Изобретение относится к электроаналитической химии и может быть использовано при определении цистеина в объектах окружающей среды.
Известны методы определения цистеина по каталитическим токам восстановления ионов различных металлов. При косвенном определении цистеина по полярографической каталитической предволне кобальта (II) (Сирко В.Н., Выскубова Н.К. Определение цистеина по полярографическому каталитическому току комплексов кобальта (II) // Журн. аналит. химии, 1977, т.32, вып. 9, с.1861-1864) в качестве рабочего электрода использовали ртутный капающий электрод (р.к.э.). Фоновым электролитом служил раствор состава: 0,19 М Н3ВО3+1,5·10-3 М Na2B4O7+0,55·10-2 М NaCI, в который вводили 1·10-2 М СоСl2. В диапазоне концентраций цистеина 2·10-6-3·10-5 М градуировочная кривая близка к линейной.
Известен способ определения этой аминокислоты, основанный на получении каталитической волны электровосстановления ионов индия (III) (Логвинов И.Н., Турьян Я.И., Кудинов П.И. Полярографическое определение цистеина на основе каталитического электровосстановления индия (III) // Изв. вузов. Пищевая технология, 1978, № 6, с.122-124 ). При этом индикаторным электродом служил р.к.э., а в качестве фонового электролита использовали раствор 0,1 М NaClO4 + 0,1 М НСlO4, в который вводили 2·10-3 М In(СlO4)3. Линейный диапазон определяемых концентраций цистеина составил 5·10-5 - 5·10-3 М.
Анодные и катодные инверсионные пики цистеина исследовали на электроде типа "висячая" ртутная капля (ВРК) в ацетатном буферном растворе с рН 4,6, содержащем 1·10-6 М Сu (II) (Forsman U. The stripping voltammetric pattern of cysteine and penicillamine at the mercury electrode in the presence of Си (II) // J. Electroanal. Chem., 1983, vol. 152, pp. 241-254). Показано, что определение аминокислоты возможно в интервале концентраций 1·10-5-1·10-3 М.
Определяли цистеин по каталитическому катодному инверсионному току восстановления Ni (II) с использованием электрода типа ВРК (Banica F.G., Moreira J.C., Fogg AJ. Application of catalytic stripping voltammetry for the determination of organic sulfur compounds at a hanging mercury drop electrode: behaviour of cysteine, cystine and N-acetyl-cysteine in the presence of nickel ion // Analyst, 1994, vol. 119, №2, рр.309-318). В качестве фоновых применяли электролиты состава: 0,025 М CH3COONa+0,025 М Na2HPO4+10% H2SO4 (с добавлением 5·10-4 М Ni (II)) и 0,05 М N- морфолинопропансульфоновая кислота + 10% NaOH (с добавлением 2,5·10-4 М Ni (II)). Калибровочная кривая линейна в области концентраций цистеина 5·10-9 - 1·10-7 М.
Перечисленные методы определения цистеина являются косвенными и имеют ряд общих недостатков: узкий диапазон определяемых концентраций цистеина; необходимость введения дополнительных реагентов, что сказывается на чистоте анализа и его метрологических характеристиках; высокие требования по технике безопасности, т.к. определение проводят на ртутно-капающем или "висячем" ртутном электроде; трудоемкость.
Наиболее близким аналогом к заявляемому изобретению является способ прямого полярографического определения цистеина на электроде типа ВРК с использованием в качестве фонового электролита ацетатного буферного раствора (рН 4,76) (Stock J.T., Larson R.E. The cathodic stripping voltammetry of certain thiols in acetate medium // Analytica Chimica Acta., 1982, vol. 138, рр.371-374). Линейный диапазон определяемых концентраций составляет 4·10-7-1,2·10-6 М.
По сравнению с косвенными методами в прототипе чистота анализа и его метрологические характеристики выше за счет исключения ввода в аналит дополнительных реагентов при сохранении остальных недостатков.
Технической задачей заявляемого изобретения является разработка способа определения цистеина, имеющего широкий линейный диапазон определяемых концентраций цистеина, экологически более безопасного и менее трудоемкого.
Для решения технической задачи предлагается предварительное формирование ртутного пленочного электрода (РПЭ) на стеклоуглеродной подложке, концентрирование электроактивного соединения на поверхности электрода в фоновом электролите состава: (0,1-0,4) М NaClO4+(0,1-1) М НСlO4, содержащем цистеин, с последующим получением аналитического сигнала при изменении потенциала.
По сравнению с прототипом в заявляемом способе концентрирование электроактивного соединения проводят на РПЭ со стеклоуглеродной подложкой, что экологически более безопасно. Использование этого электрода и фонового электролита (0,1-0,4) М NaClO4+(0,1-1) М НClO4 позволили расширить линейный диапазон определяемых концентраций цистеина.
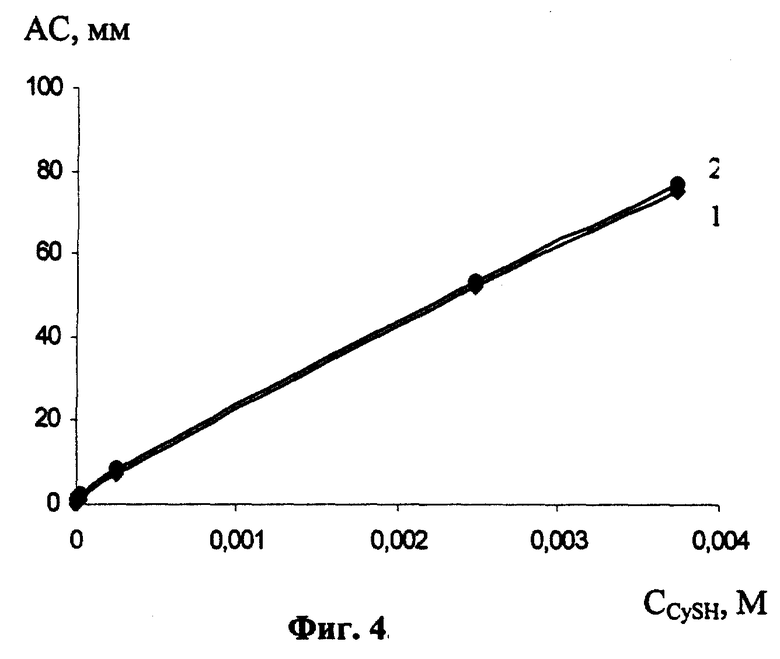
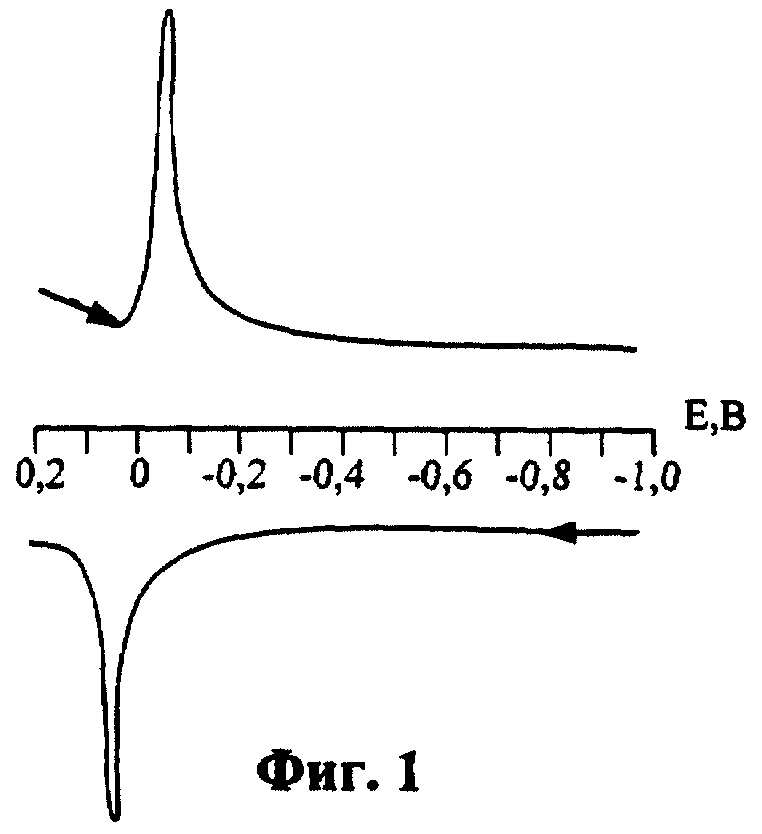
На фиг.1 изображен общий вид циклической вольтамперограммы (фон 0,1 М NaClO4+0,1 М НClO4; концентрация цистеина 2,5·10-3 М; время формирования РПЭ 180 с); на фиг.2 - зависимость аналитических сигналов цистеина (АС) от концентрации перхлората натрия ( 1 - анодный сигнал; 2 - катодный сигнал; n М NaClO4+0,1 М NaClO4; концентрация цистеина 2,5·10-3 М; время соприкосновения РПЭ с раствором цистеина 60 с; время формирования РПЭ 180 с ); фиг.3 отражает зависимость АС от концентрации хлорной кислоты (1 - анодный сигнал; 2 - катодный сигнал; суммарная концентрация перхлорат-ионов 1 М; концентрация цистеина 2,5·10-3 М; время соприкосновения РПЭ с раствором цистеина 60 с; время формирования РПЭ 180 с); фиг.4 - зависимость АС от концентрации цистеина в растворе (1 - анодный сигнал; 2 - катодный сигнал; 0,1 М NaClO4+0,1 М NaClO4; время соприкосновения РПЭ с раствором цистеина 120 с; время формирования РПЭ 180 с).
Для определения оптимального времени формирования РПЭ (τформир. РПЭ) стеклоуглеродный электрод (СУЭ) помещали в 3-электродную ячейку (вспомогательный электрод - стеклоуглерод (СУ); электрод сравнения - хлорсеребряный (нас. К.С1)) с раствором: 0,1 М КNO3+1·10-4 М Hg(NO3)2 и формировали РПЭ при потенциале электролиза -0,5 В в течение разного времени концентрирования ртути.
Полученный РПЭ промывали бидистиллированной водой, помещали его в ячейку, содержащую 0,1 М NaClO4+0,1 М НсlO4+2,5·10-3 М цистеина (CySH), проводили концентрирование электроактивного соединения ртути с цистеином при разомкнутой цепи и регистрировали катодные и анодные вольтамперограммы, получая АС с потенциалами максимумов пиков Еп=0,063 В на катодной и Еп=+0,038 В на анодной ветви, из чего следует, что регистрацию вольтамперограмм необходимо проводить в диапазоне потенциалов (от +0,2 до -0,2) В (табл.1). Из табл.1 видно, что оптимальным временем формирования РПЭ является 180 с. При меньшем τформир. РПЭ агрегаты из атомов ртути не полностью заполняют поверхность СУЭ, поэтому АС, получаемые на таком электроде, являются непригодными для определения цистеина. При большем времени накопления ртути зависимость АС цистеина от τформир. РПЭ выходит на плато, что объясняется полным заполнением поверхности СУЭ ртутью, поэтому τформир. рпэ>180 с является нецелесообразным.
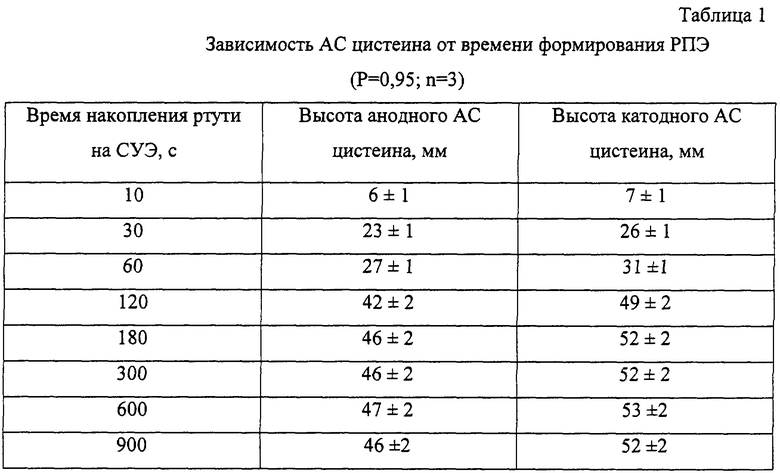
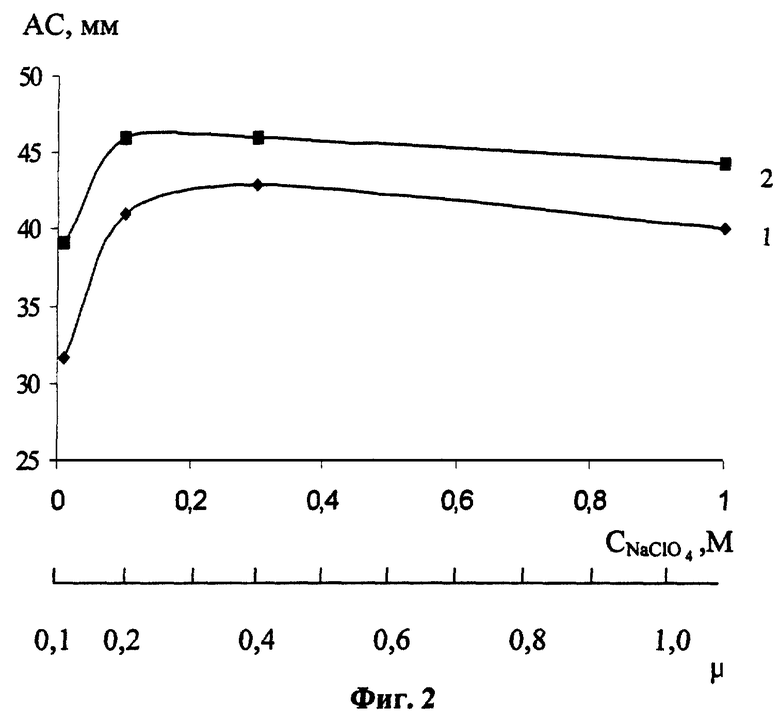
Для установления оптимальных концентраций веществ, составляющих фоновый электролит, который известен и широко применяется в аналитической практике, сформированный РПЭ помещали в ячейку, содержащую Х М NaClO4+0,1 М НClO4+2,5·10-3 М CySH, выдерживали в течение 60 с при разомкнутой цепи и регистрировали катодные и анодные вольтамперограммы в диапазоне потенциалов (от +0,2 до -0,2) В (фиг.2). Из фиг.2 видно, что АС возрастают с повышением концентрации NaClO4 за счет изменения строения двойного электрического слоя, достигая максимумов при (0,1-0,4) М NaClO4, а затем несколько уменьшаются за счет некоторого повышения вязкости среды. Таким образом, оптимальная концентрация перхлората натрия в фоновом электролите составляет (0,1-0,4) М.
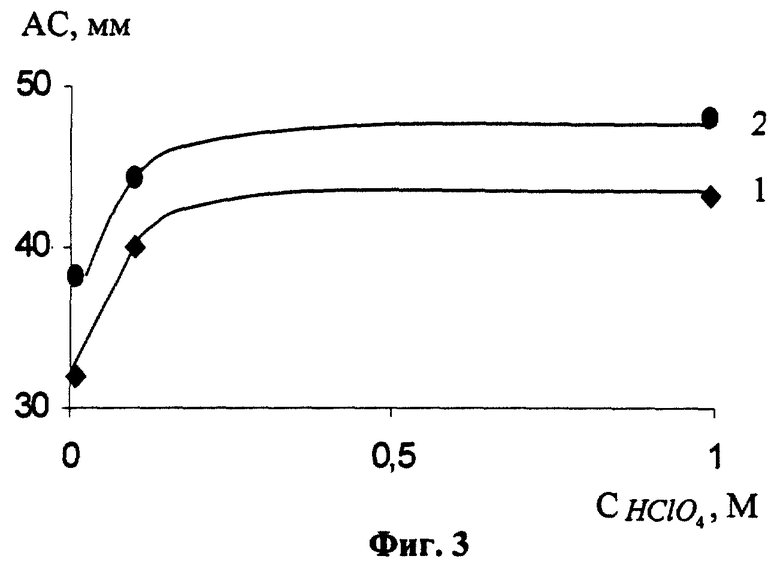
Для определения оптимальной концентрации НClO4, при условии сохранения постоянства ионной силы фонового электролита сформированный РПЭ помещали в ячейку, содержащую (1-Х) М NaClO4+Х М НСlO4+2,5·10-3 М CySH, выдерживали его в течение 60 с при разомкнутой цепи и регистрировали катодные и анодные АС (фиг.3). Из фиг.3 видно, что при С НСlO4<0,1 М АС уменьшаются, по-видимому, за счет инактивации поверхности РПЭ в связи с образованием оксидов ртути. Наиболее пригодной для аналитических целей можно считать концентрацию НСlO4, равную (0,1-1) М.
Таким образом, оптимальным можно считать состав фонового электролита (в М): перхлорат натрия 0,1-0,4; хлорная кислота 0,1-1.
В дальнейшем все зависимости получали, используя фоновый электролит состава: 0,1 М NaClO4+0,1 М НСlO4.
Поскольку ртуть химически взаимодействует с цистеином (Майрановский С.Г., Страдынь Я.П., Безуглый В.Д. Полярография в органической химии. Л., 1975, 352 с.), то необходимо изучить зависимость АС от времени соприкосновения РПЭ с раствором цистеина (τ сопр.). Для этого сформированный РПЭ помещали в ячейку, содержащую раствор состава 0,1 М NaClO4+0,1 М НСlO4+2,5·10-3 М CySH, и регистрировали катодные и анодные АС после выдерживания РПЭ в растворе цистеина при разомкнутой цепи в течение различного времени (табл. 2).
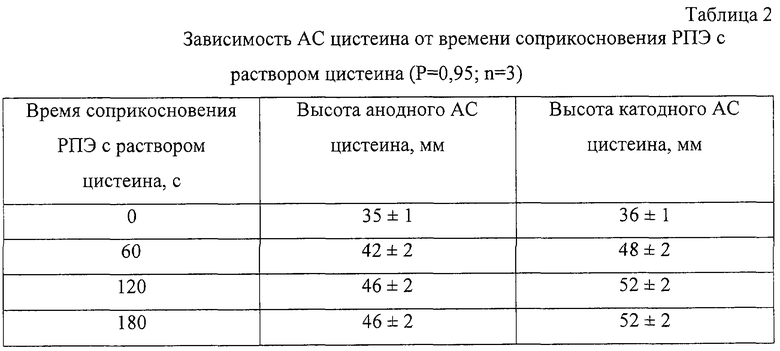
Установлено, что оптимальным τсопр является 120 с (табл.2). При этом достигается полное насыщение поверхности РПЭ электрохимически активным цистеинатом ртути, поэтому дальнейшее увеличение времени соприкосновения рабочего электрода с раствором цистеина нецелесообразно.
Для изучения влияния деаэрации на электрохимическое поведение цистеина сформированный РПЭ помещали в ячейку с раствором: 0,1 М NaClO4+0,1 М НСlO4+2,5·10-3 М CySH, через который в течение 15 мин пропускали аргон, выдерживали РПЭ в этом растворе 120 с при разомкнутой цепи и регистрировали катодные и анодные АС. Зарегистрированные АС сравнивали с АС, полученными в отсутствии деаэрации (табл.3).

Из табл.3 видно, что удаление кислорода инертным газом не влияет на электрохимическое поведение цистеина.
Для исследования линейного диапазона концентраций цистеина, в котором возможно его определение, изучали зависимость АС от концентрации цистеина (фиг.4). Для этого сформированный РПЭ помещали в ячейку, содержащую 0,1 М NaClO4+0,1 М НСlO4+Х М CySH, выдерживали в течение 120 с при разомкнутой цепи и регистрировали катодные и анодные АС (см. фиг.4). Из фиг.4 видно, что определение цистеина возможно в диапазоне 1·10-6 - 3,5·10-3 М.
Способ апробирован на модельных растворах с различными концентрациями цистеина (метод "введено - найдено") и оценена точность его определения (табл.4 ).
В результате экспериментальных исследований было выявлено, что оптимальными условиями определения являются:
- фоновый электролит состава: (0,1-0,4) М NaClO4+(0,1-1) М НСlO4;
- время формирования РПЭ на стеклоуглеродной подложке 180 с;
- концентрирование электроактивного соединения в течение 120 с;
- регистрация катодной и анодной вольтамперограммы в диапазоне потенциалов (от +0,2 до -0,2) В;
- исключение деаэрации анализируемых растворов.

Пример 1. Навеску гидрохлорида цистеина водного массой 0,00440 г помещали в колбу на 5 мл и доводили до метки бидистиллированной водой, получали 5·10-3 М раствор цистеина.
Определение цистеина проводили методом стандартной добавки в несколько этапов. Сначала формировали РПЭ. Для этого СУЭ помещали в 3-электродную ячейку (вспомогательный электрод - СУ; электрод сравнения - хлорсеребряный (нас. КС1)), содержащую 20 мл раствора состава: 0,1 М КМО3+1·10-4 М Hg(NO3)2, и формировали РПЭ при потенциале электролиза -0,5 В в течение 180 с, промывали электрод бидистиллированной водой. Затем сформированный электрод помещали в ячейку, содержащую 20 мл фонового электролита состава: 0,1 М NaClO4+0,1 М НСlO4 и 2,5·10-5 М CySH (модельная проба), выдерживали в течение 120 с при разомкнутой цепи и регистрировали катодные и анодные АС в диапазоне потенциалов (от +0,2 до -0,2) В.
После механической регенерации рабочего электрода снова формировали РПЭ, как описано выше. Затем в электрохимическую ячейку, содержащую 20 мл фонового электролита состава: 0,1 М NaClO4+0,1 М НСlO4 и 2,5·10-5 М CySH (модельная проба), добавляли 0,1 мл стандартного раствора цистеина концентрации 5·10-3 М. Раствор перемешивали мешалкой. Сформированный РПЭ опускали в электрохимическую ячейку, выдерживали в течение 120 с при разомкнутой цепи и регистрировали катодные и анодные АС в диапазоне потенциалов (от +0,2 до -0,2) В.
Найденная концентрация цистеина составила (2,5±0,4)·10-5 М при определении по катодным и анодным АС.
Пример 2. Навеску гидрохлорида цистеина водного массой 0,04400 г помещали в колбу на 5 мл и доводили до метки бидистиллированной водой, получали 5·10-2 М раствор цистеина.
Определение цистеина проводили методом стандартной добавки в несколько этапов. Сначала формировали РПЭ. Для этого СУЭ помещали в 3-электродную ячейку (вспомогательный электрод - СУ; электрод сравнения - хлорсеребряный (нас. КСl)), содержащую 20 мл раствора состава 0,1 М КМО3+1·10-4 М Hg(NO3)2, и формировали РПЭ при потенциале электролиза -0,5 В в течение 180 с, промывали электрод бидистиллированной водой. Затем сформированный электрод помещали в ячейку, содержащую 20 мл фонового электролита состава: 0,1 М NaClO4+0,1 М НСlO4 и 2,5·10-4 М CySH (модельная проба), выдерживали в течение 120 с при разомкнутой цепи и регистрировали катодные и анодные АС в диапазоне потенциалов (от +0,2 до -0,2) В.
После механической регенерации рабочего электрода снова формировали РПЭ, как описано выше. Затем в электрохимическую ячейку, содержащую 20 мл фонового электролита состава: 0,1 М NaClO4+0,1 М HClO4 и 2,5·10-4 М CySH (модельная проба), добавляли 0,1 мл стандартного раствора цистеина концентрации 5·10-2 М. Раствор перемешивали мешалкой. Сформированный РПЭ опускали в электрохимическую ячейку, выдерживали в течение 120 с при разомкнутой цепи и регистрировали катодные и анодные АС в диапазоне потенциалов (от +0,2 до -0,2) В.
Найденная концентрация цистеина составила (2,5±0,4)(10-4 М при определении по катодным АС и (2,4±0,3)(10-4 М - по анодным АС.
Пример 3. Навеску гидрохлорида цистеина водного массой 0,44000 г помещали в колбу на 5 мл и доводили до метки бидистиллированной водой, получали 5·10-1 М раствор цистеина.
Определение цистеина проводили методом стандартной добавки в несколько этапов. Сначала формировали РПЭ. Для этого СУЭ помещали в 3-электродную ячейку (вспомогательный электрод - СУ; электрод сравнения - хлорсеребряный (нас. КСl)), содержащую 20 мл раствора состава: 0,1 М КNO3+1·10-4 М Hg(NO3)2, и формировали РПЭ при потенциале электролиза -0,5 В в течение 180 с, промывали электрод бидистиллированной водой. Затем сформированный электрод помещали в ячейку, содержащую 20 мл фонового электролита состава: 0,1 М NaClO4+0,1 М НСlO4 и 1,3·10-3 М CySH (модельная проба), выдерживали в течение 120 с при разомкнутой цепи и регистрировали катодные и анодные АС в диапазоне потенциалов (от +0,2 до -0,2) В.
После механической регенерации рабочего электрода снова формировали РПЭ, как описано выше. Затем в электрохимическую ячейку, содержащую 20 мл фонового электролита состава: 0,1 М NaClO4+0,1 М НСlO4 и 1,3·10-3 М CySH (модельная проба), добавляли 0,05 мл стандартного раствора цистеина концентрации 5·10-1 М. Раствор перемешивали мешалкой. Сформированный РПЭ опускали в электрохимическую ячейку, выдерживали в течение 120 с при разомкнутой цепи и регистрировали катодные и анодные АС в диапазоне потенциалов (от +0,2 до -0,2) В.
Найденная концентрация цистеина составила (1,3±0,1)·10-3 М при определении по катодным АС и (1,2±0,1)·10-3 М - по анодным АС.
Преимуществами предлагаемого способа по сравнению с прототипом является более широкий линейный диапазон определяемых концентраций цистеина, большая экологическая безопасность и меньшая трудоемкость.
Изобретение относится к электроаналитической химии и может быть использовано при определении цистеина в объектах окружающей среды. Техническим результатом изобретения является разработка способа определения цистеина, имеющего широкий линейный диапазон определяемых концентраций цистеина, экологически более безопасного и менее трудоемкого. Сущность: предлагается предварительное формирование ртутного пленочного электрода (РПЭ) на стеклоуглеродной подложке, концентрирование электроактивного соединения на поверхности электрода в фоновом электролите состава: (0,1-0,4) М NaClO4+(0,1-1) М HClO4, содержащем цистеин, с последующим получением аналитического сигнала при изменении потенциала. 3 з.п.ф-лы, 4 табл., 4 ил.
| STOCK J.T., LARSON R.E | |||
| The catodic stripping voltammetry of certain thiols in acetate medium// Analytica Chimica Acta | |||
| Устройство для видения на расстоянии | 1915 |
|
SU1982A1 |
| ВСЕСОЮЗНАЯ fnATiHTH3-]tXf!^'^'ffA;i!БИБЛИО7 | 0 |
|
SU285072A1 |
| ВОЛЬТАМПЕРОМЕТРИЧЕСКИЙ СПОСОБ ОПРЕДЕЛЕНИЯ 5-ФТОРУРАЦИЛА | 1996 |
|
RU2106623C1 |
| ПРЯМАЯ ЭЛЕКТРОХИМИЯ ФЕРМЕНТОВ | 1997 |
|
RU2157521C2 |
| GB 1468297 А, 23.03.1977. | |||

