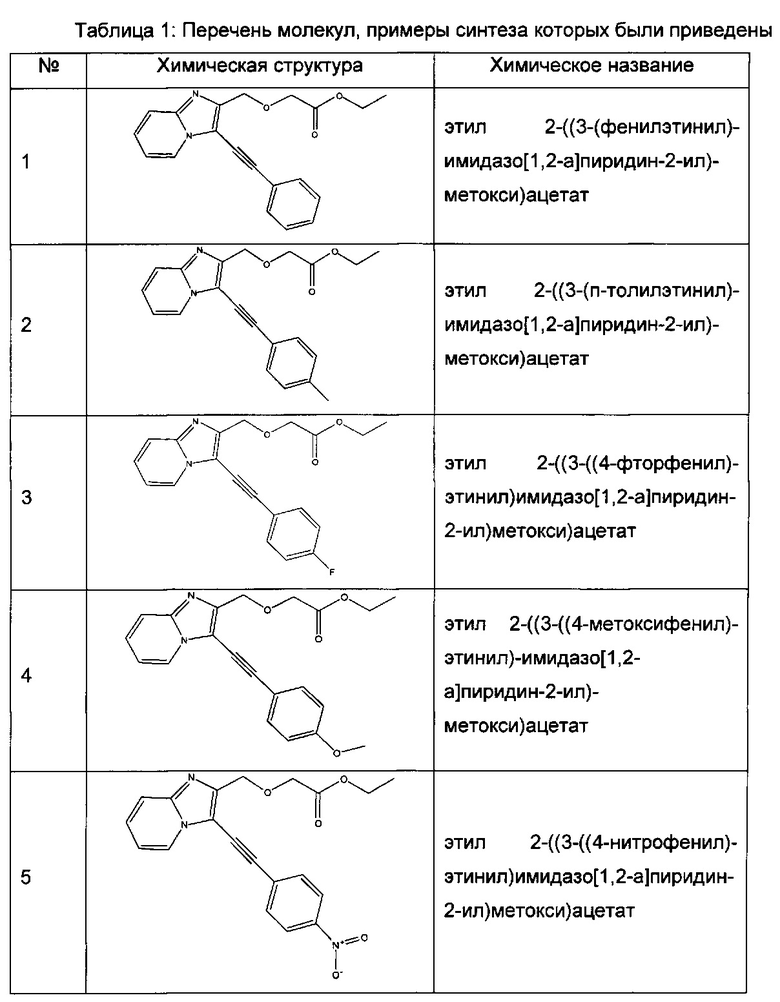
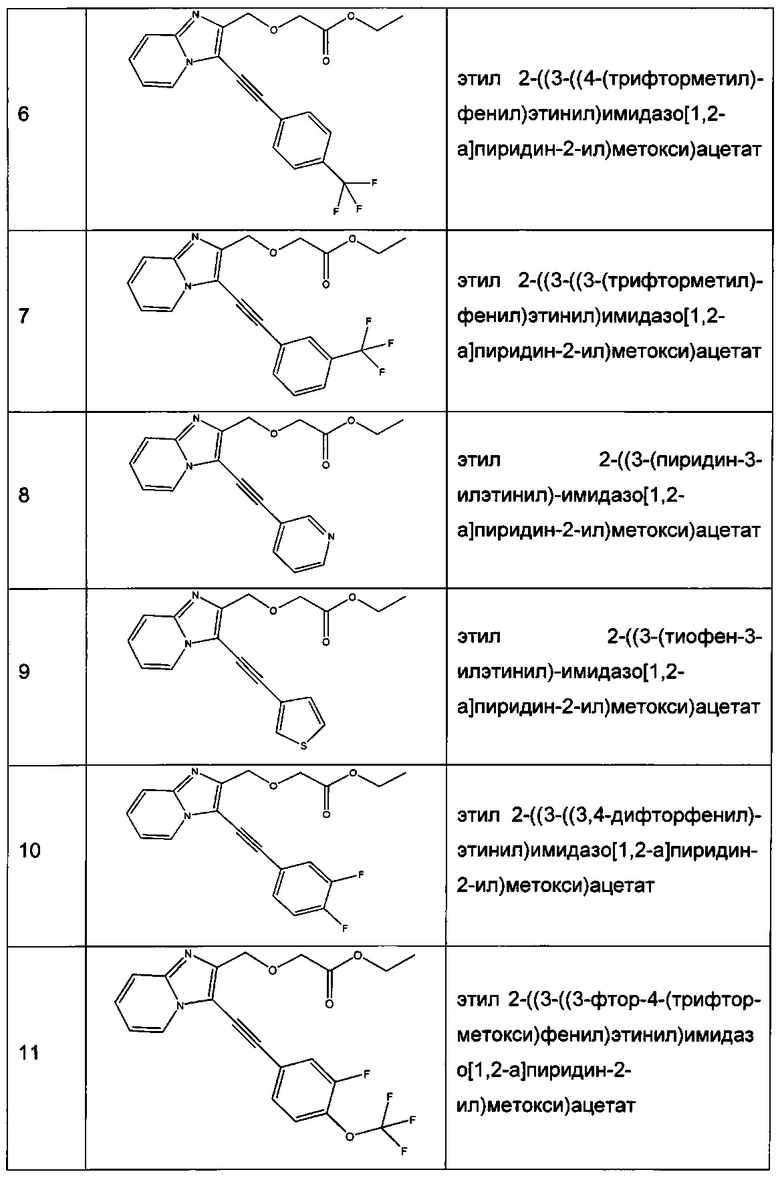















Пример 1: Приготовление производного №1: этил 2-((3-(фенилэтинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетат
Этап 1: Приготовление 2-(хлорметил)имидазо[1,2-а]пиридина

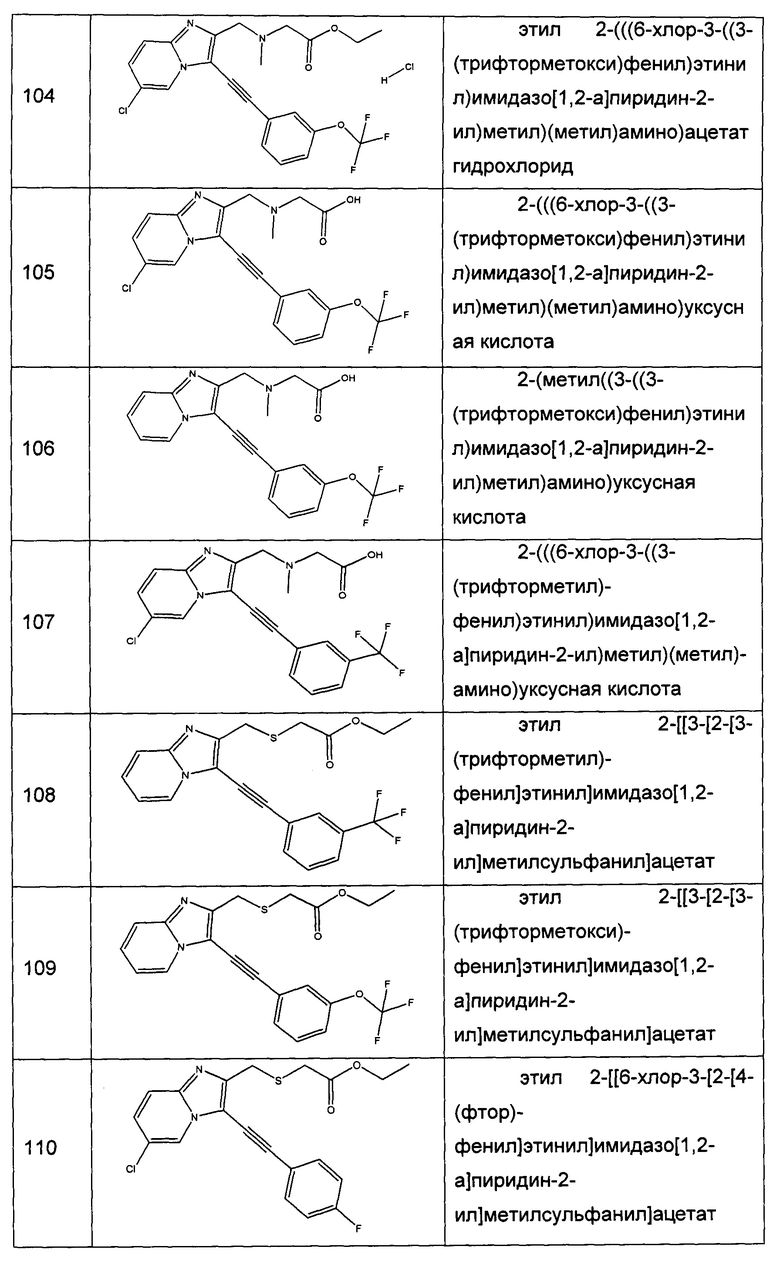
7,09 г (54,1 ммоль) 1,3-дихлорацетона солюбилизировали в 13 мл DME, к полученному раствору добавляли при перемешивании магнитной мешалкой 5 г (52,1 ммоль) 2-аминопиридина. Смесь перемешивали при к.т. в течение 16 ч. Образовавшееся твердое вещество выделяли в результате фильтрации и промывали 30 мл диизопропилового эфира. Полученное твердое вещество суспендировали в 125 мл абсолютного этанола, и полученную смесь перемешивали в колбе с обратным холодильником в течение 2 ч. Смесь концентрировали в вакууме, а затем полученный осадок переносили в 150 мл смеси, состоящей из воды и льда. Водную фазу ощелачивали до значения рН 8-9 добавлением насыщенного водного раствора NaHCO3. Водную фазу экстрагировали 3×100 мл дихлорметана. Объединенные органические фазы промывали 200 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент гептана/этилацетата, от 50% до 40% гептана, об./об. (объем/объем)). 4,77 г (выход = 55%) 2-(хлорметил)имидазо[1,2-а]пиридина получали в виде белого твердого вещества. ЖХ/МС (жидкостная хроматография/масс-спектрометрия): m/z=167 (MH+); степень чистоты (УФ-детекция при 254 нм) = 99%. 1H ЯМР (300 МГц, DMSO) δ 8,52 (д, J=6,8 Гц, 1H), 7,99 (с, 1H), 7,51 (д, J=9,1 Гц, 1H), 7,32-7,16 (м, 1H), 6,89 (т, J=6,8 Гц, 1H), 4,84 (с, 2H).
Этап 2: Приготовление 2-(имидазо[1,2-а]пиридин-2-илметокси)уксусной кислоты

4,24 г (25,4 ммоль) 2-(хлорметил)имидазо[1,2-а]пиридина растворяли при перемешивании магнитной мешалкой в 150 мл тетрагидрофурана, а затем добавляли 4,2 г (28 ммоль) иодида натрия. Смесь перемешивали при к.т. в течение 3 ч. В другой колбе 9,84 мл (101,8 ммоль) этил 2-гидроксиацетата солюбилизировали при перемешивании магнитной мешалкой в 100 мл тетрагидрофурана, после чего смесь охлаждали до температуры 0°C и добавляли по частям 4,072 г (101,8 ммоль) гидрида натрия. Полученную смесь перемешивали в течение 15 мин. при температуре 0°C, а затем добавляли по каплям свежеприготовленный раствор 2-(иодметил)имидазо[1,2-а]пиридина в тетрагидрофуране в течение 15 мин. Смесь перемешивали при к.т. в течение 24 ч, после чего фильтровали через диоксид кремния (элюент: 100% тетрагидрофуран). Фильтрат концентрировали в вакууме и непосредственно очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент дихлорметана/метанола, от 100% до 70% дихлорметана, об./об.; после чего проводили промывку картриджа 100% метанолом). 3,62 г (выход = 69%) 2-(имидазо[1,2-а]пиридин-2-илметокси)уксусной кислоты получали в виде бежевого твердого вещества. ЖХ/МС: m/z=207 (MH+); степень чистоты (УФ-детекция при 254 нм) = 98%.
Этап 3: Приготовление этил 2-(имидазо[1,2-а]пиридин-2-илметокси)ацетата

3,62 г (15,58 ммоль) 2-(имидазо[1,2-а]пиридин-2-илметокси)уксусной кислоты солюбилизировали при перемешивании магнитной мешалкой в 100 мл этанола и добавляли 0,094 мл (1,758 ммоль) серной кислоты. Полученную смесь перемешивали в колбе с обратным холодильником в течение 5 д. Реакционную среду концентрировали в вакууме и непосредственно очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: 100% этилацетат, затем дихлорметан/метанол, 9/1, об./об.). Реакционную среду промывали 2×10 мл воды, после чего экстрагировали 2×30 мл этилацетата. Объединенные органические фазы промывали 30 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. 0,162 мг (выход = 91%) изопропил 2-((3-((4-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетата получали в виде коричневого масла. MS: m/z=235 (MH+). 1H ЯМР (300 МГц, DMSO) δ 8,52 (д, J=7,9 Гц, 1H), 7,91 (с, 1H), 7,50 (д, J=9,1 Гц, 1H), 7,30-7,13 (м, 1H), 6,87 (т, J=6,7 Гц, 1H), 4,65 (с, 2H), 4,20 (с, 2H), 4,12 (к, J=7,1 Гц, 2H), 1,19 (т, J=7,2 Гц, 3H).
Этап 4: Приготовление этил 2-((3-иодимидазо[1,2-а]пиридин-2-ил)метокси)ацетата

0,45 г (1,921 ммоль) этил 2-(имидазо[1,2-а]пиридин-2-илметокси)ацетата солюбилизировали при перемешивании магнитной мешалкой в 6 мл ацетонитрила, к полученному раствору добавляли 0,475 г (2,113 ммоль) N-иодсукцинимида. Смесь перемешивали при к.т. в течение 16 ч. Твердое вещество выделяли в результате фильтрации, полученный фильтрат концентрировали в вакууме. Осадок растирали с 10 мл диизопропилового эфира, преципитат выделяли в результате фильтрации. 0,465 г (выход = 67%) этил 2-((3-иодимидазо[1,2-а]пиридин-2-ил)метокси)ацетата получали в виде желтого твердого вещества. ЖХ/МС: m/z=361 (MH+); степень чистоты (УФ-детекция при 254 нм) = 75%. 1H ЯМР (300 МГц, DMSO) δ 8,34 (д, J=6,9 Гц, 1H), 7,59 (д, J=8,1 Гц, 1H), 7,47-7,28 (м, 1H), 7,07 (д, J=5,7 Гц, 1H), 4,67 (с, 2H), 4,14 (т, J=10,7 Гц, 4Н), 1,22 (д, J=1,9 Гц, 3H).
Этап 5: Приготовление этил 2-((3-(фенилэтинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетата (производное номер 1)
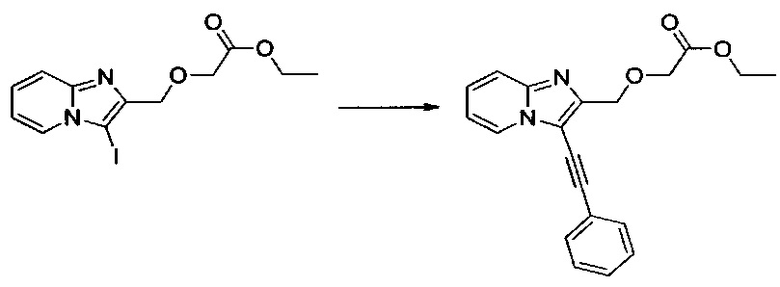
В колбе, помещенной в атмосферу с потоком аргона, 0,1 г (0,278 ммоль) этил 2-((3-иодимидазо[1,2-а]пиридин-2-ил)метокси)ацетата солюбилизировали при перемешивании магнитной мешалкой в 0,3 мл диметилформамида. К полученному раствору добавляли: 0,005 г (0,028 ммоль) иодида меди, 0,037 мл (0,333 ммоль) фенилацетилена и 0,3 мл (2,152 ммоль) триэтиламина. Реакционную среду дегазировали аргоном в течение 10 мин., после чего добавляли 0,025 г (0,028 ммоль) Pd2(dba)3. Смесь перемешивали при к.т. в течение 16 ч, после чего фильтровали через целит. Целит промывали 20 мл этилацетата, затем полученный фильтрат промывали 2×50 мл воды. После сепарации водную фазу экстрагировали 30 мл этилацетата, объединенные органические фазы промывали 30 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: гептан/этилацетат, 1/1, об./об.). 0,038 г (выход = 41%) этил 2-((3-(фенилэтинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетата получали в виде бледно-желтого твердого вещества. ЖХ/МС: m/z=335 (MH+); степень чистоты (УФ-детекция при 254 нм) = 99%. 1H ЯМР (300 МГц, DMSO) δ 8,63 (д, J=5,6 Гц, 1H), 7,68 (д, J=8,0 Гц, 3H), 7,54-7,39 (м, 4H), 7,14 (т, J=6,8 Гц, 1H), 4,79 (с, 2H), 4,24 (с, 2H), 4,09 (к, J=7,1 Гц, 2H), 1,15 (т, J=7,1 Гц, 3H).
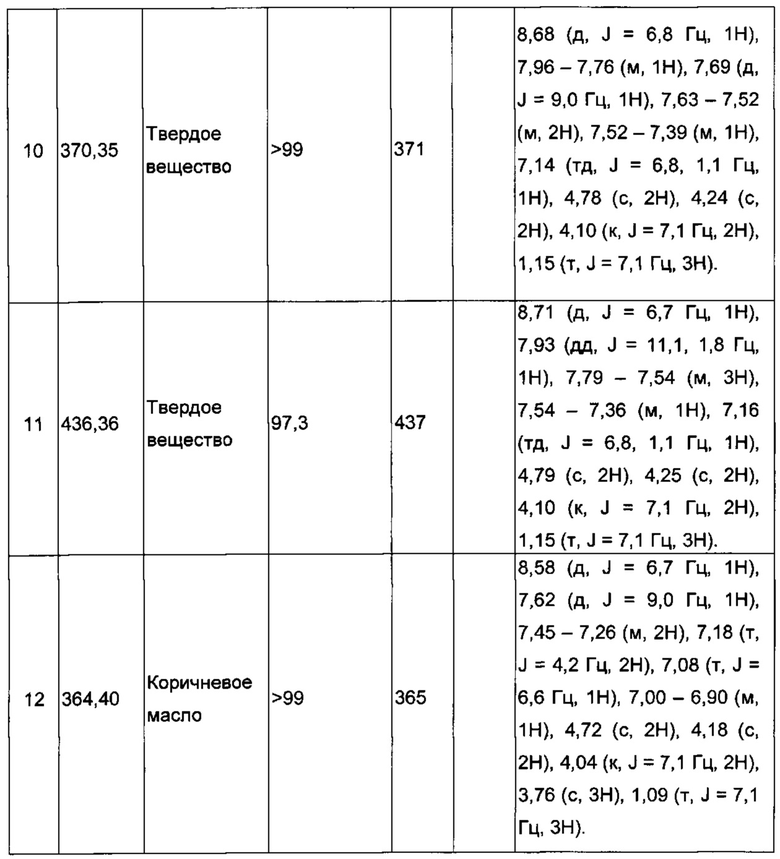
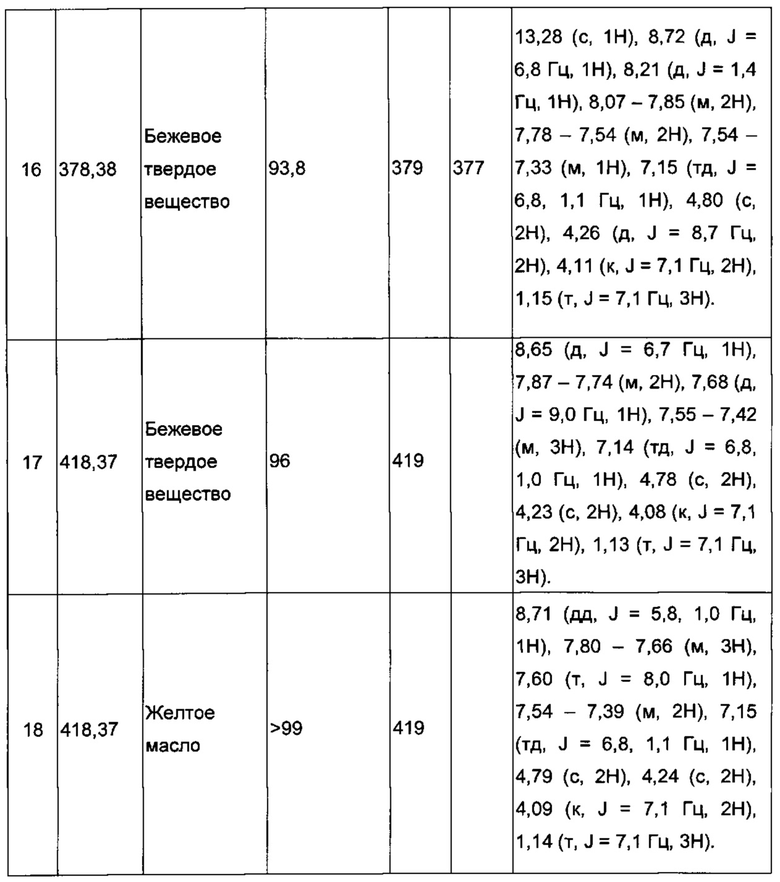
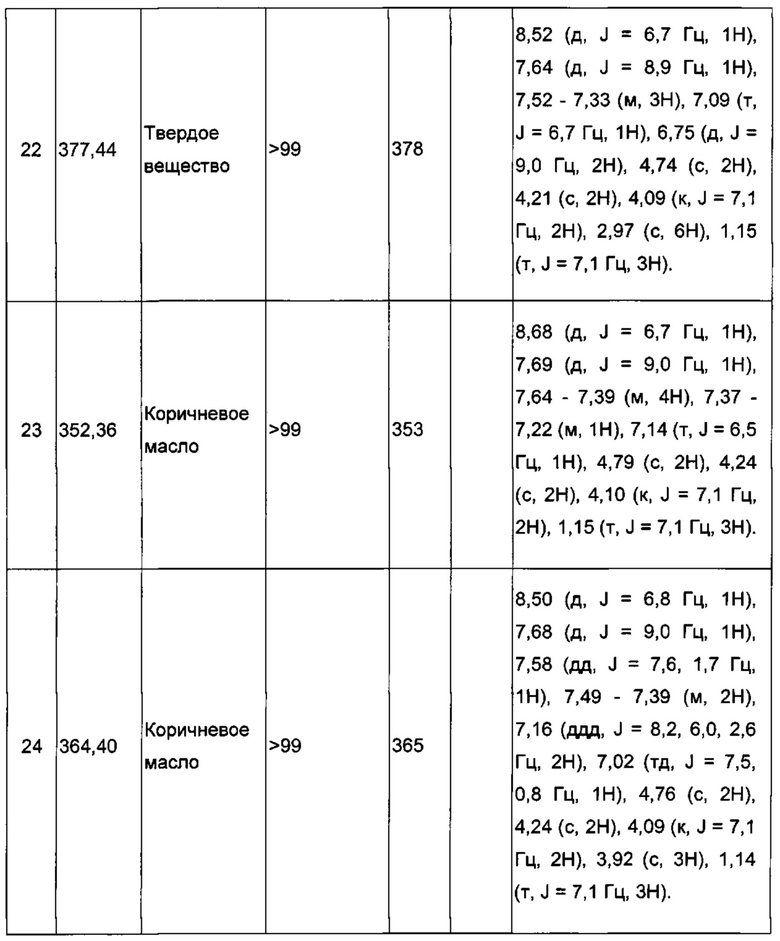
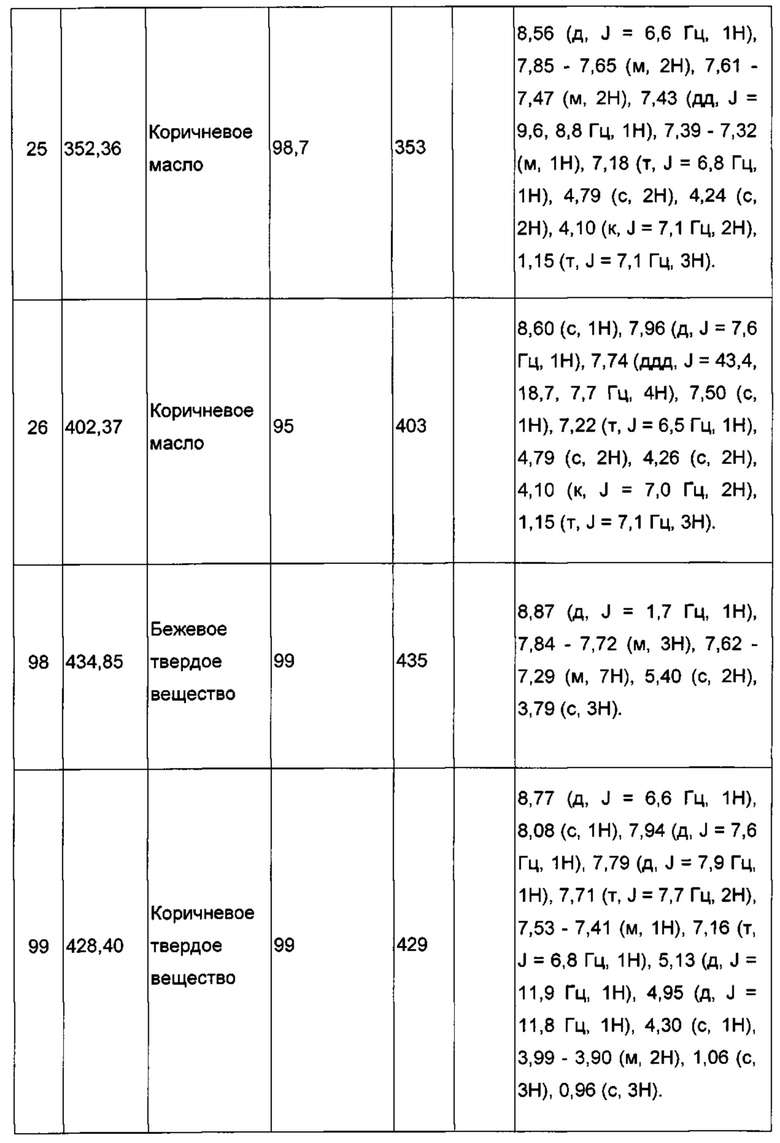
Производные под номерами с 2 по 26, 98 и 99 получали из этил 2-((3-иодимидазо[1,2-а]пиридин-2-ил)метокси)ацетата согласно аналогичной процедуре.




Пример 2: Приготовление производного №27: 2-((3-((3-циано фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)уксусная кислота
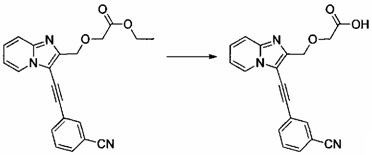
0,05 г (0,139 ммоль) этил 2-((3-((3-цианофенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетата солюбилизировали при перемешивании магнитной мешалкой в 4 мл смеси этанола/тетрагидрофурана (1/1, об./об.), а затем добавляли 0,167 мл (0,167 ммоль) 1 н водного раствора NaOH. Смесь перемешивали при к.т. в течение 16 ч, после чего добавляли 20 мл воды, а затем - 0,016 мл (0,278 ммоль) уксусной кислоты. Водную фазу экстрагировали 3×20 мл этилацетата. Объединенные органические фазы высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. 0,042 г (выход = 91%) 2-((3-((3-цианофенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)уксусной кислоты получали в виде бледно-желтого твердого вещества. ЖХ/МС: m/z=332 (MH+); степень чистоты (УФ-детекция при 254 нм) = 99%. 1H ЯМР (300 МГц, DMSO) δ 12,70 (с, 1H), 8,73 (д, J=6,7 Гц, 1H), 8,22 (т, J=1,3 Гц, 1H), 8,07-7,94 (м, 1H), 7,94-7,78 (м, 1H), 7,78-7,55 (м, 2H), 7,54-7,38 (м, 1H), 7,15 (тд, J=6,8, 0,9 Гц, 1H), 4,79 (с, 2H), 4,15 (с, 2H).
Производные под номерами с 28 по 44 были приготовлены согласно аналогичной процедуре.


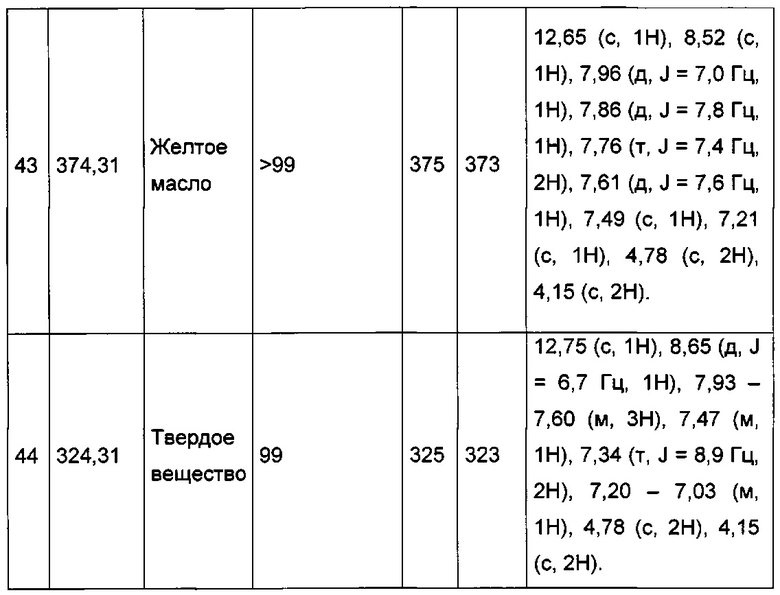
Пример 3: Приготовление производного №45: 2-((3-(фенил этинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетат натрия

0,061 г (0,182 ммоль) этил 2-((3-(фенилэтинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетата солюбилизировали при перемешивании магнитной мешалкой в 3 мл смеси, состоящей из метанола и тетрагидрофурана (1/1, об./об.), а затем добавляли 0,173 мл (0,173 ммоль) 1 н водного раствора NaOH. Смесь перемешивали при к.т. в течение 2 д, после чего концентрировали в вакууме. Неочищенный осадок растирали с 3 мл диизопропилового эфира, полученное твердое вещество выделяли в результате фильтрации и высушивали в вакуумном колпаке с получением 0,027 г (выход = 45%) 2-((3-(фенилэтинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетата натрия. ЖХ/МС: m/z=307 (MH+); степень чистоты (УФ-детекция при 254 нм) = 99%. 1H ЯМР (300 МГц, DMSO) δ 8,62 (д, J=6,7 Гц, 1H), 7,79-7,58 (м, 3H), 7,45 (дд, J=8,8, 7,2 Гц, 4H), 7,13 (т, J=6,5 Гц, 1H), 4,74 (с, 2H), 3,70 (с, 2H).
Производные под номерами с 46 по 53 были приготовлены согласно аналогичной процедуре.

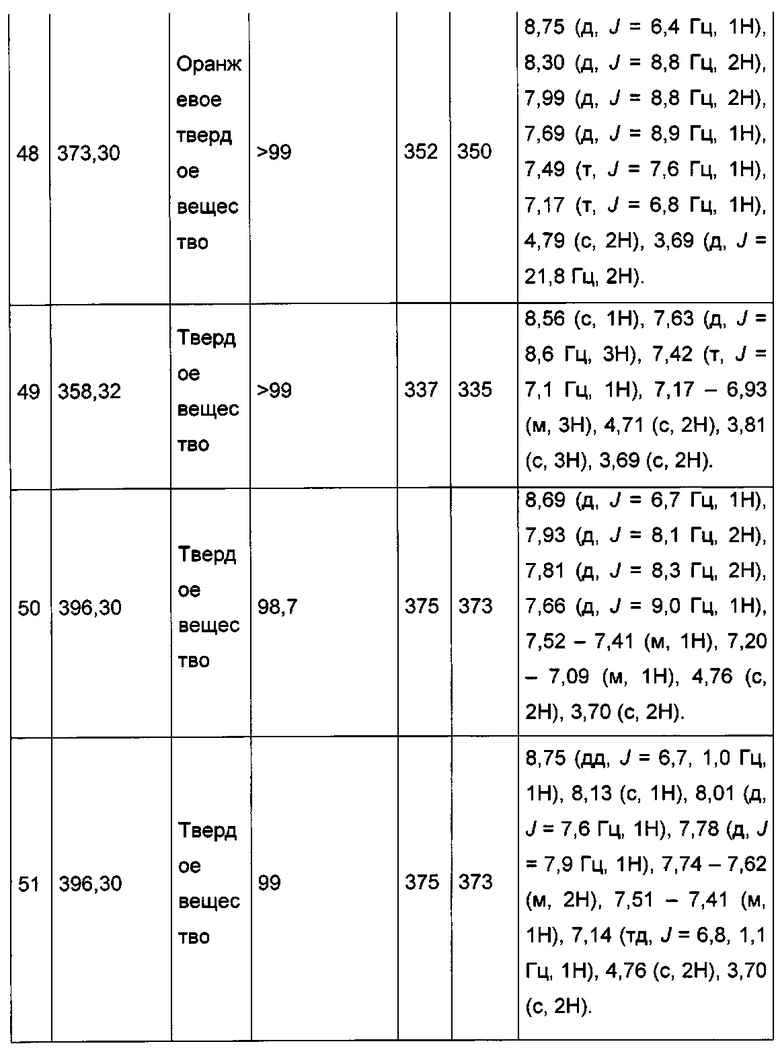

Пример 4: Приготовление производного №54: изопропил 2-((3-((4-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетат
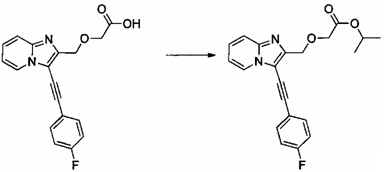
0,155 г (0,478 ммоль) 2-((3-((4-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)уксусной кислоты солюбилизировали при перемешивании магнитной мешалкой в 5 мл изопропанола и добавляли каплю серной кислоты. Полученную смесь перемешивали при температуре 60°C в течение 3 ч. Реакционную среду промывали 2×10 мл воды, а затем экстрагировали 2×30 мл этилацетата. Объединенные органические фазы промывали 30 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. 0,162 мг (выход = 91%) изопропил 2-((3-((4-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)-метокси)ацетата получали в виде коричневого масла. ЖХ/МС: m/z=367 (MH+); степень чистоты (УФ-детекция при 254 нм) = 98%. 1H ЯМР (300 МГц, DMSO) δ 8,62 (д, J=6,8 Гц, 1H), 7,74 (дд, J=8,7, 5,5 Гц, 2H), 7,67 (д, J=9,0 Гц, 1H), 7,45 (д, J=7,0 Гц, 1H), 7,33 (т, J=8,9 Гц, 2H), 7,12 (т, J=6,5 Гц, 1H), 4,93 (дт, J=12,5, 6,3 Гц, 1H), 4,78 (с, 2H), 4,19 (с, 2H), 1,15 (д, J=6,3 ГЦ, 6H).
Пример 5: Приготовление производного №55: N-(2-(диметиламино)этил)-2-((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетамид
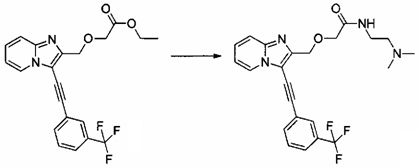
0,15 г (0,373 ммоль) этил 2-((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетата солюбилизировали при перемешивании магнитной мешалкой в 2 мл пиридина, к полученному раствору добавляли 0,409 мл (3,73 ммоль) N,N-диметилэтан-1,2-диамина и 0,249 мг (1,864 ммоль) трихлорида алюминия. Смесь перемешивали при к.т. в течение 18 ч, после чего обрабатывали 30 мл насыщенного водного раствора NaCl. Водную фазу экстрагировали 3×30 мл этилацетата. Объединенные органические фазы высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. 0,056 г (выход = 34%) N-(2-(диметиламино)этил)-2-((3-((3-(трифторметил)фенил)этинил)-имидазо[1,2-а]пиридин-2-ил)метокси)ацетамида получали в виде бежевого твердого вещества. ЖХ/МС: m/z=445 (MH+); степень чистоты (УФ-детекция при 254 нм) = 96%. 1H ЯМР (300 МГц, CDCl3) δ 7,85 (с, 1H), 7,78 (д, J=7,7 Гц, 2H), 7,70 (с, 2H), 7,58 (т, J=7,8 Гц, 2H), 7,23 (с, 1H), 5,11 (с, 2H), 4,31 (дд, J=26,6, 11,2 Гц, 2H), 3,83 (с, 2H), 3,33 (с, 2H), 2,89 (с, 6H).
Пример 6: Приготовление производного №56: 2-((3-((4-фтор фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)-1-морфолиноэтанон
Этап 1: Приготовление 2-((3-((4-фторфенил)этинил)-имидазо[1,2-а]пиридин-2-ил)метокси)ацетил хлорида
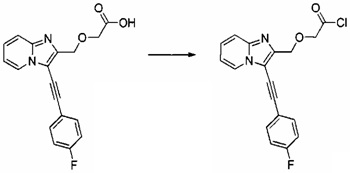
1,5 г (4,63 ммоль) 2-((3-((4-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)уксусной кислоты суспендировали в 40 мл дихлорметана при перемешивании магнитной мешалкой, смесь помещали на ледяную баню с температурой 0°C, а затем добавляли по каплям 1,215 мл (13,88 ммоль) оксалил хлорида, после чего добавляли несколько капель диметилформамида. Ледяную баню убирали, смесь перемешивали при к.т. в течение 1 ч. Реакционную среду концентрировали в вакууме и незамедлительно использовали на следующем этапе.
Этап 2: Приготовление 2-((3-((4-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)-1-морфолиноэтанона (производное №56)

0,806 мл (9,25 ммоль) морфолина солюбилизировали при перемешивании магнитной мешалкой в 4 мл дихлорметана, к смеси добавляли раствор 2-((3-((4-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетил хлорида в 4 мл дихлорметана при к.т. в течение 1 ч, после чего смесь обрабатывали 10 мл воды. Водную фазу экстрагировали 2×5 мл дихлорметана. Объединенные органические фазы промывали 5 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: 100% этилацетат). 0,211 г (выход = 58%) 2-((3-((4-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)-1-морфолиноэтанона получали в виде бледно-желтого твердого вещества. ЖХ/МС: m/z=394 (MH+); степень чистоты (УФ-детекция при 254 нм)=99%. 1H ЯМР (300 МГц, DMSO) δ 8,62 (д, J=6,8 Гц, 1H), 7,74 (дд, J=8,7, 5,5 Гц, 2H), 7,67 (д, J=9,0 Гц, 1H), 7,50-7,39 (м, 1H), 7,33 (т, J=8,9 Гц, 2H), 7,12 (т, J=6,8 Гц, 1H), 4,75 (с, 2H), 4,27 (с, 2H), 3,55-3,46 (м, 4H), 3,39 (с, 4H).
Производные под номерами с 57 по 59 получали из 2-((3-((4-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетил хлорида согласно аналогичной процедуре.


Пример 7: Приготовление производного номер 60: 2-((3-((4-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)-1-морфолиноэтанон
Этап 1: Приготовление 2-(((триизопропилсилил)окси)метил)имидазо[1,2-а]пиридина
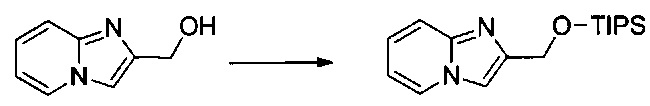 1 г (6,55 ммоль) имидазо[1,2-а]пиридин-2-илметанола солюбилизировали при перемешивании магнитной мешалкой в 25 мл дихлорметана, а затем медленно добавляли 2,145 мл (9,82 ммоль) триизопропилсилил хлорида. Смесь перемешивали при к.т. в течение 1 ч, после чего обрабатывали 100 мл воды. Водную фазу экстрагировали 3×50 мл дихлорметана. Объединенные органические фазы промывали 100 мл насыщенного водного раствора NaCl и высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: 100% дихлорметан, затем 100% этилацетат). 1,85 г (выход = 92%) 2-(((триизопропилсилил)окси)метил)имидазо[1,2-а]пиридина получали в виде бесцветного масла. ЖХ/МС: m/z=305 (MH+); степень чистоты (УФ-детекция при 254 нм) = 99%. 1H ЯМР (300 МГц, DMSO) δ 8,57-8,50 (м, 1H), 7,81 (с, 1H), 7,46 (дд, J=9,1, 0,6 Гц, 1H), 7,19 (ддд, J=9,1, 6,7, 1,3 Гц, 1H), 6,84 (тд, J=6,8, 1,1 Гц, 1H), 4,88 (д, J=0,6 Гц, 2H), 1,23-1,11 (м, 3H), 1,11-1,02 (м, 18H).
1 г (6,55 ммоль) имидазо[1,2-а]пиридин-2-илметанола солюбилизировали при перемешивании магнитной мешалкой в 25 мл дихлорметана, а затем медленно добавляли 2,145 мл (9,82 ммоль) триизопропилсилил хлорида. Смесь перемешивали при к.т. в течение 1 ч, после чего обрабатывали 100 мл воды. Водную фазу экстрагировали 3×50 мл дихлорметана. Объединенные органические фазы промывали 100 мл насыщенного водного раствора NaCl и высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: 100% дихлорметан, затем 100% этилацетат). 1,85 г (выход = 92%) 2-(((триизопропилсилил)окси)метил)имидазо[1,2-а]пиридина получали в виде бесцветного масла. ЖХ/МС: m/z=305 (MH+); степень чистоты (УФ-детекция при 254 нм) = 99%. 1H ЯМР (300 МГц, DMSO) δ 8,57-8,50 (м, 1H), 7,81 (с, 1H), 7,46 (дд, J=9,1, 0,6 Гц, 1H), 7,19 (ддд, J=9,1, 6,7, 1,3 Гц, 1H), 6,84 (тд, J=6,8, 1,1 Гц, 1H), 4,88 (д, J=0,6 Гц, 2H), 1,23-1,11 (м, 3H), 1,11-1,02 (м, 18H).
Этап 2: Приготовление 3-иод-2-(((триизопропилсилил)окси)метил)имидазо[1,2-а]пиридина

1,85 г (6,08 ммоль) 2-(((триизопропилсилил)окси)метил)имидазо[1,2-а]пиридина солюбилизировали при перемешивании магнитной мешалкой в 25 мл ацетонитрила, к полученному раствору добавляли 1,409 г (6,08 ммоль) N-иодсукцинимида. Смесь перемешивали при к.т. в течение 16 ч, после чего обрабатывали 100 мл воды. Водную фазу экстрагировали 3×50 мл дихлорметана. Объединенные органические фазы промывали 100 мл насыщенного водного раствора NaCl и высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. 2,29 г (выход = 83%) 3-иод-2-(((триизопропилсилил)окси)метил)имидазо[1,2-а]пиридина получали в виде бледно-желтого твердого вещества. ЖХ/МС: m/z=431 (MH+); степень чистоты (УФ-детекция при 254 нм) = 95%. 1H ЯМР (300 МГц, DMSO) δ 8,30 (дд, J=5,6, 2,2 Гц, 1H), 7,59 (дд, J=14,3, 9,1 Гц, 1H), 7,33 (тт (триплет триплетов), J=6,8, 4,1 Гц, 1H), 7,14-6,96 (м, 1H), 4,80 (с, 2H), 1,27-0,90 (м, 21H).
Этап 3: Приготовление 3-(фенилэтинил)-2-(((триизопропилсилил)окси)метил)имидазо[1,2-а]пиридина

В колбе, помещенной в атмосферу с потоком аргона, 2,29 г (5,32 ммоль) 3-иод-2-(((триизопропилсилил)окси)метил)имидазо[1,2-а]пиридина солюбилизировали при перемешивании магнитной мешалкой в 11 мл диметилформамида. К полученному раствору добавляли: 0,103 г (0,532 ммоль) иодида меди, 0,715 мл (6,38 ммоль) фенилацетилена и 11,12 мл (80 ммоль) триэтиламина. Реакционную среду дегазировали аргоном в течение 10 мин., после чего добавляли 0,487 г (0,532 ммоль) Pd2(dba)3. Смесь перемешивали при к.т. в течение 16 ч, после чего фильтровали через целит. Целит промывали 200 мл этилацетата. Полученный фильтрат промывали 2×100 мл воды и 100 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент гептана/этил ацетата, от 90% до 80% гептана, об./об.). 1,56 г (выход = 72%) 2 3-(фенилэтинил)-2-(((триизопропилсилил)окси)метил)имидазо[1,2-а]пиридина получали в виде коричневого твердого вещества. ЖХ/МС: m/z=405 (MH+); степень чистоты (УФ-детекция при 254 нм) = 99%. 1H ЯМР (300 МГц, DMSO) δ 8,60 (д, J=6,7 Гц, 1H), 7,72-7,55 (м, 3H), 7,52-7,36 (м, 4Н), 7,17-7,06 (м, 1H), 4,94 (с, 2H), 1,14 (м, 3H), 1,04 (д, J=6,5 Гц, 18H).
Этап 4: Приготовление (3-(фенилэтинил)имидазо[1,2-а]пиридин-2-ил)метанола (производное номер 60)
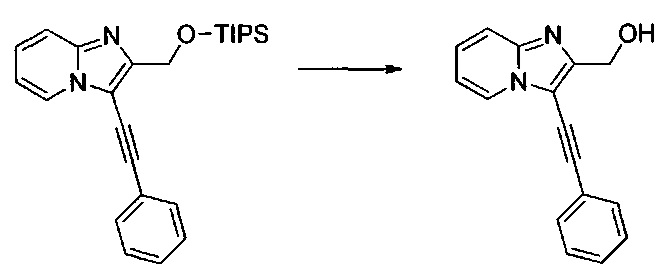
1,56 г (3,86 ммоль) 2-(((триизопропилсилил)окси)метил)имидазо[1,2-а]пиридина солюбилизировали при перемешивании магнитной мешалкой в 30 мл тетрагидрофурана, к полученному раствору добавляли 4,24 мл (4,24 ммоль) 1 М раствора тетрабутиламмония фторида в тетрагидрофуране. Смесь перемешивали при к.т. в течение 1 часа, после чего концентрировали в вакууме. Неочищенный осадок непосредственно очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: 100% этилацетат). 0,83 г (выход = 86%) (3-(фенилэтинил)имидазо[1,2-а]пиридин-2-ил)метанола получали в виде оранжевого твердого вещества. ЖХ/МС: m/z=249 (MH+); степень чистоты (УФ-детекция при 254 нм) = 99%. 1H ЯМР (300 МГц, DMSO) δ 8,59 (дт, J=6,7, 1,1 Гц, 1H), 7,77-7,56 (м, 3H), 7,56-7,32 (м, 4H), 7,11 (тд, J=6,8, 1,1 Гц, 1H), 5,33 (т, J=5,9 Гц, 1H), 4,67 (д, J=5,9 Гц, 2H).
Производное номер 100 было получено согласно аналогичной процедуре.

Пример 8: Приготовление производного №61: изобутил ((3-(фенил этинил)имидазо[1,2-а]пиридин-2-ил)метил)карбонат

0,1 г (0,403 ммоль) (3-(фенилэтинил)имидазо[1,2-а]пиридин-2-ил)метанола солюбилизировали при перемешивании магнитной мешалкой в 3 мл дихлорметана. Добавляли 0,084 мл (0,604 ммоль) триэтиламина и 0,059 мл (0,443 ммоль) изобутил хлорформата. Смесь перемешивали при к.т. в течение 15 мин., после чего концентрировали в вакууме. Осадок переносили в 10 мл диизопропилового эфира, и твердое вещество выделяли в результате фильтрации. Твердое вещество очищали методом препаративной ВЭЖХ. 0,084 г (выход = 59%) изобутил ((3-(фенилэтинил)имидазо[1,2-а]пиридин-2-ил)метил)карбоната получали в виде бледно-желтого твердого вещества. ЖХ/МС: m/z=349 (MH+); степень чистоты (УФ-детекция при 254 нм) = 99%. 1H ЯМР (300 МГц, DMSO) δ 8,70 (д, J=6,8 Гц, 1H), 7,79-7,65 (м, 3H), 7,60-7,45 (м, 4H), 7,23 (тд, J=6,8, 1,1 Гц, 1H), 5,42 (с, 2H), 3,91 (д, J=6,6 Гц, 2H), 1,95-1,80 (м, 1H), 0,86 (д, J=6,7 Гц, 6H).
Пример 9: Приготовление производного №62: (3-(фенилэтинил)имидазо[1,2-а]пиридин-2-ил)метилморфолин-4-карбоксилат

0,117 г (0,467 ммоль) (3-(фенилэтинил)имидазо[1,2-а]пиридин-2-ил)метанола солюбилизировали при перемешивании магнитной мешалкой в 4 мл тетрагидрофурана. Добавляли 0,228 г (0,7 ммоль) карбоната цезия и 0,61 мл (0,513 ммоль) морфолин-4-карбонил хлорида. Смесь перемешивали при к.т. в течение 16 ч. Дополнительно добавляли 1,22 мл (1,026 ммоль) морфолин-4-карбонил хлорида, смесь перемешивали при к.т. в течение 2 дней, после чего обрабатывали 10 мл насыщенного водного раствора NaCl. Водную фазу экстрагировали 3×10 мл этилацетата. Объединенные органические фазы высушивали над Na2SO4, который затем удаляли в результате фильтрации. Неочищенный осадок очищали методом препаративной ВЭЖХ. 0,078 г (выход = 46%) (3-(фенилэтинил)имидазо[1,2-а]пиридин-2-ил)метилморфолин-4-карбоксилата получали в виде бежевого твердого вещества. ЖХ/МС: m/z=362 (MH+); степень чистоты (УФ-детекция при 254 нм)=99%. 1H ЯМР (300 МГц, DMSO) δ 8,60 (д, J=6,7 Гц, 1H), 7,66 (дд, J=11,0, 5,4 Гц, 3H), 7,47 (дд, J=4,1, 1,3 Гц, 4H), 7,14 (т, J=6,4 Гц, 1H), 5,33 (с, 2H), 3,48 (с, 4H), 3,34 (с, 4H).
Пример 10: Приготовление производного №63: (3-(фенилэтинил)имидазо[1,2-а]пиридин-2-ил)метилдиэтилкарбамат

0,1 г (0,403 ммоль) (3-(фенилэтинил)имидазо[1,2-а]пиридин-2-ил)метанола солюбилизировали при перемешивании магнитной мешалкой в 3 мл диметилформамида. Смесь охлаждали до температуры 0°C и добавляли по частям 0,048 г (1,208 ммоль) гидрида натрия (60 масс. %). Полученную смесь перемешивали в течение 10 мин. при температуре 0°C, а затем добавляли 0,204 мл (1,611 ммоль) диэтилкарбамоил хлорида. Ледяную баню убирали, смесь перемешивали при к.т. в течение 2 ч, после чего обрабатывали 30 мл насыщенного водного раствора NaCl. Водную фазу экстрагировали 3×10 мл этилацетата. Объединенные органические фазы высушивали над Na2SO4, который затем удаляли в результате фильтрации. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: гептан/этилацетат, 6/4, об./об.). 0,101 г (выход = 72%) (3-(фенилэтинил)имидазо[1,2-а]пиридин-2-ил)метилдиэтилкарбамата получали в виде бледно-желтого твердого вещества. ЖХ/МС: m/z=348 (MH+); степень чистоты (УФ-детекция при 254 нм) = 99%. 1H ЯМР (300 МГц, DMSO) δ 8,61 (д, J=6,8 Гц, 1H), 7,74-7,59 (м, 3H), 7,53-7,38 (м, 4H), 7,13 (тд, J=6,8, 1,1 Гц, 1H), 5,30 (с, 2H), 3,22 (к, J=7,1 Гц, 4H), 1,01 (т, J=7,1 Гц, 6H).
Пример 11: Приготовление производного №64: 3-((2-((2-гидрокси этокси)метил)имидазо[1,2-а]пиридин-3-ил)этинил)бензонитрил
Этап 1: Приготовление 2-(хлорметил)-3-иодимидазо[1,2-а]пиридина

5 г (30 ммоль) 2-(хлорметил)имидазо[1,2-а]пиридина солюбилизировали при перемешивании магнитной мешалкой в 60 мл ацетонитрила, добавляли 7,31 г (31,5 ммоль) N-иодсукцинимида, смесь перемешивали при к.т. в течение 15 мин. Образовавшееся твердое вещество выделяли в результате фильтрации, промывали 30 мл воды и высушивали в вакуумном колпаке. 8,46 г (выход = 95%) 2-(хлорметил)-3-иодимидазо[1,2-а]пиридина получали в виде белого твердого вещества. ЖХ/МС: m/z=293 (MH+); степень чистоты (УФ-детекция при 254 нм) = 99%. 1H ЯМР (300 МГц, DMSO) δ 8,31 (д, J=6,9 Гц, 1H), 7,58 (д, J=9,1 Гц, 1H), 7,46-7,29 (м, 1H), 7,07 (тд, J=6,8, 1,0 Гц, 1H),4,81 (с, 2H).
Этап 2: Приготовление (3-иодимидазо[1,2-а]пиридин-2-ил)метил-3-гидроксипропаноата

В колбе, помещенной в атмосферу с потоком аргона, 0,515 мл (3,42 ммоль) 2-(хлорметил)-3-иодимидазо[1,2-а]пиридина солюбилизировали при перемешивании магнитной мешалкой в 8 мл тетрагидрофурана, затем добавляли 0,06 г (1,504 ммоль) гидрида натрия (60% масс), смесь перемешивали при к.т. в течение 30 мин. Затем добавляли 0,4 г (1,368 ммоль) 2-(хлорметил)-3-иодимидазо[1,2-а]пиридина и 0,05 мл (0,137 ммоль) тетрабутил иодида аммония. Полученную смесь перемешивали в колбе с обратным холодильником в течение 2 д. Реакционную среду обрабатывали 30 мл насыщенного водного раствора хлорида аммония. Водную фазу экстрагировали 2×30 мл этилацетата. Объединенные органические фазы промывали 30 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент гептана/этилацетата, 1/1, об./об.). 0,17 г (выход = 36%) (3-иодимидазо[1,2-а]пиридин-2-ил)метил 3-гидроксипропаноата получали в виде коричневого твердого вещества. ЖХ/МС: m/z=347 (MH+); степень чистоты (УФ-детекция при 254 нм) = 99%. 1H ЯМР (300 МГц, DMSO) δ 8,32 (д, J=6,9 Гц, 1H), 7,57 (д, J=9,1 Гц, 1H), 7,42-7,24 (м, 1H), 7,07 (т, J=6,4 Гц, 1H), 5,16 (с, 2H), 4,73 (т, J=5,3 Гц, 1H), 3,64 (дд, J=11,6, 6,0 Гц, 2H), 2,46 (м, 2H).
Этап 3: Приготовление (3-((3-цианофенил)этинил)имидазо[1,2-а]пиридин-2-ил)метил-3-гидроксипропаноата (производное №64)

В колбе, помещенной в атмосферу с потоком аргона, 0,138 г (0,397 ммоль) (3-иодимидазо[1,2-а]пиридин-2-ил)метил 3-гидроксипропаноата солюбилизировали при перемешивании магнитной мешалкой в 4 мл диметилформамида. К полученному раствору добавляли: 0,008 г (0,04 ммоль) иодида меди, 0,06 г (0,477 ммоль) 3-этинилбензонитрила и 0,831 мл (5,96 ммоль) триэтиламина. Реакционную среду дегазировали аргоном в течение 10 мин., после чего добавляли 0,036 г (0,04 ммоль) Pd2(dba)3. Смесь перемешивали при к.т. в течение 1 ч, после чего обрабатывали 50 мл насыщенного водного раствора NaCl, водную фазу экстрагировали 2×50 мл этилацетата. Объединенные органические фазы промывали 50 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: гептан/этилацетат, 2/8, об./об.). 0,12 г (выход = 82%) (3-((3-цианофенил)этинил)имидазо[1,2-а]пиридин-2-ил)метил-3-гидроксипропаноата получали в виде желтого твердого вещества. ЖХ/МС: m/z=346 (MH+); степень чистоты (УФ-детекция при 254 нм)=93%. 1H ЯМР (300 МГц, DMSO) δ 8,74 (д, J=6,8 Гц, 1H), 8,26 (с, 1H), 7,98 (дд, J=5,3, 3,9 Гц, 1H), 7,90 (дд, J=5,2, 3,8 Гц, 1H), 7,76-7,63 (м, 2H), 7,57-7,41 (м, 1H), 7,18 (тд, J=6,8, 1,0 Гц, 1H), 5,35 (с, 2H), 4,76 (т, J=5,3 Гц, 1H), 3,67 (дд, J=11,6, 6,2 Гц, 2H), 2,54-2,49 (м, 2H).
Производные под номерами с 65 по 67 были приготовлены согласно аналогичной последовательности этапов с 1 по 4.

Пример 12: Приготовление производного №68: 2-(((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метил)амино)уксусная кислота
Этап 1: Приготовление 2-(имидазо[1,2-а]пиридин-2-илметил)изоиндолин-1,3-диона
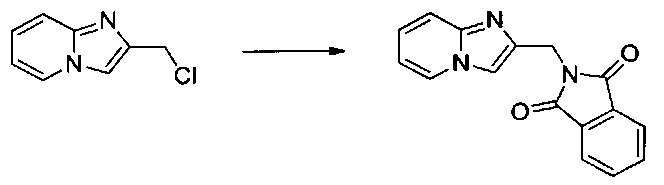
1,5 г (9 ммоль) 2-(хлорметил)имидазо[1,2-а]пиридина солюбилизировали при перемешивании магнитной мешалкой в 55 мл диметилформамида, добавляли по частям 1,834 г (9,9 ммоль) фталимида калия, после чего полученную смесь перемешивали при температуре 60°C в течение 2 ч. Полученное твердое вещество выделяли в результате фильтрации, промывали 30 мл диизопропилового эфира и высушивали в вакуумном колпаке с получением 1,81 г (выход = 67%) 2-(имидазо [1,2-а]пиридин-2-илметил)изоиндолин-1,3-диона в виде белого твердого вещества. ЖХ/МС: m/z=278 (MH+); степень чистоты (УФ-детекция при 254 нм) = 93%. 1H ЯМР (300 МГц, DMSO) δ 8,45 (д, J=6,8 Гц, 1H), 7,96-7,67 (м, 5H), 7,45 (дд, J=9,1, 0,5 Гц, 1H), 7,33-7,14(м, 1H), 6,84 (тд, J=6,8, 1,1 Гц, 1H), 4,88 (с, 2H).
Этап 2: Приготовление 2-((3-иодимидазо[1,2-а]пиридин-2-ил)метил)изоиндолин-1,3-диона
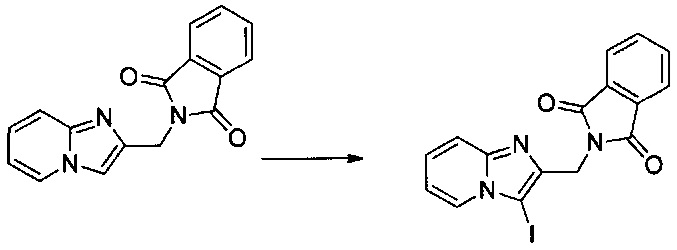
1,7 г (5,7 ммоль) 2-(хлорметил)имидазо[1,2-а]пиридина солюбилизировали при перемешивании магнитной мешалкой в 65 мл ацетонитрила, добавляли 1,389 г (5,99 ммоль) N-иодсукцинимида, смесь перемешивали при к.т. в течение 1 ч. Образовавшееся твердое вещество выделяли в результате фильтрации, промывали 20 мл диизопропилового эфира и высушивали в вакуумном колпаке. 0,105 г (выход = 87%) 2-((3-иодимидазо[1,2-а]пиридин-2-ил)метил)изоиндолин-1,3-диона получали в виде белого твердого вещества. ЖХ/МС: m/z=404 (MH+); степень чистоты (УФ-детекция при 254 нм) = 78%. 1H ЯМР (300 МГц, DMSO) δ 8,29 (д, J=6,8 Гц, 1H), 7,89 (д, J=5,6 Гц, 4H), 7,48 (д, J=9,0 Гц, 1H), 7,36-7,24 (м, 1H), 7,03 (т, J=6,4 Гц, 1H), 4,89 (с, 2H).
Этап 3: Приготовление 2-((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метил)изоиндолин-1,3-диона

В колбе, помещенной в атмосферу с потоком аргона, 2,1 г (5,21 ммоль) 2-((3-иодимидазо[1,2-а]пиридин-2-ил)метил)изоиндолин-1,3-диона солюбилизировали при перемешивании магнитной мешалкой в 11 мл диметилформамида. К полученному раствору добавляли: 0,008 г (0,04 ммоль) иодида меди, 0,921 мл (6,25 ммоль) 3-трифторметилфенил ацетилена и 10,89 мл (78 ммоль) триэтиламина. Реакционную среду дегазировали аргоном в течение 10 мин., после чего добавляли 0,477 г (0,521 ммоль) Pd2(dba)3. Смесь перемешивали при к.т. в течение 16 ч, после чего фильтровали на картридже с гелем диоксида кремния, который затем промывали 300 мл этилацетата. Органическую фазу промывали 3×150 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент гептана/этилацетата, от 50% до 40% гептана, об./об.). 1,41 г (выход = 59%) 2-((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метил)изоиндолин-1,3-диона получали в виде коричневого твердого вещества. ЖХ/МС: m/z=446 (MH+); степень чистоты (УФ-детекция при 254 нм) = 97%. 1H ЯМР (300 МГц, DMSO) δ 8,67 (д, J=6,7 Гц, 1H), 7,91 (дд, J=5,6, 3,0 Гц, 2H), 7,82 (дд, J=5,5, 3,1 Гц, 2H), 7,69 (дд, J=12,5, 8,5 Гц, 2H), 7,63-7,52 (м, 2H), 7,52-7,42 (м, 2H), 7,13 (тд, J=6,8, 1,1 Гц, 1H), 5,09 (с, 2H).
Этап 4: Приготовление (3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метанамина

1,41 г (3,08 ммоль) 2-((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метил)изоиндолин-1,3-диона солюбилизировали при перемешивании магнитной мешалкой в 30 мл этанола, добавляли 0,978 мл (30,8 ммоль) гидразина моногидрата. Смесь, которая постепенно приобретала молочно-белый цвет, перемешивали при к.т. в течение 1,5 ч. Полученный после фильтрования осадок, который представлял собой твердое вещество, выделяли в результате фильтрации и промывали 2×25 мл этанола. Фильтрат концентрировали в вакууме. Осадок растирали с 20 мл этанола, твердое вещество выделяли в результате фильтрации, и фильтрат концентрировали в вакууме с получением 0,715 г (выход = 69%) (3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метанамина в виде бледно-желтого твердого вещества. ЖХ/МС: m/z=316 (MH+); степень чистоты (УФ-детекция при 254 нм) = 94%. 1H ЯМР (300 МГц, DMSO) δ 8,73 (д, J=6,7 Гц, 1H), 8,12 (с, 1H), 7,98 (д, J=7,7 Гц, 1H), 7,82-7,63 (м, 3H), 7,49-7,40 (м, 1H), 7,13 (тд, J=6,8, 1,1 Гц, 1H), 3,96 (с, 2H).
Этап 5: Приготовление 2-(((3-((3-(трифторметил) фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метил)амино)уксусной кислоты (производное номер 68)

Реактор, оборудованный фильтром и системой опорожнения, помещали на вортекс; в реактор вносили 0,33 г (0,656 ммоль) цианоборгидридной смолы. Смола набухала в 3 мл дихлорметана, смесь перемешивали при к.т. в течение 10 мин., после чего дихлорметан удаляли в результате фильтрации. Процедуру повторяли во второй раз, а затем добавляли смесь 0,2 г (3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метанамина и 0,055 г (0,596 ммоль) гидрата 2-оксоуксусной кислоты, солюбилизированных в 3 мл метанола. Полученную смесь перемешивали на вортексе при медленном перемешивании при к.т. в течение 24 ч. Смолу удаляли в результате фильтрации, промывали 15 мл метанола, 15 мл дихлорметана, а затем снова 15 мл метанола. Полученный фильтрат концентрировали в вакууме, осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: дихлорметан/метанол/водный аммоний (20 масс. %), 80/20/2, об./об./об.). 0,049 г (выход = 22%) 2-(((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метил)амино)уксусной кислоты получали в виде белого твердого вещества. ЖХ/МС: m/z=374 (MH+); степень чистоты (УФ-детекция при 254 нм) = 98%. 1H ЯМР (300 МГц, DMSO) δ 8,76 (д, J=6,7 Гц, 1H), 8,13 (с, 1H), 8,01 (д, J=7,6 Гц, 1H), 7,78 (д, J=7,9 Гц, 1H), 7,69 (дд, J=8,3, 7,1 Гц, 2H), 7,53-7,42 (м, 1H), 7,16 (тд, J=6,8, 1,0 Гц, 1H), 4,17 (с, 2H), 3,31 (с, 6H).
Пример 13: Приготовление производного №69: N-((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метил)пропан-2-сульфонамид
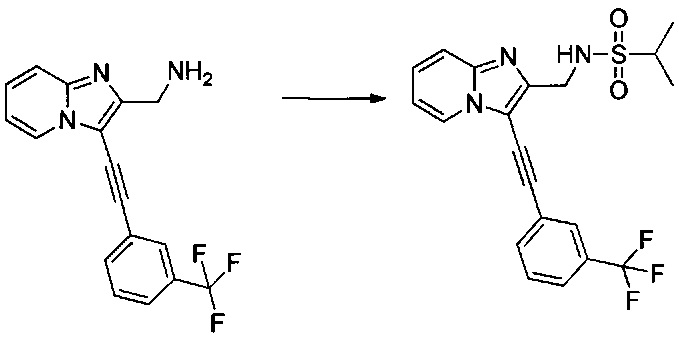
0,08 г (0,239 ммоль) (3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метанамина (приготовленного согласно этапам с 1 по 4 примера 11) солюбилизировали при перемешивании магнитной мешалкой в 2 мл дихлорметана, добавляли 0,208 мл (1,193 ммоль) диизопропилэтиламина и 0,083 мл (0,716 ммоль) изопропилсульфонил хлорида. Смесь перемешивали при к.т. в течение 16 ч. Дополнительно добавляли 0,083 мл (0,716 ммоль) изопропилсульфонил хлорида, смесь перемешивали при к.т. в течение 1 ч, после чего концентрировали в вакууме. Неочищенный осадок непосредственно очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: дихлорметан/метанол 95/5, об./об.). 0,048 г (выход = 22%) N-((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метил)пропан-2-сульфонамида получали в виде белого твердого вещества. ЖХ/МС: m/z=422 (MH+); степень чистоты (УФ-детекция при 254 нм) = 98%. 1H ЯМР (300 МГц, DMSO) δ 8,75 (д, J=6,8 Гц, 1H), 8,13 (с, 1H), 7,99 (д, J=7,5 Гц, 1H), 7,85-7,61 (м, 4H), 7,53-7,38 (м, 1H), 7,15 (т, J=6,8 Гц, 1H), 4,44 (д, J=5,9 Гц, 2H), 3,25 (дд, J=13,6, 6,8 Гц, 1H), 1,21 (д, J=6,8 Гц, 6H).
Производные под номерами с 70 и 71 были приготовлены согласно аналогичной процедуре.
Пример 14: Приготовление производного номер 72: метил 2-(метил((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метил)амино)ацетат
Этап 1: Приготовление метил 2-((имидазо[1,2-а]пиридин-2-илметил)(метил)амино)ацетата

0,303 г (1,773 ммоль) 2-(хлорметил)имидазо[1,2-а]пиридина растворяли при перемешивании магнитной мешалкой в 15 мл диметилформамида, а затем добавляли 0,253 г (1,773 ммоль) гидрохлорида метилового сложного эфира саркозина и 0,741 мл (5,32 ммоль) триэтиламина. Смесь перемешивали при к.т. в течение 16 ч, после чего обрабатывали 30 мл насыщенного водного раствора NaCl. Водную фазу концентрировали в вакууме. Осадок растирали с 75 мл изопропанола, соли удаляли в результате фильтрации, и фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: дихлорметан/метанол, 95/5, об./об.). 0,107 г (выход = 26%) метил 2-((имидазо[1,2-а]пиридин-2-илметил)(метил)амино)ацетата получали в виде бледно-желтого масла. ЖХ/МС: m/z=234 (MH+); степень чистоты (УФ-детекция при 254 нм) = 99%. 1H ЯМР (300 МГц, DMSO) δ 8,49 (д, J=6,8 Гц, 1H), 7,79 (с, 1H), 7,47 (д, J=9,0 Гц, 1H), 7,18 (ддд, J=9,0, 6,7, 1,2 Гц, 1H), 6,84 (тд, J=6,7, 1,1 Гц, 1H), 3,76 (с, 2H), 3,61 (с, 3H), 2,32 (с, 3H).
Этап 2: Приготовление метил 2-(((3-иодимидазо[1,2-а]пиридин-2-ил)метил)(метил)амино)ацетата
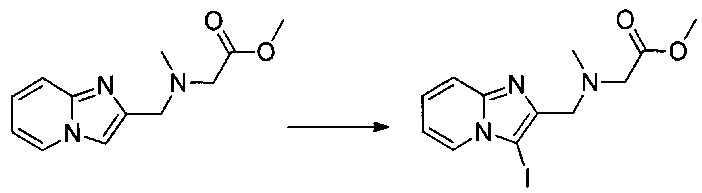
0,107 г (0,459 ммоль) метил 2-((имидазо[1,2-а]пиридин-2-илметил)(метил)амино)ацетата растворяли при перемешивании магнитной мешалкой в 4 мл ацетонитрила, к полученному раствору добавляли 0,112 г (0,482 ммоль) N-иодсукцинимида. Смесь перемешивали при к.т. в течение 16 ч. Реакционную смесь обрабатывали 10 мл воды, а затем экстрагировали 3×10 мл этилацетата. Объединенные органические фазы промывали 20 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: 100% этилацетат). 0,093 г (выход = 46%) метил 2-(((3-иодимидазо[1,2-а]пиридин-2-ил)метил)(метил)амино)ацетата получали в виде желтого масла. ЖХ/МС: m/z=360 (MH+); степень чистоты (УФ-детекция при 254 нм) = 82%. 1H ЯМР (300 МГц, DMSO) δ 8,30 (д, J=6,8 Гц, 1H), 7,54 (д, J=9,0 Гц, 1H), 7,41-7,23 (м, 1H), 7,03 (т, J=6,7 Гц, 1H), 3,79 (с, 2H), 3,62 (с, 3H), 3,37 (с, 2H), 2,34 (с, 3H).
Этап 3: Приготовление метил 2-(метил((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метил)амино)ацетата (производное номер 72)

В колбе, помещенной в атмосферу с потоком аргона, 0,093 г (0,212 ммоль) метил 2-(((3-иодимидазо[1,2-а]пиридин-2-ил)метил)(метил)амино)ацетата растворяли при перемешивании магнитной мешалкой в 0,5 мл диметилформамида. К полученному раствору добавляли 0,004 г (0,021 ммоль) иодида меди, 0,037 мл (0,255 ммоль) 3-трифторметилфенилацетилена и 0,444 мл (3,18 ммоль) триэтиламина. Реакционную среду дегазировали аргоном в течение 10 мин., после чего добавляли 0,019 г (0,021 ммоль) Pd2(dba)3. Смесь перед обработкой перемешивали при к.т. в течение 16 ч, затем разводили 15 мл этилацетата и промывали 2×15 мл насыщенного водного раствора NaCl. Водную фазу экстрагировали 20 мл этилацетата. Объединенные органические фазы высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: 100% этилацетат). 0,05 г (выход = 57%) метил 2-(метил((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метил)амино)ацетата получали в виде бледно-коричневого твердого вещества. ЖХ/МС: m/z=402 (MH+); степень чистоты (УФ-детекция при 254 нм) = 96%. 1H ЯМР (300 МГц, DMSO) δ 8,75 (д, J=6,7 Гц, 1H), 8,11 (с, 1H), 7,97 (д, J=7,6 Гц, 1H), 7,83-7,59 (м, 3H), 7,55-7,37 (м, 1H), 7,14 (тд, J=6,8, 0,9 Гц, 1H), 3,97 (с, 2H), 3,58 (с, 3H), 3,43 (с, 2H), 2,39 (с, 3H).
Производные под номерами с 101 по 104 были приготовлены согласно аналогичной процедуре.

Пример 15: Приготовление производного номер 73: 2-(метил((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метил)амино)уксусная кислота

0,22 г (0,543 ммоль) 2-(метил((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метил)амино)ацетата растворяли при перемешивании магнитной мешалкой в 6 мл смеси, состоящей из метанола и тетрагидрофурана, а затем добавляли 0,543 мл (0,543 ммоль) 1 М водного раствора NaOH. Смесь перемешивали при к.т. в течение 16 ч. Добавляли 0,163 мл (0,163 ммоль) 1 М водного раствора NaOH. Смесь перемешивали при к.т. в течение еще 24 ч, после чего обрабатывали раствором 0,04 мл (0,705 ммоль) уксусной кислоты в 10 мл воды. Смесь перемешивали при к.т. в течение 30 мин. Образовавшееся твердое вещество выделяли в результате фильтрации, промывали 5 мл воды и высушивали в вакуумном колпаке с получением 0,155 г (выход = 73%) 2-(метил((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метил)амино)уксусной кислоты в виде бежевого твердого вещества. ЖХ/МС: m/z=388 (MH+); степень чистоты (УФ-детекция при 254 нм) = 99%. 1H ЯМР (300 МГц, DMSO) δ 8,74 (д, J=6,7 Гц, 1H), 8,10 (с, 1H), 7,97 (д, J=7,6 Гц, 1H), 7,77 (д, J=7,8 Гц, 1H), 7,74-7,64 (м, 2H), 7,49-7,39 (м, 1H), 7,13 (т, J=6,4 Гц, 1H), 4,02 (с, 2H), 3,59 (с, 1H), 3,33 (с, 2H), 2,41 (с, 3H).
Производные под номерами с 105 по 107 были приготовлены согласно аналогичной процедуре.
Пример 16: Приготовление производного номер 74: метил 3-(3-((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метил)уреидо)бензоат

0,15 г (0,477 ммоль) (3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метанамина (приготовленного согласно этапам с 1 по 4 примера 11) растворяли при перемешивании магнитной мешалкой в 4 мл дихлорметана, а затем добавляли 0,09 г (0,492 ммоль) метилбензоат-3-изоцианата. Смесь перемешивали при к.т. в течение 15 мин. Образовавшееся твердое вещество выделяли в результате фильтрации и промывали 10 мл дихлорметана. Затем твердое вещество очищали методом препаративной ВЭЖХ с получением 0,057 г (выход = 25%) метил 3-(3-((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метил)уреидо)бензоата в виде белого порошка. ЖХ/МС: m/z=493 (MH+); степень чистоты (УФ-детекция при 254 нм) = 98%. 1H ЯМР (300 МГц, DMSO) δ), 8,74 (д, J=6,7 Гц, 1H), 8,11 (т, J=1,8 Гц, 1H), 8,04 (с, 1H), 7,91 (д, J=7,8 Гц, 1H), 7,70 (дд, J=13,0, 8,5 Гц, 2H), 7,58 (т, J=7,9 Гц, 2H), 7,53-7,42 (м, 2H), 7,34 (т, J=7,9 Гц, 1H), 7,15 (тд, J=6,8, 1,1 Гц, 1H), 6,81 (т, J=5,7 Гц, 1H), 4,64 (д, J=5,7 Гц, 2H), 3,83 (с, 3H).
Пример 17: Приготовление производного номер 75: 3-(3-((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метил)уреидо)бензойная кислота

0,038 г (0,078 ммоль) метил 3-(3-((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метил)уреидо)бензоата растворяли при перемешивании магнитной мешалкой в 2 мл смеси метанола/тетрагидрофурана (1/1, об./об.), затем добавляли 0,931 мл (0,931 ммоль) (1 М) водного раствора NaOH, смесь перемешивали при к.т. в течение 16 ч, после чего обрабатывали раствором 0,053 мл (0,931 ммоль) уксусной кислоты в 10 мл воды. Смесь перемешивали при к.т. в течение 30 мин. Образовавшееся твердое вещество выделяли в результате фильтрации, промывали 5 мл воды и высушивали в вакуумном колпаке с получением 0,026 г (выход = 70%) 3-(3-((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метил)уреидо)бензойной кислоты в виде белого твердого вещества. ЖХ/МС: m/z=479 (MH+); степень чистоты (УФ-детекция при 254 нм) = 99%. 1H ЯМР (300 МГц, DMSO) δ 12,83 (с, 1H), 8,96 (с, 1H), 8,73 (д, J=6,7 Гц, 1H), 8,04 (с, 2H), 7,90 (д, J=7,5 Гц, 1H), 7,69 (дд, J=13,0, 8,4 Гц, 2H), 7,58 (д, J=7,7 Гц, 2H), 7,45 (д, J=7,5 Гц, 2H), 7,29 (т, J=7,9 Гц, 1H), 7,13 (т, J=6,8 Гц, 1H), 6,80 (т, J=5,4 Гц, 1H), 4,62 (д, J=5,6 ГЦ, 2H).
Пример 18: Приготовление производного №76: метил 2-((3-((4-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)бензоат
Этап 1: Приготовление метил 2-((3-иодимидазо[1,2-а]пиридин-2-ил)метокси)бензоата

4 г (13,68 ммоль) 2-(хлорметил)-3-иодимидазо[1,2-а]пиридина растворяли при перемешивании магнитной мешалкой в диметилформамиде, а затем добавляли 2,71 мл (20,51 ммоль) метил 2-гидроксибензоата и 6,68 г (20,51 ммоль) карбоната цезия. Полученную смесь перемешивали при температуре 60°C в течение 2 ч, после чего вливали в 200 мл воды. Водную фазу экстрагировали 2×100 мл этилацетата. Объединенные органические фазы промывали 100 мл насыщенного водного раствора NaHCO3, 100 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок растирали с 50 мл диизопропилового эфира, полученное твердое вещество выделяли в результате фильтрации. Выделяли 2,55 г целевого продукта. Помимо этого, суспензию водной фазы выделяли в результате фильтрации и промывали 2×30 мл воды. Анализ подтвердил, что полученное вещество также представляло собой целевой продукт. Вследствие этого обе партии собирали и 5,58 г (выход = 76%) метил 2-((3-иодимидазо[1,2-а]пиридин-2-ил)метокси)бензоата получали в виде белого порошка. ЖХ/МС: m/z=409 (MH+); степень чистоты (УФ-детекция при 254 нм) = 98%. 1H ЯМР (300 МГц, DMSO) δ 8,33 (д, J=6,9 Гц, 1H), 7,69-7,44 (м, 3H), 7,45-7,28 (м, 2H), 7,16-6,93 (м, 2H), 5,23 (с, 2H), 3,73 (с, 3H).
Этап 2: Приготовление метил 2-((3-((4-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)бензоата (производное №76)
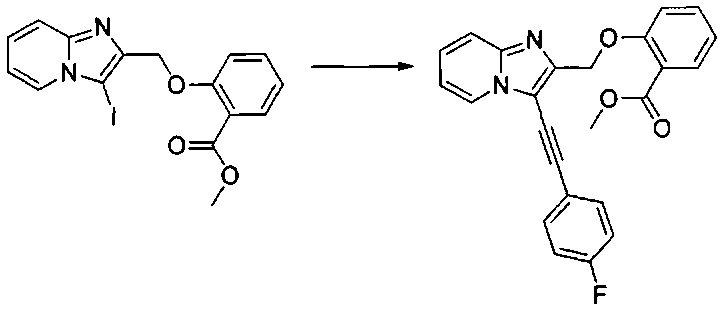
В колбе, помещенной в атмосферу с потоком аргона, 2 г (3,82 ммоль) метил 2-((3-иодимидазо[1,2-а]пиридин-2-ил)метокси)бензоата растворяли при перемешивании магнитной мешалкой в 8 мл диметилформамида. К полученному раствору добавляли: 0,074 г (0,382 ммоль) иодида меди, 0,551 г (4,59 ммоль) 1-этинил-4-фторбензена и 8 мл (57,3 ммоль) триэтиламина. Реакционную среду дегазировали аргоном в течение 10 мин., после чего добавляли 0,35 г (0,382 ммоль) Pd2(dba)3. Смесь перемешивали при к.т. в течение 16 ч, после чего обрабатывали 100 мл насыщенного водного раствора NaCl. Водную фазу экстрагировали 2×100 мл этилацетата. Объединенные органические фазы промывали 100 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: гептан/этилацетат, 1/1, об./об.). 1,36 г (выход = 84%) метил 2-((3-((4-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)бензоата получали в виде бледно-коричневого твердого вещества. ЖХ/МС: m/z=401 (MH+); степень чистоты (УФ-детекция при 254 нм) = 98%. 1H ЯМР (300 МГц, DMSO) δ 8,63 (д, J=6,7 Гц, 1H), 7,75-7,59 (м, 4Н), 7,48 (ддд, J=15,9, 11,5, 4,8 Гц, 2H), 7,40-7,26 (м, 3H), 7,13 (т, J=6,8 Гц, 1H), 7,01 (т, J=7,4 Гц, 1H), 5,40 (с, 2H), 3,68 (с, 3H).
Производные под номерами с 77 по 86 были приготовлены согласно аналогичной последовательности этапов 1 и 2.


Пример 19: Приготовление производного номер 87: метил 3-((3-(фенилэтинил)имидазо[1,2-а]пиридин-2-ил)метокси)бензоат
Этап 1: Приготовление этилимидазо[1,2-а]пиридин-2-карбоксилата
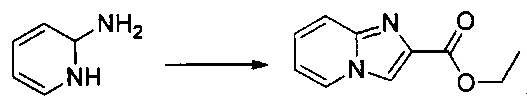
3 г (31,9 ммоль) 2-аминопиридина растворяли при перемешивании магнитной мешалкой в 65 мл тетрагидрофурана. Добавление 4,44 мл (31,9 ммоль) этил бромпирувата вызывало преципитацию твердого вещества. Гетерогенную смесь перемешивали в колбе с обратным холодильником в течение 4 ч. Смеси позволяли остыть до к.т., после чего твердое вещество выделяли в результате фильтрации, отбирали в 65 мл этанола и перемешивали в колбе с обратным холодильником в 65 мл этанола в течение 16 ч. Смеси позволяли остыть до к.т., после чего твердое вещество выделяли в результате фильтрации. Полученный фильтрат помещали на лед для облегчения кристаллизации продукта, который собирали в результате фильтрации; данную процедуру повторяли до тех пор, пока жидкая фаза не становилась прозрачной. Твердое вещество промывали 30 мл диизопропилового эфира и высушивали в вакуумном колпаке с получением 5,14 г (выход = 84%) этил имидазо[1,2-а]пиридин-2-карбоксилата в виде белого порошка. ЖХ/МС: m/z=191 (MH+); степень чистоты (УФ-детекция при 254 нм) = 99%. 1H ЯМР (300 МГц, DMSO) δ 8,95 (с, 1H), 8,84 (д, J=6,8 Гц, 1H), 7,98-7,80 (м, 2H), 7,46 (т, J=7,2 Гц, 1H), 4,43 (к, J=7,1 Гц, 2H), 1,35 (д, J=7,1 Гц, 3H).
Этап 2: Приготовление этил 3-иодимидазо[1,2-а]пиридин-2-карбоксилата

2,405 г (12,16 ммоль) этил имидазо[1,2-а]пиридин-2-карбоксилата растворяли при перемешивании магнитной мешалкой в 60 мл ацетонитрила, к полученному раствору добавляли 3,01 г (13,38 ммоль) N-иодсукцинимида. Смесь перемешивали при к.т. в течение 1 ч. Реакционную смесь обрабатывали 50 мл водного раствора Na2S2O5 концентрацией 100 мг/мл. Водную фазу экстрагировали 2×100 мл этилацетата. Водную фазу нейтрализовали добавлением по частям NaHCO3, а затем экстрагировали 3×100 мл этилацетата. Органические фазы объединяли, промывали 200 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме с получением 1,59 г (выход = 41%) этил 3-иодимидазо[1,2-а]пиридин-2-карбоксилата в виде бледно-желтого твердого вещества. ЖХ/МС: m/z=317 (MH+); степень чистоты (УФ-детекция при 254 нм) = 99%. 1H ЯМР (300 МГц, DMSO) δ 8,45 (д, J=6,9 Гц, 1H), 7,66 (д, J=9,1 Гц, 1H), 7,46 (д, J=7,0 Гц, 1H), 7,15 (д, J=6,6 Гц, 1H), 4,30 (д, J=7,1 Гц, 2H), 1,36 (д, J=7,1 Гц, 3H).
Этап 3: Приготовление этил 3-(фенилэтинил)имидазо[1,2-а]пиридин-2-карбоксилата
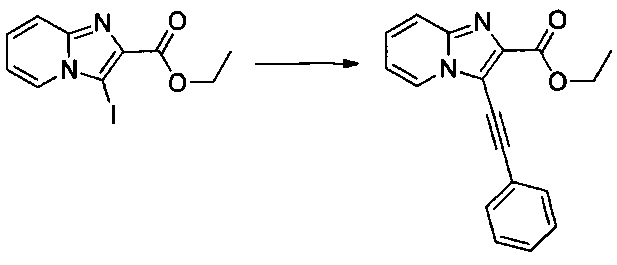
В колбе, помещенной в атмосферу с потоком аргона, 1,82 г (5,77 ммоль) этил 3-иодимидазо[1,2-а]пиридин-2-карбоксилата растворяли при перемешивании магнитной мешалкой в 6 мл диметилформамида. К полученному раствору добавляли: 0,11 г (0,577 ммоль) иодида меди, 0,776 мл (6,93 ммоль) фенилацетилена и 6 мл (43 ммоль) триэтиламина. Реакционную среду дегазировали аргоном в течение 10 мин., после чего добавляли 0,529 г (0,577 ммоль) Pd2(dba)3. Смесь перемешивали при к.т. в течение 16 ч, после чего фильтровали через целит. Целит промывали 50 мл этилацетата, а затем полученный фильтрат промывали 2×50 мл воды. После сепарации водную фазу экстрагировали 2×50 мл этилацетата; объединенные органические фазы промывали 70 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: гептан/этилацетат, 6/4, об./об.). 1,28 г (выход = 57%) этил 3-(фенилэтинил)имидазо[1,2-а]пиридин-2-карбоксилата получали в виде бледно-желтого твердого вещества. ЖХ/МС: m/z=291 (MH+); степень чистоты (УФ-детекция при 254 нм) = 93%. 1H ЯМР (300 МГц, DMSO) δ 8,70 (д, J=6,8 Гц, 1H), 7,74 (дд, J=22,1, 6,2 Гц, 3H), 7,53 (дд, J=17,6, 6,9 Гц, 4H), 7,23 (т, J=6,3 Гц, 1H), 4,36 (д, J=4,7 Гц, 2H), 1,36 (д, J=7,1 Гц, 3H).
Этап 4: Приготовление (3-(фенилэтинил)имидазо[1,2-а]пиридин-2-ил)метанола

В колбе, помещенной в атмосферу с потоком аргона, 0,078 г (1,984 ммоль) LAH растворяли в 1,4 мл тетрагидрофурана, а затем добавляли по каплям при интенсивном перемешивании магнитной мешалкой раствор 0,3 г (0,992 ммоль) этил 3-(фенилэтинил)имидазо[1,2-а]пиридин-2-карбоксилата в 3,3 мл тетрагидрофурана. Смесь перемешивали при к.т. в течение 15 мин., осторожно добавляли 0,2 мл воды, после чего добавляли раствор 0,2 мл серной кислоты в 1,5 мл воды. После сепарации органическую фазу промывали 50 мл воды. Водную фазу экстрагировали 3×50 мл этилацетата, объединенные органические фазы промывали 100 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент гептана/этилацетата, от 40% до 30% гептана, об./об.). 0,076 г (выход = 30%) (3-(фенилэтинил)имидазо[1,2-а]пиридин-2-ил)метанола получали в виде бледно-желтого твердого вещества. ЖХ/МС: m/z=249 (MH+); степень чистоты (УФ-детекция при 254 нм) = 97%. 1H ЯМР (300 МГц, DMSO) δ 8,59 (дт, J=6,7, 1,1 Гц, 1H), 7,77-7,56 (м, 3H), 7,56-7,32 (м, 4Н), 7,11 (тд, J=6,8, 1,1 Гц, 1H), 5,33 (т, J=5,9 Гц, 1H), 4,67(д, J=5,9 Гц, 2H).
Этап 5: Приготовление метил 3-((3-(фенилэтинил)имидазо[1,2-а]пиридин-2-ил)метокси)бензоата (производное №87)

0,09 г (0,594 ммоль) метил 3-гидроксибензоата растворяли при перемешивании магнитной мешалкой в 1 мл тетрагидрофурана, затем добавляли 0,312 г (1,188 ммоль) трифенилфосфина и 0,231 мл (1,188 ммоль) DIAD. Смесь перемешивали при к.т. в течение 15 мин., затем добавляли раствор 0,076 г (0,297 ммоль) (3-(фенилэтинил)имидазо[1,2-а]пиридин-2-ил)метанола в 4 мл тетрагидрофурана, и смесь перемешивали при к.т. в течение 16 ч. Добавляли смесь 0,09 г (0,594 ммоль) метил 3-гидроксибензоата, 0,312 г (1,188 ммоль) трифенилфосфина и 0,231 мл (1,188 ммоль) DIAD, растворенных в 1 мл тетрагидрофурана, и реакционную среду перемешивали при к.т. в течение еще 2 ч. Реакционную среду обрабатывали 10 мл. Водную фазу экстрагировали 3×10 мл этилацетата, затем объединенные органические фазы промывали 10 мл насыщенного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: гептан/этилацетат, от 70% до 40% гептана, об./об.). 0,048 г (выход = 42%) метил 3-((3-(фенилэтинил)имидазо[1,2-а]пиридин-2-ил)метокси)бензоата получали в виде бледно-желтого твердого вещества. ЖХ/МС: m/z=383 (MH+); степень чистоты (УФ-детекция при 254 нм) = 99%. 1H ЯМР (300 МГц. DMSO) δ 8,64 (д, J=6,8 Гц, 1H), 7,68 (дд, J=17,0, 7,2 Гц, 4H), 7,55 (д, J=8,7 Гц, 1H), 7,52-7,34 (м, 6H), 7,15 (т, J=7,3 Гц, 1H), 5,42 (с, 2H), 3,80 (с, 3H).
Пример 20: Приготовление производного №88: 2-((3-((4-фторфенил)этинил)-имидазо[1,2-а]пиридин-2-ил)метокси)бензойная кислота
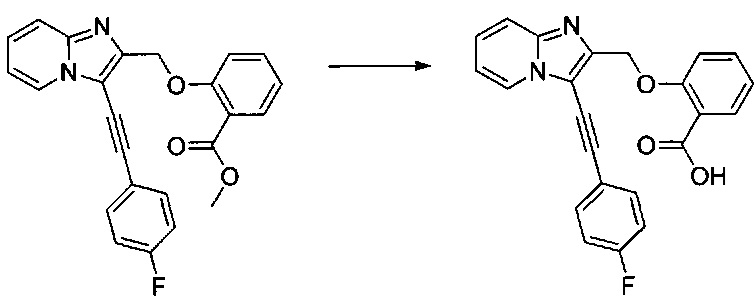
0,65 г (1,542 ммоль) метил 2-((3-((4-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)бензоата солюбилизировали при перемешивании магнитной мешалкой в 30 мл смеси, состоящей из этанола и тетрагидрофурана (1/1, об./об.), а затем добавляли 15,42 мл (15,42 ммоль) 1 н водного раствора NaOH. Смесь перемешивали при к.т. в течение 16 ч, после чего добавляли 20 мл воды и 0,883 мл (15,42 ммоль) уксусной кислоты. Образовавшийся преципитат выделяли в результате фильтрации, промывали 10 мл воды и высушивали в вакуумном колпаке с получением 0,518 г (выход = 81%) 2-((3-((4-фтор-фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)бензойной кислоты в виде бледно-желтого твердого вещества. ЖХ/МС: m/z=387 (MH+); степень чистоты (УФ-детекция при 254 нм)=99%. 1H ЯМР (300 МГц, DMSO) δ 12,72 (с, 1H), 8,64 (д, J=5,5 Гц, 1H), 7,78-7,59 (м, 4Н), 7,45 (д, J=6,2 Гц, 2H), 7,41-7,24 (м, 3H), 7,14 (т, J=6,8 Гц, 1H), 7,00 (т, J=7,3 Гц, 1H), 5,42 (с, 2H).
Производные под номерами с 89 по 93 были приготовлены согласно аналогичной процедуре.


Пример 21: Приготовление производного №94: (2-((3-((3-(трифтор метил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)фенил)метанол

В колбе, помещенной в атмосферу с потоком аргона, 0,16 г (0,352 ммоль) метил 2-((3-((3-(трифторметил)-фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)бензоата растворяли при перемешивании магнитной мешалкой в 4 мл тетрагидрофурана, смесь охлаждали до температуры 0°C, а затем добавляли 0,148 мл (0,492 ммоль) RED-Al. Полученную смесь перемешивали при температуре 0°C в течение 15 мин., после чего разводили 15 мл этилацетата и вливали в 20 мл воды. Медленно добавляли при интенсивном перемешивании 1 мл водного раствора (32% масс.) NaOH. После сепарации органическую фазу промывали 2×10 мл воды. Водные фазы экстрагировали 2×5 мл этилацетата. Объединенные органические фазы промывали 20 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали в результате растирания с 10 мл диизопропилового эфира. Твердое вещество выделяли в результате фильтрации и высушивали в вакууме. 0,091 г (выход = 58%) (2-((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)фенил)метанола получали в виде бледно-коричневого твердого вещества. ЖХ/МС: m/z=425 (M+2H+); степень чистоты (УФ-детекция при 254 нм)=94%. 1H ЯМР (300 МГц, DMSO) δ 8,77 (д, J=6,8 Гц, 1H), 8,05 (с, 1H), 7,92 (д, J=7,6 Гц, 1H), 7,73 (дт, J=14,3, 7,8 Гц, 3H), 7,55-7,43 (м, 1H), 7,38 (д, J=7,4 Гц, 1H), 7,20 (д, J=3,6 Гц, 3H), 7,03-6,86 (м, 1H), 5,39 (с, 2H), 4,98 (т, J=5,7 Гц, 1H), 4,55 (д, J=5,6 Гц, 2H).
Пример 22: Приготовление производного номер 95: N-(2-(диметиламино)этил)-2-((3-((4-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)бензамид
Этап 1: Приготовление 2-((3-((4-фторфенил)этинил)имидазо-[1,2-а]пиридин-2-ил)метокси)бензоил хлорида

0,6 г (1,553 ммоль) 2-((3-((4-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)бензойной кислоты суспендировали при перемешивании магнитной мешалкой в 20 мл дихлорметана, смесь помещали на ледяную баню с температурой 0°C, а затем добавляли по каплям 0,408 мл (4,66 ммоль) оксалил хлорида, после чего добавляли несколько капель диметилформамида. Ледяную баню убирали, смесь перемешивали при к.т. в течение 2 ч. Реакционную среду концентрировали в вакууме и незамедлительно использовали на следующем этапе.
Этап 2: Приготовление N-(2-(диметиламино)этил)-2-((3-((4-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)бензамида (производное №95)
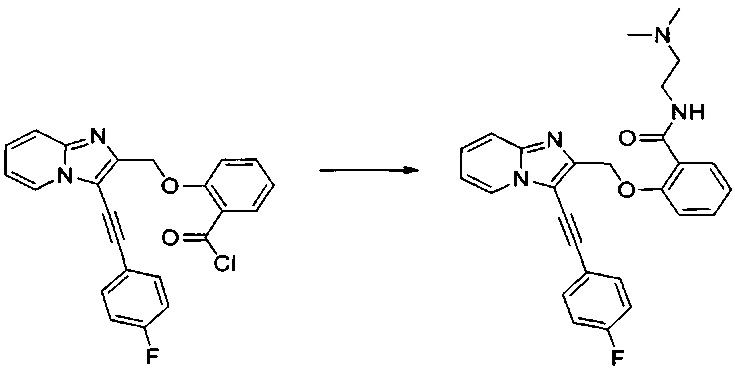
0,896 мл (7,76 ммоль) N,N-диметилэтан-1,2-диамина солюбилизировали при перемешивании магнитной мешалкой в 8 мл дихлорметана, добавляли 0,449 г (0,776 ммоль) 2-((3-((4-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)бензоил хлорида, смесь перемешивали при к.т. в течение 16 ч, после чего концентрировали в вакууме. Неочищенный осадок очищали в результате растирания с 20 мл диизопропилового эфира. Твердое вещество выделяли в результате фильтрации, промывали 10 мл воды и высушивали в вакууме. 0,178 г (выход = 50%) N-(2-(диметиламино)этил)-2-((3-((4-фторфенил)этинил)имидазо[1,2-а]-пиридин-2-ил)метокси)бензамида получали в виде бледно-желтого твердого вещества. ЖХ/МС: m/z=457 (MH+); степень чистоты (УФ-детекция при 254 нм) = 98%. 1H ЯМР (300 МГц, DMSO) δ 8,68 (д, J=6,7 Гц, 1H), 8,47 (д, J=4,8 Гц, 1H), 7,91 (дд, J=7,7, 1,5 Гц, 1H), 7,72 (дд, J=8,6, 5,2 Гц, 3H), 7,57-7,39 (м, 3H), 7,32 (т, J=8,9 Гц, 2H), 7,17 (т, J=6,6 Гц, 1H), 7,09 (т, J=7,3 Гц, 1H), 5,51 (с, 2H), 2,5 (1H, спрятан под пиком растворителя), 2,25 (т, J=6,5 Гц, 2H), 1,92 (с, 6H).
Производное номер 96 было приготовлено согласно аналогичной процедуре.

Пример 23: Приготовление производного №97: N-(3-((3-(фенил этинил)имидазо[1,2-а]пиридин-2-ил)метокси)фенил)метансульфонамид
Этап 1: Приготовление 2-(хлорметил)-3-(фенилэтинил)имидазо[1,2-а]пиридина
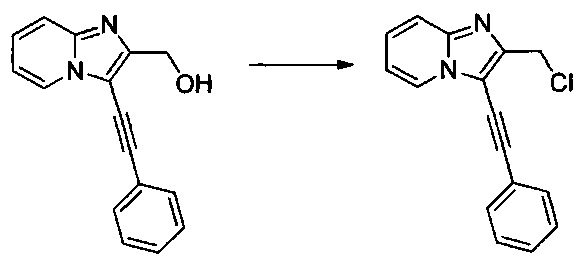
0,238 г (0,949 ммоль) (3-(фенил-этинил)имидазо[1,2-а]пиридин-2-ил)метанола растворяли при перемешивании магнитной мешалкой в 5 мл дихлорметана, а затем добавляли 0,346 мл (4,75 ммоль) тионилхлорида. Смесь перемешивали при к.т. в течение 2 ч, после чего концентрировали в вакууме. Осадок переносили в 20 мл смеси, состоящей из этилацетата и насыщенного водного раствора NaHCO3 (1/1, об./об.). После сепарации водную фазу экстрагировали 10 мл этилацетата. Объединенные органические фазы промывали 10 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. 0,246 г (выход = 96%) 2-(хлорметил)-3-(фенилэтинил)имидазо[1,2-а]пиридина получали в виде коричневого твердого вещества. ЖХ/МС: m/z=267 (MH+); степень чистоты (УФ-детекция при 254 нм) = 98%.
Этап 2: Приготовление N-(3-гидроксифенил)метансульфонамида
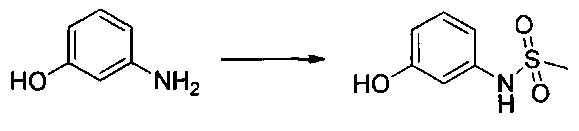
2 г (18,33 ммоль) 3-аминофенола растворяли при перемешивании магнитной мешалкой в 6 мл пиридина, смесь охлаждали до температуры 10°C, а затем добавляли 1,8 мл (23,26 ммоль) метансульфонил хлорида. Ледяную баню убирали, смесь перемешивали при к.т. в течение 18 ч, после чего разводили 100 мл насыщенного водного раствора NaCl. Водную фазу экстрагировали 100 мл этилацетата. Органическую фазу промывали 50 мл насыщенного водного раствора NaHCO3, экстрагировали 50 мл водного раствора NaOH (0,5 н). Щелочную водную фазу промывали 2×100 мл этилацетата, а затем подкисляли до значения pH 2-3 добавлением 1 н водного раствора HCl. Подкисленную водную фазу экстрагировали 2×100 мл этилацетата. Объединенные органические фазы промывали 100 мл насыщенного водного раствора NaCl, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. 3,3 г (выход = 80%) N-(3-гидроксифенил)метансульфонамида получали в виде масла. ЖХ/МС: m/z=186 (М-Н+); степень чистоты (УФ-детекция при 254 нм) = 81%.
Этап 3: Приготовление N-(3-((3-(фенилэтинил)имидазо[1,2-а]пиридин-2-ил)метокси)фенил)метансульфонамида (производное номер 97)
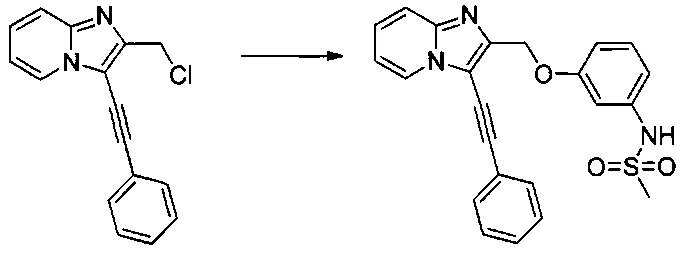
0,094 г (0,352 ммоль) 2-(хлорметил)-3-(фенилэтинил)имидазо[1,2-а]пиридина растворяли при перемешивании магнитной мешалкой в 2 мл диметилформамида, добавляли 121 мг (0,529 ммоль) N-(3-гидроксифенил)метансульфонамида и 0,172 г (0,529 ммоль) карбоната цезия. Полученную смесь перемешивали при температуре 60°C в течение 3 ч, после чего вливали в 15 мл насыщенного водного раствора NaCl. Водную фазу экстрагировали 2×20 мл этилацетата. Объединенные органические фазы высушивали над Na2SO4, который затем удаляли в результате фильтрации. Полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом препаративной ВЭЖХ. 0,028 г (выход = 18%) N-(3-((3-(фенилэтинил)имидазо[1,2-а]пиридин-2-ил)-метокси)фенил)метансульфонамида получали в виде белого твердого вещества. ЖХ/МС: m/z=418 (MH+); степень чистоты (УФ-детекция при 254 нм) = 95%. 1H ЯМР (300 МГц, DMSO) δ 9,53 (с, 1H), 8,55 (д, J=6,8 Гц, 1H), 7,76-7,57 (м, 3H), 7,54-7,33 (м, 4H), 7,17-7,02 (м, 2H), 6,89 (дд, J=8,0, 1,1 Гц, 1H), 6,79 (т, J=2,1 Гц, 1H), 6,69-6,54 (м, 1H), 5,07 (с, 2H), 3,14 (с, 3H).
Пример 24: Приготовление производного №111: этил 2-((6-хлор-3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метилтио)ацетат
Этап 1: Приготовление этил 2-((6-хлоримидазо[1,2-а]пиридин-2-ил)метилтио)ацетата
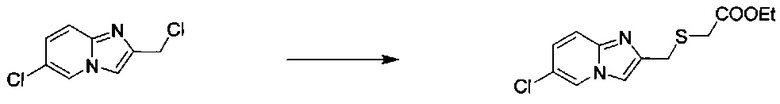
20 г (99 ммоль) 6-хлор-2-(хлорметил)имидазо[1,2-а]пиридина, 0,746 г (4,97 ммоль) иодида натрия и 64,8 г (199 ммоль) карбоната цезия вносили в 500 мл ацетонитрила. Раствор перемешивали при комнатной температуре (к.т.) в течение 3 часов. Осажденное минеральное вещество фильтровали, и растворитель выпаривали в вакууме. Полученное коричневое твердое вещество растворяли в 300 мл этилацетата, промывали 3×100 мл воды и непосредственно очищали на пробке из оксида кремния при элюировании этилацетатом. 28,74 г (выход = 100%) этил 2-((6-хлоримидазо[1,2-а]пиридин-2-ил)метилтио)ацетата получали в виде оранжевого твердого вещества. ЖХ/МС: m/z=285 (MH+); степень чистоты (УФ-детекция при 254 нм)=99%. 1H ЯМР (300 МГц, DMSO) δ 8,79 (дд, J=2,1, 0,8 Гц, 1H), 7,82 (с, 1H), 7,52 (д, J=9,6 Гц, 1H), 7,25 (дд, J=9,6, 2,1 Гц, 1H), 4,06 (к, J=7,1 Гц, 2H), 3,90 (с, 2H), 3,36 (с, 2H), 1,16 (т, J=7,1 Гц, 3H).
Этап 2: Приготовление 3-иод-2-(иодметил)имидазо[1,2-а]пиридина

К 400 мл ацетонитрила добавляли 24,5 г (86 ммоль) этил 2-((6-хлоримидазо[1,2-а]пиридин-2-ил)метилтио)ацетата, после чего добавляли 20,32 г (90 ммоль) N-иодсукцинимида. Растворитель перемешивали при к.т. в течение 1 ч. Растворитель выпаривали в вакууме, и полученный осадок интенсивно перемешивали в 250 мл воды и 200 мл диизопропилового эфира. Осажденное твердое вещество фильтровали, промывали 2×30 мл воды и 2×30 мл диизопропилового эфира. 33,93 г (выход = 88%) 3-иод-2-(иодметил)имидазо[1,2-а]пиридина получали в виде коричневого твердого вещества. ЖХ/МС: m/z=411 (MH+); степень чистоты (УФ-детекция при 254 нм) = 91,3%. 1H ЯМР 300 МГц, DMSO) δ 8,40 (дд, J=1,9, 0,7 Гц, 1H), 7,61 (дд, J=9,5, 0,6 Гц, 1H), 7,38 (дд, J=9,5, 2,0 Гц, 1H), 4,07 (к, J=7,1 Гц, 2H), 3,92 (с, 2H), 3,39 (с, 2H), 1,18 (т, J=7,1 Гц, 3H).
Этап 3: Приготовление этил 2-((6-хлор-3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метилтио)ацетата

В атмосфере аргона 27 г (59,8 ммоль) этил 2-((6-хлор-3-иодимидазо[1,2-а]-пиридин-2-ил)метилтио)ацетата, 9,51 мл (65,8 ммоль) 1-этинил-3-(трифторметил)бензена, 1,139 г (5,98 ммоль) иодида меди и 125 мл (897 ммоль) триэтиламина помещали в 100 мл DMF. В раствор барботировали аргон в течение 10 мин., затем добавляли 2,74 г (2,99 ммоль) Pd2(dba)3. Полученную смесь перемешивали в течение 1 ч при к.т. Катализатор фильтровали через сложенную бумагу, раствор вливали в 300 мл воды, экстрагировали 2×200 мл этилацетата, собранные органические фазы промывали 3×150 мл воды и один раз 150 мл солевого раствора, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Осадок очищали на пробке оксида кремния и элюировали смесью гептана/этилацетата. 23,21 г (выход = 85%) этил 2-((6-хлор-3-((3-(трифторметил)фенил)этинил)-имидазо[1,2-а]пиридин-2-ил)метилтио)ацетата получали в виде желтого твердого вещества. ЖХ/МС: m/z=453 (MH+); степень чистоты (УФ-детекция при 254 нм) = 99%. 1H ЯМР (300 МГц, DMSO) δ 8,91 (дд, J=1,9, 0,6 Гц, 1H), 8,14 (с, 1H), 8,01 (д, J=7,6 Гц, 1H), 7,71 (ддд, J=14,3, 9,4, 4,3 Гц, 3H), 7,47 (дд, J=9,5, 2,0 Гц, 1H), 4,18-3,93 (м, 4H), 3,46 (с, 2H), 1,11 (т, J=7,1 Гц, 3H).
Производные с 108 по 110 и с 112 по 114 были приготовлены согласно аналогичной процедуре.
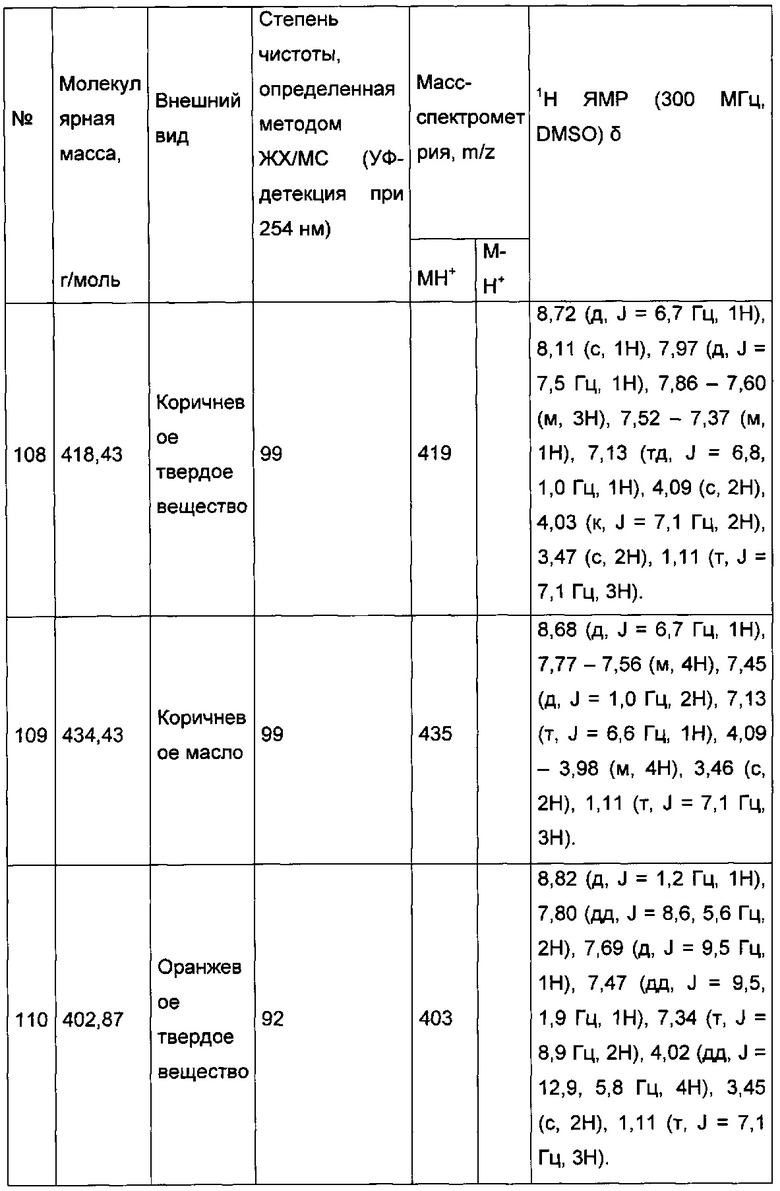

Пример 25: Приготовление производного №118: 2-((6-хлор-3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метилтио)уксусная кислота
16 г (35,3 ммоль) этил 2-((6-хлор-3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метилтио)ацетата добавляли к 75 мл этанола и 75 мл тетрагидрофурана, после чего добавляли 4,24 мл (42,4 ммоль) 32% водного раствора гидроксида натрия. Смесь перемешивали при к.т. в течение 2 ч, а затем через 30 мин. добавляли 50 мл воды для солюбилизации осажденного твердого вещества. Растворитель выпаривали в вакууме, полученный осадок разводили в 250 мл воды и 50 мл диизопропилового эфира, а затем значение pH раствора понижали до кислого добавлением 2,53 мл (44,2 ммоль) уксусной кислоты. Осажденное твердое вещество фильтровали, после чего промывали водой и диизопропиловым эфиром. 12,45 г (выход = 83%) 2-((6-хлор-3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метилтио)уксусной кислоты получали в виде желтого твердого вещества. ЖХ/МС: m/z=425 (MH+); степень чистоты (УФ-детекция при 254 нм) = 99%. 1H ЯМР (300 МГц, DMSO) δ 12,65 (с, 1H), 8,96 (с, 1H), 8,19 (с, 1H), 8,03 (д, J=7,6 Гц, 1H), 7,84-7,63 (м, 3H), 7,50 (дд, J=9,5, 1,8 Гц, 1H), 4,07 (с, 2H), 3,40 (с, 2H).
Производные с 115 по 117, 119 и 120 были приготовлены согласно аналогичной процедуре.



Пример 26: Приготовление производного №121: этил 2-((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метилсульфонил)ацетат

40 мг (0,096 ммоль) этил 2-((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]-пиридин-2-ил)метилтио)ацетата солюбилизировали в 1 мл ацетона, после чего добавляли 8,03 мг (0,096 ммоль) гидрокарбоната натрия. Раствор охлаждали до температуры 0°C, а затем добавляли раствор 118 мг (0,096 ммоль) оксона в 1 мл воды. Смесь перемешивали при к.т. в течение 4 ч. Раствор промывали 5 мл воды, полученное твердое вещество фильтровали и промывали 5 мл воды и 2 мл диизопропилового эфира. 29 мг (выход = 64%) этил 2-((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метилсульфонил)ацетата получали в виде бледно-желтого твердого вещества. ЖХ/МС: m/z=451 (MH+); степень чистоты (УФ-детекция при 254 нм)=94,6%. 1H ЯМР (300 МГц, DMSO) δ 8,80 (д, J=6,8 Гц, 1H), 8,17 (с, 1H), 8,03 (д, J=7,6 Гц, 1H), 7,76 (дт, J=13,6, 7,8 Гц, 3H), 7,62-7,45 (м, 1H), 7,21 (т, J=6,6 Гц, 1H), 4,98 (с, 2H), 4,62 (с, 2H), 4,19 (дд, J=14,1, 7,0 Гц, 2H), 1,21 (т, J=7,1 Гц, 3H).
Производное 122 было приготовлено согласно аналогичной процедуре.


Пример 27: Приготовление производного №123: 2-((6-хлор-3-((3-(трифторметокси)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метилсульфонил)уксусная кислота

100 мг (0,200 ммоль) этил 2-((6-хлор-3-((3-(трифторметокси)фенил)-этинил)имидазо[1,2-а]пиридин-2-ил)метилсульфонил)ацетата растворяли в 2 мл этанола, после чего добавляли 0,060 мл (0,599 ммоль) 32% водного раствора гидроксида натрия. Смесь перемешивали при к.т. в течение 16 ч. Раствор вливали в 10 мл воды, значение pH раствора понижали до кислого добавлением уксусной кислоты, после чего раствор экстрагировали 2×5 мл дихлорметана, объединенные органические фазы высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. 76 мг (выход = 80%) 2-((6-хлор-3-((3-(трифторметокси)фенил)этинил)-имидазо[1,2-а]пиридин-2-ил)метилсульфонил)уксусной кислоты получали в виде желтого твердого вещества. ЖХ/МС: m/z=473 (MH+); степень чистоты (УФ-детекция при 254 нм) = 99%. 1H ЯМР (300 МГц, DMSO) δ 14,72-12,20 (м, 1H), 9,00 (д, J=1,2 Гц, 1H), 7,88-7,76 (м, 3H), 7,62-7,45 (м, 3H), 4,96 (с, 2H), 4,42 (с, 2H).
Пример 28: Приготовление производного №124: N-(пиридин-3-ил)-2-((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метилтио)ацетамид

141 мг (0,358 ммоль) 2-((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]-пиридин-2-ил)метилтио)уксусной кислоты растворяли в 5 мл дихлорметана, после чего добавляли 0,094 мл (1,073 ммоль) оксалил хлорида и каплю DMF. Растворитель перемешивали при к.т. в течение 2 ч, после чего растворитель выпаривали в вакууме. Полученный осадок переносили в 5 мл тетрагидрофурана, добавляли 99 мг (0,715 ммоль) карбоната натрия, после чего добавляли 40,4 мг (0,429 ммоль) 3-аминопиридина. Раствор перемешивали при к.т. в течение 16 ч. Затем смесь вливали в 15 мл воды, трижды экстрагировали 15 мл этилацетата, объединенные органические фазы промывали 20 мл солевого раствора, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент дихлорметана/метанола, от 100% до 90% дихлорметана, об./об.). 89 мг N-(пиридин-3-ил)-2-((3-((3-(трифторметил)-фенил)-этинил)имидазо[1,2-а]пиридин-2-ил)метилтио)ацетамида получали в виде бледно-желтого твердого вещества. ЖХ/МС: m/z=467 (MH+); степень чистоты (УФ-детекция при 254 нм)=95%. 1H ЯМР (300 МГц, DMSO) δ 10,13 (с, 1H), 8,76 (дд, J=16,4, 4,5 Гц, 2H), 8,24 (дд, J=4,7, 1,5 Гц, 1H), 8,07 (ддд, J=8,3, 2,5, 1,5 Гц, 1H), 7,79-7,60 (м, 3H), 7,60-7,35 (м, 3H), 7,30 (дд, J=8,3, 4,7 Гц, 1H), 7,16 (тд, J=6,8, 1,1 Гц, 1H), 4,90 (с, 2H), 4,26 (с, 2H).
Производные с 125 по 128 были приготовлены согласно аналогичной процедуре.
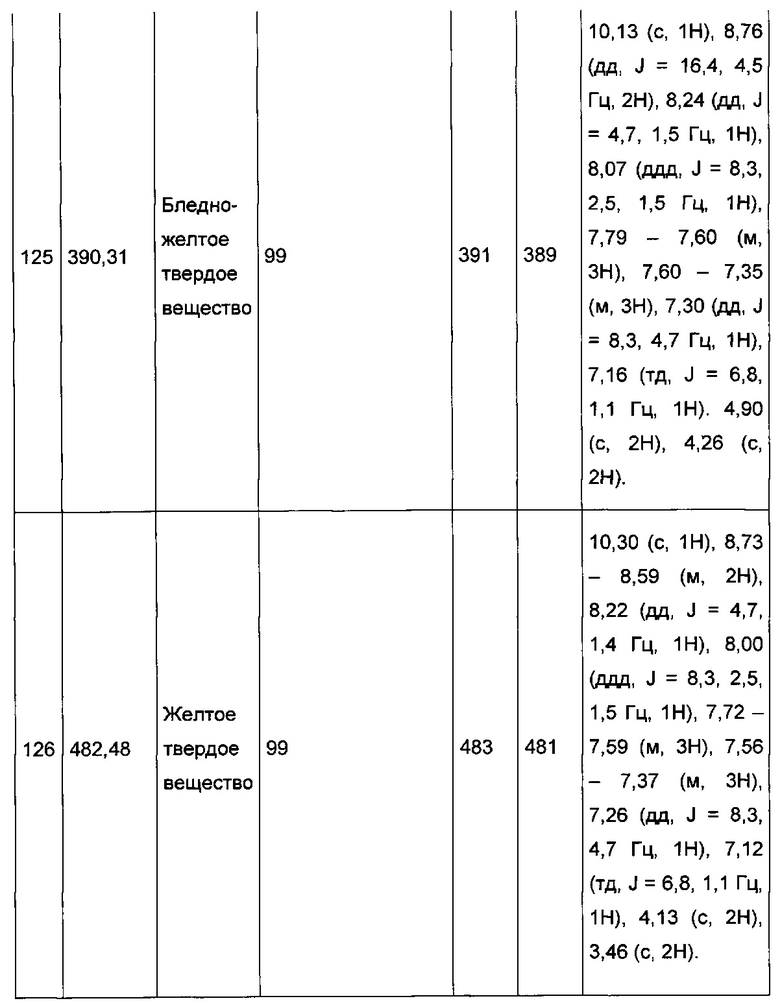

Пример 29: Приготовление производного №129: 4-гидрокси-3,3-диметил-2-((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метилтио)бутановая кислота
130 мг (0,303 ммоль) 4,4-диметил-3-((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метилтио)дигидрофуран-2(3H)-она растворяли в 2 мл метанола и 2 мл тетрагидрофурана, после чего добавляли 0,334 мл (0,334 ммоль) 1 н водного раствора гидроксида натрия. Раствор перемешивали в течение 16 ч. Раствор подкисляли до кислого значения pH уксусной кислотой и разводили водой до осаждения твердого вещества, которое затем фильтровали и промывали водой. 115 мг 4-гидрокси-3,3-диметил-2-((3-((3-(трифторметил)фенил)-этинил)имидазо[1,2-а]пиридин-2-ил)метилтио)бутановой кислоты получали в виде бледно-коричневого твердого вещества. ЖХ/МС: m/z=447 (MH+); степень чистоты (УФ-детекция при 254 нм) = 99%. 1H ЯМР (300 МГц, DMSO) δ 12,69 (с, 1H), 8,77 (д, J=6,7 Гц, 1H), 8,11 (с, 1H), 7,97 (д, J=7,5 Гц, 1H), 7,79 (д, J=7,7 Гц, 1H), 7,70 (т, J=8,2 Гц, 2H), 7,54-7,41 (м, 1H), 7,16 (т, J=6,6 Гц, 1H), 4,81 (д, J=12,3 Гц, 1H), 4,70 (бс (большой синглет), 1H), 4,58 (д, J=12,3 Гц, 1H), 3,92 (с, 1H), 3,40 (с, 1H), 3,10 (д, J=10,4 Гц, 1H), 0,86 (с, 3H), 0,82 (с, 3H).
Пример 30: Приготовление производного №130: 2-((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метилтио)этанол
В атмосфере аргона 200 мг (0,446 ммоль) этил 2-((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метилтио)ацетата растворяли в 4 мл тетрагидрофурана и добавляли 33,9 мг (0,892 ммоль) LiAlH4. Раствор нагревали до температуры 50°C в течение 1 ч. Медленно добавляли 0,030 мл воды, после чего добавляли 0,030 мл 6 н гидроксида натрия и 0,1 мл воды. Через 30 мин. перемешивания преципитат фильтровали и промывали этилацетатом. Собранные органические фазы промывали солевым раствором, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Полученное твердое вещество растирали с этилацетатом, фильтровали, а затем промывали этилацетатом. 50 мг (выход = 28%) 2-((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метилтио)этанола получали в виде белого твердого вещества. ЖХ/МС: m/z=407 (MH+); степень чистоты (УФ-детекция при 254 нм)=89%. 1H ЯМР (300 МГц, DMSO) δ 8,30 (д, 1H), 8,12 (с, 1H), 7,99 (м, 1H), 7,75 (м, 2H), 7,03 (м, 1H), 6,88 (д, 1H), 4,84 (т, 1H), 3,96 (с, 5H), 3,60 (м, 2H), 2,67 (т, 2H).
Производное 131 было приготовлено согласно аналогичной процедуре:

Пример 31: Приготовление производного №132: N-(N-(трет-бутоксикарбонил)карбамимидоил)-2-((3-((4-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетамид

200 мг 2-((3-((4-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)уксусной кислоты, 200 мг (0,617 ммоль) РуВОР и 118 мг (0,740 ммоль) гуанидин-ВОС погружали в 2 мл DMF. Затем в смесь вливали 0,258 мл (1,850 ммоль) триэтиламина, реакционную смесь перемешивали при к.т. в течение 4 ч. Полученную смесь вливали при перемешивании в 10 мл воды и 5 мл гептана. Полученное твердое вещество фильтровали, а затем промывали 2 мл воды и 1 мл гептана. 220 мг (выход = 77%) N-(N-(трет-бутоксикарбонил)карбамимидоил)-2-((3-((4-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетамида получали в виде серого твердого вещества. ЖХ/МС: m/z=466 (MH+); степень чистоты (УФ-детекция при 254 нм) = 99%. 1H ЯМР (300 МГц, DMSO) δ 11,10 (с, 1H), 8,71 (т, J=37,0 Гц, 3H), 7,86-7,62 (м, 3H), 7,51-7,37 (м, 1H), 7,30 (т, J=8,9 Гц, 2H), 7,13 (дд, J=6,7, 6,0 Гц, 1H), 4,81 (с, 2H), 4,18 (с, 2H), 1,38 (с, 9H).
Производное 133 было приготовлено согласно аналогичной процедуре.
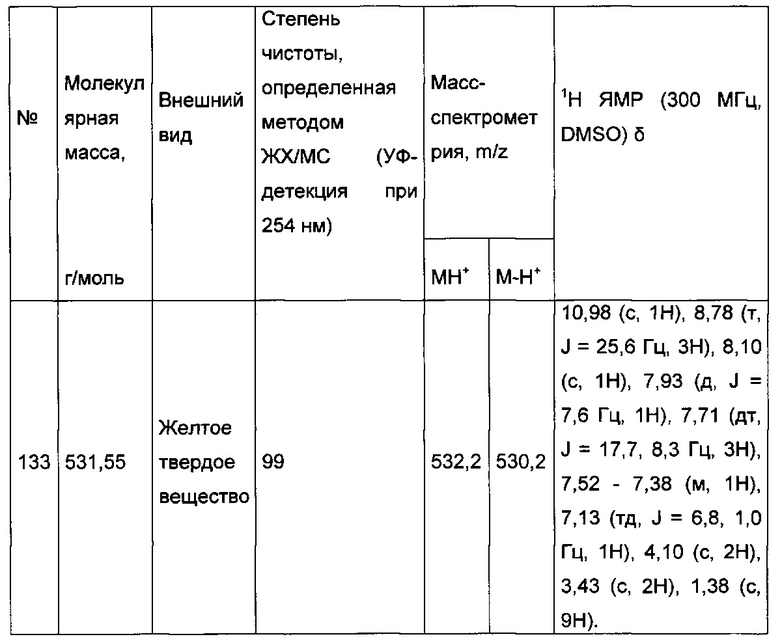
Пример 32: Приготовление производного №134: N-карбамимидоил-2-((3-((4-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)-метокси)ацетамид гидрохлорид

190 мг (0,408 ммоль) N-(N-(трет-бутоксикарбонил)карбамимидоил)-2-((3-((4-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетамида растворяли в 2 мл этанола, после чего добавляли 1,020 мл (4,08 ммоль) 4 н раствора хлористоводородной кислоты в диоксане. Раствор нагревали до температуры 60°C в течение 2 ч. Растворитель выпаривали в вакууме. После охлаждения твердое вещество, которое кристаллизовалось, фильтровали и промывали этанолом. 85 мг (выход = 52%) N-карбамимидоил-2-((3-((4-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетамид гидрохлорида получали в виде желтого твердого вещества. ЖХ/МС: m/z=366 (MH+); степень чистоты (УФ-детекция при 254 нм) = 99%. 1H ЯМР (300 МГц, DMSO) δ 11,36 (с, 1H), 8,86 (д, J=6,7 Гц, 1H), 8,55 (д, J=28,4 Гц, 4Н), 7,92 - 7,71 (м, 4Н), 7,44 - 7,31 (м, 3H), 4,99 (с, 2H), 4,40 (с, 2H).
Производное 135 было приготовлено согласно аналогичной процедуре:
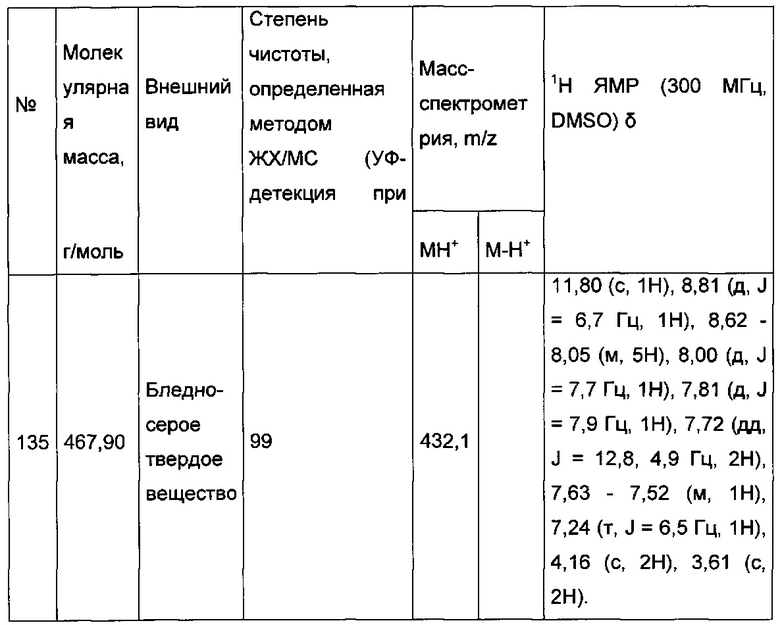
Пример 33: Приготовление производного №138: 2-((пиридин-2-илтио)метил)-3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин

150 мг (0,421 ммоль) 2-(хлорметил)-3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридина растворяли в 5 мл ацетонитрила, после чего добавляли 96 мг (0,842 ммоль) пиридин-2-тиола и 274 мг (0,842 ммоль) карбоната цезия. Растворитель перемешивали при к.т. в течение 2 ч 30 мин. Затем смесь вливали в 40 мл воды, после чего трижды экстрагировали 15 мл этилацетата. Собранные органические фазы промывали 30 мл насыщенного раствора гидрокарбоната натрия с 30 мл солевого раствора, после чего высушивали над Na2SO4, который затем удаляли в результате фильтрации, и полученный фильтрат концентрировали в вакууме. Неочищенный осадок очищали методом флэш-хроматографии на картридже с гелем диоксида кремния (элюент: градиент гептана/этилацетата, от 80% до 70% гептана, об./об.). 129 мг (выход = 74%) 2-((пиридин-2-илтио)метил)-3-((3-(трифторметил)фенил)этинил)имидазо-[1,2-а]пиридина получали в виде бежевого твердого вещества. ЖХ/МС: m/z=410 (MH+); степень чистоты (УФ-детекция при 254 нм)=99%. 1H ЯМР (300 МГц, DMSO) δ 8,70 (д, J=6,7 Гц, 1H), 8,52-8,37 (м, 1H), 8,02 (с, 1H), 7,91 (д, J=7,6 Гц, 1H), 7,71 (дд, J=41,7, 4,5 Гц, 4H), 7,43 (с, 2H), 7,13 (д, J=1,1 Гц, 2H), 4,73 (с, 2H).
Производные 136 и 137 были приготовлены согласно аналогичной процедуре:
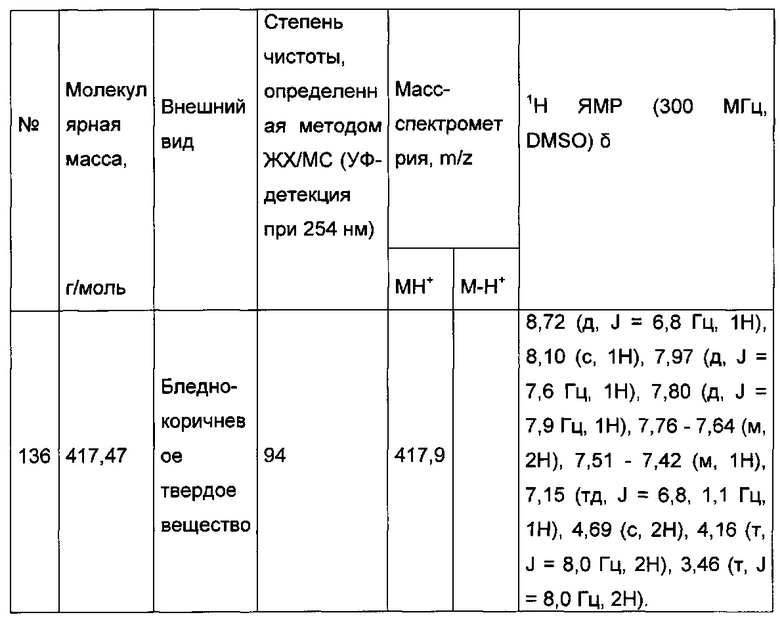

Тест стимуляции секреции инсулина клетками INS-1
Различные соединения исследовали на клетках INS-1, линии бета-клеток поджелудочной железы, для оценки способности исследуемых соединений потенциировать секрецию инсулина в ответ на поступление глюкозы. Вкратце, клетки культивировали в культуральной среде RPMI 1640 с 10 мМ глюкозы, содержащей 1 мМ пирувата натрия, 50 мкМ 2-меркаптоэтанола, 2 мМ глутамина, 10 мМ HEPES (4-(2-гидроксиэтил)-1-пиперазин этансульфоновая кислота), 100 МЕ/мл пенициллина, 100 мкг/мл стрептомицина и 10% инактивированной фетальной телячьей сыворотки (среда описана Asfari et al. [9]). Для проведения теста секреции инсулина клетки INS-1 высевали и культивировали в 96-луночных планшетах. Через 3 дня культивирования при температуре 37°C во влажной атмосфере (95% атмосферного воздуха/5% CO2) среду удаляли, и клетки инкубировали в течение 16 ч в среде, содержащей 5 мМ глюкозы и 1% инактивированной фетальной телячьей сыворотки. В день проведения теста клетки промывали буфером Кребса (pH 7,4), содержащим 0,1% бычьего альбумина, а затем предынкубировали в течение 30 мин. при температуре 37°C в том же буфере, содержащем 2,8 мМ глюкозы. Затем клетки снова промывали буфером Кребса, после чего инкубировали в течение 1 ч в буфере для проведения теста секреции (буфер Кребса, рН 7,4, содержащий 0,1% бычьего альбумина, 3,5 мМ глюкозы и исследуемые молекулы). По окончании теста супернатант клеток отбирали и измеряли количество инсулина, секретируемого клетками, с помощью набора для анализа методом ELISA (Enzyme-Linked Immuno Sorbent Assay, метод твердофазного иммуносорбентного анализа) с применением антител крысы против инсулина (ELISA Alpco, кат. №80-INSRTH-E10). Каждое условие изучали в трех повторах. В качестве положительных контролей при проведении теста использовали 3,5 мМ глюкозы, 10-7 М GLP-1 (глюкагонподобный пептид-1) и смесь 10-7 М Форсколина/10-5 М IBMX (3-изобутил-1-метилксантин). Соединение стимулирует секрецию инсулина, если данный показатель больше или равен 130% от показателя контроля для данной дозы глюкозы.

Исходя из полученных результатов, исследуемые производные формулы I оказывают значительное действие на потенциализацию секреции инсулина β-клетками поджелудочной железы INS-1 в ответ на поступление глюкозы. Полученные значения активации составляют от 131% до свыше 450%, вне зависимости от соответствующей дозы.
Исследование действия на продукцию глюкозы печенью
Оборудование и метод:
Гепатоциты выделяли из печени крыс Wistar, не получавших пищу в течение 24 ч, после перфузии коллагеназы в воротную вену. 1) Свежевыделенные гепатоциты высевали на 6-луночных планшетах, покрытых коллагеном и содержащих среду для адгезии (Williams Medium). После адгезии среду заменяли средой RPMI 1640 без добавления глюкозы, содержащей гидрокортизон (7,10-5 М), и проводили инкубацию в течение от 16 до 18 ч. На следующий день в течение 3 ч проводили тест продукции глюкозы печенью в среде Кребса. В базальных условиях клетки инкубировали исключительно с буфером Кребса, в стимулирующих условиях клетки помещали в буфер Кребса/лактат/пируват, в продуцирующих условиях клетки подвергали воздействию химических соединений в среде буфер Кребса/лактат/пируват. В случае, когда соединения растворяли в DMSO, все условия тестов выполнялись в присутствии конечной концентрации DMSO 0,1%. Положительный контроль при проведении теста представлял собой меркаптопиколинат, который, как известно, оказывает ингибирующее действие на продукцию глюкозы печенью посредством ингибирования фосфоэнолпируваткарбоксикиназы. В случае краткосрочной обработки соединения инкубировали в течение 3 часов. В случае долгосрочной обработки соединения инкубировали в течение 20 ч, пока гепатоциты культивировали в RPMI, а затем добавляли во время проведения теста продукции глюкозы печенью в течение 3 часов. По окончании 3 часов инкубации супернатант отбирали и измеряли содержание глюкозы колориметрическим методом с применением глюкозооксидазы. Клетки лизировали 0,1% водным раствором NaOH и проводили измерение количества белка методом Лоури. Результаты представлены в ммолях глюкозы на мг белка. Соединение ингибирует продукцию глюкозы в печени, если данный показатель меньше или равен 75% от показателя контроля для данной дозы глюкозы.
3 ч инкубации


Исследованные производные формулы I оказывают значительное действие на ингибирование продукции глюкозы печенью. Наиболее мощное ингибирование было достигнуто при применении производных 7, 78, 87, 92, 93, 110 и 117 в двух дозах и производных 29, 51, 59, 76, 77, 81, 88, 89, 90, 111, 112 и 115 в дозе 100 мкМ.
20 ч инкубирования

Исследованные производные формулы I оказывают значительное действие на ингибирование продукции глюкозы печенью. Наиболее мощное ингибирование было достигнуто при применении доз производного 58.
Исследование действия соединений на секрецию инсулина в ответ на поступление глюкозы на изолированных перфузируемых поджелудочных железах диабетических крыс N0STZ
Оборудование и метод:
Поджелудочные железы отбирали от крыс, у которых появление диабета было вызвано инъекцией Стрептозотоцина в день рождения [10]; животные получали анестезию Фентобарбиталом (Нембутал®: 45 мг/кг; интраперитонеальное введение). Данные крысы характеризуются специфическим дефицитом инсулина в ответ на поступление глюкозы [11], который наблюдается у человека при сахарном диабете II типа. Изолирование и перфузирование поджелудочных желез проводили согласно модификации [12] процедуры, описанной Sussman et al. [13]. Действие соединений или веществ-эталонов изучали в течение 35 минут (от t=20 мин. до t=55 мин.) в буфере Кребса, и затем в течение 20 минут (от t=55 мин. до t=75 мин.) в присутствии глюкозы в концентрации 16,5 мМ. Концентрацию инсулина, секретируемого в среду, измеряли методом Elisa (ELISA Alpco, кат. №80-INSRTH-Е10). Результаты выражали в виде среднего значения +/- COC (стандартная ошибка среднего) результатов нескольких экспериментов.

Исследованные производные формулы I оказывают значительное действие на восстановление секреции инсулина в ответ на поступление глюкозы на изолированных перфузируемых поджелудочных железах диабетических крыс N0STZ.
Исследование противодиабетической активности на крысах GK (крысах линии Гото-Какизаки)
Противодиабетическую активность соединения 118 оценивали на крысах GK - модели сахарного диабета II типа, не сопровождающегося ожирением. Данная модель была получена в результате скрещивания крыс Wistar, выбранных на основании легкого отсутствия толерантности к глюкозе [14]. Данные крысы обладают большинством нарушений функций, которые наблюдаются у человека при сахарном диабете II типа [15]: гипергликемией, отсутствием толерантности к глюкозе, резистентностью к инсулину, а также нарушенной инсулиновой реакцией на глюкозу. Данные животные были выведены в Metabrain и содержались в помещениях для животных с регулируемой температурой (22±2°C) при постоянной влажности (50±20%) с циклом день/ночь 12 ч (световая фаза с 7 ч до 19 ч) и имели неограниченный доступ к пище и воде. Условия содержания и проведения экспериментов соответствовали европейским директивам относительно здоровья и этичного обращения с лабораторными животными (ETS123). Используемые в данном исследовании крысы представляли собой самок крыс GK возрастом 16 недель, которые не получали пищи в течение 2 часов до начала исследования (состояние после всасывания, postabsorptive condition). Исследование гликемии проводили на 2 группах крыс: одна группа получала перорально соединение 118 в единичной дозе 50 мг/кг и контрольная группа получала перорально носитель. Образец крови отбирали в момент времени T0 непосредственно перед введением соединения 118 или носителя, а также в моменты времени T1 ч, T2 ч, T4 ч и T6 ч после введения. Гликемию анализировали на капле крови, отобранной из хвоста, измерение проводили с помощью глюкометра Accu-Check Performa. Результаты, представленные ниже, выражены в виде процента уменьшения гликемии в момент времени T1 ч, T2 ч, T4 ч и T6 ч для группы, получавшей соединение 118, по сравнению с контрольной группой.

Полученные результаты свидетельствуют, что соединение 118 при введении в единичной дозе 50 мг/кг способно уменьшить гипергликемию у крыс с диабетом типа II.
Перечень цитируемой литературы
[1] WO 2010089292.
[2] WO 2010070008.
[3] WO 2010034500.
[4] ЕР 2168965.
[5] WO 2009080291.
[6] WO 2009023179.
[7] Helvetica Chimica Acta (2007), 90(12), 2349-2367.
[8] Letters in Organic Chemistry (2005), 2(2), 184-187.
[9] Asfari et al., Endocrinology 130: 167-178, 1992.
[10] Portha et al., Diabetes, 23, (1974), 889-895.
[11] Giroix et al., Diabetes, 32, (1983), 445-451.
[12] Assan et al., Nature, 239, (1972), 125-126.
[13] Sussman et al., Diabetes, 15, (1966), 466-472.
[14] Goto et al., Proc. Jpn. Acad. 51, 80-85, 1975.
[15] Portha et al., Моль. Cell. Endocrinol., 297: 73-85, 2009.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКИЕ СОЕДИНЕНИЯ | 2020 |
|
RU2840815C2 |
| КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА И ПИРАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2686117C1 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2679914C9 |
| Имидазопиридиновые соединения в качестве ингибиторов PAD | 2018 |
|
RU2782743C2 |
| НОВЫЕ СОЕДИНЕНИЯ ЗАМЕЩЕННОГО N-(3-ФТОРПРОПИЛ)ПИРРОЛИДИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2742278C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ИСПОЛЬЗОВАНИЯ | 2012 |
|
RU2648997C2 |
| МОДУЛЯТОРЫ АТФ-СВЯЗЫВАЮЩИХ ТРАНСПОРТЕРОВ | 2010 |
|
RU2552353C2 |
| (АЗА)ИНДОЛ-, БЕНЗОТИОФЕН- И БЕНЗОФУРАН-3-СУЛЬФОНАМИДЫ | 2017 |
|
RU2767904C2 |
| КОНДЕНСИРОВАННОЕ ХИНОЛИНОВОЕ ПРОИЗВОДНОЕ И ЕГО ПРИМЕНЕНИЕ | 2005 |
|
RU2384571C2 |
| ИНГИБИТОРЫ КАТЕХОЛ-О-МЕТИЛТРАНСФЕРАЗЫ И ИХ ПРИМЕНЕНИЕ ПРИ ЛЕЧЕНИИ ПСИХОТИЧЕСКИХ РАССТРОЙСТВ | 2011 |
|
RU2586974C2 |
Изобретение относится к области органической химии, а именно к производному имидазопиридина формулы I или к его фармацевтически приемлемой соли, где Y представляет собой атом кислорода или серы, группу SO2 или -NR19, где R19 представляет собой атом водорода или С1-С6алкильную группу; R1, Ra, Rb и Rc представляют собой независимо друг от друга атом водорода; атом галогена; -O(С1-С6алкильную) группу; R2 представляет собой атом водорода; С1-С6алкильную группу, замещенную группой -ОН; группу (С1-С6алкил)COOR8, где R8 представляет собой атом водорода или С1-С6алкильную группу, причем указанная алкильная группа может быть замещена группой -NH2 или -ОН; группу (C1-C6алкил)CONHR9, где R9 представляет собой группу -ОН; С1-С6алкильную группу; -O(С1-С6алкильную) группу; пиридильную группу; -(C=NH)NHCOO(C1-C6алкильную) группу; группу -(C=NH)NH2 или группу -(C1-C6алкил)NR10R11, где R10 и R11 представляют собой независимо друг от друга C1-C6алкильную группу; (С1-С6алкил)СОморфолиновую группу; группу -C(=O)R12, где R12 представляет собой -O(С1-С6алкильную) группу; С1-С6алкильную группу, необязательно замещенную группой -ОН; морфолиновую группу; NH-фенильную группу, причем указанная фенильная группа необязательно замещена -СООН или -СОО(С1-С6алкильной) группой; или группу -NR13R14, где R13 и R14 представляют собой независимо друг от друга С1-С6алкильную группу; (C1-C6алкил)фенильную группу, причем указанная фенильная группа замещена -CN группой; (С1-С6алкил)тиофенильную группу; фенильную группу, причем указанная фенильная группа необязательно замещена 1 группой, которая выбрана из -СООН, -COO(C1-C6алкила), С1-С6алкила, замещенного -ОН, -CN, -CONHOH, -NHSO2(C1-C6алкилом) или группой -CONH-(C1-C6алкил)NR15R16, где R15 и R16 представляют собой независимо друг от друга С1-С6алкильную группу; пиридильную группу; дигидротиазолильную группу; тетрагидрофураноновую группу, замещенную 2 С1-С6алкильными группами; -SO2(C1-C6алкильную) группу; R3 представляет собой фенильную группу или 5-6-членную гетероарильную группу, содержащую 1 гетероатом, выбранный из азота и серы, причем указанная фенильная группа необязательно замещена 1-2 группами, которые выбраны из -C1-C6алкила, причем указанная алкильная группа необязательно замещена тремя атомами галогена, предпочтительно F, или группой -CN; атома галогена, предпочтительно F, -O(С1-С6алкила), причем указанная алкильная группа необязательно замещена 2-3 атомами галогена, предпочтительно F; -CN; -ОН; -NO2; -СООН; NR17R18, где R17 и R18 представляют собой независимо друг от друга C1-C6алкильную группу; или -NHCO-(C1-C6алкила). Также изобретение относится к фармацевтической композиции на основе соединения по п. 1. Технический результат: получены новые производные имидазопиридина, обладающие гипогликемической активностью. 2 н. и 9 з.п. ф-лы, 26 табл., 33 пр.

1. Производное имидазопиридина следующей общей формулы I:

отличающееся тем, что:
Y представляет собой атом кислорода или серы, группу SO2 или -NR19, где R19 представляет собой атом водорода или С1-С6 алкильную группу;
R1, Ra, Rb и Rc представляют собой независимо друг от друга атом водорода; атом галогена; -O(С1-С6 алкильную) группу;
R2 представляет собой
- атом водорода;
- С1-С6 алкильную группу, замещенную группой -ОН;
- группу (С1-С6 алкил)COOR8, где R8 представляет собой атом водорода или С1-С6 алкильную группу, причем указанная алкильная группа может быть замещена группой -NH2 или -ОН;
- группу (C1-C6 алкил)CONHR9, где R9 представляет собой группу -ОН; С1-С6 алкильную группу; -O(С1-С6 алкильную) группу; пиридильную группу; -(C=NH)NHCOO(C1-C6 алкильную) группу; группу -(C=NH)NH2 или группу -(C1-C6 алкил)NR10R11, где R10 и R11 представляют собой независимо друг от друга C1-C6 алкильную группу;
- (С1-С6 алкил)СОморфолиновую группу;
- группу -C(=O)R12, где R12 представляет собой -O(С1-С6 алкильную) группу; С1-С6 алкильную группу, необязательно замещенную группой -ОН; морфолиновую группу; NH-фенильную группу, причем указанная фенильная группа необязательно замещена -СООН или -СОО(С1-С6 алкильной) группой; или группу -NR13R14, где R13 и R14 представляют собой независимо друг от друга С1-С6 алкильную группу;
- (C1-C6 алкил)фенильную группу, причем указанная фенильная группа замещена -CN группой;
- (С1-С6 алкил)тиофенильную группу;
- фенильную группу, причем указанная фенильная группа необязательно замещена одной группой, которая выбрана из -СООН, -COO(C1-C6 алкила), С1-С6 алкила, замещенного -ОН, -CN, -CONHOH, -NHSO2(C1-C6 алкилом) или группой -CONH-(C1-C6 алкил)NR15R16, где R15 и R16 представляют собой независимо друг от друга С1-С6 алкильную группу;
- пиридильную группу;
- дигидротиазолильную группу;
- тетрагидрофураноновую группу, замещенную двумя С1-С6 алкильными группами;
- или -SO2(C1-C6 алкильную) группу;
R3 представляет собой фенильную группу или 5-6-членную гетероарильную группу, содержащую один гетероатом, выбранный из азота и серы, причем указанная фенильная группа необязательно замещена одной или двумя группами, которые выбраны из -C1-C6 алкила, причем указанная алкильная группа необязательно замещена тремя атомами галогена, предпочтительно F, или группой -CN; атома галогена, предпочтительно F, -O(С1-С6 алкила), причем указанная алкильная группа необязательно замещена двумя или тремя атомами галогена, предпочтительно F; -CN; -ОН; -NO2; -СООН; NR17R18, где R17 и R18 представляют собой независимо друг от друга C1-C6 алкильную группу; или -NHCO-(C1-C6 алкила);
или его фармацевтически приемлемая соль.
2. Производное имидазопиридина по п.1, отличающееся тем, что Y представляет собой атом кислорода или серы или группу -NH или -NMe, предпочтительно атом кислорода.
3. Производное имидазопиридина по п.1, отличающееся тем, что R1 Ra, Rb и Rc представляют собой атом водорода.
4. Производное имидазопиридина по п.1, отличающееся тем, что R2 представляет собой
- группу (C1-C6 алкил)COOR8, где R8 представляет собой атом водорода или C1-C6 алкильную группу;
- группу (C1-C6 алкил)CONHR9, где R9 представляет собой группу -ОН; -O(C1-С6 алкильную) группу; или группу -(C1-C6 алкил)NR10R11, где R10 и R11 представляют собой независимо друг от друга C1-C6 алкильную группу;
- фенильную группу, причем указанная арильная группа необязательно замещена одной группой, которая выбрана из -СООН, -СОО(С1-С6 алкила), -CONHOH, или -CONH-(C1-C6 алкил)NR15R16, где R15 и R16 представляют собой независимо друг от друга С1-С6 алкильную группу; или
- -СОNНфенильную группу, необязательно замещенную -СООН или -СОО(С1-С6 алкильной) группой.
5. Производное имидазопиридина по п.4, отличающееся тем, что R2 представляет собой группу -(С1-С6 алкил)СООН, предпочтительно -СН2СООН.
6. Производное имидазопиридина по п.1, отличающееся тем, что R3 представляет собой фенильную группу, необязательно замещенную одной или двумя группами, которые выбраны из -С1-С6 алкила, причем указанная алкильная группа необязательно замещена тремя атомами галогена, предпочтительно F; атома галогена, предпочтительно F, -O(C1-C6 алкила), причем указанная алкильная группа необязательно замещена двумя или тремя атомами галогена, предпочтительно F; -CN; или -NO2.
7. Производное имидазопиридина по п.6, отличающееся тем, что R3 представляет собой фенильную группу, замещенную одной или двумя группами, которые выбраны из -C1-C6 алкила, замещенного тремя атомами галогена, предпочтительно F; атома галогена, предпочтительно F, -О(C1-С6 алкила), причем указанная алкильная группа замещена двумя или тремя атомами галогена, предпочтительно F; или -CN.
8. Производное имидазопиридина по п.1, отличающееся тем, что указанное производное имидазопиридина выбрано из соединений, перечисленных ниже:
2-(((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метил)амино)уксусная кислота (68);
2-((3-((2-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)уксусная кислота (42);
2-((3-((3-(дифторметокси)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)уксусная кислота (35);
2-((3-((3-(трифторметокси)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)уксусная кислота (29);
2-((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)бензойная кислота (89);
2-((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетат натрия (51);
2-((3-((3,4-дифторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)уксусная кислота (38);
2-((3-((3-цианофенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)уксусная кислота (27);
2-((3-((3-фтор-4-(трифторметокси)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)уксусная кислота (39);
2-((3-((3-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)уксусная кислота (40);
2-((3-((3-метоксифенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)уксусная кислота (30);
2-((3-((4-(трифторметокси)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)уксусная кислота (32);
2-((3-((4-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)уксусная кислота (44);
2-((3-((4-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)бензойная кислота (88);
2-((3-((4-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)-N-гидроксиацетамид (59);
2-((3-((4-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)-N-гидроксибензамид (96);
2-((3-((4-нитрофенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетат натрия (48);
2-((3-(фенилэтинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетат натрия (45);
2-((3-(фенилэтинил)имидазо[1,2-а]пиридин-2-ил)метокси)бензоат натрия (92);
2-((3-(п-толилэтинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетат натрия (47);
2-(метил((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метил)амино)уксусная кислота (73);
3-((3-(фенилэтинил)имидазо[1,2-а]пиридин-2-ил)метокси)бензойная кислота (93);
3-(3-((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метил)уреидо)бензойная кислота (75);
4-((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)бензойная кислота (90);
этил 2-((3-((2-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетат (26);
этил 2-((3-((2-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетат (25);
этил 2-((3-((3-(дифторметокси)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетат (20);
этил 2-((3-((3-(трифторметокси)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетат (18);
этил 2-((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетат (7);
этил 2-((3-((3,4-дифторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетат (10);
этил 2-((3-((3-цианофенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетат (14);
этил 2-((3-((3-фтор-4-(трифторметокси)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетат (11);
этил 2-((3-((3-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетат (23);
этил 2-((3-((3-метоксифенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетат (12);
этил 2-((3-((4-(трифторметокси)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетат (17);
этил 2-((3-((4-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетат (3);
этил 2-((3-((4-нитрофенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетат (5);
этил 2-((3-(фенилэтинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетат (1);
этил 2-((3-(п-толилэтинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетат (2);
изопропил 2-((3-((4-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетат (54);
метил 2-((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)бензоат (77);
метил 2-((3-((3-цианофенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)бензоат (79);
метил 2-((3-((4-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)бензоат (76);
метил 2-((3-(фенилэтинил)имидазо[1,2-а]пиридин-2-ил)метокси)бензоат (78);
метил 2-(метил((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метил)амино)ацетат (72);
метил 3-((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)бензоат (81);
метил 3-((3-(фенилэтинил)имидазо[1,2-а]пиридин-2-ил)метокси)бензоат (87);
метил 3-(3-((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метил)уреидо)бензоат (74);
метил 4-((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)бензоат (83);
N-(2-(диметиламино)этил)-2-((3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетамид (55);
N-(2-(диметиламино)этил)-2-((3-((4-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетамид (57);
N-(2-(диметиламино)этил)-2-((3-((4-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)бензамид (95);
N-этокси-2-((3-((4-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетамид (58);
натрия 2-((3-((4-фторфенил)этинил)имидазо[1,2-а]пиридин-2-ил)метокси)ацетат (46);
этил 2-[метил-[[3-[2-[3-(трифторметил)фенил]этинил]имидазо[1,2-а]пиридин-2-ил]метил]амино]ацетат гидрохлорид (101);
этил 2-[[6-хлор-3-[2-[3-(трифторметил)фенил]этинил]имидазо[1,2-а]пиридин-2-ил]метил-метил-амино)ацетат (102);
этил 2-(метил((3-((3-(трифторметокси)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метил)амино)ацетат (103);
этил 2-(((6-хлор-3-((3-(трифторметокси)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метил)(метил)амино)ацетат гидрохлорид (104);
2-(((6-хлор-3-((3-(трифторметокси)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метил)(метил)амино)уксусная кислота (105);
2-(метил((3-((3-(трифторметокси)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метил)амино)уксусная кислота (106);
2-(((6-хлор-3-((3-(трифторметил)фенил)этинил)имидазо[1,2-а]пиридин-2-ил)метил)(метил)амино)уксусная кислота (107);
этил 2-[[3-[2-[3-(трифторметил)фенил]этинил]имидазо[1,2-а]пиридин-2-ил]метилсульфанил]ацетат (108);
этил 2-[[3-[2-[3-(трифторметокси)фенил]этинил]имидазо[1,2-а]пиридин-2-ил]метилсульфанил]ацетат (109);
этил 2-[[6-хлор-3-[2-[4-(фтор)фенил]этинил]имидазо[1,2-а]пиридин-2-ил]метилсульфанил]ацетат (110);
этил 2-[[6-хлор-3-[2-[3-(трифторметил)фенил]этинил]имидазо[1,2-а]пиридин-2-ил]метилсульфанил]ацетат (111);
этил 2-[[6-хлор-3-[2-[3-(трифторметокси)фенил]этинил]имидазо[1,2-а]пиридин-2-ил]метилсульфанил]ацетат (112);
этил (2R)-2-амино-3-[[3-[2-[3-(трифторметил)фенил]этинил]имидазо[1,2-а]пиридин-2-ил]метилсульфанил]пропаноат (113);
этил 2-[[8-метокси-3-[2-[3-(трифторметил)фенил]этинил]имидазо[1,2-а]пиридин-2-ил]метилсульфанил]ацетат (114);
2-[[3-[2-[3-(трифторметил)фенил]этинил]имидазо[1,2-а]пиридин-2-ил]метилсульфанил]уксусная кислота (115);
2-[[3-[2-[3-(трифторметокси)фенил]этинил]имидазо[1,2-а]пиридин-2-ил]метилсульфанил]уксусная кислота (116);
2-[[6-хлор-3-[2-(4-фторфенил)этинил]имидазо[1,2-а]пиридин-2-ил]метилсульфанил]уксусная кислота (117);
2-[[6-хлор-3-[2-[3-(трифторметил)фенил]этинил]имидазо[1,2-а]пиридин-2-ил]метилсульфанил]уксусная кислота (118);
2-[[6-хлор-3-[2-[3-(трифторметокси)фенил]этинил]имидазо[1,2-а]пиридин-2-ил]метилсульфанил]уксусная кислота (119);
2-[[8-метокси-3-[2-[3-(трифторметил)фенил]этинил]имидазо[1,2-а]пиридин-2-ил]метилсульфанил]уксусная кислота (120);
этил 2-[[3-[2-[3-(трифторметил)фенил]этинил]имидазо[1,2-а]пиридин-2-ил]метилсульфонил]ацетат (121);
этил 2-[[6-хлор-3-[2-[3-(трифторметокси)фенил]этинил]имидазо[1,2-а]пиридин-2-ил]метилсульфонил]ацетат (122);
2-[[6-хлор-3-[2-[3-(трифторметокси)фенил]этинил]имидазо[1,2-а]пиридин-2-ил]метилсульфонил]уксусная кислота (123);
N-(3-пиридил)-2-[[3-[2-[3-(трифторметил)фенил]этинил]имидазо[1,2-а]пиридин-2-ил]метилсульфанил]ацетамид (124);
N-(3-пиридил)-2-[[3-[2-[3-(трифторметокси)фенил]этинил]имидазо[1,2-а]пиридин-2-ил]метокси]ацетамид (125);
N-(3-пиридил)-2-[[3-[2-[3-(трифторметокси)фенил]этинил]имидазо[1,2-а]пиридин-2-ил]метилсульфанил]ацетамид (126);
4-гидрокси-3,3-диметил-2-[[3-[2-[3-(трифторметил)фенил]этинил]имидазо[1,2-а]пиридин-2-ил]метокси]бутановая кислота (129);
2-[[8-метокси-3-[2-[3-(трифторметил)фенил]этинил]имидазо[1,2-а]пиридин-2-ил]метилсульфанил]этанол (130);
2-[[6-хлор-3-[2-[3-(трифторметил)фенил]этинил]имидазо[1,2-а]пиридин-2-ил]метилсульфанил]этанол (131);
N-карбамимидоил-2-[[3-[2-[3-(трифторметил)фенил]этинил]имидазо[1,2-а]пиридин-2-ил]метилсульфанил]ацетамид гидрохлорид (135).
9. Фармацевтическая композиция, обладающая гипогликемической активностью, содержащая производное по любому из пп.1-8 и фармацевтически приемлемый эксципиент.
10. Производное по любому из пп.1-8 для применения в качестве лекарственного средства, обладающего гипогликемической активностью.
11. Производное по любому из пп.1-8 для применения в качестве лекарственного средства, предназначенного для лечения и/или предотвращения сахарного диабета.
| СПОСОБ ШУНТИРОВАНИЯ ПЕРЕДНЕЙ КАМЕРЫ ГЛАЗА ПРИ АНТИГЛАУКОМАТОЗНЫХ ОПЕРАЦИЯХ | 2000 |
|
RU2168965C1 |
| СПОСОБ ИЗМЕРЕНИЯ КОЭФФИЦИЕНТА ТЕМПЕРАТУРОПРОВОДНОСТИ МАТЕРИАЛА И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 1993 |
|
RU2072516C1 |
| ПРОИЗВОДНЫЕ 2-БЕНЗОИЛ-ИМИДАЗО[1,2-a]ПИРИДИНА, ИХ ПОЛУЧЕНИЕ И ИХ ПРИМЕНЕНИЕ В ТЕРАПИИ | 2008 |
|
RU2442785C1 |

