ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям, которые модулируют связывание иммуноглобулинов с Fc рецепторами и к их применению.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Fc рецепторы (FcR) являются семейством тесно связанных между собой рецепторов, которые являются специфичными для Fc фрагмента иммуноглобулина (Ig). Эти рецепторы имеют основное значение для нормального иммунитета и устойчивости к инфекции и обеспечивают гуморальную иммунную систему возможностями клеточного эффектора. Рецепторы являются специфичными для каждого из классов иммуноглобулинов и таким образом характеризуются тем классом Ig, с которым они связываются (то есть Fc гамма рецептор (FcgR) связывается с гамма иммуноглобулином (IgG), Fc эпсилон рецептор (FcgR) связывается с эпсилон иммуноглобулином (IgE), Fc альфа рецептор (FcaR) связывается с альфа иммуноглобулином (IgA)). Среди FcgR рецепторов определены члены трех специфичных подсемейств; FcgRI, являющийся высокоспецифичным рецептором по отношению к IgG; FcgRII, являющиеся рецепторами с низким сродством к IgG, который энергично связывается с агрегатами иммунных комплексов; и FcgRIII, являющиеся рецепторами с низким сродством, которые связываются с иммунными комплексами. Эти рецепторы структурно тесно связаны, но осуществляют различные функции. Представляет интерес структура и функция FcgRII ввиду его взаимодействия с иммунными комплексами и его связи с заболеванием.
FcgR экспрессируются большинством гемопоэтических клеток и, связываясь с IgG, играют ключевую роль в гомеостазе иммунной системы и защите хозяина от инфекции. FcgRII представляет собой рецептор с низким сродством к IgG и в значительной степени связывается лишь с иммунными комплексами IgG и экспрессируется клетками различных типов, включая, например, моноциты, макрофаги, нейтрофилы, эозинофилы, тромбоциты и В лимфоциты. FcgRII принимает участие в различных иммунных и воспалительных реакциях, включая антителозависимую клеточно-опосредованную цитотоксичность, клиренс иммунных комплексов, выделение медиаторов воспаления и регулирование образования антител. Связывание IgG с FcgR может приводить к показаниям при заболеваниях, которые включают в себя регуляцию FcgR. Например, аутоиммунное заболевание тромбоцитопеническая пурпура вызывает повреждение ткани (тромбоцитов), происходящее от FcgR-зависимой активации тромбоцитами иммунного комплекса IgG или их разрушения FcgR+ фагоцитами. В дополнение к этому, известны различные воспалительные заболевания, в которых участвуют иммунные комплексы IgG (например, ревматоидный артрит, эритематоз системной волчанки), включая аллергические реакции II типа и III типа. Аллергические реакции II типа и III типа осуществляются при участии IgG, который может активировать комплиментарно-опосредованный или фагоцит-эффекторный механизмы, обуславливающие повреждение ткани.
Ввиду того что FcR вовлечены в различные биологические механизмы, существует потребность в соединениях, которые влияют на связывание иммуноглобулинов с FcR. Также существует потребность в применении таких соединений для лечения различных заболеваний.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
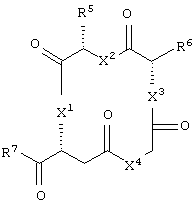
По настоящему изобретению предлагается фармацевтическая композиция, включающая в себя:
(а) соединение, выбранное из группы, включающей в себя ароматическое соединение формулы:
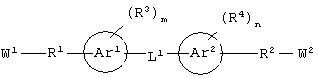
гетероароматическое соединение формулы:
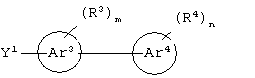
циклическое соединение формулы:
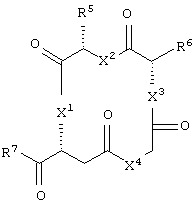
бициклическое соединение формулы:
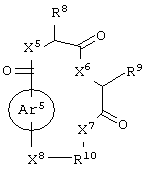
и аминокислотное производное формулы:
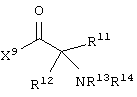
или их соли, где
каждый из W1 и W2 независимо представляет собой CO2R15, C(=NH)NH(OH), SО3R15, C(=NH)NH2, OPO(OR15)2, С(=O)СF3 или PO(OR15)2;
каждый из Аr1, Аr2, Аr4 и Аr5 независимо представляет собой C6-С20 арил или C1-С20 гетероарил;
Аr3 представляет собой C1-С20 гетероарил;
каждый из X1, X2, X3, X4, X5, X6, X7 и X8 независимо представляет собой метилен, О, S или NR16;
каждый R1 и R2 независимо представляет собой связь, C1-С6 алкилен или галогенированный C1-С6 алкилен;
каждый R3 и R4 независимо представляют собой галоген, -Z1 или C1-С6 алкил;
каждый X9, Y1 и Z1 независимо представляет собой OR17, SR17 или NR17R18;
каждый R5 и R6 независимо представляет собой аминокислотный остаток боковой цепи или частицу формулы -R19-W3;
каждый R8, R9 и R11 независимо представляет собой аминокислотный остаток боковой цепи при условии, что R11 не представляет собой Н или СН3;
R7 представляет собой OR20, NR21R22 или примерно от 1 до 10 аминокислот;
R10 представляет собой C1-С6 алкилен;
R12 представляет собой C1-С6 алкил или C6-С20 аралкил;
W3 представляет собой С(=O)Х10;
X10 представляет собой OR23 или NR24R25;
каждый R13, R15, R17. R18, R20, R21, R23 и R24 независимо представляет собой водород или C1-С6 алкил;
каждый R16 независимо представляет собой Н, C6-С20 арил или защитную группу амида;
R19 представляет собой C1-С6 алкилен;
каждый R22 и R25 независимо представляет собой Н, C1-С6 алкил или защитную группу амида;
R14 представляет собой Н, C1-С6 алкил или защитную группу амина;
L представляет собой связующую группу, включающую в себя от 1 до 20 атомов; и
каждый m и n независимо представляет собой целое число от 0 до 2; и
(b) фармацевтически приемлемый носитель.
По настоящему изобретению также предлагается способ применения соединения, выбранного из группы, включающей в себя замещенные или незамещенные бензойные кислоты; нуклеозиды и их аналоги; фолиевую кислоту и ее производные; пептиды, включающие в себя примерно от 2 до 10 аминокислотных остатков или их производных, предпочтительно трипептиды или гексапептиды; макроциклические соединения, содержащие кольцо, которое включает в себя примерно от 8 атомов до 18 атомов, предпочтительно циклические пептиды или их производные, и соединения с вышеупомянутыми формулами для модулирования, например ингибирования или усиления связывания иммуноглобулинов с Fc рецепторами пациента. В отдельном воплощении настоящего изобретения такое модулирование Fc рецепторов вышеупомянутыми соединениями применяется для лечения заболевания, при котором вырабатываются агрегаты антител или при котором вырабатываются иммунные комплексы в результате контакта антитела с внутренним или внешним антигеном. Модулирование Fc рецепторов вышеуказанными соединениями может также применяться для уменьшения IgG-зависимого повреждения ткани, для уменьшения IgE-зависимой реакции и/или для снижения воспаления у пациента.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На фиг. 1 схематически изображен сайт связывания на рецепторе FcgRIIa.
На фиг. 2 представлен боковой схематический вид бороздки, показана только одна поверхность с представляющими интерес остатками белка.
На фиг. 3 показано, как отдельный лиганд соотносится с общим строением соединения по настоящему изобретению.
Фиг. 4 отображает различные водородные связи между аминокислотами в сайте связывания рецептора FcgIIa и конкретного модулятора.
На фиг. 5А и 5В представлены некоторые соединения, модулирующие Fc рецепторы, включая те, которые соответствуют модулированию Fc рецептора, представленного на фиг. 6-9.
На фиг. 6 отображена активность модулирования некоторыми соединениями, представленными на фиг. 5А и 5В, связывания FcgRIIa с IgG1 человека.
На фиг. 7 отображена активность модулирования некоторыми соединениями, представленными на фиг. 5А и 5В, связывания FcgRIIa с IgG3 человека.
На фиг. 8 отображена активность модулирования некоторыми соединениями, представленными на фиг. 5А и 5В, связывания FcgRIIa с IgG1 человека.
На фиг. 9 отображена активность модулирования некоторыми соединениями, представленными на фиг. 5А и 5В, связывания FcgRIIa с IgG3 человека.
На фиг. 10 отображено усиление связывания sFcgRII с IgG1 и IgG3 в присутствии гексапептида.
На фиг. 11 отображено ингибирование связывания sFcgRII с IgG1 и IgG3 в присутствии трипептида.
На фиг. 12 представлен график зависимости повышения прозрачности от времени в присутствии только агониста.
На фиг. 13 представлен график зависимости повышения прозрачности от времени в присутствии агониста и соединения BRI6855.
На фиг. 14 представлен график зависимости % агрегации тромбоцитов при различных концентрациях соединения BRI6728.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
По настоящему изобретению предлагаются различные соединения, которые могут модулировать взаимодействие между Fc рецепторами и иммуноглобулинами. Вне связи с какой-либо из теорий считается, что особенно полезные соединения взаимодействуют с областью С (см. фиг. 1) Fc рецептора, например FcgRII. Таким образом, считается, что эти соединения препятствуют димеризации двух FcgRII белков, влияя, таким образом, на передачу клеточного сигнала через один или оба этих FcR белка. В частности, считается, что пептидные остатки 117-131 и 150-164 FcgRII образуют пограничную область FcgIIa димера, и предполагается, что соединения, которые имеют соответствующую структуру или способны связываться с этими областями, являются хорошими модуляторами связывания. Например, природный гексапептид Phe121 - Ser126 или более короткие сегменты перекрывают область значительного взаимодействия, обусловленного наличием водородных связей, и, таким образом, являются подходящими модуляторами димеризации двух молекул FcgRIIa. Такой фрагмент белка описан как часть SEQ ID №3 патентной заявки США №09/245764, "3-мерные структуры и модели Fc рецепторов и их применение", поданной 5 февраля 1999.
Соединения по настоящему изобретению были получены в результате произвольного скрининга, а также в результате целенаправленного создания лекарства, для модулирования Fc рецепторов. FcgR экспрессируются большинством гемопоэтических клеток и, связываясь с IgG, играют ключевую роль в гомеостазе иммунной системы и в защите хозяина от инфекции. FcgRII является рецептором с низким сродством к IgG, которые существенно связываются только с иммунными комплексами IgG и экспрессируются клетками различных типов, включая, например, моноциты, макрофаги, нейтрофилы, эозинофилы, тромбоциты и В лимфоциты. FcgRII оказывается вовлеченным в различные иммунные и воспалительные реакции, включая антителозависимую клеточно-опосредованную цитотоксичность, клиренс иммунных комплексов, выделение медиаторов воспаления и регулирование образования антител.
Связывание IgG с FcgR может приводить к возникновению заболевания, которое связано с регуляцией посредством FcgR. Например, аутоиммунное заболевание тромбоцитопеническая пурпура вызывает повреждение тканей (тромбоцитов), происходящее от FcgR-зависимой активации тромбоцитами иммунного комплекса IgG или их разрушения FcgR+ фагоцитами. В дополнение к этому, известны различные воспалительные заболевания, в которых участвуют иммунные комплексы IgG (например, ревматоидный артрит, эритематоз системной волчанки), включая аллергические реакции II типа и III типа. Аллергические реакции II типа и III типа осуществляются при участии IgG, который может активировать комплиментарно-опосредованный или фагоцитно-эффекторный механизмы, обуславливающие повреждение ткани.
Знание трехмерной структуры FcgRIIa или, разумеется, любого FcR может упростить приготовление терапевтических и диагностических реагентов для лечения заболевания. Например, зная структуру области связывания FcgRIIa, можно проектировать соединения, которые могут модулировать связывание иммуноглобулинов с FcgRIIa. Структура ряда Fc рецепторов, включая FcgRIIa, FceRI и FcgRIIIb, приводится в предварительной заявке на патент США №60/073972, поданной 6 февраля 1998, и в вышеупомянутой патентной заявке США №09/245764, "3-мерные структуры и модели Fc рецепторов и их применение", поданной 5 февраля 1999.
FcgRIIa является белковым димером и имеет ось симметрии С2. На фиг. 1 представлена схема структура области связывания FcgRIIa по данным рентгеноструктурного анализа. Вне связи с какой-либо из теорий считается, что сайты А и А1 должны быть областями взаимодействия с Fc-антителами; таким образом, соединение, которое связывается или взаимодействует с сайтами А или А1, скорее всего препятствует нормальному связыванию этого рецептора с IgG. В дополнение к этому, соединение, которое связывается с сайтами В, С и/или D, может препятствовать или упрощать связывание антитела, если данное соединение изменяет структуру этого рецептора так, что дестабилизирует связывание с антителом или способствует димеризации рецепторов соответственно.
На фиг. 2 представлен боковой схематический вид сайта В, т.е. бороздка, отображающий только одну поверхность, с представляющими интерес остатками белка при проектировании модулятора. Край бороздки содержит остатки лизина и гистидина и представляет собой мишень для взаимодействия с водородсвязывающимися и/или кислотными группами подходящего модулятора. Поверхностная часть бороздки содержит фенилаланинбензольное кольцо и может являться мишенью при гидрофобном взаимодействии, в частности при р-р взаимодействиях. "Пол" бороздки содержит остатки Phe121, Thr152, Leu159 и Ser161 вместе с Asn154, Lys117 (каркасный карбонил) и Thr119. Предполагается, что эти белки располагаются так, что образуют карман, который способен к сильному связыванию с водородом и/или к Ван-дер-Ваальсовым взаимодействиям с модулятором или лигандом.
Особенности описанной выше бороздки обуславливают конструкцию и синтез соединения, которое в целом изображается следующим образом:
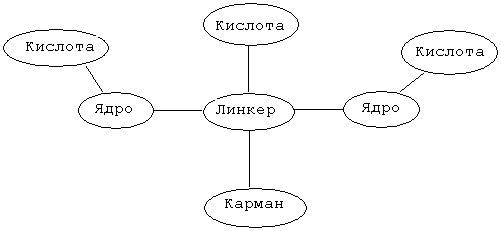
где "ядро" является липофильной группой, такой как ароматическое кольцо, и "линкер" (связующая группа) представляет собой связанную совокупность от 1 до 20 атомов, предпочтительно от 1 до 10 атомов и более предпочтительно от 2 до 8 атомов. Наличие кислоты и групп, формирующих карман, который непосредственно подсоединен к связующей группе, является необязательным. Для обеспечения благоприятного взаимодействия с основными группами, например с Lys117 и His131, на краю бороздки, кислотные группы ("кислота") могут ответвляться от "ядра" и/или "связующей группы". "Карман" представляет собой ту часть молекулы, которая заполняет карманы в нижней части бороздки. Альтернативно, модулятор может связываться или занимать только карман рецептора. Эти основные положения поясняются примером на фиг. 3, которая показывает, как отдельный модулятор соотносится с общей конструкцией, представленной выше, а также поясняются фиг. 4, которая показывает точки взаимодействия между этим модулятором и белком FcgRIIa.
Пример соединения, содержащего остатки, формирующие "карман", показан на фиг. 3, где цитозинподобное кольцо присутствует в области связующей группы соединения. Иные подходящие связующие частицы кармана включают в себя нуклеиновые кислоты и связанные с ними структуры, такие как гидразиды и амидомочевины, которые представлены ниже, или их производные.
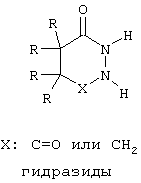
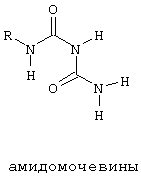
Предпочтительно, чтобы эти остатки, образующие карман, были бы представлены димером соединения, например димером нуклеиновой кислоты, гидразидами, амидомочевинами или их производными.
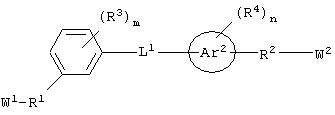
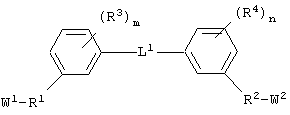
Соединения по настоящему изобретению, которые обладают вышеупомянутыми основными особенностями, включают в себя ароматическое соединение формулы:
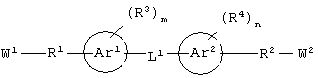
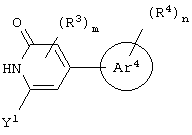
гетероароматическое соединение формулы:
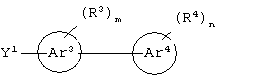
циклическое соединение формулы:
бициклическое соединение формулы:
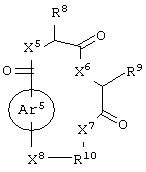
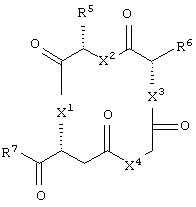
и аминокислотное производное формулы:
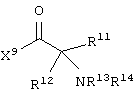
или его соли, где каждый W1 и W2 независимо представляет собой CO2R15, C(=NH)NH(OH), SО3R15, C(=NH)NH2, OPO(OR15)2, С(=O)СF3 или PO(ORI5)2; каждый Аr1, Аr2, Аr4 и Аr5 независимо представляет собой С6-С20 арил или C1-C20 гетероарил; Аr3 представляет собой C1-C20 гетероарил; каждый X1, X2, X3, X4, X5, X6, X7 и X8 независимо представляет собой метилен, О, S или NR16; каждый R1 и R2 независимо представляет собой связь, C1-С6 алкилен, или галогенированный C1-С6 алкилен; каждый R3 и R4 независимо представляют собой галоген, -Z1 или C1-С6 алкил; каждый X9, Y1 и Z1 независимо представляет собой OR17, SR17 или NR17R18; каждый R5 и R6 независимо представляет собой аминокислотный остаток боковой цепи или частицу формулы -R19-W3; каждый R8, R9 и R11 независимо представляет собой аминокислотный остаток боковой цепи при условии, что R11 не представляет собой Н или СН3; R7 представляет собой OR20, NR21R22 или примерно от 1 до 10 аминокислотных остатков; R10 представляет собой C1-С6 алкилен; R12 представляет собой C1-С6 алкил или C6-С20 аралкил; W3 представляет собой С(=O)Х10; X10 представляет собой OR23 или NR24R25; каждый R13, R15, R17, R18, R20, R21, R23 и R24 независимо представляет собой водород или C1-С6 алкил; каждый R16 независимо представляет собой Н, C6-С20 арил или защитную группу амида; R19 представляет собой C1-С6 алкилен; каждый R22 и R25 независимо представляет собой Н, C1-С6 алкил или защитную группу амида; R14 представляет собой Н, C1-С6 алкил или защитную группу амина; L представляет собой связующую группу, содержащую от 1 до 20 атомов; и каждый m и n независимо представляет собой целое число от 0 до 2.
В соответствии с настоящим изобретением "алкильные" группы представляют собой алифатические углеводороды, которые могут являться группами с прямой или с разветвленной цепью. Алкильные группы возможно могут быть замещены одним или более чем один заместителем, таким как галоген, алкенил, алкинил, арил, гидрокси, амино, тио, алкокси, карбокси, оксо или циклоалкил. Между алкильными группами может содержаться один или более чем один атом кислорода, серы или замещенные или незамещенные атомы азота. Примеры алкильных групп включают в себя метил, этил, изо-пропил, н-бутил, трет-бутил, фторметил, дифторметил, трифторметил, хлорметил, трихлорметил, метоксиэтил, аминометил и пентафторэтил.
"Арильные" группы представляют собой моноциклические или бициклические карбоциклические или гетероциклические ароматические кольцевые частицы. Арильные группы могут быть замещены одним или более чем одним заместителем, таким как галоген, алкенил, алкил, алкинил, гидрокси, амино, тио, алкокси или циклоалкил.
"Моноарил или гетероарил" относится к моноциклическому карбоциклическому или гетероциклическому ароматическому кольцу. Примеры моноарильных или гетероарильных колец включают в себя пиррол, тиофен, фуран, имидазол, пиразол, 1,2,4-триазол, пиридин, пиразин, пиримидин, пиридазин, тиазол, изотиазол, оксазол, изоксазол, s-триазин и бензол. Предпочтительной группой является фенил.
"Диарил или гетероарил" означает бициклическую кольцевую систему, содержащую два конденсированных карбоциклических и/или гетероциклических ароматических кольца. Примеры диарильных или гетероарильных колец включают в себя инден, изоинден, бензофуран, дигидробензофуран, бензотиофен, индол, 1Н-индазол, индолин, азулен, тетрагидроазулен, бензопиразол, бензоксазол, бензоимидазол, бензотиазол, 1,3-бензодиоксол, 1,4-бензодиоксан, пурин, нафталин, тетралин, кумарин, хромон, хромен, 1,2-дигидробензотиопиран, тетрагидробензотиопиран, хинолин, изохинолин, хиназолин, пиридо[3,4-b]-пиридин и 1,4-бенизоксазин.
"Аралкил" относится к алкильной группе, замещенной арильной группой. Подходящие аралкильные группы включают, но не ограничиваются бензилом, 2-фенилэтилом и пиколилом. Арильные группы могут также быть замещены другими подходящими функциональными группами. Аралкильные группы включают в себя группы с гетероциклическими и карбоциклическими ароматическими группами.
"Связующая группа" (L1) относится к цепочке атомов, которая связывает Аr1 с Аr2 определенным числом атомов. Это число, соответствующее связующей группе, относится только к числу атомов, которые непосредственно связывают Аr1 и Аr2. Группа L1 может содержать группы, которые могут участвовать в образовании водородных связей и/или во Ван-дер-Ваальсовых взаимодействиях с аминокислотными остатками в бороздке рецептора, например трифторацетил, имид, мочевина, амидин, амидоксим или их производные.
"Аминокислотный остаток боковой цепи" относится к аминокислотной боковой цепи, которая располагается у α -углеродного атома природных и имеющихся в продаже α -аминокислот. Обычные аминокислотные остатки боковой цепи включают в себя водород (глицин), метил (аланин), -CH2CH2CH2NHC(=NH)NH2 (аргинин), CH2C(=O)NH2 (аспарагин), -СН2СО2Н (аспарагиновая кислота), -СН2SН (цистеин), -CH2CH2C(=O)NH2 (глутамин), -СH2СН2СО2Н (глутаминовая кислота), -СН2-(4-имидазол) (гистидин), -СН(Еt)СН3 (изолейцин), -СН2СH(СН3) (лейцин), -(CH2)4NH2 (лизин), -(CH2)2SCH3 (метионин), -CH2Ph (фенилаланин), -СН2-СН2-СН2- (пролин), -CH2ОН (серин), -СН(ОН)СН3 (треонин), -СН2-(3-индол) (триптофан), -СН2-(4-гидроксифенил) (тирозин) и -СН(СН3)2 (валин).
рКа соответствующих кислотных групп W1 и W2 менее 9, более предпочтительно, чтобы менее 7, и наиболее предпочтительно менее 5. "Соответствующие кислотные группы W1 и W2" относятся к родительской кислотной группе W1 и W2, например, в случае, если W1 и W2 являются эфирами, соответствующая кислота соответствует карбоновой кислоте, и в случае, если W1 и W2 являются алкилфосфонатами, соответствующая кислота соответствует фосфоновой кислоте. Важно то, что рКа W1 и W2, зависит не только от вида W1 и W2, но также и от типа заместителей, имеющихся у W1 и W2 групп и/или в моно- или ди-арильных или гетероарильных группах, к которым присоединены W1 и W2. Так, например, присутствие одной или более чем одной группы, оттягивающей электронную плотность, такой как нитро, нитрозо, карбонил, циано и галогеногруппы, уменьшает значение рКа соответствующих W1 и W2 кислотных групп. рКа определяется как -log(Ka), где Ка является константой диссоциации. На силу кислоты или основания в данной среде указывает значение ее (его) константы диссоциации. Например, сильные основания являются сильными акцепторами протонов (или донорами электронных пар) и имеют высокие значения рКа. Значения рКа зависят от различных факторов, таких как природа растворителя и температура. Например, вода (Н2О), но не сопряженная воде кислота, которая представляет собой Н3О+, имеет рКа в воде 15,7 при 25° С, 16,7 при 0° С и 14,7 при 60° С. В дополнение к этому, в диметилсульфоксиде (ДМСО) при 25° С ее рКа равна 27,5. В настоящем документе значения рКа относятся к значениям рКа, которые соответствуют значению рКа воды, соответствующему примерно 15,7, если иное особо не оговорено.
В дополнение к приведенным здесь формулам:
Предпочтительно, чтобы W1 и W2 независимо представляли собой CO2R5, C(=NH)NH(OH)OPO(OR5), С(=O)СF3 или PO(OR5)2.
Предпочтительно, чтобы R1 и R2 независимо представляли собой связь, C1-С6 алкилен или фторированный C1-С6 алкилен. Более предпочтительно, чтобы R1 и R2 независимо представляли собой связь, метилен или дифторметилен.
Предпочтительно, чтобы каждый Аr1, Аr2 и Аr5 независимо представлял бы собой моноарил или гетероарил. Более предпочтительно, чтобы Аr1, Аr2 и Аr3 представлял бы собой фенил.
Предпочтительно, чтобы Аr3 представлял собой 2-пиридонил, и более предпочтительно, чтобы Ar3 представлял собой 4-Ar4-(2-пиpидoнил), т.е. 2-пиридон в 4-положении присоединен к группе Аr4.
Предпочтительно, чтобы Аr4 представлял собой C1-C20 гетероарил. Более предпочтительно, чтобы Аr4 представлял собой пиридил. Наиболее предпочтительно, чтобы Аr4 представлял собой 4-пиридил, т.е. 4-пиридин в 4-положении присоединен к группе Аr3.
Предпочтительно, чтобы Y1 представлял собой NR17R18. Более предпочтительно, чтобы Y1 представлял собой NH2. Предпочтительно, чтобы каждый R15 независимо представлял собой водород, метил или этил. Предпочтительно, чтобы группа L1 представляла собой C1-С6 алкилен; C1-C6 алкенилен, включая группы с ненасыщенной связью у α ,β -атомов углерода (например, -СН=СН-С(=O)-); или группу формулы -R33-X14-, -R34-X15-R35- или -X16-R36-Ar7-R37-X17-. Каждый из R33, R34, R35, R36 и R37 независимо представляют собой C1-С6 алкилен (включая замещенный алкилен), предпочтительно метилен. Каждый X14, X15, X16 и X17 независимо представляет собой О, S или NR38, предпочтительно О или NR38. Каждый Аr6 и Аr7 независимо представляет собой С6-С20 арил или C1-C20 гетероарил, предпочтительно 2-пиридон. И R38 представляет собой Н, C1-С6 алкил или защитную группу амина, предпочтительно -СН2СO2Н. Более предпочтительно, чтобы L1 представлял собой сульфонамид (-SO2NH-), этилен (-СН2СН2-), -СН2O-, -СН=СНС(=O)-, -СН2СН2СН(ОН)-, -СН=СН-, -СН(ОН)СН(ОН)-, -CH2N(R38)CH2-, группу формулы:
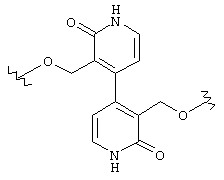
или группу формулы:
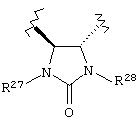
где каждый R27 и R28 независимо представляет собой Н, C1-С6 алкил, С6-С10 аралкил или защитную группу. Предпочтительные R27 и R28 независимо представляют собой Н или защитную группу. Более предпочтительно R27 и R28 независимо представляют собой Н или 4-метоксибензил.
Предпочтительно m и n равны 0.
Альтернативно, R1 и W1 и/или R2 и W2 вместе образуют -(CH2)aCH(NHR29)CO2R39 и -(CH2)bCH(NHR30)CO2R40 соответственно, где а и b независимо представляют собой целое число от 0 до 2, R29 и R30 независимо представляют собой Н или защитную группу амина, и R39 и R40 независимо представляют собой Н или C1-С6 алкил. Предпочтительно, чтобы а и b равнялись бы 1. Предпочтительно, чтобы R29 и R30 независимо представляли собой Н, C1-С6 алкил или защитную группу амина.
Предпочтительно, чтобы R5 представлял собой остаток боковой цепи аспарагина.
Предпочтительно, чтобы R6 представлял собой остаток боковой цепи глутамина.
Предпочтительно, чтобы R7 представлял собой примерно от 1 до 10 аминокислотных остатков или их производных, более предпочтительно от 1 до 6 аминокислотных остатков или их производных, еще более предпочтительно по меньшей мере 2 аминокислотных остатка или их производных и наиболее предпочтительно - частицей -lys-ser-CONHCH3, т.е. частицей формулы NHCH[(CH2)4NH2]CONHCH(CH2OH)CONHCH3.
Предпочтительно, чтобы X1, X2, X3, X4, X5, X6, X7 и X8 независимо представляли собой О или NR16. Более предпочтительно, чтобы X1, X2, X3, X4, X5, X6, X7 и X8 представляли собой NR16.
Предпочтительно, чтобы Х представлял собой OR17 или NR17R18, более предпочтительно NR17R18 и наиболее предпочтительно NH2.
Предпочтительно, чтобы R8 представлял собой остаток боковой цепи глицина (т.е. Н).
Предпочтительно, чтобы R9 представлял собой остаток боковой цепи тирозина (т.е. 4-гидроксибензил).
Предпочтительно, чтобы R10 представлял собой пропилен.
Предпочтительно, чтобы R11 представлял собой остаток боковой цепи лизина, т.е. частицу формулы -(CH2)4NH2.
Предпочтительно, чтобы R12 представлял собой С6-С20 аралкил, и более предпочтительно 2-фенилэтил.
Предпочтительно, чтобы R13 представлял собой Н.
Предпочтительно, чтобы R14 представлял собой Н или защитную группу амина, более предпочтительно защитную группу амина и наиболее предпочтительно ацетильную группу, т.е. группу формулы -С(=O)СН3.
Предпочтительно, чтобы каждый R16 независимо представлял собой Н или С6-С20 арил. Более предпочтительно, чтобы каждый R16 независимо представлял собой Н или фенил.
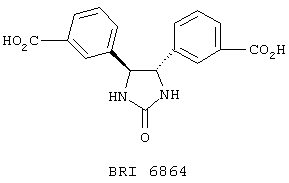
В одном частном воплощении настоящего изобретения приведенное выше ароматическое соединение является соединением формулы:
Более предпочтительно, чтобы это ароматическое соединение являлось бы соединением формулы:
В другом частном воплощении настоящего изобретения описанное выше ароматическое соединение является соединением формулы:
В одном частном воплощении настоящего изобретения описанное выше гетероароматическое соединение является соединением формулы:
Более предпочтительно, чтобы это описанное выше гетероароматическое соединение представляло собой соединение формулы:
В другом частном воплощении настоящего изобретения описанное выше циклическое соединение является соединением формулы:
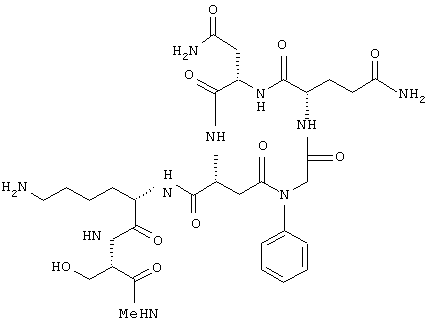
Еще в одном частном воплощении настоящего изобретения описанное выше бициклическое соединение является соединением формулы:
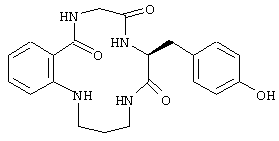
Еще в одном частном воплощении настоящего изобретения описанное выше аминокислотное производное является производным формулы:
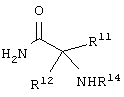
или его солями. Предпочтительно, чтобы описанное выше аминокислотное производное являлось производным формулы:
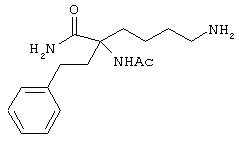
или представляло собой его соль.
Соединения по настоящему изобретению, модулирующие Fc рецепторы, могут также включать в себя нуклеозиды или их производные. Предпочтительно, чтобы нуклеозиды по настоящему изобретению имели бы формулу:
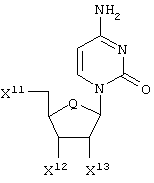
где Q представляет собой О или метилен. Предпочтительней Q является О. X11 представляет собой OR31 или OPO(OR31). Предпочтительней Х11 представляет собой ОН или ОРО3Н2. Каждый X12 и X13 независимо представляет собой Н или OR15. Предпочтительно, чтобы каждый X12 и X13 независимо представлял собой Н или ОН. Каждый R31 и R32 независимо представляет собой Н или C1-С6 алкил.
Соединения по настоящему изобретению, модулирующие Fc рецепторы, могут также включать в себя фолиевую кислоту или ее производные.
Соединения по настоящему изобретению, модулирующие Fc рецепторы могут также включать в себя пептиды, которые могут модулировать взаимодействие между Fc рецепторами и иммуноглобулинами. Вне связи с какой-либо из теорий считается, что особенно полезные пептиды взаимодействуют с областью С (см. фиг. 1) Fc рецепторов, например FcgRII. Таким образом, считается, что эти пептиды препятствуют димеризации между двумя FcgRII белками, влияя, таким образом, на передачу сигнала от клеток через один или оба FcR белка. В данном случае, остатки 117-131 и остатки 150-164 образуют пограничную область FcgIIa димера, и пептиды с такими последовательностями или с такими свойствами являются ингибиторами связывания. Например, природный гексапептид Phe121 - Ser126 или более короткие сегменты перекрывают область с большим числом водородных связей, обуславливающих значительное взаимодействие, и, таким образом, являются подходящими модуляторами димеризации двух молекул FcgRIIa. Такой фрагмент белка описан как часть SEQ ID №3 в вышеупомянутой патентной заявке США №09/245764, "3-мерные структуры и модели Fc рецепторов и их применение", поданной 5 февраля 1999 г. Таким образом, настоящие исследователи установили, что трипептид с последовательностью GKS (gly-lys-ser) или его производные и гексапептиды с последовательностью FQNGKS (phe-gln-asn-gly-lys-ser) или их производные модулируют связывание FcgRII с IgG. См. опыт 24 и фиг. 10 и 11.
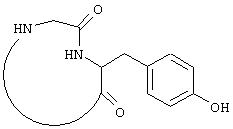
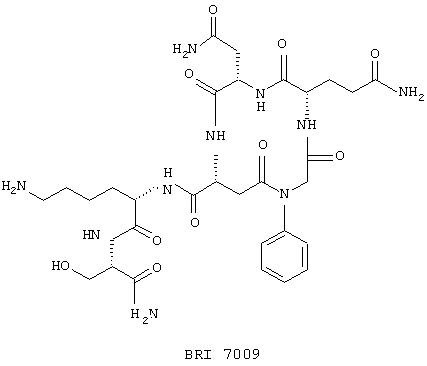
Настоящие исследователи также установили, что макроциклические соединения с определенной конформацией модулируют поведение белка FcR. Используемый в настоящем документе термин "макроциклическое соединение" относится к соединению, содержащему кольцо, которое включает в себя от 8 атомов до 18 атомов. Предпочтительно, чтобы кольцо макроциклического соединения по настоящему изобретению включало примерно от 10 до 16 атомов, более предпочтительно от 12 до 14 атомов и наиболее предпочтительно от 13 до 14 атомов. Особенно полезное макроциклическое соединение по настоящему изобретению представляет собой циклический пептид или его производные. Такой циклический пептид, имеющий формулу:
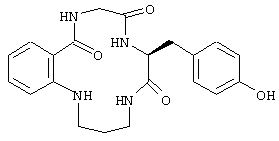
был описан выше. Верхняя часть FG цикла FcR была определена в исследованиях по мутагенезу, которые имеют значение при изучении связывания Ig. Пептидный участок FG содержит расширенную b-складку, в которой аминокислотные боковые остатки FG петли ориентируются в определенное положение. Такая ориентация Fc белка описана в вышеупомянутой патентной заявке США №09/245764, "3-мерные структуры и модели Fc рецепторов и их применение", поданной 5 февраля 1999 г. Было также установлено, что молекулы, которые могут воспроизводить витки b-спирали так, что ее боковые остатки занимают положение в верхней части FG петли, т.е. так же, как это имеет место для боковых остатков рецептора, эффективно модулируют поведение FcR рецептора. Таким образом, в еще одном воплощении настоящего изобретения соединение, модулирующее Fc рецептор, также представляет собой соединение формулы:
где макроциклический фрагмент содержит такое же число атомов, как описано выше. Одним частным воплощением такого воспроизведения витка b-спирали является соединение, описанное выше, которое имеет формулу:
Соединения по настоящему изобретению могут синтезироваться из легко доступных исходных материалов. Различные заместители в соединениях по настоящему изобретению могут присутствовать в исходных соединениях, вводиться в любое из промежуточных соединений или вводиться после образования конечных продуктов известными способами замещения или преобразования. Если сами заместители являются реакционноспособными, то данные заместители могут быть защищены в соответствии с известными способами. Известны различные защитные группы, которые могут применяться. Примеры многих возможных групп можно найти в издании "Protective Groups in Organic Synthesis" by T.W. Green, John Wiley and Sons, 1981. Например, нитрогруппы могут вводиться нитрованием, и нитрогруппа может быть превращена в другие группы, такие как аминогруппа - восстановлением и галогеногруппа - диазотированием аминогруппы и замещением этой диазогруппы галогеном. Ацильные группы могут вводиться ацилированием по Фриделю-Крафтсу. Эти ацильные группы затем могут быть преобразованы в соответствующие алкильные группы различными способами, включая восстановление по Вольфу-Кижнеру и восстановление по Клеменсону. Аминогруппы могут алкилироваться с образованием моно- и ди-алкиламиногрупп; и меркапто- и гидроксигруппы могут алкилироваться с образованием соответствующих эфиров. Первичные спирты могут окисляться известными из практики окислителями с образованием карбоновых кислот или альдегидов, и вторичные спирты могут окисляться с образованием кетонов. Таким образом, можно применять реакции замещения или изменения для ввода различных заместителей в молекулу исходного вещества, в промежуточные соединения или в конечный продукт, включая выделенные продукты. Так как соединения по настоящему изобретению могут иметь некоторые заместители, которые обязательно должны присутствовать, введение каждого заместителя, разумеется, зависит от природы участвующих заместителей и химизма реакций, необходимых для их образования. Таким образом, решение вопроса о том, как на отдельный заместитель будет влиять химическая реакция при образовании второго заместителя, будет связано со способами, которые знакомы среднему специалисту. Также это будет зависеть от природы кольца.
Следует иметь в виду, что настоящее изобретение охватывает не только различные изомеры, которые могут существовать, но также и различные смеси изомеров, которые могут образовываться.
Если соединение по настоящему изобретению содержит один или более чем один хиральный центр, его можно синтезировать энантиоселективно или в виде смеси энантиомеров, и/или могут быть получены диастереомеры с последующим разделением. Разделение соединений по настоящему изобретению, исходные материалы для их получения и/или промежуточные соединения могут быть получены известными способами, например так, как описано четырехтомном компедиуме Optical Resolution Procedures for Chemical Compounds: Optical Resolution Information Center, Manhattan College, Riverdale, N.Y., и в Enantiomers, Racemates and Resolution, Jean Jacques, Andre Collet and Samuel H. Wilen; John Wiley & Sons, Inc., New York, 1981. В целом, разделение соединений основано на различиях в физических свойствах диастереомеров при химическом или ферментативном присоединении энантиомерно чистой группы с образованием форм, которые разделяются дробной кристаллизацией, дистилляцией или хроматографией.
Если соединение по настоящему изобретению содержит олефиновую группу и эта группа может иметь либо цис-, либо транс-конфигурацию, то соединение может синтезироваться с образованием либо цис-, либо транс-олефина в виде преобладающего продукта. Альтернативно, соединение, содержащее олефиновую группу, может быть получено в виде смеси цис- и транс-олефинов и разделено при помощи известных способов, например хроматографией, как описано в W.K. Chan, et al., J. Am. Chem. Soc., 1974, 96, 3642.
Соединения по настоящему изобретению образуют соли, образованные присоединением кислот в случае, если присутствует основная аминофункция, и соли, образованные присоединением оснований, если присутствует кислотная функция, например карбоновая кислота или фосфоновая кислота. Все такие соли полезны при выделении и/или очистке новых продуктов. Особое значение имеют фармацевтически приемлемые соли как с кислотами, так и с основаниями. Подходящие кислоты включают в себя, например, хлористо-водородную, щавелевую, серную, азотную, бензолсульфоновую, толуолсульфоновую, уксусную, малеиновую, винную и им подобные кислоты, которые являются фармацевтически приемлемыми. Основные соли для фармацевтического применения включают в себя соли Na, К, Са и Mg.
В дополнение к и/или вместо целенаправленной разработки лекарства иные модуляторы Fc рецептора могут быть идентифицированы методом скрининга, при котором различные соединения тестируются для установления их активности при модулировании Fc рецептора. Этим способом были идентифицированы различные модуляторы Fc рецептора. Таким образом, соединения по настоящему изобретению включают в себя замещенные и незамещенные бензойные кислоты, в частности 4-метилбензойную кислоту и 3-метилбензойную кислоту; нуклеозиды и их аналоги; а также фолиевую кислоту и ее производные.
Соединения по настоящему изобретению являются модуляторами Fc рецептора, например, они модулируют связывание Fc рецептора с иммуноглобулинами. Предпочтительно, чтобы соединения по настоящему изобретению модулировали бы Fc рецепторы, выбираемые из группы, включающей в себя FcaR, FcgR, FcgR и их смеси, более предпочтительно из группы, включающей в себя FcgRI, FcgRII, FcgRIII и их смеси, еще более предпочтительно из группы, включающей в себя FcgRIIa, FcgRIIb, FcgRIIc и их смеси и наиболее предпочтительно FcgRIIa рецептор. Соединения по настоящему изобретению могут использоваться для разнообразных целей, включая лечение или диагностику любых заболеваний, при которых образуются агрегаты с антителами и при которых в случае контакта антитела с внутренним или с внешним антигеном образуются иммунные комплексы. Примеры применения соединений по настоящему изобретению для лечения и диагностики включают в себя иммунные комплексные заболевания; аутоиммунные заболевания, включая, но не ограничиваясь ревматоидным артритом, эритематозом системной волчанки, иммунной тромбоцитопенией, нейтропенией, гемолитической анемией; васкулитами, включая, но не ограничиваясь полиартритным утолщением, системными васкулитами; отторжением ксенотрансплантата; и инфекционными заболеваниями, при которых FcR поглощает вирусную инфекцию, включая, но не ограничиваясь флавивирусными инфекциями, такими как геморрагическая лихорадка, вызванная вирусом Денге, и вирусная инфекция кори. Соединения по настоящему изобретению также могут применяться для уменьшения IgG-опосредованного повреждения ткани и для снижения воспаления.
Соединения по настоящему изобретению могут также улучшить функцию лейкоцитов в результате улучшения FcR функции. Эти функции включают в себя антителозависимую клеточно-опосредованную цитотоксичность, фагоцитоз, высвобождение воспалительных цитокинов. Примеры лечения и диагностики при улучшении FcR функции включают любую инфекцию, при которой вырабатываются нормальные антитела для устранения патогенного фактора; и любое заболевание, требующее FcR функцию, при котором могут применяться природные или рекомбинантные антитела для лечения таких заболеваний, как рак и инфекции, например, антитела могут вводиться совместно с соединением по настоящему изобретению для улучшения эффекта при лечении антителами.
Соединения по настоящему изобретению могут вводиться пациенту для достижения желаемого физиологического эффекта. Предпочтительно, чтобы пациентом являлось животное, более предпочтительно млекопитающее и наиболее предпочтительно человек. Соединение может вводиться в различных формах, предназначенных для выбранного пути введения, т.е. перорально или парентерально. В этом отношении парентеральное введение включает в себя введение следующими путями: внутривенно; внутримышечно; подкожно; интраокулярно, интрасиновиально; трансэпителиально, в том числе, трансдермально, офтальмиально, сублингвально и трансбуккально; местным введением, в том числе офтальмиально, дермально, окулярно, ректально и назальными ингаляциями при помощи инсуффляции и аэрозоля; интраперитонеально; и ректально-систематически.
Активное соединение может вводиться перорально, например, с инертным разбавителем или с усвояемым съедобным носителем, или оно может быть заключено в твердые или мягкие желатиновые капсулы, или оно может прессоваться в таблетки, или оно может непосредственно включаться в пищу или диету. Для перорального терапевтического введения активное соединение может включаться в эксципиент и применяться в форме глотаемых таблеток, трансбуккальных таблеток, лепешек, капсул, эликсиров, суспензий, сиропов, облаток и тому подобное. Такие композиции и фармацевтические препараты могут содержать по меньшей мере 0,1% активного соединения. Процентный состав композиций и фармацевтических препаратов может, разумеется, изменяться и может составлять от примерно 1 до 10% веса формы. Количество активного соединения в подобных терапевтически полезных композициях такое, чтобы достигалась подходящая доза. Предпочтительные композиции или фармацевтические препараты в соответствии с настоящим изобретением приготавливают таким образом, чтобы лекарственная форма для перорального введения содержала бы от 1 до 1000 мг активного соединения.
Таблетки, лепешки, пилюли, капсулы и тому подобное могут также содержать следующее: связующее, такое как смола трагаканта, аравийская камедь, кукурузный крахмал или желатин; эксципиенты, такие как дикальцийфосфат; разрыхлитель, такой как кукурузный крахмал, картофельный крахмал, альгиновая кислота и тому подобное; смазывающее вещество, такое как стеарат магния; и может добавляться подсластитель, такой как сахароза, лактоза или сахарин, или корригент, такой как мятное масло, масло гаультерии, или вишневый корригент. Если лекарственной формой является капсула, то она может содержать, в дополнение к материалам вышеупомянутых типов, жидкий носитель. Различные иные материалы могут применяться в качестве покрытий или для модификации физической формы лекарственного средства. Например, таблетки, пилюли или капсулы могут быть покрыты шеллаком, сахаром или и тем и другим. Сироп или эликсир могут содержать активное соединение, сахарозу в качестве подсластителя, метил- и пропилпарабены в качестве консервантов, окрашивющее вещество и корригент, такой как вишневый или апельсиновый корригент. Разумеется, любые материалы, применяемые при изготовлении любых лекарственных форм, должны быть фармацевтически чистыми и нетоксичными в применяемых количествах. В дополнение к этому, активное соединение может быть включено в фармацевтические препараты и составы длительного действия.
Активное соединение может также вводиться парентерально. Растворы активного соединения в виде свободного основания или фармакологически приемлемой соли могут готовиться на воде, соответственно смешанной с сурфактантом, таким как гидроксипропилцеллюлоза. Дисперсии могут также готовиться на глицерине, жидких полиэтиленгликолях и их смесях и на маслах. При обычных условиях хранения и применения для предотвращения роста микроорганизмов эти фармацевтические препараты содержат консервант.
Фармацевтические формы, подходящие для применения в виде инъекций, включают в себя стерильные водные растворы или дисперсии и стерильные порошки для приготовления стерильных инъецируемых растворов или дисперсий для немедленного введения. Во всех случаях введения при помощи обычного шприца эта форма должна быть стерильной и должна быть жидкой. Она должна быть стабильной в условиях получения и хранения и должна быть защищена от контаминирующего воздействия микроорганизмов, таких как бактерии и грибы. Носитель может представлять собой растворитель в виде дисперсной среды, содержащий, например, воду, этанол, полиол (например, глицерин, пропиленгликоль, а также жидкий полиэтиленгликоль и тому подобное), подходящие их смеси и растительные масла. Надлежащая текучесть может быть достигнута, например, применением покрытия, такого как лецитин, установлением требуемого размера частиц в случае их диспергирования и применением сурфактантов. Предотвращение воздействия микроорганизмов может осуществляться при помощи различных противобактериальных и противогрибковых агентов, например парабенов, хлорбутанола, фенола, сорбиновой кислоты, тимеросала и тому подобное. Во многих случаях предпочтительно включать изотонические агенты, например сахара или хлорид натрия. Пролонгирование абсорбции инъецируемых композиций осуществляется при помощи агентов, продлевающих время абсорбции, например моностеарата аллюминия и желатина.
Стерильные инъецируемые растворы готовятся введением активного соединения в соответствующий растворитель в требуемом количестве, который содержит различные другие вышеперечисленные ингредиенты, как целесообразно, с последующей стерилизующей фильтрацией. В целом, дисперсии готовятся введением различных стерильных активных ингредиентов в стерильный носитель, который содержит основную диспергирующую среду и иные необходимые ингредиенты из числа вышеперечисленных. В случае стерильных порошков для приготовления стерильных инъецируемых растворов, предпочтительными способами получения являются методы вакуумной сушки и лиофильной сушки, которые позволяют получить порошок активного ингредиента, содержащий любой дополнительно требуемый ингредиент из их раствора, который был предварительно подвергнут стерилизующей фильтрации.
Терапевтические соединения по настоящему изобретению могут вводиться млекопитающим самостоятельно или совместно с фармацевтически приемлемыми вышеуказанными носителями, соотношения которых определяются растворимостью и химическими свойствами соединения, выбранным путем введения и стандартной фармацевтической практикой.
Лечащий врач определяет дозировку настоящих терапевтических агентов, которая будет являться наиболее подходящей для профилактики или лечения и будет изменяться в зависимости от формы введения и конкретно выбранного соединения, а также будет изменяться при лечении в зависимости от особенностей конкретного пациента. Обычно лечащий врач желает начинать лечение с малых доз, понемногу увеличивая их до тех пор, пока при данных обстоятельствах не будет достигнут оптимальный эффект. Терапевтическая доза может обычно составлять от 0,1 до 1000 мг/сутки и предпочтительно от 10 до 100 мг/сутки или от 0,1 до 50 мг/кг веса тела в сутки и предпочтительно от 0,1 до 20 мг/кг веса тела в сутки и может вводиться в виде нескольких различных дозировочных единиц. При пероральном введении могут потребоваться от 2 до 4 раз большие дозировки.
Дополнительные задачи, преимущества и новые особенности настоящего изобретения будут очевидны специалистам при изучении нижеприведенных примеров, которые его не ограничивают.
ЭКСПЕРИМЕНТАЛЬНЫЕ ДАННЫЕ
В настоящем документе используются следующие сокращения:
к.т. - комнатная температура,
Et2O - диэтиловый эфир (т.е. эфир или этиловый эфир),
МС (ХИАД) - химическая ионизация при атмосферном давлении,
ТГФ - тетрагидрофуран,
ЕtOАс - этилацетат,
ТМССl - триметилсилилхлорид,
CH3CN - ацетонитрил,
ДМФ - диметилформамид.
Опыт 1
В этом опыте представлен а синтез 1,2-бис(м-карбоксифенил)этана:
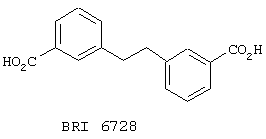
Стадия 1: 1,2-бис(м-бромфенил)этан получают по способу Lindsay et al (JACS, 1961, 83, 943) следующим образом. К раствору 3-бромбензилбромида (1,0 г, 4,0 ммоль) в Et2O (10 мл) при к.т. добавляют магний (0,05 г, 2,0 ммоль). Через 20 мин при комнатной температуре весь магний растворяется, и затем добавляют безводный хлорид трехвалентного железа (5 мг). Реакционную смесь нагревают до температуры дефлегмации в течение 1 часа, охлаждают, подкисляют до примерно рН 1 1 М водной H2SO4 и экстрагируют Et2O (3× 50 мл). Объединенные органические экстракты промывают водой (50 мл), сушат (Na2SO4), фильтруют и концентрируют под вакуумом с получением желтого твердого вещества. После перекристаллизации из петролейного эфира получают 1,2-бис(м-бромфенил)этан в виде бесцветного твердого вещества. МС (ХИАД) m/z 338 (50%), 340 (100), 342 (50). 1Н ЯМР (200 МГц, CDCl3): d 2,85, s, 2H; 7,02-7,25, m, 2H; 7,30-7,39, m, 2H.
Стадия 2: трет-бутиллитий (2,1 мл 1,7 M раствора в пентане, 3,60 ммоль) по каплям добавляют к раствору 1,2-бис(м-бромфенил)этана (305 мг, 0,90 ммоль) в ТГФ (10 мл) при -78° С. Через 20 мин при этой температуре через реакционную барботируют смесь СО2 до тех пор, пока температура реакционной смеси при отсутствии охлаждающей бани не достигнет к.т. Эту реакционную смесь распределяют между водой (50 мл) и Et2O (50 мл), водную фазу отделяют и подкисляют до примерно рН 1 концентрированной водной НСl, удерживая внутреннюю температуру ниже 25° С. Водную фазу экстрагируют ЕtOАс (3× 50 мл) и объединенные органические экстракты сушат (Na2SO4), фильтруют и концентрируют под вакуумом с получением 1,2-бис(м-карбоксифенил)этана в виде белого твердого вещества. МС (ХИАД) m/z 269 (М+1, 100%). 13С ЯМР (50 МГц, d6-ДМСО): d 38,4, 128,8, 130,3, 131,1, 132,5, 134,8, 143,5, 169,2. Температура плавления согласуется со значением, приведенным Lindsay et al. (JACS, 1961, 83, 943).
Опыт 2
В этом опыте представлен синтез 3-[(м-карбоксифенил)метокси]бензойной кислоты:
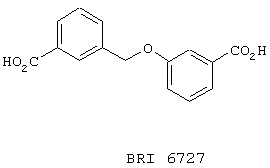
Стадия 1: смесь 3-бромфенола (13,8 г, 80 ммоль), 3-бромбензилбромида (10 г, 40 ммоль), К2СО3 (16,6 г, 120 ммоль) и NaI (300 мг, 2 ммоль) в ацетоне (100 мл) нагревают до температуры дефлегмации в течение 12 часов. Реакционную смесь охлаждают до к.т., концентрируют под вакуумом и распределяют между Et2O (300 мл) и водой (300 мл). Органическую фазу промывают водным раствором NaOH (1 M, 300 мл), сушат (Na2SO4), фильтруют и концентрируют под вакуумом с получением 3-[(м-бромфенил)метокси]бромбензола в виде прозрачного масла. МС (ХИАД) m/z 339 (М+-3,50%), 341 (M+-1, 100%), 343 (М++3,50%). 13С ЯМР (50 МГц, CDCl3); d 68,9, 113,4, 117,9, 122,5, 122,6, 124,1, 125,6, 129,9, 130,0, 130,4, 130,9, 138,4, 158,9.
Стадия 2: Используя 3-[(м-бромфенил)метокси]бромбензол и применяя способ, описанный в Примере 1, стадия 2, получают 3-[(м-карбоксифенил)метокси]-бензойную кислоту в виде белого твердого вещества. МС (ХИАД) m/z 271 (M+-1, 100%). 13С ЯМР (50 МГц, d6-ДМСО): d 68,3, 114,5, 119,3, 121,5, 127,8, 128,3, 129,3, 130,5, 131,5, 131,8, 137,0, 157,7, 166,6, 166,7.
Опыт 3
В этом опыте представлен синтез 1,2-бис(3-фосфонофенил)этана:
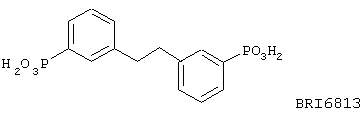
Стадия 1: 1,2-бис(3-бромфенил)этан (получен с применением способа Примера 1, стадия 1) (440 мг, 1,29 ммоль), диэтилфосфит (0,46 мл, 3,59 мл) и триэтиламин (0,5 мл, 3,59 ммоль) растворяют в толуоле и дегазируют. Затем одной порцией добавляют Pd(PPh3)4 (185 мг, 0,16 ммоль), и реакционную смесь нагревают до 90° С в течение 16 часов. Реакционную смесь охлаждают до комнатной температуры и очищают колоночной хроматографией (SiO2, 50% ЕtOАс в петролейном эфире ® 100% ЕtOАс ® 100% EtOH) с получением 1,2-бис[3-(диэтоксифосфоно)фенил]-этана в виде белого твердого вещества. МС(ХИАД) m/z 455 (M++1, 100%). 31P ЯМР (81 МГц, развязка протонов, СDСl3): d +19,5.
Стадия 2: Триметилсилилбромид (1,03 мл, 7,8 ммоль) по каплям добавляют к раствору вышеупомянутого эфира (586 мг, 1,30 ммоль) в CH2Cl2 (10 мл) при к.т. Реакционную смесь перемешивают в течение 16 часов при комнатной температуре и концентрируют в вакууме. Затем добавляют МеОН (5 мл), и этот раствор концентрируют в вакууме. Эту операцию повторяют еще два раза с получением 1,2-бис(3-фосфонофенил)этана в виде белого твердого вещества. МС (ХИАД) m/z 341 (M+-1, 100%). 31P ЯМР (81 МГц, развязка протонов, CDCl3): d +14,6.
Опыт 4
В этом опыте представлен а синтез 3,3’-дикарбокси-халкона:
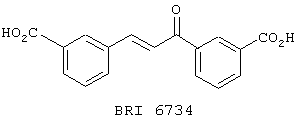
Стадия 1: 3-Цианобензальдегид (3,0 г, 23,0 ммоль), 3-цианоацетофенон (3,34 г, 23,0 ммоль) в ледяной уксусной кислоте (5 мл) и концентрированную H2SO4 (3,66 мл, 69 ммоль) перемешивают при комнатной температуре в течение 72 часов. Затем добавляют воду (200 мл) и реакционную смесь фильтруют. Осадок промывают водой (2× 200 мл) и сушат в вакууме с получением 3,3’-дицианохалкона в виде беловатого твердого вещества. МС (ХИАД) m/z 258 (M+-1, 100%). 13С ЯМР (50 МГц, d6-ДMCO): d 111,7, 117,8, 118,0, 123,0, 129,7, 131,6, 132,1, 132,4, 133,3, 133,5, 135,3, 136,1, 137,4, 142,1, 187,3.
Стадия 2: Раствор 3,3’-дицианохалкон, полученный на стадии 1 (2,0 г, 7,75 ммоль), в ледяной уксусной кислоте (30 мл) обрабатывают смесью концентрированной H2SO4 (10 мл) и воды (10 мл). Эту реакционную смесь нагревают до 130° С в течение 12 часов, охлаждают до комнатной температуры и фильтруют. Осадок промывают водой (3× 100 мл) и сушат в вакууме с получением 3,3’-дикарбоксихалкона в виде желтого твердого вещества. МС (ХИАД) m/z 295 (M+-1, 100%). 13С ЯМР (50 МГц, d6-ДМСО); d 122,5, 128,6, 128,7, 129,2, 130,8, 131,0, 131,2, 132,4, 132,5, 133,1, 134,5, 137,2, 143,1, 166,3, 166,5, 188,2.
Опыт 5
В этом опыте представлен а синтез 1,3-бис (м-карбокси-фенил)-1-пропанола:
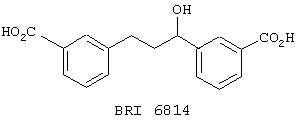
3,3’-дикарбоксихалкон (Пример 4, стадия 2) (430 мг, 1,45 ммоль) в этаноле (10 мл), содержащем водный раствор NaOH (1 М, 2,90 ммоль), гидрируют при давлении 310 кПа в течение 48 часов в присутствии катализатора Уилкинсона (67 мг, 0,07 ммоль). Реакционную смесь фильтруют и концентрируют под вакуумом. Остаток растворяют в метаноле (10 мл) и обрабатывают NaBH4 (220 мг, 5,8 ммоль) при к.т. Реакционную смесь перемешивают в течение 16 часов при к.т., гасят осторожным добавлением насыщенного водного раствора NH4Cl и распределяют между ЕtOАс (50 мл) и водным раствором НСl (1 М, 50 мл). Органический экстракт сушат (Na2SO4, фильтруют и концентрируют под вакуумом с получением 1,3-бис (м-карбоксифенил)-1-пропанола в виде вязкого масла. МС (ХИАД) m/z 299 (M+-1, 100%). 1Н ЯМР (200 МГц, CDCl3); d 1,95-2,10, m, 2H; 2,68-2,83, m, 2H; 4,62-4,78, m, 1H; 7,03-7,60, m, 4H; 7,75-8,03, m, 4H.
Опыт 6
В этом опыте представлен а синтез транс-3,3’-бис-карбоксистильбена:
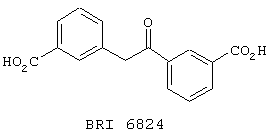
Стадия 1: Метил 3-бромбензоат (21,5 г, 100 ммоль), Pd(OAc)2 (224 мг, 1 ммоль), три-о-толилфосфин (608 мг, 2 ммоль) и трибутиламин (26,2 мл, 110 ммоль) в ДМФ (100 мл) дегазируют аргоном, нагревают до 130° С в течение 6 часов и при этом барботируют через этот раствор этилен. Реакционную смесь охлаждают до комнатной температуры и фильтруют. Осадок промывают холодной Et2O (2× 50 мл) и сушат в вакууме с получением диметилового эфира транс-3,3’-бис-карбоксистильбена в виде беловатого твердого вещества. 13С ЯМР (50 МГц, CDCl3): d 52,2, 127,5, 128,8, 130,6, 130,9, 137,2, 166,9.
Стадия 2: Вышеупомянутый диэфир (500 мг, 1,7 ммоль) в ТГФ (10 мл) обрабатывают при комнатной температуре водным раствором LiOH (1 M, 10 мл). После перемешивания в течение 16 часов при к.т. реакционную смесь распределяют между Et2O (50 мл) и водой (50 мл). Водную фазу отделяют и органическую фазу экстрагируют водой (25 мл). Объединенные водные экстракты подкисляют концентрированной водной НСl (удерживая при этом внутреннюю температуру ниже 10° С). Водную фазу экстрагируют CH2Cl2 (3× 50 мл) и объединенные органические экстракты сушат (Na2SO4), фильтруют и концентрируют в вакууме с получением транс-3,3’-бискарбоксистильбена в виде белого твердого вещества. МС (ХИАД) m/z 267 (М+-1, 100%). 1Н ЯМР (200 МГц, d6-ДМСО): d 7,28-7,56, m, 2Н; 7,78-7,90, m, 2H; 8,20, s, 1H.
Опыт 7
В этом опыте представлен а синтез (S,S)-1,2-бис-(3-карбоксифенил)этан-1,2-диола:
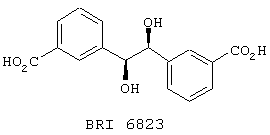
Стадия 1: Диметиловый эфир транс-3,3’-бискарбоксистильбена (Пример 6, стадия 1) (5,0 г, 16,9 ммоль) и N-метилморфолин-N-оксид (2,2 г, 18,6 ммоль) в ацетоне (50 мл) и воде (20 мл) обрабатывают при комнатной температуре водным раствором OsO4 (4,3 мл, 39,4 мМ, 0,17 ммоль). Реакционную смесь перемешивают в течение 16 часов при к.т., гасят добавлением метабисульфита натрия (3,0 г) и доводят рН до примерно рН 7 2 М водным раствором серной кислоты. Ацетон удаляют в вакууме, и оставшийся раствор подкисляют до примерно рН 2, насыщают NaCl и экстрагируют ЕtOАс (3× 100 мл). Объединенные органические экстракты сушат (Na2SO4), фильтруют и концентрируют в вакууме с получением (R,R)-1,2-бис-[3-(карбометокси)-фенил]этан-1,2-диола в виде белого твердого вещества. 1Н ЯМР (200 МГц, CDCl3): d 3,2, bs, 1H; 3,82, s, 3Н; 4,77, s 1H; 7,20-7,31, m, 2H; 7,80-7,89, m, 2H.
Стадия 2: Вышеупомянутый диэфир (500 мг, 1,5 ммоль) гидролизуют, используя методику, описанную в Примере 6, стадия 2, с получением (S,S)-1,2-бис-(3-карбоксифенил)этан-1,2-диола в виде белого твердого вещества. МС (ХИАД) m/z 301 (M+-1, 100%). 1Н ЯМР (200 МГц, d6-ДМСО): d 3,40, bs, 1H; 4,76, s, 1H; 5,56, bs, 1H; 7,20-7,29, m, 2H; 7,80-7,91, m, 2H.
Опыт 8
В этом опыте представлен а синтез 3,3'-бис-(карбокси-метил)стильбена:
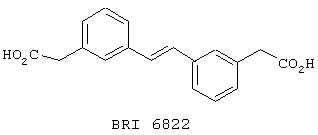
Стадия 1: Метил 3-бромфенилацетат (8,0 г, 34,9 ммоль) подвергают взаимодействию с этиленом, используя методику, описанную в Примере 6, стадия 1. Сырой продукт реакции очищают колоночной хроматографией (SiO2, 5% EtOAc в петролейном эфире) с получением 3,3’-бис-[(карбо-метокси)метил]стильбена и метил 3-(этенил)фенил-ацетата в виде белой твердой смеси.
3,3’-бис-[(карбо-метокси)метил]стильбен: 1Н ЯМР (200 МГц, СDСl3): d 3,65, s, 2H; 3,70, s, 3Н; 7,1, s, 1H, 7,15-7,50, m, 4H. 13С ЯМР (50 МГц, CDCl3): d 41,2, 52,1, 125,3, 127,4, 128,6. 128,7, 128,9, 134,4, 137,6, 171,9.
Метил 3-(этенил)фенилацетат: 1Н ЯМР (200 МГц, CDCl3): d 3,63, s, 2H; 3,68, s, 3Н; 5,28, d, J=10,9 Гц, 1H; 5,78, d, J=18,8 Гц, 1H; 6,72, d, J=10,9, 18,8 Гц, 1H; 7,18-7,41, m, 4H.
Стадия 2: 3,3’-бис-[(карбометокси)метил]стильбен гидролизуют, используя методику, описанную в Примере 6, стадия 2, с получением 3,3’-бис-[(карбокси)метил]стильбена в виде белого твердого вещества. МС (ХИАД) m/z 295 (М+-1, 100%). 1Н ЯМР (200 МГц, d6-ДMCO): d 3,60, s, 2H; 7,00-7,62, m, 5H.
Опыт 9
В этом опыте представлен синтез 1,2-бис-[м-(карбоксиметил)фенил]этана:
Стадия 1: 3,3’-бис-[(карбометокси)метил]стильбен (Пример 8, стадия 1) (500 мг, 1,5 ммоль) и палладий на углероде (10%, 200 мг) в метаноле (20 мл) гидрируют в атмосфере водорода в течение 16 часов при к.т. Реакционную смесь фильтруют и концентрируют в вакууме с получением 1,2-бис-[м-(карбометоксиметил)фенил]этана в виде бесцветного масла. 1Н ЯМР (200 МГц, CDCl3): d 2,91, s, 2H; 3,63, s, 2H; 3,72, s, 3Н; 7,08-7,31, m, 4H.
Стадия 2: Вышеупомянутый эфир гидролизуют, используя методику, описанную в Примере 6, стадия 2, с получением 1,2-бис-[м-(карбоксиметил] этана в виде белого твердого вещества. МС (ХИАД) m/z 297 (M+-1, 100%). 1Н ЯМР (200 МГц, d6-ДМСО): d 2,82, s, 2H; 3,56, s, 2H; 7,06-7,06-7,27, m, 4H; 12,25, bs, 1H.
Опыт 10
В этом опыте представлен синтез 1-[м-(карбоксиметил)фенил]-2-[м-(карбоксифенил)]этана:
Стадия 1: Метил 3-(этенил)фенилацетат (Пример 8, стадия 1) (1,1 г, 6,25 ммоль), метил 3-бромбензоат (960 мг, 4,46 ммоль), ацетат палладия (20 мг, 0,09 ммоль), N,N-диметилглицина гидрохлорид (249 мг, 1,78 ммоль) и ацетат натрия (731 мг, 8,92 ммоль) растворяют в N-метилпирролидиноне, дегазируют аргоном и нагревают до 130° С в течение 5 часов. Реакционную смесь охлаждают до к.т., разводят ЕtOАс (100 мл), и органическую фазу промывают водой (100 мл), водным раствором НСl (1 М, 100 мл) и насыщенным водным раствором NаНСО3 (100 мл). Органические экстракты сушат (Na2SO4), фильтруют и концентрируют в вакууме с получением транс-1-[м-(3-карбоксиметоксиметил)-фенил]-2-[3-(карбометокси-фенил)] этана в виде бесцветного масла. МС (ХИАД) m/z 309 (M+-1, 100%). 1Н ЯМР (200 МГц, CDCl3): d 3,64, s, 2H; 3,68, s, 3Н; 7,16-7,56, 6Н; 7,63-7,71, m, 1H; 7,90-7,98, m, 1H; 8,20, m, 1H.
Стадия 2: Вышеупомянутое соединение гидрируют в соответствии со способом, описанным в Примере 9, стадия 1, с получением 1-[м-(карбометоксиметил)-фенил]-2-[м-(карбометоксифенил)]этана в виде бесцветного масла. 1Н ЯМР (200 МГц, CDCl3): d 2,87, m, 4Н; 3,56 s, 2H; 3,60, s, 3Н; 3,84, s, 3Н; 6,95-7,36, 6Н; 7,77-7,90, m, 2H.
Стадия 3: Эфир, полученный на Стадии 2 гидролизуют, используя способ, описанный в Примере 6, стадия 2, с получением 1-[м-(карбоксиметил)фенил]-2-[м-(карбоксифенил)]этана в виде белого твердого вещества. МС (ХИАД) m/z 283 (M+-1, 100%). 1Н ЯМР (200 МГц, d6-ДMCO): d 2,92, m, 4Н; 3,55, s, 2H; 7,02-7,35, m, 4H; 7,36-7,60, m, 2H; 7,71-7,93, m, 2H. 13С ЯМР (50 МГц, d6-ДМСО): d 38,6, 38,7, 40,9, 128,5, 128,8, 130,0, 130,3, 131,0, 131,2, 132,6, 134,8, 136,7, 143,1, 143,8, 169,2, 174,5.
Опыт 11
В этом опыте представлен а синтез N,N-бис(м-карбоксибензил)глицина:
Стадия 1: м-Цианобензилбромид (2,35 г, 12,0 ммоль) медленно добавляют к раствору гидрохлорида глицинметилового эфира (0,63 г, 5,0 ммоль), NаНСО3 (1,4 г, 17,0 ммоль) и NaI (0,37 г, 2,4 ммоль) в ДМСО (5 мл) и ТГФ (20 мл). Реакционную смесь нагревают до температуры дефлегмации в течение 2 часов, охлаждают до комнатной температуры и разводят ЕtOАс (50 мл) и водой (40 мл). Органическую фазу промывают водой (3× 40 мл), насыщенным водным раствором NaCl (40 мл), сушат (Na2SO4), фильтруют и концентрируют в вакууме с получением N,N-бис(м-цианобензил)глицинметилового эфира в виде бесцветного масла достаточной чистоты для последующих реакций. Дополнительную очистку можно произвести экстракцией с разбавленным водным раствором кислоты, подщелачиванием и экстракцией органическим растворителем. 1H ЯМР (200 МГц, CDCl3): d 3,39, s, 2H; 3,71, s, 3Н; 3,86, s, 4H; 7,39-7,73, m, 10Н.
Стадия 2: Вышеупомянутый нитрил (1,5 г, 5,02 ммоль) гидролизуют в соответствии со способом, описанным в Примере 4, стадия 2, с получением N,N-биc(м-карбоксибензил)глицина (сульфатная соль) в виде беловатого твердого вещества. МС (ХИАД) m/z 342 (M+-1, 100%). 13С ЯМР (50 МГц, d4-MeOH): d 53,8, 59,0, 130,2, 131,5, 132,7, 132,8, 134,2, 136,1, 169,1, 170,7.
Опыт 12
В этом опыте представлен а синтез (3R,4R)-1,3-бис-(п-метоксибензил)-4,5-бис(м-фосфонофенил)-имидазолид-2-она:
Стадия 1: п-Метоксибензиламин (7,42 г, 54 ммоль) в СН2Сl2 (100 мл), содержащий безводный MgSO4, обрабатывают м-бромбензальдегидом (10,0 г, 54 ммоль) при 0° С. Реакционную смесь оставляют перемешиваться при комнатной температуре в течение 16 часов, фильтруют и концентрируют в вакууме с получением N-п-метоксибензилимина м-бромбензальдегида в виде бесцветного масла. 1Н ЯМР (200 МГц, CDCl3): d 3,82, s, 3Н; 4,78, s, 2H; 6,92, d, J=7,5 Гц, 2H; 7,16, d, J=7,5 Гц, 2H; 7,52-7,60, m, 1H; 7,62-7,72, m, 1H; 7,98, m, 1H; 8,29, m, 1H.
Стадия 2: 1,2-Дибромэтан (0,5 мл) добавляют к цинку (1,31 г, 20,0 ммоль) в CH3CN (5 мл), и эту смесь нагревают до температуры флегмы в течение 1 минуты. Как только реакционная смесь охладится до к.т., к ней добавляют ТМССl (1 мл), и реакционную смесь перемешивают при комнатной температуре в течение 1 часа. Вышеупомянутый имин (6,08 г, 20 ммоль) в CH3CN (20 мл) добавляют одной порцией с последующим добавлением ТМССl (3,8 мл) в течение 30 минут. Затем реакционную смесь перемешивают в течение 4 часов при 35-40° С. Реакционную смесь гасят водным раствором NH4OH (6 мл) и насыщенным водным раствором NH4Cl (14 мл) и фильтруют. Водную фазу отделяют, и эту водную фазу экстрагируют Et2O (50 мл). Объединенные органические экстракты сушат (Na2SO4), фильтруют и концентрируют в вакууме с получением оранжевого масла. После проведения колоночной хроматографии (SiO2 25% Et2O в петролейном эфире) получают (1R,2R)-N,N’-(п-метоксибензил)-1,2-(м-бромфенил)этан-1,2-диамина в виде бесцветного масла. 13С ЯМР (50 МГц, CDCl3): d 50,5, 55,3, 67,4, 113,8, 122,4, 126,7, 129,2, 129,6, 130,3, 130,7, 132,1, 143,4, 158,7.
Стадия 3: N,N’-Дисукцинимидилкарбонат (160 мг, 0,64 ммоль) добавляют к раствору вышеупомянутого диамина (260 мг, 0,43 ммоль) в CH3CN (10 мл). Реакционную смесь нагревают до температуры дефлегмации в течение 2 часов. Затем добавляют последующую порцию N,N’-дисукцинимидилкарбоната (160 мг, 0,64 ммоль), и реакционную смесь нагревают до температуры флегмы в течение последующих 2 часов. Реакционную смесь концентрируют и распределяют между ЕtOАс (40 мл) и водным раствором НСl (1 M, 40 мл). Органическую фазу промывают насыщенным водным раствором NаНСО3 (40 мл), насыщенным NaCl (40 мл), сушат (Na2SO4), фильтруют и концентрируют в вакууме с получением оранжевого масла. После проведения колоночной хроматографии (SiO2, 25% Et2O в петролейном эфире ® ЕtOАс) получают (3R,4R)-1,3-бис-(п-метоксибензил)-4,5-бис(м-бромфенил)-имидазолид-2-он в виде белого твердого вещества. 13С ЯМР (50 МГц, CDCl3): d 45,3, 55,2, 65,0, 111,4, 123,1, 125,9, 128,1, 129,8, 130,2, 130,5, 131,7, 140,7, 159,1, 159,9.
Стадия 4: Вышеупомянутую мочевину (200 мг, 0,31 ммоль) обрабатывают диэтилфосфитом в условиях, описанных в Примере 3, стадия 1, с получением (3R,4R)-1,3-бис-(п-метоксибензил)-4,5-бис[м-(диэтокси-фосфоно)фенил]-имидазолид-2-она в виде бесцветного масла. МС (ХИАД) m/z 750 (M+-1, 100%). 31P ЯМР (81 МГц, развязка протонов, CDCl3): d +18,4.
Стадия 5: Вышеупомянутый фосфонат (340 мг, 0,45 ммоль) обрабатывают триметилсилилбромидом в условиях, описанных в Примере 3, стадия 2, с получением (3R,4R)-1,3-бис-(п-метоксибензил)-4,5-бис(м-фосфонофенил)-имидазолид-2-она в виде беловатого твердого вещества. 31P ЯМР (81 МГц, развязка протонов, CDCl3): d +11,5.
Опыт 13
В этом опыте представлен синтез 6-амино-4-(4'-пиридил)-2-(1Н)-пиридона:
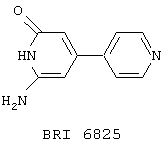
Стадия 1: при взаимодействии 4,4’-бипиридина с NaNH2 в соответствии с JOC, 1997, 62, 2774 получают, в дополнение описанному 2,2’-диамино-4,4’-бипиридину, ранее не описанный 2-амино-4,4’-бипиридин. 13С ЯМР (50 МГц, d6-ДМСО): d 105,0, 109,5, 120,9, 145,4, 148,9, 150,3, 160,5.
Стадия 2: Вышеупомянутый амино-пиридин (1,5 г, 10,5 ммоль) растворяют в уксусном ангидриде (20 мл) и нагревают до 60° С в течение 3 часов. Реакционную смесь охлаждают до комнатной температуры и фильтруют. Твердое вещество промывают Et2O (2× 50 мл) и сушат в вакууме с получением 2-(ацетиламино)-4-(4’-пиридил)-пиридина в виде светло-коричневого твердого вещества. 1H ЯМР (200 МГц, d6-ДМСО): d 2,12, s, 3Н; 7,40-7,58, m, 1H; 7,60-7,83, m, 2H; 8,30-8,58, m, 2H; 8,62-8,88, m, 2H.
Стадия 3: Вышеупомянутый пиридин (0,9 г, 4,2 ммоль) растворяют в CH2Cl2 (50 мл) и обрабатывают м-хлорпербензойной кислотой (4,86 г, 60 мас.%) и реакционную смесь нагревают до температуры флегмы в течение 16 часов. Реакционную смесь охлаждают до к.т., фильтруют и осадок промывают Et2O (2× 50 мл). Осадок добавляют к уксусному ангидриду (25 мл) и нагревают до температуры флегмы в течение 4 часов, охлаждают до комнатной температуры и собирают осадок. Этот осадок добавляют к метанолу (5 мл), обрабатывают Nа2СО3 (50 мг) и нагревают до температуры флегмы в течение 5 часов. Реакционную смесь охлаждают до к.т., фильтруют и фильтрат концентрируют в вакууме. После растирания с Et2O получают 6-амино-4-(4’-пиридил)-2-(1Н)-пиридон в виде желтого твердого вещества. МС (ХИ) m/z 188 (M+-1, 100%). 1Н ЯМР (200 МГц, d4-MeOH): d 5,83, d, J=1 Гц, 1H, 5,90, d, J=1 Гц, 1H; 7,67, d, J=7 Гц, 2Н; 8,16, d, J=7 Гц, 2H.
Опыт 14
В этом опыте отображается активность некоторых соединений по настоящему изобретению при модулировании Fc рецептора.
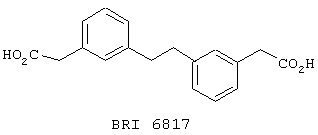
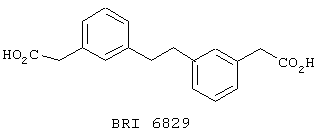
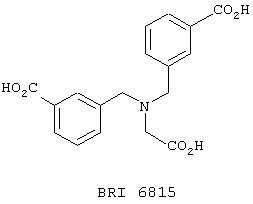
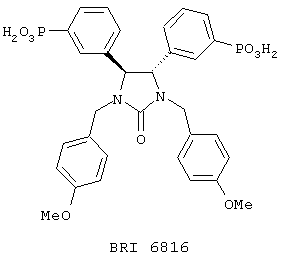
Взаимодействие между рекомбинантным растворимым FcgRIIa и иммуноглобулином человека в присутствии небольших количеств соединений, представленных на фиг. 5А и 5В, исследуют при помощи биодатчика BIAcore 2000 (Pharmacia Biotech, Uppsala, Sweden) при 22° C в Hepes буферном солевом растворе [НБР: 10 mM Hepes (рН 7,4), 150 mМ NaCl, 3,4 mМ ЭДТК, 0,005% сурфактант Р20 (Pharmacia)]. Мономерный IgG1 человека, IgG3 и IgE (50 мг/мл) (контроль на неспецифичное связывание) ковалентно связывают с поверхностью карбоксиметилированного декстрана на сенсорном чипе СМ-5 (BIAcore, Uppsala, Sweden), используя протокол связывания с аминогруппой (BIAcore, Uppsala, Sweden). Дополнительный канал химически обрабатывают, используя протокол связывания. Рекомбинантный растворимый FcgRIIa примененяют в концентрации 125 мг/мл, что эквивалентно 50% связыванию. Рекомбинантный растворимый FcgRIIa предварительно инкубируют с каждым из соединений при комнатной температуре в течение 30 минут перед нанесением на поверхность сенсорного чипа в течение 1 минуты со скоростью 10 мл/мин, с последующей 3-минутной фазой диссоциации. Все поверхности регенирируют 50 мМ диэтиламином (около рН 11,5), 1 М NaCl между анализами каждого из соединений. Затем измеряют максимальный отклик для каждого из взаимодействий. Отклики при неспецифичном связывании (канал IgE) вычитают из значений при связывании с IgG1 и IgG3. Измерения корректируют в зависимости от различий в составе буфера между соединениями и рецептором.
Пользуясь чувствительностью поверхностного плазменного резонанса, измеряют степень взаимодействия IgG1 (фиг. 6 и 8) и IgG3 (фиг. 7 и 9) с растворимым FcgRIIa в присутствии различных соединений. Соединения BRI6728, BRI6734, BRI6813, BRI6800, BRI6801, BRI6802, BRI6803, BRI6814, BRI6817, BRI6822, BRI6823 и BRI6824 ингибируют взаимодействие растворимого FcgRIIa с IgG1 (фиг. 6 и 8). При концентрации 5 мг/мл соединения BRI6798, BRI6799, BRI6815 и BRI6825 усиливают взаимодействие между растворимым FcgRIIa с IgG1 (фиг. 6 и 8). Соединения BRI6728, BRI6734, BRI6813, BRI6800, BRI6801, BRI6802, BRI6803, BRI6814, BRI6816, BRI6817, BRI6822, BRI6823 и BRI6824 ингибируют взаимодействие растворимого FcgRIIa с IgG3 (фиг. 7 и 9). Соединения BRI6727, BRI6798, BRI6815 и BRI6825 усиливают взаимодействие между растворимым FcgRIIa с IgG3 при концентрации около 5 мг/мл и 10 мг/мл.
Опыт 15
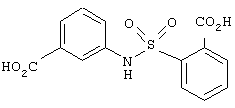
В этом опыте представлен синтез N-(3’-карбоксифенил)-2-(карбоксибензол)сульфонамида:
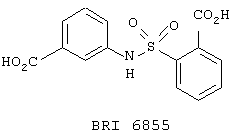
Стадия 1: Метил 2-(хлорсульфонил)-бензоат (2,25 г, 8,73 ммоль) в метиленхлориде (20 мл) по каплям добавляют к раствору этил 3-аминобензоата (1,44 г, 8,73 ммоль) и триэтиламина (1,21 мл, 8,73 ммоль) в метиленхлориде (10 мл) при 0° С. Реакционную смесь оставляют нагреваться до комнатной температуры и перемешивают в течение ночи. Реакционную смесь промывают водой (20 мл), водным раствором НСl (1 М, 20 мл) и водным раствором NaOH (1 M, 20 мл), сушат (MgSO4), фильтруют и концентрируют под вакуумом с получением оранжевого масла. После растирания с этиловым эфиром получают N-(3’-карбоэтоксифенил)-2-(карбометокси)-бензол-сульфонамид в виде белого твердого вещества. 1Н ЯМР (200 МГц, CDCl3): d 1,31, t, J=6,0 Гц, 3Н; 4,00, s, 3Н; 4,29, q, J=6,0 Гц, 2H; 7,23-7,61, m, 5Н; 7,66-7,92, m, 3H; 8,26. br s, 1H.
Стадия 2: Вышеупомянутый диэфир (1,0 г, 2,75 ммоль) гидролизуют, используя методику, описанную в Примере 6, стадия 2, с получением N-(3’-карбоксифенил)-2-(карбоксибензол)сульфонамида в виде белого твердого вещества. МС (ХИ) m/z 320 (М+-1, 100%). 13С ЯМР (50 МГц, d6-ДМСО): d 168,0, 166,3, 137,3, 135,8, 133,4, 132,6, 131,3, 130,1, 129,0, 128,8, 128,0, 124,5, 123,8 и 120,5.
Опыт 16
В этом опыте представлен а синтез транс-3,3'-бис-(N-гидроксиамидино)стильбена:
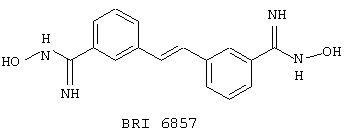
Стадия 1: транс-3,3’-Дицианостильбен получают из 3-бромбензонитрила способом Примера 6, стадия 1. МС (ХИ) m/z 230 (М+, 100%).
Стадия 2: транс-3,3’-Дицианостильбен (1,5 г, 6,52 ммоль), гидроксиламина гидрохлорид (3,26 г, 50 ммоль) и Na2CO3 (3,04 г, 30 ммоль) в ЕtOН (40 мл) и воду (15 мл) нагревают до температуры дефлегмации в течение 3 часов. Реакционную смесь охлаждают до комнатной температуры, и этанол удаляют под вакуумом. Оставшийся раствор экстрагируют EtOAc (2× 50 мл), и объединенные органические экстракты промывают водным раствором НСl (1 М, 2× 20 мл). Объединенные водные экстракты подщелачивают и экстрагируют EtOAc (3× 50 мл). Объединенные органические экстракты сушат (MgSO4), фильтруют и концентрируют под вакуумом с получением бесцветного твердого вещества. МС (ХИ) m/z 297 (M++1, 100%). 13С ЯМР (50 МГц, d6-ДМСО): d 123,3, 124,8, 127,1, 128,6, 133,8, 136,8 и 150,7.
Опыт 17
В этом опыте представлен а синтез (d,1)- и мезо-2-ацетиламино-3-(3-{2-[3-(2-ацетиламино-2-карбоксиэтил)фенил]этил}-фенил)-пропионовой кислоты:
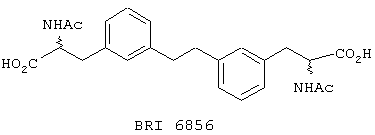
Стадия 1: 3-Бромбензальдегид (23,7 г, 128,2 ммоль), N-ацетилглицин (10,0 г, 85,5 ммоль) и ацетат натрия (5,26 г, 64,1 ммоль) в уксусном ангидриде (60 мл) нагревают до температуры дефлегмации в течение 1 часа. Реакционную смесь охлаждают до комнатной температуры и добавляют воду (100 мл). Полученную суспензию фильтруют, и твердое вещество промывают водой (2× 50 мл). Оставшееся твердое вещество растворяют в метиленхлориде (100 мл), сушат (MgSO4), фильтруют и концентрируют под вакуумом с получением желтого твердого вещества. Это твердое вещество суспендируют в сухом МеОН (200 мл) и нагревают до температуры флегмы в течение 9 часов. Реакционную смесь концентрируют под вакуумом с получением желтого твердого вещества. После перекристаллизации из EtOAc и петролейного эфира получают метил м-бром-α -ацетамидоциннамат в виде желтого твердого вещества. МС (ХИ) m/z 298 (M++1 (Br=79), 100%), 300 (M++1 (Br=81), 100%). 13С ЯМР (50 МГц, d6-ДMCO): d 23,3, 52,8, 122,5, 125,0, 128,06, 130,0, 130,2, 132,2, 132,3, 135,9, 165,4 и 168,8.
Стадия 2: транс-Метил 2-ацетиламино-3-(3-{2-[3-транс-(транс-2-ацетиламино-2-карбометоксиэтенил)фенил]этенил}фенил)проп-2-еноат получают из вышеупомянутого соединения способом Примера 6, стадия 1. МС (ХИ) m/z 461 (М+-1, 100%).
Стадия 3: Соединение со стадии 2 (380 мг, 0,82 ммоль) и Pd/C (300 мг, 10%) в МеОН (20 мл) перемешивают в атмосфере водорода при комнатной температуре в течение 16 часов. Реакционную смесь фильтруют и концентрируют под вакуумом с получением (d,1)- и мезо-метил 2-ацетиламино-3-(3-{2-[3-(2-ацетиламино-2-карбометокси-этил)-фенил]-этил}-фенил)-пропаноата в виде прозрачного вязкого масла, которое используют без последующей очистки.
Стадия 4: Соединение со стадии 3 (280 мг, 0,60 ммоль) гидролизуют, используя способ, описанный в Примере 6, стадия 2, с получением (d,1)- и мезо-2-ацетиламино-3-(3-{2-[3-(2-ацетиламино-2-карбоксиэтил)фенил]этил}-фенил)пропионовой кислоты в виде прозрачного вязкого масла. МС (ХИ) m/z 440 (М+-1, 100%).
Опыт 18
В этом опыте представлен синтез (3R,4R)-4,5-бис(м-карбоксифенил)имидазолид-2-она:
Стадия 1: К раствору (R,R)-1,2-бис-[3-(карбометокси)фенил]этан-1,2-диола (Пример 7, стадия 1) (1,5 г, 4,54 ммоль) в пиридине (10 мл) при 0° С по каплям добавляют метансульфонилхлорид (1,01 мл, 13,1 ммоль). Реакционную смесь оставляют нагреваться до комнатной температуры и перемешивают в течение ночи. Реакционную смесь разводят водой (30 мл) и 30 мл метиленхлорида, и водную фазу экстрагируют 2× 10 мл метиленхлоридом. Объединенные органические экстракты промывают 2× 20 мл 1 М водным раствором НСl, 20 мл водного раствора бикарбоната натрия, сушат над сульфатом магния, фильтруют и концентрируют под вакуумом с получением ди-метансульфоната (R,R)-1,2-бис-[3-(карбометокси)фенил]этан-1,2-диола в виде желтого вязкого масла.
Стадия 2: Раствор вышеупомянутого мезилата (505 мг, 1,0 ммоль) и NaN3 (150 мг, 2,31 ммоль) в 6 мл ДМФ нагревают до 90° С в течение 17 часов. Реакционную смесь охлаждают до к.т., разводят 50 мл диэтилового эфира и промывают 3× 50 мл водой. Органическую фазу сушат над MgSO4, фильтруют и концентрируют под вакуумом с получением (R,R)-1,2-бис-3-(карбометокси)фенил]-1,2-диазо-этана в виде желтого вязкого масла. 1Н ЯМР (200 МГц, CDCl3): d 3,93, s, 3Н; 4,73, s, 1H; 7,17-7,39, m, 2H; 7,78-8, 01, m, 2H.
Стадия 3: Вышеупомянутый диазид (611 мг, 1,61 ммоль) и Pd на углероде (10%, 50 мг) в метаноле обрабатывают концентрированной водной НСl (3,86 мл, 3,86 ммоль).
Реакционную смесь помещают в атмосферу водорода и перемешивают при комнатной температуре в течение 30 часов. Реакционную смесь фильтруют через целит и концентрируют с получением гидрохлоридной соли (R,R)-1,2-бис-[3-(карбометокси)фенил]-1,2-диамино-этана. МС (ХИ) m/z 329 (M++1 для свободного амина, 70%), 312 (100%).
Стадия 4: Вышеупомянутый диамин (в форме свободного основания) (280 мг, 0,85 ммоль) в 5 мл ацетонитрила обрабатывают ДМАП (DMAP) (104 мг, 0,85 ммоль) раствором ди-трет-бутил дикарбоната (204 мг, 0,94 ммоль) в 1 мл ацетонитрила при к.т. Реакционную смесь перемешивают в течение 25 мин при комнатной температуре и распределяют между 50 мл эфира и 50 мл 1 M HCl. Органическую фазу отделяют, сушат над сульфатом натрия, фильтруют и концентрируют под вакуумом. После колоночной хроматографии получают (3R,4R)-4,5-бис-(м-карбометоксифенил)имидазолид-2-он в виде белого твердого вещества. МС (ХИАД) m/z 355 (M++1, 100%). 1Н ЯМР (200 МГц, d6-ДМСО): d 3,86, s, 6Н; 4,57, s, 2H; 7,16, br s, 2H; 7,46-7,61, m, 4H; 7,88-8,00, m, 4H.
Стадия 5: Вышеупомянутый диэфир (68 мг, 0,19 ммоль) гидролизуют, используя методику, описанную в Примере 6, стадия 2, с получением (3R,4R)-4,5-бис(м-карбоксифенил)имидазолид-2-она в виде белого твердого вещества. МС (электроспрей) m/z 327 (M++1, 100%). 13С ЯМР (50 МГц, d6-ДМСО): d 64,3, 127,4, 129,1, 129,3, 131,1, 131,4, 142,1, 162,5, 167,3.
Опыт 19
В этом опыте представлен синтез 3-([3’-(1’’-оксо-2’’,2’’,2’’-трифторэтил)фенокси]метил)фенилтрифторметилкетона:
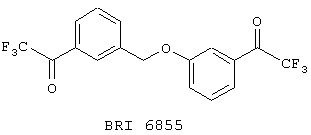
К раствору 3-[(м-бромфенил)метокси]бром-бензола (Пример 2, стадия 1) (233 мг, 3,68 ммоль) в 6 мл ТГФ при -78° С по каплям добавляют трет-бутиллитий (1,6 мл, 1,7 М в пентане, 2,72 ммоль). Через 20 мин при этой температуре этот раствор по каплям добавляют к раствору этилтрифторацетата (0,35 мл, 2,94 ммоль) в 5 мл ТГФ при -78° С. Реакционную смесь перемешивают в течение 16 часов и в течение этого времени реакционная смесь достигает к.т. Реакционную смесь распределяют между 20 мл 1 М НСl и 50 мл эфира. Органическую фазу отделяют, сушат над сульфатом натрия, фильтруют и концентрируют под вакуумом с получением 3-([3’-(1’’-оксо-2’’,2’’,2’’-трифторэтил)фенокси]метил)фенилтрифторметилкетона в виде бесцветного масла. МС (ХИ) m/z 377 (M++1, 100%), 19F ЯМР (188 МГц, CDCl3-): d-71,76 и -71,90.
Опыт 20
В этом опыте представлен а синтез Ac-Phe-Gln-Asn-Gly-Lys-Ser-NH2:
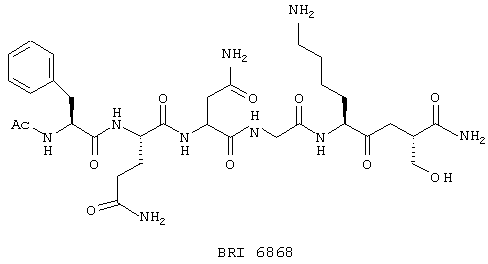
Этот пептид синтезируют методом синтеза пептида на твердой фазе. После N-ацилирования и разрыва связи со смолой получают указанное в заголовке соединение в виде белого твердого вещества. ВЭЖХ и МС анализы подтверждают чистоту и идентичность данного материала.
Опыт 21
В этом опыте представлен а синтез цикло-[N-фенилглицин-Gln-Аsn-(D)-Аsр]-Lуs-Ser-NH2:
Стадия 1: N-[(4S)-3-бензилоксикарбонил-5-оксо-оксазолидин-4-ил]-ацетилхлорид (3,00 г, 10 ммоль) в дихлорметане (20 мл) по каплям добавляют к раствору трет-бутил-N-фенилглицината (2,3 мг, 11 ммоль) в пиридине (10 мл) при 0° С. Реакционную смесь оставляют нагреваться до комнатной температуры и перемешивают в течение ночи. Реакционную смесь разводят Н2О (100 мл) и ЕtOАс (150 мл). Органическую фазу отделяют и последовательно промывают лимонной кислотой (10%, 2× 100 мл) и солевым раствором (100 мл), сушат (MgSO4), фильтруют и концентрируют в вакууме с получением желтого вязкого масла. После колоночной хроматографии (SiO2, 20-50% EtOAc в петролейном эфире) получают трет-бутил N-[(4S)-3-бензилоксикарбонил-5-оксо-оксазолидин-4-ил-ацетил]-N-фенилглицинат в виде белой пены. МС (ХИАД) m/z 467 (M+-1, 100%).
Стадия 2: Водный раствор NaOH (3 мл, 1 М, 3 ммоль) по каплям добавляют к раствору вышеупомянутого дипептида (570 мг, 1,22 ммоль) в метаноле (20 мл) при 0° С. Реакционную смесь оставляют нагреваться до комнатной температуры и контролируют при помощи ТСХ. Далее реакционную смесь концентрируют и распределяют между Et2O (30 мл) и лимонной кислотой (10%, 30 мл) при 0° С. Водную фазу экстрагируют Et2O (3× 30 мл), и объединенные органические экстракты сушат (MgSO4), фильтруют и концентрируют под вакуумом с получением белого твердого вещества. После колоночной хроматографии (SiO2, 2-5%, МеОН в дихлорметане) получают трет-бутил N-[(2S)-N-бензилоксикарбонил-аспартил]-β -N-фенилглицинат в виде белого твердого вещества. МС (ХИАД) m/z 456 (M++1, 90%).
Стадия 3: Вышеупомянутое соединение (1,35 г, 2,96 моль) в МеОН (40 мл), содержащем палладий на углероде (10%, 500 мг), помещают в атмосферу водорода и перемешивают при комнатной температуре в течение 16 часов. Реакционную смесь фильтруют и концентрируют в вакууме с получением трет-бутил N-[(2S)-аспартил])-β -N-фенилглицината в виде белого твердого вещества.
Стадия 4: Вышеупомянутое соединение (890 мг, 2,76 ммоль), Fmoc-O-Su, т.е. N-(9-фторметоксикарбонилокси)сукцинимид (932 мг, 2,76 ммоль), Na2CO3 (880 мг, 8,29 ммоль) в диоксане (15 мл) и Н2О (15 мл) перемешивают при комнатной температуре в течение 16 часов. Реакционную смесь разводят Et2O (100 мл) и H2O (100 мл). Органический слой отделяют и экстрагируют водным раствором Na2CO3 (5%, 3× 100 мл). Объединенные водные экстракты подкисляют 10% водной лимонной кислотой и экстрагируют EtOAc (3× 100 мл). Эти органические экстракты объединяют, сушат (MgSO4), фильтруют и концентрируют под вакуумом с получением трет-бутил N-[(2S)-N-Fmос-аспартил])-β -N-фенилглицината в виде белого твердого вещества. 13С ЯМР (50 МГц, d6-ДМСО): d 28,0, 36,7, 47,0, 50,4, 52,4, 67,0, 67,3, 82,3, 119,9, 125,2, 125,3, 127,1, 127,7, 127,8, 128,8, 130,1, 141,2, 142,8, 143,7, 143,9, 156,1, 167,6, 171,6, 174,5.
Стадия 5: Твердофазным аминокислотным синтезом с использованием вышеупомянутого дипептида защищенного Fmoc с последующей циклизацией на смоле и разрывом связи получают цикло-[N-фенилглицин-Gln-Аsn-(D)-Аsр]-Lуs-Sеr-NН2.
Опыт 22
В этом опыте представлен а синтез 2-(2’-фенилэтал)-α -N-ацетил-лизинамида и его гидрохлор идной соли:
Стадия 1: К смеси металлического натрия (138 мг, 5,98 ммоль) в сухом этаноле (16 мл) добавляют 2-циано-4-(фенэтил)этилбутаноат (1,0 г, 4,6 ммоль), и эту смесь перемешивают при комнатной температуре в течение 30 мин. Затем добавляют 4-бром-бут-1-ен (0,6 мл, 6 ммоль), и смесь нагревают при температуре флегмы в течение 16 часов. Полученную суспензию охлаждают до комнатной температуры, концентрируют при пониженном давлении, разводят эфиром (100 мл) и NH4Cl (100 мл насыщенного водного раствора). Водный слой отделяют и экстрагируют эфиром (3× 50 мл). Органические слои объединяют, сушат (MgSO4), фильтруют и концентрируют с получением светло-коричневого масла. После колоночной хроматографии (диоксид кремния, элюирование смесью 20% эфир/бензин) получают этил 2-циано-2-(2’-фенэтил)-гекс-5-еноат в виде прозрачного, бесцветного масла. 1Н ЯМР (200 МГц, CDCl3): d 1,35 (t, J=7,0 Гц, 3Н), 1,82-2,45 (m, 6H), 2,65 (td, J=12,4 Гц и 7,0 Гц, 1Н), 2,90 (td, J=12,4 Гц и 7,0 Гц, 1Н), 4,24 (q, J=7,0 Гц, 2Н), 5,00-5,13 (m, 2Н), 5,67-5,76 (m, 1H), 7,15-7,35 (m, 5Н).
Стадия 2: Смесь вышеупомянутого олефина (0,72 г, 2,65 ммоль), LiOH (10,6 мл, 1,0 M, 10,6 ммоль) и ТГФ (50 мл) перемешивают при комнатной температуре в течение 18 часов. Эту реакционную смесь разводят эфиром (100 мл) и водой (100 мл), и фазы разделяют. Водный слой подкисляют до примерно рН 2 2 М водным раствором НСl и переносят в делительную воронку, содержащую эфир (100 мл). Отделенный водный слой экстрагируют эфиром (3× 50 мл). Органические фракции объединяют, сушат (MgSO4), фильтруют и концентрируют при пониженном давлении с получением 2-циано-2-(2’-фенэтил)-5-гексеновой кислоты в виде вязкого, бесцветного масла. Этот материал применяют в следующей реакции без дальнейшей очистки. МС (ХИАД) m/z 244 (M++1, 55%), 242 (М+-1, 63%).
Стадия 3: Дифенилфосфорилазид (2,75 мл, 12,8 ммоль) и триэтиламин (1,75 мл, 12,6 ммоль) добавляют к раствору вышеупомянутой кислоты (2,6 г, 10,7 ммоль) в толуоле (35 мл). Этот раствор нагревают при 100° С в течение 1 часа и затем добавляют трет-бутанол (35 мл). Эту смесь нагревают при 100° С в течение последующих 2 часов, охлаждают до комнатной температуры и концентрируют при пониженном давлении. Полученное желтое масло разводят эфиром (300 мл) и водой (300 мл). Органический слой отделяют, последовательно промывают лимонной кислотой (100 мл 5% водного раствора), NаНСО3 (100 мл 5% водного раствора) и солевым раствором (100 мл), сушат (MgSO4), фильтруют и концентрируют с получением желтого масла. После колоночной хроматографии (диоксид кремния, элюирование смесью 2% этилацетат/хлороформ) получают 2-(N-бoк-aминo)-2-(2+-фeнэтил)-5-гeкceнoнитpил в виде бесцветного масла. МС (ХИАД) m/z 316 (M++1, 5%), 313 (M+-1, 2%). 13С ЯМР (50 МГц, CDCl3): d 28,4, 30,5, 36,3, 38,9, 54,9, 116,3, 119,7, 126,6, 128,4, 128,8, 136,3, 140,1, 153,5.
Стадия 4: NaOH (9,2 мл, 1,0 М) и Н2O2 (38 мл 30% (о/о) водный раствор) добавляют к раствору вышеупомянутого нитрила (523 мг, 1,93 ммоль) в этаноле (20 мл) при 0° С. Реакционную смесь перемешивают при 0° С в течение 30 мин и при комнатной температуре в течение 18 часов. Этанол удаляют при пониженном давлении, остаток разводят эфиром (100 мл) и солевым раствором (100 мл). Водный слой отделяют и экстрагируют эфиром (4× 20 мл). Органические слои объединяют, сушат (MgSO4), фильтруют и концентрируют с получением 2-(N-бок-амино)-2-(2’-фенэтил)-5-гексенамида в виде бесцветной клейкой пены. Этот материал применяют в следующей реакции без дальнейшей очистки R 0,3 (элюирование смесью 30% этилацетат/бензин). МС (ХИАД) m/z 333 М++1, 5%), 233 (100%).
Стадия 5: К раствору вышеупомянутого амида (480 мг, 1,44 ммоль) в дихлорметане (5 мл) добавляют трифторуксусную кислоту (2 мл), и эту смесь перемешивают при комнатной температуре в течение 35 мин. Эту реакционную смесь концентрируют с получением 2-амино-2-(2’-фенэтил)-5-гексанамида в виде красно-коричневого масла. Этот материал применяют в следующей реакции без дальнейшей очистки. МС (ХИАД) m/z 233 (M++1, 100%).
Стадия 6: Уксусный ангидрид (2,5 мл) добавляют к раствору вышеупомянутого амина (335 мг, 1,44 ммоль) в пиридине (2,5 мл) и перемешивают при комнатной температуре в течение 21 часа. Полученную красно-коричневую реакционную смесь концентрируют при пониженном давлении. После колоночной хроматографии (диоксид кремния, элюирование смесью 80% этилацетат/бензин, R 0,36) получают (N-ацетил-амино)-2-(2’-фенэтил)-5-гексенамид в виде пены соломенного цвета. 13С ЯМР (50 МГц, CDCl3): d 24,2, 28,3, 30,4, 35,2, 37,8, 64,0, 115,2, 126,1, 128,4, 128,5, 137,4, 141,1, 169,4, 175,3.
Стадия 7: К раствору вышеупомянутого олефина (130 мг, 0,47 ммоль) в сухом ТГФ (2 мл) по каплям добавляют 9-BBN (4,6 мл, 0,5 М раствор в ТГФ, 2,30 ммоль). Реакционную смесь перемешивают при температуре окружающей среды в течение 18 часов. Эту смесь охлаждают до 0° С, и последовательно добавляют воду (0,5 мл), NaOAc (5 мл 5,0 М водный раствор) и H2O2 (5 мл). Полученную смесь перемешивают при комнатной температуре в течение 2 часов и разводят этилацетатом (30 мл) и солевым раствором (30 мл). Водный слой отделяют и экстрагируют этилацетатом (3× 10 мл). Органические фракции объединяют, сушат (MgSO4), фильтруют и концентрируют при пониженном давлении с получением светло-желтого масла. После колоночной хроматографии (диоксид кремния, элюирование смесью 5% МеОН/этилацетат, R 0,4) получают 2-(N-ацетил-амино)-2-(2’-фенэтил)-6-гидрокси-гексанамид в виде бесцветной, липкой пены. МС (ХИАД) m/z 293 (М++1, 35%), 291 (М+-1, 35%).
Стадия 8: Триэтиламин (0,1 мл, 0,72 ммоль) и метансульфонилхлорид (0,05 мл, 0,65 ммоль) добавляют к раствору вышеупомянутого спирта (88 мг, 0,30 ммоль) в дихлорметане (2 мл) при 0° С. Полученную смесь перемешивают при температуре окружающей среды в течение 19 часов и разводят этилацетатом (30 мл) и солевым раствором (30 мл). Водный слой отделяют и экстрагируют этилацетатом (3× 10 мл). Органические слои объединяют, сушат (MgSO4), фильтруют и концентрируют при пониженном давлении с получением 2-(N-ацетил-амино)-2-(2’-фенэтил)-6-метансульфонилокси-гексанамида в виде остатка желтовато-коричневого цвета. Сырой продукт применяют в следующей реакции без последующей очистки. МС (ХИАД) m/z 371 (M++1, 45%), 369 (M+-1, 5%).
Стадия 9: Раствор вышеупомянутого мезилата (110 мг, 0,30 ммоль) и азид натрия (54 мг, 0,83 ммоль) в сухом ДМФ (2 мл) нагревают при температуре от 60° С до 65° С в течение 19,5 часов. Оранжево-окрашенную суспензию охлаждают до комнатной температуры, концентрируют и разводят этилацетатом (30 мл) и солевым раствором (30 мл). Водный слой отделяют и экстрагируют этилацетатом (4× 10 мл). Органические фракции объединяют, сушат (MgSO4), фильтруют и концентрируют с получением 2-(N-ацетиламино)-2-(2’-фенэтил)-6-азидо-гексанамида в виде масла желтовато-коричневого цвета. Этот материал применяют в следующей реакции без последующей очистки. 1Н ЯМР (200 МГц, CDCl3): d 1,20-1,78 (m, 6H), 1,92 (s, 3Н), 2,20-3,02 (m, 1H), 3,12-3,30 (m, 2H), 5,57 (s, 1H), 5,90 (s, 1H), 6,72 (s, 1H), 7,04-7,30 (m, 5H).
Стадия 10: Суспензию вышеупомянутого азида (95 мг, 0,30 ммоль) и 10% Pd на С (18,4 мг) в метаноле (2 мл) гидрируют при комнатной температуре и атмосферном давлении в течение 21 часа. Черную суспензию отфильтровывают через небольшой слой диоксида кремния-целита, который промывают несколькими порциями метанола (приблизительно 30 мл). После концентрирования фильтрата получают светлое желтовато-коричневого цвета масло. После колоночной хроматографии (диоксид кремния, элюирование смесью 10% триэтиламин/метанол, R 0,22) получают амид 2-(2’-фенилэтил)-α -N-ацетил-лизина в виде прозрачного бесцветного масла. МС (ХИАД) m/z 292 (M++1, 100%) 290 (M+-1, 30%). 1Н ЯМР (200 МГц, d4-MeOD): d 1,10-1,60 (m, 4H), 1,73-1,90 (m, 1H), 1,95 (s, 3Н), 1,95-2,35 (m, 2H), 2,40-2,80 (m, 5H), 7,10-7,35 (m, 5H).
Небольшое количество амина превращают в соответствующее производное в виде гидрохлоридной соли, добавляя к этому амину 0,5 М водный раствор НСl и концентрируя смесь при пониженном давлении.
Опыт 23
В этом опыте представлен а синтез 4,4’-бис-(3-[(м-карбоксифенокси)метил]-2-пиридона):
Стадия 1: К раствору 3-формил-4-йод-2-метоксипиридина (полученного в соответствии со способом Fang et al., J. Org. Chem., 1994, 59, 6142) (98 мг, 0,37 ммоль) в метаноле (4 мл) добавляют одной порцией твердый NaBH4 при -5° С (28 мг, 0,74 ммоль). При этом наблюдается энергичное выделение газа и желтый реакционный раствор становится бесцветным. Реакцию немедленно гасят, добавляя воду (2 мл), и метанол удаляют при пониженном давлении. Полученный остаток разводят этилацетатом (20 мл) и водой (20 мл). Водный слой отделяют и экстрагируют этилацетатом (3х10 мл). Органические фракции объединяют, сушат (MgSO4), фильтруют и концентрируют с получением 3-(гидроксиметил)-4-йод-2-метоксипиридина в виде бесцветного кристаллического твердого вещества. Этот материал применяют в следующей реакции без последующей очистки. R 0,4 (элюирование смесью 30% этилацетат/бензин). МС (ХИАД) m/z 266 (M++1, 100%). 1Н ЯМР (200 МГц, CDCl3): d 3,98 (s, 3Н), 4,80 (s, 2H), 7,34 (d, J=4,0 Гц, 1H), 7,70 (d, J=4,0 Гц, 1Н).
Стадия 2: Метансульфонил хлорид (0,5 мл, 6,4 ммоль) по каплям добавляют к раствору вышеупомянутого спирта (373 мг, 1,41 ммоль) и триэтиламина (0,95 мл, 6,8 ммоль) в дихлорметане (9,4 мл) при 0° С. Полученную смесь перемешивают при температурах окружающей среды в течение 15 часов и разводят этилацетатом (150 мл) и солевым раствором (150 мл). Водный слой отделяют и экстрагируют этилацетатом (3× 50 мл). Органические фракции объединяют, сушат (MgSO4), фильтруют и концентрируют при пониженном давлении с получением 3-(хлорметил)-4-йод-2-метоксипиридина в виде светлого желто-коричневого кристаллического твердого вещества. Этот материал применяют на следующей стадии без последующей очистки. МС (ХИАД) m/z 284 (М++1, 100%). 1Н ЯМР (200 МГц, CDCl3): d 3,95 (s, 3Н), 4,65 (s, 2H), 7,28 (d, J=4,0 Гц, 1Н), 7,65 (d, J=4,0 Гц, 1H).
Стадия 3: Натриевую соль метил-3-гидроксибензоата (372 мг, 2,14 ммоль) добавляют одной порцией к раствору вышеупомянутого хлорида (399 мг, 1,41 ммоль) в сухом ДМФ (7 мл). Оранжево-окрашенную реакционную смесь перемешивают при комнатной температуре в течение 18 часов и разводят этилацетатом (150 мл) и водой (150 мл). Водный слой отделяют и экстрагируют этилацетатом (3× 30 мл). Органические слои объединяют, сушат (MgSO4), фильтруют и концентрируют с получением коричневого масла. После колоночной хроматографии этого масла (диоксид кремния, элюирование смесью 30% эфир/бензин, R 0,35) получают 4-йод-2-метокси-3-{[(м-карбометокси)фенокси]метил}-пиридин в виде бесцветного масла. МС (ХИАД) m/z 400 (M++1, 40%). 1H ЯМР (200 МГц, CDCl3): d 3,92 (s, 3Н), 3,96 (s, 3H), 5,20 (s, 2H), 7,15-7,42 (m, 3H), 7,62-7,80 (m, 3H).
Стадия 4: Суспензию вышеупомянутого иодида (0,5 г, 1,25 ммоль), Рd(РРh3)4 (141 мг 0,13 ммоль), К2СО3 (518 мг, 3,76 ммоль), диборпинаконовый эфир (159 мг, 0,63 ммоль) в ДМФ (7,6 мл) нагревают при 80° С, защищая от воздействия света, в течение 16 часов. Темно-коричневую реакционную смесь охлаждают до комнатной температуры и разводят этилацетатом (150 мл) и водой (150 мл). Водный слой отделяют и экстрагируют этилацетатом (3× 25 мл). Органические слои объединяют, сушат (MgSO4), фильтруют и концентрируют при пониженном давлении с получением коричневого масла. После колоночной хроматографии этого масла (диоксид кремния, элюирование смесью 50% этилацетат/бензин, R 5,57) получают 4,4’-бис-2-метокси-3-{[(м-карбометокси)фенокси]метил}-пиридин в виде пены. МС (ХИАД) m/z 545 (М++1, 100%).
Стадия 5: Раствор вышеупомянутого димерного диэфира (277 мг, 0,51 ммоль) в LiOH (10 мл, 1,0 M) и ТГФ (10 мл) перемешивают при комнатной температуре в течение 18 часов. Затем сырую реакционную смесь разводят эфиром (75 мл) и фазы разделяют. Водный слой подкисляют до рН 2 2,0 М водным раствором НСl и затем экстрагируют этилацетатом (4× 50 мл). Органические фракции объединяют, сушат (MgSO4), фильтруют и концентрируют с получением 4,4’-бис-2-метокси-3-{[(м-карбокси)фенокси]метил}-пиридина в виде бесцветного твердого вещества. Этот материал применяют на следующей стадии без последующей очистки. МС (ХИАД) m/z 517 (M++1, 100%), 515 (М+-1, 100%).
Стадия 6: Гидролизом вышеупомянутого метокси-пиридина получают 4,4’-бис-{3-[(м-карбоксифенокси)метил]-2-пиридон}.
Опыт 24
В этом опыте представлена активность модулирования Fc рецептора трипептидом и гексапептидом.
Получение пептида.
Для получения ацетилированного трипептида с последовательностью GKS и гексапептида с последовательностью FQNGK-S применяют метод твердофазного пептидного синтеза (ТФПС). См., например, Merrifield, J. Am. Chem. Soc., 1963, 85, 2419, и Merrifield et al., Anal. Chem., 1966, 38, 1905. Эти пептиды были синтезированы при помощи синергичного пептидного синтезатора модели 432А. Сборка пептидов основана на химических свойствах Fmoc (Carpino et al., J. Org. Chem. 1972, 37, 3404) с использованием в качестве исходного материала амидированных смол с С-терминальными концами. После окончания сборки пептидов получали активный эфир, который взаимодействовал с пептидом с образованием ацетилированного N-конца.
Затем применяют стандартные операции по разрыву связи при помощи ТФУ (совместимы с Fmoc) и продукт очищают при помощи высокоэффективной жидкостной хроматографии с обращенной фазой (ВЭЖХ-ОФ). (См. например, Mant, C.T. and Hodges, R.S. eds, 1991, "High-Performance Liquid Chromatography of Peptides and Proteins: Separation, Analysis and Confirmation," CRC Press, Boca Raton, Florida). Две подвижные фазы представляют собой 0,1% трифторуксусную кислоту (ТФУ)/99% Н2O и 0,1% ТФУ/60% СН3СN/39,9% Н2O. Стационарная фаза представляет собой препаративную колонку С8 Brownlee Column. Конечный продукт подвергали масс-спектральному анализу, который подтверждал идентичность и степень чистоты более 95% для обоих пептидов.
Анализы на связывание FcgRIIa в присутствии гекса- или трипептидов
Анализы на взаимодействие между производным FcgRIIa бакуловируса и пептидом (трипептид: GKS, гексапептид: FQNGKS) проводят при помощи биосенсора BIAcore 2000 (Pharmacia Biotech, Uppsala, Sweden) при 22 EC в Hepes буферном солевом растворе (НБР: 10 мМ Hepes, (рН 7,4), 150 мМ NaCl, 3,4 мМ ЭДТК, 0,005% сурфактант Р20 (Pharmacia). Мономерные IgG1, IgG3 и IgE (50 мг/мл) человека ковалентно связывают с поверхностью карбоксиметилированного декстрана сенсорного чипа СМ-5 (BIAcore, Uppsala, Sweden) используя протокол связывания с аминогруппой (BIAcore, Uppsala, Sweden). Канал, с которым не связаны Ig также химически обрабатывают, используя протокол связывания. FcgRIIa в определенной концентрации (50 мг/мл, концентрация, обеспечивающая 50% связывание) смешивают с пептидами в различных концентрациях (см. фиг. 10 и 11), в течение 1 часа при 22 ЕС перед тем, как эта смесь наносится на поверхность сенсорного чипа в течение 1 мин со скоростью 20 мл/мин, с последующей 3-минутной фазой диссоциации. После измерений концентрационных зависимостей все поверхности регенерируют 50 мМ диэтиламином (рН 11,5), 1 М NaCl. Затем определяют общий отклик для каждой концентрации пептида и наносят на график против концентрации пептида. Отклики от неспецифичного связывания (канал IgE) вычитают из откликов от связывания с IgG1 или IgG3.
Результаты
Используя чувствительность поверхностного плазмонного резонанса (ППР), производят определение связывания растворимого FcgRIIa с IgG1 и IgG3 в присутствии гексапептида (FQNGKS) или трипептида (GKS). В присутствии гексапептида связывание растворимого FcgRIIa с иммобилизованным IgG1 усиливалось в четыре раза и в 1,6 раза при взаимодействии с IgG3 (фиг. 10). Однако взаимодействие растворимого FcgRIIa с IgG1 или IgG3 в присутствии трипептида ингибировалось аналогичными концентрациями этого пептида (0-4 мг/мл, фиг. 11).
Опыт 25
В этом опыте представлена активность ингибирования агрегации тромбоцитов для некоторых соединений по настоящему изобретению. В целом способ включает в себя добавление соединения к смеси тромбоцитов и гамма-глобулин, агрегированный в результате тепловой обработки (HAGG). Вне связи с какой-либо из теорий считается, что этот способ отражает способность соединения ингибировать образование агрегатов тромбоцитов, а также его способность разрушать агрегаты тромбоцитов, которые образовались до добавления этого соединения.
Тромбоциты экспрессируют FcgRIIa гамма рецепторы, относящиеся к одному классу. После сшивания FcgFIIa тромбоциты претерпевают различные биохимические и клеточные изменения, которые завершаются их агрегацией. Используя анализ, который специально предназначен для измерения агрегации тромбоцитов, производилось измерение способности соединений ингибировать активацию тромбоцитов.
Материалы и методы
Тромбоциты выделяют следующим образом: 30 мл свежей цельной крови собирают в отборные пробирки с цитратным буфером и центрифугируют их при 1000 об/мин в течение десяти минут. Плазму, обогащенную тромбоцитами, отделяют и центрифугируют при 2000 об/мин в течение пяти минут в четырех пробирках. Супернатанты удаляют и тромбоциты осторожно ресуспендируют в 2 мл Tyrodes буфера на пробирку (137 мМ NaCl, 2,7 мМ КСl, 0,36 мМ NaH2PO4, 0,1% декстроза, 30 мМ цитрат натрия, 1,0 мМ MgCl2·6H2O, pH 6,5) и вновь центрифугируют при 2000 об/мин в течение пяти минут. Супернатанты вновь удаляют и тромбоциты ресуспендируют в 0,5 мл на пробирку Hepes содержащего Tyrodes буфер (137 мМ NaCl, 2,7 мМ КСl, 0,36 мМ NaH2PO4, 0,1% декстроза, 5 мМ Hepes, 2 мМ CaCl2, 1,0 мМ MgCl2·6H2O, pH 7,35). Количество тромбоцитов определяют при помощи гематологического анализатора (Coulter) и доводят до концентрации приблизительно 100× 105 тромбоцитов/мл, используя Hepes содержащий Tyrodes буфер.
Для каждого опыта по агрегированию смесь, содержащую 50 мл агониста Fc-рецептора, гамма-глобулин, агрегированный в результате тепловой обработки ("HAGG", 200 мг/мл) или коллаген (2 мг/мл) инкубируют с 50 мл фосфатного буферного солевого раствора ("ФБР": 3,5 мМ NaH2PO4, 150 мМ NaCl) или BRI соединение (5 мг/мл в ФБР) в течение 60 минут при комнатной температуре. Затем производят анализ, используя двухклеточный агрегометр при 37° С следующим образом: стеклянные кюветы помещают в держатели и предварительно подогревают до 37° С и добавляют 400 мл суспензии тромбоцитов. После достижения стабильности базисной линией 100 мл соединения НАGG:ФБР, HAGG:BRI или соединения коллаген:ФБР, коллаген:BRI добавляют к суспензии тромбоцитов. Последующую агрегацию тромбоцитов регистрируют в течение 15 минут или до тех пор, пока агрегация не будет завершена. Скорость агрегации определяют измеряя градиент и крутизну кривой агрегации.
Результаты
Была определена способность соединений (BRI6855, BRI6803, BRI6813, BRI6864, BRI6856, BRI6868, BRI7002) ингибировать HAGG-индуцируемую FcgRIIa-зависимую агрегацию. Скорость агрегации тромбоцитов, измеренная как отношение приращения прозрачности (у) ко времени (х), см., например, фиг. 12 и 13, в присутствии соединения BRI6855, BRI6803, BRI6813, BRI6864 и BRI6856 уменьшается по сравнению со скоростью, достигнутой при использовании агониста FcgRIIa, и гамма-глобулина, агрегированного в результате тепловой обработки (100%), см. Таблицу 1. Соединения BRI6868 и BRI7002 не проявляют способность значительно ингибировать скорость активации тромбоцитов. Таблица 1. Соединения BRI6855 и BRI6803 понижают HAGG-индуцируемую агрегацию тромбоцитов, но не ингибируют в значительной степени агрегацию тромбоцитов, индуцируемую коллагеном. Это указывает на то, что действие BRI6855 и BRI6803 специфично по отношению к HAGG.
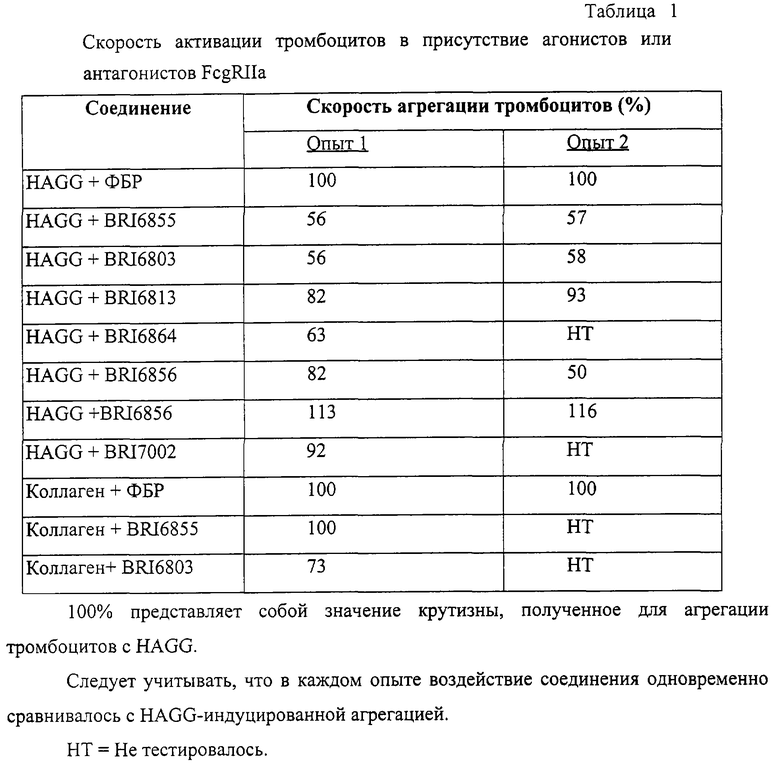
Опыт 26
В этом опыте представлена активность ингибирования агрегации тромбоцитов для некоторых соединений по настоящему изобретению. В целом способ включает в себя добавление соединения к смеси тромбоцитов и ГАТО. Вне связи с какой-либо из теорий считается, что в отличие от Опыта 25, этот способ лишь отображает способность соединения ингибировать образование агрегатов тромбоцитов
Материалы и методы
Для выделения тромбоцитов и определения числа тромбоцитов применялась экспериментальная методика Опыта 25.
В отличие от Опыта 25, анализ на агрегацию тромбоцитов проводят, добавляя 50 мл ФБР или соединения BPI к суспензии тромбоцитов. Спустя примерно одну минуту, к суспензии тромбоцитов добавляют 50 мл агониста (ГАТО, коллаген или АДФ). Последующую агрегацию тромбоцитов регистрируют в течение 10-15 минут или до тех пор, пока агрегация не будет завершена. Скорость агрегации определяют измеряя градиент и крутизну кривой агрегации.
Результаты
Была определена способность соединения BRI6728 ингибировать HAGG-индуцируемую FcgRIIa-зависимую агрегацию. Скорость агрегации тромбоцитов, измеренная как отношение приращения прозрачности (у) ко времени (х), см. например, фиг. 14, в присутствии титруемых количеств соединения BRI6728 уменьшается по сравнению со скоростью, достигнутой при использовании агониста FcgRIIa, и гамма-глобулина, агрегированного в результате тепловой обработки (100%), см. фиг. 14. Результаты агрегации тромбоцитов при использовании других соединений представлены в Таблице 2.
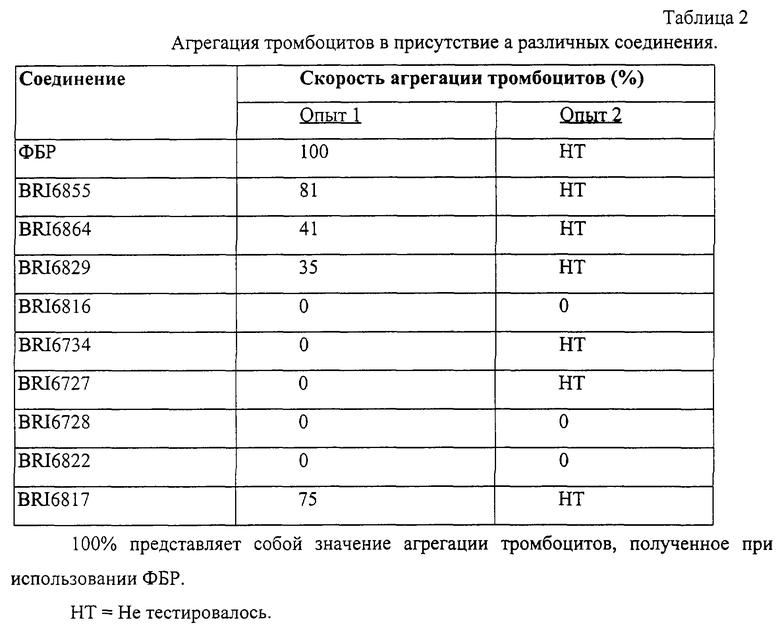
Специалистам в данной области понятно, что предпочтительные воплощения настоящего изобретения допускают многочисленные изменения и модификации и что подобные изменения могут быть произведены не выходя за пределы настоящего изобретения. Таким образом, предполагается, что прилагаемая формула изобретения охватывает все такие эквивалентные изменения, которые отвечают духу и пределам настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ СОЕДИНЕНИЯ | 2001 |
|
RU2419608C2 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2001 |
|
RU2298550C2 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ В КАЧЕСТВЕ МИМЕТИКОВ ТРО | 2006 |
|
RU2385865C2 |
| СОЕДИНЕНИЯ 4-ОКСО-3,4-ДИГИДРОХИНАЗОЛИНОНА ДЛЯ ЛЕЧЕНИЯ BRAF-АССОЦИИРОВАННЫХ ЗАБОЛЕВАНИЙ И НАРУШЕНИЙ | 2021 |
|
RU2814662C1 |
| АЛКИЛСУЛЬФОНАМИДХИНОЛИНЫ С АФФИННОСТЬЮ К РЕЦЕПТОРАМ NK-3 | 2006 |
|
RU2421447C2 |
| ПРОЛЕКАРСТВА ИНГИБИТОРОВ ТРОМБИНА | 1996 |
|
RU2176644C2 |
| МОДУЛЯТОРЫ ИНТЕГРИРОВАННОГО СИГНАЛЬНОГО ПУТИ СТРЕССА | 2017 |
|
RU2769327C2 |
| ПРОИЗВОДНЫЕ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ N-АДАМАНТИЛМЕТИЛА В КАЧЕСТВЕ ФАРМАЦЕВТИЧЕСКИХ КОМПОЗИЦИЙ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ | 2002 |
|
RU2300525C2 |
| СОЕДИНЕНИЯ АЗАЛАКТАМА В КАЧЕСТВЕ ИНГИБИТОРОВ HPK1 | 2021 |
|
RU2819642C1 |
| ПРОИЗВОДНЫЕ АЗОТСОДЕРЖАЩИХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБЫ ЛЕЧЕНИЯ ВОСПАЛИТЕЛЬНЫХ ЗАБОЛЕВАНИЙ И ЗАБОЛЕВАНИЙ ДЫХАТЕЛЬНЫХ ПУТЕЙ | 2001 |
|
RU2265011C2 |
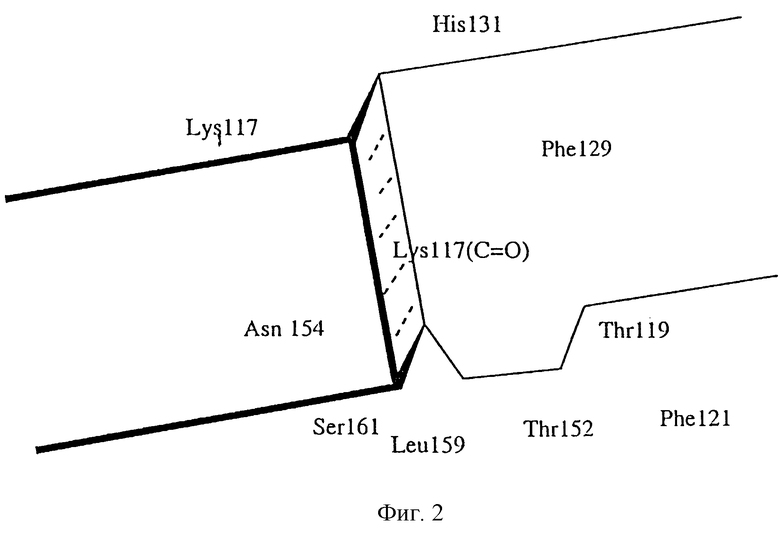
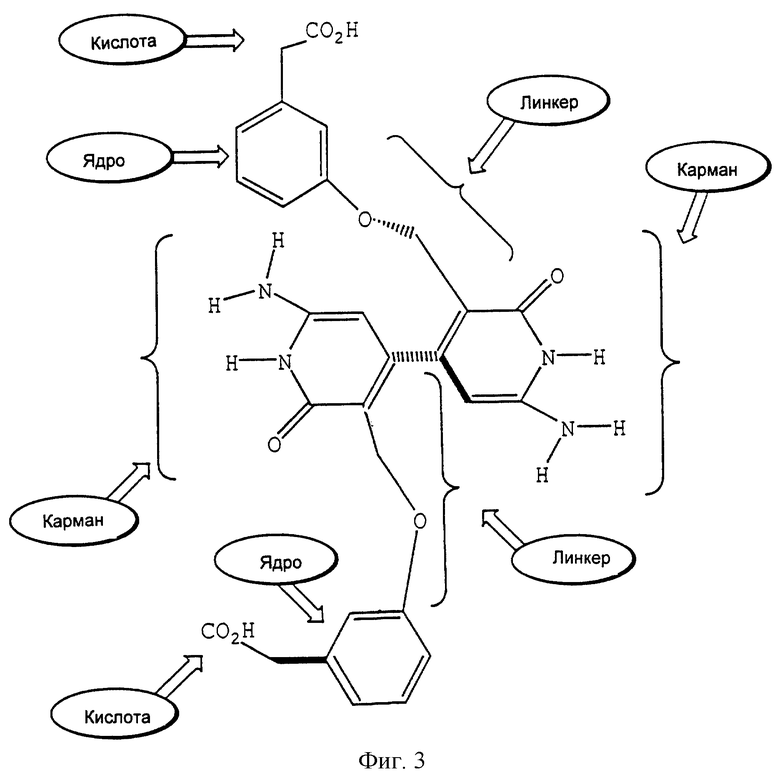
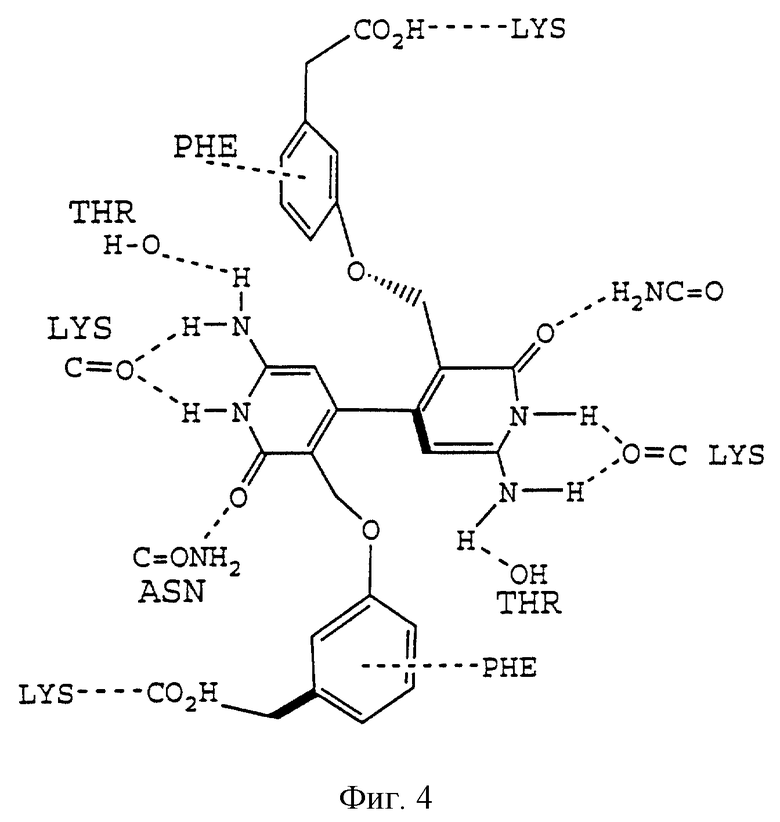
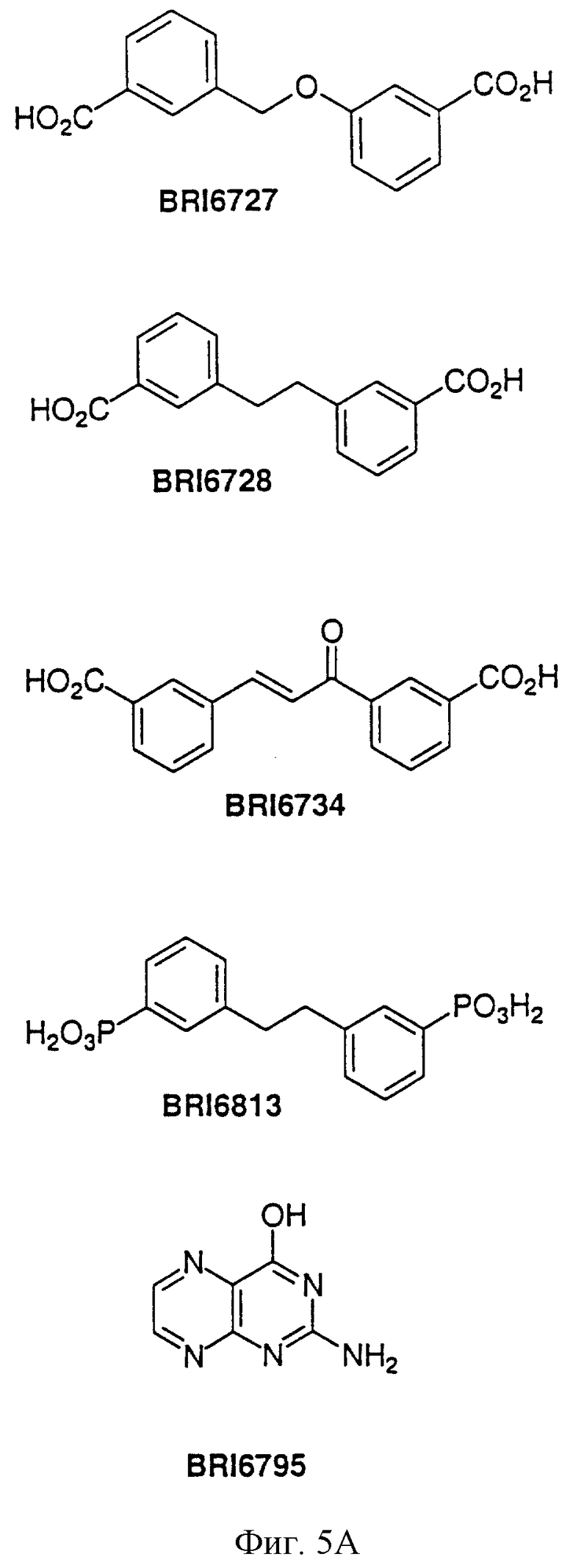
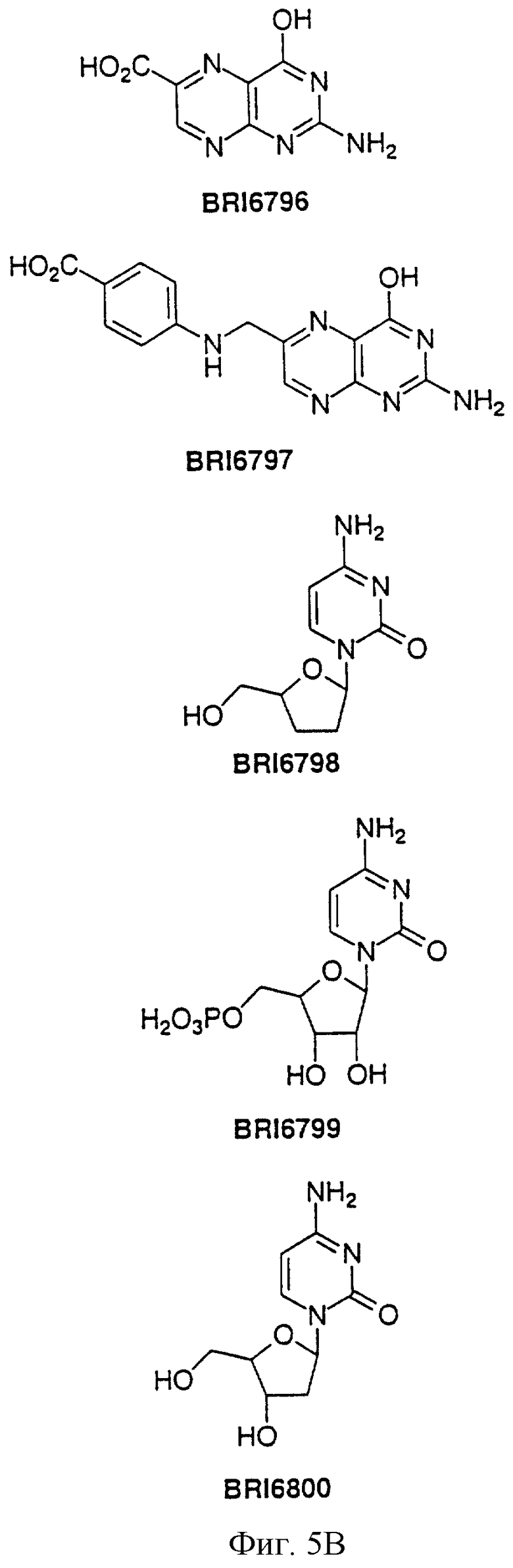
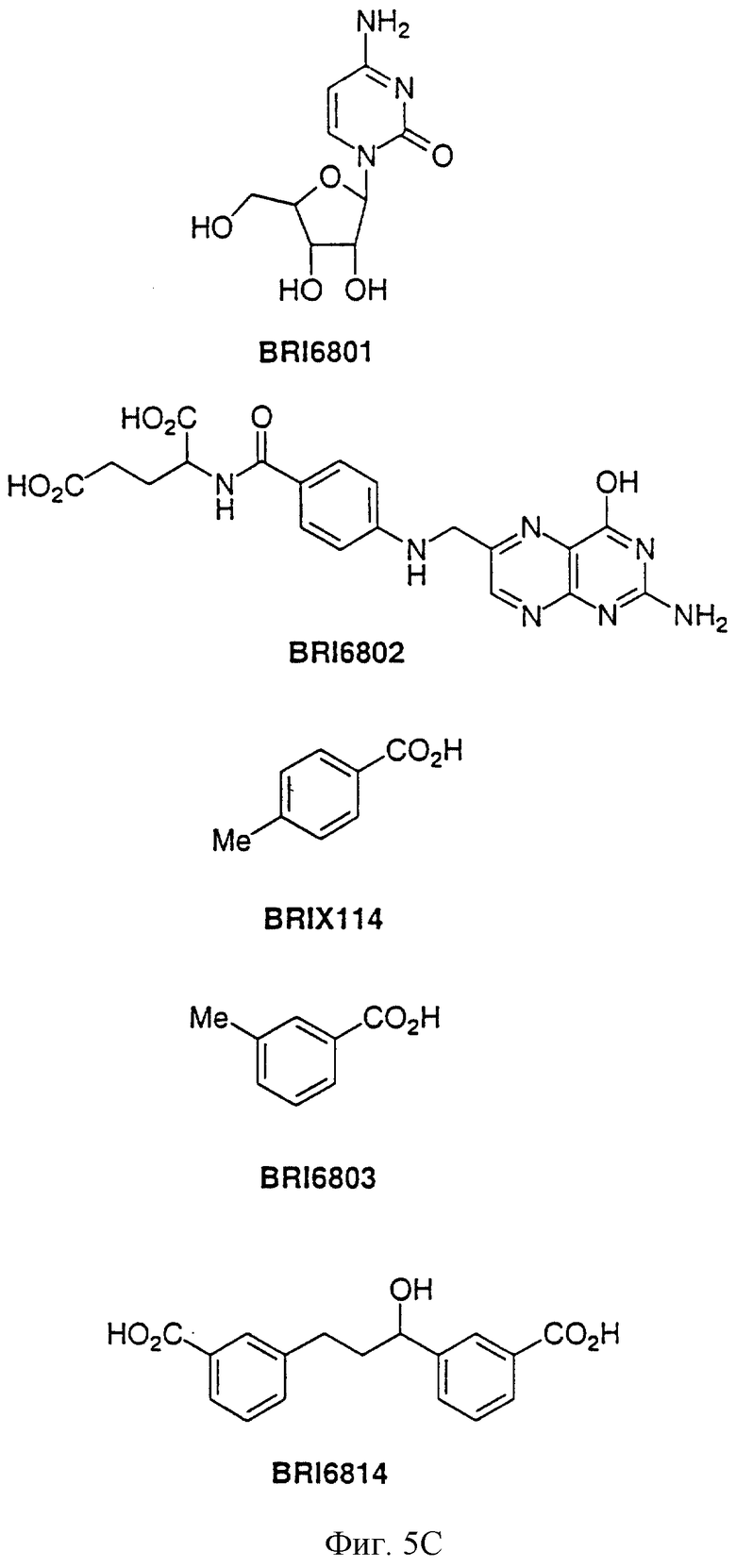
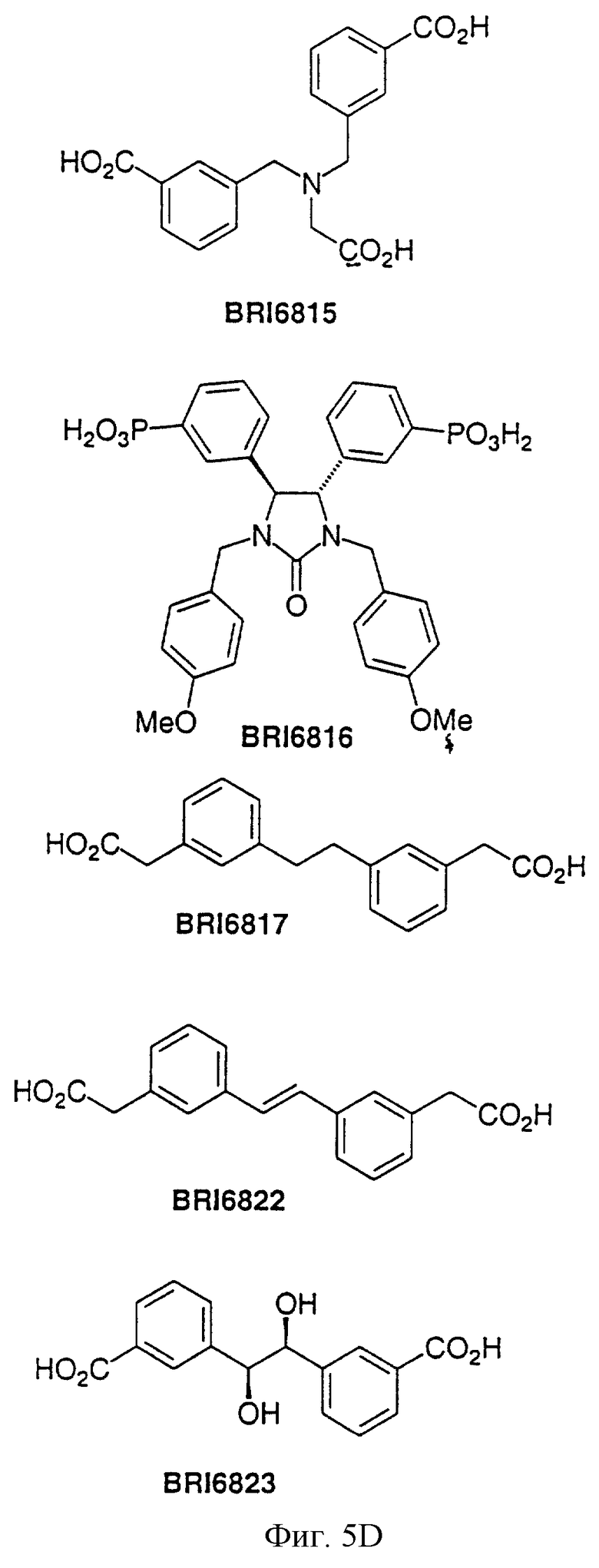
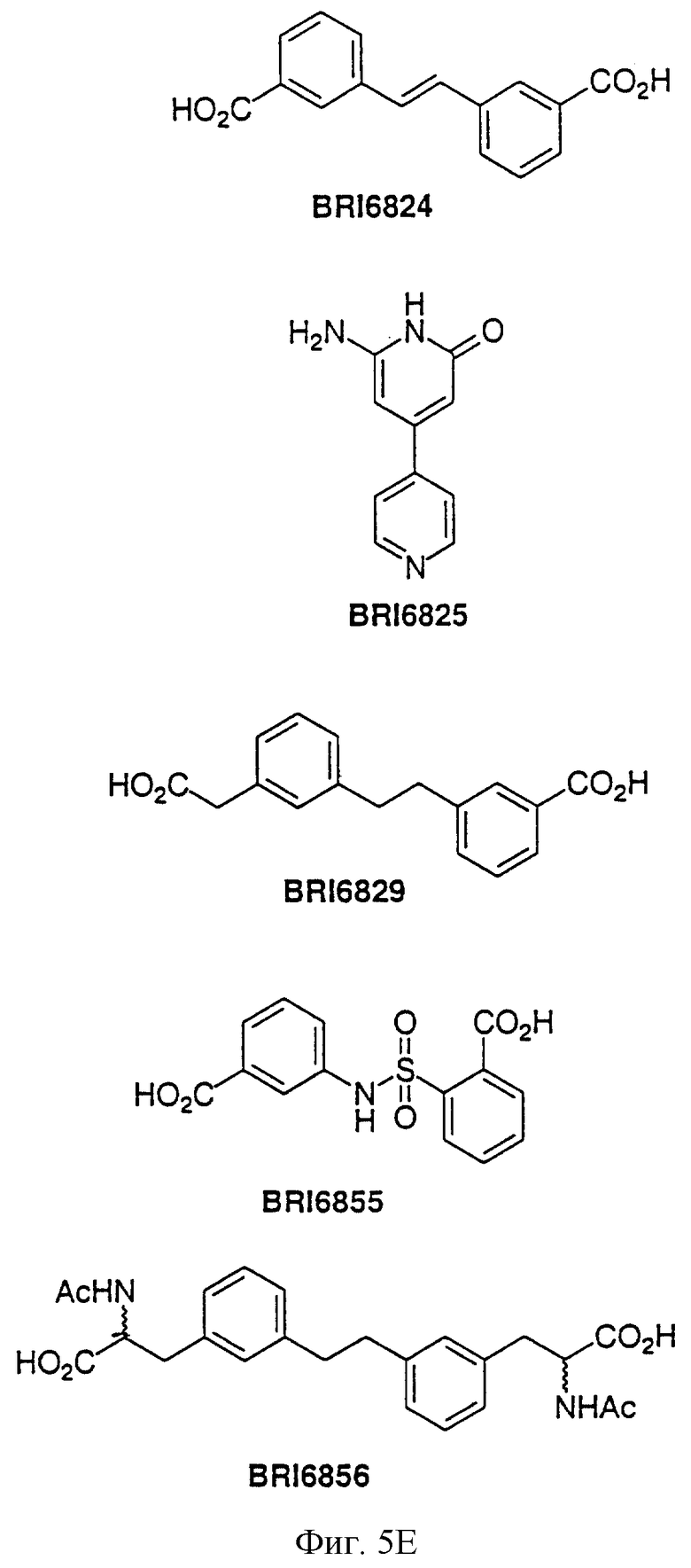
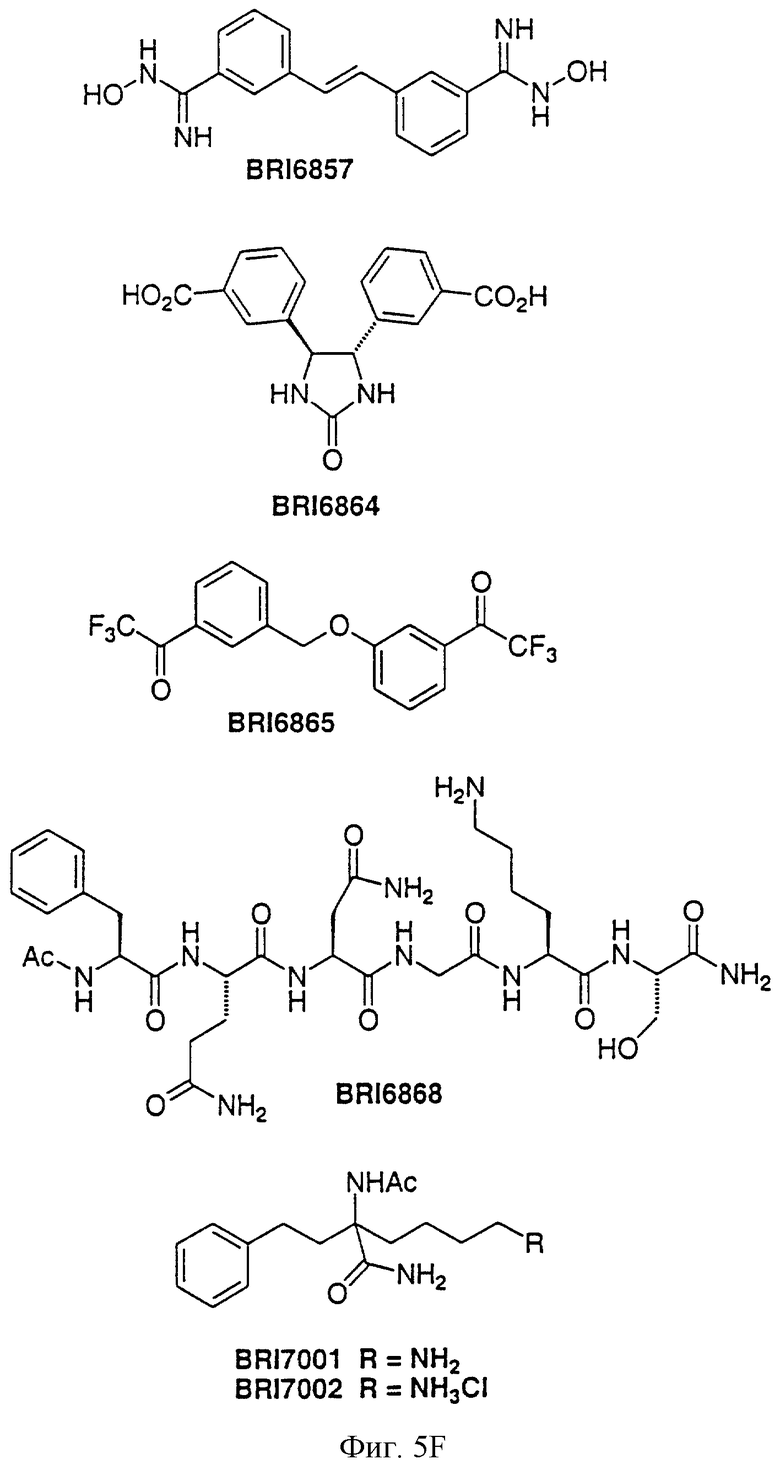
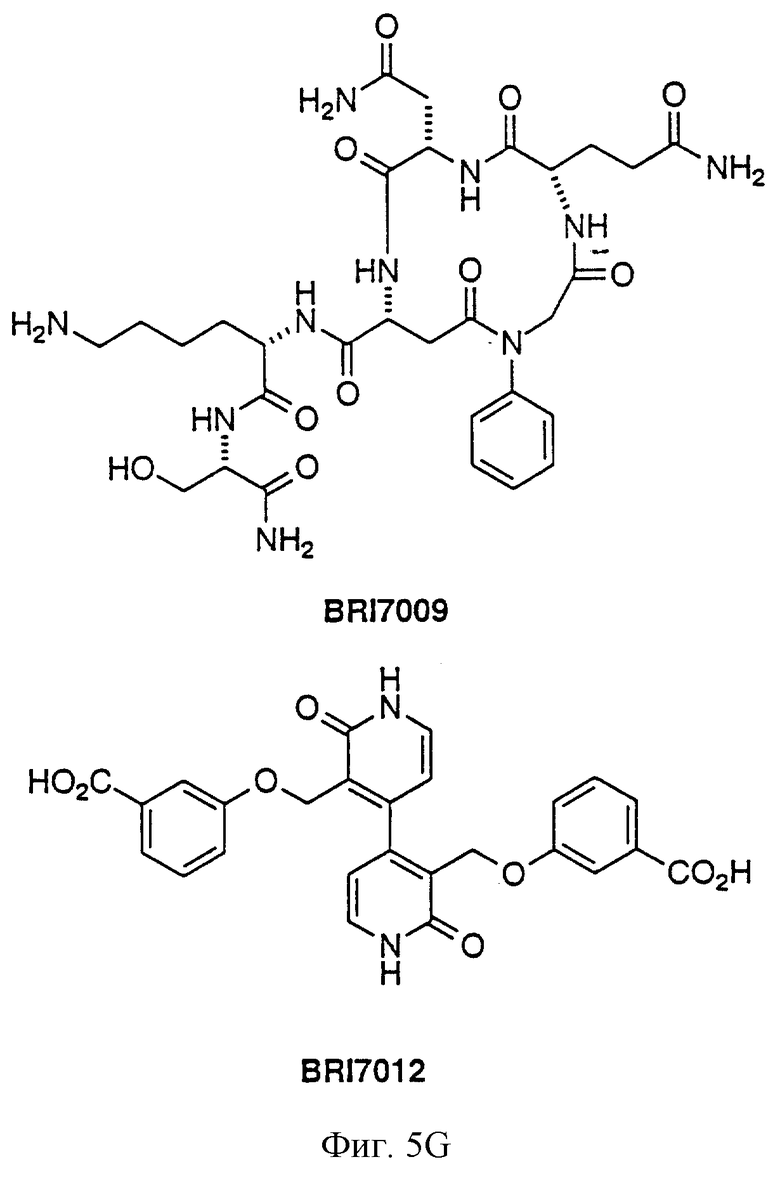
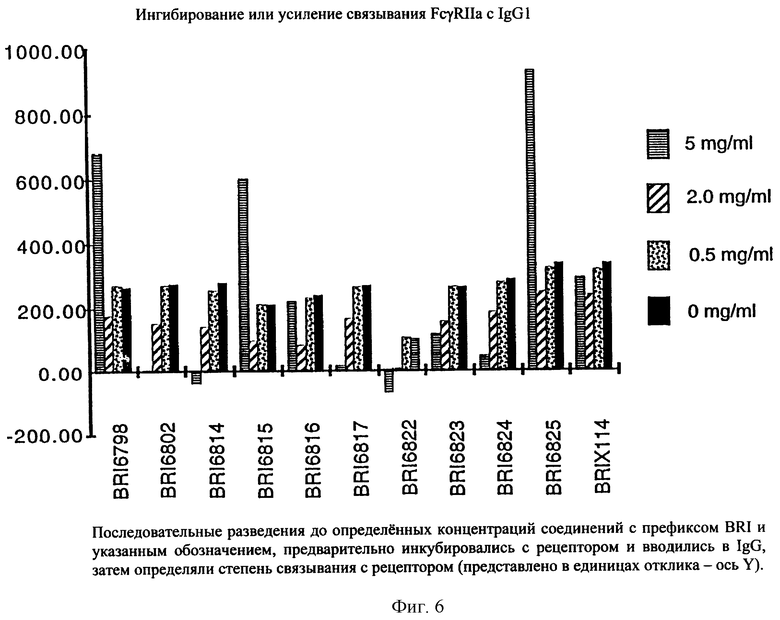
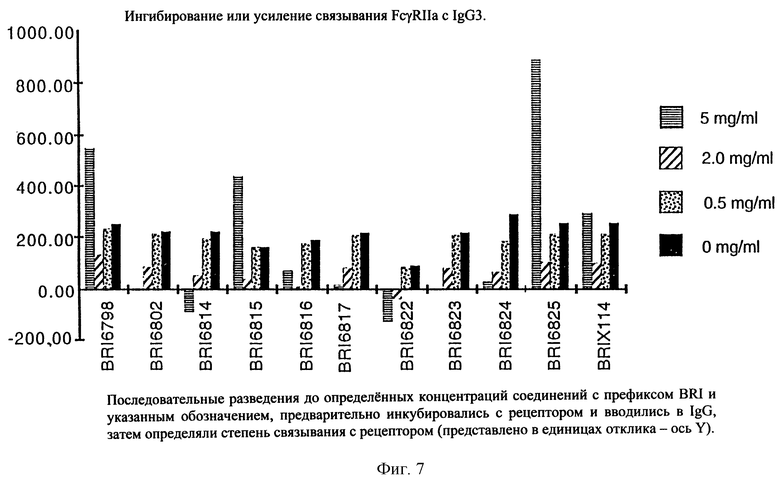
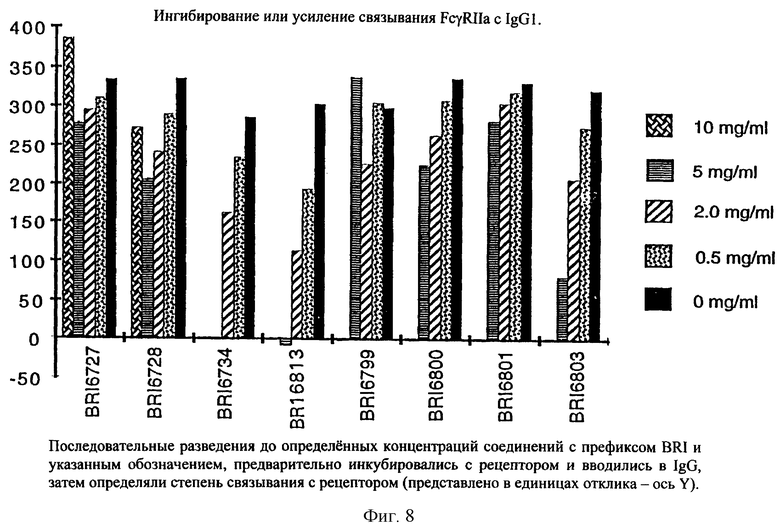
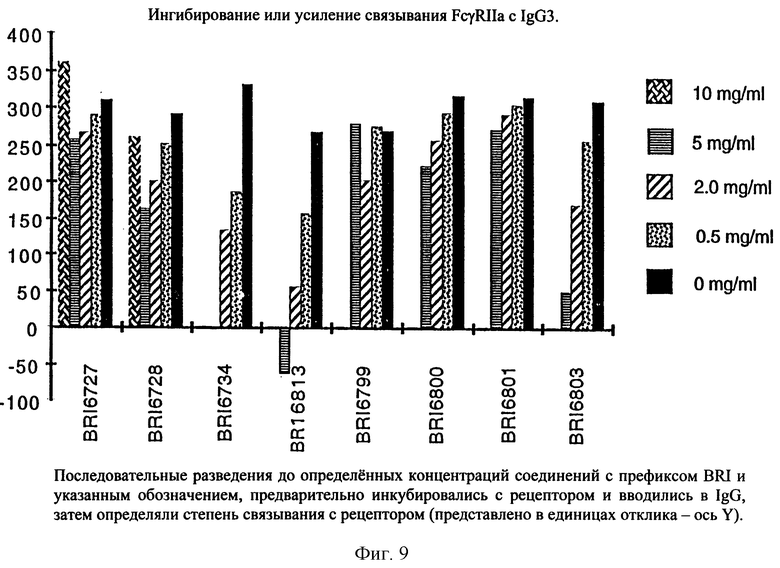
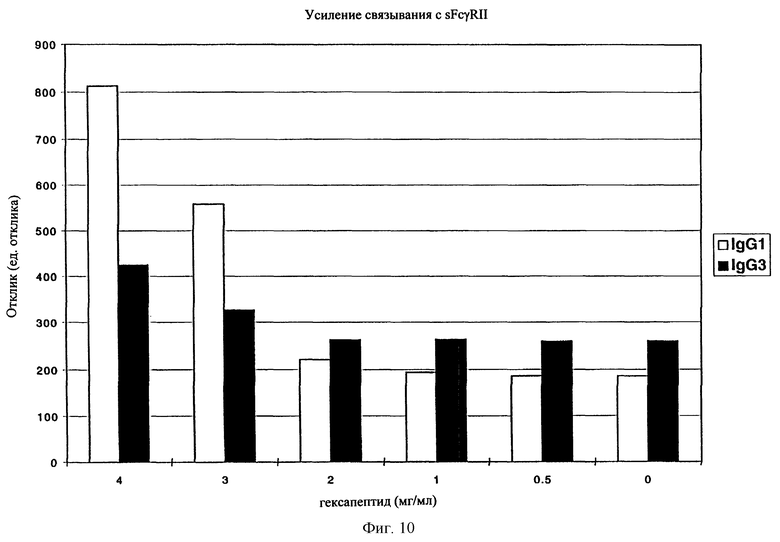
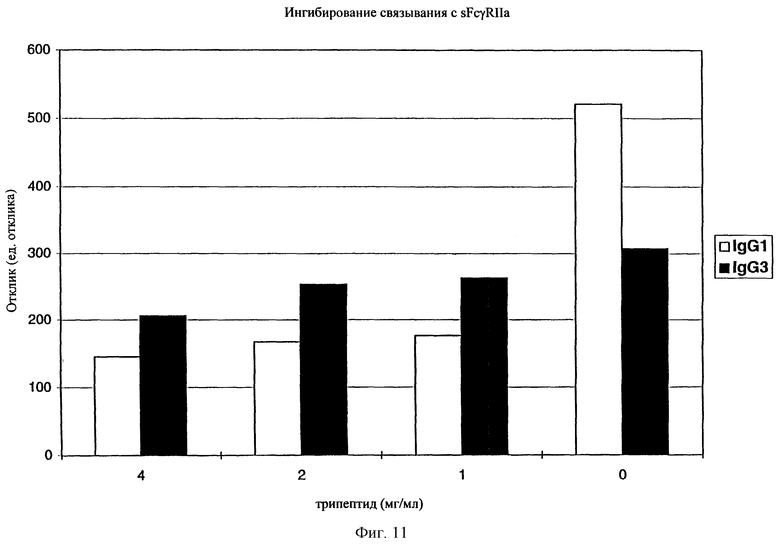
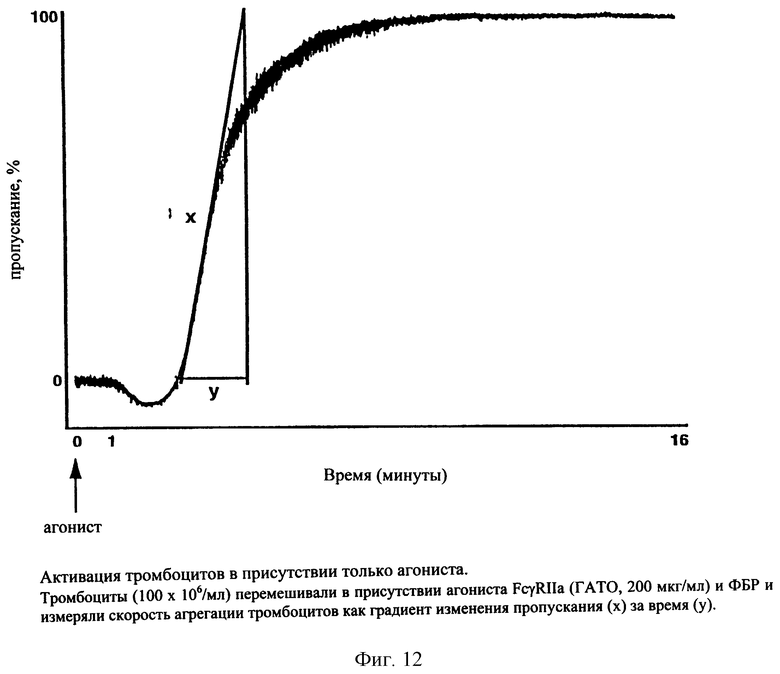
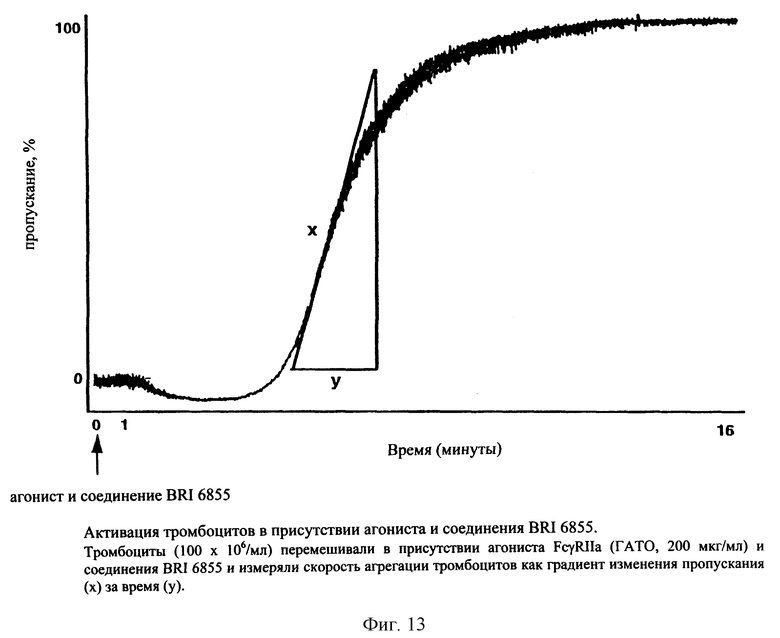
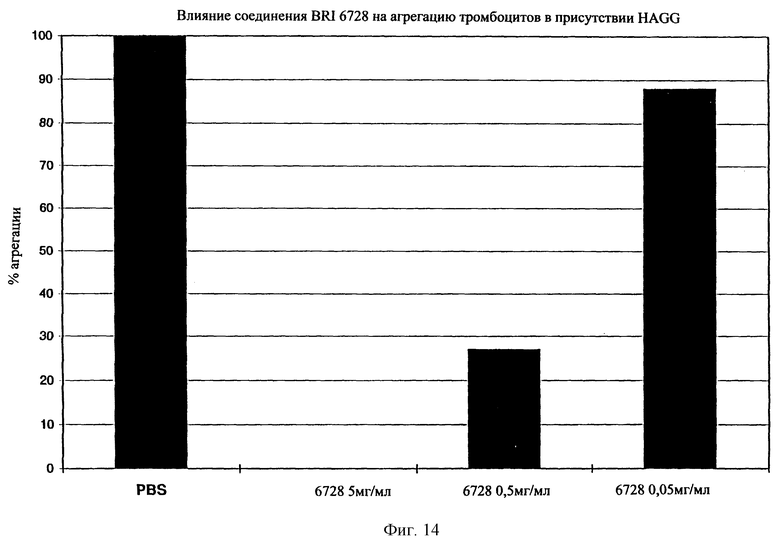
Настоящее изобретение относится к области медицины и касается фармацевтической композиции, включающей в себя соединение, модулирующее Fc рецептор, и фармацевтически приемлемый носитель. Настоящее изобретение также относится к способу лечения различных заболеваний при помощи соединения, модулирующего Fc рецептор. 2 н. и 96 з.п. ф-лы, 20 ил., 2 табл.
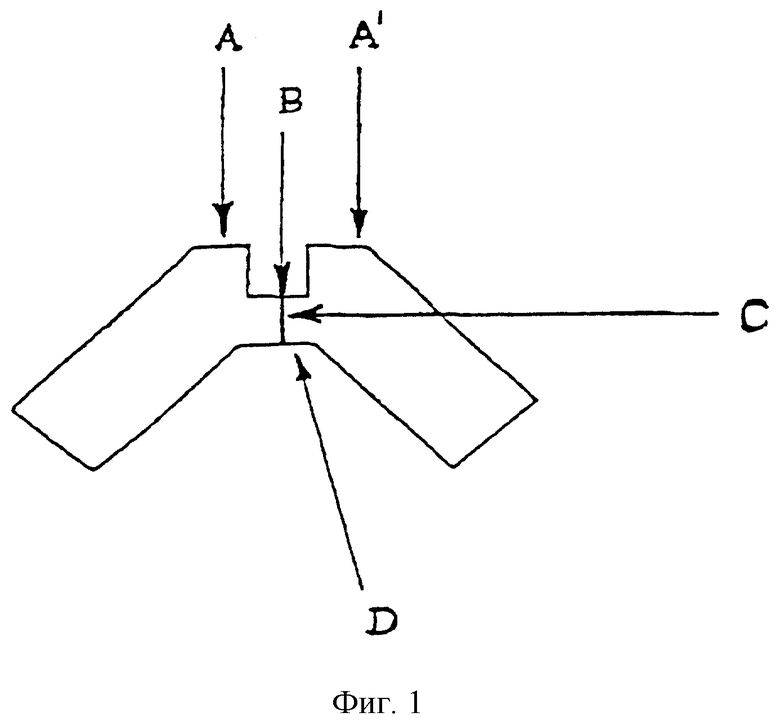
(а) соединение, модулирующее активность Fc-рецептора, выбранное из группы, включающей в себя ароматическое соединение формулы
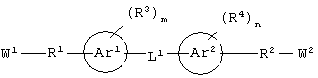
гетероароматическое соединение формулы
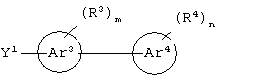
циклическое соединение формулы
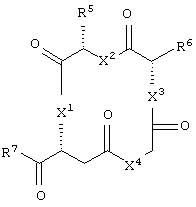
бициклическое соединение формулы
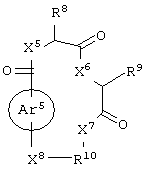
и аминокислотное производное формулы
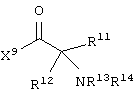
или их соли,
где каждый из W1 и W2 независимо представляет собой CO2R15, C(=NH)NH(OH), SO3R15, C(=NH)NH2, OPO(OR15)2, С(=O)СF3 или PO(OR15)2;
каждый из Аr1, Аr2, Аr4 и Аr5 независимо представляет собой С6-С20 арил или C1-С20 гетероарил;
Аr3 представляет собой C1-C20 гетероарил;
каждый из X1, X2, X3, X4, X5, X6, X7 и X8 независимо представляет собой метилен, О, S или NR16;
каждый из R1 и R2 независимо представляет собой связь, C1-С6 алкилен или галогенированный C1-С6 алкилен;
каждый из R3 и R4 независимо представляют собой галоген, -Z1 или C1-С6 алкил;
каждый из X9, Y1 и Z1 независимо представляет собой OR17, SR17 или NR17R18;
каждый из R5 и R6 независимо представляет собой аминокислотный остаток боковой цепи или группу формулы -R19-W3;
каждый из R8, R9 и R11 независимо представляет собой аминокислотный остаток боковой цепи при условии, что R11 не является Н или СН3;
R7 представляет собой OR20, NR21R22 или примерно от 1 до 10 аминокислот;
R10 представляет собой C1-С6 алкилен;
R12 представляет собой C1-С6 алкил или C6-С20 аралкил;
W3 представляет собой С(=O)Х10;
X10 представляет собой OR23 или NR24R25
каждый из R13, R15, R17, R18, R20, R21, R23 и R24 независимо представляет собой водород или C1-С6алкил;
R16 представляет собой Н, C6-С20 арил или защитную группу амида;
R19 представляет собой C1-С6 алкилен;
каждый из R22 и R25 независимо представляет собой Н, C1-С6 алкил или защитную группу амида;
R14 представляет собой Н, C1-С6 алкил или защитную группу амина;
L представляет собой связующую группу, включающую в себя от 1 до 20 атомов; и
каждый из m и n независимо представляет собой целое число от 0 до 2,
и (b) фармацевтически приемлемый носитель.
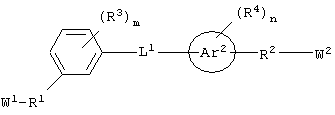
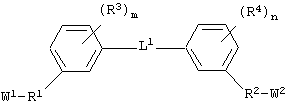
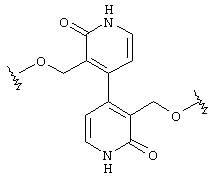
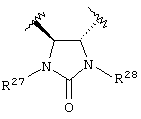
где каждый из R27 и R28 независимо представляют собой Н, C1-С6 алкил, C6-С10 аралкил или защитную группу.
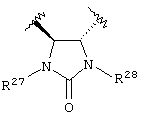
где каждый из R27 и R28 независимо представляет собой Н, C1-С6 алкил, C6-С10 аралкил или защитную группу.
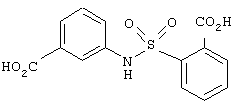
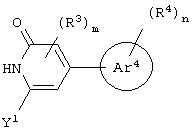
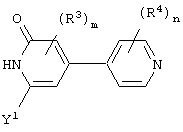
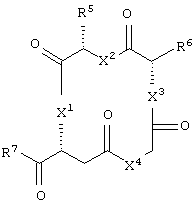
где X1, X2, X3, X4 представляют собой NR16.
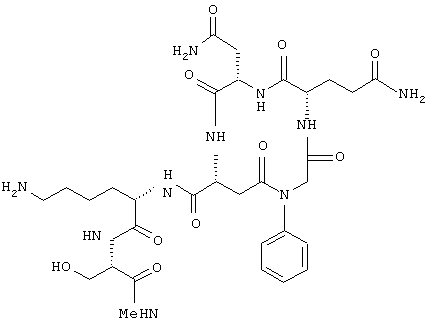
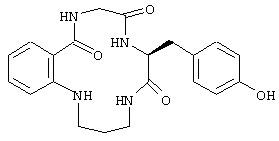
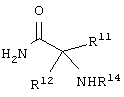
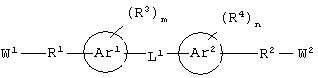
гетероароматическое соединение формулы
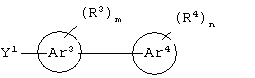
циклическое соединение формулы
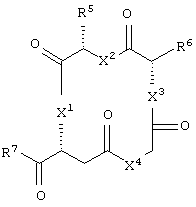
бициклическое соединение формулы
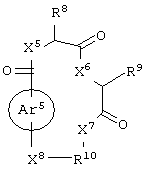
и аминокислотное производное формулы
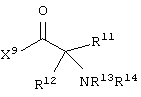
или их соли,
где каждый из W1 и W2 независимо представляет собой CO2R15, C(=NH)NH(OH), SO3R15, C(=NH)NH2, OPO(OR15)2, С(=O)СF3 или PO(OR15)2;
каждый из Ar1, Ar2, Ar4 и Ar5 независимо представляет собой C6-С20 арил или C1-С20 гетероарил;
Ar3 представляет собой C1-С20 гетероарил;
каждый из X1, X2, X3, X4, X5, X6, X7 и X8 независимо представляет собой метилен, О, S или NR16;
каждый из R1 и R2 независимо представляет собой связь, C1-С6 алкилен или галогенированный C1-С6 алкилен;
каждый из R3 и R4 независимо представляют собой галоген, -Z1 или C1-С6 алкил;
каждый из X9, Y1 и Z1 независимо представляет собой OR17, SR17 или NR17R18;
каждый из R5 и R6 независимо представляет собой аминокислотный остаток боковой цепи или группу формулы -R19-W3;
каждый из R8, R9 и R11 независимо представляет собой аминокислотный остаток боковой цепи при условии, что R11 не представляет собой Н или СН3;
R7 представляет собой OR20, NR21R22 или примерно от 1 до 10 аминокислот;
R10 представляет собой C1-С6 алкилен;
R12 представляет собой C1-С6 алкил или C6-С20 аралкил;
W3 представляет собой С(=O)Х10;
X10 представляет собой OR23 или NR24R25;
каждый из R13, R15, R17, R18, R20, R21, R23 и R24 независимо представляет собой водород или C1-С6 алкил;
R16 представляет собой Н, C6-С20 арил или защитную группу амида;
R19 представляет собой C1-С6 алкилен;
каждый из R22 и R25 независимо представляет собой Н, C1-С6 алкил или защитную группу амида;
R14 представляет собой Н, C1-С6 алкил или защитную группу амина;
L представляет собой связующую группу, включающую в себя от 1 до 20 атомов;
каждый m и n независимо представляет собой целое число от 0 до 2.
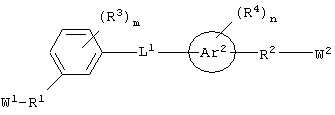
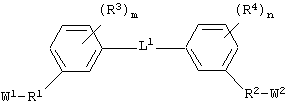
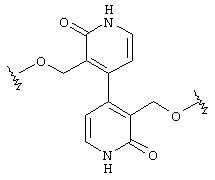
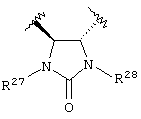
где каждый из R27 и R28 независимо представляют собой Н, C1-С6 алкил, C6-С10 аралкил или защитную группу.
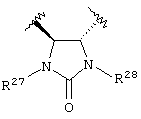
где каждый из R27 и R28 независимо представляет собой Н, C1-С6 алкил, C6-С10 аралкил или защитную группу.
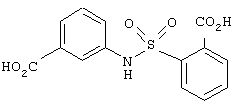
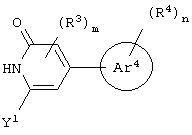
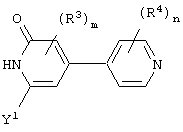
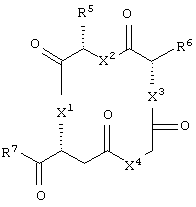
где X1, X2, X3, X4 представляют собой NR16.
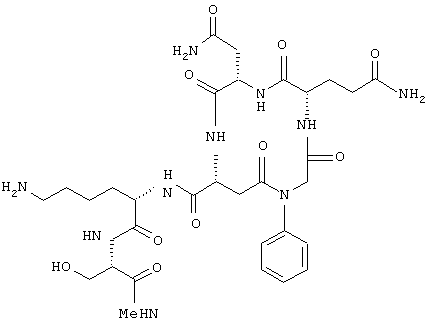
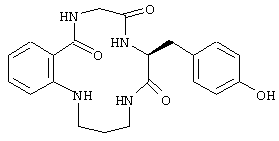
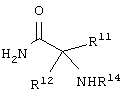
Приоритет по пунктам:
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ ИММУННЫХ ЗАБОЛЕВАНИЙ | 2000 |
|
RU2203682C2 |
| КОНДЕНСИРОВАННЫЕ 5-ЧЛЕННЫЕ ГЕТЕРОЦИКЛЫ ИЛИ ИХ СОЛИ, ПРОЯВЛЯЮЩИЕ АКТИВНОСТЬ ПО ТОРМОЖЕНИЮ АГРЕГАЦИИ | 1992 |
|
RU2041211C1 |
| Пюпитр для работы на пишущих машинах | 1922 |
|
SU86A1 |
| US 4686282 А, 11.08.1987. | |||

