Данное изобретение было осуществлено при частичной правительственной поддержке Медицинской Исследовательской Службы Департамента по делам ветеранов. Правительство имеет определенные права на данную заявку.
Настоящее изобретение относится к новым синтетическим пептидам, которые ингибируют высвобождение гормона роста из гипофиза у млекопитающих, и к терапевтическим композициям, содержащим такие новые пептиды.
Предпосылки создания изобретения
Гормон роста (GH) является пептидом, имеющим 191 аминокислоту, который стимулирует продуцирование многочисленных различных факторов роста, например инсулиноподобного фактора роста I (IGF-I), и, таким образом, стимулирует рост многочисленных тканей (скелет, соединительная ткань, мышцы и внутренние органы) и физиологическую активность (повышение синтеза нуклеиновых кислот и белков и липолиза и понижение секреции мочевины).
Высвобождение GH находится под контролем высвобождающих и ингибирующих факторов, выделяемых гипоталамусом. Основным рилизинг-фактором является рилизинг-гормон гормона роста (GH-RH); рилизинг-гормон человеческого гормона роста (hGH-RH) является пептидом, состоящим из 44 аминокислот. Новые пептиды по настоящему изобретению относятся к аналогам hGH-RH, имеющим только остатки от 1 до 29 (hGH-RH(1-29)NH2), то есть к аналогам пептида, имеющего аминокислотную последовательность:
Tyr-Ala-Asp-Ala-Ile5-Phe-Thr-Asn-Ser-Tyr10-Arg-Lys-Val-Leu-Gly15-
Gln-Leu-Ser-Ala-Arg20-Lys-Leu-Leu-Gln-Asp25-Ile-Met-Ser-Arg29-NH2
GH причастен к некоторым заболеваниям. Одним из заболеваний, в которое вовлечен GH, является акромегалия, при которой присутствуют избыточные уровни GH. Патологически гипертрофированные лицевые кости и кости конечностей при данном заболевании могут подвергаться лечению путем введения антагониста GH-RH.
Другими заболеваниями, в которые вовлечен GH, являются диабетическая ретинопатия и диабетическая нефропатия. Полагают, что соответственно повреждение сетчатки и почек при таких заболеваниях, приводящее к слепоте или снижению функции почек, связано с GH. Такое поражение может быть предотвращено или замедлено при введении эффективного антагониста GH-RH.
Однако главное применение антагонистов GH-RH предполагается в области злокачественных новообразований (A.V. Schally et al. in Growth Hormone Secretagogues in Clinical Practice, eds. Bercu, B.B. & Walker, R.F., Dekker, New York, pp. 145-162, 1998). IGF-I и -II являются сильнодействующими митогенами для различных злокачественных опухолей. Подавляя секрецию GH, антагонисты GH-RH уменьшают синтез IGF-I в печени и других тканях. Антагонисты GH-RH также понижают аутокринное и паракринное продуцирование IGF-I и/или IGF-II различными опухолями. При некоторых экспериментальных злокачественных опухолях лечение антагонистами GH-RH приводит к уменьшению IGF-I и -II, сопутствующему ингибированию роста опухоли.
При попытке воздействовать на развитие этих заболеваний и других состояний некоторые исследователи попытались контролировать уровни GH, используя соматостатин, один из ингибиторов высвобождения GH. Однако соматостатин, введенный в отдельности, не подавляет уровни GH или IGF-I до желаемой степени. При введении в комбинации с антагонистом GH-RH соматостатин намного бы улучшил подавление уровней IGF-I.
Исследовались различные модификации GH-RH для выяснения связи структуры GH-RH с его активностью в попытке обеспечить синтетические родственные соединения с улучшенными агонистическими или антагонистическими свойствами. Так, ранее было установлено, что фрагмент GH-RH, включающий остатки 1-29, или GH-RH (1-29), является минимальной последовательностью, необходимой для биологической активности. Этот фрагмент сохраняет 50% или более активности природного GH-RH.
Было найдено, что первый описанный антагонист GH-RH, [Ас-Туr1, D-Arg2]hGH-RH (1-29) NH2, который обычно называют в литературе "стандартным антагонистом", предотвращает активацию аденилатциклазы передней доли гипофиза крысы с помощью hGH-RH(l-29)NH2. Тот же пептид блокировал действие GH-RH на его рецепторах в гипофизе и гипоталамусе и ингибировал пульсирующую секрецию гормона роста.
В значительном числе патентов и статей в открытой литературе описываются аналоги GH-RH, которые действуют либо как агонисты GH-RH (то есть, действуют как стимуляторы высвобождения GH), либо как антагонисты GH-RH (то есть, действуют как ингибиторы высвобождения GH). Большинство этих пептидов происходит от пептидной последовательности GH-RH(1-29) со специфическими структурными модификациями, которые отвечают за их повышенные агонистические или антагонистические свойства.
Так, патент США №4659693 раскрывает антагонистические аналоги GH-RH, которые содержат определенные N,N'-диалкил-α-гуанидино-α-аминоацильные остатки в положении 2 последовательности GH-RH(1-29).
Опубликованная международная заявка на патент WO 91/16923 рассматривает ранние попытки изменить вторичную структуру hGH-RH путем модификации его аминокислотной последовательности. Такие предпринятые ранее попытки включают замену Туr1, Ala2, Asp3 или Asn8 их D-изомерами; замену Asn8 на L- или D-Ser, D-Arg, Asn, Thr, Gln или D-Lys; замену Ser9 на Ala для повышения амфифильности участка и замену Gly15 на Ala или Aib. Когда R2 в аналогах является D-Arg и R8, R9 и R15 замещены как указано выше, в результате наблюдают антагонистическую активность. Такие антагонистические пептиды считаются подходящими для введения в качестве фармацевтических композиций для лечения состояний, связанных с избыточными уровнями GH, например акромегалии.
Антагонистическая активность аналога hGH-RH [Ser9-ψ[СН2-NH]-Туг10]hGH-RH(1-29) по патенту США №5084555 была определена как результат псевдопептидной связи (то есть, пептидной связи, восстановленной до [CH2-NH]-связи) между остатками R9 и R10. Однако было указано, что антагонистические свойства [Sеr9-ψ[СH2-NН]-Туr10] hGH-RH(1-29) были ниже, чем у стандартного антагониста, [N-Ac-Tyr1, D-Arg2]GH-RH(1-29)-NH2.
Патент США №5550212 и заявка на патент США 08/642472, представляющие такие же объекты, что и настоящая заявка, описывают аналоги hGH-RH(1-29)NH2, характеризующиеся повышенными антагонистическими свойствами и продолжительным временем действия. Считается, что такие свойства являются результатом замены различных аминокислот и ацилирования ароматическими или неполярными кислотами по N-концу GH-RH(1-29)NH2. Необходимо заметить, что в вышеупомянутых патенте США и заявке на патент США R9 всегда является Ser, R16 является Gln или аминокислотой, образующей лактамный мостик (то есть, Glu), R28 является Ser, Asn, Asp, Ala или Abu и R29 означает Agm, Arg-NH2, Arg-OH, Cit-NH2, Cit-OH, Har-NH2, Har-OH или аминокислоту, образующую лактамный мостик (то есть, Lys или Orn).
Краткое описание изобретения
Представлен новый ряд синтетических аналогов hGH-RH(1-29)NH2. Данные аналоги ингибируют активность эндогенного hGH-RH и тем самым предотвращают высвобождение гормона роста. Более сильные ингибирующие эффекты новых аналогов по сравнению с описанными ранее являются результатом замены различных аминокислот.
Конкретно, изобретение относится к пептидам, которые охватываются формулами:
Х-R1-R2-Аsр-Аla-R5-R6-Тhr-R8-R9-R10-Аrg-R12-R13-R14-R15-R16-Leu-R18-R19-Arg-R2l-R22-Leu-Gln-Asp-Ile-R27-R28-R29-NH2,
где Х означает PhAc, IndAc, Ibu, Nac, 1- или 2-Npr или
Fpr,
R1 означает Туr или His,
R2 означает D-Arg или D-Cit,
R5 означает Ilе или Val,
R6 означает Phe, Nal или Phe(Y), в котором Y=F, Cl, Br,
R8 означает Asn, Gln, Ser, Thr, Ala, D-Asn, D-Gln, D-Ser, D-Thr, Abu, D-Abu или Aib,
R9 означает Arg, Har, Lys, Orn, D-Arg, D-Har, D-Lys, D-Orn, Cit, Nle, Tyr(Me), Ser, Ala или Aib,
R10 означает Туr или Phe(Y), в котором Y=H, F, Cl, Br или
ОСН3,
R12 означает Lys, D-Lys или Orn,
R13 означает Val или Nle,
R14 означает Leu или Nle,
R15 означает Gly, Ala, Abu, Nle или Gln,
R16 означает Gln или Arg,
R18 означает Ser или Nle,
R19 означает Ala или Abu,
R21 означает Lys или Orn,
R22 означает Leu, Ala или Aib,
R27 означает Met, Leu, Nle, Abu или D-Arg,
R28 означает Arg, D-Arg, Ser, Asn, Asp, Ala или Abu,
R29 означает Arg, D-Arg, Наг или D-Har, и к их фармацевтически приемлемым солям.
В предпочтительном варианте осуществления изобретения рассматриваются пептиды, в которых Х означает PhAc, IndAc или Nac, R1 означает Туr или His, R2 означает D-Arg, R5 означает Ile, R6 означает Phe(pCl), R8 означает Asn или Abu, R9 означает Аrg или Наr, Lys, Orn, D-Arg, D-Har, D-Lys, D-Orn, Cit, Nle или Тyr(Me), R10 означает Тyr или Тyr(Me), R12 означает Lys, R13 означает Val или Nle, R14 означает Leu или Nle, R15 означает Abu, Ala или Nle, R16 означает Gln или Arg, R18 означает Ser или Nle, R19 означает Ala или Abu, R21 означает Lys, R27 означает Nle или D-Arg, R28 означает D-Arg, Arg или Ser, R29 означает D-Arg, Наr или D-Har.
Отмечено, что аминокислотные остатки от 30 до 44 природной молекулы GH-RH, по-видимому, не являются существенными для активности; также не является критичной их идентичность. Поэтому похоже, что присоединение некоторых или всех из этих дополнительных аминокислотных остатков к С-концу аналогов hGH-RH(1-29)-NH2 по настоящему изобретению не повлияет на эффективность этих аналогов в качестве антагонистов GH-RH. Если некоторые или все из этих аминокислот присоединяли к С-концу аналогов hGH-RH(1-29)-NH2, то присоединенные аминокислотные остатки могли бы быть такими же, как и остатки от 30 до 44 в последовательности природного hGH-RH, или приемлемыми эквивалентами.
Синтетические методы.
Синтетические пептиды получают подходящим способом, как, например, исключительно твердофазным способом, частично твердофазным способом, путем конденсации фрагментов или путем классического синтеза в растворе.
Когда аналоги по изобретению синтезируют твердофазным способом, С-концевой остаток (здесь R29) подходящим образом связывается (закрепляется) с инертным твердым носителем (смола), сохраняя в то же время защитные группы для своей α-аминогруппы (и, соответственно, при необходимости, для функциональной группы боковой цепи). После завершения этой стадии защитную группу α-аминогруппы удаляют с закрепленного аминокислотного остатка и прибавляют следующий аминокислотный остаток, R28, имеющий подходящим образом замещенную α-аминогруппу (как и любую соответствующую функциональную группу боковой цепи), и так далее. Защитные группы N-конца удаляют после присоединения каждого остатка, но в то же время не удаляют защитные группы боковой цепи. После того как присоединили все желаемые аминокислоты в надлежащей последовательности, пептид отделяют от носителя и освобождают от всех защитных групп боковой цепи в условиях, которые являются минимально деструктивными по отношению к остаткам в последовательности. Затем следует тщательная очистка и точная идентификация синтетического продукта для гарантии того, что действительно получена желаемая структура.
Особо предпочтительно защитить α-аминофункциональную группу аминокислот во время стадии сочетания с помощью чувствительной к кислоте или основанию защитной группы. Такие защитные группы должны обладать свойствами сохранения стабильности в условиях образования пептидных связей, в то же время должны легко удаляться без разрушения растущей пептидной цепи и без рацемизации любого из содержащихся там хиральных центров. Подходящими защитными группами α-аминогруппы являются Воc(трет-бутоксикарбонил) и Fmoc(9-флуоренилметилоксикарбонил).
Применения в медицине.
Являющиеся антагонистами hGH-RH пептиды или соли этих пептидов могут быть приготовлены в виде фармацевтических лекарственных форм, содержащих эффективные количества указанных соединений, и введены людям или животному с лечебными или диагностическими целями. Пептиды могут быть использованы для подавления уровней GH и для лечения состояний, связанных с избыточными уровнями GH, например диабетическая ретинопатия и нефропатия, и акромегалия. Также обеспечены методы лечения этих заболеваний путем введения композиции по изобретению индивидууму, нуждающемуся в таком лечении. Основное применение антагонистов GH-RH относится, однако, к области злокачественных новообразований, например при раке молочной железы, легкого, ободочной кишки, мозга, поджелудочной железы и предстательной железы человека, где присутствуют рецепторы IGF-I или IGF-II.
Краткое описание чертежей
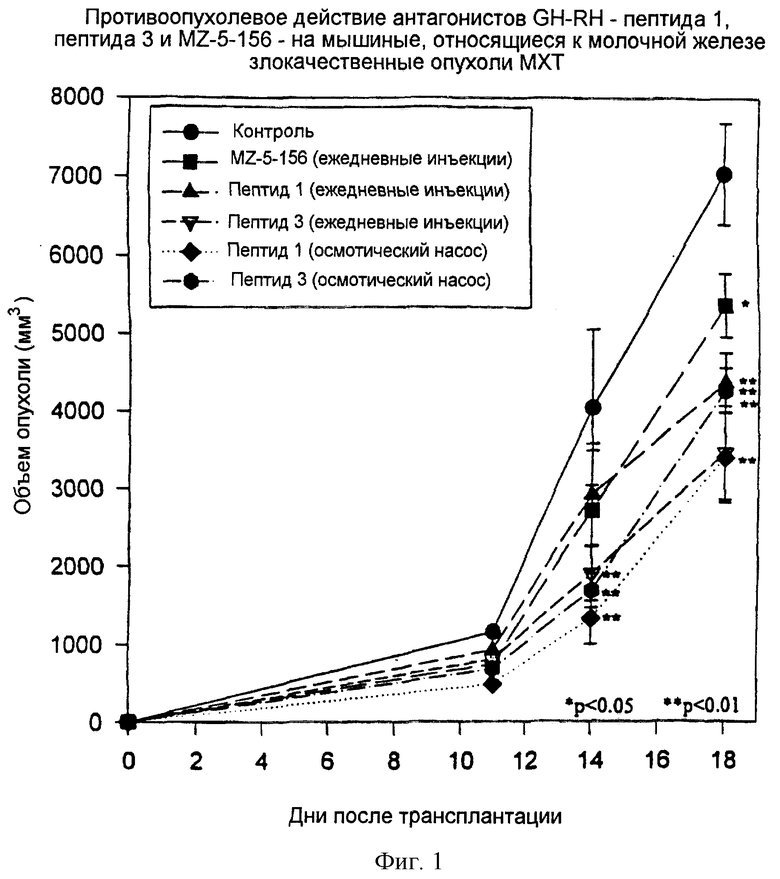
Фиг.1 представляет графическую зависимость изменений объема мышиных, относящихся к молочной железе злокачественных опухолей МХТ от дней обработки при обработке определенными антагонистами GH-RH.
Фиг.2 представляет графическую зависимость изменений объема человеческих злокачественных опухолей молочной железы MDA-MB-468 у бестимусных мышей от дней обработки при обработке определенными антагонистами GH-RH.
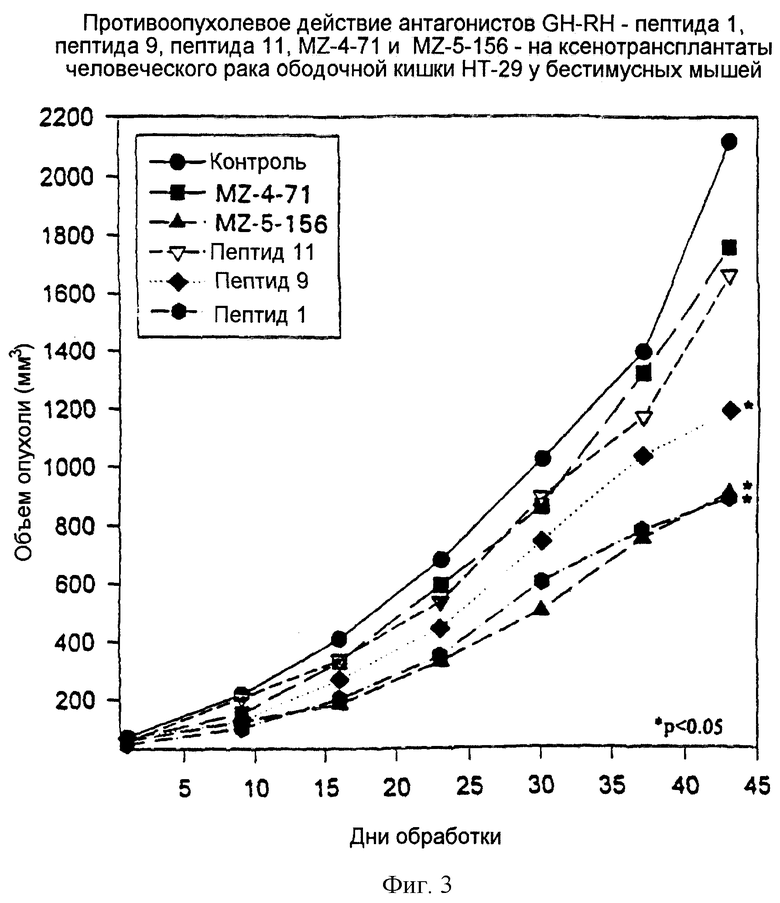
Фиг.3 представляет графическую зависимость изменений объема человеческих злокачественных опухолей ободочной кишки НТ-29 у бестимусных мышей от дней обработки при обработке определенными антагонистами GH-RH.
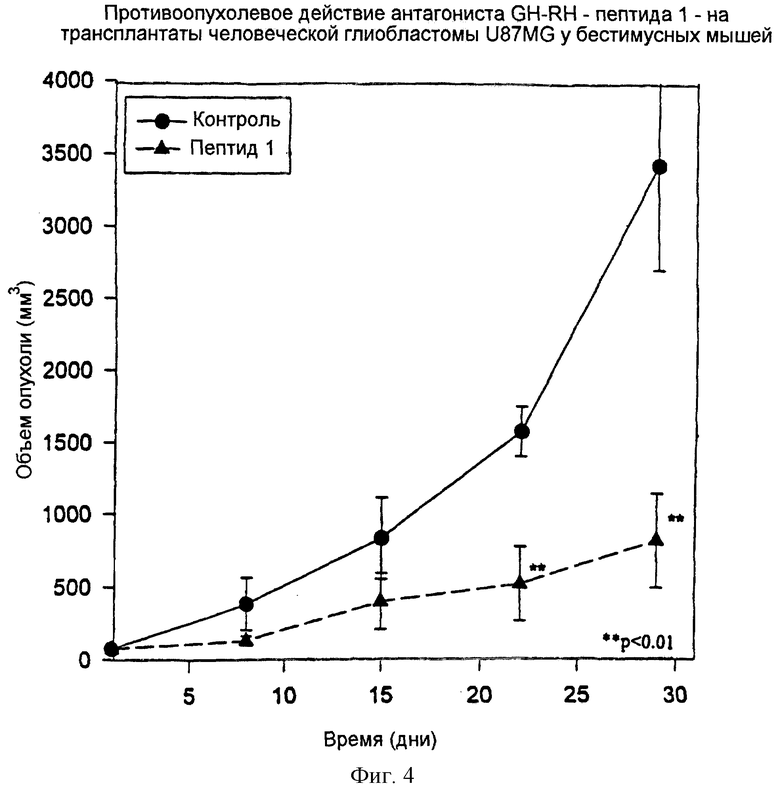
Фиг.4 представляет графическую зависимость изменений объема человеческих глиобластом U87MG у бестимусных мышей от дней обработки при обработке антагонистом GH-RH.
Фиг.5 представляет графическую зависимость изменений объема человеческих злокачественных опухолей предстательной железы РС-3 у бестимусных мышей от дней обработки при обработке определенными антагонистами GH-RH.
Подробное описание предпочтительных вариантов осуществления изобретения
А. Сокращения
Номенклатура, используемая для определения пептидов, является такой, которая установлена специальным уполномоченным ИЮПАК-МБС (МБС - Международный биохимический союз) по биохимической номенклатуре, где в соответствии с общепринятым представлением аминогруппа при N-конце находится слева и карбоксильная группа при С-конце находится справа. Термин "природная аминокислота", используемый здесь, означает одну из обычных, встречающихся в природе L-аминокислот, находящихся в природных белках: Gly, Ala, Val, Leu, Ile, Ser, Thr, Lys, Arg, Asp, Asn, Glu, Gln, Cys, Met, Phe, Tyr, Pro, Trp и His, Когда остаток природной аминокислоты существует в изомерных формах, именно L-форма аминокислоты представлена здесь, если специально не указано иначе.
Некодированные аминокислоты или аминокислотные аналоги также включены в антагонисты GH-RH. ("Некодированные" аминокислоты представляют те аминокислоты, которые не находятся среди примерно 20 природных аминокислот, встречающихся в природных пептидах). К некодированным аминокислотам или аминокислотным аналогам, которые могут быть использованы в пептидах, являющихся антагонистами hGH-RH, относятся следующие: под Abu имеют ввиду α-аминомасляную кислоту, под Aib имеют ввиду α-аминоизомасляную кислоту, под Наr имеют ввиду гомоаргинин, под Nal имеют ввиду 2-нафтилаланин, под Nle имеют ввиду норлейцин и под Оrn имеют ввиду орнитин. Когда эти некодированные аминокислоты или аналоги аминокислот существуют в изомерных формах, именно L-форма аминокислоты представлена здесь, если специально не указано иначе. Используемые здесь сокращения:
Abu - α-аминомасляная кислота;
Ас - ацетил;
АсОН - уксусная кислота;
Ас2O - уксусный ангидрид;
Aib - α-аминоизомасляная кислота;
Воc - трет-бутоксикарбонил;
Воm - бензилоксиметил;
2BrZ - 2-бромбензилоксикарбонил;
сНх - циклогексил;
Cit - цитруллин (2-амино-5-уреидовалериановая кислота);
2CIZ - 2-хлорбензилоксикарбонил;
DCM - дихлорметан (ДХМ);
DIC - N,N'-диизопропилкарбодиимид (ДИКД);
DIEA - диизопропилэтиламин (ДИЭА);
DMF - диметилформамид (ДМФА);
Fmoc - флуоренилметилоксикарбонил;
Fpr - 3-фенилпропионил;
GH - гормон роста;
GH-RH - рилизинг-гормон гормона роста;
Наr - гомоаргинин;
HBTU - 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметил-урония гексафторфосфат;
hGH-RH - человеческий GH-RH;
HOBt - 1-гидроксибензотриазол;
HPLC - высокоэффективная жидкостная хроматография (ВЭЖХ);
Ibu - изобутирил;
IndAc - индол-3-ацетил;
МВНА - п-метилбензгидриламин (МБГА);
МеОН - метанол;
MeCN - ацетонитрил;
Nac - 1-нафтилацетил;
Nal - 2-нафтилаланин;
NMM - N-метилморфолин;
Npr - нафтилпропионил;
РАМ - фенилацетамидометил;
Phe(pCl) - п-хлорфенилаланин;
PhAc - фенилацетил;
rGH-RH - крысиный GH-RH;
RP-HPLC - ВЭЖХ с обращенной фазой;
TFA - трифторуксусная кислота (ТФК);
Tos - п-толуолсульфонил;
Тyr(Me) - простой тирозинметиловый эфир;
Z - бензилоксикарбонил.
В. Аналоги GH-RH
Аналоги hGH-RH по настоящему изобретению были получены с целью повышения сродства пептидов к рецептору, повышения метаболической устойчивости и максимизирования амфифильности вторичной структуры молекул. Многие из этих аналогов вызывают очень эффективное и продолжительное ингибирование высвобождения GH, стимулированного hGH-RH(1-29)NH2 in vitro и in vivo.
Следующие варианты осуществления изобретения особенно предпочтительны как обладающие прекрасной биологической активностью:
[PhAc0,D-Arg2, Phe(pCl)6, Arg9, Abu15, Nle27, D-Arg28, Har29]hGH-RH(1-29)NH2 пептид 1;
[IndAc0, D-Arg2, Phe(pCl)6, Arg9, Abu15, Nle27, D-Arg28, Har29]hGH-RH(1-29)NН2 пептид 2;
[PhAc0, D-Arg2, Phe(pCl)6, Наr9, Tyr(Me)10, Abu15, Nle27, D-Arg28, Har29]hGH-RH(1-29)NH2 пептид 3;
[PhAc0, D-Arg2, Phe(pCl)6, Har9, Abu15, Nle27, D-Arg28, Har29]hGH-RH(1-29)NH2 пептид 4;
[Nac0, D-Arg3, Phe(pCl)6, Arg9, Abu15, Nle27, D-Arg28, Har29]hGH-RH(1-29)NH2 пептид 5;
[PhAc0, D-Arg2, Phe(pCl)6, Arg9, Tyr(Me)10, Abu15, Nle27, D-Arg28, Har29]hGH-RH(1-29)NH2 пептид 6;
[PhAc0, His1, D-Arg2, Phe(pCl)6, Arg9, Abu15, Nle27, D-Arg28, Har29]hGH-RH(1-29)NH2 пептид 7;
[Nac0, His1, D-Arg2, Phe(pCl)6, Arg9, Abu15, Nle27, D-Arg28, Har29]hGH-RH(1-29)NH2 пептид 8;
[PhAc0, D-Arg2, Phe(pCl)6, Arg9, Abu15, Nle27, D-Arg29]hGH-RH(1-29)NH2 пептид 9;
[PhAc0, D-Arg2, Phe(pCl)6, Abu15, Аrg16, Nle27, D-Arg29]hGH-RH(1-29)NH2 пептид 10;
[PhAc0, D-Arg2, Phe(pCl)6, Abu15, Nle27, D-Arg28, Har29]hGH-RH(1-29)NH2 пептид 11;
[PhAc0, D-Arg2, Phe(pCl)6, Nle9, Abu15, Nle27, D-Arg29]hGH-RH(1-29)NH2 пептид 12;
[PhAc0, D-Arg2, Phe(pCl)6, Nle13, Nle14, Abu15, Nle27, D-Arg29]hGH-RH(1-29)NH2 пептид 13;
[PhAc0, D-Arg2, Phe(pCl)6, Nle15, Nle27, D-Arg29]hGH-RH(1-29)NH2 пептид 14;
[PhAc0, D-Arg2, Phe(pCl)6, Abu15, Nle18, Nle27, D-Arg29]hGH-RH(1-29)NH2 пептид 15;
[PhAc0, D-Arg2, Phe(pCl)6, Тyr(Ме)10, Abu15, Nle27, D-Arg29]hGH-RH(1-29)NH2 пептид 16;
[PhAc0, D-Arg2, Phe(pCl)6, Abu8, Tyr(Me)10, Abu15, Nle27, D-Arg29]hGH-RH(1-29)NH2 пептид 17;
[PhAc0, D-Arg2, Phe(pCl)6, D-Abu8, Tyr(Me)10, Abu15, Nle27, D-Arg29]hGH-RH(1-29)NH2 пептид 18;
[PhAc0, D-Arg2, Phe(pCl)6, Tyr(Me)10, Abu15, D-Arg27, Arg28, D-Arg29]hGH-RH(1-29)NH2 пептид 19;
[PhAc0, D-Arg2, Phe(pCl)6, Tyr(Me)3, Abu15, D-Arg27, Arg28, D-Arg29]hGH-RH(1-29)NH2 пептид 20;
[PhAc0, D-Arg2, Phe(pCl)6, Abu15, D-Arg27, Arg28, D-Arg29]hGH-RH(1-29)NH2 пептид 21;
[PhAc0, D-Arg2, Phe(pCl)6, Abu8, Tyr(Me)10, Abu15, D-Arg27, Arg28, D-Arg29]hGH-RH(1-29)NH2 пептид 22;
[PhAc0, D-Arg2, Phe(pCl)6, D-Abu8, Туr(Ме)10, Abu15, D-Arg27, Arg28, D-Arg29]hGH-RH(1-29)NH2 пептид 23;
[PhAc0, D-Arg2, Phe(pCl)6, Lys9, Abu15, Nle27, D-Arg28, Har29]hGH-RH(1-29)NH2 пептид 24;
[PhAc0, D-Arg2, Phe(pCl)6, Orn9, Abu15, Nle27, D-Arg28, Har29]hGH-RH(1-29)NH2 пептид 25;
[PhAc0, D-Arg2, Phe(pCl)6, D-Аrg9, Abu15, Nle27, D-Arg28, Har29]hGH-RH(1-29)NH2 пептид 26;
[PhAc0, D-Arg2, Phe(pCl)6, D-Har9, Abu15, Nle27, D-Arg28, Har29]hGH-RH(1-29)NH2 пептид 27;
[PhAc0, D-Arg2, Phe(pCl)6, D-Lys9, Abu15, Nle27, D-Arg28, Har29]hGH-RH(1-29)NH2 пептид 28;
[PhAc0, D-Arg2, Phe(pCl)6, D-Orn9, Abu15, Nle27, D-Arg28, Har29]hGH-RH(1-29)NH2 пептид 29;
(PhAc0, D-Arg2, Phe(pCl)6, Cit9, Abu15, Nle27, D-Arg28, Har29]hGH-RH(1-29)NH2 пептид 30.
Шесть очень предпочтительных вариантов осуществления изобретения имеют формулы:
PhAc0-Tyr1-D-Arg2-Asp3-Ala4-Ile5-Phe(pCl)6-Thr7-Asn8-Arg9-Tyr10-Arg11-Lys12-Val13-Leu14-Abu15-Gln16-Leu17-Ser18-Ala19-Arg20-Lys21-Leu22-Leu23-Gln24-Asp25-Ile26-Nle-27-D-Arg28-Har29-NH2 пептид 1;
lndAc0-Tyr1-D-Arg2-Asp3-Ala4-Ile5-Phe(pCl)6-Thr7-Asn8-Arg9-Tyr10-Arg11-Lys12Val13-Leu14-Abu15-Gln16-Leu17-Ser18-Ala19-Arg20-Lys21-Leu22-Leu23-Gln24-Asp25-Ile26-Nle27-D-Arg28-Har29-NH2 пептид 2;
PhAc0-Tyr1-D-Arg2-Asp3-Ala4-Ile5-Phe(pCl)6-Thr7-Asn8-Har9-Tyr(Me)10-Arg11-Lys12-Var13-Leu14-Abu15-Gln16-Leu17-Ser18-Ala19-Arg20-Lys21-Leu22-Leu23-Gln24-Asp25-Ile26-Nle27-D-Arg28-Har29-NH2 пептид 3;
PhAc0-Tyr1-D-Arg2-Asp3-Ala4-Ile5-Phe(pCl)6-Thr7-Asn8-Arg9-Tyr(Me)10-Arg11-Lys12-Val13-Leu14-Abu15-Gln16-Leu17-Ser18-Ala19-Arg20-Lys21-Leu22-Leu23-Gln24-Asp25-Ile26-Nle27-D-Arg28-Har29-NH2 пептид 6;
PhAc0-His1-D-Arg2-Asp3-Ala4-Ile5-Phe(pCl)6-Thr7-Asn8-Arg9-Tyr10-Arg11-Lys12-Val13-Leu14-Abu15-Gln16-Leu17-Ser18-Ala19-Arg20-Lys21-Leu22-Leu23-Gln24-Asp25-Ile26-Nle27-D-Arg28-Har29-NH2 пептид 7;
Nac0-His1-D-Arg2-Asp3-Ala4-Ile5-Phe(pCl)6-Thr7-Asnв-Arg9-Tyr10-Arg11-Lys12-Val13-Leu14-Abu15-Gln16-Leu17-Ser18-Ala19-Arg20-Lys21-Leu22-Leu23-Gln24-Asp25-Ile26-Nle27-D-Arg28-Har29-NH2 пептид 8.
Согласно общепринятому правилу такие соединения могут быть представлены с сокращенным наименованием, как следует ниже:
[PhAc0, D-Arg2, Phe(pCl)6, Arg9, Abu15, Nle27, D-Arg28, Har29]hGH-RH(1-29)NH2 пептид 1;
[IndAc0, D-Arg2, Phe(pCl)6, Arg9, Abu15, Nle27, D-Arg28, Har29]hGH-RH(1-29)NH2 пептид 2;
[PhAc0, D-Arg2, Phe(pCl)6, Har9, Tyr(Me)10, Abu15, Nle27, D-Arg28, Har29]hGH-RH(1-29)NH2 пептид 3;
[PhAc0, D-Arg2, Phe(pCl)6, Arg9, Tyr(Me)10, Abu15, Nle27, D-Arg28, Har29]hGH-RH(1-29)NH2 пептид 6;
[PhAc0, His1, D-Arg2, Phe(pCl)6, Arg9, Abu15, Nle27, D-Arg28, Har29]hGH-RH(1-29)NH2 пептид 7;
[Nac0, His1, D-Arg2, Phe(pCl)6, Arg9, Abu15, Nle27, D-Arg28, Har29]hGH-RH(1-29) NH2 пептид 8.
С. Способ получения
1. Обзор синтеза.
Пептиды синтезируют подходящими способами, как, например, исключительно путем твердофазного пептидного синтеза, частично путем твердофазного синтеза, путем конденсации фрагментов или путем классического синтеза в растворе. Например, методики исключительно твердофазного синтеза представлены в учебнике "Solid Phase Peptide Synyhesis", J.M. Stewart and J.D. Young, Pierce Chem. Company, Rockford, 111, 1984 (2nd. ed.), and M. Bodanszky, "Principles of Peptide Synthesis", Springer Verlag, 1984. Являющиеся антагонистами hGH-RH пептиды предпочтительно получают при применении твердофазного синтеза, как, например, описанного Merrifield, J. Am. Chem. Soc., 85, р. 2149 (1963), хотя другие эквивалентные химические синтезы, известные в данной области, могут быть также применены, как указывалось выше.
Синтез осуществляют с аминокислотами, которые защищены по α-аминогруппе. Предпочтительно для защиты α-аминогруппы используют защитные группировки типа уретана (Воc или Fmoc). Предпочтительной защитной группой является Воc.
При твердофазном синтезе защищенный по атому азота α-аминогруппы участок аминокислоты, который образует аминоацильную группу конечного пептида при С-конце, присоединяют химической связью к носителю из полимерной смолы. После завершения реакции связывания защитную группу α-аминогруппы для проведения последующих реакций связывания по аминоконцу селективно удаляют, предпочтительно с помощью 50% трифторуксусной кислоты (ТФК) в дихлорметане (ДХМ), когда N-α-защитной группой является Воc. Остальные аминокислоты с подобным же образом Вос-защищенными α-аминогруппами постадийно присоединяют к свободной аминогруппе предыдущей аминокислоты на смоле для получения желаемой пептидной последовательности. Так как аминокислотные остатки связываются с α-аминогруппой С-терминального остатка, рост синтетических пептидных аналогов hGH-RH начинается при С-конце и продвигается в направлении к N-концу. Когда получена желаемая последовательность, пептид ацилируют по N-концу и его удаляют с полимерного носителя.
Каждую защищенную аминокислоту применяют в избытке (2,5 или 3 эквивалента), и реакции сочетания обычно проводят в ДХМ, ДМФА или в их смесях. Степень завершения реакции связывания контролируется на каждой стадии с помощью реакции с нингидрином. В случаях, когда определяют неполное связывание, процедуру связывания повторяют или реакцию завершают путем ацилирования непрореагировавших аминогрупп до удаления защитной группы с α-аминогруппы перед связыванием следующей аминокислоты.
Типичный цикл синтеза представлен в таблице 1.

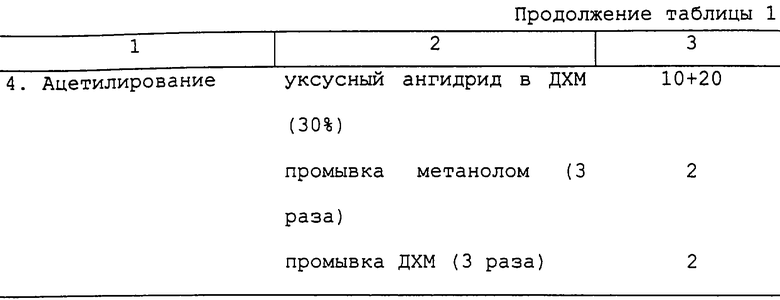
После завершения синтеза отщепление пептида от смолы может быть осуществлено с использованием методик, хорошо известных в химии пептидов.
Некоторые из аминокислотных остатков пептидов имеют такие функциональные группы в боковой цепи, которые реакционноспособны по отношению к реагентам, использующимся при связывании или снятии защиты. Когда в боковой цепи присутствуют такие группы, подходящие защитные группы присоединяют к этим функциональным группам для предотвращения нежелательных химических реакций, происходящих при реакциях, используемых для образования пептидов. При выборе соответствующей защитной группы для боковой цепи придерживаются следующих общих правил: (а) защитная группа предпочтительно сохраняет свои защитные свойства и не отщепляется в условиях связывания, (b) защитная группа должна быть устойчивой в условиях удаления защитной группы с α-аминогруппы на каждой стадии синтеза, (с) защитная группа боковой цепи должна быть способной после завершения синтеза желаемой аминокислотной последовательности удаляться в реакционных условиях, которые не изменят нежелательным образом пептидную цепь.
Реакционноспособные функциональные группы боковой цепи предпочтительно защищают следующим образом: бензил для Thr и Ser; 2-бромбензилоксикарбонил для Тyr; п-толуолсульфонил или нитрогруппа для Аrg и Наr; 2-хлорбензилоксикарбонил или флуоренилметилоксикарбонил для Lys, Orn; бензилоксиметил для His и циклогексил или флуоренилметил для Asp и Glu. Боковые цепочки Asn и Gln не защищаются.
3. Постадийное связывание аминокислотных остатков с полимерным носителем.
Пептиды, являющиеся антагонистами hGH-RH, могут быть синтезированы на ряде полимерных носителей, то есть, на смоле на основе п-метилбензгидриламина (МБГА), на смоле Меррифилда, на смоле на основе фенилацетамидометила или на смоле Wang. Когда для синтеза используются N-α-Boc-защищенные аминокислоты, предпочтительной смолой является смола на основе МБГА. В этом случае получают пептиды с амидированным С-концом при отщеплении от фазы-носителя.
Сначала С-концевая аминокислота присоединяется к нейтрализованной МБГА-смоле и затем проводятся последующие связывания аминокислот. Каждая защищенная аминокислота связывается примерно при трехкратном молярном избытке по отношению к связанным смолой остаткам со свободными аминогруппами, и связывание может быть проведено в среде, как, например, ДМФА: CH2Cl2 (1:1) или только в ДМФА, или только в CH2Cl2. Выбор подходящего связывающего реагента является компетенцией специалиста в данной области. Особенно подходящими в качестве реагентов связывания являются N,N-диизопропилкарбодиимид (ДИКД) или гексафторфосфат 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония, смешанный с 1-гидроксибензотриазолом. Успех прохождения реакции связывания на каждой стадии синтеза предпочтительно контролируется с помощью нингидриновой реакции. В случаях, когда происходит неполное связывание, либо процесс связывания повторяют, либо связанные смолой непрореагировавшие остатки с аминогруппами ацетилируют с использованием уксусного ангидрида в ДХМ до удаления защитной группы α-аминогруппы.
Конечное ацилирование N-конца пептида проводят таким же образом, как и предыдущие связывания, с той разницей, что вместо аминокислоты используют соответствующую карбоновую кислоту.
4. Удаление пептида с полимерного носителя.
Когда синтез завершен, пептид отщепляют от фазы-носителя. Удаление пептида со смолы осуществляют при обработке реагентом, как, например, жидкий фтористый водород, который также отщепляет все оставшиеся защитные группы боковой цепи.
Соответственно, высушенный и защищенный пептид-смола обрабатывают смесью, состоящей из 1,0 мл м-крезола и 10 мл безводного фтористого водорода на грамм пептида-смолы, в течение 60 мин при 0°С для отщепления пептида со смолы, а также для удаления всех защитных групп боковой цепи. После удаления фтористого водорода в токе азота и в вакууме свободные пептиды осаждают диэтиловым эфиром, фильтруют, промывают диэтиловым эфиром и этилацетатом, экстрагируют 50% уксусной кислотой и подвергают лиофилизации.
5. Очистка.
Очистку технических пептидов можно осуществлять, используя методики, хорошо известные в химии пептидов. Например, очистка может быть проведена с использованием системы MacRabbit для ВЭЖХ (Rainin Instrument Co. Inc., Woburn, MA) с УФ-фотометром Knauer и регистрирующим устройством Kipp и Zonen BD40, используя колонку Vydac 218TP5010 с обращенной фазой (10×250 мм, заполненную силикагелем С 18, размер пор 300 , размер частиц 5 мкм) (The Separations Group Inc., Hesperia, CA). Колонку элюируют системой растворителей, состоящей из (А) 0,1% водной ТФК и (В) 0,1% ТФК в 70% водном MeCN с линейным градиентом (например, 30-55% В в 120 мин). Элюент контролируется при 220 нм и фракции исследуются с помощью аналитической ВЭЖХ при использовании жидкостного хроматографа модели HP-1090 фирмы Hewlett-Packard и объединяются для обеспечения максимальной чистоты. Аналитическую ВЭЖХ осуществляют на колонке Vydac 218TP52 с обращенной фазой (2×250 мм, С 18, 300, 5 мкм), применяя изократное элюирование системой растворителей, состоящей из (А) и (В), обозначенных выше. Пики регистрируются при 220 и 280 нм. Пептиды по данным аналитической ВЭЖХ получают в значительной степени чистыми (> 95%). Ожидаемый аминокислотный состав также подтверждается аминокислотным анализом.
, размер частиц 5 мкм) (The Separations Group Inc., Hesperia, CA). Колонку элюируют системой растворителей, состоящей из (А) 0,1% водной ТФК и (В) 0,1% ТФК в 70% водном MeCN с линейным градиентом (например, 30-55% В в 120 мин). Элюент контролируется при 220 нм и фракции исследуются с помощью аналитической ВЭЖХ при использовании жидкостного хроматографа модели HP-1090 фирмы Hewlett-Packard и объединяются для обеспечения максимальной чистоты. Аналитическую ВЭЖХ осуществляют на колонке Vydac 218TP52 с обращенной фазой (2×250 мм, С 18, 300, 5 мкм), применяя изократное элюирование системой растворителей, состоящей из (А) и (В), обозначенных выше. Пики регистрируются при 220 и 280 нм. Пептиды по данным аналитической ВЭЖХ получают в значительной степени чистыми (> 95%). Ожидаемый аминокислотный состав также подтверждается аминокислотным анализом.
D. Фармацевтическая композиция
Пептиды по изобретению могут вводиться в виде фармацевтически приемлемых нетоксичных солей, как, например, аддитивные соли кислоты. Примерами таких аддитивных солей кислот являются гидрохлорид, гидробромид, сульфат, фосфат, фумарат, глюконат, таннат, малеат, ацетат, цитрат, бензоат, сукцинат, альгинат, памоат, малат, аскорбат, тартрат и им подобные. Особенно предпочтительными антагонистами являются соли с малой растворимостью, например памоаты и им подобные. Они проявляют продолжительную активность.
Соединения по настоящему изобретению соответственно вводят людям или животным подкожно, внутримышечно или внутривенно, через нос или путем легочной ингаляции, или в виде формы с замедленным выделением (депо-форма) (например, микрокапсулы, микрогранулы или цилиндрическая палочка, подобная имплантатам), приготовленной из биораспадающегося подходящего полимера (как, например, D,L-лактид-согликолид), причем первые две депо-формы являются предпочтительными. Другие эквивалентные формы введения также представлены в рамках данного изобретения, то есть, продолжительное капельное внутривенное вливание, инъекции веществ замедленного всасывания, инфузионный насос и формы с высвобождением во времени, как, например, микрокапсулы, и им подобные. Введение допустимо в любом физиологически приемлемом инъецируемом носителе, физиологическом растворе, хотя другие носители, известные в данной области, могут также использоваться.
Пептиды предпочтительно вводят парентерально, внутримышечно, подкожно или внутривенно с фармацевтически приемлемым носителем, как, например, изотонический солевой раствор. Альтернативно пептиды могут вводиться в виде интраназального аэрозоля с помощью подходящего носителя или путем легочной ингаляции. Подходящим путем введения является депо-форма, приготовленная из биораспадающегося соответствующего полимера, например поли-D,L-лактид-согликолида, в виде микрокапсул, микрогранул или цилиндрических имплантатов, содержащих диспергированные антагонистические соединения.
Необходимое количество пептида зависит от способа введения и предполагаемого результата. Обычно диапазон доз составляет 1-100 мкг/кг массы тела организма-хозяина в день.
Е. Терапевтическое применение антагонистов GH-RH.
Антагонисты hGH-RH могут быть применены при лечении состояний, вызванных избытком гормона роста, например акромегалии, которая проявляется в виде аномального разрастания костей лица и конечностей. Антагонисты GH-RH могут также применяться для лечения диабетической ретинопатии (главная причина слепоты у диабетиков) и диабетической нефропатии, при которых повреждения глаз и почек, соответственно, связывают с GH.
Антагонисты hGH-RH предназначаются для блокирования связывания и, следовательно, действия GH-RH, который стимулирует выделение GH, который, в свою очередь, стимулирует продуцирование IGF-I. Антагонисты GH-RH могут вводиться отдельно или вместе с аналогами соматостатина, как комбинация, которая более полно понижает уровни IGF-I. Полезнее вводить антагонисты-GH-RH, чем соматостатин, благодаря тому факту, что антагонисты GH-RH могут быть применены в ситуациях, когда сайты мишеней не имеют рецепторов соматостатина.
Однако, главным образом, антагонисты GH-RH применяются в области злокачественных заболеваний. Это основывается на следующих соображениях: антагонисты GH-RH предназначаются для блокирования связывания и поэтому действия GH-RH, который стимулирует выделение GH, который, в свою очередь, стимулирует продуцирование инсулиноподобного фактора роста I (IGF-I), который также называется соматомедин-С. Хорошо установлено вовлечение IGF-I (соматомедин-С) в злокачественные заболевания молочной железы, предстательной железы, ободочной кишки, в опухоли костей и другие злокачественные заболевания, но одни аналоги соматостатина адекватно не подавляют уровни GH и IGF-I. Для лучшего ингибирования роста опухоли требуется полное подавление уровней IGF-I или его выделения. Аутокринное продуцирование IGF-I различными опухолями могло бы быть также под контролем GH-RH и поэтому могло бы ингибироваться антагонистами GH-RH. Антагонисты GH-RH могли бы также ингибировать продуцирование IGF-I. Более детальная теоретическая предпосылка применений GH-RH в области онкологии (рака) следующая: рецепторы IGF-I присутствуют в первичных человеческих злокачественных опухолях молочной железы, злокачественных опухолях предстательной железы, злокачественных опухолях легких, злокачественных опухолях ободочной кишки, в опухолях мозга, в злокачественных опухолях поджелудочной железы и в гипернефроидных опухолях почки.
Присутствие рецепторов IGF-I в этих опухолях связано с злокачественной трансформацией и пролиферацией этих злокачественных опухолей. IGF-I может действовать как эндокринный, паракринный или аутокринный фактор роста для различных злокачественных опухолей человека, именно рост этих новообразований зависит от IGF-I. Антагонисты GH-RH, подавляя выделение GH, понизят продуцирование IGF-I. Поскольку IGF-I стимулирует рост таких различных новообразований (рака), то понижение уровней циркулирующего IGF-I должно привести к ингибированию опухолевого роста. Возможно, что антагонисты GH-RH смогли бы также понизить паракринное или аутокринное продуцирование IGF-I опухолями, что должно также привести к ингибированию пролиферации злокачественных новообразований. Эти взгляды находятся в соответствии с современными концепциями клинической онкологии. Антагонисты GH-RH следует давать отдельно или вместе с аналогами соматостатина, и комбинация привела бы к достижению более полного подавления уровней IGF-I, к ликвидации уровней IGF-I в тканях, например, в человеческих остеосаркомах, а также при раке молочной железы, раке ободочной кишки, раке предстательной железы и немелкоклеточном раке легкого (non-SCLC).
Преимущество антагонистов GH-RH над аналогами соматостатина основывается на факте, что антагонисты GH-RH могут быть использованы для подавления опухолей, которые не имеют соматостатиновых рецепторов, например человеческие остеогенные саркомы.
Было показано, что антагонистические аналоги GH-RH подавляют рост различных опухолей in vivo. Этот эффект проявляется частично через ингибирование системы GH-RH-IGF-I. Тем не менее, аутокринный/паракринный контроль пролиферации с помощью IGF-II также является главным фактором во многих опухолях. Нарушение такого аутокринного, стимулирующего рост метаболического пути предлагает подход к контролю опухолей. Антагонистические аналоги GH-RH, MZ-4-71 {[Ibu0, Tyr1, D-Arg2, Abu15, Nle27]hGH-RH(1-28) Agm} и MZ-5-156 {[PhAc0, D-Arg2, Abu15, Nle27]hGH-RH(1-28) Agm} значительно ингибировали скорость пролиферации линий клеток рака молочной железы (MDA-MB-468, ZR-75-1), предстательной железы (РС-3 и DU-145) и поджелудочной железы (MiaPaCa-2, SW-1990 и Capan-2) in vitro, как показано с помощью колориметрических тестов и тестов по включению [3H]-тимидина, понижали экспрессию мРНК IGF-II в клетках и концентрацию IGF-II, выделяемого в культуральную среду. Те же антагонисты GH-RH давали похожие результаты in vivo (ингибирование пролиферации и уменьшение продуцирования IGF-II) в случае опухолей предстательной железы (РС-3, DU-145), аденокарциномы почки (Caki-I) и немелкоклеточного рака легкого (Н157). Эти результаты предполагают, что антагонистические аналоги GH-RH могут ингибировать рост опухоли не только путем ингибирования системы GHRH-GH-IGF-I, но также за счет уменьшения выработки IGF-II в некоторых опухолевых клетках, тем самым нарушая ее аутокринный регуляторный метаболический путь.
Настоящее изобретение описывается следующими примерами, которые далее представлены только с целью иллюстрации. В примерах используются оптически активные защищенные аминокислоты в L-конфигурации, за исключением специальных указаний.
Следующие примеры далее представляют подходящие способы синтеза новых антагонистов GH-RH с помощью твердофазного синтеза.
Пример 1
PhAc0-Tyr1-D-Arg2-Asp3-Ala4-Ile5-Phe(pCl)6-Thr7-Asn8-Arg9-Tyr10-Arg11-Lys12-Val13-Leu14-Abu15-Gln16-Leu17-Ser18-Ala19-Arg20-Lys21-Leu22-Leu23-Gln24-Asp25-Ile26-Nle27-D-Arg28-Har29-NH2 (пептид 1) {[PhAc0, D-Arg2, Phe(pCl)6, Arg9, Abu15, Nle27, D-Arg28, Наr29] hGH-RH (1-29) NH2}
Синтез осуществляют постадийно, используя оборудование для твердофазного пептидного синтеза с ручным управлением. Вкратце, смолу на основе МБГА (Bachem, California) (720 мг, 0,5 ммоль) нейтрализуют 5% ДИЭА в CH2Cl2 и промывают в соответствии с протоколом, описанным в таблице 1. Раствор Boc-Har(NO2)-ОН (500 мг, 1,5 ммоль) в ДМФА-СН2Сl2 (1:1) встряхивают с нейтрализованной смолой и ДИКД (235 мкл, 1,5 ммоль) в аппарате с ручным управлением для твердофазного пептидного синтеза в течение 1 часа. После завершения реакции связывания, что подтверждается отрицательным нингидриновым тестом, снятия защиты с помощью 50% ТФК в CH2Cl2 и нейтрализации с помощью 5% ДИЭА в CH2Cl2 пептидную цепь строят постадийно путем связывания на смоле следующих защищенных аминокислот в указанном порядке для получения желаемой пептидной последовательности: Boc-D-Arg(Tos)-ОН, Boc-Nle-OH, Boc-Ile-OH, Boc-Asp(OcHx)-OH, Boc-Gln-OH, Boc-Leu-OH, Boc-Leu-OH, Boc-Lys(2CIZ)-OH, Boc-Arg(Tos)-OH, Boc-Ala-OH, Boc-Ser(Bzl)-OH, Boc-Leu-OH, Boc-Gln-OH, Boc-Abu-OH, Boc-Leu-OH, Boc-Val-OH, Boc-Lys(2CIZ)-OH, Boc-Arg(Tos)-OH, Boc-Tyr(2BrZ)-OH, Boc-Arg(Tos)-OH, Boc-Asn-OH, Boc-Thr(Bzl)-OH, Boc-Phe(pCl)-OH, Boc-Ile-OH, Boc-Ala-OH, Boc-Asp(OcHx)-OH, Boc-D-Arg(Tos)-OH, Boc-Tyr(2BrZ)-OH.
Эти защищенные аминокислотные остатки (также обычно получаемые от фирмы Bachem Co.) обозначены выше в соответствии с хорошо известным правилом. Подходящая защитная группа для функциональной группы боковой цепи определенных аминокислот указывается в скобках. Гидроксильные группы в представленных выше формулах указывают, что карбоксильный конец каждого остатка свободен.
Защищенные аминокислоты (1,5 ммоль каждой) связывают с ДИКД (235 мкл, 1,5 ммоль) за исключением Boc-Asn-OH и Вос-Gln-ОН, которые связываются с их ранее образованными сложными эфирами с 1-гидроксибензотриазолом. После удаления Nα-Boc-защитной группы от Туr1 пептид ацилируют с помощью фенилуксусной кислоты (PhAc) (272 мг, 2 ммоль), применяя ДИКД (313 мкл, 2 ммоль).
Для того чтобы отщепить пептид от смолы и снять с него защиту, высушенную смолу с пептидом (2,18 г) перемешивают с 2 мл м-крезола и 20 мл фтористого водорода (HF) при 0° в течение 1 часа. После упаривания HF в вакууме остаток промывают безводным диэтиловым эфиром и этилацетатом. Отщепленный и лишенный защиты пептид растворяют в 50% уксусной кислоте и отделяют от смолы путем фильтрации. После разбавления водой и лиофилизации получают 1,51 г неочищенного продукта.
Неочищенный пептид исследуют с помощью аналитической ВЭЖХ, используя жидкостной хроматограф модели HP-1090 фирмы Hewlett-Packard с колонкой Vydac 218TP52 с обращенной фазой (2×250 мм, заполнена силикагелем С18, размер пор 300 , размер частиц 5 мкм) (The Separation Group Inc., Hesperia, CA) и линейный градиент элюции (например, 40-70% В) с помощью системы растворителей, состоящей из (А) 0,1% водной ТФК и (В) 0,1% ТФК в 70% водном MeCN. Растворяют 500 мг неочищенного пептида в AcOH/H2O, перемешивают, фильтруют и наносят на колонку Beckman Ultraprep ODS (21,2×150 мм, заполнена силикагелем С18, размер пор 300
, размер частиц 5 мкм) (The Separation Group Inc., Hesperia, CA) и линейный градиент элюции (например, 40-70% В) с помощью системы растворителей, состоящей из (А) 0,1% водной ТФК и (В) 0,1% ТФК в 70% водном MeCN. Растворяют 500 мг неочищенного пептида в AcOH/H2O, перемешивают, фильтруют и наносят на колонку Beckman Ultraprep ODS (21,2×150 мм, заполнена силикагелем С18, размер пор 300 , размер частиц 10 мкм). Колонку элюируют системой растворителей, описанной выше, с линейным градиентом (например, 30-55% В за 120 мин); скорость потока 6 мл/мин. Элюент контролируют при 220 нм и фракции исследуют с помощью аналитической ВЭЖХ. Фракции с чистотой выше 95% объединяют и лиофилизуют, получая 98 мг чистого продукта. Аналитическая ВЭЖХ проводится на колонке Vydac C18 с обращенной фазой, описанной выше, с применением изократной элюции системой растворителей, описанной выше, со скоростью потока 0,2 мл/мин. Пики анализируются при 220 и 280 нм. Судя по данным аналитической ВЭЖХ, продукт в значительной степени является чистым (> 95%). Молекулярную массу определяют с помощью масс-спектрометрии с электродиспергированием, и ожидаемый аминокислотный состав подтверждают аминокислотным анализом.
, размер частиц 10 мкм). Колонку элюируют системой растворителей, описанной выше, с линейным градиентом (например, 30-55% В за 120 мин); скорость потока 6 мл/мин. Элюент контролируют при 220 нм и фракции исследуют с помощью аналитической ВЭЖХ. Фракции с чистотой выше 95% объединяют и лиофилизуют, получая 98 мг чистого продукта. Аналитическая ВЭЖХ проводится на колонке Vydac C18 с обращенной фазой, описанной выше, с применением изократной элюции системой растворителей, описанной выше, со скоростью потока 0,2 мл/мин. Пики анализируются при 220 и 280 нм. Судя по данным аналитической ВЭЖХ, продукт в значительной степени является чистым (> 95%). Молекулярную массу определяют с помощью масс-спектрометрии с электродиспергированием, и ожидаемый аминокислотный состав подтверждают аминокислотным анализом.
Пример 2
PhAc0-Tyr1-D-Arg2-Asp3-Ala4-Ile5-Phe(pCl)6-Thr7-Asn8-Har9-Tyr(Me)10-Arg11-Lys12-Val13-Leu14-Abu15-Gln16-Leu17-Ser18-Ala19-Arg20-Lys21-Leu22-Leu23-Gln24-Asp25-Ile26-Nle27-D-Arg28-Har29-NH2 (пептид 3)
{[PhAc0, D-Arg2, Phe(pCl)6, Har9, Tyr(Me)10, Abu15, Nle27, D-Arg28, Har29]hGH-RH(1-29)NH2}
Синтез осуществляют постадийно, используя оборудование для твердофазного пептидного синтеза с ручным управлением, Вкратце, смолу на основе МБГА (Bachem, California) (100 мг, 0,070 ммоль) нейтрализуют 5% ДИЭА в CH2Cl2 и промывают в соответствии с протоколом, описанным в таблице 1. Раствор Вос-Har(NO2)-OH (83 мг, 0,25 ммоль) в ДМФА-СН2Сl2 (1:1) встряхивают с нейтрализованной смолой и ДИКД (44 мкл, 0,275 ммоль) в аппарате с ручным управлением для твердофазного пептидного синтеза в течение 1 часа. После завершения реакции связывания, что подтверждается отрицательным нингидриновым тестом, снятия защиты с помощью 50% ТФК в CH2Cl2 и нейтрализации с помощью 5% ДИЭА в CH2Cl2 пептидную цепь строят постадийно путем связывания на смоле следующих защищенных аминокислот в указанном порядке для получения желаемой пептидной последовательности: Boc-D-Arg(Tos)-ОН, Boc-Nle-OH, Boc-Ile-OH, Вос-Asp(OcHx)-ОН, Boc-Gln-OH, Boc-Leu-OH, Boc-Leu-OH, Boc-Lys(2CIZ)-OH, Boc-Arg(Tos)-OH, Boc-Ala-OH, Boc-Ser(Bzl)-OH, Boc-Leu-OH, Boc-Gln-OH, Boc-Abu-OH, Boc-Leu-OH, Boc-Val-OH, Boc-Lys(2CIZ)-OH, Boc-Arg(Tos)-OH, Boc-Tyr(Me)-OH, Boc-Har(NO2)-OH, Boc-Asn-OH, Boc-Thr(Bzl)-OH, Boc-Phe(pCl)-OH, Boc-Ile-OH, Boc-Ala-OH, Boc-Asp(OcHx)-OH, Boc-D-Arg(Tos)-OH, Boc-Tyr(2BrZ)-OH.
Эти защищенные аминокислотные остатки (также обычно поставляемые фирмой Bachem Co.) обозначены выше в соответствии с хорошо известным правилом. Подходящая защитная группа для функциональной группы боковой цепи определенных аминокислот указывается в скобках. Гидроксильные группы в представленных выше формулах указывают, что карбоксильный конец каждого остатка свободен.
Защищенные аминокислоты (0,25 ммоль каждой) связывают с ДИКД (44 мкл, 0,275 ммоль) за исключением Boc-Asn-OH и Boc-Gln-OH, которые связываются с их ранее образованными сложными эфирами с 1-гидроксибензотриазолом. После удаления Nα-Вос-защитной группы от Тyr1 пептид ацилируют с помощью фенилуксусной кислоты (PhAc) (54 мг, 0,4 ммоль), применяя ДИКД (70 мкл, 0,44 ммоль).
Для того чтобы отщепить пептид от смолы и снять с него защиту, высушенную смолу с пептидом (206 мг) перемешивают с 0,5 мл м-крезола и 5 мл фтористого водорода (HF) при 0° в течение 1 часа. После упаривания HF в вакууме остаток промывают безводным диэтиловым эфиром и этилацетатом. Отщепленный и лишенный защиты пептид растворяют в 50% уксусной кислоте и отделяют от смолы путем фильтрации. После разбавления водой и лиофилизации получают 112 мг неочищенного продукта.
Неочищенный пептид исследуют с помощью аналитической ВЭЖХ, используя жидкостной хроматограф модели HP-1090 фирмы Hewlett-Packard с колонкой Vydac 218TP52 с обращенной фазой (2×250 мм, заполнена силикагелем С18, размер пор 300 , размер частиц 5 мкм) (The Separation Group Inc., Hesperia, CA) и линейный градиент элюции (например, 40-70% В) с системой растворителей, состоящей из (А) 0,1% водной ТФК и (В) 0,1% ТФК в 70% водном MeCN. Растворяют 80 мг неочищенного пептида в АсОН/Н2O, перемешивают, фильтруют и наносят на колонку Vydac 218ТР5010 (10×250 мм), заполненную силикагелем С 8. Колонку элюируют системой растворителей, описанной выше, с линейным градиентом (например, 30-55% В за 120 мин); скорость потока 2 мл/мин. Элюент контролируют при 220 нм и фракции исследуют с помощью аналитической ВЭЖХ. Фракции с чистотой выше 95% объединяют и лиофилизуют, получают 9,6 мг чистого продукта. Аналитическая ВЭЖХ осуществляется на колонке Vydac C18 с обращенной фазой, описанной выше, с применением изократной элюции системой растворителей, описанной выше, со скоростью потока 0,2 мл/мин. Пики анализируются при 220 и 280 нм. Судя по данным аналитической ВЭЖХ, продукт в значительной степени является чистым (>95%). Молекулярную массу определяют с помощью масс-спектрометрии с электродиспергированием, и ожидаемый аминокислотный состав подтверждают аминокислотным анализом.
, размер частиц 5 мкм) (The Separation Group Inc., Hesperia, CA) и линейный градиент элюции (например, 40-70% В) с системой растворителей, состоящей из (А) 0,1% водной ТФК и (В) 0,1% ТФК в 70% водном MeCN. Растворяют 80 мг неочищенного пептида в АсОН/Н2O, перемешивают, фильтруют и наносят на колонку Vydac 218ТР5010 (10×250 мм), заполненную силикагелем С 8. Колонку элюируют системой растворителей, описанной выше, с линейным градиентом (например, 30-55% В за 120 мин); скорость потока 2 мл/мин. Элюент контролируют при 220 нм и фракции исследуют с помощью аналитической ВЭЖХ. Фракции с чистотой выше 95% объединяют и лиофилизуют, получают 9,6 мг чистого продукта. Аналитическая ВЭЖХ осуществляется на колонке Vydac C18 с обращенной фазой, описанной выше, с применением изократной элюции системой растворителей, описанной выше, со скоростью потока 0,2 мл/мин. Пики анализируются при 220 и 280 нм. Судя по данным аналитической ВЭЖХ, продукт в значительной степени является чистым (>95%). Молекулярную массу определяют с помощью масс-спектрометрии с электродиспергированием, и ожидаемый аминокислотный состав подтверждают аминокислотным анализом.
Пептид 2 и пептиды от 4 до 30 синтезируют таким же образом, как пептид 1 и пептид 3, за исключением того, что эти пептиды также содержат другие замещения, получая:
[IndAc0, D-Arg2, Phe(pCl)6, Arg9, Abu15, Nle27, D-Arg28, Har29]hGH-RH(1-29)NH, пептид 2;
[PhAc0, D-Arg2, Phe(pCl)6, Наr9, Abu15, Nle27, D-Arg28, Har29]hGH-RH(1-29)NH2 пептид 4;
[Nac0, D-Arg2, Phe(pCl)6, Arg9, Abu15, Nle27, D-Arg28, Har29]hGH-RH(1-29)NH2 пептид 5;
[PhAc0, D-Arg2, Phe(pCl)6, Arg9, Tyr(Me)10, Abu15, Nle27, D-Arg28, Har29]hGH-RH(1-29)NH2 пептид 6;
[PhAc0, His1, D-Arg2, Phe(pCl)6, Arg9, Abu15, Nle27, D-Arg28, Har29]hGH-RH(1-29)NH2 пептид 7;
[Nac0, His1, D-Arg2, Phe(pCl)6, Arg9, Abu15, Nle27, D-Arg28, Har29]hGH-RH(1-29)NH2 пептид 8;
[PhAc0, D-Arg2, Phe(pCl)6, Arg9, Abu15, Nle27, D-Arg29]hGH-RH(1-29)NH2 пептид 9;
[PhAc0, D-Arg2, Phe(pCl)6, Abu15, Arg16, Nle27, D-Arg29]hGH-RH(1-29)NH2 пептид 10;
[PhAc0, D-Arg2, Phe(pCl)6, Abu15, Nle27, D-Arg28, Har29]hGH-RH(1-29)NH2 пептид 11;
[PhAc0, D-Arg2, Phe(pCl)6, Nle9, Abu15, Nle27, D-Arg29]hGH-RHC(1-29)NH2
пептид 12;
[PhAc0, D-Arg2, Phe(pCl)6, Nle13, Nle14, Abu15, Nle27, D-Arg29]hGH-RH(1-29)NH2 пептид 13;
(PhAc0, D-Arg2, Phe(pCl)6, Nle15, Nle27, D-Arg29]hGH-RH(1-29)NH2 пептид 14;
[PhAc0, D-Arg2, Phe(pCl)6, Abu15, Nle18, Nle27, D-Arg29]hGH-RH(1-29)NH2 пептид 15;
[PhAc0, D-Arg2, Phe(pCl)6, Tyr(Me)10, Abu15, Nle27, D-Arg29]hGH-RH(1-29)NH2 пептид 16;
[PhAc0, D-Arg2, Phe(pCl)6, Abu8, Tyr(Me)10, Abu15, Nle27, D-Arg29]hGH-RH(1-29)NH2 пептид 17;
[PhAc0, D-Arg2, Phe(pCl)6, D-Abu8, Тyr(Ме)10, Abu15, Nle27, D-Arg29]hGH-RH(1-29)NH2 пептид 18;
[PhAc0, D-Arg2, Phe(pCl)6, Тyr(Ме)10, Abu15, D-Arg27, Arg28, D-Arg29]hGH-RH(1-29)NH2 пептид 19;
[PhAc0, D-Arg2, Phe(pCl)6, Tyr(Me)9, Abu15, D-Arg27, Arg28, D-Arg29]hGH-RH(1-29)NH2 пептид 20;
[PhAc0, D-Arg2, Phe(pCl)6, Abu15, D-Arg27, Arg28, D-Arg29]hGH-RH(1-29)NH2 пептид 21;
[PhAc0, D-Arg2, Phe(pCl)8, Abu8, Tyr(Me)10, Abu16, D-Arg27, Arg28, D-Arg29]hGH-RH(1-29)NH2 пептид 22;
[PhAc0, D-Arg2, Phe(pCl)6, D-Abu8, Tyr(Me)10, Abu15, D-Arg27, Arg28, D-Arg29]hGH-RH(1-29)NH2 пептид 23;
[PhAc0, D-Arg2, Phe(pCl)6, Lys9, Abu15, Nle27, D-Arg28, Har29]hGH-RH(1-29)NH2 пептид 24;
[PhAc0, D-Arg2, Phe(pCl)6, Orn9, Abu15, Nle27, D-Arg28, Har29]hGH-RH(1-29)NH2 пептид 25;
[PhAc0, D-Arg2, Phe(pCl)6, D-Arg9, Abu15, Nle27, D-Arg28, Har29]hGH-RH(1-29)NH2 пептид 26;
[PhAc0, D-Arg2, Phe(pCl)6, D-Har9, Abu15, Nle27, D-Arg28, Har29]hGH-RH(1-29)NH2 пептид 27;
[PhAc0, D-Arg2, Phe(pCl)6, D-Lys9, Abu15, Nle27, D-Arg28, Har29]hGH-RH(1-29)NH2 пептид 28;
[PhAc0, D-Arg2, Phe(pCl)6, D-Orn9, Abu15, Nle27, D-Arg28, Har29]hGH-RH(1-29)NH2 пептид 29;
[PhAc0, D-Arg2, Phe(pCl)6, Cit9, Abu15, Nle27, D-Arg28, Har29]hGH-RH(1-29)NH2 пептид 30.
Пример 3
Биологическая активность
Пептиды по настоящему изобретению исследовались в in vitro и in vivo тестах на способность ингибировать индуцированное hGH-RH(1-29)NН3 высвобождение GH.
Суперслитая гипофизарная система крысы.
Аналоги исследовались in vitro в тесте, описанном ранее (S. Vigh and A.V. Schally, Peptides 5: 241-347, 1984) с модификацией (Z. Rekasi and A.V. Schally, P.N.A.S. 90:2146-2149, 1993).
Вкратце, клетки предварительно инкубируют с пептидами в течение 9 минут (3 мл) при различных концентрациях. Сразу после инкубирования вводят 1 нМ hGH-RH(1-29)NH2 в течение 3 минут (1 мл) [ответная реакция 0 минут]. Для проверки продолжительности антагонистического эффекта аналога применяют 1 нМ hGH-RH(1-29)NH2 через 30, 60, 90 и 120 минут в течение 3 минут [ответные реакции 30, 60, 90, 120 мин]. Оцениваются результирующие суммарные значения ответов GH. Ответы GH сравниваются и выражаются как процент от первоначального ответа GH, вызванного 1 нМ GH-RH(1-29)NH2. Эффект новых антагонистов сравнивают с от действия [Ас-Туr1, D-Arg2]hGH-RH(1-29)NH2, "стандартного антагониста".
Радиоиммунный анализ гормона роста.
Уровни крысиного GH в аликвотных пробах неразбавленных и разбавленных суперслитых образцов измеряли с помощью радиоиммунного анализа с двойным антителом, используя материалы, предоставляемые Национальной Программой по гормонам и гипофизу (National Hormone and Pituitary Program), Baltimore, Maryland. Результаты радиоиммунного анализа анализировали с помощью компьютерной программы, разработанной в институте, где работают авторы (V. Csernus and A.V. Schally, in Neuroendocrine Research Methods, Harwood Academic (Greenstein, B.D. ed., London, pp. 71-109, 1991), включенной сюда в виде ссылки. Разброс между анализами был менее 15% и разброс внутри серии анализов был менее 10%.
Анализ связывания GH-RH. Для определения характеристик связывания антагонистов GH-RH был проведен анализ связывания чувствительного радиорецептора (G. Halmos, A.V. Schally et al., Receptor 3, 87-97, 1993), на что здесь имеется ссылка. Анализ основывается на связывании меченого [His1, Nle27] hGH-RH (1-32) NH2 с гомогенатами мембран передней доли гипофиза крысы. Иодированные производные [His1, Nle27]hGH-RH(1-32)NH2 получают по методу с хлорамином Т (F.C. Greenwood et al., Biochemistry 89; 114-123, 1963), на что здесь ссылаются. Гипофизы самцов крыс линии Sprague-Dawley (250-300 г) используются для приготовления неочищенных мембран. Для анализов насыщенности связывания гомогенаты мембран инкубируют с, как минимум, 6 концентрациями [His1, 125I-Tyr10, Nle27]hGH-RH(1-32)NH2 в диапазоне от 0,005 до 0,35 нМ в присутствии или отсутствии избытка немеченого пептида (1 мкМ).
Радиоактивность осадка подсчитывают в счетчике гамма-квантов. Сродство пептидов антагониста, исследуемое по отношению к рецепторам GH-RH гипофиза крысы, определяют в экспериментах по конкурентному связыванию. Конечное сродство к связыванию оценивают с помощью Ki (константа диссоциации комплекса ингибитор-рецептор) и определяют с помощью компьютерных программ Ligand PC и McPherson, разработанных Munson и Rodbard (P.J. Munson and D. Rodbard, Anal. Biochem. 107, 220-239, 1980). Относительное сродство по сравнению с [Ас-Туr1, D-Arg2]hGH-RH(l-29)NH2, стандартным антагонистом, рассчитывают как отношение Ki исследуемого антагониста GH-RH к Ki стандартного антагониста.
Тесты in vivo.
Антагонистический эффект аналогов в отношении GH-RH исследовали на молодых самцах крыс линии Sprague-Dawley (200-250 г). В каждом эксперименте использовали 5 групп, из 7 животных каждая. Соединения (80 мкг/кг) и GH-RH(1-29)NH2 (3 мкг/кг) растворяли в 5,5% манните и вводили внутривенно в шейную вену крыс при анестезии нембуталом. Время, протекающее между введениями антагониста и GH-RH, варьировалось между группами в соответствии со следующим графиком. Первая группа животных получала инъекцию GH-RH через 5 мин после введения антагониста; для второй, третьей и четвертой групп животных временные интервалы между инъекцией антагониста и инъекцией GH-RH составляли 15, 30 и 60 мин соответственно. Контрольная группа была сначала инъецирована только растворителем вместо антагониста, после чего через 5 мин делали инъекцию GH-RH.
Образцы крови в 0,4 мл отбирали для радиоиммунного анализа GH перед введением антагониста ("кровь 0") и через 5 мин после инъекции GH-RH ("кровь 1"). Ответ GH в каждой группе рассчитывали как rСН=(СНкровь1/СНкровь0), среднее значение ± стандартная ошибка измерения индивидуальных разностей. Относительное ингибирование ответа GH (%) в каждой подвергшейся обработке группе рассчитывали как 100×(rСНобравотанная-1)/(rСНконтрольная-1).
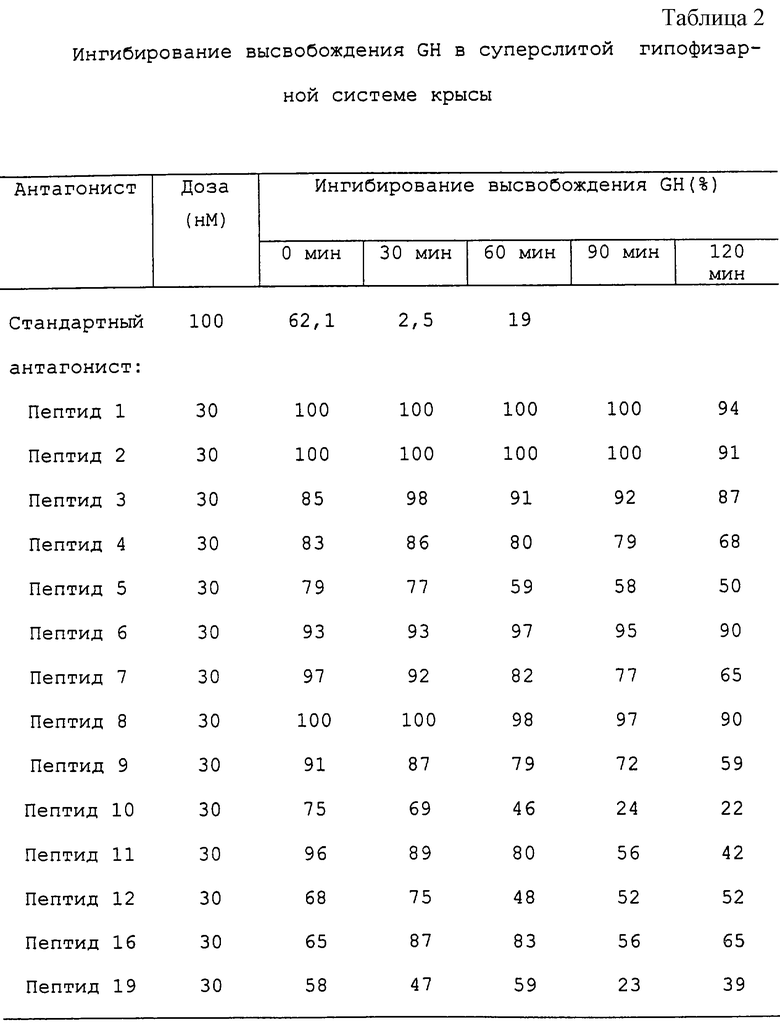
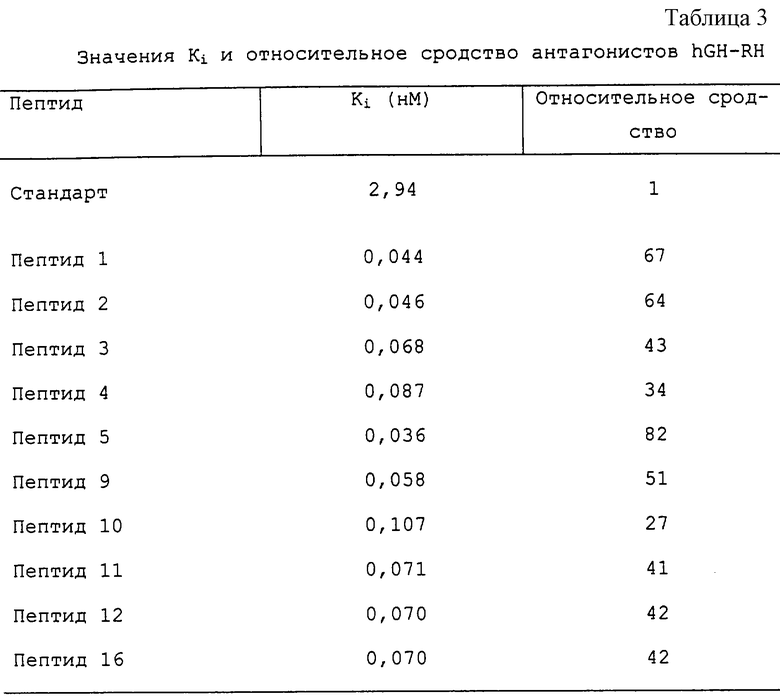
Результаты исследований in vitro.
Результаты антагонистической активности in vitro, исследованной в суперслитой крысиной гипофизарной системе, и анализ связывания подытожены в таблице 2 и таблице 3 соответственно. Как можно видеть из этих данных, замещения, имеющие место в молекулах, вызывают огромное увеличение связывания рецептора, а также ингибирования высвобождения GH in vitro по сравнению со стандартным антагонистом. Наиболее сильный антагонист in vitro, пептид 1, вызывал полное ингибирование вызванного GH-RH высвобождения GH за 90 мин в стандартных условиях анализа. Первый признак восстановления реактивности GH-RH определяли через 120 мин после воздействия данного аналога.
Результаты исследования in vivo.
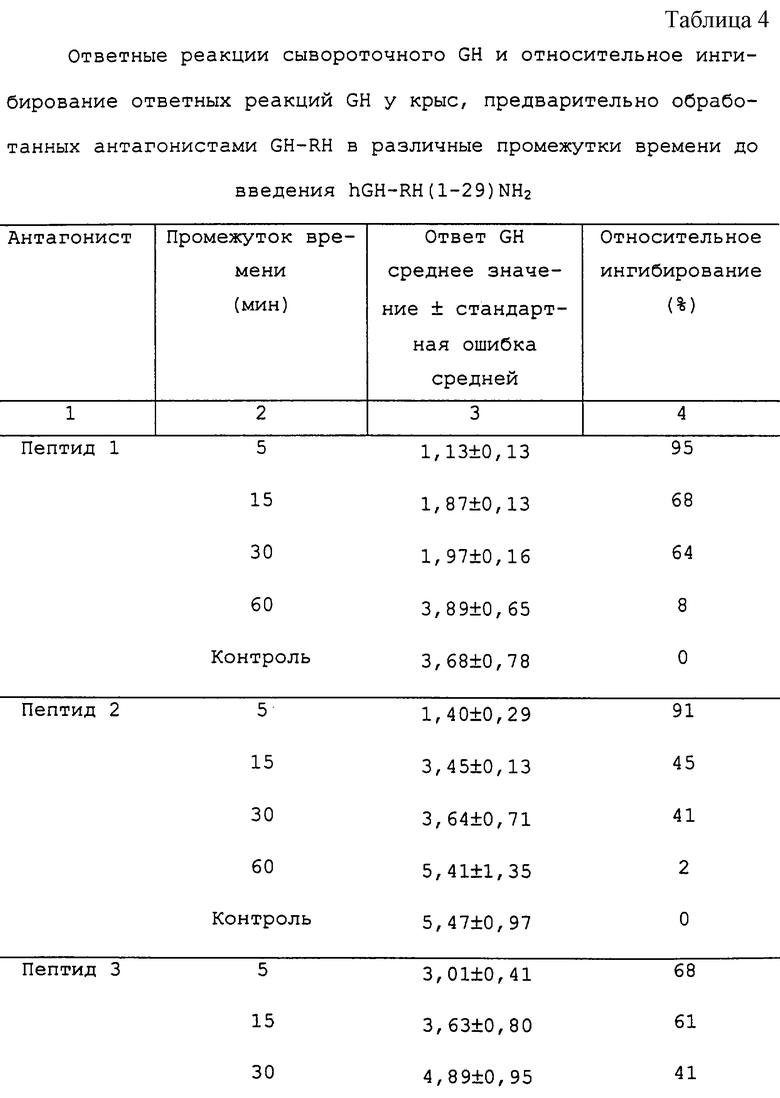
В таблице 4 представлены ответные реакции сывороточного GH и их относительное ингибирование у крыс, предварительно обработанных антагонистами GH-RH. Все из исследуемых аналогов (пептид 1, пептид 2, пептид 3, пептид 4, пептид 8, пептид 9, пептид 11 и пептид 16) вызывают сильное и продолжительное ингибирование высвобождения GH, стимулированного hGH-RH(1-29)NН2. Пептид 1 и пептид 2 являются наиболее сильнодействующими в течение короткого времени, ингибируя ответы GH на 95% и 91%, когда их вводят за 5 мин до hGH-RH(1-29)NH2. Действие этих двух пептидов длится, как минимум, 30 мин. С другой стороны, пептид 11 и пептид 3, которые немного менее активны в течение короткого времени, являются чрезвычайно длительно действующими: их действие длится, как минимум, 60 мин.

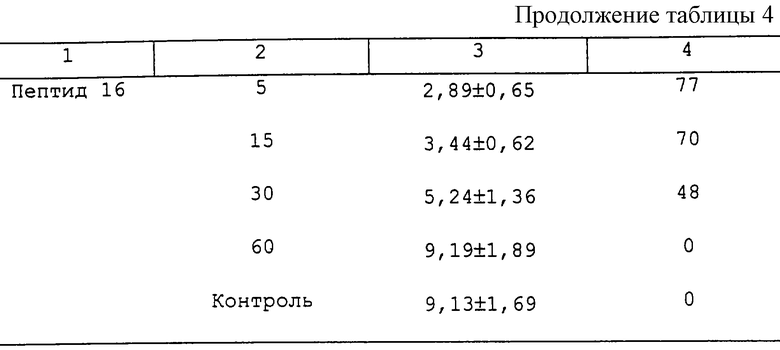
Пример 4
Онкологические исследования.
Противоопухолевую активность пептидов по настоящему изобретению исследовали на различных моделях рака. Противоопухолевые эффекты этих новых пептидов сравнивали с таковыми для ранних аналогов (MZ-4-71 и MZ-5-156, объект патента США №5550212 и заявки на патент США 08/642472).
Действие антагонистов GH-RH на мышиные, относящиеся к молочной железе опухоли МХТ.
Эстроген-независимые опухоли МХТ трансплантировали подкожно самкам мышей BDF. Через день после трансплантации мышей делили на группы из 10 животных каждая и начинали обработку. Мышам в группах 1, 2, 3 и 4 делали ежедневно одноразовые инъекции различных антагонистов GH-RH подкожно в дозе 20 мкг в день в течение 18 дней. В группах 5 и 6 пептиды вводили с помощью осмотических насосов Alzet, высвобождающих ежедневное количество пептида в 20 мкг. Опухоли регулярно измеряли и рассчитывали объем опухоли. Мышей умерщвляли на 18 день и измеряли массы опухолей.
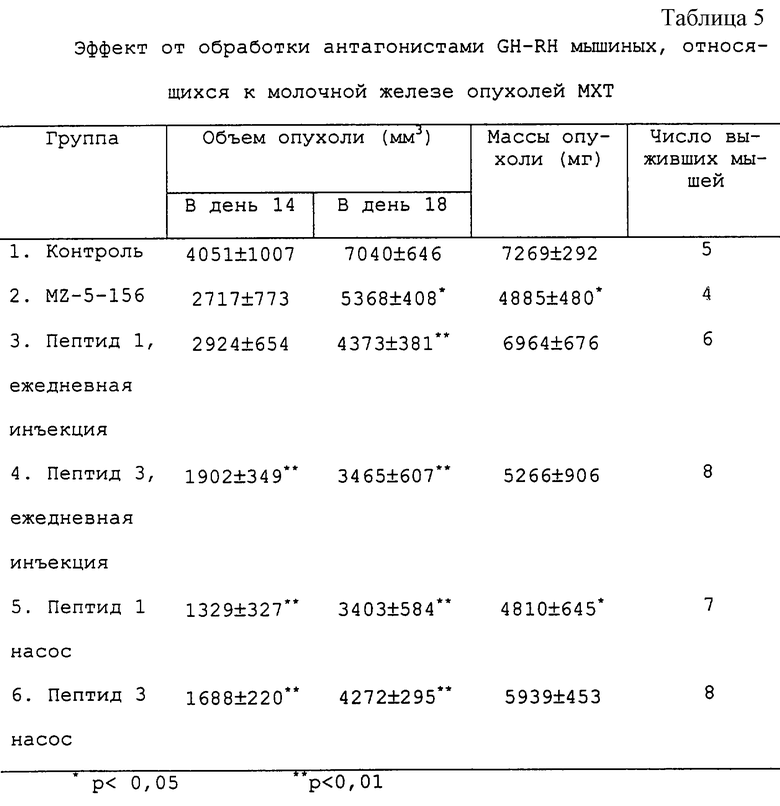
Результаты
Пептиды, пептид 1 и пептид 3, оказывали похожий сильный ингибирующий эффект на мышиные, относящиеся к молочной железе опухоли МХТ. Обработка с помощью MZ-5-156 также приводила к значительному ингибированию роста опухоли, но в этом случае эффект был слабее, чем эффект пептида 1 или пептида 3 (см. таблицу 5 и фиг.1).
Воздействие антагонистов GH-RH на ксенотрансплантаты человеческого рака молочной железы MDA-MB-468 у бестимусных мышей.
Бестимусных мышей с ксенотрансплантатами гормононезависимого рака молочной железы человека MDA-MB-468 делили на группы из 10 животных каждая. Подвергающимся обработке группам делали ежедневно подкожные инъекции из 20 мкг антагонистов GH-RH. Одну группу обрабатывали пептидом 1, вторую группу обрабатывали MZ-5-156 для сравнения. Контрольную группу инъецировали носителем в виде растворителя. Обработку продолжали 5 недель. Опухоли измеряли раз в неделю и рассчитывали объем опухоли. В конце эксперимента мышей умерщвляли и измеряли массы опухолей.
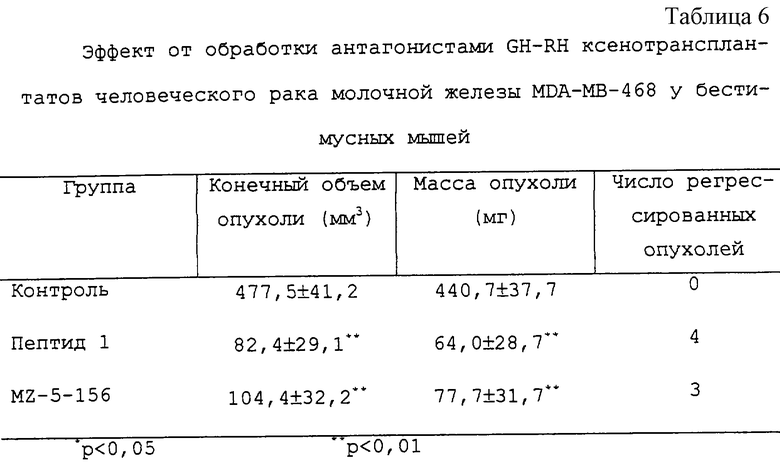
Результаты
Оба пептида обладали значительными ингибирующими опухоль эффектами на ксенотрансплантаты MDA-MB-468. В группе, инъецируемой пептидом 1, 4 опухоли обнаруживали постоянную регрессию во время эксперимента. Подобным же образом MZ-5-156 вызывал регрессию 3 опухолей. Через 5 недель обработки у этих злокачественных опухолей отмечали регрессию до небольших, напоминающих шрам остатков ткани. Гистологическое исследование этих тканей обнаружило недифференцированные эпителиальные опухоли с обширным некрозом и только узкую краевую линию живой опухолевой ткани. По контрасту все опухоли у контрольных животных стабильно прогрессировали. Конечные объемы опухолей и массы в группах, подвергшихся обработке, существенно уменьшались (см. таблицу 6 и фиг.2), пептид 1 проявлял более сильный эффект.
Действие антагонистов GH-RH на ксенотрансплантаты человеческого рака ободочной кишки НТ-29 у бестимусных мышей
Человеческие злокачественные опухоли ободочной кишки НТ-29 трансплантировали подкожно самцам бестимусных мышей. Через 19 дней после трансплантации мышей делили на группы в каждой по 10 животных и начинали обработку. Мышам делали ежедневно одноразовые инъекции различных антагонистов GH-RH подкожно в дозе 20 мкг в день в течение 6 недель. Регулярно измеряли опухоли и рассчитывали объем опухоли. В конце эксперимента мышей умерщвляли и измеряли массы опухолей.
Результаты
Пептид 1 и MZ-5-156 оказывали одинаково сильный ингибирующий эффект на человеческие злокачественные опухоли ободочной кишки НТ-29. Обработка пептидом 9 приводила к меньшему, но еще существенному ингибированию роста опухоли. Пептид 11 и MZ-4-71 проявляли только малый незначительный эффект. (Результаты обобщены в таблице 7 и на фиг.3).

Действие антагониста GH-RH, пептида 1, на ксеноттрансплантаты человеческой глиобластомы U87MG у бестимусных мышей.
Мышам имплантировали подкожно глиобластомы U87MG и, когда опухоли достигали объема примерно 70 мм3, мышей путем случайной выборки делили на 2 экспериментальные группы. Одну группу обрабатывали пептидом 1 в виде одноразовых ежедневных подкожных инъекций в 20 мкг в течение 28 дней, в то время как другая группа служила контролем.
Результаты
Обработка пептидом 1 приводила к ингибированию роста опухоли на 77% через четыре недели обработки против контрольной группы (см. таблицу 8 и фиг.4).

Действие антагонистов GH-RH на ксенотрансплантаты человеческого рака предстательной железы РС-3 у бестимусных мышей.
Самцам бестимусных мышей имплантировали подкожно в оба бока фрагменты ткани в 3 мм3 человеческого гормононезависимого рака предстательной железы РС-3. Когда опухоли достигали объема примерно в 40-50 мм3, мышей делили на 3 экcпepимeнтальные группы. Первую и вторую группы обрабатывали пептидами, пептидом 3 и MZ-5-156 соответственно, ежедневно путем одноразовых подкожных инъекций в 20 мкг в течение 21 дня, в то время как третья группа служила контролем. Объемы опухолей измеряли с интервалами в неделю, массы опухолей измеряли в конце эксперимента.
Результаты
Оба антагониста GH-RH ингибировали рост опухолей РС-3 (см. таблицу 9 и фиг.5). Пептид 3 вызывал более сильное ингибирование роста (65% ингибирования в объеме опухоли и 62% в массе опухоли), чем MZ-5-156 (52% и 46% соответственно).

| название | год | авторы | номер документа |
|---|---|---|---|
| ПЕПТИДНЫЕ АНАЛОГИ GH-RH С АНТАГОНИСТИЧЕСКИМ ДЕЙСТВИЕМ, СПОСОБ СНИЖЕНИЯ УРОВНЯ GH, СПОСОБ СНИЖЕНИЯ УРОВНЯ IGF-I И IGF-II, ПРИМЕНЕНИЕ ДЛЯ ИНГИБИРОВАНИЯ РОСТА РАКОВЫХ КЛЕТОК, ФАРМАКОЛОГИЧЕСКИ ПРИЕМЛЕМАЯ КОМПОЗИЦИЯ (ВАРИАНТЫ) | 2004 |
|
RU2335506C2 |
| Способ получения пептидов | 1984 |
|
SU1477248A3 |
| ЛИПОФИЛЬНЫЕ ПРОИЗВОДНЫЕ ПЕПТИДНЫХ ГОРМОНОВ | 1996 |
|
RU2171261C2 |
| Способ получения пептидов | 1985 |
|
SU1530097A3 |
| Блокаторы рецептора гормона роста при предупреждении заболеваний и лечении | 2016 |
|
RU2726254C2 |
| Способ получения пептидов | 1984 |
|
SU1435157A3 |
| СЛИТОЙ БЕЛОК И СПОСОБ ВЫДЕЛЕНИЯ СЛИТОГО БЕЛКА | 1993 |
|
RU2114119C1 |
| Способ получения пептидов | 1988 |
|
SU1598881A3 |
| ПРОИЗВОДНЫЕ ПЕПТИДОВ - АНАЛОГИ GRF ИЛИ ИХ НЕТОКСИЧНЫЕ СОЛИ | 1990 |
|
RU2096416C1 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ НА ОСНОВЕ α-ЦИКЛОДЕКСТРИНА ДЛЯ ПЕРОРАЛЬНОГО ВВЕДЕНИЯ АНАЛОГОВ РИЛИЗИНГ-ГОРМОНА ЛЮТЕИНИЗИРУЮЩЕГО ГОРМОНА (LH-RH) | 1999 |
|
RU2214270C2 |

Изобретение относится к пептиду, выбранному из группы, имеющей формулу: X-R1-R2-Asp-Ala-R5-R6-Thr-R8-R9-R10-Arg-R12-R13-R14-R15-R16-Leu-R18-R19-Arg-R21-R22-Leu-Gln-Asp-Ile-R27-R28-R29-NH2, где X означает PhAc, IndAc или Nac, R1 означает Tyr или His, R2 означает D-Arg, R5 означает Ile или Val, R6 означает Phe или Phe(Cl), R8 означает Asn, Gln, Ala или D-Asn, R9 означает Arg, Har, Lys, Orn, D-Arg, D-Har, D-Lys, D-Orn, Cit, Nle, Tyr(Me), Ser, Ala или Aib, R10 означает Tyr или Tyr(Me), R12 означает Lys, R13 означает Val или Nle, R14 означает Leu или Nle, R15 означает Gly, Ala, Abu, Nle или Gln, R16 означает Gln или Arg, R18 означает Ser или Nle, R19 означает Ala, R21 означает Lys, R22 означает Leu, Ala или Aib, R27 означает Met, Leu, Nle, Abu или D-Arg, R28 означает Arg, D-Arg или Ser, R29 означает Arg, D-Arg, Har или D-Har, при условии, что когда R9 и R28 означают Ser, R29 не является Arg или Har, и его фармацевтически приемлемым солям. Соединения по изобретению являются антагонистами рилизинг-гормона гормона роста и обладают повышенным сродством к рецептору. Описан также способ подавления избыточных уровней гормона роста у больного, способ лечения злокачественной опухоли и способ ингибирования уровней IGF-II в опухолях. 4 с. и 7 з.п. ф-лы, 9 табл., 5 ил.
X-R1-R2-Asp-Ala-R5-R6-Thr-R8-R9-R10-Arg-R12-R13-R14-R15-R16-Leu-R18-R19-Arg-R21-R22-Leu-Gln-Asp-Ile-R27-R28-R29-NH2,
где X означает PhAc, IndAc или Nac;
R1 означает Туr или His;
R2 означает D-Arg;
R5 означает Ilе или Val;
R6 означает Phe или Phe(Cl);
R8 означает Asn, Gln, Ala или D-Asn;
R9 означает Arg, Har, Lys, Orn, D-Arg, D-Har, D-Lys, D-Orn, Cit, Nle, Tyr(Me), Ser, Ala или Aib;
R10 означает Туr или Туr(Me);
R12 означает Lys;
R13 означает Val или Nle;
R14 означает Leu или Nle;
R15 означает Gly, Ala, Abu, Nle или Gln;
R16 означает Gln или Arg;
R18 означает Ser или Nle;
R19 означает Ala;
R21 означает Lys;
R22 означает Leu, Ala или Aib;
R27 означает Met, Leu, Nle, Abu или D-Arg;
R28 означает Arg, D-Arg или Ser;
R29 означает Arg, D-Arg, Наr или D-Har при условии, что когда R9 и R28 означают Ser, R29 не является Arg или Наr,
и его фармацевтически приемлемые соли.
[PhAc0, D-Arg2, Phe(pCl)6, Arg9, Abu15, Nle27, D-Arg28, Har29]hGH-RH(l-29)NH2 пептид 1
[IndAc0, D-Arg2, Phe(pCl)6, Arg9, Abu15, Nle27, D-Arg28, Har29]hGH-RH(l-29)NH2 пептид 2
[PhAc0, D-Arg2, Phe(pCl)6, Har9, Tyr(Me)10, Abu15, Nle27, D-Arg28, Har29] hGH-RH(1-29)NH2 пептид 3
[PhAc0, D-Arg2, Phe(pCl)6, Arg9, Tyr(Me)10, Abu15, Nle27, D-Arg28, Har29] hGH-RH(1-29)NH2 пептид 6
[PhAc0, His1, D-Arg2, Phe(pCl)6, Arg9, Abu15, Nle27, D-Arg28, Har29]hGH-RH(l-29)NH2 пептид 7
[Nac0, His1, D-Arg2, Phe(pCl)6, Arg9, Abu15, Nle27, D-Arg28, Har29] hGH-RH(1-29)NH2 пептид 8.
| Бесколесный шариковый ход для железнодорожных вагонов | 1917 |
|
SU97A1 |
| АНАЛОГ ПЕПТИДА ФАКТОРА ГОРМОНА РОСТА (ФГР), СПОСОБ СТИМУЛИРОВАНИЯ СЕКРЕЦИИ ГОРМОНА РОСТА И КОМПОЗИЦИЯ ДЛЯ СТИМУЛИРОВАНИЯ СЕКРЕЦИИ | 1991 |
|
RU2119800C1 |
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Огнетушитель | 0 |
|
SU91A1 |

