Изобретение относится к искусственным белкам-иммуногенам, имеющим свойства антигенов меланомы, используемым для создания синтетических полиэпитопных иммунотерапевтических вакцин против меланомы и может быть использовано в молекулярной биологии, генетической инженерии и медицине.
В настоящее время показано, что иммунотерапевтические вакцины могут быть использованы в качестве адъювантной терапии после удаления опухоли для индукции ответа CD4+ и CD8+ Т-лимфоцитов, препятствующих возникновению метастатических очагов при меланоме и других онкологических заболеваниях (Maverakis et al., 2015) [1].
В последнее десятилетие были достигнуты значительные успехи в понимании молекулярных основ взаимодействия опухоли и организма человека. В частности, большой интерес вызвала высокая иммуногенность клеток меланомы и обнаружение специфических поверхностных антигенов опухоли. Были отмечены случаи спонтанной полной ремиссии злокачественной меланомы, ассоциированные со спонтанной индукцией как гуморального, так и Т-клеточного иммунного ответа (Halama et al., 2010) [2]. Высокая иммуногенность меланомных опухолей дает основания полагать, что создание терапевтической вакцины против меланомы возможно. Несмотря на некоторые успехи в предотвращении и лечении онкологических заболеваний, эта проблема все еще далека от решения.
Разрабатываемые в настоящее время противоопухолевые терапевтические вакцины создаются с использованием различных антигенов, включая пептиды, белки, нуклеиновые кислоты или рекомбинантные вирусы, кодирующие целевые антигены, а также лизаты опухолей или целые облученные опухолевые клетки. В качестве альтернативного подхода разрабатываются вакцины на основе дендритных клеток (ДК) пациентов, которые нагружаются целевым антигеном in vitro и затем после созревания ex vivo вводятся обратно пациенту. В 2010 году FDA одобрило первую ДК-вакцину Sipuleucel-T для лечения рака простаты (Beer et al., 2011) [3]. В случае меланомы клинические испытания фазы I – II ДК-вакцины проводили у пациентов на IV стадии заболевания. В этих испытаниях использовались аутологичные дендритные клетки, нагруженные коктейлем из пяти синтетических меланома-ассоциированных антигенов (gp100, тирозиназы, MAGE-A2, MAGE-A3 и MART-1 или MAGE-A1), рестриктированных молекулами HLA-A2 или HLA-A24 (Oshita et al., 2012) [4]. Вакцина вызывала увеличение общей выживаемости до 13,6 месяцев против 7,3 месяцев у невакцинированных. В других клинических исследованиях использовали аутологичные ДК, нагруженные либо пептидами, либо опухолевыми клеточными лизатами (Bercovici et al., 2008) [5], либо убитыми клетками меланомы (Palucka et al., 2006) [6], либо путем использования векторов, кодирующих опухолевые антигены (Benteyn et al., 2013) [7].
Несмотря на безопасность и высокую иммуногенность ДК-вакцин in vivo их клинический эффект по-прежнему остается низким (Breckpot, Escors, 2009 [8]; Escors et al., 2013 [9]; Anguille et al., 2014) [10]. Эффективность ДК-вакцинации может зависеть от нескольких причин, в том числе от типа ДК, стадии их созревания, антигена, которым их нагружают, а также от дозы, частоты и способа введения ДК (Van Lint et al., 2014) [11].
За последнее десятилетие были предложены многие вакцинные стратегии, учитывающие современные фундаментальные знания об иммунной системе. Один из наиболее обещающих подходов к созданию эффективных и безопасных вакцин нового поколения основан на дизайне мозаичных полиэпитопных антигенов на основе широкого спектра протективных В- и/или Т-клеточных детерминант (Karpenko et al., 2014 [12]; Cardinaud et al., 2009 [13]; Iglesias et al., 2007 [14]; Tine et al., 2005) [15]. В частности, одним из таких подходов является создание искусственных Т-клеточных полиэпитопных иммуногенов, включающих большое количество консервативных Т-клеточных эпитопов, взаимодействующих с широким спектром аллельных вариантов молекул MHC.
Такие вакцины, по сравнению с вакцинами, создаваемыми на основе традиционных подходов, обладают следующими преимуществами, а именно:
1) они не содержат полноразмерных молекулярных структур патогенов, которые могут являться факторами патогенности и/или могут ингибировать формирование протективного иммунитета или стимулировать развитие аутоиммунных реакций;
2) полиэпитопы могут включать в свой состав большое количество CTL- и Т-хелперных (Th) эпитопов из раковых антигенов;
3) они могут быть спроектированы с учетом распространенности различных алломорф молекул HLA I класса в целевой человеческой популяции либо с учетом генетических особенностей конкретного пациента;
4) полиэпитопы могут быть сконструированы так, чтобы максимизировать эффективность процессинга и презентации большинства целевых эпитопов (Ishioka et al., 1999 [16]; Livingston et al., 2001 [17]; Uebel, Tampe, 1999 [18]; Peters et al., 2003 [19]; Cardinaud et al 2009) [20];
5) для увеличения эффективности индукции ответа Т-лимфоцитов целевые полиэпитопы могут содержать дополнительные сигнальные последовательности (например, N-концевой убиквитин, N-концевой лидерный пептид, и C-концевой фрагмент белка LAMP-1 человека) (Varshavsky, 2000 [21]; Rowell et al., 1995 [22]; Ruff et al., 1997) [23].
Данный подход использовался в организации-заявителе (ФБУН ГНЦ ВБ «Вектор») при конструировании ДНК-вакцины против меланомы (Боробова и др., 2012) [24] и рака молочной железы (Назаркина и др., 2015) [25], кодирующих полиэпитопные Т-клеточный иммуногены, спроектированные с использованием CTL- и Th-эпитопов опухолевых антигенов меланомы и рака молочной железы.
Известна полиэпитопная конструкция для иммунотерапии меланомы, содержащая десять HLA-A2 эпитопов из пяти антигенов меланомы [Mateo L., Gardner J., Chen Q., Schmidt C., Down M., Elliott S.L., Pye S.J., Firat H., Lemonnier F.A., Cebon J., Suhrbier A. An HLA-A2 polyepitope vaccine for melanoma immunotherapy. J Immunol. 1999; 163:4058-63) [26]. Было показано, что данная искусственная полиэпитопная вакцинная конструкция является иммуногенной, при этом линии CTL, извлеченные из вакцинированных HLA-A2 трансгенных мышей были способны лизировать клетки меланомы iv vitro. Полученные данные свидетельствуют о применимости полиэпитопных вакцин для иммунотерапии меланомы.
Известна другая полиэпитопная конструкция для иммунотерапии рака, в частности миеломы (международная заявка WO2017060360, МПК C07K14/47; C12N15/85, опубл. 13.04.2017 г.) [27]. Полиэпитопная конструкция относится к вектору экспрессии ДНК или смеси векторов экспрессии ДНК, которые кодируют по меньшей мере два CD4-эпитопа обратной транскриптазы теломеразы (TERT) и по меньшей мере один опухолевый CD8-эпитоп.
Однако указанные полиэпитопные конструкции недостаточно оптимизированы, что снижает индуцирование специфического Т-клеточного иммунного ответа против антигенов меланомы.
Наиболее близким аналогом (прототипом) заявляемого изобретения является ДНК-вакцина рMEL-TCI-A0201, кодирующая полиэпитопный Т-клеточный иммуноген для иммунотерапии меланомы, (Патент РФ № 2522830, опубл. 21.05.2014, прототип) [28]. Объектами патентной защиты являются ген MEL-TCI-A0201, кодирующий полиэпитопный белок-иммуноген MEL-TCI-A0201, рекомбинантная плазмидная ДНК рMEL-TCI-A0201, обеспечивающая экспрессию искусственного гена MEL-TCI-A0201, и искусственный белок-иммуноген MEL-TCI-A0201, содержащий множественные CTL- и Th-эпитопы антигенов меланомы. Было показано, что ДНК-вакцинная конструкция, кодирующая полиэпитопный иммуноген меланомы, вызывают не только наработку искусственных антигенов в трансфицированных клетках но и индуцируют Т-клеточный ответ на множественные эпитопы в системах in vitro и in vivo.
Однако использование ДНК-вакцины рMEL-TCI-A0201 имеет определенные ограничения, связанные с тем, что плазмида рMEL-TCI-A0201, кодирует «аллелеспецифический» иммуноген, в состав которого входят CTL-эпитопы, рестриктированные одним аллелельным вариантом HLA I класса – A*0201. Следовательно, аллелеспецифическая конструкция сама по себе должна индуцировать меланома специфический Т-клеточный ответ только в ограниченной популяции, В данном случае ее следует рассматривать в качестве одной из составляющей комбинированной вакцины, предназначенной для индивидуализированной иммунотерапии, при которой лечение должно осуществляться с использованием набора аллелеспецифических поли-CTL-эпитопных конструкций, несущих CTL-эпитопы, рестриктированные аллельными вариантами молекул HLA, присутствующими у конкретного пациента.
Техническим результатом настоящего изобретения является создание с использованием компьютерного дизайна нового искусственного полиэпитопного Т-клеточного белка-иммуногена MEL-TCI, в состав которого входят CTL-эпитопы, рестриктированные множественными аллелельными вариантами молекул HLA I класса, способного индуцировать специфический Т-клеточный иммунный ответ против меланомы у пациентов с различными генотипами HLA, что обеспечивает более высокую иммуногенную активность указанного белка и его более универсальный спектр действия.
Указанный технический результат достигается получением искусственного гена, кодирующего полиэпитопный белок-иммуноген MEL-TCI со свойствами антигенов меланомы, имеющий последовательность SEQ ID NO:2 длиной 3428 п.н., представленную на фиг. 2 и в приложении (перечень последовательностей), и содержащий на 5'-конце сайт эндонуклеазы рестрикции NheI, последовательность Козак и на 3'-конце – сайт Hind III и три стоп-кодона.
Указанный технический результат достигается также созданием рекомбинантной плазмидной ДНК pMEL-TCI, имеющей размер 8845 п.н., молекулярную массу 5.8×103 кДа, содержащей в соответствии с физической и генетической картой, представленной на фиг. 3, целевой ген по п. 1, кодирующий искусственный полиэпитопный белок MEL-TCI, находящийся под контролем промотора CMV, обеспечивающего его экспрессию в клетках млекопитающих, и состоящая из следующих фрагментов:
- NheI-HindIII – векторного фрагмента ДНК плазмиды рсDNA3.1(-) размером 5423 п.н., содержащего промотор CMV и последовательность BGH poly A, обеспечивающие экспрессию гена MEL- TCI в клетках млекопитающих; ген устойчивости к ампициллину (bla) и pMB1 ori, обеспечивающие селекцию и размножение целевой плазмиды в клетках бактерий Escherichia coli;
- NheI-HindIII – фрагмента размером 3428 п.н., содержащего ген MEL-TCI и последовательность Козак с инициирующим кодоном ATG, полученного путем обработки рестриктазами NheI–HindIII плазмиды pAL-TA- MEL- TCI;
- уникальных сайтов рестрикции: NruI-209, NheI-896, HindIII-4329, NarI-5692 , SmaI-5505, и имеющая следующее положение генов:
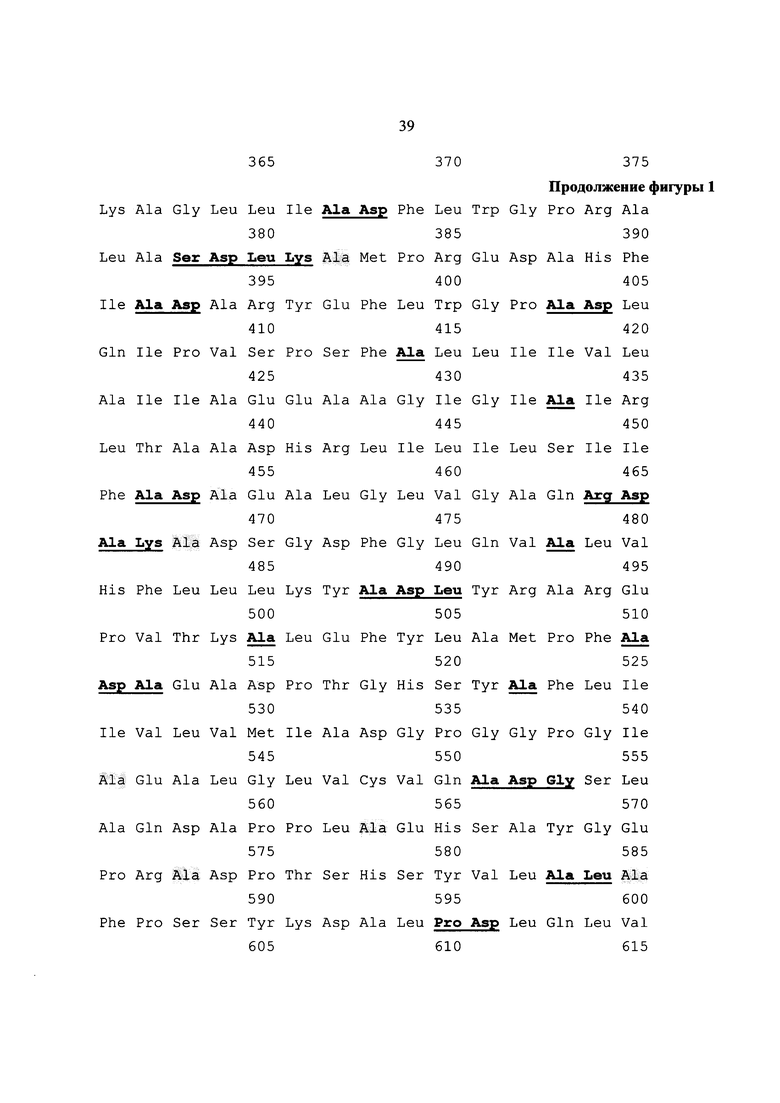
Указанный технический результат достигается также созданием искусственного белка-иммуногена MEL-TCI, имеющего аминокислотную последовательность SEQ ID NO:1 и содержащего CTL-эпитопы, рестриктированные множественными аллельными вариантами молекул HLA I класса (HLA-A*0101, A*0201, A*0301, A*1101, A*2402, A*6801, B*0702, B*0801, B*3501, B*1801, B*4402, B*2705) и Т-хелперные эпитопы антигенов меланомы (NY-ESO-1, MART1, MAGE-A1, MAGE- A3, MAGE-A11, MAGE-C1), рестриктированные множественными аллельными вариантами молекул HLA II класса, объединенные с использованием спейсерных аминокислотных остатков, причем, N-конец полиэпитопа содержит сигнальный пептид белка HER2 (P04626), а С-конец включает 11 последних аминокислотных остатков белка LAMP-1 человека.
Для того чтобы пояснить принцип конструирования искусственного полиэпитопного белка-иммуногена MEL-TCI, приведем следующие пояснения, представленные ниже.
Вначале проводился выбор оптимального набора Т-клеточных эпитопов и проектирования на их основе целевого полиэпитопного иммуногена.
Дизайн целевого иммуногена проводился с использованием современных данных о механизмах процессинга и презентации иммунной системе Т-клеточных антигенов. Учитывалось, что при индукции Т-клеточного ответа Т-лимфоциты узнают короткие пептидные фрагменты процессированных антигенов, представленные на поверхности клеток в ассоциации с определенными молекулами главного комплекса гистосовместимости (Main Histocompatibility Complex – MHC, или Human Leukocyte Antigen – HLA для обозначения молекул MHC человека) (Goldberg, Rock, 1992 [29]; Pamer, Cresswell, 1998) [30]. Учитывалось также, что для индукции ответа CD8+ Т-лимфоцитов процессинг полиэпитопа и презентация CTL-эпитопов должны осуществляться по пути MHC I класса, а для индукции ответа CD4+ Т-лимфоцитов – по пути MHC II класса. При этом молекулы MHC I класса презентируют, в основном, эндогенно синтезированные антигены (т.е. внутри клетки), процессинг которых происходит в протеасоме, а молекулы MHC II класса презентируют, в основном, экзогенные антигены, процессинг которых происходит в лизосоме. Молекулы MHC человека чрезвычайно полиморфны: известно несколько тысяч аллельных вариантов молекул HLA I и II класса; частоты их встречаемости сильно различаются в разных географических популяциях и у разных этнических групп (URL: http://www.ncbi.nlm.nih.gov/gv/mhc/) [31]. Поэтому поиск пептидов, способных связываться с достаточно высокой аффинностью с широким спектром аллельных вариантов молекул HLA, имеет важное значение.
Важную роль в процессинге эндогенных антигенов играет протеасома – внутриклеточный мультисубъединичный протеолитический комплекс (Rock, Goldberg, 1999) [32]; Niedermann et al., 1999) [33]. Показано, что протеасома генерирует C-конец пептидов, связывающихся с молекулами МНС класса I (Craiu et al., 1997 [34]; Stoltze et al., 1998) [35], таким образом, при выборе эпитопов необходимо учитывать специфичность протеасомного расщепления целевых антигенов. Пептиды, полученные в результате расщепления белков протеасомой, избирательно связываются гетеродимерным комплексом TAP (транспортеры, ассоциированные с процессингом) и транспортируются в эндоплазматический ретикулум (ЭПР), где связываются с молекулами MHC класса I и образованные комплексы транспортируются на поверхность клетки (Goldberg, Rock, 1992; Pamer, Cresswell, 1998; Lundegaard et al., 2007) [36].
В результате проведенного анализа литературы для дизайна полиэпитопной конструкции, предназначенной для иммунотерапии меланомы, целевые (оптимальные) эпитопы выбирались из шести наиболее иммуногенных антигенов:
1. Cancer/testis antigen 1 (CTAG1A, NY-ESO-1) (P78358), размером 180 аминокислот.
2. MART1, Melanoma antigen recognized by T-cells 1 (Q16655), размером 118 а.к.
3. MAGE-A1 (P43355), размером 309 а.к.
4. MAGE-A11 (CT1.11) (P43364), размером 429 а.к.
5. MAGE-A3 (P43357), размером 314 а.к.
6. MAGE-C1 (O60732), размером 1142 а.к.
Предсказание эпитопов в указанных антигенах проводилось с помощью созданного нами ранее оригинального программного обеспечения TEpredict (Antonets, Maksiutov, 2010) [37], позволяющего учесть (иммуно)протеасомный процессинг, аффинность связывания олигопептидов с TAP, а также провести выбор эпитопов с учетом частот встречаемости алломорф молекул HLA в целевой человеческой популяции.
При создании полиэпитопной вакцинной конструкции необходимо обеспечить максимально эффективную индукцию Т-клеточного иммунного ответа на включенные в ее состав антигенные пептиды. Для этого необходимо обеспечить адекватную доставку и эффективный процессинг искусственного иммуногена. Наиболее эффективный путь представления CTL-эпитопов иммунной системе обеспечивается при использовании ДНК-вакцин или вирусных векторов, которые могут доставлять ген, кодирующий целевой иммуноген, в антиген-презентирующие клетки, что необходимо для эндогенной экспрессии антигена, его процессинга и представления освободившихся эпитопов CD8+ клеткам в комплексе с молекулами MHC I класса. При этом необходимо обеспечить эффективный протеасомный процессинг продукта экспрессии целевого гена для освобождения антигенных пептидов с целью их последующего связывания с молекулами MHC I класса.
Поставленная задача решалась путем оптимального размещения целевых Т-клеточных эпитопов в составе полиэпитопного иммуногена и выбора оптимальных спейсерных аминокислотных последовательностей, а также с помощью выбора специальных сигнальных последовательностей, оптимизирующих MHC I- и/или MHC II-зависимую презентацию антигена (Ishioka et al., 1999 [16]; Livingston et al., 2001 [17]; Uebel, Tampe, 1999 [18]; Peters et al., 2003 [19]; Cardinaud et al 2009 [20]; Varshavsky, 2000 [21]; Rowell et al., 1995 [22]; Ruff et al., 1997 [23]). Дизайн искусственного полиэпитопного Т-клеточного иммуногена проведен с использованием разработанного нами ранее оригинального программного обеспечения PolyCTLDesigner (Antonets, Bazhan, 2013 [38]; Bazhan et al., 2010) [39].
Ниже приведен перечень графических материалов, иллюстрирующих заявляемое изобретение.
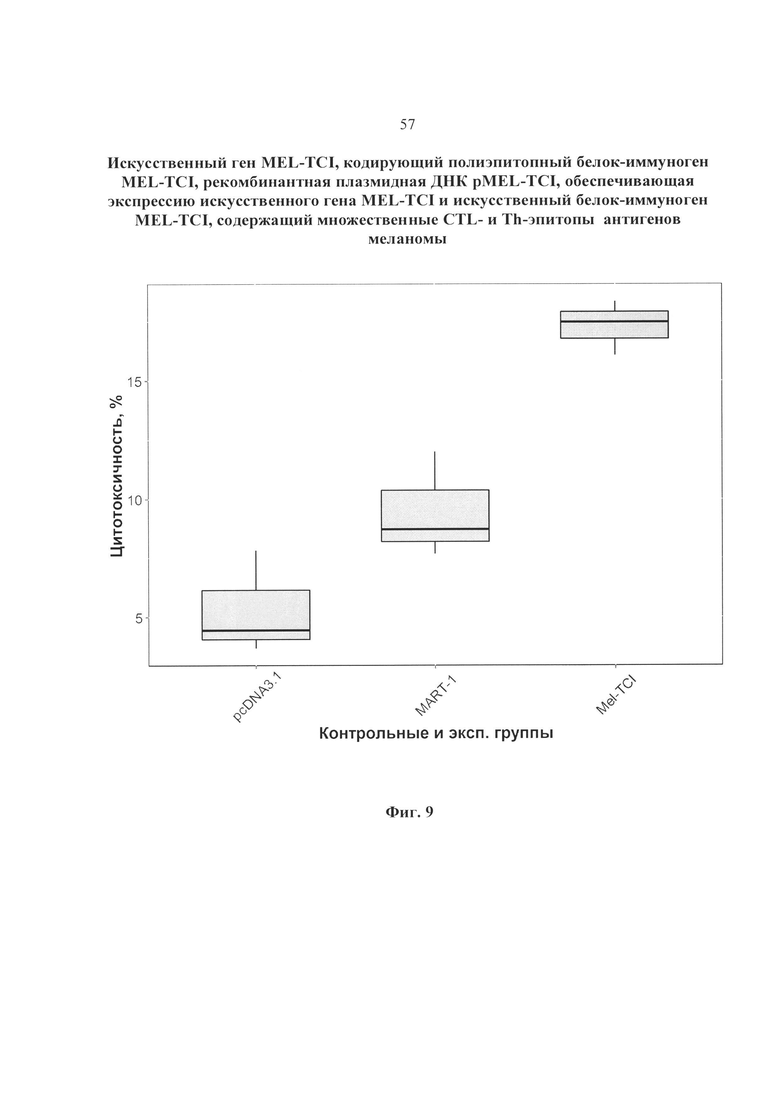
На фиг. 1 (см. приложение) приведена аминокислотная последовательность SEQ ID NO:1 полиэпитопного белка иммуногена MEL-TCI. На фиг. 2 (см. приложение) представлены нуклеотидная и аминокислотная последовательности SEQ ID NO:2 искусственного гена, кодирующего полиэпитопный иммуноген MEL-TCI. На фиг. 3 изображена физическая и генетическая карта рекомбинантной плазмиды рMEL-TCI. На фиг. 4 представлена электрофореграмма в 1% агарозном геле. Фрагменты ДНК после гидролиза плазмид рестриктазами NheI и HindIII. На фиг. 5 изображена электрофореграмма в 1% агарозном геле. Фрагменты ДНК после гидролиза плазмиды pMEL-TCI рестриктазами BglII и PsiI. На фиг. 6 представлена электрофореграмма продуктов ОТ-ПЦР в 1 % агарозном геле. На фиг. 7 представлена гистограмма, подтверждающая факт синтеза целевых иммуногенов в эукариотических клетках. На фиг. 8 (А, Б, В и Г) представлены результаты исследования количества IFN-γ-продуцирующих клеток (спотов) в совместной культуре МНК условно-здоровых доноров в реакции IFNγ -ELISpot. На диаграмме фиг. 9 приведены результаты исследования цитотоксической активности эффекторных Т-лимфоцитов, индуцированных в совместной культуре МНК условно-здоровых доноров в опытной и контрольных группах, против линии клеток меланомы человека Mel Is.
Пример 1. Дизайн и конструирование заявляемых объектов изобретения методом компьютерного моделирования
1.1. Дизайн поли-CTL-эпитопного фрагмента иммуногена MEL-TCI
С помощью новой версии программы TEpredict в выбранных антигенах были предсказаны CTL-эпитопы для наиболее распространенных в мировой популяции аллельных вариантов молекул HLA I класса (HLA-A*0101, A*0201, A*0301, A*1101, A*2402, A*6801, B*0702, B*0801, B*3501, B*1801, B*4402, B*2705). При этом при предсказании аффинности связывания пептида с молекулой HLA в качестве порогового было выбрано значение pIC50=6.8; пептиды, для которых была предсказана более низкая аффинность связывания с молекулой MHC, исключались из дальнейшего анализа. Кроме того, предсказывался протеасомный и иммунопротеасомный процессинг антигенов, а также аффинность связывания пептидов с транспортерами, ассоциированными с процессингом антигенов – и только те пептиды, для которых была предсказана достаточная аффинность связывания с TAP и на С-конце которых был предсказан сайт расщепления, были отобраны для конструирования полиэпитопного антигена. С помощью программы PolyCTLDesigner из каждого антигена были выбраны потенциальные Т-клеточные эпитопы, покрывающие выбранный репертуар молекул MHC I класса с двукратной избыточностью. В результате было отобрано 74 пептида, приведенные ниже.
LEFYLAMPF, QDAPPLPVP, IRLTAADHR, ESRLLEFYL, LMWITQCFL, LTAADHRQL, LAQDAPPLP, AASGLNGCC, ADGPGGPGI, FTVSGNILT, SLAQDAPPL, EAAGIGILT, AEEAAGIGI, LTVILGVLL, GILTVILGV, RRRNGYRAL, YRALMDKSL, ALMDKSLHV, EPVVPNAPP, MPREDAHFI, ARYEFLWGP, FLIIVLVMI, EADPTGHSY, SLFRAVITK, LVTCLGLSY, VADLVGFLL, YVKVLEYVI, LEYVIKVSA, QAATSSSSP, RVRFFFPSL, YRAREPVTK, FLWGPRALA, EALGLVCVQ, EHSAYGEPR, GHSYVLVTC, YKHCFPEIF, LIIVLGVIF, DSGDFGLQV, TQIFPTVRP, EAAFFSSTL, VCMQLLFGI, CKPEEGLQA, DPTSHSYVL, EVMWEVLSI, DPTSYPSLY, LIDPESFSQ, LVGAQALQA, QRITGGEQV, YPSLYEDAL, YEDYFPEIF, HLLLRKYRV, AQALQAEEQ, LQLVFGIEL, ACYEFLWGP, IMPKAGLLI, SVLEVFEGR, EALGLVGAQ, QAALSRKVA, YPPLHEWVL, SKASSSLQL, LVHFLLLKY, LLIIVLAII, YFFPVIFSK, LPTTMNYPL, KAGLLIIVL, IPVSSSFSY, SSTLLSIFQ, LILILSIIF, VIWDVLSGI, LQIPVSPSF, SSTLLSILQ, IEILFGISL, FPSSYKDAL, FFSSALLSI
Было проведено предсказание аффинности связывания этих пептидов с TAP и при необходимости для увеличения эффективности взаимодействия к пептидам на N-конец добавлялись остатки аланина (отмечены цветом):

Далее с помощью программы PolyCTLDesigner (Antonets, Bazhan, 2013) [38] проводился дизайн поли-CTL-эпитопного фрагмента с использованием вырожденного спейсерного мотива [ARSP][DLIT][LGA][VKA] с оптимизацией протеасомного расщепления и при строгости протеасомного фильтра 4%. При этом при выборе наилучших спейсеров для каждой пары эпитопов проводилась минимизация количества нецелевых эпитопов, образующихся при стыковке пептидов, рестриктированных 12 мажорными аллелельными вариантами молекул HLA I класса (HLA-A*0101, A*0201, A*0301, A*1101, A*2402, A*6801, B*0702, B*0801, B*3501, B*1801, B*4402, B*2705).
В результате был спроектирован поли-CTL-эпитопный фрагмент. Длина полученной конструкции 843 а.к.о., доля спейсерных последовательностей 19.22 %:
1.2. Дизайн поли-Т-хелперного фрагмента иммуногена MEL-TCI
Для наиболее эффективной индукции Т-клеточного иммунного ответа необходимо стимулировать ответ не только CD8+, но и CD4+ Т-лимфоцитов, поэтому следующей задачей было конструирование поли-Th-эпитопного фрагмента. Для этого с помощью TEpredict проводилось предсказание в составе выбранных раковых антигенов Th эпитопов с наиболее широкой специфичностью по отношению к HLA II класса. С помощью PolyCTLDesigner было выбрано 6 фрагментов длиной от 20 до 30 а.к.о., содержавших наибольшее количество Th эпитопов с наиболее широкой специфичностью по отношению к различным алломорфам HLA II класса (Табл. 1). N-концы выбранных фрагментов были продлены на 5 а.к.о. относительно начала первого эпитопа, а C-концы – на 5 а.к.о. относительно окончания последнего эпитопа.
Таблица 1
Последовательности, выбранные для конструирования поли-Т-хелперного фрагмента целевого иммуногена
Кроме того, было решено включить в состав поли-Th-эпитопной конструкции универсальный Т-хелперный эпитоп PADRE (Pan-DR epitope). Для подтверждения экспрессии целевого полипептида в его состав был включен маркерный В-клеточный эпитоп белка Gag ВИЧ-1 (Фиг. 2). Фрагменты были объединены с использованием мотива [KR][KR] – формирующего сайты расщепления для лизосомных катепсинов B и L, принимающих участие в MHC-II-зависимом процессинге антигенов.
Таким образом, был спроектирован поли-Th-эпитопный фрагмент иммуногена MEL-TCI, содержащий 45 T-хелперных эпитопов (рестриктированных не менее чем 48 аллельными вариантами молекул HLA II класса) из шести наиболее иммуногенных меланомных антигенов.
1.3. Проектирование итоговой полиэпитопной конструкции MEL-TCI
С помощью программы PolyCTLDesigner поли-CTL и поли-Th-эпитопные фрагменты были объединены в одну конструкцию посредством выбранной спейсерной аминокислотной последовательности. Чтобы увеличить индукцию ответа CD4+ и CD8+ Т-лимфоцитов, дополнительно на N-конец полиэпитопа был добавлен сигнальный пептид белка HER2 (P04626), направляющий образующийся полипептид в эндоплазматический ретикулум, а на С-конец было добавлено 11 последних аминокислотных остатков белка LAMP-1 человека, обеспечивающих перенаправление полипептида из секреторного пути на деградацию в лизосомы, где его пептидные фрагменты, образующиеся в результате протеолиза, могут связываться с рециркулирующими молекулами MHC II. Подбор последовательности сигнального пептида был проведен с использованием сервера SignalP 3.0 (Dyrlov Bendtsen et al., 2004) [40]. Общая длина полученной конструкции составляет 1133 п.о. Последовательность SEQ ID NO:1 и общий вид итогового полиэпитопного иммуногена представлены на Фиг. 1 и в приложении (см. перечень последовательностей).
1.4. Проектирование искусственного гена, кодирующего иммуноген MEL-TCI. Была спроектирована последовательность искусственного гена, кодирующего целевой полиэпитопный антиген MEL-TCI, оптимизированного для экспрессии в клетках человека. Для дизайна последовательности синтетического гена использовались различные программы и on-line сервисы (DNASTER, VectorNTI, NCBI-Dlast и др.), позволяющие оптимизировать последовательности генов для их высокой экспрессии в клетках человека. На фиг. 2 и в приложении (см. перечень последовательностей) представлены нуклеотидная последовательность SEQ ID NO:2 искусственного гена, кодирующего полиэпитопный иммуноген MEL-TC. Последовательность искусственного гена имеет длину 3428 п.н. На 5'-конце добавлен сайт эндонуклеазы рестрикции NheI (Bmt I) (выделен черным цветом с подчеркиванием). Серым цветом на рисунке выделена последовательность Козак. Инициирующий кодон ATG выделен полужирным шрифтом и подчеркнут. Стоп-кодоны показаны полужирным курсивом отмечены серым цветом. На 3' – сайт HindIII (выделен черным цветом с подчеркиванием).
1.5. Конструирование рекомбинантной плазмиды, содержащей искусственный ген, кодирующий белок–иммуногены и MEL-TCI
Ген, кодирующий белок MEL-TC-, был получен химическим синтезом и клонирован в экспрессионный вектор pcDNA3.1 (Invitrogen,США) по сайтам рестрикции NheI/HindIII. После трансформации клеток E.coli DH5α F’ полученная рекомбинантная плазмида pMEL-TCI, содержащая целевой ген, была отобрана стандартными процедурами скрининга, используя рестрикционный анализ. Структура клонированной последовательности подтверждена секвенированием.
Сконструированная рекомбинантная плазмида ДНК pMEL-TCI имеет размер 8845 п.н. (Фиг. 3). В составе этой плазмиды целевой ген, кодирующий искусственный полиэпитопный белок MEL-TCI, находится под контролем промотора CMV, обеспечивающего его экспрессию в клетках млекопитающих. Плазмидная ДНК pMEL-TCI состоит из следующих фрагментов:
NheI-HindIII – векторного фрагмента ДНК плазмиды рсDNA 3.1(-) [Invitrogen, США] размером 5423 п.н., содержащего промотор CMV и последовательность BGH poly A, обеспечивающие экспрессию гена MEL- TCI в клетках млекопитающих; ген устойчивости к ампициллину (bla) и pMB1 ori, обеспечивающие селекцию и размножение целевой плазмиды в клетках бактерий Escherichia;
NheI-HindIII – фрагмента размером 3428 п.н., содержащего ген MEL-TCI и последовательность Козак с инициирующим кодоном ATG.
Кроме того, плазмида рсDNA-MEL-TCI содержит уникальные сайты рестрикции: NruI-209, NheI-896, HindIII-4329, NarI-5692, SmaI-5505.
Молекулярная масса целевой рекомбинантной плазмидной ДНК равна 5.8 ×103 кДа.
Положение генов в плазмиде pMEL-TCI-
Для того, чтобы доказать способность полученной плазмиды индуцировать синтез соответствующего целевого полипептида в эукариотических клетках была проведена трансфекция 293Т-клеток выделенной плазмидной ДНК. Наличие целевого полипептида в трансфецированных клетках было подтверждено по синтезу специфической мРНК с помощью ОТ-ПЦР (Фиг. 6) и по окрашиванию клеток, трансфицированных плазмидой pMel-TCI, с помощью моноклональных антител (МАТ) 29F2 к эпитопу р24, входящему в состав белка Mel-TCI.
Таким образом, спроектирован искусственный белок-иммуноген MEL-TCI, содержащий множественные цитотоксические и хелперные Т-клеточные эпитопы меланомных антигенов (NY-ESO-1, MART1, MAGE-A1, MAGE-A3, MAGE-A11, MAGE-C1). На основе спроектированной аминокислотной последовательности проведен дизайн и синтезирован искусственный ген, кодирующий полиэпитопный белок-иммуноген MEL-TCI. Полученные данные подтверждают, что продукт экспрессии гена MEL-TCI, соответствует целевому иммуногену MEL-TCI.
Ниже приведены примеры 2-5 конкретного выполнения заявленных объектов изобретения.
Пример 2. Синтез искусственного гена, кодирующего иммуноген MEL-TCI и получение рекомбинантной плазмиды pMEL-TCI, кодирующей целевой полиэпитопный иммуноген MEL-TCI
2.1. Синтез искусственного гена, кодирующего иммуноген MEL-TCI, и получение рекомбинантной плазмиды pMEL-TCI, кодирующей целевой полиэпитопный иммуноген MEL-TCI
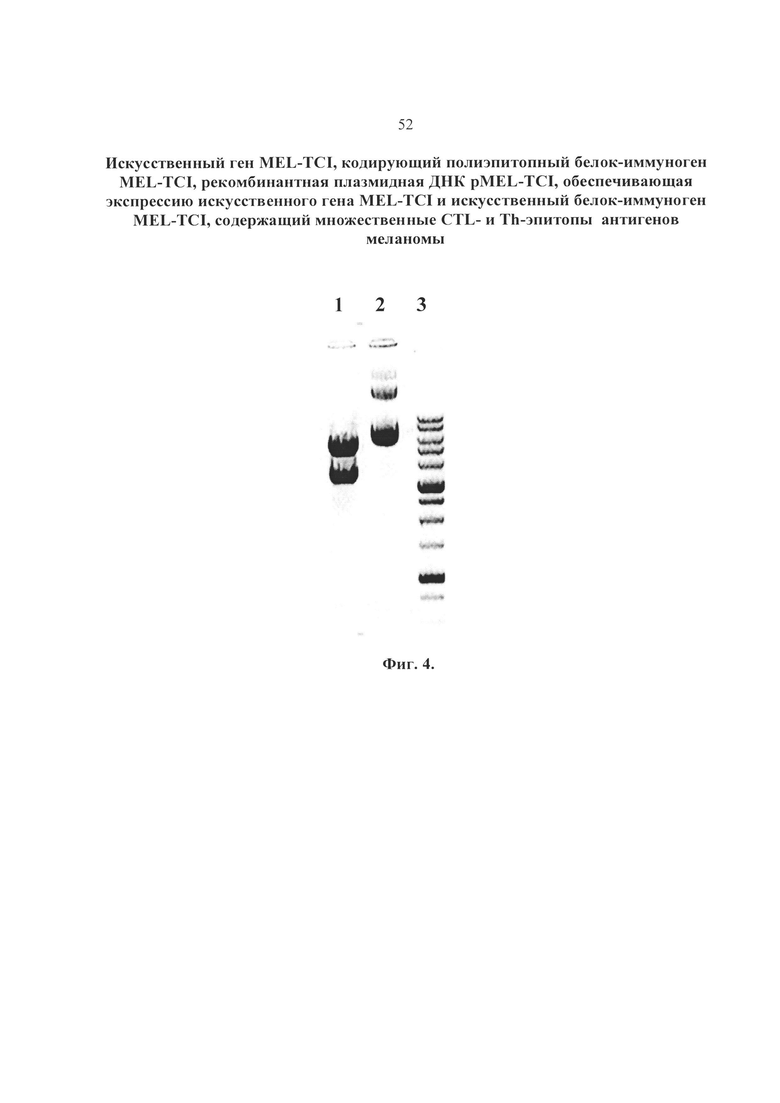
Выбор метода синтеза был продиктован соображениями эффективности и точности. Наиболее оптимальным оказался метод, основанный на использовании термостабильной РНК-лигазы (NEB, USA) с последующей ПЦР и клонированием в вектор pcDNA3.1. Синтез олигонуклеотидов осуществлялся на синтезаторе ABI3900 (AppliedBiosystems, USA). При синтезе использовался модифицированный протокол, позволяющий получать олигонуклеотиды длинной до 90 звеньев с чистотой более 95 %. Очищенные и обессоленные олигонуклеотиды прямой цепи кинировались, затем добавлялись праймеры-подставки и проводилась реакция лигирования. Полученный лигат клонировался в вектор pcDNA3.1. После скрининга отбиралось по три клона, несущих вставку, из них выделяли плазмиды и подвергали рестрикционному анализу с помощью эндонуклеаз рестрикции NheI, HindIII (Фиг. 4), и BglII, PsiI (Фиг. 5). Правильность синтезированной последовательности подтверждали после последующего ее секвенирования на автоматическом секвенаторе ABI3730x1.
2.2. Подтверждение структуры плазмиды pMEL-TCI рестрикционным анализом
Согласно теоретически рассчитанной нуклеотидной последовательности при гидролизе плазмиды pMEL-TCI с помощью эндонуклеаз рестрикции NheI (сайт узнавания GCTAGC) и HindIII (сайт узнавания AAGCTT) должны появиться фрагменты 3421 и 5329 п.н. При гидролизе плазмиды pMEL-TCI рестриктазами BglII (сайт узнавания AGATCT) и PsiI (сайт узнавания ACATGT) должны появляться наборы фрагментов (в п.н.) 2320, 2150, 1675, 1468, 1232 . Из данных, представленных на фиг. 4 и 5 видно, что подвижность фрагментов гидролизата плазмиды pMEL-TCI в 1% агарозном геле относительно маркеров молекулярной массы совпадает с теоретически рассчитанной.
На фиг. 4 представлена электрофореграмма в 1% агарозном геле. Фрагменты ДНК после гидролиза плазмид рестриктазами NheI и HindIII, где:
1) pMEL-TCI – гидролиз NheI и HindIII, фрагменты 3421, 5329 п.н.
2) pMEL-TCI – нативная
3) Маркер М12 фирмы Сибэнзим. (фрагменты ДНК 10000, 8000, 6000, 5000, 4000, 3000, 2500, 2000, 1500, 1000, 750, 500, 250 п.н.)
1) На фиг. 5 представлена электрофореграмма в 1 % агарозном геле. Фрагменты ДНК после гидролиза плазмиды pMEL-TCI рестриктазами BglII и PsiI, где:
2) Маркер М12 фирмы СибЭнзим (фрагменты ДНК 10000, 8000, 6000, 5000, 4000, 3000, 2500, 2000, 1500, 1000, 750, 500, 250 п.н.)
3) pMEL-TCI – нативная
4) pMEL-TCI – гидролиз BglII и PsiI, фрагменты 2320, 2150, 1675, 1468, 1232
2.3. Подтверждение структуры плазмиды секвенированием
Структура гена MEL-TCI в составе полученной рекомбинантной плазмиды рMEL-TCI, помимо рестрикционного анализа, была также подтверждена секвенированием. Показано, что нуклеотидные последовательности гена в образцах совпадает с теоретически рассчитанной последовательностью.
Пример 3. Исследование экспрессии гена MEl-TCI в эукариoтических клетках
3.1. Трансфекция клеток 293Т рекомбинантными плазмидами
Трансфекцию клеток 293Т плазмидой рMEL-TCI проводили, когда количество клеток в монослое составляло 60-70 %. В пробирку типа Eppendorf помещали 200 мкл среды ДМЕМ, 5 мкл плазмидной ДНК (1 мг/мл) и 5 мкл FuGENE® HD Transfection Reagent (Roshe). Смесь тщательно перемешивали и инкубировали при комнатной температуре в течение 15 мин. После инкубации смесь вносили к культуре клеток помещали в культуральный планшет в СО2-инкубатор на 4 часа. Затем в каждую лунку добавляли 1 мл среды ДМЕМ, содержащей 10 % фетальную бычью сыворотку, и помещали в СО2-инкубатор на 48 часов.
Оценку экспрессии целевого гена проводили двумя способами: 1) по определению наличия синтеза специфических мРНК в клетках 293Т, трансфицированных плазмидой pMEL-TCI или pcDNA-mart1; 2) по окрашиванию клеток, трансфицированных плазмидой pMEL-TCI, с помощью моноклональных антител (МАТ) 29F2 к эпитопу р24, входящему в состав белка MEL-TCI.
3.2. Анализ экспрессии целевого гена по определению синтеза специфической мРНК с помощью ОТ-ПЦР
Выделение суммарной РНК проводили из 0,8×106 клеток 293Т, трансфецированных плазмидой pMEL-TCI с использованием набора для выделения суммарной РНК фирмы Promega (cat. NZ3100) по прилагаемой инструкции. Методика выделения включает в себя стадии лизиса клеток, ДНКазного гидролиза для удаления примесей ДНК, и конечную очистку РНК на микроколонках.
Получение кДНК на РНК проводили в реакции обратной транскрипции: по 10 мкл РНК добавляли к 40 мкл реакционной смеси, содержащей в конечном объеме 50 мкл буфер REVx1, по 100 мкг/мл соответствующего для данного гена прямого праймера для ревертазы, по 0.5 мМ каждого из четырех дезоксинуклеозидтрифосфатов и по 800 ед/мл фермента – обратной транскриптазы M-MuLV (ревертазы) фирмы СибЭнзим. Реакцию синтеза кДНК проводили в течение 2-х часов при 42°С, после этого содержимое пробирок прогревали в течение 3-х мин при 95°С.
Далее полученную кДНК использовали для проведения ПЦР с использованием специфических праймеров к гену MEL-TCI (Табл. 2.) Амплификацию проводили в 25 мкл реакционной смеси, содержащей 5–10 нг кДНК, 0.2 мМ dNTPs, по 200 нг каждого из двух праймеров к гену, 1×буфер для ПЦР и 1 единицу Taq-ДНК-полимеразы фирмы ООО «Сибэнзим».
Таблица 2
Праймеры для ОТ-ПЦР
5’-CGTCTGCGAAAATCACGCCGAGT
62
Смесь помещали в амплификатор с заданной программой температурно-временных циклов: 95°C – 1 мин, 60°C, 72°C – 1 мин 20 сек; всего - 30 циклов. В конце последнего цикла ПЦР проводили завершающий синтез при 72°С в течение 5 мин. Продукты амплификации анализировали электрофорезом в 1 %-ном агарозном геле. На фиг. 6 представлена электрофореграмма продуктов ОТ-ПЦР в 1 % агарозном геле, где:
1 – продукт ОТ-ПЦР с использованием в качестве матрицы исходной плазмиды рMEL-TCI;
2 – маркер молекулярных весов М12 фирмы СибЭнзим;
3 – продукт ОТ-ПЦР, полученный с использованием в качестве матрицы кДНК рMEL-TCI трансфецированных 293Т-клеток.
Результаты, представленные на фиг. 6, показывают, что размеры амплифицированных фрагментов соответствую теоретически рассчитанным продуктам амплификации. Эти данные подтверждают наличие в суммарной фракции кДНК последовательности, кодирующей белок MEL-TCI, и, следовательно, указывает на экспрессию гена и синтез специфической мРНК.
3.3. Анализ экспрессии целевого гена по окрашиванию клеток, трансфицированных плазмидой pMEL-TCI, с помощью моноклональных антител (МАТ) 29F2 к эпитопу р24, входящему в состав белка MEL-TCI
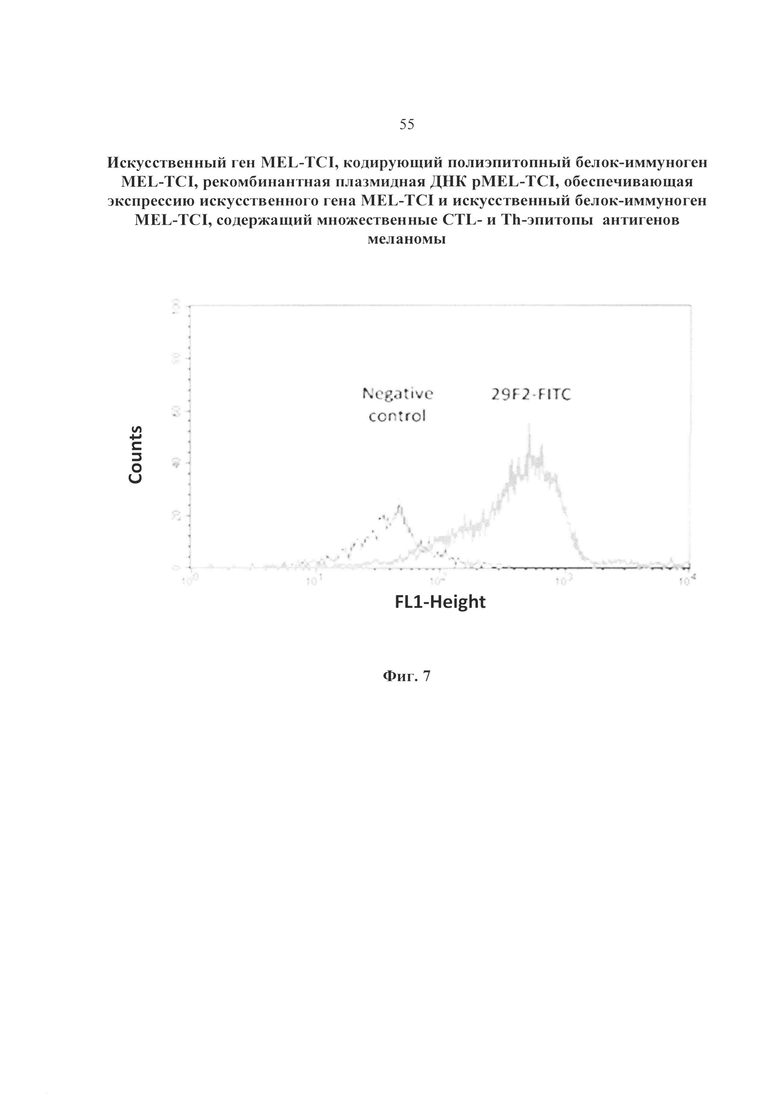
Экспрессию целевого гена оценивали также с помощью проточной цитофлуориметрии по выявлению окрашенных клеток с использованием FITC-МАТ к эпитопу-маркеру в составе белка MEL-TCI, который является продуктом экспрессии целевого гена. С этой целью предварительно проводили фиксацию и пермеабилизацию 293Т клеток, трансфицированных плазмидой pMEL-TCI. Антитела специфически связывались с экспрессируемым в составе целевого иммуногена эпитопом белка р24. Анализ образцов осуществляли на проточном цитофлуориметре BD FACSCalibur™ с использованием предложенного производителем программного обеспечения. Согласно полученным данным клетки, трансфицированные плазмидами, экспрессировали целевой иммуноген, что подтверждает факт синтеза целевых иммуногенов в эукариотических клетках (фиг. 7). На фиг. 7 представлена гистограмма, отражающая степень интенсивности свечения по FL1 окрашенных антителами 29F2-FITC клеток 293Т, трансфицированных плазмидой pMEL-TCI. Negative control – клетки 293Т, трансфицированные векторной плазмидой pcDNA3.1. 29F2-FITC – клетки 293Т, трансфицированные плазмидой pMEL-TCI (положительный пик демонстрирует наличие белка MEL-TCI в эукариотических клетках по связыванию эпитопа р24 с МКА 29F2).
Пример 4. Исследование специфической активности ДНК-вакцинной конструкции pMEL-TCI. Узким местом в создании Т-клеточных вакцин является оценка их биологической (специфической) активности. Дело в том, что при создании вакцин, индуцирующих Т-клеточный ответ у человека, нужна релевантная система для оценки их иммуногенности и противоопухолевой активности. Чтобы избежать проблем, связанных с несовместимостью лабораторных животных и разрабатываемых для человека Т-клеточных вакцин по антигенам MHC I класса, специфическую активность созданной ДНК-вакцинной конструкции pMEL-TCI оценивали с использованием мононуклеарных клеток (МНК), выделенных из периферической крови здоровых доноров, генотипированных по антигенам MHC I класса, для индукции Т-клеточного ответа в системе ex vivo. В данной системе иммунокомпетентные эффекторные Т-лимфоциты генерируются в результате совместного культивирования их предшественников с аутологичными дендритными клетками (ДК) моноцитарного или миелоидного происхождения, презентирующими целевые антигены. Эта система позволяет изучить иммуногенность и оценить противораковую целевых вакцинных конструкций, предназначенных для иммунотерапии или иммунопрофилактики онкологических заболеваний человека в наиболее релевантной системе [Anguille et al., 2014 [41]; Datta J. et al. 2014 [42]; Datta J. et al. 2015 [43]; Podrazil M. et al. 2015 [44]; Vacchelli E. et al. 2013 [45]; Jähnisch H. et al. 2010) [46].
4.1. Исследование иммуногенности ДНК-вакцинной конструкции pMEL-TCI в системе индукции Т-клеточного ответа ex vivo
Способность целевой ДНК- вaкцинной конструкции индуцировать специфический Т-клеточный иммунный ответ изучали в реакции IFNγ -ELISpot. Метод ELISpot основан на определении цитокинов, продуцируемых одиночными клетками после стимуляции их митогенами или антигенами. Секретируемые цитокины определяются моноклональными антителами и проявляются в виде отдельных спотов, отражающих количество цитокин-продуцирующих клеток. Анализ проводили с использованием наборов фирмы “Becton Dickinson” (США) согласно инструкции производителя. Для стимуляции клеток использовали смесь пептидов (20 мкг/мл каждого пептида), входящих в состав целевых Т- и В-клеточных антигенов.
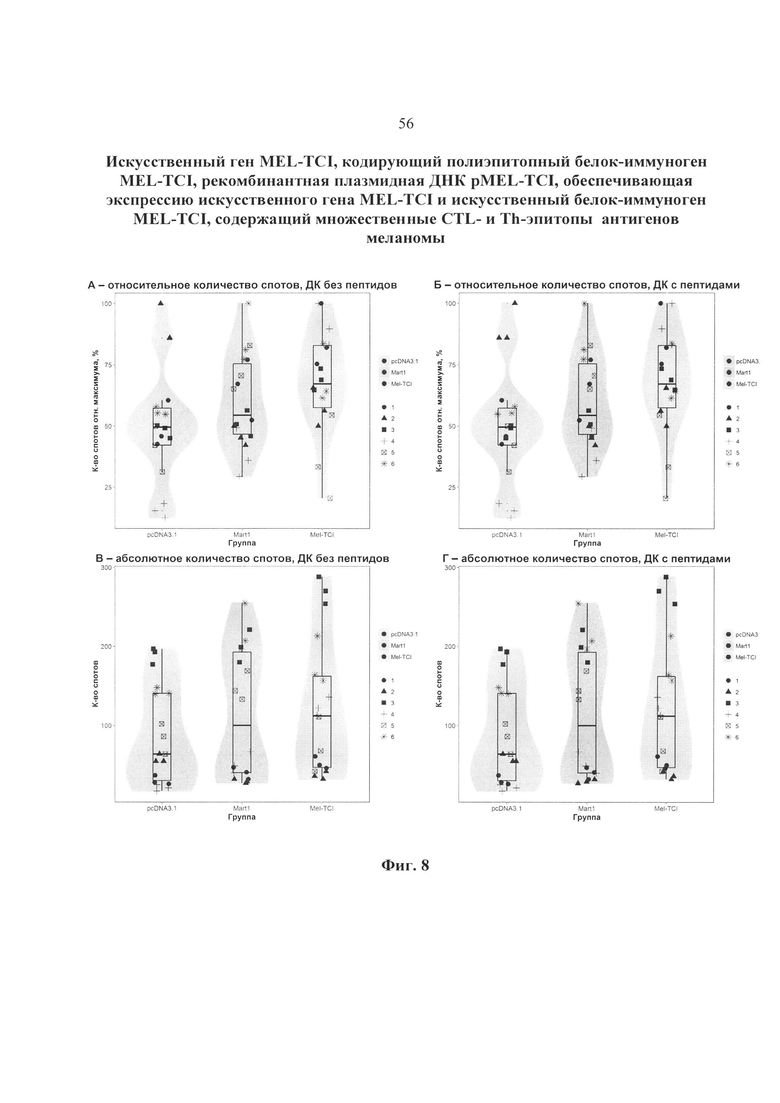
Результаты исследования количества IFN-γ -продуцирующих клеток (спотов) в совместной культуре МНК условно-здоровых доноров в реакции IFNγ -ELISpot представлены на фиг. 8. Показаны медианные значения, межквартильные интервалы и разброс значений. На верхних графиках (А и Б) приведены относительные количества спотов (пятен). Здесь для каждого донора определялось максимальное количество спотов и для каждого измерения был определен процент от максимального сигнала (для каждого донора). На нижних графиках (В и Г) приведены фактически измеренные (абсолютные) значения количества пятен (количество спотов на 2×105 клеток). Слева (А и В) показаны результаты, полученные без добавления пептидов, справа (Б и Г) – с пептидами. Различными цветами выделены результаты, полученные для лимфоцитов, стимулированных с помощью ДК, трансфицированных целевой и различными контрольными ДНК-вакцинными конструкциями: pcDNA3.1 – совместная культура МНК и ДК, трансфецированных контрольной плазмидой; MART-1 – совместная культура МНК и ДК, трансфецированных плазмидой pcDNA-mart1, кодирующей полноразмерный белок MART-1; MEL-TCI – совместная культура МНК и ДК, трансфецированных плазмидой pMEL-TCI. Маркеры различной формы соответствуют различным донорам.
Статистический анализ проводили с помощью двух методов: 1) парного однонаправленного теста Уэлча (используется для сравнения двух выборочных средних при неизвестных неравных дисперсиях) и 2) регрессионного анализа (используется для оценки влияния отдельных факторов.
4.2. Результаты, полученные с помощью парного однонаправленного теста Уэлча. При сравнении относительных значений сигналов (количество спотов в отношении к максимальному количеству спотов для данного донора, фиг. 8А) в отсутствие пептидов иммуностимулирующая активность ДНК-вакцины, кодирующих целевой полиэпитопный антиген MEL-TCI, достоверно отличалась от отрицательного контроля pcDNA3.1 (p=0,031).
В присутствии пептидов (фиг. 8Б) целевая ДНК-вакцинная конструкция показала достоверно более высокую иммуностимулирующую активность, чем отрицательный контроль (p=0,039) и чем положительный контроль – pcDNA-mart1 (p= 0,033). Ответ, индуцируемый плазмидой pcDNA-mart1, кодирующей полноразмерный белок MART1 (положительный контроль) был выше, чем при стимуляции векторной плазмидой pcDNA3.1 (отрицательный контроль). Однако, достоверных отличий между положительным и отрицательным контролем в данном случае обнаружено не было.
Для фактически измеренных значений количества спотов в отсутствие пептидов (фиг. 8В) наблюдались достоверные отличия в ответах, наблюдаемых в группах MART-1 и MEL-TCI от отрицательного контроля (p=0,006 и 0,006, соответственно). При этом достоверных отличий целевой ДНК-вакцинной конструкции pMEL-TCI от pcDNA-mart1 не наблюдалось. При добавлении пептидов (рис. 8Г) наблюдались достоверные отличия в группе MEL-TCI от отрицательного контроля (p=0,016). Отличие MART-1 от pcDNA3.1 было недостоверным (p=0.071).
Из этих результатов мы можем сделать вывод, что целевая ДНК-вакцинная конструкция pMEL-TCI вызывает стимуляцию секреции IFNγ эффекторными Т-лимфоцитами, особенно в присутствие антигенных пептидов. При этом Т-клеточный ответ, индуцированный pMEL-TCI, достоверно превосходит ответ в группе pcDNA3.1 (отрицательный) и не уступает ответу в группе pcDNA-mart1 (положительный контроль).
4.3. Результаты, полученные с помощью регрессионного анализа
Регрессионный анализ проводился с целью оценить влияние отдельных факторов, таких как «вакцинные конструкции», «доноры», и «специфические пептиды», на величину Т-клеточного ответа, индуцируемого различными конструкциями. Анализ проводился для относительных и фактически измеренных значений количества IFNγ -продуцирующих клеток (спотов), полученных в опытных и контрольных группах.
Регрессионный (факторный) анализ с использованием относительных значений (фиг. 8А и Б). Результаты анализа модели зависимости относительного значения секреции IFNγ от фактора «вакцинные конструкции» без добавления пептидов и без учета особенностей доноров показали, что целевая вакцинная конструкция обладает достоверным иммуностимулирующим эффектом. Величина эффекта для pMEL-TCI равна 15.65±6.58 (p = 0.02). При построении модели зависимости относительного значения секреции IFNγ от фактора «вакцинные конструкции» и от фактора, учитывающего добавление пептидов, иммуностимулирующий эффект pMEL-TCI становится заметней. Величина эффекта в данном случае составила 16.78 (p < 0.00061). При этом добавление пептидов обладает достоверной иммуностимулирующей активностью (величина эффекта 7.56±3.39 (p = 0.027). Включение в состав модели зависимости относительного значения секреции IFNγ от фактора «вакцинные конструкции» и от факторов, учитывающих добавление пептидов и особенности доноров, увеличивает качество модели (доля объясненной дисперсии возрастает с 14 до 20%).
Регрессионный (факторный) анализ с использованием фактически измеренных значений (фиг. 8В и фиг. 8Г). В случае использования фактически измеренных значения количества IFNγ -продуцирующих клеток анализ моделей зависимости наблюдаемого количества спотов (IFNγ -секретирующих клеток) от фактора «вакцинные конструкции» без учета особенностей доноров доля объясненной дисперсии оказалась очень низкой и составила 2,4 %. При добавлении факторов, учитывающих индивидуальных доноров, доля объясненной дисперсии увеличивается до 83%. Достоверным эффектом обладают факторы доноров 3, 4, 5 и 6 (p ≤ 0.0055). Достоверным положительным эффектом обладают конструкции pcDNA-mart1 (p = 0.031) и pMEL-TCI (p = 0.001). Стоит отметить, что в данном случае эффект от добавления пептидов не был достоверным. По-видимому, это связано с тем, что количество IFNγ -продуцирующих эффекторных Т-лимфоцитов в большей степени обусловлено активирующим действием дендритных клеток, трансфицированных исследуемыми вакцинными конструкциями, презентирующими главным образом пептиды, входящими в состав эндогенно экспрессируемых антигенов, и в меньшей степени – добавлением экзогенных пептидов.
Из данного анализа можно сделать следующий вывод: ДНК-вакцинная конструкция pMEL-TCI, кодирующая целевой полиэпитопный иммуноген, обладают иммуностимулирующей активностью; ответ CD8+ T-лимфоцитов, эффекторов Т-клеточного ответа, индуцированный плазмидой pMEL-TCI, достоверно превышает ответы, индуцированные в контрольных группах pcDNA3.1 (отрицательный контроль) и pcDNA-mart1 (положительный контроль).
Пример 5. Исследование цитотоксической активности активированных эффекторных клеток по отношению к клеткам-мишеням (культуре клеток меланомы человека Mel-Is) в системе индукции Т-клеточного ответа ex vivo
Наиболее важной характеристикой противоопухолевого иммунного ответа является способность специфических эффекторных CD8+ Т-лимфоцитов распознавать и убивать раковые клетки, в данном случае клетки меланомы. Основным механизмом элиминации опухолевых клеток является их непосредственный лизис, поэтому исследование цитотоксической активности МНК, активированных дендритными клетками, трансфецированными целевыми ДНК-вакцинами, проводилось против клеток линии меланомы человека, экспрессирующей на своей поверхности антиген MART-1. Цитотоксическая активность эффекторных клеток определялась колориметрическим методом, позволяющим провести количественное определение содержания лактат-дегидрогеназы – цитозольного фермента, выделяющегося из лизированных клеток (согласно протоколу производителя набора CytoTox 96® Non-Radioactive Cytotoxicity Assay, Promega).
Результаты исследования цитотоксической активности эффекторных Т-лимфоцитов, индуцированных в совместной культуре МНК условно-здоровых доноров в опытной и контрольных группах, против линии клеток меланомы человека Mel Is представлены на (фиг. 9). Результаты представлены, как процент цитотоксичности для эффекторных Т-лимфоцитов, стимулированных с помощью ДК, трансфицированных целевой и различными контрольными ДНК-вакцинными конструкциями: pcDNA3.1 – совместная культура МНК и ДК, трансфецированных контрольной плазмидой; MART-1 – совместная культура МНК и ДК, трансфецированных плазмидой pcDNA-mart1, кодирующей полноразмерный белок MART-1; Mel-TCI – совместная культура МНК и ДК, трансфецированных плазмидой pMEL-TCI. Маркеры различной формы соответствуют различным донорам.
Статистический анализ проводился с помощью однонаправленного парного теста Уэлча. Полученные результаты показали, что:
- цитотоксическая активность эффекторных Т-лимфоцитов, индуцированных вакцинными конструкциями pMEL-TCI и pcDNA-mart1 (положительный контроль), достоверно превосходит по эффективности индукции цитотоксический ответ, индуцированный плазмидой pcDNA3.1 (p ≤ 0.01).
- цитотоксический ответ, индуцированный целевой вакцинной конструкцией pMEL-TCI достоверно превосходят по эффективности индукции цитотоксический ответ, индуцированный плазмидой pcDNA-mart1, кодирующей полноразмерный белок MART-1 (p = 0.028).
Таким образом, исследование специфической активности ДНК-вакцины pMEL-TCI позволяет сделать следующие выводы:
1) ДНК-вакцина pMEL-TCI индуцирует формирование специфических CD8+ T-лимфоцитов в системе индукции Т-клеточного ответе ex vivo, ответ которых достоверно превышают ответы, индуцированные в контрольных группах pcDNA3.1 (отрицательный контроль) и pcDNA-mart1 (положительный контроль).
2) цитотоксическая активность эффекторных Т-лимфоцитов, индуцированных ДНК-вакциной (pMEL-TCI, достоверно превосходит цитотоксический ответ, индуцированный контрольными плазмидами pcDNA3.1 и pcDNA-mart1;
Из выше приведенных результатов следует, что создана генетическая конструкция – кандидат ДНК-вакцины против меланомы, содержащая ген, кодирующий искусственный полиэпитопный Т-клеточный белок-иммуноген, который за счет более оптимального подбора и размещения эпитопов обеспечивает индукцию CD8+ Т-лимфоцитов, обладающих цитотоксической активностью в отношении клеток меланомы.
Полученные результаты позволяют надеяться, что созданная ДНК-вакцинная конструкция может применяться для иммунотерапии меланомы с использованием трех различных протоколов: либо в качестве ДНК-вакцины; либо с использованием аутологичных зрелых ДК, трансфицированных полученной ДНК-вакцинной конструкцией ex vivo; либо на основе адаптивного переноса аутологичных эффекторных Т-лимфоцитов, полученных ex vivo.
Источники научно-технической и патентной информации
1. Maverakis E., Cornelius L.A., Bowen G.M., et al. Metastatic melanoma – a review of current and future treatment options // Acta Derm. Venereol. 2015. Vol. 95. No. 5. P. 516–524.
2. Halama N., Zoernig I., Jaeger D. Advanced malignant melanoma: immunologic and multimodal therapeutic strategies // J. Oncol. 2010. Vol. 2010. P. 1–8. Doi: 10.1155/2010/689893.
3. Beer T.M., Bernstein G.T., Corman J.M., Glode L.M., Hall S.J., Poll W.L., Schellhammer P.F., Jones L.A., Xu Y., Kylstra J.W., Frohlich M.W. Randomized trial of autologous cellular immunotherapy with sipuleucel-T in androgen-dependent prostate cancer // Clin. Cancer Res. –2011. Vol. 17. No. 13. P. 4558-4567. doi: 10.1158/1078-0432.CCR-10-3223.
4. Oshita C., Takikawa M., Kume A., Miyata H., Ashizawa T., Iizuka A., Kiyohara Y., Yoshikawa S., Tanosaki R., Yamazaki N., Yamamoto A, Takesako K, Yamaguchi K, Akiyama Y. Dendritic cell-based vaccination in metastatic melanoma patients: phase II clinical trial // Oncol. Rep. 2012. Vol. 28. No. 4. P. 1131-1138. doi: 10.3892/or.2012.1956.
5. Bercovici N., Haicheur N., Massicard S., Vernel-Pauillac F., Adotevi O., Landais D., Gorin I., Robert C., Prince H.M., Grob J.J., Leccia M.T., Lesimple T., Wijdenes J., Bartholeyns J., Fridman W.H., Salcedo M., Ferries E., Tartour E. Analysis and characterization of antitumor T-cell response after administration of dendritic cells loaded with allogeneic tumor lysate to metastatic melanoma patients // J. Immunother. 2008. Vol. 31. No. 1. P. 101-112.
6. Palucka A.K., Ueno H., Connolly J., Kerneis-Norvell F., Blanck J.P., Johnston D.A., Fay J., Banchereau J. Dendritic cells loaded with killed allogeneic melanoma cells can induce objective clinical responses and MART-1 specific CD8+ T-cell immunity // J. Immunother. –2006. Vol. 29. No. 5. P. 545-557.
7. Benteyn D., Van Nuffel A.M., Wilgenhof S., Corthals J., Heirman C., Neyns B., Thielemans K., Bonehill A. Characterization of CD8+ T-cell responses in the peripheral blood and skin injection sites of melanoma patients treated with mRNA electroporated autologous dendritic cells (TriMixDC-MEL) // Biomed Res. Int. 2013. doi: 10.1155/2013/976383.
8. Breckpot K., Escors D. Dendritic cells for active anti-cancer immunotherapy: targeting activation pathways through genetic modification // Endocr. Metab. Immune Disord. Drug Targets. 2009. Vol. 9. No. 4. P. 328-343.
9. Escors D., Liechtenstein T., Perez-Janices N., Schwarze J., Dufait I., Goyvaerts C., Lanna A., Arce F., Blanco-Luquin I., Kochan G., Guerrero-Setas D., Breckpot K. Assessing T-cell responses in anticancer immunotherapy: Dendritic cells or myeloid-derived suppressor cells? // Oncoimmunology. 2013. Vol. 2. No. 10. P. 26148.
10. Anguille S., Smits E.L., Lion E., van Tendeloo V.F., Berneman Z.N. Clinical use of dendritic cells for cancer therapy // Lancet Oncol. 2014. Vol. 15. No. 7. P. 257-267. doi: 10.1016/S1470-2045(13)70585-0.
11. Van Lint S., Wilgenhof S., Heirman C., Corthals J., Breckpot K., Bonehill A., Neyns B., Thielemans K. Optimized dendritic cell-based immunotherapy for melanoma: the TriMix-formula // Cancer Immunol. Immunother. 2014. Vol. 63. No. 9. P. 959-967. doi: 10.1007/s00262-014-1558-3.
12. Karpenko L.I. et al. Novel approaches in polyepitope T-cell vaccine development against HIV-1 // Expert Review of Vaccine. 2014. Vol. 13. No. 1. P. 155–173.
13. Cardinaud S., Bouziat R., Rohrlich P.S., Tourdot S., Weiss L., Langlade-Demoyen P., Burgevin A., Fiorentino S., van Endert P. and Lemonnier F.A. Design of a HIV-1-derived HLA-B07.02-restricted polyepitope construct // AIDS. 2009. Vol. 23. No. 15. P. 1945-1954.
14. Iglesias M.C., Mollier K., Beignon A.S., Souque P., Adotevi O., Lemonnier F. and Charneau P. Lentiviral vectors encoding HIV-1 polyepitopes induce broad CTL responses in vivo // Mol. Ther. 2007. Vol. 15. No. 6. P. 1203-1210.
15. Tine J.A., Firat H., Payne A., Russo G., Davis S.W., Tartaglia J., Lemonnier F.A., Demoyen P.L. and Moingeon P. Enhanced multiepitope-based vaccines elicit CD8+ cytotoxic T cells against both immunodominant and cryptic epitopes // Vaccine. 2005. –Vol. 23. No. 8. –P. 1085-1091.
16. Ishioka G.Y., Fikes J., Hermanson G., Livingston B., Crimi C., Qin M., del Guercio M.F., Oseroff C., Dahlberg C., Alexander J., Chesnut R.W., Sette A. Utilization of MHC class I transgenic mice for development of minigene DNA vaccines encoding multiple HLA-restricted CTL epitopes. J Immunol. 1999. 162(7):3915-25.
17. Livingston B.D., Newman M., Crimi C., McKinney D., Chesnut R., Sette A. Optimization of epitope processing enhances immunogenicity of multiepitope DNA vaccines. Vaccine. 2001. 19(32):4652-60.
18. Uebel S., Tampe R. Specificity of the proteasome and the TAP transporter. Curr Opin Immunol. 1999. 11(2):203-8.
19. Peters B., Bulik S., Tampe R., Van Endert P.M., Holzhutter H.G. Identifying MHC class I epitopes by predicting the TAP transport efficiency of epitope precursors. J Immunol. 2003. 171(4):1741-9.
20. Cardinaud S., Bouziat R., Rohrlich P.S., Tourdot S., Weiss L., Langlade-Demoyen P., Burgevin A., Fiorentino S., van Endert P., Lemonnier F.A. Design of a HIV-1-derived HLA-B07.02-restricted polyepitope construct. AIDS. 2009. 23(15):1945-54.
21. Varshavsky A. Ubiquitin fusion technique and its descendants. Methods Enzymol. 2000. 327:578-93.
22. Rowell J.F., Ruff A.L., Guarnieri F.G., Staveley-O'Carroll K., Lin X., Tang J., August J.T., Siliciano R.F. Lysosome-associated membrane protein-1-mediated targeting of the HIV-1 envelope protein to an endosomal/lysosomal compartment enhances its presentation to MHC class II-restricted T cells. J Immunol. 1995. 155(4):1818-28.
23. Ruff A.L., Guarnieri F.G., Staveley-O'Carroll K., Siliciano R.F.. The enhanced immune response to the HIV gp160/LAMP chimeric gene product targeted to the lysosome membrane protein trafficking pathway. J Biol Chem. 1997. 272(13):8671-8.
24. Боробова Е.А., Антонец Д.В., Старостина Е.В., Смирнова О.Ю., Щербаков Д.Н., Волкова О.Ю., Орешкова С.Ф, Карпенко Л.И., Ильичев А.А., Бажан С. И. Кандидаты ДНК-вакцин против меланомы: дизайн, конструирование и оценка экспрессии целевых генов в эукариотических клетках // Вестник НГУ: - 2012. - №5. – С.23-30.
25. Назаркина Ж.К., Харькова М.В., Антонец Д.В., Морозкин Е.С., Бажан С.И., Карпенко Л.И., Власов В.В., Ильичев А.А. Лактионов П.П. Конструирование полиэпитопной ДНК-вакцины против клеток опухолей молочной железы и исследование ее экспрессии в дендритных клетках. Бюллетень экспериментальной биологии и медицины. 2015, Том 160, № 10, С. 492-496.
26. Mateo L., Gardner J., Chen Q., Schmidt C., Down M., Elliott S.L., Pye S.J., Firat H., Lemonnier F.A., Cebon J., Suhrbier A. An HLA-A2 polyepitope vaccine for melanoma immunotherapy. J Immunol. 1999; 163:4058-63.
27. Международная заявка WO2017060360, «Полиэпитопная конструкция для иммунотерапии рака», опубл. 13.04.2017 г.
28. Антонец Д.В., Бажан С.И., Ильичёв А.А., Карпенко Л.И., Боробова Е.А., Старостина Е.В., Смирнова О.Ю., Орешкова С.Ф. Искусственный ген MEL-TCI-A0201, кодирующий полиэпитопный белок-иммуноген MEL-TCI-A0201, рекомбинантная плазмидная ДНК рMEL-TCI-A0201, обеспечивающая экспрессию искусственного гена MEL-TCI-A0201 и искусственный белок-иммуноген MEL-TCI-A0201, содержащий множественные CTL- и Th-эпитопы антигенов меланомы. Патент РФ № 2522830, опубл. 21.05.2014, (прототип).
29. Goldberg A.L., Rock K.L. 1992. Proteolysis, proteasomes and antigen presentation. Nature. 357, 375–379.
30. Pamer E., Cresswell P. 1998. Mechanisms of MHC class Irestricted antigen processing. Ann. Rev. Immunol. 16, 323–358.
31. URL: http://www.ncbi.nlm.nih.gov/gv/mhc/
31. Rock K.L., Goldberg A.L. 1999. Degradation of cell proteins and the generation of MHC class Ipresented peptides. Annu. Rev. Immunol. 17, 739–779.
32. Niedermann G., Geier E., LucchiariHartz M., Hitziger N., Ramsperger A., Eichmann K. 1999. The specificity of proteasomes: impact on MHC class I processing and presentation of antigens. Immunol. Rev. 172, 29–48.
33. Craiu A., Akopian T., Goldberg A., Rock K.L. 1997. Two distinct proteolytic processes in the generation of a major histocompatibility complex class Ipresented peptide. Proc. Natl. Acad. Sci. USA. 94, 10850–10855.
34. Stoltze L., Dick T.P., Deeg M., Pommerl B., Rammensee H.G., Schild H. 1998. Generation of the vesicular stomatitis virus nucleoprotein cytotoxic T lymphocyte epitope requires proteasomedependent and independent proteolytic activities. Eur. J. Immunol. 28, 4029–4036.
35. Lundegaard C., Lund O., Kesmir C., Brunak S., Nielsen M. 2007. Modeling the adaptive immune system: predictions and simulations. Bioinformatics. 23, 3265–3275.
36. Antonets DV, Maksiutov AZ. TEpredict: software for T-cell epitope prediction. Mol Biol (Mosk). 2010. 44(1):130-9.
37. Antonets D.V., Bazhan S.I. PolyCTLDesigner: a computational tool for constructing polyepitope T-cell antigens. BMC Res. Notes. 2013 Oct 10;6:407. doi: 10.1186/1756-0500-6-407.
38. Bazhan S.I., Karpenko L.I., Ilyicheva T.N., Belavin P.A., Seregin S.V., Danilyuk N.K., Antonets D.V., Ilyichev A.A. Rational design based synthetic polyepitope DNA vaccine for eliciting HIV-specific CD8+ T cell responses. Mol Immunol. 2010. 47(7-8):1507-15.
39. Dyrlov Bendtsen J., Nielsen H., von Heijne G., Brunak S. 2004. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol., Vol.340, pp.783-795.
40. Anguille S., Smits E.L., Lion E., van Tendeloo V.F., Berneman Z.N. Clinical use of dendritic cells for cancer therapy // Lancet Oncol. 2014. Vol. 15. No. 7. P. 257-267. doi: 10.1016/S1470-2045(13)70585-0.
41. Datta J. et al. Optimizing dendritic cell-based approaches for cancer immunotherapy // Yale J. Biol. Med. 2014. Vol. 87(4). P. 491-518.
42. Datta J. et al. Rationale for a Multimodality Strategy to Enhance the Efficacy of Dendritic Cell-Based Cancer Immunotherapy // Front. Immunol. 2015. № 6. P. 271. doi: 10.3389/fimmu.2015.00271.
43. Podrazil M. et al. Phase I/II clinical trial of dendritic-cell based immunotherapy (DCVAC/PCa) combined with chemotherapy in patients with metastatic, castration-resistant prostate cancer // Oncotarget. 2015. Vol. 6(20). P. 18192-18205.
44. Vacchelli E. et al. Trial watch: Dendritic cell-based interventions for cancer therapy // Oncoimmunology. 2013. Vol. 2(10). P. e25771.
45. Jähnisch H. et al. Dendritic cell-based immunotherapy for prostate cancer // Clin. Dev. Immunol. 2010. Vol. 2010. P. 517493. doi: 10.1155/2010/517493.

























Группа изобретений относится к области биотехнологии. Заявлен искусственный ген, кодирующий полиэпитопный белок-иммуноген MEL-TCI со свойствами антигенов меланомы, длиной 3428 п.н. Заявлена рекомбинантная плазмидная ДНК pMEL-TCI, обеспечивающая экспрессию указанного гена, имеющая размер 8845 п.н. и молекулярную массу 5.8×103 кДа. Заявлен искусственный белок-иммуноген MEL-TCI, содержащий CTL-эпитопы, рестриктированные множественными аллельными вариантами молекул HLA I класса (HLA-A*0101, A*0201, A*0301, A*1101, A*2402, A*6801, B*0702, B*0801, B*3501, B*1801, B*4402, B*2705), и Т-хелперные эпитопы антигенов меланомы (NY-ESO-1, MART1, MAGE-A1, MAGE-A3, MAGE-A11, MAGE-C1), рестриктированные множественными аллельными вариантами молекул HLA II класса. Группа изобретений позволяет получать искусственный полиэпитопный Т-клеточный белок-иммуноген, который за счет более оптимального подбора и размещения эпитопов обеспечивает индукцию CD8+ Т-лимфоцитов, обладающих улучшенной цитотоксической активностью в отношении клеток меланомы. 3 н.п. ф-лы, 9 ил., 2 табл., 5 пр.
1. Искусственный ген, кодирующий полиэпитопный белок-иммуноген MEL-TCI со свойствами антигенов меланомы, имеющий последовательность SEQ ID NO:2 длиной 3428 п.н. и содержащий на 5'-конце сайт эндонуклеазы рестрикции NheI, последовательность Козак и на 3'-конце - сайт Hind III и три стоп-кодона.
2. Рекомбинантная плазмидная ДНК pMEL-TCI, обеспечивающая экспрессию искусственного гена MEL-TCI, имеющая размер 8845 п.н., молекулярную массу 5.8×103 кДа, содержащая в соответствии с физической и генетической картой, представленной на фиг.3, целевой ген по п.1, кодирующий искусственный полиэпитопный белок MEL-TCI, находящийся под контролем промотора CMV, обеспечивающего его экспрессию в клетках млекопитающих, и состоящая из следующих фрагментов:
- NheI-HindIII - векторного фрагмента ДНК плазмиды рсDNA3.1(-) размером 5423 п.н., содержащего промотор CMV и последовательность BGH poly A, обеспечивающие экспрессию гена MEL-TCI в клетках млекопитающих; ген устойчивости к ампициллину (bla) и pMB1 ori, обеспечивающие селекцию и размножение целевой плазмиды в клетках бактерий Escherichia coli;
- NheI-HindIII - фрагмента размером 3428 п.н., содержащего ген MEL-TCI и последовательность Козак с инициирующим кодоном ATG, полученного путем обработки рестриктазами NheI-HindIII плазмиды pAL-TA-MEL-TCI;
- уникальных сайтов рестрикции: NruI-209, NheI-896, HindIII-4329, NarI-5692, SmaI-5505, и имеющая следующее положение генов:
3. Искусственный белок-иммуноген MEL-TCI, имеющий аминокислотную последовательность SEQ ID NO:1 и содержащий CTL-эпитопы, рестриктированные множественными аллельными вариантами молекул HLA I класса (HLA-A*0101, A*0201, A*0301, A*1101, A*2402, A*6801, B*0702, B*0801, B*3501, B*1801, B*4402, B*2705), и Т-хелперные эпитопы антигенов меланомы (NY-ESO-1, MART1, MAGE-A1, MAGE-A3, MAGE-A11, MAGE-C1), рестриктированные множественными аллельными вариантами молекул HLA II класса, объединенные с использованием спейсерных аминокислотных остатков, причем N-конец полиэпитопа содержит сигнальный пептид белка HER2 (P04626), а С-конец включает 11 последних аминокислотных остатков белка LAMP-1 человека.
| ИСКУССТВЕННЫЙ ГЕН MEL-TCI-A0201, КОДИРУЮЩИЙ ПОЛИЭПИТОПНЫЙ БЕЛОК-ИММУНОГЕН MEL-TCI-A0201, РЕКОМБИНАНТНАЯ ПЛАЗМИДНАЯ ДНК pMEL-TCI-A0201, ОБЕСПЕЧИВАЮЩАЯ ЭКСПРЕССИЮ ИСКУССТВЕННОГО ГЕНА MEL-TCI-A0201 И ИСКУССТВЕННЫЙ БЕЛОК-ИММУНОГЕН MEL-TCI-A0201, СОДЕРЖАЩИЙ МНОЖЕСТВЕННЫЕ CTL- И Th-ЭПИТОПЫ АНТИГЕНОВ МЕЛАНОМЫ | 2012 |
|
RU2522830C2 |
| АНТОНЕЦ Д.В | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| дис | |||
| канд | |||
| биол | |||
| наук | |||
| ФБУН "Государственный научный центр вирусологии и биотехнологии "Вектор", Кольцово, 2013 | |||
| БОРОБОВА Е.А | |||
| и др | |||
| Кандидаты ДНК-вакцины против меланомы: дизайн, конструирование и оценка экспрессии целевых генов в эукариотических клетках | |||
| Вестник НГУ, т | |||
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| MATEO L | |||
| et al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| The Journal of Immunology Vol | |||
| Деревянное стыковое устройство | 1920 |
|
SU163A1 |
| CN102030829 B, 21.11.2012. | |||

