Изобретение относится к области органической химии, а именно к способу получения перфторированного диацилпероксида - перфтор-2-метил-3-оксагексаноилпероксида, используемого в качестве инициатора радикальной сополимеризации фторированных олефинов.
Известен способ получения фторсодержащего диацилпероксида (ДАП) реакцией фторангидрида полифторкарбоновой кислоты с пероксидом натрия в среде четыреххлористого углерода, фтортрихлорметана (хладон 11) или 1,1,2-трифтортрихлорэтана (хладон 113) при температуре от -10° до -40°С. Например, к раствору 9,75 г пероксида натрия в 100 г хладона 113 при температуре -20°С добавляют по каплям 16,3 г фторангидрида 2,2,3-трифторпропионовой кислоты, перемешивают 3 ч, вводят 1 г воды и еще перемешивают 1 ч, дважды промывают ледяной водой. Получают 2,1 г фторсодержащего ДАП формулы (CH2FCF2COO-)2 в виде раствора в хладоне 113. Полученный пероксид используют в процессе сополимеризации тетрафторэтилена с перфторалкилвиниловым эфиром в среде хладона 113 [заявка Японии №61-152653, кл. С 07 С 179/15, С 08 F 4/36, опубл. 11.07.86]. Указанный способ имеет недостатки. Известно, что пероксид натрия наиболее активен в момент получения. В данном способе применяют готовый пероксид натрия, поэтому его берут в 100%-ном избытке от стехиометрии по отношению к исходному фторангидриду, а поскольку пероксид натрия плохо растворим в хладоие 113, то в результате получают целевой пероксид с низким выходом (12,9%) и очень низкой концентрацией раствора (2,6 мас.%), что делает этот способ непроизводительным при промышленном применении.
Известен способ получения фторированных диацилпероксидов, в том числе перфтор-2-метил-3-оксагексаноилпероксида, в среде жидкого или суперкритического диоксида углерода путем контактирования фторангидрида с пероксидным комплексом, взятым в 2-4-кратном избытке по отношению к фторангидриду, причем пероксидный комплекс вводят в полимеризационную среду в виде твердой фазы, а охлаждение ведут от температуры +40°С до -40°С, с последующей фильтрацией органической фазы для отделения избытка пероксидного комплекса (пат. ЕР №1157988, опубл. 28.11.2001).
В спецстальной автоклав вместимостью 300 мл, предварительно высушенный азотом в течение нескольких часов при температуре 100°С, помещают 2 г (12,7 ммол) сухого перкарбоната натрия. Автоклав закрывают, вакуумируют и охлаждают до температуры -20°С. В литровый спецстальной цилиндр помещают 5,2 мл (24,7 ммол) фторангидрида димера окиси гексафторпропилена (ФА), охлаждают сухим льдом, вакуумируют и вводят 220 г диоксида углерода. Цилиндр соединяют с автоклавом спецстальной трубкой диаметром 3,2 мм, переворачивают и выливают приблизительно 199 г жидкого ФА в автоклав. Содержимое автоклава размешивают со скоростью 5000 об/мин в течение 4 ч при температуре 0°С, при этом температуру в автоклаве поддерживают в интервале от -2°С до +0,5°С, а давление изменяется от 3,29 МПа до 3,59 МПа. Затем содержимое автоклава при перемешивании охлаждают до -27°С, при этом давление снижается до 1,27 МПа, и перемещают в отвакуумированный и охлажденный жидким азотом цилиндр, после этого давление смеси в цилиндре составляет 0,2 атм (20 кПа), Цилиндр открывают и добавляют 100 мл 2,3-дигидроперфторпентана (СF3СFНСНFСF2СF3), чтобы облегчить контроль выхода целевого пероксида. Цилиндр вынимают из ванны с азотом, медленно нагревают до комнаткой температуры и испаряют СО2 по падению давления (в течение 30-45 мин). Содержимое цилиндра в жидком виде сливают в охлажденную сухим льдом полиэтиленовую бутылку. Полученную жидкость три раза промывают водой при комнатной температуре. В результате получают 85 мл жидкости, содержащей 41% перфтор-2-метил-3-оксагексаноилпероксида. После вскрытия на стенках автоклава обнаруживается твердый белый осадок, вес которого по объему примерно соответствует количеству добавленного вначале перкарбоната натрия. Указанный способ имеет недостатки: наличие дополнительных стадий предварительной просушки автоклава и отмывки полученного перфтор-2-метил-3-оксагексаноилпероксида от использованных в избытке твердых реагентов, которые усложняют и затягивают процесс получения готового продукта. Высокая концентрация полученного раствора перфтор-2-метил-3-оксагексаноилпероксида (41% по массе) осложняет его использование. Известно, что чем выше концентрация полученного раствора пероксида, тем меньше устойчивость этого раствора при хранении. Концентрированные растворы перфтор-2-метил-3-оксагексаноилпероксида взрывоопасны и работа с ними требует соблюдения особых мер предосторожности. К тому же получение перфтор-2-метил-3-оксагексаноилпероксида в среде жидкого диоксида углерода при давлении 3,29-3,59 МПа требует использования специальной аппаратуры и повышает опасность метода.
Наиболее близким по совокупности существенных признаков к предлагаемому является способ получения перфтор-2-метил-3-оксагексаноилпероксида в апротонном органическом растворителе, в том числе во фторированном эфире - 2Н-перфтор-5-метил-3,6-диоксанонане, при охлаждении, включающий контактирование фторангидрида с пероксидным комплексом, взятым в избытке по отношению к фторангидриду, причем пероксидный комплекс вводят в полимеризационную среду в виде твердой фазы, с последующей фильтрацией органической фазы для отделения избытка оставшегося пероксидного комплекса (пат. ЕР №1164130, опубл. 28.11.2001). В круглодонную колбу помещают 50 мл растворителя - 2Н-перфтор-5-метил-3,6-диоксанонана и 2,0 г (13 ммол) сухого перкарбоната натрия, что эквивалентно 19 ммол Н2О2. После охлаждения до 0°С к содержимому колбы добавляют 5,2 мл (25 ммол) фторангидрида. Полученную смесь перемешивают магнитной мешалкой в течение 3 ч. Реакционную массу фильтруют через осушитель - сульфат кальция на стекловате для удаления остатков непрореагировавшего перкарбоната натрия. В фильтрате определили 18 ммоль перфтор-2-метил-3-оксагексаноилпероксида, допуская, что объем продукта равен 55 мл, это составляет 79%-ный выход целевого пероксида по отношению к исходному органическому фторангидриду.
Известный способ имеет следующие недостатки. Известно, что пероксиды наиболее активны в момент их получения. В данном способе применяют готовый твердый пероксидный комплекс - пероксидикарбонат натрия и берут его в избытке от стехиометрии по отношению к исходному фторангидриду, а поскольку предлагается использовать пероксидные комплексы, нерастворимые в апротонных растворителях, в твердой фазе, то в результате для контактирования пероксидного комплекса и фторангидрида требуется довольно длительное время. Кроме того, возникает необходимость введения дополнительной стадии фильтрации получаемого перфтор-2-метил-3-оксагексаноилпероксида от остатков непрореагировавшего пероксида натрия, что усложняет известный способ, повышает его опасность. Возникает необходимость утилизации осушителя - сульфата кальция на стекловате с остатками целевого пероксида и растворителя, что приводит к увеличению отходов.
Техническая задача настоящего изобретения состоит в сокращении времени контактирования пероксидного комплекса с фторангидридом и в упрощении процесса получения перфтор-2-метил-3-оксагексаноилпероксида.
Поставленная задача решается способом получения перфтор-2-метил-3-оксагексаноилпероксида, включающим контактирование фторангидрида перфтор-2-метил-3-оксагексановой кислоты с водным раствором пероксида водорода при охлаждении в среде жидкого фторсодержащего галогензамещенного углеводорода, согласно изобретению контактирование проводят путем одновременной раздельной подачи фторангидрида перфтор-2-метил-3-оксагексановой кислоты и водного раствора гидроксида натрия в контактную массу, образованную предварительным смешением водного раствора пероксида водорода с фторсодержащим галогензамещенным углеводородом.
Контактирование проводят при отношении суммарного объема водных растворов к объему органической фазы (0,28-0,85):1.
Для образования контактной массы водный раствор пероксида водорода берут с массовой концентрацией 5-10% в объемном отношении к фторсодержащему галогензамещенному углеводороду (0,15-0,46):1.
Водный раствор гидроксида натрия подают на контактирование с массовой концентрацией 5-10% при мольном отношении гидроксида натрия к пероксиду водорода (0,87-1,2):1.
В качестве фторсодержащего галогензамещенного углеводорода используют предпочтительно перфторметилциклогексан или 1-гидро-4-хлороктафторбутан.
Контактирование ведут в интервале температуры от -5°С до +5°С.
Осуществление способа подтверждено лабораторными примерами.
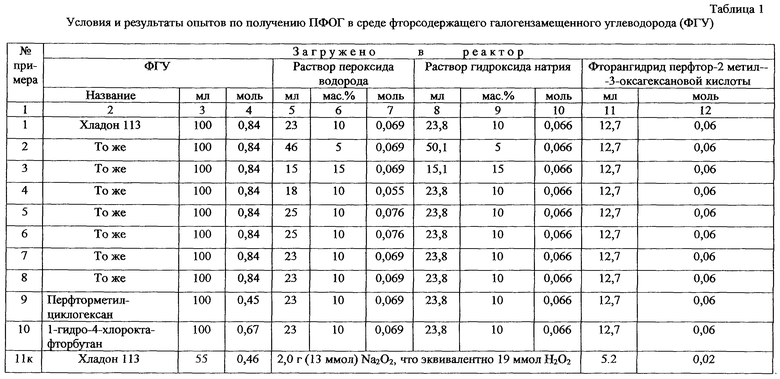
Пример 1. Перфтордиацилпероксид, а именно перфтор-2-метил-3-оксагексаноилпероксид (ПФОГ), получают в круглодонной четырехгорлой колбе, снабженной мешалкой, термометром и двумя капельными воронками для раздельной подачи реагентов. В колбу загружают 100 мл (0,84 моль) хладона 113, охлаждают его до температуры 0°С и добавляют 23 мл (0,069 моль) пероксида водорода в виде 10%-ного (по массе) раствора в воде. Водный раствор пероксида водорода берут в объемном отношении к фторсодержащему галогензамещенному углеводороду 0,23:1. В полученную контактную массу с температурой 0°С при интенсивном перемешивании одновременно из двух дозаторов подают 12,7 мл (19,9 г или 0,06 моль) фторангидрида перфтор-2-метил-3-оксагексановой кислоты CF3CF2CF2OCF(CF3)C(O)F и 23,8 мл (0,066 моль) гидроксида натрия в виде 10%-ного (по массе) раствора, при этом мольное отношение пероксида водорода к фторангидриду составляет 1,1:1. Подачу реагентов ведут с такой скоростью, чтобы температура реакционной массы не превышала 0°С. После окончания подачи реагентов перемешивание реакционной массы продолжают в течение 1 ч, выдерживая ту же температуру. Затем реакционную массу переносят в делительную воронку, отстаивают и отделяют нижний органический слой, представляющий собой раствор полученного ПФОГ в хладоне 113, от водного раствора минеральных веществ. При отстое расслоение слоев происходит практически мгновенно, граница слоев четкая, промывки органического перекисного слоя не требуется. Получают 16,9 г ПФОГ в виде 9,5%-ного (по массе) раствора в хладоне 113 (анализируют иодометрическим титрованием). Полученный раствор используют в качестве инициатора в процессе сополимеризации тетрафторэтилена (ТФЭ) с гексафторпропиленом (ГФП), которую проводят следующим образом.
Используют реактор вместимостью 1,6 л из хромоникелевой стали, снабженный высокоскоростной пропеллерной мешалкой (1500 об/мин), рубашкой для обогрева, капельницей и шприцевым устройством для ввода раствора инициатора и ввода регулятора молекулярной массы. Реактор герметизируют, охлаждают до температуры -25°С и вакуумируют до остаточного давления 0,001 МПа. В охлажденную до той же температуры капельницу помещают 1 г полученного инициатора ПФОГ в виде 8%-ного (по массе) раствора в хладоне 113, капельницу герметизируют и содержимое ее сливают в реактор. Затем в реактор загружают 600 г сжиженного ГФП. Содержимое реактора при перемешивании нагревают до заданной температуры, соответствующей выбранному инициатору, в данном примере 36°С. В реактор подают 55 г ТФЭ и проводят сополимеризацию мономеров при постоянной температуре и установившемся давлении, добавляя в реактор подпиточную смесь, содержащую 10 мол.% ГФП и 90 мол.% ТФЭ, до исходного давления каждый раз при снижении давления на 0,02 МПа. По израсходовании 50 г подпиточной смеси в реактор добавляют 14 мг (в виде раствора в хладоне 113) метанола в качестве регулятора молекулярной массы сополимера. Процесс сополимеризации ведут до израсходования 200 г подпиточной смеси в течение 4,5 ч. Затем реакцию прекращают путем остановки мешалки и сдувки непрореагировавших мономеров в предварительно охлажденную и отвакуумированную емкость. Полимер дегазируют путем прогрева под вакуумом, реактор охлаждают и вскрывают. Полученный сополимер прогревают еще в течение 24 ч при 120°С и определяют его свойства. Получают 190 г белого порошка сополимера ТФЭ с ГФП.
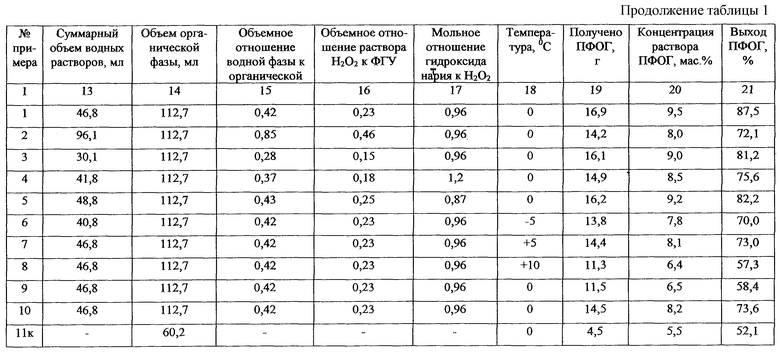
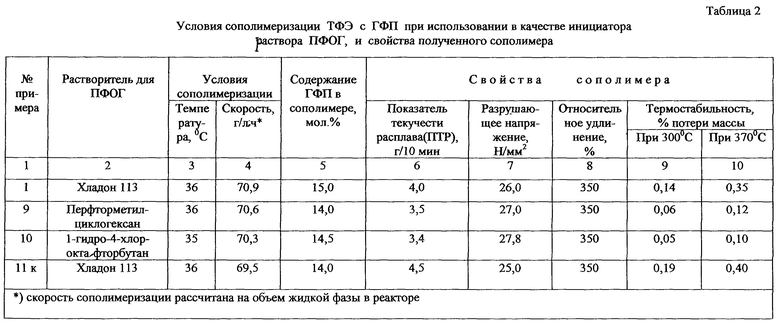
Условия получения раствора перфтордиацилпероксида и результаты по этому и последующим примерам сведены в таблицу 1, а условия и результаты сополимеризации с использованием полученного инициатора - в таблицу 2.
Пример 2. ПФОГ получают, как описано в примере 1, но в колбу загружают 46 мл (0,069 моль) пероксида водорода и 50,1 мл (0,066 моль) гидроксида натрия, которые используют в виде 5%-ных (по массе) растворов. При отстое расслоение слоев происходит достаточно быстро, граница слоев четкая и промывки органического перекисного слоя не требуется. Получают 14,2 г ПФОГ в виде 8,0%-ного (по массе) раствора в хладоне 113.
Пример 3. ПФОГ получают, как описано в примере 1, но в колбу загружают 15 мл (0,069 моль) пероксида водорода и 15,1 мл (0,066 моль) гидроксида натрия, которые используют в виде 15%-ных (по массе) растворов. При отстое расслоение слоев происходит удовлетворительно, но граница слоев нечеткая, появляется легкая муть, которую убирают промывкой органического перекисного слоя холодной водой при температуре 0°С. Получают 16,1 г ПФОГ в виде 9,0%-ного (по массе) раствора в хладоне 113.
Пример 4. ПФОГ получают, как описано в примере 1, но используют 18 мл (0,055 моль) пероксида водорода. При отстое расслоение слоев происходит достаточно быстро, граница слоев четкая, промывки органического перекисного слоя не требуется. Получают 14,9 г ПФОГ в виде 8,5%-ного (по массе) раствора в хладоне 113.
Пример 5. ПФОГ получают, как описано в примере 1, но используют 25 мл (0,076 моль) пероксида водорода. При отстое расслоение слоев происходит достаточно быстро, граница слоев четкая, промывки органического перекисного слоя не требуется. Получают 16,2 г ПФОГ в виде 9,15%-ного (по массе) раствора в хладоне 113.
Пример 6. ПФОГ получают, как описано в примере 1, но используют 25 мл (0,076 моль) пероксида водорода. Кроме того, в контактную массу при температуре -5°С последовательно подают 12,7 мл (0,06 моль) фторангидрида перфтор-2-метил-3-оксагексановой кислоты и 23,8 мл (0,066 моль) гидроксида натрия в виде 10%-ного (по массе) раствора. При отстое расслоение слоев происходит достаточно быстро, граница слоев четкая, промывки органического перекисного слоя не требуется. Получают 13,8 г ПФОГ в виде 7,8%-ного (по массе) раствора в хладоне 113.
Пример 7. ПФОГ получают, как описано в примере 1, но подачу фторангидрида и гидроксида натрия ведут с такой скоростью, чтобы температура реакционной массы не превышала +5°С. После окончания подачи реагентов при перемешивании выдерживают ту же температуру. При отстое расслоение слоев происходит достаточно быстро, граница слоев четкая, и промывки органического перекисного слоя не требуется. Получают 14,4 г ПФОГ в виде 8,1%-ного (по массе) раствора в хладоне 113.
Пример 8. ПФОГ получают, как описано в примере 1, но подачу фторангидрида и гидроксида натрия ведут с такой скоростью, чтобы температура реакционной массы не превышала +10°С. После окончания подачи реагентов при перемешивании выдерживают ту же температуру. При отстое расслоение слоев происходит достаточно быстро, граница слоев четкая и промывки органического перекисного слоя не требуется. Получают 11,3 г ПФОГ в виде 6,4%-ного (по массе) раствора в хладоне 113.
Пример 9. ПФОГ получают, как описано в примере 1, но в качестве фторсодержащего галогензамещенного углеводорода берут озонобезопасный растворитель - перфторметилциклогексан С6F11СF3 (хладон 350) в количестве 100 мл (0,45 моль). При отстое расслоение слоев происходит практически мгновенно, граница слоев очень четкая и промывки органического перекисного слоя не требуется. При этом получают 11,5 г ПФОГ в виде 6,5%-ного (по массе) раствора в указанном растворителе.
Сополимеризацию ТФЭ с ГФП проводят, как описано в примере 1, но с использованием в качестве инициатора полученного раствора, при этом получают 200 г белого порошка сополимера.
Пример 10. ПФОГ получают, как описано в примере 1, но в качестве фторсодержащего галогензамещенного углеводорода используют озонобезопасный растворитель - 1-гидро-4-хлороктафторбутан С4F8СlН (хладон 328) в количестве 100 мл (0,67 моль). При отстое расслоение слоев происходит практически мгновенно, граница слоев четкая, промывки органического перекисного слоя не требуется. При этом получают 14,5 г ПФОГ в виде 8,2%-ного (по массе) раствора в хладоне 328.
Сополимеризацию ТФЭ с ГФП проводят, как описано в примере 1, но с использованием в качестве инициатора полученного раствора, при этом получают 250 г белого порошка сополимера.
Пример 11 (контрольный). Синтез ПФОГ осуществляют в условиях прототипа. В круглодонную колбу помещают 50 мл растворителя - хладона 113 и 2,0 г (13 ммол) сухого перкарбоната натрия, что эквивалентно 19 ммол H2O2. После охлаждения до 0°С к содержимому колбы добавляют 5,2 мл (25 ммол) фторангидрида. Полученную смесь перемешивают магнитной мешалкой в течение 3 ч. Получают реакционную массу, представляющую собой взвесь непрореагировавшего перкарбоната натрия в растворе ПФОГ в хладоне 113, которую фильтруют через осушитель - сульфат кальция на стекловате для удаления твердой фазы. Полученный фильтрат вновь помещают в колбу, добавляют осушитель и выдерживают при перемешивании еще 1 ч (дополнительная сушка ПФОГ от образовавшейся в синтезе воды). Смесь вновь фильтруют от осушителя. Получают 4,5 г перфтор-2-метил-3-оксагексаноилпероксида в виде 5,5%-ного (по массе) раствора в хладоне 113, это составляет 52%-ный выход целевого пероксида по отношению к исходному органическому фторангидриду.
Сополимеризацию ТФЭ с ГФП проводят, как описано в примере 1, но с использованием в качестве инициатора полученного раствора, при этом получают 150 г белого порошка сополимера.
Из представленных примеров видно, что предлагаемый способ обеспечивает получение ПФОГ, пригодного для получения фторсополимера, при этом позволяет в отличие от прототипа сократить время контактирования пероксидного комплекса с фторангидридом, поскольку характеризуется более высокой скоростью реакции (реакция проходит за 1ч, а по прототипу за 3 ч).
Предлагаемый способ проще, чем прототип, поскольку он не требует сложного разделения фаз и дополнительной промывки продукта. Это достигается особым порядком ввода реагентов в процесс, изменением соотношения объемов водной и органической фаз, подбором концентраций реагентов в водных растворах и температурного интервала процесса (см. табл.1, примеры 1 и 11к).
Поскольку пероксид натрия взят в виде водного раствора и в недостатке по отношению к ФГУ, это позволяет устранить образование твердой фазы и при его реализации в отличие от прототипа отсутствует необходимость фильтрации получаемого ПФОГ (от остатков непрореагировавшего пероксида натрия), что упрощает известный способ и снижает его опасность при промышленной реализации. Кроме того, в прототипе существует необходимость дополнительной утилизации осушителя - сульфата кальция на стекловате с остатками целевого пероксида и растворителя, это приводит к появлению твердых неорганических отходов сульфата кальция и стекловаты, что усложняет известный метод.
Предлагаемая в прототипе сушка раствора пероксида в динамическом режиме (во время фильтрации) неэффективна и при реализации требует длительного времени и дополнительной стадии последующей фильтрации раствора целевого пероксида от осушителя. Все это приводит к потерям (до 20 мас.%) растворителя и получаемого пероксида, снижает производительность известного метода и повышает стоимость конечного продукта.
Предлагаемый способ позволяет получать ПФОГ с требуемой концентрацией раствора, удобной для использования (6,4-9,5 мас.%), по сравнению с прототипом он экономичнее, проще и безопаснее, характеризуется более высоким выходом целевого продукта.
При синтезе ПФОГ в среде озонобезопасных растворителей - перфтор-метилциклогексана или 1-гидро-4-хлороктафторбутана получаемый раствор инициатора наиболее перспективен для использования в процессе сополимеризации ТФЭ с ГФП, т.к. получаемый в этом случае сополимер имеет лучшую термостабильность (см. табл.2, примеры 9 и 10).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ПЕРФТОР-2-МЕТИЛ-3-ОКСАГЕКСАНОИЛПЕРОКСИДА | 2002 |
|
RU2213730C1 |
| СПОСОБ ПОЛУЧЕНИЯ СОПОЛИМЕРА ТЕТРАФТОРЭТИЛЕНА С ГЕКСАФТОРПРОПИЛЕНОМ | 2001 |
|
RU2195466C1 |
| СПОСОБ ПОЛУЧЕНИЯ СОПОЛИМЕРА ТЕТРАФТОРЭТИЛЕНА С ПЕРФТОРПРОПИЛВИНИЛОВЫМ ЭФИРОМ | 2001 |
|
RU2195465C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПЕРФТОРДИАЦИЛПЕРОКСИДА | 2001 |
|
RU2203273C1 |
| СПОСОБ ПОЛУЧЕНИЯ СОПОЛИМЕРОВ ТЕТРАФТОРЭТИЛЕНА С 12-15 МОЛ.% ГЕКСАФТОРПРОПИЛЕНА | 1996 |
|
RU2109761C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПЕРФТОРИРОВАННЫХ СОПОЛИМЕРОВ С ФУНКЦИОНАЛЬНЫМИ СУЛЬФОНИЛФТОРИДНЫМИ ГРУППАМИ | 2002 |
|
RU2230075C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТЕРМОПЕРЕРАБАТЫВАЕМЫХ СОПОЛИМЕРОВ ТЕТРАФТОРЭТИЛЕНА С ГЕКСАФТОРПРОПИЛЕНОМ | 2011 |
|
RU2463312C1 |
| ЛАТЕКС ФТОРПОЛИМЕРА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФТОРПОЛИМЕР | 2005 |
|
RU2376332C2 |
| ВОДОЭМУЛЬСИОННАЯ ПОЛИМЕРИЗАЦИЯ ФТОРИРОВАННЫХ МОНОМЕРОВ С ИСПОЛЬЗОВАНИЕМ ФТОРСОДЕРЖАЩЕГО ПОВЕРХНОСТНО-АКТИВНОГО ВЕЩЕСТВА | 2006 |
|
RU2406731C2 |
| СПОСОБ ПОЛУЧЕНИЯ СОПОЛИМЕРОВ ТЕТРАФТОРЭТИЛЕНА С ГЕКСАФТОРПРОПИЛЕНОМ | 2001 |
|
RU2206580C1 |
Изобретение относится к органической химии, а именно к синтезу перфорированного диацилпероксида, конкретно перфтор-2-метил-3-оксагексаноилпероксида, используемого в качестве инициатора радикальной сополимеризации фторированных олефинов. Фторангидрид перфтор-2-метил-3-оксагексановой кислоты и водный раствор гидроксида натрия одновременно раздельными потоками подают в контактную массу, образованную смешением водного раствора НрОр с фторсодержащим галогензамещенным углеводородом (ФГУ), предпочтительно перфторметилциклогексаном или 1-гидро-4-хлороктафторбутаном. Используют 5-10%-ные водные растворы реагентов. Предпочтительный режим: объемное отношение водной фазы к органической 0,28-0,80:1, Н2О2 к ФГУ 0,15-0,46:1, мольное отношение гидроксида натрия к Н2О2 0,87-1,2:1. Технический результат - сокращение расхода реагентов, повышение безопасности, улучшение термостабильности полученного сополимера с данным инициатором. 6 з.п. ф-лы, 2 табл.
| Шасси прицепа | 1983 |
|
SU1164130A1 |
| СПОСОБ ПОЛУЧЕНИЯ ПЕРФТОРДИПРОПИОНИЛПЕРОКСИДА | 1996 |
|
RU2154057C2 |
| US 4654444 A, 31.03.1987 | |||
| JP 61152653 A, 11.07.1986. | |||

