Существующий уровень техники
Данное изобретение относится к низкомолекулярным ингибиторам реакции IgE (иммуноглобулина Е) на аллергены, полезным при лечении аллергии и/или астмы или любых заболеваний, для которых IgE является патогенным.
По оценкам астмой страдают 10 миллионов человек в Соединенных Штатах, т.е. приблизительно 5% населения. Оцениваемые расходы на астму в Соединенных Штатах превышают 6 миллиардов долларов. Приблизительно 25% пациентов с астмой, обращающихся за скорой помощью, нуждаются в госпитализации, а большая часть общих прямых медицинских расходов идет на стационарное больничное лечение (скорую помощь) на сумму более 1,6 миллиарда долларов. Расходы на предписанные медикаменты, увеличившиеся между 1985 и 1990 годами на 54%, приблизились к 1,1 миллиарда долларов (Kelly, Pharmacotherapy 12:13 S-21 S (1997)).
В соответствии с Национальным обзором амбулаторного здравоохранения на астму приходится 1% всех амбулаторных посещений, и это заболевание продолжает оставаться важной причиной пропусков школы у детей. Несмотря на улучшение понимания процесса заболевания и лучшие лекарства, заболеваемость астмой и смертность от астмы в США и во всем мире продолжает расти (Министерство здравоохранения и гуманитарных служб США; 1991, публикация №91-3042). Таким образом, астма представляет собой серьезную угрозу здоровью общества.
Патофизиологические процессы, вызывающие возникновение астматического эпизода, могут быть разделены на две фазы, обе отличает бронхоконстрикция, вызывающая затруднение дыхания, сдавливание груди и одышку. Астматическая реакция первой, ранней фазы вызывается аллергенами, раздражителями или физической нагрузкой. Аллергены поперечно связывают молекулы иммуноглобулина Е (IgE), связанные с рецепторами на тучных клетках, заставляя их высвобождать определенное количество ранее сформированных медиаторов воспаления, в том числе гистамина. К дополнительным инициаторам относятся осмотические изменения в тканях дыхательных путей после физической нагрузки или вдыхания холодного сухого воздуха. Реакция второй, поздней фазы характеризуется инфильтрацией активированных эозинофилов и других воспалительных клеток в ткани дыхательных путей, эпителиальным шелушением и присутствием в дыхательных путях сильно вязкой слизи. Нарушение, вызванное этой воспалительной реакцией, оставляет дыхательные пути подвергнутыми первичному воздействию или сенсибилизированными, так что для вызывания последующих симптомов астмы требуются меньшие инициализаторы.
Для паллиативного лечения астмы доступно несколько лекарств; однако, их эффективность сильно варьируется. Короткодействующие β 2-адренергические агонисты, тербуталин и альбутерол, долгое время являющиеся основой лечения астмы, действуют прежде всего в ходе ранней фазы в качестве бронходилататоров. Более новые долгодействующие β 2-агонисты, салметерол и формотерол, могут уменьшать бронхоконстрикторный компонент поздней реакции. Однако, поскольку β 2-агонисты не обладают значительной противовоспалительной активностью, они не оказывают воздействия на бронхиальную гиперреактивность.
Многочисленные другие лекарства нацелены на специфические аспекты ранней или поздней астматической реакции. Например, антигистамины, такие как лоратадин, ингибируют ранние опосредованные гистамином воспалительные реакции. Некоторые из более новых антигистаминов, такие как азеластин и кетотифен, могут иметь и противовоспалительный, и слабый бронходилататорный эффект, но они в настоящий момент не имеют установленной эффективности при лечении астмы. Такие ингибиторы фосфодиэстеразы, как теофиллин/ксантины, могут ослаблять поздние воспалительные реакции, но нет очевидных подтверждений того, что эти соединения уменьшают бронхиальную гиперреактивность. Такие антихолинергики, как ипратопиум бромид, используемые в случаях острой астмы для ингибирования тяжелого сужения бронхов, не воздействуют на воспаления ранней и поздней фазы, не воздействуют на бронхиальную гиперреактивность, а потому не играют никакой роли в хронической терапии.
Кортикостероидные лекарства, такие как будесонид, являются наиболее сильными противовоспалительными агентами. Ингибиторы высвобождения медиатора воспаления, такие как кромолин и недокромил, действуют путем стабилизации тучных клеток и, тем самым, ингибирования воспалительной реакции на аллерген в поздней стадии. Таким образом, кромолин и недокромил, как и другие кортикостероиды, уменьшают бронхиальную гиперреактивность путем минимизации сенсибилизирующего эффекта воспалительного нарушения дыхательных путей. К сожалению, эти противовоспалительные агенты не дают бронхорасширяющего эффекта.
Разрабатываются несколько новых агентов, ингибирующих специфические аспекты астматического воспаления. Например, антагонисты лейкотриенового рецептора (ICI-204, 219, accolate) специфически ингибируют опосредованные лейкотриенами воздействия. Лейкотриены участвуют и в возникновении воспаления дыхательных путей, и в бронхоконстрикции.
Таким образом, несмотря на то, что в настоящее время доступно множество лекарств для лечения астмы, эти соединения прежде всего паллиативны и/или имеют значительные побочные эффекты. Следовательно, были бы в высшей степени желательны новые терапевтические подходы, нацеленные на причину, лежащую в основании, а не на каскад симптомов. Астма и аллергия имеют общую зависимость от опосредованных IgE событий. Разумеется, известно, что избыточное вырабатывание IgE является основной причиной аллергий в целом и аллергической астмы в частности (Duplantier and Cheng, Ann. Rep. Med. Chem. 29:73-81 (1994)). Таким образом, соединения, понижающие уровни IgE, могут быть эффективными при лечении причины, лежащей в основе астмы и аллергии.
Ни одна из современных терапий не устраняет избыточный циркулирующий IgE. Гипотеза о том, что понижение IgE плазмы может уменьшить аллергическую реакцию, была подтверждена недавними клиническими результатами с химерическим анти-IgE антителом CGP-51901 и рекомбинантным очеловеченным моноклональным антителом rhuMAB-E25. Действительно, три компании, Tanox Biosystems Inc., Genentech Inc. и Novartis AG совместно работают над разработкой очеловеченного анти-IgE антитела (BioWorld Today, February 26, 1997, р.2), которое будет лечить аллергию и астму путем нейтрализации избыточного IgE. Компания Tanox уже успешно протестировала анти-lgE антитело CGP-51901, уменьшавшее тяжесть и продолжительность назальных симптомов аллергического ринита в фазе II теста на 155 пациентах (Scrip №2080, Nov.24, 1995, р.26). Компания Genentech недавно раскрыла положительные результаты тестов фаз II/III на 536 пациентах рекомбинантного очеловеченного моноклонального антитела rhuMAB-E25 (BioWorld Today, November 10, 1998, р.1). Это антитело rhuMAB-E25, вводимое путем инъекции (высшая доза 300 мг каждые 2-4 недели по необходимости), обеспечивало 50%-ное снижение количества дней, когда пациенту требовались дополнительные неотложные медикаменты (антигистамины и противоотечные средства), по сравнению с плацебо. Получение недостающих данных по этому продукту предполагается в 2000 году. Положительные результаты тестов анти-IgE антитела показывают, что терапевтические стратегии, направленные на понижающее регулирование IgE, могут быть эффективными.
Раскрытие изобретения
Настоящее изобретение рассматривает семейство родственных соединений для использования при лечении состояния, связанного с избыточным уровнем IgE. Бензимидазоловые ингибиторы IgE в соответствии с настоящим изобретением представлены родовой формулой
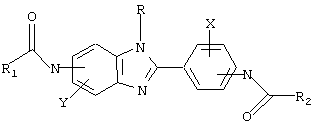
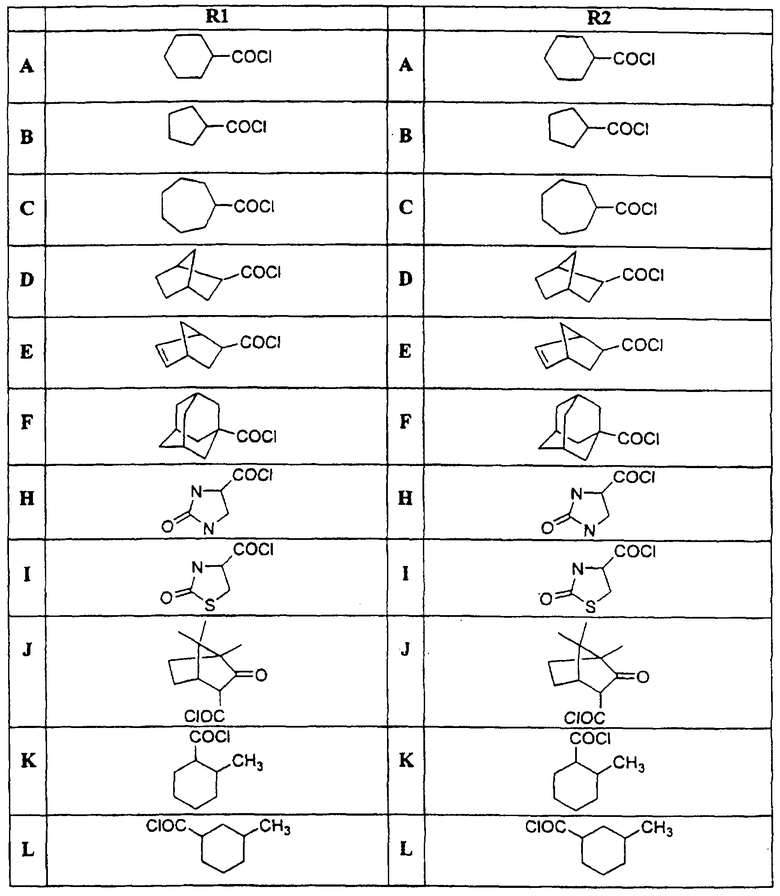
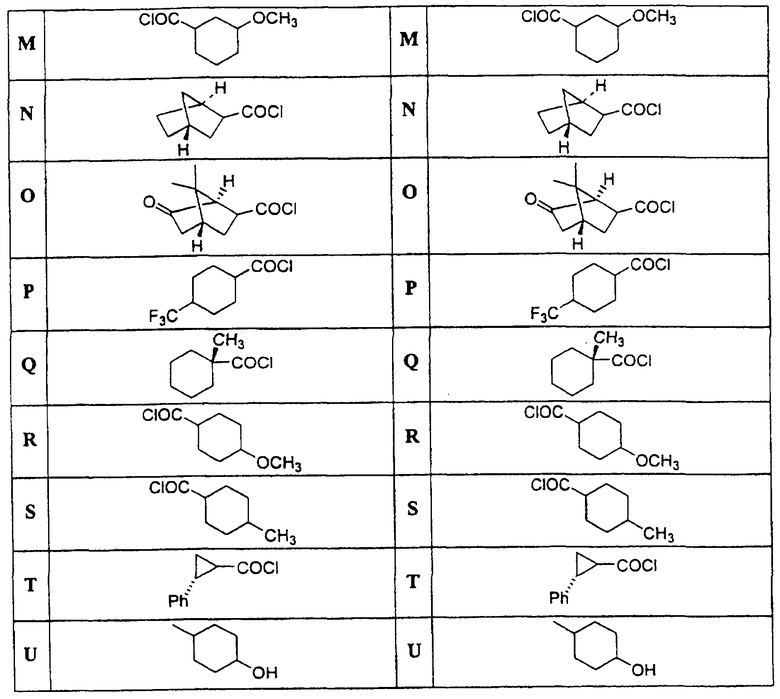
в которой Х и Y независимо выбираются из группы, состоящей из Н, алкила, алкоксигруппы, арила, замещенного арила, гидроксигруппы, галогена, аминогруппы, алкиламиногруппы, нитрогруппы, цианогруппы, СF3, ОСF3, CONH2, CONHR и NHCOR1. R выбирается из группы, состоящей из Н, СН3, С2Н5, С3Н7, С4Н9, CH2Ph и СН2С6Н4-F(р-). R1 и R2 выбираются независимо из группы, состоящей из алкила, циклоалкила, замещенного циклоалкила, многокольцевого циклоалкила, алифатических соединений со сцепленными кольцами, циклопропила, замещенного циклопропила, циклобутила, замещенного циклобутила, циклопентила, замещенного циклопентила, циклогексила, замещенного циклогексила, циклогептила, замещенного циклогептила, бициклогептила, бициклооктила, бициклононила, замещенного бициклоалкила, адамантила, замещенного адамантила и тому подобного. Замещениями являются алкил, арил, СF3, СН3, ОСН3, ОН, CN, COOR, СООН и тому подобное.
В соответствии с еще одним аспектом изобретения рассматривается состав для использования при лечении аллергического состояния, содержащий диациловый бензимидазоловый ингибитор IgE, раскрытый выше, и по меньшей мере один дополнительный активный ингредиент, объединенные в фармацевтически приемлемом разбавителе. Дополнительные активные ингредиенты могут выбираться из группы, состоящей из короткодействующих b2-адренергических агонистов, таких как тербуталин и альбутерол, долгодействующих b2-адренергических агонистов, таких как салметерол и формотерол, антигистаминов, таких как лоратадин, азеластин и кетотифен, ингибиторов фосфодиэстеразы, антихолинергических агентов, кортикостероидов, ингибиторов высвобождения воспалительного медиатора или антагонистов лейкотриенового рецептора.
В соответствии с еще одним аспектом изобретения рассматривается семейство диациловых бензимидазоловых соединений для использования при лечении аллергического состояния, содержащее следующие виды:
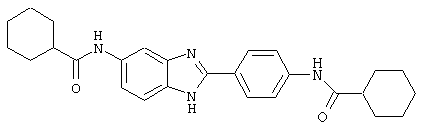
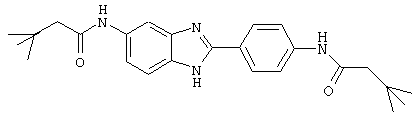
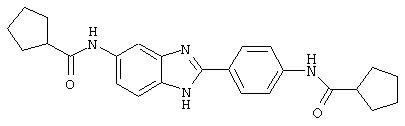
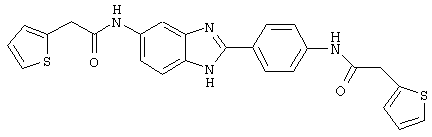
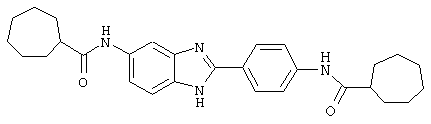
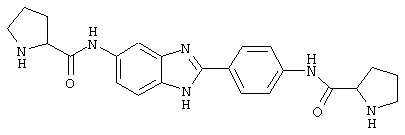
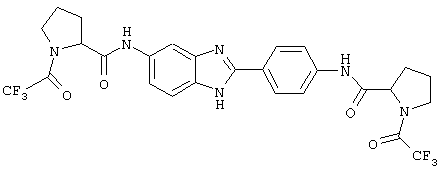
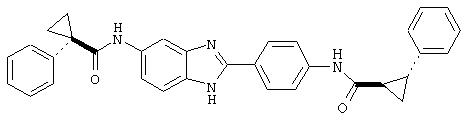
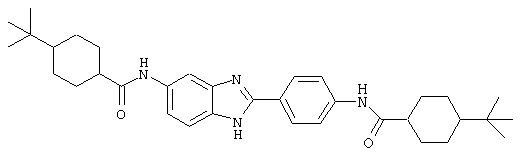
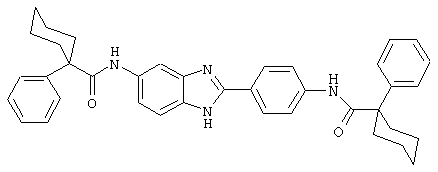
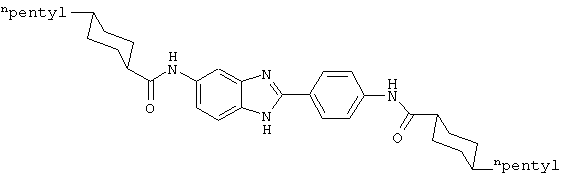
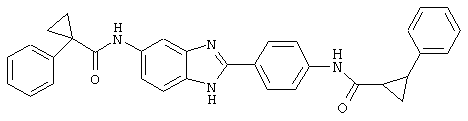
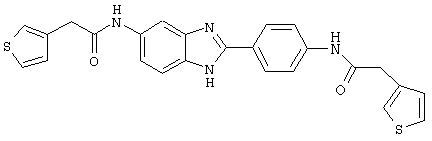
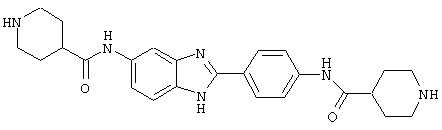
В соответствии с еще одним аспектом настоящего изобретения рассматривается моноацилированный вариант рассмотренного рода. Рассматриваются несколько видов асимметричных моноациловых бензимидазоловых соединений. Эти виды имеют следующие формулы
В соответствии с еще одним аспектом настоящего изобретения рассматривается способ приготовления медикамента для лечения состояния, связанного с избыточным уровнем IgE. Это соединение имеет формулу:
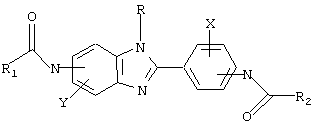
в которой Х и Y независимо выбираются из группы, состоящей из Н, алкила, алкоксигруппы, арила, замещенного арила, гидроксигруппы, галогена, аминогруппы, алкиламиногруппы, нитрогруппы, цианогруппы, СF3, ОСF3, CONH2, CONHR и NHCOR1. R выбирается из группы, состоящей из Н, СН3, С2Н5, С3Н7, С4Н9, CH2Ph и СН2С6Н4-Р(р-). R1 и R2 выбираются независимо из группы, состоящей из алкила, циклоалкила, замещенного циклоалкила, многокольцевого циклоалкила, алифатических соединений со сцепленными кольцами, циклопропила, замещенного циклопропила, циклобутила, замещенного циклобутила, циклопентила, замещенного циклопентила, циклогексила, замещенного циклогексила, циклогептила, замещенного циклогептила, бициклогептила, бициклооктила, бициклононила, замещенного бициклоалкила, адамантила и замещенного адамантила, в которой замещения выбираются из группы, состоящей из алкила, арила, СF3, СН3, ОСН3, ОН, CN, COOR и СООН.
В соответствии с еще одним аспектом настоящего изобретения рассматривается способ лечения млекопитающего, имеющего состояние, связанное с избыточным уровнем IgE. Способ содержит прием млекопитающим количества соединения, достаточного для уменьшения уровней IgE у млекопитающего. Это соединение имеет формулу:
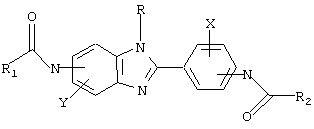
в которой Х и Y независимо выбираются из группы, состоящей из Н, алкила, алкоксигруппы, арила, замещенного арила, гидроксигруппы, галогена, аминогруппы, алкиламиногруппы, нитрогруппы, цианогруппы, СF3, ОСF3, CONH2, CONHR и NHCOR1. R выбирается из группы, состоящей из Н, СН3, С2Н5, С3Н7, С4Н9, CH2Ph и СН2С6Н4-F(р-). R1 и R2 выбираются независимо из группы, состоящей из алкила, циклоалкила, замещенного циклоалкила, многокольцевого циклоалкила, алифатических соединений со сцепленными кольцами, циклопропила, замещенного циклопропила, циклобутила, замещенного циклобутила, циклопентила, замещенного циклопентила, циклогексила, замещенного циклогексила, циклогептила, замещенного циклогептила, бициклогептила, бициклооктила, бициклононила, замещенного бициклоалкила, адамантила и замещенного адамантила, в которой замещения выбираются из группы, состоящей из алкила, арила, СF3, СН3, ОСН3, ОН, CN, COOR и СООН.
В варианте раскрытого выше способа по меньшей мере один дополнительный активный ингредиент может приниматься в сочетании с приемом соединения. Дополнительный активный ингредиент может объединяться с упомянутым соединением в фармацевтически приемлемом разбавителе и совместно приниматься млекопитающим. Дополнительным активным ингредиентом может быть короткодействующий b2-адренергический агонист, выбранный из группы, состоящей из тербуталина и альбутерола. В одном из вариантов дополнительным активным ингредиентом может быть долгодействующий b2-адренергический агонист, выбранный из группы, состоящий из салметерола и формотерола, или антигистамин, выбранный из группы, состоящий из лоратадина, азеластина и кетотифена. В еще одном варианте дополнительным активным ингредиентом может быть ингибитор фосфодиэстеразы, антихолинергический агент, кортикостероид, ингибитор высвобождения воспалительного медиатора или антагонист лейкотриенового рецептора.
Соединение предпочтительно принимается дозами величиной от приблизительно 0,01 мг до приблизительно 100 мг на килограмм массы тела в день разделенными дозами упомянутого соединения в течение по меньшей мере двух последовательных дней с регулярными периодическими интервалами.
Прочие варианты в рамках объема настоящего изобретения могут быть более полно поняты по последующему подробному описанию.
Подробное описание предпочтительного выполнения
Настоящее изобретение направлено на низкомолекулярные ингибиторы IgE (синтеза и/или высвобождения), которые полезны при лечении аллергии и/или астмы или любых заболеваний, в которых IgE является патогенным. Конкретные рассматриваемые здесь соединения были идентифицированы по их способности к подавлению уровней IgE как в ex vivo тестах, так и в in vivo тестах. Разработка и оптимизация режимов клинического лечения может быть прослежена специалистами по описанным ниже тестам ех vivo и in vivo.
Тест Ех Vivo
Этот тест начинается с инициирования антигена in vivo и измеряет вторичные реакции антитела in vitro. Основной протокол был задокументирован и оптимизирован для диапазона параметров, в том числе доза антигена для инициирования и временной промежуток после инициирования, количество культивированных in vitro клеток, концентрации антигена для выявления вторичной реакции IgE (и прочих иммуноглобулинов) in vitro, проба фетальной бычьей сыворотки (ФБС), которая обеспечивает оптимальную реакцию IgE in vitro, важность CD4+ инициированных Т-клеток и гаптен-специфических В-клеток, а также специфичность теста ELISA для IgE (Marcelletti and Katz, Cellular Immunology 135:471-489 (1991); включено сюда посредством ссылки).
Реальный протокол, использовавшийся для данного проекта, был адаптирован для анализа с большей производительностью. Мыши BALB/cByj были иммунизированы внутрибрюшинно с помощью 10 мкг DNP-KLH, абсорбированного на 4 мг квасцов, и убивались через 15 дней. Селезенки изымались и гомогенизировались в тканевом дефибрере, дважды промывались и содержались в DMEM, дополненном 10% ФБС, 100 ед/мл пенициллина, 100 мкг/мл стрептомицина и 0,0005% 2-меркаптоэтанола. Были установлены культуры клеток селезенки (2-3 миллиона клеток/мл, 0,2 мл/ячейка в четырех экземплярах, 96-ячеечные пластины) в присутствии или отсутствии DNP-KLH (10 нг/мл). Тестовые соединения (2 мкг/мл и 50 нг/мл) добавлялись к культурам клеток селезенки, содержащим антиген, и инкубировались при 37° С в течение 8 дней в атмосфере 10% СО2.
Поверхностный слой культуры был собран через 8 дней и были измерены иммуноглобулины путем модификации специфического изотипселективного теста ELISA, описанного в Marcelletti and Katz (см. выше). Тест был модифицирован для облегчения высокой производительности. Пластины ELISA были подготовлены путем покрывания DNP-KLH в течение ночи. После блокирования с помощью альбумина бычьей сыворотки (АБС), аликвотное количество поверхностного слоя каждой культуры разводилось (1:4 в фосфатно-буферном солевом растворе (ФБСР) с АБС, азидом натрия и Tween 20), добавлялось на пластины ELISA и инкубировалось в течение ночи в увлажняемом ящике при 4° С. Уровни IgE были подсчитаны после последовательных инкубации с биотинилированным антимышиным IgE козла (b-GAME), АР-стрептавидином и субстратом.
Подобным же образом был измерен антигенно-специфический IgGI, за исключением того, что поверхностный слой культуры был разведен 200-кратно, а b-GAME был заменен на биотинилированный антимышиный IgGI козла (b-GAMGl). IgG2a был измерен в пластинах ELISA, которые были покрыты DNP-KLH с последующим разведением 1:20 поверхностного слоя культуры и инкубацией с биотинилированным антимышиным IgG2a козла (b-GAMG2a). Количество каждого изотипа определялось путем сравнения со стандартной кривой. Уровень определяемости всего антитела был приблизительно равен 200-400 пг/мл, и существовала кросс-реактивность в объеме менее 0,001%, с любым другим изотипом Ig в ELISA для IgE.
Тест In Vivo
Соединения, активность которых была обнаружена в тесте ex vivo (выше), были дополнительно протестированы на активность при подавлении реакций IgE in vivo. Мыши, получившие облучение низкой дозы до иммунизации с помощью носителя, проявляли увеличенную реакцию IgE на сенсибилизацию антигеном 7 дней спустя. Прием тестовых соединений производился непосредственно перед и после сенсибилизации антигеном, измерялась способность данного лекарства подавлять реакцию IgE. Сравнивались уровни IgE, IgGI и IgG2a в сыворотке.
Самки мышей BALB/cByj облучались дозой 250 рад через 7 часов после начала дневного светового цикла. Два часа спустя мыши иммунизировались внутрибрюшинно с помощью 2 мкг KJLH в 4 мг квасцов. Через 6 дней начинались инъекции лекарства в течение от 2 до 7 дней, раз или два в день. Обычно внутрибрюшинные инъекции и пероральные принудительные введения производились в виде суспензий (150 мкл/инъекция) в солевом растворе с 10% этанола и 0,25% метилцеллюлозы. Каждая группа состояла из 5-6 мышей. На второй день приема лекарства вводилось 2 мкг DNP-KLH внутрибрюшинно в 4 мг квасцов, непосредственно после утренней инъекции лекарства. Мышам пускалась кровь через 7-21 дней после введения DNP-KLH.
Антиген-специфические антитела IgE, IgGI и IgG2a измерялись посредством ELISA. Периорбитальные пробы крови центрифугировались при 14000 об/мин в течение 10 минут, поднявшиеся на поверхность элементы разводились пятикратно в солевом растворе и снова центрифугировались. Концентрации антитела каждой пробы крови определялись посредством ELISA четырех растворений (в трех экземплярах) и сравнивались со стандартной кривой: анти-DNP IgE (1:100 до 1:800), анти-DNP IgG2a (1:100 до 1:800) и анти-DNP IgG 1 (1:1600 до 1:12800).
Диациловые бензимидазоловые ингибиторы IgE
Было синтезировано несколько описываемых родовой формулой видов и была оценена их эффективность при понижающем регулировании IgE в ex vivo и в in vivo тестах
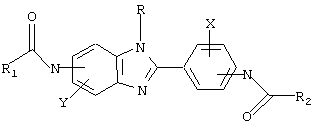
в которой Х и Y независимо выбираются из группы, состоящей из H, алкила, алкоксигруппы, арила, замещенного арила, гидроксигруппы, галогена, аминогруппы, алкиламиногруппы, нитрогруппы, цианогруппы, СF3, ОСF3, CONH2, CONHR и nhcor1. R выбирается из группы, состоящей из Н, СН3, С2Н5, С3Н7 и С4Н9, СН2Рh и СН2С6Н4-F(р-). R1 и R2 выбираются независимо из группы, состоящей из алкила, циклоалкила, замещенного циклоалкила, многокольцевого циклоалкила, алифатических соединений со сцепленными кольцами, циклопропила, замещенного циклопропила, циклобутила, замещенного циклобутила, циклопентила, замещенного циклопентила, циклогексила, замещенного циклогексила, циклогептила, замещенного циклогептила, бициклогептила, бициклооктила, бициклононила, замещенного бициклоалкила, адамантила, замещенного адамантила и тому подобного. Замещениями являются алкил, арил, СF3, СН3, ОСН3, ОН, CN, COOR, СООН и тому подобное.
Еще одним связанным родом является проиллюстрированный ниже моноацилированный вариант
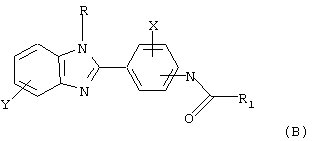
X выбирается из группы, состоящей из Н, алкила, алкоксигруппы, арила, замещенного арила, гидроксигруппы, галогена, аминогруппы, алкиламиногруппы, нитрогруппы, цианогруппы, СF3, ОСF3, CONH2, CONHR и NHCOR1. Y выбирается из групп, состоящей из моно-, ди- и тризамещенного Н, алкила, алкоксигруппы, арила, замещенного арила, гидроксигруппы, галогена, аминогруппы, алкиламиногруппы, нитрогруппы, цианогруппы, СF3, ОСF3, CONH2, CONHR и NHCOR1. R выбирается из группы, состоящей из Н, СН3, С2Н5, С3Н7 и С4Н9, CH2Ph и СН2С6Н4-F(р-). R1 выбирается из группы, состоящей из алкила, циклоалкила, замещенного циклоалкила, многокольцевого циклоалкила, алифатических соединений со сцепленными кольцами, циклопропила, замещенного циклопропила, циклобутила, замещенного циклобутила, циклопентила, замещенного циклопентила, циклогексила, замещенного циклогексила, циклогептила, замещенного циклогептила, бициклогептила, бициклооктила, бициклононила, замещенного бициклоалкила, адамантила, замещенного адамантила и тому подобного. Замещениями являются алкил, арил, СF3, СН3, ОСН3, ОН, CN, COOR, СООН и тому подобное.
Синтез комбинаторной библиотеки
Диациловые бензимидазоловые соединения по настоящему изобретению были приготовлены с помощью следующих реакций синтеза, в которых желательные кислые хлориды выбираются из групп r1 и R2, приведенных в Таблице.
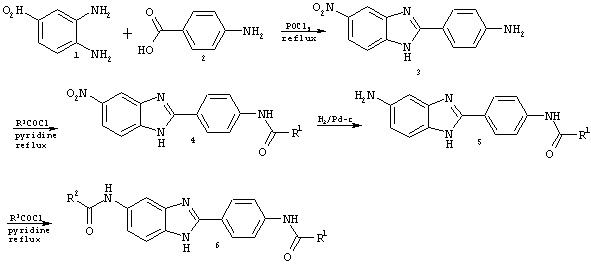
Синтез соединения 3: 4-нитро-1,2-фенилендиамин (10 г, 65,3 ммоль) и 4-аминобензойная кислота (8,95 г, 65,3 ммоль) помещались в колбу с круглым дном и медленно добавлялся оксихлорид фосфора (95 мл). Реакционной смеси позволяли перемешиваться в условиях оттока. Через 18 часов реакции давали возможность охладиться, а затем медленно выливали на водно-ледяную смесь в колбе Эрленмайера при энергичном перемешивании. Выпадал зеленовато-желтый осадок, который затем фильтровался и промывался обильным количеством воды. Остаток затем высушивался, давая 16,9 г сырого желательного продукта. Масс-спектральный анализ (положительный ион) показал наличие соединения 3.
Синтез соединения 4: Бензимидазол 3 (800 мг, 3,14 ммоль) растворялся в сухом пиридине (5 мл) в сцинтилляционном сосуде и медленно добавлялись желательные кислые хлориды (1,1 экв). Реакции проводились в печи при 60° С. Через 16 часов реакция охлаждалась до комнатной температуры, и добавлялась дистиллированная вода. Происходило выпадение осадка, который отфильтровывался, промывался водой и высушивался на воздухе. Водный слой экстрагировался с помощью ЕtOАс (6× 50 мл), высушивался на безводном Na2SO4, а растворитель удалялся в вакууме, давая в результате окрашенное твердое вещество. Путем позитивно-ионной масс-спектрографии было обнаружено, что желательный моноацилированный продукт присутствует в начальном осадке, а также в органическом слое. Поэтому полученные твердые остатки комбинировались и использовались как таковые для шага восстановления.
Восстановление соединения 4: Сырой моноацилированный нитробензимидазол 4 (1,22 г, 3,40 ммоль) растворялся в МеОН (20 мл), и добавлялось минимальное количество THF для полного растворения. Добавлялось каталитическое количество 10% Pd на С, и раствор дегазировался и оставлялся перемешиваться под давлением 3,4 атм в атмосфере Н2 на 4 часа. После завершения реакции по наблюдениям посредством тонкослойной хроматографии реакционная смесь фильтровалась через селит, и растворитель удалялся при пониженном давлении до получения 979 мг сырого остатка.
Общие органические анализы
HPLC/масс-спектрографические данные были получены с помощью Gilson semi-prep HPLC с УФ-детектором Gilson 170 Diode Array и масс-спектрографического детектора РЕ Sciex API 100LC. Детектор Waters 600Е с УФ Waters 490E также использовался для записи HPLC-данных. Соединения элюировались градиентом СН3СN (с 0,0035% TFA) и Н2О (с 0,01% TFA). Оба HPLC-инструмента использовали колонны Advantage С 18 60 А 5 μ 1 50 мм × 4,6 мм компании Thomson Instrument Compamy. Масс-спектры были получены путем прямой инъекции и электроаэрозольной ионизации на масс-спектрографическом детекторе РЕ Sciex API 100LC. Тонкослойная хроматография производилась с помощью алюминиевых прокаленных покрытых пластин Merck 60F-254. Флэш-хроматография выполнялась на силикагеле Merck 60 (230-400 меш), купленном в фирме ЕМ Scientific.
Синтез симметричных диамидов
Симметричные диациловые бензимидазоловые соединения по настоящему изобретению в общем случае приготавливались из 2-(4-аминофенил)-5-аминобензимидазола, который получался путем восстановления 2-(4-нитрофенил)-6-нитробензимидазола
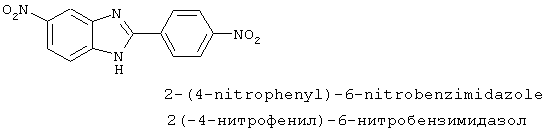
Динитробензимидазол приготавливался следующим образом: смесь 4-нитрофенилендиамина (6,4 г, 41,83 ммоль) и 4-нитробензойной кислоты (7,86 г, 47 ммоль) растворялась в РОCl3 (250 мл) и нагревалась для оттока в течение 2 часов. Реакционная смесь охлаждалась, выливалась на лед и перемешивалась в течение 30 минут. Получающееся твердое вещество фильтровалось, промывалось метанолом и бикарбонатом натрия для удаления непрореагировавшей кислоты и оставлялось высыхать на ночь, давая желательный продукт в виде коричневого твердого вещества (5,8 г). Продукт определялся посредством электроаэрозольной масс-спектрографии (температура плавления >300° С).
2-(4-Аминофенил)-5-аминобензимидазол был приготовлен путем суспендирования вышеупомянутого твердого вещества (75 г) в THF (75 мл), к которой добавлялся Pd-C (10% Pd по весу). Колба продувалась водородом и перемешивалась в течение ночи под стеклянным колпаком с водородом. Тонкослойная хроматография и масс-спектрография показали, что начальный материал все еще наличествовал, поэтому реакция была оставлена продолжаться в течение выходных. Тонкослойная хроматография показала завершение реакции, реакция фильтровалась через селит и промывалась метанолом. Растворитель удалялся при пониженном давлении, при этом получалось темно-коричневое твердое вещество (0,37 г), которое использовалось без дополнительной очистки
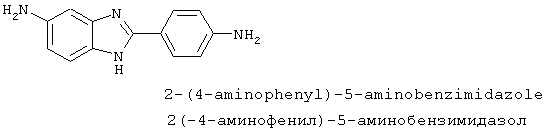
Альтернативно 2-(4-аминофенил)-5-аминобензимидазол был приготовлен путем следующего восстановления: 2-(4-нитрофенил)-6-нитробензимидазол (8,9 г, 31 ммоль) суспендировался в концентрированной НСl (100 мл), к которой был добавлен хлорид олова (42,3 г, 180 ммоль). Реакционная смесь нагревалась для оттока в течение 5 часов. Смесь охлаждалась до комнатной температуры и соль соляной кислоты желательного продукта осаждалась посредством добавления этанола. Полученное твердое вещество фильтровалось, заново растворялось в воде, и раствор защелачивался путем добавления концентрированного гидроксида аммония. Полученный осадок фильтровался и высушивался в течение ночи под вакуумом, давая желательный продукт в виде серого твердого вещества (6,023 г, 26,9 ммоль, 87%). Продукт определялся путем электроаэрозольной масс-спектрографии и HPLC (температура плавления 222-227° С).
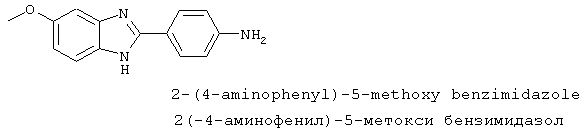
2-(4-Аминофенил)-5-метокси бензимидазол был синтезирован из 2-(4-нитрофенил)-5-метокси бензимидазола, который был приготовлен следующим образом: 1,2-диамино-4-метоксибензол (1,26 г, 10,0 ммоль) смешивался с 4-нитробензойной кислотой (1,67 г, 9,8 ммоль), растворялся в РОСl3 (10 мл) и нагревался для оттока в течение 2,5 часов. Реакционная смесь охлаждалась и осторожно выливалась на лед. Полученное твердое вещество фильтровалось, промывалось NаНСО3 и использовалось без дальнейшей очистки
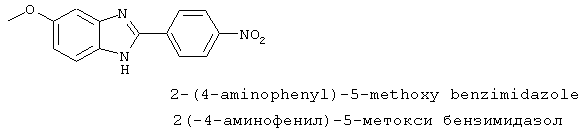
2-(4-Аминофенил)-5-метокси бензимидазол был получен путем растворения 1 г вышеупомянутого нитробензимидазола в 30% Na2S· 9H2O (20 мл) с перемешиванием при комнатной температуре в течение 21 часа. Реакционная смесь разбавлялась водой и экстрагировалась с помощью ЕtOАс. Комбинированные органические экстракты сушились на сульфате натрия и концентрировались в вакууме. Продукт определялся посредством масс-спектроскопии
2-(4-Аминофенил)-5,6-дихлор бензимидазол был синтезирован из 2-(4-нитрофенил)-5-дихлор бензимидазола, который был приготовлен следующим образом: 1,2 диамино-4,5-дихлорбензол (1,68 г, 10,0 ммоль) смешивался с 4-нитробензойной кислотой (1,58 г, 9,3 ммоль), растворялся в РОСl3 (10 мл) и нагревался для оттока в течение 2,5 часов. Реакционная смесь охлаждалась и осторожно выливалась на лед. Полученное твердое вещество фильтровалось, промывалось NаНСО3 и использовалось без дальнейшей очистки

2-(4-Аминофенил)-5,6-дихлор бензимидазол был получен путем растворения 1 г вышеупомянутого нитробензимидазола в 30% Na2S· 9H2O (20 мл) с перемешиванием при комнатной температуре в течение 21 часа. Реакционная смесь разбавлялась водой и экстрагировалась с помощью EtOAc. Комбинированные органические экстракты сушились на сульфате натрия и концентрировались в вакууме. Продукт определялся посредством масс-спектроскопии

2-(4-Аминофенил)-7-метил бензимидазол был синтезирован из 2-(4-нитрофенил)-7-метил бензимидазола, который был приготовлен посредством смешивания 1,2-диамино-3-метилбензола (1,24 г, 10,0 ммоль) с 4-нитробензойной кислотой (1,69 г, 9,8 ммоль), растворения в РОСl3 (10 мл) и нагревания для оттока в течение 2,5 часов. Реакционная смесь охлаждалась и осторожно выливалась на лед. Полученное твердое вещество фильтровалось, промывалось NaHCO3 и использовалось без дальнейшей очистки

2-(4-Аминофенил)-7-метил бензимидазол был синтезирован путем растворения 1 г вышеупомянутого нитробензимидазола в 30% Na2S· 9H2O (20 мл) с перемешиванием при комнатной температуре в течение 4,5 часов. Реакционная смесь разбавлялась водой и экстрагировалась с помощью EtOAc. Комбинированные органические экстракты сушились на сульфате натрия и концентрировались в вакууме. Продукт определялся посредством масс-спектроскопии

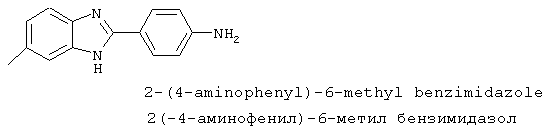
2-(4-Аминофенил)-6-метил бензимидазол был синтезирован из 2-(4-нитрофенил)-6-метил бензимидазола, который был приготовлен посредством смешивания 1,2-диамино-4-метилбензола (1,24 г, 9,8 ммоль) с 4-нитробензойной кислотой (1,6 г, 9,9 ммоль), растворения в РОСl3 (10 мл) и нагревания для оттока в течение 2,5 часов. Реакционная смесь охлаждалась и осторожно выливалась на лед. Полученное твердое вещество фильтровалось, промывалось NаНСО3 и использовалось без дальнейшей очистки

2-(4-Аминофенил)-6-метил бензимидазол был синтезирован путем растворения 1 г вышеупомянутого нитробензимидазола в 30% Na2S· 9H2O (20 мл) с перемешиванием при комнатной температуре в течение 4,5 часов. Реакционная смесь разбавлялась водой и экстрагировалась с помощью EtOAc. Комбинированные органические экстракты сушились на сульфате натрия и концентрировались в вакууме. Продукт определялся посредством масс-спектроскопии
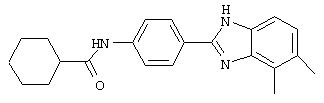
2-(4-Аминофенил)-5,6-диметил бензимидазол был синтезирован из 2-(4-нитрофенил)-5,6-диметил бензимидазола, который был приготовлен посредством смешивания 1,2-диамино-4,5-диметилбензол (1,38 г, 10,1 ммоль) с 4-нитробензойной кислотой (1,69 г, 9,9 ммоль), растворения в РОСl3 (10 мл) и нагревания для оттока в течение 2,5 часов. Реакционная смесь охлаждалась и осторожно выливалась на лед. Полученное твердое вещество фильтровалось, промывалось NаНСО3 и использовалось без дальнейшей очистки

2-(4-Аминофенил)-5,6-диметил бензимидазол был синтезирован путем растворения 1 г вышеупомянутого нитробензимидазола (31.1) в 30% Na2S· 9H2O (20 мл) с перемешиванием при комнатной температуре в течение 4,5 часов. Реакционная смесь разбавлялась водой и экстрагировалась с помощью ЕtOАс. Комбинированные органические экстракты сушились на сульфате натрия и концентрировались в вакууме. Продукт определялся посредством масс-спектроскопии

Последующее приготовление симметричных диамидов выполнялось одним из следующих способов:
Способ А: 2-(4-аминофенил)-6-аминобензимидазол (1 ммоль) суспендировался в THF (5 мл), к которой добавлялся DIEA (2,5 ммоль), и смесь охлаждалась до -78° С. К вышеупомянутой охлажденной смеси добавлялся кислый хлорид (2,5 ммоль), и смесь оставлялась нагреваться до комнатной температуры на ночь. К реакции добавлялась вода (2 мл) и экстрагировалась с помощью EtOAc. Объединенные органические экстракты комбинированно промывались NаНСО3 (вод.) и концентрировались при пониженном давлении. Полученный остаток очищался на силикагеле (гексаны/ЕtOАс или MeOH/CH2Cl2) или с помощью обратно-фазового HPLC (CH3CN/H2O).
Способ Б: 2-(4-аминофенил)-6-аминобензимидазол (1 ммоль) и DMAP (кат.) растворялись в пиридине (5 мл). К вышеупомянутому раствору был добавлен кислый хлорид (2,5 ммоль), и реакция перемешивалась в течение ночи при 60° С. Реакция охлаждалась до комнатной температуры, и для осаждения продукта добавлялась вода. Полученное твердое вещество собиралось путем фильтрации с промыванием твердого вещества гексанами и водой и NaHCO3 (вод.). Полученный остаток очищался на силикагеле (гексаны/ЕtOАс или МеОН/СН2Сl2) или с помощью обратно-фазового HPLC (CH3CN/H2O).
Способ В: 2-(4-аминофенил)-6-аминобензимидазол (1 ммоль) суспендировался в THF (10 мл), к которой добавлялся К2СО3 (2,5 ммоль) в воде (0,5 мл), и смесь охлаждалась до -78° С. К вышеупомянутой охлажденной смеси добавлялся кислый хлорид (2,5 ммоль), и смесь оставлялась нагреваться до комнатной температуры на ночь. К реакции добавлялась вода (10 мл) и экстрагировалась с помощью ЕtOАс. Комбинированные органические экстракты комбинированно промывались NаНСО3 (вод.) и концентрировались при пониженном давлении. Полученный остаток очищался на силикагеле (гексаны/ЕtOАс или МеОН/СН2Сl2) или с помощью обратно-фазового HPLC (CH3CN/H2O).
Способ Г: Карбоксиловая кислота (2,2 ммоль), EDC (2,2 ммоль) и DMAP (кат.) растворялись в горячем пиридине. К вышеупомянутому раствору был добавлен 2-(4-аминофенил)-6-аминобензимидазол (1 ммоль), и смесь нагревалась в течение ночи до 60° С. Охлажденная реакционная смесь разделялась между водой и ЕtOАс. Органический слой промывался NаНСО3, высушивался на Na2SO4 и концентрировался под вакуумом. Полученный остаток очищался на силикагеле (гексаны/EtOAc или МеОН/СН2Сl2) или с помощью обратно-фазового HPLC (CH3CN/H2O).
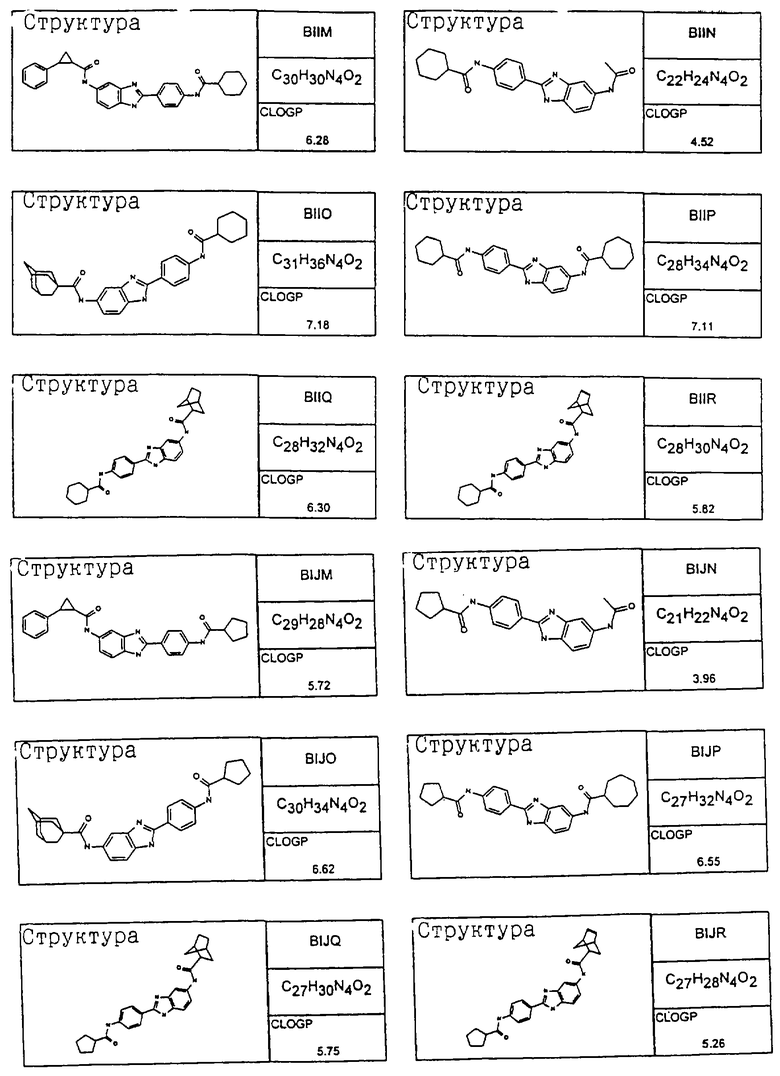
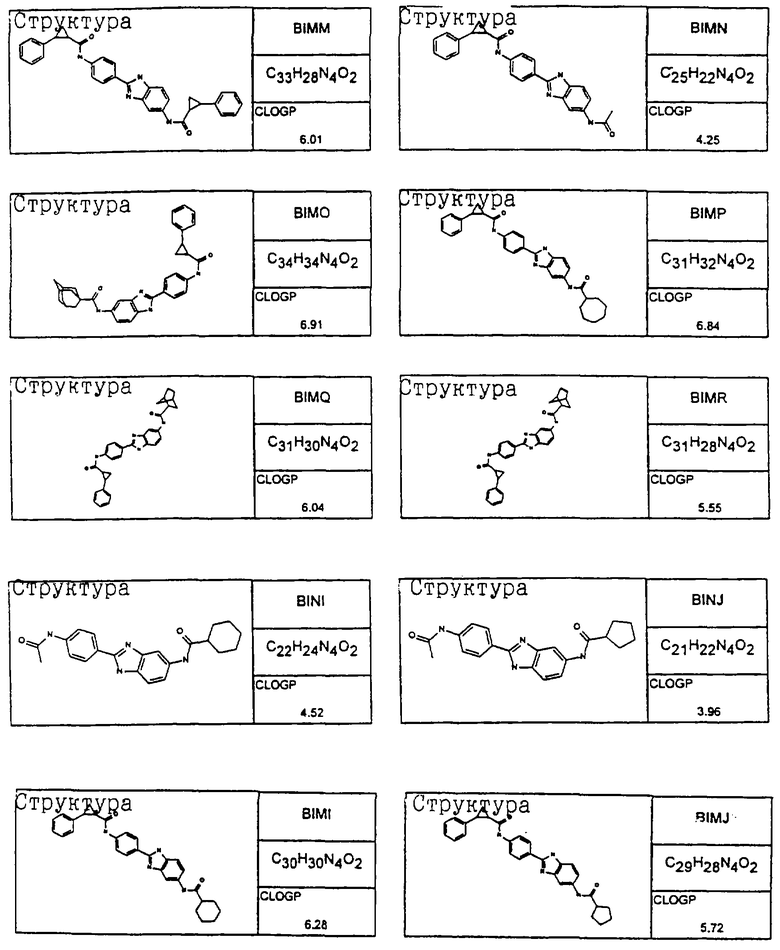
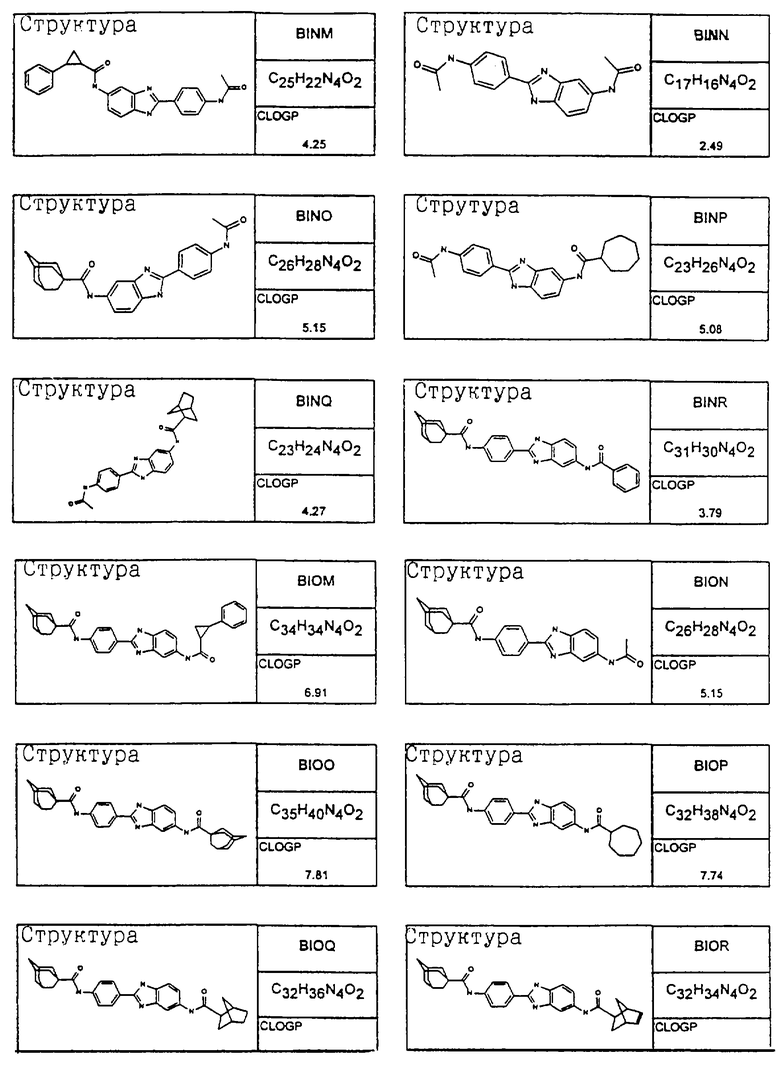
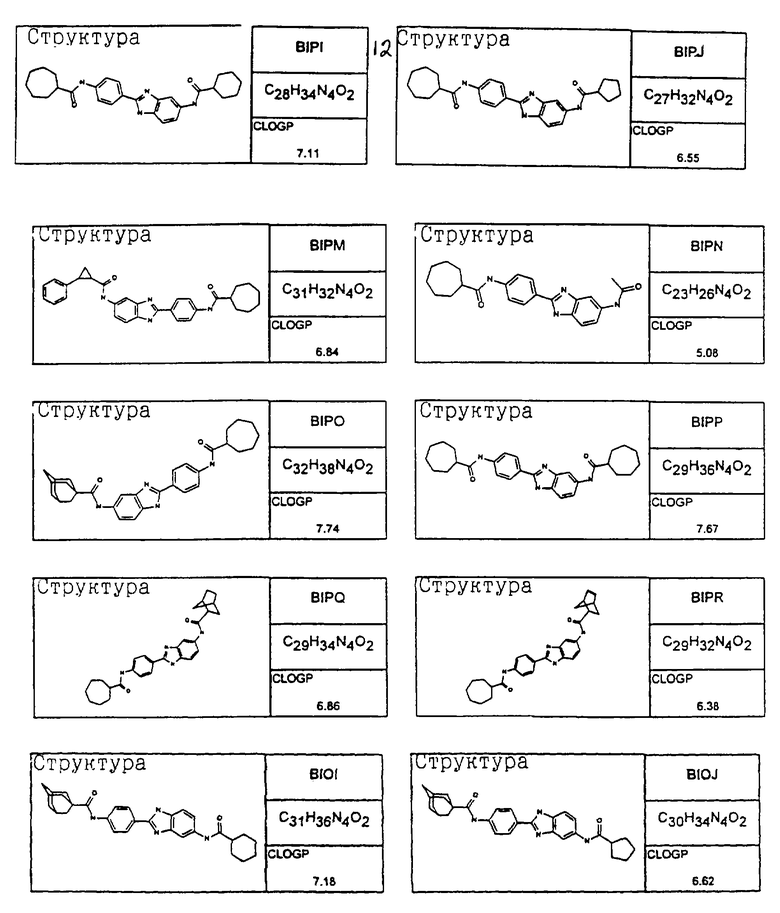
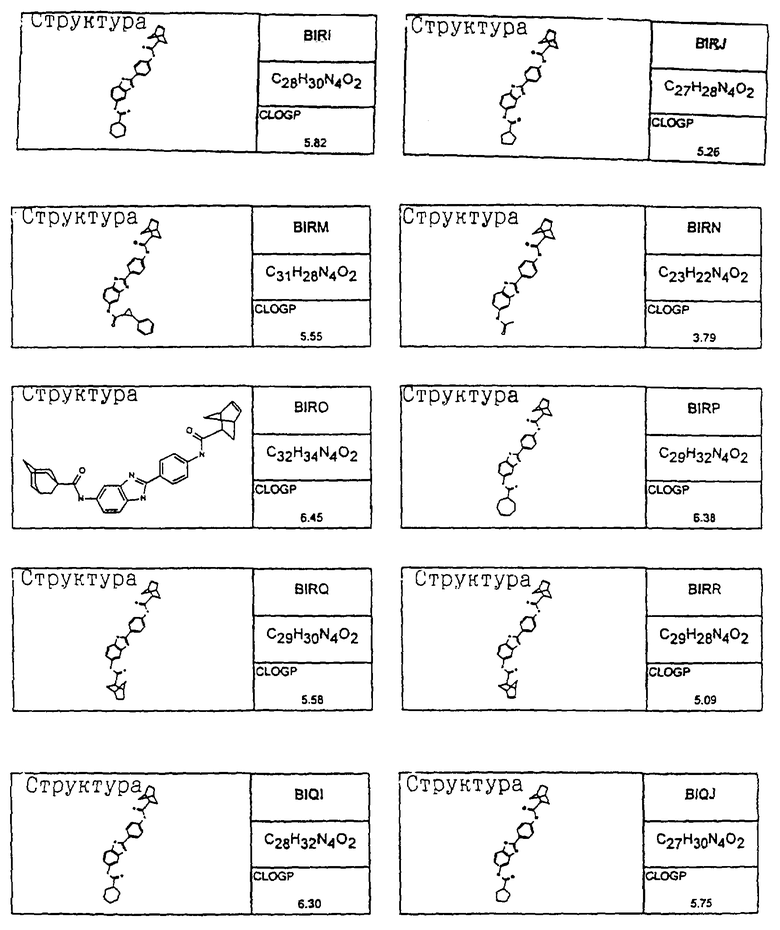
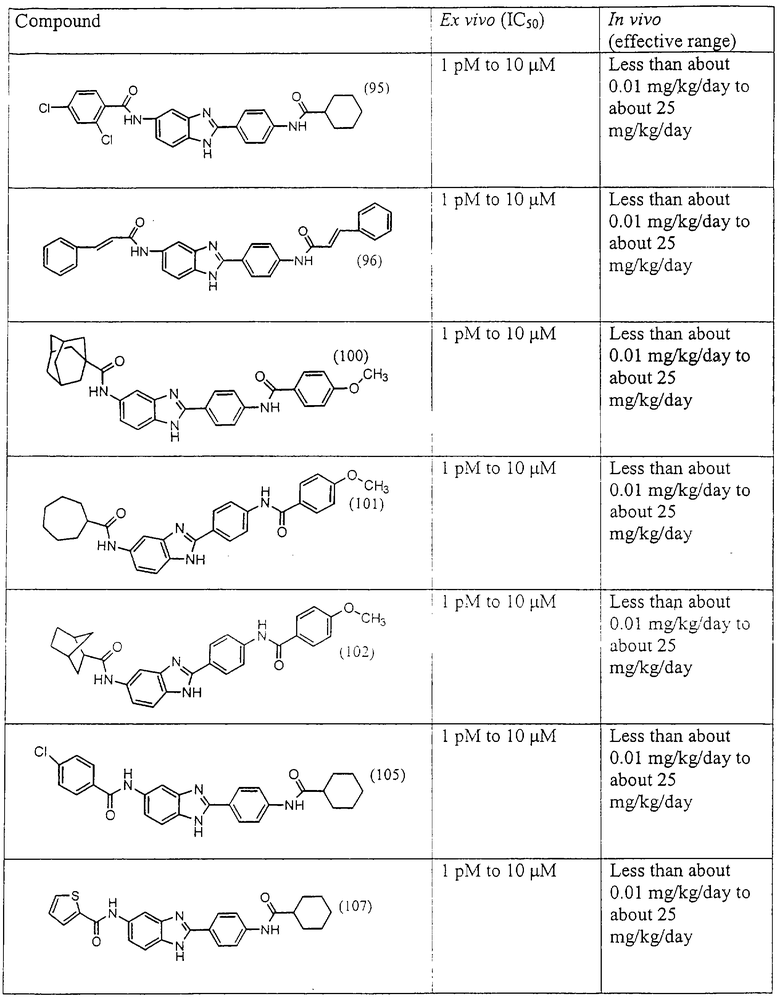
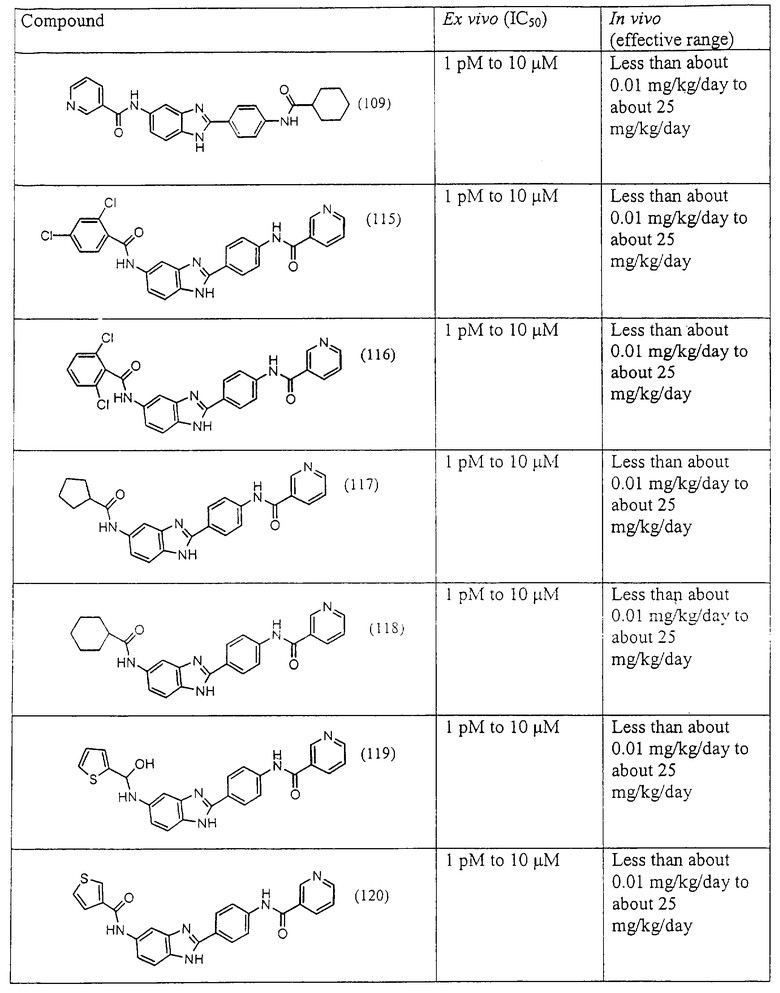
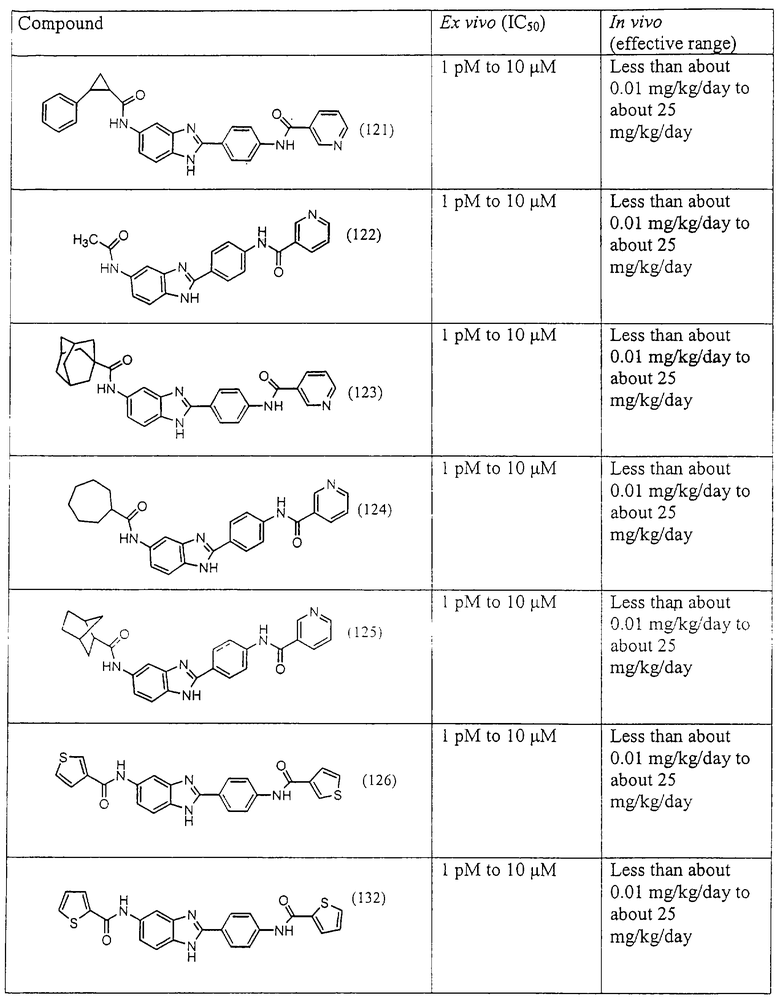
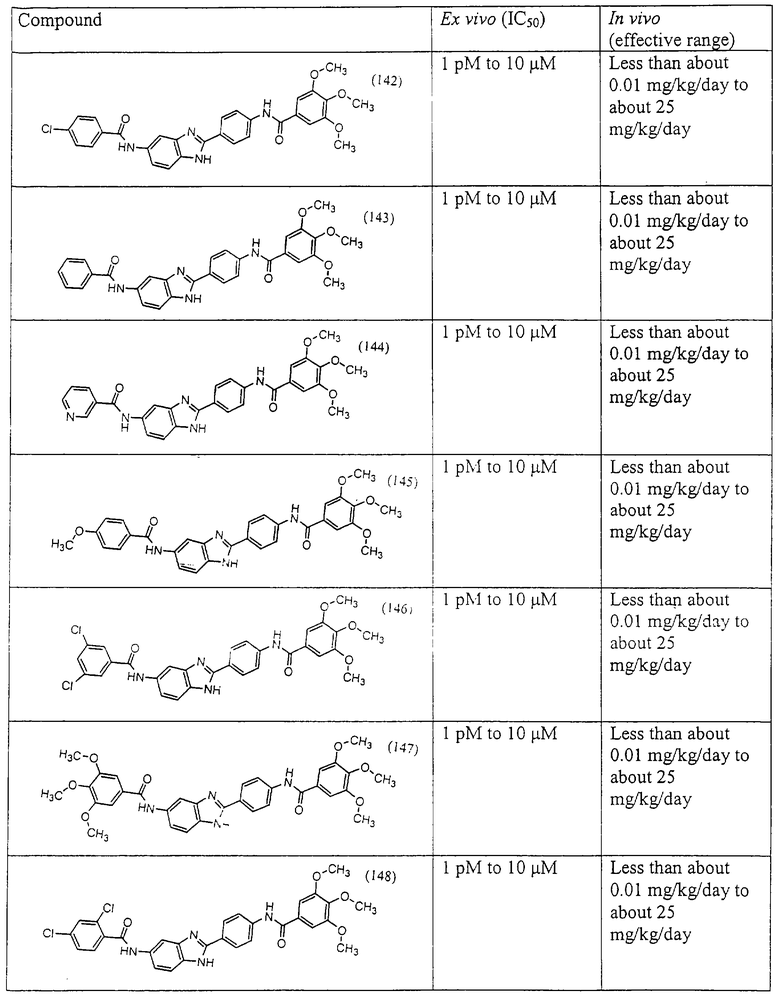
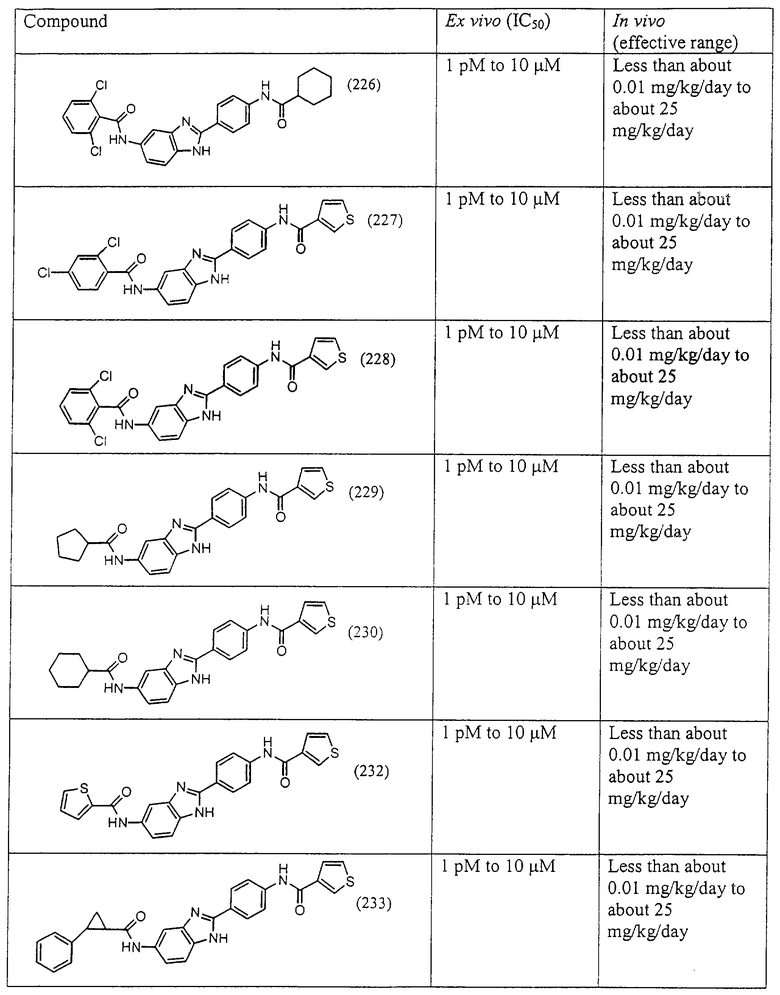
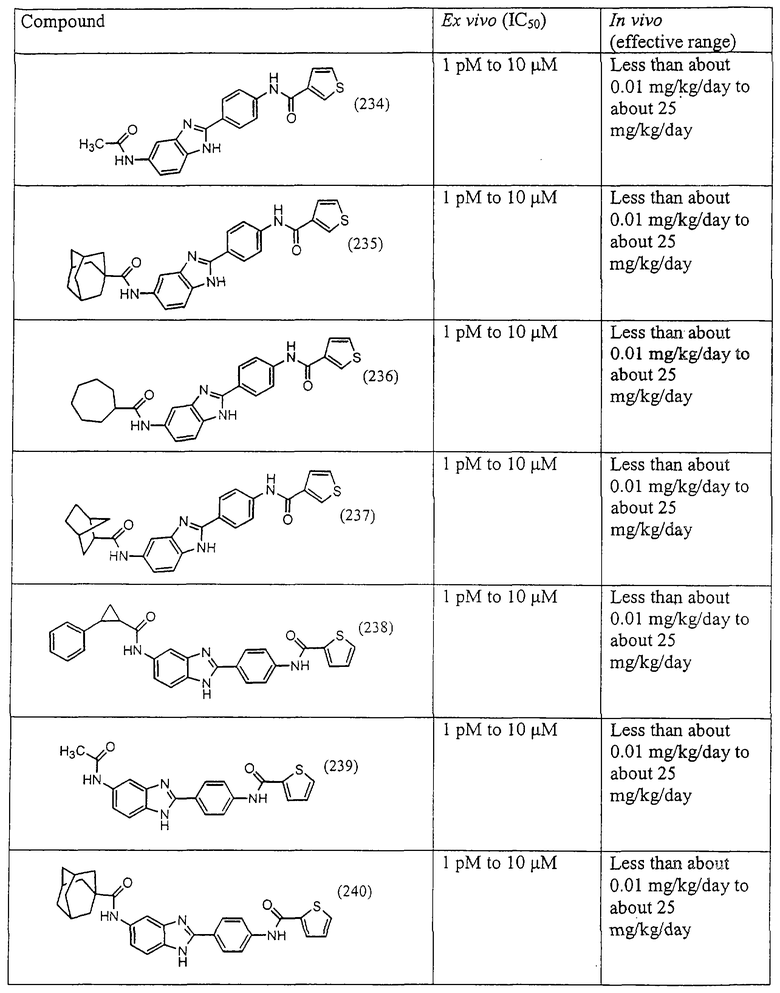
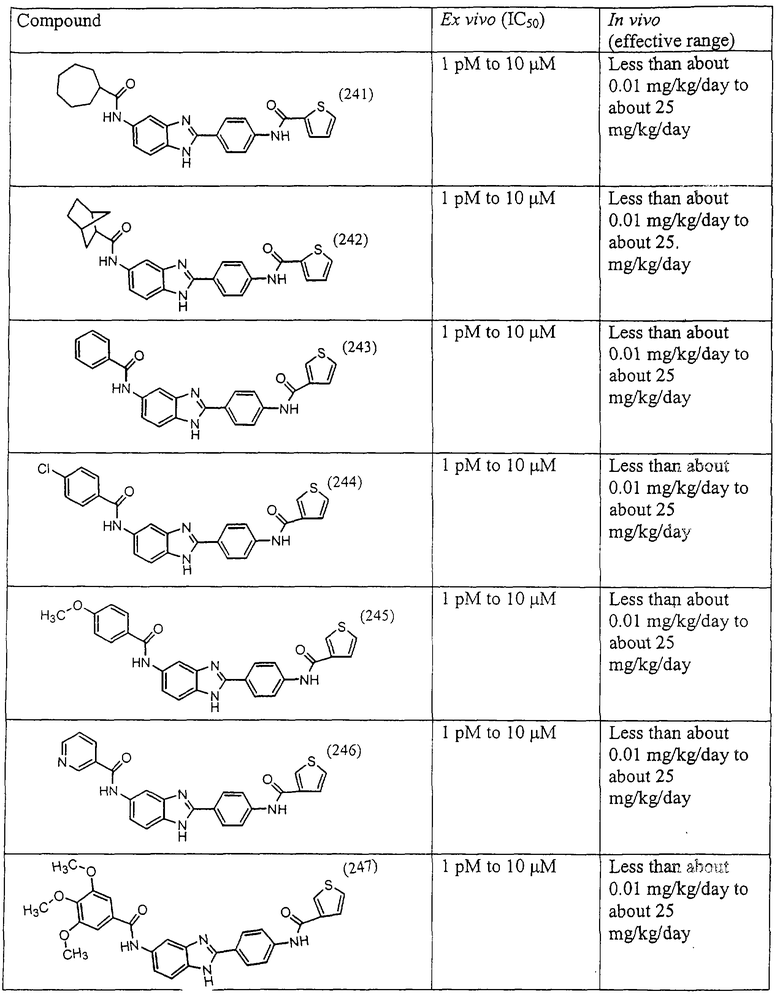
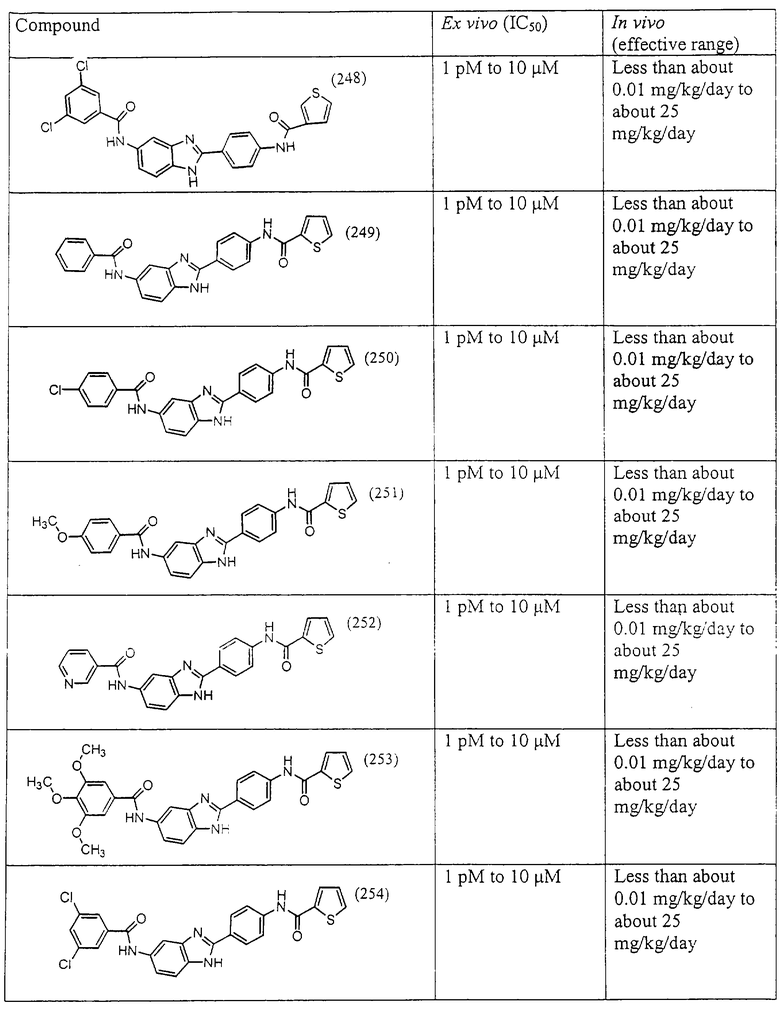
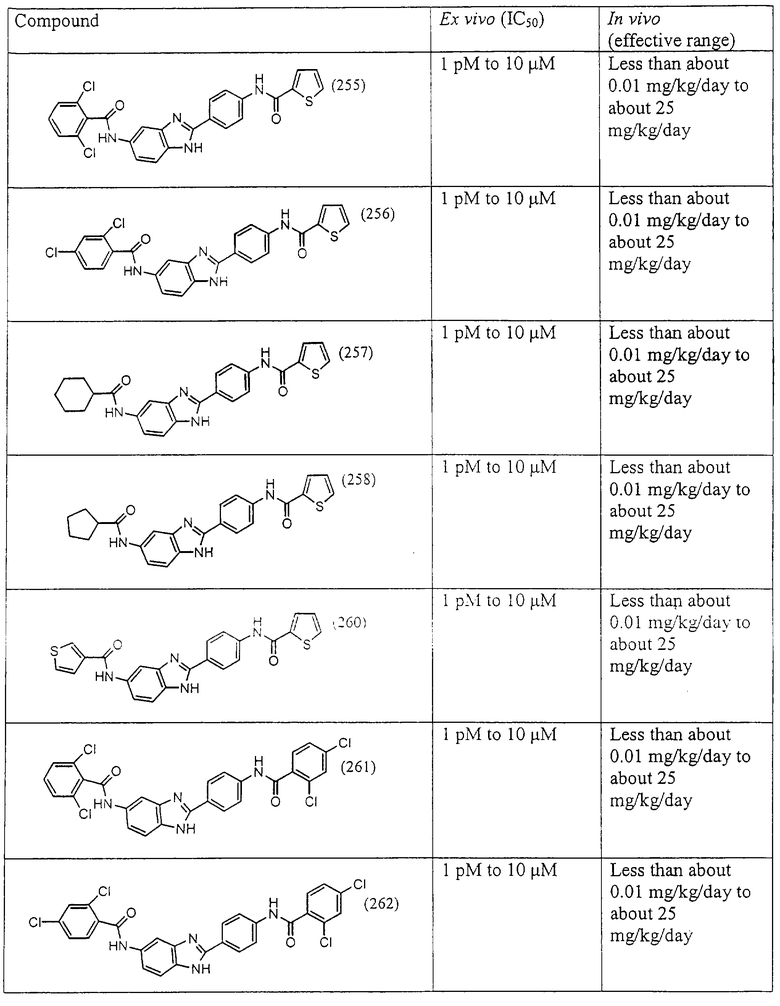
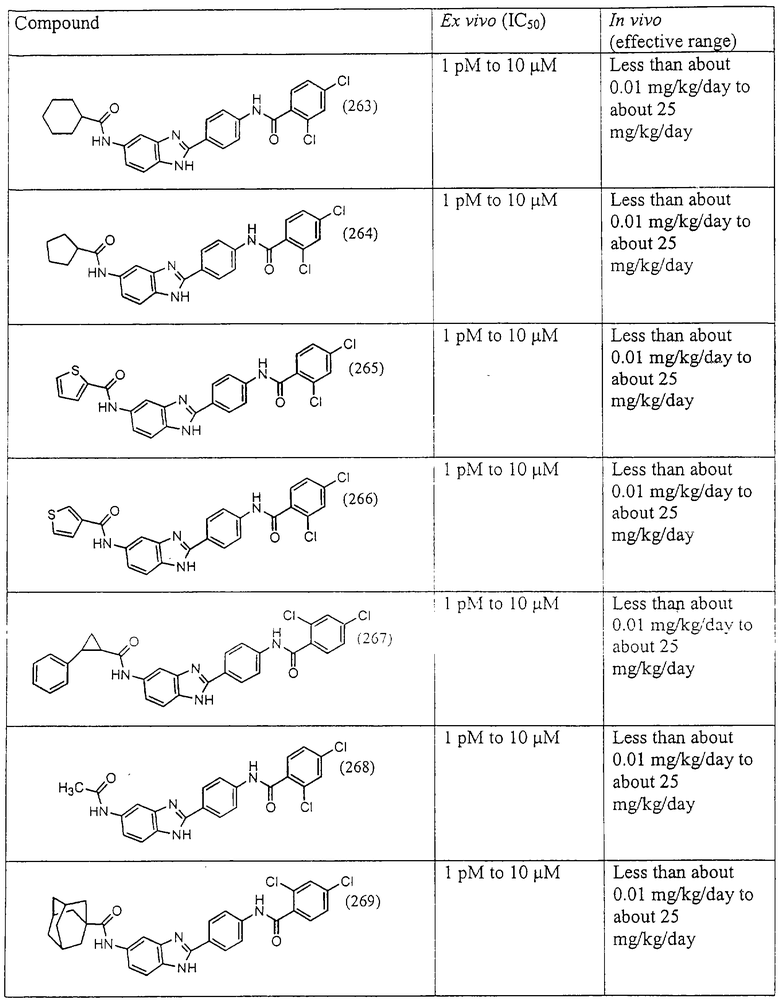
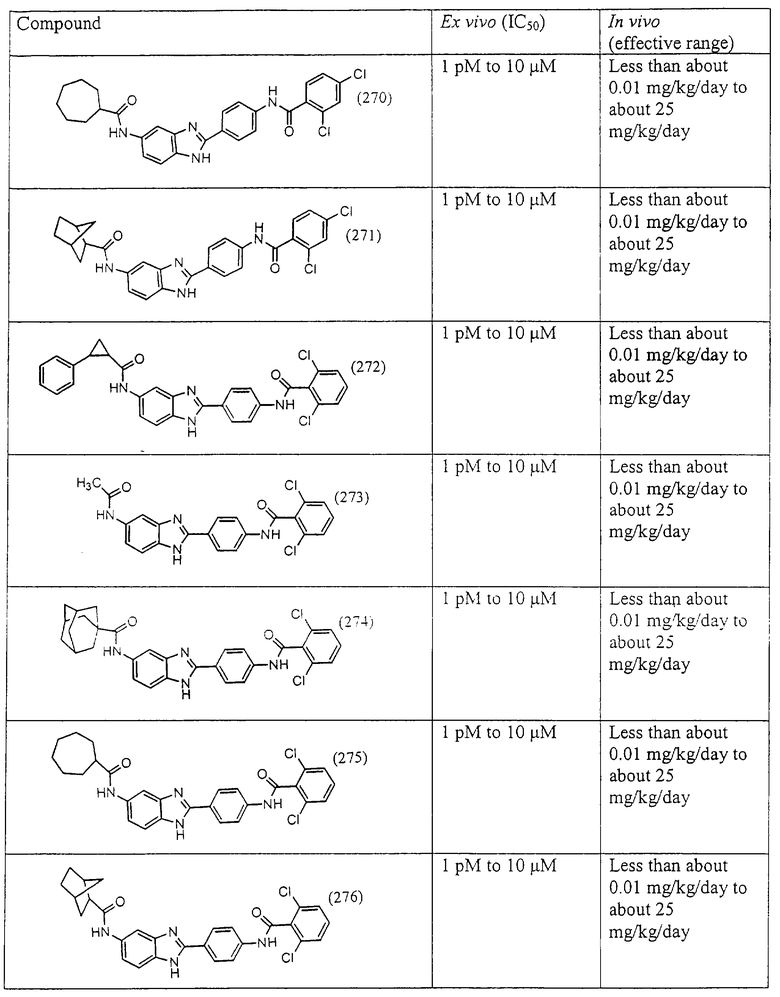
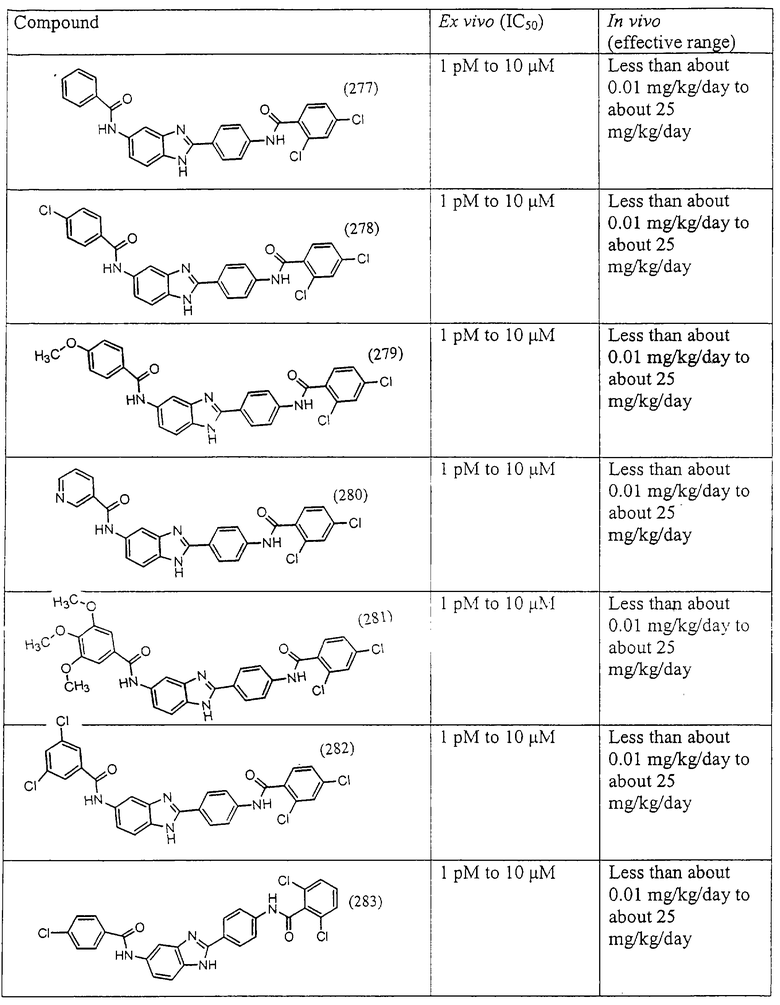
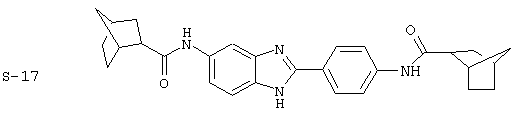
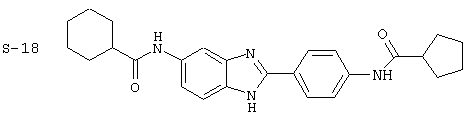
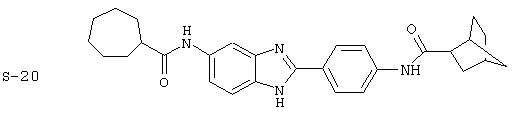
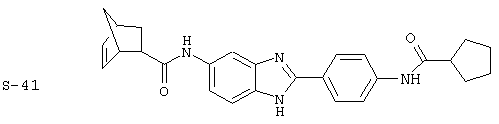
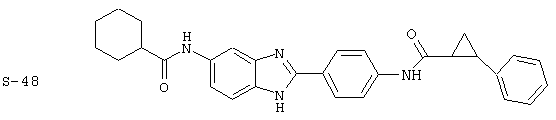
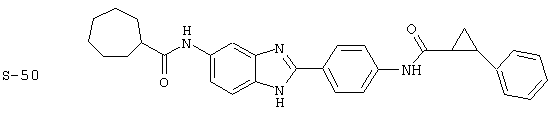
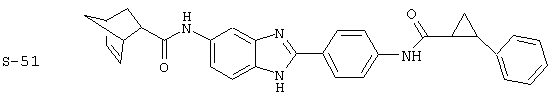
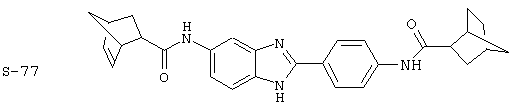
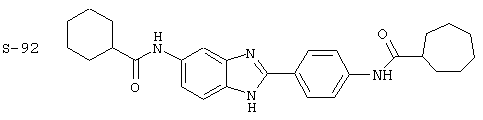
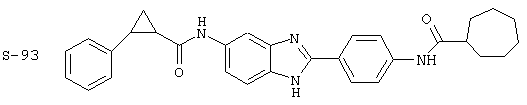
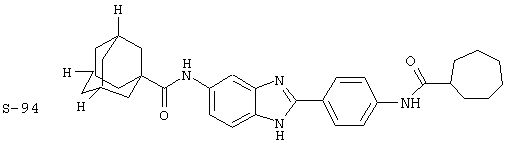
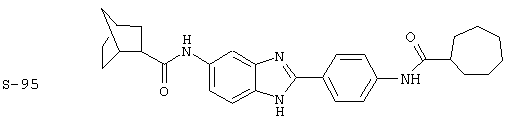
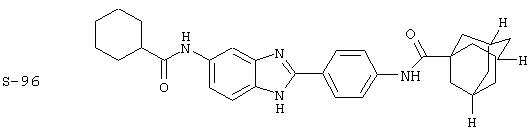
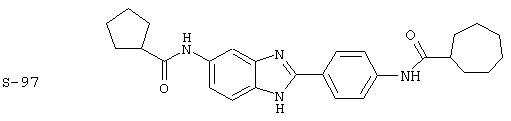
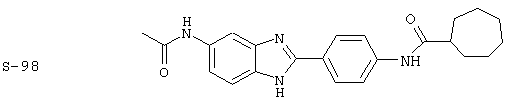
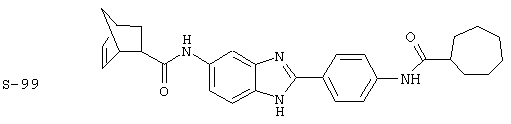
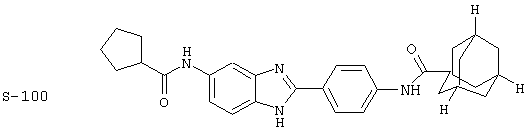
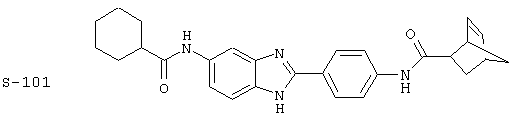
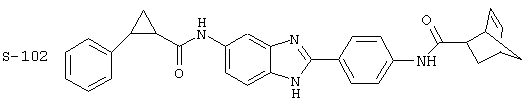
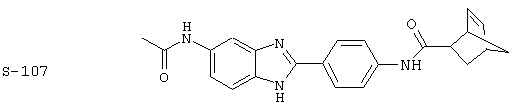
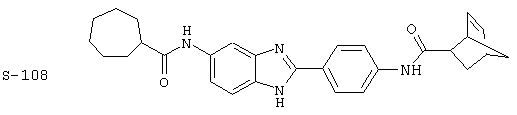
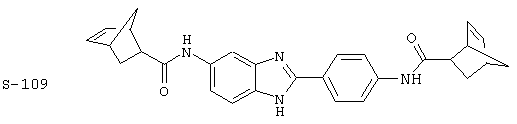
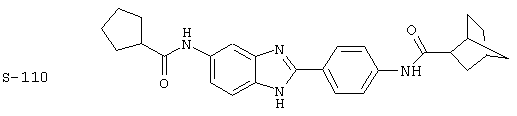
Виды диациловых бензимидазолов
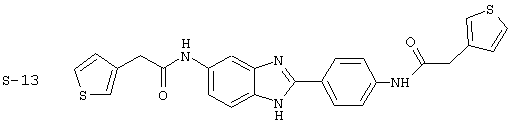
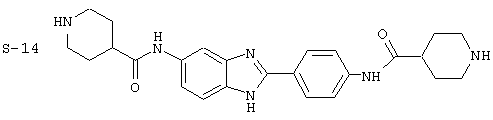
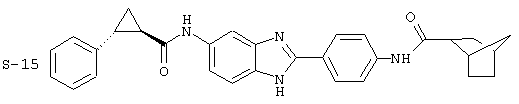
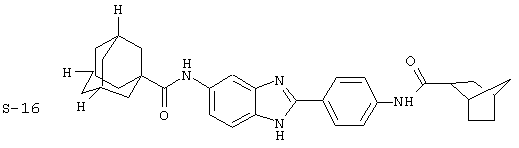
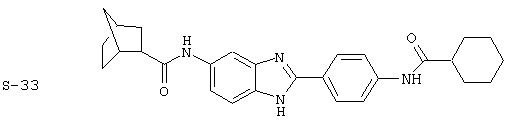
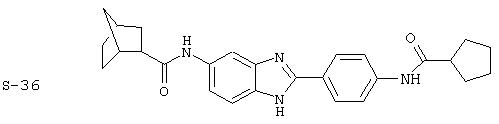
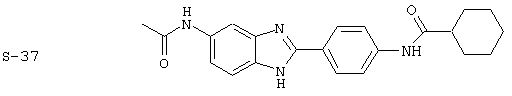
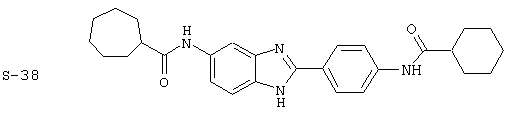
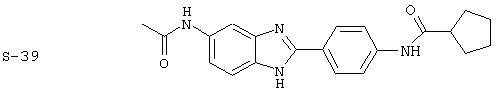
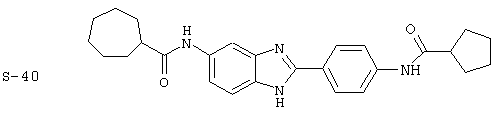
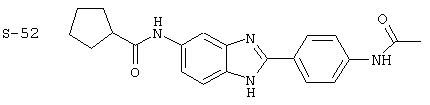
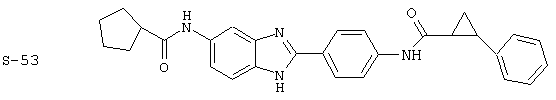
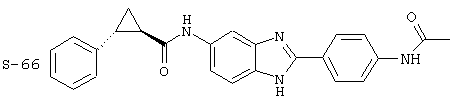
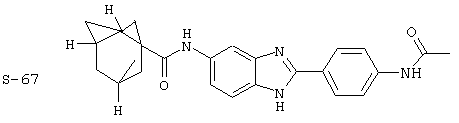
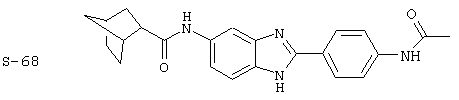
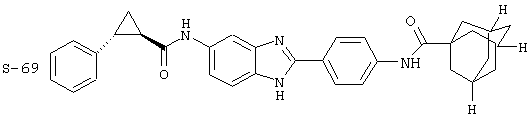
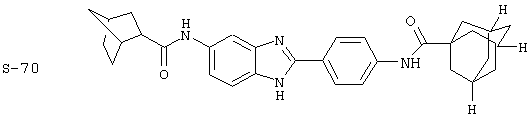
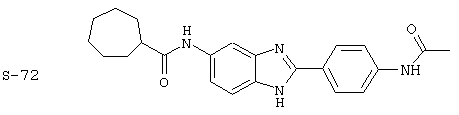
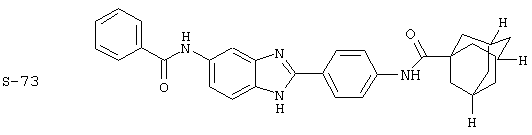
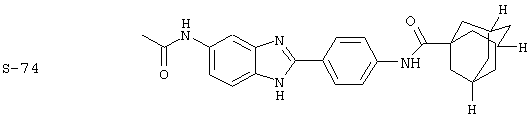
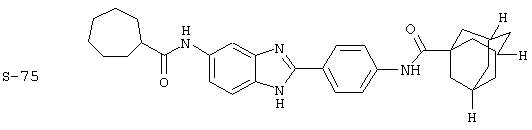
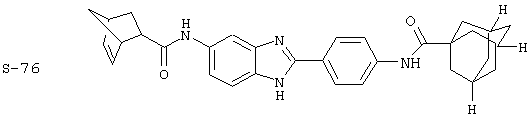
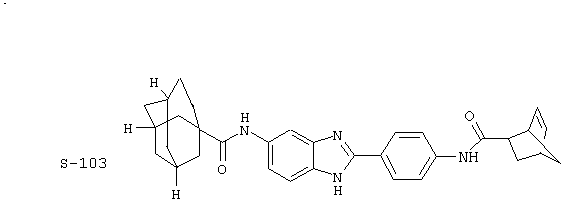
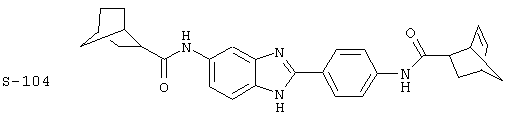
Были синтезированы и проверены на предмет своей способности подавлять IgE следующие виды, охватываемые раскрытой родовой формулой. Ниже эти виды пронумерованы.
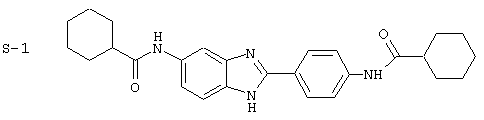
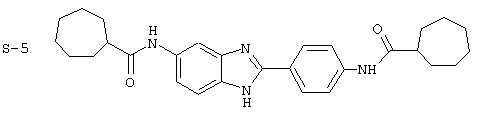
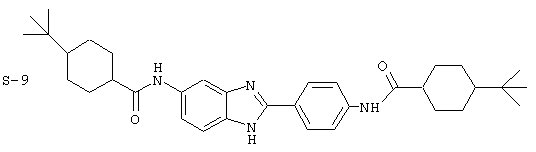
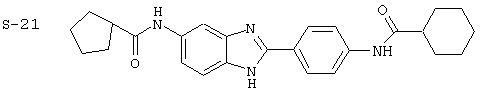
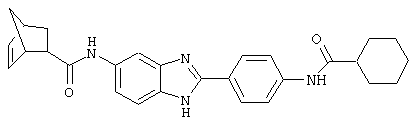
(1) 2-(N-циклогексилкарбанойл-4’-аминофенил)-6-(циклогексилкар-банойлаино)-бензиминдазол был приготовлен по Способу А из 2-(4-аминофенил)-6-аминобензимидазола (0,195 г, 0,87 ммоль) и хлорида циклогексилкарбонила (0,291 мл, 0,319 г, 2,175 ммоль). Полученное твердое вещество (76,7 мг) очищалось посредством препаратного HPLC
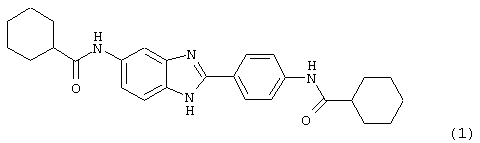
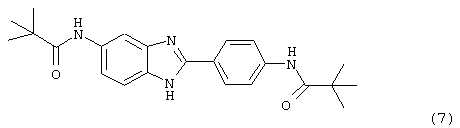
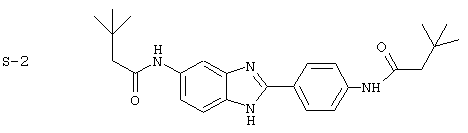
(2) Бис-t-бутилацетил бензиминдазол был приготовлен по Способу А из 2-(4-аминофенил)-6-аминобензимидазола (0,195 г, 0,87 ммоль) и хлорида t-бутилацетила (0,302 мл, 0,292 г, 2,175 ммоль). Полученное твердое вещество (42,3 мг) очищалось посредством препаратного HPLC
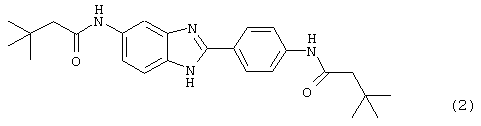
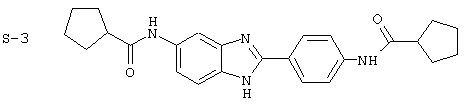
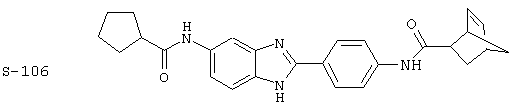
(3) Бис-циклопентилкарбонил бензиминдазол был приготовлен по Способу А из 2-(4-аминофенил)-6-аминобензимидазола (0,195 г, 0,87 ммоль) и хлорида циклопентилкарбонила (0,227 мл, 0,228 г, 2,175 ммоль). Полученное твердое вещество (42,3 мг) очищалось посредством препаратного HPLC
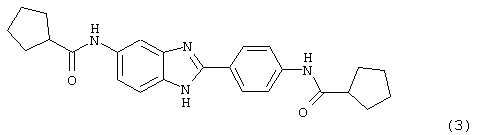
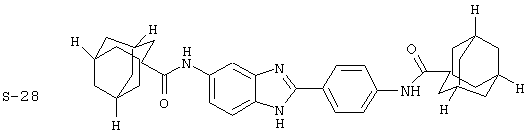
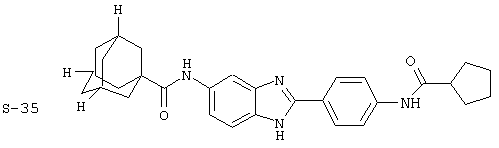
(4) Бис-адамантилкарбонил бензиминдазол был приготовлен по Способу В из 2-(4-аминофенил)-6-аминобензимидазола (0,500 г, 2,23 ммоль) и хлорида адамантилкарбонила (1,063 г, 5,35 ммоль). Полученное твердое вещество очищалось посредством препаратного HPLC, давая приблизительно 100 мг материала с чистотой 97%
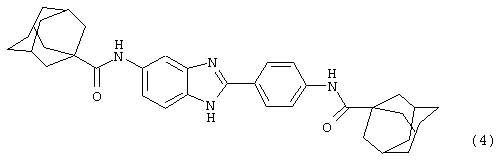
(5) Бис-циклопропилкарбонил бензиминдазол был приготовлен по Способу В из 2-(4-аминофенил)-6-аминобензимидазола (0,500 г, 2,23 ммоль) и хлорида циклопропилкарбонила (0,485 мл, 0,559 г, 5,35 ммоль). Полученное твердое вещество очищалось на силикагеле (5% МеОН в СH2Сl2). По данным HPLC продукт имел чистоту 94%
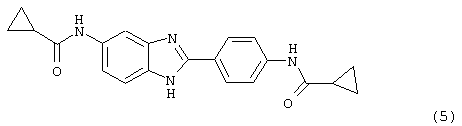
(6) Бис-циклобутилкарбонил бензиминдазол был приготовлен по Способу В из 2-(4-аминофенил)-6-аминобензимидазола (0,500 г, 2,23 ммоль) и хлорида циклобутилкарбонила (0,610 мл, 0,634 г, 5,35 ммоль). Полученное твердое вещество очищалось на силикагеле (5% МеОН в СН2Сl2). По данным HPLC продукт имел чистоту 97,4%
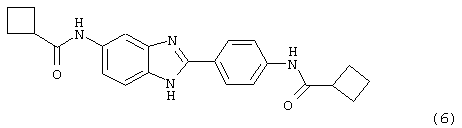
(7) Бис-триметилацетил бензиминдазол был приготовлен по Способу В из 2-(4-аминофенил)-6-аминобензимидазола (0,500 г, 2,23 ммоль) и хлорида триметилацетила (0,610 мл, 0,634 г, 5,35 ммоль). Полученное твердое вещество очищалось путем рекристаллизации (ацетон/гексан). По данным HPLC продукт имел чистоту 95%
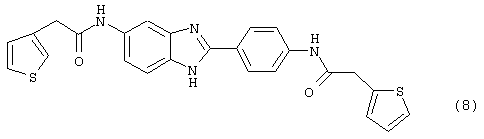
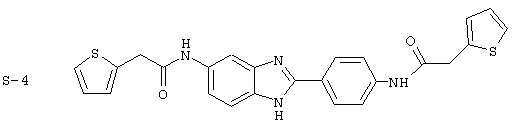
(8) Бис-2-тиофенацетил бензиминдазол был приготовлен по Способу В из 2-(4-аминофенил)-6-аминобензимидазола (0,500 г, 2,23 ммоль) и хлорида тиофенацетила (0,660 мл, 0,860 г, 5,35 ммоль). Полученное твердое вещество очищалось на силикагеле (5% МеОН в СН2С12). По данным HPLC продукт имел чистоту 92%
(9) Бис-2-циклогептанкарбонил бензиминдазол был приготовлен по Способу В из 2-(4-аминофенил)-6-аминобензимидазола (0,500 г, 2,23 ммоль) и хлорида циклогептанкарбонила (0,610 мл, 0,634 г, 5,35 ммоль). Полученное твердое вещество очищалось посредством препаратного HPLC, давая твердое вещество с чистотой 98,8%. Хлорид циклогептанкарбонила был синтезирован следующим образом: циклогептан карбоксиловая кислота (1,37 мл, 1,42 г, 10 ммоль) добавлялась в сухую 25-миллилитровую колбу с круглым дном и продувалась с помощью Na. В колбу добавлялся оксалил хлорид (7,5 мл, 2 М в СН2Сl2) через шприц, затем одна капля DMF. Реакция перемешивалась в течение ночи при комнатной температуре и концентрировалась под вакуумом. Добавлялся метиленхлорид (5 мл) и производилась концентрация под вакуумом для удаления остаточного оксалил хлорида (повторялось 5 раз)
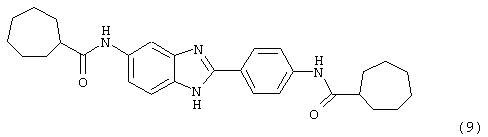
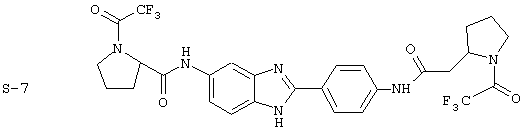
(10) Бис-(N-трифторацетилпролин) бензиминдазол был приготовлен по Способу А, за исключением того, что в качестве растворителя использовался СН2Сl2, из 2-(4-аминофенил)-6-аминобензимидазола (0,448 г, 2,0 ммоль) и хлорида (s)-(-)-N-трифторацетилпролина (42,0 мл, 0,1 М в СН2Сl2). Полученное твердое вещество очищалось на силикагеле (5% МеОН в СН2Сl2). По данным HPLC продукт имел чистоту 98,5%
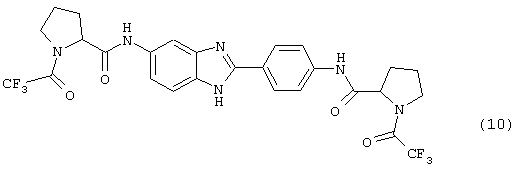
(11) Бис-пролин бензимидазол был синтезирован путем растворения производного бис-трифторацетила в МеОН (5 мл), к которому добавлялся раствор LiOH (0,210 г, в 5 мл воды). Вышеупомянутая смесь нагревалась до 42° С в течение 2 часов. Реакционная смесь экстрагировалась с помощью СН2Сl2 (5× 15 мл). Комбинированные органические экстракты концентрировались под вакуумом, давая твердое вещество, которое по данным HPLC имело чистоту 95,6%
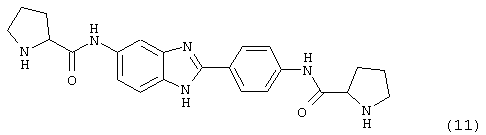
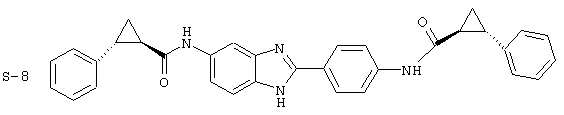
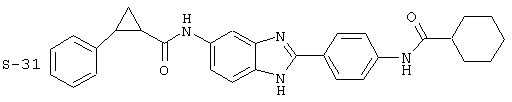
(12) Бис-транс-2-фенил-циклопропанкарбонил бензиминдазол был приготовлен по Способу В из 2-(4-аминофенил)-6-аминобензимидазола (0,500 г, 2,23 ммоль) и хлорида транс-2-фенил-1-циклопропанкарбонила (0,831 мл, 0,966 г, 5,35 ммоль). Полученное твердое вещество очищалось на силикагеле (5% МеОН в СН2Сl2). По данным HPLC продукт имел чистоту 95,5%
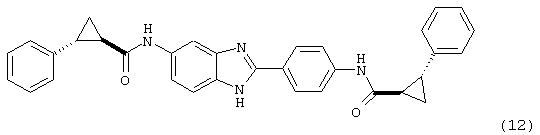
(13) Бис-4-t-бутилциклогексил карбонил бензиминдазол был приготовлен по Способу В из 2-(4-аминофенил)-6-аминобензимидазола (0,425 г, 1,89 ммоль) и хлорида 4-t-бутилциклогексилкарбонил (0,814 г, 4,25 ммоль). Полученное твердое вещество очищалось на силикагеле (5% МеОН в СН2Сl2). По данным HPLC продукт имел чистоту 90%
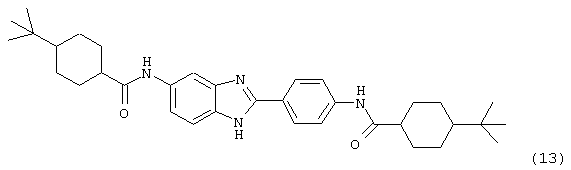
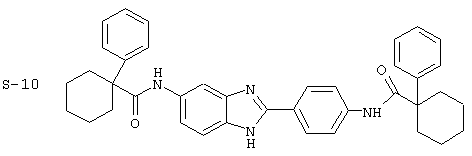
(14) Бис-1-фенилциклогексил карбонил бензиминдазол был приготовлен по Способу В из 2-(4-аминофенил)-6-аминобензимидазола (0,467 г, 2,08 ммоль) и хлорида 1-фенилциклогексилкарбонила (1,046 г). Полученное твердое вещество очищалось на силикагеле (5% МеОН в СН2Сl2). По данным HPLC продукт имел чистоту 93,3%
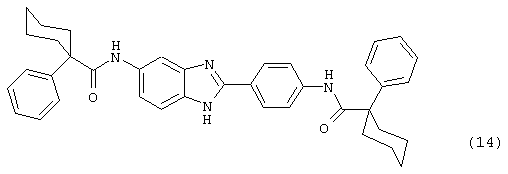
(15) Бис-транс-4-пентилциклогексил карбонил бензиминдазол был синтезирован следующим образом: оксалил хлорид (1,07 мл, 2 М в СН2Сl2) добавлялся к транс-4-пентилциклогексил карбоксиловой кислоте (0,424 г, 2,14 ммоль), после чего добавлялась капля DMF. Смесь оставляли реагировать при комнатной температуре на 1 час. К вышеупомянутому раствору добавляли 2-(4-аминофенил)-6-аминобензимидазол (0,200 г, 0,89 ммоль) в пиридине (2 мл). Реакция нагревалась до 60° С в течение ночи. Реакция охлаждалась, а осадок фильтровался и промывался NаНСО3 и гексанами. Полученное твердое вещество очищалось посредством препаратного HPLC, давая твердое вещество чистотой >99%
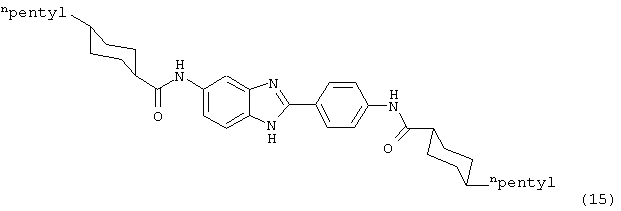
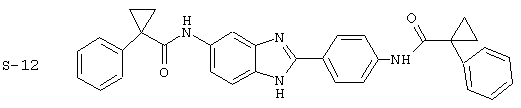
(16) Бис-1-фенилциклопропан карбонил бензиминдазол был приготовлен по Способу В из 2-(4-аминофенил)-6-аминобензимидазола (0,530 г, 2,36 ммоль) и хлорида 1-фенилциклопропанкарбонила (0,9625 г, 5,3 ммоль). Полученное твердое вещество очищалось на силикагеле (5% МеОН в СН2Сl2). По данным HPLC продукт имел чистоту 93,4%
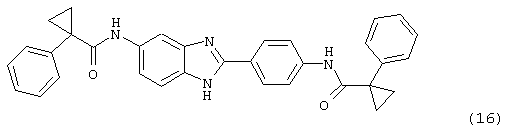
(17) Бис-(2,2,3,3-тетраметилциклопропан)карбонил бензиминдазол был синтезирован следующим образом: оксалил хлорид (1,07 мл, 2 М в СН2Сl2) добавлялся к 2,2,3,3-тетраметилциклопропан карбоксиловой кислоте (0,305 г, 2,14 ммоль), после чего добавлялась капля DMF. Смесь оставляли реагировать при комнатной температуре на 1 час. К вышеупомянутому раствору добавляли 2-(4-аминофенил)-6-аминобензимидазол (0,200 г, 0,89 ммоль) в пиридине (2 мл). Реакция нагревалась до 60° С в течение ночи. Реакция охлаждалась, а осадок фильтровался и промывался NaHCO3 и гексанами. Полученное твердое вещество очищалось посредством препаратного HPLC, давая твердое вещество чистотой >99%
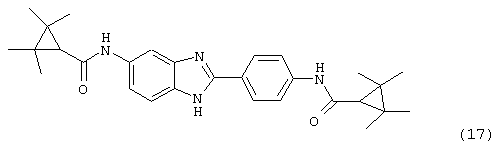
(18) Бис-4-метилциклогексил карбонил бензиминдазол был приготовлен по Способу Г из 2-(4-аминофенил)-6-аминобензимидазола (0,100 г, 0,44 ммоль) и хлорида 4-метилциклогексилкарбоксиловой кислоты (0,138 г, 0,96 ммоль). Полученное твердое вещество очищалось на силикагеле (5% МеОН в СН2Сl2). По данным HPLC продукт имел чистоту 94,5%
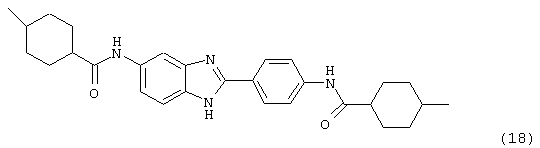
(19) Бис-1-фенилциклогексил карбонил бензиминдазол был синтезирован следующим образом: оксалил хлорид (1,07 мл, 2 М в CH2Cl2) добавлялся к 1-метилциклогексанкарбоксиловой кислоте (0,305 г, 2,14 ммоль), после чего добавлялась капля DMF. Смесь оставляли реагировать при комнатной температуре на 1 час. К вышеупомянутому раствору добавляли 2-(4-аминофенил)-6-аминобензимидазол (0,200 г, 0,89 ммоль) в пиридине (2 мл). Реакция нагревалась до 60° С в течение ночи. Реакция охлаждалась, а осадок фильтровался и промывался NаНСО3 и гексанами. Полученное твердое вещество очищалось посредством препаратного HPLC, давая твердое вещество чистотой >99%
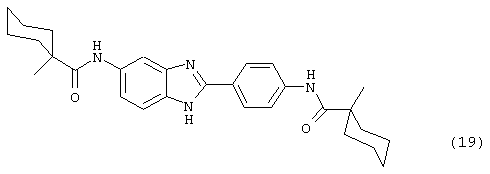
(20) Бис-бицикло[2,2,1]гептан-2-карбонил бензиминдазол был синтезирован следующим образом: оксалил хлорид (1,07 мл, 2 М в CH2Cl2) добавлялся к бицикло[2,2,1]гептанкарбоксиловой кислоте (0,305 г, 2,14 ммоль), после чего добавлялась капля DMF. Смесь оставляли реагировать при комнатной температуре на 1 час. К вышеупомянутому раствору добавляли 2-(4-аминофенил)-6-аминобензимидазол (0,200 г, 0,89 ммоль) в пиридине (2 мл). Реакция нагревалась до 60° С в течение ночи. Реакция охлаждалась, а осадок фильтровался и промывался NаНСО3 и гексанами. Полученное твердое вещество очищалось посредством препаратного HPLC, давая твердое вещество чистотой 68%
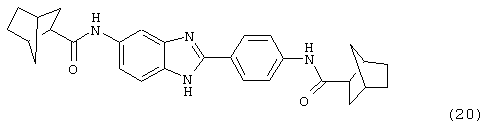
(21) Бис-4-метоксициклогексил карбонил бензиминдазол был синтезирован следующим образом: оксалил хлорид (1,07 мл, 2 М в СН2Сl2) добавлялся к 4-метокси-1-циклогексан карбоксиловой кислоте (0,338 г, 2,14 ммоль), после чего добавлялась капля DMF. Смесь оставляли реагировать при комнатной температуре на 1 час. К вышеупомянутому раствору добавляли 2-(4-аминофенил)-6-аминобензимидазол (0,200 г, 0,89 ммоль) в пиридине (2 мл). Реакция нагревалась до 60° С в течение ночи. Реакция охлаждалась, а осадок фильтровался и промывался NaHCO3 и гексанами
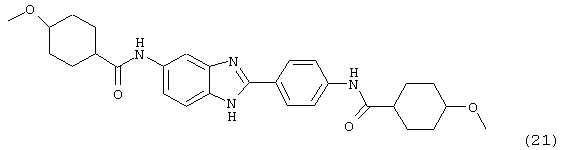
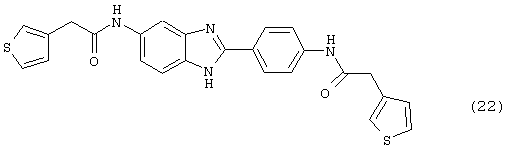
(22) Бис-3-тиофенацетил бензиминдазол был получен следующим образом: оксалил хлорид (1,07 мл, 2 М в СН2Сl2) добавлялся к 3-тиофенуксусной кислоте (0,338 г, 2,14 ммоль), после чего добавлялась капля DMF. Смесь оставляли реагировать при комнатной температуре на 1 час. К вышеупомянутому раствору добавляли 2-(4-аминофенил)-6-аминобензимидазол (0,200 г, 0,89 ммоль) в пиридине (2 мл). Реакция нагревалась до 60° С в течение ночи. Реакция охлаждалась, а осадок фильтровался и промывался NаНСО3 и гексанами
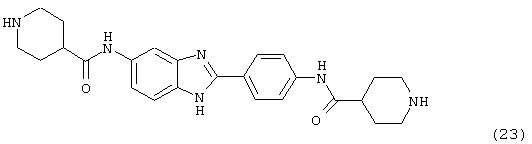
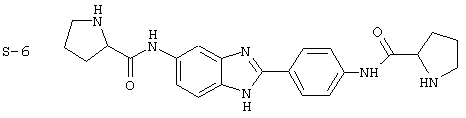
(23) Бис-4-нипекотамид бензиминдазол был получен следующим образом: бис-N-бок-4-нипекотамид бензиминдазол (0,400 г) был растворен в 1:1 ТРА:СН2Сl2 (4 мл) при -20° С в течение ночи. Растворитель был удален под вакуумом, добавлялась вода, замораживалась на сухом льду и лиофилизировалась до сухого состояния. Бок-защищенный бензиминдазол был синтезирован следующим образом: оксалил хлорид (2,82 мл, 2 М в СН2Сl2) добавлялся к N-бок-нипекотовой кислоте (1,293 г, 5,64 ммоль), после чего добавлялась капля DMF. Смесь оставляли реагировать при комнатной температуре на 1 час. К вышеупомянутому раствору добавляли 2-(4-аминофенил)-6-аминобензимидазол (0,500 г, 2,24 ммоль) в пиридине (5 мл). Реакция нагревалась до 60° С в течение ночи. Реакция охлаждалась, а осадок фильтровался, промывался NаНСО3 и гексанами. Полученное твердое вещество по данным HPLC имело чистоту >99%
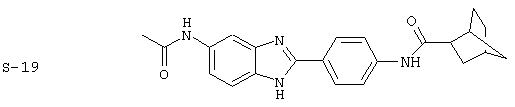
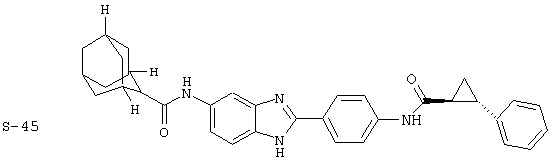
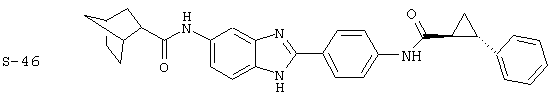
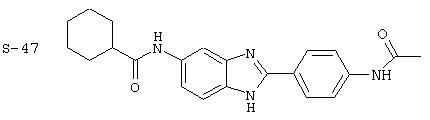
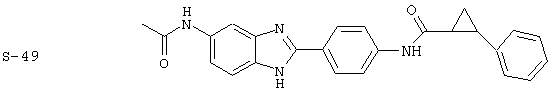
Моноациловые бензимидазоловые ингибиторы IgE
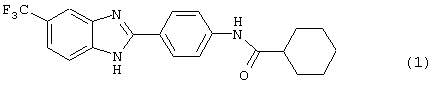
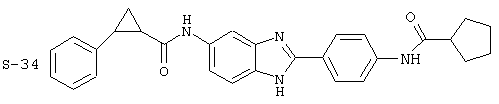
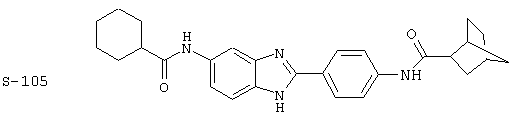
Вышеописанное семейство ингибиторов, связанных с диациловыми соединениями, является асимметричными моноацилированными бензимидазоловыми соединениями. Было синтезировано несколько моноациловых вариантов, которые описаны ниже:
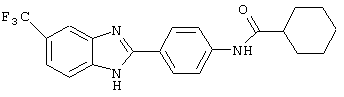
(1) 2-(N-циклогексанкарбонил-4-аминофенил)-5-трифторметил бензимидазол был синтезирован из следующих серий бензимидазоловых посредников: 1) 2-(4-нитрофенил)-5-трифторметил бензимидазол (обозначен 1.1) и 2) 2-(4-аминофенил)-5-трифторметил бензимидазол (обозначен 1.2);
(1.1) 2-(4-нитрофенил)-5-трифторметил бензимидазол был синтезирован следующим образом: 1,2-диамино-4-трифторметилбензол (1,76 г, 10,0 ммоль) смешивался с 4-нитробензойной кислотой (1,67 г, 9,8 ммоль), растворялся в РОСl3 (12 мл) и нагревался для оттока в течение 2,5 часов. Реакционная смесь охлаждалась и осторожно выливалась на лед. Полученное твердое вещество фильтровалось, промывалось NaHCO3 и использовалось без дальнейшей очистки
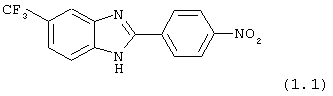
(1.2) 2-(4-аминофенил)-5-трифторметил бензимидазол был получен из 2-(4-нитрофенил)-5-трифторметил бензимидазола (см. выше (1.1)). Сырой фильтрат 2-(4-нитрофенил)-5-трифторметил бензимидазола растворялся в концентрированной НСl (15 мл), к которой добавлялся SnCl2·H2O (13,5 г, 59 ммоль) и нагревался для оттока в течение 16 часов. Реакция охлаждалась, и соль НСl осаждалась посредством добавления ЕtOН (75 мл).
Твердое вещество фильтровалось, промывалось этанолом и растворялось в воде. Соль нейтрализовалась путем добавления концентрированного гидроксида аммония, и свободное основание изолировалось посредством фильтрации. Продукт определялся путем масс-спектрографии
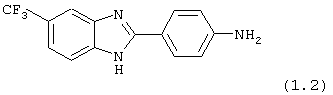
(1) 2-(N-циклогексанкарбонил-4-аминофенил)-5-трифторметил бензимидазол был приготовлен из амина, 2-(4-аминофенил)-5-трифторметил бензимидазола (см. выше (1.2)). Этот амин (0,239 г, 0,86 ммоль) растворялся в ТНF:Н2О (5 мл, 1:1), затем растворялись К2СО3 (0,1213 г, 0,88 ммоль) и хлорид циклогексилкарбонила (130 мкл, 0,95 ммоль). Реакционная смесь встряхивалась при комнатной температуре в течение 23 часов. К реакции добавлялся хлорид натрия, и смесь экстрагировалась с помощью EtOAc. Объединенные органические экстракты промывались водой, высушивались на Na2SO4 и концентрировались под вакуумом. Полученное твердое вещество очищалось с помощью препаратной тонкослойной хроматографии (10% МеОН в СН2Сl2)
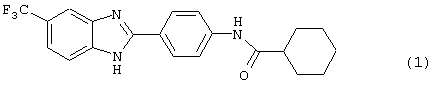
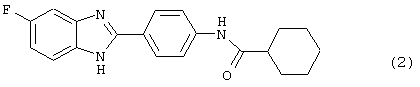
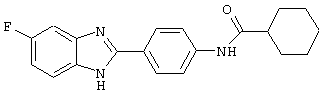
Следующий вид, (2), 2-(N-циклогексанкарбонил-4-аминофенил)-5-фтор бензимидазол был синтезирован из следующих серий бензимидазоловых посредников: 1) 2-(4-нитрофенил)-5-фтор бензимидазол (обозначен 2.1) и 2) 2-(4-аминофенил)-5-фтор бензимидазол (обозначен 2.2).
(2.1) 2-(4-нитрофенил)-5-фтор бензимидазол был синтезирован следующим образом: 1,2-диамино-4-фторбензол (1,26 г, 10,0 ммоль) смешивался с 4-нитробензойной кислотой (1,67 г, 9,8 ммоль), растворялся в РОСl3 (10 мл) и нагревался для оттока в течение 2,5 часов. Реакционная смесь охлаждалась и осторожно выливалась на лед. Полученное твердое вещество фильтровалось, промывалось NaHCO3 и использовалось без дальнейшей очистки
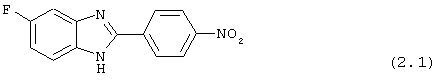
(2.2) 2-(4-аминофенил)-5-фтор бензимидазол был получен путем
растворения 1 г вышеупомянутого нитробензимидазола (2.1) в 30% Na2S· 9H2O (20 мл) с перемешиванием при комнатной температуре в течение 24 часов. Реакционная смесь разбавлялась водой и экстрагировалась с помощью EtOAc. Объединенные органические экстракты высушивались на сульфате натрия и концентрировались под вакуумом. Продукт определялся посредством масс-спектроскопии
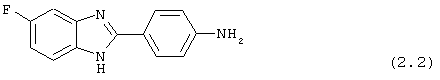
(2) 2-(N-циклогексанкарбонил-4-аминофенил)-5-фтор бензимидазол был получен путем растворения 0,100 г (0,44 ммоль) вышеупомянутого амина (2.2) в пиридине (1 мл), затем растворения хлорида циклогексанкарбонила (63,2 мкл) и нагревания до 60° С в течение ночи. Реакция разбавлялась водой (8 мл) и экстрагировалась с помощью EtOAc. Комбинированные органические фракции объединялись, высушивались (Na2SO4) и концентрировались под вакуумом. Полученное твердое вещество очищалось с помощью флэш-хроматографии (5% МеОН/СН2Сl2)
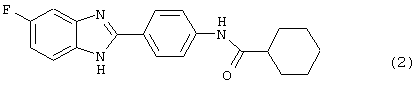
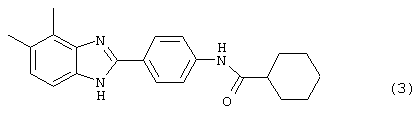
Следующий вид, (3), 2-(N-3',4'-дихлорбензойл-4-аминофенил)-3,4-диметил бензимидазол был синтезирован из следующих серий бензимидазоловых посредников: 1) 2-(4-нитрофенил)-4,5-диметил бензимидазол (обозначен 3.1) и 2) 2-(4-аминофенил)-4,5-диметил бензимидазол (обозначен 3.2).
(3.1) 2-(4-нитрофенил)-4,5-диметил бензимидазол был синтезирован путем смешивания 1,2-диамино-3,4-диметилбензола (1,36 г, 9,8 ммоль) с 4-нитробензойной кислотой (1,67 г, 9,8 ммоль), растворения в РОСl3 (10 мл) и нагревания для оттока в течение 2,5 часов. Реакционная смесь охлаждалась и осторожно выливалась на лед. Полученное твердое вещество фильтровалось, промывалось NaHCO3 и использовалось без дальнейшей очистки
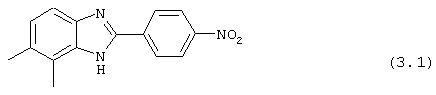
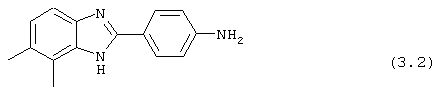
(3.2) 2-(4-аминофенил)-4,5-диметил бензимидазол был получен путем растворения 1 г вышеупомянутого нитробензимидазола (3.1) в 30% Na2S· 9H2O (20 мл) с перемешиванием при комнатной температуре в течение 2,5 часов. Реакционная смесь разбавлялась водой и экстрагировалась с помощью EtOAc. Объединенные органические экстракты высушивались на сульфате натрия и концентрировались под вакуумом. Продукт определялся посредством масс-спектроскопии
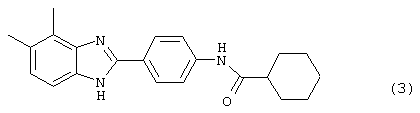
(3) 2-(N-циклогексанкарбонил-4-аминофенил)-3,4-диметил бензимидазол был получен путем растворения 0,0954 г (0,402 ммоль) вышеупомянутого амина (3.2) в 1 мл пиридина, затем растворения хлорида циклогексанкарбонила (57,6 мкл) и нагревания до 60° С в течение ночи. Реакция разбавлялась водой (8 мл) и экстрагировалась с помощью EtOAc. Объединенные органические фракции объединялись, высушивались (Na2SO4) и концентрировались под вакуумом. Полученное твердое вещество очищалось с помощью флэш-хроматографии (5% MeOH/СН2Сl2)
Активность понижающего регулирования IgE
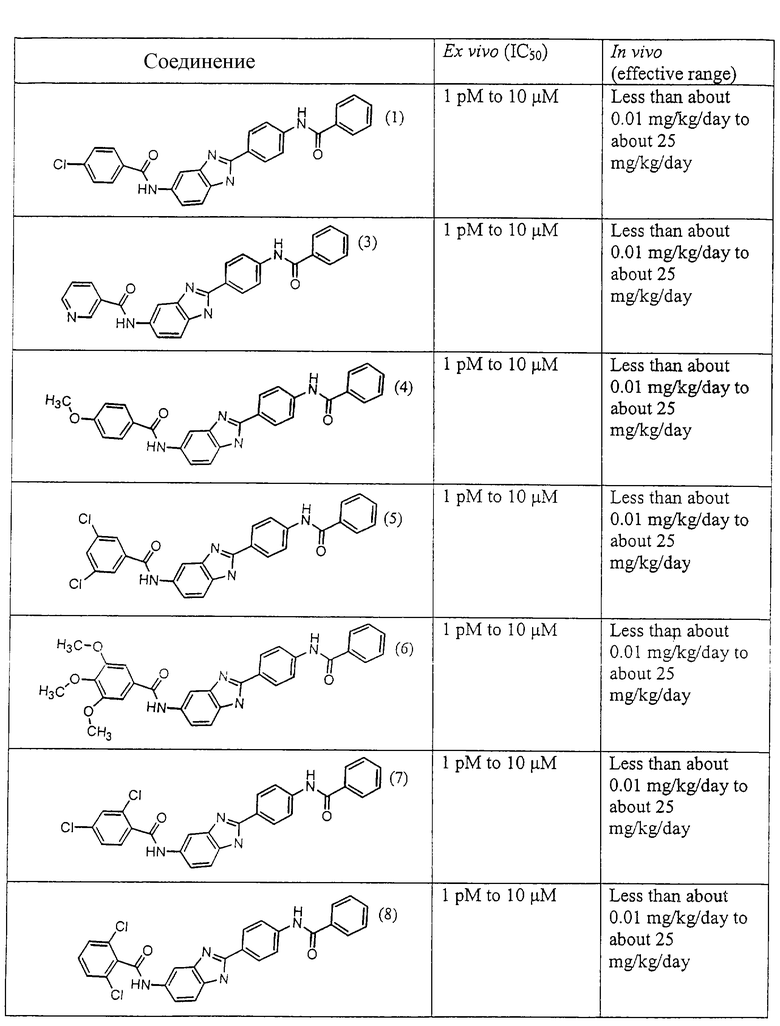
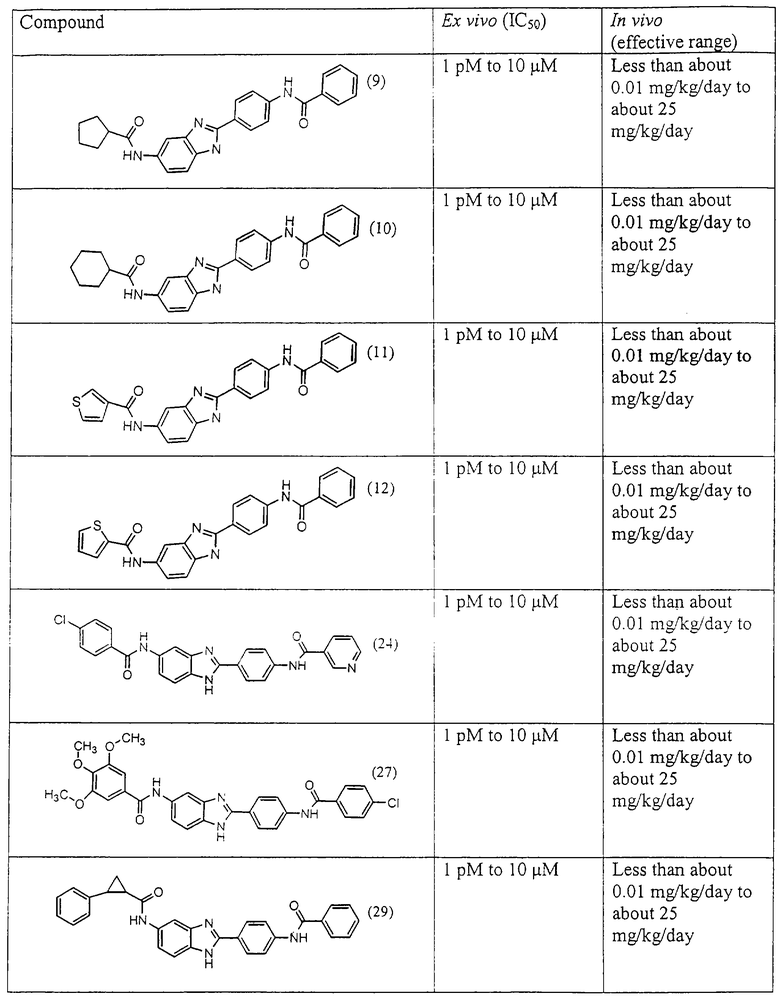
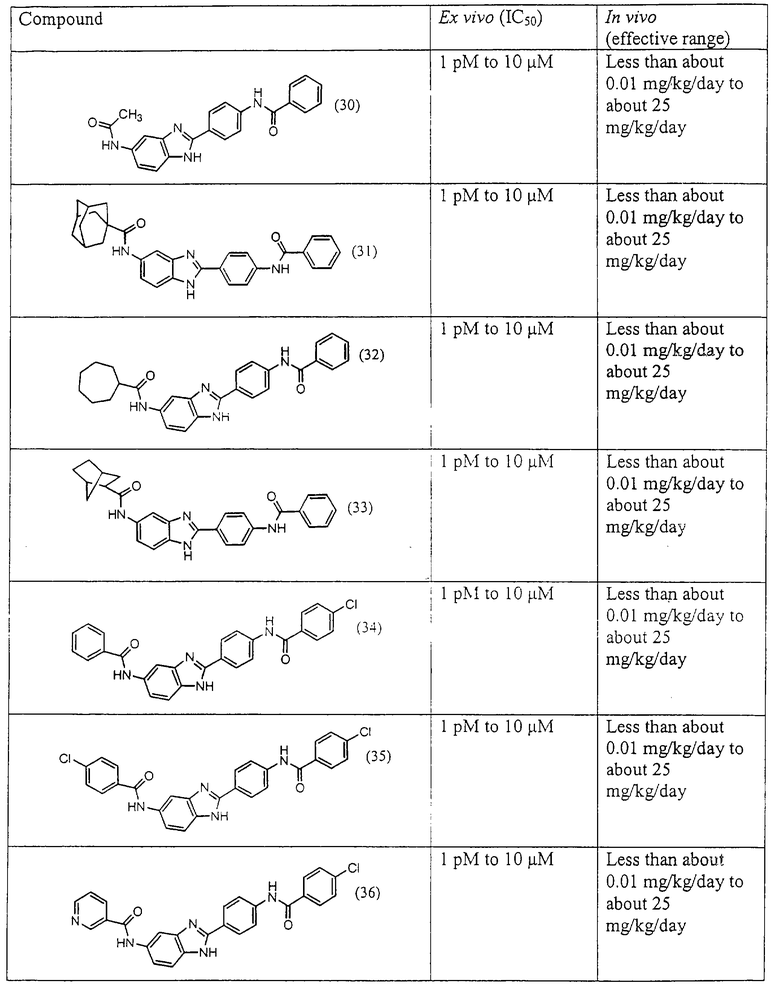
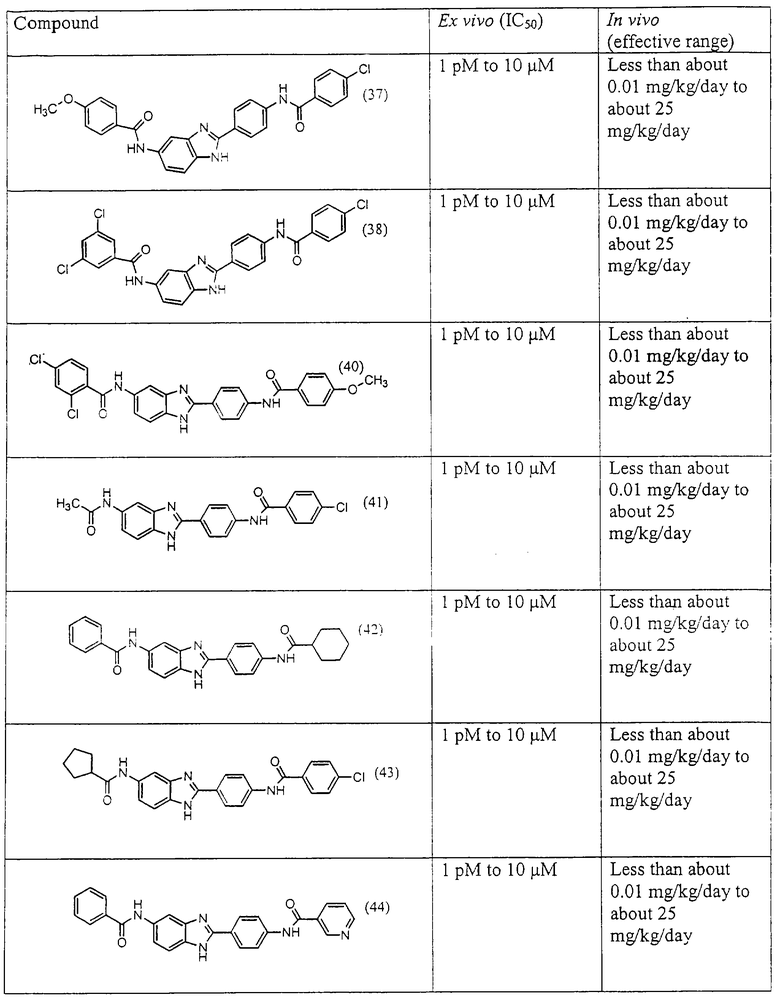
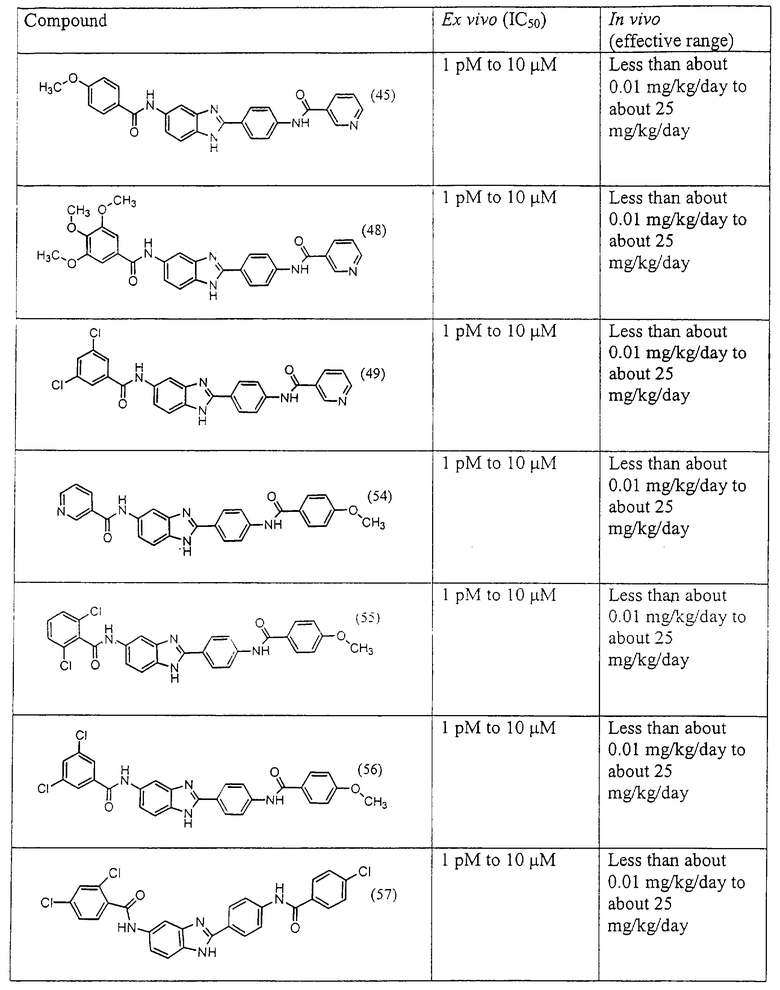
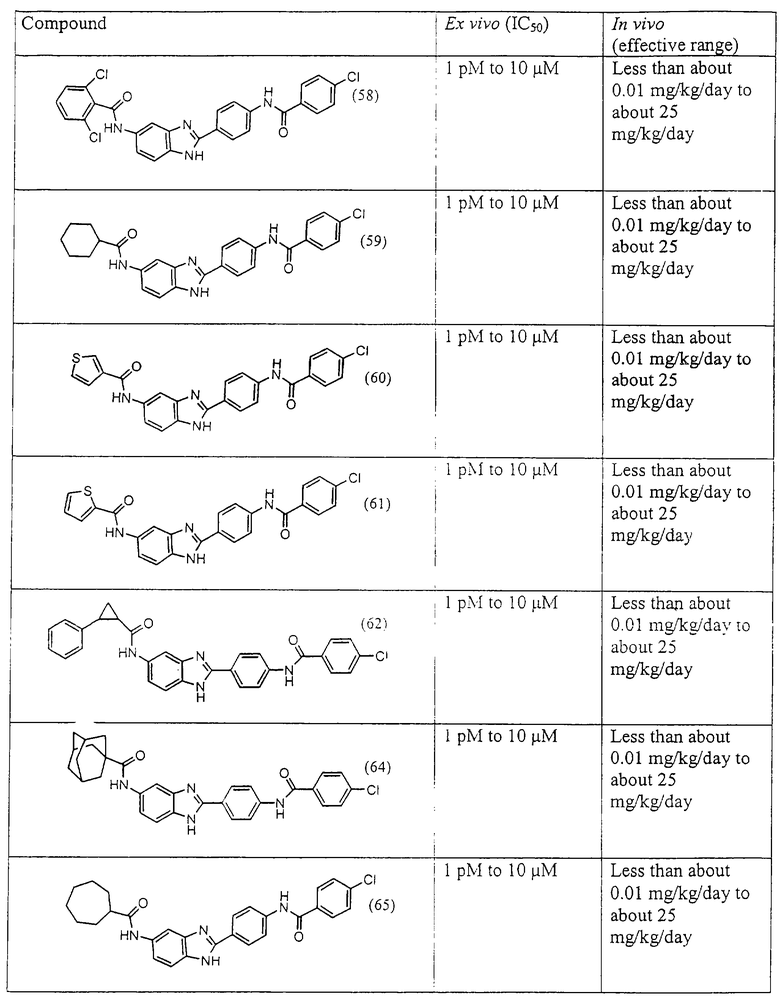
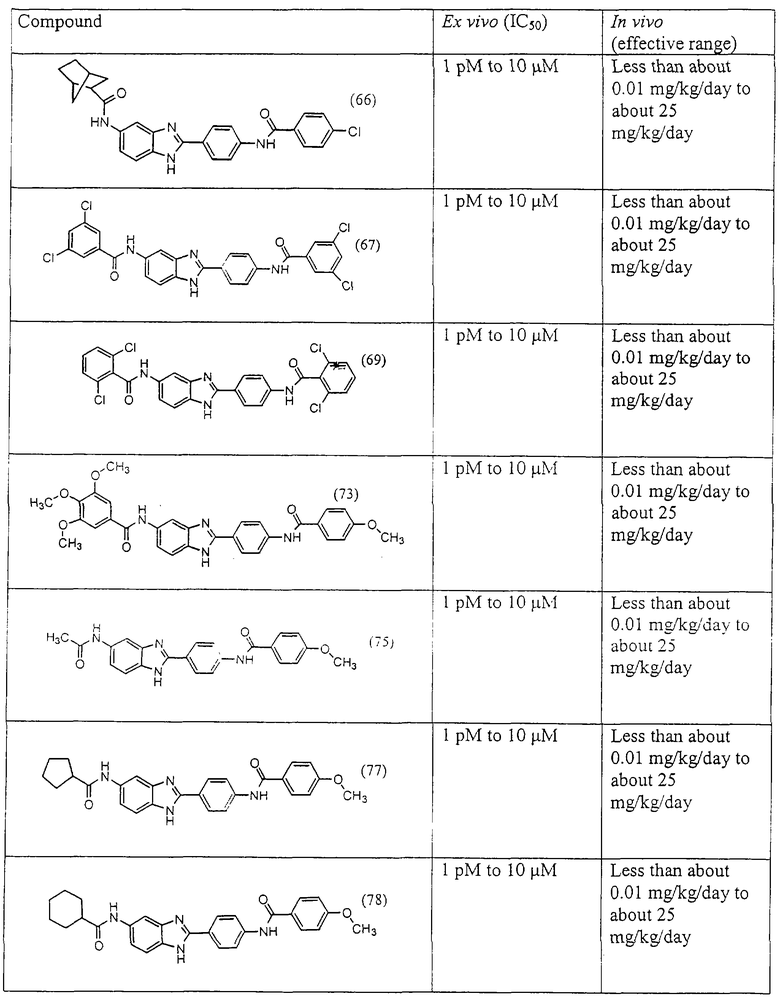
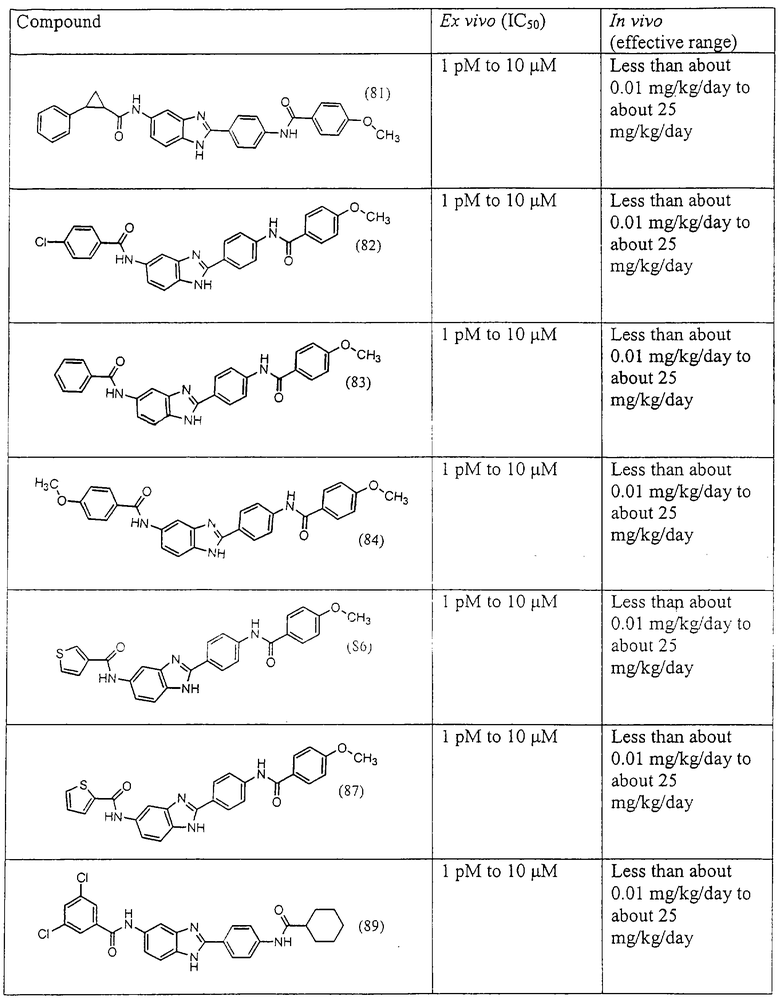
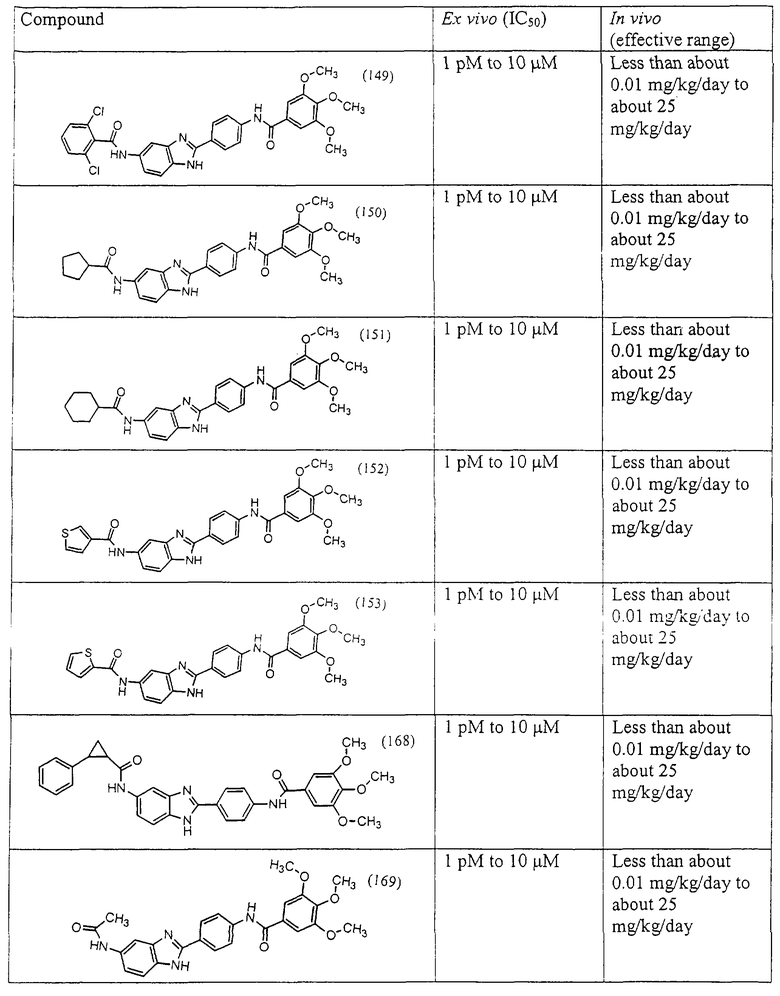
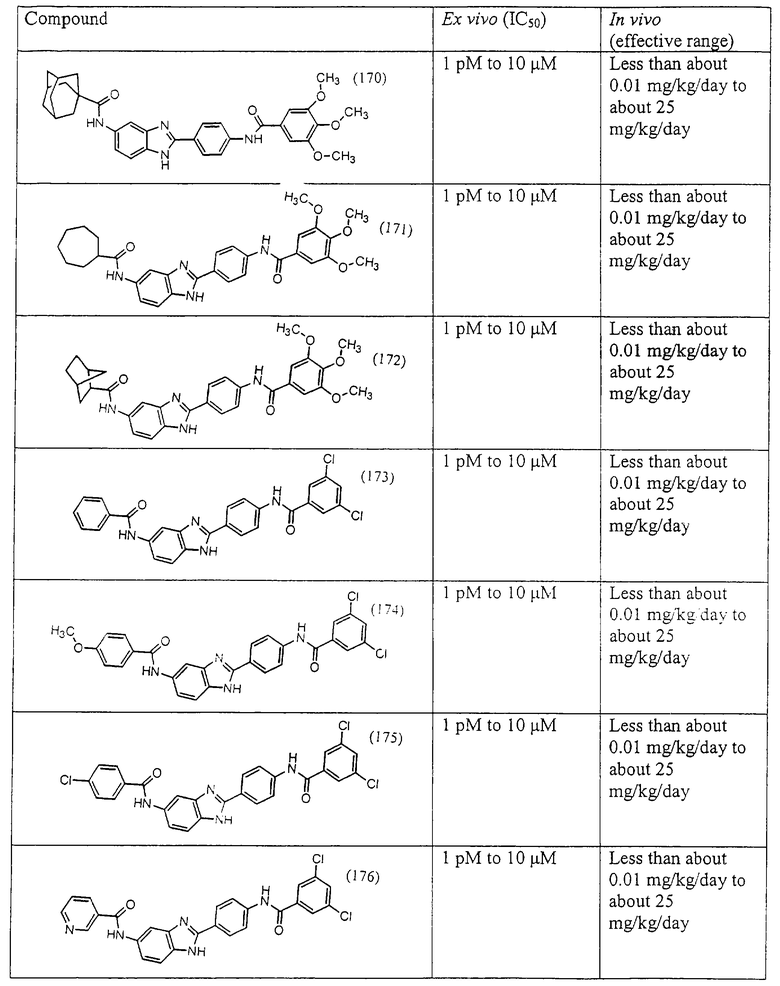
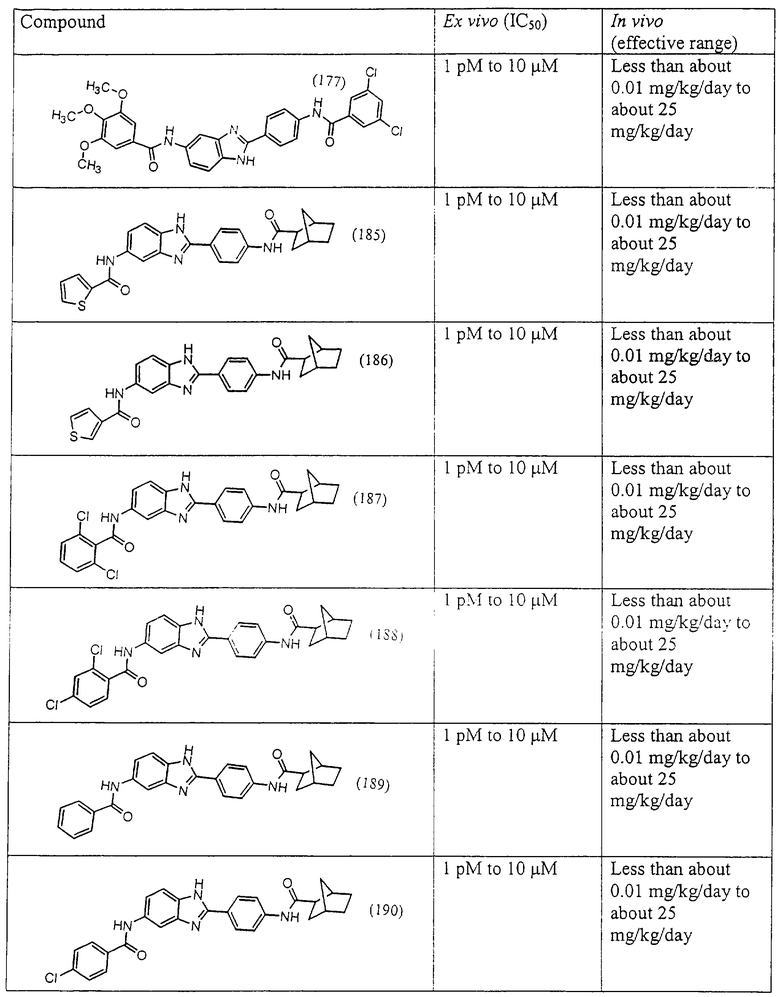
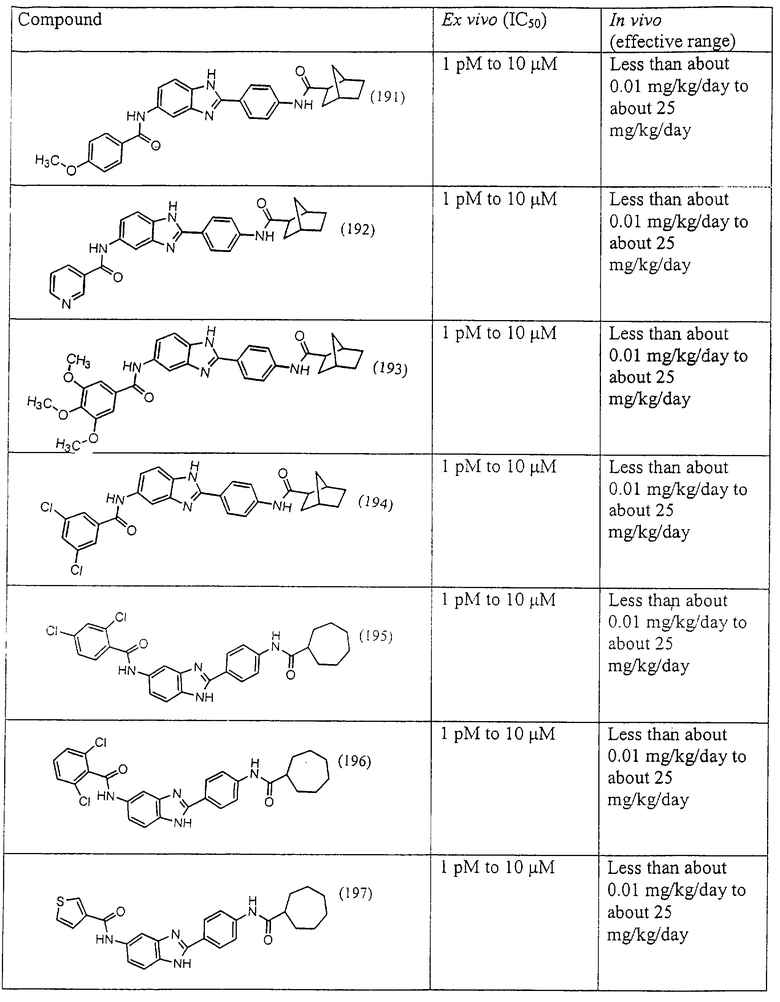
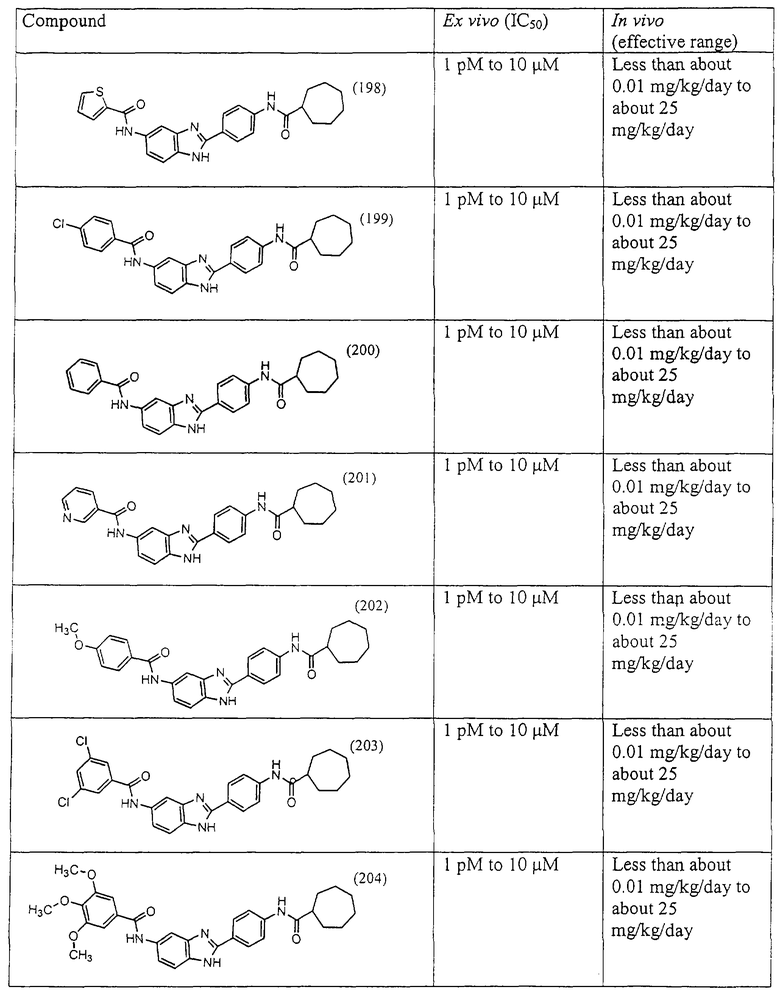
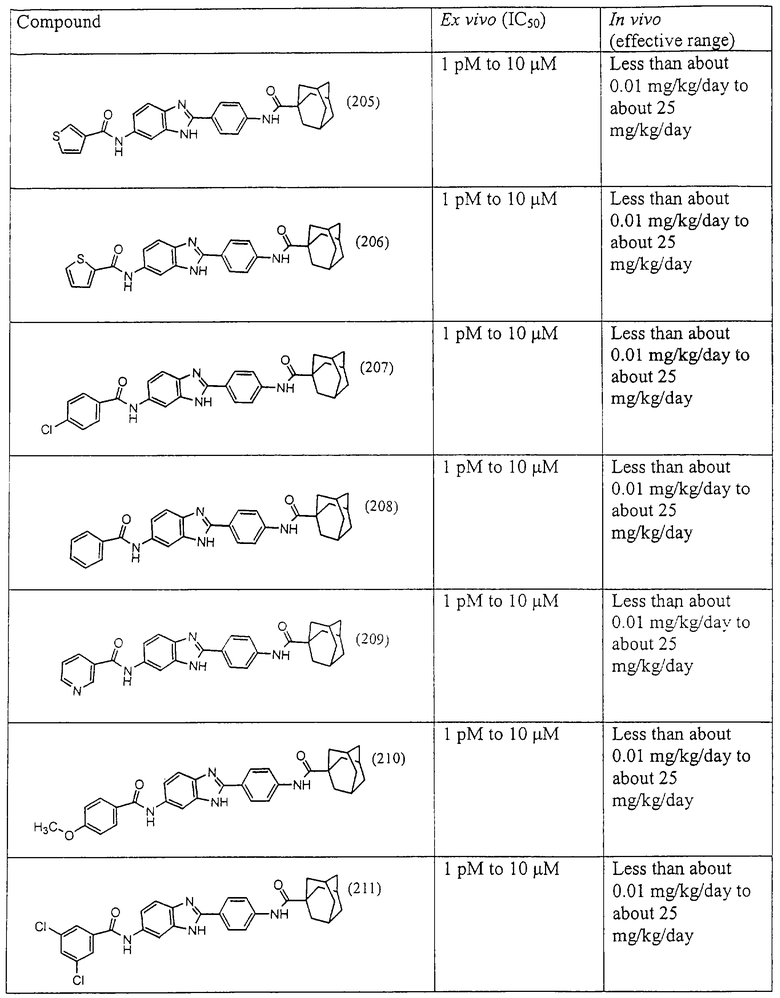
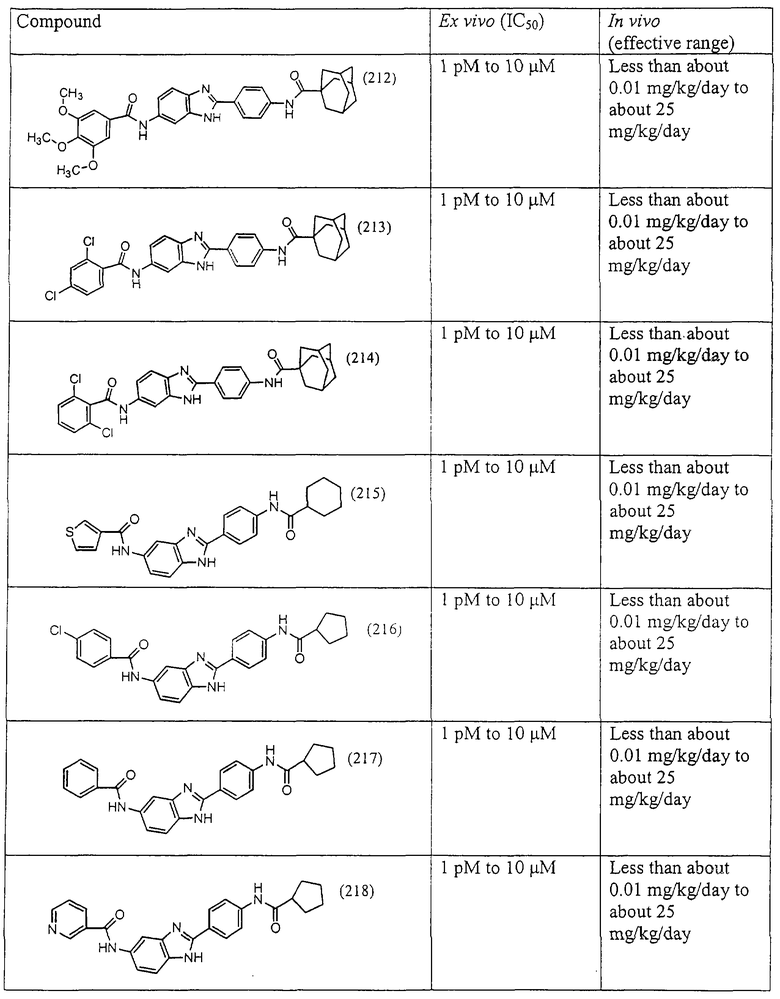
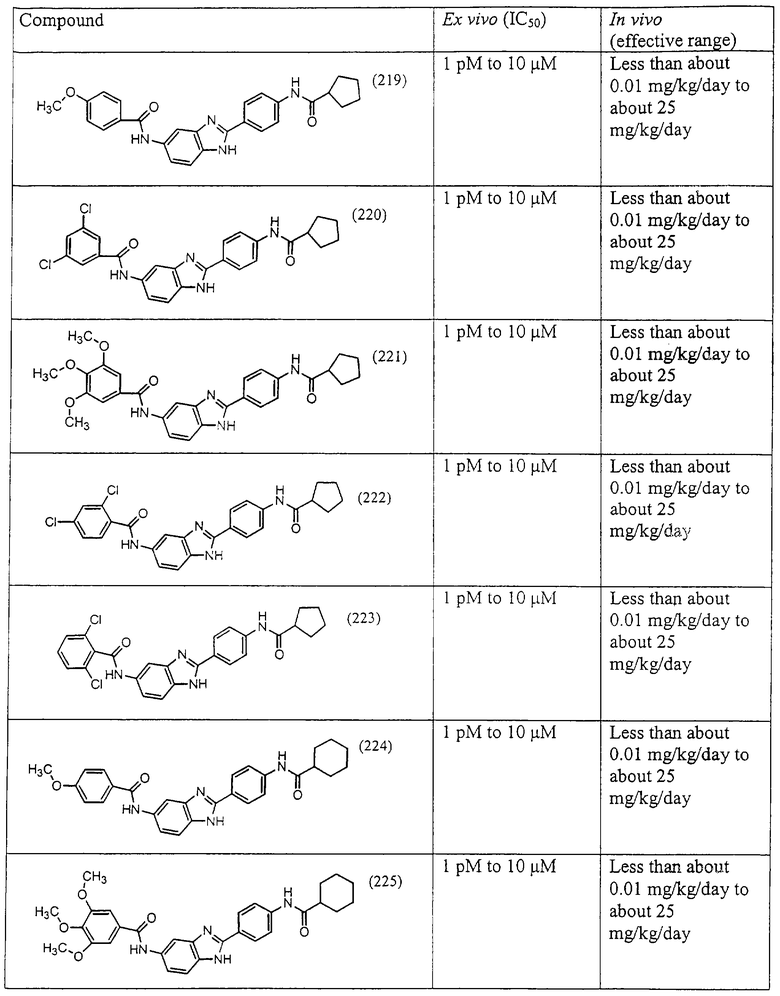
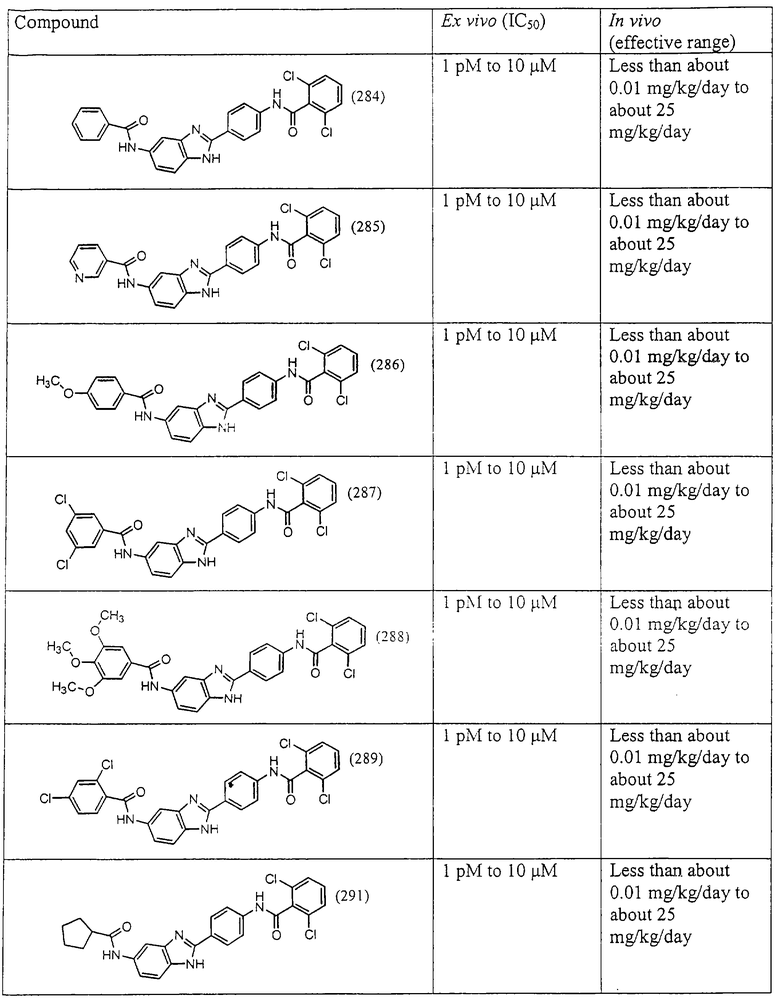
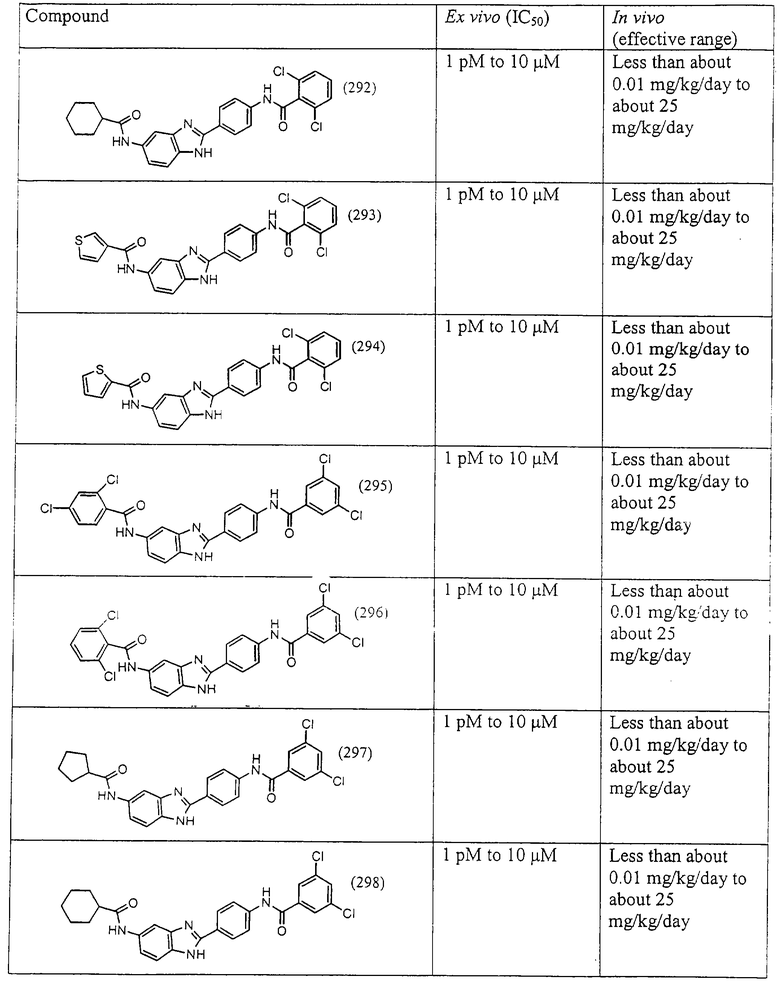
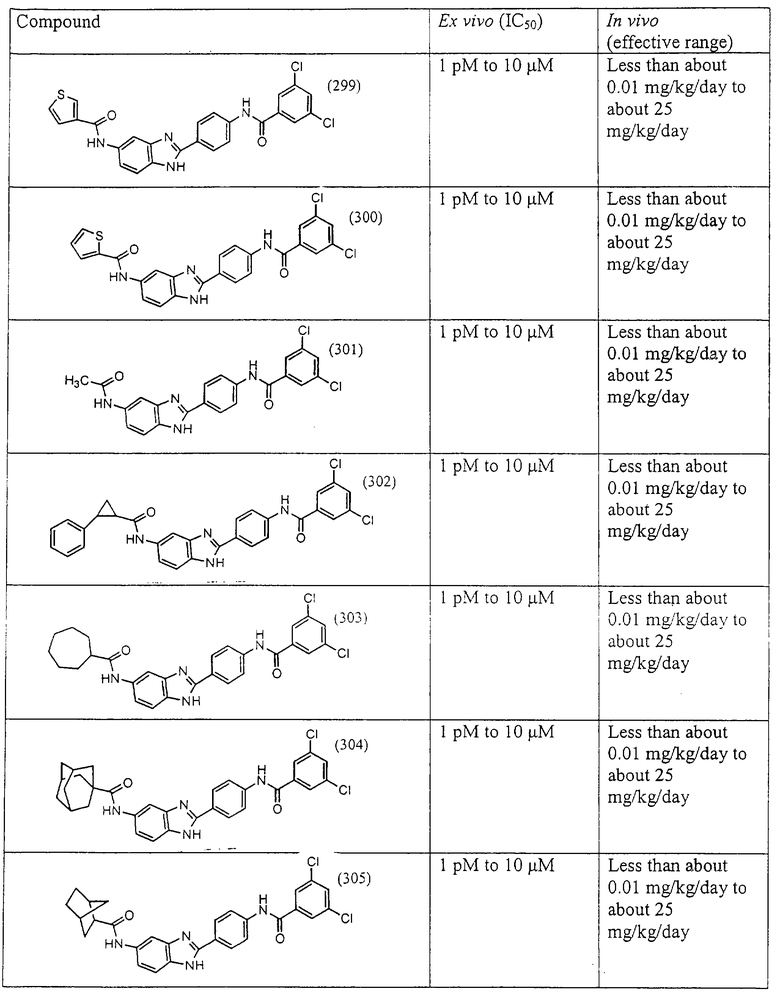
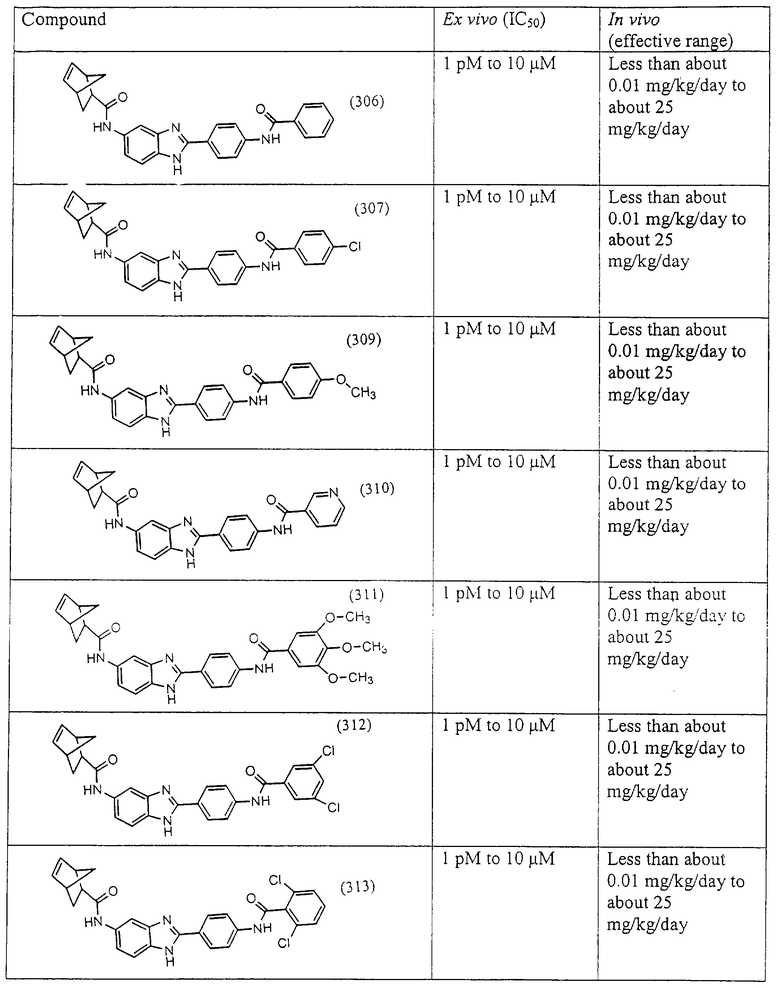
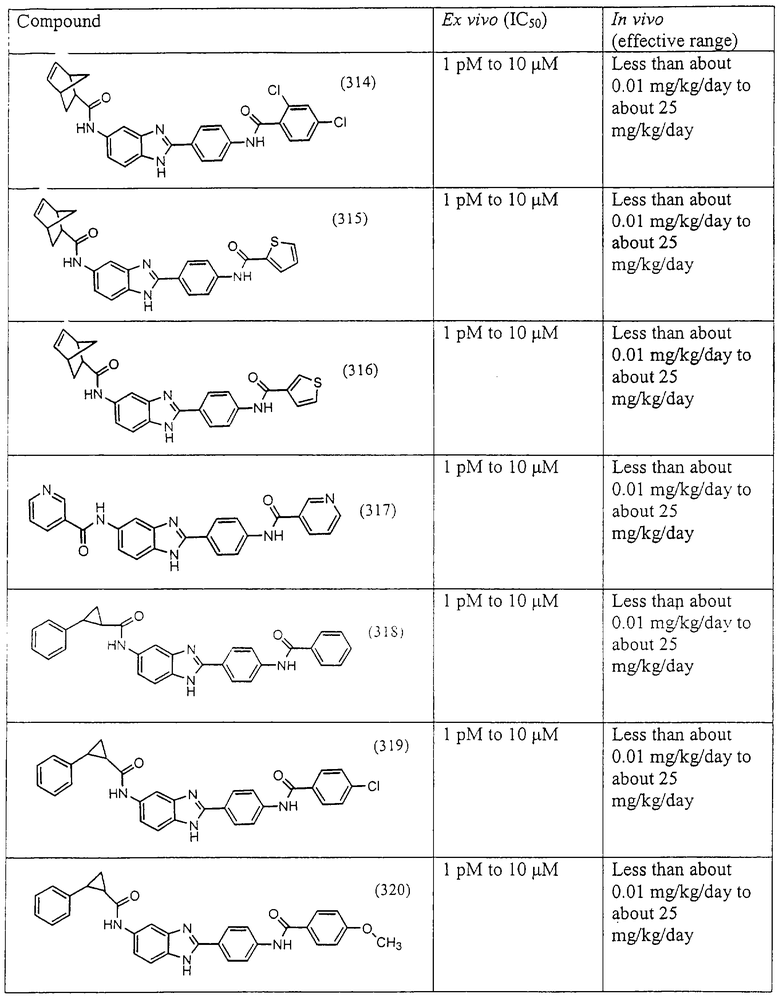
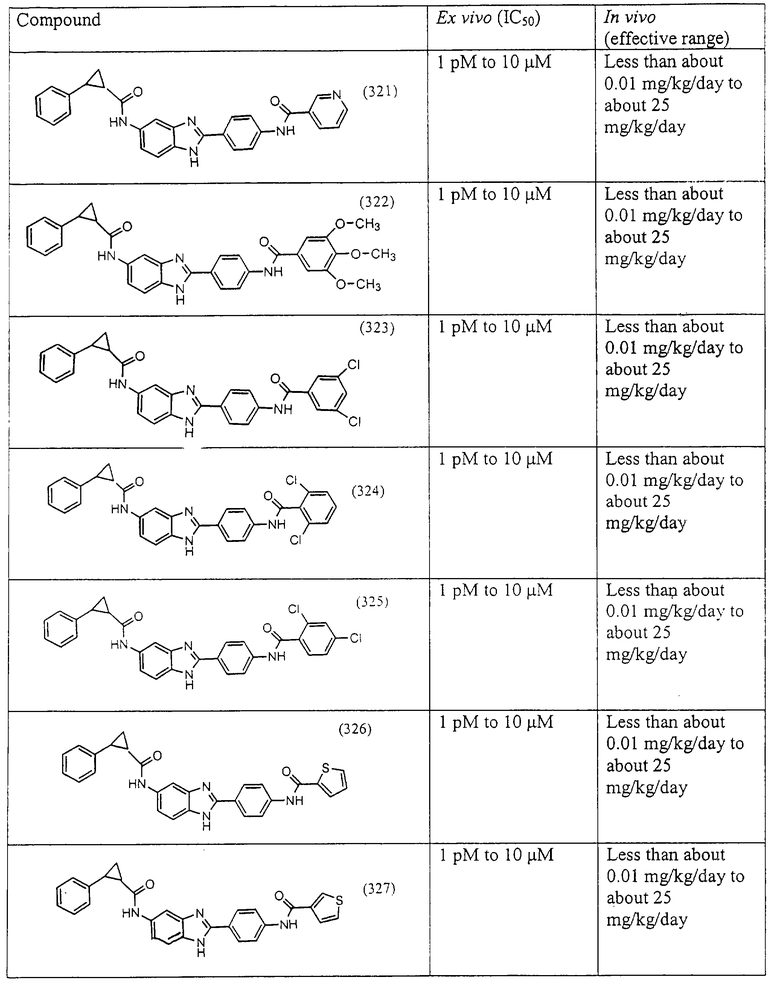
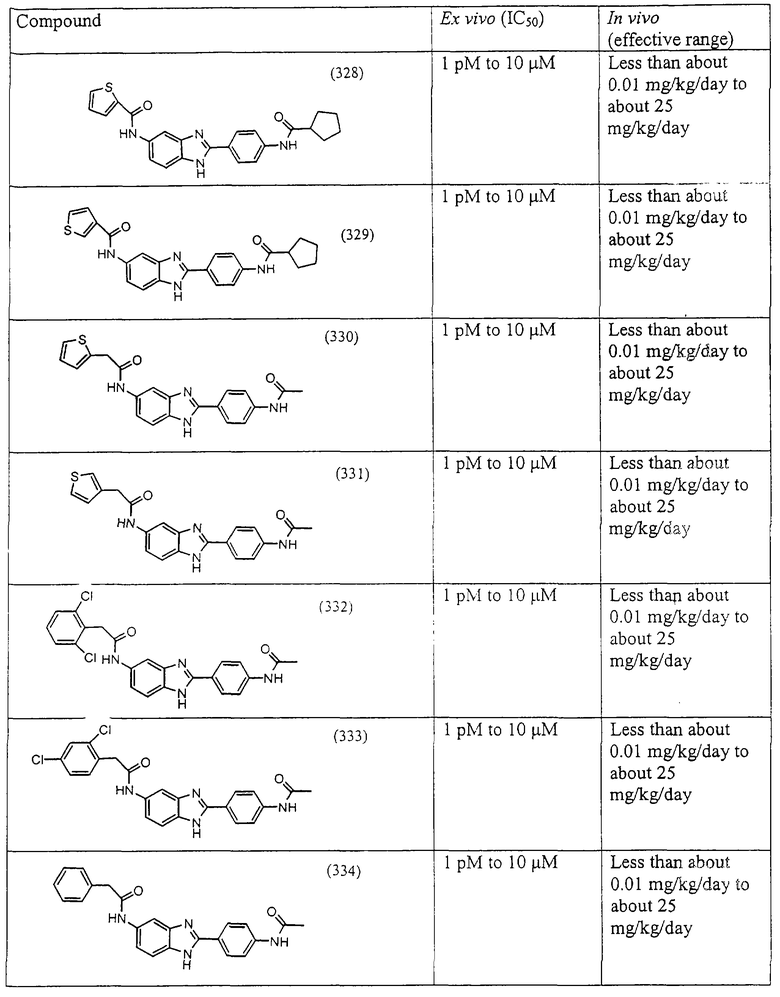
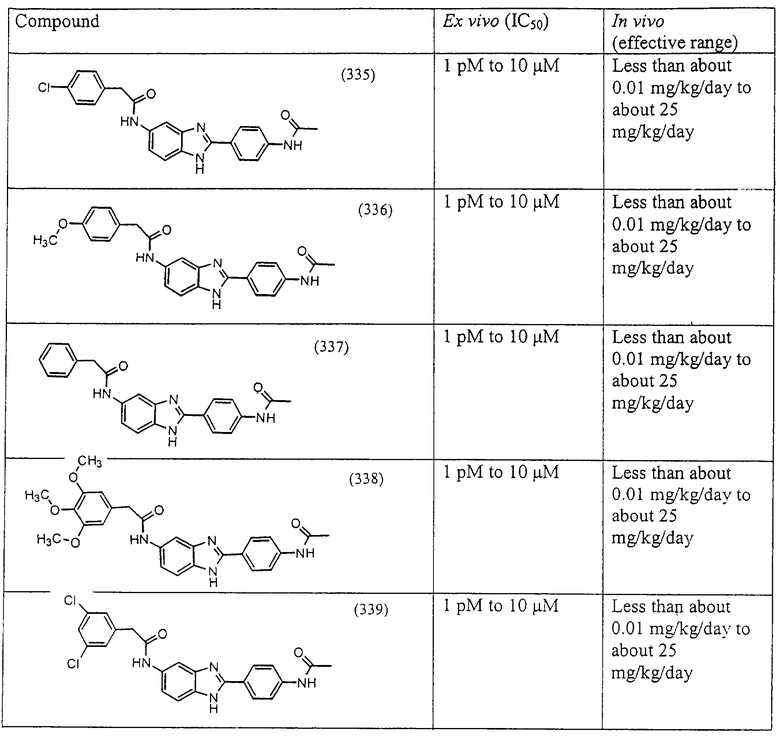
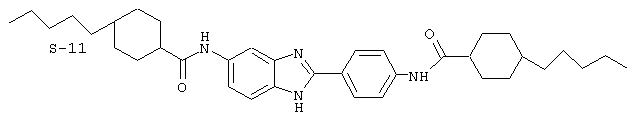
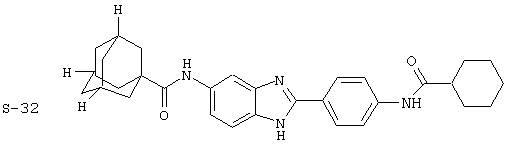
Все раскрытые виды были протестированы на предмет своей способности подавлять IgE как в тесте ex vivo, так и в тесте in vivo. Все они были активны в обоих тестах. Активности (IC50) видов в тесте ех vivo находились в диапазоне от приблизительно 100 пМ до 1 нМ. В тесте in vivo доза IC50 находилась в пределах от приблизительно 100 мкг/кг веса тела в день до приблизительно 10 мг/кг веса тела в день. Диациловые бензимидазоловые соединения в целом были более мощными, чем моноациловые соединения.
Подавление реакции IgE
Ингибиторная активность малых молекул по настоящему изобретению была протестирована в описанных выше ex vivo и in vivo тестах. Все представленные выше соединения были активны при подавлении реакции IgE. В тесте ех vivo соединения родов I-XI давали 50%-ное ингибирование при концентрациях от 1 пМ до 10 мкМ. В тесте in vivo соединения были эффективны при концентрациях от менее чем приблизительно 0,01 мг/кг/день до приблизительно 25 мг/кг/день, при принятии разделенными дозами (например, от двух до четырех раз в день) в течение по меньшей мере 2-7 последовательных дней. Таким образом, низкомолекулярные ингибиторы по настоящему изобретению рассматриваются как полезные при понижении вызванного антигеном повышения концентрации IgE и, следовательно, при лечении зависящих от IgE процессов, таких как аллергии в целом и аллергическая астма в частности.
Режимы лечения
Количество соединения ингибитора IgE, которое может быть эффективным при лечении частной аллергии или состояния, будет зависеть от природы нарушения и может быть определено посредством стандартных клинических методов. Точная доза, которую следует использовать в данной ситуации, также будет зависеть от выбора соединения и серьезности состояния и должна выбираться в соответствии с мнением практикующего врача и состоянием каждого пациента. Подходящие дозировки могут быть определены и скорректированы практикующим врачом на основе зависимостей реакции на дозу между уровнями IgE пациента, а также стандартными индексами легочных и гемодинамических изменений. Кроме того, специалисты поймут, что вариации дозы могут быть определены без лишних экспериментов путем следования протоколу (протоколам), рассматриваемым здесь для ех vivo и in vivo скрининга (см., например, Hasegawa et al., J.Med.Chem. 40:395-407 (1997) и Ohmori et al. Int.J. Immunopharmacol. 15:573-579 (1993); используя подобные ex vivo и in vivo тесты для определения зависимостей доза - реакция для подавления IgE производными нафталина; включенные сюда посредством ссылки).
Сначала приемлемые дозировки соединений в общем случае варьируются от приблизительно 0,001 мг до приблизительно 300 мг на килограмм веса тела в день разделенными дозами, более предпочтительно между приблизительно 0,01 мг и 100 мг на килограмм веса тела в день разделенными дозами. Соединения предпочтительно вводятся системно в виде фармацевтических рецептур, пригодных для таких путей, как пероральный, аэрозольный, внутривенный, подкожный, либо любым другим путем, который может быть эффективным для обеспечения системного дозирования активного соединения. Составы фармацевтических рецептур хорошо известны из уровня техники. Режим лечения предпочтительно содержит периодический прием. Более того, может быть назначена долгосрочная терапия, если оказывается, что аллергические реакции инициируются посредством продолжительного воздействия аллергена (аллергенов). Прием раз в день или дважды в день был эффективен для подавления реакции IgE на появление одного антигена у животных, если этот прием производился непрерывно в течение 2-7 последовательных дней. Таким образом, в предпочтительном выполнении соединение принимается в течение по меньшей мере двух следующих друг за другом дней с регулярными периодическими интервалами. Однако, режим лечения, в том числе частота приема дозы и продолжительность лечения, могут определяться специалистом-практиком и модифицироваться так, как требуется для обеспечения оптимального понижающего регулирования IgE, в зависимости от природы аллергена, дозы, частоты и продолжительности воздействия аллергена и от стандартных клинических показателей.
В одном из выполнений настоящего изобретения подавляющее IgE соединение может приниматься в сочетании с одним или несколькими другими раскрытыми низкомолекулярными ингибиторами для выполнения оптимального понижающего регулирования реакции IgE пациента. Дополнительно предусматривается, что одно или несколько соединений по настоящему изобретению могут приниматься в сочетании с другими, уже известными или раскрытыми позже лекарствами для лечения как причины, лежащей в основе аллергии или астмы, так и их острых симптомов. Такие комбинированные терапии, предусмотренные в объеме настоящего изобретения, содержат смешивание одного или нескольких низкомолекулярных ингибиторов с одним или несколькими дополнительными ингредиентами, эффективность которых при снижении по меньшей мере одного из симптомов болезненного состояния известна. В качестве варианта раскрытые здесь низкомолекулярные ингибиторы могут приниматься отдельно от дополнительных лекарств, но в течение того же болезненного состояния, при этом и ингибитор(ы) IgE, и паллиативные соединения принимаются в соответствии со своими независимыми эффективными режимами лечения.
Хотя подробно описаны несколько предпочтительных выполнении изобретения и их вариантов, другие модификации и способы использования сразу станут понятны специалистам. Соответственно, следует понимать, что различные применения, модификации и замены могут производиться эквивалентами без отхода от сущности изобретения или объема формулы изобретения.
В таблице указаны диапазоны активности для каждого соединения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ IGE | 1999 |
|
RU2236221C2 |
| СОЕДИНЕНИЯ И СПОСОБЫ ДЛЯ ИНГИБИРОВАНИЯ ВЗАИМОДЕЙСТВИЯ BCL БЕЛКОВ С КОМПОНЕНТАМИ ПО СВЯЗЫВАНИЮ | 2005 |
|
RU2416606C2 |
| БЕНЗАМИДНЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ДЕАЦЕТИЛАЗЫ ГИСТОНОВ | 2006 |
|
RU2448965C2 |
| 6-АЛКОКСИПИРИДОПИРИМИДИНЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ АКТИВНОСТЬЮ ИНГИБИТОРОВ КИНАЗЫ МАР р38 | 2003 |
|
RU2324695C2 |
| СПИРОИНДОЛИНОВЫЕ СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ РЕСПИРАТОРНО-СИНЦИТИАЛЬНОГО ВИРУСА (RSV) | 2015 |
|
RU2734248C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕАЗЫ, АКТИВИРУЮЩЕЙ КАНАЛЫ | 2007 |
|
RU2419625C2 |
| ПРОИЗВОДНЫЕ 2-АМИНО-БЕНЗИМИДАЗОЛА И ИХ ИСПОЛЬЗОВАНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ 5-ЛИПОКСИГЕНАЗЫ И/ИЛИ ПРОСТАГЛАНДИН Е-СИНТАЗЫ | 2015 |
|
RU2732416C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ ДЛЯ ОПОСРЕДОВАНИЯ АКТИВНОСТИ ТИРОЗИНКИНАЗЫ 2 | 2020 |
|
RU2826012C2 |
| БИАРИЛЗАМЕЩЕННЫЕ ТРИАЗОЛЫ КАК БЛОКАТОРЫ НАТРИЕВЫХ КАНАЛОВ | 2004 |
|
RU2356897C2 |
| МАКРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ПИРИДАЗИНОНА | 2014 |
|
RU2660435C2 |
Изобретение относится к области медицины и касается фармацевтического состава для лечения аллергической реакции, связанной с повышением уровня IgE, содержащего одно или несколько соединений общей формулы (А) или (В). Состав обладает повышенной активностью. 1 табл.
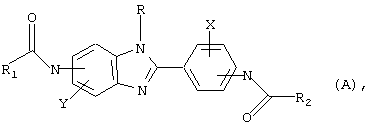
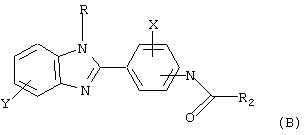
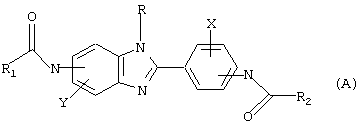
где Х, Y и R представляет водород и
R1 и R2 выбираются независимо из группы, состоящей из алкила, циклоалкила, замещенного циклоалкила, например циклопропила, замещенного циклопропила, циклобутила, замещенного циклобутила, циклопентила, замещенного циклопентила, циклогексила, замещенного циклогексила, циклогептила, замещенного циклогептила, адамантила, замещенного адамантила или незамещенных гетероциклических колец, содержащих в качестве гетероатома атом азота или серы, причем R1 и R2 не могут оба быть метильными группами;
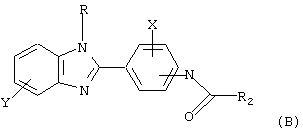
где Х и R представляет Н;
Y выбирается из группы, состоящей из арила, замещенного арила, галогена, CF3;
R2 означает циклоалкил, например циклогексил.
| СПОСОБ ПОЛУЧЕНИЯ 2-ФЕНИЛБЕНЗИМИДАЗОЛА | 1995 |
|
RU2084451C1 |
| Способ изготовления резьбовых деталей | 1978 |
|
SU719765A1 |
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| Устройство для возбуждения синхронного двигателя | 1978 |
|
SU700906A1 |

