В развитых странах повсеместно признано, что ожирение является серьезной проблемой для здоровья. В Соединенных Штатах этой проблеме придан эпидемический статус. Считается, что более 50% населения США обладают избыточным весом, больше 25% клинически диагностируют как страдающих ожирением и подверженных значительному риску заболевания сердца, инсулиннезависимым сахарным диабетом (NIDDM), гипертонии и определенным формам рака. Эта эпидемия оказывает значительную нагрузку на систему страховой медицины, поскольку только в США прогнозируемые ежегодные затраты на лечение ожирения должны превысить 70 миллиардов долларов. Стратегия лечения ожирения включает снижение аппетита или усиление затрат энергии.
На мышах было продемонстрировано, что когда в третий желудочек головного мозга или внутрибрюшинно вводили обладающий агонистической активностью к рецептору меланокортина-4 (MC4-R) циклический гептапептидный аналог гормона, стимулирующего образование α -меланоцитов (α -MSH), наступало продолжительное подавление аппетита. Этот эффект был обратимым, когда одновременно вводили антагонист MC4-R (Fan и др., Nature (1997), 385: 165-168). Следовательно, активность агонистов MC4-R должна быть полезна при лечении или профилактике ожирения.
Известны пять рецепторов меланокортина, сходных по гомологии их последовательностей, степень которой для членов данного семейства варьируется от 35 до 60% (Cone и др., Rec. Prog. Hormone Res. (1996), 51: 287-318), но эти рецепторы различаются по своим функциям. Например, MC1-R представляет собой связанный с G-белком рецептор, регулирующий пигментацию в ответ на α -MSH, являющийся сильнодействующим агонистом MC1-R (Cone и др., там же). Агонизм рецептора MC1-R приводит к стимуляции меланоцитов, которые влияют на эумеланин и увеличивают риск возникновения рака кожи. Агонизм MC1-R может также оказывать нейрологическое действие. Стимуляция активности MC2-R может приводить к карциноме тканей надпочечников. Эффекты агонизма MC3-R и MC5-R еще до сих пор не выяснены. Все рецепторы меланокортина, отвечающие на действия пептидных гормонов, классифицируются как гормоны, стимулирующие меланоциты (MSH). Эти пептиды являются производными проопиомеланокортина (РОМС), прогормона, состоящего из 131 аминокислоты, которые затем трансформируются в три класса гормонов: меланокортины (α -, β - и γ -), адренокортикотропный гормон (АСТН) и различные эндорфины (например, липотропин) (Cone и др., там же). Из-за их различных функций одновременный агонизм активности многочисленных рецепторов меланокортина способен вызвать нежелательные побочные эффекты. Поэтому важно, чтобы агонист MC4-R был бы более селективен к MC4-R, чем к одному или нескольким рецепторам меланокортина.
Haskell-Luevano и др. [Peptides (1996), 17(6): 995-1002] описали пептиды, которые содержат трипептид (D) Phe-Arg-Trp и проявляют меланотропную (потемнение кожи) активность в биоиспытаниях на коже лягушки (Rana pipiens). Haskell-Luevano и др. (там же) не упомянуты какие-либо соединения формулы I или II, которые представлены ниже.
Bednarek и др. [Peptides (1999), 20: 401-409] и Bednarek и др. (Biochem.Biophys.Res.Comm. (1999), 261: 209-213) описали аналоги циклических пептидов МТ-II. Какие-либо соединения формулы I или II, которые представлены ниже, ими не упомянуты.
По настоящему изобретению предлагается соединение формулы:
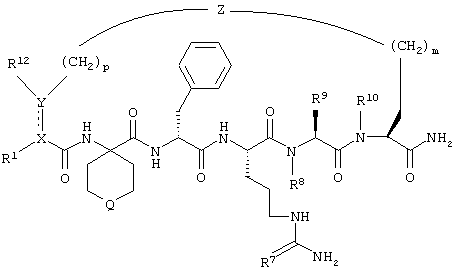
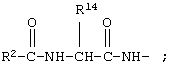
В соединениях формулы I R1 и R12 совместно с Х и Y образуют фенильное кольцо, Х обозначает С, Y обозначает С или R1 обозначает водородный атом или группу

R12 обозначает водородный атом, причем либо каждый из Х и Y обозначает С и связь между Х и Y является двойной связью, либо каждый из Х и Y обозначает СН, а связь между Х и Y является одинарной связью; R2 обозначает алкил, содержащий от 1 до 5 углеродных атомов, алкенил, содержащий от 2 до 5 углеродных атомов, или алкинил, содержащий от 2 до 5 углеродных атомов; R14 обозначает алкил, содержащий от 1 до 5 углеродных атомов, а n обозначает 0 или 1, Q обозначает группу

у которой каждый из R3, R4 и R5 независимо обозначает водородный атом, гало, алкил, содержащий от 1 до 4 углеродных атомов, гидрокси- или алкоксирадикал, содержащий от 1 до 4 углеродных атомов, причем когда R4 не обозначает водородный атом, как R3, так и R5 обозначают водородный атом; R6 обозначает водородный атом, алкил, содержащий от 1 до 3 углеродных атомов, алкоксирадикал, содержащий от 1 до 3 углеродных атомов, фенокси или гало; каждый из R11 и R13 независимо обозначает водородный атом, алкил, содержащий 3 или 4 углеродных атома, или циклоалкил, содержащий 5 или 6 углеродных атомов, или как R11, так и R13 обозначают фенил; R обозначает О или NH; R8 обозначает водородный атом или метил. R9 обозначает группу
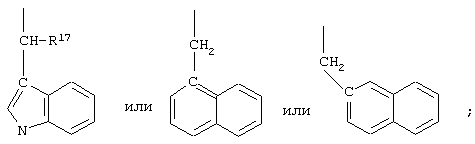
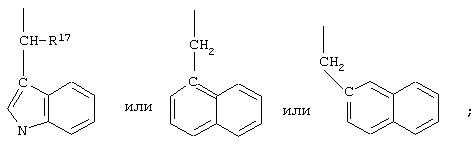
R10 обозначает водородный атом или метил; р обозначает 0 или 1; m обозначает 0, 1, 2 или 3; Z обозначает группу

R17 обозначает водородный атом или низший алкил, соответственно (низш.)алкил, предпочтительно метил.
Предлагаются также их фармацевтически приемлемые соли.
Когда каждый из Х и Y обозначает -СН-, изображенная пунктирными линиями связь в соединении формулы I является гидрированной. С другой стороны, когда имеется изображенная пунктирными линиями связь, Y и X, взятые совместно с R1 и R12, фенильного кольца не образуют, и как X, так и Y обозначают четырехвалентные атомы С.
По настоящему изобретению предлагается также соединение формулы
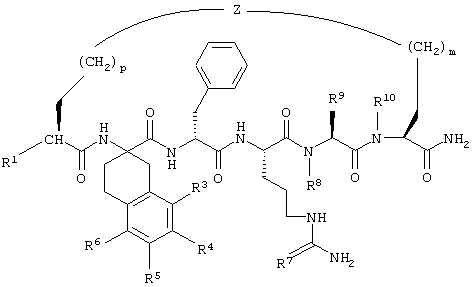
В соединениях формулы II R1 обозначает водородный атом или группу
или R2 обозначает алкил, содержащий от 1 до 5 углеродных атомов, алкенил, содержащий от 2 до 5 углеродных атомов, или алкинил, содержащий от 2 до 5 углеродных атомов; R14 обозначает алкил, содержащий от 1 до 5 углеродных атомов; а n обозначает 0 или 1, один из R3, R4, R5 и R6 обозначает водородный атом, гало, алкил, содержащий от 1 до 3 углеродных атомов, или алкоксирадикал, содержащий от 1 до 3 углеродных атомов, а каждый из остальных обозначает водородный атом. R7 обозначает О или NH. R8 обозначает водородный атом или метил. R9 обозначает группу
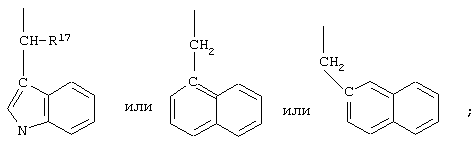
R10 обозначает водородный атом или метил; р обозначает 0 или 1; m обозначает 0, 1, 2 или 3; Z обозначает группу
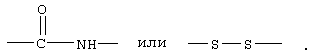
R17 обозначает водородный атом или (низш.)алкил, предпочтительно метил. Предлагаются также их фармацевтически приемлемые соли.
Соединения формул I и II, а также Penta-цикло(Asp-Lys)-Asp-Apc-(D)Phe-Ala-Trp-Lys-NH2 и Penta-цикло(Asp-Lys)-Asp-Apc-(D)Phe-Arg-(2S,3S)-бета-метил-Trp-Lys-NH2 являются агонистами MC4-R. Известно, что у мышей, на которых проводили эксперименты как на модели ожирения человека, агонисты активости MC4-R вызывали снижение аппетита. Следовательно, эти соединения полезны при лечении и профилактике ожирения.
Все соединения формул I и II, представленные ниже в примерах, также как и Penta-цикло(Asp-Lys)-Asp-Apc-(D)Phe-Ala-Trp-Lys-NH2 и Penta-цикло(Аsр-Lys)-Asp-Apc-(D)Phe-Arg-(2S,3S)-бета-метил-Trp-Lys-NH2 испытывали в опытах in vitro, описанных ниже в примере А раздела "Пример биологической активности" на агонистическую активность к MC4-R и на агонистическую активность к MC1-R. Все тестируемые соединения показывали значение ЕС50 для агонистической активности к MC4-R меньше 500 нМ и все проявляли по крайней мере в 10 раз более высокую агонистическую активность к MC4-R, чем к MC1-R. В противоположность этому соединение Ac-Nle-цикло(Asp-Lys)-Asp-His-(D)Phe-Arg-Trp-Lys-NH2 проявляло примерно одинаковую агонистическую активность в отношении к MC1-R и MC4-R.
Номенклатура и аббревиатуры
Понятие "алкил" означает прямоцепочечную или разветвленную алкильную группу, а понятие "(низш.)алкил" означает алкильную группу, содержащую от 1 до 6 углеродных атомов. Понятие "алкенил" означает прямоцепочечную или разветвленную алкенильную группу. Понятие "алкинил" относится к прямоцепочечной или разветвленной алкинильной группе.
Понятие "алкокси" означает группу формулы алкил-O-, в которой, алкил является представленной выше группой. Понятие "фенокси" означает группу формулы фенил-O-. Во всех случаях, если не указано иное, понятие "фенил" относится к незамещенному фенильному кольцу, а понятие "фенокси" относится к незамещенной феноксигруппе.
Понятие "гало" означает группу, выбранную из атомов фтора, хлора, брома и иода.
Понятие "фармацевтически приемлемая соль" относится к тем солям, которые сохраняют биологическую эффективность и свойства свободных оснований или свободных кислот, которые ни с биологической, ни с какой-либо другой точки зрения не являются нежелательными. Эти соли образуются неорганическими кислотами, такими, как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п., и органическими кислотами, такими, как уксусная кислота, пропионовая кислота, гликолевая кислота, пировиноградная кислота, оксиловая кислота, малеиновая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, п-толуолсульфоновая кислота, салициловая кислота, N-ацетилцистеин и т.п. Кроме того, эти соли могут быть получены в результате присоединения неорганического основания или органического основания к свободной кислоте. Соли, дериватизированные из неорганического основания, включают, хотя ими их список не ограничен, натриевые, калиевые, литиевые, аммониевые, кальциевые, магниевые соли и т.п. Соли, дериватизированные из органических оснований, включают, хотя ими их список не ограничен, соли первичных, вторичных и третичных аминов, замещенных аминов, включающих встречающиеся в природе замещенные амины, циклические амины и основные ионообменные смолы, такие, как изопропиламиновая, триметиламиновая, диэтиламиновая, триэтиламиновая, трипропиламиновая, этаноламиновая, лизиновая, аргининовая, н-этилпиперидиновая, пиперидиновая, полиминовая смолы и т.п.
Соединения формулы IA представлены следующим образом:
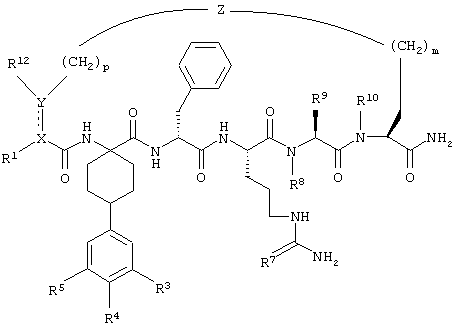
в которой R1, R3, R4, R5, R7, R8, R9, R10, R12, X, Y, Z, m и p имеют значения, указанные выше,
и их фармацевтически приемлемые соли.
В соединениях формулы IA R1 и R12 совместно с Х и Y образуют фенильное кольцо или R1 обозначает водородный атом или группу
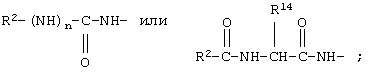
R12 обозначает водородный атом, причем либо каждый из Х и Y обозначает С и связь между Х и Y является двойной связью, либо каждый из Х и Y обозначает СН, а связь между Х и Y является одинарной связью; R2 обозначает алкил, содержащий от 1 до 5 углеродных атомов, алкенил, содержащий от 2 до 5 углеродных атомов, или алкинил, содержащий от 2 до 5 углеродных атомов; R14 обозначает алкил, содержащий от 1 до 5 углеродных атомов, а n обозначает 0 или 1. Каждый из R3, R4 и R5 независимо обозначает водородный атом, гало, алкил, содержащий от 1 до 4 углеродных атомов, гидрокси или алкоксирадикал, содержащий от 1 до 4 углеродных атомов, причем когда R4 не обозначает водородный атом, как R3, так и R5 обозначают водородный атом. R7 обозначает О или NH. R8 обозначает водородный атом или метил. R9 обозначает группу
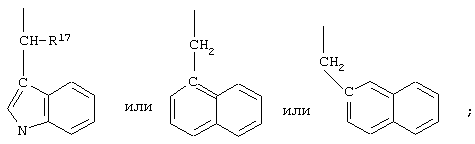
R10 обозначает водородный атом или метил; р обозначает 0 или 1; m обозначает 0, 1, 2 или 3; Z обозначает группу
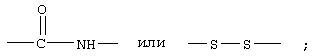
R17 обозначает водородный атом или (низш.)алкил, предпочтительно метил.
Обозначенная пунктирными линиями связь в формуле IA может быть гидрированной. Когда пунктирная линия обозначает гидрированную связь, как X, так и Y обозначают -СН-. С другой стороны, когда имеется обозначенная пунктирными линиями связь, Y и X, взятые совместно с R1 и R12, не образуют фенильного кольца, как X, так и Y обозначают четырехвалентные С атомы.
В одном варианте соединения формулы IA каждый из Х и Y обозначает СН, и связь между Х и Y гидрирована до одинарной связи; Z обозначает группу
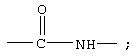
R7 обозначает О; R1 обозначает группу
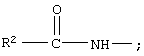
R2 обозначает алкил, содержащий от 1 до 5 углеродных атомов; как R10, так и R12 обозначают водородный атом.
Примеры таких соединений включают Penta-цикло(Asp-Lys)-Asp-Apc-(D)Phe-Cit-Trp-Lys-NH2.
В другом, предпочтительном варианте, соединения формулы IA Z обозначает группу
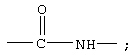
R7 обозначает NH; R1 обозначает водородный атом или группу
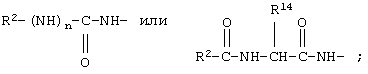
R2 обозначает алкил; как R10, так и R12 обозначают водородный атом.
В другом варианте соединения формулы IA Z обозначает группу
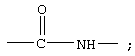
R7 обозначает NH; R1 обозначает группу
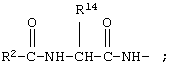
R2 обозначает алкил, как R10, так и R12 обозначают водородный атом.
В более конкретном варианте каждый из Х и Y обозначает СН, а связь между Х и Y гидрирована до одинарной связи; n обозначает 0; а R9 обозначает группу

Примеры таких соединений включают Penta-цикло(Asp-Lys)-Asp-Apc-(D)Phe-Arg-(2)Nal-Lys-NH2 и Penta-цикло(Asp-Lys)-Asp-Apc-(D)Phe-Arg-N-метил(2)Nal-Lys-NH2.
В другом, более конкретном варианте, соединения формулы IA Z обозначает группу
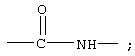
R7 обозначает NH; R1 обозначает группу
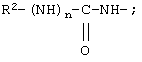
R2 обозначает алкил, как R10, так и R12 обозначают водородный атом, a R9 обозначает группу
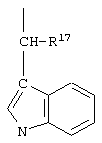
а R17 обозначает водородный атом или (низш.)алкил, предпочтительно метил.
Эти соединения включают те, у которых каждый из Х и Y обозначает СН, связь между Х и Y представляет собой одинарную связь, один из R3, R4 и R5 обозначает водородный атом, гало или алкил, а каждый из остальных обозначает водородный атом, например Penta-цикло(Asp-Lys)-Asp-Apc-(D)Phe-Arg-Trp-Lys-NH2, Penta-цикло(Asp-Lys)-Asp-4-MeApc-(D)Phe-Arg-Trp-Lys-NH2, Penta-цикло(Glu-Lys)-Glu-Apc-(D)Phe-Arg-Trp-Lys-NH2, Penta-цикло(Asp-Orn)-Asp-Apc-(D)Phe-Arg-Trp-Orn-NH2, Penta-цикло(Asp-Dbr)-Asp-Apc-(D)Phe-Arg-Trp-Dbr-NH2, Penta-цикло(Asp-Dpr)-Asp-Apc-(D)Phe-Arg-Trp-Dpr-NH2 или Ас-цикло(Asp-Dpr)-Asp-Apc-(D)Phe-Arg-Trp-Dpr-NH2.
По еще одному конкретному варианту соединений формулы IA предлагаются те соединения, у которых каждый из Х и Y обозначает СН, связь между Х и Y представляет собой одинарную связь, а R1 обозначает группу
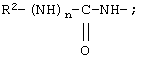
один из R3, R4 и R5 обозначает алкокси, а каждый из остальных обозначает водородный атом; n обозначает 0, например Penta-цикло(Asp-Lys)-Asp-4-MeOApc-(D)Phe-Arg-Trp-Lys-NH2, Penta-цикло(Asp-Lys)-Asp-4-EtOApc-(D)Phe-Arg-Trp-Lys-NH2, Penta-цикло(Asp-Lys)-Asp-4-изо-PrOApc-(D)Phe-Arg-Trp-Lys-NH2, Penta-цикло(Asp-Lys)-Asp-3-MeOApc-(D)Phe-Arg-Trp-Lys-NH2, Penta-цикло(Asp-Lys)-Asp-4-OHApc-(D)Phe-Arg-Trp-Lys-NH2 или Penta-цикло(Аsp-Lys)-Asp-4-ClApc-(D)Phe-Arg-Trp-Lys-NH2.
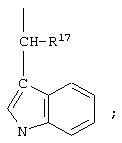
Варианты соединений формулы IA включают те соединения, у которых каждый из R1, R3, R4, R5, R8 и R10 обозначает водородный атом, R7 обозначает NH, R9 обозначает группу
R17 обозначает водородный атом или (низш.)алкил, предпочтительно метил, а p обозначает 0, например цикло(янтарная кислота-Lys)-янтарная кислота-Арс-(D)Phe-Arg-Trp-Lys-NH2, цикло(малеиновая кислота-Lys)-малеиновая кислота-Apc-(D)Phe-Arg-Trp-Lys-NH2, цикло(янтарная киcлoтa-Dpr)-янтapнaя кислота-Apc-(D)Phe-Arg-Trp-Dpr-NH2, цикло(малеиновая кислота-Dpr)-малеиновая кислота-Apc-(D)Phe-Arg-Trp-Dpr-NH2.
В другом варианте соединения формулы IA R1 и R12 совместно с Х и Y образуют фенильное кольцо. Примеры таких соединений включают цикло(фталевая кислота-Lys)-фталевая кислота-Apc-(D)Phe-Arg-Trp-Lys-NH2, цикло(фталевая кислота-Dpr)-фталевая кислота-Apc-(D)Phe-Arg-Trp-Dpr-NH2 и Ac-Nle-цикло(Cys-Cys)-Cys-Apc-(D)Phe-Arg-Trp-Cys-NH2.
Соединения формулы IB представлены формулой
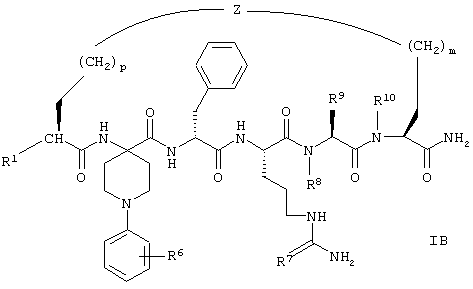
В соединениях формулы IB, включая их фармацевтически приемлемые соли, R1 обозначает водородный атом или группу

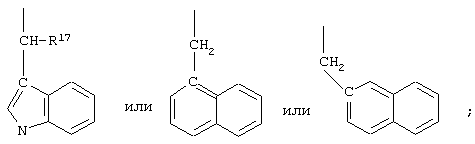
R2 обозначает алкил, содержащий от 1 до 5 углеродных атомов, алкенил, содержащий от 2 до 5 углеродных атомов, или алкинил, содержащий от 2 до 5 углеродных атомов. R14 обозначает алкил, содержащий от 1 до 5 углеродных атомов. n обозначает 0 или 1; R6 обозначает водородный атом, алкил, содержащий от 1 до 3 углеродных атомов, алкоксирадикал, содержащий от 1 до 3 углеродных атомов, фенокси или гало; R7 обозначает О или NH; R8 обозначает водородный атом или метил; R9 обозначает группу
R10 обозначает водородный атом или метил, р обозначает 0 или 1, m обозначает 0, 1, 2 или 3; Z обозначает группу
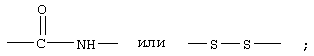
R17 обозначает водородный атом или (низш.)алкил, предпочтительно метил.
В одном из вариантов соединений формулы IB, т.е. соединений формулы IB1, Z обозначает группу
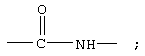
R7 обозначает NH, R1 обозначает группу
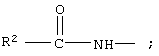
R2 обозначает алкил, каждый из R8 и R10 обозначает водородный атом, а R9 обозначает группу
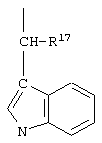
и R17 имеет указанные выше значения.
В более конкретном варианте таких соединений формулы IB1 R6 обозначает водородный атом или алкил. Примеры таких соединений включают Penta-цикло(Asp-Lys)-Asp-Appc-(D)Phe-Arg-Trp-Lys-NH2, Penta-цикло(Asp-Lys)-Asp-2-MeAppc-(D)Phe-Arg-Trp-Lys-NH2, Penta-цикло(Аsp-Lys)-Аsp-2-изо-PrАррс-(D)Phe-Arg-Trp-Lys-NH2, Penta-цикло(Asp-Lys)-Asp-3-MeAppc-(D)Phe-Arg-Trp-Lys-NH2 и Penta-цикло(Asp-Lys)-Asp-4-MeAppc-(D)Phe-Arg-Trp-Lys-NH2.
B другом более конкретном варианте таких соединений формулы IB1 R6 обозначает гало. Примеры таких соединений включают Penta-цикло(Asp-Lys)-Asp-4-ClAppc-(D)Phe-Arg-Trp-Lys-NH2.
В еще одном более конкретном варианте таких соединений формулы IB1 R6 обозначает алкокси или фенокси. Примеры таких соединений включают Penta-цикло(Asp-Lys)-Asp-4-PhOAppc-(D)Phe-Arg-Trp-Lys-NH2 и Penta-(Asp-Lys)-Asp-3-MeO-Appc-(D)Phe-Arg-Trp-Lys-NH2.
Соединения формулы IC представлены следующим образом:
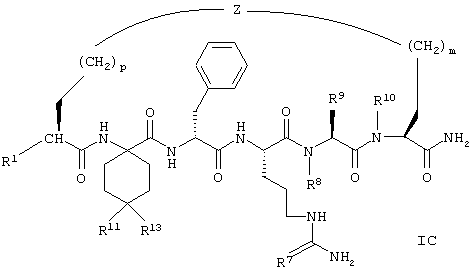
В соединениях формулы IC, включая их фармацевтически приемлемые соли, R1 обозначает водородный атом или группу
R2 обозначает алкил, содержащий от 1 до 5 углеродных атомов, алкенил, содержащий от 2 до 5 углеродных атомов, или алкинил, содержащий от 2 до 5 углеродных атомов; R14 обозначает алкил, содержащий от 1 до 5 углеродных атомов; n обозначает 0 или 1, каждый из R11 и R13 независимо обозначает водородный атом, алкил, содержащий 3 или 4 углеродных атома, циклоалкил, содержащий 5 или 6 углеродных атомов, или как R11, так и R13 обозначают фенил; R7 обозначает О или NH; R8 обозначает водородный атом или метил; R9 обозначает группу
R10 обозначает водородный атом или метил, p обозначает 0 или 1, m обозначает 0, 1, 2 или 3, a Z обозначает группу
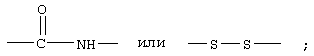
R17 обозначает водородный атом или (низш.)алкил, предпочтительно метил.
В одном из вариантов соединения формулы IC, соединения формулы IC1 Z обозначает группу
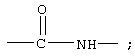
R7 обозначает NH, R1 обозначает группу
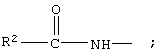
R2 обозначает алкил, каждый из R8 и R10 обозначает водородный атом, а R9 обозначает группу
В одном более конкретном варианте таких соединений формулы IC1 один из R11 и R13 обозначает алкил или циклоалкил, а другой обозначает водородный атом. Примеры таких соединений включают Penta-(Asp-Lys)-Asp-Achc-(D)Phe-Arg-Trp-Lys-NH2 и Penta-цикло(Asp-Lys)-Asp-Abc-(D) Phe-Arg-Trp-Lys-NH2.
В другом более конкретном варианте таких соединений формулы IC1 один из R11 и R13 обозначает фенил, а другой обозначает водородный атом или фенил. Примеры таких соединений включают Penta-(Asp-Lys)-Asp-4-Adpc-(D)Phe-Arg-Trp-Lys-NH2.
В одном варианте соединения формулы II, соединения формулы IIА Z обозначает группу
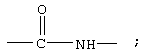
R1 обозначает группу
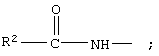
R2 обозначает алкил;
R3, R4, R5, R8 и R10 каждый обозначает водородный атом; R6 обозначает водородный атом, гало, алкил, содержащий от 1 до 3 углеродных атомов, или алкоксирадикал, содержащий от 1 до 3 углеродных атомов; R9 обозначает группу
а R17 имеет указанные выше значения.
В одном варианте соединений формулы IIA, как это изложено в предыдущем абзаце, R7 обозначает NH. В более конкретном варианте R7 обозначает NH, a R6 обозначает водородный атом или алкил. Примеры таких соединений включают Penta-цикло(Asp-Lys)-Asp-(D,L)-Atc-(D)Phe-Arg-Trp-Lys-NH2, Penta-(Asp-Lys)-Asp-5-Me-(D,L)Atc-(D)Phe-Arg-Trp-Lys-NH2, Penta-(Asp-Lys)-Asp-5-Et-(D,L)Atc-(D)Phe-Arg-Trp-Lys-NH2 и Penta-(Asp-Lys)-Asp-5-изо-Pr-(D,L)Atc-(D)Phe-Arg-Trp-Lys-NH2.
B другом конкретном варианте соединения формулы IIA R7 обозначает NH, а R6 обозначает гало. Примеры таких соединений включают Penta-(Asp-Lys)-Asp-5-BrAtc-(D)Phe-Arg-Trp-Lys-NH2 и Penta-(Asp-Lys)-Asp-5-ClAtc-(D)Phe-Arg-Trp-Lys-NH2.
В еще одном конкретном варианте соединения формулы IIA R7 обозначает NH, a R6 обозначает алкокси. Примерами служат Penta-(Asp-Lys)-Asp-5-MeO-(D,L)Atc-(D)Phe-Arg-Trp-Lys-NH2, Penta-(Asp-Lys)-Asp-5-EtO-(D,L)Atc-(D)Phe-Arg-Trp-Lys-NH2, Penta-(Asp-Lys)-Asp-5-изо-PrO-(D,L)Atc-(D)Phe-Arg-Trp-Lys-NH2.
Другой вариант соединений формулы II составляют соединения формулы II, в которой Z, R1-R5 и R8-R10 имеют значения, указанные выше, R7 обозначает О, а R6 обозначает гало. Примеры таких соединений включают Penta-(Asp-Lys)-Asp-5-BrAtc-(D)Phe-Cit-Trp-Lys-NH2 и Penta-(Asp-Lys)-Asp-5-ClAtc-(D)Phe-Cit-Trp-Lys-NH2.
В другом варианте соединений формулы II соединения формулы IIB представляют собой те соединения, у которых Z обозначает -S-S-, R1 обозначает группу

R3, R4, R5, R8 и R10 каждый обозначает водородный атом, R6 обозначает водородный атом или гало; R7 обозначает NH, R9 обозначает группу
а R17 имеет указанные выше значения.
Примеры таких соединений формулы IIВ включают Ac-Nle-цикло(Cys-Cys)-Cys-(D,L)Atc-(D)Phe-Arg-Trp-Cys-NH2 и Penta-цикло(Cys-Cys)-Cys-5-Br(D,L)Atc-(D)Phe-Arg-Trp-Cys-NH2.
По настоящему изобретению предлагаются также следующие соединения:
Penta-цикло(Asp-Lys)-Asp-Apc-(D)Phe-Ala-Trp-Lys-NH2 и Penta-цикло(Asp-Lys)-Asp-Apc-(D)Phe-Arg-(2S,3S)-бета-метил-Trp-Lys-NH2.
Для определения таких пептидов используют ту номенклатуру, которую, как правило, применяют в данной области техники, в соответствии с которой аминогруппа на N-конце находится слева, а карбоксильная группа на С-конце находится справа. Под природными аминокислотами подразумевают встречающиеся в природе аминокислоты, которые содержатся в белках, т.е. Gly, Ala, Val, Leu, Ile, Ser, Thr, Lys, Arg, Asp, Asn, Glu, Gln, Cys, Met, Phe, Tyr, Pro, Trp и His. Когда у аминокислоты имеются изомерные формы, во всех случаях, если не указано иное, представлена именно L-форма аминокислоты.
Для обозначения аминокислот, защитных групп, растворителей, реагентов и т.п. использованы следующие аббревиатуры или символы.
β -Ala - бета-Alanine,
(2)-Nal - (2)-нафтилаланин,
Atс-2 - аминотетралин-2-карбоновая кислота,
5-BrAtc - 5-бром-2-аминотетралин-2-карбоновая кислота,
5-ClAtc - 5-хлор-2-аминотетралин-2-карбоновая кислота,
5-MeOAtc - 5-метокси-2-аминотетралин-2-карбоновая кислота,
5-EtOAtc - 5-этокси-2-аминотетралин-2-карбоновая кислота,
5-изо-Рr - 5-изопропокси-2-аминотетралин-2-карбоновая кислота,
5-MeAtc - 5-метил-2-аминотетралин-2-карбоновая кислота,
5-EtAtc - 5-этил-2-аминотетралин-2-карбоновая кислота,
5-изо-Рr - 5-изопропил-2-аминотетралин-2-карбоновая кислота,
5-DmaAtc - 5-диметиламино-2-аминотетралин-2-карбоновая кислота,
DBr - D-2,4-диаминомасляная кислота,
DPr - D-2,3-диаминопропионовая кислота,
Sar - саркозин (N-метилглицин),
Cit - цитрулин,
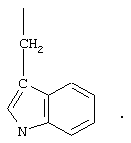
Арс - 1-амино-4-фенилциклогексан-1-карбоновая кислота,
4-НОАрс - 1-амино-4-(4-гидроксифенил)циклогексан-1-карбоновая кислота,
4-МеОАрс - 1-амино-4-(4-метоксифенил)циклогексан-1-карбоновая кислота,
3-МеОАрс - 1-амино-4-(3-метоксифенил)циклогексан-1-карбоновая кислота,
4-ЕtOАрс - 1-амино-4-(4-этоксифенил)циклогексан-1-карбоновая кислота,
4-изо-Рr - 1-амино-4-(4-изопропоксифенил)циклогексан-1-карбоновая кислота,
4-МеАрс - 1-амино-4-(4-метилфенил)циклогексан-1-карбоновая кислота,
4-ClApc - 1-амино-4-(4-хлорфенил)циклогексан-1-карбоновая кислота,
Аррс - 4-амино-1-фенилпиперидин-4-карбоновая кислота,
2-МеАррс - 4-амино-1-(2-метилфенил)пиперидин-4-карбоновая кислота,
2-изо-ProAppc - 4-амино-1 -(2-изопропоксифенил)пиперидин-4-карбоновая кислота,
3-МеАррс - 4-амино-1-(3-метилфенил)пиперидин-4-карбоновая кислота,
3-МеОАррс - 4-амино-1-(3-метоксифенил)пиперидин-4-карбоновая кислота,
4-МеАррс - 4-амино-1-(4-метилфенил)пиперидин-4-карбоновая кислота,
4-ClAppc - 4-амино-1-(4-хлорфенил)пиперидин-4-карбоновая кислота,
4-PhOAppc - 4-амино-1-(4-феноксифенил)пиперидин-4-карбоновая кислота,
Ache - 1-амино-4-циклогексилциклогексан-1-карбоновая кислота,
Adpc - 1-амино-4-дифенилциклогексан-1-карбоновая кислота,
Abс - 1-амино-4-трет-бутилциклогексан-1-карбоновая кислота,
3-Amb - 3-аминометилбензойная кислота,
4-Amb - 4-аминометилбензойная кислота,
2-Аbа - 2-аминобензойная кислота,
Вu - бутил,
Penta - пентаноил,
ФМОК - 9-флуоренилметоксикарбонил,
Пмх - 2,2,5,7,8-пентаметилхроман-6-сульфонил,
Trt - тритил (трифенилметил),
CH2Cl2 - метиленхлорид,
CH3CN - ацетонитрил,
ДМФ - диметилформамид,
ДИПЭА - N,N-диизопропилэтиламин,
ТФК - трифторуксусная кислота,
ГОБТ - N-гидроксибензотриазол,
ДИК - N,N’-диизопропилкарбодиимид,
БОФ - бензотриазол-1-илокси-трис-(диметиламино)фосфонийгексафторфосфат,
ПиБроФ - бром-трис-пирролидинофосфонийгексафторфосфат,
БТУГ - 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилуронийгексафторфосфат,
МС-БУА - масс-спектроскопия с бомбардировкой ускоренными атомами,
МС-ПЭ - масс-спектроскопия с ионизацией пучком электронов.
Замещенные аминокислоты, расположенные в круглых скобках, указывают на аналоги в пептидной последовательности. Замещение N-концевой аминогруппы указывают слева от N-концевого остатка, отделяя его разделительной черточкой. Другими словами, например, формула Ac-His-(D)Phe-Arg-Trp-Gly-NH2 означает пептид, в аминокислотной последовательности которого на N-конце ацетильная группа замещает водородный атом. Суффиксами "-ОН" и "-NH2", которые следуют за разделительными черточками или скобками, обозначают соответственно свободную кислотную и амидную группы полипептида.
Линейные пептиды, используемые в качестве предшественников для типичных предлагаемых соединений, могут быть легко синтезированы по любому обычному из известных методов формирования пептидной связи между аминокислотами. Такие обычные методы включают, например, любой метод в растворе, осуществление которого позволяет проводить реакцию конденсации между свободной альфа-аминогруппой аминокислоты или ее остатком, у которого карбоксильная группа или другие реакционноспособные группы защищены, и свободной первичной карбоксильной группой другой аминокислоты или ее остатка, у которого ее аминогруппа или другие реакционноспособные группы защищены.
Процесс синтеза линейных пептидов можно проводить по методу, при осуществлении которого каждую аминокислоту по одной последовательно присоединяют к другой аминокислоте или ее остатку, или по методу, при осуществлении которого вначале обычным путем синтезируют пептидные фрагменты, а затем проводят реакцию их конденсации с получением целевого пептида.
Такие обычные методы синтеза линейных пептидов-предшественников включают, например, любой твердофазный метод синтеза пептидов. В таком методе синтез новых соединений можно проводить последовательно поочередным введением в растущую пептидную цепь целевых остатков аминокислот в соответствии с основными принципами твердофазных методов синтеза [Merrifield R.B., J. Amer.Chem.Soc., 1963, 85, 2149-2154; Ваrаnу и др. The peptides, Analysis, Synthesis and Biology, том 2, Gross E. и Meienhofer J., Eds.Academic Press, 1-284 (1980)].
Общим для химических синтезов пептидов является защита реакционноспособных групп боковых цепей аминокислотных остатков приемлемыми защитными группами, которые обычно препятствуют химическому взаимодействию на данных центрах до тех пор, пока защитная группа не будет удалена. Обычно также общей является защита альфа-аминогруппы аминокислоты или ее фрагмента до завершения реакции по карбоксильной группе, после чего селективно удаляют защитную группу альфа-аминогруппы и тем самым создают возможность протекания дальнейшей реакции на данном центре. Несмотря на то, что эти конкретные защитные группы представлены применительно к твердофазному методу синтеза, необходимо отметить, что любая аминокислота может быть защищена защитной группой, которую обычно используют для соответствующей аминокислоты при синтезе в растворе.
Альфа-аминогруппы можно защищать приемлемой защитной группой, выбранной из ароматических защитных групп уретанового типа, таких, как бензилоксикарбонил (Z) и замещенный бензилоксикарбонил, в частности п-хлорбензилоксикарбонил, п-нитробензилоксикарбонил, п-бромбензилоксикарбонил, п-дифенилизопропоксикарбонил, 9-флуоренилметоксикарбонил (ФМОК) и п-метоксибензилоксикарбонил (МОБ); алифатических защитных групп уретанового типа, таких, как трет-бутилоксикарбонил (БОК), диизопропилметоксикарбонил, изопропоксикарбонил и аллилоксикарбонил. При этом для защиты альфа-аминогруппы наиболее предпочтительна группа ФМОК.
Гуанидиновые группы могут быть защищены приемлемой защитной группой, выбранной из нитрогруппы, п-толуолсульфонила (Toc), Z, пентаметилхромансульфонила (Пмх), адамантилоксикарбонила и БОК. Самой предпочтительной для аргинина (Arg) является группа Пмх.
Все растворители, изопропанол (изо-РrОН), метиленхлорид (CH2Cl2), диметилформамид (ДМФ) и N-метилпирролидинон (N-МПН) приобретали на фирме Fisher или Burdick & Jackson и использовали без дополнительной перегонки. Трифторуксусную кислоту приобретали на фирме Halocarbon или Fluka и использовали без дополнительной очистки. Диизопропилкарбодиимид (ДИК) и диизопропилэтиламин (ДИПЭА) приобретали на фирме Fluka или Aldrich и использовали без дополнительной очистки. Гидроксибензотриазол (ГОБТ), диметилсульфид (ДМС) и 1,2-этандитиол (ЭДТ) приобретали на фирме Sigma Chemical Co. и использовали без дополнительной очистки. Обычно использовали защищенные аминокислоты L-конфигурации, которые получали промышленным путем на фирме Bachem, Advanced ChemTech или Neosystem. Степень чистоты этих реагентов перед применением проверяли тонкослойной хроматографией, ЯМР-анализом и по температуре плавления. Бензгидриламиновая смола (БГА) представляла собой сополимер стирола и 1% дивинилбензола (частицы размером от 100 до 200 или от 200 до 400 меш), полученный на фирме Bachem или Advanced ChemTech. Общее содержание азота в этих смолах обычно находилось в пределах от 0,3 до 1,2 мэкв/г.
Высокоэффективную жидкостную хроматографию (ВЭЖХ) осуществляли в приборе LDC, включавшем насосы Constametric I и III, программатор Gradient Master для растворителя и смеситель, а также УФ-детектор с варьируемой длиной волны устройства Spectromonitor III. Процедуры ВЭЖХ анализа проводили по методу с обращенной фазой с применением колонок Vydac C18 (0,4× 30 см). Процессы разделения препаративной ВЭЖХ проводили в колонках Vydac (2× 25 см).
В предпочтительном варианте линейные пептиды получали с использованием твердофазного синтеза по методу, который в общем описан в работе Merrifield [J. Amer.Chem.Soc., 1963, 85, 2149], хотя, как упомянуто выше, можно было бы использовать и другой эквивалентный химический синтез, известный в данной области техники. Твердофазный синтез начинают с С-концевого фрагмента пептида реакцией конденсации защищенной альфа-аминокислоты с приемлемой смолой. Такой исходный материал может быть приготовлен присоединением аминокислоты с защищенной альфа-аминогруппой с помощью сложноэфирной связи к смоле п-бензилоксибензилового спирта (по Wang) или посредством амидной связи между ФМОК-мостиком, таким, как п-[(R,S)-α -[1-(9Н-флуорен-9-ил)метоксиформамидо]-2,4-диметилоксибензил]феноксиуксусная кислота (мостик Rink), и бензгидриламиновой (БГА) смолой. Процесс получения гидроксиметиловой смолы в данной области техники известен хорошо. Подложки ФМОК-мостик-БГА смола технически доступны, их обычно используют, когда у целевого синтезируемого пептида при С-концевом фрагменте имеется незамещенный амид.
Поскольку соединения по настоящему изобретению представляют собой циклические пептиды, получаемые с образованием лактамной или дисульфидной связи, линейные пептиды-предшественники конструируют таким образом, чтобы разместить приемлемые аминокислоты или миметики, несущие соответствующие остатки боковых цепей в линейных пептидах в таких положениях, благодаря которым в конечном счете создается возможность принудительного образования внутримолекулярной амидной связи или дисульфидной связи. Лактамы образуются вследствие реакции конденсации функциональной аминогруппы боковой цепи С-концевого аминокислотного остатка с периферическим карбоксильным остатком при одновременном образовании дисульфидной связи вследствие окислительной реакции конденсации двух цистеиновых остатков, соответствующим образом введенных по месту С-концевого фрагмента и по месту или вблизи N-концевого фрагмента линейного пептида-предшественника. Так, например, в процессе получения лактамовых пептидов в линейных пентапептидах-предшественниках N-концевой фрагмент может быть использован в качестве матрицы для введения карбоксильного остатка, в частности в Х структуре, или в случае гексапептидов, пептид планируют таким образом, чтобы в качестве N-концевого аминокислотного остатка иметь возможность выбрать одну из аминокислот, содержащих подходящим образом защищенную карбоксильную группу боковой цепи, например аспарагиновую кислоту, глутаминовую кислоту. В линейных гептапептидах аспарагиновую кислоту или глутаминовую кислоту вводят в виде предпоследнего остатка относительно N-концевого остатка. Во всех линейных пептидах-предшественниках, т.е. в гепта-, гекса- и пентапептидах, С-концевой остаток выбирают из остатков природных или неприродных аминокислот, несущих соответствующим образом защищенный основный остаток боковой цепи, способный при удалении защитной группы образовывать амидную связь, например лизина, орнитина, 2,3-диаминопропионовой кислоты, 2,4-диаминомасляной кислоты. Для образования циклического пептида, содержащего дисульфидную связь, когда предшественник представляет собой линейный гексапептид, этот пептид конструируют таким путем, чтобы ввести соответствующим образом S-защищенный цистеиновый остаток как в С-, так и N-концевые остатки, тогда как если предшественник представляет собой линейный гептапептид, соответствующим образом S-защищенные цистеиновые остатки вводят как в С-концевой остаток, так и в предпоследний относительно N-концевого остатка, как, например, в X.
Обычно для получения линейных пептидов аминокислоты или миметики конденсируют с образованием системы ФМОК-мостик-БГА смола, используя защищенную ФМОК форму аминокислоты или миметика с применением от 2 до 5 экв. аминокислоты и приемлемого конденсирующего реагента. После конденсации смолу можно промывать и сушить под вакуумом. Содержание аминокислоты в смоле можно установить аминокислотным анализом аликвоты ФМОК-аминокислотной смолы или УФ-анализом содержания группы ФМОК. Все непрореагировавшие аминогруппы можно заблокировать реакцией смолы с уксусным ангидридом и диизопропилэтиламином в метиленхлориде.
Для последовательного присоединения аминокислот смолы проводят через несколько повторных циклов. Защитные группы ФМОК для альфа-аминогрупп удаляют в основных условиях. С этой целью можно использовать пиперидин, пиперазин или морфолин (от 20 до 40 об. %) в ДМФ. В предпочтительном варианте используют 40%-ный пиперидин в ДМФ.
После удаления защитной группы у альфа-аминогруппы в дальнейшем защищенные аминокислоты постадийно в целевом порядке вводят в реакцию конденсации с получением промежуточного продукта, защищенной пептид-смолы. Активирующие реагенты, используемые для реакции конденсации аминокислот в твердофазном синтезе пептидов, в данной области техники известны хорошо. Так, например, соответствующими реагентами для таких синтезов служат бензотриазол-1-илокситри(диметиламино)фосфонийгексафторфосфат (БОФ), бром-трис-пирролидинфосфонийгексафторфосфат (ПиБроФ), 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилуронийгексафторфосфат (БТУГ) и диизопропилкарбодиимид (ДИК). При этом предпочтительны БТУГ и ДИК. Могут быть использованы и другие активирующие агенты, как они представлены в работе Ваrаnу и Merrifield [The Peptides, том 2, J. Meienhofer, ed., Academic Press, 1979, сс. 1-284]. Для оптимизации циклов синтеза в смеси реакций конденсации можно добавлять различные реагенты, такие, как 1-гидроксибензотриазол (ГОБТ), N-гидроксисукцинимид (ГОСу) и 3,4-дигидро-3-гидрокси-4-оксо-1,2,3-бензотриазин (ГООБТ). При этом предпочтителен ГОБТ. В таблице 1 приведен протокол 1 типичного цикла синтеза.

Растворители для всех промывных жидкостей и реакций конденсации приводили в соответствие с объемами от 10 до 20 мл/г смол. Во время всего синтеза для определения степени завершения за ходом реакций конденсации следили с помощью нингидринового теста Kaiser [Kaiser и др. Anal.Biochem., 1970, 34, 595-598]. В случаях ФМОК-Arg (Пмх) и в случаях реакций конденсации вторичных аминов с пространственно затрудненными кислотами отмечали низкую скорость реакций. В случаях всех незавершенных реакций конденсации либо их проводили повторно с использованием свежеполученной активированной аминокислоты, либо непрореагировавшие группы блокировали обработкой пептид-смол уксусным ангидридом, как это представлено выше. Полностью прореагировавшие пептид-смолы сушили под вакуумом в течение нескольких часов.
У каждого соединения удаляли защитные группы и линейный пептид отщепляли от смолы согласно следующему методу. В общем пептид-смолу при комнатной температуре в течение 120 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 300 мкл анизола и 9,5 мл трифторуксусной кислоты на грамм смолы. Смолу отфильтровывали и в фильтратах проводили осаждение в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O и повторно центрифугировали. При необходимости сырые линейные пептиды очищали препаративной ВЭЖХ. Пептиды вводили в колонки в минимальном объеме либо АсОН/Н2О, либо 0,1% ТФК/Н2О. Градиентное элюирование обычно начинали при 10% буфера Б, затем содержание буфера Б в течение 90 мин повышали от 10 до 60% (буфер А: 0,1% ТФК/Н2O, буфер Б: 0,1% ТФК/СН3СN) при скорости потока 8 мл/мин. УФ-анализ осуществляли при 280 нм. Фракции собирали через интервалы от 1,0 до 2,5 мин и исследовали аналитической ВЭЖХ. Фракции, которые оценивали как высокочистые, объединяли и лиофилизировали.
Для получения лактамов соответствующий неочищенный линейный пептид растворяли в приемлемом инертном растворителе, например в N-метилпирролидоне или ДМФ, предпочтительно в ДМФ, и добавлением третичного аминового основания, например N-метилморфолина, рН доводили до явного значения 8,0, а затем обрабатывали реагентом, образующим амиднуго связь, предпочтительно БОФ. Эту реакцию удобно проводить при температуре в пределах 40 и 0° С, предпочтительно при примерно комнатной температуре. Очистку сырых циклических пептидов проводили препаративной ВЭЖХ. Градиентное элюирование обычно начинали при 20% буфера Б, затем его содержание в течение 90 мин повышали от 20 до 60% (буфер А: 0,1% ТФК/Н2О, буфер Б: 0,1% ТФК/СН3СN при скорости потока 8 мл/мин. УФ-анализ осуществляли при 280 нм. Фракции собирали и анализировали аналитической ВЭЖХ. Фракции, которые оценивали как высокочистые, объединяли и лиофилизировали.
Для получения циклических дисульфидных пептидов очищенный с помощью ВЭЖХ линейный пептид, содержавший два находившихся в соответствующих положениях цистеиновых остатка, растворяли с довольно высокой степенью разбавления в смеси приемлемых инертных растворителей, например в водном ДМСО, и осторожным добавлением гидроксида аммония рН раствора доводили до 8,0. Затем через перемешиваемый раствор барботировали кислород. Эту реакцию было удобно проводить при температуре в пределах 40 и 0° С, предпочтительно при примерно комнатной температуре, и посредством аналитической ВЭЖХ следили за ходом циклизации. После того как реакцию считали завершенной, раствор лиофилизировали и сырой циклический пептид очищали препаративной ВЭЖХ. Градиентное элюирование обычно начинали при 20% буфера Б, затем его содержание в течение 90 мин повышали от 20 до 60% (буфер А: 0,1% ТФК/Н2О, буфер Б: 0,1% ТФК/CH3CN) при скорости потока 8 мл/мин. УФ-анализ осуществляли при 280 нм. Фракции, которые оценивали как высокочистые, объединяли и лиофилизировали.
Очистку сырых пептидов проводили препаративной ВЭЖХ. Пептиды вводили в колонки в минимальном объеме либо АсОН/Н2О, либо 0,1% ТФК/Н2О. Градиентное элюирование обычно начинали при 10% буфера Б, затем его содержание в течение 90 мин повышали от 10 до 60% (буфер А: 0,1% ТФК/Н2О, буфер Б: 0,1% ТФК/СН3СN) при скорости потока 8 мл/мин. УФ-анализ осуществляли при 280 нм. Фракции собирали через интервалы от 1,0 до 2,5 мин и исследовали аналитической ВЭЖХ. Фракции, которые оценивали как высокочистые, объединяли и лиофилизировали.
Чистоту конечных продуктов проверяли, как сказано выше, аналитической ВЭЖХ в колонке с обращенной фазой. Чистоту всех продуктов оценивали как составлявшую приблизительно от 95 до 99%. Все конечные продукты подвергали также масс-спектроскопии с бомбардировкой ускоренными атомами (МС-БУА) или масс-спектроскопии с ионизацией пучком электронов (МС-ПЭ). Все продукты в приемлемых пределах образовывали ожидаемые исходные М+Н ионы.
Процесс синтеза линейных пептидов можно проводить по методу, при осуществлении которого каждую аминокислоту по одной последовательности присоединяют к другой аминокислоте или ее остатку, или по методу, при осуществлении которого вначале обычным путем синтезируют пептидные фрагменты, а затем проводят реакцию их конденсации с получением целевого пептида.
Такие обычные методы синтеза линейных пептидов-предшественников включают, например, любой твердофазный метод синтеза пептидов. В таком методе синтез новых соединений можно проводить последовательно поочередным введением в растущую пептидную цепь целевых остатков аминокислот в соответствии с основными принципами твердофазных методов синтеза [Merrifield R.B., J.Amer.Chem.Soc., 1963, 85, 2149-2154; Barany и др. The peptides, Analysis, Synthesis and Biology, том 2, Gross E. и Meienhofer J., Eds.Academic Press, 1-284 (1980)].
Общим для химических синтезов пептидов является защита реакционноспособных групп боковых цепей аминокислотных остатков приемлемыми защитными группами, которые обычно препятствуют химическому взаимодействию на данных центрах до тех пор, пока защитная группа не будет удалена. Обычно также общей является защита альфа-аминогруппы аминокислоты или ее фрагмента до завершения реакции по карбоксильной группе, после чего селективно удаляют защитную группу альфа-аминогруппы и тем самым создают возможность протекания дальнейшей реакции на данном центре. Хотя применительно к твердофазному методу синтеза конкретные защитные группы уже описаны, необходимо отметить, что каждая аминокислота может быть защищена защитной группой, которую обычно используют для соответствующей аминокислоты в синтезе в растворной фазе.
Альфа-аминогруппы могут быть защищены приемлемой защитной группой, выбранной из ароматических защитных групп уретанового типа, таких, как бензилоксикарбонил (Z) и замещенный бензилоксикарбонил, в частности п-хлорбензилоксикарбонил, п-нитробензилоксикарбонил, п-бромбензилоксикарбонил, п-дифенилизопропоксикарбонил, 9-флуоренилметоксикарбонил (ФМОК) и п-метоксибензилоксикарбонил (МОЕ); алифатических защитных групп уретанового типа, таких, как трет-бутилоксикарбонил (БОК), диизопропилметоксикарбонил, изопропоксикарбонил, аллилоксикарбонил. При этом для защиты альфа-аминогруппы наиболее предпочтительной является группа ФМОК.
Гуанидиновые группы могут быть защищены приемлемой защитной группой, выбранной из нитрогруппы, п-толуолсульфонила (Toc), Z, пентаметилхромансульфонила (Пмх), адамантилоксикарбонила, БОК. Для аргинина (Arg) наиболее предпочтителен Пмх.
Все растворители, изопропанол (изо-РrОН), метиленхлорид (CH2Cl2), диметилформамид (ДМФ) и N-метилпирролидинон (N-МПН) приобретали на фирме Fisher или Burdick & Jackson и использовали без дополнительной перегонки. Трифторуксусную кислоту приобретали на фирме Halocarbon или Fluka и использовали без дополнительной очистки. Диизопропилкарбодиимид (ДИК) и диизопропилэтиламин (ДИПЭА) приобретали на фирме Fluka или Aldrich и использовали без дополнительной очистки. Гидроксибензотриазол (ГОБТ), диметилсульфид (ДМС) и 1,2-этандитиол (ЭДТ) приобретали на фирме Sigma Chemical Co. и использовали без дополнительной очистки. Обычно использовали защищенные аминокислоты L-конфигурации, которые получали промышленным путем на фирме Bachem, Advanced ChemTech или Neosystem. Степень чистоты этих реагентов перед применением проверяли тонкослойной хроматографией, ЯМР-анализом и по температуре плавления. Бензгидриламиновая смола (БГА) представляла собой сополимер стирола и 1% дивинилбензола (частицы размером от 100 до 200 или от 200 до 400 меш), полученный на фирме Bachem или Advanced ChemTech. Общее содержание азота в этих смолах обычно находилось в пределах от 0,3 до 1,2 мэкв/г.
Высокоэффективную жидкостную хроматографию (ВЭЖХ) осуществляли в приборе LDC, включавшем насосы Constametric I и III, программатор Gradient Master для растворителя и смеситель, а также УФ-детектор с варьируемой длиной волны устройства Spectromonitor III. Процедуры ВЭЖХ анализа проводили по методу с обращенной фазой с применением колонок Vydac C18 (0,4× 30 см). Процессы разделения препаративной ВЭЖХ проводили в колонках Vydac (2× 25 см).
В предпочтительном варианте пептиды получали с использованием твердофазного синтеза по методу, который в общем описан в работе Merrifield [J. Amer.Chem.Soc., 1963, 85, 2149], хотя, как упомянуто выше, можно было бы использовать и другой эквивалентный химический синтез, известный в данной области техники. Твердофазный синтез начинали с С-концевого фрагмента пептида реакцией конденсации защищенной альфа-аминокислоты с приемлемой смолой. Такой исходный материал может быть приготовлен присоединением аминокислоты с защищенной альфа-аминогруппой с помощью сложноэфирной связи к смоле п-бензилоксибензилового спирта (по Wang) или посредством амидной связи между ФМОК мостиком, таким, как п-[(R,S)-α -[1-(9Н-флуорен-9- ил)метоксиформамидо]-2,4-диметилоксибензил]феноксиуксусная кислота (мостик Rink), и бензгидриламиновой (БГА) смолой. Процесс получения гидроксиметиловой смолы в данной области техники известен хорошо. Подложки ФМОК-мостик-БГА смола технически доступны, их обычно используют, когда у целевого синтезируемого пептида при С-концевом фрагменте имеется незамещенный амид.
Обычно аминокислоты или миметики конденсировали с получением системы ФМОК-мостик-БГА смола, используя защищенную ФМОК-форму аминокислоты или миметика с применением от 2 до 5 экв. аминокислоты и приемлемого конденсирующего реагента. После конденсации смолу можно промывать и сушить под вакуумом. Содержание аминокислоты в смоле можно установить аминокислотным анализом аликвоты ФМОК-аминокислотной смолы или УФ-анализом содержания группы ФМОК. Все непрореагировавшие аминогруппы можно заблокировать реакцией смолы с уксусным ангидридом и диизопропилэтиламином в метиленхлориде.
Для последовательного присоединения аминокислот смолы проводили через несколько повторных циклов. Защитные группы ФМОК для альфа-аминогрупп удаляли в основных условиях. С этой целью можно использовать пиперидин, пиперазин или морфолин (от 20 до 40 об. %) в ДМФ. В предпочтительном варианте использовали 40% пиперидин в ДМФ.
После удаления защитной группы у альфа-аминогруппы в дальнейшем защищенные аминокислоты постадийно в целевом порядке вводили в реакцию конденсации с получением промежуточного продукта, защищенной пептид-смолы. Активирующие реагенты, используемые для реакции конденсации аминокислот в твердофазном синтезе пептидов, в данной области техники известны хорошо. Так, например, соответствующими реагентами для таких синтезов служат бензотриазол-1-илокситри(диметиламино)фосфонийгексафторфосфат (БОФ), бромтрис-пирролидинофосфонийгексафторфосфат (ПиБроФ), 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилуронийгексафторфосфат (БТУГ) и диизопропилкарбодиимид (ДИК). При этом предпочтительны БТУГ и ДИК. Могут быть использованы и другие активирующие агенты, как они представлены в работе Ваrаnу и Merrifield [The Peptides, том 2, J. Meienhofer, ed., Academic Press, 1979, ее. 1-284]. Для оптимизации циклов синтеза в смеси реакций конденсации можно добавлять различные реагенты, такие, как 1-гидроксибензотриазол (ГОБТ), N-гидроксисукцинимид (ГОСу) и 3,4-дигидро-3-гидрокси-4-оксо-1,2,3- бензотриазин (ГООБТ). При этом предпочтителен ГОБТ.
В таблице 2 приведен протокол 2 типичного цикла синтеза.
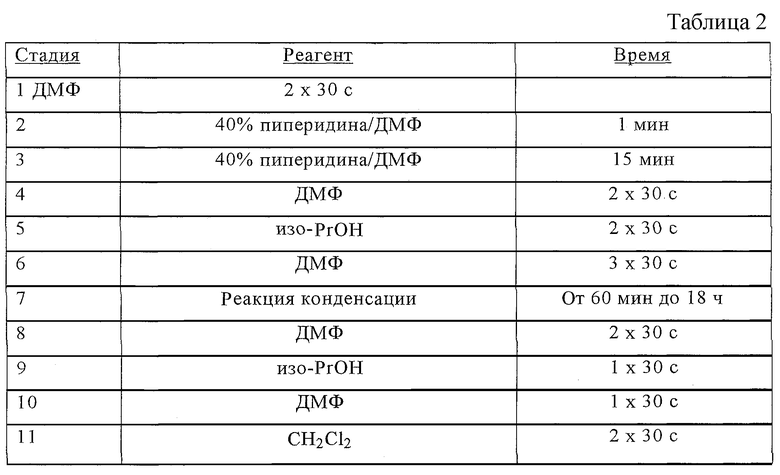
Растворители для всех промывных жидкостей и реакций конденсации приводили в соответствие с объемами от 10 до 20 мл/г смол. Во время всего синтеза для определения степени завершения за ходом реакций конденсации следили с помощью нингидринового теста Kaiser [Kaiser и др. Anal.Biochem., 1970, 34, 595-598]. В случаях ФМОК-Arg (Пмх) и в случаях реакций конденсации вторичных аминов с пространственно затрудненными кислотами отмечали низкую скорость реакций. В случаях всех незавершенных реакций конденсации либо их проводили повторно с использованием свежеполученной активированной аминокислоты, либо непрореагировавшие группы блокировали обработкой пептид-смол уксусным ангидридом, как это представлено выше. Полностью прореагировавшие пептид-смолы сушили под вакуумом в течение нескольких часов.
У каждого соединения удаляли защитные группы и пептид отщепляли от смолы согласно следующему методу. Пептид-смолу при комнатной температуре в течение 120 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 300 мкл анизола и 9,5 мл трифторуксусной кислоты на грамм смолы. Смолу отфильтровывывали и в фильтратах проводили осаждение в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O и повторно центрифугировали. Сырые продукты сушили под вакуумом.
Очистка сырых пептидных препаратов
Очистку сырых пептидов проводили препаративной ВЭЖХ. Пептиды вводили в колонки в минимальном объеме либо АсОН/Н2О, либо 0,1% ТФК/Н2О. Градиентное элюирование обычно начинали при 10% буфера Б, затем его содержание в течение 90 мин повышали от 10 до 60% (буфер А: 0,1% ТФК/Н2О, буфер Б: 0,1% TOK/CH3CN) при скорости потока 8 мл/мин. УФ-анализ осуществляли при 280 нм. Фракции собирали через интервалы от 1,0 до 2,5 мин и исследовали аналитической ВЭЖХ. Фракции, которые оценивали как высокочистые, объединяли и лиофилизировали.
Чистоту конечных продуктов проверяли, как сказано выше, аналитической ВЭЖХ в колонке с обращенной фазой. Чистоту всех продуктов оценивали как составлявшую приблизительно от 95 до 99%. Все конечные продукты подвергали также масс-спектроскопии с бомбардировкой ускоренными атомами (МС-БУА) или масс-спектроскопии с ионизацией пучком электронов (МС-ПЭ). Все продукты в приемлемых пределах образовывали ожидаемые исходные М+Н-ионы.
С помощью вышеописанной технологии соединения по настоящему изобретению могут быть синтезированы в соответствии со схемами реакций, приведенными далее.
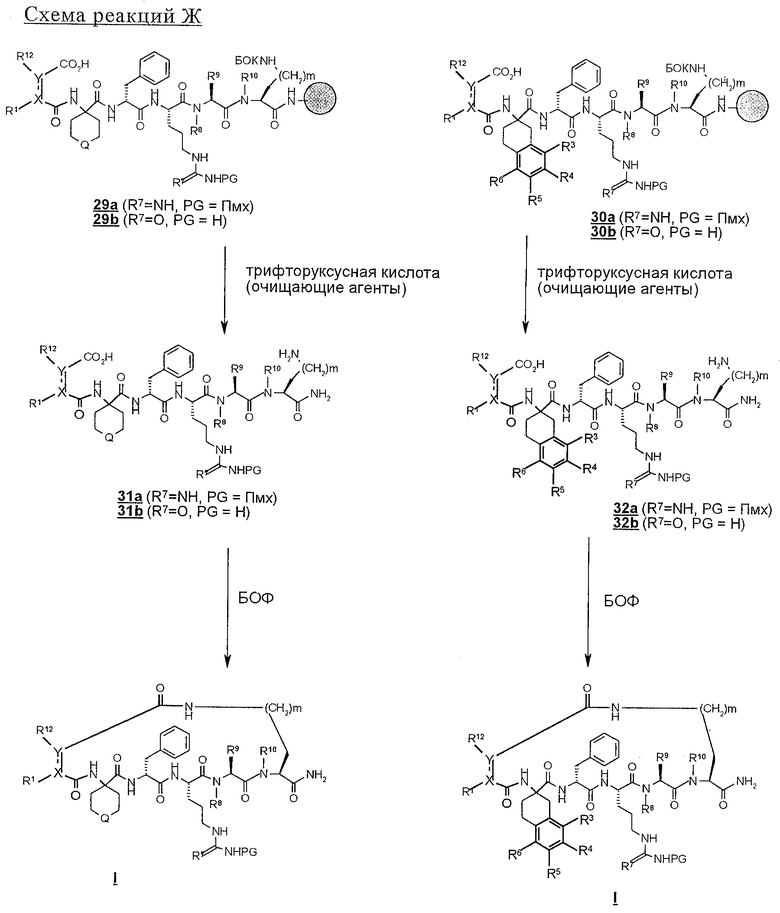

Линейные пептиды, используемые при этом в качестве предпоследних промежуточных продуктов в синтезах циклических пептидов по настоящему изобретению (строения 1), получают с применением обычной методологии твердофазного синтеза пептидов, которая обсуждалась в предыдущем разделе.
Каждый цикл состоит из двух процессов: вначале от концевого атома азота в связанной со смолой цепи отщепляют защитную группу ФМОК с последующим ацилированием аминовой функциональной группы посредством защищенной ФМОК аминокислоты. Этот цикл обычно осуществляют в соответствии с постадийными методами, представленными в общих чертах в протоколе 1. Удаление защитной группы осуществляют с использованием органического основания, например пиперазина, морфолина или пиперидина, предпочтительно пиперидина, в приемлемом инертном растворителе, например в N,N-диметилформамиде (ДМФ) или N-метилпирролидоне (N-МП), Реакцию конденсации можно проводить при одном из многих условий, подобранных для образования амидной связи, например с использованием О-бензотриазол-1-ил-N,N,N’,N’-тетраметилуронийгексафторфосфата (БТУГ) в присутствии органического основания, например диизопропилэтиламина (ДИПЭА), в инертном растворителе, в частности в ДМФ. По другому варианту в данном случае амидная группа может быть получена с использованием карбодиимида, например диизопропилкарбодиимида (ДИК), совместно с активирующим агентом, таким, как 1-гидроксибензотриазол (ГОБТ), в приемлемом инертном растворителе, таком, как ДМФ.
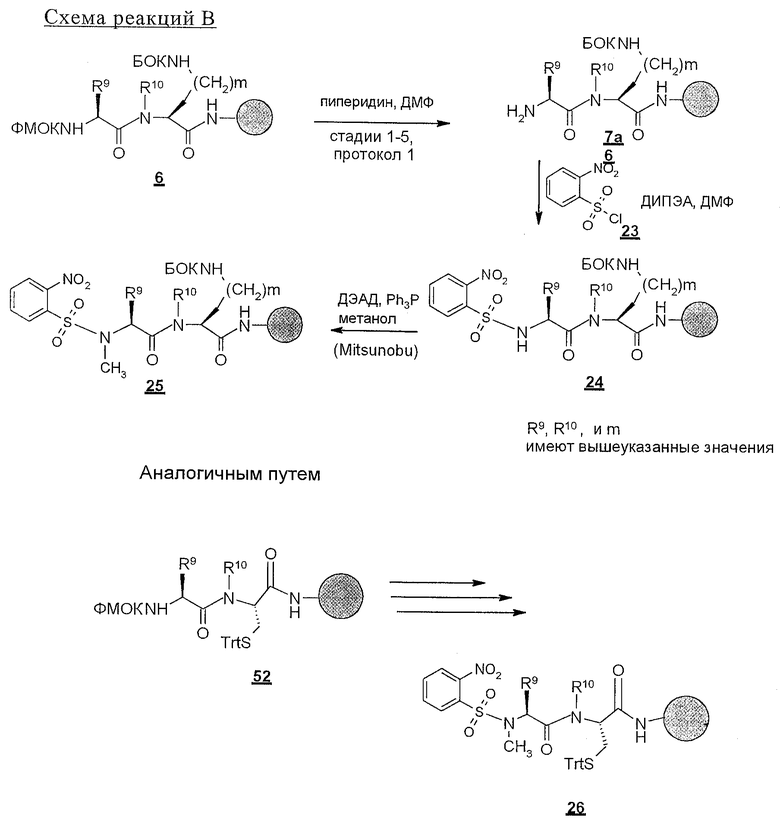
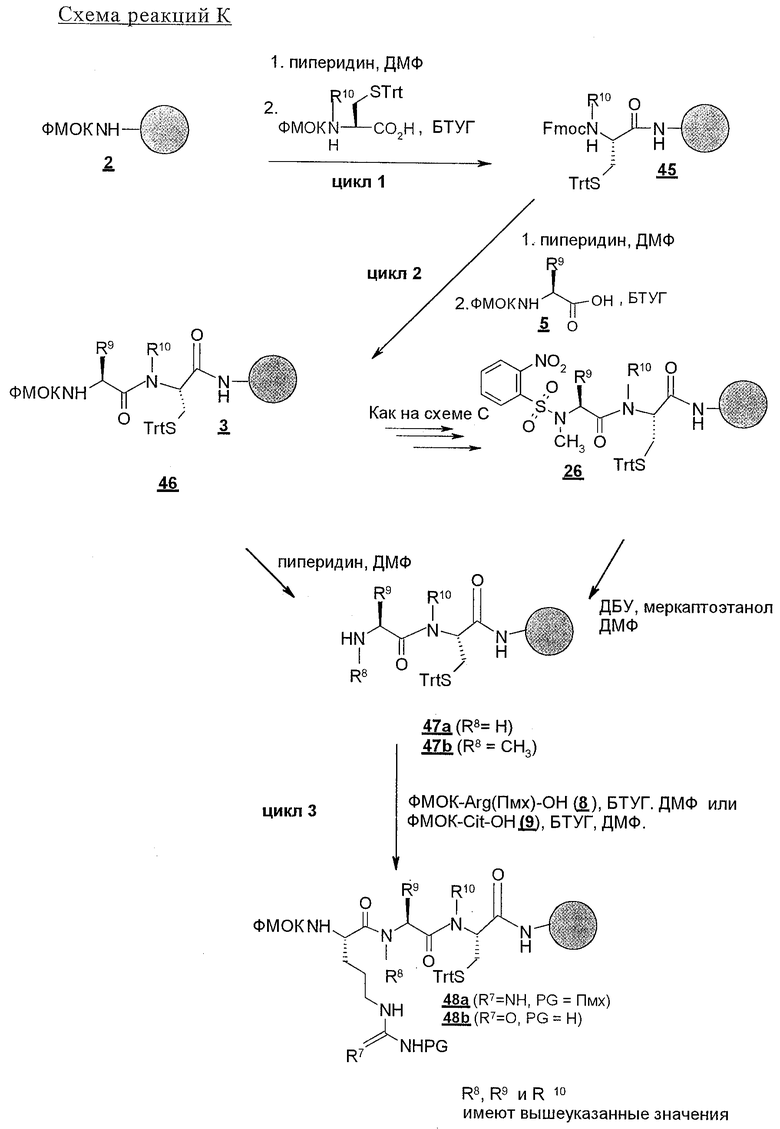
В соответствии со схемой реакций А в первом цикле получения линейных полипептидов-предшественников для циклических пептидов строения 1, где Z обозначает NHCO, у ФМОК-мостик-БГА смолы, представленной структурой 2, удаляют защитную группу и проводят реакцию конденсации с ФМОК-аминокислотами строения 3, получая связанные со смолой соединения 4. В этих синтезах циклических пептидов необходимо, чтобы ФМОК-аминокислота 3 содержала требуемый ключевой структурный компонент, который представляет собой соответствующим образом защищенную основную боковую цепь, которая, когда удаляют защитную группу, может принимать участие в образовании внутримолекулярной амидной связи. Для увеличения растущей пептидной цепи во втором цикле вводят ФМОК-аминокислоту 5 с получением соединений строения 6. В третьем цикле обработка связанного со смолой пептида 6 обеспечивает образование промежуточных продуктов строения 7а, где R8 обозначает водородный атом. Промежуточные продукты строения 7b, где R8 обозначает метил, синтезируют так, как показано на схеме С.
Промежуточные продукты строения 7b получают из соединений строения 7а так, как показано на схеме С. В этом методе проводят реакцию соединений строения 7а, полученных обработкой соединений строения 6 так, как изложено в описании стадий от 1 до 5 протокола 1, с арилсульфонилхлоридом, предпочтительно с 2-нитробензолсульфонилхлоридом 23, с получением соединений строения 24. Эту реакцию проводят в присутствии акцептора протонов, например пиридина, триэтиламина (ТЭА) или ДИПЭА, предпочтительно ДИПЭА, в приемлемом инертном растворителе, предпочтительно в ДМФ. N-Метилирование образовавшейся сульфонамидной группы в промытых связанных со смолой соединениях строения 24 осуществляют в условиях реакции Mitsunobu с получением соединения строения 25. В ходе проведения этой реакции сульфонамиды строения 24 взаимодействуют с метанолом в присутствии диэтилазодикарбоксилата (ДЭАД) и трифенилфосфина, причем в качестве растворителя используют метанол. После завершения реакции связанный со смолой N-метилсульфонамид строения 25 промывают с целью освободить от остаточных реагентов и побочных продуктов.
На последующих стадиях, как это представлено на схеме реакций А, 2-нитробензолсульфонильный остаток из структуры 25 удаляют реакцией 25 с 2-меркаптоэтанолом и сильным органическим основанием, 1,8-диазабицикло[5.4.0]ундец-7-еном (ДБУ), в приемлемом растворителе, предпочтительно в ДМФ, с получением связанного со смолой промежуточного продукта строения 7b. Третий цикл схемы реакций А завершают реакцией конденсации соединений либо строений 7а и 7b с ФMOK-Arg(Пмx)-OH (8), либо ФМОК-Cit-OH (9) с получением связанных со смолой соединений строения 10.
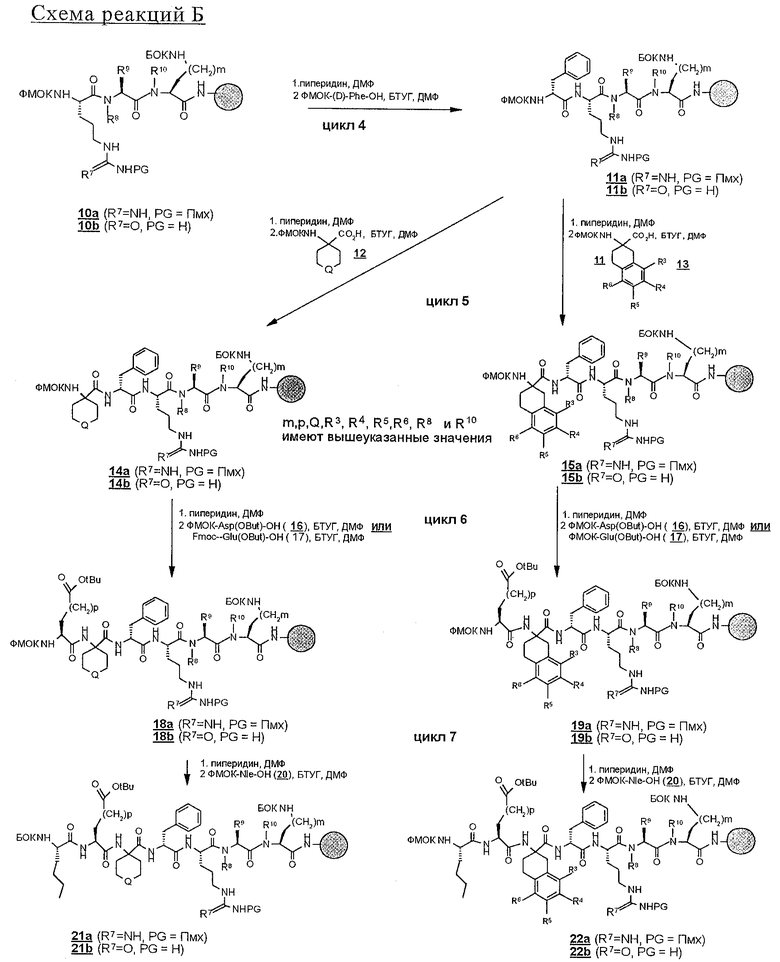
Два следующих цикла, представленных на схеме реакций В, осуществляют вначале реакцией пептидов строения 10 с аминокислотой-ФМОК-(D)Phe-ОН с получением соединения строения 11, а затем реакцией этого соединения строения 11 с любым одним из аминокислотных миметиков строения 12 или 13 вводят эти аминокислоты в связанный со смолой пептид и получают связанные со смолой пентапептиды одного из строений 14 и 15 в зависимости от того, какие используют аминокислотные миметики. Введение дополнительной аминокислоты, содержащей карбоксильные боковые цепи, в линейные пентапептиды, приемлемые для участия в конечном счете в образовании циклических пептидов по настоящему изобретению, осуществляют двумя путями.
а) Как показано на схеме реакций В, ФМОК-аминокислоту с соответственно защищенной кислотной боковой цепью вводят в связанные со смолой пентапептиды 14 и 15. Таким образом, в цикле 6 (схема реакций В) ФМОК-Asp(OtBu)-OH (16) или ФMOK-Glu(OtBu)-OH (17) вводят в растущую пептидную цепь с получением связанных со смолой гексапептидов строений соответственно 18 и 19 или, по другому варианту,
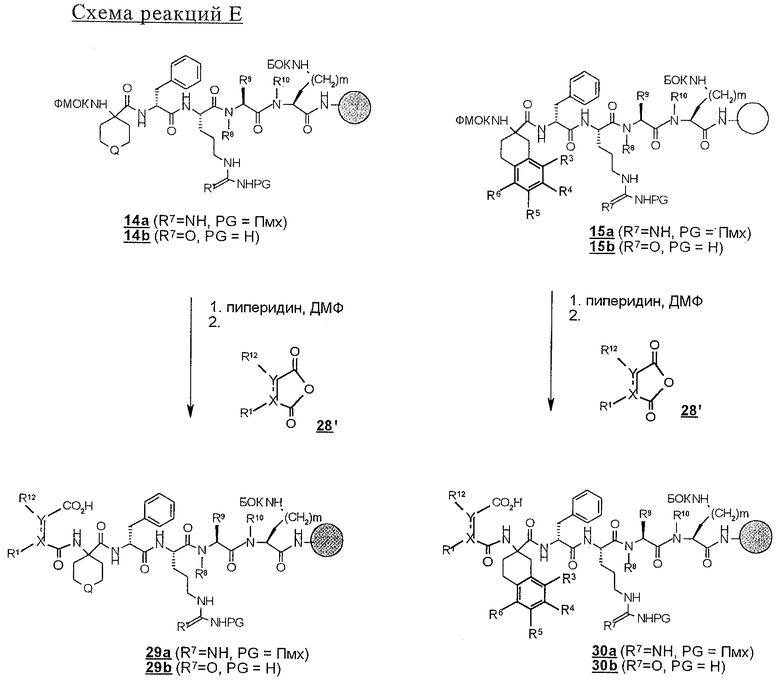
б) связанные со смолой пентапептиды 14 и 15 N-блокируют циклическим ангидридом строения 28' (схема реакций F), например малеиновым ангидридом или фталевым ангидридом, с получением соединений строения 29 или 30 или, по другому варианту,
в) как показано на схеме реакций В, связанные со смолой гексапептиды 18 и 19 перед N-блокированием вводят в дальнейшую реакцию с дополнительной аминокислотой с образованием гептапептида. Это осуществляют введением обычным путем остатка аминокислоты, предпочтительно ФМОК-Nle-OH, с получением 21 и 22.
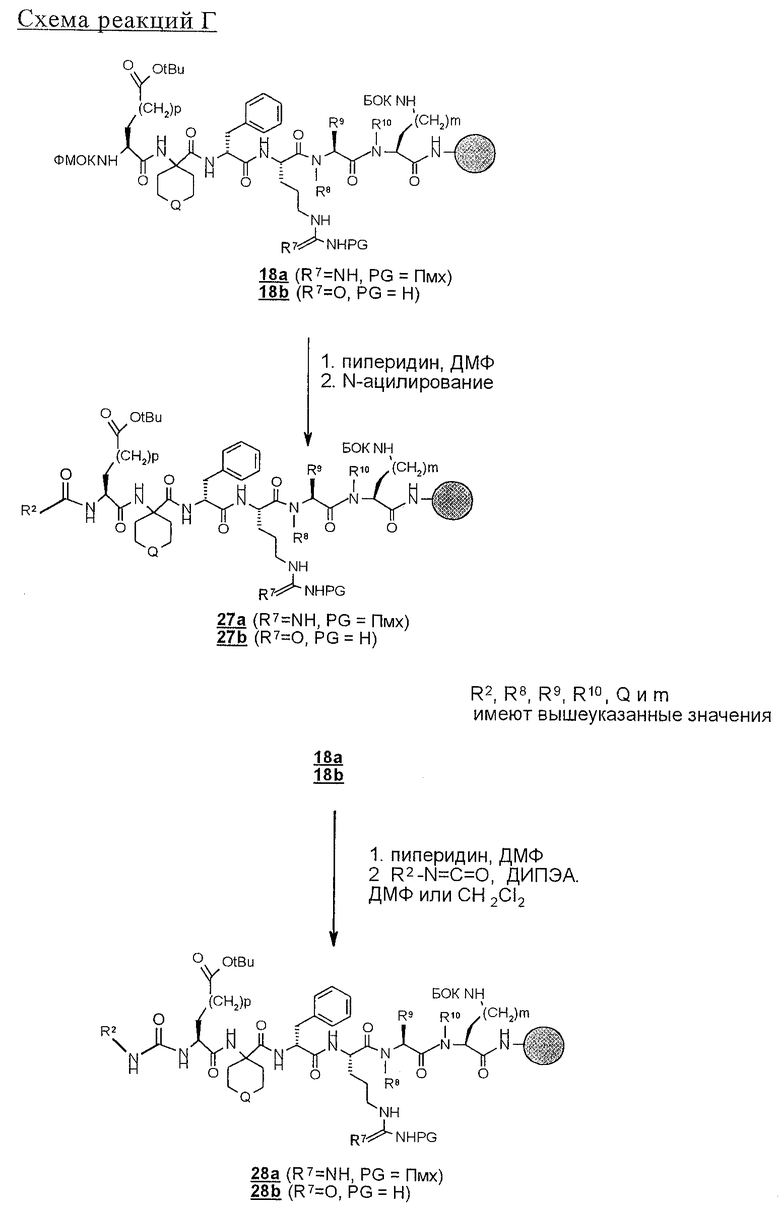
В результате N-блокирования гептапептида или гексапептида образуется концевая амидная функциональная группа соединения 1. Таким путем получают заместители X, Y, R12 и R1. Для N-блокирования связанных со смолой гексапептидов (18, 19) или гептапептидов (21, 22) полипептид вначале обрабатывают пиперидином в ДМФ с удалением защитной группы ФМОК и затем вводят в реакцию с ацилирующим агентом. Как представлено на схеме реакций D, для получения соединения строения 1 перед образованием группы Z, где каждый из Х и Y обозначает СН, а R1 обозначает группу
у связанного со смолой полипептида строения 18 удаляют защитную группу и его N-ацилируют с получением связанного со смолой амида строения 27 или после удаления защитной группы проводят реакцию с изоцианатом с получением производных мочевины строения 28. N-Ацилирование проводят по самым разнообразным методам, хорошо известным специалисту в данной области техники. Среди применяемых методов
(I) реакция концевой функциональной аминогруппы с карбоновой кислотой R2-СО2Н в приемлемом растворителе, таком, как ДМФ, в присутствии БТУГ и органического основания, предпочтительно ДИПЭА;
(II) реакция концевой функциональной аминогруппы с хлорангидридом карбоновой кислоты R2-COCl в приемлемом растворителе, таком, как дихлорметан, в присутствии органического основания, такого, как пиридин, ТЭА и ДИПЭА, предпочтительно ДИПЭА, и
(III) реакция концевой функциональной аминогруппы с ангидридом карбоновой кислоты строения (2) 28', как это представлено на схеме реакций F; эту реакцию проводят в приемлемом растворителе, таком, как дихлорметан или ДМФ, в присутствии органического основания, предпочтительно ДИПЭА.
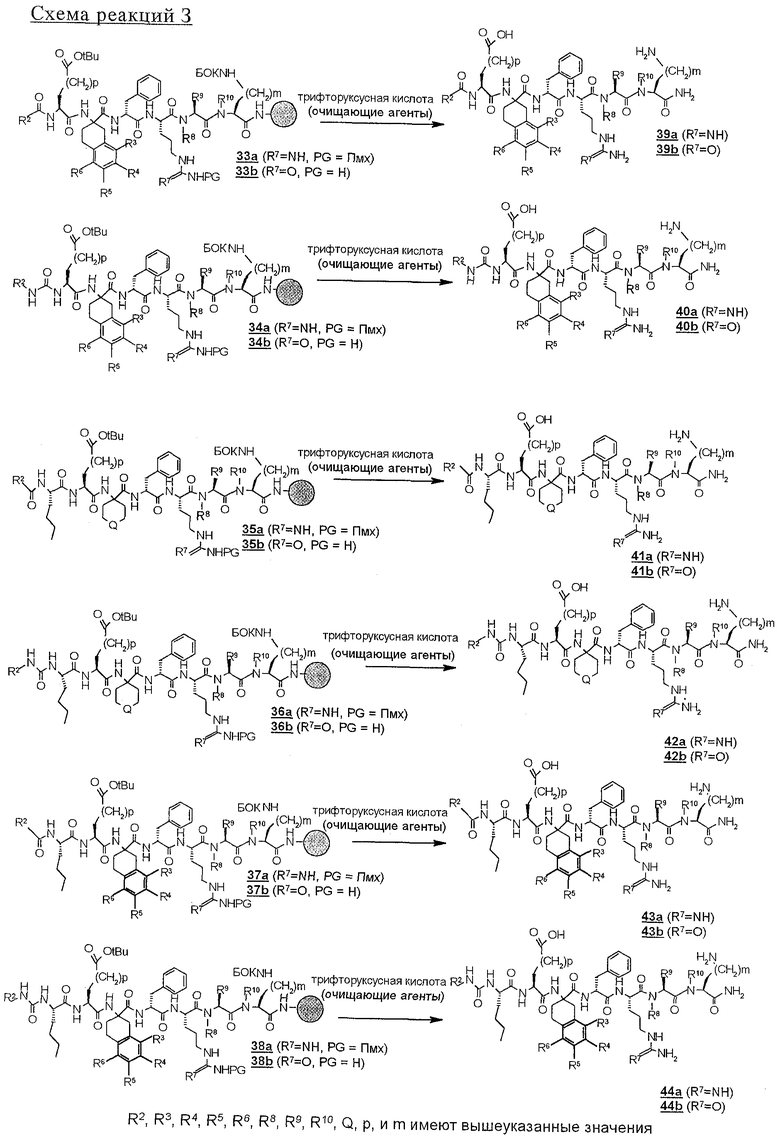
Реакцию N-блокирования в соответствии со схемой реакций D, где соединения строения 18 превращают в производные мочевины строения 28, осуществляют введением концевой аминогруппы в соединениях строения 28 во взаимодействие с изоцианатом R2-NCO. Эту реакцию проводят в приемлемом растворителе, таком, как дихлорметан или ДМФ, в присутствии органического основания, предпочтительно ДИПЭА. Когда реакции ацилирования и образования мочевины завершают, связанные со смолой продукты 27 и 28 промывают с целью освободить от остаточных реагентов и побочных продуктов. В аналогичных условиях реакцию N-блокирования связанных со смолой полипептидов строения 19, 21 и 22 проводят с получением N-ацилированных соединений строений 33, 35, 37 и производных мочевины строений 34, 36 и 38 (схема реакций Е). Однако схема реакций Е может быть модифицирована для получения групп R14, отличных от тех, которые модифицированы из Nle, с использованием известных аминокислот, отличных от соединения строения 20, в соответствии со схемой реакций В с получением соединений строений 21 и 22.
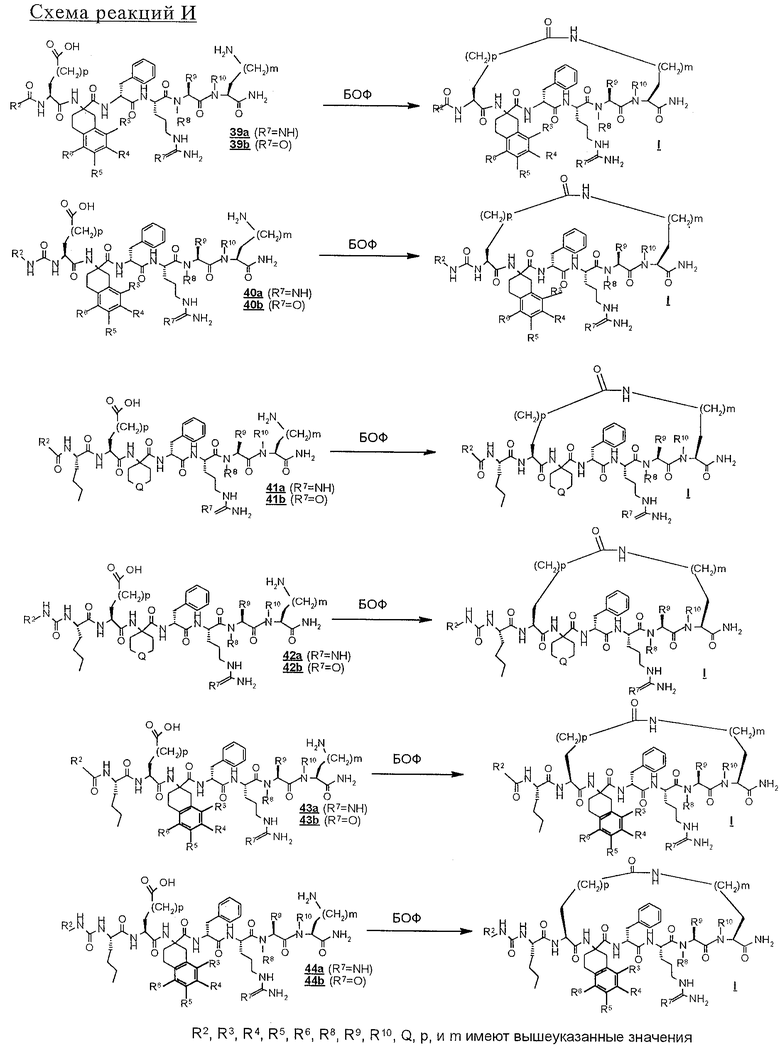
На схемах реакций G и Н проиллюстрировано отщепление оставшихся защитных групп у N-блокированных полипептидов 29, 30 и с 33 по 38 и параллельное отщепление пептидов от твердой подложки. Эту реакцию проводят с использованием сильной органической кислоты, предпочтительно трифторуксусной кислоты, необязательно в присутствии инертного растворителя, такого, как дихлорметан, и следов (1%) воды, и необязательно в присутствии одного или нескольких акцепторов карбкатионов, например этандитиола, диметилсульфида, триэтилсилана и анизола. Раствор для отщепления полипептидов фильтруют с целью освободить от твердой подложки, а затем разбавляют подходящим растворителем, предпочтительно диэтиловым эфиром, и полученные твердые материалы собирают фильтрованием. Твердые полипептиды строений с 39 по 44, полученные в соответствии со схемой реакций Н, можно очищать хроматографией с обращенной фазой с использованием колонки С 18 для препаративной хроматографии.
Теперь при наличии соответствующих функциональных групп, с помощью которых возможно образование внутримолекулярной амидной связи, N-блокированные линейные полипептиды подвергают обработке в условиях реакции образования амида, которые хорошо известны в данной области техники. Соответственно каждый из линейных пептидов 31, 32 (схема реакции G) и с 39 по 44 (схема реакции I) поочередно растворяют в инертном растворителе, например в ДМСО, и перед введением образующего амид реагента, например БОФ, добавлением третичного аминового основания, например N-метилморфолина, рН раствора доводят до явного значения 8. Эту реакцию удобно проводить при температуре в пределах от 0 до 40° С, предпочтительно при примерно комнатной температуре. Реакции дают возможность протекать до тех пор, пока ее не посчитают завершенной. Общепринятыми методами, используемыми обычными специалистами с целью следить за протеканием реакции, являются, например, ТСХ и аналитическая ВЭЖХ. После удаления под вакуумом реакционных растворителей сырые циклические пептиды строения I, где Z обозначает NHCO, как это представлено на схеме реакции I, можно очищать хроматографией с обращенной фазой с применением колонки С18 для препаративной хроматографии. Таким путем получают соединение строения 1, где Z обозначает мостик NHCO.
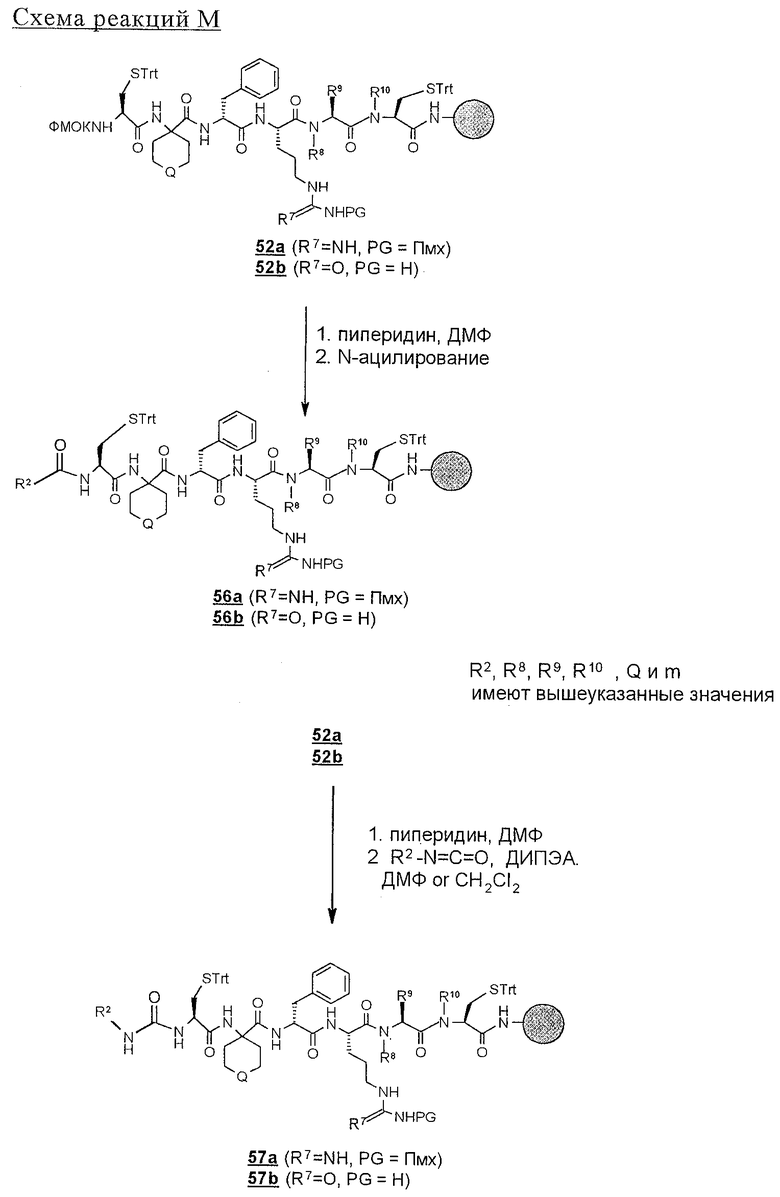
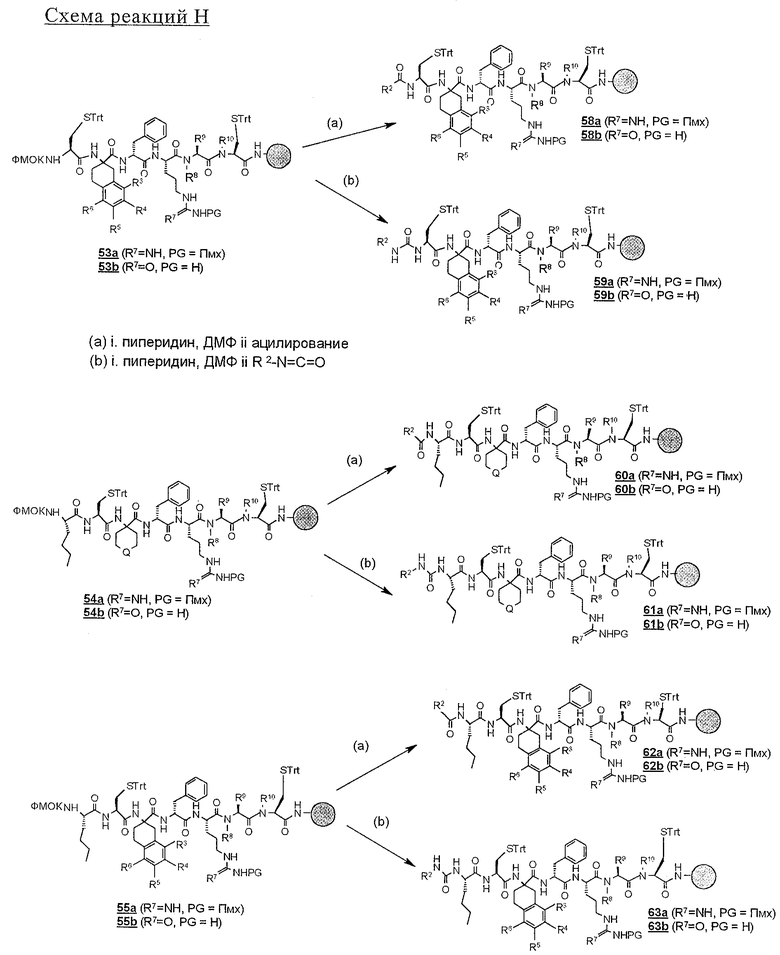
Циклические дисульфидные пептиды строения 1, где Z обозначает S-S, получают по методологиям, в общих чертах представленным на схемах реакций и аналогичным тем, которые представлены выше на схемах реакций с А по Н для получения лактамов строения 1, где Z обозначает NHCO. Предпоследние линейные полипептиды собирают идентичным образом, за исключением того, что аминокислоты, которые содержат защищенные остатки тиоловых боковых цепей, например ФMOK-Cys(Trt)-OH, вводят в соответствующие положения растущего связанного со смолой полипептида, предпочтительно в циклах 1 и 6. Такое получение связанных со смолой линейных полипептидов проиллюстрировано на схемах реакций J и К. Как ранее изложено и показано на схеме L, после удаления защитной группы ФМОК связанный со смолой линейный гексапептид 52 можно N-блокировать либо ацилированием с получением 56, либо реакцией с изоцианатом с получением мочевины 57. Аналогичным путем связанный со смолой гексапептид 53 и связанные со смолой гептапептиды 54 и 55 превращают в соответствующие N-ацилированные производные 58, 60 и 62 и производные мочевины 59, 61 и 63 (схема реакции М).
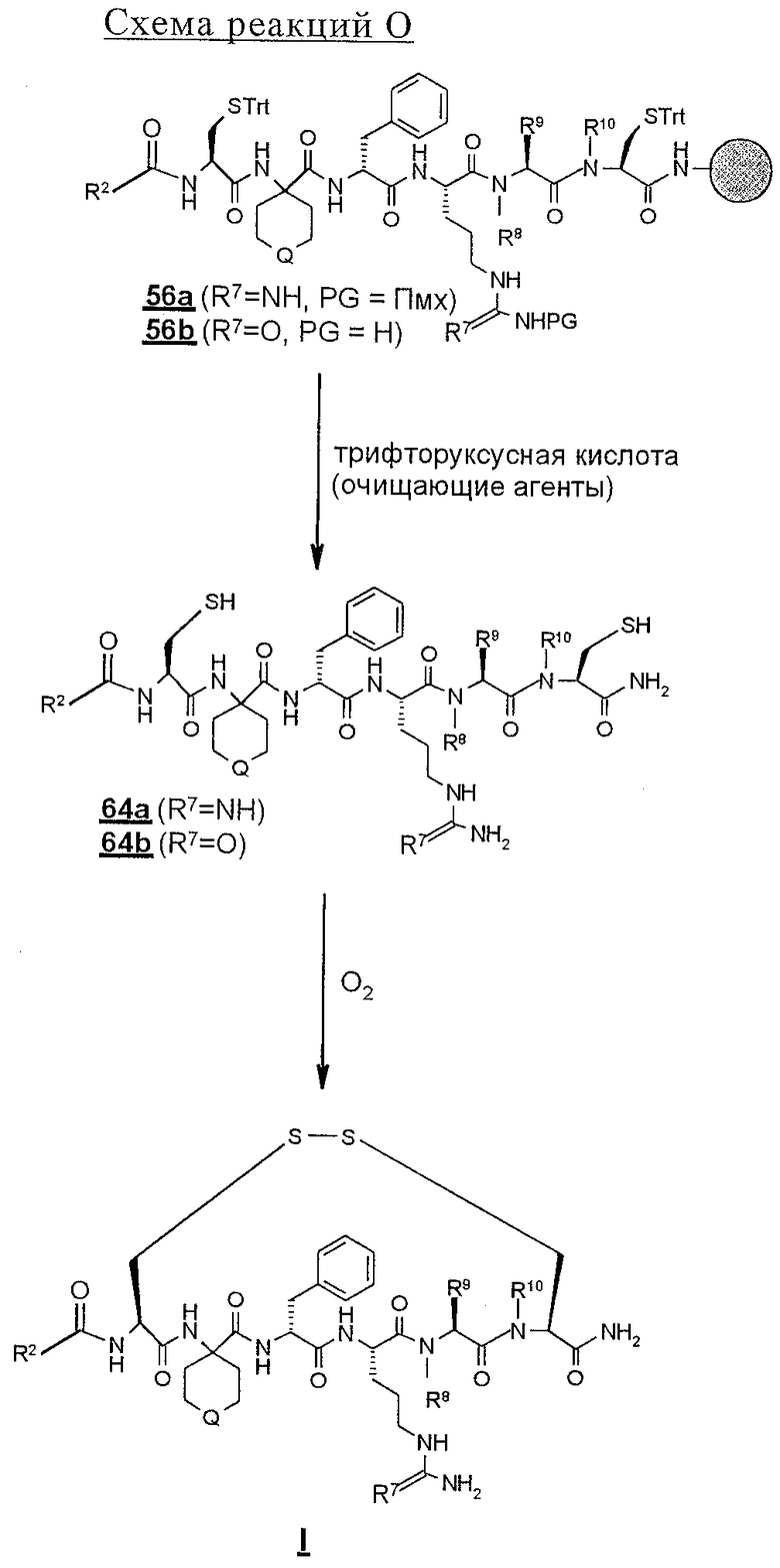
В соответствии со схемой реакций N N-блокированный связанный со смолой линейный гексапептид 56 обрабатывают сильной кислотой, предпочтительно трифторуксусной кислотой, необязательно в присутствии инертного растворителя, такого, как дихлорметан, и необязательно в присутствии одного или нескольких акцепторов карбкатионов, например этандитиола, диметилсульфида, триэтилсилана или анизола. Это вызывает отщепление всех защитных групп боковых цепей, а также отщепление линейного пептида от твердой подложки. Эту реакцию удобно проводить при температуре в пределах 0 и 35° С, предпочтительно при комнатной температуре. Раствор для отщепления полипептида фильтруют с целью освободить от твердой подложки, а затем разбавляют подходящим растворителем, предпочтительно диэтиловым эфиром, и полученные твердые материалы собирают фильтрованием. Выделенный таким образом твердый полипептид строения 64 можно, но необязательно, очищать хроматографией с обращенной фазой с использованием колонки С18 для препаративной хроматографии. Далее линейный гексапептид 64 обрабатывают в окислительных условиях, которые хорошо известны специалисту в данной области техники и которые способны заставить тиолы образовывать дисульфидную связь. Соответственно с использованием умеренно слабого неорганического основания, предпочтительно гидроксида аммония, рН разбавленного водного раствора продукта 64 доводят до 8,0, а затем через раствор барботируют кислород до тех пор, пока циклизацию не посчитают завершенной, основываясь на стандартных методах, например ТСХ или ВЭЖХ. После удаления реакционных растворителей лиофилизацией сырой циклический пептид (I; Z обозначает S-S), полученный и выделенный таким путем, можно очищать хроматографией с обращенной фазой с применением колонки С18 для препаративной хроматографии.
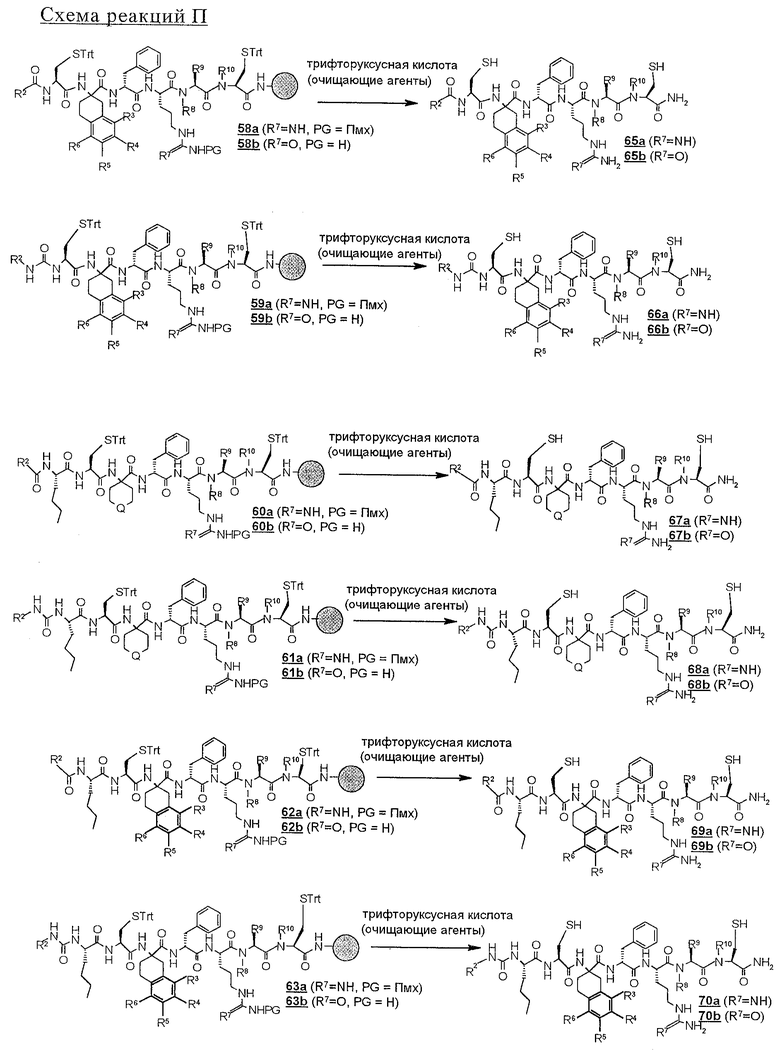
В условиях, подобных тем, которые описаны выше в связи со схемами реакций Н и I, удаляют защитные группы у связанных со смолой линейных полипептидов с 58 по 63, которые представлены на схемах реакций О и Р, и их отщепляют от твердой подложки с получением линейных пептидов с 65 по 70 (схема реакций О), которые затем циклизуют в окислительных условиях, как это представлено выше, с получением соответствующих соединений строения 1 (схема реакций Р).
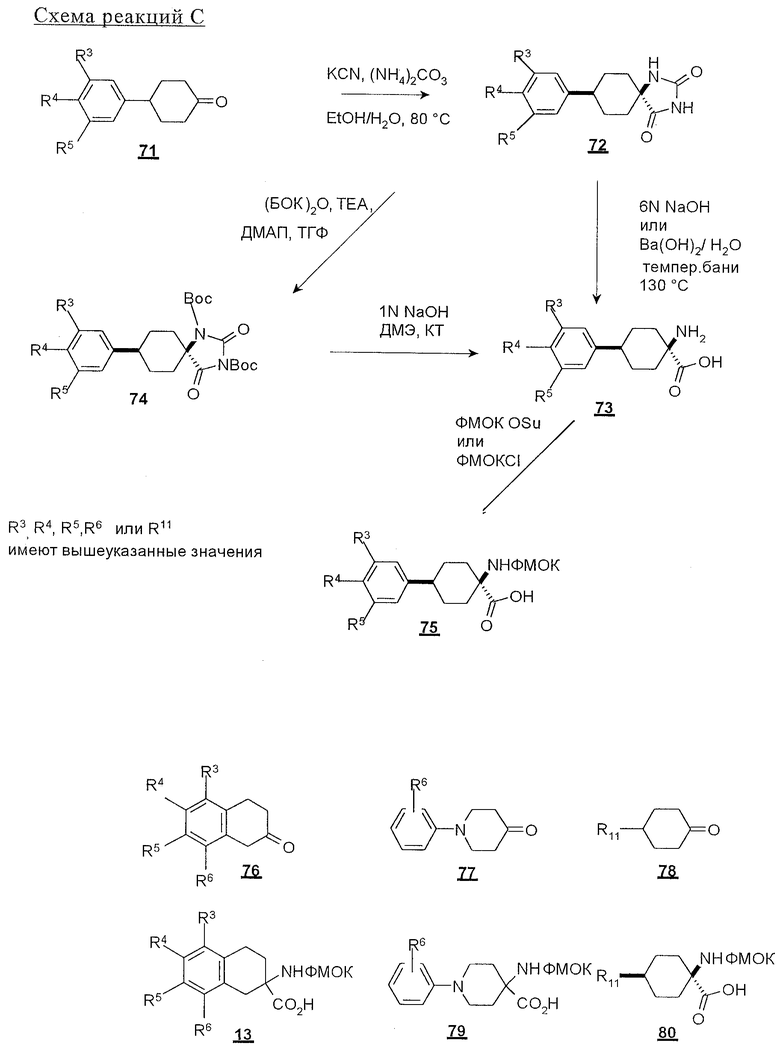
ФМОК-аминокислоты, используемые при получении описанных выше пептидов, а также ацилирующие агенты и изоцианаты, применяемые для N-блокирования полипептидов, являются известными соединениями, которые технически доступны. ФМОК-аминокислоты 12, включая материалы для них, соединение строения 13, используемое в соответствии со схемой реакций В, получают так, как изложено в настоящем описании, по методам, которые хорошо известны обычным специалистам в практике органической химии. На схеме Q в общих чертах представлено получение ФМОК-аминокислотных материалов соединения строения 12 из циклических кетонов. Такие материалы, которые соответствуют строению 12 и строению 13, 79 и 80, получают точно так же, как материалы строения 75, как это в общих чертах представлено в процессе на схеме Q. 4-Фенилциклогексаноны строения 71 превращают в гидантоины строения 72 обработкой карбонатом аммония и цианидом калия. Эту реакцию обычно проводят в водно-этанольной смеси при температуре от 50 до 90° С, предпочтительно в пределах 80 и 90° С. Прямой гидролиз гидантоинов до аминокислот строения 73 требует длительной обработки сильным основанием, например 6 н. раствором гидроксида натрия или гидроксида бария, при температуре кипения с обратным холодильником. По другому варианту соединения строения 72 можно превращать в бис-БОК-производные строения 74. Эту реакцию проводят с использованием трет-бутилдикарбоната [(БОК)2О] в инертном растворителе, предпочтительно в тетрагидрофуране (ТГФ), в присутствии органического аминового основания, предпочтительно ТЭА, и в качестве катализатора 4-диметиламинопиридина (ДМАП) при температуре от нуля градусов до комнатной температуры, предпочтительно при комнатной температуре. Такие бис-БОК гидантоины строения 74 легко превращают в аминокислоты строения 73. Реакцию осуществляют с использованием 1 н. гидроксида натрия в инертном растворителе, предпочтительно в диметоксиэтане (ДМЭ), при температуре от нуля градусов до 50° С, предпочтительно при примерно комнатной температуре. Защиту аминовой функциональной группы в соединении строения 73 группой ФМОК проводят во многих реакционных условиях с получением соединения строения 75, которое представляет собой разновидность ФМОК-аминокислоты соединения строения 12. Такую реакцию можно эффективно проводить обработкой раствора аминокислоты 73 в смеси ТГФ или диоксана, предпочтительно диоксана, и водного карбоната натрия, 9-флуоренилметоксихлорформиатом (ФМОК-С1) при температуре от нуля градусов до комнатной температуры, предпочтительно при комнатной температуре. По другому варианту N-(9-флуоренилметоксикарбонилокси)сукцинимид (ФМОК-OSu) добавляют в раствор аминокислоты 73 в водном ацетонитриле, содержащем органическое третичное аминовое основание, предпочтительно ТЭА. Реакцию проводят при температуре от нуля градусов до комнатной температуры, предпочтительно при комнатной температуре. В другом варианте этого метода ДМЭ выпаривают из гидролизной смеси в процессе превращения соединения 74 в соединение 73 и значение рН реакционной смеси доводят до ~11. Далее образовавшийся раствор натриевой соли соединения 73 in situ обрабатывают с использованием ФМОК-OSu или ФМОК-С1 в диоксане при температуре от нуля градусов до комнатной температуры, предпочтительно при комнатной температуре. Таким же путем, как в соответствии со схемой реакций Q, тетралоновые 76, N-арил-4-кетопиперидиновые 77 и циклогексаноновые производные 78 можно превращать в соответствующие ФМОК-аминокислоты строений 13, 79 и 80, каждая из которых совместно с 73 образует подгруппу соединений строения 12, используемых в соответствии со схемой реакций В.
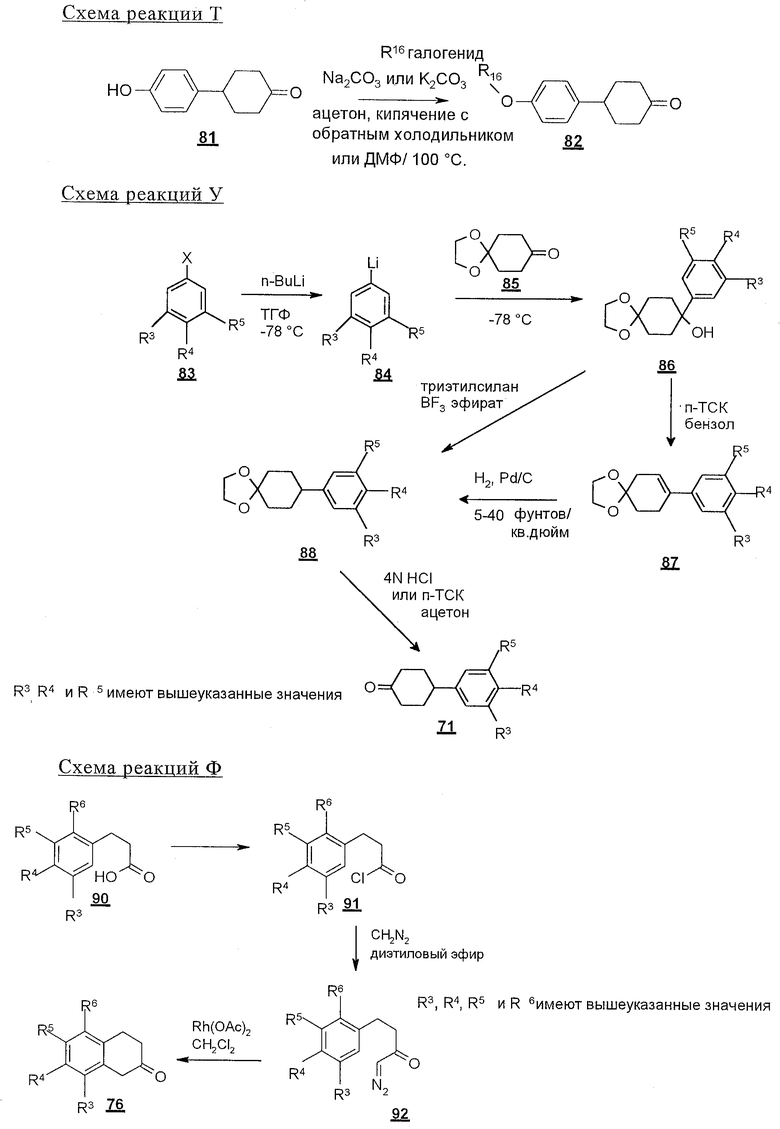
В соответствии со схемой реакций Q соединения строения 73, где R4 обозначает линейный или разветвленный (низш.)алкокси, и как R2, так и R3 каждый обозначает водородный атом в подгруппе соединений строения 82, могут быть получены O-алкилированием соединения строения 81, как представлено на схеме реакции R. Когда R16 обозначает неразветвленный (низш.)алкильный остаток, алкилирование проводят с использованием первичного алкилгалогенида строения R16 в присутствии карбоната щелочного металла, например карбоната натрия или калия. Алкилгалогенид может представлять собой хлор-, бром- или иодпроизводное, предпочтительно алкилиодид. Эту реакцию можно эффективно проводить в инертном растворителе, который ускоряет реакции замещения SN2, например в ацетоне, 2-бутаноне или N,N-диметилформамиде, предпочтительно в ацетоне, при температуре от комнатной температуры до температуры кипения раствора, предпочтительно при температуре кипения. Когда R16 обозначает разветвленную (низш.)алкильную группу, например 2-пропил, алкилирование этого соединения строения 81 с получением соединения строения 82 проводят с использованием вторичного алкилгалогенида строения R16 в присутствии карбоната щелочного металла, например карбоната калия. Предпочтительный вторичный алкилгалогенид представляет собой вторичный алкилиодид, например 2-иодпропан. Эту реакцию можно эффективно проводить в инертном растворителе, предпочтительно в N,N-диметилформамиде, при температуре от комнатной до температуры кипения раствора, предпочтительно при примерно 100° С.
4-Арилциклогексаноны строения 71, которые представляют собой исходные материалы в соответствии со схемой реакций Q, могут быть получены по методам, которые хорошо известны любому специалисту в практике органической химии. Как представлено в общих чертах на схеме S, в результате обработки арилгалогенидов строения 83, где Х1 обозначает атом брома или иода, металлалкильным реагентом, предпочтительно трет-бутиллитием, протекает реакция переметаллирования с получением соответствующего ариллития строения 84. Эту реакцию удобно проводить при -78° С добавлением раствора алкиллития в раствор соединения строения 83 в инертном безводном растворителе, таком, как диэтиловый эфир или тетрагидрофуран, предпочтительно в тетрагидрофуране. Затем in situ проводят реакцию ариллития строения 84, полученного таким путем, с раствором монокеталя циклогексан-1,4-диона (85) в приемлемом инертном растворителе, например в тетрагидрофуране, одновременно поддерживая реакционную температуру ниже -60° С, предпочтительно при примерно -78° С, с получением карбинолов строения 86. Соединения строения 87 получают дегидратацией карбинолов строения 86. Эту реакцию удобно проводить с использованием в качестве катализатора сильной органической кислоты, предпочтительно п-толуолсульфоновой кислоты, в инертном растворителе, например в бензоле или толуоле, предпочтительно в бензоле, при температуре кипения растворителя. Образовавшуюся воду удаляют из реакционной смеси с помощью прибора Дина-Старка, что дает возможность реакции протекать до завершения. Соединения строения 88 получают гидрогенизацией олефинов строения 87. Эту реакцию удобно проводить с использованием катализатора на основе благородного металла, например палладия на угле, в водородной атмосфере в инертном растворителе, например в этаноле или этилацетате. Гидрогенизацию обычно проводят при комнатной температуре и под давлением водорода 40 фунтов/кв.дюйм, однако если арильное кольцо в соединении строения 87 содержит группу, склонную к гидрогенолизу, например если R3, R4 или R5 обозначает атом хлора, реакционное давление поддерживают на уровне примерно 5 фунтов/кв.дюйм. Соединения строения 88 могут быть также получены непосредственно из карбинолов строения 86 восстановительным элиминированием гидроксильной группы. В ходе проведения такой реакции раствор соединения строения 86 (каждый из R3 и R4 обозначает Н, а R5 обозначает ОМе) в инертном растворителе, например в дихлорметане, обрабатывают кислотой Льюиса, такой, как эфират трифторида бора, и восстановителем, например триэтилсиланом, при температуре от нуля градусов до комнатной температуры. В результате удаления кеталевой защитной группы в соединениях строения 88 получают кетон формулы 71, который представляет собой исходный материал для схемы реакций Q при получении ФМОК-аминокислоты строения 75 соединения строения 12. Такую реакцию удобно проводить в ацетоне или 2-бутаноне, предпочтительно в ацетоне, с кислотным катализом, например 4 н. соляной кислотой или п-толуолсульфоновой кислотой, при температуре от комнатной до температуры кипения реакционной смеси, предпочтительно при температуре кипения.
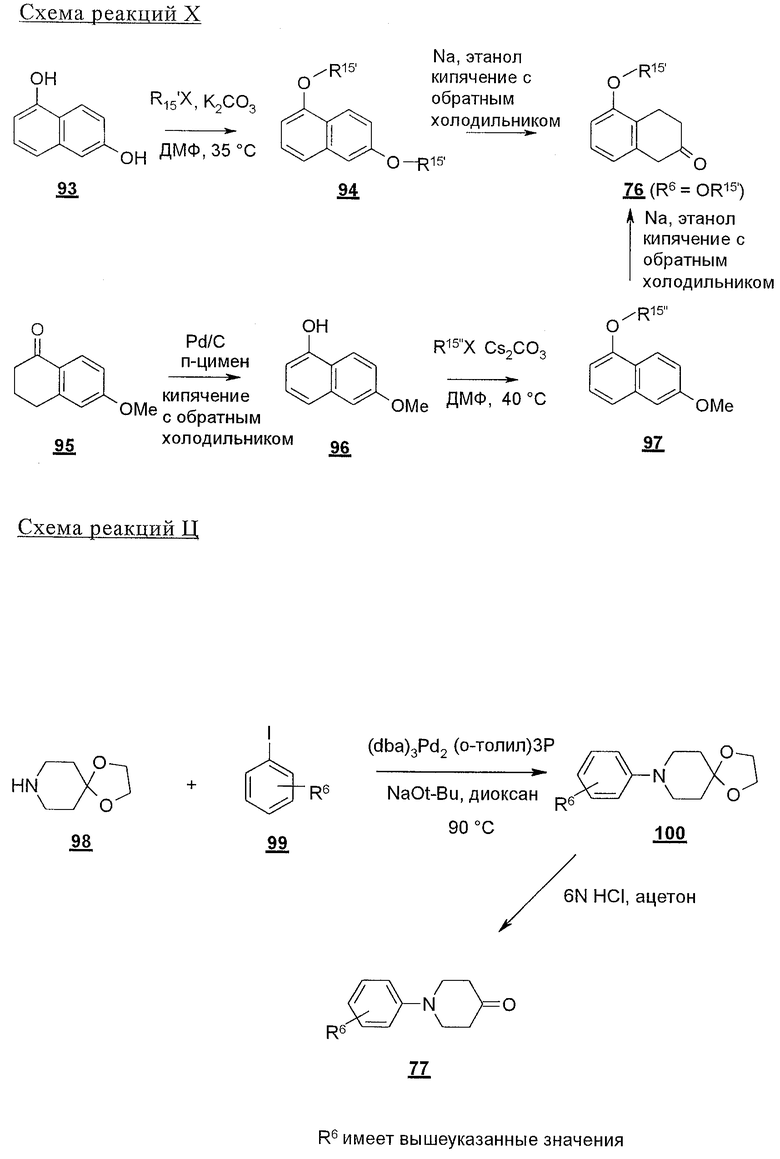
5-Замещенные бета-тетралоны строения 76, приведенные на схеме реакций Q, которые представляют собой исходный материал для получения соединения строения 13, являются известными соединениями, или, если они неизвестны, их можно получить по методам, которые хорошо известны любому специалисту в области органической химии. В данном случае соединения строения 76 получают в основном по двум методам, представленным в общих чертах на схемах реакций Т и U. Как показано на схеме Т, 2-замещенную гидрокоричную кислоту строения 90 превращают в соответствующий хлорангидрид карбоновой кислоты строения 91. Это превращение можно осуществлять по нескольким методам, например обработкой гидрокоричной кислоты оксалилхлоридом, необязательно в присутствии каталитически эффективного количества N,N-диметилформамида, в инертном растворителе, таком, как бензол или дихлорметан, предпочтительно в дихлорметане. Эту реакцию можно эффективно проводить при температуре от нуля градусов до комнатной температуры, предпочтительно при комнатной температуре. По другому варианту проводят реакцию соединения строения 90 с образующим ацилхлорид реагентом, таким, как сульфурилхлорид, в инертном растворителе, например в бензоле или толуоле, предпочтительно в толуоле, при температуре в пределах от комнатной до температуры кипения раствора, предпочтительно при температуре кипения. Диазокетон строения 92 готовят обработкой полученного таким образом ацилгалогенида строения 91, в инертном растворителе, например в дихлорметане, избытком свежеприготовленного эфирного раствора диазометана. Такое объединение реагентов удобно осуществлять при температуре ледяной бани, и затем реакции дают возможность протекать при температуре от нуля градусов до комнатной температуры, предпочтительно при комнатной температуре. Как представлено на схеме реакций Т, циклизации диазокетона строения 92 с получением тетралона строения 76 содействует димер ацетата родия II в инертном растворителе, например в дихлорметане. Эту реакцию, как правило, проводят при температуре от комнатной до температуры кипения раствора, предпочтительно при температуре кипения.
Соединения строения 76, которые представляет собой исходный материал в соответствии со схемой реакций Q, в которой R6 обозначает линейную или разветвленную (низш.)алкоксигруппу, могут быть получены так, как представлено на схеме реакций U, из соединений строения 93. В соответствии со схемой реакций U соединения строения 94, где R15’ представляет собой неразветвленный (низш.)алкильный остаток, получают пер-O-алкилированием нафталиндиола строения 93 первичным алкилиодидом или -бромидом, предпочтительно -иодидом, в присутствии основания, такого, как карбонат щелочного металла, например карбонат натрия или калия. Такую реакцию можно проводить в инертном растворителе, предпочтительно в N,N-диметилформамиде, при температуре от комнатной до 100° С, предпочтительно при 35° С. Соединения строения 97, где R15’ обозначает разветвленный (низш.)алкил, получают из 2-тетралона строения 94 в две стадии. Тетралон строения 95 подвергают дегидрогенизации в присутствии катализатора на основе благородного металла, такого, как металлический палладий (10%-ный на угле), в подходящем высококипящем растворителе, таком, как п-цимен, с получением ароматизированного соединения строения 96. Затем вторичным алкилиодидом в присутствии основания, такого, как карбонат щелочного металла, предпочтительно карбоната цезия, O-алкилируют нафтол строения 96 с получением соединения строения 97. Эту реакцию можно эффективно проводить в инертном растворителе, предпочтительно в N,N-диметилформамиде, при температуре от комнатной до 100° С, предпочтительно при примерно 40° С.
Тетралоны строения 76 получают восстановлением соединений строений 94 и 97 в условиях растворения металла с последующим катализируемым кислотой гидролизом промежуточных енольных эфиров. Такое превращение удобно осуществлять добавлением отдельными порциями большого избытка щелочного металла, такого, как натрий или калий, предпочтительно натрия, в кипящий раствор обрабатываемого вещества в низшем спирте, предпочтительно в этаноле, до тех пор, пока не израсходуется исходный материал. Тетралоны строения 76 получают обработкой раствора выделенных промежуточных енольных эфиров сильной кислотой в качестве катализатора, предпочтительно п-толуолсульфоновой кислотой. Гидролиз можно эффективно проводить в смеси низшего спирта, предпочтительно этанола, и воды при температуре в пределах от комнатной до температуры кипения раствора, предпочтительно при температуре кипения.
При получении соединений строения 77, используемых в соответствии со схемой реакций Q в качестве исходных материалов при получении соединений формулы 79, которые представляют собой ФМОК-аминокислоты соединения строения 12, в качестве исходного материала используют соединение строения 98. Это показано на схеме реакций V. В соответствии со схемой реакций V соединения строения 100 могут быть получены проведением реакций, которые известны. Так, например, их можно получить реакцией конденсации вторичного амина строения 98 с арилбромидом или -иодидом, предпочтительно с арилиодидом строения 99 (схема реакций V). Такую реакцию конденсации катализируют катализатором на основе благородного металла, предпочтительно три(дибензилиденацетон)дипалладием, в присутствии хелатообразующего фосфинового лиганда, предпочтительно три-о-толилфосфина, и затрудненного алкоксидного основания, такого, как трет-бутоксид натрия. Эту реакцию удобно проводить в инертной атмосфере с использованием безводного растворителя, такого, как диоксан или толуол, предпочтительно диоксана, при температуре от 60° С до температуры кипения, предпочтительно при 90° С. Удаление карбонильной защитной группы в соединении 100 с получением соединений строения 77 можно осуществлять по самым разнообразным методам, хорошо известным в области органической химии. Так, например, удаление защитной группы может быть достигнуто обработкой раствора соединения 100 в низкокипящем кетоне, таком, как ацетон или 2-бутанон, водным раствором минеральной кислоты, например 6 н. соляной кислоты, при температуре от комнатной до температуры кипения смеси, предпочтительно при температуре кипения. Когда соединение строения 100 обрабатывают таким путем водной минеральной кислотой в соответствии со схемой реакций V образуется соединение строения 77. Соединение строения 77, как это продемонстрировано в обсуждении схемы реакций Q, представляет собой промежуточный продукт для получения соединения строения 79, которое является материалом для кислоты строения 12, у которой Q обозначает группу
Используемые в соответствии со схемой реакций А аминокислоты строения 5 являются известными соединениями, где R9 обозначает
С другой стороны, аминокислоты строения 5, где R17 в R9 обозначает (низш.)алкил, т.е. где R9 обозначает группу
получают из соединения формулы 101 в соответствии со схемой реакций V следующим образом.
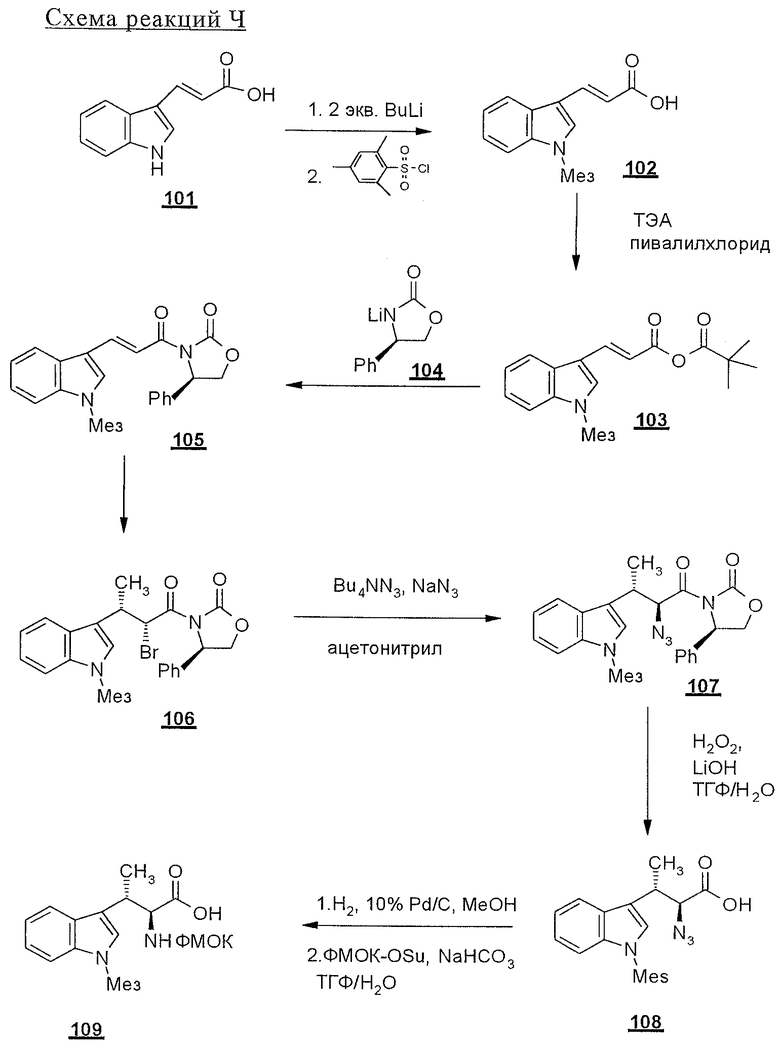
Как представлено на схеме реакций W, β -метил(Nin-Mes)триптофан 109 получают с использованием метода, описанного ранее в работе Boteju L.W., Wenger К. и Hruby V.J. Tet. Lett., 33, 7491 (1992). На первой стадии атом азота транс-индол-3-акриловой кислоты 101 защищают превращением в соответствующий мезитиленсульфонамид 102. Эту реакцию проводят обработкой индолкислоты 101 избытком (>2 экв.) раствора алкил- или ариллитиевого реагента, например фениллития или н-бутиллития, предпочтительно н-бутиллития, в инертном растворителе, предпочтительно в тетрагидрофуране, при температуре от -40 до примерно -100° С, наиболее удобно при -78° С. Далее при одновременном поддержании реакционной температуры примерно -78° С проводят реакцию образовавшегося дилитийсодержащего материала с мезитиленсульфонилхлоридом, получая мезитиленсульфонамид 102. Затем проводят реакцию конденсации N-защищенной индолакриловой кислоты 102 с хиральным вспомогательным (R)-4-фенил-2-оксазолидиноном (см. описание получения в работе Nicolas и др., J.Org.Chem., 1993, 58, 766-770) в качестве его N-литийсодержащей разновидности 104 с получением хирального акриламида 105. Такую реакцию конденсации проводят через смешанный ангидрид, образующийся из соединения 102. Для получения смешанного ангидрида 103 проводят реакцию N-защищенной индолакриловой кислоты 102 с приемлемым ацилхлоридом, например с трет-бутилхлорформиатом, 2,4,6-трихлорбензоилхлоридом или пивалоилхлоридом, предпочтительно с пивалоилхлоридом в присутствии третичного аминового основания, например триэтиламина или диизопропилэтиламина, предпочтительно триэтиламина. Критерии, благодаря которым возможен выбор ацилхлорида, приемлемого для получения ангидрида 103, хорошо определены и обычному специалисту в области органической химии известны. Процесс получения ангидрида проводят в инертном растворителе, например в тетрагидрофуране, при начальной температуре в пределах -100 и 0° С, предпочтительно при примерно -78° С. Реакции дают возможность протекать до завершения при температуре в пределах -78 и 0° С, предпочтительно при примерно 0° С. Далее полученный таким образом смешанный ангидрид 103 in situ вводят в реакцию с раствором N-литийсодержащего (R)-4-фенил-2-оксазолидинона 104, полученного по вышеизложенному обработкой раствора (R)-4-фенил-2-оксазолидинона в инертном растворителе, например в тетрагидрофуране, эквимолярным количеством раствора алкил- или ариллитиевого реагента, например фениллития или н-бутиллития, предпочтительно н-бутиллития, в инертном растворителе, например в гексане, при температуре в пределах -100 и 0° С, предпочтительно при примерно -78° С. Такую реакцию конденсации, в результате которой получают хиральный акриламид 105, проводят при начальной температуре в пределах -100 и 0° С, предпочтительно при примерно -78° С, а после того как реагенты объединяют, реакции дают возможность протекать при температуре в пределах от -78° С до комнатной, предпочтительно при примерно комнатной температуре.
Основной момент превращения хирального акриламида 105 в соединение 106 заключается в регулируемом образовании двух новых смежных хиральных центров, имеющихся у соединения 106. Эта реакция включает стереоселективное присоединение по сопряженным 1,4-положениям (реакция Михаэля) метилкупрата, образующегося in situ из комплекса бромиддиметилсульфида одновалентной меди и метилмагнийбромида, к акцептору Михаэля, к α ,β -ненасыщенной карбонильной системе, имеющейся у соединения 105. Затем полученный металл-хелатированный енолят непосредственно галоидируют галоидирующим агентом, предпочтительно N-бромсукцинимидом, с получением соединения 106. Вновь, как в случае реакции Михаэля, введение атома брома является стереоселективным, регулируемым объемистой фенильной группой хирального вспомогательного вещества, которое эффективно защищает реакционноспособные участки (si face) как α ,β -ненасыщенной ацилоксазолидиноновой системы, так и промежуточного металл-хелатированного енолята от атаки участвующих в процессе реагентов. Для получения метилкупрата раствор метилмагнийбромида в диэтиловом эфире добавляют в раствор комплекса бромиддиметилсульфида одновалентной меди (1:1) в инертном растворителе, например в диметилсульфиде или тетрагидрофуране, предпочтительно в их смеси. Эту реакцию проводят при температуре в пределах от -78° С до комнатной температуры, предпочтительно при -4° С. Для получения R17, где R17 обозначает (низш.)алкильную группу, отличную от метила, вместо метилмагнийбромида можно добавлять любой (низш.)алкилбромид. В этот приготовленный раствор метилкупрата in situ добавляют раствор α ,β -ненасыщенного ацилоксазолидинона 105 в инертном растворителе, предпочтительно в тетрагидрофуране. Присоединение метилкупрата осуществляют первоначально при температуре в пределах от -30° С до комнатной температуры, предпочтительно при -4° С, и затем реакции дают возможность протекать при комнатной температуре. Когда полагают, что реакция завершена (например, по данным ТСХ или ВЭЖХ анализа), смесь охлаждают до температуры в пределах -100 и -40° С, предпочтительно до примерно -78° С, после чего добавляют раствор галоидирующего агента, предпочтительно N-бромсукцинимида, в инертном растворителе, например в тетрагидрофуране. Затем реакции дают возможность протекать при температуре в пределах от 0° С до комнатной температуры, предпочтительно при примерно комнатной температуре, с получением после выделения бромида 106. Этот бромид замещают азидным ионом с сопутствующим обращением конфигурации. Такую перегруппировку осуществляют реакцией бромида 106 с тетрабутиламмонийазидом в присутствии избытка натрийазида в инертном растворителе, например в ацетонитриле, с получением азида 107. Эту реакцию удобно проводить при температуре в пределах от 80° С до комнатной температуры, предпочтительно при примерно комнатной температуре. В результате обработки соединения 107 гидроксидом щелочного металла, например гидроксидом натрия, калия или лития, предпочтительно гидроксидом лития, в присутствии пероксида водорода происходит гидролиз хирального вспомогательного вещества с образованием азидокислоты 108. Реакцию гидролиза проводят в инертном растворителе, предпочтительно в воде, при температуре в пределах от 0° С до комнатной температуры, предпочтительно при примерно 0° С. Гидрогенизацией α -азидокислоты 108 получают β -метил(Nin-Мез)триптофан, который сразу же превращают в соответствующий N(α )-ФMOK-β -метил(Nin-Мез)триптофан 109. Гидрогенизацию соединения 108 проводят над катализатором на основе благородного металла, предпочтительно над 10%-ным Pd/C, в инертном растворителе, например в (низш.)алканоле, предпочтительно в метаноле, под низким давлением (<2 ат) и при комнатной температуре. После удаления катализатора фильтрованием удаляют летучие компоненты и продукт растворяют в инертном растворителе, например в тетрагидрофуране или воде, предпочтительно в их смеси, и обрабатывают мягким неорганическим основанием, например бикарбонатом щелочного металла, предпочтительно бикарбонатом натрия, и образующим N-защитную группу ФМОК реагентом, например 9-флуоренилметилхлорформиатом (ФМОК-С1) или 9-флуоренилметил-N-сукцинимидилкарбонатом (ФМОК-OSu), предпочтительно ФМОК-Osu, с получением соединения 109. Такую реакцию удобно проводить при температуре в пределах от 0° С до комнатной температуры, предпочтительно при примерно комнатной температуре. Соединение формулы 109 в схеме реакций А представляет собой разновидность аминокислоты строения 5.
Объектом настоящего изобретения является также способ получения соединения формулы
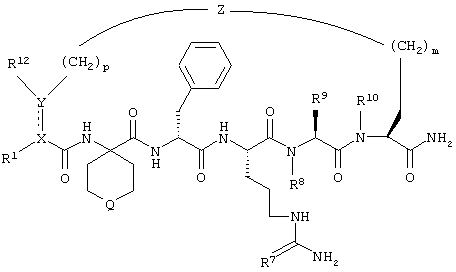
в которой символы с R1 по R12, m, p, Q, X, Y и Z имеют указанные выше значения,
посредством образования лактамовой связи или дисульфидной связи в Z-положении линейных пептидов-предшественников.
Фармацевтические композиции
Кроме того, объектом настоящего изобретения являются фармацевтические композиции, включающие соединения, как они представлены выше, и терапевтический инертный носитель.
Из соединений формул I и II, а также из Penta-цикло(Asp-Lys)-Asp-Apc-(D)Phe-Ala-Trp-Lys-NH2 и Penta-цикло(Asp-Lys)-Asp-Apc-(D)Phe-Arg-(2S,3S)-бета-метил-Тrp-Lys-NН2, полученных в соответствии с настоящим изобретением, фармацевтические композиции, приемлемые для введения в организм или ингаляции с подходящим носителем или наполнителем, могут быть приготовлены по методам, известным в данной области техники.
Соединения, как они представлены выше, могут быть использованы в качестве лекарственных средств, например в форме фармацевтических препаратов, в частности для парентерального введения. Их можно вводить, например, парентерально, в частности в форме растворов для инъекций или растворов для вливаний.
Процессы приготовления фармацевтических препаратов можно проводить по методам, которые знакомы любому специалисту в данной области техники, совмещением соединений, как они представлены выше, необязательно в сочетании с другими терапевтически ценными веществами, в форме композиции для введения в организм вместе с приемлемыми, нетоксичными, инертными, терапевтически совместимыми твердыми или жидкими материалами-носителями и, если необходимо, эффективными фармацевтическими адъювантами.
Приемлемыми материалами-носителями являются не только неорганические материалы-носители, но также органические материалы-носители.
В качестве фармацевтических адъювантов можно рассматривать обычные стабилизаторы, консерванты, смачивающие агенты и эмульгаторы, добавки для улучшения консистенции, корригенты, соли для варьирования осмотического давления, буферные добавки, солюбилизаторы, красители и маскирующие добавки, а также антиоксиданты.
Дозу соединений, как они представлены выше, можно варьировать в широких интервалах в зависимости от заболевания, при котором требуется лечение, возраста, индивидуального состояния пациента и пути введения в организм, и в каждом конкретном случае она, что очевидно, должна соответствовать индивидуальным потребностям. Для взрослого пациента следует принимать во внимание ежедневную дозу, которая составляет от примерно 1 до примерно 1000 мг, преимущественно от примерно 10 до примерно 500 мг. В зависимости от величины ежедневной дозы препарат удобно вводить в организм в виде нескольких дробных доз.
Целесообразное содержание в фармацевтических препаратах соединений, как они представлены выше, составляет примерно от 1 до 500 мг, предпочтительно от 5 до 200 мг.
Лечение ожирения
Соединения, полученные согласно настоящему изобретению, проявляют in vitro селективную агонистическую активность к рецептору МС-4. Известно, что у мышей, на которых моделировали ожирение человека, агонисты активности MC4-R вызывали снижение аппетита. Следовательно, введение этих соединений повышает активность MC4-R, что очень важно при регулировании массы тела. Фармацевтические композиции, содержащие соединения по настоящему изобретению, могут быть приготовлены в концентрации, эффективной для введения различными путями человеку или животному, страдающему либо только заведомо нежелательной, избыточной массой тела, либо и другими неблагоприятными медицинскими состояниями или заболеваний, например такими, как сахарный диабет II типа. При этом может быть использована разнообразная техника введения препаратов. Среднее количество активного соединения можно варьировать, основываясь, в частности, на рекомендациях и предписаниях квалифицированного врача или ветеринара.
Таким образом, объектом настоящего изобретения является применение описанных выше соединений при приготовлении лекарственных препаратов для лечения и/или профилактики заболеваний, связанных с активностью рецептора меланокортина-4. Кроме того, объектом настоящего изобретения является метод лечения и/или профилактики заболеваний, связанных с активностью рецептора меланокортина-4, который включает введение человеку или животному вышеописанного соединения. Такие соединения особенно эффективны при лечении и/или профилактике ожирения. Объектом изобретения является также соединение, как оно представлено выше, когда его получают согласно описанному выше способу. Более того, объектом изобретения являются описанные выше соединения для применения в качестве терапевтически активных веществ, в частности терапевтически активных веществ при лечении и/или профилактике заболеваний, которые связаны с рецептором меланокортина-4, напрмер ожирения.
Настоящее изобретение более понятно, если обратиться к следующим примерам, которые иллюстрируют представленное в настоящем описании изобретение, не ограничивая его объема. Для удобства в структурные формулы конкретных соединений, приведенных в разделе примеров, водородные атомы как правило не включены.
Пример 1
Получение ФМОК-1-амино-4-фенилциклогексан-1-карбоновой кислоты (ФМОК-Арс)
Стадия 1
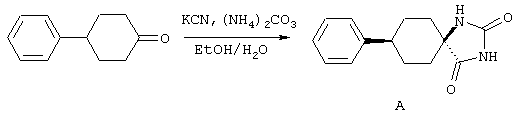
В раствор 4-фенилциклогексанона (10,0 г, 57,5 ммоля) в этаноле (100 мл) и воде (33 мл) в стеклянной толстостенной колбе вводили карбонат аммония (33 г, 344 ммоля, 6 экв.) и цианид калия (5,6 г, 86,2 ммоля, 1,5 экв.). Смесь выдерживали при температуре от 80 до 90° С в течение 24 ч. Охлажденную реакционную смесь добавляли в смесь воды со льдом (400 мл) и интенсивно перемешивали в течение 30 мин. Полученный осадок отфильтровывали под вакуумом, тщательно промывали водой и сушили с получением в виде белого твердого вещества гидантоина А (14,0 г, 100%-ный выход). 1Н-ЯМР (ДМСО-d6): 8,63 (s, 1H), 7,23-7,36 (m, 4Н), 7,15 (m, 1Н), 2,50 (m, 1H), 2,10 (m, 1H), 1,85 (d, 1H) и 1,55-1,80 (m, 6H).
Стадия 2
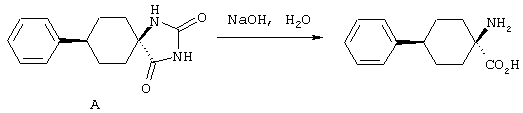
Гидантоин А (10,0 г) суспендировали в водном NaOH (6 н., 350 мл) и выдерживали при 130° С в течение 2-3 дней. По завершении гидролиза реакционную смесь нейтрализовали конц. НСl до слабокислой реакции (рН ~6). Образовавшуюся суспензию фильтровали, промывали водой и сушили с получением в виде белого твердого вещества 1-амино-4-фенилциклогексанкарбоновой кислоты (Арc) (25 г, >100%-ный выход, загрязнена неорганической солью), которую использовали непосредственно на следующей стадии. Небольшую порцию сырого продукта очищали ВЭЖХ. 1Н-ЯМР (ДМСО-d6): 7,23~ 7,35 (m, 2), 7,10-7,19 (m, 3H), 2,45 (m, 1H), 1,92-2,18 (m, 3Н), 1,56-1,78 (m, 4H) и 1,20 (m, 1H). MCHP (пучок электронов) m/e: 220 (М+1)+, рассч. для C13H17NO2 219.
Стадия 3
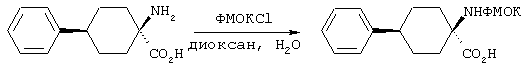
Сырую 1-амино-4-фенилциклогексанкарбоновую кислоту (Apc) с последней стадии (25 г) суспендировали в диоксане (300 мл) и водном 10%-ном Nа2СО3 (150 мл) и интенсивно перемешивали в течение ночи. Реакционную смесь концентрировали для удаления диоксана, нейтрализовали 6 н. НСl до слабокислой реакции (рН 5 до 6) и экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом и сушили над Na2SO4. Удалением растворителя получали сырой продукт, который затем очищали экспресс-хроматографией (с переходом от гексана/ЕtOАс, к CH2Cl2/MeOH), получая чистый ФМОК-цис-Арс (18,2 г, 72%-ный общий выход за две стадии) и ФМОК-транс-Арс (2,1 г, выход 8%). ФМОК-цис-Арс, 1H-ЯМР (CD3OD): 7,79 (d, 2H), 7,72 (d, 2Н), 7,37 (t, 2), 7,24-7,32 (m, 4), 7,14-7,23 (m, 3), 4,37 (d, 2H), 4,24 (t, 1H), 2,55 (m, 1H), 2,28 (m, 2H), 1,84-1,96 (m, 2H) и 1,64-1,73 (m, 4H).
Пример 2
Получение ФМОК-1-амино-4-(4-метоксифенил)циклогексан-1-карбоновой кислоты (ФМОК-4-МеО-Арс-ОН)
Стадия 1

Раствор 4-(4-гидроксифенил)циклогексанона (5,0 г, 26,3 ммоля) в ацетоне (100 мл) обрабатывали К2СО3 (14,5 г, 105 ммоля, 4 экв.) и метаниодидом (4,9 мл, 11,2 г, 78,6 ммоля, 3 экв.). Реакционную смесь выдерживали при 65° С в течение ночи. После удаления растворителя остаток обрабатывали Н2О и экстрагировали EtOAc. Органические экстракты объединяли и промывали рассолом, сушили над Na2SO4 и концентрировали под вакуумом с получением спектроскопически чистого 4-(4-метоксифенил)циклогексанона (5,34 г, 100%-ный выход). 1H-ЯМР (CDCl3): 7,16 (dt, 2H), 6,87 (dt, 2H), 3,78 (s, 3Н), 2,99 (tt, 1H), 2,47-2,53 (m, 4H), 2,20 (m, 2H) и 1,83-1,98 (m, 2H). МС (пучок электронов) m/e: 205 (М+1)+ рассч. для C13H16O2 204.
Стадия 2
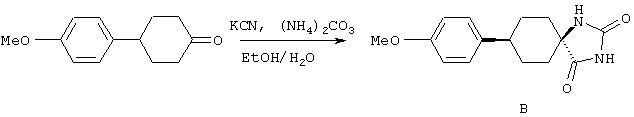
В раствор 4-(4-метоксифенил)циклогексанона (3,86 г, 18,9 ммоля) в этаноле (50 мл) и воде (15 мл) в стеклянной толстостенной колбе добавляли карбонат аммония (14,5 г, 151 ммоль, 8 экв.) и цианид калия (2,0 г, 30,7 ммоля, 1,6 экв.).
Смесь выдерживали при 80-90° С в течение 24 ч. Охлажденную реакционную смесь добавляли в смесь воды со льдом (300 мл) и интенсивно перемешивали в течение 30 мин. Полученный осадок отфильтровывали под вакуумом, тщательно промывали водой и сушили с получением в виде белого твердого вещества гидантоина В (4,75 г, 91%-ный выход). МС (пучок электронов) m/е: 273 (М-Н), рассч. для C15H18N2O3 274.
Стадия 3
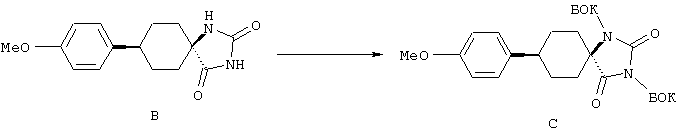
В суспензию гидантоина В (18,7 г, 68,25 ммоля) в сухом ТГФ (450 мл) последовательно добавляли ди-трет-бутилдикарбонат (37,2 г, 170,5 ммоля, 2,5 экв.), триэтиламин (10,5 мл, 7,59 г, 75,0 ммоля, 1,1 экв.) и ДМАП (460 мг, 3,65 ммоля). По прошествии примерно 15 мин после добавления реакционная смесь превращалась в прозрачный желтый раствор, и ее перемешивали в течение ночи при комнатной температуре. Реакционную смесь концентрировали под пониженным давлением с получением твердого вещества, которое затем растворяли в ЕtOАс (800 мл), промывали 1 н. НСl (3 порции по 50 мл), насыщенным водным Nа2СО3 (2 порции по 50 мл) и рассолом (2 порции по 50 мл), сушили над безводным Na2SO4 и концентрировали под пониженным давлением. Сырой светло-желтый продукт очищали экспресс-хроматографией (гексан/ЕtOАс, 90/10 → 70/30) с получением в виде белого твердого вещества чистого бис-БОК-гидантоина С (27,6 г, 87%-ный выход). 1H-ЯМР (CDCl3): 7,28 (dt, 2Н), 6,88 (dt, 2H), 3,79 (s, 3H), 2,14-2,24 (m, 2H), 1,59 (s, 9H) и 1,38 (s, 9H). МС (пучок электронов) m/е: 538 (M+MeCN+Na)+, рассч. для C25H34N2O7 474.
Стадия 4
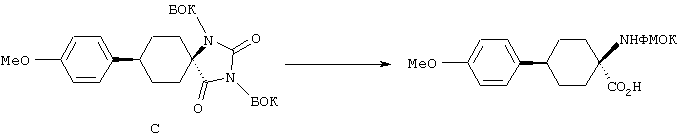
Бис-БОК-гидантоин С (15,08 г, 31,78 ммоля) растворяли в ДМЭ (500 мл) с получением прозрачного раствора. В этот раствор добавляли 1 н. NaOH (290 мл, 290 ммолей) и реакционную смесь перемешивали в течение ночи при комнатной температуре, получая слегка мутную смесь. Данные ВЭЖХ указывали на завершение реакции. Реакционную смесь концентрировали под пониженным давлением для удаления ДМЭ и экстрагировали Et2O. Без очистки полученный водный слой, содержавший 1-амино-4-(4-метоксифенил)циклогексанкарбоновую кислоту (4-МеОАрс), обрабатывали 6 н. НСl для доведения рН до 11-12. В этот раствор (-300 мл) добавляли ДМЭ (300 мл) и раствор ФМОК-OSu (16,7 г, 49,42 ммоля) в ДМЭ (200 мл) и реакционную смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь концентрировали под пониженным давлением для удаления ДМЭ, подкисляли 3 н. НСl и экстрагировали ЕtOАс. Объединенные органические экстракты промывали рассолом, сушили над безводным Na2SO4 и концентрировали. Сырой продукт очищали экспресс-хроматографией (CH2Cl2/MeOH, 98/2 → 90/10) с получением в виде белого твердого вещества в качестве чистого продукта ФМОК-4-МеОАрс (12,4 г, 83%-ный выход из бис-БОК-гидантоина С). 1H-ЯМР (ДМСО-d6): 7,88 (d, 2H), 7,76 (d, 2Н), 7,40 (t, 2H), 7,30 (t, 2H), 7,11 (d, 2H), 6,85 (d, 2H), 3,71 (s, 3H). МС (пучок электронов) m/е: 470 (М-Н), рассч. для C29H29NO5 471.
Пример 3
Получение ФМОК-1-амино-4-(4-этоксифенил)циклогексан-1-карбоновой кислоты (ФМОК-4-EtOApc-OH)
Стадия 1

Раствор 4-(4-гидроксифенил)циклогексанона (5,0 г, 26,3 ммоля) в ацетоне (100 мл) обрабатывали К2СО3 (14,5 г, 105 ммоля, 4 экв.) и этаниодидом (10,5 мл, 20,5 г, 131 ммолей, 5 экв.). Реакционную смесь выдерживали при 65° С в течение ночи. После того как растворитель удаляли, остаток обрабатывали Н2О и экстрагировали EtOAc. Органические экстракты объединяли и промывали рассолом, сушили над Na2SO4 и концентрировали под вакуумом с получением спектроскопически чистого 4-(4-этоксифенил)циклогексанона (5,74 г, 100%-ный выход). 1Н-ЯМР (CDCl3): 7,15 (dt, 2H), 6,86 (dt, 2H), 4,02 (q, 2H), 2,99 (tt, 1H), 2,46-2,54 (m, 4H), 2,16-2,24 (m, 2H), 1,83-2,00 (m, 2H) и 1,41 (t, 3H). МС (пучок электронов) m/e: 219 (М+1)+, рассч. для C14H18O2 218.
Стадия 2
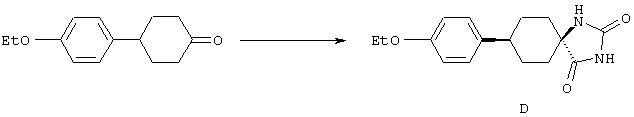
В раствор 4-(4-этоксифенил)циклогексанона (4,15 г, 19,01 ммоля) в этаноле (50 мл) и воде (15 мл) в стеклянной толстостенной колбе добавляли карбонат аммония (14,5 г, 151 ммоль, 8 экв.) и цианид калия (2,05 г, 31,42 ммоля, 1,6 экв.). Смесь выдерживали при 80-90° С в течение 19 ч. Охлажденную реакционную смесь вводили в смесь воды со льдом (300 мл) и интенсивно перемешивали в течение 30 мин. Полученный осадок отфильтровывали под вакуумом, тщательно промывали водой и сушили с получением в виде белого твердого вещества гидантоина D (5,17 г, 94%-ный выход). МС (пучок электронов) m/е: 287 (М-Н), рассч. для C16H20N2O3 288.
Стадия 3
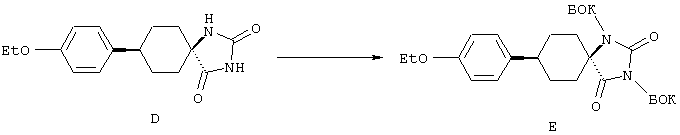
В суспензию гидантоина D (4,22 г, 14,65 ммоля) в сухом ТГФ (100 мл) последовательно вводили ди-трет-бутилдикарбонат (7,98 г, 36,60 ммоля, 2,5 экв.), триэтиламин (2,3 мл, 1,63 г, 16,11 ммоля, 1,1 экв.) и ДМАП (89,4 мг, 0,73 ммоля). По прошествии примерно 15 мин после добавления реакционная смесь превращалась в прозрачный желтый раствор, и ее перемешивали в течение ночи при комнатной температуре. Реакционную смесь концентрировали под пониженным давлением с получением твердого вещества, которое затем растворяли в ЕtOАс (300 мл), промывали 1 н. НСl (3 порции по 20 мл), насыщенным водным Nа2СО3 (2 порции по 20 мл) и рассолом (2 порции по 20 мл), сушили над безводным Na2SO4 и концентрировали под пониженным давлением. Сырой светло-желтый продукт очищали экспресс-хроматографией (гексан/EtOAc, 90/10 → 70/30) с получением в виде белого твердого вещества чистого бис-БОК-гидантоина Е (7,01 г, 98%-ный выход). 1H-ЯМР (CDCl3): 7,27 (dt, 2Н), 6,87 (dt, 2H), 4,02 (q, 2Н), 1,59 (s, 9H), 1,43 (t, 3H) и 1,38 (s, 9Н). МС (пучок электронов) m/е: 999 (2M+Na)+, рассч. для C26H36N2O7 488.
Стадия 4
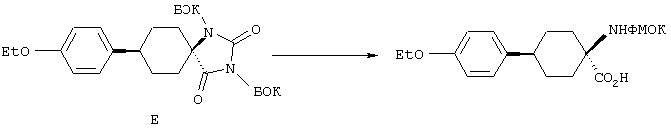
Бис-БОК-гидантоин Е (6,58 г, 13,46 ммоля) растворяли в ДМЭ (200 мл) с получением прозрачного раствора. В этот раствор добавляли 1 н. NaOH (121 мл, 121 ммоля) и реакционную смесь перемешивали в течение ночи при комнатной температуре, получая слегка мутную смесь. Данные ВЭЖХ указывали на завершение реакции. Реакционную смесь концентрировали под пониженным давлением для удаления ДМЭ и экстрагировали Et2O. Без очистки полученный водный слой, содержавший 1-амино-4-(4-этоксифенил)циклогексанкарбоновую кислоту (4-ЕtOАрс), обрабатывали 6 н. НСl для доведения рН до 11-12. В этот раствор (~130 мл) добавляли ДМЭ (100 мл) и раствор ФМОК-OSu (6,83 г, 20,24 ммоля) в ДМЭ (30 мл) и реакционную смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь концентрировали под пониженным давлением для удаления ДМЭ, подкисляли 3 н. НСl и экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, сушили над безводным Na2SO4 и концентрировали. Сырой продукт очищали экспресс-хроматографией (СН2Сl2/МеОН, 98/2 → 90/10) с получением в виде белого твердого вещества в качестве чистого продукта ФМОК-4-EtOApc (5,56 г, 85%-ный выход из бис-БОК-гидантоина Е). 1H-ЯМР (ДМСО-d6): 7,88 (d, 2H), 7,74 (d, 2H), 7,40 (td, 2H), 7,30 (td, 2H), 7,11 (d, 2H), 6,84 (d, 2H), 3,97 (q, 2H) и 1,29 (t, 3H). МС (пучок электронов) m/е: 484 (М-Н), рассч. для С30Н31NO5 485.
Пример 4
Получение ФМОК-1-амино-4-(4-гидроксифенил)циклогексан-1-карбоновой кислоты (ФМОК-4-НОАрс-ОН)
Стадия 1
В раствор 4-(4-гидроксифенил)циклогексанона (2,00 г, 10,52 ммоля) в этаноле (30 мл) и воде (10 мл) в стеклянной толстостенной колбе вводили карбонат аммония (6,17 г, 64,2 ммоля, 6 экв.) и цианид калия (1,07 г, 15,8 ммоля, 1,5 экв.). Смесь выдерживали при 80-90° С в течение ночи. Охлажденную реакционную смесь добавляли в смесь воды со льдом (200 мл) и интенсивно перемешивали в течение 30 мин. Полученный осадок отфильтровывали под вакуумом, тщательно промывали водой и сушили с получением в виде белого твердого вещества гидантоина F (2,56 г, 94%-ный выход). МС (пучок электронов) m/е: 261 (М+Н)+, рассч. для С14Н16N2О3 260.
Стадия 2
Гидантоин F (2,10 г, 8,06 ммоля) суспендировали в водном NaOH (6 н., 100 мл) и выдерживали при 130° С в течение 2-3 дней. По завершении гидролиза реакционную смесь нейтрализовали конц. НСl до слабокислой реакции (рН ~6). Образовавшуюся суспензию фильтровали, промывали водой и сушили с получением в виде белого твердого вещества 1-амино-4-(4-гидроксифенил)циклогексанкарбоновой кислоты (4-НОАрс) (3,1 г, >100%-ный выход, загрязнена неорганической солью). МС (пучок электронов) m/е: 236 (М+Н)+, рассч. для C13H17NO3 235.
Стадия 3

Сырую 1-амино-4-(4-гидроксифенил)циклогексанкарбоновую кислоту (4-НОАрс) с последней стадии (3,1 г) суспендировали в диоксане (100 мл) и водном 10%-ном Na2CO3 (50 мл) и интенсивно перемешивали в течение ночи. Реакционную смесь концентрировали для удаления диоксана, нейтрализовали 6 н. НСl до слабокислой реакции (рН от 5 до 6) и экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом и сушили над Na2SO4. Удалением растворителя получали сырой продукт, который очищали экспресс-хроматографией (с переходом от гексана/EtOAc к CH2Cl2/MeOH) с выделением чистого ФМОК-4-НОАрс (2,76 г, 75%-ный общий выход за две стадии). 1Н-ЯМР (CD3OD): 7,78 (d, 2H), 7,72 (d, 2H), 7,38 (t, 2H), 7,30 (td, 2H), 7,04 (d, 2H), 6,72 (dt, 2H), 4,38 (d, 2H), 4,25 (t, 1H), 2,46 (m, 1H), 2,24-2,34 (m, 2H) и 1,81-1,92 (m, 6H). МС (пучок электронов) m/е: 456 (М-Н), рассч. для C28H27NO5 457.
Пример 5
Получение ФМОК-1-амино-4-(4-изопропоксифенил)циклогексан-1-карбоновой кислоты (ФМОК-4-изо-РrОАрс-ОН)
Стадия 1
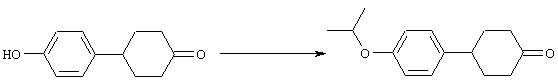
Раствор 4-(4-гидроксифенил)циклогексанона (6,0 г, 31,6 ммоля) в ДМФ (90 мл) обрабатывали К2СО3 (21 г, 158 ммолей, 5 экв.) и 2-иодпропаном (15 мл, 26,8 г, 158 ммолей, 5 экв.). Реакционную смесь выдерживали при 100° С в течение ночи. После того как растворитель удаляли, остаток обрабатывали Н2О и экстрагировали EtOAc. Органические экстракты объединяли и промывали рассолом, сушили над Na2SO4 и концентрировали под вакуумом с получением спектроскопически чистого 4-(4-изопропоксифенил)циклогексанона (7,02 г, 95%-ный выход). 1H-ЯМР (CDCl3): 7,14 (dt, 2H), 6,84 (dt, 2H), 4,3 (септет, 1H), 2,97 (tt, 1H), 2,46-2,52 (m, 4H), 2,16-2,24 (m, 2H), 1,83-1,98 (m, 2H) и 1,33 (d, 6H).
Стадия 2

В раствор 4-(4-изопропоксифенил)циклогексанона (5,1 г, 21,98 ммоля) в этаноле (90 мл) и воде (30 мл) в стеклянной толстостенной колбе вводили карбонат аммония (12,6 г, 131 ммоля, 6 экв.) и цианид калия (2,14 г, 32,9 ммоля, 1,5 экв.). Смесь выдерживали при 80-90° С в течение 24 ч. Охлажденную реакционную смесь добавляли в смесь воды со льдом (400 мл) и интенсивно перемешивали в течение 30 мин. Полученный осадок отфильтровывали под вакуумом, тщательно промывали водой и сушили с получением в виде белого твердого вещества гидантоина G (6,60 г, 99%-ный выход). 1Н-ЯМР (ДМСО-d6): 10,60 (s, 1Н), 8,65 (s, 1H), 7,18 (d, 2H), 6,80 (d, 2Н), 4,52 (септет, 1Н). 2,43 (m, 1H), 1,85-2,15 (m, 2H), 1,56-1,80 (m, 6H) и 1,22 (d, 6H). МС (пучок электронов) m/e: 301 (М-Н), рассч. для C17H22N2O3 302.
Стадия 3
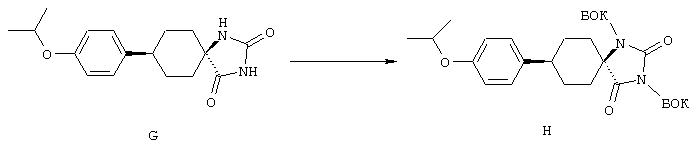
В суспензию гидантоина G (5,8 г, 19,20 ммоля) в сухом ТГФ (180 мл) последовательно добавляли ди-трет-бутилдикарбонат (10,46 г, 48,0 ммоля, 2,5 экв.), триэтиламин (2,9 мл, 2,13 г, 21,12 ммоля, 1,1 экв.) и ДМАП (140 мг, 1,15 ммоля). По прошествии примерно 15 мин после добавления реакционная смесь превращалась в прозрачный желтый раствор, и ее перемешивали в течение ночи при комнатной температуре. Реакционную смесь концентрировали под пониженным давлением с получением твердого вещества, которое затем растворяли в EtOAc (600 мл), промывали 1 н. НСl (3 порции по 40 мл), насыщенным водным Na2CO3 (2 порции по 40 мл) и рассолом (2 порции по 40 мл), сушили над безводным Na2SO4 и концентрировали под пониженным давлением. Сырой светло-желтый продукт очищали экспресс-хроматографией (гексан/EtOAc, 90/10 → 80/20) с получением в виде белого твердого вещества в качестве чистого продукта бис-БОК-гидантоина Н (9,4 г, 98%-ный выход). 1Н-ЯМР (CDCl3): 7,27 (dt, 2H), 6,87 (dt, 2H), 4,02 (q, 2H), 2,98 (t, 1H), 2,26-2,56 (m, 4H), 2,14-2,24 (m, 2H), 1,76-1,86 (m, 2H), 1,59 (s, 9H), 1,43 (t, 3H) и 1,38 (s, 9H). МС (пучок электронов) m/е: 999 (2M+Na)+, рассч. для C26H36N2O7 488.
Стадия 4
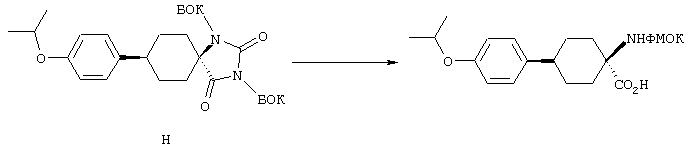
Бис-БОК-гидантоин Н (4,34 г, 8,64 ммоля) растворяли в ДМЭ (100 мл) с получением прозрачного раствора. В этот раствор добавляли 1 н. NaOH (78 мл, 78 ммолей) и реакционную смесь перемешивали в течение ночи при комнатной температуре, получая довольно прозрачную смесь. Данные ВЭЖХ указывали на завершение реакции. Реакционную смесь концентрировали под пониженным давлением для удаления ДМЭ и экстрагировали Et2O. Без очистки полученный водный слой, содержавший 1-амино-4-(4-изопропоксифенил)циклогексанкарбоновую кислоту (4-изо-РrОАрс), обрабатывали 6 н. НСl для доведения рН до 11-12. В этот раствор (~90 мл) добавляли ДМЭ (120 мл) и раствор ФМОК-OSu (3,49 г, 10,34 ммоля, 1,2 экв.) в ДМЭ (20 мл) и реакционную смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь концентрировали под пониженным давлением для удаления ДМЭ, подкисляли 3 н. НСl и экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, сушили над безводным Na2SO4 и концентрировали. Сырой продукт очищали экспресс-хроматографией (гексан/EtOAc → CH2Cl2/MeOH) с получением в качестве чистого продукта в виде белого твердого вещества ФМОК-4-изо-РrОАрс (3,23, 75%-ный выход из бис-БОК-гидантоина Н). 1Н-ЯМР (ДМСО-d6): 7,76 (d, 2H), 7,60 (d, 2H), 7,39 (t, 2H), 7,31 (t, 2H), 7,08 (d, 2H), 6,84 (d, 2H), 4,24 (m, 1H) и 1,34 (d, 6H). МС (пучок электронов) m/e: 498 (М-Н), рассч. для С31Н33NO5 499.
Пример 6
Получение ФМОК-1-амино-4-(4-метилфенил)циклогексан-1-карбоновой кислоты (ФМОК-4-МеApc-OH)
Стадия 1

В раствор 4-иодтолуола (10,9 г, 50,0 ммоля) в сухом ТГФ (180 мл) при -78° С в течение 20 мин добавляли раствор н-BuLi (1,6 М, 31,0 мл, 50 ммолей) в гексане. Реакционную смесь перемешивали в течение дополнительных 20 мин перед добавлением по каплям раствора 1,4-циклогександионмоноэтиленкеталя (6,0 г, 38,46 ммоля) в сухом ТГФ (100 мл). После перемешивания в течение 2 ч при -78° С реакцию гасили водным NH4Cl и экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, сушили над Na2SO4 и концентрировали под вакуумом с получением в виде белого твердого вещества спектроскопически чистого продукта I (9,34 г, 98%-ный выход). 1Н-ЯМР (CDCl3): 7,41 (m, 2Н), 7,16 (d, 2H), 3,98 (m, 4Н), 2,34 (s, 3H). MC (EI) m/e: 248(M+), рассч. для С15H20О3 248.
Стадия 2
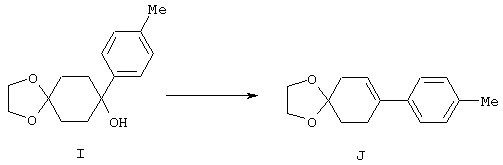
В раствор спирта I (9,10 г, 36,65 ммоля) в сухом бензоле (200 мл) в колбе, снабженной ловушкой Дина-Старка, вводили моногидрат п-толуолсульфоновой кислоты (650 мг) и реакционную смесь выдерживали при 100° С в течение 3 ч. Реакционную смесь охлаждали до КТ, разбавляли EtOAc (500 мл) и промывали водным Na2CO3 (50 мл), рассолом (3 порции по 50 мл), сушили над Na2SO4 и концентрировали под пониженным давлением с получением спектроскопически чистого продукта J (8,36 г, 100%-ный выход), который без очистки использовали на следующей стадии. МС (EI) m/е: 230 (М+), 190 (М-ОСН2СН2О), рассч. для C15H18O2 230.
Стадия 3

В раствор олефина J (7,49 г) в EtOAc (180 мл) вводили Pd/C (5 мас.% на угле, 800 мг) и реакцию проводили под давлением водорода 40 фунтов/кв.дюйм при комнатной температуре в течение 3 ч. Катализатор отфильтровывали и фильтрат концентрировали с получением в виде бесцветного масла спектроскопически чистого продукта К (7,40 г, 100%-ный выход). МС (EI) m/е: 232 (М+), 188 (М-ОСН2СН2), рассч. для C15H20O2 232.
Стадия 4
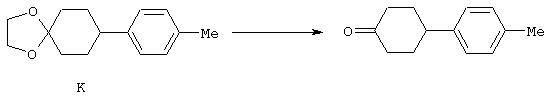
Раствор кеталя К (6,90 г) в ацетоне (140 мл) обрабатывали 4 н. НСl (60 мл) и выдерживали при 65° С в течение 4 ч. Растворитель удаляли, остаток разбавляли EtOAc и нейтрализовали 4 н. НСl. Водную фазу экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, сушили и концентрировали. Полученный сырой 4-(4-метилфенил)циклогексанон использовали на следующей стадии без очистки (5,57 г, количественный выход). МС (EI) m/е: 188 (М+), рассч. для C13H16O 188.
Стадия 5
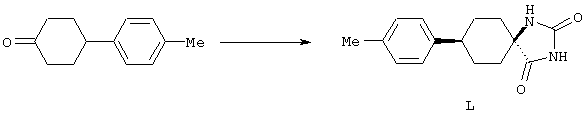
В раствор 4-(4-метилфенил)циклогексанона (5,32 г, 28,3 ммоля) в этаноле (90 мл) и воде (30 мл) в стеклянной толстостенной колбе вводили карбонат аммония (16,3 г, 169,8 ммоля, 6 экв.) и цианид калия (3,68 г, 56,5 ммоля, 2 экв.).
Смесь выдерживали при 80-90° С в течение ночи. Охлажденную реакционную смесь добавляли в смесь воды со льдом (400 мл) и интенсивно перемешивали в течение 30 мин. Полученный осадок отфильтровывали под вакуумом, тщательно промывали водой и сушили с получением в виде белого твердого вещества гидантоина L (6,3 г, 86%-ный выход). МС (пучок электронов) m/е: 517 (2М+Н), рассч. для C15H18ClN2O2 258
Стадия 6
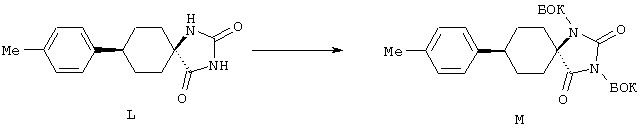
В суспензию гидантоина L (5,82 г, 22,55 ммоля) в сухом ТГФ (250 мл) последовательно вводили ди-трет-бутилдикарбонат (12,3 г, 56,4 ммоля, 2,5 экв.), триэтиламин (3,5 мл, 2,5 г, 24,7 ммоля, 1,1 экв.) и ДМАП (275 мг, 2,25 ммоля). Реакционная смесь превращалась в прозрачный желтый раствор, и его перемешивали в течение ночи при комнатной температуре. Реакционную смесь концентрировали под пониженным давлением с получением твердого вещества, которое затем растворяли в EtOAc (500 мл), промывали 1 н. НСl (3 порции по 50 мл), насыщенным водным Na2CO3 (2 порции по 50 мл) и рассолом (2 порции по 50 мл), сушили над безводным Na2SO4 и концентрировали под пониженным давлением. Сырой светло-желтый продукт очищали экспресс-хроматографией (гексан/EtOAc, 90/10 → 70/30) с получением в виде белого твердого вещества чистого бис-БОК-гидантоина М (10,03 г, 100%-ный выход). 1H-ЯМР (CDCl3): 7,26 (d, 2Н), 6,87 (d, 2H), 3,00 (m, 1H), 2,32 (s, 3H), 1,59 (s, 9H) и 1,37 (s, 9H).
Стадия 7

Бис-БОК-гидантоин М (6,40 г, 13,97 ммоля) растворяли в ДМЭ (200 мл) с получением прозрачного раствора. В этот раствор добавляли 1 н. NaOH (120 мл, 120 ммоля) и реакционную смесь перемешивали в течение ночи при комнатной температуре, получая слегка мутную смесь. Данные ВЭЖХ указывали на завершение реакции. Реакционную смесь концентрировали под пониженным давлением для удаления ДМЭ и экстрагировали Et2O. Без очистки полученный водный слой, содержавший 1-амино-4-(4-метилфенил)циклогексанкарбоновую кислоту (4-МеАрс), обрабатывали 6 н. НСl для доведения рН до 11-12. В этот раствор (~140 мл) добавляли ДМЭ (240 мл) и раствор ФМОК-OSu (5,10 г, 15,13 ммоля, 1,1 экв.) в ДМЭ (40 мл) и реакционную смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь концентрировали под пониженным давлением для удаления ДМЭ, подкисляли 3 н. НСl и экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, сушили над безводным Na2SO4 и концентрировали. Сырой продукт очищали экспресс-хроматографией (CH2Cl2/MeOH, 98/2 → 90/10) с получением в виде белого твердого вещества в качестве чистого продукта ФМОК-4-МеАрс (4,35 г, 69%-ный выход из бис-БОК-гидантоина М). 1Н-ЯМР (ДМСО-d6): 7,88 (d, 2Н), 7,75 (d, 2H), 7,24-7,43 (m, 4H), 7,02-7,14 (m, 4H), 4,25 (m, 3H), 2,24 (s, 3H).
Пример 7
Получение ФМОК-1-амино-4-(4-хлорфенил)циклогексан-1-карбоновой кислоты (ФМОК-4-ClApc-OH)
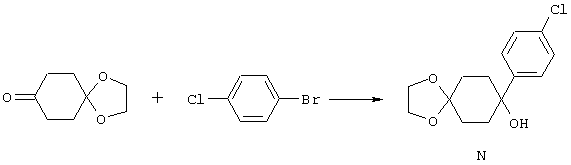
Стадия 1
Раствор 4-хлорфенилбромида (7,5 г, 39,2 ммоля) в сухом ТГФ (180 мл) охлаждали до -78° С и обрабатывали добавляемым по каплям раствором н-BuLi (1,6 М, 25 мл, 40 ммолей) в гексане в течение 20 мин. Реакционную смесь перемешивали в течение дополнительных 30 мин перед добавлением по каплям раствора 1,4-циклогександионмоноэтиленкеталя (6,0 г, 38,46 ммоля) в сухом ТГФ (100 мл). После перемешивания в течение 1 ч при -78° С реакцию гасили водным NH4Cl и экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, сушили над Na2SO4 и концентрировали под вакуумом с получением в виде белого твердого вещества спектроскопически чистого продукта N (9,40 г, 91%-ный выход). 1H-ЯМР (CDCl3): 7,45 (m 2H), 7,31 (m, 2H), 3,99 (m, 4Н), 2,02-2,20 (m, 4H), 1,75-1,82 (m, 2H), 1,66-1,73 (m, 2H), 1,54 (s, 1H). MC (EI) m/e: 268 (М+), 251 (М-ОН), 250 (М-Н2O), рассч. для C14H17ClO3 268.
Стадия 2
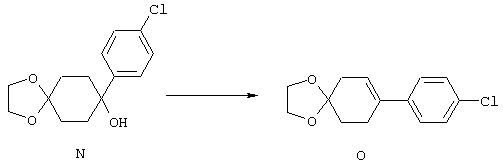
В раствор спирта N (6,78 г, 25,30 ммоля) в сухом бензоле (120 мл) в колбе, снабженной ловушкой Дина-Старка, вводили моногидрат п-толуолсульфоновой кислоты (960 мг) и реакционную смесь кипятили с обратным холодильником в течение 3 ч. Реакционную смесь охлаждали до КТ, разбавляли EtOAc (500 мл) и промывали водным Na2СО3 (50 мл), рассолом (3 порции по 50 мл), сушили над Na2SO4 и концентрировали под пониженным давлением с получением спектроскопически чистого продукта О (6,30 г, 100%-ный выход), который без очистки использовали на следующей стадии. MC (EI) m/e: 250 (M+), 190 (М-ОСН2СН2O), рассч. для C14H15ClO2 250.
Стадия 3
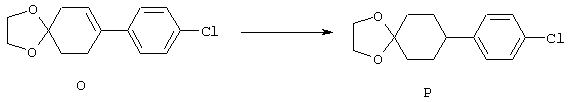
В раствор олефина О (6,11 г) в EtOAc (120 мл) вводили Pd/C (5 мас.% на угле, 600 мг) и реакцию проводили при комнатной температуре в течение 3 ч под давлением водорода 5 фунтов/кв.дюйм. Катализатор отфильтровывали и фильтрат концентрировали с получением в виде бесцветного масла спектроскопически чистого продукта Р (6,10 г, 100%-ный выход). MC (EI) m/e: 252 (М+), рассч. для C14H17ClO2 252.
Стадия 4
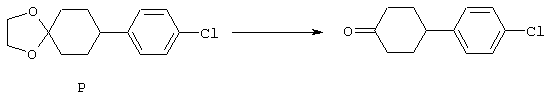
Раствор кеталя Р (5,81 г, 23,06 ммоля) в ацетоне (200 мл) обрабатывали моногидратом п-толуолсульфоновой кислоты (876 мг) и выдерживали при 60° С в течение ночи. Растворитель удаляли и остаток растворяли в EtOAc, промывали водным раствором Na2CO3, рассолом, сушили и концентрировали с получением в виде желтого масла сырого продукта (5,38 г, >100%-ный выход). Очисткой экспресс-хроматографией (гексан/EtOAc, 80/20 → 60/40) в виде светло-желтого масла получали 4-(4-хлорфенил)циклогексанон (4,54 г, 95%-ный выход). МС (EI) m/e: 208 (М+), рассч. для C12H13ClO2 208.
Стадия 5
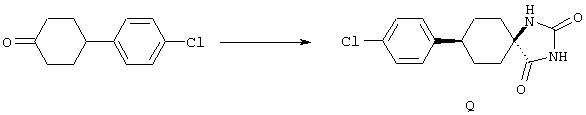
В раствор 4-(4-хлорфенил)циклогексанона (4,26 г, 20,48 ммоля) в этаноле (90 мл) и воде (30 мл) в стеклянной толстостенной колбе вводили карбонат аммония (13,8 г, 144 ммоля, 7 экв.) и цианид калия (3,56 г, 54,77 ммоля, 2,5 экв.). Смесь выдерживали при 80-90° С в течение ночи. Охлажденную реакционную смесь добавляли в смесь воды со льдом (400 мл) и интенсивно перемешивали в течение 30 мин. Полученный осадок отфильтровывали под вакуумом, тщательно промывали водой и сушили с получением в виде белого твердого вещества гидантоина Q (5,58 г, 98%-ный выход). МС (пучок электронов) m/e: 277 (М-Н), рассч. для C14H15ClN2O2 278.
Стадия 6
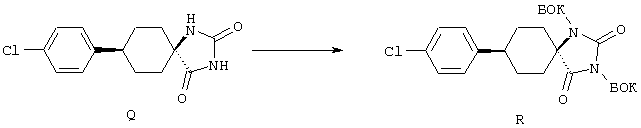
В суспензию гидантоина Q (5,15 г, 18,5 ммоля) в сухом ТГФ (250 мл) последовательно добавляли ди-трет-бутилдикарбонат (10,1 т, 46,3 ммоля, 2,5 экв.), триэтиламин (2,8 мл, 2,07 г, 20,45 ммоля, 1,1 экв.) и ДМАП (226 мг, 1,85 ммоля). Реакционная смесь превращалась в прозрачный желтый раствор, и его перемешивали в течение ночи при комнатной температуре. Реакционную смесь концентрировали под пониженным давлением с получением твердого вещества, которое затем растворяли в EtOAc (500 мл), промывали 1 н. НСl (3 порции по 50 мл), насыщенным водным Nа2СО3 (2 порции по 50 мл) и рассолом (2 порции по 50 мл), сушили над безводным Na2SO4 и концентрировали под пониженным давлением. Сырой светло-желтый продукт очищали экспресс-хроматографией (гексан/EtOАс, 90/10 → 70/30) с получением в виде белого твердого вещества чистого бис-БОК-гидантоина R (8,05 г, 91%-ный выход). МС (пучок электронов) m/e: 542 (M+Na+MeCN), рассч. для С24Н31СlN2O6 478.
Стадия 7
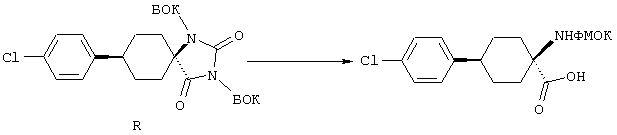
Бис-БОК-гидантоин R (6,41 г, 13,97 ммоля) растворяли в ДМЭ (200 мл) с получением прозрачного раствора. В этот раствор добавляли 1 н. NaOH (120 мл, 120 ммолей) и реакционную смесь перемешивали в течение ночи при комнатной температуре, получая слегка мутную смесь. Данные ВЭЖХ указывали на завершение реакции. Реакционную смесь концентрировали под пониженным давлением для удаления ДМЭ и экстрагировали Et2O. Без очистки полученный водный слой, содержавший 1-амино-4-(4-хлорфенил)циклогексанкарбоновую кислоту (4-ClApc), обрабатывали 6 н. НСl для доведения рН до 11-12. В этот раствор (~180 мл) добавляли ДМЭ (240 мл) и раствор ФМОК-OSu (5,31 г, 15,74 ммоля, 1,1 экв.) в ДМЭ (30 мл) и реакционную смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь концентрировали под пониженным давлением для удаления ДМЭ, подкисляли 3 н. НСl и экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, сушили над безводным Na2SO4 и концентрировали. Сырой продукт очищали экспресс-хроматографией (CH2Cl2/MeOH, 98/2 → 90/10) с получением в виде белого твердого вещества в качестве чистого продукта ФМОК-4-ClApc (5,04 г, 76%-ный выход из бис-БОК-гидантоина). 1Н-ЯМР (ДМСО-d6): 7,88 (d, 2Н), 7,74 (d, 2Н), 7,19-7,42 (m, 8Н), 4,20-4,31 (m, 3H). МС (пучок электронов) m/e: 474 (М-Н), рассч. для C28H26ClNO4 475.
Пример 8
Получение ФМОК-1-амино-4-(3-метоксифенил)циклогексан-1-карбоновой кислоты (ФМОК-3-МеОАрс-ОН)
Стадия 1
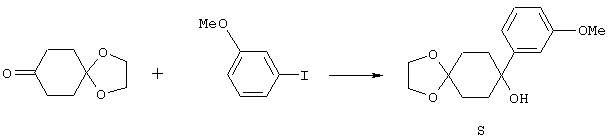
В раствор 3-иоданизола (11,7 г, 50,0 ммоля, 1,3 экв.) в сухом ТГФ (180 мл) при -78° С в течение 25 мин вводили раствор н-BuLi (1,6 M, 31,0 мл, 50 ммолей, 1,3 экв.) в гексане.
Реакционную смесь перемешивали в течение дополнительных 30 мин перед добавлением по каплям раствора 1,4-циклогександионмоноэтиленкеталя (6,0 г, 38,46 ммоля) в сухом ТГФ (100 мл). После перемешивания в течение 2 ч при -78° С реакцию гасили водным NH4Cl и экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, сушили над Na2SO4 и концентрировали под вакуумом с получением в виде белого твердого вещества спектроскопически чистого продукта S (9,34 г, 98%-ный выход). 1H-ЯМР (CDCl3): 7,26 (dd, 1H), 7,06-7,11 (m, 2H), 6,79 (dd, 1H), 3,98 (m, 4H), 3,81 (s, 3H).
Стадия 2
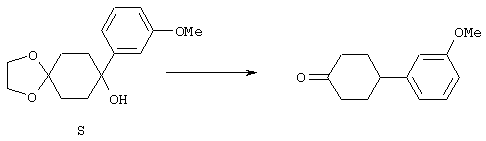
В перемешиваемый раствор спирта S (5,6 г, 21,21 ммоля) в сухом СН2Сl2 (200 мл) в азотной атмосфере при температуре бани из соли со льдом последовательно добавляли триэтилсилан (10,2 мл, 7,4 г, 63,63 ммоля, 3 экв.) и эфират трифторида бора (21,5 мл, 24,1 г, 169,7 ммоля, 8 экв.). Далее реакционной смеси давали нагреться до комнатной температуры и ее перемешивали в течение 3 ч перед промывкой 10%-ным водным раствором К2СО3 и Н2О, сушили над Na2SO4 и концентрировали под вакуумом с получением в виде масла дезоксигенированного соединения (4,91 г), которое было достаточно чистым для прямого применения. Этот сырой промежуточный продукт растворяли в ацетоне (130 мл), обрабатывали 4 н. НСl (60 мл) и выдерживали при 65° С в течение 4 ч. Растворитель удаляли под пониженным давлением, остаток разбавляли EtOAc и нейтрализовали 4 н. раствором NaOH. Водный слой экстрагировали EtOAc и объединенные органические экстракты промывали рассолом, сушили и концентрировали. Полученный остаток очищали экспресс-хроматографией на силикагеле (80/20 → 60/40) с получением в виде желтого масла 4-(3-метоксифенил)циклогексанона (3,67 г, 85%-ный общий выход). 1H-ЯМР (CDCl3): 7,25 (dt, 1H), 6,75-6,86 (m, 3H), 3,81 (s, 3H), 3,00 (tt, 1H). MC (EI) m/e: 204 (М+), рассч. для C13H16O2 204.
Стадия 3
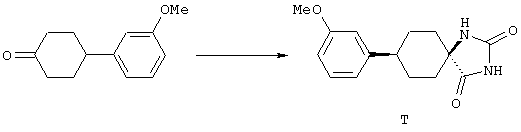
В раствор 4-(3-метоксифенил)циклогексанона (3,10 г, 15,20 ммоля) в этаноле (60 мл) и воде (20 мл) в стеклянной толстостенной колбе вводили карбонат аммония (8,75 г, 91,20 ммоля, 6 экв.) и цианид калия (1,98 г, 30,40 ммоля, 2 экв.). Смесь выдерживали при 80-90° С в течение ночи. Охлажденную реакционную смесь добавляли в смесь воды со льдом (300 мл) и интенсивно перемешивали в течение 30 мин. Полученный осадок отфильтровывали под вакуумом, тщательно промывали водой и сушили с получением в виде белого твердого вещества гидантоина Т (4,08 г, 98%-ный выход). 1Н-ЯМР (ДМСО-d6): 7,11 (d, 1H), 6,70-6,94 (m, 3H), 3,72 (s, 3H). MC (пучок электронов) m/e: 316 (M+MeCN+H), рассч. для C15H18N2O3 274.
Стадия 4
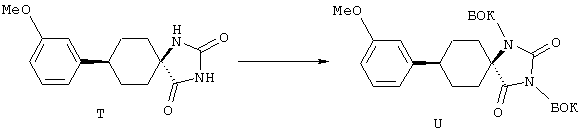
В суспензию гидантоина Т (5,29 г, 19,30 ммоля) в сухом ТГФ (250 мл) последовательно вводили ди-трет-бутилдикарбонат (10,5 г, 48,16 ммоля, 2,5 экв.), триэтиламин (3,0 мл, 2,17 г, 21,52 ммоля, 1,1 экв.) и ДМАП (235 мг, 1,92 ммоля). Реакционная смесь превращалась в прозрачный желтый раствор, и ее перемешивали в течение ночи при комнатной температуре. Реакционную смесь концентрировали под пониженным давлением с получением твердого вещества, которое затем растворяли в EtOAc (500 мл), промывали 1 н. НСl (3 порции по 50 мл), насыщенным водным Nа2СО3 (2 порции по 50 мл) и рассолом (2 порции по 50 мл), сушили над безводным Na2SO4 и концентрировали под пониженным давлением. Сырой светло-желтый продукт очищали экспресс-хроматографией (гексан/EtOAc, 80/20 → 60/40) с получением в виде белого твердого вещества чистого бис-БОК-гидантоина U (8,70 г, 95%-ный выход). МС (пучок электронов) m/e: 538 (M+MeCN+Na), рассч. для C25H24N2O7 474.
Стадия 5
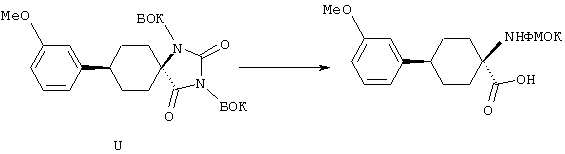
Бис-БОК-гидантоин U (2,30 г, 4,84 ммоля) растворяли в ДМЭ (80 мл) с получением прозрачного раствора. В этот раствор добавляли 1 н. NaOH (44 мл, 44 ммоля) и реакционную смесь перемешивали в течение ночи при комнатной температуре, получая слегка мутную смесь. Данные ВЭЖХ указывали на завершение реакции. Реакционную смесь концентрировали под пониженным давлением для удаления ДМЭ и экстрагировали Et2O. Без очистки полученный водный слой, содержавший 1-амино-4-(3-метоксифенил)циклогексанкарбоновую кислоту (3-МеОАрс), обрабатывали 6 н. НСl для доведения рН до 11-12. В этот раствор (~40 мл) добавляли диоксан (80 мл) и ФМОК-С1 (1,73 г, 6,76 ммоля, 1,4 экв.) и реакционную смесь перемешивали в течение ночи при комнатной температуре. Далее реакционную смесь концентрировали под пониженным давлением для удаления ДМЭ, нейтрализовали 3 н. НСl и экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, сушили над безводным Na2SO4 и концентрировали. Сырой продукт очищали экспресс-хроматографией на силикагеле (СН2Сl2/МеОН, 98/2 → 90/10) с получением в виде белого твердого вещества ФМОК-3-МеОАрс (1,98 г, 87%-ный выход из бис-БОК-гидантоина U). 1Н-ЯМР (ДМСО-d6): 7,88 (d, 2H), 7,75 (d, 2H), 7,40 (td, 2H), 7,30 (td, 2H), 7,21 (m, 1H), 6,71-6,80 (m, 3H), 3,72 (s, 3H). МС (пучок электронов) m/e: 494 (M+Na), рассч. для C29H29NO5 471.
Пример 9
Получение ФМОК-(D,L)-5-бром-2-аминотетралин-2-карбоновой кислоты (ФMOK-(D,L) 5-Br-Atc-OH)
Стадия 1
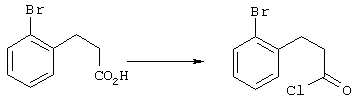
Смесь 3-(2-бромфенил)пропионовой кислоты (полученной на 2-й стадии из 2-бромбензилбромида, 2,0 г, 8,73 ммоля), оксалилхлорида (1,14 мл, 13,1 ммоля) и метиленхлорида (20 мл) охлаждали на ледяной бане и по каплям добавляли N,N-диметилформамид (34 мкл, 0,44 ммоля). Смесь перемешивали при комнатной температуре в течение 3 ч. Концентрированием под вакуумом получали 3-(2-бромфенил)пропаноилхлорид, который растворяли в метиленхлориде и в неочищенном виде использовали на следующей стадии.
Стадия 2
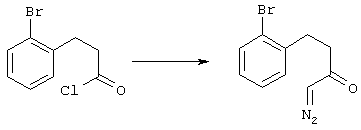
Раствор вышеупомянутого хлорангидрида кислоты (сырой, 8,73 ммоля) в метиленхлориде медленно добавляли в раствор диазометана (получен из 5,70 г 1-метил-3-нитро-1-нитрозогуанидина) в диэтиловом эфире (40 мл), охлажденный на ледяной бане. Затем смесь нагревали до комнатной температуры и перемешивали в течение ночи. Смесь концентрировали под вакуумом и очищали хроматографией в колонке (10 → 20% этилацетата/гексаны) с получением 1-диазо-4-(2-бромфенил)бутан-2-она (1,88 г, 85%-ный выход за 2 стадии). 1Н-ЯМР (CDCl3, δ ): 7,50 (1Н, d, фенил), 7,24 (2Н, m, фенил), 7,06 (1Н, m, фенил), 5,21 (1Н, широкий s, диазо), 3,05 (2Н, t, бензиловый), 2,62 (2Н, m).
Стадия 3
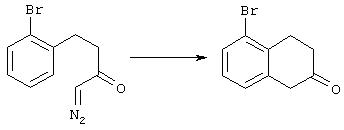
В смесь димера ацетата родия(II) (15 мг, 0,068 ммоля) в метиленхлориде (120 мл) при кипячении с обратным холодильником медленно добавляли раствор 1-диазо-4-(2-бромфенил)бутан-2-она (1,74 г, 6,85 ммоля) в метиленхлориде (30 мл). После того как завершали добавление, смесь кипятили с обратным холодильником в течение больше двадцати минут. Смесь охлаждали до комнатной температуры, добавляли трифторуксусной кислоты (1,5 мл) и смесь перемешивали при комнатной температуре в течение одного часа. Реакцию гасили насыщенным раствором бикарбоната натрия. Слои разделяли и метиленхлоридный слой еще раз промывали насыщенным раствором бикарбоната натрия. Объединенные водные слои подвергали обратной экстракции метиленхлоридом. Объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали под вакуумом с получением продукта в виде коричневого масла. Очисткой хроматографией в колонке (10 → 15% этилацетата/гексаны) в виде бесцветного масла получали 5-бром-β -тетралон (1,18 г, 77%-ный выход). 1H-ЯМР (CDCl3, δ ): 7,46 (1Н, t, фенил), 7,05-7,09 (2Н, m, фенил), 3,58 (2Н, s, бензиловый), 3,22 (2Н, t, бензиловый), 2,54 (2Н, t).
Стадия 4
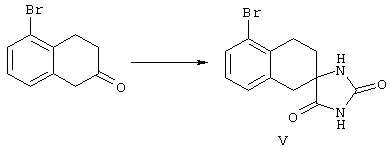
Смесь 5-бром-β -тетралона (1,18 г, 5,24 ммоля), цианида калия (512 мг, 7,86 ммоля), карбоната аммония (3,0 г, 31,22 ммоля), этанола (25 мл) и воды (5 мл) в герметично закрытой толстостенной колбе выдерживали при 80° С на масляной бане в течение 4 дней. После охлаждения до комнатной температуры белую суспензию выливали в смесь воды со льдом и перемешивали при комнатной температуре в течение пары часов. Фильтрованием с последующей сушкой на воздухе получали гидантоин V (1,31 г, 85%-ный выход). 1Н-ЯМР (ДМСО-d6, δ ): 10,71 (1Н, широкий, NH), 8,28 (1Н, широкий s, NH), 7,0-7,5 (3Н, m, фенил). МСНР (пучок электронов): C12H11BrN2O2, рассч. 294; обнаруж. 293 (М-Н), 295 (М-Н).
Стадия 5
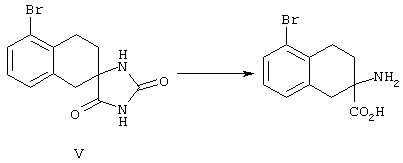
Смесь гидантоина V (1,287 г, 4,36 ммоля) и Ва(ОН)2·Н2O (4,20 г, 22,2 ммоля) в воде (25 мл) в герметично закрытой толстостенной колбе выдерживали при 125° С на масляной бане в течение 4 дней. Реакционную смесь охлаждали до комнатной температуры и с использованием 4 н. серной кислоты подкисляли до рН ~3 при одновременном интенсивном перемешивании. Суспензию перемешивали на бане с кипящей водой в течение одного часа и охлаждали до комнатной температуры. Белую суспензию фильтровали и осадки промывали водой. Объединенные фильтрат и промывные жидкости концентрировали под вакуумом до ~20 мл. Нейтрализацией концентрированным раствором гидроксида аммония получали белый осадок, который отфильтровывали, промывали водой и сушили под вакуумом в течение ночи с получением рацемической 5-бром-2-аминотетралин-2-карбоновой кислоты (893 мг, 76%-ный выход). МСНР (пучок электронов): C11H12BrNO2, рассч. 269; обнаруж. 270 (М+Н), 272 (М+Н), 268 (М-Н), 270 (М-Н).
Стадия 6
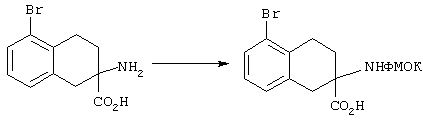
Смесь рацемической 5-бром-2-аминотетралин-2-карбоновой кислоты (882 мг, 3,27 ммоля), триэтиламина (0,60 мл, 4,30 ммоля) и 9-флуоренилметилсукцинимидилкарбоната (ФМОК-OSu, 1,32 г, 3,91 ммоля) в ацетонитриле (30 мл) и воде (30 мл) в течение ночи перемешивали при комнатной температуре. Данные ТСХ-анализа реакционной смеси на следующий день указывали на наличие исходного материала, аминокислоты. Добавляли 9-флуоренилметилсукцинимидилкарбоната (0,25 г), триэтиламина (0,6 мл) и ацетонитрила (5 мл) и при комнатной температуре смесь перемешивали в течение еще одного дня. Реакционную смесь концентрировали под вакуумом для удаления большей части ацетонитрила, 10%-ным водным раствором лимонной кислоты подкисляли до рН ~3 и белую эмульсию дважды экстрагировали метиленхлоридом. Объединенные органические слои промывали водой и рассолом и сушили над сульфатом магния. В результате фильтрования и концентрирования в виде масла получали сырой продукт, который очищали хроматографией в колонке (элюировали 2 → 5 → 10% метанола/метиленхлоридом) с получением в виде белого твердого вещества рацемической ФМОК-5-бром-2-аминотетралин-2-карбоновой кислоты (1,09 г, 68%-ный выход). МСВР (БУА): для C26H22BrNNaO4 (M+Na) рассч. 514,0630; обнаруж. 514,0643.
Пример 10
Получение ФМОК-(D,L)-5-хлор-2-аминотетралин-2-карбоновой кислоты (ФMOK-(D,L) 5-ClAtc-OH)
Стадия 1
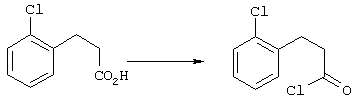
Смесь 3-(2-хлорфенил)пропионовой кислоты (5,0 г, 27,1 ммоля), тионилхлорида (10,9 мл, 149 ммолей) и толуола (75 мл) кипятили с обратным холодильником в течение двух часов. Концентрированием под вакуумом получали 3-(2-хлорфенил)пропаноилхлорид, который растворяли в метиленхлориде и без дополнительной очистки использовали на следующей стадии.
Стадия 2
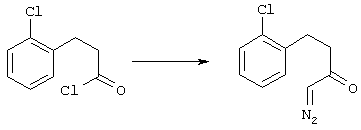
Раствор вышеупомянутого хлорангидрида кислоты (сырой, 27,1 ммоля) в метиленхлориде медленно добавляли в раствор диазометана (получен из 17,8 г 1-метил-3-нитро-1-нитрозогуанидина) в диэтиловом эфире (120 мл), охлажденный на ледяной бане. Затем смесь нагревали до комнатной температуры и перемешивали в течение ночи. Смесь концентрировали под вакуумом с получением в виде ярко-желтого масла 1-диазо-4-(2-хлорфенил)бутан-2-она (5,87 г, >100%-ный выход за 2 стадии). Это соединение без дополнительной очистки использовали на следующей стадии. 1Н-ЯМР (СDСl3, δ ): 7,05-7,32 (4Н, m, фенил), 5,13 (1Н, широкий s, диазо), 3,00 (2Н, t, бензиловый), 2,57 (2Н, m).
Стадия 3
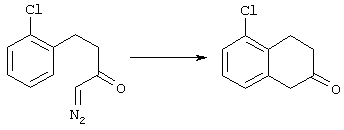
В смесь димера ацетата родия (II) (60 мг, 0,27 ммоля) в метиленхлориде (400 мл) при кипячении с обратным холодильником медленно добавляли раствор сырого 1-диазо-4-(2-бромфенил)бутан-2-она (5,87 г, 27,1 ммоля, теоретическое количество) в метиленхлориде (50 мл). После того как завершали добавление, смесь кипятили с обратным холодильником в течение больше двадцати минут. Смесь охлаждали до комнатной температуры, добавляли трифторуксусной кислоты (6,0 мл) и смесь перемешивали при комнатной температуре в течение двух часов. Реакцию гасили насыщенным раствором бикарбоната натрия. Слои разделяли и метиленхлоридный слой еще раз промывали насыщенным раствором бикарбоната натрия. Объединенные водные слои подвергали обратной экстракции метиленхлоридом. Объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали под вакуумом с получением продукта в виде коричневого масла. Очисткой хроматографией в колонке (10 → 15% этилацетата/гексаны) в виде светло-коричневого масла получали 5-хлор-β -тетралон (3,32 г, 68%-ный выход на стадиях с 1 по 3). 1Н-ЯМР (СDСl3, δ ): 7,30 (1H, m, фенил), 7,15 (1H, t, фенил), 7,05 (1H, d, фенил), 3,60 (2Н, s, бензиловый), 3,22 (2Н, t, бензиловый), 2,56 (2Н, t).
Стадия 4
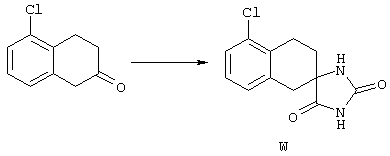
Смесь 5-хлор-β -тетралона (880 мг, 4,87 ммоля), цианида калия (500 мг, 7,67 ммоля), карбоната аммония (2,85 г, 29,7 ммоля), этанола (24 мл) и воды (6 мл) в герметично закрытой толстостенной колбе выдерживали при 80° С на масляной бане в течение 66 ч. После охлаждения до комнатной температуры суспензию выливали в смесь воды со льдом и перемешивали при комнатной температуре в течение пары часов. В результате фильтрования с последующей сушкой на воздухе в виде светло-бежевого твердого вещества получали гидантоин W (0,92 г, 75%-ный выход). 1H-ЯМР (ДМСО-d6, δ ): 10,70 (1H, широкий, NH), 8,25 (1H, широкий s, NH), 7,0-7,3 (3Н, m, фенил). МСНР (пучок электронов): C12H11ClN2O2, рассч. 250; обнаруж. 249 (М-Н), 251 (М-Н).
Стадия 5
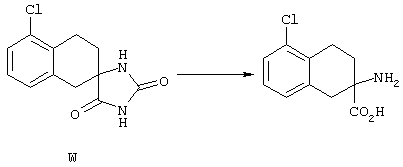
Смесь гидантоина W (880 мг, 3,51 ммоля) и Ва(ОН)2·Н2O (3,40 г, 18,0 ммоля) в воде (50 мл, избыточное разбавление) в герметично закрытой толстостенной колбе в течение 2 дней выдерживали на масляной бане при 125° С. Реакционную смесь охлаждали до комнатной температуры и при одновременном интенсивном перемешивании с использованием 4 н. серной кислоты подкисляли до рН ~3. Суспензию перемешивали на бане с кипящей водой в течение двух часов и охлаждали до комнатной температуры. Белую суспензию фильтровали и осадки промывали водой. Объединенные фильтрат и промывные жидкости концентрировали под вакуумом до ~50 мл. Нейтрализацией концентрированным раствором гидроксида аммония получали белый осадок, который отфильтровывали, промывали водой и сушили под вакуумом в течение ночи с получением рацемической 5-хлор-2-аминотетралин-2-карбоновой кислоты (788 мг, 99%-ный выход). МСНР (пучок электронов): C11H12ClNO2, рассч. 225; обнаруж. 226 (М+Н), 228 (М+Н), 224 (М-Н), 226 (М-Н).
Стадия 6
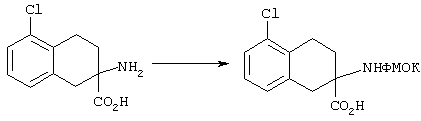 Смесь рацемической 5-хлор-2-аминотетралин-2-карбоновой кислоты (402 мг, 1,78 ммоля), триэтиламина (0,38 мл, 2,73 ммоля) и 9-флуоренилметилсукцинимидилкарбоната (ФМОК-OSu, 904 мг, 2,68 ммоля) в ацетонитриле (20 мл) и воде (20 мл) при комнатной температуре перемешивали в течение двух дней. Данные ТСХ-анализа реакционной смеси после двух дней указывали на наличие исходного материала, аминокислоты. Добавляли 9-флуоренилметилсукцинимидилкарбоната (0,12 г) и триэтиламина (0,1 мл) и при комнатной температуре смесь перемешивали в течение еще одного дня. Реакционную смесь концентрировали под вакуумом для удаления большей части ацетонитрила, 10%-ным водным раствором лимонной кислоты подкисляли до рН ~3 и белую эмульсию три раза экстрагировали этилацетатом. Объединенные органические слои промывали водой, рассолом и сушили над сульфатом магния. В результате фильтрования и концентрирования в виде масла получали сырой продукт, который очищали хроматографией в колонке (элюировали 3 → 6 → 8% метанола/метиленхлоридом) с получением в виде белого твердого вещества рацемической ФМОК-5-хлор-2-аминотетралин-2-карбоновой кислоты (540 мг, 68%-ный выход). МСВР (EI): С26Н22СlNO4 (М), рассч. 447,1237; обнаруж. 447,1234.
Смесь рацемической 5-хлор-2-аминотетралин-2-карбоновой кислоты (402 мг, 1,78 ммоля), триэтиламина (0,38 мл, 2,73 ммоля) и 9-флуоренилметилсукцинимидилкарбоната (ФМОК-OSu, 904 мг, 2,68 ммоля) в ацетонитриле (20 мл) и воде (20 мл) при комнатной температуре перемешивали в течение двух дней. Данные ТСХ-анализа реакционной смеси после двух дней указывали на наличие исходного материала, аминокислоты. Добавляли 9-флуоренилметилсукцинимидилкарбоната (0,12 г) и триэтиламина (0,1 мл) и при комнатной температуре смесь перемешивали в течение еще одного дня. Реакционную смесь концентрировали под вакуумом для удаления большей части ацетонитрила, 10%-ным водным раствором лимонной кислоты подкисляли до рН ~3 и белую эмульсию три раза экстрагировали этилацетатом. Объединенные органические слои промывали водой, рассолом и сушили над сульфатом магния. В результате фильтрования и концентрирования в виде масла получали сырой продукт, который очищали хроматографией в колонке (элюировали 3 → 6 → 8% метанола/метиленхлоридом) с получением в виде белого твердого вещества рацемической ФМОК-5-хлор-2-аминотетралин-2-карбоновой кислоты (540 мг, 68%-ный выход). МСВР (EI): С26Н22СlNO4 (М), рассч. 447,1237; обнаруж. 447,1234.
Пример 11
Получение ФМОК-(D,L)-5-метокси-2-аминотетралин-2-карбоновой кислоты (ФMOK-(D,L) 5-MeOAtc-OH)
Стадия 1
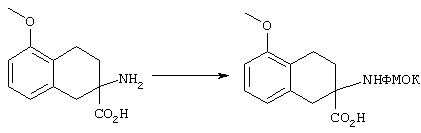
Смесь рацемической 5-метокси-2-аминотетралин-2-карбоновой кислоты (полученной в соответствии с работой Obrecht D. и др., Helv.Chim.Acta., 1992, 75, 1666) (802 мг, 3,62 ммоля), триэтиламина (0,62 мл, 4,45 ммоля) и 9-флуоренилметилсукцинимидилкарбоната (ФМОК-OSu, 1,47 г, 4,36 ммоля) в ацетонитриле (25 мл) и воде (25 мл) при комнатной температуре перемешивали в течение 30 ч. Данные ТСХ-анализа реакционной смеси указывали на наличие исходного материала, аминокислоты. Добавляли 9-флуоренилметилсукцинимидилкарбоната (370 мг) и триэтиламина (0,6 мл) и при комнатной температуре смесь перемешивали в течение дополнительных 24 ч. Реакционную смесь концентрировали под вакуумом для удаления большей части ацетонитрила, 10%-ным водным раствором лимонной кислоты подкисляли до рН ~3 и белую эмульсию три раза экстрагировали этилацетатом. Объединенные органические слои промывали водой, рассолом и сушили над сульфатом магния. В результате фильтрования и концентрирования в виде масла получали сырой продукт, который очищали хроматографией в колонке (элюировали 1 → 3 → 5 → 10% метанола/метиленхлоридом) с получением в виде не совсем белого твердого вещества рацемической ФМОК-5-метокси-2-аминотетралин-2-карбоновой кислоты (1,14 г, 71%-ный выход). МСВР (БУА): C27H26NO5 (M+H), рассч. 444,1812; обнаруж. 444,1814.
Пример 12
Получение ФМОК-(D,L)-5-этокси-2-аминотетралин-2-карбоновой кислоты (ФMOK-(D,L) 5-EtOAtc-OH)
Стадия 1
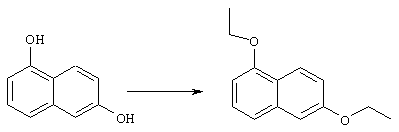
Смесь 1,6-дигидроксинафталина (5,02 г, 31,3 ммоля), безводного карбоната калия (52,0 г, 376 ммолей), N,N-диметилформамида (50 мл) и этаниодида (15 мл, 188 ммолей) перемешивали на масляной бане при 35° С в течение 24 ч.
Реакционную смесь фильтровали и твердый остаток тщательно промывали диэтиловым эфиром. Фильтрат и промывные жидкости объединяли и концентрировали под вакуумом для удаления большинства растворителей. Коричневый остаток делили между водой и диэтиловым эфиром и слои разделяли. Эфирный слой промывали водой. Объединенные водные слои подвергали обратной экстракции диэтиловым эфиром. Эфирные экстракты объединяли, промывали рассолом и сушили над сульфатом магния. В результате фильтрования и концентрирования получали сырой коричневый твердый продукт (6,74 г, 99%-ный выход). Перекристаллизацией этого сырого продукта из горячего метанола в виде светло-коричневого твердого вещества получали 1,6-диэтоксинафталин (4,36 г, 64%-ный выход, первая порция продукта). 1Н-ЯМР (СDСl3, δ ): 8,20 (1Н, d, фенил), 7,06-7,36 (4Н, m, фенил), 6,66 (1Н, dd, фенил), 4,10-4,23 (4Н, 2 совокупности q, 2 СН2), 1,45-1,56 (6Н, 2 совокупности t, 2 СН3).
Стадия 2
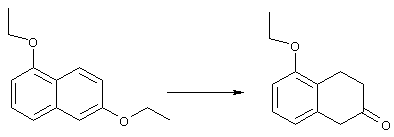
В кипящий раствор 1,6-диэтоксинафталина (4,15 г, 19,2 ммоля) в абсолютном этаноле (100 мл) осторожно, в течение 60 мин добавляли небольшие кусочки металлического натрия (6,8 г, 296 ммолей). Смесь кипятили с обратным холодильником в течение еще 90 мин. Данные ТСХ указывали на наличие непрореагировавшего исходного материала. Добавляли дополнительное количество металлического натрия (1,0 г, 43,5 ммоля) и реакционную смесь кипятили с обратным холодильником в течение дополнительных 60 мин. Реакционную смесь охлаждали до комнатной температуры, реакцию гасили водой и смесь подкисляли концентрированной соляной кислотой. Смесь концентрировали под вакуумом для удаления большей части этанола. Водную смесь три раза экстрагировали диэтиловым эфиром. Объединенные органические слои промывали водой и сушили над сульфатом натрия. В результате фильтрования и концентрирования получали коричневый твердый продукт, который растворяли в этаноле/воде в соотношении 1:1 (200 мл), затем добавляли п-толуолсульфоновой кислоты (400 мг). Смесь кипятили с обратным холодильником в течение 210 мин. Добавляли дополнительное количество п-толуолсульфоновой кислоты (100 мг) и эту смесь кипятили с обратным холодильником в течение еще 60 мин. После охлаждения до комнатной температуры под пониженным давлением удаляли большую часть этанола. Водную смесь три раза экстрагировали диэтиловым эфиром и объединенные органические слои промывали водой, насыщенным раствором хлорида натрия и сушили над сульфатом натрия. В результате фильтрования и концентрирования в виде коричневого масла получали продукт, который очищали хроматографией в колонке (7% этилацетата/гексаны) с получением в виде светло-желтого масла 5-этокси-β -тетралона (2,43 г, 67%-ный выход). 1Н-ЯМР (СDСl3, δ ): 7,15 (1Н, t, фенил), 6,76 (1Н, d, фенил), 6,72 (1Н, d, фенил), 4,05 (2Н, q, CH2), 3,56 (2Н, s, бензиловый), 3,10 (2Н, t, бензиловый), 2,53 (2Н, t), 1,44 (3Н, t, СН3).
Стадия 3
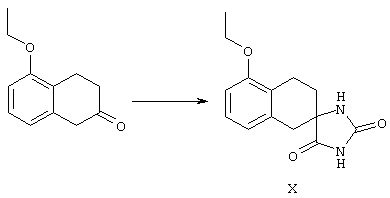
Смесь 5-этокси-β -тетралона (2,23 г, 11,7 ммоля), цианида калия (1,20 г, 18,4 ммоля), карбоната аммония (6,75 г, 70,2 ммоля), этанола (80 мл) и воды (20 мл) в герметично закрытой толстостенной колбе в течение 3 дней выдерживали на масляной бане при 80° С. После охлаждения до комнатной температуры суспензию выливали в смесь воды со льдом и в течение пары часов перемешивали при комнатной температуре. В результате фильтрования с последующей сушкой на воздухе в виде бежевого твердого вещества получали гидантоин Х (2,69 г, 88%-ный выход). 1H-ЯМР (ДМСО-d6, δ ): 10,65 (1Н, широкий s, NH), 8,22 (1Н, широкий s, NH), 7,06 (1Н, t, фенил), 6,75 (1Н, d, фенил), 6,65 (1Н, d, фенил), 3,98 (2Н, q, СН2), 1,32 (3Н, t, СН3). МСНР (пучок электронов): C14H16N2O3, рассч. 259; обнаруж. 258 (М-Н).
Стадия 4
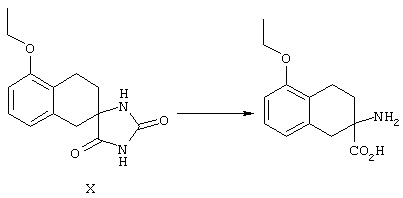
Смесь гидантоина Х (2,57 г, 9,87 ммоля) и Ва(ОН)2·Н2O (9,40 г, 49,6 ммоля) в воде (200 мл, избыточное разбавление) в герметично закрытой толстостенной колбе выдерживали на масляной бане при 105° С в течение 39 ч. Добавляли дополнительное количество Ва(ОН)2·Н2O (9,40 г, 49,6 ммоля) и смесь выдерживали на масляной бане при 125° С в течение еще 21 ч. Реакционную смесь охлаждали до комнатной температуры и с использованием 4 н. серной кислоты при одновременном интенсивном перемешивании подкисляли до рН ~3. Суспензию перемешивали на бане с кипящей водой в течение одного часа и охлаждали до комнатной температуры. Белую суспензию фильтровали и осадки промывали водой. Объединенные фильтрат и промывные жидкости концентрировали под вакуумом до ~75 мл. Нейтрализацией концентрированным раствором гидроксида аммония получали белый осадок, который отфильтровывали, промывали водой и сушили на воздухе с получением в виде светло-бежевого твердого вещества рацемической 5-этокси-2-аминотетралин-2-карбоновой кислоты (2,34 г, количественный выход). МСНР (пучок электронов): C13H17NO3, рассч. 235; обнаруж. 236 (М+Н), 234 (М-Н).
Стадия 5
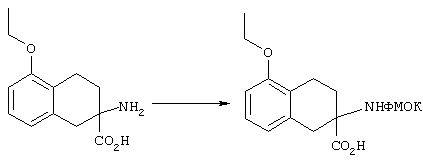
Смесь рацемической 5-этокси-2-аминотетралин-2-карбоновой кислоты (2,22 г, 9,44 ммоля), триэтиламина (2,00 мл, 14,3 ммоля) и 9-флуоренилметилсукцинимидилкарбоната (ФМОК-OSu, 4,81 г, 14,3 ммоля) в ацетонитриле (75 мл) и воде (75 мл) при комнатной температуре перемешивали в течение двух дней. Данные ТСХ-анализа реакционной смеси указывали на наличие исходного материала, аминокислоты. Добавляли 9-флуоренилметилсукцинимидилкарбоната (645 мг) и триэтиламина (1,0 мл) и смесь перемешивали при комнатной температуре в течение еще одного дня. Реакционную смесь концентрировали под вакуумом для удаления большей части ацетонитрила, 10%-ным водным раствором лимонной кислоты подкисляли до рН ~3 и белую эмульсию три раза экстрагировали этилацетатом. Объединенные органические слои промывали водой, рассолом и сушили над сульфатом магния. В результате фильтрования и концентрирования в виде масла получали сырой продукт, который очищали хроматографией в колонке (элюировали 3 → 5 → 10% метанола/метиленхлоридом) с получением в виде белого твердого вещества рацемической ФМОК-5-этокси-2-аминотетралин-2-карбоновой кислоты (4,66 г, больше чем количественный выход). МСВР (БУА): C28H28NO5 (M+H), рассч. 458,1967; обнаруж. 458,1985.
Пример 13
Получение ФМОК-(D,L)-5-изопропокси-2-аминотетралин-2-карбоновой кислоты (ФMOK-(D,L) 5-изо-PrOAtc-OH)
Стадия 1
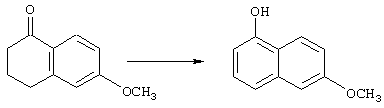
Смесь 6-метокси-1-тетралона (5,07 г, 28,8 ммоля) и 10% Pd/C (3,53 г, 3,32 ммоля) в сухом п-цимене (250 мл) кипятили в аргоновой атмосфере с обратным холодильником в течение 38 ч. Реакционную смесь охлаждали до комнатной температуры, фильтровали через броунмиллерит и остаток тщательно промывали п-цименом. Фильтрат и промывные жидкости объединяли и дважды экстрагировали 1 н. раствором гидроксида натрия (2 порции по 70 мл). Объединенные водные экстракты 6 н. соляной кислотой подкисляли до рН ~3 и три раза экстрагировали диэтиловым эфиром. Объединенные органические слои промывали водой и сушили над безводным сульфатом натрия. В результате фильтрования и концентрирования в виде светло-коричневого твердого вещества получали сырой 5-гидрокси-6-метоксинафталин (2,31 г, 46%-ный выход), который без дополнительной очистки использовали на следующей стадии. МСНР (пучок электронов): С11H10О2, рассч. 174; обнаруж. 173 (М-Н).
Стадия 2
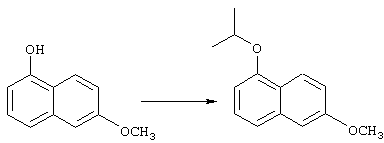
Смесь 5-гидрокси-6-метоксинафталина (2,10 г, 12,1 ммоля), карбоната цезия (19,7 г, 60,5 ммоля), N,N-диметилформамида (12 мл) и 2-бромпропана (3,50 мл, 36,9 ммоля) в течение ночи перемешивали на масляной бане при 40° С. Реакционную смесь фильтровали и твердый остаток тщательно промывали диэтиловым эфиром. Фильтрат и промывные жидкости объединяли и концентрировали под вакуумом для удаления большинства растворителей. Коричневый остаток делили между водой и диэтиловым эфиром и слои разделяли. Эфирный слой промывали водой. Объединенные водные слои подвергали обратной экстракции диэтиловым эфиром. Эфирные экстракты объединяли, промывали рассолом и сушили над сульфатом натрия. В результате фильтрования и концентрирования получали сырой продукт, который очищали хроматографией в колонке (2,5 → 5% этилацетата/гексаны) с выделением в виде светло-коричневого масла 1-изопропокси-6-метоксинафталина (2,23 г, 86%-ный выход). 1Н-ЯМР (СDСl3, δ ): 8,17 (1Н, d, фенил), 7,05-7,38 (4Н, m, фенил), 6,72 (1H, dd, фенил), 4,73 (1Н, m, СН изо-Рr), 3,92 (3Н, s, ОСН3), 1,42 (6Н, d, 2 СН3 изо-Рr).
Стадия 3
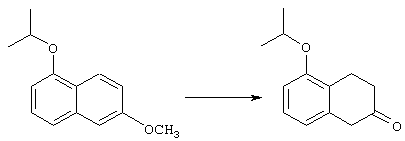
В кипящий раствор 1-изопропокси-6-метоксинафталина (2,23 г, 10,3 ммоля) в абсолютном этаноле (50 мл) осторожно, в течение 45 мин добавляли небольшие кусочки металлического натрия (3,6 г, 157 ммолей). Смесь кипятили с обратным холодильником в течение дополнительных 120 мин. Реакционную смесь охлаждали до комнатной температуры, реакцию гасили водой и подкисляли концентрированной соляной кислотой. Смесь концентрировали под вакуумом для удаления большей части этанола. Водную смесь три раза экстрагировали диэтиловым эфиром. Объединенные органические слои промывали водой и сушили над сульфатом натрия. Фильтрованием и концентрированием в виде красноватого масла получали продукт, который растворяли в этаноле/воде в соотношении 1:1 (90 мл), а затем добавляли п-толуолсульфоновой кислоты (200 мг). Смесь кипятили с обратным холодильником в течение 60 мин. После охлаждения до комнатной температуры под пониженным давлением удаляли большую часть этанола. Водную смесь дважды экстрагировали диэтиловым эфиром и объединенные органические слои промывали водой, насыщенным раствором хлорида натрия и сушили над сульфатом натрия. В результате фильтрования и концентрирования в виде красноватого масла получали продукт, который очищали хроматографией в колонке (8 → 15% этилацетата/гексаны) с выделением в виде бесцветного масла 5-изопропокси-β -тетралона (1,37 г, 65%-ный выход). 1Н-ЯМР (СDСl3, δ ): 7,16 (1Н, t, фенил), 6,78 (1Н, d, фенил), 6,71 (1Н, d, фенил), 4,53 (1Н, m, CH изо-Pr), 3,56 (2Н, s, бензиловый), 3,08 (2Н, t, бензиловый), 2,50 (2Н, t), 1,37 (6Н, d, 2 СН3 изо-Pr).
Стадия 4
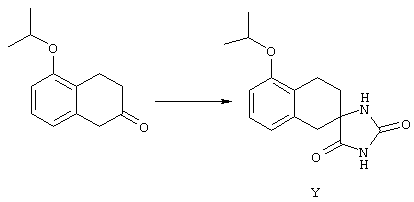
Смесь 5-изопропокси-β -тетралона (1,37 г, 6,71 ммоля), цианида калия (660 мг, 10,1 ммоля), карбоната аммония (3,87 г, 40,3 ммоля), этанола (44 мл) и воды (9 мл) в герметично закрытой толстостенной колбе на масляной бане выдерживали при 80° С в течение 42 ч. После охлаждения до комнатной температуры суспензию выливали в смесь воды со льдом и перемешивали при комнатной температуре в течение пары часов. В результате фильтрования с последующей сушкой на воздухе получали гидантоин Y (1,64 г, 89%-ный выход).
Стадия 5
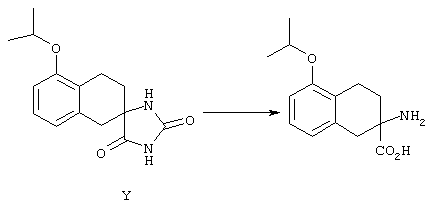
Смесь гидантоина Y (1,64 г, 5,98 ммоля) и Ва(ОН)2·Н2O (5,66 г, 29,9 ммоля) в воде (25 мл) в герметично закрытой толстостенной колбе выдерживали на масляной бане при 100° С в течение 70 ч. Реакционную смесь охлаждали до комнатной температуры и с использованием 4 н. серной кислоты при одновременном интенсивном перемешивании нейтрализовали до рН ~7. Суспензию в течение одного часа перемешивали на бане с кипящей водой, охлаждали до комнатной температуры и подщелачивали 1 н. раствором гидроксида натрия, белую суспензию фильтровали и осадки промывали водой. Объединенные фильтрат и промывные жидкости концентрировали под вакуумом до ~75 мл. Нейтрализацией концентрированным раствором соляной кислоты получали белый осадок, который отфильтровывали, промывали водой и сушили на воздухе с получением рацемической 5-изопропокси-2-аминотетралин-2-карбоновой кислоты (3,48 г, влажная и содержавшая неорганическую соль, больше чем количественный выход). МСНР (пучок электронов): C14H19NO3, рассч. 249; обнаруж. 248 (М-Н).
Стадия 6
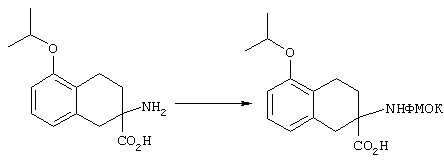
Смесь рацемической 5-изопропокси-2-аминотетралин-2-карбоновой кислоты (3,48 г, 5,98 ммоля, теоретическое количество), триэтиламина (1,10 мл, 7,89 ммоля) и 9-флуоренилметилсукцинимидилкарбоната (ФМОК-OSu, 2,62 г, 7,77 ммоля) в ацетонитриле (30 мл) и воде (30 мл) при комнатной температуре перемешивали в течение одного дня. Данные ТСХ анализа реакционной смеси указывали на наличие исходного материала, аминокислоты. Добавляли 9-флуоренилметилсукцинимидилкарбоната (500 мг) и при комнатной температуре смесь перемешивали в течение еще одного дня. Реакционную смесь концентрировали под вакуумом для удаления большей части ацетонитрила, 10%-ным водным раствором лимонной кислоты подкисляли до рН ~3, белую эмульсию три раза экстрагировали метиленхлоридом. Объединенные органические слои промывали водой и рассолом и сушили над сульфатом магния. В результате фильтрования и концентрирования в виде масла получали сырой продукт, который очищали хроматографией в колонке (элюировали 1 → 2 → 5 → 8% метанола/метиленхлоридом) с получением в виде белого твердого вещества рацемической ФМОК-5-изопропокси-2-аминотетралин-2-карбоновой кислоты (0,50 г, 18%-ный выход за 2 стадии). МСВР (БУА): C29H30NO5 (M+H), рассч. 472,2124; обнаруж. 472,2117.
Пример 14
Получение ФМОК-(D,L)-5-метил-2-аминотетралин-2-карбоновой кислоты (ФMOK-(D,L) 5-MeAtc-OH)
Стадия 1
Смесь 2-метилгидрокоричной кислоты (3,0 г, 18,3 ммоля), оксалилхлорида (3,19 мл, 36,6 ммоля) и метиленхлорида (30 мл) охлаждали на ледяной бане и по каплям добавляли N,N-диметилформамид (0,14 мл, 1,81 ммоля). При комнатной температуре смесь перемешивали в течение ночи. Концентрированием под вакуумом получали 3-(2-метилфенил)пропаноилхлорид, который растворяли в метиленхлориде и в неочищенном виде использовали на следующей стадии.
Стадия 2
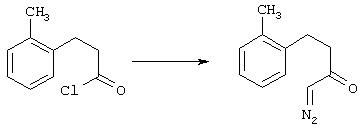
Раствор вышеупомянутого хлорангидрида кислоты (сырой, 18,3 ммоля) в метиленхлориде медленно добавляли в охлажденный на ледяной бане раствор диазометана (получен из 11,9 г 1-метил-3-нитро-1-нитрозогуанидина) в диэтиловом эфире (80 мл). Затем смесь нагревали до комнатной температуры и перемешивали в течение ночи. Смесь концентрировали под вакуумом и очищали хроматографией в колонке (10 → 20% этилацетата/гексаны) с получением в виде ярко-желтого масла 1-диазо-4-(2-метилфенил)бутан-2-она (2,08 г, 60%-ный выход за 2 стадии).
Стадия 3
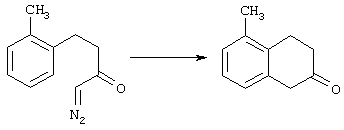
В смесь димера ацетата родия (II) (24 мг, 0,109 ммоля) в метиленхлориде (200 мл) при кипячении с обратным холодильником медленно, в течение 180 мин добавляли раствор 1-диазо-4-(2-метилфенил)бутан-2-она (2,08 г, 11,1 ммоля) в метиленхлориде (50 мл). После того как завершали добавление, смесь кипятили с обратным холодильником в течение больше двадцати минут. Смесь охлаждали до комнатной температуры, добавляли трифторуксусной кислоты (2,40 мл) и затем перемешивали при комнатной температуре в течение одного часа. Реакцию гасили насыщенным раствором бикарбоната натрия. Слои разделяли и метиленхлоридный слой еще раз промывали насыщенным раствором бикарбоната натрия. Объединенные водные слои подвергали обратной экстракции метиленхлоридом. Объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали под вакуумом с получением сырого продукта в виде коричневого масла. Очисткой хроматографией в колонке (15% этилацетат/гексаны) в виде светло-коричневого масла выделяли 5-метил-β -тетралон (1,48 г, 84%-ный выход). 1Н-ЯМР (СDСl3, δ ): 6,90-7,20 (3Н, m, фенил), 3,58 (2Н, s, бензиловый), 3,03 (2Н, t, бензиловый), 2,55 (2Н, t), 2,34 (3Н, s, СН3).
Стадия 4
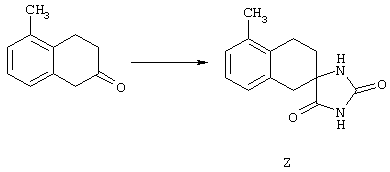
Смесь 5-метил-β -тетралона (1,48 г, 9,24 ммоля), цианида калия (902 мг, 13,9 ммоля), карбоната аммония (5,33 г, 55,5 ммоля), этанола (45 мл) и воды (9 мл) в герметично закрытой толстостенной колбе в течение 3 дней выдерживали на масляной бане при 80° С. После охлаждения до комнатной температуры суспензию выливали в смесь воды со льдом и перемешивали при комнатной температуре в течение пары часов. В результате фильтрования с последующей сушкой на воздухе в виде бежевого твердого вещества получали сырой гидантоин Z (1,81 г, 85%-ный выход). 1H-ЯМР (ДМСО-d6, δ ): 10,66 (1Н, широкий s, NH), 8,22 (1H, широкий s, NH), 6,85-7,05 (3Н, m, фенил), 2,17 (3Н, s, СН3).
Стадия 5
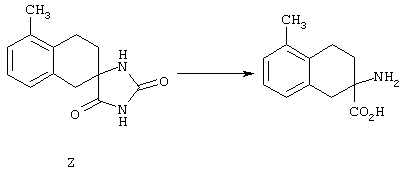
Смесь гидантоина Z (1,80 г, 7,82 ммоля) и Ва(ОН)2·Н2O (7,40 г, 39,1 ммоля) в воде (28 мл) в герметично закрытой толстостенной колбе на масляной бане выдерживали при 125° С в течение 88 ч. Реакционную смесь охлаждали до комнатной температуры и с использованием 4 н. серной кислоты при одновременном интенсивном перемешивании подкисляли до рН ~3. Суспензию в течение одного часа перемешивали на бане с кипящей водой и охлаждали до комнатной температуры. Белую суспензию фильтровали и осадки промывали водой. Объединенные фильтрат и промывные жидкости концентрировали под вакуумом до ~50 мл. Нейтрализацией концентрированным раствором гидроксида аммония получали белый осадок, который отфильтровывали, промывали водой и сушили на воздухе с получением в виде бежевого твердого вещества рацемической 5-метил-2-аминотетралин-2-карбоновой кислоты (1,05 г, 65%-ный выход). МСНР (пучок электронов): C12H15NO2, рассч. 205; обнаруж. 206 (М+Н).
Стадия 6
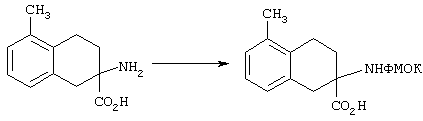
Смесь рацемической 5-метил-2-аминотетралин-2-карбоновой кислоты (1,05 г, 5,12 ммоля), триэтиламина (0,93 мл, 6,67 ммоля) и 9-флуоренилметилсукцинимидилкарбоната (ФМОК-OSu, 2,24 г, 6,64 ммоля) в ацетонитриле (30 мл) и воде (30 мл) при комнатной температуре перемешивали в течение 2 дней. Данные ТСХ-анализа реакционной смеси указывали на наличие исходного материала, аминокислоты. Добавляли 9-флуоренилметилсукцинимидилкарбоната (520 мг) и при комнатной температуре смесь перемешивали в течение дополнительных 24 ч. Реакционную смесь концентрировали под вакуумом для удаления большей части ацетонитрила, 10%-ным водным раствором лимонной кислоты подкисляли до рН ~3 и белую эмульсию дважды экстрагировали метиленхлоридом. Объединенные органические слои промывали водой, рассолом и сушили над сульфатом магния. В результате фильтрования и концентрирования в виде масла получали сырой продукт, который очищали хроматографией в колонке (элюировали 2 → 5 → 8% метанола/метиленхлоридом) с получением в виде светло-коричневого твердого вещества рацемической ФМОК-5-метил-2-аминотетралин-2-карбоновой кислоты (1,62 г, 74% выход). МСВР (БУА): C27H26NO4 (М+Н), рассч. 428,1862; обнаруж. 428,1844.
Пример 15
Получение ФМОК-(D,L)-5-этил-2-аминотетралин-2-карбоновой кислоты (ФMOK-(D,L) 5-EtAtc-OH)
Стадия 1
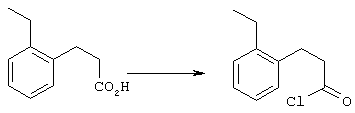
Смесь 3-(2-этилфенил)пропионовой кислоты (полученной в 3 стадии из 1-этил-2-иодбензола, 4,24 г, 23,8 ммоля), тионилхлорида (9,50 мл, 130 ммолей) и толуола (100 мл) кипятили с обратным холодильником в течение 2 ч. Концентрированием под вакуумом получали 3-(2-этилфенил)пропаноилхлорид, который растворяли в метиленхлориде и в неочищенном виде использовали на следующей стадии.
Стадия 2

Раствор 3-(2-этилфенил)пропаноилхлорида (сырой, 23,8 ммоля) в метиленхлориде медленно добавляли в охлажденный на ледяной бане раствор диазометана (получен из 15,6 г 1-метил-3-нитро-1-нитрозогуанидина) в диэтиловом эфире (100 мл). Затем смесь нагревали до комнатной температуры и перемешивали в течение ночи. Смесь концентрировали под вакуумом и очищали хроматографией в колонке (10 → 20% этилацетата/гексаны) с получением 1-диазо-4-(2-этилфенил)бутан-2-она (3,47 г, 72%-ный выход за 2 стадии). 1H-ЯМР (CDCl3, δ ): 7,1-7,25 (4Н, m, фенил), 5,21 (1Н, широкий s, диазо), 2,97 (2Н, m, СН2 этила), 1,20 (3H, t, СН3).
Стадия 3
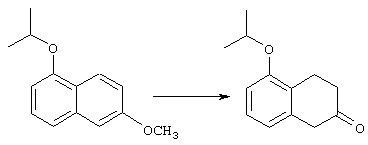
В смесь димера ацетата родия (II) (38 мг, 0,172 ммоля) в метиленхлориде (300 мл) при кипячении с обратным холодильником медленно, в течение 90 мин вводили раствор 1-диазо-4-(2-этилфенил)бутан-2-она (3,47 г, 17,2 ммоля) в метиленхлориде (50 мл). После того как. добавление завершали, смесь кипятили с обратным холодильником в течение больше двадцати минут. Смесь охлаждали до комнатной температуры, добавляли трифторуксусной кислоты (3,75 мл) и при комнатной температуре смесь перемешивали в течение одного часа. Реакцию гасили насыщенным раствором бикарбоната натрия. Слои разделяли и метиленхлоридный слой еще раз промывали насыщенным раствором бикарбоната натрия. Объединенные водные слои подвергали обратной экстракции метиленхлоридом. Объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали под вакуумом с получением в виде красновато-коричневого масла сырого 5-этил-β -тетралона (3,09 г, больше чем количественный выход). Это соединение без дополнительной очистки использовали на следующей стадии. 1H-ЯМР (CDCl3, δ ): 6,9-7,2 (3Н, m, фенил), 3,58 (2Н, s, бензиловый), 3,08 (2Н, s, бензиловый), 2,70 (2Н, q, CH2 этила), 2,52 (2Н, t, бензиловый), 1,20 (3Н, t, СН3 этила).
Стадия 4
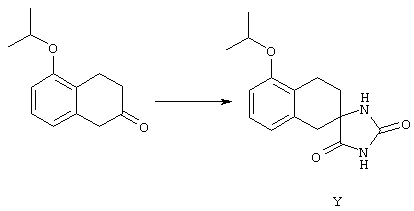
Смесь 5-этил-β -тетралона (3,09 г, 17,7 ммоля), цианида калия (1,73 г, 26,6 ммоля), карбоната аммония (10,2 г, 106 ммолей), этанола (80 мл) и воды (16 мл) в герметично закрытой толстостенной колбе выдерживали на масляной бане при 80° С в течение 48 ч. После охлаждения до комнатной температуры белую суспензию выливали в смесь воды со льдом и в течение пары часов перемешивали при комнатной температуре. Фильтрованием с последующей сушкой на воздухе в виде светло-бежевого твердого вещества получали гидантоин АА (3,85 г, 92%-ный выход за 2 стадии). 1H-ЯМР (ДМСО-d6, δ ): 10,67 (1Н, широкий s, NH), 8,26 (1Н, широкий s, NH), 6,8-7,1 (3Н, m, фенил), 1,13 (3Н, t, СН3). МСНР (пучок электронов): С14Н16N2О2, рассч. 244; обнаруж. 243 (М-Н).
Стадия 5

Смесь гидантоина АА (1,00 г, 4,09 ммоля) и Ва(ОН)2·Н2О (4,00 г, 21,1 ммоля) в воде (20 мл) в герметично закрытой толстостенной колбе выдерживали при 125° С на масляной бане в течение 48 ч. Реакционную смесь охлаждали до комнатной температуры и с использованием 4 н. серной кислоты при одновременном интенсивном перемешивании подкисляли до рН ~3. Суспензию перемешивали на бане с кипящей водой в течение двух часов и охлаждали до комнатной температуры. Белую суспензию фильтровали и осадки промывали водой. Объединенные фильтрат и промывные жидкости концентрировали под вакуумом до ~50 мл. Нейтрализацией концентрированным раствором гидроксида аммония получали белый осадок, который отфильтровывали, промывали водой и сушили под вакуумом в течение ночи с получением рацемической 5-этил-2-аминотетралин-2-карбоновой кислоты (796 мг, 89%-ный выход). МСНР (пучок электронов): C13H17NO2, рассч. 219; обнаруж. 220 (М+Н).
Стадия 6
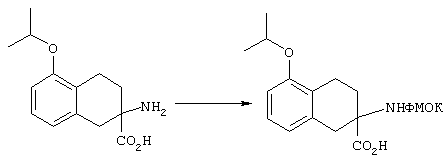
Смесь рацемической 5-этил-2-аминотетралин-2-карбоновой кислоты (765 мг, 3,49 ммоля), триэтиламина (1,0 мл, 7,17 ммоля) и 9-флуоренилметилсукцинимидилкарбоната (ФМОК-OSu, 1,79 г, 5,31 ммоля) в ацетонитриле (40 мл) и воде (40 мл) перемешивали при комнатной температуре в течение 2 дней. Реакционную смесь концентрировали под вакуумом для удаления большей части ацетонитрила и 10%-ным водным раствором лимонной кислоты подкисляли до рН ~3, белую эмульсию дважды экстрагировали метиленхлоридом и дважды этилацетатом. Метиленхлоридные экстракты промывали водой, рассолом и сушили над сульфатом магния. Этилацетатные экстракты промывали водой, рассолом и сушили над сульфатом магния. В результате фильтрования и концентрирования в виде масла получали сырой продукт, который очищали хроматографией в колонке (элюировали 2 → 5 → 8% метанола/метиленхлоридом) с получением в виде белого твердого вещества рацемической ФМОК-5-этил-2-аминотетралин-2-карбоновой кислоты (330 мг, 21%-ный выход). МСВР (БУА): C28H28NO4 (М+Н), рассч. 442,2018; обнаруж. 442,2010.
Пример 16
Получение ФМОК-(D,L)-5-изопропил-2-аминотетралин-2-карбоновой кислоты (ФMOK-(D,L) 5-изо-РrАtс-ОН)
Стадия 1
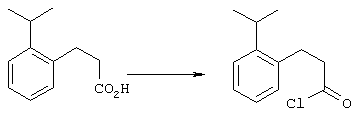
Смесь 3-(2-изопропилфенил)пропионовой кислоты (полученной на 3-й стадии из 1-изопропил-2-иодбензола, 2,01 г, 10,5 ммоля), тионилхлорида (4,30 мл, 59,0 ммоля) и толуола (40 мл) кипятили с обратным холодильником в течение 2 ч. Концентрированием под вакуумом получали 3-(2-изопропилфенил)пропаноилхлорид, который растворяли в метиленхлориде и в неочищенном виде использовали на следующей стадии.
Стадия 2
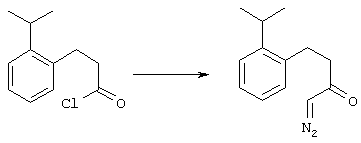
Раствор 3-(2-изопропилфенил)пропаноилхлорида (сырой, 10,5 ммоля) в метиленхлориде медленно добавляли в охлажденный на ледяной бане раствор диазометана (получен из 6,95 г 1-метил-3-нитро-1-нитрозогуанидина) в диэтиловом эфире (50 мл). Затем смесь нагревали до комнатной температуры и перемешивали в течение ночи. Смесь концентрировали под вакуумом и очищали хроматографией в колонке (20% этилацетата/гексаны) с получением в виде ярко-желтого масла 1-диазо-4-(2-изопропилфенил)бутан-2-она (1,87 г, 82%-ный выход за 2 стадии). 1Н-ЯМР (СDСl3, δ ): 7,10-7,30 (4Н, m, фенил), 5,21 (1Н, широкий s, диазо), 3,15 (1H, m, CH изо-Pr), 3,00 (2Н, t, бензиловый), 2,57 (2Н, m), 1,24 (6Н, d, 2 СН3 изо-Pr).
Стадия 3
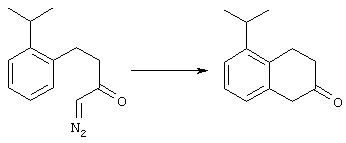
В смесь димера ацетата родия(II) (20 мг, 0,091 ммоля) в метиленхлориде (160 мл) при кипячении с обратным холодильником медленно, в течение 60 мин добавляли раствор 1-диазо-4-(2-бромфенил)бутан-2-она (1,87 г, 8,65 ммоля) в метиленхлориде (25 мл). После того как добавление завершали, смесь кипятили с обратным холодильником в течением дополнительных пятнадцати минут. Смесь охлаждали до комнатной температуры, добавляли трифторуксусной кислоты (1,90 мл) и при комнатной температуре смесь перемешивали в течение 45 мин. Реакцию гасили насыщенным раствором бикарбоната натрия. Слои разделяли и метиленхлоридный слой еще раз промывали насыщенным раствором бикарбоната натрия. Объединенные водные слои подвергали обратной экстракции метиленхлоридом. Объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали под вакуумом с получением сырого продукта в виде коричневого масла. Очисткой хроматографией в колонке (5% этилацетата/гексаны) в виде светло-желтого масла выделяли 5-изопропил-β -тетралон (1,57 г, 96%-ный выход). 1Н-ЯМР (СDСl3, δ ): 6,93-7,22 (3Н, m, фенил), 3,59 (2Н, s, бензиловый), 3,24 (1Н, m, СН изо-Рr), 3,12 (2Н, t, бензиловый), 2,52 (2Н, t), 1,27 (6Н, d, 2 СН3 изо-Рr).
Стадия 4
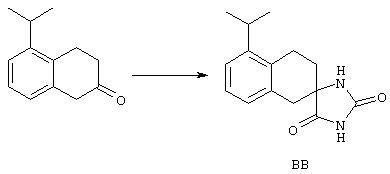
Смесь 5-изопропил-β -тетралона (1,57 г, 8,34 ммоля), цианида калия (0,82 г, 12,6 ммоля), карбоната аммония (4,81 г, 50,1 ммоля), этанола (40 мл) и воды (10 мл) в герметично закрытой толстостенной колбе выдерживали на масляной бане при 80° С в течение 48 ч. После охлаждения до комнатной температуры коричневую суспензию выливали в смесь воды со льдом и в течение пары часов перемешивали при комнатной температуре. В результате фильтрования с последующей сушкой на воздухе в виде бежевого твердого вещества получали сырой гидантоин ВВ, который без дополнительной очистки использовали на следующей стадии. 1H-ЯМР (ДМСО-d6, δ ): 10,69 (1Н, широкий s, NH), 8,30 (1Н, широкий s, NH), 6,85-7,32 (3Н, m, фенил), 1,15 (6Н, t, СН3). МСНР (пучок электронов): C15H18N2O2, рассч. 258; обнаруж. 539 (2M+Na).
Стадия 5
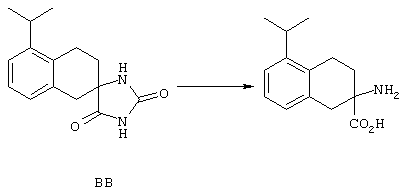
Смесь гидантоина ВВ (сырой, 8,34 ммоля, теоретическое количество) и Ва(ОН)2·Н2O (7,90 г, 41,7 ммоля) в воде (40 мл) в герметично закрытой толстостенной колбе выдерживали на масляной бане при 125° С в течение 38 ч. Реакционную смесь охлаждали до комнатной температуры и с использованием 4 н. серной кислоты при одновременном интенсивном перемешивании подкисляли до рН ~3. Суспензию в течение двух часов перемешивали на бане с кипящей водой и охлаждали до комнатной температуры. Белую суспензию фильтровали и осадки промывали водой. Объединенные фильтрат и промывные жидкости концентрировали под вакуумом до ~50 мл. Нейтрализацией концентрированным раствором гидроксида аммония получали белый осадок, который отфильтровывали, промывали водой и сушили под вакуумом в течение ночи с получением в виде бежевого твердого вещества рацемической 5-изопропил-2-аминотетралин-2-карбоновой кислоты (1,23 г, 63%-ный выход за 2 стадии). МСНР (пучок электронов): С14Н19NО2, рассч. 233; обнаруж. 232 (М-Н).
Стадия 6
Смесь рацемической 5-изопропил-2-аминотетралин-2-карбоновой кислоты (250 мг, 1,07 ммоля), триэтиламина (1,2 мл, 8,61 ммоля) и 9-флуоренилметилсукцинимидилкарбоната (ФМОК-OSu, 2,70 г, 8,00 ммоля) в ацетонитриле (30 мл) и воде (30 мл) перемешивали при комнатной температуре в течение 2 дней. Реакционную смесь концентрировали под вакуумом для удаления большей части ацетонитрила, 10%-ным водным раствором лимонной кислоты подкисляли до рН ~3 и белую эмульсию экстрагировали этилацетатом. Органический слой промывали водой, рассолом и сушили над сульфатом натрия.
В результате фильтрования и концентрирования в виде масла получали сырой продукт, который очищали хроматографией в колонке (элюировали 2 → 5 → 8% метанола/метиленхлоридом) с получением в виде не совсем белой пены рацемической ФМОК-5-изопропил-2-аминотетралин-2-карбоновой кислоты (208 мг, 43%-ный выход). МСВР (БУА): C29H30NO4 (М+Н), рассч. 456,2175; обнаруж. 456,2184.
Пример 17
Получение ФМОК-4-амино-1-фенилпиперидин-4-карбоновой кислоты (ФМОК-Аррс-ОН)
Стадия 1
В раствор иодбензола (6,37 г, 3,5 мл, 31,2 ммоля), 1,4-диокса-8-азаспиро[4.5]декана (10,32 г, 9,3 мл, 72,2 ммоля, 2,3 экв.) и трет-бутоксида натрия (8,0 г, 83,3 ммоля, 2,7 экв.) в сухом диоксане (120 мл) вводили трис(дибензилиденацетон)дипалладий(0) (91 мг, 0,1 ммоля) и три-о-толилфосфин (180 мг, 0,591 ммоля). Реакционную смесь выдерживали при 90° С в течение 26 ч. Образовавшуюся реакционную смесь концентрировали для удаления растворителя. Остаток обрабатывали водой и экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, сушили над Na2SO4 и концентрировали с получением продукта в виде коричневого масла. Этот сырой продукт очищали экспресс-хроматографией (гексан/EtOAc в соотношении от 95/5 до 75/25) с получением в виде слегка желтого твердого вещества чистого продукта СС (6,08 г, 89%-ный выход). 1Н-ЯМР (СDСl3): 7,25 (ddt, 2Н), 6,95 (dd, 2H), 6,84 (t, 1H), 4,00 (s, 4Н), 3,32 (t, 4H) и 1,84 (t, 4H). МС (пучок электронов) m/e: 220 (М+Н), рассч. для С13Н17NО2 219.
Стадия 2
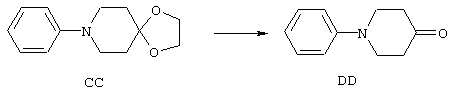
В раствор кеталя СС (3,22 г, 15,16 ммоля) в ацетоне (100 мл) вводили 6 н. соляную кислоту (50 мл) и реакционную смесь кипятили с обратным холодильником в течение ночи. Образовавшуюся реакционную смесь концентрировали для удаления растворителя. Остаток растворяли в EtOAc и нейтрализовали 6 н. водным раствором NaOH. Слои разделяли и водный слой экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, сушили над Na2SO4 и концентрировали. Сырой продукт очищали экспресс-хроматографией (гексан/EtOAc, 80/20 → 60/40) с получением продукта DD в виде желтого масла (2,58 г, 97%-ный выход). МС (пучок электронов) m/e: 176 (М+Н), рассч. для C11H13NO 175.
Стадия 3
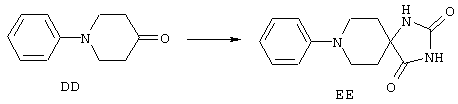
В раствор кетона DD (2,53 г, 14,46 ммоля) в этаноле (75 мл) и воде (25 мл) в стеклянной толстостенной колбе вводили карбонат аммония (12,9 г, 134,3 ммоля, 9 экв.) и цианид калия (2,11 г, 32,5 ммоля, 2 экв.). Смесь выдерживали при температуре от 80 до 90° С в течение 18 ч. Охлажденную реакционную смесь концентрировали под вакуумом, остаток обрабатывали водой и экстрагировали EtOAc (4 раза). Объединенные органические экстракты промывали водой, сушили над безводным Na2SO4 и концентрировали с получением в виде белого твердого вещества спектроскопически чистого гидантоина ЕЕ (3,36 г, 95%-ный выход). МС (пучок электронов) m/e: 246 (М+Н), рассч. для C13H15N3O2 245.
Стадия 4
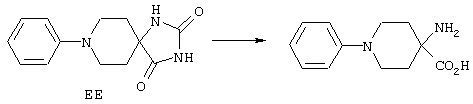
Гидантоин ЕЕ (3,36 г) суспендировали в водном NaOH (6 н., 100 мл) и выдерживали при 130° С в течение от 2 до 3 дней. По завершении (по данным ВЭЖХ) гидролиза реакционную смесь нейтрализовали конц. НСl до слабокислой реакции (рН ~6). Образовавшуюся суспензию фильтровали, промывали водой и сушили с получением в виде белого твердого вещества 4-амино-1-фенилпиперидин-4-карбоновой кислоты (Аррс) (5,26 г, >100%-ный выход, мокрая и загрязненная неорганической солью), которая характеризовалась единственным пиком ВЭЖХ и которую использовали непосредственно на следующей стадии. МС (пучок электронов) m/e: 221 (М+Н), рассч. для C12H16N2O2 220.
Стадия 5
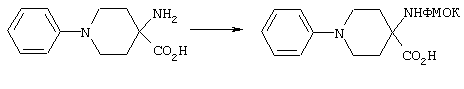
Сырую 4-амино-1-фенилпиперидин-4-карбоновую кислоту (Аррс) с последней стадии суспендировали в диоксане (80 мл) и водном 10%-ном Nа2СО3 (40 мл), обрабатывали ФМОК-С1 (5,3 г, 20,57 ммоля, 1,5 экв.) и интенсивно перемешивали в течение ночи. Далее реакционную смесь концентрировали для удаления диоксана, нейтрализовали 6 н. НСl до слабокислой реакции (рН 6) и экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом и сушили над Na2SO4. Удалением растворителя получали сырой продукт, который очищали экспресс-хроматографией (с переходом от гексана/EtOAc к CH2Cl2/MeOH) с выделением чистого ФМОК-Аррс (4,91 г, 81%-ный общий выход за две стадии). 1H-ЯМР (ДМСО-d6): 7,88 (d, 2H), 7,74 (d, 2H), 7,19-7,42 (m, 8H), 4,20-4,31 (m, 3H). MSBP m/z: 465,1788, рассч. для C27H26N2O4Na 465,1791.
Пример 18
Получение ФМОК-4-амино-1-(4-метилфенил)пиперидин-4-карбоновой кислоты (ФМОК-4-МеАррс-ОН)
Стадия 1
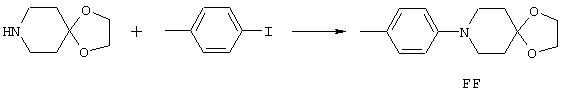
В раствор 4-иодтолуола (2,12 г, 9,7 ммоля), 1,4-диокса-8-азаспиро[4.5]декана (2,8 мл, 3,12 г, 21,82 ммоля, 2,2 экв.) и трет-бутоксида натрия (2,6 г, 27,08 ммоля, 2,8 экв.) в сухом диоксане (40 мл) добавляли трис(дибензилиденацетон)дипалладия(0) (44,4 мг, 0,0485 ммоля) и три-о-толилфосфина (59,0 мг, 0,194 ммоля). Реакционную смесь выдерживали при 90° С в течение 26 ч. Образовавшуюся реакционную смесь концентрировали для удаления растворителя. Остаток обрабатывали водой и экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, сушили над Na2SO4 и концентрировали с получением продукта в виде коричневого масла. Этот сырой продукт очищали экспресс-хроматографией (гексан/ЕtOАс в соотношении от 95/5 до 75/25) с получением в виде слегка желтого твердого вещества чистого продукта FF (1,937 г, 85%-ный выход). 1H-ЯМР (CDCl3): 7,06 (d, 2Н), 6,87 (d, 2H), 3,99 (s, 4Н), 3,26 (t, 4H), 2,26 (s, 3H) и 1,85 (t, 4H).
Стадия 2
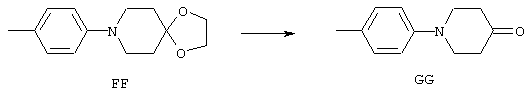
В раствор кеталя FF (1,58 г, 6,79 ммоля) в ацетоне (50 мл) вводили 6 н. соляную кислоту (25 мл) и реакционную смесь кипятили с обратным холодильником в течение ночи. Образовавшуюся реакционную смесь концентрировали для удаления растворителя. Остаток растворяли в EtOAc и нейтрализовали 6 н. водным раствором NaOH. Слои разделяли и водный слой экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, сушили над Na2SO4 и концентрировали. Сырой продукт очищали экспресс-хроматографией (гексан/EtOAc, 90/10 → 70/30) с получением в виде желтого масла продукта GG (1,27 г, 98%-ный выход). МС (пучок электронов) m/е: 190 (М+Н), рассч. для C12H15NO 189.
Стадия 3
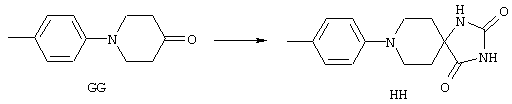
В раствор кетона GG (1,17 г, 6,18 ммоля) в этаноле (60 мл) и воде (20 мл) в стеклянной толстостенной колбе вводили карбонат аммония (4,74 г, 49,44 ммоля, 8 экв.) и цианид калия (1,01 г, 15,54 ммоля, 2,5 экв.). Смесь выдерживали при 90° С в течение 22 ч. Охлажденную реакционную смесь концентрировали под вакуумом, остаток обрабатывали водой и экстрагировали EtOAc (4 раза). Объединенные органические экстракты промывали водой, сушили над безводным Na2SO4 и концентрировали с получением в виде белого твердого вещества спектроскопически чистого гидантоина НН (1,554 г, 97%-ный выход). МС (пучок электронов) m/e: 260 (М+Н), рассч. для С14Н17N3О2 259.
Стадия 4
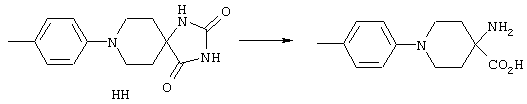
Гидантоин НН (1,502 г) суспендировали в водном NaOH (6 н., 40 мл) и выдерживали при 130° С в течение 4 дней. По завершении (по данным ВЭЖХ) гидролиза реакционную смесь нейтрализовали конц. НСl до слабокислой реакции (рН ~6). Образовавшуюся суспензию фильтровали, промывали водой и сушили с получением в виде белого твердого вещества 4-амино-1-(4-метилфенил)пиперидин-4-карбоновой кислоты (4-МеАррс) (2,10 г, >100%-ный выход, мокрая и загрязненная неорганической солью), которая характеризовалась единственным пиком ВЭЖХ и которую непосредственно использовали на следующей стадии. МС (пучок электронов) m/e: 235 (М+Н), рассч. для C13H18N2O2 234.
Стадия 5
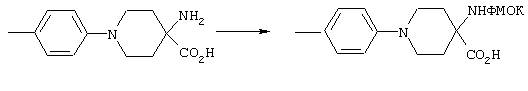
Сырую 4-амино-1-(4-метилфенил)пиперидин-4-карбоновую кислоту (4-МеАррс) с последней стадии суспендировали в диоксане (80 мл) и водном 10%-ном Na2CO3 (40 мл), обрабатывали ФМОК-С1 (2,2 г, 8,59 ммоля, 1,5 экв.) и интенсивно перемешивали в течение ночи. Далее реакционную смесь концентрировали для удаления диоксана, нейтрализовали 6 н. НСl до слабокислой реакции (рН 6) и экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом и сушили над Na2SO4. Удалением растворителя получали сырой продукт, который очищали экспресс-хроматографией (с переходом от гексана/EtOAc к СН2Сl2/МеОН) с выделением чистого ФМОК-4-МеАррс (2,16 г, 82%-ный общий выход за две стадии). 1H-ЯМР (ДМСО-d6): 7,88 (d, 2H), 7,72 (d, 2H), 7,39 (t, 2H), 7,30 (td, 2H), 6,99 (d, 2H), 6,82 (d, 2H), 2,18 (s, 3Н). МС (пучок электронов) m/e: 457 (М+Н), рассч. для C28H28N2O4 456.
Пример 19
Получение ФМОК-4-амино-1-(4-хлорфенил)пиперидин-4-карбоновой кислоты (ФМОК-4-ClAppc-OH)
Стадия 1
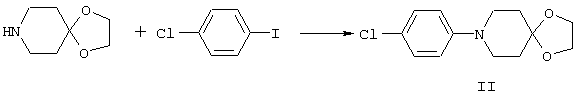
В раствор 1-хлор-4-иодбензола (2,38 г, 10,0 ммоля), 1,4-диокса-8-азаспиро[4.5]декана (3,1 мл, 3,44 г, 24,0 ммоля, 2,4 экв.) и трет-бутоксида натрия (2,68 г, 28,0 ммоля, 2,8 экв.) в сухом диоксане (40 мл) вводили трис(дибензилиденацетон)дипалладий(0) (45,5 мг, 0,0497 ммоля) и три-о-толилфосфин (61 мг, 0,20 ммоля). Реакционную смесь в течение 9 ч выдерживали при 90° С. Образовавшуюся реакционную смесь концентрировали для удаления растворителя. Остаток обрабатывали водой и экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, сушили над Na2SO4 и концентрировали с получением продукта в виде коричневого масла. Этот сырой продукт очищали экспресс-хроматографией (гексан/EtOAc в соотношении от 95/5 до 75/25) с получением в виде слегка желтого твердого вещества чистого продукта II (2,17 г, 86%-ный выход). 1H-ЯМР (CDCl3): 7,18 (dt, 2Н), 6,85 (dt, 2H), 3,98 (s, 4H), 3,28 (t, 4H) и 1,82 (t, 4H).
Стадия 2
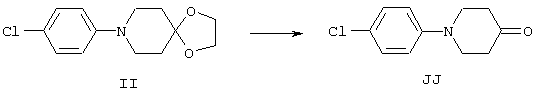
В раствор кеталя II (2,123 г, 8,39 ммоля) в ацетоне (75 мл) вводили 6 н. соляную кислоту (30 мл) и реакционную смесь кипятили с обратным холодильником в течение ночи. Образовавшуюся реакционную смесь концентрировали для удаления растворителя. Остаток растворяли в EtOAc и нейтрализовали 6 н. водным раствором NaOH. Слои разделяли и водный слой экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, сушили над Na2SO4 и концентрировали. Сырой продукт очищали экспресс-хроматографией (гексан/EtOAc, 95/5 → 70/30) с получением в виде желтого твердого вещества продукта JJ (1,515 г, 86%-ный выход). МС (пучок электронов) m/e: 210 (М+Н), рассч. для C11H12ClNO 209.
Стадия 3
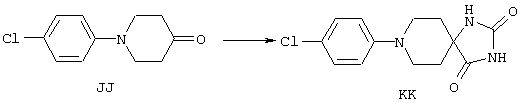
В раствор кетона JJ (1,465 г, 6,986 ммоля) в этаноле (75 мл) и воде (25 мл) в стеклянной толстостенной колбе вводили карбонат аммония (5,36 г, 55,88 ммоля, 8 экв.) и цианид калия (1,135 г, 17,46 ммоля, 2,5 экв.). Смесь выдерживали при температуре от 80 до 90° С в течение 18 ч. Охлажденную реакционную смесь концентрировали под вакуумом, остаток обрабатывали водой и экстрагировали EtOAc (4 раза). Объединенные органические экстракты промывали водой, сушили над безводным Na2SO4 и концентрировали с получением в виде белого твердого вещества спектроскопически чистого гидантоина КК (1,817 г, 93%-ный выход). МС (пучок электронов) m/e: 280 (М+Н), рассч. для С13Н14СlN3O2 279.
Стадия 4
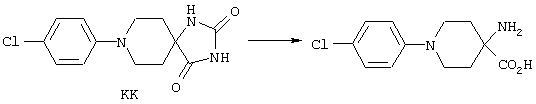
Гидантоин КК (1,768 г) суспендировали в водном NaOH (6 н., 50 мл) и выдерживали при 130° С в течение 4 дней. По завершении (по данным ВЭЖХ) гидролиза реакционную смесь нейтрализовали конц. НСl до слегка кислой реакции (рН ~6). Образовавшуюся суспензию фильтровали, промывали водой и сушили с получением в виде белого твердого вещества 4-амино-1-(4-хлорфенил)пиперидин-4-карбоновой кислоты (4-ClAppc) (2,05 г, >100% выход, мокрая и загрязненная неорганической солью), которая характеризовалась единственным пиком ВЭЖХ и которую использовали непосредственно на следующей стадии. МС (пучок электронов) m/e: 253 (М-Н), рассч. для C12H15ClN2O2 254.
Стадия 5
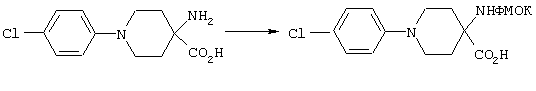
Сырую 4-амино-1-(4-хлорфенил)пиперидин-4-карбоновую кислоту (4-ClAppc) с последней стадии суспендировали в диоксане (100 мл) и водном 10%-ном Na2CO3 (50 мл), обрабатывали ФМОК-С1 (2,0 г, 7,75 ммоля, 1,2 экв.) и интенсивно перемешивали в течение ночи. Далее реакционную смесь концентрировали для удаления диоксана, нейтрализовали 6 н. НСl до слабокислой реакции (рН 6) и экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом и сушили над Na2SO4. Удалением растворителя получали сырой продукт, который очищали экспресс-хроматографией (с переходом от гексана/EtOAc к СН2Сl2/МеОН) с выделением чистого ФМОК-4-ClAppc (1,18 г, 81%-ный общий выход за две стадии). 1H-ЯМР (ДМСО-d6): 7,87 (d, 2H), 7,71 (d, 2H), 7,39 (td, 2H), 7,30 (td, 2H), 7,20 (d, 2H), 6,92 (d, 2H), 3,44 (d, 2H), 2,93 (t, 2H). МС (пучок электронов) m/e: 477 (М+Н), рассч. для C27H25N2O4 476.
Пример 20
Получение ФМОК-4-амино-1-(4-феноксифенил)пиперидин-4-карбоновой кислоты (ФМОК-4-PhOAppc-OH)
Стадия 1
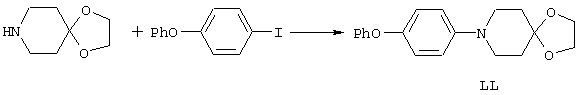
В раствор 1-иод-4-феноксибензола (3,15 г, 10,6 ммоля), 1,4-диокса-8-азаспиро[4.5]декана (3,3 мл, 3,66 г, 25,6 ммоля, 2,4 экв.) и трет-бутоксида натрия (2,85 г, 29,7 ммоля, 2,8 экв.) в сухом диоксане (40 мл) вводили трис(дибензилиденацетон)дипалладий(0) (48,5 мг, 0,053 ммоля) и три-о-толилфосфин (64 мг, 0,4 ммоля). Реакционную смесь в течение 9 ч выдерживали при 90° С. Образовавшуюся реакционную смесь концентрировали для удаления растворителя. Остаток обрабатывали водой и экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, сушили над Na2SO4 и концентрировали с получением продукта в виде коричневого масла.
Этот сырой продукт очищали экспресс-хроматографией (гексан/EtOAc в соотношении от 95/5 до 80/20) с получением в виде слегка желтого твердого вещества чистого продукта LL (2,805 г, 85%-ный выход). 1H-ЯМР (CDCl3): 7,26-7,32 (m, 2Н), 7,03 (t, 1H), 6,92 - 6,97 (m, 6Н), 4,00 (s, 4H), 3,26 (t, 4H), 1,86 (t, 4H).
Стадия 2
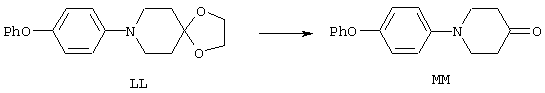
В раствор кеталя LL (2,755 г, 8,86 ммоля) в ацетоне (90 мл) вводили 6 н. соляную кислоту (45 мл) и реакционную смесь кипятили с обратным холодильником в течение ночи. Образовавшуюся реакционную смесь концентрировали для удаления растворителя. Остаток разбавляли EtOAc и нейтрализовали 6 н. водным раствором NaOH. Слои разделяли и водный слой экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, сушили над Na2SO4 и концентрировали с получением сырого продукта, который очищали экспресс-хроматографией (гексан/EtOAc в соотношении от 90/10 до 70/30) с получением продукта ММ в виде желтого масла (2,21 г, 93%-ный выход). МС (пучок электронов) m/e: 268 (М+Н), рассч. для C17H17ClNO2 267.
Стадия 3

В раствор кетона MM (2,01 г, 7,52 ммоля) в этаноле (80 мл) и воде (25 мл) в стеклянной толстостенной колбе вводили карбонат аммония (5,78 г, 60,0 ммоля, 8 экв.) и цианид калия (1,22 г, 18,80 ммоля, 2,5 экв.). Смесь выдерживали при температуре от 80 до 90° С в течение 18 ч. Охлажденную реакционную смесь концентрировали под вакуумом, остаток обрабатывали водой и экстрагировали EtOAc (4 раза). Объединенные органические экстракты промывали водой, сушили над безводным Na2SO4 и концентрировали с получением в виде белого твердого вещества спектроскопически чистого гидантоина NN (2,34 г, 95%-ный выход). МС (пучок электронов) m/e: 338 (М+Н), рассч. для C19H19N3O3 337.
Стадия 4
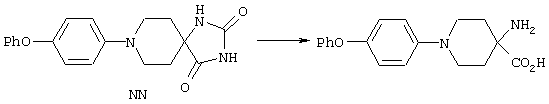
Гидантоин NN (2,28 г, 6,76 ммоля) суспендировали в водном NaOH (6 н., 60 мл) и выдерживали при 130° С в течение 4 дней. По завершении (по данным ВЭЖХ) гидролиза реакционную смесь нейтрализовали конц. НСl до слабокислой реакции (рН ~6). Образовавшуюся суспензию фильтровали, промывали водой и сушили с получением в виде белого твердого вещества 4-амино-1-(4-феноксифенил)пиперидин-4-карбоновой кислоты (4-PhOAppc) (2,53 г, >100%-ный выход, мокрая и загрязненная неорганической солью), которая характеризовалась единственным пиком ВЭЖХ и которую использовали непосредственно на следующей стадии. МС (пучок электронов) m/e: 313 (М+Н), рассч. для C18H20N2O3 312.
Стадия 5
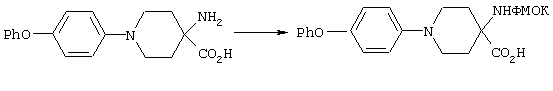
Сырую 4-амино-1-(4-феноксифенил)пиперидин-4-карбоновую кислоту (4-PhOAppc) с последней стадии суспендировали в диоксане (50 мл) и водном 10%-ном Na2CO3 (50 мл) и интенсивно перемешивали в течение ночи. Реакционную смесь концентрировали для удаления диоксана, нейтрализовали 6 н. НСl до слабокислой реакции (рН 6) и экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом и сушили над Na2SO4. Удалением растворителя получали сырой продукт, который очищали экспресс-хроматографией (с переходом от гексана/EtOAc к СН2Сl2/МеОН) с выделением чистого ФМОК-4-PhOAppc (2,18 г, 60%-ный общий выход за две стадии). 1H-ЯМР (ДМСО-d6): 7,87 (d, 2Н), 7,72 (d, 2Н), 7,38 (t, 2H), 7,30 (td, 4H), 7,02 (dt, 1H), 6,86-6,96 (m, 6H), 3,35 (m, 2H), 2,94 (t, 2H). МС (пучок электронов) m/e: 535 (М+Н), рассч. для С33Н30Н2O5 534.
Пример 21
Получение ФМОК-4-амино-1-(2-метилфенил)пиперидин-4-карбоновой кислоты (ФМОК-2-МеАррс-ОН)
Стадия 1
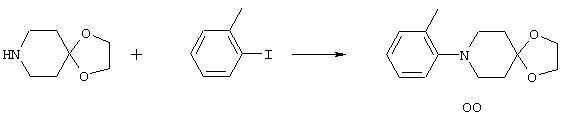
В раствор 2-иодтолуола (4,36 г, 2,5 мл, 20,0 ммоля), 1,4-диокса-8-азаспиро[4.5]декана (6,88 г, 6,2 мл, 48,1 ммоля, 2,4 экв.) и трет-бутоксида натрия (5,3 г, 55,2 ммоля, 2,8 экв.) в сухом диоксане (80 мл) вводили трис(дибензилиденацетон)дипалладий(0) (91 мг, 0,1 ммоля) и три-о-толилфосфин (122 мг, 0,4 ммоля). Реакционную смесь выдерживали при 90° С в течение 26 ч. Образовавшуюся реакционную смесь концентрировали для удаления растворителя. Остаток обрабатывали водой и экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, сушили над Na2SO4 и концентрировали с получением продукта в виде коричневого масла. Этот сырой продукт очищали экспресс-хроматографией (гексан/EtOAc в соотношении от 95/5 до 75/25) с получением в виде слегка желтого твердого вещества чистого продукта OO (2,66 г, 57%-ный выход). 1H-ЯМР (CDCl3): 7,12-7,18 (m, 2Н), 6,94-7,06 (m, 2H), 4,01 (s, 4Н), 2,98 (t, 4H) и 1,88 (t, 4H).
Стадия 2
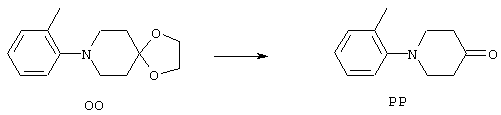
В раствор кеталя OO (2,66 г, 11,4 ммоля) в ацетоне (70 мл) вводили 6 н. соляную кислоту (35 мл) и реакционную смесь выдерживали при 85° С в течение ночи. Образовавшуюся реакционную смесь концентрировали для удаления растворителя. Остаток разбавляли EtOAc и нейтрализовали водным NaOH (6 н.). Слои разделяли и водный слой экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, сушили над Na2SO4 и концентрировали. Сырой продукт очищали экспресс-хроматографией (гексан/EtOAc в соотношении от 90/10 до 70/30) с получением в виде желтого масла продукта РР (2,04 г, 95%-ный выход). МС (пучок электронов) m/e: 190 (М+Н), рассч. для C12H15NO 189.
Стадия 3
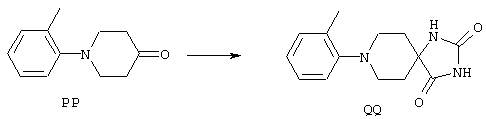
В раствор кетона РР (1,54 г, 8,15 ммоля) в этаноле (60 мл) и воде (20 мл) в стеклянной толстостенной колбе вводили карбонат аммония (4,69 г, 48,9 ммоля, 6 экв.) и цианид калия (800 мг, 12,2 ммоля, 1,5 экв.). Смесь выдерживали при температуре от 80 до 90° С в течение 18 ч. Охлажденную реакционную смесь добавляли в смесь воды со льдом (300 мл) и интенсивно перемешивали в течение 30 мин. Полученный осадок отфильтровывали под вакуумом, тщательно промывали водой и сушили с получением в виде белого твердого вещества гидантоина QQ (2,01 г, 95%-ный выход). МС (пучок электронов) m/e: 260 (М+Н), рассч. для C14H17N3O2 259.
Стадия 4
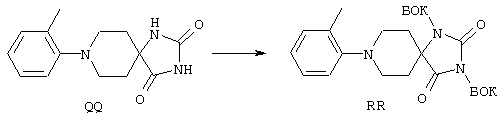
В суспензию гидантоина QQ (1,07 г, 4,13 ммоля) в сухом ТГФ (25 мл) последовательно вводили ди-трет-бутилдикарбонат (2,25 г, 10,32 ммоля, 2,5 экв.), триэтиламин (0,63 мл, 460 мг, 4,54 ммоля, 1,1 экв.) и ДМАП (36 мг, 0,29 ммоля). По прошествии примерно 15 мин после добавления реакционная смесь превращалась в прозрачный желтый раствор, и ее перемешивали в течение ночи при комнатной температуре. Реакционную смесь концентрировали под пониженным давлением с получением твердого вещества, которое затем растворяли в EtOAc (300 мл), промывали 1 н. НСl (3 порции по 30 мл), насыщенным водным Na2CO3 (2 порции по 30 мл) и рассолом (2 порции по 30 мл), сушили над безводным Na2SO4 и концентрировали под пониженным давлением. Сырой светло-желтый продукт очищали экспресс-хроматографией (гексан/EtOAc, 90/10 → 80/20) с получением в виде белого твердого вещества чистого бис-БОК-гидантоина RR (1,71 г, 90%-ный выход). МС (пучок электронов) m/e: 460 (М+Н), рассч. для C24H33N3O6 459.
Стадия 5
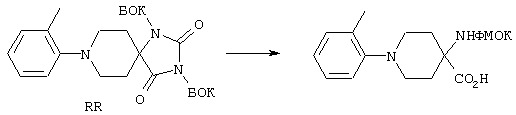
Бис-БОК-гидантоин RR (1,71 г, 3,72 ммоля) растворяли в ДМЭ (23 мл) с получением прозрачного раствора. В этот раствор добавляли 1 н. NaOH (33 мл, 33 ммоля) и реакционную смесь перемешивали в течение ночи при комнатной температуре, получая слегка мутную смесь. Данные ВЭЖХ указывали на завершение реакции. Реакционную смесь концентрировали под пониженным давлением для удаления ДМЭ и экстрагировали Et2O. Без очистки полученный водный слой, содержавший 4-амино-1-(2-метилфенил)пиперидин-4-карбоновую кислоту (2-МеАррс), обрабатывали 6 н. НСl для доведения рН до 11-12. Далее этот раствор (30 мл) разбавляли 1,4-диоксаном (30 мл) и обрабатывали ФМОК-С1 (1,28 г, 4,96 ммоля, 1,3 экв.) и смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь концентрировали под пониженным давлением для удаления диоксана, нейтрализовали 1 н. НСl и экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, сушили над безводным Na2SO4 и концентрировали. Сырой продукт очищали экспресс-хроматографией (гексан/EtOAc → CH2Cl2/MeOH) с получением в виде белого твердого вещества в качестве чистого продукта ФМОК-2-МеАррс (1,09 г, 64%-ный выход из бис-БОК-гидантоина RR). 1H-ЯМР (ДМСО-d6): 7,87 (d, 2H), 7,74 (d, 2H), 7,40 (td, 2H), 7,31 (td, 2H), 7,12 (m, 2H), 6,97 (d, 1H), 6,92 (td, 1H), 2,72-2,88 (m, 4H) и 2,22 (s, 3H). МС (пучок электронов) m/e: 457 (М+Н), рассч. для C28H28N2O4 456.
Пример 22
Получение ФМОК-4-амино-1-(2-изопропилфенил)пиперидин-4-карбоновой кислоты (ФМОК-2-изо-РrАррс-ОН)
Стадия 1
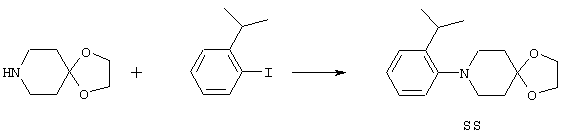
В раствор 1-иод-2-изопропилбензола (10,0 г, 40,7 ммоля), 1,4-диокса-8-азаспиро[4.5]декана (12,0 мл, 13,3 г, 93,0 ммоля, 2,3 экв.) и трет-бутоксида натрия (10,0 г, 104,2 ммоля, 2,6 экв.) в сухом диоксане (160 мл) вводили трис(дибензилиденацетон)дипалладий(0) (180 мг, 0,197 ммоля) и три-о-толилфосфин (244 мг, 0,80 ммоля) и реакционную смесь выдерживали при 90° С в течение 26 ч. Образовавшуюся реакционную смесь концентрировали для удаления растворителя, обрабатывали водой и экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, сушили над Na2SO4 и концентрировали с получением продукта в виде коричневого масла. Этот сырой продукт очищали экспресс-хроматографией (гексан/EtOAc, 95/5 → 75/25) с получением в виде слегка желтого твердого вещества чистого продукта SS (3,61 г, 35%-ный выход). МС m/z: 262 (М+Н), рассч. для C16H23NO2 261.
Стадия 2
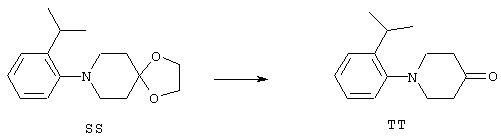
В раствор кеталя SS (3,24 г, 12,4 ммоля) в ацетоне (90 мл) вводили 6 н. соляную кислоту (45 мл) и реакционную смесь кипятили с обратным холодильником в течение ночи. Образовавшуюся реакционную смесь концентрировали для удаления растворителя, остаток разбавляли EtOAc и нейтрализовали водным NaOH (6 н.). Слои разделяли и водный слой экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, сушили над Na2SO4 и концентрировали. Сырой продукт очищали экспресс-хроматографией (гексан/EtOAc, 90/10 → 70/30) с получением продукта ТТ в виде желтого масла (2,42 г, 89%-ный выход). 1H-ЯМР (CDCl3): 7,27 (m, 1H), 7,04-7,19 (m, 3H), 3,58 (m, 1H), 3,20 (t, 4H), 2,60 (t, 4H) и 1,25 (d, 6H). МС m/z 218 (М+Н), рассч. для C14H19NO 217.
Стадия 3
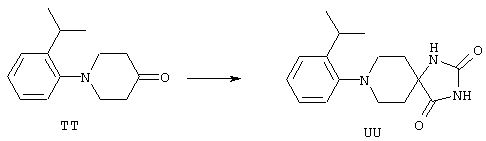
В раствор кетона ТТ (2,30 г, 10,6 ммоля) в этаноле (90 мл) и воде (20 мл) в стеклянной толстостенной колбе вводили карбонат аммония (8,1 г, 84,3 ммоля, 8 экв.) и цианид калия (1,72 г, 26,5 ммоля, 2,5 экв.). Смесь выдерживали при температуре от 80 до 90° С в течение 18 ч. Охлажденную реакционную смесь добавляли в смесь воды со льдом (400 мл) и интенсивно перемешивали в течение 30 мин. Полученный осадок отфильтровывали под вакуумом, тщательно промывали водой и сушили с получением в виде белого твердого вещества гидантоина UU (2,78 г, 91%-ный выход). МС m/z: 288 (М+Н), рассч. для C16H21N3O2 287.
Стадия 4
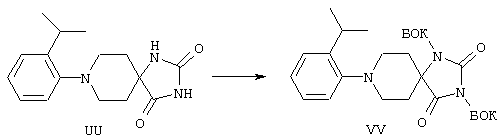
В суспензию гидантоина UU (2,74 г, 9,54 ммоля) в сухом ТГФ (100 мл) последовательно вводили ди-трет-бутилдикарбонат (5,2 г, 24,24 ммоля, 2,5 экв.), триэтиламин (1,5 мл, 1,07 г, 10,5 ммоля, 1,1 экв.) и ДМАП (46 мг, 0,29 ммоля). По прошествии примерно 15 мин после добавления реакционная смесь превращалась в прозрачный желтый раствор, и ее перемешивали в течение ночи при комнатной температуре. Реакционную смесь концентрировали под пониженным давлением с получением твердого вещества, которое затем растворяли в EtOAc (300 мл), промывали рассолом (3 порции по 30 мл), сушили над безводным Na2SO4 и концентрировали под пониженным давлением. Сырой светло-желтый продукт очищали экспресс-хроматографией (гексан/EtOAc, 90/10 → 80/20) с получением в виде белого твердого вещества чистого бис-БОК-гидантоина VV (4,39 г, 94%-ный выход). МС m/z: 488 (М+Н), рассч. для С26Н37N3О6 487.
Стадия 5

Бис-БОК-гидантоин VV (2,34 г, 4,8 ммоля) растворяли в ДМЭ (30 мл) с получением прозрачного раствора. В этот раствор добавляли 1 н. NaOH (45 мл, 45 ммоля) и реакционную смесь перемешивали в течение ночи при комнатной температуре, получая довольно прозрачную смесь. Данные ВЭЖХ указывали на завершение реакции. Реакционную смесь концентрировали под пониженным давлением для удаления ДМЭ и экстрагировали Et2O. Без очистки полученный водный слой, содержавший 4-амино-1-(2-изопропилфенил)пиперидин-4-карбоновую кислоту (2-изо-РrАррс), обрабатывали 6 н. НСl для доведения рН до 11-12. Далее этот раствор (~45 мл) разбавляли 1,4-диоксаном (45 мл), обрабатывали ФМОК-С1 (1,78 г, 6,89 ммоля, 1,5 экв.) и перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали под пониженным давлением для удаления диоксана, нейтрализовали 1 н. НСl и экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, сушили над безводным Na2SO4 и концентрировали. Сырой продукт очищали экспресс-хроматографией (гексан/EtOAc → CH2Cl2/MeOH) с получением в виде белого твердого вещества в качестве чистого продукта ФМОК-2-изо-РrАррс (1,46 г, 63%-ный выход из бис-БОК-гидантоина). МСВР m/z: 507,2263, рассч. для С30Н32N2O4Nа 507,2260.
Пример 23
Получение ФМОК-4-амино-1-(3-метилфенил)пиперидин-4-карбоновой кислоты (ФМОК-3-МеАррс-ОН)
Стадия 1
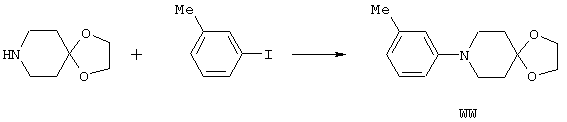
В раствор 3-иодтолуола (4,36 г, 2,6 мл, 20,0 ммоля), 1,4-диокса-8-азаспиро[4.5]декана (6,88 г, 6,2 мл, 48,1 ммоля, 2,4 экв.) и трет-бутоксида натрия (5,3 г, 55,2 ммоля, 2,8 экв.) в сухом диоксане (80 мл) вводили трис(дибензилиденацетон)дипалладий(0) (91 мг, 0,1 ммоля) и три-о-толилфосфин (122 мг, 0,4 ммоля). Реакционную смесь выдерживали при 90° С в течение 26 ч. Образовавшуюся реакционную смесь концентрировали для удаления растворителя. Остаток обрабатывали водой и экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, сушили над Na2SO4 и концентрировали с получением продукта в виде коричневого масла. Этот сырой продукт очищали экспресс-хроматографией (гексан/EtOAc в соотношении от 95/5 до 75/25) с получением в виде слегка желтого твердого вещества чистого продукта WW (3,21 г, 69%-ный выход).
Стадия 2

В раствор кеталя WW (1,25 г, 5,36 ммоля) в ацетоне (20 мл) вводили 6 н. соляную кислоту (10 мл) и реакционную смесь кипятили с обратным холодильником в течение ночи. Образовавшуюся реакционную смесь концентрировали для удаления растворителя. Остаток разбавляли EtOAc и нейтрализовали водным NaOH (6 н.). Слои разделяли и водный слой экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, сушили над Na2SO4 и концентрировали. Сырой продукт очищали экспресс-хроматографией (гексан/EtOAc в соотношении от 90/10 до 70/30) с получением в виде желтого масла продукта XX (843 мг, 83%-ный выход). МС m/z: 190 (М+Н), рассч. для C12H15NO 189.
Стадия 3

В раствор кетона XX (763 г, 4,03 ммоля) в этаноле (45 мл) и воде (15 мл) в стеклянной толстостенной колбе вводили карбонат аммония (3,09 г, 32,21 ммоля, 8 экв.) и цианид калия (675 мг, 10,38 ммоля, 2,5 экв.). Смесь выдерживали при температуре от 80 до 90° С в течение 18 ч. Охлажденную реакционную смесь добавляли в смесь воды со льдом (200 мл) и интенсивно перемешивали в течение 30 мин. Полученный осадок отфильтровывали под вакуумом, тщательно промывали водой и сушили с получением в виде белого твердого вещества гидантоина YY (930 мг, 89%-ный выход). МС m/z: 260 (М+Н), рассч. для C14H17N3O2 259.
Стадия 4
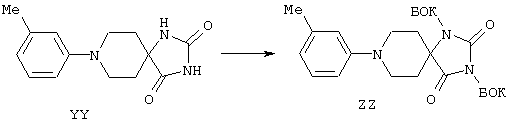
В суспензию гидантоина YY (780 мг, 3,012 ммоля) в сухом ТГФ (22 мл) последовательно вводили ди-трет-бутилдикарбонат (1,64 г, 7,52 ммоля, 2,5 экв.), триэтиламин (0,42 мл, 305 мг, 3,01 ммоля, 1,0 экв.) и ДМАП (20 мг, 0,164 ммоля). По прошествии примерно 5 мин после добавления реакционная смесь превращалась в прозрачный желтый раствор, и его перемешивали в течение ночи при комнатной температуре. Реакционную смесь концентрировали под пониженным давлением с получением твердого вещества, которое затем растворяли в EtOAc (300 мл), промывали рассолом (3 порции по 30 мл), сушили над безводным Nа2SO4 и концентрировали под пониженным давлением. Сырой светло-желтый продукт очищали экспресс-хроматографией (гексан/EtOAc, 90/10 → 80/20) с получением в виде белого твердого вещества чистого бис-БОК-гидантоина ZZ (1,37 г, количественный). МСВР m/z: 482,2261 (M+Na), рассч. для С24Н33N3О6Nа 482,2267.
Стадия 5
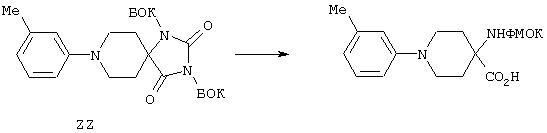
Бис-БОК-гидантоин ZZ (1,29 г, 2,818 ммоля) растворяли в ДМЭ (20 мл) с получением прозрачного раствора. В этот раствор добавляли 1 н. NaOH (25 мл, 25 ммоля) и реакционную смесь перемешивали в течение ночи при комнатной температуре, получая довольно прозрачную смесь. Данные ВЭЖХ указывали на завершение реакции. Реакционную смесь концентрировали под пониженным давлением для удаления ДМЭ и экстрагировали Et2O. Без очистки полученный водный слой, содержавший 4-амино-1-(3-метилфенил)пиперидин-4-карбоновую кислоту (3-МеАррс), обрабатывали 6 н. НСl для доведения рН до 11-12. Далее этот раствор (30 мл) разбавляли 1,4-диоксаном (30 мл), обрабатывали ФМОК-С1 (1,46 мг, 5,65 ммоля, 2,0 экв.) и перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали под пониженным давлением для удаления диоксана, нейтрализовали 1 н. НСl и экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, сушили над безводным Na2SO4 и концентрировали. Сырой продукт очищали экспресс-хроматографией (гексан/EtOAc → СН2Сl2/МеОН) с получением в виде белого твердого вещества в качестве чистого продукта ФМОК-3-МеАррс (1,002 г, 78%-ный выход из бис-БОК-гидантоина). МСВР m/z: 479,1940 (M+Na), рассч. для C28H28N2O4Na 479,1947.
Пример 24
Получение ФМОК-4-амино-1-(3-метоксифенил)пиперидин-4-карбоновой кислоты (ФМОК-3-МеОАррс-ОН)
Стадия 1

В раствор 3-иоданизола (4,68 г, 2,4 мл, 20,0 ммоля), 1,4-диокса-8-азаспиро[4.5]декана (6,2 мл, 6,88 г, 48,1 ммоля, 2,4 экв.) и трет-бутоксида натрия (5,3 г, 55,2 ммоля, 2,8 экв.) в сухом диоксане (80 мл) вводили трис(дибензилиденацетон)дипалладий(0) (91 мг, 0,1 ммоля) и три-о-толилфосфин (122 мг, 0,4 ммоля) и реакционную смесь выдерживали при 90° С в течение 26 ч. Образовавшуюся реакционную смесь концентрировали для удаления растворителя, остаток обрабатывали водой и экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, сушили над Na2SO4 и концентрировали с получением продукта в виде коричневого масла. Этот сырой продукт очищали экспресс-хроматографией (гексан/EtOAc в соотношении от 95/5 до 75/25) с получением в виде слегка желтого твердого вещества чистого продукта ААА (3,10 г, 62%-ный выход). МС m/z: (M+H), 250 (М+Н), рассч. для C14H19NO3 249.
Стадия 2
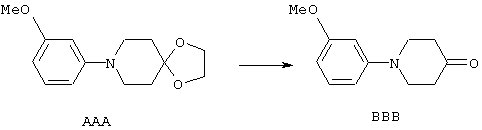
В раствор кеталя ААА (3,10 г, 12,45 ммоля) в ацетоне (90 мл) вводили 6 н. соляную кислоту (45 мл) и реакционную смесь кипятили с обратным холодильником в течение ночи. Образовавшуюся реакционную смесь концентрировали для удаления растворителя. Остаток разбавляли EtOAc и нейтрализовали водным NaOH (6 н.). Слои разделяли и водный слой экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, сушили над Na2SO4 и концентрировали. Сырой продукт очищали экспресс-хроматографией (гексан/EtOAc в соотношении от 90/10 до 70/30) с получением в виде желтого масла продукта ВВВ (2,53 г, 99%-ный выход). 1H-ЯМР (CDCl3): 7,20 (m, 1H), 6,58 (d, 1H), 6,39-6,56 (m, 2H), 3,80 (s, 3H), 3,59 (m, 4Н) и 2,58 (m, 4Н).
Стадия 3
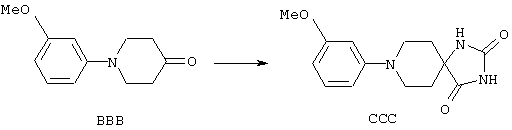
В раствор кетона ВВВ (1,81 г, 8,82 ммоля) в этаноле (60 мл) и воде (20 мл) в стеклянной толстостенной колбе вводили карбонат аммония (6,77 г, 70,52 ммоля, 8 экв.) и цианид калия (1,14 г, 17,6 ммоля, 2,0 экв.). Смесь выдерживали при температуре от 80 до 90° С в течение 18 ч. Охлажденную реакционную смесь добавляли в смесь воды со льдом (200 мл) и интенсивно перемешивали в течение 30 мин. Полученный осадок отфильтровывали под вакуумом, тщательно промывали водой и сушили с получением в виде белого твердого вещества гидантоина ССС (2,23 г, 92%-ный выход). МС m/z: 276 (М+Н), рассч. для C14H17N3O3 275.
Стадия 4
В суспензию гидантоина ССС (1,10 г, 4,00 ммоля) в сухом ТГФ (50 мл) последовательно вводили ди-трет-бутилдикарбонат (2,18 г, 10,0 ммоля, 2,5 экв.), триэтиламин (0,62 мл, 445 мг, 4,4 ммоля, 1,1 экв.) и ДМАП (20 мг, 0,164 ммоля). По прошествии примерно 15 мин после добавления реакционная смесь превращалась в прозрачный желтый раствор, и ее перемешивали в течение ночи при комнатной температуре. Реакционную смесь концентрировали под пониженным давлением с получением твердого вещества, которое затем растворяли в EtOAc (300 мл), раствор промывали рассолом (3 порции по 30 мл), сушили над безводным Na2SO4 и концентрировали под пониженным давлением. Сырой светло-желтый продукт очищали экспресс-хроматографией (гексан/EtOAc, 90/10 → 80/20) с получением в виде белого твердого вещества чистого бис-БОК-гидантоина DDD (1,90 г, количественный выход). 1H-ЯМР (CDCl3): 7,16 (t, 1Н), 6,57 (d, 1H), 6,24 (s, 1H), 6,19 (d, 1H), 3,77 (s, 3H), 1,58 (s, 9Н), 1,42 (s, 9H). MC m/z: 476 (М+Н), рассч. для С24Н33N3O7 475.
Стадия 5

Бис-БОК-гидантоин DDD (1,06 г, 2,23 ммоля) растворяли в ДМЭ (20 мл) с получением прозрачного раствора. В этот раствор добавляли 1 н. NaOH (20 мл, 20 ммолей) и реакционную смесь перемешивали при комнатной температуре в течение ночи, получая довольно прозрачную смесь. Данные ВЭЖХ указывали на завершение реакции. Реакционную смесь концентрировали под пониженным давлением для удаления ДМЭ и экстрагировали Et2O. Без очистки полученный водный слой, содержавший 4-амино-1-(3-метоксифенил)пиперидин-4-карбоновую кислоту (3-МеОАррс), обрабатывали 6 н. НСl для доведения рН до 11-12. Далее этот раствор (35 мл) разбавляли 1,4-диоксаном (35 мл), обрабатывали ФМОК-С1 (755 мг, 2,93 ммоля, 1,3 экв.) и перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали под пониженным давлением для удаления диоксана, нейтрализовали 1 н. НСl и экстрагировали EtOAc. Объединенные органические экстракты промывали рассолом, сушили над безводным Na2SO4 и концентрировали. Сырой продукт очищали экспресс-хроматографией (гексан/EtOAc → CH2Cl2/MeOH) с получением в виде белого твердого вещества в качестве чистого продукта ФМОК-3-МеОАррс (668 мг, 63%-ный выход из бис-БОК-гидантоина DDD). 1H-ЯМР (CDCl3): 7,83 (d, 2Н), 7,72 (d, 2H), 7,41 (td, 2Н), 7,34 (dt, 2H), 7,16 (t, 1H), 6,52 (d, 1H), 6,42 (s, 1H), 6,36 (d, 1H), 4,25 (m, 3H), 3,68 (s, 3H), 3,23-3,40 (m, 2H), 2,96 (t, 2H) и 1,86-2,18 (m, 4H). MCBP m/z: 495,1901 (M+Na), рассч. для C28H28N2O5Na 495,1896.
Пример 25
Получение ФМОК-1-амино-4-циклогексилциклогексан-1-карбоновой кислоты (ФМОК-Achc-OH)
Стадия 1
Смесь 4-циклогексилциклогексанона (3,00 г, 16,6 ммоля), цианида калия (1,63 г, 25,0 ммоля), карбоната аммония (9,59 г, 99,8 ммоля), этанола (75 мл) и воды (15 мл) в герметично закрытой толстостенной колбе выдерживали на масляной бане при 80° С в течение 15 ч. После охлаждения до комнатной температуры белую суспензию выливали в смесь воды со льдом и в течение пары часов перемешивали при комнатной температуре. В результате фильтрования и сушки на воздухе в виде белого твердого вещества получали гидантоин ЕЕЕ (6,10 г, в мокром состоянии, >100%-ный выход). 1H-ЯМР (ДМСО-d6, δ ): 10,52 (1Н, широкий, NH), 8,43 (1Н, широкий s, NH), 0,80-1,80 (20Н, m). MCHP (APCI): C14H22N2O2, рассч. 250; обнаруж. 249 (М-Н), 251 (М+Н).
Стадия 2
Смесь гидантоина ЕЕЕ (1,39 г, 5,55 ммоля) и 6 н. раствора гидроксида натрия (50 мл) в герметично закрытой толстостенной колбе выдерживали на масляной бане при 130° С в течение 2 дней. Реакционную смесь охлаждали на ледяной бане и с использованием концентрированной соляной кислоты нейтрализовали до рН ~7. Белую суспензию фильтровали и осадки промывали водой с получением сырой 1-амино-4-циклогексилциклогексан-1-карбоновой кислоты (48,3 г, мокрая и содержавшая неорганические соли, >100%-ный выход). MCHP (пучок электронов): C13H23NO2, рассч. 225; обнаруж. 226 (М+Н).
Стадия 3
Смесь сырой 1-амино-4-циклогексилциклогексан-1-карбоновой кислоты (48,3 г, 5,55 ммоля, теоретическое количество), триэтиламина (1,0 мл, 7,17 ммоля) и 9-флуоренилметилсукцинимидилкарбоната (ФМОК-OSu, 2,43 г, 7,20 ммоля) в ацетонитриле (75 мл) и воде (75 мл) при комнатной температуре перемешивали в течение 24 ч. Реакционную смесь концентрировали под вакуумом для удаления большей части ацетонитрила, подкисляли 10%-ным водным раствором лимонной кислоты до рН ~3 и белую эмульсию три раза экстрагировали метиленхлоридом. Объединенные органические слои промывали водой, рассолом и сушили над сульфатом магния. В результате фильтрования и концентрирования в виде масла получали сырой продукт, который очищали хроматографией в колонке (элюировали 1 → 5 → 8% метанола/метиленхлоридом) с получением ФМОК-1-амино-4-транс-циклогексилциклогексан-1-карбоновой кислоты (250 мг, 10%-ный выход за две стадии). МСВР (БУА): С28Н34NO4, (М+Н) рассч. 448,2488; обнаруж. 448, 2497.
Пример 26
Получение ФМОК-1-амино-4,4-дифенилциклогексан-1-карбоновой кислоты (ФМОК-Adpc-OH)
Стадия 1
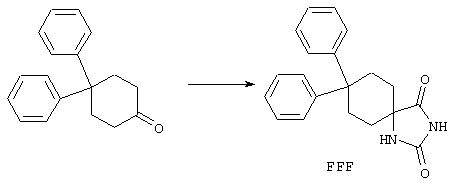
Смесь 4,4-дифенилциклогексанона (получен гидрогенизацией 4,4-дифенилциклогексанона в соответствии с методами, описанными в работе Freeman Р.К. и др. в J.Org.Chem., 1989, 54, 782-789) (1,55 г, 6,19 ммоля), цианида калия (0,65 г, 9,97 ммоля), карбоната аммония (3,60 г, 37,5 ммоля), этанола (48 мл) и воды (12 мл) в герметично закрытой толстостенной колбе выдерживали на масляной бане при 80° С в течение 24 ч. После охлаждения до комнатной температуры белую суспензию выливали в смесь воды со льдом и в течение пары часов перемешивали при комнатной температуре. В результате фильтрования и сушки на воздухе в виде белого твердого вещества получали гидантоин FFF (1,89 г, 95%-ный выход). 1H-ЯМР (ДМСО-d6, δ ): 10,57 (1Н, широкий, NH), 8,59 (1Н, широкий s, NH), 7,00-7,50 (10Н, m, фенил). МСНР (пучок электронов): C20H20N2O2, рассч. 320; обнаруж. 319 (М-Н).
Стадия 2
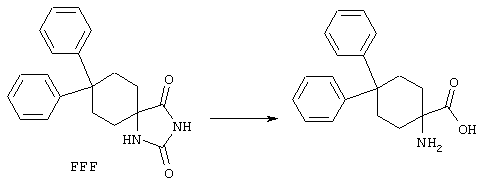
Смесь гидантоина FFF (1,88 г, 5,87 ммоля), моногидрата гидроксида бария (5,60 г, 29,6 ммоля) и воды (100 мл, избыточное разбавление) в герметично закрытой толстостенной колбе выдерживали на масляной бане при 105° С в течение 2 дней. Дополнительно добавляли моногидрата гидроксида бария (5,60 г, 29,6 ммоля) и смесь выдерживали на масляной бане при 105° С в течение дополнительных 24 ч. Реакционную смесь охлаждали до комнатной температуры и с использованием 4 н. серной кислоты при одновременном интенсивном перемешивании подкисляли до рН ~3. Суспензию в течение двух часов перемешивали на бане с кипящей водой и охлаждали до комнатной температуры. Белую суспензию фильтровали и осадки промывали водой. Объединенные фильтрат и промывные жидкости концентрировали под вакуумом до ~30 мл. Нейтрализацией концентрированным раствором гидроксида аммония получали белые осадки, которые отфильтровывали, промывали водой и сушили под вакуумом в течение ночи с получением в виде белого твердого вещества сырой 1-амино-4,4-дифенилциклогексан-1-карбоновой кислоты (0,52 г, 30%-ный выход). МСНР (пучок электронов): C19H21NO2, рассч. 295; обнаруж. 294 (М-Н), 296 (М+Н).
Стадия 3
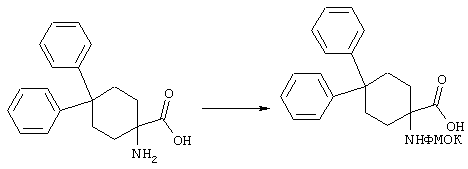
Смесь сырой 1-амино-4,4-дифенилциклогексан-1-карбоновой кислоты (510 мг, 1,73 ммоля), триэтиламина (0,37 мл, 2,65 ммоля) и 9-флуоренилметилсукцинимидилкарбоната (ФМОК-OSu, 880 мг, 2,61 ммоля) в ацетонитриле (25 мл) и воде (25 мл) при комнатной температуре перемешивали в течение ночи. Данные ТСХ-анализа реакционной смеси указывали на наличие исходного материала, аминокислоты. Добавляли 9-флуоренилметилсукцинимидилкарбоната (200 мг) и ацетонитрила (5 мл) и при комнатной температуре смесь перемешивали в течение дополнительных 24 ч. Реакционную смесь концентрировали под вакуумом для удаления большей части ацетонитрила, 10%-ным водным раствором лимонной кислоты подкисляли до рН ~3 и белую эмульсию три раза экстрагировали этилацетатом. Объединенные органические слои промывали водой, рассолом и сушили над сульфатом натрия. В результате фильтрования и концентрирования в виде масла получали сырой продукт, который очищали хроматографией в колонке (элюировали 1 → 4 → 8% метанола/метиленхлоридом) с получением в виде белого твердого вещества ФМОК-1-амино-4,4-дифенилциклогексан-1-карбоновой кислоты (350 мг, 39%-ный выход). МСВР (БУА): С34Н32NO4, (М+Н) рассч. 518,2331; обнаруж. 518, 231.
Пример 27
Получение ФМОК-1-амино-4-транс-трет-бутилциклогексан-1-карбоновой кислоты (ФМОК-Аbс-ОН)
Стадия 1
Смесь 4-трет-бутилциклогексанона (2,00 г, 13,0 ммоля), цианида калия (1,27 г, 19,5 ммоля), карбоната аммония (7,48 г, 77,8 ммоля), этанола (60 мл) и воды (12 мл) в герметично закрытой толстостенной колбе выдерживали на масляной бане при 80° С в течение 15 ч. После охлаждения до комнатной температуры белую суспензию выливали в смесь воды со льдом и в течение пары часов перемешивали при комнатной температуре. Фильтрованием в виде белого твердого вещества получали гидантоин GGG (2,78 г, 96%-ный выход), который в неочищенном виде использовали на следующей стадии. 1H-ЯМР (ДМСО-d6, δ ): 10,52 (1Н, широкий, NH), 8,50 (1Н, широкий s, NH), 0,81 (9Н, s, трет-Bu).
Стадия 2

Смесь гидантоина GGG (2,78 г, 12,4 ммоля), моногидрата гидроксида бария (11,74 г, 62,0 ммоля) и воды (50 мл) в герметично закрытой толстостенной колбе выдерживали на масляной бане при 120° С в течение 2 дней. Реакционную смесь охлаждали до комнатной температуры и с использованием 4 н. серной кислоты при одновременном интенсивном перемешивании подкисляли до рН ~3. Суспензию в течение одного часа перемешивали на бане с кипящей водой и охлаждали до комнатной температуры. Белую суспензию фильтровали и осадки промывали водой. Объединенные фильтрат и промывные жидкости концентрировали под вакуумом до ~30 мл. Нейтрализацией концентрированным раствором гидроксида аммония получали белые осадки, которые отфильтровывали, промывали водой и сушили под вакуумом в течение ночи с получением в виде белого твердого вещества 1-амино-4-транс-трет-бутилциклогексан-1-карбоновой кислоты (2,10 г, 85%-ный выход).
Стадия 3
Смесь сырой 1-амино-4-транс-трет-бутилциклогексил-1-карбоновой кислоты (2,10 г, 10,54 ммоля) и 9-флуоренилметилсукцинимидилкарбоната (ФМОК-OSu, 6,33 г, 7,20 ммоля) в диоксане (150 мл) и 10%-ном растворе карбоната натрия (120 мл) при комнатной температуре перемешивали в течение 24 ч. Реакционную смесь концентрировали под вакуумом для удаления большей части диоксана, 3 н. НСl подкисляли до рН ~3 и белую эмульсию дважды экстрагировали метиленхлоридом. Объединенные органические слои промывали водой и рассолом и сушили над сульфатом магния. В результате фильтрования и концентрирования получали сырой продукт, который очищали хроматографией в колонке (элюировали 1 → 4 → 5% метанола/метиленхлоридом) с получением ФМОК-1-амино-4-транс-трет-бутилциклогексан-1-карбоновой кислоты (1,42 г, 32%-ный выход). МСВР (БУА): С26Н32NO4, (M+H) рассч. 422,2331; обнаруж. 422,23.
Пример 28
Получение 3S,2S-ФМОК-(L)-бета-метил(Nin-Мез)триптофана, ФМОК-(L)-β -Ме(Nin-Мез)Тrр-ОН)
Стадия 1
В раствор транс-3-индолариловой кислоты (15,0 г, 0,08 моля) в 350 мл сухого ТГФ при -78° С медленно добавляли 125 мл 1,6 М н-BuLi в гексане. Образовавшуюся суспензию перемешивали при -78° С в течение 1 ч. Затем медленно добавляли раствор 2-мезитиленсульфонилхлорида (21,9 г, 0,1 моля) в 50 мл сухого ТГФ. Смесь нагревали до КТ и перемешивали в течение ночи. Смесь выливали в насыщенный водный раствор NH4Cl. Слои разделяли и водный слой экстрагировали EtOAc. Объединенный органический слой сушили над сульфатом натрия. Удалением растворителей получали 14,1 г сырого продукта ННН, который без дополнительной очистки использовали на следующей стадии. 1Н-ЯМР-анализ указывал на то, что он содержал 2,8 г 2-мезитиленсульфоновой кислоты. 1Н-ЯМР (CD3OD, δ ): 7,57 (s, 1H), 7,42 (d, 1H), 7,15-7,30 (m, 3H), 7,02 (s, 2H), 6,54 (d, 1H), 6,36 (d, 1H), 2,52 (s, 9H), 2,30 (s, 3H).
Стадия 2
В раствор N-2-мезитиленсульфонил-транс-3-индолариловой кислоты (3,26 г, 8,8 ммоля) в 140 мл сухого ТГФ при -78° С вводили 3,7 мл (3 экв.) триэтиламина и 2,17 мл (2 экв.) Ме3ССОСl. Образовавшуюся смесь при -78° С перемешивали в течение 15 мин и при 0° С в течение 1,5 ч. Смесь охлаждали до -78° С, после чего добавляли 5,5 мл 1,6 М н-BuLi в гексане, затем посредством полой иглы добавляли смесь (R)-4-фенил-2-оксазолидинона и н-BuLi в ТГФ (готовили добавлением при -78° С 11 мл 1,6 М н-BuLi в гексане в раствор (R)-4-фенил-2-оксазолидинона (2,87 г, 17,6 ммоля) в 70 мл сухого ТГФ). Образовавшуюся смесь при -78° С перемешивали в течение 2 ч и при КТ в течение ночи. Реакцию гасили водным раствором NH4Cl (100 мл). После удаления под вакуумом органических растворителей водный остаток экстрагировали EtOAc. Объединенный органический слой сушили над сульфатом натрия. В результате фильтрования и концентрирования получали сырой продукт, который очищали экспресс-хроматографией (EtOAc/гексан в соотношении 1:4) с получением в виде светло-коричневой смолы с 63%-ным выходом продукта III (2,86 г). LR-Электроспрей (LR-Electrospray): C29H26N2O5S, рассч. 514, обнаруж. m/z 515 (М+Н).
Стадия 3
В смесь CuBr· Me2S (0,84 г, 4,08 ммоля) и 5 мл диметилсульфида в 10 мл сухого ТГФ при -4° С вводили 1,36 мл 3 М CH3MgBr в диэтиловом эфире. После перемешивания в течение 10 мин добавляли упомянутый продукт (1,4 г, 2,72 ммоля) в 8 мл сухого ТГФ. Образовавшуюся смесь перемешивали при -4° С в течение 1 ч и при КТ в течение 6 ч. После охлаждения до -78° С в смесь добавляли 1,45 г (8,16 ммоля) N-бромсукцинимида в 15 мл сухого ТГФ. Смесь перемешивали при -78° С в течение 30 мин и при КТ в течение ночи. Смесь выливали в 100 мл рассола и экстрагировали EtOAc (2 порции по 100 мл). Органический слой сушили над сульфатом натрия. В результате фильтрования и концентрирования получали сырой продукт, который очищали экспресс-хроматографией (EtOAc/гексан в соотношении 1:4) с выделением в виде светло-коричневой смолы с 46%-ным выходом продукта JJJ (0,77 г). 1H-ЯМР (CDCl3, δ ): 7,63 (d, 1H), 7,47 (s. 1H), 7,20-7,37 (m, 8H), 6,98 (s, 2H), 6,16 (d, 1H), 5,13 (dd, 1H), 4,49 (t, 1H), 4,17 (dd, 1H), 3,75 (dt, 1H), 2,54 (s, 9H), 2,31 (s, 3H), 1,59 (d, 3H).
Стадия 4
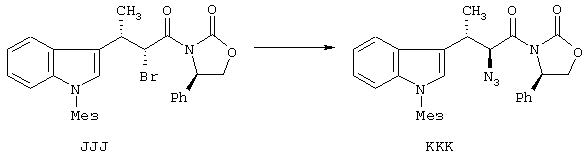
Упомянутый бромид JJJ (0,72 г, 1,18 ммоля) в 10 мл ацетонитрила смешивали с азидом тетра-н-бутиламмония (1,68 г, 5,9 ммоля) и азидом натрия (77 мг, 1,18 ммоля) и при КТ перемешивали в течение 6 ч. Смесь выливали в 100 мл водного раствора NH4Cl и экстрагировали EtOAc (2 порции по 100 мл). Органический слой сушили над сульфатом натрия. В результате фильтрования и концентрирования получали сырой продукт, который очищали экспресс-хроматографией (EtOAc/СН2Сl2/гексан в соотношении 1:2:5) с получением в виде светло-коричневой смолы с 82%-ным выходом продукта KKK (0,55 г). 1H-ЯМР (CDCl3, δ ): 7,65 (d, 1H), 7,59 (s, 1H), 7,18-7,29 (m, 8H), 6,90 (s, 2H), 5,54 (d, 1H), 5,50 (dd, 1H), 4,78 (t, 1H), 4,35 (dd, 1H), 3,62 (квинтет, 1H), 2,43 (s, 9H), 2,28 (s, 3H), 1,28 (d, 3H).
Стадия 5
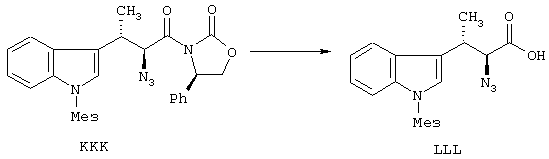
В смесь упомянутого азида ККК (0,55 г, 0,96 ммоля), воды (4 мл) и ТГФ (12 мл) при 0° С вводили 0,65 мл 30%-ного Н2О2, затем добавляли 48 мг (2 экв.) LiOH в 1 мл воды. Образовавшуюся смесь перемешивали при 0° С в течение 2 ч. Реакцию гасили Na2SO4 (1 г) в 6 мл воды. Смесь перемешивали при КТ в течение дополнительных 30 мин. После удаления органического растворителя водный раствор разбавляли 10 мл насыщенного раствора NаНСО3 и экстрагировали EtOAc (2 порции по 30 мл). В результате фильтрования и концентрирования получали сырой продукт, который очищали экспресс-хроматографией (HOAc/MeOH/EtOAc в соотношении 1:10:100) с получением в виде не совсем белого твердого вещества с 83%-ным выходом продукта LLL (0,34 г). LR-Electrospray: C21H22N4O4S, рассч. 426 обнаруж. m/z 425 (М-Н).
Стадия 6
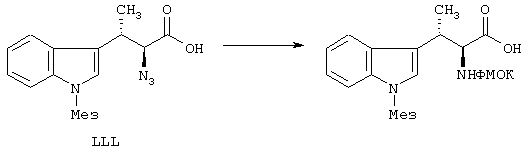
Упомянутую азидокислоту LLL (0,34 г, 0,8 ммоля) растворяли в 20 мл метанола. В раствор добавляли 170 мг 10%-ного Pd на угле. Образовавшуюся смесь в течение 3 ч при КТ перемешивали в атмосфере Н2 (баллон). После фильтрования и концентрирования сырой продукт растворяли в смешанном растворителе из ТГФ (12 мл) и воды (4 мл). В смесь добавляли NaHCO3 (254 мг, 3 ммоля) и ФМОК-OSu (540 мг, 1,6 ммоля). Образовавшуюся смесь перемешивали при КТ в течение 18 ч, разбавляли 30 мл насыщенного раствора NH4Cl и экстрагировали EtOAc (2 порции по 30 мл). В результате фильтрования и концентрирования получали сырой продукт, который очищали экспресс-хроматографией (НОАс/МеОН/EtOAc в соотношении 1:10:100) с выделением в виде не совсем белого твердого вещества с 50%-ным выходом в качестве продукта 3S,2S-ФМОК-(L)-бета-метил(Nin-Мез)триптофана (0,25 г). LR-Electrospray: C36H34N2O6S, рассч. 622; обнаруж. m/z 621 (М-Н).
Пример 29
Получение ФМОК-мостик-БГА смолы
Сополимерную бензгидриламин-стирольную сшитую 1% дивинилбензола смолу (10,0 г, 9,3 мэкв., с размерами частиц от 100 до 200 меш по стандарту ASTM, фирма Advanced ChemTech) подвергали набуханию в 100 мл CH2Cl2, отфильтровывали и последовательно промывали с использованием по 100 мл каждого из СН2Сl2, 6% ДИПЭА/СН2Сl2 (два раза) и CH2Cl2 (два раза). Эту смолу в течение 24 ч при комнатной температуре обрабатывали п-[(R,S)-α -[1-(9Н-флуорен-9-ил)метоксиформамидо]-2,4-диметоксибензил]феноксиуксусной кислотой (ФМОК-мостик) (7,01 г, 13,0 ммоля), N-гидроксибензотриазолом (2,16 г, 16,0 ммоля) и диизопропилкарбодиимидом (2,04 мл, 13,0 ммоля) в 100 мл 25% ДМФ/СН2Сl2. Смолу отфильтровывали и последовательно промывали с использованием по 100 мл каждого из СН2Сl2 (два раза), изопропанол (два раза), ДМФ и CH2Cl2 (три раза). Нингидриновый тест Kaiser был отрицательным. Эту смолу сушили под вакуумом с получением 16,12 г ФМОК-мостик-БГА смолы. У части этой смолы (3,5 мг) удаляли защитную ФМОК группу, и количественный УФ-анализ указывал на содержание 0,56 ммоля/г.
Пример 30
Получение Ac-Nle-цикло(Asp-Lys)-Asp-His-(D)Phe-Arg-Trp-Lys-NH2
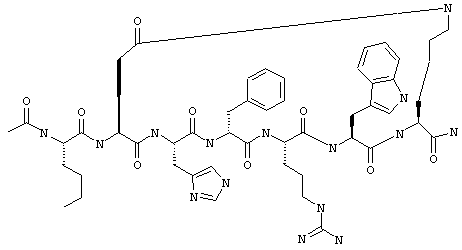
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Семь циклов реакций конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-His (Trt) (600 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Nle (430, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали CH2Сl2 (три раза) и в течение 30 мин обрабатывали 1 мл уксусного ангидрида в 6% ДИПЭА/СН2Сl2. Эту смолу отфильтровывали и последовательно промывали с использованием по 50 мл каждого из СН2Сl2 (два раза), изопропанол и CH2Cl2 (три раза). Эту смолу сушили под вакуумом с получением 1,2 г Ас-гептапептидной смолы. Ас-гептапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Эту смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали, и сырой линейный продукт сушили под вакуумом с получением 250 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
250 мг сырых линейных пептидов растворяли в 250 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 600 мкл N-метилморфолина. Добавляли 300 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac C18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2O, буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции выделяли с помощью аналитической ВЭЖХ собранных фракций, объединяли и лиофилизировали с получением 60 мг (15%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C50H69N15O9, рассч. 1024; обнаруж. m/z 1025 (М+Н).
Пример 31
Получение Penta-цикло(Asp-Lys)-Asp-Apc-(D)Phe-Arg-Trp-Lys-NH2
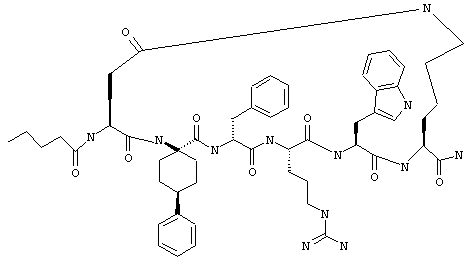
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Арс (550 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали CH2Cl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/СН2Сl2. Эту смолу отфильтровывали и последовательно промывали 50 мл каждого из CH2Cl2 (два раза), изопропанол, CH2Cl2 (три раза). Смолу сушили под вакуумом с получением 1,0 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Эту смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O и повторно центрифугировали, и сырой линейный продукт сушили под вакуумом с получением 220 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации. 220 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ, а образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac C18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2О, буфер Б: 0,1% ТФК/СН3CN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции выделяли с помощью аналитической ВЭЖХ собранных фракций, объединяли и лиофилизировали с получением 53 мг (13%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C54H72N12O8, рассч. 1017, обнаруж. m/z 1018 (М+Н).
Пример 32
Получение Penta-цикло(Asp-Lys)-Asp-Apc-(D)Phe-Arg-(2)Nal-Lys-NH2
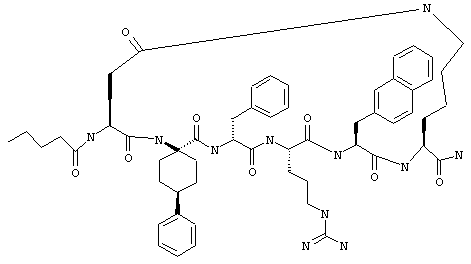
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(2)Наl (530 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Арс (550 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали CH2Cl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/СН2Сl2. Эту смолу отфильтровывали и последовательно промывали 50 мл каждого из CH2Cl2 (два раза), изопропанол и СН2Сl2 (три раза). Смолу сушили под вакуумом с получением 1,1 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Эту смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 220 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
240 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac C18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2O, буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. С помощью аналитической ВЭЖХ собранных фракций соответствовавшие основному пику фракции выделяли, объединяли и лиофилизировали с получением 55 мг (14%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C56H73N11O8, рассч. 1028, обнаруж. m/z 1029 (М+Н).
Пример 33
Получение Penta-цикло(Asp-Lys)-Asp-Apc-(D)Phe-Arg-N-метил(2)Nal-Lys-NH2
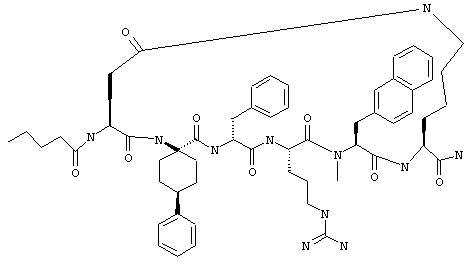
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Два цикла реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФMOK-(2)Nal (530 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля).
После удаления ФМОК у остатка 2-Nal полученный амин с использованием 2-нитробензолсульфонилхлорида (5 экв., 426 мг, 1,93 ммоля) и ДИПЭА (5 экв.) в качестве основания в ДМФ превращали в его 2-нитробензолсульфонильное производное. Промывки осуществляли с использованием ДМФ (6 порций по 30 мл) с последующим CH2Cl2 (3 порции по 30 мл) и смолу сушили под вакуумом. С использованием трифенилфосфина (5 экв., 505 мг, 1,93 ммоля), N,N-диэтилазодикарбоксилата (5 экв., 303 мкл, 1,93 ммоля) и метанола (10 экв., 156 мкл, 3,85 ммоля) в ТГФ полученный сульфонамид подвергали метилированию.
Промывки осуществляли с использованием ТГФ (6 порций по 30 мл), а затем СН2Сl2 (5 порций по 30 мл) и смолу сушили под вакуумом. Далее с использованием 1,8-диазабицикло[5.4.0]ундец-7-ена (3 экв., 173 мкл, 1,16 ммоля) и 2-меркаптоэтанола (5 экв., 135 мкл, 1,93 ммоля) в ДМФ удаляли 2-нитробензолсульфонильную группу. Промывки осуществляли с использованием ДМФ (3 порции по 30 мл), изопропанола (3 порции по 30 мл), после чего диэтилового эфира (3 порции по 30 мл) и смолу сушили под вакуумом. Полученный остаток N-Me-2-Nal использовали в четырех циклах реакций конденсации, причем каждый цикл осуществляли с применением одного из следующих сочетаний: ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Арс (550 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали СH2Сl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/СН2Сl2. Эту смолу отфильтровывали и промывали последовательно с использованием по 50 мл каждого из CH2Cl2 (два раза), изопропанол и CH2Cl2 (три раза). Эту смолу сушили под вакуумом с получением 1,2 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Эту смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 235 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
235 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac C18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2O, буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 43 мг (10%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C57H75N11O9, рассч. 1042, обнаруж. m/z 1043 (М+Н).
Пример 34
Получение цикло(янтарная кислота-Lys)янтарная кислота-Арс-(D)Рhе-Аrg-Trp-Lys-NH2
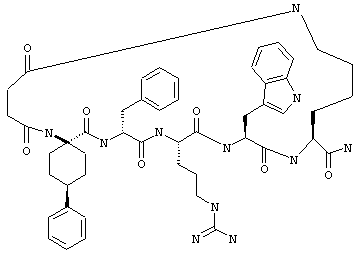
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Арс (550 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля) и янтарного ангидрида (600 мг, 6 ммоля) в ДМФ с 1,1 мл ДИПЭА.
Смолу отфильтровывали и последовательно промывали 50 мл каждого из CH2Cl2 (два раза), изопропанол и CH2Cl2 (три раза). Эту смолу сушили под вакуумом с получением 1,0 г пентапептидной смолы. Пентапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Эту смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O и повторно центрифугировали, сырой линейный продукт сушили под вакуумом с получением 220 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
220 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac С18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2О, буфер Б: 0,1% ТФК/CH3CN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 40 мг (11%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C49H63N11O7, рассч. 918, обнаруж. m/z 919 (М+Н).
Пример 35
Получение цикло(малеиновая кислота-Lys)малеиновая кислота-Арс-(D)Рhе-Arg-Trp-Lys-NH2
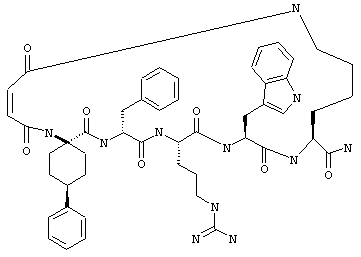
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Арс (550 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), малеиновый ангидрид (600 мг, 6 ммоля) в ДМФ с добавлением ГОБТ (800 мг, 6 ммоля), без ДИПЭА. Смолу отфильтровывали и последовательно промывали 50 мл каждого из CH2Cl2 (два раза), изопропанол и CH2Cl2 (три раза). Эту смолу сушили под вакуумом с получением 1,0 г пентапептидной смолы. Пентапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Эту смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой продукт сушили под вакуумом с получением 230 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
230 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac C18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2О, буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 38 мг (11%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray С49Н61N11O7, рассч. 916, обнаруж. m/z 917 (М+Н).
Пример 36
Получение цикло(фталевая кислота-Lys)фталевая кислота-Apc-(D)Phe-Arg-Trp-Lys-NH2
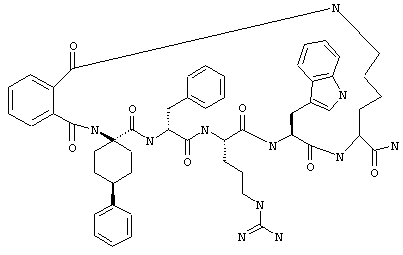
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Арс (550 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), фталевый ангидрид (660 мг, 6 ммоля) в ДМФ с 1,1 мл ДИПЭА.
Смолу отфильтровывали и последовательно промывали 50 мл каждого из CH2Cl2 (два раза), изопропанол и СН2Сl2 (три раза). Эту смолу сушили под вакуумом с получением 1,0 г пентапептидной смолы. Эту пентапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O и повторно центрифугировали, сырой линейный продукт сушили под вакуумом с получением 220 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
220 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac C18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2О, буфер Б: 0,1% ТФК/CH3CN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 35 мг (10%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C53H63N11O7, рассч. 966, обнаруж. m/z 967 (М+Н).
Пример 37
Получение Penta-цикло(Asp-Lys)-Asp-4-OHApc-(D)Phe-Arg-Trp-Lys-NH2
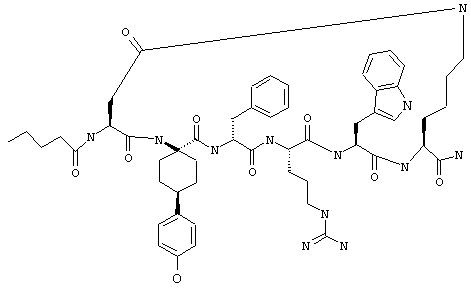
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-4-ОНАрс (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали CH2Cl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/СН2Сl2. Эту смолу отфильтровывали и последовательно промывали 50 мл каждого из CH2Cl2 (два раза), изопропанол и CH2Cl2 (три раза). Смолу сушили под вакуумом с получением 1,1 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 225 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
225 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac C18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2О, буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 55 мг (13%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C54H72N12O9, рассч. 1033, обнаруж. m/z 1034 (М+Н).
Пример 38
Получение Penta-цикло(Asp-Lys)-Asp-4-Me-OApc-(D)Phe-Arg-Trp-Lys-NH2
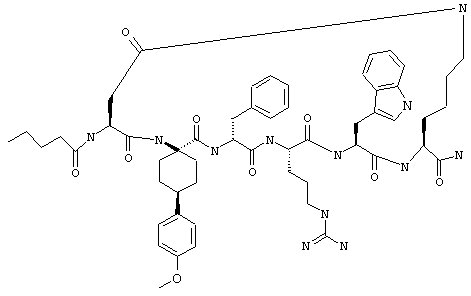
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), OMOK-(D)Phe (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-4-МеОАрс (600 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали CH2Cl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/СН2Сl2. Эту смолу отфильтровывали и последовательно промывали 50 мл каждого из СН2Сl2 (два раза), изопропанол и CH2Cl2 (три раза). Смолу сушили под вакуумом с получением 1,1 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали, сырой линейный продукт сушили под вакуумом с получением 235 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
235 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac C18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2О, буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 49 мг (12%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C55H74N12O9, рассч. 1047, обнаруж. m/z 1048 (М+Н).
Пример 39
Получение Penta-цикло(Asp-Lys)-Asp-4-EtO-Apc-(D)Phe-Arg-Trp-Lys-NH2
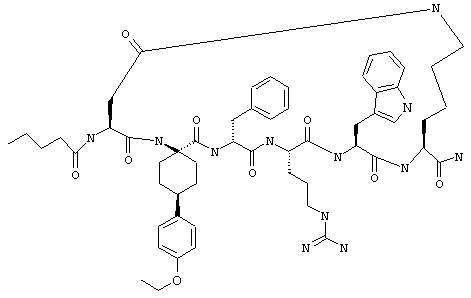
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-4-EtOApc (640 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали СН2Сl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/СН2Сl2. Эту смолу отфильтровывали и последовательно промывали 50 мл каждого из CH2Cl2 (два раза), изопропанол и CH2Cl2 (три раза). Смолу сушили под вакуумом с получением 1,2 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 235 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
235 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac C18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2O, буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 60 мг (14%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C56H76N12O9, рассч. 1061, обнаруж. m/z 1062 (М+Н).
Пример 40
Получение Penta-цикло(Asp-Lys)-Asp-4-изо-PrO-Apc-(D)Phe-Arg-Trp-Lys-NH2
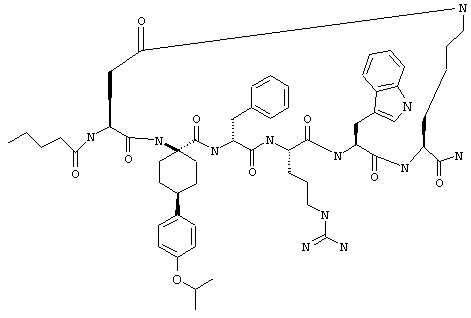
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФMOK-(D)Phe (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-4-изо-РrОАрс (660 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали CH2Cl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/СН2Сl2. Эту смолу отфильтровывали и последовательно промывали 50 мл каждого из CH2Cl2 (два раза), изопропанол и СН2Сl2 (три раза). Смолу сушили под вакуумом с получением 1,2 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 260 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
260 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac С18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2О, буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 63 мг (15%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C57H78N12O9, рассч. 1075, обнаруж. m/z 1076 (М+Н).
Пример 41
Получение Penta-цикло(Asp-Lys)-Asp-3-Me-OApc-(D)Phe-Arg-Trp-Lys-NH2
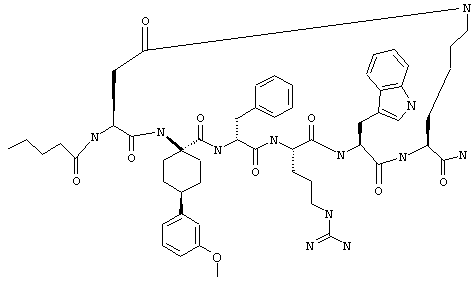
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-3-МеОАрс (600 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали CH2Cl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/СН2Сl2. Эту смолу отфильтровывали и последовательно промывали 50 мл каждого из CH2Cl2 (два раза), изопропанол и СН2Сl2 (три раза). Смолу сушили под вакуумом с получением 1,1 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире.
Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 235 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
235 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac C18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2O, буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 49 мг (12%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C55H74N12O9, рассч. 1047, обнаруж. m/z 1048 (М+Н).
Пример 42
Получение Penta-цикло(Asp-Lys)-Asp-4-ClApc-(D)Phe-Arg-Trp-Lys-NH2
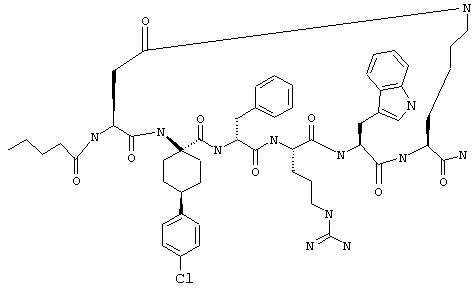
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве
агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФMOK-(D)Phe (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-4-ClApc (560 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали CH2Cl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/СН2Сl2. Эту смолу отфильтровывали и последовательно промывали 50 мл каждого из CH2Cl2 (два раза), изопропанол и CH2Cl2 (три раза). Смолу сушили под вакуумом с получением 1,0 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 230 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
230 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac С18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/H2O буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 49 мг (12%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C54H71N12O8Cl, рассч. 1051, обнаруж. m/z 1052 (М+Н).
Пример 43
Получение Penta-цикло(Asp-Lys)-Asp-4-Me-Apc-(D)Phe-Arg-Trp-Lys-NH2
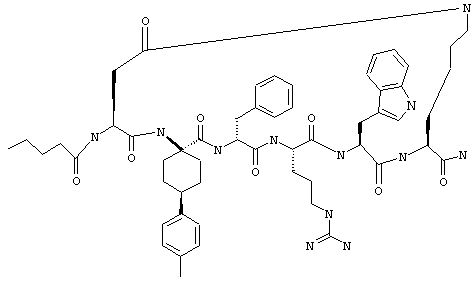
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-4-МеАрс (590 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали CH2Cl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/СН2Сl2. Эту смолу отфильтровывали и последовательно промывали 50 мл каждого из CH2Cl2 (два раза), изопропанол и CH2Cl2 (три раза). Смолу сушили под вакуумом с получением 1,2 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 240 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
240 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac C18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2О, буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 55 мг (14%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C55H74N12O8, рассч. 1031, обнаруж. m/z 1032 (М+Н).
Пример 44
Получение Penta-цикло(Glu-Lys)-Glu-Apc-(D)Phe-Arg-Trp-Lys-NH2
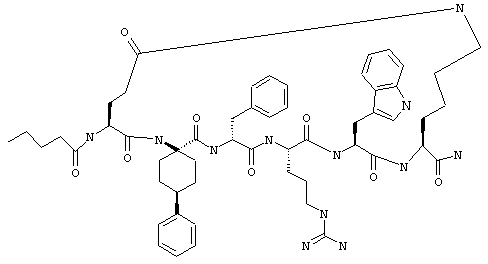
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФMOK-(D)Phe (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Арс (550 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Glu (OBut) (510 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали СН2Сl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/СН2Сl2. Эту смолу отфильтровывали и последовательно промывали 50 мл каждого из CH2Cl2 (два раза), изопропанол и CH2Cl2 (три раза). Смолу сушили под вакуумом с получением 1,1 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 255 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
255 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac C18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2О, буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 60 мг (15%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C55H74N12O8, рассч. 1031, обнаруж. m/z 1032 (М+Н).
Пример 45
Получение Penta-цикло(Asp-Orn)-Asp-Apc-(D)Phe-Arg-Trp-Orn-NH2
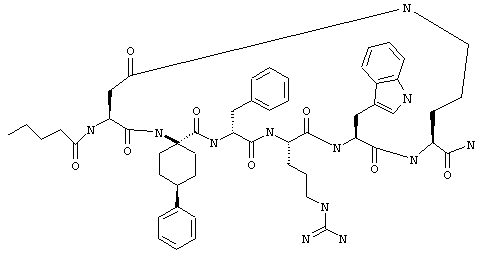
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Оrn (БОК) (550 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Арс (550 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали СН2Сl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/СН2Сl2. Эту смолу отфильтровывали и последовательно промывали 50 мл каждого из CH2Cl2 (два раза), изопропанол и CH2Cl2 (три раза). Смолу сушили под вакуумом с получением 1,15 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире.
Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 240 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
240 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac C18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/H2О, буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 53 мг (13%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C53H70N12O8, рассч. 1003, обнаруж. m/z 1004 (М+Н).
Пример 46
Получение Penta-цикло(Asp-Dbr)-Asp-Apc-(D)Phe-Arg-Trp-Dbr-NH2
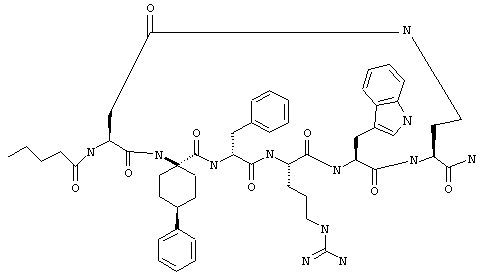
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Dbr (БОК) (540 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФMOK-(D)Phe (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Арс (550 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали CH2Cl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/СН2Сl2. Эту смолу отфильтровывали и последовательно промывали 50 мл каждого из СН2Сl2 (два раза), изопропанол и CH2Cl2 (три раза). Смолу сушили под вакуумом с получением 1,10 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 220 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
220 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac С 18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2О, буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 35 мг (9%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C52H68N12O8, рассч. 989, обнаруж. m/z 990 (М+Н).
Пример 47
Получение Penta-цикло(Asp-Dpr)-Asp-Apc-(D)Phe-Arg-Trp-Dpr-NH2
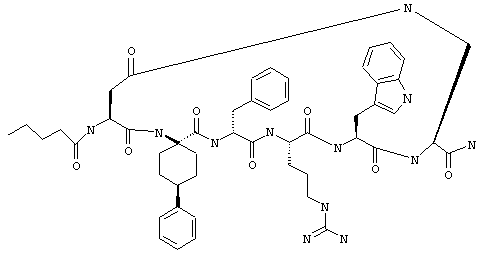
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Dpr (БОК) (530 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Арс (550 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали CH2Cl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/СН2Сl2. Эту смолу отфильтровывали и последовательно промывали 50 мл каждого из СН2Сl2 (два раза), изопропанол и CH2Cl2 (три раза). Смолу сушили под вакуумом с получением 1,0 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире.
Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 200 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
200 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac C18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2О, буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 30 мг (8%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C51H66N12O8, рассч. 975, обнаруж. m/z 976 (М+Н).
Пример 48
Получение Ac-цикло(Asp-Dpr)-Asp-Apc-(D)Phe-Arg-Trp-Dpr-NH2
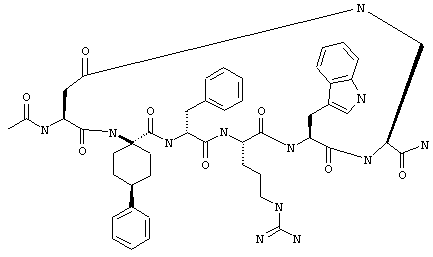
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Dpr (БОК) (530 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФMOK-(D)Phe (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Арс (550 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали CH2Cl2 (три раза) и в течение 30 мин обрабатывали 2 мл уксусного ангидрида в 6% ДИПЭА/СН2Сl2. Эту смолу отфильтровывали и последовательно промывали 50 мл каждого из CH2Cl2 (два раза), изопропанол и CH2Cl2 (три раза). Смолу сушили под вакуумом с получением 1,1 г ацетилгексапептидной смолы.
Ацетилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 200 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
210 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac C18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2О. буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 28 мг (8%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C48H60N12O8, рассч. 933, обнаруж. m/z 934 (М+Н).
Пример 49
Получение цикло(фталевая кислота-Dpr)фталевая кислота-Apc-(D)Phe-Arg-Trp-Dpr-NH2
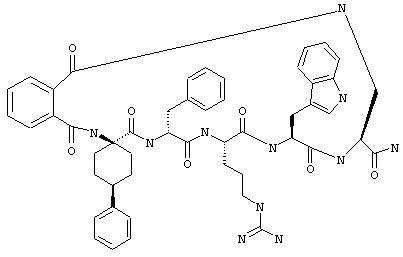
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Dpr (БОК) (530 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Арс (550 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), фталевый ангидрид (660 мг, 6 ммоля) в ДМФ с 1,1 мл ДИПЭА. Смолу отфильтровывали и последовательно промывали 50 мл каждого из СН2Сl2 (два раза), изопропанол и CH2Cl2 (три раза). Эту смолу сушили под вакуумом с получением 1,0 г пентапептидной смолы.
Пентапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 220 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
220 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac C18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2О, буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 30 мг (8%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C50H57N11O7, рассч. 924, обнаруж. m/z 925 (М+Н).
Пример 50
Получение цикло(янтарная кислота-Dpr)янтарная кислота-Apc-(D)Phe-Arg-Trp-Dpr-NH2
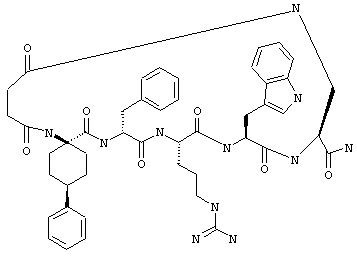
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Dpr (БОК) (530 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), OMOK-(D)Phe (480 мг. 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Арс (550 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля) и янтарный ангидрид (600 мг, 6 ммоля) в ДМФ с 1,1 мл ДИПЭА. Смолу отфильтровывали и последовательно промывали 50 мл каждого из CH2Cl2 (два раза), изопропанол и CH2Cl2 (три раза). Эту смолу сушили под вакуумом с получением 1,0 г пентапептидной смолы.
Пентапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 220 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
220 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac С 18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2О, буфер Б: 0,1% ТФК/CH3CN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 31 мг (8%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C46H57N11O7, рассч. 876, обнаруж. m/z 877 (М+Н).
Пример 51
Получение цикло(малеиновая кислота-Dpr)малеиновая кислота-Apc-(D)Phe-Arg-Trp-Dpr-NH2
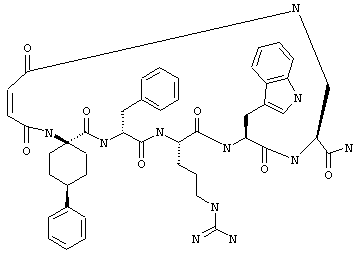
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Dpr (БОК) (530 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Арс (550 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), малеиновый ангидрид (600 мг, 6 ммолей) в ДМФ с добавлением ГОБТ (800 мг, 6 ммолей) и без ДИПЭА. Смолу отфильтровывали и последовательно промывали 50 мл каждого из CH2Cl2 (два раза), изопропанол и CH2Cl2 (три раза). Эту смолу сушили под вакуумом с получением 1,0 г пентапептидной смолы.
Пентапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой продукт сушили под вакуумом с получением 230 мг не совсем белого твердого вещества.
Сырой пептид без очистки подвергали циклизации.
230 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac C18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2O, буфер Б: 0,1% ТФК/CH3CN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 28 мг (8%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C46H55N11O7, рассч. 874, обнаруж. m/z 875 (М+Н).
Пример 52
Получение Penta-цикло(Asp-Lys)-Asp-Apc-(D)Phe-Cit-Trp-Lys-NH2

ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Cit (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Арс (550 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали CH2Cl2 (три раза) и обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/СН2Сl2 в течение 30 мин. Смолу отфильтровывали и последовательно промывали 50 мл каждого из СН2Сl2 (два раза), изопропанол и CH2Cl2 (три раза). Эту смолу сушили под вакуумом с получением 1,3 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 300 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
300 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac C18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2О, буфер Б: 0,1% ТФК/CH3CN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 80 мг (20%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C54H71N11O9, рассч. 1018, обнаруж. m/z 1019 (М+Н).
Пример 53
Получение Penta-цикло(Asp-Lys)-Asp-Apc-(D)Phe-Ala-Trp-Lys-NH2
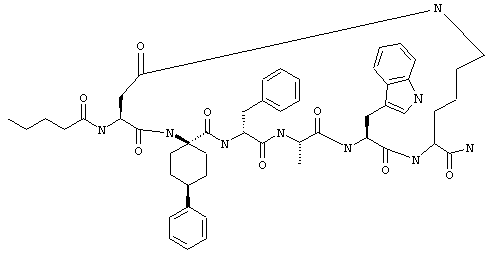
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Ala (380 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Арс (550 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали CH2Cl2 (три раза) и обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/СН2Сl2 в течение 30 мин. Смолу отфильтровывали и последовательно промывали 50 мл каждого из CH2Cl2 (два раза), изопропанол и CH2Cl2 (три раза). Эту смолу сушили под вакуумом с получением 1,4 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 330 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
330 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac C18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2О, буфер Б: 0,1% ТФК/CH3CN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 87 мг (20%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C51H65N9O8, рассч. 932, обнаруж. m/z 933 (М+Н).
Пример 54
Получение Ac-Nle-цикло(Cys-Cys)-Cys-Apc-(D)Phe-Arg-Trp-Cys-NH2
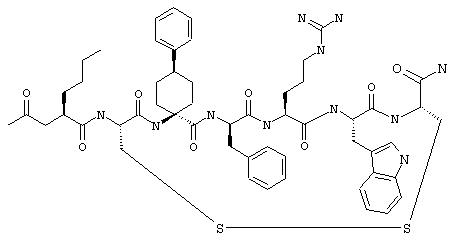
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Семь циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Cys (Trt) (710 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Арс (550 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Cys (Trt) (710 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Nle (430, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали CH2Cl2 (три раза) и в течение 30 мин обрабатывали 1 мл уксусного ангидрида в 6% ДИПЭА/СН2Сl2. Смолу отфильтровывали и последовательно промывали 50 мл каждого из CH2Cl2 (два раза), изопропанол и CH2Cl2 (три раза). Эту смолу сушили под вакуумом с получением 1,2 г Ас-гептапептидной смолы.
Ас-гептапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 250 мг не совсем белого твердого вещества.
Этот сырой линейный пептид очищали препаративной ВЭЖХ в колонке Vydac С18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2O, буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 45 мг очищенного линейного пептида.
Очищенный линейный пептид растворяли в 2 мл ДМСО, разбавляли 500 мл воды и с использованием NH4OH pH доводили до 8,0. Через раствор барботировали О2 и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 24 до 48 ч. Раствор лиофилизировали, материал растворяли в СН3СООН и подвергали препаративной ВЭЖХ в колонке Vydac C18 (2,5× 20 см), элюируя с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2О, буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции выделяли с помощью аналитической ВЭЖХ собранных фракций, объединяли и лиофилизировали с получением 20 мг (4,7%-ный выход) очищенного циклического пептида. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C53H70N12O8S2, рассч. 1067, обнаруж. m/z 1068 (М+Н).
Пример 55
Получение Penta-цикло(Asp-Lys)-Asp-(D,L)-Atc-(D)Phe-Arg-Trp-Lys-NH2
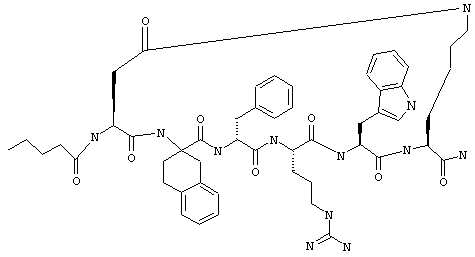
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих совокупностей: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D,L)-Аtс (510 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали CH2Cl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/СН2Сl2. Смолу отфильтровывали и последовательно промывали 50 мл каждого из СН2Сl2 (два раза), изопропанол и CH2Cl2 (три раза). Эту смолу сушили под вакуумом с получением 1,15 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2О, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 245 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
250 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac С18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2O, буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 55 мг (14%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C52H68N12O8, рассч. 989, обнаруж. m/z 990 (М+Н).
Пример 56
Получение Penta-(Asp-Lys)-Asp-5-BrAtc-(D)Phe-Arg-Trp-Lys-NH2 (пик 1)
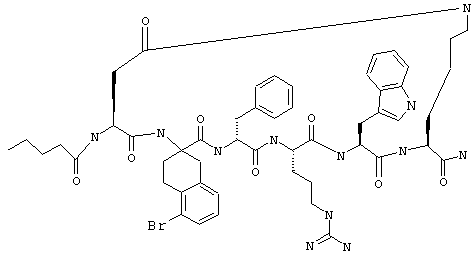
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих совокупностей: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФMOK-5-Br-(D,L)Atc (620 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали СН2Сl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/СН2Сl2. Смолу отфильтровывали и последовательно промывали 50 мл каждого из СН2Сl2 (два раза), изопропанол и CH2Cl2 (три раза). Эту смолу сушили под вакуумом с получением 1,1 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 240 мкл анизола и 10 мл трифторуксусной кислоты. Эту смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 240 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
240 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac С18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2О, буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Фракции, которые соответствовали первому основному пику, с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 26 мг (6%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C52H67N12O8Br, рассч. 1068, обнаруж. m/z 1069 (М+Н).
Пример 57
Получение Penta-(Asp-Lys)-Asp-5-BrAtc-(D)Phe-Arg-Trp-Lys-NH2 (пик 2)
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФMOK-(D)Phe (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-5-Br-(D,L)Atc (620 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали СН2Сl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/СН2Сl2. Смолу отфильтровывали и последовательно промывали 50 мл каждого из СН2Сl2 (два раза), изопропанол и СН2Сl2 (три раза). Эту смолу сушили под вакуумом с получением 1,1 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 240 мкл анизола и 10 мл трифторуксусной кислоты. Эту смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 240 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
240 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac C18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2О, буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Фракции, которые соответствовали второму основному пику, с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 20 мг (5%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C52H67N12O8Br, рассч. 1068, обнаруж. m/z 1069 (М+Н).
Пример 58
Получение Penta-(Asp-Lys)-Asp-5-ClAtc-(D)Phe-Arg-Trp-Lys-NH2 (пик 1)
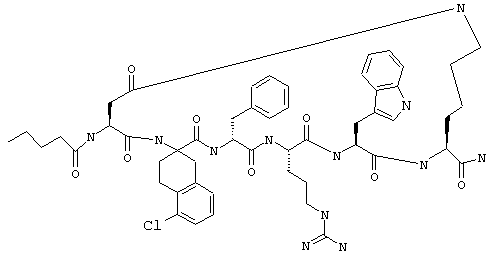
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-5-Cl(D,L)Atc (560 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали СН2Сl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/СН2Сl2. Смолу отфильтровывали и последовательно промывали 50 мл каждого из СН2Сl2 (два раза), изопропанол и СН2Сl2 (три раза). Эту смолу сушили под вакуумом с получением 1,2 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 250 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
250 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac С18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2O, буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 им. Фракции, которые соответствовали первому основному пику, с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 24 мг (6%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray С52Н67N12O8Сl, рассч. 1024, обнаруж. m/z 1025 (М+Н).
Пример 59
Получение Penta-(Asp-Lys)-Asp-5-ClAtc-(D)Phe-Arg-Trp-Lys-NH2 (пик 2)
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФMOK-(D)Phe (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-5-Cl-(D,L)Atc (560 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали СН2Сl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/СН2Сl2. Смолу отфильтровывали и последовательно промывали 50 мл каждого из СН2Сl2 (два раза), изопропанол и СН2Сl2 (три раза). Эту смолу сушили под вакуумом с получением 1,2 г пентилгексапептидной смолы. Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 250 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
250 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac C18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2O, буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Фракции, которые соответствовали второму основному пику, с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 20 мг (4%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C52H67N12O8Cl, рассч. 1024, обнаруж. m/z 1025 (М+Н).
Пример 60
Получение Penta-(Asp-Lys)-Asp-5-MeO-(D,L)Atc-(D)Phe-Arg-Trp-Lys-NH2
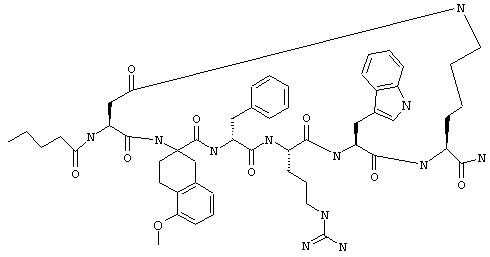
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-5-MeO-(D,L)Atc (600 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали СН2Сl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/СН2Сl2. Смолу отфильтровывали и последовательно промывали 50 мл каждого из CH2Cl2 (два раза), изопропанол и CH2Cl2 (три раза). Эту смолу сушили под вакуумом с получением 1,2 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 250 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
250 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac C18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2O, буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 55 мг (13%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C53H70N12O9, рассч. 1019, обнаруж. m/z 1020 (М+Н).
Пример 61
Получение Penta-(Asp-Lys)-Asp-5-EtO-(D,L)Atc-(D)Phe-Arg-Trp-Lys-NH2
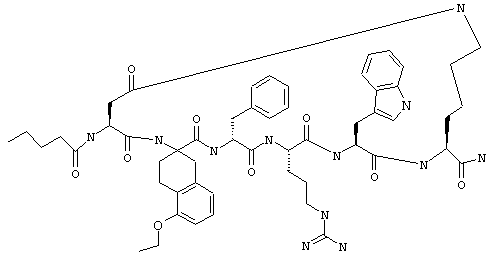
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), OMOK-(D)Phe (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-5-EtO-(D,L)Atc (620 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали СН2Сl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/СН2Сl2. Смолу отфильтровывали и последовательно промывали 50 мл каждого из СН2Сl2 (два раза), изопропанол и СН2Сl2 (три раза). Эту смолу сушили под вакуумом с получением 1,3 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 260 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
260 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac С18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2O, буфер Б: 0,1% ТФК/CH3CN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 58 мг (13%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C54H72N12O9, рассч. 1033, обнаруж. m/z 1034 (М+Н).
Пример 62
Получение Penta-(Asp-Lys)-Asp-5-изо-PrO-(D,L)Atc-(D)Phe-Arg-Trp-Lys-NH2
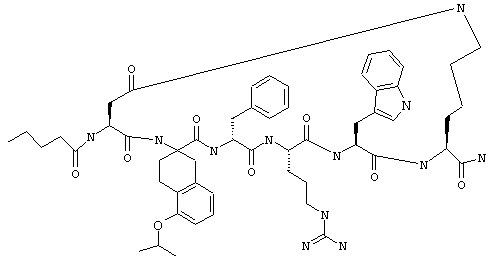
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-5-изо-PrO-(D,L)Atc (620 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали СН2Сl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/СН2Сl2. Смолу отфильтровывали и последовательно промывали 50 мл каждого из СН2Сl2 (два раза), изопропанол и СН2Сl2 (три раза). Эту смолу сушили под вакуумом с получением 1,2 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 250 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
250 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac С18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2O, буфер Б: 0,1% ТФК/CH3CN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 58 мг (13%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C55H74N12O9, рассч. 1047, обнаруж. m/z 1048 (М+Н).
Пример 63
Получение Penta-(Asp-Lys)-Asp-5-Me-(D,L)Atc-(D)Phe-Arg-Trp-Lys-NH2
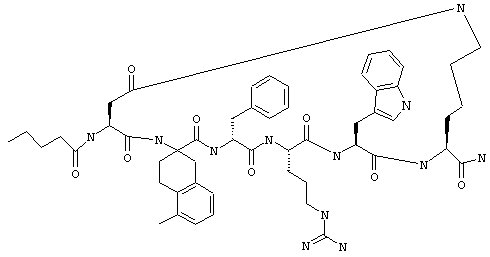
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1.
Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-5-Me-(D,L)Atc (590 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали СН2Сl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/СН2Сl2. Смолу отфильтровывали и последовательно промывали 50 мл каждого из СН2Сl2 (два раза), изопропанол и СН2Сl2 (три раза). Эту смолу сушили под вакуумом с получением 1,2 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 260 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
260 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac C18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2О, буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 62 мг (13%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C53H70N12O8, рассч. 1003, обнаруж. m/z 1004 (М+Н).
Пример 64
Получение Penta-(Asp-Lys)-Asp-5-Et-(D,L)Atc-(D)Phe-Arg-Trp-Lys-NH2
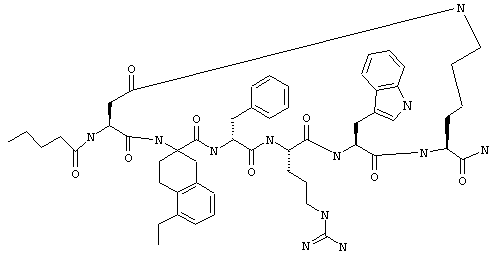
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФMOK-(D)Phe (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-5-Et-(D,L)Atc (600 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали СН2Сl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/СН2Сl2. Смолу отфильтровывали и последовательно промывали 50 мл каждого из СН2Сl2 (два раза), изопропанол и СН2Сl2 (три раза). Эту смолу сушили под вакуумом с получением 1,3 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 245 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
245 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac С18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2О, буфер Б: 0,1% ТФК/CH3CN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 55 мг (12%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C54H72N12O8, рассч. 1017, обнаруж. m/z 1018 (М+Н).
Пример 65
Получение Penta-(Asp-Lys)-Asp-5-изо-Pr-(D,L)Atc-(D)Phe-Arg-Trp-Lys-NH2
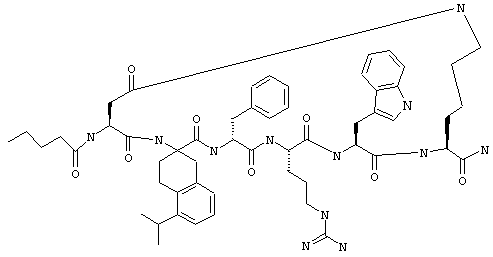
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-5-изо-Pr-(D,L)Atc (600 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали СН2Сl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/СН2Сl2. Смолу отфильтровывали и последовательно промывали 50 мл каждого из СН2Сl2 (два раза), изопропанол и СН2Сl2 (три раза). Эту смолу сушили под вакуумом с получением 1,2 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 245 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
245 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac C18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2О, буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 54 мг (13%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C55H74N12O8, рассч. 1031, обнаруж. m/z 1032 (М+Н).
Пример 66
Получение Penta-(Asp-Lys)-Asp-5-BrAtc-(D)Phe-Cit-Trp-Lys-NH2 (пик 1)
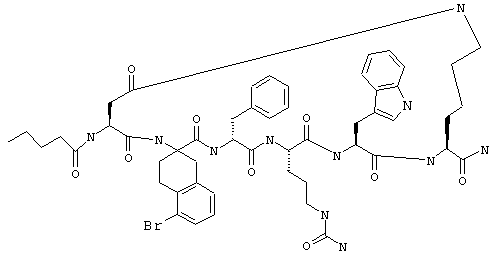
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Cit (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-5-Br-(D,L)Atc (620 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали CH2Cl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/СН2Сl2. Смолу отфильтровывали и последовательно промывали 50 мл каждого из CH2Cl2 (два раза), изопропанол и СН2Сl2 (три раза). Эту смолу сушили под вакуумом с получением 1,2 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 240 мкл анизола и 10 мл трифторуксусной кислоты. Эту смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 260 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
240 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac C18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2О, буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Фракции, которые соответствовали первому основному пику, с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 24 мг (5,6%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C52H66N11O9Br, рассч. 1069, обнаруж. m/z 1070 (М+Н).
Пример 67
Получение Penta-(Asp-Lys)-Asp-5-BrAtc-(D)Phe-Cit-Trp-Lys-NH2 (пик 2)

ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Cit (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-5-Br-(D,L)Atc (620 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали СН2Сl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/СН2Сl2. Смолу отфильтровывали и последовательно промывали 50 мл каждого из СН2Сl2 (два раза), изопропанол и СН2Сl2 (три раза). Эту смолу сушили под вакуумом с получением 1,1 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 240 мкл анизола и 10 мл трифторуксусной кислоты. Эту смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 240 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
240 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac C18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2О, буфер Б: 0,1% ТФК/CH3CN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Фракции, которые соответствовали второму основному пику, с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 22 мг (4,8%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C52H66N11O9Br, рассч. 1069, обнаруж. m/z 1070 (М+Н).
Пример 68
Получение Penta-(Asp-Lys)-Asp-5-ClAtc-(D)Phe-Cit-Trp-Lys-NH2 (пик 1)
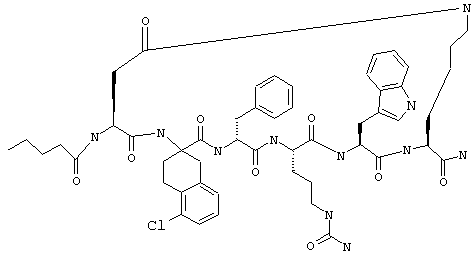
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Cit (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-5-Сl-(D,L)Atc (560 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали СН2Сl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/СН2Сl2. Смолу отфильтровывали и последовательно промывали 50 мл каждого из СН2Сl2 (два раза), изопропанол и СН2Сl2 (три раза). Эту смолу сушили под вакуумом с получением 1,2 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 245 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
245 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac С18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2О, буфер Б: 0,1% ТФК/CH3CN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Фракции, которые соответствовали первому основному пику, с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 22 мг (5,8%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C52H66N11O9Cl, рассч. 1024, обнаруж. m/z 1025 (М+Н).
Пример 69
Получение Penta-(Asp-Lys)-Asp-5-ClAtc-(D)Phe-Cit-Trp-Lys-NH2 (пик 2)
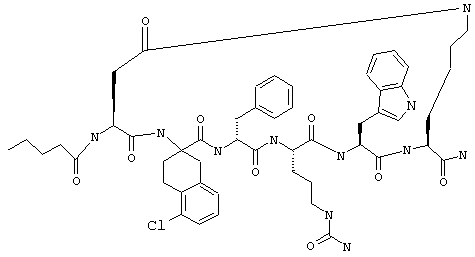
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1.
Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Cit (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-5-Сl-(D,L)Atc (560 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали СН2Сl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/СН2Сl2. Смолу отфильтровывали и последовательно промывали 50 мл каждого из СН2Сl2 (два раза), изопропанол и СН2Сl2 (три раза). Эту смолу сушили под вакуумом с получением 1,2 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 250 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
250 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac C18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2O, буфер Б: 0,1% ТФК/CH3CN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Фракции, которые соответствовали второму основному пику, с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 20 мг (5,4%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C52H66N11O9Cl, рассч. 1024, обнаруж. m/z 1025 (М+Н).
Пример 70
Получение Ac-Nle-цикло(Cys-Cys)-Cys-(D,L)Atc-(D)Phe-Arg-Trp-Cys-NH2
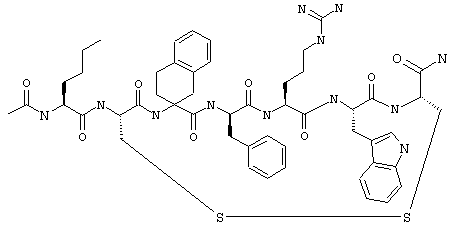
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Семь циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Cys (Trt) (710 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D,L)Atc (550 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Cys (Trt) (710 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Nle (430 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали СН2Сl2 (три раза) и в течение 30 мин обрабатывали 1 мл уксусного ангидрида в 6% ДИПЭА/СН2Сl2. Смолу отфильтровывали и последовательно промывали 50 мл каждого из СН2Сl2 (два раза), изопропанол и СН2Сl2 (три раза). Эту смолу сушили под вакуумом с получением 1,0 г Ас-гептапептидной смолы.
Ацетилгептапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 240 мг не совсем белого твердого вещества.
Этот сырой линейный пептид очищали препаративной ВЭЖХ в колонке Vydac C18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2O, буфер Б: 0,1% ТФК/CH3CN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 55 мг очищенного линейного пептида.
Очищенный линейный пептид растворяли в 2 мл ДМСО, разбавляли 500 мл воды и с использованием NH4OH рН доводили до 8,0. Через раствор барботировали О2 и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 24 до 48 ч. Раствор лиофилизировали, материал растворяли в СН3СООН и подвергали препаративной ВЭЖХ в колонке Vydac C18 (2,5× 20 см), элюируя с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2O, буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции выделяли с помощью аналитической ВЭЖХ собранных фракций, объединяли и лиофилизировали с получением 20 мг (5,0%-ный выход) очищенного циклического пептида. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C51H66N12O8S2, рассч. 1039, обнаруж. m/z 1040 (М+Н).
Пример 71
Получение Penta-цикло(Cys-Cys)-Cys-5-Br(D,L)Atc-(D)Phe-Arg-Trp-Cys-NH2
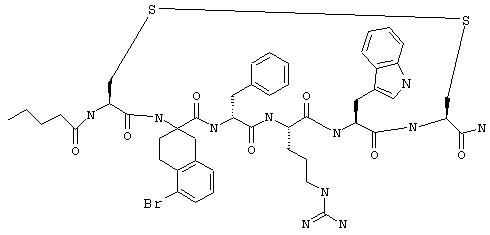
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Семь циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Cys (Trt) (710 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrp (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-5-BrAtc (620 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Cys (Trt) (710 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали СН2Сl2 (три раза) и в течение 30 мин обрабатывали 1 мл валерианового ангидрида в 6% ДИПЭА/СН2Сl2. Смолу отфильтровывали и последовательно промывали 50 мл каждого из СН2Сl2 (два раза), изопропанол и СН2Сl2 (три раза). Смолу сушили под вакуумом с получением 1,1 г Ас-гептапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 240 мг не совсем белого твердого вещества.
Этот сырой линейный пептид очищали препаративной ВЭЖХ в колонке Vydac С18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2O, буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 50 мг очищенного линейного пептида.
Очищенный линейный пептид растворяли в 2 мл ДМСО, разбавляли 500 мл воды и с использованием NH4OH рН доводили до 8,0. Через раствор барботировали О2 и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 24 до 48 ч. Раствор лиофилизировали, материал растворяли в СН3СООН и подвергали препаративной ВЭЖХ в колонке Vydac С18 (2,5× 20 см), элюируя с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2О, буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции выделяли с помощью аналитической ВЭЖХ собранных фракций, объединяли и лиофилизировали с получением 22 мг (5,2%-ный выход) очищенного циклического пептида. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C48H60N11O7S2Br, рассч. 1047, обнаруж. m/z 1048 (M+H).
Пример 72
Получение Penta-цикло(Asp-Lys)-Asp-Appc-(D)Phe-Arg-Trp-Lys-NH2
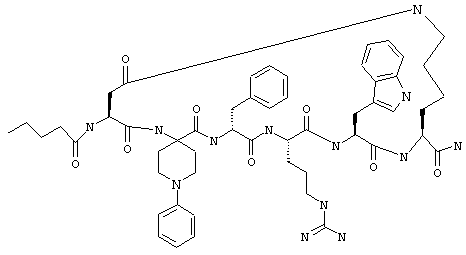
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Аррс (550 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали СН2Сl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/СН2Сl2. Смолу отфильтровывали и последовательно промывали 50 мл каждого из СН2Сl2 (два раза), изопропанол и СН2Сl2 (три раза). Смолу сушили под вакуумом с получением 1,2 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 245 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
245 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac С18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/H2O, буфер Б: 0,1% ТФК/CH3CN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 57 мг (14%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray: C53H71N13O8, рассч. 1018, обнаруж. m/z 1019 (М+Н).
Пример 73
Получение Penta-цикло(Asp-Lys)-Asp-2-Me-Appc-(D)Phe-Arg-Trp-Lys-NH2
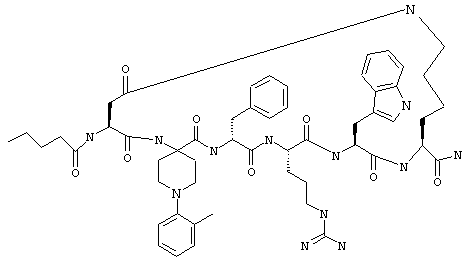
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве 25 агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-2-МеАррс (570 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали СН2Сl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/СН2Сl2. Смолу отфильтровывали и последовательно промывали 50 мл каждого из СН2Сl2 (два раза), изопропанол и СН2Сl2 (три раза). Смолу сушили под вакуумом с получением 1,2 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 250 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
250 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac С18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2О, буфер Б: 0,1% ТФК/CH3CN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 61 мг (15%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray: C54H73N13O8, рассч. 1032, обнаруж. m/z 1033 (М+Н).
Пример 74
Получение Penta-цикло(Asp-Lys)-Asp-2-изо-PrAppc-(D)Phe-Arg-Trp-Lys-NH2
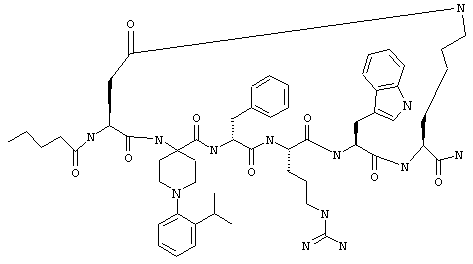
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-2-изо-РrАррс (600 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали СН2Сl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/СН2Сl2. Смолу отфильтровывали и последовательно промывали 50 мл каждого из СН2Сl2 (два раза), изопропанол и СН2Сl2 (три раза). Эту смолу сушили под вакуумом с получением 1,2 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире.
Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 245 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
245 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac C18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2О, буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 52 мг (14%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray: C56H77N13O8, рассч. 1060 обнаруж. m/z 1061 (М+Н).
Пример 75
Получение Penta-цикло(Asp-Lys)-Asp-3-Me-Appc-(D)Phe-Arg-Trp-Lys-NH2
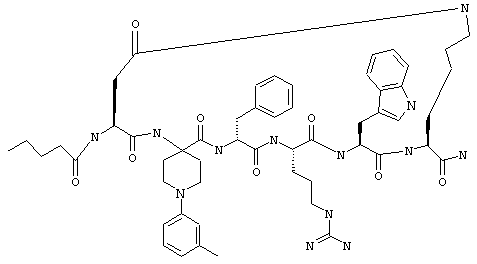
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-3-МеАррс (570 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали CH2Cl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/CH2Cl2. Смолу отфильтровывали и последовательно промывали 50 мл каждого из CH2Cl2 (два раза), изопропанол и CH2Cl2 (три раза). Смолу сушили под вакуумом с получением 1,2 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 248 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
248 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac С18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2О, буфер Б: 0,1% ТФК/CH3CN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 55 мг (14%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray: C54H73N13O8, рассч. 1032, обнаруж. m/z 1033 (М+Н).
Пример 76
Получение Penta-цикло(Asp-Lys)-Asp-4-Me-Appc-(D)Phe-Arg-Trp-Lys-NH2
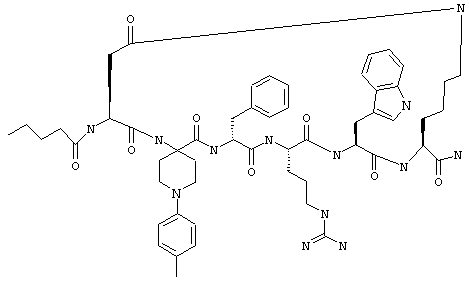
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-4-МеАррс (570 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали CH2Cl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/СН2Сl2. Смолу отфильтровывали и последовательно промывали 50 мл каждого из CH2Cl2 (два раза), изопропанол и CH2Cl2 (три раза). Смолу сушили под вакуумом с получением 1,2 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 254 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
254 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac C18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2O, буфер Б: 0,1% ТФК/CH3CN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 57 мг (14%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray: C54H73N13O8, рассч. 1032, обнаруж. m/z 1033 (М+Н).
Пример 77
Получение Penta-цикло(Asp-Lys)-Asp-4-ClAppc-(D)Phe-Arg-Trp-Lys-NH2
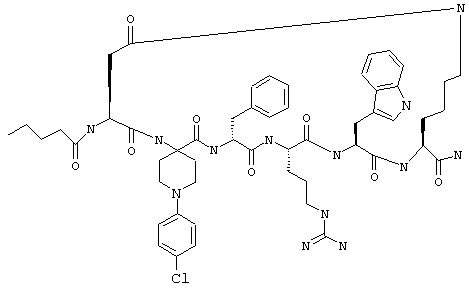
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-4-ClAppc (580 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали CH2Cl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/CH2Cl2. Смолу отфильтровывали и последовательно промывали 50 мл каждого из CH2Cl2 (два раза), изопропанол и CH2Cl2 (три раза). Смолу сушили под вакуумом с получением 1,2 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 250 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
250 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac C18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2О, буфер Б: 0,1% ТФК/CH3CN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм.
Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 55 мг (14%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray: С33Н70N13О8Сl, рассч. 1032, обнаруж. m/z 1033 (М+Н).
Пример 78
Получение Penta-цикло(Asp-Lys)-Asp-4-PhO-Appc-(D)Phe-Arg-Trp-Lys-NH2
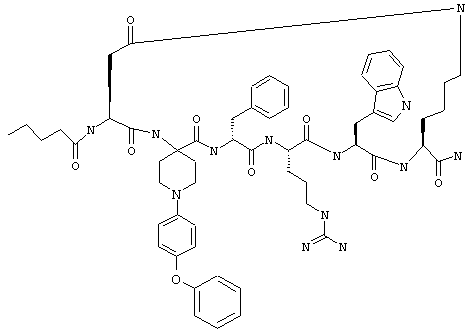
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-4-PhOAppc (650 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали CH2Cl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/CH2Cl2. Смолу отфильтровывали и последовательно промывали 50 мл каждого из CH2Cl2 (два раза), изопропанол и CH2Cl2 (три раза). Эту смолу сушили под вакуумом с получением 1,2 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 270 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
270 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac С18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2O, буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 58 мг (13%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray: C59H75N13O9, рассч. 1110, обнаруж. m/z 1111 (М+Н).
Пример 79
Получение Penta-(Asp-Lys)-Asp-3-MeO-Appc-(D)Phe-Arg-Trp-Lys-NH2

ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-3-МеОАррс (580 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали CH2Cl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/CH2Cl2. Смолу отфильтровывали и последовательно промывали 50 мл каждого из CH2Cl2 (два раза), изопропанол и CH2Cl2 (три раза). Эту смолу сушили под вакуумом с получением 1,2 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 250 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
250 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac C18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2O, буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 54 мг (13%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray: C54H73N13O9, рассч. 1048, обнаруж. m/z 1049 (М+Н).
Пример 80
Получение Penta-цикло(Asp-Lys)-Asp-4-ADpc-(D)Phe-Arg-Trp-Lys-NH2
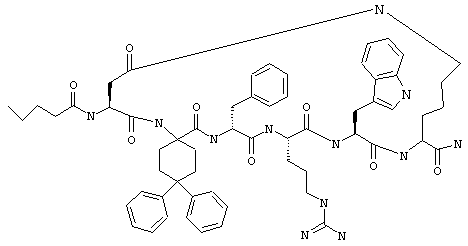
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-ADpc (620 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали CH2Cl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/CH2Cl2. Смолу отфильтровывали и последовательно промывали 50 мл каждого из CH2Cl2 (два раза), изопропанол и CH2Cl2 (три раза). Смолу сушили под вакуумом с получением 1,1 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 242 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
242 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac С18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2O, буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 48 мг (11%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray: C60H76N12O8, рассч. 1093, обнаруж. m/z 1094 (М+Н).
Пример 81
Получение Penta-цикло(Asp-Lys)-Asp-Achc-(D)Phe-Arg-Trp-Lys-NH2
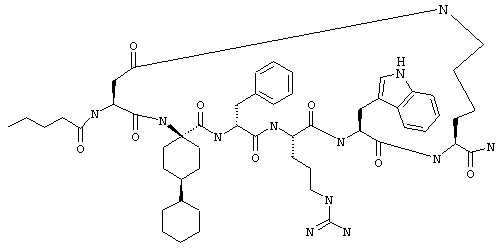
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1.
Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Achc (560 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали CH2Cl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/CH2Cl2. Смолу отфильтровывали и последовательно промывали 50 мл каждого из CH2Cl2 (два раза), изопропанол и CH2Cl2 (три раза). Смолу сушили под вакуумом с получением 1,1 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали, промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 250 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
250 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac С18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2О, буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 52 мг (13%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray: C54H78N12O8, рассч. 1023, обнаруж. m/z 1024 (М+Н).
Пример 82
Получение Penta-цикло(Asp-Lys)-Asp-Abc-(D)Phe-Arg-Trp-Lys-NH2
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Тrр (520 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Аbс (530 мг, 1,2 ммоля) и БТУГ (452 мг, 0,6 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали CH2Cl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/CH2Cl2. Смолу отфильтровывали и последовательно промывали 50 мл каждого из CH2Cl2 (два раза), изопропанол и CH2Cl2 (три раза). Смолу сушили под вакуумом с получением 13 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при комнатной температуре в течение 180 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл трифторуксусной кислоты. Далее смолу отфильтровывали,
промывали ~2 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 255 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
255 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac C18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2О, буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 58 мг (14%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C52H76N12O8, рассч. 997, обнаруж. m/z 998 (М+Н).
Пример 83
Получение Penta-цикло(Asp-Lys)-Asp-Apc-(D)Phe-Arg(2S,3S)-бета-метил-Trp-Lys-NH2
ФМОК-мостик-БГА смолу (720 мг, 0,4 ммоля) из примера 29 использовали в твердофазном синтезе в соответствии с представленным выше протоколом 1. Все реакции конденсации проводили с применением БТУГ в ДМФ в качестве агента реакции конденсации и ДИПЭА (3 экв.) в качестве основания. Шесть циклов реакции конденсации осуществляли с поочередным использованием в этих циклах соответственно следующих сочетаний: ФМОК-Lys (БОК) (565 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(2S,3S)-бета-метил(н-Мез)Тrр (616 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Arg (Пмх) (800 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-(D)Рhе (480 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Арс (550 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля), ФМОК-Asp (OBut) (500 мг, 1,2 ммоля) и БТУГ (452 мг, 1,2 ммоля). Пептидную смолу проводили через стадии с 1 по 5 протокола 1, промывали CH2Cl2 (три раза) и в течение 30 мин обрабатывали 2 мл валерианового ангидрида в 6% ДИПЭА/CH2Cl2. Смолу отфильтровывали и последовательно промывали 50 мл каждого из CH2Cl2 (два раза), изопропанол и CH2Cl2 (три раза). Эту смолу сушили под вакуумом с получением 1,0 г пентилгексапептидной смолы.
Пентилгексапептидную смолу при 0° С в течение 60 мин обрабатывали 100 мкл этандитиола, 100 мкл диметилсульфида, 250 мкл анизола и 10 мл HF. HF выпаривали, смолу промывали этилацетатом, отфильтровывали, промывали ~5 мл ТФК и фильтраты осаждали в охлажденном диэтиловом эфире. Осадки центрифугировали и эфирный слой декантировали. Остаток промывали двумя или тремя объемами Et2O, повторно центрифугировали и сырой линейный продукт сушили под вакуумом с получением 180 мг не совсем белого твердого вещества. Сырой пептид без очистки подвергали циклизации.
180 мг сырых линейных пептидов растворяли в 220 мл ДМФ и для достижения гарантированного значения рН 8,0 добавляли 500 мкл N-метилморфолина. Добавляли 280 мг БОФ и за циклизацией следили посредством ВЭЖХ. Циклизацию, как правило, завершали в течение от 18 до 24 ч. Для остановки реакции добавляли 10 мл воды, под вакуумом выпаривали ДМФ и образовавшуюся реакционную смесь очищали ВЭЖХ.
Этот сырой циклический материал очищали препаративной ВЭЖХ в колонке Vydac С18 (2,5× 20 см) и элюировали с линейным градиентом буфера Б от 20 до 60% (буфер А: 0,1% ТФК/Н2O, буфер Б: 0,1% ТФК/СН3СN) в течение 90 мин при скорости потока 8 мл/мин и с обнаружением при 280 нм. Соответствовавшие основному пику фракции с помощью аналитической ВЭЖХ собранных фракций выделяли, объединяли и лиофилизировали с получением 40 мг (10%-ный выход) белого аморфного порошка. Это соединение гомогенизировали посредством ВЭЖХ. LR-Electrospray C55H74N12O8, рассч. 1031, обнаруж. m/z 1032 (M+H).
Пример биологической активности
Пример А: испытание агонистов
Метод
Описание: клетки НЕК 295, трансфектированные либо рецептором МС-4, либо рецептором МС-1, выращивали в 96-луночных планшетах. Клетки стимулировали либо 100 нМ NDP-α -MSH, либо скринирующими соединениями. Циклический АМР экстрагировали из клеток и его концентрацию определяли, используя метод Biotrak-cAMP SPA. Агонисты идентифицировали как соединения, вызывающие увеличение образования сАМР.
Культура клеток: клетки НВК 295, трансфектированные либо рецептором 2 МС-4, либо рецептором МС-1, выращивали в сосудах на 75 см2 в D-MEM, в который добавляли 10% FCS (фетальная телячья сыворотка) и 500 мкг/мл G418. Клетки обрабатывали трипсином и расщепляли 1:3 в 96-луночном планшете с лунками с плоским дном, специально обработанными для культуры тканей. Клетки стимулировали при слиянии (от 2 до 4 дней).
Ответ сАМР: соединения, разбавленные серийно в 100%-ном ДМСО, далее разбавляли 1:200 (2,5 мкл раствора соединения + 500 мкл сред) в D-MEM, содержавшем 10% FBS и 0,1 мМ IBMX. Для нестимулированных клеток 2,5 мкл ДМСО прибавляли к 500 мкл сред. Для NDP-α -MSH стимулированных клеток 2,5 мкл 20 мкМ NDP-α -MSH в 100% ДМСО добавляли к 500 мкл сред (конечная конц. 100 нМ). Конечная концентрация ДМСО во всех лунках составляла 0,5%.
Примечание: эксперименты с каждым образцом дублировали на отдельных планшетах.
Культуральную среду отделяли от слившихся 96-луночных культуральных планшетов и замещали 200 мкл вышеописанных растворов в соответствующих лунках. Планшеты инкубировали в течение 1 ч при КТ. Среды удаляли, а планшеты промывали 1 раз 200 мкл ФБР (фосфатно-буферный раствор) на лунку. сАМР экстрагировали добавлением 60 мкл 70%-ного этанола (хранили в холодильнике). После 30 мин экстракции 10 мкл этанольного экстракта переносили в планшет для анализа на сАМР или образцы хранили при -20° С до проведения анализа на сАМР.
Анализ на сАМР: экстрагированные образцы и все реагенты, включенные в данный набор, нагревали до комнатной температуры. В 96-луночный планшет OptiPlate вводили по 10 мкл этанольного экстракта, 40 мкл буфера для анализа, 50 мкл [125I]сАМР, 50 мкл антисыворотки и 50 мкл SPA гранул на лунку. Суммарный объем в лунке после добавления составлял 200 мкл. Планшеты герметизировали и инкубировали в течение от 15 до 20 ч при комнатной температуре. Количество [125I]сАМР, связанного с гранулами SPA, определяли по величине радиоактивности за 2 мин, анализируя каждый планшет на счетчике типа Packard TopCountтм.
Примечание: каждый планшет содержал контрольные образцы для нестимулированных клеток и NDP-α -MSH для стимулированных клеток.
Результаты показаны в таблице 3.

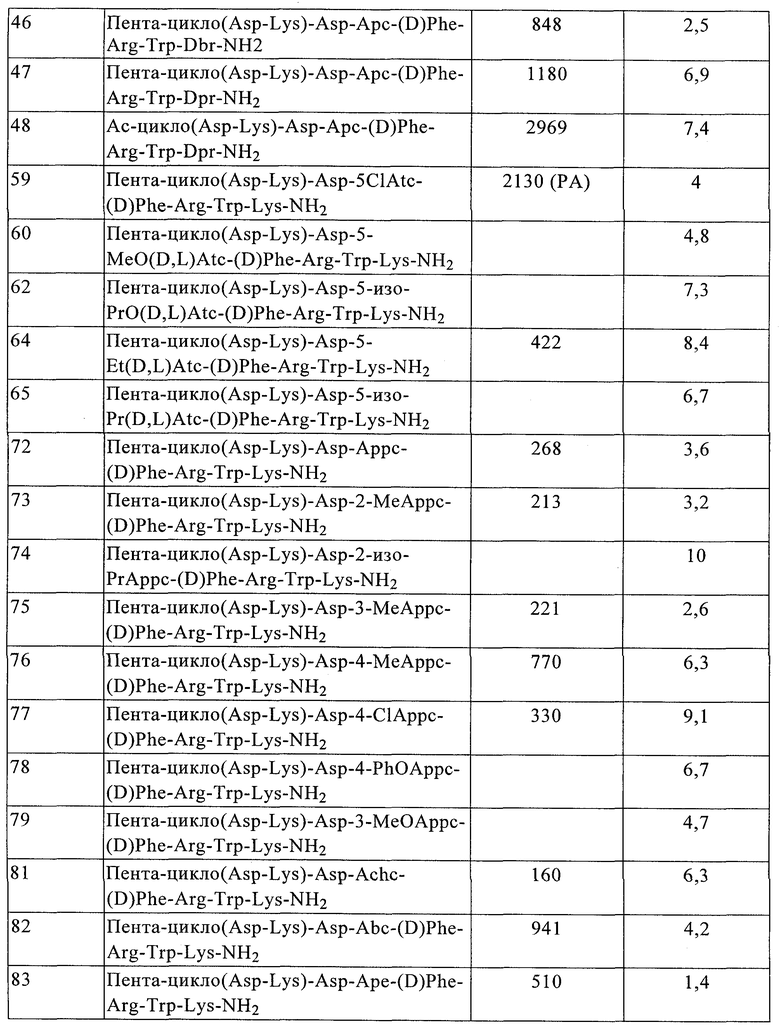
Пример Б: растворы для инъекций могут обладать следующим составом, мг:
Соединение формулы I 3,0
Желатина 150,0
Фенол 4,7
Вода для раствора для инъекций До 1,0 мл
| название | год | авторы | номер документа |
|---|---|---|---|
| ПЕПТИДЫ, ОБЛАДАЮЩИЕ АГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ РЕЦЕПТОРА НЕЙРОПЕПТИДА-2-(Y2R) | 2006 |
|
RU2383553C2 |
| ЦИКЛИЧЕСКИЕ АЗАПЕПТИДЫ С АНГИОГЕННЫМ ДЕЙСТВИЕМ | 1998 |
|
RU2188205C2 |
| ЦИКЛОПЕПТИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ, СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1994 |
|
RU2130030C1 |
| C26-СВЯЗАННЫЕ АНАЛОГИ РАПАМИЦИНА В КАЧЕСТВЕ ИНГИБИТОРОВ mTOR | 2019 |
|
RU2826559C2 |
| С40-, С28- И С-32-СВЯЗАННЫЕ АНАЛОГИ РАПАМИЦИНА В КАЧЕСТВЕ ИНГИБИТОРОВ MTOR | 2019 |
|
RU2805211C2 |
| ЦИКЛОПЕПТИДЫ ИЛИ ИХ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И СПОСОБ ИХ ПОЛУЧЕНИЯ, СПОСОБ ЛЕЧЕНИЯ | 1993 |
|
RU2129563C1 |
| ГЛИКОЗИЛИРОВАННЫЙ ПЕПТИД GLP-1 | 2009 |
|
RU2543157C2 |
| ЦИКЛОГЕКСАПЕПТИДЫ, ИХ СМЕСИ, СПОСОБ ИХ ПОЛУЧЕНИЯ | 1995 |
|
RU2163242C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПОЛИПЕПТИДОВ | 1990 |
|
RU2037498C1 |
| АНТАГОНИСТИЧЕСКИЕ АНАЛОГИ РИЛИЗИНГ-ГОРМОНА ГОРМОНА РОСТА (GH-RH), ИНГИБИРУЮЩИЕ ИНСУЛИНОПОДОБНЫЕ ФАКТОРЫ РОСТА (IGF-I И-II) | 1999 |
|
RU2235099C2 |
Описаны соединения формулы (I), в которой R1 и R12 совместно с X и Y образуют фенильное кольцо, Х обозначает С и Y обозначает С, или R1 обозначает водородный атом или группу формулы, указанную в п.1, R12 обозначает водородный атом, причем либо каждый из Х и Y обозначает С и связь между Х и Y является двойной связью, либо каждый из Х и Y обозначает СН, а связь между Х и Y является одинарной связью; R2 обозначает алкил, содержащий от 1 до 5 углеродных атомов, алкенил, содержащий от 2 до 5 углеродных атомов, или алкинил, содержащий от 2 до 5 углеродных атомов; R14 обозначает алкил, содержащий от 1 до 5 углеродных атомов; n обозначает 0 или 1, Q обозначает группу формулы, указанную в п.1, у которой каждый из R3, R4 и R5 независимо обозначает водородный атом, гало, алкил, содержащий от 1 до 4 углеродных атомов, гидрокси или алкоксирадикал, содержащий от 1 до 4 углеродных атомов, причем когда R4 не обозначает водородный атом, как R3, так и R5 обозначают водородный атом; R6 обозначает водородный атом, алкил, содержащий от 1 до 3 углеродных атомов, алкоксирадикал, содержащий от 1 до 3 углеродных атомов, фенокси или гало; каждый из R11 и R13 независимо обозначает водородный атом, алкил, содержащий 3 или 4 углеродных атома, циклоалкил, содержащий 5 или 6 углеродных атомов, или как R11, так и R13 обозначают фенил; R7 обозначает О или NH; R8 обозначает водородный атом или метил; R9 обозначает группу формулы, указанную в п.1, R10 обозначает водородный атом или метил; р обозначает 0 или 1; m обозначает 0, 1, 2 или 3; а Z обозначает группу формулы, указанную в п.1; R17 обозначает водородный атом или (низш.)алкил, и их фармацевтически приемлемые соли. Соединения по изобретению являются селективными агонистами в отношении меланокортина-4. Описаны также способ получения соединений и содержащие их фармацевтические композиции. 7 н. и 40 з.п. ф-лы, 3 табл.
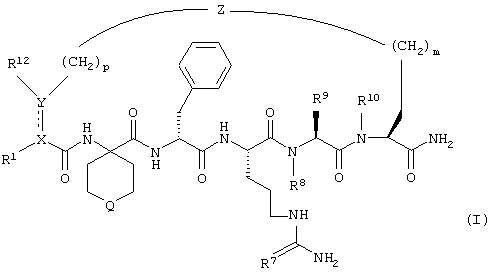
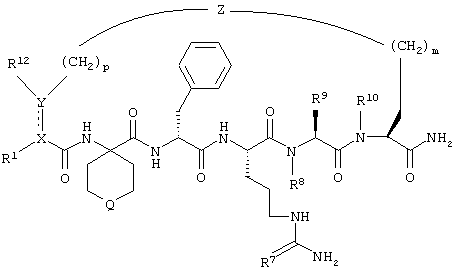
в которой R1 и R12 совместно с X и Y образуют фенильное кольцо, Х обозначает С и Y обозначает С; или
R1 обозначает водородный атом или группу

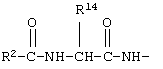
R12 обозначает водородный атом; причем либо каждый из Х и Y обозначает С и связь между Х и Y является двойной связью, либо каждый из Х и Y обозначает СН, а связь между Х и Y является одинарной связью;
R2 обозначает алкил, содержащий от 1 до 5 углеродных атомов, алкенил, содержащий от 2 до 5 углеродных атомов, или алкинил, содержащий от 2 до 5 углеродных атомов;
R14 обозначает алкил, содержащий от 1 до 5 углеродных атомов;
n обозначает 0 или 1;
Q обозначает группу
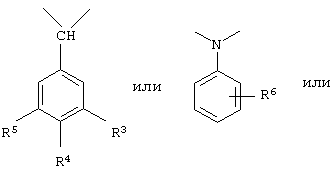
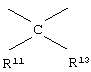
у которой каждый из R3, R4 и R5 независимо обозначает водородный атом, гало, алкил, содержащий от 1 до 4 углеродных атомов, гидрокси или алкоксирадикал, содержащий от 1 до 4 углеродных атомов, причем когда R4 не обозначает водородный атом, как R3, так и R5 обозначают водородный атом;
R6 обозначает водородный атом, алкил, содержащий от 1 до 3 углеродных атомов, алкоксирадикал, содержащий от 1 до 3 углеродных атомов, фенокси или гало;
каждый из R11 и R13 независимо обозначает водородный атом, алкил, содержащий 3 или 4 углеродных атома, циклоалкил, содержащий 5 или 6 углеродных атомов, или как R11, так и R13 обозначают фенил;
R7 обозначает О или NH;
R8 обозначает водородный атом или метил;
R9 обозначает группу
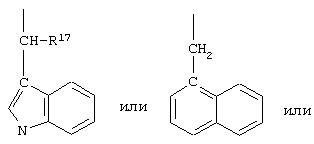
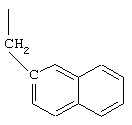
R10 обозначает водородный атом или метил;
р обозначает 0 или 1;
m обозначает 0, 1, 2 или 3;
Z обозначает группу

R17 обозначает водородный атом или (низш.)алкил,
и его фармацевтически приемлемые соли.
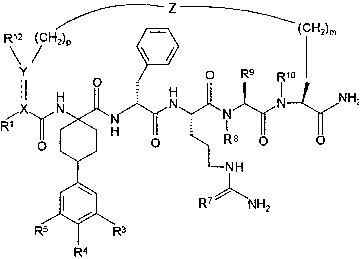
в которой R1 и R12 совместно с X и Y образуют фенильное кольцо, Х обозначает С, Y обозначает С или
R1 обозначает водородный атом или группу
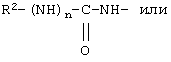
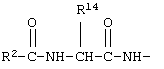
R12 обозначает водородный атом, причем либо каждый из Х и Y обозначает С и связь между Х и Y является двойной связью, либо каждый из Х и Y обозначает СH, а связь между Х и Y является одинарной связью;
R2 обозначает алкил, содержащий от 1 до 5 углеродных атомов, алкенил, содержащий от 2 до 5 углеродных атомов, или алкинил, содержащий от 2 до 5 углеродных атомов;
R14 обозначает алкил, содержащий от 1 до 5 углеродных атомов;
n обозначает 0 или 1;
R3, R4 и R5 каждый независимо обозначает водородный атом, гало, алкил, содержащий от 1 до 4 углеродных атомов, гидрокси или алкоксирадикал, содержащий от 1 до 4 углеродных атомов; причем когда R4 не обозначает водородный атом, как R3, так и R5 обозначают водородный атом;
R7 обозначает О или NH;
R8 обозначает водородный атом или метил;
R9 обозначает группу
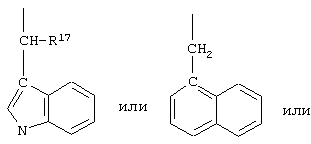
R10 обозначает водородный атом или метил;
р обозначает 0 или 1;
m обозначает 0, 1, 2 или 3;
Z обозначает группу
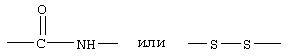
R17 обозначает водородный атом или (низш.)алкил,
и его фармацевтически приемлемые соли.
Z обозначает группу

R7 обозначает О;
R1 обозначает группу
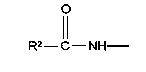
R2 обозначает алкил, содержащий от 1 до 5 углеродных атомов;
как R10, так и R12 обозначают водородный атом.
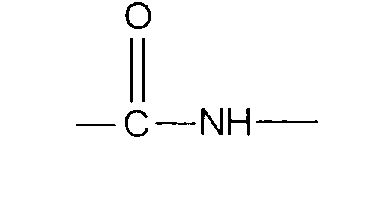
R7 обозначает NH;
R1 обозначает водородный атом или группу
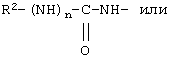
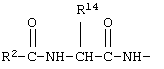
R2 обозначает алкил;
как R10, так и R12 обозначают водородный атом;
n и R14 имеют значения, указанные в п.2.
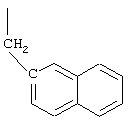
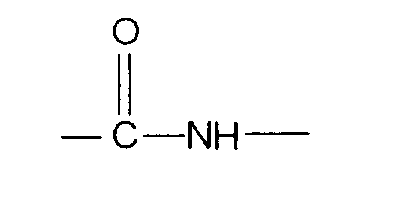
R7 обозначает NH;
R1 обозначает группу
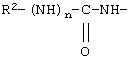
R2 обозначает алкил;
как R10, так и R12 обозначают водородный атом;
R9 обозначает группу
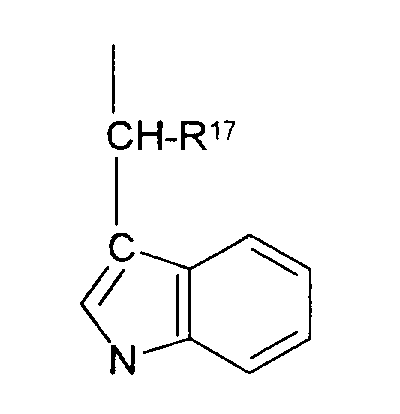
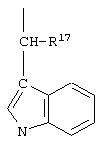
R17 имеет значения, указанные в п.2.
R1 обозначает группу
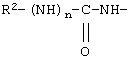
один из R3, R4 и R5 обозначает алкокси, каждый из остальных обозначает водородный атом;
n обозначает 0.
R7 обозначает NH;
R9 обозначает группу
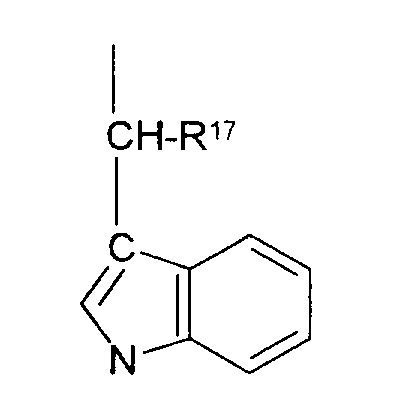
р обозначает 0;
R17 имеет значения, указанные в п.2.
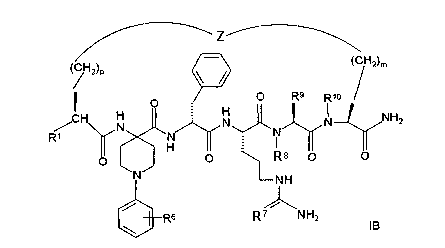
в которой R1 обозначает водородный атом или группу
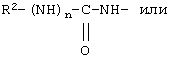
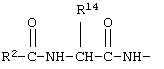
R2 обозначает алкил, содержащий от 1 до 5 углеродных атомов, алкенил, содержащий от 2 до 5 углеродных атомов, или алкинил, содержащий от 2 до 5 углеродных атомов;
R14 обозначает алкил, содержащий от 1 до 5 углеродных атомов;
n обозначает 0 или 1;
R6 обозначает водородный атом, алкил, содержащий от 1 до 3 углеродных атомов, алкоксирадикал, содержащий от 1 до 3 углеродных атомов, фенокси или гало;
R7 обозначает О или NH;
R8 обозначает водородный атом или метил;
R9 обозначает группу
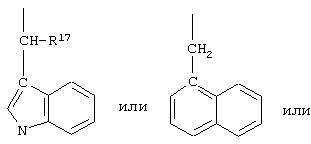
R10 обозначает водородный атом или метил;
р обозначает 0 или 1;
m обозначает 0, 1, 2 или 3, a Z обозначает группу
R17 обозначает водородный атом или (низш.)алкил,
и его фармацевтически приемлемые соли.
R7 обозначает NH;
R1 обозначает группу
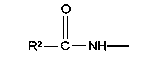
R2 обозначает алкил;
R8 и R10 каждый обозначает водородный атом;
R9 обозначает группу
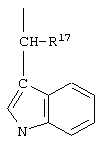
R17 имеет значения, указанные в п.2.
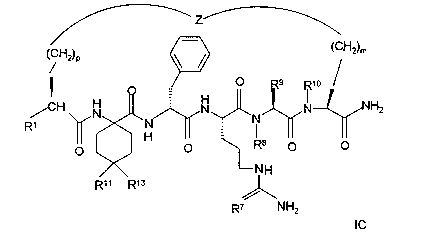
в которой R1 обозначает водородный атом или группу
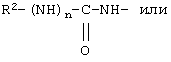
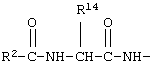
R2 обозначает алкил, содержащий от 1 до 5 углеродных атомов, алкенил, содержащий от 2 до 5 углеродных атомов, или алкинил, содержащий от 2 до 5 углеродных атомов;
R14 обозначает алкил, содержащий от 1 до 5 углеродных атомов;
n обозначает 0 или 1;
R11 и R13 каждый независимо обозначает водородный атом, алкил, содержащий 3 или 4 углеродных атома, или циклоалкил, содержащий 5 или 6 углеродных атомов, или как R11, так и R13 обозначают фенил;
R7 обозначает О или NH;
R8 обозначает водородный атом или метил;
R9 обозначает группу
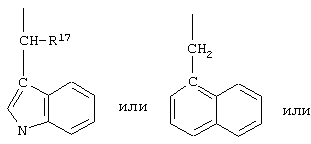
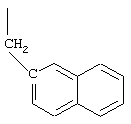
R10 обозначает водородный атом или метил;
р обозначает 0 или 1;
m обозначает 0, 1, 2 или 3;
Z обозначает группу
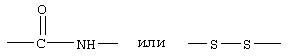
R17 обозначает водородный атом или (низш.)алкил,
и его фармацевтически приемлемые соли.

R7 обозначает NH;
R1 обозначает группу
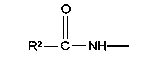
R2 обозначает алкил;
R8 и R10 каждый обозначает водородный атом;
R9 обозначает группу
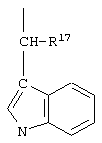
R17 обозначает водородный атом или (низш.)алкил.
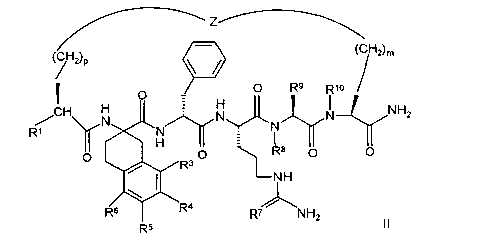
в которой R1 обозначает водородный атом или группу
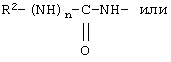
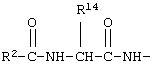
R2 обозначает алкил, содержащий от 1 до 5 углеродных атомов, алкенил, содержащий от 2 до 5 углеродных атомов, или алкинил, содержащий от 2 до 5 углеродных атомов;
R14 обозначает алкил, содержащий от 1 до 5 углеродных атомов;
n обозначает 0 или 1;
один из R3, R4, R5 и R6 обозначает водородный атом, гало, алкил, содержащий от 1 до 3 углеродных атомов, или алкоксирадикал, содержащий от 1 до 3 углеродных атомов, а каждый из остальных обозначает водородный атом;
R7 обозначает О или NH;
R8 обозначает водородный атом или метил;
R9 обозначает группу
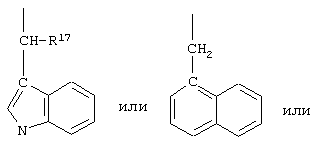
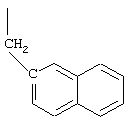
R10 обозначает водородный атом или метил;
р обозначает 0 или 1;
m обозначает 0, 1, 2 или 3;
Z обозначает группу

R17 обозначает водородный атом или (низш.)алкил,
и его фармацевтически приемлемые соли.

R1 обозначает группу
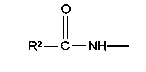
R2 обозначает алкил;
R3, R4, R5, R8 и R10 каждый обозначает водородный атом;
R6 обозначает водородный атом, гало, алкил, содержащий от 1 до 3 углеродных атомов, или алкоксирадикал, содержащий от 1 до 3 углеродных атомов;
R9 обозначает группу
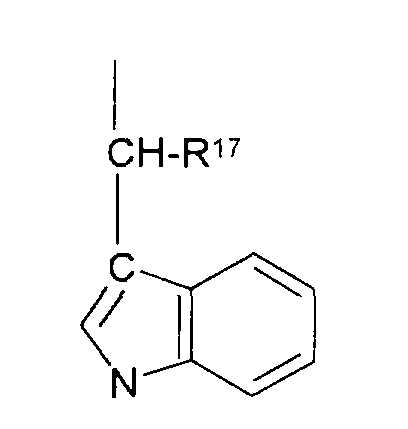
R17 имеет значения, указанные в п.31.
R1 обозначает группу
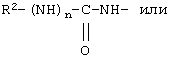
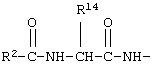
R3, R4, R5 и R10 каждый обозначает водородный атом;
R6 обозначает водородный атом или гало;
R7 обозначает NH;
R9 обозначает группу
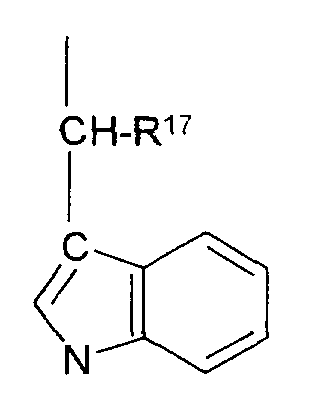
в которой R17 имеет указанные выше значения.
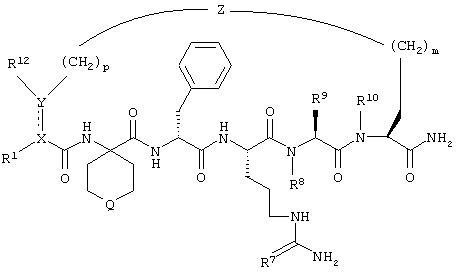
в которой R1-R12, m, p, Q, X, Y и Z имеют значения, указанные в пп.1-43, формированием лактамовой связи или дисульфидной связи по месту Z линейных пептидов-предшественников.
| ЦИКЛОПЕПТИДЫ ИЛИ ИХ СОЛИ, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И СПОСОБ ИХ ПОЛУЧЕНИЯ, СПОСОБ ЛЕЧЕНИЯ | 1993 |
|
RU2129563C1 |
| Одновальный, снабженный дробителем, торфяной пресс | 1919 |
|
SU261A1 |
| Трансляция, предназначенная для телефонирования быстропеременными токами | 1921 |
|
SU249A1 |
| NEUROSIENCE, 2000, V.20, no 9, 3442-3448. | |||

