Область изобретения
Данное изобретение относится к новым замещенным 3-пиридил-4-арилпирролам и их терапевтическому и профилактическому использованию. Расстройства, которые лечат и/или предотвращают с использованием данных соединений, включают воспалительные и связанные со СПИД расстройства.
Предпосылки к созданию изобретения
TNF-α и р38-связанные расстройства
Воспалительные цитокины, такие как TNF-α, продуцируются в результате активности киназ. Такие киназы включают цитокин, подавляющий противовоспалительный связывающий лекарственный препарат протеин (CSBP)/p38 киназу, митоген-активированную протеин (MAP) киназу семейства серин-треонин протеинкиназ. Воспалительные цитокины играют важную роль в ряде воспалительных расстройств [1], нейродегенеративных расстройств [10] и связанных со СПИД расстройств [11-14]. Хотя точный механизм киназ, таких как р38, не известен, р38 вовлечена как в продуцирование TNF-α, так и в сигнальные ответные реакции, связанные с рецептором TNF-α.
Артрит представляет основной пример воспалительного расстройства и является, таким образом, воспалительным расстройством, которому, главным образом, уделено внимание в данном разделе. Миллионы людей страдают от артрита, и он может поразить любой сустав в теле человека. Его симптомы меняются от слабой боли и воспаления в пораженных суставах до сильной и ослабляющей боли и воспаления. Хотя данное расстройство связано, главным образом, со стареющими взрослыми, оно не ограничивается взрослыми людьми.
Наиболее обычная терапия ревматического артрита включает использование нестероидных противовоспалительных лекарственных средств (НСПВА (NSAID's)) для облегчения симптомов.
Однако, несмотря на широко распространенное использование НСПВА, многие индивидуумы не могут переносить дозы, необходимые для лечения данного расстройства в течение продолжительного периода времени. Кроме того, НСПВА лечат лишь симтомы нарушения, не затрагивая лежащую в их основе причину.
Когда пациенты не проявляют реакции на НСПВА, часто используют другие лекарственные средства, такие как метотрексат, соли золота, D-пенициламин и преднизон. Данные лекарственные средства также обладают существенной токсичностью, а механизм их действия остается невыясненным. Было показано, что моноклональные антитела к TNF-α, и антагонисты рецептора интерлейкина 1β (IL-1β) уменьшают симптомы ревматического артрита при клинических испытаниях на людях в небольшом масштабе [2].
Помимо основанной на белке терапии, существуют небольшие молекулярные агенты, ингибирующие продуцирование данных цитокинов, которые показали активность на моделях ревматического артрита на животных [3]. Из данных небольших молекулярных агентов SB 203580 оказались эффективными в снижении продуцирования TNF-α и IL-1β в липополисахарид (LPS)-стимулированных моноцитарных клеточных линиях человека со значениями IC50 порядка 50-100 нМ [4].
В дополнение к результатам испытаний in vitro было показано, что SB 203580 ингибируют продуцирование воспалительных цитокинов у крыс и мышей при значениях IC50 15-25 мг/кг [5]. SB 203580 снижает продуцирование воспалительных цитокинов, ингибируя активность CSBP/p38 киназы при IC50 200 нМ [6]. Вследствие пероральной активности и эффективности SB 203580 на моделях животных исследователи предположили, что любое соединение с таким профилем активности имеет потенциал в качестве жизнеспособного лечебного средства против ревматоидного артрита [5].
Пиридилпирролы и их аналоги также были получены в качестве ингибиторов цитокинов и антагонистов глюкагона [7] и, в частности, в качестве ингибиторов IL-1β, TNF-α и других цитокинов. В качестве ингибиторов цитокинов были также получены арилпирролы [8] и триарилпирролы [9].
Роль CSBP/p38 была отмечена недавно в различных нейродегенеративных и связанных со СПИД расстройствах. Что касается нейродегенеративных расстройств, было показано, что роль р38 состоит в определении того, выживает ли клетка, или подвергается нейронной программированной клеточной гибели, или апоптозу [10, 11].
Было показано, что относящийся также к СПИД'у связанный с саркомой вирус герпеса HHV 8 Капоши (Kaposi) кодирует G-белоксвязанный рецептор, активирующий р38. Было высказано предположение о том, что данная активация способствует онкогенезу и ангиогенезу, приводящим к саркоме Капоши [12].
Со СПИД'ом связана быстрая активация р38, индуцируемая заражением CCR5* Т клеточной линии лимфоцитов человека ВИЧ, предполагая, что р38 может играть роль в ранней вирусной инфекции [13]. Кроме того, было показано, что ингибиторы р38 блокируют репликацию ВИЧ in vitro способом, который может быть независимым от TNF-α [14].
Отсутствие клинически эффективных агентов
В целом, артрит - в частности, ревматический артрит - и большинство других воспалительных и связанных со СПИД расстройств уносит много жизней пораженных ими людей. Существует огромная потребность в небольших молекулярных агентах для лечения данных расстройств. Однако на настоящее время не идентифицирован ни один из таких агентов и не показано, что он клинически эффективен на людях.
Краткое описание изобретения
Данное изобретение предоставляет соединения, имеющие структуру
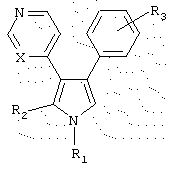
или их фармацевтически приемлемые соли, причем в указанной формуле:
(a) R1 выбран из группы, состоящей из (i) водорода, (ii) C1-5 алкила, (iii) замещенного или незамещенного C1-5 алкиламино, (iv) N-содержащего C1-5 алкилгетероцикла, выбранного из тиазолидина, пиперидина, морфолина, пиперазина, тиоморфолина, пирролидина, тиазина, пиррола и имидазола, (v) фенила, (vi) фенила, независимо замещенного одним или более C1-5 алкилом, амино, замещенным амино, нитро, нитрилом и сульфоном, и (vii) пиридина;
(b) R2 выбран из группы, состоящей из (i) водорода, (ii) (СН2)3ОН, (iii) замещенного или незамещенного C1-5 алкилфенила, и (iv) N-содержащего C1-5 алкилгетероцикла, выбранного из тиазолидина, пиперидина, морфолина, пиперазина, тиоморфолина, пирролидина, тиазина, пиррола и имидазола;
(c) R3 представляет один или более заместителей, независимо выбранных из группы, состоящей из водорода, галогена, метокси, нитро, трифторметила, гидрокси, диметиламино и метилсульфоксида; и
(d) X представляет либо С, либо N.
Данное изобретение предоставляет также еще одну группу соединений, имеющих структуру:
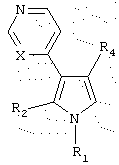
или их фармацевтически приемлемые соли, в которых:
(а) R1 выбран из группы, состоящей из (i) водорода, (ii) C1-5 алкила, (iii) замещенного или незамещенного C1-5 алкиламино, (iv) N-содержащего C1-5 алкилгетероцикла, выбранного из тиазолидина, пиперидина, морфолина, пиперазина, тиоморфолина, пирролидина, тиазина, пиррола и имидазола, (v) фенила, (vi) фенила, независимо замещенного одним или более C1-5 алкилом, амино, замещенным амино, нитро, нитрилом и сульфоном, и (vii) пиридина;
(b) R2 выбран из группы, состоящей из (i) водорода, (ii) (СН2)3ОН, (iii) замещенного или незамещенного C1-5 алкилфенила, и (iv) N-содержащего C1-5 алкилгетероцикла, выбранного из тиазолидина, пиперидина, морфолина, пиперазина, тиоморфолина, пирролидина, тиазина, пиррола и имидазола;
(c) R4 представляет замещенный или незамещенный гетероцикл, выбранный из пиридина, пиримидина, фурана, или тиофена; и
(d) X представляет либо С, либо N.
Кроме того, данное изобретение предоставляет фармацевтическую композицию, содержащую одно из настоящих соединений и фармацевтически приемлемый носитель.
Кроме того, данное изобретение предоставляет также способ лечения субъекта, имеющего расстройство, улучшаемое путем снижения продуцирования TNF-α и/или активности р38 в соответствующих клетках, который включает введение данному субъекту терапевтически эффективной дозы настоящей фармацевтической композиции.
Наконец, данное изобретение предоставляет способ предотвращения воспалительной ответной реакции у субъекта, включающий введение данному субъекту профилактически эффективного количества настоящей фармацевтической композиции либо до, либо после события, которое, как ожидают, вызовет у субъекта воспалительную ответную реакцию.
Подробное описание изобретения
Данное изобретение предоставляет новые замещенные 3-пиридил-4-арилпирролы. Данные соединения удивительно полезны при лечении расстройств, улучшаемых путем снижения продуцирования TNF-α и/или активности р38 и, следовательно, полезны для лечения воспалительных расстройств, таких как ревматический артрит, а также связанных со СПИД расстройств.
Конкретно, данное изобретение предоставляет первую группу соединений, имеющих структуру
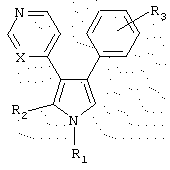
или их фармацевтически приемлемые соли, в которых:
(a) R1 выбран из группы, состоящей из (i) водорода, (ii) C1-5 алкила, (iii) замещенного или незамещенного C1-5 алкиламино, (iv) N-содержащего C1-5 алкилгетероцикла, выбранного из тиазолидина, пиперидина, морфолина, пиперазина, тиоморфолина, пирролидина, тиазина, пиррола и имидазола, (v) фенила, (vi) фенила, независимо замещенного одним или более C1-5 алкилом, амино, замещенным амино, нитро, нитрилом и сульфоном, и (vii) пиридина;
(b) R2 выбран из группы, состоящей из (i) водорода, (ii) (СН2)3ОН, (iii) замещенного или незамещенного C1-5 алкилфенила, и (iv) N-содержащего C1-5 алкилгетероцикла, выбранного из тиазолидина, пиперидина, морфолина, пиперазина, тиоморфолина, пирролидина, тиазина, пиррола и имидазола;
(c) R3 представляет один или более заместителей, независимо выбранных из группы, состоящей из водорода, галогена, метокси, нитро, трифторметила, гидрокси, диметиламино и метилсульфоксида; и
(d) X представляет либо С, либо N.
В одном из вариантов воплощения первой группы соединений:
(a) R1 выбран из группы, состоящей из (i) водорода, (ii) C1-5 алкила, (iii) замещенного или незамещенного C1-5 алкиламино, (iv) N-содержащего C1-5 алкилгетероцикла, выбранного из пиперидина, морфолина и пирролидина, и (v) фенила, замещенного заместителем, выбранным из группы, состоящей из амино, замещенного амино, нитро и нитрила.
(b) r2 выбран из группы, состоящей из водорода и (СН2)3 фенила;
(c) R3 выбран из группы, состоящей из галогена, нитро и трифторметила; и
(d) Х представляет С.
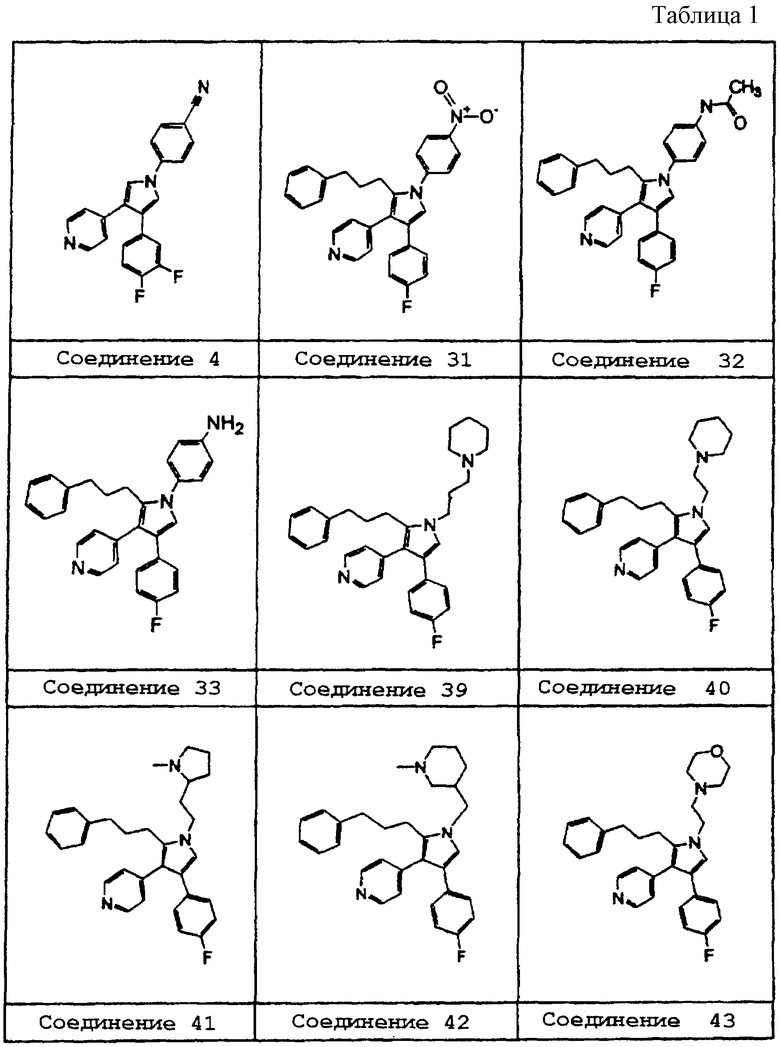
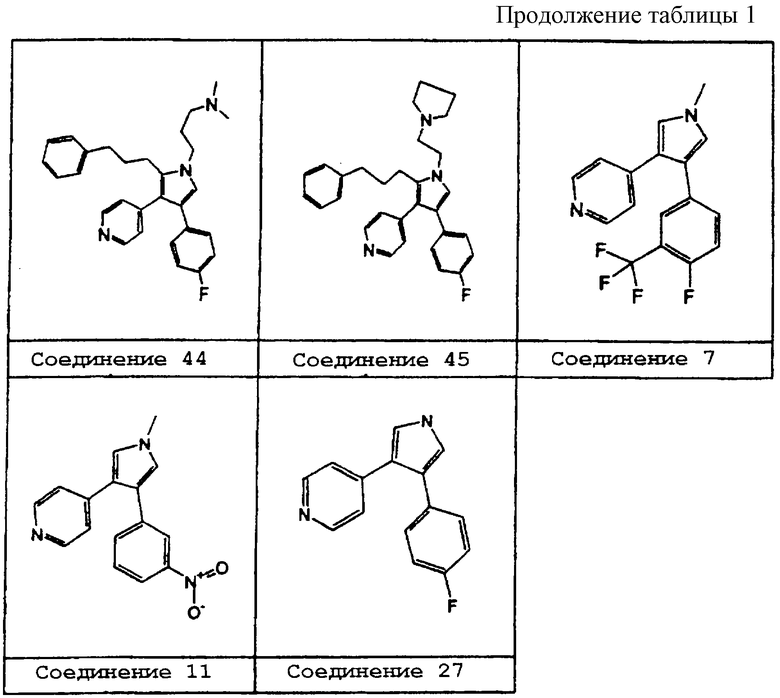
В предпочтительном воплощении первое соединение выбрано из группы соединений, представленных в Таблице 1.
Данное изобретение предоставляет также вторую группу соединений, имеющих структуру
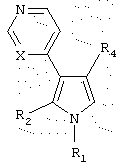
или их фармацефтически приемлемые соли, в которых:
(а) R1 выбран из группы, состоящей из (i) водорода, (ii) С1-5 алкила, (iii) замещенного или незамещенного С1-5 алкиламино, (iv) N-содержащего С1-5 алкилгетероцикла, выбранного из тиазолидина, пиперидина, морфолина, пиперазина, тиоморфолина, пиролидина, тиазина, пиррола и имидазола, (v) фенила, (vi) фенила, независимо замещенного одним или более C1-5 алкилом, амино, замещенным амино, нитро, нитрилом и сульфоном, и (vii) пиридина;
(b) R2 выбран из группы, состоящей из (i) водорода, (ii) (СН2)3ОН, (iii) замещенного или незамещенного C1-5 алкилфенила, и (iv) N-содержащего C1-5 алкилгетероцикла, выбранного из тиазолидина, пиперидина, морфолина, пиперазина, тиоморфолина, пирролидина, тиазина, пиррола и имидазола;
(c) R4 представляет замещенный или незамещенный гетероцикл, выбранный из пиридина, пиримидина, фурана, или тиофена; и
(d) X представляет либо С, либо N.
В одном из воплощений второй группы соединений:
(a) R1 выбран из группы, состоящей из (i) C1-5 алкила, (ii) замещенного или незамещенного C1-5 алкиламино, (iii) замещенного или незамещенного C1-5 алкилгетероциклического амино, (iv) фенила, и (v) фенила, независимо замещенного одним или более амино, замещенным амино, нитро, или нитрилом.
(b) R2 выбран из группы, состоящей из водорода и (СН2)3 фенила; и
(c) Х представляет С.
Настоящие соединения можно выделить и использовать в виде свободных оснований. Их можно также выделить и использовать в виде фармацевтически приемлемых солей. Примеры таких солей включают соли бромисто-водородной, йодисто-водородной, хлористо-водородной, хлорной, серной, малеиновой, фумаровой, яблочной, винной, лимонной, бензойной, миндальной, метансульфокислоты, гидроэтансульфокислоты, бензолсульфокислоты, щавелевой, памовой, 2-нафталинсульфокислоты, п-толуолсульфокислоты, циклогексансульфамовой и сахарной кислот.
Кроме того, настоящее изобретение предоставляет фармацевтическую композицию, включающую одно из настоящих соединений и фармацевтически приемлемый носитель.
Фармацевтически приемлемые носители хорошо известны специалистам в данной области и включают, но не ограничиваются, от около 0,01 до около 0,1 М, а предпочтительно 0,05 М фосфатный буфер или 0,8% солевой раствор. Такие фармацевтически приемлемые носители могут представлять водные или неводные растворы, суспензии и эмульсии. Примерами неводных растворителей являются пропиленгликоль, полиэтиленгликоль, растительные масла, такие как оливковое масло, и инъецируемые органические сложные эфиры, такие как этилолеат. Водные носители включают воду, этанол, спиртово-водные растворы, глицерин, эмульсии или суспензии, включая солевую или буферную среды. Пероральные носители могут представлять эликсиры, сиропы, желатиновые капсулы, таблетки и тому подобные. Типичный твердый носитель является инертным веществом, таким как лактоза, крахмал, глюкоза, метилцеллюлоза, стеарат магния, дикальцийфосфат, маннит и тому подобные. Парентеральные носители включают раствор хлорида натрия, декстрозу Ринджера (Ringer's декстрозу), декстрозу и хлорид натрия, лактированные Ринджеровы и густые масла. Внутривенные носители включают наполнители жидких и питательных веществ, пополнители электролита, такие как на основе декстрозы Ринджера и аналогичные. Могут также присутствовать консерванты и другие добавки, например, такие как антимикробные агенты, антиоксиданты, хелатирующие агенты, инертные газы и аналогичные. Все носители можно смешивать при необходимости с разрыхлителями, разбавителями, гранулирующими агентами, смазывающими веществами, связывающими веществами и тому подобным с использованием обычных методик, известных в данной области.
Кроме того, настоящее изобретение предоставляет способ лечения субъекта, имеющего расстройство, улучшаемое снижением продуцирования TNF-α и/или активности р38 в соответствующих клетках, который включает введение субъекту терапевтически эффективной дозы настоящей фармацевтической композиции.
В одном из вариантов воплощения расстройством является воспалительное расстройство. В еще одном варианте воплощения расстройством является связанное со СПИД расстройство. Примеры расстройств, которые можно лечить настоящей фармацевтической композицией, включают, без ограничения, ревматический артрит, остеопороз, остеоартрит, аллергическое воспаление, периодонтальное расстройство, воспалительное кишечное расстройство, септический шок, инсулин-зависимый сахарный диабет, инсулин-независимый диабет, кахексию, легочный фиброз, миастению, болезнь Крона, гепатит, первичный билиарный цирроз печени, острый панкреатит, отторжение аллотрансплантата, глиобластому, гнездную алопецию, псориаз, ишемию, застойную сердечную недостаточность, рестеноз, атеросклероз, системную эритемную волчанку, нефрит, синдром Гиллена-Барра (Guillain-Barre), вирусный миокардит, репликацию ВИЧ, истощение Т-лимфоцитов при заражении ВИЧ, нарушение познавательной способности, вызванное нейронным воспалением, рассеянный склероз, удар или шок, нейропатическую боль, ВИЧ слабоумие и болезнь Aльцгеймера. В предпочтительном воплощении расстройством является ревматический артрит.
Используемый здесь термин “субъект” включает, без ограничений, любое животное, или искусственно модифицированное животное с расстройством, улучшаемым снижением продуцирования TNF-α и/или активности р38 в соответствующих клетках. В предпочтительном воплощении субъектом является человек.
Используемый здесь термин “соответствующие клетки” включают, например, клетки, которые выделяют или способны выделять TNF-α, и клетки, в которых активирована р38. Конкретные примеры соответствующих клеток включают, без ограничения, моноциты, макрофаги, Т лимфоциты, фибробласты, дендритные клетки, клетки Лангеранса (Langerhans), клетки Купфера (Kuppfer) и астроглиальные клетки.
Введение настоящей фармацевтической композиции можно выполнять или осуществлять с использованием любого из различных способов, известных специалистам в данной области. Настоящие соединения можно вводить, например, внутривенно, внутримышечно, перорально и подкожно. В предпочтительном способе воплощения настоящую фармацевтическую композицию вводят перорально. Кроме того, введение может предусматривать предоставление субъекту для приема множества доз на протяжении подходящего периода времени. Такие режимы введения можно определять обычными способами.
Используемой здесь “терапевтически эффективной дозой” фармацевтической композиции является количество, достаточное для того, чтобы остановить, привести в норму или снизить развитие расстройства. “Профилактически эффективная доза” фармацевтической композиции представляет количество, достаточное для предотвращения расстройства, то есть прекращения, улучшения и/или замедления наступления расстройства. В данной области известны способы определения терапевтически и профилактически эффективных доз для настоящей фармацевтической композиции. Эффективную дозу для введения фармацевтической композиции человеку, например, можно определить математически по результатам исследований на животных.
В одном из воплощений терапевтически и/или профилактически эффективная доза представляет дозу, достаточную для доставки настоящей фармацевтической композиции в количестве от около 0,05 мг/кг массы тела до около 200 мг/кг массы тела. В другом воплощении терапевтически и/или профилактически эффективная доза представляет дозу, достаточную для доставки от около 0,5 мг/кг массы тела до около 50 мг/кг массы тела. Более конкретно, в одном из воплощений пероральные дозы меняются в интервале от около 0,05 мг/кг до около 100 мг/кг в день. В еще одном воплощении пероральная доза составляет в интервале от около 0,05 мг/кг до около 50 мг/кг в день и в следующем воплощении от около 0,05 мг/кг до около 20 мг/кг в день. В еще одном воплощении дозы для вливания составляют в интервале от около 1,0 мкг/кг/мин до около 1,0×104 мкг/кг/мин ингибитора в смеси с фармацевтическим носителем на протяжении периода времени в интервале от нескольких минут до примерно нескольких дней. Согласно еще одному воплощению для локального введения настоящее соединение можно объединять с фармацевтическим носителем в соотношении лекарственное средство/носитель от около 0,001 до около 0,1.
Далее настоящее изобретение предоставляет также способ предотвращения воспалительной ответной реакции у субъекта, предусматривающий введение субъекту профилактически эффективного количества настоящей фармацевтической композиции либо до, либо после события, которое предположительно вызовет у субъекта воспалительную ответную реакцию. Данным событием может быть укус насекомого или укус животного.
Используемые здесь следующие химические термины имеют значения, изложенные в данном абзаце: “независимо”, применительно к химическим заместителям, означает, что при наличии более чем одного заместителя данные заместители могут быть одинаковыми или различными; “алкил” означает алкил с линейной, циклической или разветвленной цепью; “алкокси” означает O-алкил; “галоген” представляет фтор, хлор, бром или йод; “Ph” означает фенил; “ТХК” (ТСА) означает трихлоруксусную кислоту; “ФТС” (FCS) означает фетальную телячью сыворотку; и “RPMI” означает среду из Roswell Park Memorial Institute (Sigma cat # R0833).
Данное изобретение будет более понятно при обращении к следующим экспериментальным подробностям, но специалисты в данной области очевидно легко понимают, что они являются лишь иллюстрацией изобретения, описанного более полно в формуле изобретения, которая следует ниже. Кроме того, в данной заявке приводятся различные публикации. Раскрытие этих публикаций включено для сведения в данную заявку для более полного описания состояния области техники, к которой относится данное изобретение.
Экспериментальные подробности
I. Общие схемы синтеза
Характерные представители соединений настоящего изобретения можно получать в соответствии с общими методами синтеза, описанными ниже и иллюстрируемыми общими схемами 1-6, приведенными в конце описания. Продукты некоторых схем можно использовать в качестве промежуточных соединений для получения более чем одного из настоящих соединений. В этих случаях выбор промежуточных соединений, используемых для получения последующих соединений настоящего изобретения, представляется на усмотрение и находится в компетенции специалистов в данной области.
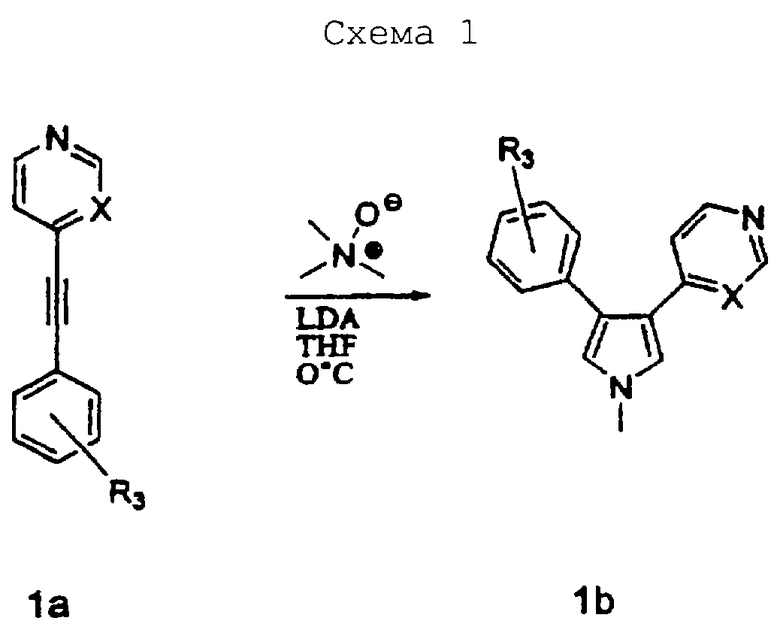
Схему 1 можно использовать для получения соединений изобретения, в которых R1 представляет метил. В качестве исходного материала в схеме 1 можно использовать соединение 1a, такое как 1,2-дизамещенный алкин. 1,2-Дизамещенные алкины можно получить с помощью известных процедур. Заместители Х и R3 соединений изобретения определяются заместителями соединения 1а. Соединение 1а объединяют с N-оксидом триметиламина и растворяют в сухом растворителе, таком как ТГФ, и охлаждают до 0°С. Добавляют основание, такое как диизопропиламид лития, и реакционную смесь перемешивают при 0°С в течение 1 ч, получая соединение 1b в качестве продукта.
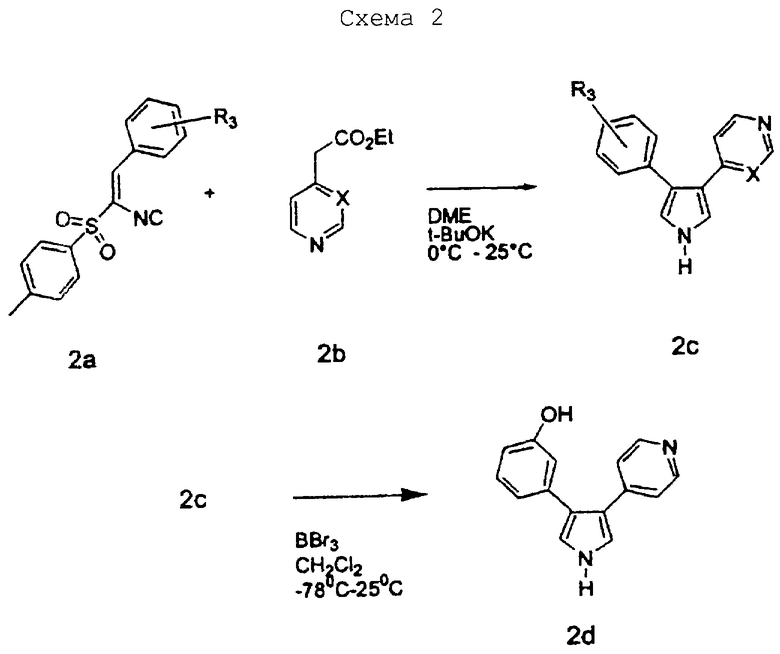
Схему 2 можно использовать для получения соединений изобретения, в которых R1 представляет водород. В качестве исходного в схеме 2 можно использовать соединение 2а типа 1-[(1-изоциано-2-(3-метоксифенил)этенил)сульфонил]-4-метилбензол. Соединение 2а можно получить по известным методикам. Заместитель R3 соединений изобретения обычно определяется заместителями у фенила этенильной группы соединения 2а; атом Х определяется гетероароматическим заместителем ацетатной группы в соединении 2b. Соединение 2а растворяют в сухом растворителе, таком как диметиловый эфир этиленгликоля, и добавляют по каплям к смеси соединения 2b, такого как этил 4-пиридилацетат, и основания, такого как трет-бутилат калия, в сухом растворителе, таком как диметиловый эфир этиленгликоля, при 0°С. По завершении добавления реакционную смесь подогревают до 25°С и перемешивают в течение 3 часов, получая промежуточное соединение 2с. Когда R3 представляет метокси, промежуточное соединение 2с может обрабатываться деметилирующим агентом, таким как ВВr3, в инертном растворителе, таком как СН3Сl2, при -78°С, давая соединение 2d.
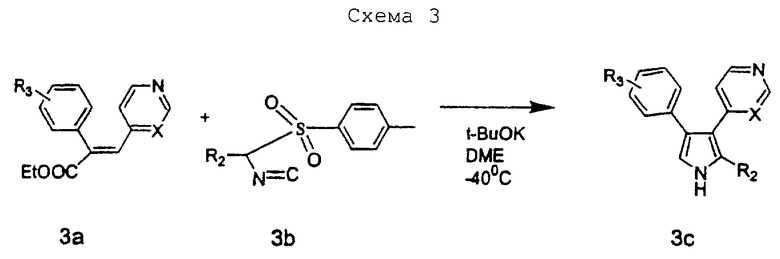
Схему 3 можно использовать для получения соединений изобретения, в которых R1 представляет водород, a R2 является замещенным или незамещенным. В качестве исходного вещества в схеме 3 можно использовать соединение 3а, диарилзамещенный α,β-ненасыщенный сложный эфир. Соединения данного типа можно получать по известным методикам. Соединением 3b может быть другой исходный материал, замещенное тозилметилизоцианидное производное (tosMIC), которое можно получить с помощью известных процедур.
Например, соединением 3а может быть этиловый эфир 4-фтор-α-[(4-пиридил)метилен ]бензолуксусной кислоты, а соединением 3b может быть 1-(4-толилсульфонил)-1-(3-фенилпропил)метилизоцианид. Соединение 3а и соединение 3b растворяют в сухом растворителе, таком как диметиловый эфир этиленгликоля, и добавляют по каплям к -40°С-ной смеси основания, такого как третбутилат калия, в сухом растворителе, таком как диметиловый эфир этиленгликоля. Перемешивание при -40°С продолжают в течение 1 часа перед тем, как дают температуре подняться до -20°С. Соответственно, получающееся соединение 3с представляет 4-(4-фторфенил)-2-(3-фенилпропил)-3-(4-пиридил)-пиррол (соединение 27).
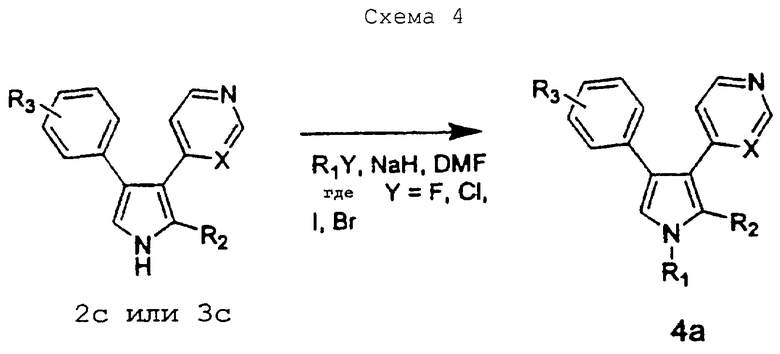
Как показано на схеме 4, соединение 2с и соединение 3с можно использовать в качестве промежуточных соединений для получения других соединений настоящего изобретения.
Схему 4 можно использовать для получения соединений изобретения, в которых R1 замещен. Промежуточное соединение 2с или соединение 3с добавляют порциями к основанию, такому как гидрид натрия, в растворителе, таком как диметилформамид, при 0°С. По завершении добавления реакционную смесь перемешивают при 0°С еще в течение 15 минут, после чего добавляют порциями алкилирующий агент, такой как гидрохлорид 4-(2-хлорэтил)морфолина. Реакционную смесь нагревают до 60°С в течение 16 часов перед тем, как дают температуре понизиться до 25°С с получением соединения 4а.
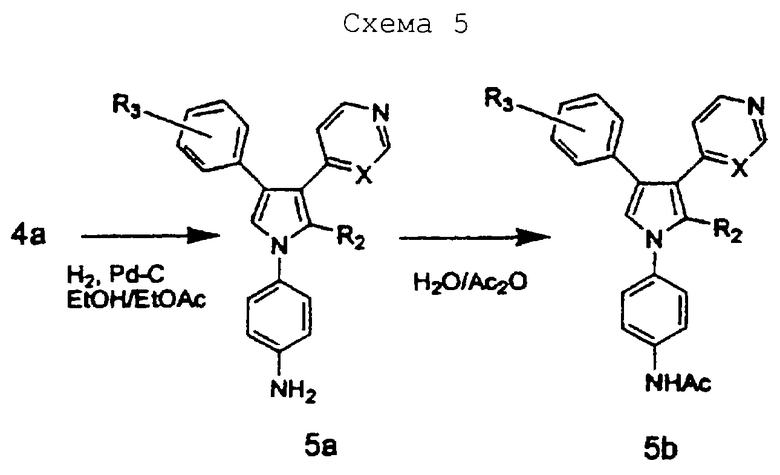
Схему 5 можно использовать для получения соединений изобретения, в которых R1 замещен. Когда R1 представляет 4-нитрофенил, промежуточное соединение 4а может восстанавливаться с использованием катализатора, такого как палладий на угле, в растворителе, таком как этилацетат, давая соединение 5а. Аминовое соединение 5а может обрабатываться ацилирующим агентом, таким как уксусный ангидрид, в растворителе, таком как вода, давая продукт - соединение 5b.
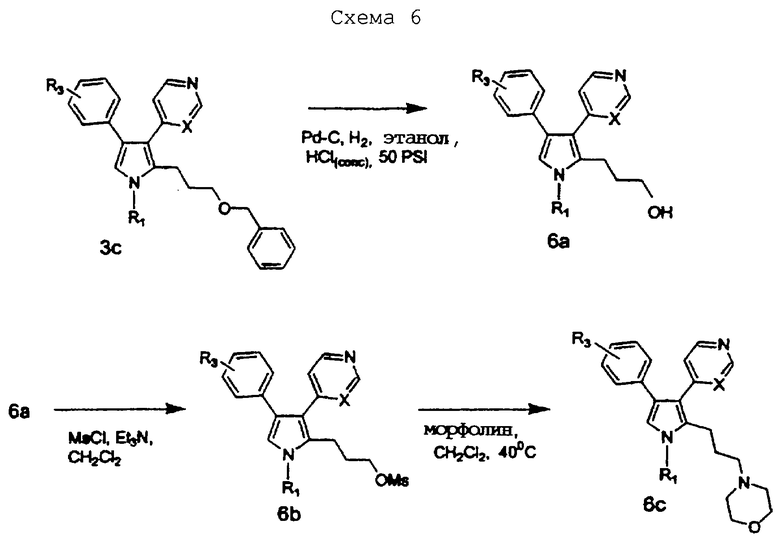
Схему 6 можно использовать для получения соединений изобретения, в которых R2 представляет алкильную цепь, содержащую гетероатомы. Промежуточное соединение 3с подвергают действию условий восстановления с использованием катализатора, такого как палладий на угле, в растворителе, таком как этанол, к которому добавляют каталитическое количество кислоты, такой как концентрированная соляная кислота, для получения соединения 6а. Спиртовое соединение 6а можно обрабатывать сульфонирующим агентом, таким как мезилхлорид, и основанием, таким как триэтиламин, в растворителе, таком как CH2Cl2, для получения соединения 6b. Соединение 6b можно затем нагревать с нуклеофилом, таким как морфолин, в растворителе, таком как CH2Cl2, для получения соединения 6с.
II. Синтез конкретных соединений
Конкретные соединения, являющиеся характерными представителями данного изобретения, могут получаться, как описано в следующих примерах. Никаких попыток оптимизировать выходы, полученные в данных реакциях, не делалось. Однако, исходя из нижеследующего, специалист знает, как повысить выходы с помощью обычного варьирования продолжительности реакции, температур, растворителей и/или реагентов.
Продукты некоторых способов синтеза можно использовать в качестве промежуточных соединений для получения более чем одного из настоящих соединений. В данных случаях выбор промежуточных соединений, используемых для получения последующих соединений настоящего изобретения, предоставляется на усмотрение специалистов в данной области и находится в их компетенции.
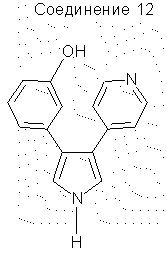
Гидробромид 3-(3-гидроксифенил)-4-(4-пиридил)пиррола
Соединение 13 (0,15 г, 0,006 моль) растворяли в СН2Сl2 (50 мл) и охлаждали до -78°С. Смесь перемешивали в течение 16 часов, давая температуре повыситься до 25°С. Реакционную смесь гасили МеОН (20 мл) и упаривали до твердого вещества. Твердое вещество растирали с эфиром и фильтровали, получая соединение 12 (0,15 г, выход 79%). 1H ЯМР (ДМСО-d6) δ 11,91 (1Н, с, NH), 9,47 (1Н, шир.с., ОН), 8,66 (2Н, д, J=8,6 Гц), 7,86-7,86 (3Н, м), 7,25-7,14 (1Н, м), 7,06 (1Н, с), 6,80-6,66 (3Н, м).
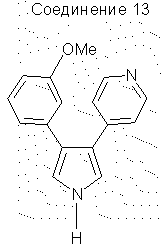
3-(3-метоксифенил)-4-(4-пиридил)пиррол
1-[(1-Изоциано-2-(3-метоксифенил)этенил)сульфонил]-4-метил-бензол (9,0 г, 0,0285 моль) растворяли в сухом ДМЭ (200 мл) и добавляли по каплям к охлажденной (0°С) смеси этил 4-пиридилацетата (9,0 г, 0,545 моль) и трет-бутилата калия (7,1 г, 0,0633 моль) в сухом ДМЭ (100 мл). По завершении добавления реакционную смесь подогревали до 25°С и перемешивали в течение 3 ч. Затем реакционную смесь выливали в ледяную воду (1200 мл) и экстрагировали СН2Сl2 (3×500 мл). Объединенные органические экстракты сушили над Na2SO4, фильтровали и упаривали в вакууме, получая твердое вещество. Данное твердое вещество затем растирали с эфиром и фильтровали, получая 3,0 г чистого 3-(3-метоксифенил)-4-(4-пиридил)пиррола. Фильтрат снова упаривали в вакууме и затем растирали со смесью 50/50 CH2Cl2 и эфира, получая дополнительно 1,5 г чистого продукта. Наконец фильтрат упаривали в вакууме и очищали на SiO2 с элюированием этилацетатом, получая еще 0,5 г чистого продукта, получая в результате 70% общий выход. 1H ЯМР (ДМСО-d6) δ 11,38 (1Н, шир.с., NH), 8,37 (2Н, д, J=6,1 Гц), 7,24-7,17 (4Н, м), 7,02-7,00 (1H, м), 6,80-6,77 (3Н, м), 3,68 (3Н, с).
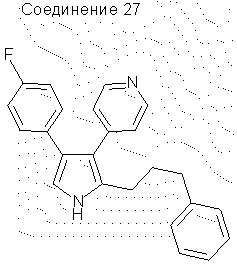
4-(4-фторфенил)-2-(3-фенилпропил)-3-(4-пиридил)пиррол
1-(4-Толилсульфонил)-1-(3-фенилпропил)метилизоцианид (10,9 г, 0,0346 моль) и этил 4-фтор-α-[(4-пиридил)метилен]-бензолуксусную кислоту (9,4 г, 0,0346 моль) растворяли в сухом ДМЭ (250 мл) и добавляли по каплям к охлажденной (-40°С) смеси трет-бутилата калия (9,5 г, 0,0847 моль) в сухом ДМЭ (50 мл). Смесь перемешивали в течение 1 часа, давая температуре повыситься до -20°С. Смесь выливали в Н2O (1800 мл) и экстрагировали СН2Сl2 (3×500 мл). Объединенные органические экстракты сушили над Na2SO4, фильтровали и упаривали в вакууме, получая твердое вещество. Растирание твердого вещества с ацетонитрилом давало чистое соединение 27 (6,0 г, выход 49%). 1H ЯМР (ДМСО-d6) δ 11,17 (1Н, с, NH), 8,38 (2Н, д, J=5,8 Гц), 7,27-6,92 (12Н, м), 2,61-2,51 (4Н, м), 1,93-1,83 (2Н, м).
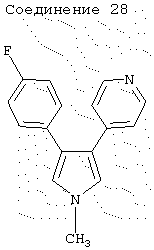
1-метил-3-(4-фторфенил)-4-(4-пиридил)пиррол
4-[(4-Фторфенил)этинил]пиридин (2,0 г, 0,0101 моль) и N-оксид триметиламина (1,0 г, 0,0133 моль) растворяли в сухом ТГФ (200 мл) и охлаждали до 0°С. Добавляли диизопропиламид лития (1,5 М в ТГФ, 14 мл) и перемешивали реакционную смесь при 0°С в течение 1 ч. Затем реакционную смесь гасили H2O (20 мл) и экстрагировали CH2Cl2 (2×100 мл). Органические экстракты сушили над Na2SO4 и упаривали в вакууме, получая масло. Очистка на SiO2 с элюированием EtOAc давала 0,356 г (выход 14%) соединения 28. 1H ЯМР (СDСl3) δ 8,41 (2Н, д, J=5,4 Гц), 7,19 (2Н, дд, J=5,7, 6,0 Гц), 7,11 (2Н, д, J=5,4 Гц), 6,99 (2Н, дд, J=8,7, 8,5 Гц), 6,87 (1Н, д J=2,1 Гц), 6,69 (1H, д, J=2,4 Гц), 3,71 (3Н, с).
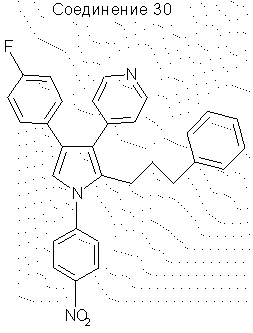
4-(4-фторфенил)-1-(4-нитрофенил)-2-(3-фенилпропил)-3-(4-пиридил)пиррол
Гидрид натрия (60%-ная суспензия в минеральном масле, 0,80 г, 0,0209 моль) промывали 3 раза гексаном и затем растворяли в ДМФА (15 мл). Затем порциями добавляли соединение 27 (6,0 г, 0,01683 моль) при 0°С при перемешивании. По завершении добавления реакционную смесь перемешивали при 0°С еще в течение 15 минут, после чего добавляли по каплям 4-фторнитробензол (2,4 г, 0,170 моль). Реакционную смесь перемешивали при 0°С в течение 1 ч перед тем, как дать температуре подняться до 25°С. Реакционную смесь выливали в 1,5 л воды и экстрагировали CH2Cl2 (3×500 мл). Объединенные органические экстракты промывали водой (4×500 мл) и сушили над Na2SO4. Упаривание в вакууме давало желтое твердое вещество, которое растирали с ацетонитрилом, фильтровали и сушили на воздухе, получая 6,6 г (выход 82,5%) чистого соединения 30. 1H ЯМР (CDCl3) δ 8,52 (2Н, д, J=5,8 Гц), 8,26 (2Н, д, J=8,9 Гц), 7,46 (2Н, д, J=8,9 Гц), 7,16-7,13 (3Н, м), 7,09-7,03 (4Н, м), 6,95-6,82 (5Н, м), 2,73-2,67 (2Н, м), 2,35-2,32 (2Н, м), 1,53-1,43 (2Н, м).
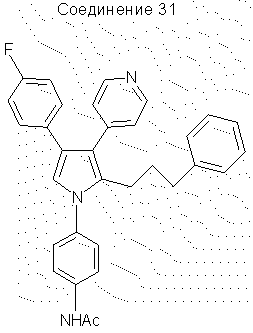
1-(4-ацетоаминофенил)-4-(4-фторфенил)-2-(3-фенилпропил)-3-(4-пиридил)пиррол
Соединение 32 (0,85 г, 0,0019 моль) перемешивали в течение 16 часов в уксусном ангидриде (20 мл) и воде (50 мл). Раствор экстрагировали этилацетатом (100 мл), промывали водой (3×50 мл), затем промывали насыщенным гидрокарбонатом натрия (3×50 мл) и затем снова промывали Н2О (2×50 мл). Органические вещества сушили над Na2SO4 и упаривали в вакууме, получая соединение 31 (0,89 г, выход 96%), выделенное в виде масла. 1H ЯМР (СОСl3) δ 8,41 (2Н, д, J=5,6 Гц), 8,05 (1Н, с), 7,73 (2Н, д, J=8,6 Гц), 7,28 (2Н, д, J=8,7 Гц), 7,17-7,04 (7Н, м), 6,91-6,85 (4Н, м), 6,79 (1Н, с), 2,66-2,61 (2Н, м), 2,35-2,30 (2Н, м), 2,22 (3Н, с), 1,59-1,51 (2Н, м).
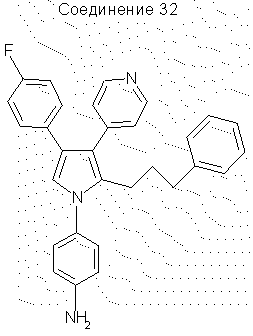
1-(4-аминофенил)-4-(4-фторфенил)-2-(3-фенилпропил)-3-(4-пиридил)пиррол
Соединение 30 (6,6 г, 0,0138 моль) суспендировали в этаноле (200 мл) и этилацетате (50 мл) и подвергали воздействию условий восстановления в течение 16 часов в гидрогенизаторе Парра при 50 фунт./кв. дюйм (3,515 кг/см2). Смесь фильтровали через целит и упаривали в вакууме, получая масло. Растирание данного масла с ацетонитрилом и фильтрование полученного твердого вещества давало 3,2 г соединения 32. Фильтрат упаривали в вакууме и очищали на SiO2 с элюированием 50%-ным этилацетатом в гексане, получая дополнительно 1,75 г продукта (общий выход 79%). 1H ЯМР (СDСl3) δ 8,44 (2Н, д, J=5,9 Гц), 7,22-6,71 (16Н, м), 3,85 (2Н, с), 2,63-2,57 (2Н, м), 2,38-2,33 (2Н, м), 1,61-1,49 (2Н, м).
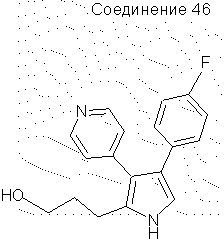
4-(4-фторфенил)-2-(3-гидроксипропил)-3-(4-пиридил)пиррол
Раствор 2-(3-бензилоксипропил)-4-(4-фторфенил)-3-(4-пиридил) пиррола (0,95 г, 0,0025 моль) в этаноле (125 мл), содержащий концентрированную НСl (0,2 мл), добавляли к Pd на угле (0,2 г). Данную смесь помещали в атмосферу водорода на 16 часов в гидрогенизаторе Парра при 50 фунт./кв. дюйм (3,515 кг/см2). Смесь фильтровали через целит и добавляли к полученному раствору триэтиламин (0,5 мл), после чего упаривали в вакууме, получая твердое вещество. Твердое вещество экстрагировали этилацетатом (100 мл) и промывали водой (3×50 мл). Органические фракции сушили над Na2SO4 и упаривали в вакууме, получая светло-желтое твердое вещество (0,7 г, выход 96%). 1H ЯМР (ДМСО-d6) δ 11,10 (1H, с, NH), 8,44 (2Н, д), 7,05 (6Н, м), 6,91 (1Н, д), 4,53 (1H, шир.с, ОН), 3,49 (2Н, шир.с), 2,64 (2Н, т), 1,72 (2Н, м).
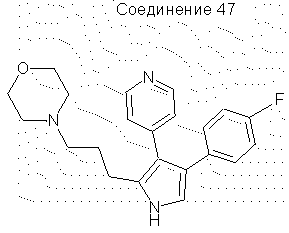
4-(4-фторфенил)-2-(3-морфолинопропил)-3-(4-пиридил)пиррол
4-(4-Фторфенил)-2-(3-мезилоксипропил)-3-(4-пиридил)пиррол (0,25 г, 0,0007 моль) нагревали с обратным холодильником в течение 16 часов в CH2Cl2 (50 мл) и морфолине (0,25 мл). Раствор охлаждали и разбавляли CH2Cl2 (~100 мл), затем промывали H2O (3×50 мл). Органические вещества сушили над Na2SO4 и упаривали в вакууме, получая масло. Данное масло очищали на SiO2, элюируя 10%-ным МеОН в CH2Cl2, получая 4-(4-фторфенил)-2-(3-морфолинопропил)-3-(4-пиридил)пиррол, выделенный в виде твердого вещества (0,088 г, выход 36%). 1H ЯМР (ДМСО-d6) δ 11,12 (1Н, с, NH), 8,42 (2Н, д), 7,05 (6Н, м), 6,95 (1Н, д), 3,55 (4Н, т), 2,62 (2Н, т), 2,29 (6Н, м), 1,71 (2Н, м).
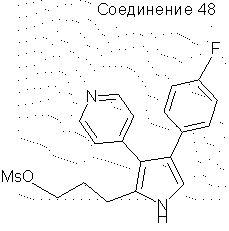
4-(4-фторфенил)-2-(3-мезилоксипропил)-3-(4-пиридил)пиррол
4-(4-Фторфенил)-2-(3-гидроксипропил)-3-(4-пиридил)пиррол (0,55 г, 0,0019 моль) объединяли с триэтиламином (0,52 мл, 0,0037 моль) в CH2Cl2 (50 мл) и охлаждали до 10°С. Добавляли по каплям метансульфонилхлорид (0,16 мл, 0,0020 моль) и давали полученой смеси подогреться до комнатной температуры. Данную смесь разбавляли CH2Cl2 (50 мл) и промывали водой (30 мл). Органические вещества сушили над Na2SO4 и упаривали в вакууме, получая масло. Данное масло растворяли в этилацетате и очищали на слое SiO2 (~ 20 мл), элюируя EtOAc. Упаривание растворителя в вакууме давало желтое твердое вещество (0,63 г, выход 91%). 1H ЯМР (ДМСО-d6) δ 11,25 (1Н, с, NH), 8,45 (2Н, д), 7,09 (6Н, м), 6,94 (1Н, д), 4,19 (2Н, т), 3,17 (3Н, с), 2,71 (2Н, т), 1,98 (2Н, м).
III. Биологические анализы и активность
А. Ингибирование р38 в ферментном анализе in vitro
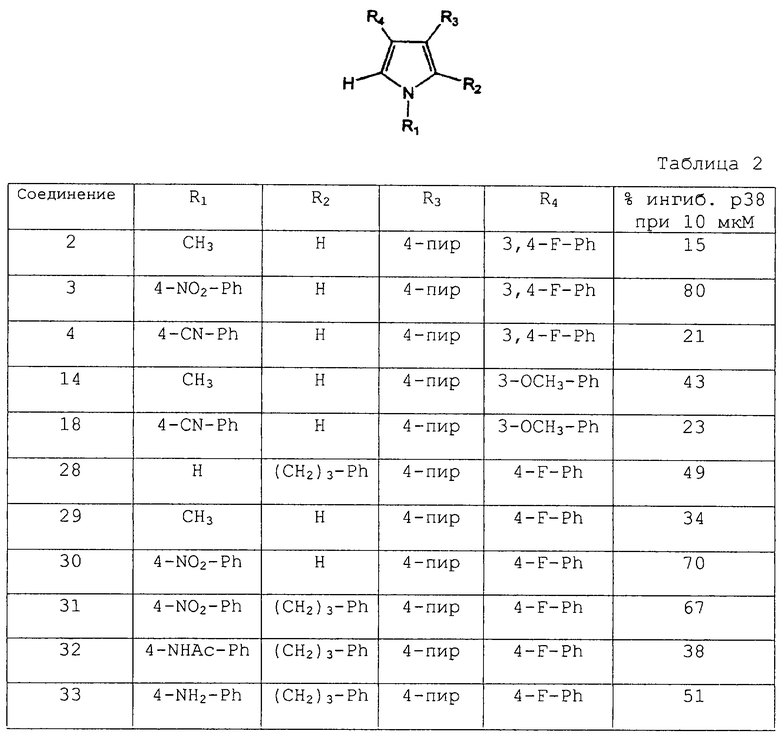
Раствор (38 мкл) очищенного рекомбинанта р38 (6xHis-p38, экспрессированного в E.coli), миелинового основного белкового субстрата (определенного эмпирически) и буфера с рН 7,5 (Hepes: 25 мМ, MgCl2: 10 мМ, MnCl2: 10 мМ) добавляли в 92 лунки 96-луночного круглодонного полипропиленового планшета. Количество фермента определяли эмпирически на основании линейного интервала анализа и приемлемого соотношения сигнала и шума. Оставшиеся лунки использовали для контроля (“КОНТР” CTRL) и фона (“ФОН” BKG). КОНТР готовили с использованием фермента, субстратного буфера и 2% ДМСО, а ФОН готовили при помощи субстратного буфера и 2% ДМСО.
Раствор (12 мкл) тестируемого соединения в ДМСО добавляли в испытываемые лунки. Соединения разбавляли до 125 мкМ смесью 10% ДМСО/Н2О и анализировали при 25 мкМ, когда конечная концентрация ДМСО составляла 2%. Во все лунки добавляли раствор АТР/33P АТР (10 мкл, содержащих 50 мкМ немеченого АТР и 1 мкCi 33P-АТР) и планшеты смешивали и инкубировали при 30°С в течение 30 мин. В каждую лунку добавляли охлажденную льдом 5%-ную ТХК/10 мМ фосфат натрия (60 мкл) и планшеты хранили на льду в течение 15 мин. Содержимое каждой лунки переносили в лунки 96-луночного фильтрпланшета (Millipore, MultiScreen-DP) и фильтрпланшет помещали в вакуумный коллектор, снабженный лотком для сбора отходов. Лунки пять раз промывали смесью 10% ТХК/10 мМ фосфата натрия (200 мкл) в вакууме. Добавляли сцинтиллятор MicroScint-20 и планшеты герметизировали с использованием листов Topseal-S и производили подсчет в сцинтилляционном счетчике Packard TopCount с использованием 33Р жидкостной программы с поправкой на гашение цвета, где выходной сигнал представляет удельное число импульсов в минуту (срm) с учетом поправки на гашение цвета. % Ингибирования каждого тестируемого соединения, приведенный в таблице 2, рассчитывали по следующей формуле: % ингибирования = [1-(образец - ФОН)/(КОНТР - ФОН)]×100.
Хотя соединения первоначально тестировали при 20 мкМ, для надежности соединения также тестировали при концентрациях в 4 раза больших и в 4 раза меньших, чем указанная концентрация. Кроме того, для некоторых соединений рассчитывали IС50 с использованием 4-параметрической программы получения кривой на Deltagraph.
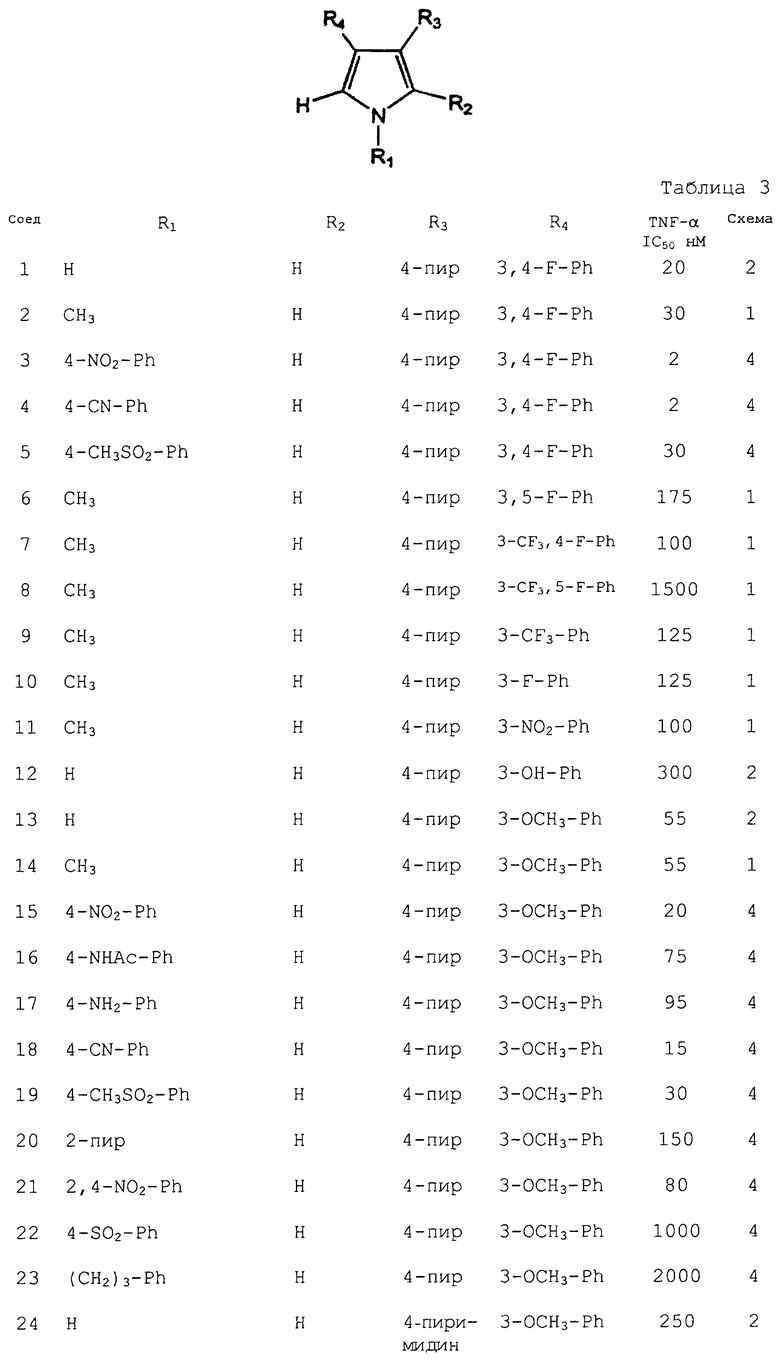
В. Полный клеточный анализ на ингибирование TNF-α in vitro
Свежеполученную венозную кровь антикоагулировали гепарином, разбавляли равным объемом фосфатно-солевого буферного раствора (“ФСБР”, PBS) и помещали в стерильную пробирку или другой резервуар. Аликвоты (30 мл) данной смеси переносили в центрифужные пробирки с внесенным в них Ficoll-Hypaque (15 мл). Подготовленные таким образом пробирки центрифугировали при 400×g без перерыва в течение 30 мин при комнатной температуре. Приблизительно от 1/2 до 2/3 тромбоцитарного слоя поверх слоя мононуклеарных клеток удаляли пипеткой. Большую часть данного слоя мононуклеарных клеток осторожно удаляли с помощью пипетки, и данные РВМС разбавляли ФСБР и центрифугировали при 600×g в течение 15 мин. Полученные РВМС промывали еще одной порцией ФСБР и центрифугировали при 400×g в течение 10 мин при комнатной температуре. Полученные осадки разбавляли питательной средой RPMI/1% FCS (эмбриональная, бычья сыворотка) с низким содержанием эндотоксина, и получали концентрацию клеток 0,5-2,0×106 РВМС/мл. Небольшой объем данной суспензии отбирали для подсчета на гемоцитометре, а оставшийся препарат центрифугировали при 200×g в течение 15 мин при комнатной температуре. Полученные осадки РВМС повторно суспендировали в RPMI/1% FCS до концентрации 1,67×106/мл.
Для проведения данного анализа суспензию РВМС (180 мкл) переносили в двойные лунки 96-луночного плоскодонного микротитровочного планшета и инкубировали в течение 1 ч при 37°С. Раствор тестируемого соединения (10 мкл: приготовленный в 20-кратной концентрации от требуемой конечной концентрации) добавляли в каждую лунку, и инкубировали планшет в течение 1 ч при 37°С. Добавляли раствор (10 мкл) LPS в RPMI/1% FCS (200 нг/мл), и инкубировали лунки в течение ночи при 37°С. Надосадочную жидкость (100 мкл) удаляли из каждой лунки и разбавляли RPMI/1% FCS (400 мкл). Образцы анализировали на TNF-α с использованием набора ELISA (твердофазный иммунноферментный анализ) (Genzyme).
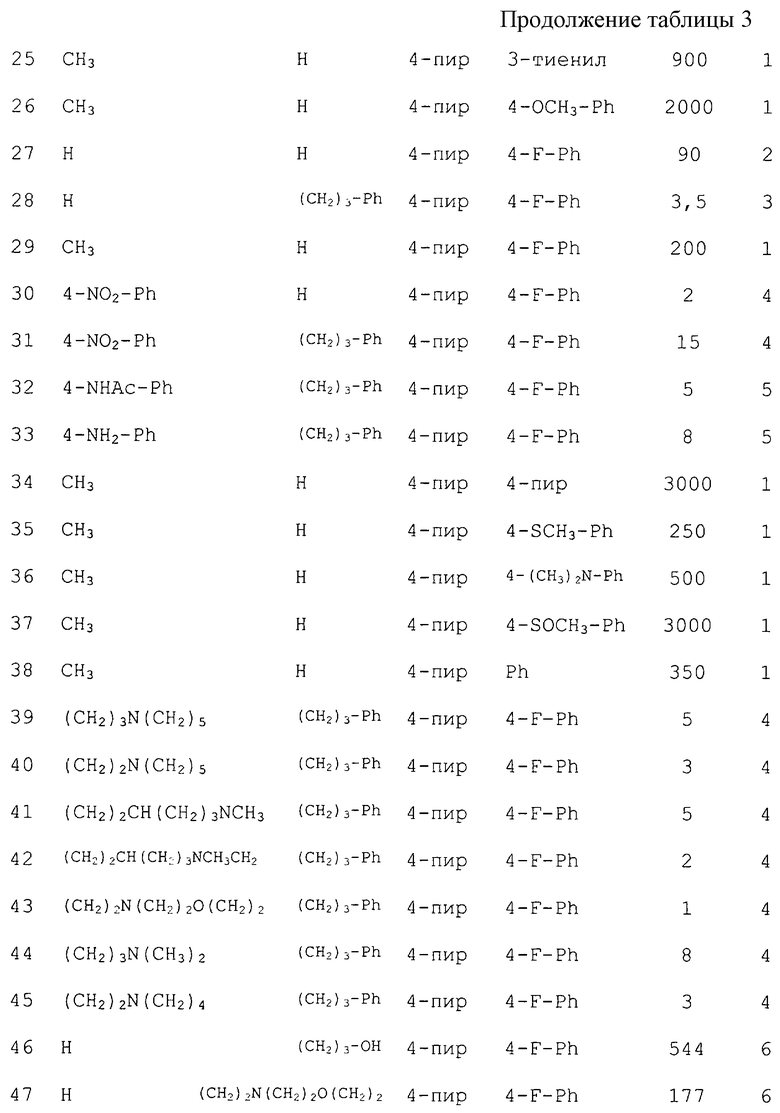
Некоторые соединения согласно изобретению перечислены в таблице 3. Данные соединения проверяли на их способность ингибировать продуцирование TNF-α. Для указанных соединений приведены результаты IC50, нМ.
С. Анализ ингибирования продуцирования TNF-α in vivo на грызунах
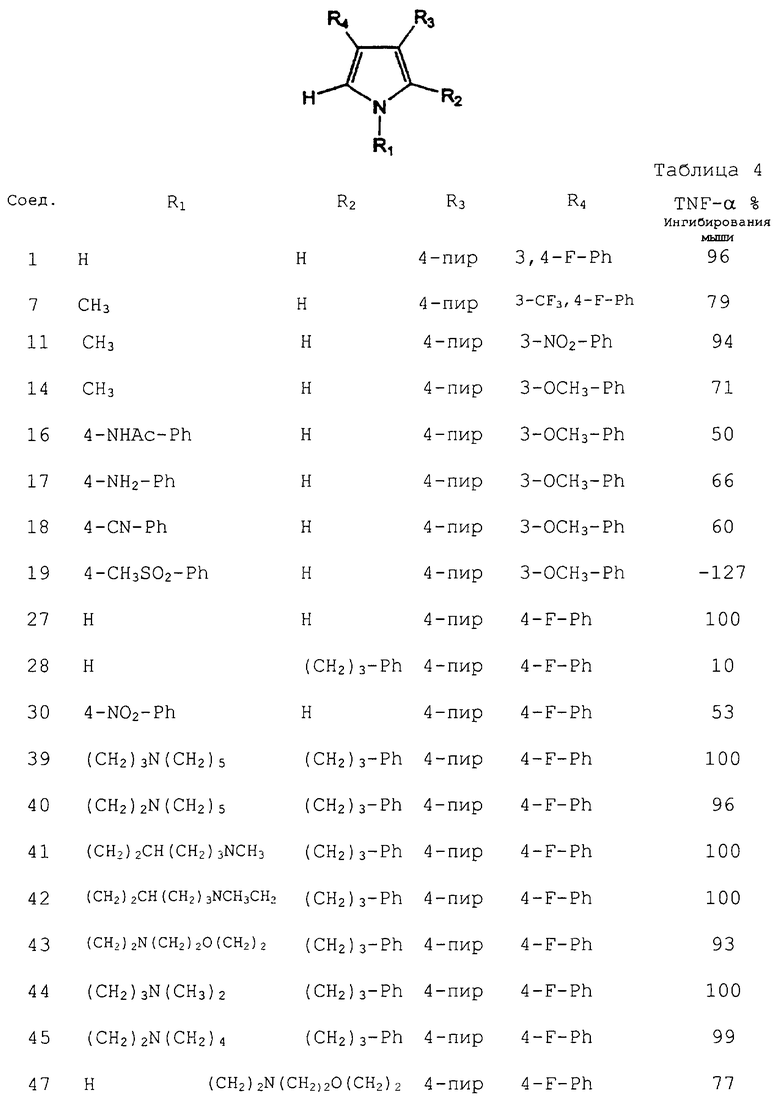
Способность настоящих соединений ингибировать вызванное LPS продуцирование TNF-α было показано следующими анализами in vivo на грызунах. Мышей (BALB/cJ самки, Jackson Laboratories), или крыс (самцы, Lewis, Charles River) подвергали голоданию в течение 30 мин перед введением пероральной дозы 5-10 мл/кг тестируемого соединения при 5-50 мг/кг. Через тридцать минут после дозировки животным делали внутрибрюшинную инъекцию LPS в расчете 1 мг/кг и возвращали их в клетки на 1 ч. Животных анестезировали CO2, обескровливали путем кардиальной пункции и собирали цельную кровь (0,1-0,7 мл). Крови давали свернуться, и сыворотку переносили в центрифужную пробирку. Данный образец центрифугировали, а сыворотку собирали, делили на аликвоты и замораживали при -80°С. Образцы тестировали при помощи промышленного набора ELISA на TNF-α (эндоген для TNF-α у мышей и биоресурс для TNF-α у крыс). Результаты теста in vivo для некоторых соединений согласно изобретению приведены в таблице 4. Данные соединения тестировали на их способность ингибировать продуцирование TNF-α у мышей, и данные приведены в виде % ингибирования при 25 мг/кг.
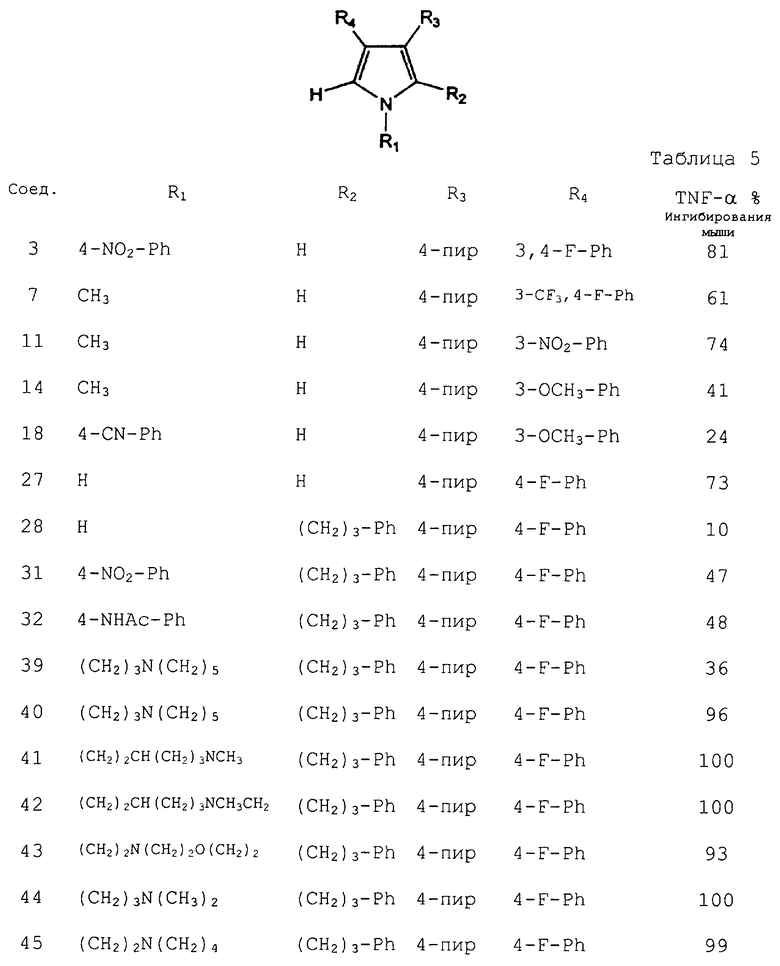
Результаты испытания in vivo для некоторых соединений согласно изобретению приведены в таблице 5. Соединения исследовали на их способность ингибировать продуцирование TNF-α у мышей, и данные приведены в виде % ингибирования при 10 мг/кг.
ССЫЛКИ
1. C. Dinarello, et al., Inflammatory Cytokines: Interleukin-1 and Tumor Necrosis Factor as Effector Molecules in Autoimmune Diseases. Curr. Opin. Immunol. 1991, 3, 941-48.
2. M.J. Elliot, et al., Treatment of Rheumatoid Arthritis with Chimeric Monoclonal Antibodies to Tumor Necrosis Factor α. Arthritis Rheum. 1993, 36, 1681-90.
3. J.C. Boehm, et al., 1-Substituted 4-Aryl-5-pyridinyl-imidazoles: A New Class of Cytokine Suppressive Drugs with Low 5-Lipoxygenase and Cyclooxygenase Inhibitory Potency, J. Med. Chem., 1996, 39, 3929-37.
4. International Publication No. WO 93/14081.
5. A.M. Badger, et al., Pharmacological Profile of SB 203580, A Selective Inhibitor of Cytokine Suppressive Binding Protein p38 Kinase, in Animal Models of Arthritis, Bone Resorption, Endotoxin Shock and Immune Function, The Journal of Pharmacology and Experimental Therapeutics, 1996, 279, 1453-61.
6. D. Griswold, et al., Pharmacology of Cytokine Suppressive Anti-Inflammatory Drug Binding Protein (CSBP), A Novel Stress-Induced Kinase, Pharmacology Communications, 1996, 7, 323-29.
7. U.S. Patent No.5776954.
8. International Publication No. WO 97/05877.
9. International Publication No. WO 97/05878.
10. Davis, Roger J., et al., Opposing effects of ERK and JNK-p38 MAP Kinases on Apoptosis, Science, 1995, 270 (5240), 1326-31.
11. Heidenreich, Kim A., et al., Inhibition of p38 Mitogen-Activated Protein Kinase by Insulin in Cultured Fetal Neurons. J. Biol. Chem., 1996, 271 (17), 9891-4.
12. Arvanitakis L., et al., G-Protein-Coupled Receptor of Kaposi's Sarcoma-Associated Herpesvirus is a Viral Oncogene and Angiogenesis Activator. Nature, 1998, 391 (6662), 86-89.
13. Pitha, Paula M., et al., Eariy Activation of Mitogen-Activated Protein Kinase Kinase, Extracellular Signal-Regulated Kinase, p38 Mitogen-Activated Protein Kinase, and c-Jun N-terminal Kinase in Response to Binding of Simian Immunodeficiency Virus to Jurkat Т Cells Expressing CCR5 Receptor, Virology, 1998, 252 (1), 210-217.
14. Bukrinsky, M., The Critical Role of p38 MAP Kinase in Т Cell HIV-1 Replication, Mol. Med., 1997, 3 (5), 339-346.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ КОНДЕНСИРОВАННЫЕ ПИРИДИНОВЫЕ ПРОИЗВОДНЫЕ, ПРИМЕНИМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ТИРОЗИНКИНАЗЫ с-MET | 2013 |
|
RU2619130C2 |
| ЗАМЕЩЕННЫЕ 2-АРИЛ-3-(ГЕТЕРОАРИЛ) ИМИДАЗО [1,2-А] ПИРИМИДИНЫ, СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И СВЯЗАННЫЕ С НИМИ СПОСОБЫ | 2000 |
|
RU2264403C2 |
| ПРОИЗВОДНЫЕ АМИДА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ЛЕЧЕНИЯ | 2000 |
|
RU2284187C2 |
| ПРОИЗВОДНЫЕ АМИНОПИРРОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ИХ ПРИМЕНЕНИЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ ПРОТИВОВОСПАЛИТЕЛЬНОЙ АКТИВНОСТЬЮ | 2002 |
|
RU2315045C2 |
| АЗАИНДОЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ФАКТОРА Xa | 2004 |
|
RU2330853C2 |
| ПРОИЗВОДНЫЕ 5-ПИРИДИЛ-1,3-АЗОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ИХ ПРОЛЕКАРСТВО, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ АНТАГОНИЗАЦИИ АДЕНОЗИНОВОГО А3-РЕЦЕПТОРА, СПОСОБ ИНГИБИРОВАНИЯ Р38 МАР-КИНАЗЫ, СПОСОБ ИНГИБИРОВАНИЯ ПРОДУЦИРОВАНИЯ TNF-АЛЬФА И СПОСОБ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ РЯДА ЗАБОЛЕВАНИЙ С ИХ ИСПОЛЬЗОВАНИЕМ | 2000 |
|
RU2237062C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2005 |
|
RU2390522C2 |
| ЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ИНДЕНО[ [1,2-с] ИЗОХИНОЛИНА, КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ НА ИХ ОСНОВЕ | 2002 |
|
RU2300523C2 |
| АЗАИНДОЛИЛПИРИДОНЫ И ДИАЗАИНДОЛИЛПИРИДОНЫ | 2018 |
|
RU2788659C2 |
| НОВЫЕ ГЕТЕРОЦИКЛЫ | 2009 |
|
RU2572616C2 |
Данное изобретение относится к новым замещенным 3-пиридил-4-арилпирролам формулы I
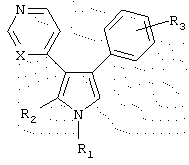
где R1 - Н, С1-5 алкила, С1-5 алкиламино, аминогруппы которого замещены диметилом, N-содержащего С1-5 алкилгетероцикла, выбранного из пиперидина, замещенного метилом, морфолина, пирролидина, тиазина, пиррола, замещенного метилом, фенила, фенила, замещенного одним или более С1-5 алкилом, амино, NHAc, нитро, нитрилом и сульфоном, и пиридина; R2 - Н, (СН2)3ОН, незамещенного С1-5 алкилфенила, и морфолина; R3 представляет один или более заместителей, независимо выбранных из группы, состоящей из водорода, галогена, метокси, трифторметила, гидрокси, диметиламино; и Х представляет либо С, либо N, или его фармацевтически приемлемая соль. Соединения I ингибируют продуцирование TNF-α, что позволяет использовать их в фармацевтической композиции для лечения противовоспалительных расстройств, опосредуемых ингибированием продуцирования TNF-α и/или активности р38. 4 н. и 15 з.п. ф-лы, 5 табл.
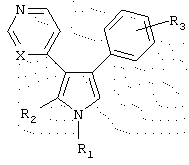
в которой R1 выбран из группы, состоящей из (i) водорода, (ii) C1-5алкила, (iii) C1-5алкиламино, аминогруппы которого замещены диметилом, (iv) N-содержащего C1-5алкилгетероцикла, выбранного из пиперидина, замещенного метилом, морфолина, пирролидина, тиазина, пиррола, замещенного метилом, (v) фенила, (vi) фенила, замещенного одним или более C1-5алкилом, амино, NHAc, нитро, нитрилом и сульфоном, и (vii) пиридина;
R2 выбран из группы, состоящей из (i) водорода, (ii) (СН2)3ОН, (iii) незамещенного C1-5 алкилфенила, и (iv) морфолина;
R3 представляет один или более заместителей, независимо выбранных из группы, состоящей из водорода, галогена, метокси, трифторметила, гидрокси, диметиламино;
Х представляет либо С, либо N,
или его фармацевтически приемлемая соль.
R2 выбран из группы, состоящей из водорода и (СH2)3 фенила;
R3 выбран из группы, состоящей из галогена и трифторметила;
Х представляет С.
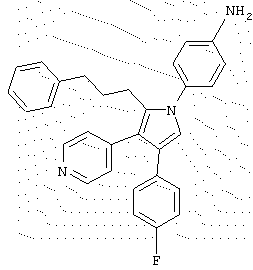
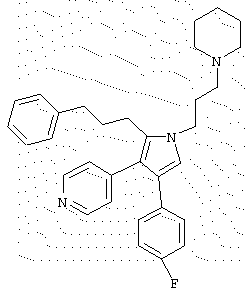
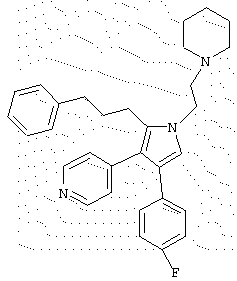
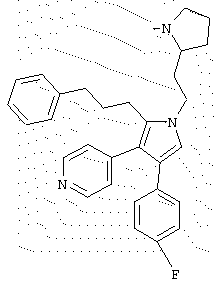
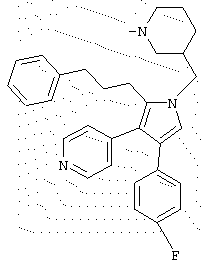
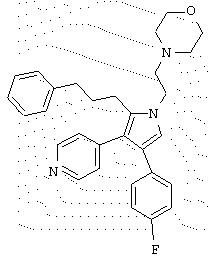
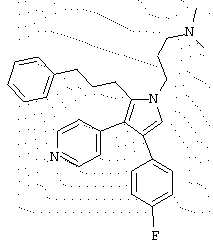
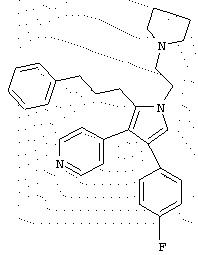
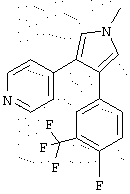
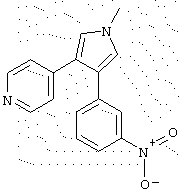
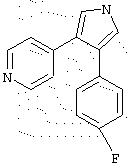
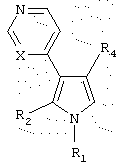
в которой R1 означает С1-5 алкил;
R2 означает водород;
R4 представляет гетероцикл, выбранный из пиридила или тиофена;
Х означает С,
или его фармацевтически приемлемая соль.
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |
| Способ получения производных пиридина или их солей | 1976 |
|
SU731898A3 |

