Изобретение относится к молекулярной биологии и генетической инженерии, конкретно к созданию синтетических полиэпитопных вакцин против ВИЧ-1.
Известные стратегии создания вакцины против ВИЧ-1 основаны на использовании различных форм вирусного антигена, включая инактивированный вирус, модифицированный вирус, аттенуированный живой вирус, нативные белки, генно-инженерные пептиды, а также рекомбинантные бактерии, вирусы и плазмидные ДНК [1]. Каждая из этих стратегий была оценена, однако проблемы создания анти-ВИЧ-1-вакцины до сих пор не решены.
Один из обещающих подходов к созданию новой генерации эффективных и безопасных вакцин основан на идентификации в вирусных белках Т- и В-клеточных эпитопов и создании на их основе синтетических полиэпитопных вакцин [2, 3]. Такие вакцины должны быть свободны от многих дефектов, которые свойственны вакцинам, создаваемым на основе живого аттенуированного и целого инактивированного вируса или на основе нативных вирусных белков. В частности, такие вакцины не должны содержать нежелательные эпитопы, которые индуцируют иммунопатологию или ингибируют протективный иммунитет. Поэтому генно-инженерные конструкции имеют потенциал, позволяющий улучшить иммунный ответ против ВИЧ относительно ответа, который индуцируется естественной ВИЧ-инфекцией.
Предложенный подход был использован для конструирования четырех-α-спирального искусственного белка TBI (Т and В Cell Epitopes Containing Immunogen), кандидата молекулярной полиэпитопной вакцины [2, 3]. Сконструированный белок TBI содержит четыре Т-клеточных эпитопа и пять В-клеточных нейтрализующих эпитопов из белков ВИЧ-1 Env и Gag. Мыши, иммунизированные синтетическим белком TBI, показали как гуморальный, так и клеточный (пролиферативный) ответы к ВИЧ-1. Анти-TBI-антитела обладали ВИЧ-нейтрализующей активностью.
Полученные результаты свидетельствуют о перспективности данного подхода. Вместе с этим предложенная конструкция имеет некоторые ограничения. Во-первых, она имеет сравнительно ограниченный антигенный потенциал (всего 9 эпитопов). Во-вторых, не показано, что данная конструкция обладает вирус-нейтрализующей активностью в отношении первичных изолятов. Действительно, было показано, что в случае ВИЧ-инфекции создание вакцины, индуцирующей нейтрализующие антитела, является весьма сложной задачей, поскольку такие вакцины оказались эффективны только против лабораторных штаммов ВИЧ и были не способны нейтрализовать первичные (полевые) вирусные изоляты [4]. При этом ускользание вируса от нейтрализации не было обусловлено возникновением escape-мутантов. Объяснение этого феномена стало возможным благодаря анализу кристаллической структуры белка gp120, который выявил ряд механизмов, посредством которых вирус предотвращает эффективную индукцию антител [5].
Поэтому в последнее время акцент многих вакцинных дизайнеров сместился в сторону индукции клеточного иммунитета. И это не случайно, так как появились убедительные доказательства того, что ответы цитотоксических Т-лимфоцитов (CD8+ CTL), ассоциированные с ВИЧ-инфекцией, являются важными медиаторами противовирусного иммунитета и, следовательно, ВИЧ-специфические CTL могут быть важным компонентом эффективной вакцины против ВИЧ-1 [6, 7]. В пользу этого свидетельствуют данные о том, что в плазме ВИЧ-инфицированных индивидуумов обнаружена достоверная обратная корреляция между частотой ВИЧ-специфических CTL и нагрузкой вирусной РНК [8]. Предполагается, что CTL могут защищать от ВИЧ-инфекции, так как CTL убивают ВИЧ-инфицированные клетки до того, как они нарабатывают новые вирионы [9], и, кроме того, CTL освобождают хемокины, которые ингибируют ВИЧ-инфекцию [10, 11]. Поэтому основные усилия направлены на разработку вакцины, индуцирующей CTL-ответы.
В настоящее время известно несколько искусственных вакцинных конструкций, содержащих множественные CTL-эпитопы ВИЧ-1. В частности, описана полиэпитопная конструкция, содержащая семь смежных минимальных HLА-А2-рестриктированных CTL-эпитопов [12]. HLA-A2-трансгенные мыши, иммунизированные рекомбинантным вирусом осповакцины, кодирующим CTL-полиэпитоп, генерируют CTL-специфические ответы на каждый включенный эпитоп. Кроме того, эпитоп-специфические линии CTL, полученные от ВИЧ-1-инфицированных индивидуумов, опознают каждый эпитоп внутри полиэпитопной конструкции. Эти данные иллюстрируют пригодность предложенного подхода для конструирования ВИЧ-вакцины.
Более сложная вакцинная конструкция, содержащая 20 перекрывающихся CTL-эпитопов человека из белков ВИЧ-1 Gag, Pol, Env и Nef, рестриктированных двенадцатью HLA молекулами I класса, была предложена Hanke et al. [13, прототип]. В данную конструкцию были включены также один мышиный и три обезьяньих эпитопа для тестирования иммуногенности, а также выбора оптимальных доз и режимов вакцинации на экспериментальных животных. Для доставки и экспрессии полученной вакцинной конструкции использовались ДНК-плазмида и модифицированный вирус Анкара (MVA). Полученные вакцинные кандидаты индуцировали CD8+ CTL и продукцию IFN-y после однократной иммунизации мышей [13, 14]. Кроме того, было показано, что комбинированный режим вакцинации, при котором мыши были сначала прамированы ДНК-вакциной, а затем бустированы MVA, был наиболее оптимальным (эффективным) протоколом для индукции как IFN-γ, так и CD8+ CTL [15]. Комбинированный режим вакцинации (ДНК-MVA) оказался также эффективньм при иммунизации обезьян Macaque rhesus, которые показали высокую частоту циркулирующих CTL, сравнимую с той, которая наблюдалась у SIV-инфицированных обезьян [16, 17]. Эти результаты позволяют предположить, что подход, основанный на разработке поли-СTL-эпитопных ДНК и MVA вакцин, может оказаться эффективньм для индукции высокого уровня Т-клеточного иммунитета у человека.
Вместе с этим эффективная вакцина кроме индукции высокого уровня ответов цитотоксических Т-лимфоцитов на отобранные эпитопы должна также обеспечить иммунный ответ против достаточно широкого спектра CTL-эпитопов. Рассмотренный выше прототип поли-СTL-эпитопной ДНК вакцины содержит 20 CTL-эпитопов ВИЧ-1, что составляет не более 10% от всех идентифицированных в настоящее время CTL-эпитопов (см. Los Alamos HIV Molecular Immunology Database), что не может гарантировать высокую эффективность этой вакцины.
Исходя из этого технической задачей изобретения является создание (дизайн и конструирование) нового искусственного поли CTL-эпитопного Т-клеточного иммуногена, содержащего CTL-эпитопы как можно большего числа основных антигенов ВИЧ-1, для использования в качестве эффективной и безопасной ДНК-вакцины против ВИЧ-1.
Поставленная задача решается путем разработки стратегии выбора эпитопов, вовлеченных в индукцию ВИЧ-1 -специфичных CTL ответов у инфицированных индивидуумов.
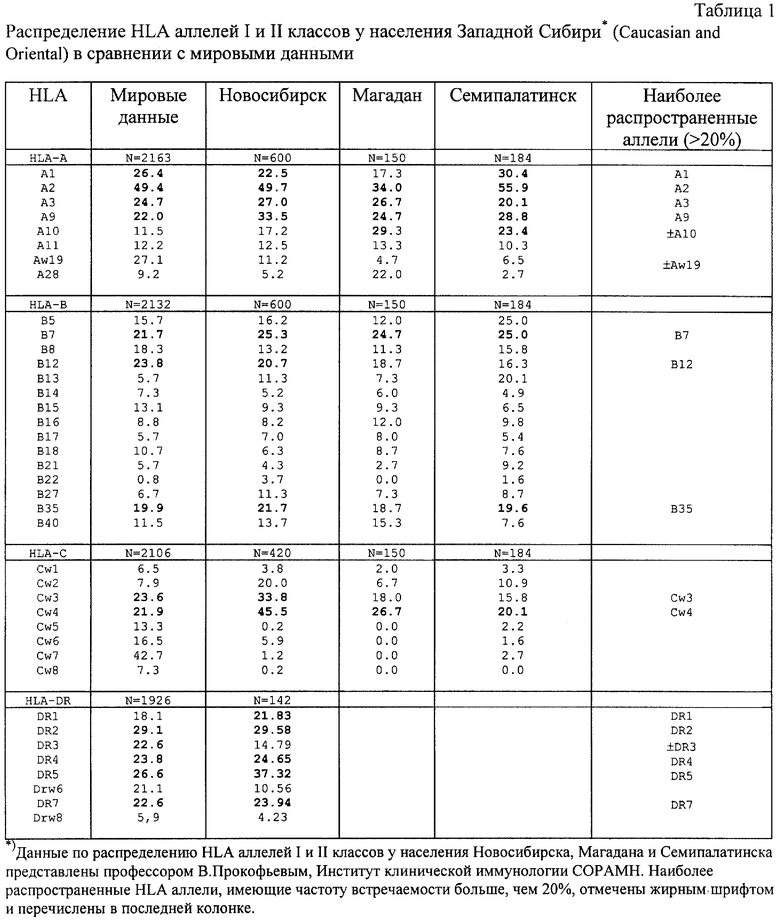
Известно, что вирусная инфекция индуцирует CD8+ CTL ответы путем представления вирусных антигенов совместно с молекулами МНС I класса (Major Hisrocopatibility Complex) на поверхности инфицированных клеток. При этом вирусные антигены распознаются специфическими Т-лимфоцитами не как полноразмерные белки, а как короткие пептиды (8-12 а.к.), ассоциированные со специфическими аллелями МНС I класса [18, 19]. Эти короткие антигенные эпитопы появляются из вирусных цитоплазматических белков в результате протеасом-опосредуемого процессинга [20, 21]. Так как распределение HLA аллелей варьирует для популяций различных географических регионов, то эффективная вакцина против СПИДа должна содержать эпитопы, рестриктированные различными HLA-аллелями, чтобы покрыть генетическое разнообразие молекул главного комплекса гистосовместимости (МНС) I класса для населения конкретного географического региона.
Эффективные полиэпитопные CTL-вакцины, кроме определенной HLA-специфичности, должны содержать также большое число консервативных эпитопов, вовлеченных в индукцию как CD8+ CTL и CD4+ Th-лимфоцитов. В настоящее время уже идентифицированно достаточно много таких эпитопов. Их аминокислотные последовательности суммированы в Los Alamos HI V Molecular Immunology Database. Для конструирования CTL-иммуногена, кандидата в ДНК-вакцину, были выбраны те из них, которые удовлетворяют следующим критериям:
1. Были выбраны эпитопы, представленные в Los Alamos HIV Molecular Immunology Database, которые индуцируют как CD8+ CTL, так и CD4+ Th и являются высоко консервативными среди подтипов А, В и С ВИЧ-1, циркулирующих на территории России, США и Западной Европы.
2. Эпитопы выбирались из основных вирусных белков-антигенов Env, Gag, Pol и Nef, которые индуцируют CTL ответы, являющиеся важными компонентами защиты от ВИЧ.
3. В тех случаях, когда было известно, в рассмотрение принимались эпитопы, которые индуцируют CTL, опознающие соответствующие естественно процессируемые эпитопы.
4. Выбирались, по-возможности, так называемые оптимальные эпитопы, которые в исследованиях по титрованию перекрывающихся усеченных пептидов определены, как минимальные эпитопы, вызывающие наиболее эффективную сенсибилизацию CTLs, специфичных к определенным молекулам МНС I класса.
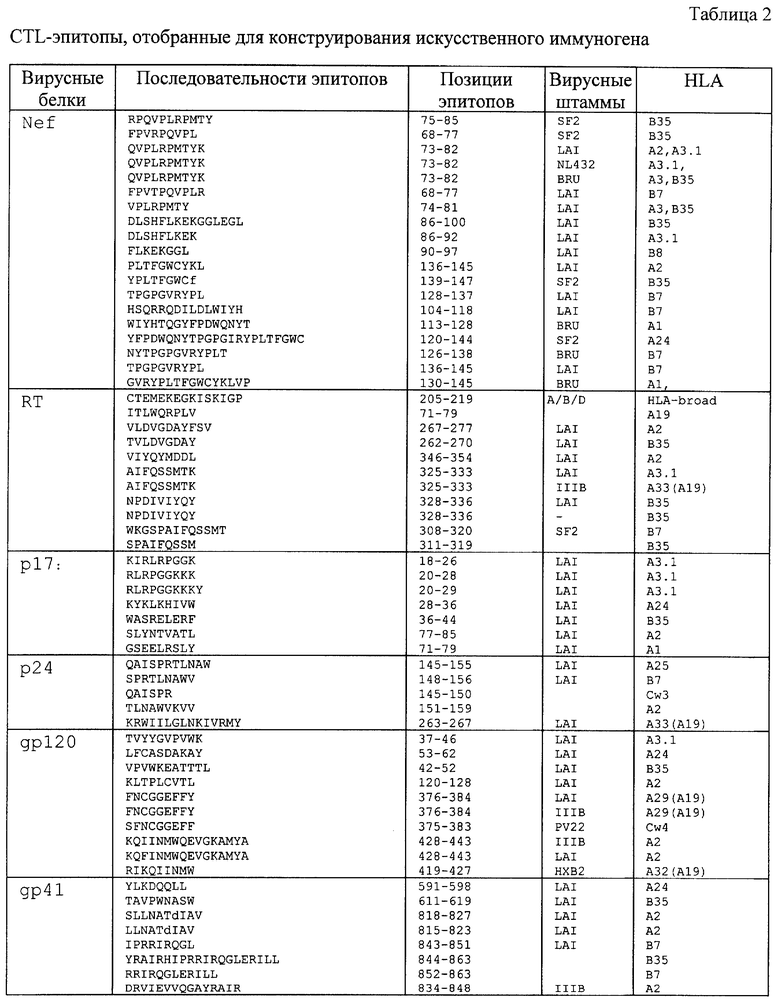
5. Учитывались CD8+ CTL эпитопы, которые в совокупности рестриктированы десятью различными оптимально подобранными аллелями HLA I класса. Как известно, этого достаточно, чтобы покрыть генетическое разнообразие антигенов МНС I класса в популяции практически любого географического района [13, 22, 23]. При этом выбирались эпитопы, специфичные к HLA аллелям, которые наиболее распространены в популяции России. К таким отнесены те антигены HLA I и II классов, которые встречаются в 20% и более случаев, а именно А1, А2, A3, А9, А10, Aw19, В7, В12, В35, Cw3, Cw4, DR1, DR2, DR3, DR4, DR5, DR7 (см. табл. 1).
В результате проведенного анализа для конструирования CTL-иммуногена, кандидата в ДНК-вакцину, были выбраны эпитопы, которые удовлетворяют перечисленным выше критериям. Такие эпитопы перечислены в табл.2 и 3. Необходимо отметить, что некоторые из перечисленных в табл.2 эпитопов способны связываться с дополнительными HLA молекулами, которые первоначально не были приняты к рассмотрению, так как их аллели имеют низкую частоту встречаемости в популяции. Можно предположить, что такие эпитопы, узнаваемые несколькими HLA аллелями, могут повысить потенциал создаваемой на их основе вакцины.
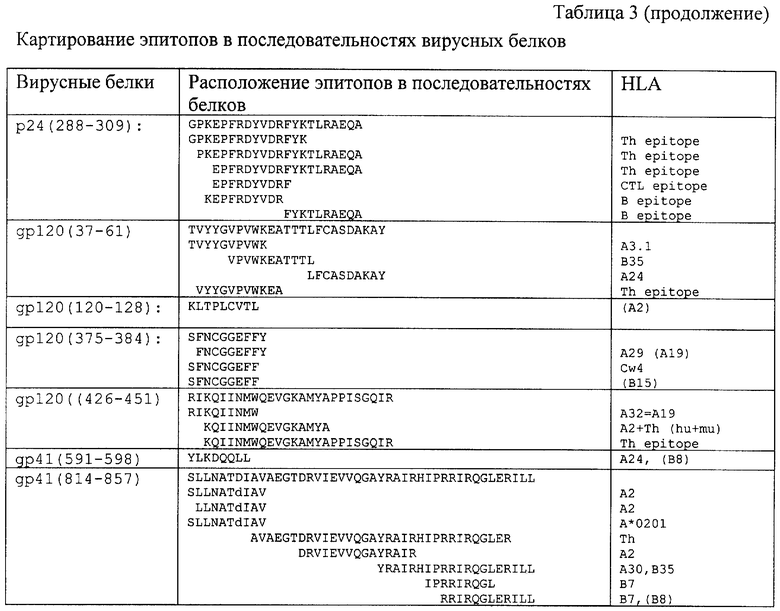
В качестве стратегии включения (объединения) множества отобранных из разных вирусных белков эпитопов в одну полипептидную цепь был выбран подход, основанный на объединении эпитопов как перекрывающихся пептидов. В данном случае перекрывающиеся эпитопы располагаются друг относительно друга также как в нативных белках. При этом перекрывание эпитопов позволяет минимизировать общую длину иммуногена. Оказалось, что в рассматриваемом случае большинство выбранных эпитопов (табл. 2) картируется в последовательностях нативных вирусных белков как частично или полностью перекрывающиеся пептиды и локализованы в нескольких непрерывных областях (табл. 3). Такие антиген-активные районы, содержащие перекрывающиеся эпитопы, были идентифицированы и использовались для дизайна поли-эпитопной ДНК-вакцины.
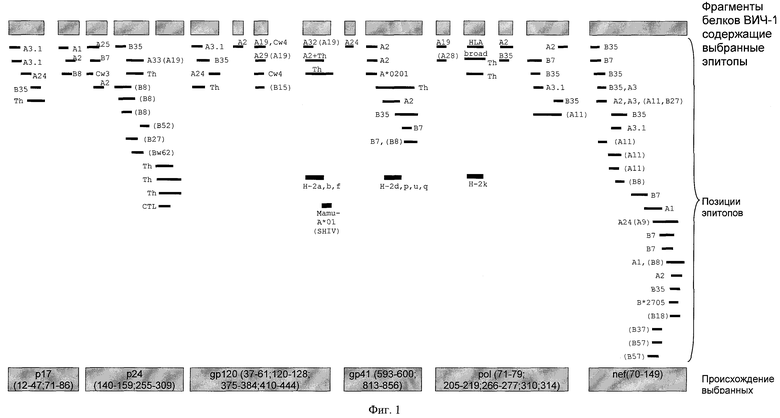
Таким образом, в предложенной разработке проведен дизайн искусственного Т-клеточного иммуногена, кандидата для использования в качестве ДНК-вакцины против ВИЧ-1. В дальнейшем для обозначения этого иммуногена будет использоваться аббревиатура TCI (T Cell Imunogen). Общий дизайн TCI-иммуногена представлен на фиг 1. Длина искусственного белка составляет 392 аминокислотных остатка (последовательность приведена на фиг.2). Сконструированный белок содержит около восьмидесяти эпитопов (как CD8+ CTL, так и CD4+ Th), многие из которых перекрываются и в совокупности рестриктированы десятью различными HLA-аллелями. Чтобы исследовать CTL-ответы, индуцируемые ДНК-вакциной на экспериментальных животных, в состав целевого иммуногена включены дополнительные эпитопы, представляемые молекулами МНС I класса мышей и обезьян Macaque rhesus.
Поставленная задача решается также путем конструирования искусственного гена, кодирующего TCI-иммуноген.
Отметим, что последовательность гена, кодирующая белок TCI (392 а.к.) является достаточно протяженной и составляет 1176 нуклеотидов (нуклеотидная последовательность приведена на фиг.3). Поэтому полный химический синтез гена TCI является достаточно трудоемкой задачей. Альтернативный и более экономичный подход к синтезу гена TCI может быть основан на использовании метода полимеразной цепной реакции (ПЦР) для амплификации фрагментов генома ВИЧ, содержащих непрерывные районы белка TCI. Действительно, как уже отмечалось, CTL-иммуноген содержит фрагменты, которые соответствуют достаточно протяженным и непрерывным аминокислотным последовательностям нативных вирусных белков. Именно поэтому идея синтеза целевого гена TCI методом ПЦР является достаточно привлекательной, так как позволяет использовать для амплификации фрагментов целевого гена нативные последовательности генома ВИЧ-1. Наиболее подходящим кандидатом для этой цели оказалась последовательность генома штамма ВН-10 ВИЧ-1 [24], так как соответствующие аминокислотные последовательности у вирусных белков и TCI полностью совпадают. Обнаруженное совпадение позволяет использовать для амплификации фрагментов целевого гена TCI везде, где возможно, последовательность геномной кДНК ВИЧ-1 (штамм ВН-10).
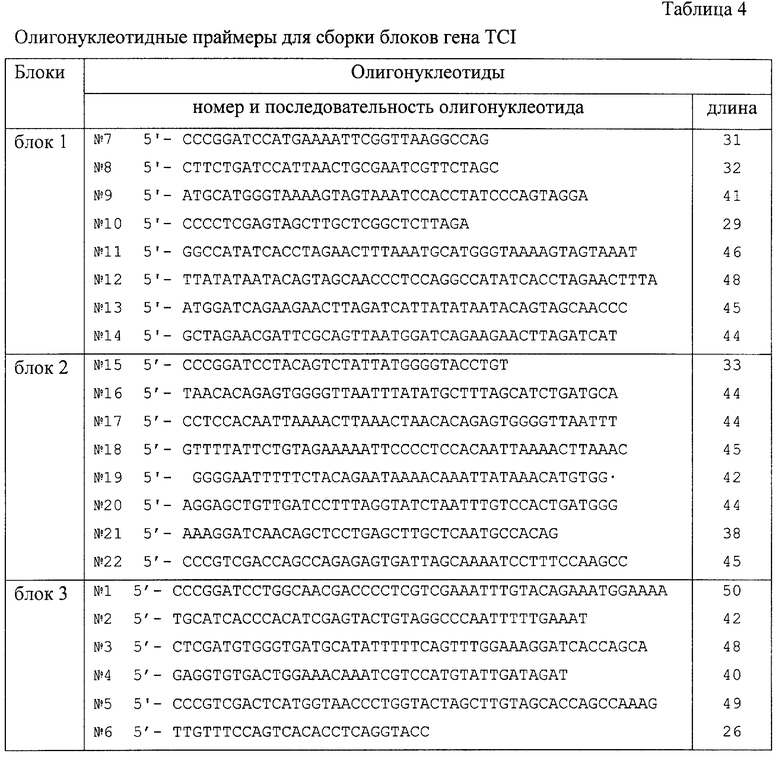
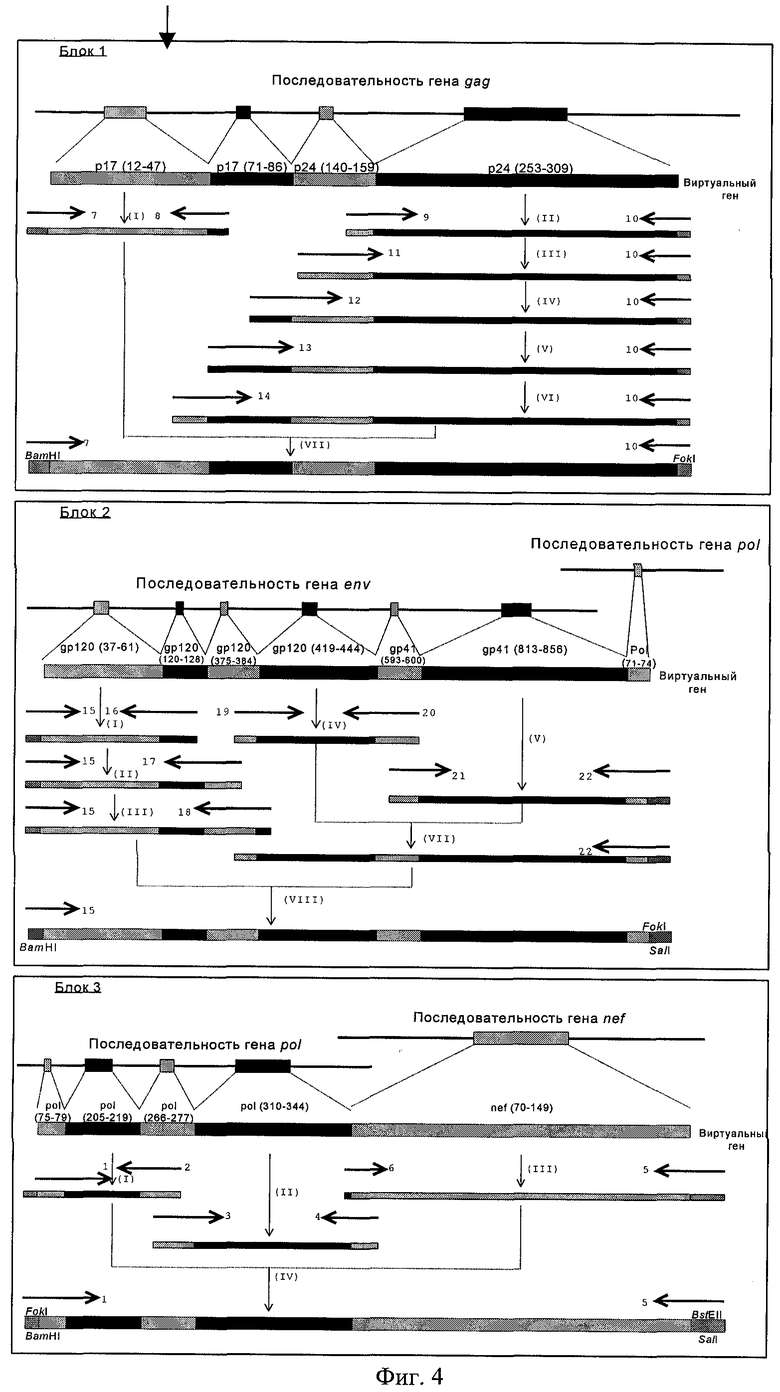
В соответствии с общим дизайном TCI иммуногена (фиг.1) последовательность гена, кодирующая белок TCI, была разбита на три блока (фиг.4). Блок 1 содержит фрагменты, кодирующие CTL-эпитопы гена gag, блок 2 - эпитопы гена env и блок 3 - эпитопы генов pol и nef. Была разработана уникальная схема блочной амплификации гена TCI, согласно которой фрагменты целевого гена синтезируются с использованием набора перекрывающихся праймеров для безматричного синтеза участков целевого гена, классических пар праймеров, позволяющих получать фрагменты гена на матрице - геномной кДНК и термофильных полимераз. Требуемые праймеры были рассчитаны синтезированы (табл. 4), после чего фрагменты (блоки 1, 2 и 3) были получены согласно предложенной схеме (фиг.4).
Для клонирования блоков 1-3 гена TCI в клетках E.coli использовалась ранее разработанная векторная плазмида pFH123 [25]. Эта плазмида несет специфический полилинкер, содержащий сайты для SII-эндонуклеаз рестрикции, позволяющие получать клонированные фрагменты ДНК с запланированными уникальными липкими концами. Клонирование блока 1 в плазмиду pFH123 осуществлялось по сайтам ВаmHI и XhoI/SalI, a блоков 2 и 3 - по сайтам BamHI, SalI. Использование плазмиды pFH123 для клонирования фрагментов целевого иммуногена обеспечивает однозначную сборку полноразмерного гена TCI. С этой целью блоки целевого гена были выделены из рекомбинантных плазмид pFH123 (блок 1 по сайтам рестрикции BamHI и FokI, блок 2 по сайтам рестрикции FokI и блок 3 по сайтам рестрикции FokI и SalI). Чтобы обеспечить однозначное “вырезание” клонированных блоков, в праймеры (табл. 4) были заложены синонимичные замены нуклеотидов, приводящие к исчезновению сайтов рестрикции ВаmН, и SalI I и FokI. После выделения блоки 1-3 и SalI были одновременно клонированы в векторной плазмиде pFH123 по участкам рестрикции ВаmHI и SalI, в результате чего была получена рекомбинантная плазмида pFH-TCI.
Рекомбинантные плазмиды, содержащие блоки и полную последовательность целевого гена, были отобраны стандартными процедурами скрининга, используя ПЦР и рестрикционный анализ. Структура всех клонированных последовательностей подтверждалась секвенированием. Исправление мутаций, введенных Taq-полимеразой, осуществлялось на основе ступенчатой амплификации последовательности гена TCI с использованием корректирующих олигонуклеотидов-праймеров. Таким образом, предложенная схема позволила синтезировать целевой ген, соответствующий заданной структуре, направляющий синтез синтетического рекомбинантного иммуногена TCI.
Отметим, что технология блочной сборки имеет ряд преимуществ:
1) независимый синтез и клонирование трех разных фрагментов гена TCI фактически приводит к трем разным CTL-иммуногенам, иммуногенные и протективные свойства которых могут быть исследованы индивидуально;
2) комбинации блоков позволяют достаточно быстро синтезировать целый набор новых генов (в том числе и целевой ген TCI);
3) введение в одну из исходных последовательностей специальных сайтов позволяет получать новые конструкции генов путем добавления последовательностей, кодирующих новые эпитопы.
Чтобы доказать, что искусственный белок TCI является иммуноактивным в отношении ВИЧ-1-позитивных сывороток и тем самым подтвердить, что целевой ген экспрессирует эпитопы ВИЧ-1, генно-инженерная конструкция, кодирующая полный набор эпитопов, была клонирована в клетках E.coli в составе серии векторных экспрессионных плазмид. Продукция рекомбинантного белка TCI составила 10-20% от общего клеточного белка. Присутствие фрагментов белков ВИЧ-1 в структуре целевого иммуногена подтверждено с помощью ИФА с использованием панелей ВИЧ-1-позитивных сывороток и моноклональных антител к белку р24.
Таким образом, полученные данные подтверждают, что по иммунохимическим свойствам продукт экспрессии гена TCI соответствует целевому иммуногену TCI.
Перечень графических материалов
Фиг.1. Общий дизайн TCI-иммуногена, кандидата в вакцину против ВИЧ-1. Прямоугольниками обозначены последовательности белков ВИЧ-1, содержащие выбранные эпитопы. Позиции индивидуальных эпитопов обозначены линиями. Ограничения эпитопов по антигенам главного комплекса гистосовместимости человека, мышей и обезьян отмечены соответствующими буквами (HLA-A, В, Cw - human, H-2a, b, d, f, k, p, u, q - mouse, Mamu - A*01 - Macaca mulatta). Th - хелперные эпитопы.
Фиг.2. Аминокислотная последовательность искусственного белка-иммуногена TCI.
Фиг.3. Нуклеотидная последовательность искусственного гена TCI.
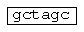 - сайт рестрикции NheI;
- сайт рестрикции NheI; 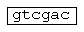 - сайт рестрикции SalI;
- сайт рестрикции SalI;  - сайт рестрикции BstEII (PspEI);
- сайт рестрикции BstEII (PspEI);  - последовательность Kozak;
- последовательность Kozak; 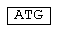 - инициирующий кодон;
- инициирующий кодон;  - стоп-кодон.
- стоп-кодон.
Фиг.4. Схема синтеза и сборки гена TCI.
Этапы ПЦР (вертикальные стрелки) пронумерованы арабскими цифрами. Олигонуклеотиды-праймеры (горизонтальные стрелки) пронумерованы римскими цифрами. Чередующиеся черные и серые прямоугольники внутри блоков гена TCI соответствуют последовательностям генома ВИЧ-1, кодирующим непрерывные районы белка TCI. В олигонуклеотиды 1, 5, 7, 10, 15, 22 заложены сайты гидролиза рестриктаз для клонирования в векторе pFH, а в олигонуклеотиды 2, 3, 5 введены “молчащие” мутации, приводящие к исчезновению сайта FokI. Кроме того, в олигонуклеотид 7 введен инициирующий кодон ATG, а в олигонуклеотид 5 - стоп-кодон.
Для лучшего понимания сущности предлагаемого изобретения ниже следуют примеры его осуществления.
Пример 1. Конструирование (синтез) искусственного гена TCI
Перечень всех олигонуклеотидных праймеров для сборки блоков гена TCI представлен в табл.4. Предварительно были приготовлены растворы всех олигонуклеотидных праймеров в концентрации 10 пмоль/мкл, т.е. 10 мМ, раствор смешанных четырех дезоксинуклеотидтрифосфатов (dNTP) в концентрации 5 мМ каждого, а также кДНК ВИЧ-1 в концентрации 100 нг/мкл (далее кДНК). На всех этапах использовали термофильную Tte-ДНК-полимеразу с активностью 10 ед. акт./мкл (далее полимераза) и буфер для Tte-ДНК-полимеразы х10 (далее буфер). На всех этапах использовали дистиллированную деионизованную стерильную воду (далее вода).
1.1. Получение блока 1 гена TCI
Были приготовлены реакционные смеси для проведения ПЦР следующего состава:
Вода - 70 мкл; буфер - 10 мкл; кДНК - 1 мкл; dNTP - 16 мкл; праймер 7 - 1 мкл; праймер 8 - 1 мкл; полимераза - 1 мкл. Смесь для получения фрагмента А.
Вода - 70 мкл; буфер - 10 мкл; кДНК - 1 мкл; dNTP - 16 мкл; праймер 9 - 1 мкл; праймер 10 - 1 мкл; полимераза - 1 мкл. Смесь для получения фрагмента В.
Условия проведения реакций:
92°С - 0,8 мин; 48°С - 0,6 мин; 70°С - 0,5 мин - 2 цикла;
92°С - 0,6 мин; 52°С - 0,6 мин; 70°С - 0,2 мин - 13 циклов;
92°С - 1,2 мин; 56°С - 1,2 мин; 70°С - 1,2 мин - 1 цикл.
По окончании реакции в каждую реакционную смесь добавляли по 100 мкл хлороформа и 100 мкл фенола, интенсивно встряхивали и центрифугировали на центрифуге типа Eppendorf (12000 g) в течение 1 мин. Водную фазу переносили в другую пробирку, добавляли 10 мкл ацетата калия 3 М и 250 мкл этанола, перемешивали, выдерживали 1 ч при - 20°С и ДНК осаждали центрифугированием, как указано выше. Осадок промывали этанолом, центрифугировали и высушивали при комнатной температуре в течение 20 мин. ДНК-фрагменты разделяли в 6% полиакриламидном геле (ПААГ), полоски геля, содержащие ПЦР-продукты расчетной длины, промывали в воде при комнатной температуре 1 час, затем тщательно измельчали, добавляли 500 мкл воды и выдерживали при 37°С в течение 16 ч. Суспензию геля центрифугировали при 12000 g в течение 3 мин и верхнюю фазу, содержащую ПЦР-фрагменты А и В, осторожно переносили в другую пробирку.
Для получения ПЦР фрагмента С была приготовлена реакционная смесь следующего состава:
вода - 57 мкл; буфер - 10 мкл; dNTP - 16 мкл; фрагмент В - 5 мкл; праймер 10 - 6 мкл; праймер 11 - 1 мкл; праймер 12 - 1 мкл; праймер 13 - 1 мкл; праймер 14 - 2 мкл; полимераза - 1 мкл.
Условия проведения реакции:
92°С - 1,2 мин; 45°С - 0,6 мин; 70°С - 0,5 мин - 2 цикла;
92°С - 0,6 мин; 50°С - 0,6 мин; 70°С - 0,3 мин - 22 цикла;
92°С - 1,2 мин; 52°С - 1,2 мин; 70°С - 1,2 мин - 1 цикл.
ПЦР - фрагмент С выделяли из реакционной смеси по схеме, описанной выше для ПЦР-фрагмента А.
Для ПЦР сборки блока 1 гена TCI смешивали:
вода - 62 мкл; буфер - 10 мкл; dNTP - 16 мкл; фрагмент А - 5 мкл; фрагмент С - 5 мкл; полимераза - 1 мкл.
Условия проведения реакции:
92°С - 1,2 мин; 45°С - 1,5 мин; 50°С - 1,5 мин; 70°С - 1,5 мин - 1 цикл; затем в реакционную смесь добавляли праймер 7 и праймер 10 по 1 мкл и проводили ПЦР в следующих условиях:
92°С - 0,8 мин; 50°С - 0,8 мин; 70°С - 0,6 мин - 19 циклов;
92°С - 1,3 мин; 52°С - 1,2 мин; 70°С - 1,5 мин - 1 цикл.
Целевой ПЦР-фрагмент обрабатывали смесью фенол-хлороформ, осаждали и высушивали, как описано выше, затем гидролизовали рестриктазами BamHI и XhoI в 100 мкл реакционной смеси при 37°С в течение 16 ч, осаждали и разделяли электрофорезом в 4% ПААГ аналогично вышеописанному. После элюции фрагмента ДНК из геля материал осаждали, промывали и высушивали, как описано выше. ДНК растворяли в 20 мкл ТЕ-буфера до конечной концентрации 0,1 пмоль/мкл. Хранили при -20°С.
1.2. Получение блока 2 гена TCI
Для ПЦР - реакции с праймерами II блока смешивали:
Вода - 70 мкл; буфер - 10 мкл; кДНК - 1 мкл; dNTP - 16 мкл; праймер 15 - 1 мкл; праймер 16 - 1 мкл; полимераза - 1 мкл. Смесь для получения фрагмента D.
Вода - 70 мкл; буфер - 10 мкл; кДНК - 1 мкл; dNTP - 16 мкл; праймер 19 - 1 мкл; праймер 20 - 1 мкл; полимераза - 1 мкл. Смесь для получения фрагмента Е.
Вода - 70 мкл; буфер - 10 мкл; кДНК - 1 мкл; dNTP - 16 мкл; праймер 21 - 1 мкл; праймер 22 - 1 мкл; полимераза - 1 мкл. Смесь для получения фрагмента F.
Условия проведения реакций:
92°С - 0,8 мин; 48°С - 0,6 мин; 70°С - 0,5 мин - 2 цикла;
92°С - 0,6 мин; 52°С - 0,6 мин; 70°С - 0,2 мин - 13 циклов;
92°С - 1,2 мин; 56°С - 1,2 мин; 70°С - 1,2 мин - 1 цикл.
Очищенные фрагменты D, Е, F получали аналогично фрагменту А.
Для получения фрагмента G смешивали:
Вода - 62 мкл; буфер - 10 мкл; dNTP - 16 мкл; фрагмент Е - 5 мкл; фрагмент F - 5 мкл; полимераза - 1 мкл.
Условия проведения реакции:
92°С - 1,2 мин; 45°С - 1,2 мин; 50°С - 1,2 мин; 70°С - 1,2 мин - 1 цикл; затем в реакционную смесь добавляли праймер 19 и праймер 22 по 1 мкл и проводили ПЦР в следующих условиях:
92°С - 0,8 мин; 50°С - 0,8 мин; 70°С - 0,4 мин - 19 циклов;
92°С - 1,2 мин; 55°С - 1,2 мин; 70°С - 1,5 мин - 1 цикл.
Для получения фрагмента Н смешивали:
вода - 63 мкл; буфер - 10 мкл; dNTP - 16 мкл; фрагмент D - 5 мкл; праймер 15 - 2 мкл; праймер 17 - 1 мкл; праймер 18 - 2 мкл; полимераза - 1 мкл.
Условия проведения реакции:
92°С - 1,2 мин; 45°С - 0,6 мин; 70°С - 0,4 мин - 1 цикл;
92°С - 0,6 мин; 50°С - 0,6 мин; 70°С - 0,2 мин - 18 циклов;
92°С - 1,2 мин; 52°С - 1,2 мин; 70°С - 0,8 мин - 1 цикл.
Получение целевого ПЦР-фрагмента, содержащего II блок гена TCI, осуществляли аналогично сборке блока 1, с использованием соответствующих праймеров блока 2.
Выделение ПЦР-фрагмента блока II гена TCI проводили по схеме, описанной для блока 1. Для формирования 5 - выступающих липких концов вместо рестриктазы XhoI использовали SalI.
1.3. Получение блока 3 гена TCI
Для ПЦР-реакции с праймерами блока 3 смешивали:
Вода - 70 мкл; буфер - 10 мкл; кДНК - 1 мкл; dNTP - 16 мкл; праймер 1 - 1 мкл; праймер 2 - 1 мкл; полимераза - 1 мкл. Смесь для получения фрагмента К.
Вода - 70 мкл; буфер - 10 мкл; кДНК - 1 мкл; dNTP - 16 мкл; праймер 3 - 1 мкл; праймер 4 - 1 мкл; полимераза - 1 мкл. Смесь для получения фрагмента L.
Условия проведения реакций:
92°С - 0,8 мин; 48°С - 0,6 мин; 70°С - 0,4 мин - 2 цикла;
92°С - 0,6 мин; 52°С - 0,6 мин; 70°С - 0,2 мин - 13 циклов;
92°С - 1,2 мин; 56°С - 1,2 мин; 70°С - 0,8 мин - 1 цикл.
Для реакционной смеси фрагмента М смешивали:
Вода - 70 мкл; буфер - 10 мкл; кДНК - 1 мкл; dNTP - 16 мкл; праймер 5 - 1 мкл; праймер 6 - 1 мкл; полимераза - 1 мкл.
Условия проведения реакции:
92°С - 1,2 мин; 48°С - 0,6 мин; 70°С - 0,8 мин - 2 цикла;
92°С - 0,6 мин; 52°С - 0,6 мин; 70°С - 0,4 мин - 13 циклов;
92°С - 1,2 мин; 56°С - 1,2 мин; 70°С - 0,8 мин - 1 цикл.
Очищенные фрагменты К, L, М выделяли аналогично фрагментам А, В, С, D, Е.
Блок 3 конструировали, используя полученные фрагменты К, L и М, для чего смешивали следующие компоненты:
вода - 57 мкл; буфер - 10 мкл; dNTP - 16 мкл; фрагмент К - 5 мкл; фрагмент L - 5 мкл; фрагмент М - 5 мкл; полимераза - 1 мкл.
Условия проведения реакции:
92°С - 1,2 мин; 45°С - 1,5 мин; 50°С - 1,5 мин; 70°С - 1,2 мин - 2 цикла; затем в реакционную смесь добавляли праймер 1 и праймер 5 по 1 мкл и проводили ПЦР в следующих условиях:
92°С - 0,8 мин; 50°С - 0,8 мин; 70°С - 0,6 мин - 19 циклов;
92°С - 1,3 мин; 52°С - 1,2 мин; 70°С - 1,5 мин - 1 цикл.
Дальнейшую обработку ПЦР-фрагмента с блоком 3 проводили аналогично схеме, описанной для блока 2.
1.4. Клонирование ПЦР-фрагмента, содержащего блоки 1,2 и 3 гена TCI
10 мкг плазмиды pFH123 расщепляли рестриктазами BamHI и SalI в 100 мкл реакционной смеси в течение 6 ч при 37°С, затем векторную часть плазмиды выделяли по схеме, описанной для готовых блоков гена TCI. Концентрацию вектора доводили до 0,1 пмоль/мкл.
Для сшивки каждого выделенного фрагмента и вектора смешивали три лигазных смеси следующего состава:
Вода - 38 мкл; лигазный буфер (×10) - 5 мкл; вектор - 1 мкл; блок 1 - 5 мкл; Т4-ДНК-лигаза (20 ед. акт./мкл) - 1 мкл. Смесь выдерживали 16 часов при 5°С.
Для второго и третьего блоков лигазные смеси получали по схеме, описанной для блока 1.
10 мкл лигазной смеси использовали для трансформации компетентных клеток E.coli Xlbluel с селекцией на среде, содержащей 50 мкг/мл ампициллина. Рекомбинантную ДНК выделяли стандартным методом [26], затем расщепляли рестриктазами ВаmHI и PstI и анализировали электрофоретически на наличие блока (1,2 или 3 соответственно) гена TCI в составе плазмиды. Структуру клонированного фрагмента определяли секвенированием по методу Сенгера. Плазмиды, содержащие целевые последовательности гена TCI, соответствующие расчетньм, были обозначены как плазмиды pFHblockl, pFHblock2, pFHblock3.
Пример 2. Конструирование рекомбинантной плазмиды pFH-TCI
Для получения плазмиды pFH-TCI, содержащей полный целевой ген TCI, использовали вектор, описанный в примере 1.4, а также три фрагмента гена TCI, которые были выделены из плазмид pFHblockl, pFHblock2, pFHblock3. Для этого плазмиду pFHblockl расщепляли рестриктазой ВаmHI, а затем рестриктазой FokI и выделяли фрагмент block 1/BamHI-FokI аналогично описанному в примере 1.1. Плазмиду pFHblock2 расщепляли рестриктазой FokI и выделяли фрагмент block2/FokI-FokI. Плазмиду pFHblock3 расщепляли рестриктазой SalI, а затем FokI и выделяли фрагмент block3/SalGI-FokI. Концентрацию выделенных блоков приводили к величине, равной 1 пмоль/мкл.
Для получения рекомбинантной плазмиды с полным геном TCI смешивали:
вода - 26 мкл; лигазный буфер (×10) - 5 мкл; вектор - 2 мкл; блок I/BamHI-FokI - 5 мкл; блок II/FokI-FokI - 5 мкл; блок III/FokI-SalI - 5 мкл; Т4-ДНК-лигаза (20 ед. акт./мкл) - 2 мкл.
Условия проведения реакции и отбор рекомбинантных клонов были аналогичны описанным для плазмид pFH1block, pFH2block, pFH3block в примере 1.4.
Структуру последовательности гена TCI, сшитой из 3-х блоков в рекомбинантной плазмиде pFH-TCI, подтверждали секвенированием по методу Сенгера.
Пример 3. Конструирование экпрессирующей плазмиды pET-TCI, экспрессия гена TCI в клетках E.coli
Для получения рекомбинантной плазмиды pET-TCI плазмиду рЕТ-32а (Novagen) гидролизовали последовательно эндонуклеазами рестрикции BamHI и SalGI в стандартных условиях. Вектор pET32a/BamHI-SalGI выделяли по схеме, описанной для вектора pFH123/BamHI-SalGI в примере 1.4. Bam-Sal фрагмент гена с 5'-выступающими липкими концами получали путем гидролиза плазмиды pFH-TCI теми же рестриктазами аналогично описанному в примере 2.
Готовили следующую лигазную смесь:
вектор - 1 мкл (0,05 nмоль); фрагмент - 5 мкл (0,5 nмоль); лигазный буфер (×10) - 2 мкл; вода - 11 мкл; Т4-ДНК лигаза (20 ед. акт./мкл); общий объем - 20 мкл. Лигазную смесь выдерживали ночь при 5°С. 10 мкл смеси использовали для трансформации Rb-компетентных клеток JM109. Анализ рекомбинантных клонов проводили методом амплификации их ДНК с использованием пары праймеров, входящих в состав 2-го блока гена TCI, а также подтверждающей рестрикцией рекомбинантных ДНК указанными рестриктазами. 10 нг полученной плазмиды pET-TCI использовали для трансформации компетентных клеток E.coli АД 494 (pLys).
Для экспрессии рекомбинантного гена TCI 5 мл ночной культуры клеток E.coli АД 494 (pLys), трансформированных плазмидой pET-TCI, ресуспендировали в 50 мл LB среды с ампициллином (50 мкг/мл) и выращивали при 37°С с интенсивной аэрацией до плотности ОД550 0,8-0,9. В культуру добавляли ИПТГ до 1 мМ концентрации индуктора и культивировали еще 4 часа. Клеточную биомассу собирали центрифугированием при 3000 об./мин в течение 10 мин. Затем клетки ресуспендировали в 5 мл буфера PBS×1 (0,02 М Na-фосфатный буфер рН 7,5; 0,15 М NaCl) с добавлением раствора MgCl2, бензамидина и лизоцима до концентрации 5 мМ, 0,5 мМ и 1 мг/мл соответственно. Смесь выдерживали 15 мин при комнатной температуре, а клетки разрушали трехкратным замораживанием в азоте с последующим быстрым оттаиванием на водяной бане при 65°С. Клеточный гидролизат центрифугировали 20 мин при 12 тыс. об/мин, супернатант удаляли, а осадок ресуспендировали в 3-х мл раствора 7 М мочевины, 10 мМ Трис рН 8, 0,1 М NaCl, 5 мМ ДТТ и мягко перемешивали 5 час при комнатной температуре. Экстракт центрифугировали 30 мин при 16 тыс. об/мин. Супернатант использовали для определения специфической иммунохимической активности находящегося в нем рекомбинантного белка TCI, содержащего детерминанты ВИЧ-1.
Пример 4. Определение специфической иммунохимической активности рекомбинантного белка TCI
Полученный раствор, содержащий TCI-иммуноген, был сорбирован по 100 мкл в лунку на полистирольную планшету с разведением 1:100, 1:200, 1:500 в буфере PBS×1 и инкубирован ночь при 25°С. В качестве нескольких положительных контролей (К+) были использованы растворы р24 белка ВИЧ-1, р41 белка оболочки ВИЧ-1, а также лизата эукариотических клеток, инфицированных ВИЧ-1, с конечной концентрацией 2-4 мкг/мл. В качестве двух отрицательных контролей (К-) были использованы раствор лизата белков нетрансформированных клеток E.coli, полученных аналогично фракции рекомбинантного белка TCI, а также лизат неинфицированных эукариотических клеток. Сорбцию положительных и отрицательных контролей антигенного материала проводили в условиях, описанных выше.
По окончании сорбции планшету встряхивали и в лунки добавляли по 150 мкл блокирующего раствора (0.2% казеин, 0.1 М натрий фосфатный буфер, рН 7.5, 0.05% твина), выдерживали 1 час при 25°С и планшету затем тщательно промывали 4 раза водой. Реакцию иммунохимического связывания с иммобилизованными антигенами проводили по стандартной методике ИФА - анализа с использованием растворов лиофилизованной поликлональной сыворотки на р24 белок, белок оболочки р41, высокотитражной сыворотки на вирусные белки ВИЧ-1, а также асцитной жидкости мышиных моноклонов, полученных на (288-309) детерминанту белка р24 (инструкция тест-системы “РекомбиБест анти ВИЧ 1+2”, производство ЗАО “Вектор-Бест”, ФС42-3847-99). Все разведения рекомбинантного антигена, а также контрольные образцы K+ показали устойчивый положительный сигнал с поликлональными сыворотками на белки вируса ВИЧ-1. IgG антитела двух моноклональных сывороток 29F2 и 30А6 устойчиво взаимодействовали на планшете с антигенами р24 белка, лизатом эукариотических клеток, инфицированных ВИЧ-1, а также с рекомбинантным белком TCI.
ИФА-активность приведенных отрицательных контролей была низкой и не превышала значений фона.
Таким образом, впервые успешно проведены дизайн и конструирование искусственного белка-иммуногена TCI, содержащего набор из 80 CTL-эпитопов белков Env, Gag, Pol, Nef ВИЧ-1, который вызывает развитие цитотоксического иммунного ответа.
Источники информации
1. Cease K.B., Berzofsky J.A, Annu. Rev. Immunol., 12 (1994) 923.
2. Eroshkin A.M., Karginova E.A., Gileva I.P., Lomakin A.S., Lebedev L.R., Kamyinina T.P., Pereboev A.V., Ignat'ev G.M. 1995. Design of four-helical protein as a candidate for HIV vaccine. Protein Engineering, Vol. 8, No.2, pp. 167-173.
3. Loktev V.B., Ilyichev A.A., Eroshkin A.M., Karpenko L.I., Pokrovsky A.G., Svyatchenko V.A., Ignat’ev G.M., Smolina M.I., Melamed N.V., Lebedeva C.D., Sandakhchiev L.S. 1996. Design of immunogens as components of a new generation of molecular vaccines. J. Biotechnology, Vol.44, pp.129-137.
4. Hanke Т, McMichael A.J., 2000. Design and construction of an experimental HIV-1 vaccine for a year- 2000 clinical trial in Kenya. Nature Med, Vol. 6, pp.951.
5. Wyatt R., Kwong P.D., Desjardins E., Sweet R.W., Robinson J., Hendrickson W.A., Sodroski J.G. 1998. The antigenic structure of the HIV gp l20 envelope glycoprotein. Nature, Vol.393, No. 6686, pp.705-11.
6. Rowland-Jones S., Sutton J., Ariyoshi K., Dong Т., Gotch F., McAdam S., Whitby D., Sabally S., Gallimore A., Corrah Т., Takiguchi M., Schultz Т., McMichael A., Whittle H. 1995. HIV-specific cytotoxic T-cells in HIV-exposed but uninfected Gambian women. Nature Med., Vol. 1, p.59.
7. Letvin N.L. 1998. Progress in the development of an HIV-1 vaccine. Science, Vol. 280, No. 5371,pp.1875-1880.
8. Ogg G.S., Jin X., Bonhoeffer S., Dunbar P.R., Nowak M.A., Monard S., Segal J.P., Cao Y., Rowland-Jones S.L., Cerundolo V., Hurley A., Markowitz M., Ho D.D., Nixon D.F., McMichael A.J. 1998 Quantitation of HIV-1-specific cytotoxic Т lymphocytes and plasma load of viral RNA. Science, Vol.279, No.5359, pp.2103-6.
9. Yang O.O., Kalams S.A., Trocha A., Cao H., Luster A., Johnson R.P., Walker B.D. 1997. Suppression of human immunodeficiency virus type 1 replication by CD8+ cells: evidence for HLA class I-restricted triggering of cytolytic and noncytolyticmechanisms. J.Virol., Vol.71, pp.3120-8.
10. Wagner E. et al, 1998. Beta-chemokines are release from HIN-1-specific Т cells granules complexed to proteoglycans. Nature, Vol.391, pp.908-911.
11. Price D.A., Sewell A.K., Dong Т., Tan R., Goulder P.J., Rowland-Jones S.L., Phillips R.E. 1998. Antigen-specific release of beta-chemokines by anti-HIV-1 cytotoxic Т lymphocytes. Cur. Biol., Vol.8, 355-8.
12. Woodberry Т, Gardner J, Mateo L, Eisen D, Medveczky J, Ramshaw IA, Thomson SA, Ffrench RA, Elliott SL, Firat H, Lemonnier FA, Suhrbier A. 1999. Immunogenicity of a human immunodeficiency virus (HIV) polytope vaccinecontaining multiple HLA A2 HIV CD8(+) cytotoxic T-cell epitopes. J. Virol. Vol.73, No.7, pp.5320-5.
13. Hanke Т., Schneider J., Gilbert S.C., Hill A.V.S., McMichael A. 1998a. DNA multi-CTL epitope vaccines for HIV and Plasmodium falciparum: immunogeicity in mice. Vaccine, Vol. 16, No.4, pp.426-436.
14. Hanke Т., Blanchard T.J., Schneider J., Ogg G.S., Tan R., Becker M., Gilbert S.C., Hill A.V.S., Smith G.L., McМichael A. 1998b. Immunogenecities of intravenous and intramuscular administration of modified vaccinia virus Ankara-based multi-CTL epitope vaccine for human immunodeficiency virus type 1 in mice. J. Gen. Virol., Vol.79, pp.83-90.
15. Hanke Т., Blanchard T.J., Schneider J., Hannan C.M., Becker M., Gilbert S.C., Hill A.V.S., Smith G.L., McМichael А. 1998с. Enhancement of MHC class I-restricted peptide-specific Т cell induction by a DNA prime MVA boost vaccination regime. Vaccine, Vol. 16, No.5, pp.439-445.
16. Hanke T, Samuel RV, Blanchard TJ, Neumann VC, Alien TM, Boyson JE, Sharpe SA, Cook N, Smith GL, Watkins DI, Cranage MP, McMichael AJ. 1999.Effective induction of simian immunodeficiency virus-specific cytotoxic T lymphocytes in macaques by using a multiepitope gene and DNA prime-modified vaccinia virus Ankara boost vaccination regimen. J Virol. Vol.73, No.9, pp.7524-32.
17. Hanke T, McMichael A. 1999. Pre-clinical development of a multi-CTL epitope-based DNA prime MVA boost vaccine for AIDS. Immunol Lett. Vol.66(l-3), pp.177-81.
18. Oldstone MBA. Viral persistence. (1989), Cell 56:517-520.
19. Goldberg A.L., Rock K.L. Proteolysis, proteasomes and antigen presentation. (1992), Nature 357:375-379.
20. York LA., Rock K.L. Antigen processing and presentation by the class I major histocompatibility complex. (1996),Ann.Rev.Immunol. 14:369-396.
21. Rammensee H.G., Falk K., Roetzschke О. МНС molecules as peptide receptors. (1993), Curr.Opin.Immunol. 5:35-44.
22. Lalvani A., Aidoo M., Allsopp C.E., Plebanski M., Whittle H.C., Hill A.V. 1994. An HLA-based approach to the design of a CTL-inducing vaccine against Plasmodium falciparum. Research in Immunology, Vol.145, pp.461-468.
23. Sidney J., Grey H.M., Kubo R.T., Sette A. 1996. Practical, biochemical and evolutionary implication of the discovery of HLA class I supermotifs. Immunology today, Vol.17, pp.261-266.
24. Ratner L., Haseltine W., Patarca R., Livak K.J., Starcich В., Josephs S.F., Josephs S.J., Doran E.R., Rafalski J.A., Whitehorn E.A., Baumeister K., Ivanoff L., Petteway S.R.J.r, Pearson M.L., Lautenberger J.A., Papas T.S., Ghrayeb, J., Chang N.T., Gallo R.C., Haseltine W.A., Wong-Staal F. 1985. Complete nucleotide sequence of the AIDS virus, HTLV-III. Nature, Vol.313, pp.277-284.
25. Данилюк Н.К., Серегин С.В., Синяков А.Н., Серпинский О.И., Бабкина И.Н., Урманова М.А., Рябинин В.А., Поздняков С.Г. Эффективный синтез и клонирование гена интерлейкина-2 человека и его аналога; экспрессия в клетках E.coli гена интерлейкина-2. Биоорганическая химия. 1991. Т.17. N 6. С.779-788.
26. Маниатис Т., Фрич Э., Сэмбук Д. Молекулярное клонирование. M.: Мир, 1984.



Изобретение относится к молекулярной биологии и генетической инженерии, конкретно к созданию синтетических полиэпитопных вакцин против ВИЧ-1. Искусственный белок-иммуноген TCI содержит CTL-эпитопы как можно большего числа основных антигенов ВИЧ-1 для использования в качестве эффективной и безопасной ДНК-вакцины ВИЧ-1. Длина искусственного белка составляет 392 аминокислотных остатка (фиг.2). Сконструированный белок содержит около 80 эпитопов белков Env, Gag, Pol, Nef ВИЧ-1. Синтез искусственного гена TCI (фиг.3) проводят с использованием метода ПЦР для амплификации фрагментов генома ВИЧ, кодирующих непрерывные районы белка TCI. Генно-инженерная конструкция, кодирующая полный набор эпитопов, клонирована в составе векторных экспрессионных плазмид в клетки E.coli. Продукция рекомбинантного белка TCI составила 10-20% от общего клеточного белка. Присутствие фрагментов белков ВИЧ-1 в структуре целевого иммуногена подтверждено с помощью ИФА с использованием панелей ВИЧ-1 позитивных сывороток и МАТ к белку р24 (фиг.2 и 3 представлены в описании). 2 с.п. ф-лы, 4 ил., 4 табл.
| WO 9511701, 04.05.1995 | |||
| US 6130082, 10.10.2000 | |||
| WO 9109625, 11.07.1991. |

