Область техники, к которой относится изобретение
Настоящее изобретение относится к низкомолекулярным соединениям и их применению при лечении заболеваний, в частности, воспалительных заболеваний, рака, приступов и/или болезни Альцгеймера.
Уровень техники
Было показано, что путь арахидонат 5-липоксигеназы (5-ЛОГ, 5-ЛО, 5-липоксигеназы или АЛОГ5) играет важную роль в патофизиологии многих воспалительных заболеваний, таких как астма, хроническая обструктивная болезнь легких (ХОБЛ), аллергический ринит, атеросклероз, атопический дерматит и боль, управляя выработкой основных медиаторов воспаления. Также есть сообщения о том, что он связан с раком и болезнью Альцгеймера.
Фермент 5-ЛОГ необходим при выработке ЛТВ4 (лейкотриена В4), важного хемоаттрактанта и активатора лейкоцитов. В основном, ЛТВ4 вырабатывается в нейтрофилах и макрофагах, где фермент ЛТА4-гидролаза преобразует ЛТА4 в ЛТВ4. 5-ЛОГ также участвует в синтезе ЛТС4, D4 и Е4 (цистеинил-лейкотриены; цис-ЛТ), которые являются сильными бронхоконстрикторами и провоспалительными медиаторами. Цис-ЛТ также состоят из ЛТА4, 5-ЛОГ метаболита арахидоновой кислоты (АК). Другой фермент, ЛТС4-синтаза, присутствующий в нескольких клетках, включая эозинофилы, базофилы и тучные клетки, связывает ЛТА4 с глутатионом с получением ЛТС4. Далее ЛТС4 метаболизируется в ЛТD4 и ЛТЕ4. Активность 5-ЛОГ также приводит к выработке биоактивных метаболитов 5-гидроксиэйкозатетраеновой кислоты (ГЭТЕ) и 5-оксо-6,8,11,14-эйкозатетраеновой кислоты (5-оксо-ЭТЕ). Было показано, что 5-оксо-ЭТЕ индуцирует тканевую эозинофилию; таким образом, она может играть роль при астме и других заболеваниях.

Клиническая значимость лейкотриенового пути для воспалительных заболеваний дыхательных путей была продемонстрирована с помощью эффективности различных агентов при лечении астмы и аллергических ринитов. Антагонисты цис-ЛТ рецепторов 1 типа (например, монтелукаст, зафирлукаст и пранлукаст) показали свою эффективность при лечении астмы и аллергических ринитов, а ингибитор 5-ЛОГ (зилеутон) показал свою эффективность при лечении астмы. Ингибиторы 5-ЛОГ, блокирующие выработку и цис-ЛТ, и ЛТВ4, имеют потенциал повышения эффективности лечения астмы и аллергических ринитов по сравнению с антагонистами лейкотриеновых рецепторов. Ингибиторы 5-ЛОГ будут блокировать провоспалительную активность ЛТВ4 и цис-ЛТ, а также других лейкотриенов, таких как 5-ГЭТЕ и 5-оксо-ЭТЕ. Мета-анализ нескольких клинических исследований позволяет предположить улучшение показателей объема форсированного выдоха за 1 секунду (ОФВ1) при использовании зилеутона по сравнению с антагонистами рецептора цис-ЛТ у пациентов, страдающих тяжелой астмой.
Единственным имеющимся на рынке ингибитором 5-ЛОГ является зилеутон, представляющий собой окислительно-восстановительное (редокс) соединение гидроксимочевины, которое хелатно связывает железный остаток критического активного сайта в ферменте 5-ЛОГ. Его эффективность и медицинскую приемлемость ослабляет неудобство схемы применения (т.е. четыре раза в день), квазиоптимальный фармакокинетический и фармакодинамический профиль, а также риск гепатотоксичности. Кроме того, усилия по разработке не-редокс ингибиторов 5-ЛОГ потерпели неудачу из-за недостаточной эффективности для людей. Таким образом, в медицине существует потребность в ингибиторах 5-ЛОГ, обладающих повышенной активностью и переносимостью и меньшей гепатотоксичностью, которые позволят максимально использовать достоинства ингибирования лейкотриенового пути и повысить эффективность по сравнению с зилеутоном и антагонистами рецептора цис-ЛТ.
Простагландин E-синтаза (ПГЕС) - фермент, участвующий в метаболизме эйкозаноида и глутатиона, ускоряющий реакцию из простагландина Н2 в простагландин Е. В качестве важного кофактора для его активности требуется глутатион. Он имплицирован при остеоартрите, ревматоидном артрите, атеросклерозе и воспалительной боли.
Кроме того, микросомальная простагландин E-синтаза-1 (мПГЕС), индуцированная воспалением, является конечным ферментом, который синтезирует простагландин Е2 (ПГЕ2) после циклооксигеназы-2 (ЦОГ-2). мПГЕС-1 повышается в ответ на различные провоспалительные стимулы с сопутствующей повышенной экспрессией ЦОГ-2. Повышение координированной экспрессии ЦОГ-2 и мПГЕС-1 можно обратить с помощью глюкокортикоида. Ингибирование ПГЕС является целесообразным вариантом для исследования ингибирования мПГЕС-1 в качестве потенциального нового способа лечения вышеуказанных заболеваний. В соответствии с данными о мышах с нокаутированным мПГЕС-1, они оказались менее чувствительными к воспалительным и невропатическим болям при моделировании артрита у грызунов, таким образом, мПГЕС-1 была выбрана в качестве цели разработок лекарственных препаратов для лечения воспалительных заболеваний, боли, рака, атеросклероза и апоплексического удара.
В настоящем описании раскрыты соединения с противовоспалительной активностью, а также активностью против 5-ЛОГ и/или ПГЕС.
Целью настоящего изобретения является идентификация соединений, взаимодействующих и создающих препятствия на пути 5-ЛОГ и/или пути ПГЕС, в частности, соединений, оказывающих ингибирующее действие против арахидонат 5-липоксигеназы и/или простагландин E-синтазы.
Еще одной целью настоящего изобретения является идентификация соединений с противовоспалительной активностью.
Еще одной целью настоящего изобретения является идентификация соединений, эффективно помогающих при воспалительных заболеваниях, в частности, астме, атеросклерозе, боли, ХОБЛ, аллергическом рините, постинфекционном воспалении, артрите, дерматите, боли, аллергиях, таких как сенная лихорадка, аутоиммунных заболеваниях, таких как эритематоз, воспалительных заболеваниях кишечника, таких как болезнь Крона, целиакия, угревая сыпь, а также при других заболеваниях, связанных с патологией пути 5-ЛОГ и/или пути ПГЕС, или заболеваниях, связанных с путями 5-ЛОГ и/или путями ПГЕС, таких как рак и/или болезнь Альцгеймера, и/или апоплексический удар.
1. Expert Opin. Ther. Patents (2010) 20(3), 335-375
2. Eur Respir J 2012; 40: 724, 741
3. CurrOpin Allergy ClinImmunol. 2010 February; 10(1): 60-66
4. Biochemical Pharmacology 70 (2005) 327-333
5. Int J ClinPract, April 2007, 61, 4, 663-676
Раскрытие изобретения
В соответствии с одним аспектом настоящее изобретение относится к соединению, имеющему общую формулу I:

где
n равно 0 или 1;
X1 и X2 независимо, в каждом случае, представляют собой CR5 или N;
Y представляет собой C1-C6 алкилен, где алкилен необязательно замещен от одной до двух групп C1-С3 алкила;
R1 выбран из группы, состоящей из водорода, галогена, C1-C6 алкокси, -NH2, -NHR6, -NR7R8 и группы -NH-(R9)n-R10, где n равно 0 или 1;
R2 выбран из группы, состоящей из водорода, галогена, С1-C6 алкила, -NH2, -NHR6, -NR7R8 и группы -NH-(R9)n-R10, где n равно 0 или 1;
R3 выбран из группы, состоящей из водорода, гидроксила, OR11, -NR7R8, C1-C6 алкокси, C1-C6 алкила, С3-С10 циклоалкила, C1-С3 галогеналкила, -C(O)NHR11, арила, гетероарила и гетероциклила, где каждый из указанных циклоалкила, арила, гетероарила и гетероциклила, необязательно и независимо замещен от одной до четырех групп Ra;
R4 выбран из группы, состоящей из -NH2, -N(R12)(V)pR13, -NH(V)p-OR14, -NHC(O)R15, и групп формулы Ia, приведенных ниже,

где
p равно 0 или 1,
V представляет собой C1-C6 алкилен, где алкилен необязательно замещен от одной до трех групп C1-С3 алкила, C3-C6 циклоалкила или фенила, или где атом углерода указанного алкилена образует часть группы C3-C6 циклоалкила;
R5 выбран из группы, состоящей из водорода, галогена, группы C1-C6 алкила, C1-C6 галогеналкила и C1-C6 алкокси;
R6 выбран из группы, состоящей из C1-C6 алкила, С3-С10 циклоалкила, C1-C6 галогеналкила, арила, гетероарила и гетероциклила, где каждый из указанных алкила, циклоалкила, галогеналкила, арила, гетероарила и гетероциклила необязательно и независимо замещен от одной до четырех групп Ra;
R7 и R8 независимо, в каждом случае, представляют собой C1-C6 алкил или гетероциклил; или
R7 и R8 соединены друг с другом, образуя четырех-, пяти- или шестичленный гетероциклил или гетероарил, где каждый из указанных гетероциклила и гетероарила необязательно и независимо замещен от одной до четырех групп Ra;
R9 представляет собой C1-C4 алкилен, где указанный алкилен необязательно замещен от одной до трех групп C1-C3 алкила;
R10 выбран из группы, состоящей из гидроксила, -OR11, -CN, -C(O)OR18, -C(O)NH2, -C(NH)NH2, арила, гетероарила и гетероциклила, где каждый из указанных арила, гетероарила и гетероциклила необязательно и независимо замещен от одной до четырех групп Ra;
R11 независимо в каждом случае выбран из группы, состоящей из арила, например, фенила или бензила, гетероарила и гетероциклила, где каждый из указанных арила, гетероарила и гетероциклила необязательно и независимо замещен от одной до четырех групп Ra;
R12 выбран из группы, состоящей из водорода, C1-C6 алкила, C1-C4 алкил-гидроксила и C1-C4 алкил-алкокси;
R13 выбран из группы, состоящей из водорода, C1-C6 алкила, С3-С10 циклоалкила, C1-C4 алкил-гидроксила, -ОН, -C(O)NH2, -C(O)OR18, -CN, C1-C3 галогеналкила и гетероциклила, и групп формулы Ib, указанных ниже,

где
n равно 0 или 1;
А1 представляет собой -О-, -CH2O-, -ОСН2-, -S-, -SO2-, -SO2NH-, -С(О)-, -C(O)NH-, -C(O)N(R7)-, -СН(ОН)-, -CH(OR7)-, -NH-, -N(CH3)- или -N(CH2COOR7)-;
А2 представляет собой -О- или NH-;
В выбран из группы, состоящей из арила, например, фенила или бензила, гетероарила и гетероциклила, где каждый из указанных арила, гетероарила и гетероциклила необязательно и независимо замещен от одной до четырех групп Ra;
R14 выбран из группы, состоящей из водорода, C1-C6 алкила и арила, где каждый из указанных алкила и арила необязательно и независимо замещен от одной до четырех групп галогена или C1-C3 галогеналкокси;
R15 представляет собой арил, где арил необязательно замещен от одной до четырех групп галогена;
R16 выбран из группы, состоящей из водорода, галогена, C1-C6 алкила и гидроксила;
R17 выбран из группы, состоящей из водорода, галогена, С1-С6 алкила, -C(O)R11, -C(O)NHR11, -OR11 и арила, где каждый из указанных алкила и арила необязательно и независимо замещен от одной до четырех групп Ra;
R18 представляет собой водород или C1-С6 алкил;
R19 выбран из группы, состоящей из водорода, -OR22 и -CH2OR22;
R20 выбран из группы, состоящей из водорода, галогена, С1-С6 алкила, C1-C6 алкокси, -C(O)Ra и C1-C6 галогеналкокси;
R21 выбран из группы, состоящей из водорода, галогена и C1-C6 алкила;
R22 выбран из группы, состоящей из C1-C6 галогеналкила, арила, например, фенила или бензила, и гетероарила, где каждый из указанных галогеналкила, арила и гетероарила необязательно и независимо замещен от одной до четырех групп Ra;
Ra независимо в каждом случае выбран из группы, состоящей из водорода, галогена, C1-C3 алкила, C3-C10 циклоалкила, C1-C4 алкокси, C1-C4 галогеналкила, C1-C6 галогеналкокси, гидроксила, C1-C3 алкилгидроксила, -CH2ORc, -OCH2Rc, -ORc, -CN, NO2, -NRbRc, -C(O)NRbRc, -C(NH)NH2, -C(O)Rc, -C(O)ORc, сульфонила, сульфоксида, гетероциклила, гетероарила и арила, например, фенила, бензила, где каждый из указанных алкила, галогеналкила, галогеналкокси, алкокси, циклоалкила, арила, гетероарила и гетероциклила необязательно и независимо замещен от одной до четырех групп C1-С3 алкила, C1-C4 алкокси, галогена, C1-C3 галогеналкила, -CN, -C(O)NH2, -СООН, -CO2Et и гетероарила;
Rb и Rc независимо друг от друга в каждом случае выбраны из группы, состоящей из водорода, C1-C10 алкила, C3-C10 циклоалкила, C1-C6 алкил-О-алкила, C2-C10 алкенила, C1-C4 алкокси, C1-C3 алкилгидроксила, C3-C10 циклоалкенила, C2-C10 алкинила, С1-С6 галогеналкила, арила, например, фенила или бензила, гетероарила и гетероциклила, где каждый из указанных алкила, циклоалкила, алкил-О-алкила, алкенила, алкокси, циклоалкенила, алкинила, галогеналкила, арила, гетероарила и гетероциклила необязательно и независимо замещен от одной до четырех групп C1-С3 алкила, C1-C4 алкокси, галогена, C1-C6 галогеналкила, C1-C6 галогеналкокси, гидроксила, -C(O)NH2, -СООН, -СООМе, -COOEt, -CN, -NO2, -NH2; или
Rb и Rc соединены друг с другом, образуя четырех-, пяти- или шестичленное насыщенное или ненасыщенное циклическое или гетероциклическое кольцо;
и его фармацевтически приемлемым солям.
В соответствии с другим аспектом настоящее изобретение относится к соединению, имеющему общую формулу II:

где
n равно 0 или 1;
X1 представляет собой CR5 или N;
Y представляет собой C1-C6 алкилен, где алкилен необязательно замещен от одной до двух групп C1-C3 алкила;
R5 выбран из группы, состоящей из водорода, галогена, C1-C6 алкила, C1-C6 галогеналкила и C1-C6 алкокси;
R23 выбран из группы, состоящей из водорода, C1-C6 алкила, C1-C3 галогеналкила, арила, гетероарила и гетероциклила, где каждый из указанных арила, гетероарила и гетероциклила необязательно и независимо замещен от одной до четырех групп Ra;
R24 выбран из группы, состоящей из водорода, C3-C10 циклоалкила, арила, гетероарила и гетероциклила, где каждый из указанных циклоалкила, арила, гетероарила и гетероциклила необязательно и независимо замещен от одной до четырех групп Ra;
R25 выбран из группы, состоящей из водорода, -(V)pR26 и -(V)p-OR14;
при этом
p равно 0 или 1,
V представляет собой C1-С6 алкилен, где алкилен необязательно замещен от одной до трех групп C1-С3 алкила или фенила, или где атом углерода алкилена образует часть группы C3-C6 циклоалкила;
R14 выбран из группы, состоящей из водорода, C1-C6 алкила и арила, где каждый из указанных алкила и арила необязательно и независимо замещен от одной до четырех групп галогена или C1-С3 галогеналкокси;
R26 выбран из группы, состоящей из водорода, C1-C6 алкила, -CN и C1-С3 галогеналкила, и групп формулы IIa, указанных ниже,
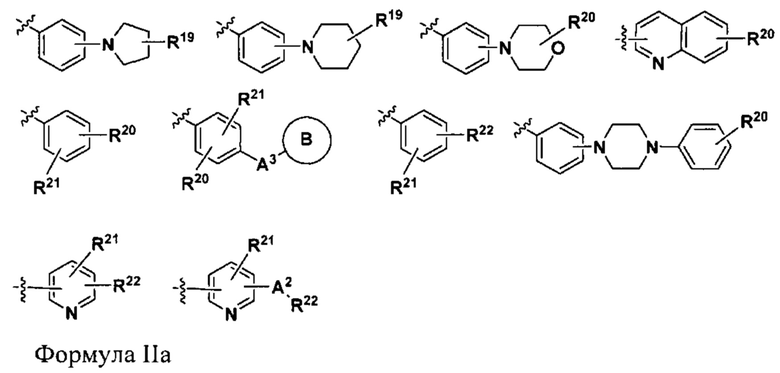
где
А2 представляет собой -О- или NH-;
А3 представляет собой -О-, -CH2O-, -ОСН2-- или -NH-;
В выбран из группы, состоящей из арила, например, фенила или бензила, гетероарила и гетероциклила, где каждый из указанных арила, гетероарила и гетероциклила необязательно и независимо замещен от одной до четырех групп Ra;
R19 выбран из группы, состоящей из водорода, -OR22 и -CH2OR22;
R20 выбран из группы, состоящей из водорода, галогена, С1-C6 алкила, C1-С6 алкокси и C1-C6 галогеналкокси;
R21 выбран из группы, состоящей из водорода, галогена и C1-C6 алкила;
R22 выбран из группы, состоящей из арила, например, фенила или бензила, и гетероарила, где каждый из указанных арила и гетероарила необязательно и независимо замещен от одной до четырех групп Ra;
Ra независимо в каждом случае выбран из группы, состоящей из водорода, галогена, C1-С3 алкила, С3-С10 циклоалкила, C1-C4 алкокси, C1-C4 галогеналкила, C1-C6 галогеналкокси, гидроксила, C1-C3 алкилгидроксила, -CH2ORc, -OCH2Rc, -ORc, -CN, NO2, -NRbRc, -C(O)NRbRc, -C(NH)NH2, -C(O)Rc, -C(O)ORc, сульфонила, сульфоксида, гетероциклила, гетероарила и арила, например, фенила или бензила, где каждый из указанных алкила, галогеналкила, галогеналкокси, алкокси, циклоалкила, арила, гетероарила и гетероциклила необязательно и независимо замещен от одной до четырех групп C1-C3 алкила, C1-C4 алкокси, галогена, C1-С3 галогеналкила, -CN, -C(O)NH2, -СООН, -CO2Et и гетероарила;
Rb и Rc независимо в каждом случае выбраны из группы, состоящей из водорода, C1-C10 алкила, С3-С10 циклоалкила, C1-C6 алкил-О-алкила, C2-C10 алкенила, C1-C4 алкокси, C1-C3 алкилгидроксила, С3-С10 циклоалкенила, C2-C10 алкинила, C1-C6 галогеналкила, арила, например, фенила или бензила, гетероарила, и гетероциклила, где каждый из указанных алкила, алкил-О-алкила, алкенила, алкокси, циклоалкенила, алкинила, галогеналкила, циклоалкила, арила, гетероарила и гетероциклила необязательно и независимо замещен от одной до четырех групп C1-С3 алкила, C1-C4 алкокси, галогена, C1-C6 галогеналкила, C1-C6 галогеналкокси, гидроксила, -C(O)NH2, -СООН, -СООМе, -COOEt, -CN, -NO2, -NH2; или
Rb и Rc соединены друг с другом, образуя четырех-, пяти- или шестичленное насыщенное или ненасыщенное циклическое или гетероциклическое кольцо;
и его фармацевтически приемлемым солям.
В соответствии с еще одним аспектом настоящее изобретение относится к соединению, имеющему общую формулу III:

где
n равно 0 или 1;
X1 представляет собой CR5 или N;
Y представляет собой С1-С6 алкилен, где алкилен необязательно замещен от одной до двух групп С1-С3 алкила;
R5 выбран из группы, состоящей из водорода, галогена, С1-C6 алкила, C1-C6 галогеналкила и C1-C6 алкокси;
R23 выбран из группы, состоящей из водорода, C1-C6 алкила, арила, гетероарила и гетероциклила, где каждый из указанных арила, гетероарила и гетероциклила необязательно и независимо замещен от одной до четырех групп Ra;
R27 выбран из группы, состоящей из водорода, -R6 и -R9-R10;
где
R6 выбран из группы, состоящей из С1-С6 алкила, С3-С10 циклоалкила, C1-C6 галогеналкила, арила, гетероарила и гетероциклила, где указанные алкил, циклоалкил, галогеналкил, арил, гетероарил и гетероциклил необязательно и независимо замещены от одной до четырех групп Ra;
R9 представляет собой C1-C4 алкилен, где алкилен необязательно замещен от одной до трех групп С1-С3 алкила;
R10 выбран из группы, состоящей из гидроксила, -OR11, -C(O)OR18, -C(O)NH2, арила, гетероарила и гетероциклила, где каждый из указанных арила, гетероарила и гетероциклила необязательно и независимо замещен от одной до четырех групп Ra;
R11 независимо в каждом случае выбран из группы, состоящей из арила, например, фенила или бензила, гетероарила и гетероциклила, где указанные арил, гетероарил и гетероциклил необязательно и независимо замещены от одной до четырех групп Ra;
R18 представляет собой водород или С1-С6 алкил;
R28 выбран из группы, состоящей из водорода, -(V)pR29 и -(V)p-OR14;
где
p равно 0 или 1,
V представляет собой C1-C6 алкилен, где алкилен необязательно замещен от одной до трех групп С1-С6 алкила или фенила;
R14 выбран из группы, состоящей из водорода, C1-C6 алкила и арила, где каждый из указанных алкила и арила необязательно и независимо замещен от одной до четырех групп галогена или C1-C3 галогеналкокси;
R29 выбран из группы, состоящей из водорода, C1-C6 алкила и C1-С3 галогеналкила, и групп формулы IIIa, указанных ниже,

где
А2 представляет собой -О- или NH-;
А3 представляет собой -О-, -CH2O-, -ОСН2- или -NH-;
В выбран из группы, состоящей из арила, например, фенила или бензила, гетероарила и гетероциклила, где каждый из указанных арила, гетероарила и гетероциклила необязательно и независимо замещен от одной до четырех групп Ra;
R19 выбран из группы, состоящей из водорода, -OR22 и -CH2OR22;
R20 выбран из группы, состоящей из водорода, галогена, C1-С6 алкила, C1-C6 алкокси и C1-C6 галогеналкокси;
R21 выбран из группы, состоящей из водорода, галогена и С1-С6 алкила;
R22 выбран из группы, состоящей из арила, например, фенила или бензила, и гетероарила, где указанные арил и гетероарил необязательно и независимо замещен от одной до четырех групп Ra;
Ra независимо в каждом случае выбран из группы, состоящей из водорода, галогена, C1-С3 алкила, С3-С10 циклоалкила, C1-C4 алкокси, C1-C4 галогеналкила, C1-C6 галогеналкокси, гидроксила, C1-C3 алкилгидроксила, -CH2ORc, -OCH2Rc, -ORc, -CN, NO2, -NRbRc, -C(O)NRbRc, -C(NH)NH2, -C(O)Rc, -C(O)ORc, сульфонила, сульфоксида, гетероциклила, гетероарила и арила, например, фенила, бензила, где каждый из указанных алкила, галогеналкила, галогеналкокси, алкокси, циклоалкила, арила, гетероарила и гетероциклила необязательно и независимо замещен от одной до четырех групп C1-С3 алкила, C1-C4 алкокси, галогена, С1-С3 галогеналкила, -CN, -C(O)NH2, -СООН, -CO2Et и гетероарила;
Rb и Rc независимо друг от друга в каждом случае выбраны из группы, состоящей из водорода, C1-C10 алкила, С3-С10 циклоалкила, С1-С6 алкил-О-алкила, C2-C10 алкенила, C1-C4 алкокси, C1-C3 алкилгидроксила, C3-C10 циклоалкенила, C2-C10 алкинила, C1-С6 галогеналкила, алкил-О-алкила, алкенила, алкокси, циклоалкенила, алкинила, галогеналкила, арила, например, фенила или бензила, гетероарила, и гетероциклила, при этом каждый элемент из группы алкила, циклоалкила, арила, гетероарила и гетероциклила необязательно и независимо замещен от одной до четырех групп C1-C3 алкила, C1-C4 алкокси, галогена, C1-C6 галогеналкила, C1-C6 галогеналкокси, гидроксила, -C(O)NH2, -СООН, -СООМе, -COOEt, -CN, -NO2, -NH2; или
Rb и Rc соединены друг с другом, образуя четырех-, пяти- или шестичленное насыщенное или ненасыщенное циклическое или гетероциклическое кольцо;
и его фармацевтически приемлемым солям.
В одном варианте осуществления соединение по изобретению имеет одну из формул 1-556, приведенных в таблицах 1-6 и/или в примере 7, и/или в таблице 7, или их фармацевтически приемлемые соли, предпочтительно имеющие одну из формул 1-12, 14-16, 19-21, 24, 28-34, 36-40, 44, 48, 51-54, 56-68, 70, 71, 73-77, 79-81, 84, 86-173, 175-192, 194-234, 236-241, 243, 244, 246-261, 263-278, 280-321, 323-354, 356-385, 387-428, 430-440, 442-446, 449-463, 465-471, 473-487, 489-492, 495-496, 499, 501, 503, 505, 507, 509, 512-514, 525, 529-544, 546-556, приведенную в таблицах 1 и 7, или формулу 14, приведенную в таблицах 2 и 7, или одну из формул 53, 54, 86, 90, 91, 95, 99, 103, 226, приведенную в таблицах 5-7; или одну из формул 14, 17, 24, 29, 30, 32, 33, 38, 43-46, 48, 49, 51-60, 62, 65-70, 73,74, 79-81, 84, 86-88, 90-107, 109-113, 116, 118-132, 134-138, 140, 145, 147-150, 152, 153, 155, 160-162, 164-166, 168, 169, 172, 175-177, 179, 180, 184-187, 190-197, 199, 200, 202-220, 223, 224, 226, 227, 229-231, 233-236, 238-255, 257-262, 264, 265, 267-302, 304-306, 313, 316, 322-333, 335, 340, 342, 346, 347, 349, 350, 352, 353, 357-359, 361, 365-369, 372-375, 377-380, 382-384, 387, 389-393, 395-403, 405-407, 410, 412-415, 419-428, 430-432, 434-437, 440, 442, 445-453, 455, 457-458, 460-461, 463-482, 486-487, 489-496, 499, 501-553, 556, приведенную в таблицах 3 и 7, или одну из формул 11, 12, 14, 24, 30, 32, 48, 52-54, 62, 65, 77, 79-81, 86-88, 92, 94, 97, 98, 101-103, 106, 109-111, 113, 119, 120, 122, 123, 125, 130, 136-138, 145, 147, 149, 155, 165, 166, 176, 177, 184, 193, 195, 199, 204, 211, 226, 227, 229, 231, 233, 234, 238, 239, 243, 249, 251, 253, 256, 268-271, 275, 277, 279, 281, 284, 288, 289, 296, 306, 311, 324, 328, 336, 341, 345, 350, 351, 358, 360, 362, 367-369, 373, 374, 378, 381, 392, 412-415, 430, 431, 433, 447-450, 461, 464-466, 468-471, 473-477, 479, 481, 482, 486, 487, 489-496, 498, 499, 501-506, 508, 509, 512-514, 516-519, 525-538, 540-547, 550, 553-554, приведенную в таблицах 4 и 7; или имеющих формулу 211, приведенную на Фиг. 1 и в таблице 7; или их фармацевтически приемлемые соли.
В одном варианте соединения, имеющего формулу I, R4 представляет собой -N(R12)(V)pR13, R12 представляет собой Н, V представляет собой C1-алкилен, p равно 1, R13 представляет собой

а В представляет собой гетероарил, который необязательно замещен от одной до четырех групп Ra.
В одном варианте соединения, имеющего формулу I, R1 выбран из группы, состоящей из -NH2 и -NHR6, где R6 это гетероарил или гетероциклил, где каждый из указанных гетероарила и гетероциклила необязательно и независимо замещен от одной до четырех групп Ra.
В соответствии с другим аспектом настоящее изобретение относится к соединению, указанному выше, или его фармацевтически приемлемой соли для использования при лечении заболеваний, связанных с путем 5ЛОГ и/или путем простагландин E-синтазы (ПГЕС), при этом заболевание выбрано из группы, состоящей из воспалительных заболеваний, например, астмы, атеросклероза, боли или ХОБЛ, рака, апоплексического удара и болезни Альцгеймера.
В одном варианте соединение оказывает ингибирующее действие на фермент, участвующий в одном или нескольких воспалительных путях, например, пути арахидонат 5-липоксигеназы и/или пути простагландин E-синтазы, предпочтительно на арахидонат 5-липоксигеназу (5-липоксигеназу, 5-ЛО, 5-ЛОГ, АЛОГ5), при концентрации соединения 0,001-50 мкМ, особенно предпочтительно имеющего ИК50 менее 1 мкМ на арахидонат 5-липоксигеназу и/или имеющего ЭК50 менее 10 мкМ на выработку лейкотриена В4 (ЛТВ4) в базофильных клетках крыс (БКК) и/или цельной крови крыс (ЦКК), и/или имеющего 40-70% ингибирующего действия, предпочтительно >70% ингибирующего действия на выработку простагландина Е2 в клетках HeLaS3, стимулированных с помощью ФНО-α, при концентрации соединения, равной 10 мкМ.
Настоящее изобретение также относится к композиции, содержащей соединение или его фармацевтически приемлемую соль, определенные выше, и фармацевтически приемлемый носитель.
Настоящее изобретение также относится к такой композиции, предназначенной для использования при лечении заболевания, связанного с путем 5ЛОГ и/или путем простагландин E-синтазы (ПГЕС), при этом заболевание выбирается из группы, состоящей из воспалительных заболеваний, например, астмы, атеросклероза, боли или ХОБЛ, рака, апоплексического удара и болезни Альцгеймера.
В одном варианте воспалительное заболевание представляет собой одно или несколько из следующих заболеваний: астма, аллергический ринит, дерматит, хроническая обструктивная болезнь легких (ХОБЛ), постинфекционное воспаление, артрит, атеросклероз, аллергии, такие как сенная лихорадка, аутоиммунные заболевания, такие как эритематоз, воспалительные заболевания кишечника, такие как болезнь Крона, целиакия, угревая сыпь или боль, например, воспалительная и/или невропатическая боль.
В одном варианте лечение предусматривает введение подходящего количества соединения, определенного в любом из пп. 1-6 формулы изобретения, или композиции, определенной в п. 9 формулы изобретения, нуждающемуся в этом пациенту, который страдает от воспалительного заболевания и/или рака, и/или апоплексического удара, и/или болезни Альцгеймера.
Настоящее изобретение также относится к способу лечения заболевания, связанного с путем 5-ЛОГ и/или путем простагландин E-синтазы (ПГЕС), при этом заболевание выбирается из группы, состоящей из воспалительных заболеваний, рака и болезни Альцгеймера, где способ подразумевает введение подходящего количества соединения, определенного выше, или композиции, определенной выше, нуждающемуся в этом пациенту, который страдает от заболевания, связанного с путем 5-ЛОГ и/или путем простагландин Е синтазы (ПГЕС).
В одном варианте осуществления воспалительное заболевание выбирается из группы, включающей в себя астму, аллергический ринит, дерматит, хроническую обструктивную болезнь легких (ХОБЛ), постинфекционное воспаление, артрит, атеросклероз, аллергии, такие как сенная лихорадка, аутоиммунные заболевания, такие как эритематоз, воспалительные заболевания кишечника, такие как болезнь Крона, целиакию, угревую сыпь, и боль, например, воспалительную и/или невропатическую боль.
В одном варианте осуществления вводимое подходящее количество составляет от 0,01 мг/кг до 1 г/кг веса тела пациента.
Настоящее изобретение также относится к соединению, которое полностью ингибирует специфическое связывание соединения, определенного выше, с арахидонат 5-липоксигеназой (5-липоксигеназой, 5-ЛО, 5-ЛОГ, АЛОГ5) или с простагландин E-синтазой (ПГЕС). Такое конкурентно ингибирующее соединение в некоторых случаях будет называться «конкурентным соединением» или конкурентно ингибирующим соединением.
Настоящее изобретение также относится к способу лечения воспалительного заболевания, в частности, астмы, атеросклероза, боли, хронической обструктивной болезни легких (ХОБЛ), постинфекционного воспаления, аллергического ринита, артрита, боли, например, воспалительной или невропатической боли, аллергии, такой как сенная лихорадка, аутоиммунных заболеваний, таких как эритематоз, воспалительных заболеваний кишечника, таких как болезнь Крона, целиакии, угревой сыпи и/или дерматита, или способу лечения рака, и/или болезни Альцгеймера, при этом данный способ включает в себя введение подходящего количества конкурентно ингибирующего соединения, определенного выше, нуждающемуся в этом пациенту.
В одном варианте осуществления пациент страдает от воспалительного заболевания и/или рака, и/или апоплексического удара, и/или болезни Альцгеймера.
В соответствии с одним аспектом настоящее изобретение также относится к использованию соединения по настоящему изобретению или композиции по настоящему изобретению для изготовления лекарственного средства для лечения заболевания, связанного с путем 5ЛОГ или путем простагландин E-синтазы (ПГЕС), при этом заболевание выбрано из группы, состоящей из воспалительных заболеваний, например, астмы, атеросклероза, боли или ХОБЛ, рака, апоплексического удара и болезни Альцгеймера, при этом воспалительное заболевание предпочтительно выбрано из группы, включающей в себя астму, атеросклероз, боль, хроническую обструктивную болезнь легких (ХОБЛ), постинфекционное воспаление, аллергический ринит, артрит, боль, например, воспалительную или невропатическую боль, аллергию, такую как сенная лихорадка, аутоиммунные заболевания, такие как эритематоз, воспалительные заболевания кишечника, такие как болезнь Крона, целиакию, угревую сыпь и/или дерматит, при этом лечение включает в себя введение подходящего количества соединения или композиции по настоящему изобретению или определенной выше композиции по настоящему изобретению нуждающемуся в этом пациенту. Соединение и состав по настоящему изобретению представляют собой соединение с формулой I, II или III, в частности, соединение по любому из пп. 1-6 формулы изобретения, или соединение, которое представляет собой конкурентное соединение или конкурентно ингибирующее соединение, определенное выше.
Термин «замещенный», используемый в настоящем документе, например, в словосочетании «необязательно замещенный», означает, что атом водорода, присоединенный к атому члена группы, («необязательно») заменяется группой, включающей в себя галоген, таких как фтор, хлор, бром, C1-C10 алкил, C1-C3 галогеналкил, C3-C7циклоалкил, оксо, -ОН, -OR23, -OC(O)R23, -CN, NO2, -N(R23)2, -N(R23)C(O)R23, -R23N(R23)C(O)R23, -C(O)R23, -R23C(O)R23, -C(O)OR23, -R23C(O)OR23, -C(O)N(R23)2, -R23C(O)N(R23)2, -S(O)R23, -S(O)2R23, -S(O)2N(R23)2, фенил, бензил, арил, гетероарил или гетероциклил, любой из которых сам может быть «необязательно замещен»; следует отметить, что при использовании в данном документе термин «замещенный» также может относиться к нескольким таким замещениям в одном фрагменте.
R23 независимо в каждом случае выбран из группы, состоящей из водорода, галогена, C1-C10 алкила, C1-C3 галогеналкила, C3-C7 циклоалкила, гидроксила, оксо, -OR24, -C(O)OR24, -C(O)R24, -C(O)N(R24)2, -CN, -NO2, -NH2, -N(R24)2, -OR24HetA, -OR24N(R24)2, -C(O)N(R24)HetA, -C(O)HetA, -C(O)N(R24)R24S(O)2R24; -S(O)2N(R24)2, -S(O)2R24, -N(R24)C(O)R24SR24, -N(R24)R24S(O)2R24 или -N(R24)S(O)2R24, арила, например, фенила или бензила, гетероарила и гетероциклила, каждый из которых может быть необязательно замещен.
R24 независимо в каждом случае выбран из группы, состоящей из водорода, C1-C8 алкила, необязательно замещенного гидроксилом и/или галогеном; C3-C7 циклоалкила, арила, например, фенила или бензила, и гетероциклила, каждый из которых необязательно замещен.
В одном варианте осуществления настоящее изобретение также относится к фармацевтически приемлемым солям соединения по настоящему изобретению и к таким фармацевтически приемлемым солям для использования при лечении воспалительного заболевания и/или рака, и/или апоплексического удара, и/или болезни Альцгеймера.
Термин «алкил» относится к одновалентному насыщенному алифатическому углеводородному радикалу с прямой или разветвленной цепью, с количеством атомов углерода в указанном диапазоне, каждый из которых необязательно замещен. Таким образом, например, «C1-C6 алкил» относится к любому из изомеров гексила и пентила, а также к н-, изо-, втор- и трет-бутилу, н- и изопропилу, этилу и метилу, каждый из которых необязательно замещен.
Термин «алкокси» относится к группе, имеющей формулу -О-алкил, в которой алкильная группа, определенная выше, присоединена к исходной молекуле через атом кислорода. Алкильная часть алкокси-группы может иметь от 1 до 20 атомов углерода (т.е. C1-C20 алкокси), от 1 до 12 атомов углерода (т.е. C1-C12 алкокси) или от 1 до 6 атомов углерода (т.е. С1-С6 алкокси). Примеры подходящих алкокси-групп включают в себя, помимо прочего, метокси (-O-СН3 или ОМе), этокси- (-ОСН2СН3 или -OEt), трет-бутокси (-O-С(СН3)3 или -OtBu) и аналогичные группы. Такая алкокси-группа может быть замещена один или несколько раз.
Термин «алкенил» относится к одновалентному алифатическому углеводородному радикалу с прямой или разветвленной цепью, с одной или несколькими двойными связями углерод-углерод и с количеством атомов углерода в указанном диапазоне (необязательно замещенных). Таким образом, например, «С2-С6 алкенил» относится ко всем изомерам гексенила и пентенила, а также 1-бутенилу, 2-бутенилу, 3-бутенилу, изобутенилу, 1-пропенилу, 2-пропенилу и этенилу (или винилу).
Термин «алкинил» относится к одновалентному алифатическому углеводородному радикалу с прямой или разветвленной цепью, с одной тройной связью углерод-углерод и с количеством атомов углерода в указанном диапазоне. Таким образом, например, «C2-C6 алкинил» относится ко всем изомерам гексинила и пентинила, а также 1-бутинилу, 2-бутинилу, 3-бутинилу, 1-пропинилу, 2-пропинилу и этинилу.
Термин «алкилен» относится к насыщенному углеводородному радикалу с разветвленной или прямой цепью или циклическому, имеющему два одновалентных радикальных центра, полученных посредством удаления двух атомов водорода от одного или двух разных атомов углерода, от 1 до 10 атомов углерода или от 1 до 6 атомов углерода. Типичные алкиленовые радикалы включают в себя, помимо прочего, метилен (-СН2-), 1,1-этил (-СН(СН3)-), 1,2-этил (-СН2СН2-), 1,1-пропил (-СН(СН2СН3)-), 1,2-пропил (-СН2СН(СН3)-), 1,3-пропил (-СН2СН2СН2-), 1,4-бутил (-СН2СН2СН2СН2-) и т.д.
Термин «алкенилен» относится к ненасыщенному углеводородному радикалу с разветвленной или прямой цепью или циклическому, имеющему два одновалентных радикальных центра, полученных посредством удаления двух атомов водорода от одного или двух разных атомов углерода исходного алкена. Например, алкениленовая группа может иметь от 1 до 20 атомов углерода, от 1 до 10 атомов углерода или от 1 до 6 атомов углерода. Типичные алкениленовые радикалы включают, помимо прочего, 1,2-этенил (-СН=СН-).
Термин «алкинилен» относится к ненасыщенному углеводородному радикалу с разветвленной или прямой цепью или циклическому, имеющему два одновалентных радикальных центра, полученных посредством удаления двух атомов водорода от одного или двух разных атомов углерода исходного алкина. Например, алкиниленовая группа может иметь от 1 до 20 атомов углерода, от 1 до 10 атомов углерода или от 1 до 6 атомов углерода. Типичные алкиниленовые радикалы включают, помимо прочего, ацетилен (-С≡С-), пропаргил (-СН2С≡С-) и 4-пентинил (-СН2СН2СН2С≡СН-).
Термин «циклоалкил» отдельно или в комбинации с любым другим термином относится к группе, например, необязательно замещенному или незамещенному циклическому углеводороду, имеющей от трех до восьми атомов углерода, если не определено иное. Таким образом, например, «С3-C8 циклоалкил» относится к циклопропилу, циклобутилу, циклопентилу, циклогексилу, циклогептилу и циклооктилу.
Термин «галогеналкил» относится к алкильной группе, как определено в настоящем описании, которая замещена по меньшей мере одним галогеном. Примеры «галогеналкильных» групп с прямой или разветвленной цепью, применяемых в настоящем изобретении, включают в себя, помимо прочего, метил, этил, пропил, изопропил, н-бутил и трет-бутил, замещенные независимо друг от друга одним или несколькими галогенами. Следует понимать, что термин «галогеналкил» следует интерпретировать как включающий в себя такие заместители, как, например, -CHF2, -CF3, -CH2-CH2-F, -CH2-CF3, и т.д.
Термин «гетероалкил» относится к алкильной группе, в которой один или несколько атомов углерода замещены гетероатомом, например, О, N или S. Например, если атом углерода алкильной группы, присоединенный к исходной молекуле, замещен гетероатомом (например, О, N или S), полученные гетероалкильные группы представляют собой соответственно алкоксигруппу (например, -ОСН3 и т.д.), амин (например, -NHCH3, -N(CH3)2 и т.д.) или тиоалкильную группу (например, -SCH3 и т.д.). Если неконцевой атом углерода алкильной группы, не присоединенный к исходной молекуле, замещен гетероатомом (например, О, N или S), то полученные гетероалкильные группы представляют собой, соответственно, алкиловый эфир (например, -СН2СН2-O-СН3, и т.д.), алкиламин (например, -CH2NHCH3, -CH2N(CH3)2 и т.д.) или тиоалкиловый эфир (например, -CH2-S-CH3).
Термин «галоген» относится к фтору, хлору, брому или йоду.
Термин «арил» относится к (i) необязательно замещенному фенилу, (ii) необязательно замещенным 9- или 10-членным бициклическим системам конденсированных карбоциклических колец, в которых по меньшей мере одно кольцо является ароматическим, и (iii) необязательно замещенным 11-14-членным трициклическим системам конденсированных карбоциклических колец, в которых по меньшей мере одно кольцо является ароматическим. Подходящие арилы включают в себя, например, фенил, бифенил, нафтил, тетрагидронафтил (тетралинил), инденил, антраценил и флуоренил.
Термин «фенил», как используется в настоящем описании, обозначает необязательно замещенную или незамещенную фенильную группу.
Термин «бензил», как используется в настоящем описании, обозначает необязательно замещенную или незамещенную бензильную группу.
Термин «гетероарил» (сокращенно называемый в настоящем документе как «HetA») относится к (i) необязательно замещенным 5- и 6-членным гетероароматическим кольцам и (ii) необязательно замещенным 9- и 10-членным бициклическим системам конденсированных колец, в которых по меньшей мере одно кольцо является ароматическим, при этом гетероароматическое кольцо или бициклическая система конденсированных колец содержит от 1 до 4 гетероатомов, независимо выбранных из N, О и S, где каждый N необязательно представлен в форме оксида, а каждый S в кольце, которое не является ароматическим, необязательно представляет собой S(O) или S(O)2. Подходящие 5- и 6-членные гетероароматические кольца включают в себя, например, пиридил, пирролил, пиразинил, пиримидинил, пиридазинил, триазинил, тиенил, фуранил, имидазолил, пиразолил, триазолил, тетразолил, оксазолил, изооксазолил, оксадиазолил, тиазолил, изотиазолил и тиадиазолил. Подходящие 9- и 10-членные гетеробициклические системы конденсированных колец включают в себя, например, бензофуранил, индолил, индазолил, нафтиридинил, изобензофуранил, бензопиперидинил, бензизоксазолил, бензоксазолил, хроменил, хинолинил, изохинолинил, циннолинил, хиназолинил, тетрагидрохинолинил, тетрагидроизохинолинил, изоиндолил, бензодиоксолил, бензофуранил, имидазо[1,2-а]пиридинил, бензотриазолил, дигидроиндолил, дигидроизоиндолил, индазолил, индолинил, изоиндолинил, хиноксалинил, хиназолинил, 2,3-дигидробензофуранил и 2,3-дигидробензо-1,4-диоксинил.
Термин «гетероциклил» относится к (i) необязательно замещенным 4-8-членным насыщенным и ненасыщенным, но неароматическим моноциклическим кольцам, содержащим по меньшей мере один атом углерода и от 1 до 4 гетероатомов, (ii) необязательно замещенным бициклическим системам колец, содержащим от 1 до 6 гетероатомов, и (iii) необязательно замещенным трициклическим системам колец, при этом каждое кольцо в (ii) или (iii) независимо конденсировано или соединено мостиком с другим кольцом или кольцами, и каждое кольцо является насыщенным или ненасыщенным, но неароматическим, и при этом каждый гетероатом в (i), (ii) и (iii) независимо выбран из N, О и S, где каждый N необязательно представлен в форме оксида, а каждый S необязательно окислен до S(O) или S(O)2. Подходящие 4-8-членные насыщенные гетероциклилы включают в себя, например, азетидинил, пиперидинил, морфолинил, тиоморфолинил, тиазолидинил, изотиазолидинил, оксазолидинил, изоксазолидинил, пирролидинил, имидазолидинил, пиперазинил, тетрагидрофуранил, тетрагидротиенил, пиразолидинил, гексагидропиримидинил, тиазинанил, тиазепанил, азепанил, диазепанил, тетрагидропиранил, тетрагидротиопиранил, диоксанил и азациклооктил. Подходящие ненасыщенные гетероциклические кольца включают в себя кольца, которые соответствуют насыщенным гетероциклическим кольцам, перечисленным в предложении выше, в которых одинарная связь заменена двойной связью. Следует понимать, что конкретные кольца и системы колец, подходящие для применения в настоящем изобретении, не ограничены перечисленными в данном и предыдущем абзацах. Такие кольца и системы колец приведены исключительно в качестве примера.
В одном варианте осуществления соединение оказывает ингибирующее действие на фермент, участвующий в воспалительном пути (путях), предпочтительно арахидонат 5-липоксигеназу (5-липоксигеназу, 5-ЛО, 5-ЛОГ, АЛОГ 5), при концентрации соединения 0,001-50 мкМ, особенно предпочтительно имеющего ИК50 менее 1 мкМ на арахидонат 5-липоксигеназу и/или имеющего ЭК50 менее 10 мкМ на выработку лейкотриена В4 (ЛТВ4) в базофильных клетках крыс (БКК) или цельной крови крыс (ЦКК).
Термин «путь 5-ЛОГ» относится к пути, начинающемуся от арахидоновой кислоты к ЛТА4, ЛТВ4, ЛТС4, ЛТD4, ЛТЕ4 и 5-оксоЭТЕ (5-оксо-6,8,11,14-эйкозатетраеновой кислоте). Путь 5-ЛОГ вовлекает один или несколько из следующих ферментов: арахидонат-5-липоксигеназа (5-ЛОГ), ЛТА4-гидролаза, ЛТС4-синтаза, глутамилтранспептидаза и дегидрогеназа 5-гидрокси-6,8,11,14-эйкозатетраеновой кислоты. В таком пути 5-ЛОГ может произойти нарушение функции одного или нескольких ферментов, что приведет к патологии, которая во многих случаях проявляется в виде заболевания или болезненного состояния. Подобное нарушение функции фермента может выражаться в отклонении активности по сравнению с нормальным здоровым состоянием (на которое не повлияла такая патология). Подобное отклонение может представлять собой чрезмерную активность соответствующего фермента или недостаточная активность такого фермента по сравнению с нормальной активностью у здорового человека. Термин «заболевание, связанное с путем 5-ЛОГ» может относиться к заболеванию, при котором один или несколько из вышеуказанных ферментов демонстрируют чрезмерную активность. Предпочтительно, это 5-липоксигеназа (5-ЛОГ), которая демонстрирует отклонение активности, предпочтительно чрезмерную активность по сравнению со здоровым, непатологическим состоянием. В одном варианте осуществления 5-липоксигеназа (5-ЛОГ) представляет собой единственный фермент пути 5-ЛОГ, демонстрирующий пониженную активность. В соответствии с другим вариантом осуществления другой фермент (ферменты) отдельно или в сочетании с 5-липоксигеназой демонстрирует пониженную или повышенную активность. Специалистам в данной области техники известно, как определить, демонстрирует ли один или несколько ферментов пути 5-ЛОГ отклонение активности. Например, это можно легко определить путем измерения одного или нескольких лейкотриенов, которые являются продуктами соответствующих реакций в пути 5-ЛОГ. Измерение этих лейкотриенов, например, одного или нескольких из группы ЛТА4, ЛТВ4, ЛТС4, ЛТD4, ЛТЕ4, 5-ГпЭТЕ, 5-ГЭТЕ и 5-оксо-ЭТЕ, может быть выполнено в соответствующей жидкости организма пациента, такой как кровь, носовые секреты, содержимое бронхов, моча, сперма, спинномозговая жидкость, лимфа, тканевая жидкость, слизистая жидкость, жидкость, полученная из клеток или гомогената клеток, влагалищное отделяемое, слезы, синовиальная жидкость, пот, гной, плевральная жидкость, жидкость брюшной полости, перикардиальная жидкость, слизь, хилус, грудное молоко, цереброспинальная жидкость, серозная жидкость и околоплодная жидкость. Измерение таких лейкотриенов может быть выполнено различными способами, например путем хроматографии отдельно или в сочетании с масс-спектрометрией, например, согласно публикациям Knapp et al. (N. Engl. J. Med. 1989; 320: 1037-1043), Knapp, Prostaglandins 1990; 39: 407-423; и Reilly et al., J. Clin. Pathol. 1988; 41: 1163-1167; ЖХВД с обратной фазой (Antonelli et al.) Intensive Care Med. 1989; 15(5): 296-301, ЖХВД согласно публикациям Otila et al. Acta Derm. Venereol. 1986; 66(5): 381-385, фермент-связанного иммуносорбентного исследования, выполняемого, например, с помощью имеющегося в продаже набора для лейкотриенов компании Oxford Biomedical Research, Assay Designs Inc. или Cayman (кат. номер 520111), например, согласно публикациям Chu et al., American Journal of Pathology, 2011, Volume 178, No. 4, pp. 1762-1769, или Tardif et al. Circ. Cardiovasc. Imaging, 2010, 298-307. Таким образом, способы определения уровней лейкотриенов и других продуктов реакции/промежуточных продуктов пути 5-ЛОГ доступны специалистам в данной области техники, следовательно, специалисты в данной области техники могут определить, связано ли заболевание с путем 5-ЛОГ, определив, имеет ли активность одного или нескольких ферментов из пути 5-ЛОГ отклонения по сравнению с нормальным/здоровым состоянием.
Термин «путь ПГЕС» относится к пути, участвующему в выработке простагландина Е. Путь ПГЕС содержит один или несколько ферментов, включая циклооксигеназу-2 (ЦОГ-2) и простагландин E-синтазу (ПГЕС). В таком пути может произойти нарушение функции одного или нескольких ферментов, что приведет к патологии, которая во многих случаях проявляется в виде заболевания или болезненного состояния. Подобное нарушение функции фермента может выражаться в отклонении активности по сравнению с нормальным здоровым состоянием (на которое не повлияла такая патология). Подобное отклонение может представлять собой чрезмерную активность соответствующего фермента или недостаточная активность такого фермента по сравнению с нормальной активностью у здорового человека. Термин «заболевание, связанное с путем ПГЕС» может относиться к заболеванию, при котором один или несколько из вышеуказанных ферментов демонстрируют чрезмерную активность. Предпочтительно, это простагландин E-синтаза (ПГЕС), которая демонстрирует отклонение активности, предпочтительно чрезмерную активность по сравнению со здоровым, непатологическим состоянием.
В соответствии с предпочтительным вариантом осуществления такое «заболевание, связанное с путем 5-ЛОГ» или «заболевание, связанное с путем ПГЕС» представляет собой заболевание, которое выбрано из группы, включающей в себя воспалительные заболевания, рак, апоплексический удар и болезнь Альцгеймера. В предпочтительном варианте такое заболевание связано с повышенным уровнем одного или нескольких продуктов реакции или промежуточных продуктов пути 5-ЛОГ, таких как вышеуказанные лейкотриены или 5-ГпЭТЕ, 5-ГЭТЕ или 5-оксо-ЭТЕ, или одного или нескольких продуктов реакции или промежуточных продуктов из пути ПГЕС. В одном варианте воспалительное заболевание представляет собой заболевание, которое выбрано из группы, включающей в себя воспалительные заболевания дыхательной системы, такие как астма, аллергический ринит, хроническая обструктивная болезнь легких (ХОБЛ), синусит, бронхит, сенная лихорадка, воспалительные заболевания кожи, такие как дерматит, псориаз, угревая сыпь, воспалительные заболевания кишечника, такие как болезнь Крона, целиакию, язвенный колит, аутоиммунные заболевания, такие как эритематоз, артрит, в частности, ревматоидный артрит, воспалительные заболевания сердечно-сосудистой системы, такие как атеросклероз, васкулит, гранулематоз Вегенера, постинфекционное воспаление и боль, например воспалительная или невропатическая боль.
Путь 5-липоксигеназы вовлечен при различных заболеваниях, а продукты/интермедиаты 5-липоксигеназы были измерены для различных заболеваний, как показано на примере атеросклероза в публикации Tardif et al., Circ. Cardiovasc. Imaging, 2010; 3: 298-307; болезни Альцгеймера согласно публикации Chu et al., Am. J. Cathol. 2011 April; 178(4); 1762-1769; рака согласно публикации Lim et al., J. Neurooncol. 2010, May; 97(3); 339-346; астмы согласно публикации Berger et al., Int. J. Clin. Pract, April 2007, 61, 4, 663-676; ХОБЛ согласно публикации Cazzola et al., European Respiratory Journal 2012; 40(3); 724-741, аллергического ринита согласно публикации Knapp, N. Engl. J. Med. 1990; 323; 1745-1748, атопического дерматита согласно публикации Woodmansee et al., Ann. Allergy Asthma Immunol. 1999; 83: 548-552, крапивницы согласно публикации Spector et al., J. Allergy Clinc. Immunol. 1998; 101(4)572, хронического полипоза носа/синусита согласно публикации Ravikumar, J. Allergy Clin. Immunol. 2005; 115(2) Supplemental S201; 801, артрита согласно публикации Lewis et al., N. Engl. J. Med. 1990; 323: 645-655, боли согласно публикации Noguchi et al., Biol. Pharm. Bull. 2011; 34(8): 1163-1169, аутоиммунных заболеваний согласно публикации в журнале J. Rheumatol. 1995 (March); 22(3): 462-468, болезни Крона согласно публикации Lewis et al., N. Engl. J. Med. 1990; 323: 645-655, угревой сыпи согласно публикации Zouboulis, Dermatoendocrinology 2009 May/June; 1(3): 188-192.
В соответствии с одним аспектом настоящее изобретение относится к соединениям для использования при лечении заболевания, связанного с путем 5-ЛОГ и/или простагландин E-синтазы (ПГЕС), например, воспалительного заболевания или рака, или болезни Альцгеймера, при этом данное соединение имеет одну из формул 1-415, приведенную в таблицах 1-6 и/или примере 7, и таблице 7, предпочтительно имеет одну из формул 1-12, 14-16, 19-21, 24, 28-34, 36-40, 44, 48, 51-54, 56-68, 70, 71, 73-77, 79-81, 84, 86-173, 175-192, 194-234, 236-241, 243, 244, 246-261, 263-278, 280-321, 323-354, 356-385, 387-428, 430-440, 442-446, 449-463, 465-471, 473-487, 489-492, 495-496, 499, 501, 503, 505, 507, 509, 512-514, 525, 529-544, 546-556, приведенную в таблицах 1 и 7, или формулу 14, приведенную в таблицах 2 и 7, или одну из формул 53, 54, 86, 90, 91, 95, 99, 103, 226, приведенную в таблицах 5-7; или одну из формул 14, 17, 24, 29, 30, 32, 33, 38, 43-46, 48, 49, 51-60, 62, 65-70, 73, 74, 79-81, 84, 86-88, 90-107, 109-113, 116, 118-132, 134-138, 140, 145, 147-150, 152, 153, 155, 160-162, 164-166, 168, 169, 172, 175-177, 179, 180, 184-187, 190-197, 199, 200, 202-220, 223, 224, 226, 227, 229-231, 233-236, 238-255, 257-262, 264, 265, 267-302, 304-306, 313, 316, 322-333, 335, 340, 342, 346, 347, 349, 350, 352, 353, 357-359, 361, 365-369, 372-375, 377-380, 382-384, 387, 389-393, 395-403, 405-407, 410, 412-415, 419-428, 430-432, 434-437, 440, 442, 445-453, 455, 457-458, 460-461, 463-482, 486-487, 489-496, 499, 501-553, 556, приведенную в таблицах 3 и 7, или одну из формул 11, 12, 14, 24, 30, 32, 48, 52-54, 62, 65, 77, 79-81, 86-88, 92, 94, 97, 98, 101-103, 106, 109-111, 113, 119, 120, 122, 123, 125, 130, 136-138, 145, 147, 149, 155, 165, 166, 176, 177, 184, 193, 195, 199, 204, 211, 226, 227, 229, 231, 233, 234, 238, 239, 243, 249, 251, 253, 256, 268-271, 275, 277, 279, 281, 284, 288, 289, 296, 306, 311, 324, 328, 336, 341, 345, 350, 351, 358, 360, 362, 367-369, 373, 374, 378, 381, 392, 412-415, 430, 431, 433, 447-450, 461, 464-466, 468-471, 473-477, 479, 481, 482, 486, 487, 489-496, 498, 499, 501-506, 508, 509, 512-514, 516-519, 525-538, 540-547, 550, 553-554, приведенную в таблицах 4 и 7; или формулу 211, приведенную на Фиг. 1 и в таблице 7; или их фармацевтически приемлемым солям.
Предпочтительно, соединения, определенные выше, оказывают ингибирующее действие на фермент, участвующий в воспалительном пути (путях), например, в пути арахидонат 5-липоксигеназы и/или пути простагландин E-синтазы (ПГЕС), предпочтительно на арахидонат 5-липоксигеназу (5-липоксигеназу, 5-ЛО, 5-ЛОГ, АЛОГ5), при концентрации соединения 0,001-50 мкМ, особенно предпочтительно имеющего ИК50 менее 1 мкМ на арахидонат 5-липоксигеназы и/или имеющего ЭК50 менее 10 мкМ на выработку лейкотриена В4 (ЛТВ4) в базофильных клетках крыс (БКК) или цельной крови крыс (ЦКК), и/или имеющего 40-70% ингибирующего действия, предпочтительно >70% ингибирующего действия на выработку простагландина Е2 в клетках HeLaS3, стимулированных с помощью ФНО-α, при концентрации соединения, равной 10 мкМ.
В одном аспекте настоящее изобретение относится к композиции, содержащей соединение по настоящему изобретению и фармацевтически приемлемый носитель для использования при лечении заболевания, связанного с путем 5-ЛОГ и/или путем ПГЕС, при этом заболевание выбрано из группы, включающей в себя воспалительное заболевание, например, астму или ХОБЛ, рак, апоплексический удар и болезнь Альцгеймера.
В одном варианте осуществления воспалительное заболевание представляет собой астму или аллергический ринит, или дерматит, или хроническую обструктивную болезнь легких (ХОБЛ), или постинфекционное воспаление, или артрит, или атеросклероз, или боль, например, воспалительную и/или невропатическую боль.
В одном варианте осуществления указанное лечение включает в себя введение подходящего количества соединения или композиции, определенных выше, нуждающемуся в этом пациенту, который страдает от заболевания, связанного с путем 5-ЛОГ и/или путем ПГЕС, при этом заболевание выбрано из группы, включающей в себя воспалительное заболевание, рак, апоплексический удар и болезнь Альцгеймера.
В соответствии с другим аспектом настоящее изобретение относится к способу лечения заболевания, связанного с путем 5-ЛОГ и/или путем ПГЕС, при этом заболевание выбрано из группы, включающей в себя воспалительное заболевание, рак, апоплексический удар и болезнь Альцгеймера, причем способ включает в себя введение подходящего количества соединения или композиции, определенных выше, нуждающемуся в этом пациенту. В одном варианте осуществления воспалительное заболевание представляет собой астму или аллергический ринит, или дерматит, или хроническую обструктивную болезнь легких (ХОБЛ), или постинфекционное воспаление, или артрит, или атеросклероз, или боль, например, воспалительную и/или невропатическую боль.
В одном варианте, подходящее количество составляет от 0,01 мг/кг до 1 г/кг веса тела пациента.
Настоящее изобретение также относится к использованию соединения или композиции по настоящему изобретению, определенных выше, для производства лекарственного препарата для лечения заболевания, связанного с путем 5-ЛОГ и/или путем ПГЕС, при этом заболевание выбрано из группы, включающей в себя воспалительное заболевание, рак, апоплексический удар и болезнь Альцгеймера, а также лечению, включающему в себя введение подходящего количества соединения или композиции, определенных выше, нуждающемуся в этом пациенту. Воспалительное заболевание и подходящее количество определены выше.
В соответствии с другим аспектом настоящее изобретение относится к определенному выше соединению, которое конкурентно ингибирует специфическое связывание соединения по настоящему изобретению с арахидонат 5-липоксигеназой (5-липоксигеназой, 5-ЛО, 5-ЛОГ, АЛОГ5) и/или простагландин E-синтазой (ПГЕС).
В соответствии с еще одним аспектом настоящее изобретение относится к способу лечения заболевания, связанного с путем 5-ЛОГ и/или путем ПГЕС, при этом заболевание выбрано из группы, включающей в себя воспалительное заболевание, в частности, астму, атеросклероз, боль, ХОБЛ, постинфекционное воспаление, аллергический ринит и/или атопический дерматит, или рак, или апоплексический удар, или болезнь Альцгеймера, при этом данный способ включает в себя введение подходящего количества соединения, определенного выше, т.е. соединения, которое конкурентно ингибирует специфическое связывание соединения по настоящему изобретению с арахидонат 5-липоксигеназой или с простагландин E-синтазой, нуждающемуся в этом пациенту.
Соединение, которое конкурентно ингибирует специфическое связывание соединения по настоящему изобретению с 5-ЛОГ или с ПГЕС, в настоящем документе в некоторых случаях будет обозначаться как «конкурентно ингибирующее соединение».
В одном варианте осуществления такой пациент страдает от воспалительного заболевания, предпочтительно такого, как было определено выше.
Настоящее изобретение также относится к использованию конкурентно ингибирующего соединения производителем лекарственного препарата для лечения заболевания, связанного с путем 5-ЛОГ или путем ПГЕС, при этом заболевание выбрано из группы, включающей в себя воспалительное заболевание, в частности, астму, атеросклероз, боль или ХОБЛ, постинфекционное воспаление, аллергический ринит и/или артрит, или рак, или апоплексический удар, или болезнь Альцгеймера, лечение, включающее в себя введение подходящего количества соединения, которое конкурентно ингибирует специфическое связывание, как было определено выше, нуждающемуся в этом пациенту.
Термины «ИК50» и «ЭК50» относятся соответственно к концентрации полумаксимального ингибирования и полумаксимальной эффективной концентрации соединения с учетом заданной активности, например, ингибирования фермента с помощью соединения или выработки вещества, стимулированной соединением. В одном примере ИК50 составляет половину концентрации соединения для максимального ингибирования активности арахидонат 5-липоксигеназы. В одном примере ЭК50 составляет половину максимальной эффективной концентрации соединения на выработку и/или секрецию лейкотриена В4 (ЛТВ4) в клетке или цельной крови, например, базофильной клетке крыс (БКК) или цельной крови крыс (ЦКК).
Фармацевтические композиции
Фармацевтически приемлемые соли
Примеры фармацевтически приемлемых солей присоединения включают в себя, помимо прочего, нетоксичные неорганические и органические соли присоединения кислот, например, ацетат, получаемый из уксусной кислоты, аконат, получаемый из аконитовой кислоты, аскорбат, получаемый из аскорбиновой кислоты, бензолсульфонат, получаемый из бензолсульфоновой кислоты, бензоат, получаемый из бензойной кислоты, циннамат, получаемый из коричной кислоты, цитрат, получаемый из лимонной кислоты, эмбонат, получаемый из эмбоновой кислоты, энантат, получаемый из энантовой кислоты, формиат, получаемый из муравьиной кислоты, фумарат, получаемый из фумаровой кислоты, глутамат, получаемый из глутаминовой кислоты, гликолят, получаемый из гликолевой кислоты, гидрохлорид, получаемый из соляной кислоты, гидробромид, получаемый из бромистоводородной кислоты, лактат, получаемый из молочной кислоты, малеат, получаемый из малеиновой кислоты, малонат, получаемый из малоновой кислоты, манделат, получаемый из миндальной кислоты, метансульфонат, получаемый из метансульфокислоты, нафталин-2-сульфонат, получаемый из нафталин-2-сульфокислоты, нитрат, получаемый из азотной кислоты, перхлорат, получаемый из перхлорной кислоты, фосфат, получаемый из фосфорной кислоты, фталат, получаемый из фталиевой кислоты, салицилат, получаемый из салициловой кислоты, сорбат, получаемый из сорбиновой кислоты, стеарат, получаемый из стеариновой кислоты, сукцинат, получаемый из янтарной кислоты, сульфат, получаемый из серной кислоты, тартрат, получаемый из винной кислоты, толуол-п-сульфонат, получаемый из п-толуолсульфокислоты, и т.д. Такие соли могут быть получены способами, хорошо известными и описанными в данной области техники.
Другие кислоты, которые нельзя считать фармацевтически приемлемыми, могут быть полезными для получения солей, используемых в качестве интермедиатов при получении химического соединения по изобретению и его фармацевтически приемлемой соли присоединения кислоты, для использования при лечении воспалительного заболевания.
В соответствии с другим вариантом осуществления соединения по изобретению используют в их соответствующей свободной основной форме для применения при лечении воспалительного заболевания, согласно настоящему изобретению.
Соли металлов химического соединения по изобретению включают в себя соли щелочных металлов, например, натриевую соль химического соединения по изобретению, содержащего карбоксигруппу.
Химические соединения по изобретению могут быть представлены в несольватированной или сольватированной форме вместе с фармацевтически приемлемым растворителем (растворителями), например, водой, этанолом и тому подобным. Сольватированные формы могут также включать в себя гидратные формы, такие как моногидрат, дигидрат, полугидрат, тригидрат, тетрагидрат и т.д. В общем, для целей настоящего изобретения считается, что сольватированные формы равноценны несольватированным формам.
Введение и приготовление состава
Приготовление лекарственных средств, содержащих соединения по настоящему изобретению, их активные метаболиты или изомеры и соли по настоящему изобретению, и их применение можно осуществлять в соответствии с хорошо известными фармацевтическими способами.
Хотя соединения по изобретению, пригодные в соответствии с изобретением для применения в терапии, можно вводить в форме сырого химического соединения, активный ингредиент предпочтительнее включать, по возможности, в форме физиологически приемлемой соли, в лекарственный состав вместе с одним или несколькими адъювантами, наполнителями, носителями, буферами, разбавителями и/или другими обычными фармацевтическими вспомогательными веществами. Такие соли соединений по настоящему изобретению могут быть безводными или сольватированными.
В предпочтительном варианте настоящее изобретение предлагает лекарственные средства, содержащие соединение, пригодное для использования в соответствии с настоящим изобретением, или его фармацевтически приемлемую соль или производное вместе с одним или несколькими фармацевтически приемлемыми их носителями и, необязательно, другими терапевтическими и/или профилактическими ингредиентами. Носитель (носители) должен быть «приемлемым» в смысле его совместимости с другими ингредиентами состава и безвредным для его реципиента.
Лекарственным средством по изобретению могут быть лекарственные средства, подходящие для перорального, ректального, бронхиального, назального, местного, буккального, подъязычного, чрескожного, вагинального или парентерального введения (включая кожную, подкожную, внутримышечную, внутрибрюшинную, внутривенную, внутриартериальную, внутримозговую, внутриглазную инъекцию или инфузию), или выпускаемые в форме, подходящей для введения посредством ингаляции или инсуфляции, включая введение порошков и жидкого аэрозоля, или посредством систем с замедленным высвобождением. Подходящие примеры систем с замедленным высвобождением включают в себя полупроницаемые матрицы из твердых гидрофобных полимеров, содержащие соединение по изобретению, и такие матрицы могут быть выполнены в виде изделий определенной формы, например, пленок или микрокапсул.
Соединения, пригодные для использования в соответствии с изобретением, вместе с традиционным адъювантом, носителем или разбавителем, таким образом, могут быть введены в форму лекарственного средства и его единичные дозировки. Такие формы включают в себя твердые вещества, и, в частности, таблетки, заполненные капсулы, порошковые и гранулированные формы; и жидкости, в частности, водные или неводные растворы, суспензии, эмульсии, эликсиры и капсулы, заполненные такими жидкостями, все такие формы предназначены для перорального применения; суппозитории для ректального введения; и стерильные инъекционные растворы для парентерального применения. Такое лекарственное средство и его формы, содержащие единичные дозы, могут содержать традиционные ингредиенты в традиционных соотношениях, с дополнительными активными соединениями или действующими началами или без них, и такие формы, содержащие единичные дозы, могут содержать любое подходящее эффективное количество активного ингредиента, соразмерно назначенному для ежедневного применения диапазону дозировок.
Соединения, пригодные для использования в соответствии с настоящим изобретением, можно вводить в виде широкого диапазона пероральных и парентеральных лекарственных форм. Специалистам в данной области техники очевидно, что следующие лекарственные формы могут содержать в качестве активного компонента как соединение (соединения), пригодное для использования в соответствии с настоящим изобретением, так и фармацевтически приемлемую соль соединения (соединений), пригодного для использования в соответствии с настоящим изобретением.
Фармацевтически приемлемые носители для приготовления лекарственного средства из соединения, пригодного для использования в соответствии с настоящим изобретением, могут быть твердыми или жидкими. Составы в твердой форме включают в себя порошки, таблетки, драже, капсулы, крахмальные капсулы, суппозитории и диспергируемые гранулы. Твердый носитель может представлять собой одно или несколько веществ, которые могут также действовать в качестве разбавителей, ароматизаторов, сжижающих реагентов, скользящих веществ, суспендирующих веществ, связывающих веществ, консервантов, веществ для улучшения распадаемости таблеток или материала для капсулирования.
В порошках носитель представляет собой мелкоизмельченное твердое вещество, которое находится в смеси с мелкоизмельченным активным компонентом. В таблетках активный компонент в подходящем соотношении смешан с носителем, имеющим необходимую связывающую способность, и спрессован в желаемые форму и размер. Подходящими носителями являются карбонат магния, стеарат магния, тальк, сахар, лактоза, пектин, декстрин, крахмал, желатин, трагакант, метилцеллюлоза, карбоксиметилцеллюлоза натрия, низкоплавкий воск, масло какао и т.д. Предполагается, что термин «приготовление» включает в себя смешение активного соединения с материалом для инкапсулирования в качестве носителя, образующего капсулу, в которой активный компонент, с носителями или без носителей, окружен носителем, который, таким образом, связывается с ним. Аналогичным образом, в изобретение включены крахмальные капсулы и леденцы для рассасывания. Таблетки, порошки, капсулы, драже, крахмальные капсулы и леденцы для рассасывания можно применять в качестве твердых форм, подходящих для перорального введения.
Для приготовления суппозиториев сначала расплавляют низкоплавкий воск, например, смесь глицерида жирных кислот или масла какао, и растворяют в нем активный компонент до образования однородной смеси путем перемешивания. Затем расплавленную гомогенную смесь выливают в формы подходящего размера, оставляют остыть и соответственно, затвердеть. Составы, подходящие для вагинального введения, могут быть представлены в виде пессариев, тампонов, кремов, гелей, паст, пенок или спреев, содержащих в дополнение к активному ингредиенту такие носители, которые, известны в данной области техники как подходящие. Жидкие составы включают в себя растворы, суспензии и эмульсии, например, водные или водно-пропиленгликолевые растворы. Например, жидкие составы для парентерального введения могут быть приготовлены в виде растворов в водно-полиэтиленгликолевом растворе.
Химические соединения по изобретению, следовательно, можно приготовить для парентерального введения (например, посредством инъекции, например, болюсной инъекции или непрерывной инфузии), и они могут быть представлены в виде формы, содержащей единичные дозы, в ампулах, предварительно заполненных шприцах, малообъемных контейнерах для инфузии или в многодозных контейнерах с добавленным консервантом. Составы могут быть выполнены в таких формах, как суспензии, растворы или эмульсии на масляных или водных основах, и могут содержать вещества для приготовления составов, такие как суспендирующие, стабилизирующие и/или диспергирующие вещества. В качестве варианта, активный ингредиент может иметь форму порошка, полученную путем асептического выделения стерильного твердого вещества или путем лиофилизации из раствора, для объединения с подходящей основой, например, стерильной апирогенной водой, перед применением.
Водные растворы, подходящие для перорального применения, могут быть получены растворением активного компонента в воде и добавлением, при желании, подходящих красителей, ароматизаторов, стабилизаторов и загустителей. Водные суспензии, подходящие для перорального применения, могут быть получены посредством распределения мелкоизмельченного активного компонента в воде с вязким материалом, таким как натуральные или синтетические камеди, смолы, метилцеллюлоза, натрий карбоксиметилцеллюлоза или иные широко известные суспендирующие вещества.
Также включены составы в твердой форме, которые необходимо растворить непосредственно перед применением с получением жидких составов для перорального введения. Такие жидкие формы включают в себя растворы, суспензии и эмульсии. В добавление к активному компоненту такие составы могут содержать красители, ароматизаторы, стабилизаторы, буферы, искусственные и натуральные подсластители, диспергирующие вещества, загустители, солюбилизирующие вещества и т.д.
В одном варианте настоящего изобретения лекарственное средство применяют местно или системно, или посредством комбинации этих двух путей.
Для целей введения в организм, соединения по настоящему изобретению в одном варианте можно вводить в составе, содержащем от 0,001% до 70% соединения по весу, предпочтительно, от 0,01% до 70% соединения по весу, еще более предпочтительно от 0,1% до 70% соединения по весу. В одном варианте осуществления вводимое подходящее количество составляет от 0,01 мг/кг до 1 г/кг веса тела.
Подходящие для введения композиции также включают в себя леденцы для рассасывания, содержащие активное вещество в ароматизированной основе, как правило, сахарозе и акациевой или трагакантовой камеди; пастилки, содержащие активный ингредиент в инертной основе, такой как желатин и глицерин или сахароза и акациевая камедь; и жидкости для полоскания полости рта, содержащие активный ингредиент в подходящем жидком носителе.
Растворы или суспензии помещают непосредственно в носовую полость при помощи традиционных средств, например, с помощью пипетки или распылителя. Составы могут быть предоставлены в однодозовой или многодозовой форме. В последнем случае при использовании пипетки этого можно достичь путем введения пациенту соответствующего предварительно определенного объема раствора или суспензии. В случае распылителя этого можно достичь, например, за счет использования дозирующего распыляющего насоса.
Введение в дыхательные пути также может быть выполнено путем использования аэрозольной формы, где активный ингредиент поставляется в упаковке под давлением с подходящим пропеллентом, таким как хлорфторуглерод (ХФУ), например, дихлордифторметан, трихлорфторметан или дихлортетрафторэтан, углекислый газ или иной подходящий газ. Аэрозоль также может для удобства содержать поверхностно-активное вещество, например, лецитин. Дозировкой лекарственного средства можно управлять при помощи установки дозирующего клапана.
В качестве варианта активные ингредиенты могут быть предоставлены в форме сухого порошка, например, порошковой смеси соединения в подходящей порошковой основе, такой как лактоза, крахмал, производные крахмала, такие как гидроксипропилметилцеллюлоза и поливинилпирролидон (ПВП). В носовой полости порошковый носитель обычно преобразуется в гель. Порошковый состав может быть представлен в форме, содержащей единичные дозы, например, в капсулах или картриджах, например, из желатина, или в блистерных упаковках, из которых порошок может быть введен посредством ингалятора.
В составах, предназначенных для введения в дыхательные пути, включая интраназальные составы, частицы соединения, как правило, имеют небольшие размеры, например, порядка 5 микрон или менее. Такой размер частиц может быть получен с помощью средств, известных в данной области техники, например, микронизации.
При необходимости можно применять составы, адаптированные для замедленного высвобождения активного ингредиента.
Предпочтительнее выпускать фармацевтические препараты в формах, содержащих единичные дозы. В такой форме препарат разделен на единичные дозы, содержащие необходимые количества активного компонента. Единичная дозированная форма может представлять собой упакованный препарат, причем упаковка содержит дискретные количества препарата, такие как упакованные таблетки, капсулы и порошки во флаконах или ампулах. Также единичная дозированная форма сама может представлять собой капсулу, таблетку, крахмальную капсулу или леденец для рассасывания, или она может представлять собой соответствующее количество любых из них в упакованной форме. Предпочтительными формами являются таблетки или капсулы для перорального введения и жидкости для внутривенного введения и непрерывной инфузии.
Дополнительные сведения о способах приготовления и введения приведены в последнем издании Remington's Pharmaceutical Sciences (Maack Publishing Co., г. Истон, шт. Пенсильвания) и Remington: science и practice of pharmacy», Lippincott Williams and Wilkins.
Соответствующие формулы и способы производства, например, также раскрыты в публикации «Arzneiformenlehre, Paul Heinz List, Ein Lehrbuch  Pharmazeuten, Wissenschaftliche Verlagsgesellschaft Stuttgart, 4. Auflage, 1985», или «The theory and practice of industrial pharmacy» авторов Lachman et al., Varghese Publishing House, 1987, или «Modern Pharmaceutics», под редакцией James Swarbrick, 2-е издание.
Pharmazeuten, Wissenschaftliche Verlagsgesellschaft Stuttgart, 4. Auflage, 1985», или «The theory and practice of industrial pharmacy» авторов Lachman et al., Varghese Publishing House, 1987, или «Modern Pharmaceutics», под редакцией James Swarbrick, 2-е издание.
Краткое описание чертежей и таблиц
Далее приведены ссылки на таблицы и сопроводительные чертежи.
на Фиг. 1 изображен противовоспалительный эффект соединения 211 в модели воспаления дыхательных путей, индуцированных овальбумином, у серых крыс;
в Таблице 1 приведены результаты исследования ингибирующей активности ИК50 (мкМ) 5-ЛОГ, измеренной с помощью флуоресцентного анализа, при этом номер соединения относится к соединениям, перечисленным в примере 7;
в Таблице 2 приведены результаты исследования ингибирующей активности ИК50 (мкМ) 5-ЛОГ, измеренной с помощью фермент-связанного иммуносорбентного исследования (ELISA), при этом номер соединения относится к соединениям, перечисленным в примере 7;
в Таблице 3 приведены результаты исследовании секреции ЛТВ4 (ЭК50, мкМ) в клетках БКК (базофильных клетках крыс), при этом номер соединения относится к соединениям, перечисленным в примере 7;
в Таблице 4 приведены результаты исследования секреции ЛТВ4 (ЭК50, мкМ) в ЦКК (цельной крови крыс), при этом номер соединения относится к соединениям, перечисленным в примере 7;
в Таблице 5 приведены результаты исследования ингибирования ПГЕ2 (в %) при концентрации 10 мкМ, измеренного с помощью ГФВР (гомогенной флуоресценции с временным разрешением), при этом номер соединения относится к соединениям, перечисленным в примере 7;
в Таблице 6 приведены результаты исследования ингибирования ЦОГ-2 (циклооксигеназы 2) (в %) при концентрации 10 мкМ, измеренного с помощью ИФА (иммуноферментный анализ), при этом номер соединения относится к соединениям, перечисленным в примере 7 и таблице 7;
в Таблице 7 приведены характеристики соединений 1-556, синтез которых описан в примере 7.
Осуществление изобретения
Настоящее изобретение далее описано со ссылкой на следующие примеры, которые имеют иллюстративную цель, но не ограничивают объем настоящего изобретения.
Пример 1: Активность соединений против фермента 5-ЛОГ
Активность соединения против 5-ЛОГ была определена путем измерения уровней ЛТВ4 (лейкотриена В4) и/или флуоресцентным методом. Оба подхода более подробно описаны ниже;
Измерение ЛТВ4 с помощью ELISA:
Человеческую 5-липоксигеназу (5-ЛОГ) (Cayman, кат. №60402), продуцированную в клетках насекомых, предварительно инкубировали с соединениями в течение 5 минут при комнатной температуре в инкубационном буфере (50 мМ трис-Cl, pH 7,4, 2 мМ CaCl2, 0,1 мМ АТФ, 2% ДМСО). Соединения тестировали на дозозависимый эффект от 0,5 нМ до 10 мкМ. Запускали ферментативную реакцию путем добавления арахидоновой кислоты до конечной концентрации 3 мкМ. После 5 минут инкубации при температуре +25°C реакцию останавливали добавлением H2O2 до конечной концентрации 1 мМ. Уровни ЛТВ4 выражали количественно при помощи набора для определения ЛТВ4 методом ФИА (Cayman, кат. №520111) согласно инструкциям.
Флуоресцентный метод:
Помимо вышеуказанного способа количественного выражения уровней ЛТВ4, выполняли флуоресцентный анализ для измерения 5-гидропероксиэйкозатетраеновой кислоты (5-ГПЭТЕ) в целях высокопроизводительного скрининга в 384-луночных микротитрационных планшетах (Pufahl et al., 2007) Development of Fluorescence-based enzyme assay of human 5-lipoxygenase. ANALYTICAL BIOCHEMISTRY 364, 204-212. Для неспецифического сложноэфирного расщепления ацетатных групп в H2DCFDA (2',7'-дихлордигидрофлуоресцеин диацетат), лизат клеток насекомых, экспрессирующих человеческую 5-ЛОГ (Cayman, кат. №60402) инкубировали с H2DCFDA (50 мМ трис-Cl, pH 7,5, 2 мМ CaCl2, 20 мкМ H2DCFDA, 600 миллиединиц 5-ЛОГ на реакцию) в течение 5 минут. Соединение, имеющее концентрацию для проверки эффективности дозы (от 0,5 нМ до 10 мкМ), и смесь ферментов предварительно инкубировали в течение 5 минут, а ферментативную реакцию инициировали путем добавления АТФ и арахидоновой кислоты до конечной концентрации 100 мкМ и 3 мкМ соответственно. После 5 минут инкубации измеряли флуоресценцию с помощью молекулярного устройства Spectramax М5 (Ex/Em=485 нм/530 нм). Все этапы выполняли при комнатной температуре.
Пример 2: Исследование секреции ЛТВ4 в БКК (базофильные клетки крыс)
Базофильные клетки крыс (БКК) (АТСС, кат. № CRL-2256) выдерживали в минимальной питательной среде Игла (ЕМЕМ, АТСС, кат. №30-2003) с добавлением 15% фетальной телячьей сыворотки (FBS). За день до проверки БКК клетки высеивали при концентрации 2 X 104 клеток на лунку в 96-луночные планшеты. Среду заменили на 100 мкл минимальной питательной среды Игла с добавлением 0,5% FBS и соединения с концентрацией для проверки эффективности дозы (от 5 нМ до 100 мкМ). После 15 минут предварительной инкубации при температуре +37°C добавляли арахидоновую кислоту и кальций-ионофор (А23187) до получения конечной концентрации 2,5 мкМ и 5 мкМ соответственно. После следующих 10 минут инкубации при температуре +37°C культурный супернатант переносили, а затем количественно измеряли ЛТВ4 с помощью набора для определения лейкотриена В4 методом ФИА (Cayman, кат. №520111) согласно инструкциям.
Пример 3: Исследование секреции ЛТВ4 в ЦКК (цельной крови крыс)
Крысиную кровь отбирали из каудальной полой вены у мужской особи крысы линии Спрег-Доули (6-9 недель) в пробирке, покрытой гепарином натрия (Greiner bio-one, кат. №455051). Кровь смешивали и разбавляли RPMI-раствором (WelGENE, кат. № LM 011-05) до получения соотношения 1:1. Разбавленную кровь разделяли на аликвоты в 96-луночном планшете (200 мкл на лунку), затем добавляли соединения с концентрацией для проверки эффективности дозы до достижения нужного диапазона концентраций от 2,5 нМ до 50 мкМ. После 15 минут предварительной инкубации при температуре +37°C добавляли кальций-ионофор (А23187) до конечной концентрации 10 мкМ. После 10 минут дальнейшей инкубации при температуре +37°C выработку ЛТВ4 останавливали путем разбавления реакционной смеси охлажденного льдом ССБ, (разбавление 1:4). Клетки удаляли центрифугированием при 1000Xg, +4°C в течение 10 минут, переносили супернатант, а затем определяли количество ЛТВ4 с помощью набора для определения лейкотриена В4 методом ФИА (Cayman, кат. №520111) согласно инструкциям.
Пример 4: Воспалительная эффективность в модели воспаления дыхательных путей, индуцированных овальбумином, у серых крыс
Целью данного исследования является определение эффективности соединения в легких индуцированных овальбумином (OA) серых крыс. Как и в большинстве моделей индуцированной аллергической астмы, животных систематически делали более чувствительными к специфическим антигенам, а затем по истечении определенного периода времени подвергали воздействию того же аллергена, введенного воздушным путем. Для сенсибилизации и провокации животных в рамках настоящего исследования в качестве стандартного аллергена использовали овальбумин.
Тридцать (30) мужских особей серых крыс разделяли случайным образом на 5 групп по весу тела. Для сенсибилизации 1% овальбумин в натрий-фосфатном буферном растворе смешивали с раствором квасцов (с объемным соотношением 1:9). Все крысы в группах 1-5 были сенсибилизированы путем внутрибрюшинного введения (1 мл/крыса) 1% сенсибилизирующего раствора овальбумина и квасцов на 1, 2 и 3 дни. На 21 день крыс в группах 1-5 аллергизировали 1% овальбумином в натрий-фосфатном буферном растворе с помощью аэрозольной системы дозирования в течение 20 минут.
Тестируемые соединения вводили перорально на 19-21 день исследования согласно следующему графику:
Группа 1: сенсибилизированный носитель,
Группа 2: контрольный препарат дексаметазон, 0,3 мг/кг, перорально, 19-21 день, 2 раза в день,
Группа 3: соединение 211, 25 мг/кг, перорально, 19-21 день, 2 раза в день,
Группа 4: соединение 211, 50 мг/кг, перорально, 19-21 день, 2 раза в день,
Группа 5: соединение 211, 100 мг/кг, перорально, 19-21 день, 2 раза в день,
На 22 день отбирали бронхоальвеолярный лаваж и легочную ткань. Общее количество клеток и результаты дифференцированного подсчета клеток в жидкости бронхоальвеолярного лаважа, лейкотриена В4 (ЛТВ4) в жидкости бронхоальвеолярного лаважа и легочной ткани, гистопатологии легких оценивали путем окрашивания гематоксилином и эозином.
Результаты представлены на Фиг. 1. Лечение крыс с использованием соединения 211 значительно снизило рост воспалительных клеток, включая общие клетки и эозинофилы, снизило количество ЛТВ4 в жидкости бронхоальвеолярного лаважа и легких, а также значительно снизило инфильтрацию воспалительных клеток. Эти данные свидетельствуют о том, что соединение 211 демонстрирует противовоспалительную активность при астме, индуцированной овальбумином.
Пример 5: Исследование ингибирования ПГЕ2
Клетки эпителиоидного рака шейки матки человека HeLa S3 (2X106 клеток/мл) высеивали с модифицированной буферной средой F-12K с pH 7,4 в планшет для тканевых культур и инкубировали в течение ночи при температуре +37°C и 5% CO2. Затем среду заменили на новую среду, содержащую 10 мкМ тестируемого соединения или 0,1% ДМСО, и инкубировали в течение ночи. После инкубации клетки стимулировали при помощи 30 нМ ФНО-α (фактор некроза опухолей-α) в течение 16 часов. Для определения ПГЕ2 анализировали супернатант с помощью набора реактивов для ПГЕ2 методом ГФВР (гомогенная флуоресценция с временным разрешением). Ингибирование ПГЕ2 соединением рассчитывали в виде процента активности в присутствии препарата по сравнению с активностью в стимуляторе ФНО-α.
Пример 6: Исследование ингибирования ЦОГ-2
Использовали рекомбинантую циклооксигеназу-2 человека, экспрессированную в клетках насекомых Sf21. Тестируемое соединение и контрольное соединение (Rofecoxib) растворяли в ДМСО (Sigma, США) и тестировали при 10 мкМ. Пробы предварительно инкубировали с помощью 0,11 единиц фермента в 100 мМ модифицированного буфера трис-HCl с pH 7,7 в течение 15 минут при температуре +37°C. Реакции инициировали добавлением 0,3 мкМ арахидоновой кислоты в течение следующего 5-минутного инкубационного периода, затем прекращали их последующим добавлением 1N HCl. Затем аликвоту объединяли с набором для спектрофотометрического определения количества образовавшегося ПГЕ2 методом ФИА. Процент ингибирования определяли путем сравнения с относительными количествами ПГЕ2, индуцированными арахидоновой кислотой. Полученные результаты показали, что соединения по настоящему изобретению, в частности, соединения 90 и 95, не оказали ингибирующего воздействия на циклооксигеназу-2 (ЦОГ-2), тем самым подтверждая, что их ингибирующее действие на пути ПГЕС происходило на уровне «ПГЕС».
Пример 7: Приготовление соединений
Синтез соединений, описанных в настоящем изобретении, может быть выполнен с помощью средств, описанных в химической литературе, и/или методик, описанных в настоящем документе.
Соединения, описанные в настоящем документе, могут быть синтезированы с помощью стандартных синтетических методик, известных специалистам в данной области техники, или с помощью методов, известных специалистам в данной области техники, в сочетании с методами, описанными в настоящем документе. При этом растворители, температуры и другие условия реакции, указанные в настоящем документе, могут отличаться от известных из уровня техники. Исходный материал, использованный для синтеза соединений, описанных в настоящем документе, может быть синтезирован или может быть получен из коммерческих источников, включая, помимо прочего, Aldrich Chemical Со. (г. Милуоки, шт. Висконсин), или Sigma Chemical Со. (г. Сент-Луис, шт. Миссури). Соединения, описанные в настоящем документе, и другие соответствующие соединения с другими заместителями могут быть синтезированы с помощью технологий и материалов, описанных в настоящем документе и известных специалистам в данной области техники. Общие способы приготовления соединения, раскрытые в настоящем документе, могут быть выведены из известных реакций в данной области техники, при этом реакции могут быть модифицированы использованием пригодных реагентов и условий, очевидных для специалистов в данной области техники, с целью введения различных остатков из описанных формул. В качестве руководства для получения описанного соединения можно использовать синтетический метод согласно нижеприведенным Схемам.
Соединения (каркас I, II и III; см. таблицы 1-7), полученные согласно приведенным ниже способам (схемы 1-37), и синтезированные соединения, а также соответствующие характеристические данные ЯМР приведены в таблице 7. Полученные производные были проверены на ингибиторную активность (ИК50, ЭК50, in vivo), используя исследования, описанные выше (примеры 1, 2, 3, 4, 5 и 6), а полученные результаты были обобщены в таблицах 1, 2, 3, 4, 5 и 6 и на Фиг. 1.
Схема 1. Общий синтез 1 аминосоединения

Общая процедура синтеза А1
К перемешиваемому раствору 4-фторбензонитрила (5,00 г, 41,3 ммоль) и 4-трифторметокси-фенола (8,10 г, 45,5 ммоль) в ДМА (30 мл) добавляли K2CO3 (6,30 г, 45,6 ммоль). Полученную смесь перемешивали при температуре 120°C в течение 16 часов. Смесь охлаждали до 50°C и по каплям добавляли 100 мл воды при сильном перемешивании. Смесь экстрагировали с помощью этилацетата (100 мл × 2), объединенную органическую фазу промывали водой (100 мл) и солевым раствором (100 мл), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением А1.
Общая процедура для синтеза А2
К перемешиваемому раствору А1 (3,00 г, 10,7 ммоль) в безводном ТГФ (30 мл) при температуре 0°C частями добавляли LiAlH4 (1,63 г, 43,0 ммоль), реакционную смесь перемешивали при температуре 0°C в течение 3 часов, полученную смесь перемешивали при температуре 50°C в течение следующих 16 часов. Смесь охлаждали водой (1,6 мл) по каплям при температуре 0°C, с последующим добавлением в смесь NaOH (10%, 3,2 мл) по каплям и воды (1,6 мл). Смесь фильтровали, фильтрационный кек промывали с этилацетатом (30 мл), а фильтрат концентрировали при пониженном давлении с получением А2.
Схема 2. Общий синтез 2 аминосоединения

Общая процедура для синтеза В1
К перемешиваемому раствору 2-(4-гидроксифенил)ацетонитрила (10,0 г, 75,2 ммоль) в ДМА (120 мл) добавляли 2,5-дихлорпиридин (12,2 г, 82,7 ммоль), K2CO3 (15,6 г, 113 ммоль) и ТБАФ (589 мг, 2,26 ммоль), затем смесь перемешивали при температуре 115°C в течение 16 часов. Реакционную смесь вливали в воду (600 мл), энергично перемешивая в течение 1 часа. Осадок собирали фильтрацией, высушивали на воздухе в течение 2 часов и очищали с помощью Combi Flash (PE : EtOAc = от 5:1 до 2:1) с получением В1.
Общая процедура для синтеза В2
К перемешиваемому раствору В1 (500 мг, 2,04 ммоль) в МеОН (15 мл) добавляли никель Ренея (400 мг) и NH3.H2O (0,5 мл, 28%). Реакционный раствор перемешивали при температуре 15°C в атмосфере Н2 (45 psi) в течение 6 часов. Затем раствор фильтровали через подушку из целита. Фильтрат концентрировали и разбавляли ДХМ (30 мл), затем высушивали над безводным Na2SO4 и выпаривали с получением В2.
Схема 3. Общий синтез 3 аминосоединения

Общая процедура для синтеза С1
Смесь 4'-гидроксиацетофенона (5,00 г, 36,7 ммоль), 5-хлор-2-фторпиридина (5,79 г, 44,0 ммоль) и K2CO3 (10,1 г, 73,4 ммоль) в ДМФА (150 мл) перемешивали при температуре 100°C в течение 6 часов. После охлаждения до комнатной температуры реакционную смесь разбавляли этилацетатом (200 мл) и фильтровали, а фильтрат промывали солевым раствором (150 мл × 3), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением осадка. Осадок промывали с помощью РЕ/EtOAc = 20/1 (40 мл) с получением С1.
Общая процедура для синтеза С2
Смесь С2 (4,50 г, 18,2 ммоль) и NH2OH.HCl (1,26 г, 18,2 ммоль) в МеОН/пиридине (25 мл/25 мл) перемешивали при температуре 8°C - 10°C в течение 16 часов. Полученную смесь концентрировали при пониженном давлении, подкисляли HCl (2 М, 120 мл), экстрагировали с помощью ДХМ (50 мл × 4), объединенный органический слой промывали водой (200 мл × 3), солевым раствором (200 мл), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением С2.
Общая процедура для синтеза С3
Смесь С2 (4,70 г, 17,9 ммоль) и порошкового цинка (11,6 г, 179 ммоль) в АсОН (120 мл) перемешивали при температуре 65°C - 75°C в течение 28 часов. После охлаждения до комнатной температуры реакционную смесь фильтровали, промывали этилацетатом (10 мл × 3), а объединенный фильтрат концентрировали при пониженном давлении с получением осадка. Осадок разбавляли насыщенным NaHCO3 (150 мл), экстрагировали этилацетатом (100 мл × 3), объединенный органический слой промывали солевым раствором (200 мл × 2), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением С3.
Схема 4. Общий синтез 4 аминосоединения

Общая процедура для синтеза D1
В перемешиваемую смесь 5-хлор-2-гидроксипиридина (15,0 г, 76,5 ммоль) в безводном ТГФ (200 мл) добавляли 4-цианобензил бромид (8,26 г, 63,8 ммоль) и Ag2CO3 (10,5 г, 38,3 ммоль). Полученную смесь дефлегмировали в течение 16 часов. Смесь охлаждали до комнатной температуры и фильтровали. Фильтрационный кек промывали ТГФ (100 мл). Объединенный фильтрат концентрировали при пониженном давлении с получением осадка, который очищали с помощью Combi Flash (РЕ/EtOAc = от 92/8 до 70/30) с получением D1.
Общая процедура для синтеза D2
К перемешиваемому раствору D1 (5,00 г, 2,04 ммоль) в МеОН (100 мл) добавляли никель Ренея (1,00 г) и NH3.H2O (0,5 мл, 28%) в атмосфере N2. Суспензию дегазировали при вакуумировании и продували Н2 три раза. Раствор перемешивали при температуре 12°C в атмосфере Н2 (45 psi) в течение 2 часов. Смесь фильтровали через подушку из целита, а подушку промывали МеОН (200 мл). Фильтрат концентрировали при пониженном давлении с получением D2.
Схема 5. Общий синтез 5 аминосоединения

Общая процедура для синтеза Е1
К перемешиваемому раствору циклопентанкарбоновой кислоты (5,00 г, 44,2 ммоль) в безводном ДХМ (100 мл) частями добавляли КДИ (8,10 г, 57,5 ммоль) при температуре 28°C. Реакционный раствор перемешивали при температуре 28°C в течение 0,5 часа. Затем добавляли ДИЭА (8,00 г, 61,9 ммоль) и N,O-диметил-гидроксиламингидрохлорид (5,13 г, 61,9 ммоль). Реакционный раствор перемешивали при температуре 28°C в течение 16 часов. Реакционный раствор разбавляли насыщенным NaHCO3 (50 мл) и отделяли. Органический слой высушивали над безводным Na2SO4 и концентрировали при пониженном давлении. Осадок очищали с помощью Combi Flash (РЕ : EtOAc = от 5:1 до 2:1) с получением Е1.
Общая процедура для синтеза Е2
К перемешиваемому раствору 4-бромбензонитрила (1,40 г, 7,69 ммоль) в безводном ТГФ (15 мл) по каплям добавляли i-PrMgBr (19,2 мл, 19,2 ммоль, 1 М в ТГФ) при температуре -30°C в атмосфере N2. Затем смесь перемешивали при температуре -30°C в атмосфере N2 в течение 30 минут. Раствор Е1 (1,00 г, 6,37 ммоль) в безводном ТГФ (5 мл) добавляли по каплям при температуре -30°C. Реакционному раствору давали нагреться до 30°C и перемешивали его при температуре 30°C в течение 16 часов. Реакционный раствор охлаждали насыщенным раствором NH4Cl (15 мл) при температуре 0°C. Органическую фазу отделяли и концентрировали при пониженном давлении. Осадок очищали с помощью Combi Flash (РЕ : EtOAc = 10:1) с получением Е2.
Общая процедура для синтеза Е3
Смесь Е2 (250 мг, 1,26 ммоль), BF3.Et2O (0,5 мл), этиленгликоля (1 мл) в толуоле (50 мл) дефлегмировали с азеотропным удалением воды в течение 48 часов. Реакционный раствор охлаждали до комнатной температуры и разбавляли насыщенным Na2CO3 (30 мл). Органический слой отделяли, высушивали над безводным Na2SO4 и концентрировали при пониженном давлении. Осадок очищали с помощью препаративной ТСХ (РЕ : EtOAc = 5:1) с получением Е3.
Общая процедура для синтеза Е4
К перемешиваемому раствору Е3 (100 мг, 0,410 ммоль) в МеОН (5 мл) добавляли никель Ренея (100 мг) и аммиачную воду (0,5 мл, 28%). Реакционный раствор перемешивали при температуре 25°C в атмосфере Н2 (45 psi) в течение 2 часов. Затем раствор фильтровали через подушку из целита. Фильтрат концентрировали при пониженном давлении с получением Е4.
Схема 6. Общий синтез 6 аминосоединения
Общая процедура для синтеза F1
К перемешиваемому раствору 4-хлоранилина (634 мг, 4,96 ммоль) и пиридина (1,17 г, 14,9 ммоль) в ДХМ (10 мл) добавляли раствор 4-цианобензолсульфонилхлорида (1,00 г, 4,96 ммоль) в ДХМ (5 мл) на ледяной бане, затем смесь перемешивали в атмосфере N2 при температуре 25°C в течение 12 часов. Смесь вливали в воду (30 мл), экстрагировали ДХМ (10 мл × 3). Объединенные экстракты высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением остатка, который очищали с помощью колоночной флэш-хроматографии (элюент : ДХМ) с получением F1.
Общая процедура для синтеза F2
К перемешиваемому раствору F2 (500 мг, 1,71 ммоль) в безводном ТГФ (2,5 мл) добавляли BH3/Me2S (1,35 мл, 13,5 ммоль, 10 М) на ледяной бане, смесь перемешивали в атмосфере N2 при температуре 25°C в течение 12 часов. Осторожно по каплям добавляли МеОН (5 мл) для охлаждения реакции, затем смесь разбавляли водой (50 мл) регулировали pH до 8 насыщенным Na2CO3, экстрагировали этилацетатом (30 мл × 3). Объединенный экстракт высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением остатка, который очищали с помощью Combi Flash (элюент : РЕ/EtOAc = от 1/1 до EtOAc) с получением F2.
Схема 7. Общий синтез 7 аминосоединения
Общая процедура для синтеза G1
К перемешиваемому раствору 4-бромбензонитрила (1,00 г, 5,50 ммоль), LiCl (252 мг, 6,00 ммоль) в безводном ТГФ (10 мл) по каплям добавляли i-PrMgCl (3 мл, 6,0 ммоль, 2 M в ТГФ) при температуре -15°C в атмосфере N2. Затем смесь перемешивали при температуре -15°C в атмосфере N2 в течение 2 часов. Затем по каплям добавляли пиколинальдегид (640 мг, 6,00 ммоль) при температуре -15°C. Реакционному раствору давали нагреться до 30°C. Тонкослойная хроматография показала, что реакция завершена. Реакционный раствор охлаждали насыщенным раствором NH4Cl (10 мл) при температуре 0°C. Органический слой отделяли и концентрировали при пониженном давлении. Осадок очищали с помощью Combi Flash (РЕ : EtOAc = от 5:1 до 2:1) с получением G1.
Общая процедура для синтеза G2
К перемешиваемому раствору G2 (400 мг, 1,90 ммоль) в безводном ТГФ (6 мл) добавляли NaH (298 мг, 2,10 ммоль, 60% дисперсия в минеральном масле) при температуре 0°C, затем смесь перемешивали при температуре 0°C в течение 15 минут. Затем в смесь добавляли MeI (298 мг, 2,10 ммоль) при температуре 0°C. Смесь перемешивали в атмосфере N2 при температуре 25°C в течение 1 часа. Реакционную смесь вливали в воду (10 мл), экстрагировали этилацетатом (10 мл × 3). Объединенный органический слой промывали солевым раствором (10 мл), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали с помощью Combi Flash (PE : EtOAc = от 5:1 до 1:1) с получением G2.
Общая процедура для синтеза G3
К перемешиваемому раствору G2 (1,50 г, 6,70 ммоль) в МеОН (40 мл) добавляли никель Ренея (2,0 г) и концентрированную аммиачную воду (4 мл, 28%). Реакционный раствор перемешивали при температуре 25°C в атмосфере Н2 (45 psi) в течение 3 часов. Затем раствор фильтровали через подушку из целита. Фильтрат концентрировали при пониженном давлении с получением 1,53 г остатка, который очищали колоночной хроматографией с силикагелем (ДХМ : МеОН = 10:1) с получением G3.
Схема 8. Общий синтез 8 аминосоединения

Общая процедура для синтеза H1
К перемешиваемому раствору 2-амино-1-фенилэтанола (5,00 г, 36,4 ммоль) и ТЭА (5,51 г, 54,6 ммоль) в ДХМ (100 мл) добавляли Boc2O (9,54 г, 43,7 ммоль), затем полученную смесь перемешивали при температуре 27°C - 28°C в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении для удаления большей части ДХМ. Остаток разбавляли этилацетатом (100 мл), затем промывали 1% раствором HCl (50 мл), солевым раствором (50 мл × 2), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток промывали РЕ/EtOAc (50 мл/5 мл) с получением H1.
Общая процедура для синтеза Н2
К перемешиваемому раствору H1 (4,00 г, 16,9 ммоль), 4-хлорфенола (4,33 г, 33,7 ммоль) и PPh3 (6,63 г, 25,3 ммоль) в безводном ТГФ (150 мл) по каплям добавляли раствор ДЭАД (4,40 г, 25,3 ммоль) в ТГФ (50 мл) при температуре 0°C - 5°C. Полученную смесь перемешивали при температуре 26°C - 30°C в течение 16 часов. Реакционную смесь разбавляли этилацетатом (500 мл), затем промывали солевым раствором (300 мл × 3), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали колоночной флэш-хроматографией (PE/EtOAc = от 20/1 до 5/1), разбавляли МТБЭ (100 мл), затем промывали 12 М раствором NaOH (50 мл × 3), солевым раствором (50 мл × 3), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением Н2.
Общая процедура для синтеза Н3
Смесь Н2 (2,00 г, 5,75 ммоль) в HCl/диоксане (30 мл, 4,0 М) перемешивали при температуре 27°C в течение 20 минут. Реакционную смесь концентрировали при пониженном давлении с получением Н3.
Схема 9. Общий синтез 9 аминосоединения

Общая процедура для синтеза I1
К перемешиваемому раствору 1-Вос-пирролидин-3-карбоксиловой кислоты (5,00 г, 23,3 ммоль) и 4-(трифторметокси)анилина (3,91 г, 22,1 ммоль) в безводном ДМФА (50 мл) добавляли ДИПЭА (9,0 г, 69,8 ммоль). Затем порциями добавляли HATU (13,2 г, 34,7 ммоль) при температуре 0°C. Реакционную смесь перемешивали при температуре 25°C в течение 4 часов. Смесь охлаждали водой (100 мл), экстрагировали этилацетатом (100 мл × 2). Объединенную органическую фазу промывали насыщенным NaHCO3 (100 мл), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением I1.
Общая процедура для синтеза I2
К перемешиваемому раствору I1 (6,00 г, 16,0 ммоль) в ДХМ (120 мл) по каплям добавляли ТФА (40 мл) при температуре 0°C. После добавления смесь перемешивали при температуре 25°C в течение 12 часов. Смесь концентрировали при пониженном давлении, а остаток разбавляли 100 мл воды. Твердый K2CO3 добавляли до pH 10 и экстрагировали этилацетатом (200 мл × 2). Объединенную органическую фазу высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением I2.
Схема 10. Общий синтез 10 аминосоединения

Общая процедура для синтеза J1
К перемешиваемому раствору трет-бутил-3-гидроксипирролидин-1-карбоксилата (10,0 г, 53,4 ммоль) в ТГФ (50 мл) по каплям добавляли водный раствор NaOH (5,30 г/134 ммоль в 5 мл воды) на ледяной бане (ниже 10°C), затем TosCl (13,2 г, 68,4 ммоль) добавляли порциями при температуре ниже 10°C. Затем смесь нагревали при температуре 30°C в течение 12 часов. ТГФ концентрировали при пониженном давлении с получением остатка, остаток разбавляли водой (100 мл), экстрагировали этилацетатом (50 мл × 3). Объединенный экстракт промывали водой (50 мл × 2), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением J1.
Общая процедура для синтеза J2
Смесь соединения 2 (5,00 г, 14,6 ммоль), 4-(трифторметокси)фенола (2,73 г, 15,3 ммоль) и Cs2CO3 (9,50 г, 29,2 ммоль) в ДМФА (25 мл) перемешивали при температуре 80°C - 90°C в течение 2 часов. Смесь разбавляли водой (150 мл), экстрагировали этилацетатом (50 мл × 4). Объединенный экстракт высушивали над безводным Na2SO4 и концентрировали с получением сырого продукта, который очищали колоночной флэш-хроматографией (элюент : EtOAc/РЕ = от 1/100 до 1/5) с получением J2
Общая процедура для синтеза J3
К перемешиваемому раствору J2 (3,80 г, 10,9 ммоль) в МеОН (20 мл) добавляли водный раствор HCl (5 мл, 12 М) при температуре 10°C, смесь перемешивали при температуре 10°C в течение 2 часов. Смесь концентрировали при пониженном давлении с получением остатка. Остаток разбавляли водой (50 мл), водным раствором NaOH (2 М) доподили до pH 9, экстрагировали этилацетатом (30 мл × 3). Объединенный экстракт высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением J3.
Схема 11. Общий синтез 11 аминосоединения

Общая процедура для синтеза K1
К перемешиваемому раствору 4-(4-(трифторметокси) фенил)пиперидина (86,0 г, 0,351 моль) в ДМСО (1 л) добавляли K2CO3 (121 г, 0,878 моль) и 4-фторбензонитрил (63,8 г, 0,527 моль) при температуре 18°C. Реакционную смесь нагревали до 120°C в течение 16 часов в атмосфере N2. После охлаждения до комнатной температуры смесь концентрировали примерно до 400 мл под высоким вакуумом. H2O (2 л) вливали в смесь, а полученный осадок собирали фильтрацией. Сырой продукт растирали в порошок с H2O (1,5 л), высушивали под высоким вакуумом, затем растирали в порошок с EtOAc/PE (800 мл/4 л), высушивали под высоким вакуумом с получением K1.
Общая процедура для синтеза K2
К перемешиваемому раствору K1 (60,0 г, 0,173 моль) в безводном ТГФ (1,0 л) добавляли LiAlH4 (32,9 г, 0,865 моль) при температуре 0°C. Реакционную смесь нагревали до температуры рефлюкса в течение 3 часов в атмосфере N2. После охлаждения до комнатной температуры смесь осторожно охлаждали последовательным добавлением H2O (33 мл), 10% раствора NaOH (33 мл), H2O (100 мл). Смесь фильтровали, фильтрационный кек промывали ТГФ (300 мл), а фильтрат концентрировали под высоким вакуумом с получением K2.
Схема 12. Общий синтез алкилгалида
Общая процедура для синтеза L1
К перемешиваемому раствору 4-(трифторметокси)анилина (2,00 г, 17,7 ммоль), ДИПЭА (2,51 г, 19,3 ммоль) в ДХМ (80 мл) по каплям добавляли раствор хлорангидрида хлоруксусной кислоты (2,00 г, 17,7 ммоль) в ДХМ (8 мл) при температуре 0°C. Реакционную смесь перемешивали при температуре 0°C в течение 0,5 часа и добавляли в воду (50 мл). Смесь отделяли, а органический слой промывали водой (20 мл), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали рекристаллизацией из РЕ/EtOAc (1/1) с получением L1.
Схема 13. Общий синтез 1 арилальдегида

Общая процедура для синтеза M1
К перемешиваемому раствору 2-хлор-5-гидроксипиридина (3,00 г, 23,3 ммоль) в безводном ДМА (40 мл) добавляли 4-фторбензальдегид (2,88 г, 23,3 ммоль) и K2CO3 (6,40 г, 46,6 ммоль). Полученную смесь нагревали до 130°C в течение 16 часов. Воду (30 мл) вливали в смесь, затем экстрагировали этилацетатом (40 мл × 3). Объединенную органическую фазу промывали солевым раствором (40 мл × 2), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением сырого продукта. Осадок очищали с помощью Combi Flash (РЕ : EtOAc = от 20:1 до 15:1) с получением M1.
Схема 14. Общий синтез 2 арилальдегида

Общая процедура для синтеза N1
К перемешиваемому раствору 1-(4-бромфенил)этан-1-ола (30,0 г, 150 ммоль) в ДМФА (18 мл) добавляли ДМФА-ДМА (43,5 мл, 300 ммоль), полученную смесь перемешивали при температуре 110°C в течение 4 часов. После охлаждения до комнатной температуры в смесь добавляли МТБЭ (150 мл), затем в вышеуказанную смесь добавляли MeNHNH2 (78,9 г, 600 ммоль), полученную смесь перемешивали при температуре 25°C в течение 17 часов. Смесь разбавляли этилацетатом (100 мл), фильтровали, а фильтрационный кек промывали этилацетатом (150 мл). Объединенный органический слой промывали водой (200 мл × 3) и солевым раствором (200 мл), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением остатка. Остаток очищали колоночной флэш-хроматографией (элюент : РЕ/EtOAc = от 8/1 до 4/1) с получением N1.
Общая процедура для синтеза N2
К перемешиваемому раствору N1 (1,20 г, 5,10 ммоль) в безводном ТГФ (10 мл) добавляли NaH (240 мг, 6,00 ммоль, 60% дисперсия в минеральном масле), полученную смесь перемешивали при температуре 0°C в течение 1 часа, затем н-BuLi (2,3 мл, 5,75 ммоль, 2,5 М в гексане) добавляли в вышеуказанную смесь при температуре -70°C и перемешивали при температуре -70°C в течение 1 часа, в вышеуказанную смесь добавляли свежеразбавленный ДМФА (1,85 г, 25,3 ммоль) и перемешивали при температуре -70°C в течение 1 часа, затем реакционной смеси давали нагреться до 0°C. Тонкослойная хроматография показала, что реакция завершилась. Реакцию охлаждали водой (30 мл) при температуре 0°C, экстрагировали этилацетатом (30 мл × 3). Объединенную органическую фазу промывали солевым раствором (50 мл), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением остатка. Остаток очищали колоночной флэш-хроматографией (элюент : РЕ/EtOAc = 2/1) с получением N2.
Схема 15. Общий синтез 3 арилальдегида

Общая процедура для синтеза O1
К перемешиваемому раствору 2-бром-6-гидроксипиридина (100 мг, 0,57 ммоль) и 4-формилфенилборовой кислоты (72 мг, 0,48 ммоль) в ДМЭ (2 мл) добавляли Pd(PPh3)4 (55 мг, 0,05 ммоль) и раствор Na2CO3 (0,63 мл, 2 М). Смесь перемешивали в атмосфере N2, осторожно поддерживая температуру рефлюкса в течение 16 часов. Смесь разбавляли водой (10 мл) и экстрагировали этилацетатом (20 мл × 3). Объединенные экстракты промывали солевым раствором (20 мл), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением сырого продукта, который очищали с помощью Combi Flash (элюент : EtOAc) с получением O1.
Схема 16. Общий синтез 4 арилальдегида

Общая процедура для синтеза Р1
К перемешиваемому раствору 4,4,4-трифтор-2-бутанола (450 мг, 3,51 ммоль) в безводном ТГФ (10 мл) добавляли 4-гидроксибензальдегида (390 мг, 3,19 ммоль), PPh3 (1,26 г, 4,28 ммоль) и ДИАД (967 мг, 4,78 ммоль). Затем смесь перемешивали при температуре 27°C - 30°C в течение 16 часов. Затем смесь разбавляли водой (50 мл), экстрагировали этилацетатом (30 мл × 2). Объединенные экстракты промывали солевым раствором (50 мл), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением неочищенного остатка, который очищали с помощью Combi Flash (РЕ/EtOAc=10/1) с получением P1.
Схема 17. Общий синтез 5 арилальдегида

Общая процедура для синтеза Q1
К перемешиваемому раствору гидрохлорида метилового сложного эфира DL-серина (11,1 г, 71,4 ммоль), MgSO4 (8,64 г, 71,4 ммоль) в безводном ТГФ (350 мл) добавляли 4-хлор-бензальдегид (10,0 г, 71,4 ммоль) и ТЭА (14,4 г, 143 ммоль), затем смесь перемешивали при температуре 25°C - 30°C в течение 12 часов. Полученную смесь фильтровали, промывали МТБЭ (100 мл × 2). Объединенный фильтрат концентрировали при пониженном давлении с получением неочищенного Q1.
Общая процедура для синтеза Q2
К перемешиваемому раствору Q1 (13,8 г, сырой) в безводном ДХМ (220 мл) добавляли BrCCl3 (16,4 мл, 166 ммоль) и ДБУ (25 мл, 166 ммоль) при температуре 0°C. Полученную смесь перемешивали при температуре 0°C в течение 2 часов, а затем при температуре 25°C - 30°C в течение 10 часов. Смесь концентрировали при пониженном давлении до сухого состояния. Остаток растворяли в EtOAc (250 мл), поочередно промывали водой (140 мл × 2) и солевым раствором (80 мл), высушивали над безводным Na2SO4 и концентрировали с получением остатка. Остаток промывали этилацетатом (20 мл) с получением Q2.
Общая процедура для синтеза Q3
К перемешиваемому раствору Q2 (3,46 г, 14,6 ммоль) в безводном ДХМ (65 мл) добавляли DIBAL-H (16,1 мл, 16,1 ммоль, 1 М в толуоле) при температуре -78°C в атмосфере N2. Полученную смесь перемешивали при температуре 0°C в течение 4 часов. Реакцию охлаждали насыщенным NH4Cl (25 мл) при температуре 0°C. Затем смесь нагревали до комнатной температуры и доводили до pH 2 с помощью HCl (1 М). Смесь экстрагировали ДХМ (50 мл × 3), объединенный органический слой высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением остатка. Остаток промывали ДХМ (15 мл) с получением Q3.
Общая процедура для синтеза Q4
Смесь Q3 (1,49 г, 7,13 ммоль) и MnO2 (6,20 г, 71,3 ммоль) в ДХМ (65 мл) перемешивали в атмосфере N2 при температуре 25°C - 30°C в течение 18 часов. Смесь фильтровали, а фильтрационный кек промывали ДХМ (10 мл × 2), фильтрат концентрировали при пониженном давлении до сухого состояния с получением Q4.
Схема 18. Общий синтез 6 арилальдегида
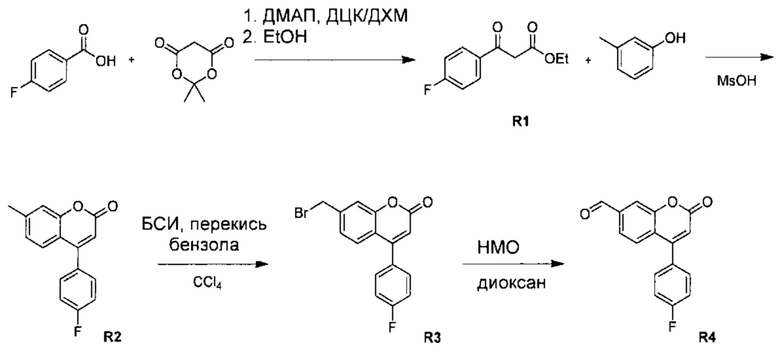
Общая процедура для синтеза R1
К перемешиваемому раствору 4-фторбензойной кислоты (50,0 г, 357 ммоль), 2,2-диметил-1,3-диоксан-4,6-диона (61,7 г, 428 ммоль) и ДМАП (65,5 г, 536 ммоль) в ДХМ (1,5 л) по каплям добавляли ДЦК (97,5 г, 464 ммоль) при температуре 0°C и перемешивали при температуре 0°C в течение 30 минут, затем перемешивали при температуре 25°C в течение 17 часов. Смесь фильтровали, фильтрат промывали водным раствором HCl (1 М, 800 мл × 3), промывали солевым раствором (800 мл), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением остатка. Остаток растворяли в EtOH (600 мл) и перемешивали при температуре 100°C в течение 17 часов. Избыток EtOH удаляли при пониженном давлении с получением остатка, который очищали колоночной флэш-хроматографией (элюент : PE/EtOAc = 20/1) с получением R1.
Общая процедура для синтеза R2
Смесь R1 (7,50 г, 35,7 ммоль) и м-крезола (3,86 г, 35,7 ммоль) в MsOH (9,5 мл) перемешивали при температуре 40°C в течение 17 часов. После охлаждения до комнатной температуры реакционную смесь вливали в охлаждающий EtOH (-30°C, 60 мл) и перемешивали при температуре -30°C в течение 1 часа. Осадок отфильтровывали. Твердое вещество растворяли в EtOAc (50 мл), а затем промывали насыщенным NaHCO3 (50 мл × 2), солевым раствором (50 мл), высушивали над Na2SO4 и концентрировали при пониженном давлении с получением R2.
Общая процедура для синтеза R3
К перемешиваемому раствору R2 (1,00 г, 3,94 ммоль) в CCl4 (30 мл) добавляли БСИ (842 мг, 4,73 ммоль) и перекись бензоила (195 мг, 0,806 ммоль), полученную смесь перемешивали при температуре рефлюкса в течение 48 часов. Затем смесь фильтровали, фильтрат концентрировали при пониженном давлении с получением остатка, который очищали колоночной флэш-хроматографией (РЕ/EtOAc = 30/1) с получением R3.
Общая процедура для синтеза R4
К перемешиваемому раствору R4 (1,00 г, 3,00 ммоль) в диоксане (15 мл) добавляли НМО (882 мг, 7,53 ммоль), затем полученную смесь перемешивали при температуре рефлюкса в течение 6 часов. После охлаждения до комнатной температуры смесь разбавляли водой (50 мл), экстрагировали этилацетатом (30 мл × 3). Объединенный органический слой промывали солевым раствором (50 мл), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением остатка, который очищали колоночной флэш-хроматографии (PE/EtOAc = 20/1) с получением R4.
Схема 19. Общий синтез 7 арилальдегида

Общая процедура для синтеза S1
К перемешиваемому раствору 4-фторанилина (360 мг, 3,24 ммоль), 4-бромбензолальдегида (500 мг, 2,70 ммоль), XantPhos (60 мг, 0,108 ммоль) и Pd2(dba)3 (25 мг, 0,027 ммоль) в безводном толуоле (15 мл) добавляли Cs2CO3 (1,32 г, 4,05 ммоль) в атмосфере N2. Реакционную смесь очищали в атмосфере N2 три раза, затем нагревали до температуры рефлюкса (масляная баня при температуре 120°C) в атмосфере N2 в течение 16 часов, в смесь вливали 40 мл воды, затем экстрагировали этилацетатом (20 мл × 2), объединенную органическую фаз высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением сырого продукта. Остаток очищали с помощью Combi Flash (РЕ : EtOAc = от 20:1 до 9:1) с получением S1.
Общая процедура для синтеза S2
К перемешиваемому раствору S1 (200 мг, 0,929 ммоль) в безводном ДМА (20 мл) добавляли NaH (74 мг, 1,86 ммоль, 60% дисперсия в минеральном масле) при температуре 0°C в течение 0,5 часа, затем добавляли этилбромацетат (186 мг, 1,12 ммоль), после 1 часа полученную смесь перемешивали при температуре 60°C в течение 16 часов. Смесь охлаждали водой (20 мл), затем экстрагировали этилацетатом (30 мл × 2). Объединенную органическую фазу промывали солевым раствором (20 мл × 2), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением S2.
Схема 20. Общий синтез производного имидазопиридина

Общая процедура для синтеза Т1
Смесь 5-бромпиридина-2, 3-диамина (5,00 г, 26,6 ммоль) в муравьиной кислоте (80 мл) нагревали до 110°C в течение 16 часов. Реакционный раствор концентрировали при пониженном давлении. Остаток разделяли между 2N NaOH (200 мл) и ДХМ (100 мл). Органическую фазу отделяли, а слой водного раствора охлаждали до 0°C в течение 1 часа. Остаток собирали фильтрацией, высушивали под высоким вакуумом с получением Т1.
Общая процедура для синтеза Т2
К перемешиваемому раствору Т1 (200 мг, 1,01 ммоль) в ДМФА (4 мл) добавляли 1-(2-бромэтокси)-4-(трифторметокси)бензол (432 мг, 1,52 ммоль) и Cs2CO3 (823 мг, 2,53 ммоль) и перемешивали при температуре 50°C в течение 16 часов. Реакционную смесь вливали в воду (200 мл) и экстрагировали этилацетатом (20 мл × 3). Объединенный органический слой промывали водой (10 мл × 3) и солевым раствором (10 мл), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали с помощью Combi Flash (PE : EtOAc = 1:1) с получением Т2.
Общая процедура для синтеза Т3
К перемешиваемому раствору диизопропиламина (0,125 мл, 0,894 ммоль) в безводном ТГФ (1,5 мл) по каплям добавляли н-BuLi (0,37 мл, 2,5 М в гексане) при температуре -78°C. Раствор перемешивали при температуре -78°C в течение 30 минут. Затем раствор Т2 (200 мг, 0,447 ммоль) в безводном ТГФ (0,5 мл) по каплям добавляли при температуре -78°C, потом раствор перемешивали при температуре -78°C в течение 0,5 часа. Затем раствор йода (226 мг, 0,894 ммоль) добавляли по каплям в безводный ТГФ (0,5 мл). Реакционный раствор перемешивали при температуре -78°C в течение 1,5 часа и нагревали до комнатной температуры. Реакционную смесь охлаждали насыщенным Na2S2O3 (1,5 мл) и разбавляли этилацетатом (20 мл). Органический слой промывали солевым раствором (10 мл), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток перекристаллизовывали из РЕ/EtOAc (3 мл/3 мл) с получением Т3.
Общая процедура для синтеза T4
Смесь T3 (70 мг, 0,133 ммоль), пирролидина (13 мкг, 0,159 ммоль) и Na2CO3 (21 мг, 0,200 ммоль) в EtOH (1 мл) перемешивали при температуре 80°C в течение 16 часов. Реакционную смесь разбавляли этилацетатом (20 мл), промывали солевым раствором (10 мл), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали препаративной тонкослойной хроматографией (РЕ : EtOAc = 1:1) с получением Т4.
Общая процедура для синтеза Т5
К перемешиваемому раствору XantPhos (5 мг, 0,00847 ммоль), Pd2(dba)3 (8 мг, 0,00847 ммоль) и NaOt-Bu (20 мг, 0,211 ммоль) в безводном толуоле (1 мл) добавляли Т4 (40 мг, 0,0847 ммоль) и бензофенонимин (18 мг, 0,101 ммоль). Реакционную смесь перемешивали при температуре 120°C в атмосфере N2 в течение 1,5 часа. В реакционную смесь добавляли HCl (1 мл, 4 N), после чего смесь перемешивали при температуре 16°C в течение 5 минут. Слой водного раствора подщелачивали NaOH (4 мл, 2 N) и экстрагировали этилацетатом (10 мл × 2). Объединенный экстракт промывали солевым раствором (10 мл), высушивали над безводным Na2SO4, испаряли при пониженном давлении. Полученное масло очищали предварительной тонкослойной хроматографией (ДХМ/МеОН, 15/1) с получением Т5.
Схема 21. Общий синтез N4-замещенного производного бензимидазола

Общая процедура для синтеза U1
Смесь 4-бром-2-нитроанилина (10,0 г, 0,046 моль) и N-ХСИ (6,15 г, 0,046 моль) в ДМФА (100 мл) перемешивали при температуре 120°C в течение 16 часов. Смесь вливали в воду (100 мл), остаток отфильтровывали, промывали водой (50 мл) и высушивали при пониженном давлении с получением U1.
Общая процедура для синтеза U2
К перемешиваемому раствору U1 в МеОН (100 мл) добавляли концентрированную HCl (9 мл, 108 ммоль, 12 М) и железный порошок (10,0 г, 179 ммоль). Реакционную смесь перемешивали при температуре 8°C - 16 С в течение 16 часов. После завершения реакции смесь фильтровали. Фильтрат нейтрализовывали до pH 8 водным раствором NaHCO3 и фильтровали. Фильтрат экстрагировали этилацетатом (100 мл × 2). Объединенные экстракты промывали солевым раствором (200 мл), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением U2.
Общая процедура для синтеза U3
Смесь U2 (7,2 г, 32,5 ммоль) и КДИ (6,32 г, 39,0 ммоль) в безводном ТГФ (100 мл) дефлегмировали в течение 16 часов. После охлаждения до комнатной температуры белый осадок собирали фильтрацией и высушивали при пониженном давлении с получением U3.
Общая процедура для синтеза U4
Смесь U3 (4,90 г, 19,8 ммоль) в POCl3 (30 мл) дефлегмировали в течение 16 часов. После этого смесь охлаждали до комнатной температуры, смесь вливали в воду (100 мл) при перемешивании. Белый осадок собирали фильтрацией, промывали водой (50 мл) и высушивали при пониженном давлении с получением U4.
Общая процедура для синтеза U5
Смесь U4 (2,10 г, 7,90 ммоль) в пирролидине (20 мл) дефлегмировали в течение 16 часов. Смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (50 мл × 2). Объединенные экстракты промывали солевым раствором (100 мл), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением сырого остатка, который очищали колоночной флэш-хроматографией (РЕ/EtOAc = от 5/1 до 2/1) с получением U5.
Общая процедура для синтеза U6
К перемешиваемому раствору U5 (850 мг, 2,83 ммоль) в безводном ДМФА (10 мл) добавляли NaH (170 мг, 4,24 ммоль, 60% в минеральном масле) при температуре 0°C. Смесь перемешивали при температуре 0°C в течение 30 минут, добавляли раствор 1-(2-бромэтокси)-4-(трифторметокси)бензола (1,61 г, 5,66 ммоль) в безводном ДМФА (5 мл) и перемешивали при температуре 60°C в течение 16 часов. Смесь охлаждали водой (50 мл) и экстрагировали этилацетатом (50 мл × 2). Объединенные экстракты промывали солевым раствором (50 мл), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением сырого остатка, который очищали с помощью Combi Flash с получением U6.
Общая процедура для синтеза U7
К перемешиваемому раствору U6 (680 мг, 1,35 ммоль), бензофенонимина (488 мг, 2,69 ммоль), Pd2(dba)3 (124 мг, 0,135 ммоль) и XantPhos (156 мг, 0,270 ммоль) в безводном толуоле (10 мл) добавляли NaOt-Bu (324 мг, 3,38 ммоль) в атмосфере N2. Реакционную смесь очищали в атмосфере N2 три раза. Смесь нагревали до 80°C - 100°C в течение 16 часов. Смесь концентрировали, растворяли в МеОН (2 мл), добавляли HCl (3 М, 2 мл) и перемешивали при температуре 20°C - 25°C в течение 2 часов. Затем смесь нейтрализовывали до pH водным раствором NaHCO3 и экстрагировали этилацетатом (30 мл × 2), объединенные экстракты промывали солевым раствором (50 мл), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением неочищенного остатка, который очищали с помощью подготовительной ЖХВД (0,05% HCl) с получением U7.
Схема 22. Общий синтез 1 N1-замещенного производного бензимидазола
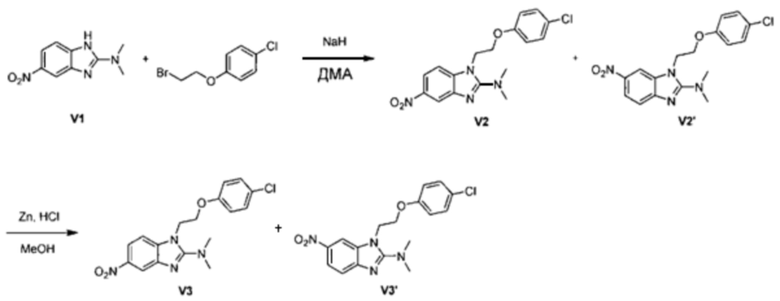
Общая процедура для синтеза V2
К перемешиваемому раствору соединения 2 (538 мг, 2,61 ммоль) в ДМА (10 мл) порциями добавляли NaH (125 мг, 3,13 ммоль, 60% дисперсия в минеральном масле). Смесь перемешивали при температуре 0°C в атмосфере N2 в течение 5 минут. Затем раствор 1-(2-бромэтокси)-4-хлорбензола (908 мг, 3,90 ммоль) в ДМА (8 мл) добавляли в смесь, потом раствор перемешивали при температуре 60°C в атмосфере N2 в течение 24 часов. Смесь разбавляли водой (80 мл) и экстрагировали этилацетатом (40 мл × 3), объединенные экстракты промывали водой (20 мл × 2) и солевым раствором (10 мл), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением сырого продукта, который перекристаллизовывали из EtOAc (5 мл) с получением смеси V2 и V2'.
Общая процедура для синтеза V3
К перемешиваемому раствору смеси V2 и V2' (100 мг, 0,302 ммоль) в МеОН добавляли цинковый порошок (98,2 мг, 1,51 ммоль) и NH4Cl (326 мг, 6004 ммоль) при температуре 19°C. Смесь перемешивали при температуре 45°C в течение 16 часов. Реакционный раствор разбавляли этилацетатом (10 мл) и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученное масло очищали препаративной тонкослойной хроматографией (ДХМ/МеОН, 20/1) с получением V3.
Схема 23. Общий синтез 2 N1-замещенного производного бензимидазола

Общая процедура для синтеза W2
Смесь соединения W1 (300 мг, 0,843 ммоль), оксетан-3-амина (74 мг, 1,0 ммоль), K2CO3 (175 мг, 1,26 ммоль) в ДМФА (3 мл) перемешивали при температуре 10°C в течение 16 часов. Реакционный раствор разбавляли водой (15 мл) и экстрагировали этилацетатом (20 мл × 2). Объединенный экстракт промывали водой (10 мл × 2) и солевым раствором (10 мл), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением W2.
Общая процедура для синтеза W4
К перемешиваемому раствору W2 (500 мг, 1,22 ммоль) в МеОН (20 мл) добавляли Pd/C (50 мг, 5%, 60% веса сырого вещества) в атмосфере N2. Суспензию дегазировали при вакуумировании и продували Н2 несколько раз. Смесь перемешивали в атмосфере Н2 (1 атм.) в течение 1,5 часов. Реакционную смесь фильтровали через подушку из целита. В фильтрат (W3 в МеОН) добавляли BrCN (261 мг, 2,44 ммоль). Реакционный раствор перемешивали при температуре 10°C в течение 16 часов. Реакционный раствор концентрировали при пониженном давлении. Остаток разбавляли водой (30 мл) и EtOAc (30 мл). Затем Na2CO3 в твердом виде добавляли при перемешивании до подщелачивания слоя водного раствора до pH 9. Органический слой отделяли и высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением W4.
Схема 24. Общий синтез 3 N1-замещенного производного бензимидазола

Общая процедура для синтеза X1
Смесь 3-фтор-4-нитроанилина (1,00 г, 6,40 ммоль), (4-(4-хлорфенокси)фенил)метанамина (1,79 г, 7,28 ммоль) и ДИПЭА (1,65 г, 12,8 ммоль) в ацетонитриле (15 мл) дефлегмировали 16 часов. Смесь охлаждали до комнатной температуры и разбавляли водой (40 мл). Смесь экстрагировали этилацетатом (40 мл × 2). Объединенные экстракты высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением 3,42 г неочищенного остатка, который очищали с помощью Combi Flash с получением X1.
Общая процедура для синтеза Х2
К перемешиваемому раствору X1 (1,50 г, 4,06 ммоль), ди-трет-бутил бикарбоната (1,15 г, 5,27 ммоль) в ДХМ (20 мл) добавляли ДМАП (248 мг, 2,03 ммоль) и ТЭА (413 мг, 4,06 ммоль). Смесь перемешивали при температуре 20°C в течение 16 часов. Смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (40 мл × 2). Объединенные экстракты промывали солевым раствором (100 мл × 2), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением 3,00 г неочищенного остатка, который очищали с помощью Combi Flash (PE/EtOAc = от 25/1 до 20/1 до 15/1) с получением Х2.
Общая процедура для синтеза Х3
К перемешиваемому раствору Х2 (100 мг, 0,213 ммоль) в МеОН (5 мл) добавляли никель Ренея (20 мг) в Ar. Суспензию дегазировали при вакуумировании и продували Н2 три раза. Раствор перемешивали при температуре 28°C в атмосфере Н2 (50 фунт/дюйм) в течение 16 часов. Смесь фильтровали через подушку из целита, а подушку промывали МеОН (200 мл). Фильтрат концентрировали при пониженном давлении с получением Х3.
Общая процедура для синтеза Х4
Смесь Х3 (80 мг, 182 ммоль), цианогенбромида (29 мг, 0,273 ммоль) в EtOH (4 мл) перемешивали при температуре 25°C в течение 16 часов. Смесь подщелачивали до pH 9 водным раствором Na2CO3, разбавляли водой (20 мл) и экстрагировали этилацетатом (20 мл × 2). Объединенные экстракты промывали солевым раствором (40 мл × 2), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением Х4.
Общая процедура для синтеза Х5
К перемешиваемому раствору Х4 (80 мг, 0,172 ммоль) в МеОН (2 мл) добавляли раствор HCl в МеОН (4М, 2 мл). Полученный раствор перемешивали при температуре 25°C в течение 2 часов. Смесь разбавляли водой (20 мл), подщелачивали до pH 9 водным раствором Na2CO3 и экстрагировали этилацетатом (20 мл × 2). Объединенные экстракты промывали солевым раствором (40 мл × 2), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением сырого остатка, который очищали с помощью подготовительной ЖХВД (0,01% NH3.H2O) с получением Х5.
Схема 25. Общий синтез 1 N2-замещенного производного бензимидазола

Общая процедура для синтеза Y1
Смесь 5-бром-1,3-дигидро-2H-бензо[d]имидазол-2-ола (5,00 г, 23,5 ммоль) и POCl3 (80 мл) дефлегмировали в течение 16 часов. После охлаждения до комнатной температуры смесь вливали в воду (300 мл) и добавляли водный раствор NaOH (5 М, 80 мл). Осадок фильтровали и промывали водой. Фильтрат высушивали при сильном вакуумировании с получением Y1.
Общая процедура для синтеза Y2
К перемешиваемому раствору Y1 (500 мг, 2,16 ммоль) и ДИПЭА (558 мг, 4,32 ммоль) в ТГФ (5 мл) добавляли SEMCl (540 мг, 3,24 ммоль) при температуре 0°C, а полученную смесь перемешивали при температуре 35°C в течение 16 часов. Смесь разбавляли водой (50 мл), экстрагировали этилацетатом (50 мл × 2). Объединенные экстракты промывали солевым раствором (50 мл), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением остатка. Остаток очищали колоночной флэш-хроматографией с получением Y2.
Общая процедура для синтеза Y3
Смесь Y2 (480 мг, 1,33 ммоль), 4-метил-1H-пиразола (109 мг, 1,33 ммоль) и K2CO3 (275 мг, 1,99 ммоль) в ДМФА (5 мл) перемешивали при температуре 100°C в течение 16 часов. Смесь разбавляли водой (50 мл), экстрагировали этилацетатом (50 мл × 2). Объединенные экстракты промывали солевым раствором (50 мл), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением осадка. Осадок очищали с помощью колоночной флэш-хроматографии с получением Y3.
Общая процедура для синтеза Y4
Смесь Y3 (500 мг, 1,23 ммоль), 1-(4-фторфенил)пиперазина (243 мг, 1,35 ммоль), NaOt-Bu (236 мг, 2,45 ммоль), Pd2(dba)3 (112 мг, 0,123 ммоль) и Xantphos (71 мг, 0,12 ммоль) в толуоле (5 мл) перемешивали при температуре 120°C в течение 16 часов. Смесь разбавляли водой (50 мл), экстрагировали этилацетатом (50 мл × 2). Объединенные экстракты промывали солевым раствором (50 мл), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением остатка. Остаток очищали колоночной флэш-хроматографией с получением Y4.
Общая процедура для синтеза Y5
Смесь Y4 (160 мг, 0,316 ммоль) и концентрированной HCl (2 мл) в МеОН (6 мл) перемешивали при температуре 32°C в течение 16 часов. Смесь разбавляли водой (50 мл), нейтрализовывали водным раствором NaOH и экстрагировали этилацетатом (50 мл × 2). Объединенные экстракты промывали солевым раствором (50 мл), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением Y5.
Схема 26. Общий синтез 2 N2-замещенного производного бензимидазола

Общая процедура для синтеза Z1
К перемешиваемому раствору диметиламина в ТГФ (18 мл, 2 N) добавляли 2-хлор-5-нитро-1H-бензо[d]имидазола (1,80 г, 9,1 ммоль) при температуре 20°C. Реакционный раствор перемешивали при температуре 150°C в запаянной трубке в течение 3 часов. Реакционный раствор вливали в воду (20 мл) и экстрагировали этилацетатом (10 мл × 3). Объединенный органический слой промывали солевым раствором (10 мл), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением Z1.
Схема 27. Общая реакция сочетания 1 бензимидазола N2 амина

Общая процедура для синтеза АА1
Смесь 5-нитро-1H-бензо[d]имидазол-2-амина (200 мг, 1,12 ммоль), (4-фтор-фенил)-ацетальдегида (388 мг, 2,80 ммоль) и Ti(Oi-Pr)4 (637 мг, 2,24 ммоль) в безводном ТГФ (5 мл) перемешивали при температуре 60°C в течение 30 минут, затем охлаждали до 20°C и выдерживали при данной температуре в течение 30 минут, постоянно перемешивая. Затем NaBH4 (213 мг, 5,60 ммоль) добавляли в смесь при температуре 0°C и перемешивали при температуре 20°C в течение 3 часов, а затем при температуре 60°C в течение 16 часов. Смесь остужали и охлаждали водным раствором NH3.H2O (2 М, 10 мл). Осадок фильтровали и промывали ТГФ (20 мл). Фильтрат разбавляли водой (20 мл) и экстрагировали этилацетатом (20 мл × 2). Объединенные экстракты промывали солевым раствором (50 мл × 2), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением сырого продукта, который очищали препаративной ЖХВД (0,01% NH3.H2O) с получением АА1.
Схема 28. Общая реакция связывания 2 бензимидазол N2 амина

Общая процедура для синтеза АВ1
Смесь 5-нитро-1H-бензо[d]имидазол-2-амина (1,00 г, 5,60 ммоль), 4-фторбензальдегида (2,78 г, 22,4 ммоль) и Ti(Oi-Pr)4 (3,18 г, 11,2 ммоль) в ТГФ (6 мл) перемешивали при температуре 60°C в течение 30 минут, смесь охлаждали до 20°C и выдерживали при данной температуре в течение 30 минут. Затем NaBH4 (851 мг, 22,4 ммоль) добавляли в смесь при температуре 0°C и перемешивали при 20°C в течение 3 часов и при температуре 60°C в течение 16 часов. Смесь охлаждали до 20°C и охлаждали водным раствором NH3.H2O (2 М, 15 мл). Осадок отфильтровывали, промывали ТГФ (50 мл). Фильтрат разбавляли водой (50 мл) и экстрагировали этилацетатом (50 мл × 2). Объединенные экстракты промывали солевым раствором (100 мл × 2), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением сырого продукта, который очищали препаративной ЖХВД (0,01% NH3.H2O) с получением АВ1.
Схема 29. Общая реакция связывания 3 бензимидазола N2 амина

Общая процедура для синтеза АС1
К перемешиваемому раствору 1-метил-6-нитро-1H-бензо[d]имидазол-2-амина (900 мг, 3,63 ммоль) и ацетофенона (700 мг, 3,64 ммоль) в безводном толуоле (150 мл) добавляли Ti(Oi-Pr)4 (3,13 г, 10,9 ммоль). Полученную смесь перемешивали при температуре рефлюкса в атмосфере N2 в течение 24 часов. После охлаждения до комнатной температуры в смесь добавляли NaBH4 (290 мг, 7,26 ммоль), а затем МеОН (5 мл). Полученную смесь перемешивали при температуре 12°C в течение 2 часов. Смесь разбавляли этилацетатом (200 мл), промывали солевым раствором (200 мл) и водой (200 мл × 2), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением сырого продукта, который очищали колоночной флэш-хроматографией (элюент : РЕ/EtOAc = от 50/1 до 5/1) с получением АС1.
Схема 30. Общая реакция связывания 4 бензимидазола N2 амина

Общая процедура для синтеза AD1
Смесь 5-нитро-1H-бензо[d]имидазол-2-амина (3,00 г, 16,8 ммоль) и 4-фторбензоилхлорида (8,10 г, 53,6 ммоль) в пиридине (30 мл) перемешивали при температуре 20°C в течение 10 минут. Смесь разбавляли водой (150 мл) и фильтровали. Фильтрационный кек промывали водой (100 мл) и высушивали при сильном вакуумировании с получением неочищенного остатка, который очищали колоночной флэш-хроматографией с получением AD1.
Схема 31. Общая реакция связывания 5 бензимидазол N2 амина
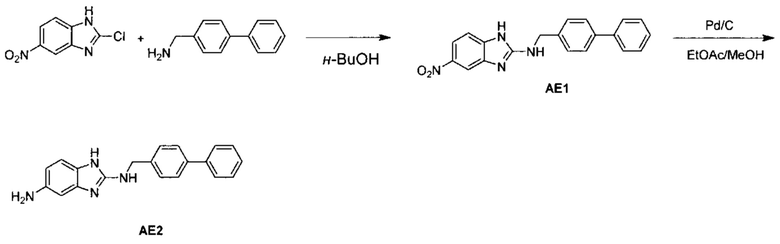
Общая процедура для синтеза АЕ1
Смесь 2-хлор-5-нитро-1H-бензо[d]имидазола (500 мг, 2,53 ммоль), 4-фенилбензиламина (696 мг, 3,80 ммоль) в н-BuOH (8 мл) дефлегмировали в течение 36 часов. После завершения реакции смесь разбавляли водой (50 мл) и экстрагировали этилацетатом (50 мл × 2). Объединенные экстракты промывали солевым раствором (100 мл × 2), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением сырого остатка, который очищали с помощью Combi Flash (PE/EtOAc = от 3/1 до 2/1 до 1/1) с получением АЕ1.
Общая процедура для синтеза АЕ2
К перемешиваемому раствору АЕ1 (150 мг, 0,436 ммоль) в EtOAc/MeOH (8 мл/2 мл) добавляли Pd/C (30 мг, 10%) в N2. Суспензию дегазировали при вакуумировании и продували Н2 три раза. Раствор перемешивали при температуре 22°C в бутыли Н2 в течение 16 часов. Смесь фильтровали через подушку из целита и промывали этилацетатом (30 мл). Фильтрат концентрировали при пониженном давлении с получением сырого остатка, который очищали промыванием этилацетатом/МеОН (5 мл/1 мл) с получением АЕ2.
Схема 32. Общая реакция связывания 1 бензимидазол N6 амина

Общая процедура для синтеза AF2
Смесь А1 (200 мг, 0,488 ммоль), АсОН (0,3 мл) и 3-оксетанона (42 мг, 0,59 ммоль) в абсолютном EtOH (3 мл) перемешивали при температуре 18°C - 22°C в течение 16 часов. Затем в реакционную смесь добавляли NaBH3CN (92 мг, 1,5 ммоль), полученную смесь перемешивали при температуре 18°C - 20°C в течение 4 часов. Реакционную смесь разбавляли этилацетатом (50 мл), затем промывали водой (40 мл) и солевым раствором (40 мл × 3), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали препаративной ЖХВД (0,01% NH3.H2O в качестве добавки) с получением AF2.
Общая процедура для синтеза AF3
Смесь AF2 (100 мг, 0,229 ммоль) и формальдегида (86 мг, 1,15 ммоль, 40% водного раствора в воде) в EtOH (5 мл) добавляли АсОН (доводили реакционную смесь до pH 4). Полученную смесь перемешивали при температуре 10°C в течение 16 часов. В смесь добавляли NaBH3CN (72 мг, 1,15 ммоль) и реакционную смесь перемешивали при температуре 10°C в течение 4 часов. Смесь разбавляли водой (20 мл) и экстрагировали этилацетатом (20 мл × 2), Объединенные экстракты промывали солевым раствором (40 мл × 2), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением остатка, который очищали препаративной ЖХВД (0,01% NH3.H2O) с получением AF3.
Схема 33. Общая реакция связывания 2 бензимидазол N6 амина

Общая процедура для синтеза AG2
К перемешиваемому раствору AG1 (150 мг, 0,583 ммоль), 1Н-имидазол-5-карбальдегида (39 мг, 0,416 ммоль) в EtOH (3 мл) добавляли АсОН (реакционную смесь доводили до pH 4). Полученную смесь перемешивали при температуре 20°C в течение 16 часов. Затем добавляли NaBH3CN (73 мг, 1,17 ммоль), а смесь перемешивали при температуре 20°C в течение 16 часов. Смесь нейтрализовывали Na2CO3, разбавляли водой (20 мл) и экстрагировали этилацетатом (20 мл × 2). Объединенные экстракты промывали солевым раствором (40 мл × 2), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением сырого остатка, который очищали препаративной ЖХВД (0,01% NH3.H2O) с получением AG2.
Схема 34. Общая реакция связывания 3 бензимидазол N6 амина

Общая процедура для синтеза АН2
К перемешиваемому раствору АН1 (200 мг, 0,69 ммоль) и этил 2-бромизобутирата (537 мг, 2,75 ммоль) в EtOH (10 мл) добавляли NaOAc (226 мг, 2,75 ммоль). Смесь дефлегмировали в N2 в течение 16 часов. Смесь разбавляли водой (10 мл) и экстрагировали этилацетатом (10 мл × 2). Объединенные экстракты промывали солевым раствором (10 мл × 2), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением неочищенного АН2.
Общая процедура для синтеза АН3
К перемешиваемому раствору соединения АН2 (200 мг, сырой) в безводном ТГФ (5 мл) добавляли LiAlH4 (75 мг, 1,98 ммоль) при температуре 0°C. После перемешивания при температуре 0°C в течение 2 часов смесь перемешивали при температуре 25°C в атмосфере N2 в течение 16 часов. Смесь охлаждали водным раствором NaOH (5 М, 0,2 мл) и фильтровали. Фильтрат разбавляли водой (10 мл) и экстрагировали этилацетатом (10 мл × 2), объединенные экстракты промывали солевым раствором, высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением сырого продукта, который очищали препаративной тонкослойной хроматографией (МеОН : ДХМ = 1:10) с получением АН3.
Схема 35. Общий синтез N6-замещенного производного бензимидазола

Общая процедура для синтеза AI2
Смесь AI1 (1,00 г, 4,05 ммоль), 5-фтор-2-нитроанилина (631 мг, 4,05 ммоль) и ДИПЭА (1,55 г, 12,2 ммоль) в ацетонитриле (10 мл) нагревали до температуры рефлюкса в течение 12 часов. Смесь концентрировали при пониженном давлении с получением остатка, который очищали колоночной хроматографией с силикагелем (элюент : EtOAc/РЕ = от 1/10 до ДХМ до ДХМ/EtOAc = 1/1) с получением AI2.
Общая процедура для синтеза AI3
Смесь соединения AI2 (1,30 г, 3,39 ммоль) и Pd-C (200 мг, 10%) в МеОН (30 мл) перемешивали при температуре 10°C в Н2 (1 атм.) в течение 2 часов. Смесь фильтровали, фильтрат концентрировали при пониженном давлении с получением сырого AI3.
Общая процедура для синтеза AI4
К перемешиваемому раствору соединения AI3 (100 мг, 0,283 ммоль) в МеОН (5 мл) добавляли BrCN (31 мг, в 0,5 мл ацетонитриле) на водяной бане, смесь перемешивали при температуре 10°C в течение 1 часа. Реакционный раствор очищали препаративной ЖХВД (0,01% HCl) с получением AI4.
Схема 36. Общая реакция связывания 4 бензимидазола N6 амина
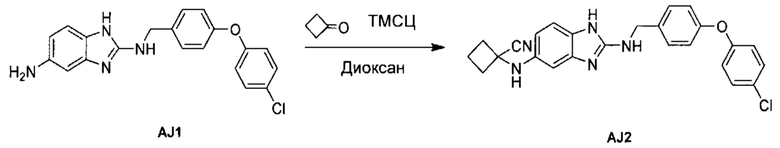
Общая процедура для синтеза AJ2
К перемешиваемому раствору AJ1 (100 мг, 0,274 ммоль) в безводном диоксане (3 мл) добавляли циклобутанон (23 мг, 0,329 ммоль) и ТМСЦ (38 мг, 0,384 ммоль), полученную смесь перемешивали при температуре 50°C в атмосфере N2 в течение 16 часов. Смесь разбавляли водой (15 мл) и экстрагировали этилацетатом (15 мл × 2), объединенные экстракты промывали солевым раствором (30 мл × 2), высушивали над безводным Na2SO4 и концентрировали при пониженном давлении с получением 90 мг сырого остатка, который очищали препаративной ЖХВД (0,05% HCl) с получением AJ2.
Схема 37. Общая реакция связывания 5 бензимидазол N6 амина

Общая процедура для синтеза AK2
К перемешиваемому раствору AK1 (100 мг, 0,244 ммоль) в безводном толуоле (5 мл) добавляли RuPhos (12 мг, 0,0244 ммоль), Pd2(dba)3 (23 мг, 0,0244 ммоль), 3-бром-1-метил-1H-пиразол (47 мг, 0,293 ммоль) и NaOt-Bu (58 мг, 0,610 ммоль). Смесь перемешивали при температуре 110°C в N2 в течение 16 часов. Реакцию разбавляли солевым раствором (5 мл) и фильтровали. Фильтрат экстрагировали этилацетатом (5 мл × 3). Объединенный экстракт промывали солевым раствором (5 мл), высушивали над Na2SO4, концентрировали при пониженном давлении. Полученное масло очищали препаративной ЖХВД (0,01% NH3⋅H2O в качестве добавки) с получением AK2.
С помощью вышеуказанных процедур были синтезированы соединения 1-556, приведенные в таблице 7 (см. ниже).
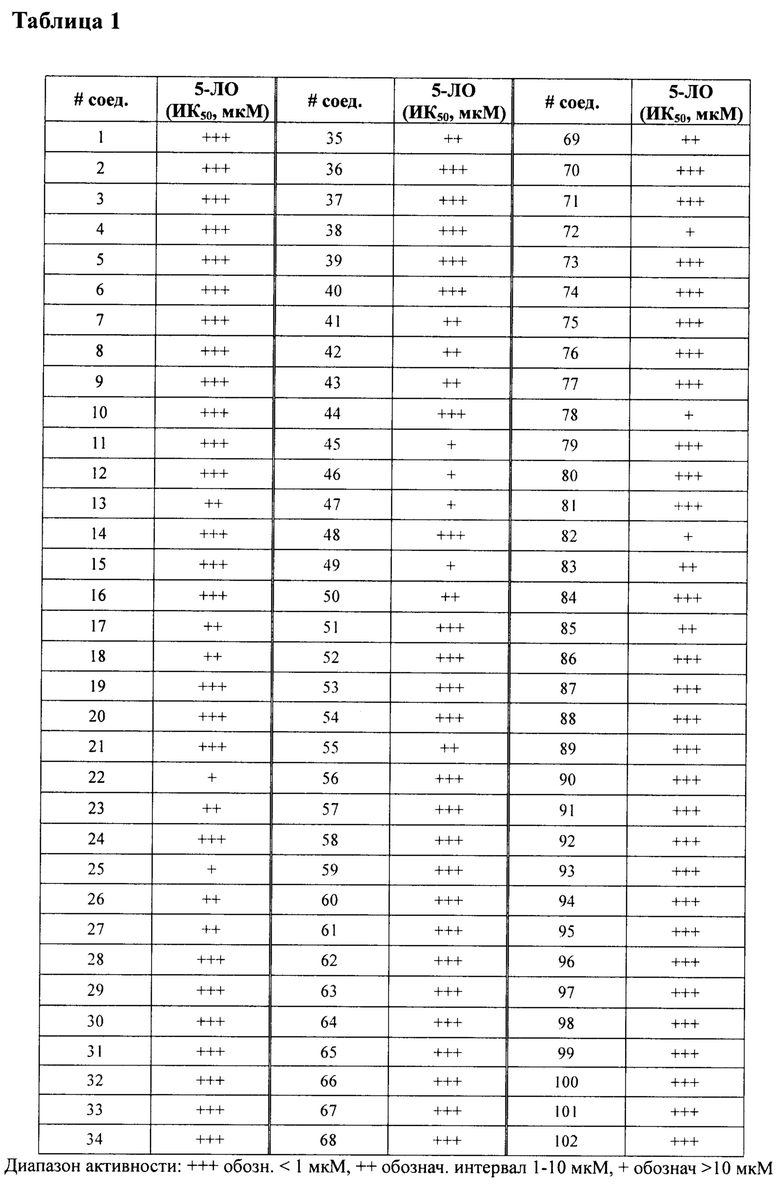

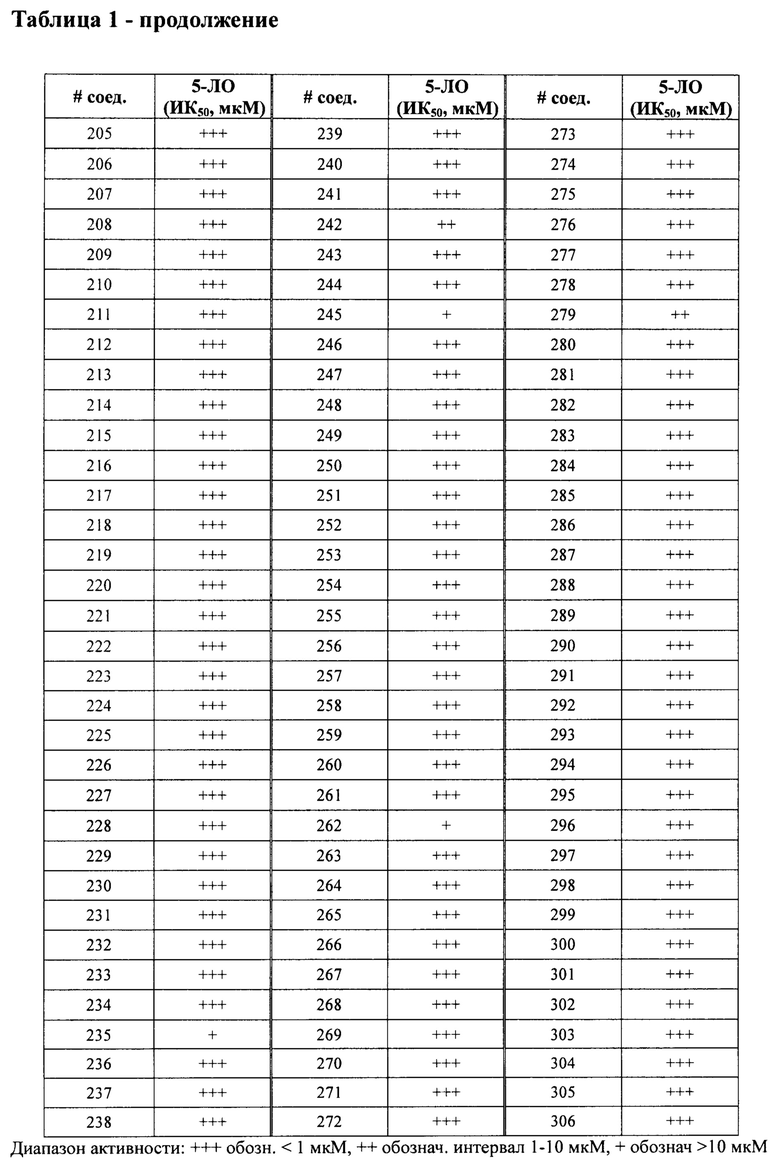


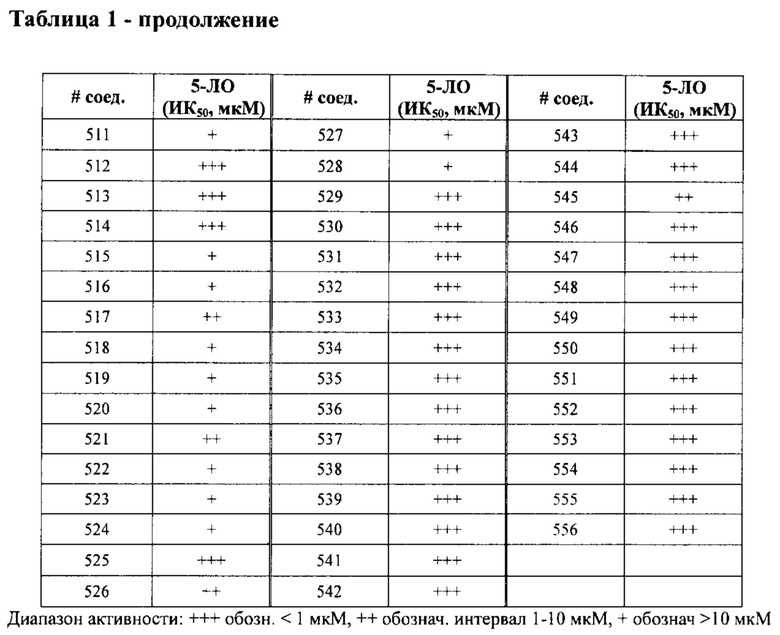

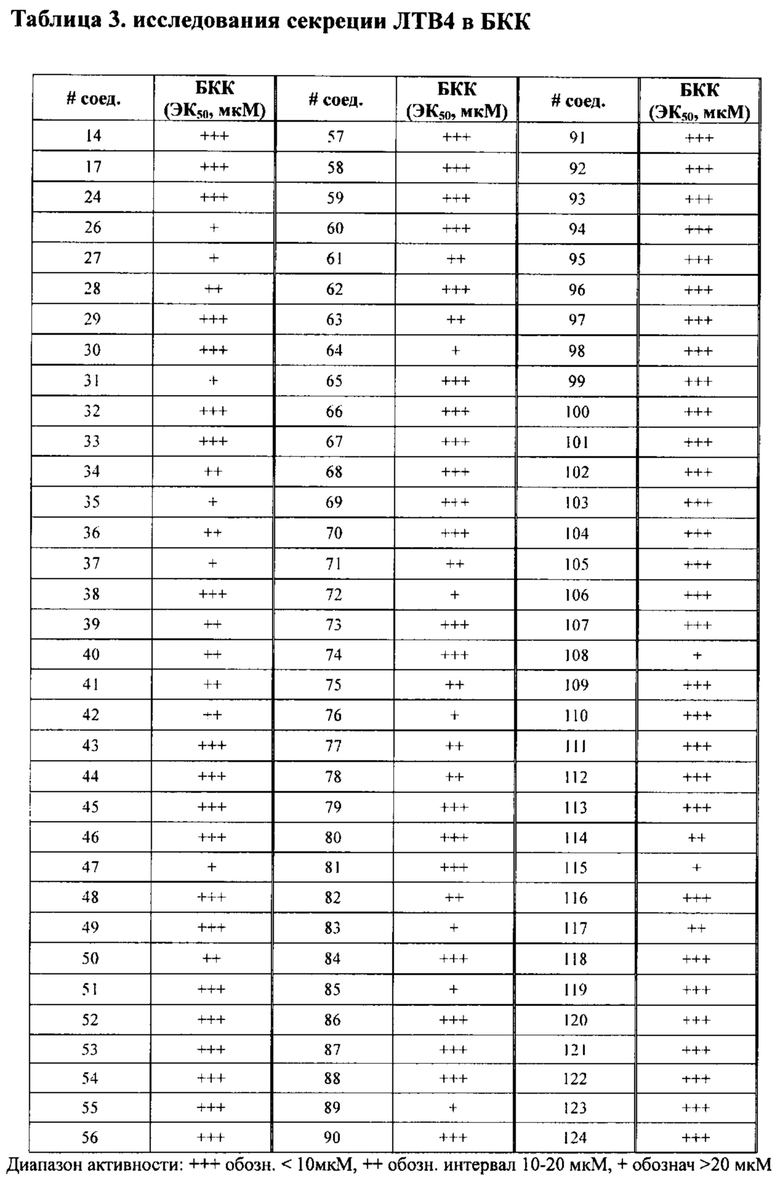






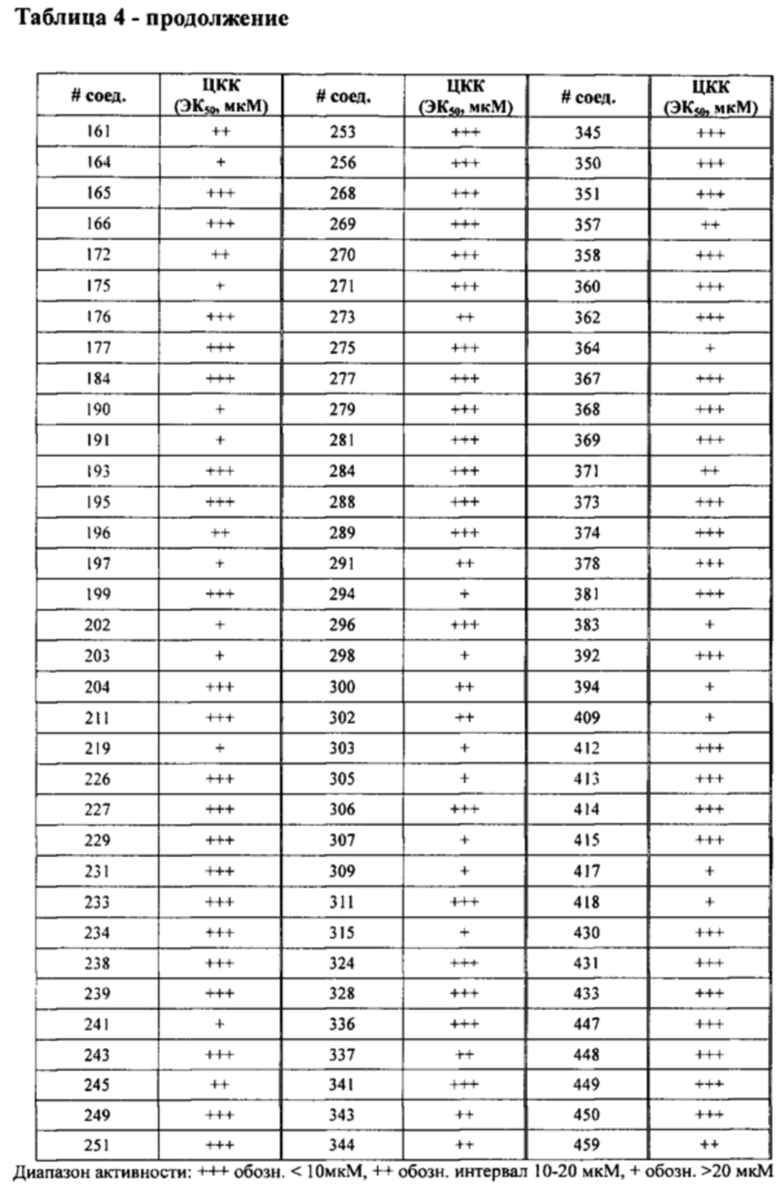
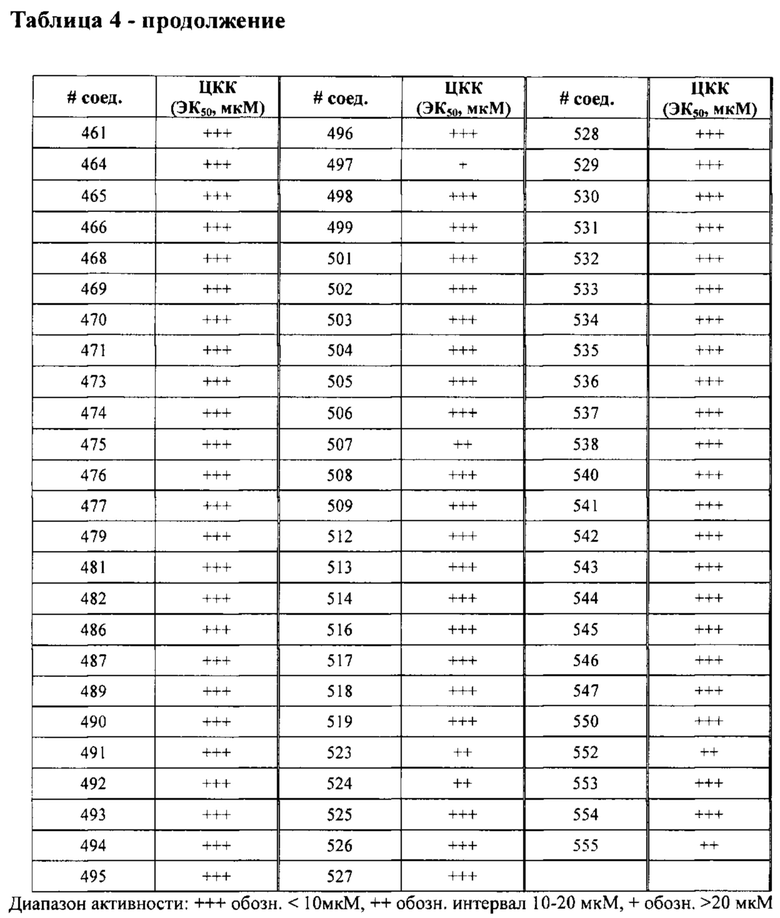



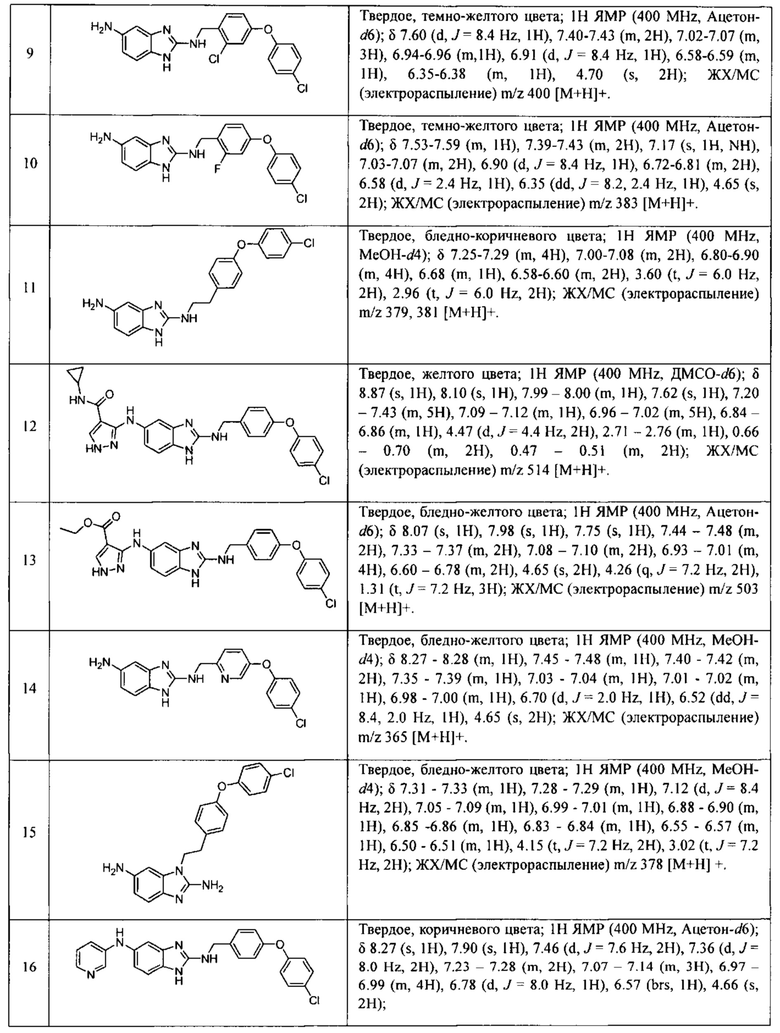

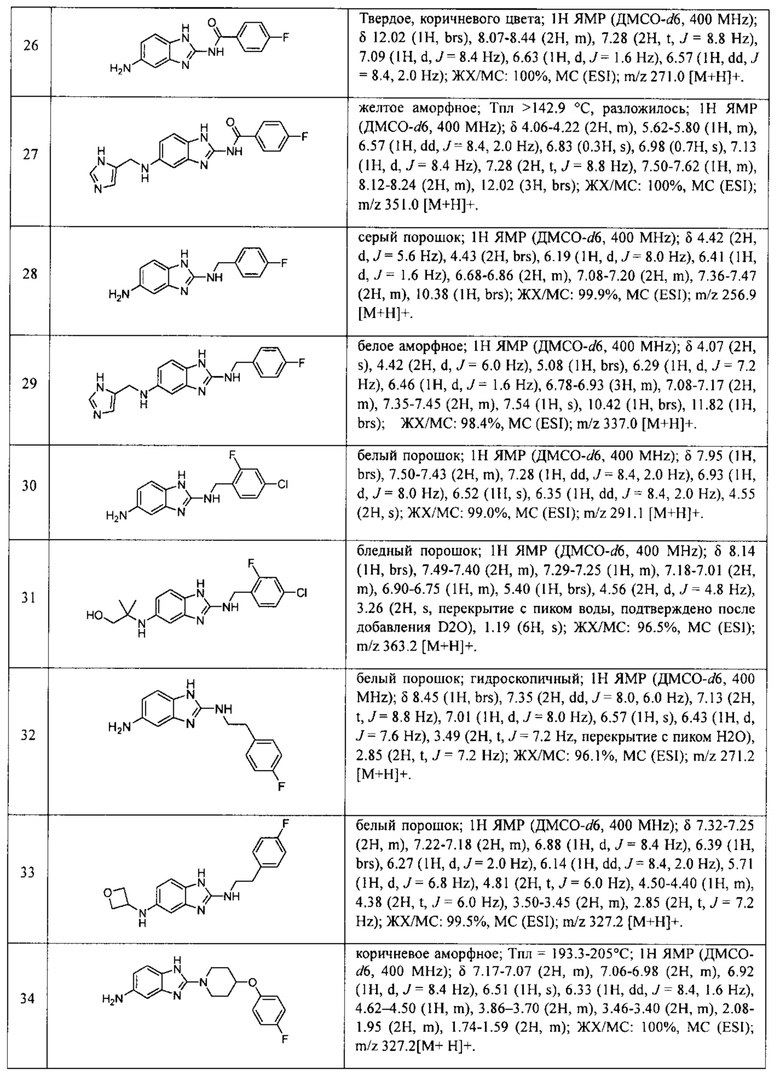




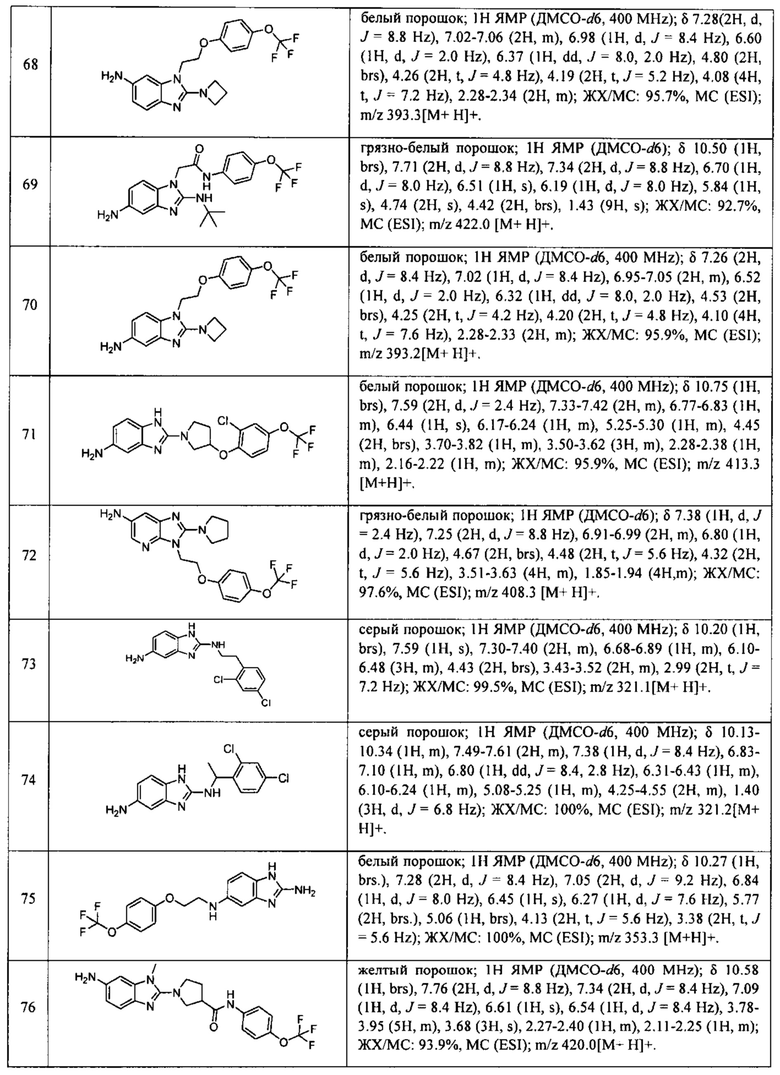


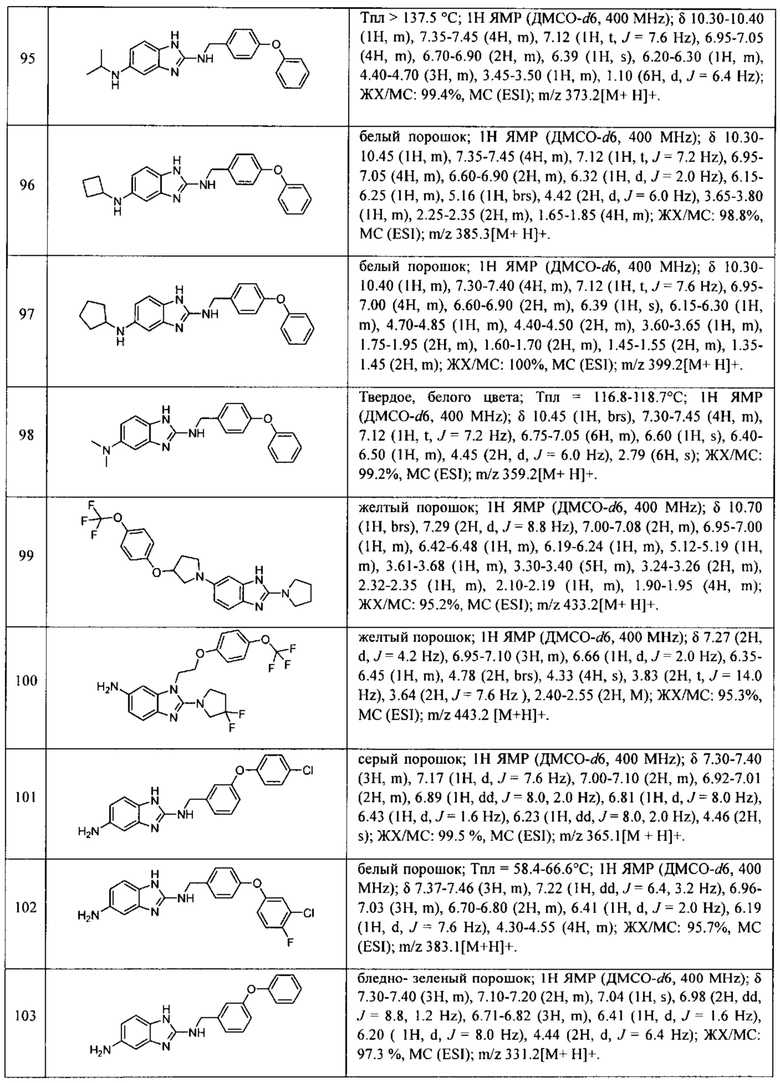


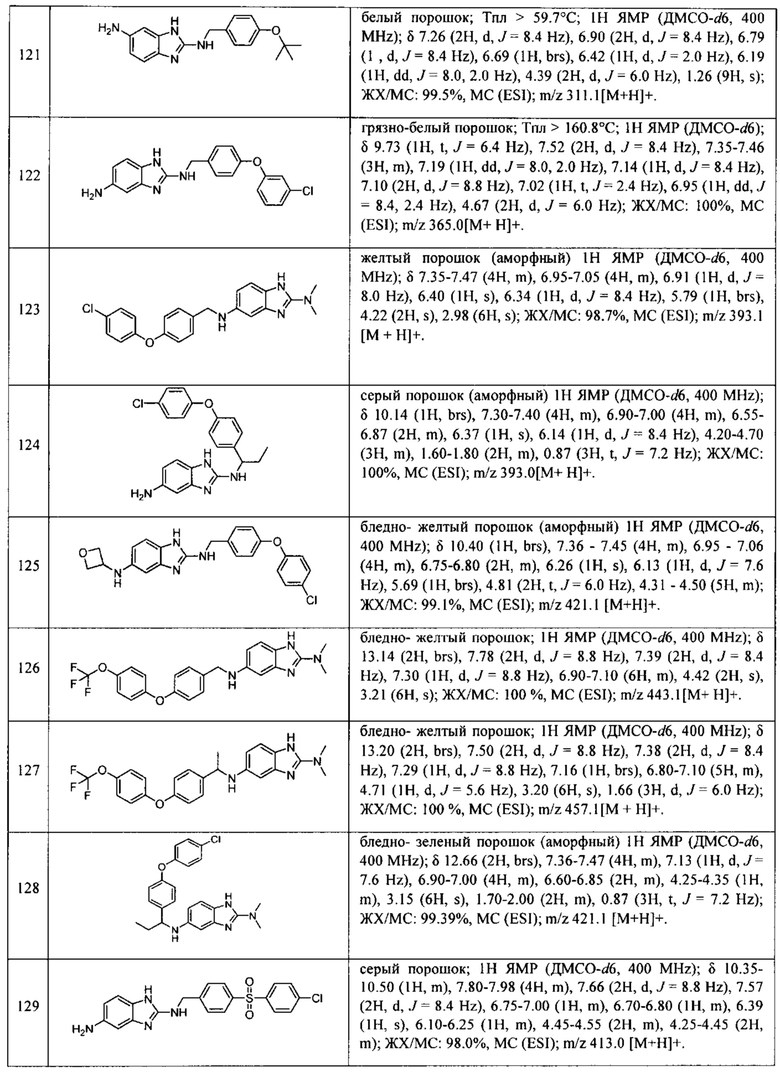

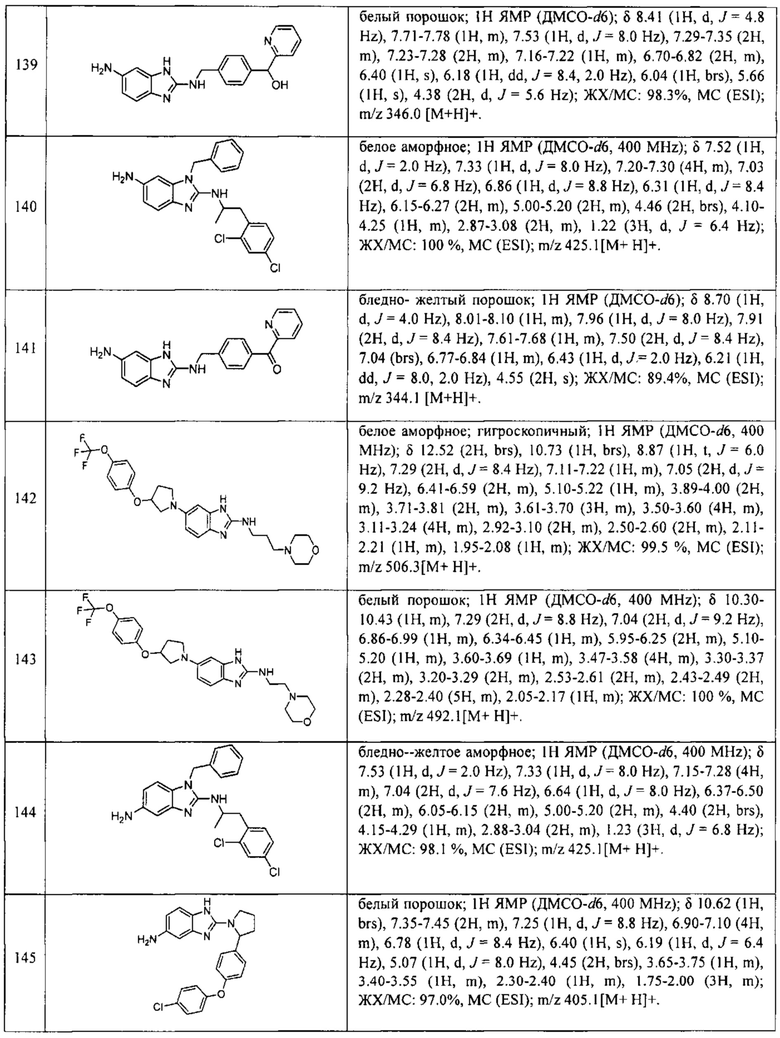


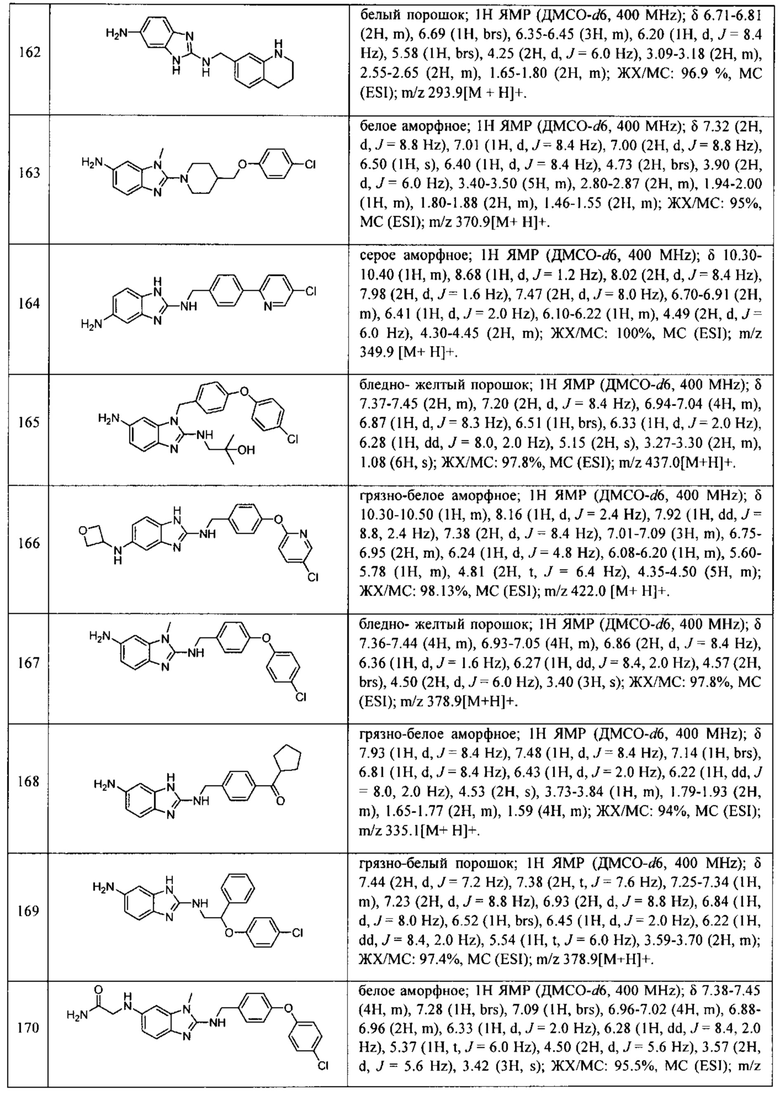



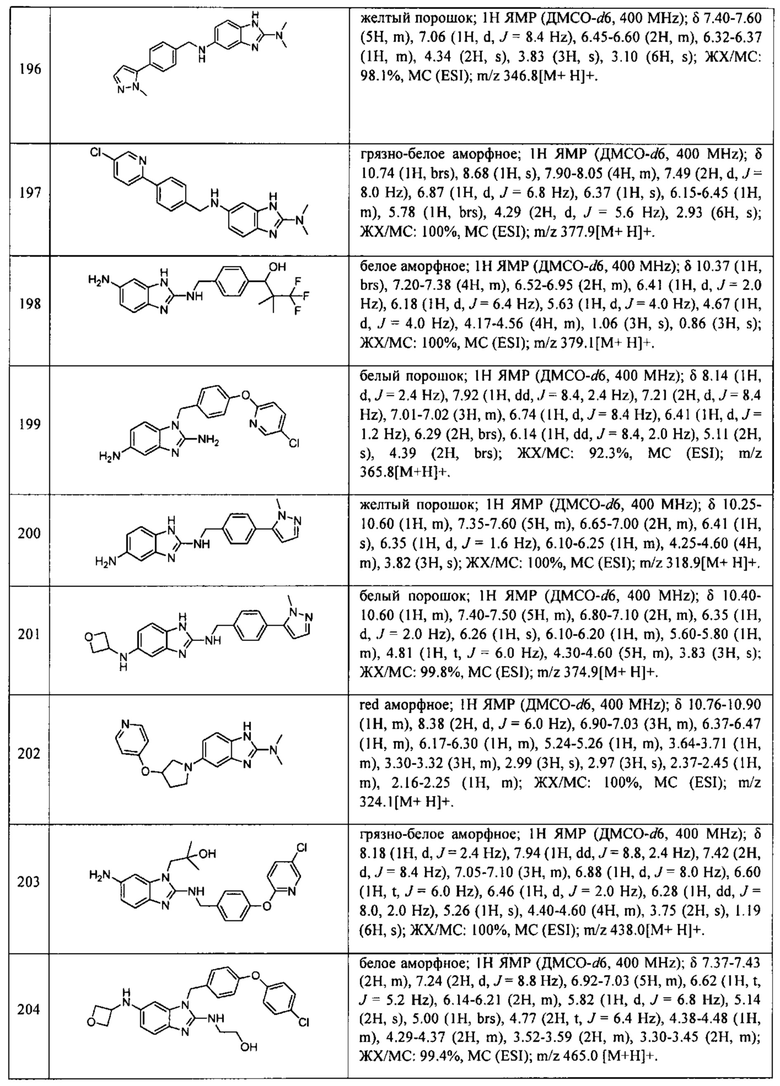
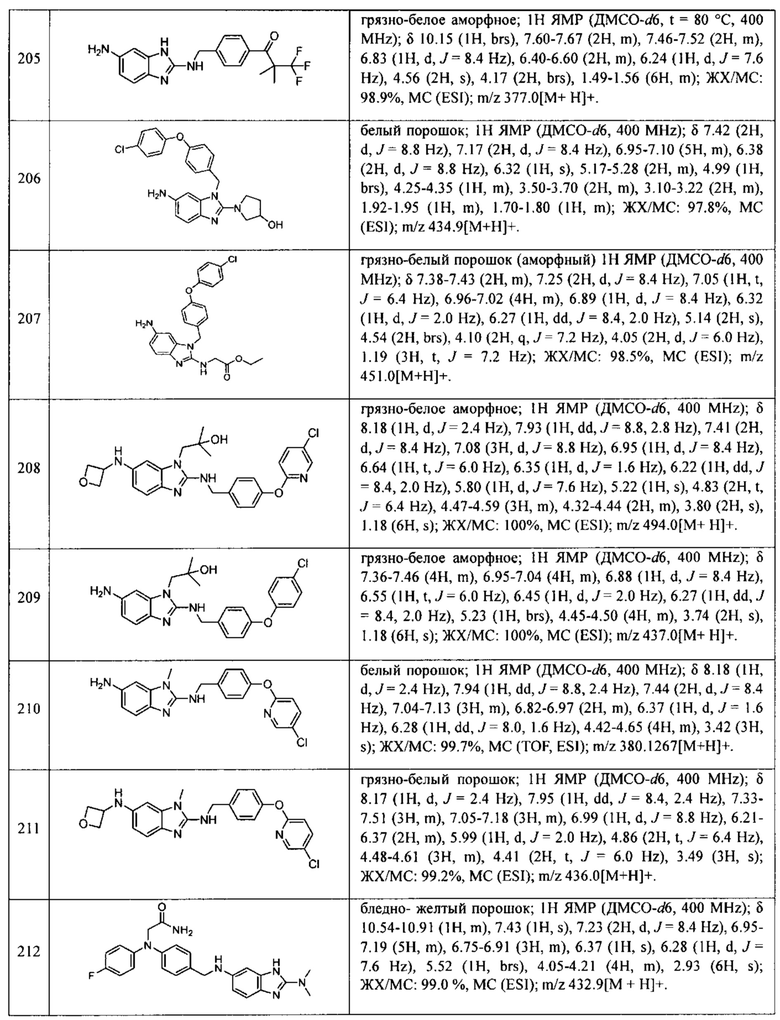








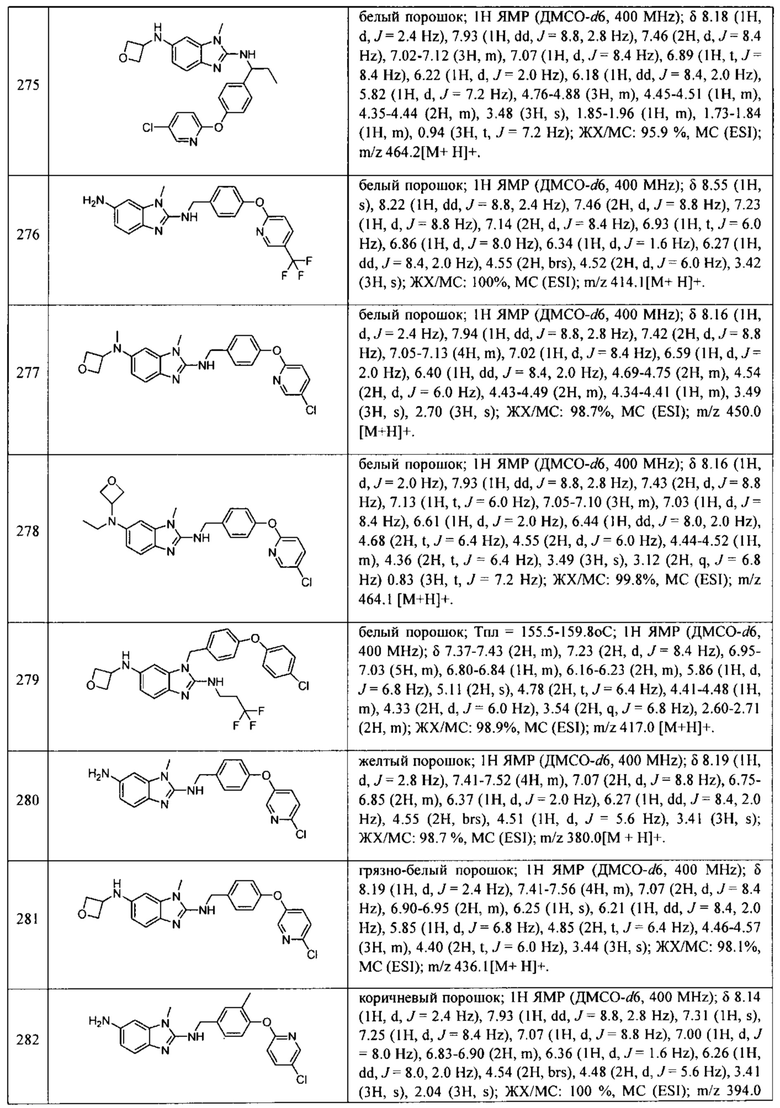














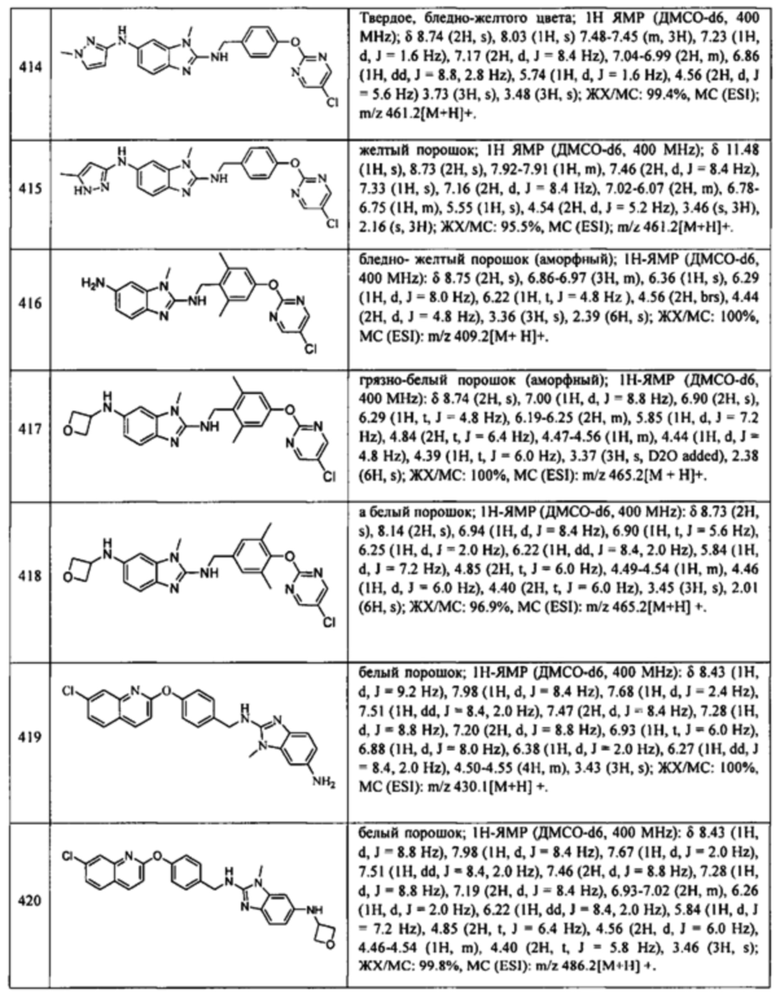
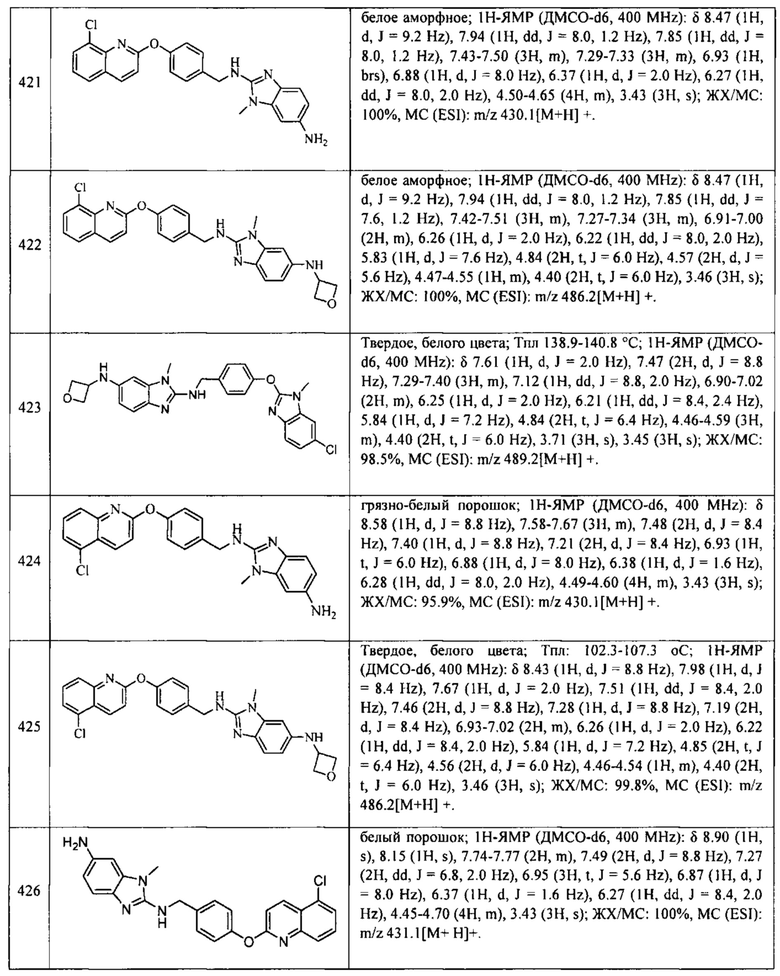
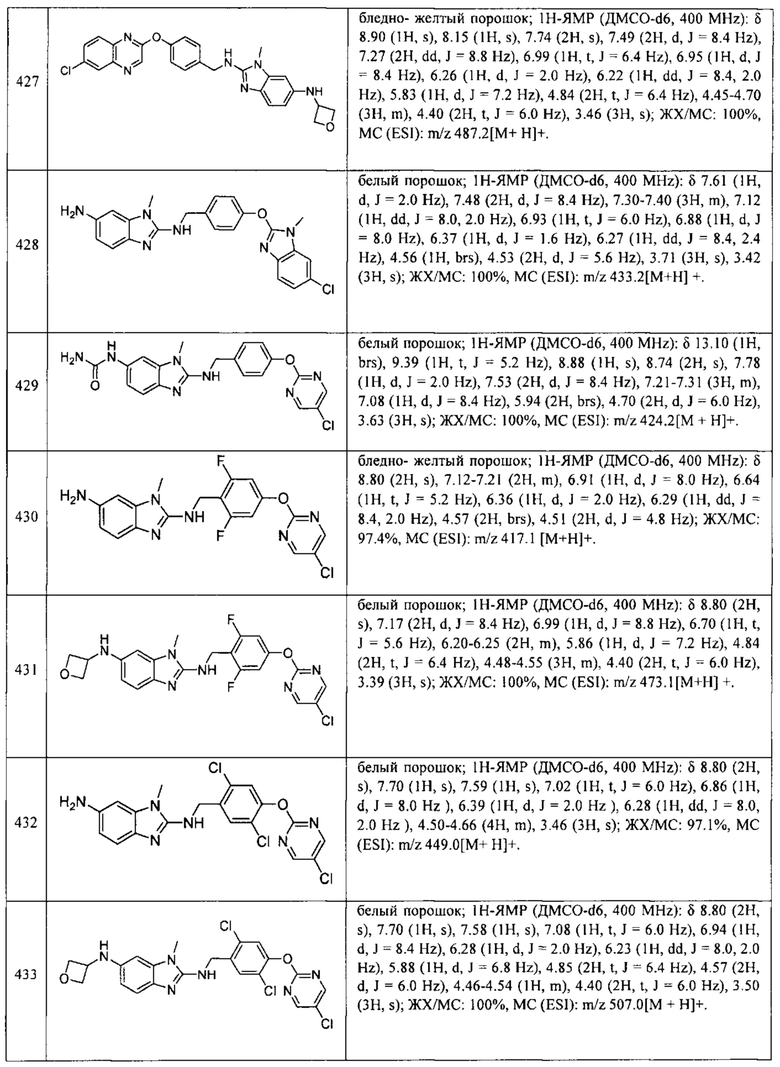





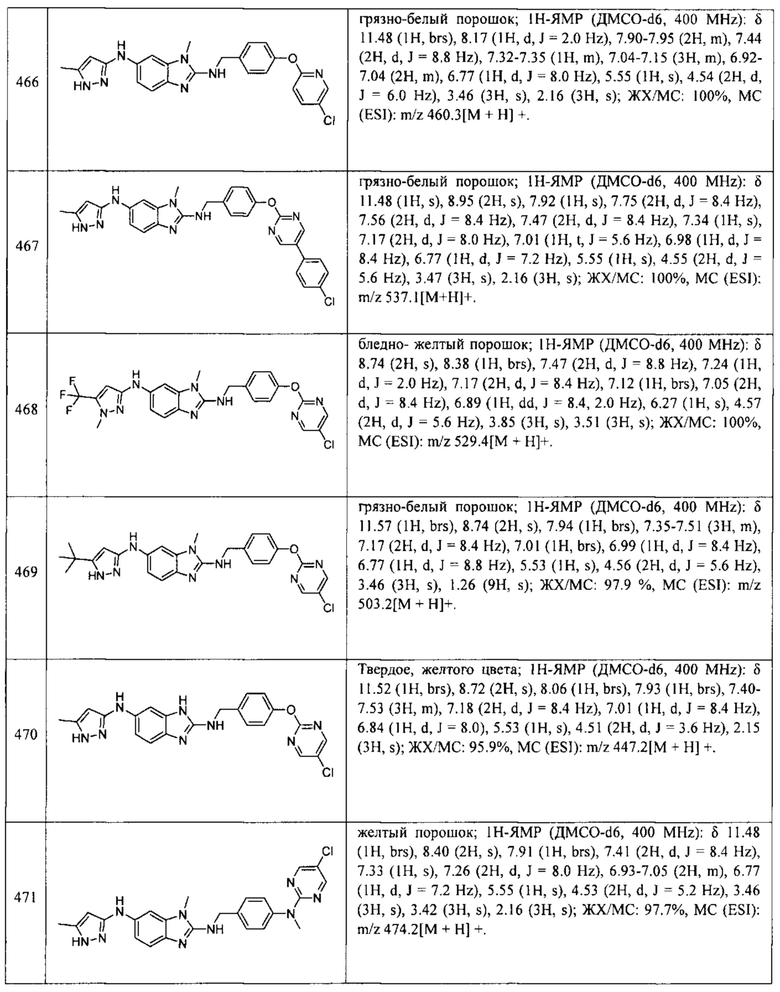



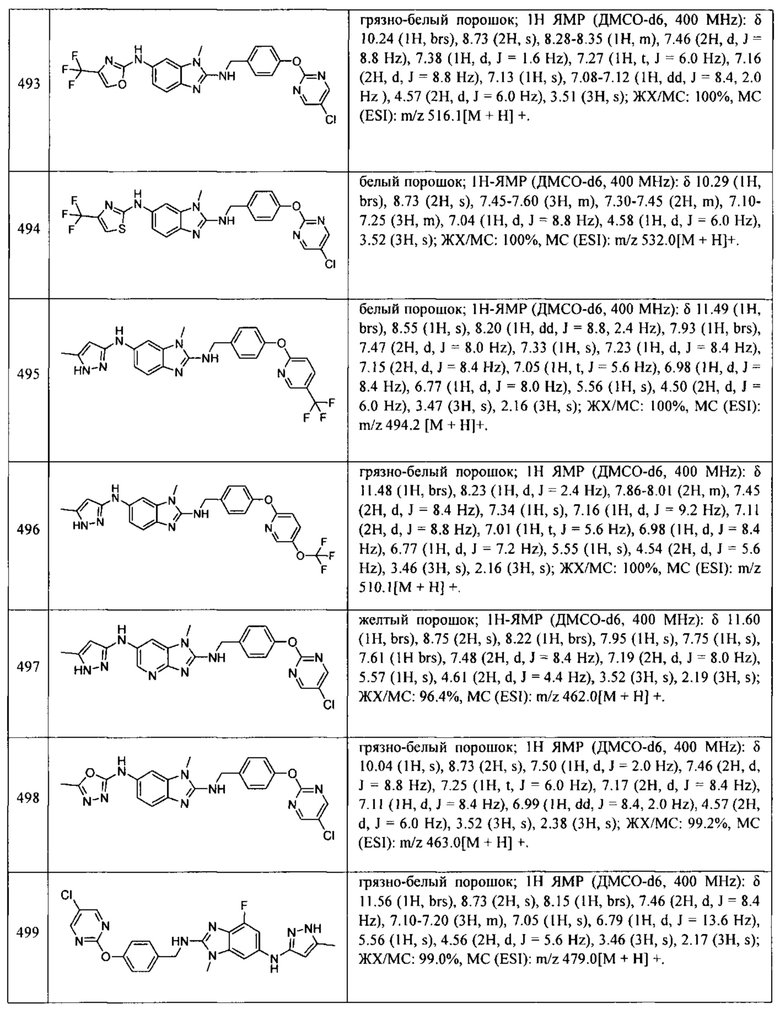





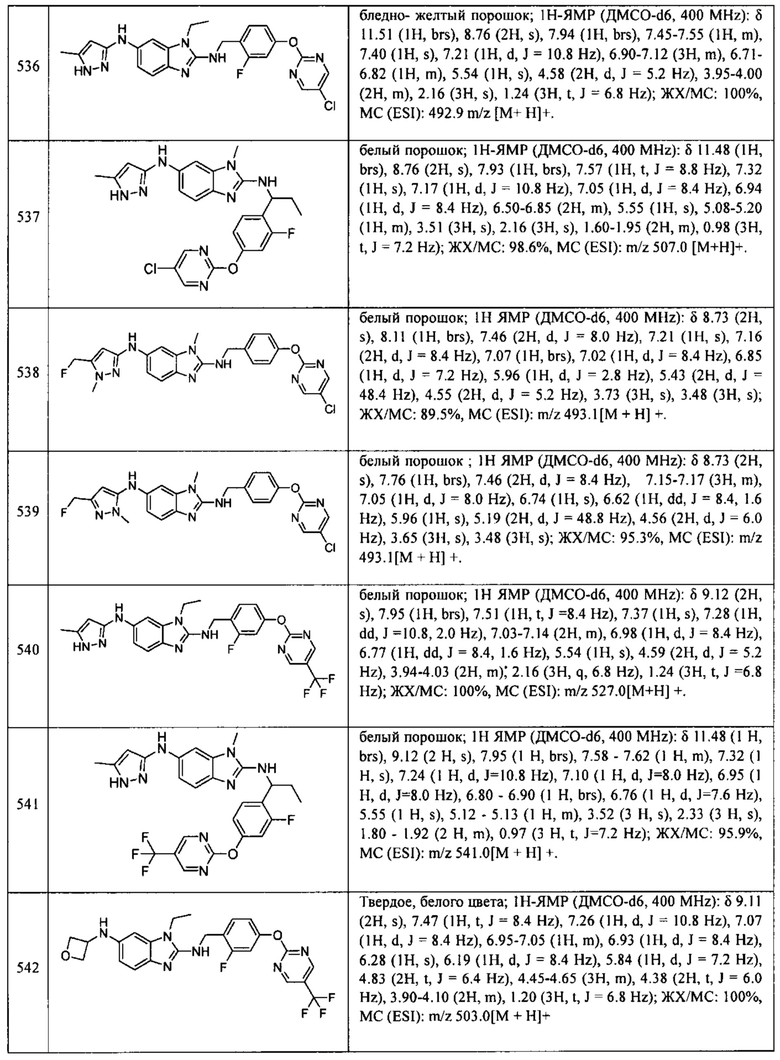
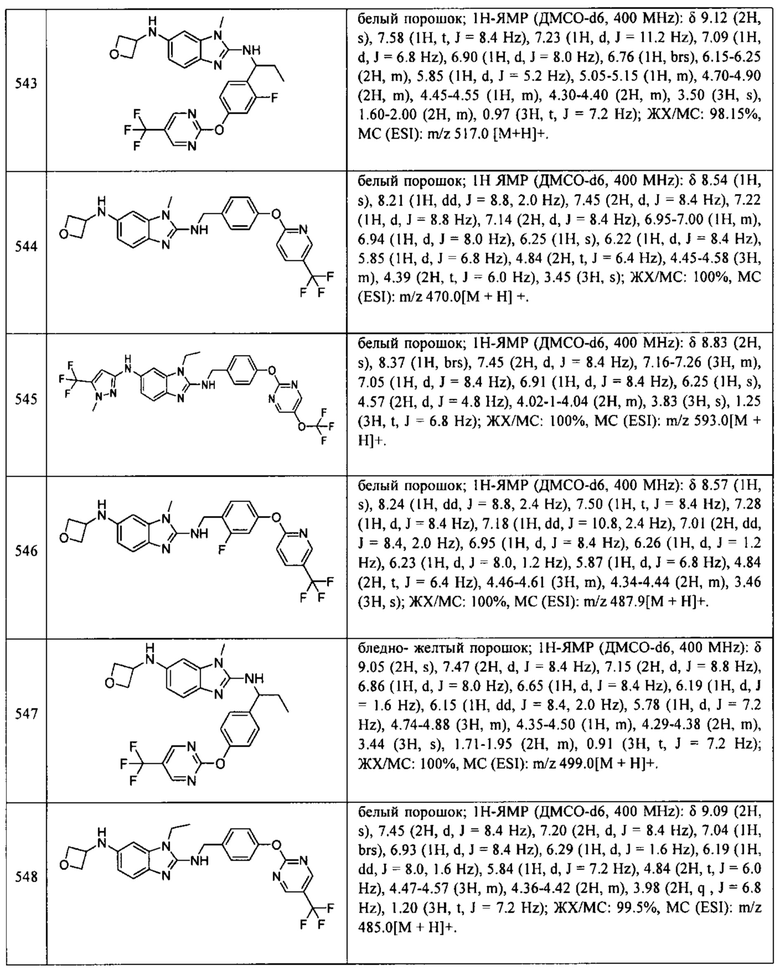

| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКИ АКТИВНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ РЕЦЕПТОРНОЙ ТИРОЗИНКИНАЗЫ СЕМЕЙСТВА ТАМ | 2016 |
|
RU2750727C2 |
| ФАРМАЦЕВТИЧЕСКИ АКТИВНЫЕ ПРОИЗВОДНЫЕ ПИРАЗОЛОТРИАЗИНА И/ИЛИ ПИРАЗОЛОПИРИМИДИНА | 2019 |
|
RU2818563C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛОТРИАЗИНА И/ИЛИ ПИРАЗОЛОПИРИМИДИНА В КАЧЕСТВЕ СЕЛЕКТИВНОГО ИНГИБИТОРА ЦИКЛИНЗАВИСИМОЙ КИНАЗЫ | 2019 |
|
RU2809779C2 |
| ТРИЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ЯНУС-КИНАЗЫ 1, ИХ КОМПОЗИЦИИ, СПОСОБЫ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2019 |
|
RU2824349C2 |
| ПРОИЗВОДНЫЕ ХИНОЛИНА В КАЧЕСТВЕ ИНГИБИТОРОВ РЕЦЕПТОРНОЙ ТИРОЗИНКИНАЗЫ AXL/MER И CSF1R | 2019 |
|
RU2812631C2 |
| СОЕДИНЕНИЕ, НАЦЕЛЕННОЕ НА БЕЛОК И ЕГО ДЕГРАДАЦИЮ, И СПОСОБ ЕГО ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2021 |
|
RU2829459C1 |
| АНАЛОГИ РАПАМИЦИНА КАК ИНГИБИТОРЫ mTOR | 2018 |
|
RU2828109C2 |
| СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ РАСЩЕПЛЯЕМЫЙ ЛИНКЕР, И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2018 |
|
RU2795168C2 |
| НЕЙРОАКТИВНЫЕ СТЕРОИДЫ, ЗАМЕЩЕННЫЕ В ПОЛОЖЕНИИ 10 ЦИКЛИЧЕСКОЙ ГРУППОЙ, ДЛЯ ИСПОЛЬЗОВАНИЯ ПРИ ЛЕЧЕНИИ РАССТРОЙСТВ ЦНС | 2019 |
|
RU2796006C2 |
| ХИРАЛЬНЫЕ ДИАРИЛЬНЫЕ МАКРОЦИКЛЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ ПРОТЕИНКИНАЗ | 2016 |
|
RU2732405C2 |
Изобретение относится к новым гетероциклическим соединениям, а именно производным бензоимидазола, имеющим общую формулу II, где n равно 0 или 1; X1 представляет собой CR5 или N; Y представляет собой C1-С6 алкилен, где алкилен необязательно замещен одной или двумя группами С1-С3 алкила; R5 выбран из группы, состоящей из водорода и галогена; R23 выбран из группы, состоящей из гидроксила, OR11, -NR7R8, C1-С6 алкила, C1 галогеналкила, -C(O)NHR11, фенила, пиридинила и оксетанила, где каждый из указанных фенила, пиридинила и оксетанила необязательно и независимо замещен одной или двумя группами Ra; R24 выбран из группы, состоящей из водорода, C1-С6 алкила, гетероарила, выбранного из пиразолила, тиазолила, оксазолила, оксадиазолила и пиридинила, и гетероциклила, выбранного из оксетанила, тиетанила, тиетанилоксида и азетидинила, где каждый из указанных алкила, гетероарила и гетероциклила необязательно и независимо замещен одной или двумя группами Ra; R25 выбран из группы, состоящей из -(V)pR26 и -(V)p-OR14; при этом р равно 1, V представляет собой C1-С6 алкилен, где алкилен необязательно замещен 1-3 группами C1-С3 алкила; R14 выбран из группы, состоящей из водорода и фенила, где фенил необязательно замещен одной или двумя группами галогена или группами C1 галогеналкокси; R26 выбран из группы, состоящей из водорода, С5 циклоалкила, -ОН, -C(O)NH2, -C(O)OR18, -CN, C1 галогеналкила, и групп формулы IIa, где А2 представляет собой -О-; А3 представляет собой -О-, -CH2O-, -ОСН2-, -C(O)NH-, -C(O)N(R7)-, -N(CH3)- или -NH-; В выбран из группы, состоящей из фенила, гетероарила, выбранного из пиридила, пиримидинила, пиразинила, пиридазинила, оксазолила, бензимидазолила, хинолинила, бензоксазолила, хиноксалинила, хиназолинила, имидазопиридинила, где каждый из указанных фенила и гетероарила необязательно и независимо замещен одной или двумя группами Ra; R7 и R8 независимо представляют собой C1-С6 алкил; R11 представляет собой фенил, замещенный одной группой Ra; R18 представляет собой водород или C1-С6 алкил; R20 выбран из группы, состоящей из водорода, галогена, C1-С6 алкила и C1-С6 алкокси; R21 выбран из группы, состоящей из водорода, галогена и C1-С6 алкила; R22 выбран из группы, состоящей из арила, представляющего собой фенил, и гетероарила, представляющего собой пиридинил, где указанный арил необязательно замещен одной группой Ra; Ra независимо, в каждом случае, выбран из группы, состоящей из галогена, C1-С3 алкила, С3-С6 циклоалкила, C1 алкокси, C1 галогеналкила, C1 галогеналкокси, -CH2ORc, -OCH2Rc, -ORc, -CN, -NRbRc, -C(O)NRbRc, -C(O)ORc, гетероарила, выбранного из имидазолила, и арила, представляющего собой фенил, где каждый из указанных алкила, арила и гетероарила необязательно и независимо замещен одной группой C1-С3 алкила, галогена; Rb и Rc независимо, в каждом случае, выбраны из группы, состоящей из водорода, C1-С10 алкила, С3 и С5 циклоалкила, фенила, гетероарила, выбранного из пиридила и бензоксазолила, где каждый из указанных фенила и гетероарила необязательно и независимо замещен одной группой галогена, C1 галогеналкокси. Также описывается соединение, имеющее общую формулу III, где n равно 1; X1 представляет собой CR5; Y представляет собой C1-С6 алкилен; R5 представляет собой водород; R23 выбран из группы, состоящей из OR11, -C(O)NHR11, C1 алкила и фенила, необязательно и независимо замещенного одной группой Ra; R27 представляет собой водород или оксетанил; R11 представляет собой фенил, замещенный группой Ra; R28 выбран из группы, состоящей из -(V)pR29 и -(V)p-OR14; причем р равно 1, V представляет собой C1-С6 алкилен, где алкилен необязательно замещен одной или двумя группами C1-С6 алкила; R14 выбран из группы, состоящей из водорода и фенила, замещенного одной или двумя группами галогена или C1-С3 галогеналкокси; R29 выбран из группы, состоящей из водорода и фенила, замещенного двумя группами Ra; Ra независимо, в каждом случае, выбран из группы, состоящей из галогена и -ORc; Rc выбран из группы, состоящей из C1-С10 алкила и пиридила, замещенного 1-3 группами галогена. Также раскрывается фармацевтическая композиция на основе указанных соединений и способы лечения. Технический результат: получены новые гетероциклические соединения, полезные в качестве ингибиторов 5-липоксигеназы и/или простагладин Е-синтазы. 5 н. и 23 з.п. ф-лы, 7 пр., 7 табл.
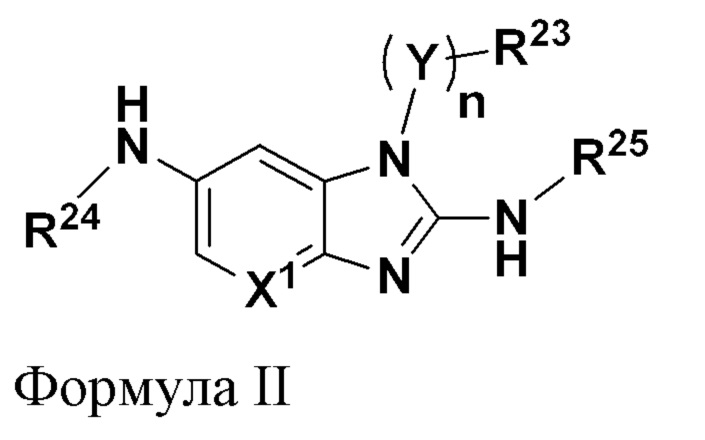


1. Соединение, имеющее общую формулу II
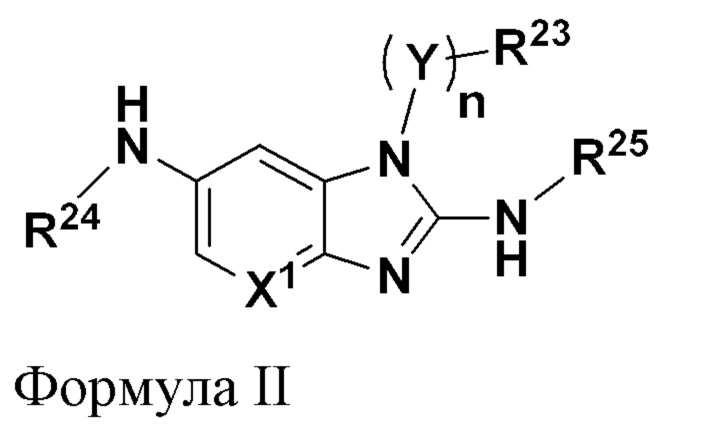
где n равно 0 или 1;
X1 представляет собой CR5 или N;
Y представляет собой C1-С6 алкилен, где алкилен необязательно замещен одной или двумя группами С1-С3 алкила;
R5 выбран из группы, состоящей из водорода и галогена;
R23 выбран из группы, состоящей из гидроксила, OR11, -NR7R8, C1-С6 алкила, C1 галогеналкила, -C(O)NHR11, фенила, пиридинила и оксетанила, где каждый из указанных фенила, пиридинила и оксетанила необязательно и независимо замещен одной или двумя группами Ra;
R24 выбран из группы, состоящей из водорода, C1-С6 алкила, гетероарила, выбранного из пиразолила, тиазолила, оксазолила, оксадиазолила и пиридинила, и гетероциклила, выбранного из оксетанила, тиетанила, тиетанилоксида и азетидинила, где каждый из указанных алкила, гетероарила и гетероциклила необязательно и независимо замещен одной или двумя группами Ra;
R25 выбран из группы, состоящей из -(V)pR26 и -(V)p-OR14;
при этом р равно 1,
V представляет собой C1-С6 алкилен, где алкилен необязательно замещен 1-3 группами C1-С3 алкила;
R14 выбран из группы, состоящей из водорода и фенила, где фенил необязательно замещен одной или двумя группами галогена или группами C1 галогеналкокси;
R26 выбран из группы, состоящей из водорода, С5 циклоалкила, -ОН, -C(O)NH2, -C(O)OR18, -CN, C1 галогеналкила, и групп формулы IIa, приведенных ниже,

где А2 представляет собой -О-;
А3 представляет собой -О-, -CH2O-, -ОСН2-, -C(O)NH-, -C(O)N(R7)-, -N(CH3)- или -NH-;
В выбран из группы, состоящей из фенила, гетероарила, выбранного из пиридила, пиримидинила, пиразинила, пиридазинила, оксазолила, бензимидазолила, хинолинила, бензоксазолила, хиноксалинила, хиназолинила, имидазопиридинила, где каждый из указанных фенила и гетероарила необязательно и независимо замещен одной или двумя группами Ra;
R7 и R8 независимо представляют собой C1-С6 алкил;
R11 представляет собой фенил, замещенный одной группой Ra;
R18 представляет собой водород или C1-С6 алкил;
R20 выбран из группы, состоящей из водорода, галогена, C1-С6 алкила и C1-С6 алкокси;
R21 выбран из группы, состоящей из водорода, галогена и C1-С6 алкила;
R22 выбран из группы, состоящей из арила, представляющего собой фенил, и гетероарила, представляющего собой пиридинил, где указанный арил необязательно замещен одной группой Ra;
Ra независимо, в каждом случае, выбран из группы, состоящей из галогена, C1-С3 алкила, С3-С6 циклоалкила, C1 алкокси, C1 галогеналкила, C1 галогеналкокси, -CH2ORc, -OCH2Rc, -ORc, -CN, -NRbRc, -C(O)NRbRc, -C(O)ORc, гетероарила, выбранного из имидазолила, и арила, представляющего собой фенил, где каждый из указанных алкила, арила и гетероарила необязательно и независимо замещен одной группой C1-С3 алкила, галогена;
Rb и Rc независимо, в каждом случае, выбраны из группы, состоящей из водорода, C1-С10 алкила, С3 и С5 циклоалкила, фенила, гетероарила, выбранного из пиридила и бензоксазолила, где каждый из указанных фенила и гетероарила необязательно и независимо замещен одной группой галогена, C1 галогеналкокси.
2. Соединение по п. 1, которое имеет одну из формул 52, 56, 58, 60, 140, 155, 165, 167, 170, 171, 184, 187, 203, 204, 207-211, 217, 223-227, 229, 250-252, 256-262, 264-268, 270-276, 279-316, 319-328, 330-332, 334-337, 340-364, 366-378, 380-398, 401-428, 430-437, 440, 442, 444, 447-450, 452-456, 458-469, 471, 473-477, 479, 481-483, 486, 488-499, 501-508, 510-512, 515-556, приведенных ниже:






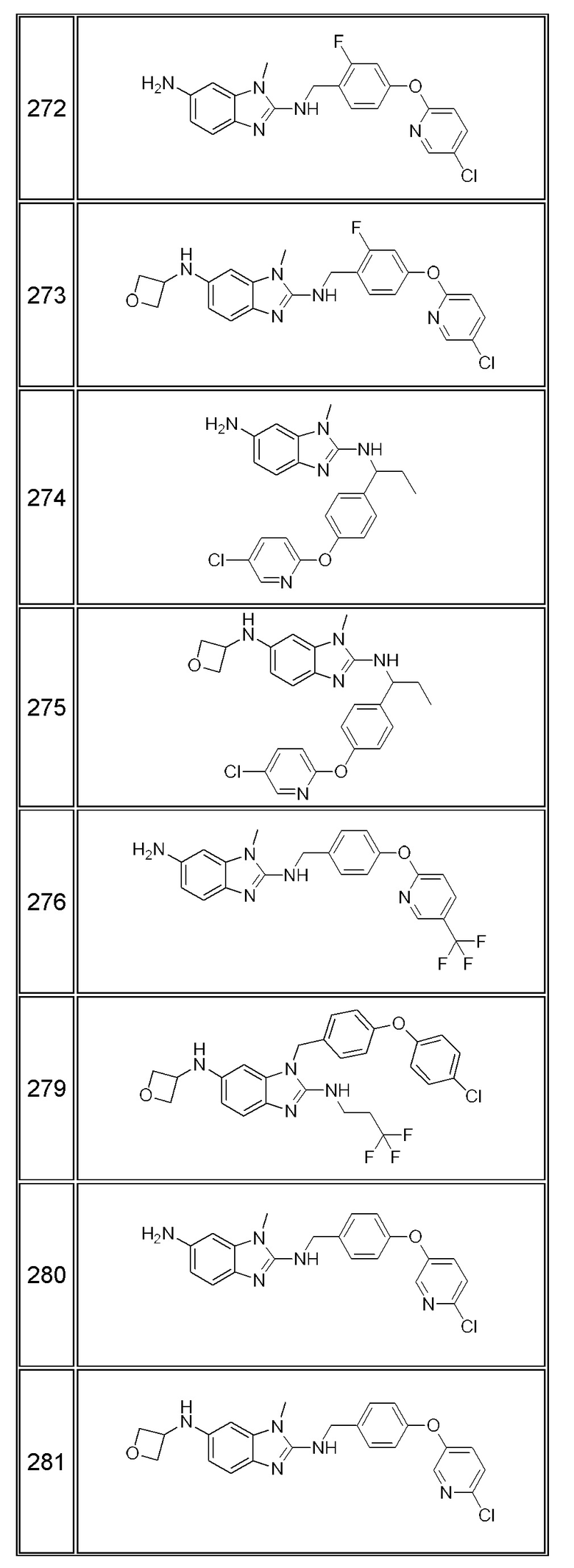








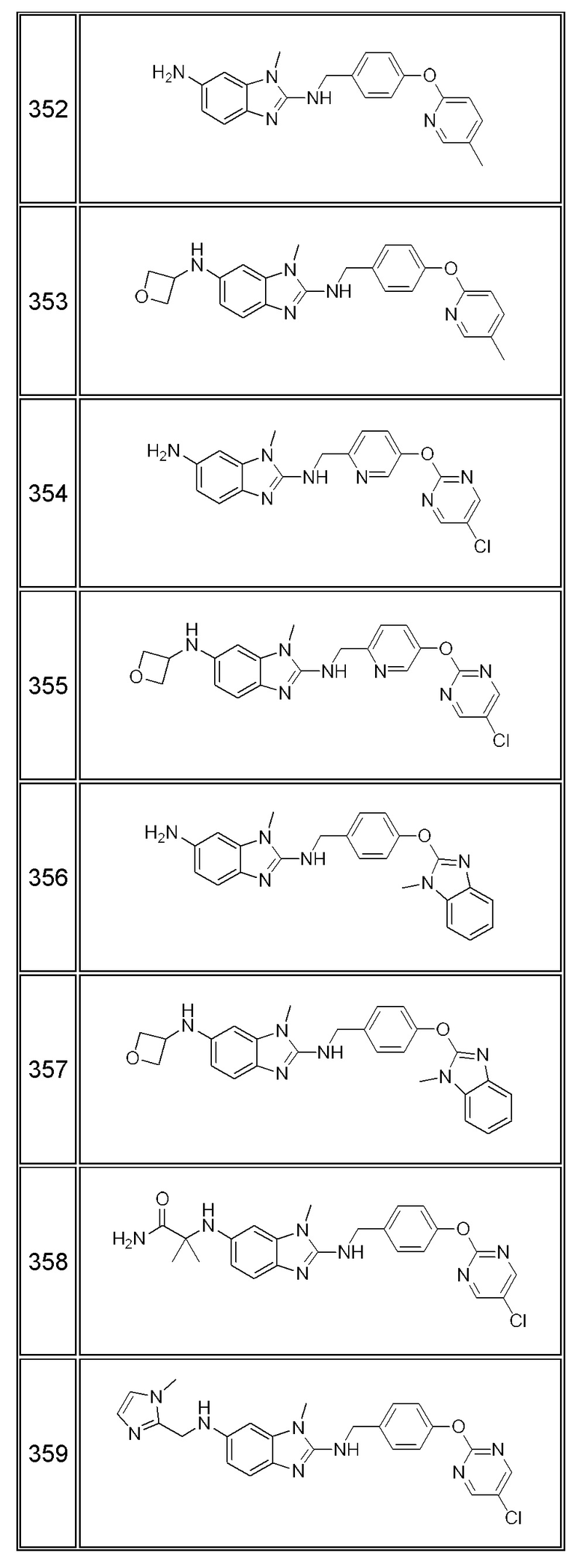
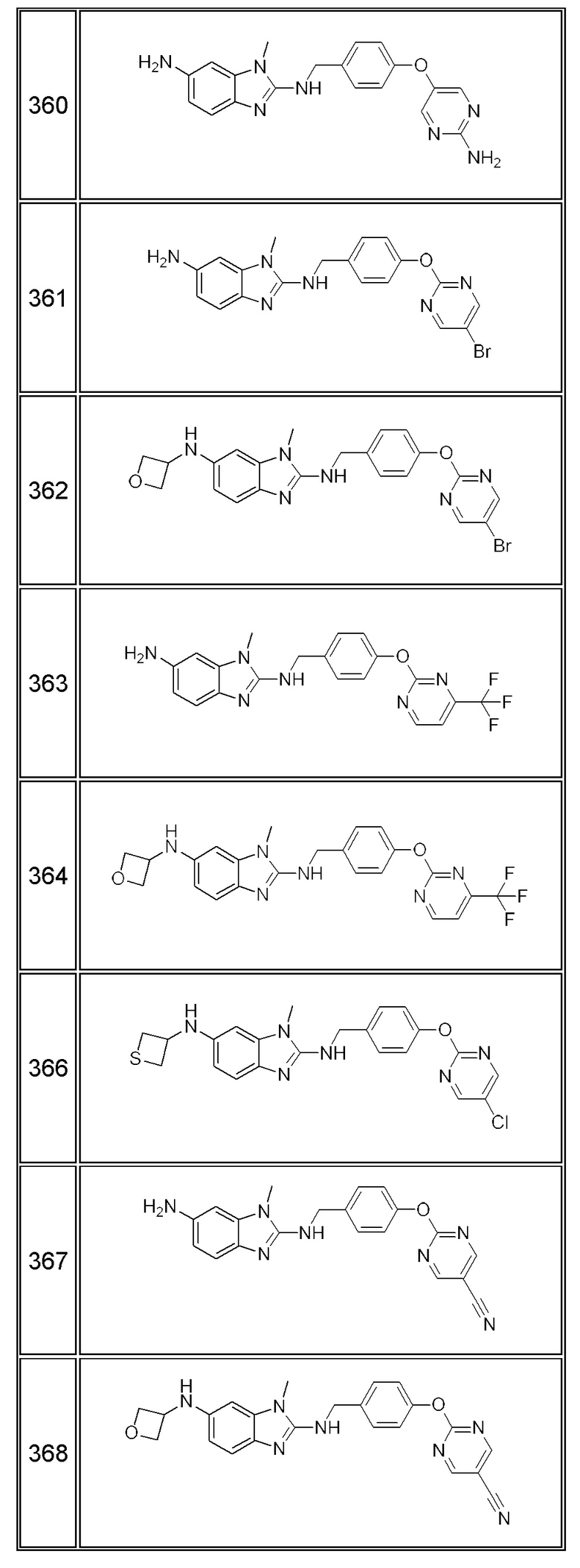
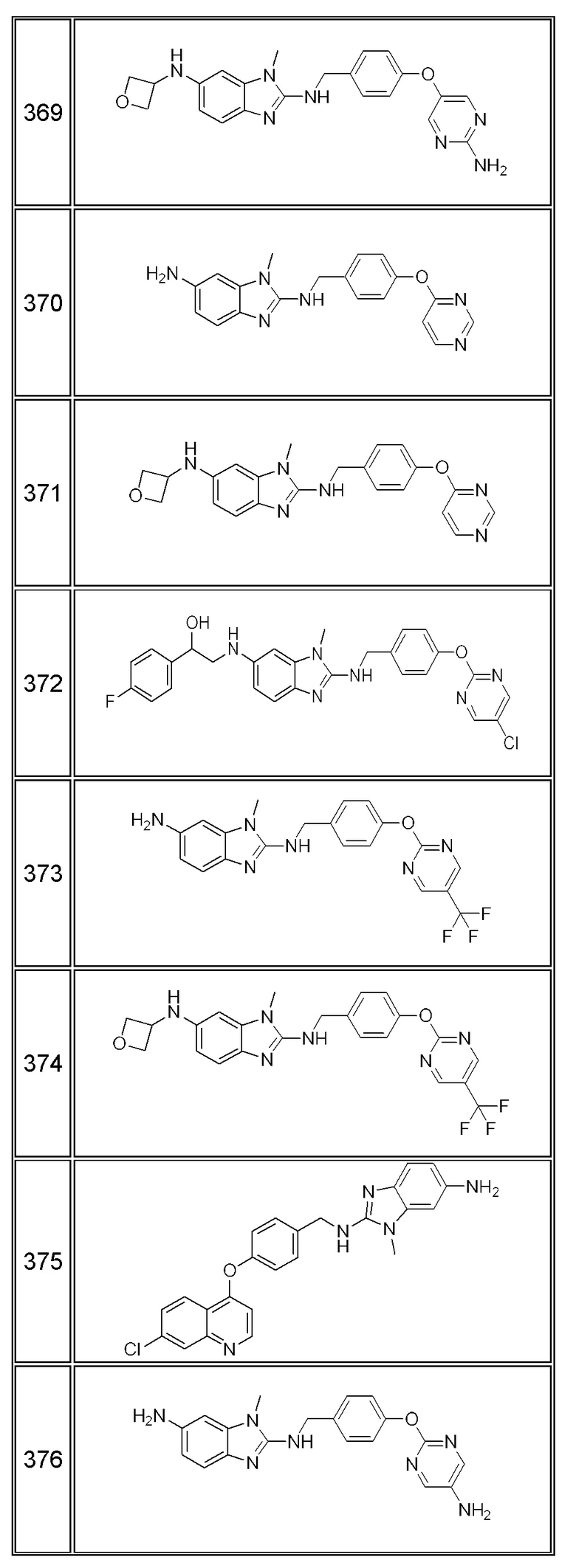







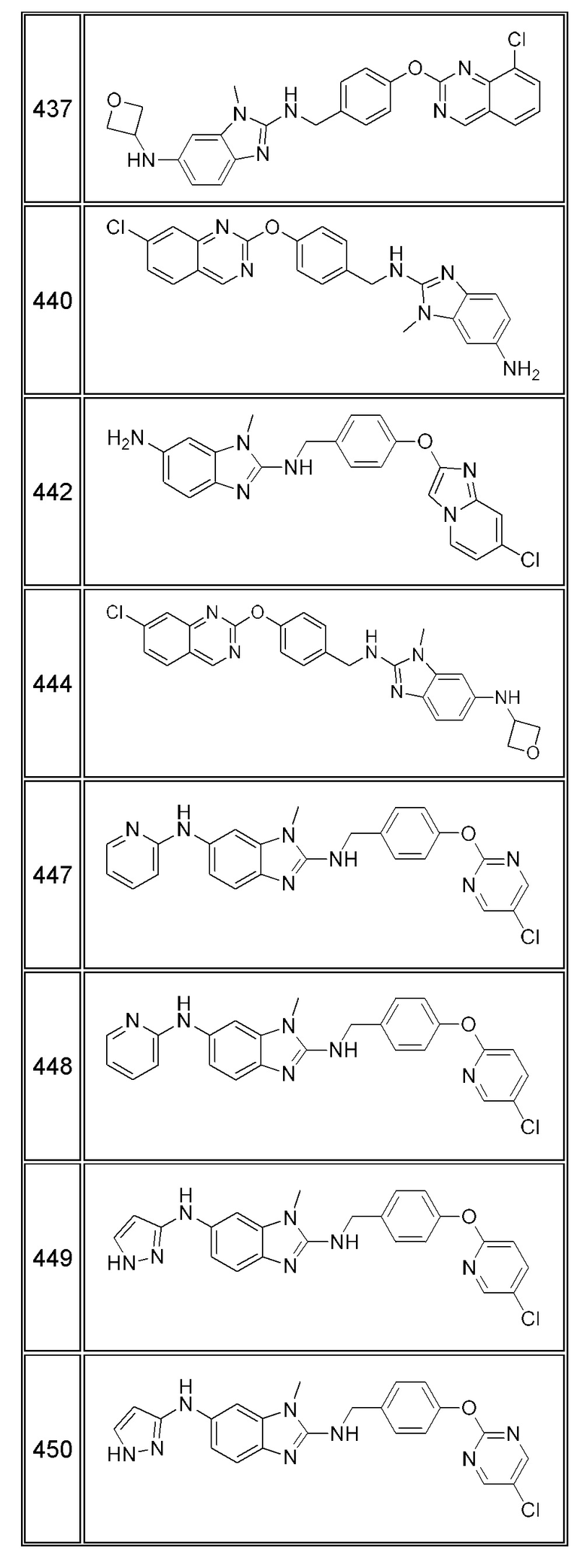









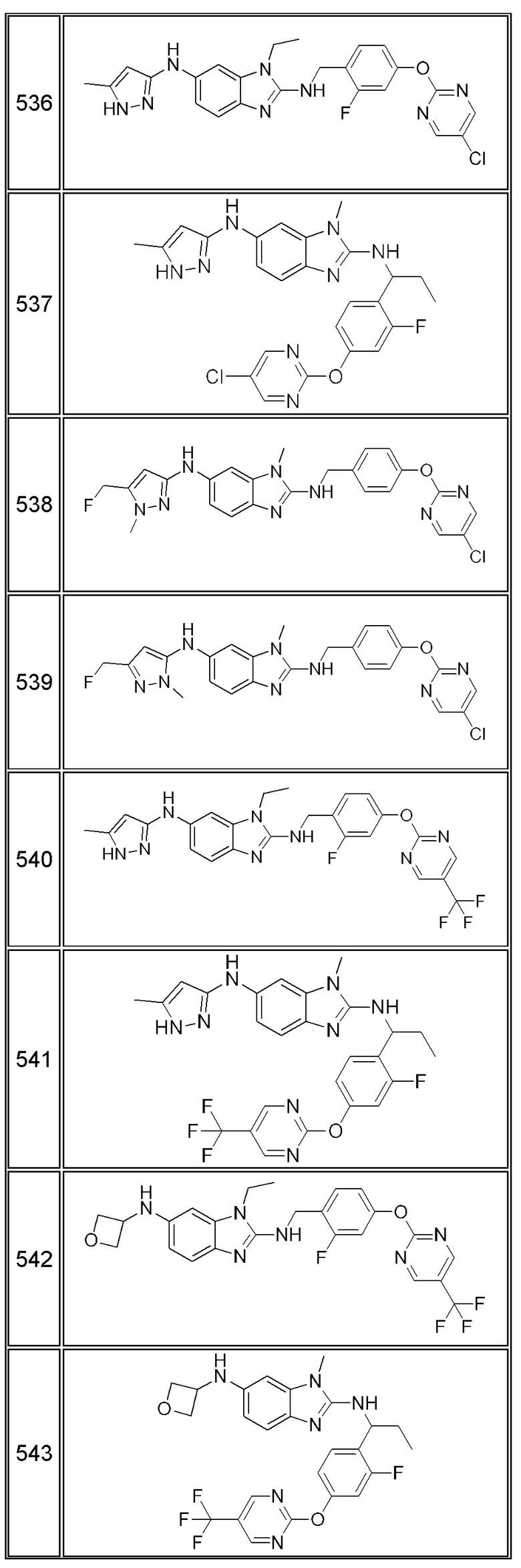

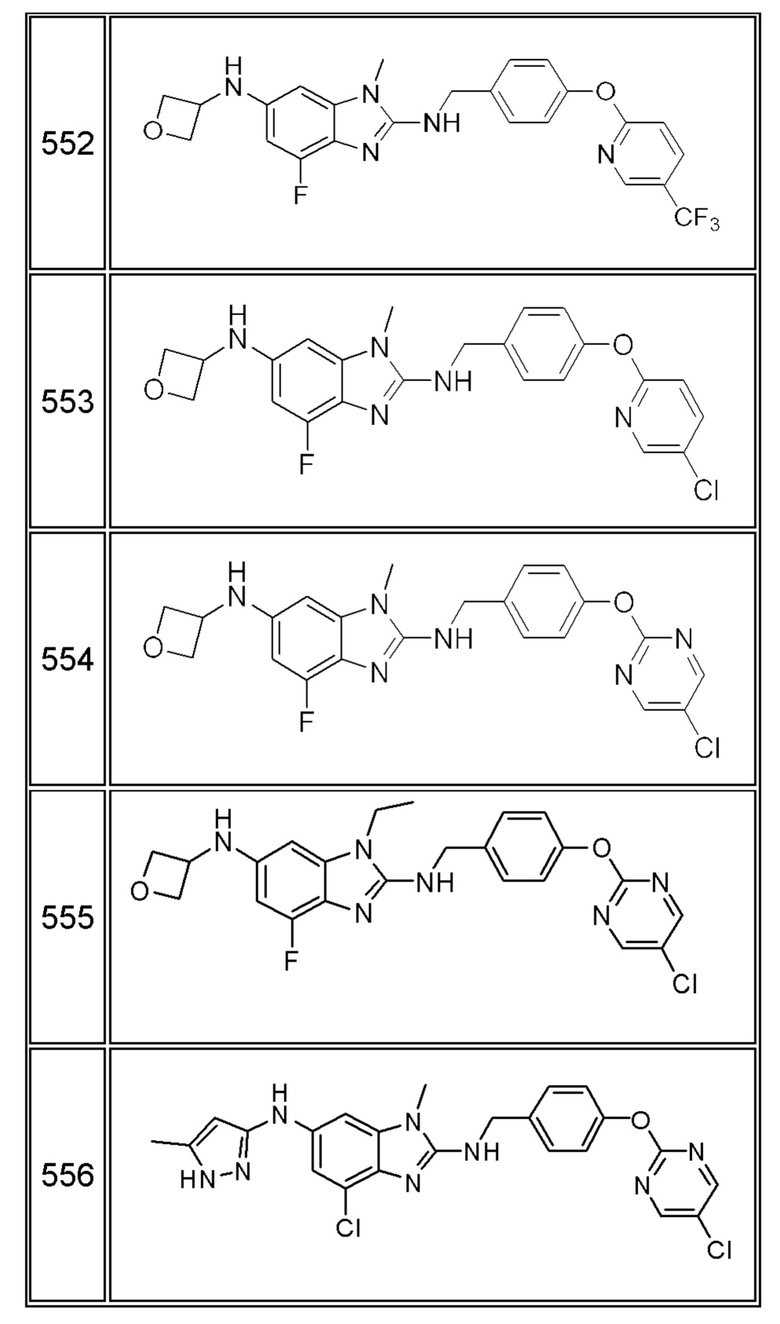
3. Соединение по п. 2, которое имеет одну из формул 52, 56, 58, 60, 140, 155, 165, 167, 170, 171, 184, 187, 203, 204, 207-211, 217, 223-227, 229, 250-252, 256-261, 264-268, 270-276, 280-316, 319-321, 323-328, 330-332, 334-337, 340-354, 356-364, 366-378, 380-385, 387-398, 401-428, 430-437, 440, 442, 444, 449, 450, 452-456, 458-463, 465-469, 471, 473-477, 479, 481-483, 486, 489-492, 495, 496, 499, 501, 503, 505, 507, 512, 525, 529-544 и 546-556.
4. Соединение по п. 2, которое имеет формулу 226.
5. Соединение по п. 2, которое имеет одну из формул 52, 56, 58, 60, 140, 155, 165, 184, 187, 203, 204, 207-211, 217, 223, 224, 226, 227, 229, 250-252, 257-262, 264, 265, 267, 268, 270-276, 279-302, 304-306, 313, 316, 322-328, 330-332, 335, 340, 342, 346, 347, 349, 350, 352, 353, 357-359, 361, 366-369, 372-375, 377, 378, 380, 382-384, 387, 389-393, 395-398, 401-403, 405-407, 410, 412-415, 419-428, 430-432, 434-437, 440, 442, 447-450, 452, 453, 455, 458, 460, 461, 463-469, 471, 473-477, 479, 481, 482, 486, 489-496, 499, 501-508, 510-512, 515-553 и 556.
6. Соединение по п. 2, которое имеет одну из формул 52, 155, 165, 184, 204, 211, 226, 227, 229, 251, 256, 268, 270, 271, 275, 279, 281, 284, 288, 289, 296, 306, 311, 324, 328, 336, 341, 345, 350, 351, 358, 360, 362, 367-369, 373, 374, 378, 381, 392, 412-415, 430, 431, 433, 447-450, 461, 464-466, 468, 469, 471, 473-477, 479, 481, 482, 486, 489-496, 498, 499, 501-506, 508, 512, 516-519, 525-538, 540-547, 550, 553 и 554.
7. Соединение, имеющее общую формулу III

где n равно 1;
X1 представляет собой CR5;
Y представляет собой C1-С6 алкилен;
R5 представляет собой водород;
R23 выбран из группы, состоящей из OR11, -C(O)NHR11, C1 алкила и фенила, необязательно и независимо замещенного одной группой Ra;
R27 представляет собой водород или оксетанил;
R11 представляет собой фенил, замещенный группой Ra;
R28 выбран из группы, состоящей из -(V)pR29 и -(V)p-OR14;
причем р равно 1,
V представляет собой C1-С6 алкилен, где алкилен необязательно замещен одной или двумя группами C1-С6 алкила;
R14 выбран из группы, состоящей из водорода и фенила, замещенного одной или двумя группами галогена или C1-С3 галогеналкокси;
R29 выбран из группы, состоящей из водорода и фенила, замещенного двумя группами Ra;
Ra независимо, в каждом случае, выбран из группы, состоящей из галогена и -ORc;
Rc выбран из группы, состоящей из C1-С10 алкила и пиридила, замещенного 1-3 группами галогена.
8. Соединение по п. 7, которое имеет одну из формул 66, 69, 82 и 144, приведенных ниже:

9. Соединение по п. 8, имеющее одну из формул 66 и 144.
10. Соединение по п. 8, имеющее одну из формул 66 и 69.
11. Соединение по любому из пп. 1-10, предназначенное для использования при лечении заболеваний, связанных с путем 5-ЛОГ и/или путем простагландин Е-синтазы (ПГЕС), где заболевание выбрано из группы, включающей в себя воспалительные заболевания, рак, апоплексический удар и болезнь Альцгеймера, где воспалительные заболевания выбраны из группы, включающей в себя астму, аллергический ринит, дерматит, хроническую обструктивную болезнь легких (ХОБЛ), постинфекционное воспаление, артрит, атеросклероз, аллергии, аутоиммунные заболевания, воспалительные заболевания кишечника, целиакию, угревую сыпь или боль.
12. Соединение по п. 11, в котором воспалительное заболевание выбрано из группы, включающей в себя астму, атеросклероз, боль или ХОБЛ.
13. Соединение по п. 11, где аллергия представляет собой сенную лихорадку, и/или аутоиммунное заболевание представляет собой эритематоз, и/или заболевание кишечника представляет собой болезнь Крона, и/или боль представляет собой воспалительную и/или невропатическую боль.
14. Соединение по любому из пп. 1-10, предназначенное для использования при лечении, которое включает в себя введение подходящего количества соединения, определенного в любом из пп. 1-10, нуждающемуся в этом пациенту, который страдает от воспалительного заболевания, и/или рака, и/или апоплексического удара, и/или болезни Альцгеймера.
15. Соединение по п. 14, для которого вводимое пациенту количество составляет от 0,01 мг/кг до 1 г/кг веса тела пациента.
16. Фармацевтическая композиция, оказывающая ингибирующее действие на фермент, участвующий в таких воспалительных путях, как 5-липоксигеназа, 5-ЛО, 5-ЛОГ, АЛОГ5, которая содержит эффективное количество соединения по любому из пп. 1-10 и фармацевтически приемлемый носитель.
17. Фармацевтическая композиция по п. 16, предназначенная для использования при лечении заболевания, ассоциированного с путем 5-ЛОГ и/или путем простагландин Е-синтазы (ПГЕС), где указанное заболевание выбрано из группы, состоящей из воспалительных заболеваний, рака, апоплексического удара и болезни Альцгеймера.
18. Фармацевтическая композиция по п. 17, где указанное воспалительное заболевание представляет собой одно или несколько из следующих заболеваний: астма, аллергический ринит, дерматит, хроническая обструктивная болезнь легких (ХОБЛ), постинфекционное воспаление, артрит, атеросклероз, аллергии, аутоиммунные заболевания, воспалительные заболевания кишечника, целиакия, угревая сыпь или боль.
19. Фармацевтическая композиция по п. 18, где аллергия представляет собой сенную лихорадку, и/или аутоиммунное заболевание представляет собой эритематоз, и/или воспалительное заболевание кишечника представляет собой болезнь Крона, и/или боль представляет собой воспалительную и/или невропатическую боль.
20. Фармацевтическая композиция по любому из пп. 17-19, где указанное лечение включает в себя введение эффективного количества композиции нуждающемуся в этом пациенту, который страдает от воспалительного заболевания, и/или рака, и/или апоплексического удара, и/или болезни Альцгеймера.
21. Фармацевтическая композиция по п. 20, в которой вводимое пациенту количество составляет от 0,01 мг/кг до 1 г/кг веса тела пациента.
22. Способ лечения заболевания, ассоциированного с путем 5-ЛОГ и/или путем простагландин Е-синтазы (ПГЕС), где указанное заболевание выбрано из группы, состоящей из воспалительных заболеваний, рака и болезни Альцгеймера, причем способ включает в себя введение подходящего количества соединения, определенного в любом из пп. 1-10, или композиции, определенной в п. 16, нуждающемуся в этом пациенту, который страдает от заболевания, ассоциированного с путем 5-ЛОГ и/или путем простагландин Е-синтазы (ПГЕС).
23. Способ лечения по п. 22, в котором указанное воспалительное заболевание выбрано из группы, включающей в себя астму, аллергический ринит, дерматит, хроническую обструктивную болезнь легких (ХОБЛ), постинфекционное воспаление, артрит, атеросклероз, аллергии, аутоиммунные заболевания, воспалительные заболевания кишечника, целиакию, угревую сыпь и боль.
24. Способ лечения по п. 23, в котором аллергия представляет собой сенную лихорадку, и/или аутоиммунное заболевание представляет собой эритематоз, и/или воспалительное заболевание кишечника представляет собой болезнь Крона, и/или боль представляет собой воспалительную и/или невропатическую боль.
25. Способ лечения по любому из пп. 22-24, в котором вводимое пациенту подходящее количество составляет от 0,01 мг/кг до 1 г/кг веса тела пациента.
26. Способ лечения воспалительного заболевания, выбранного из астмы, атеросклероза, боли, хронической обструктивной болезни легких (ХОБЛ), постинфекционного воспаления, аллергического ринита, артрита, воспалительной или невропатической боли, аллергий, аутоиммунных заболеваний, воспалительных заболеваний кишечника, целиакии, угревой сыпи и/или дерматита, или способ лечения рака и/или болезни Альцгеймера, при этом данный способ включает в себя введение подходящего количества соединения по любому из пп. 1-10 нуждающемуся в этом пациенту.
27. Способ лечения по п. 26, где аллергия представляет собой сенную лихорадку, и/или аутоиммунное заболевание представляет собой эритематоз, и/или воспалительное заболевание кишечника представляет собой болезнь Крона, и/или боль представляет собой воспалительную и/или невропатическую боль.
28. Способ лечения по п. 26, в котором пациент страдает от воспалительного заболевания, и/или рака, и/или апоплексического удара, и/или болезни Альцгеймера.
| ЭЛЕКТРИФИЦИРОВАННЫЙ КОРМОРАЗДАТЧИК | 1972 |
|
SU419210A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| WO 00/31067 A1, 02.06.2000 | |||
| Fu, R | |||
| et al.: "Design, synthesis and bioevaluation of dihydropyrazolo[3,4-b]pyridine and benzo[4,5]imidazo[1,2-a]pyrimidine compounds as dual KSP and Aurora-A kinase | |||

