Настоящее изобретение относится к способам идентификации генов, участвующих в адаптации микроорганизма в среде его обитания, особенно к способам идентификации генов, ответственных за вирулентность патогенного микроорганизма.
Область техники, к которой относится изобретение
Антибиотическая резистентность бактериальных и других патогенов становится все более важной проблемой. В связи с этим важны новые терапевтические подходы к борьбе с патогенными микроорганизмами.
Патогенные микроорганизмы способны избегать воздействия защитных механизмов хозяина и могут расти в плохой питательной среде, вызывая заражение. Для этого микроорганизм должен содержать ряд "вирулентных" генов.
Вирулентные гены могут быть определены с использованием приемов классической генетики. С помощью транспозонного мутагенеза осуществляют идентификацию бактериальных вирулентных генов. Так, например, мутанты подвергали скринингу на определенные физиологические дефекты, такие как потеря железорегулируемых белков (Holland с сотр., 1992), или использовали в анализах для изучения пенетрации эпителиальных клеток (Finlay с сотр., 1988) и выживания в макрофагах (Fields с сотр., 1989; Miller с сотр., 1989а; Groisman с сотр., 1989). Транспозонные мутанты также тестировали на измененную вирулентность живых животных инфицированных моделей (Miller с сотр., 1989b). Такой подход имеет преимущество в том, что могут идентифицироваться гены, играющие важную роль на различных стадиях заражения, но имеет ограничения, связанные с необходимостью индивидуального тестирования большого числа мутантов на изменения вирулентности. Miller с сотр. (1989b) использовали группы из 8-10 мышей и инфицировали орально 95 отдельных групп различными мутантами, вследствие чего использовали 760-950 мышей. В связи с тем, что требуется чрезвычайно большое количество животных, полный скрининг бактериального генома на вирулентные гены не представляется возможным.
Недавно была описана генетическая система (технология экспрессии in vivo (IVET)), которая позволяет осуществлять положительную селекцию специфически инфицированных генов Salmonella (Mahan с сотр., 1993). Такая техника позволяет идентифицировать гены, которые экспрессируются на конкретной стадии процесса инфицирования. Однако эта техника не позволяет идентифицировать вирулентные гены, которые регулируются посттранскрипционно и, что более важно, не обеспечивает информацией о том, действительно ли идентифицированные гены требуются для процесса инфицирования или способствуют ему.
Lee и Falkow (1994) в Methods Enzymol. 236, 531-545 описывают способ идентификации факторов, влияющих на инвазию Salmonella в клетки млекопитающих in vitro, путем выделения гиперинвазивных мутантов.
Walsh и Серко (1992) Science 255, 434-440 описывают способ отслеживания пространственного расположения недифференцированных клеток коры головного мозга в ходе развития коры головного мозга крыс. В методе Walsh и Серко используется метка, содержащая уникальную последовательность нуклеиновой кислоты и IacZ ген, однако не имеется указаний о возможности детекции этим методом ценных мутантов или генов.
В WO 94/26933 и в статье Smith с сотр., (1995) Proc.Natl. Acad.Sci.USA 92, 6479-6483 описываются методы, нацеленные на идентификацию функциональных участков известного гена или, по крайней мере, молекулы ДНК, о последовательности которой имеется некоторая информация.
Groisman с сотр. (1993) в Proc-Natl.Acad.Sci, США 90, 1033-1037 описывают молекулярный, функциональный и эволюционный анализ последовательностей специфичный на Salmonella.
Hensel с сотр. (1995) в Science 269, 400-403 описывают одновременную идентификацию бактериальных вирулентных генов путем негативного отбора. Эта статья была опубликована в июле 1995 г.
Slauch с сотр. (1994) в Methods in Enzymol. 235, 481-492 описывают технологию экспрессии in vivo для селекции бактериальных генов специфически индуцированных в тканях хозяина.
Pascopella с сотр. (1994) в Inf. Immun. 62, 1313-1319 описывают использование in vivo комплементации в Mycobacterium tuberculosis для идентификации геномного фрагмента, связанного с вирулентностью.
В патенте США 5397697 описывается идентификация растительно-чувствительных генов бактерий.
Некоторые вирулентные гены уже известны для таких патогенных микроорганизмов, как Escherichia coli, Salmonella typhimurium, Salmonella typhi, Vibrio cholerae, Clostridium botulinum, Versinia pestis, Shigella flexneri и Listeria monocytogenes, однако во всех случаях идентифицировано лишь небольшое их число.
Заболевание, которое Salmonella typhimurium вызывает у мышей, обеспечивает хорошую экспериментальную модель тифоидной лихорадки (Carter и Collins, 1974). К настоящему времени идентифицировано примерно сорок два гена, влияющих на вирулентность Salmonella (Groisman и Ochman, 1994). Это составляет примерно одну треть от общего числа предсказанных вирулентных генов (Groisman и Saier, 1990).
Цель настоящего изобретения заключается в идентификации генов, принимающих участие в адаптации микроорганизма к среде его обитания, особенно в идентификации дополнительных вирулентных генов в патогенных микроорганизмах, с повышенной эффективностью. Другая цель изобретения состоит в уменьшении числа экспериментальных животных, используемых для идентификации генов. Другие цели настоящего изобретения состоят в разработке вакцин и методов скрининга лекарственных средств, уменьшающих вирулентность.
Краткое изложение сущности изобретения
Первый аспект изобретения относится к разработке способа идентификации микроорганизма с пониженной адаптацией к конкретной среде, включающему стадии:
(1) обеспечения множества микроорганизмов, каждый из которых независимо мутирован инсерционной инактивацией гена с помощью нуклеиновой кислоты, включающей уникальную маркерную последовательность так, что каждый мутант содержит различную маркерную последовательность, или клонов, указанных микроорганизмов;
(2) индивидуального получения хранимого образца каждого мутанта, полученного на стадии (1) и обеспечения индивидуально накопленной нуклеиновой кислоты, включающей особую маркерную последовательность от каждого индивидуального мутанта;
(3) введения множества мутантов, полученных на стадии (1), в указанную конкретную среду и культивирования микроорганизмов в указанной среде;
(4) извлечение микроорганизмов из указанной среды или ее выбранной части и выделение нуклеиновой кислоты из извлеченных микроорганизмов;
(5) сравнения любой маркерной последовательности нуклеиновой кислоты, выделенной на стадии (4), с особой маркерной последовательностью каждого индивидуального мутанта, полученного на стадии (2) и
(6) отбора индивидуального мутанта, не содержащего какую-либо из маркерных последовательностей, выделенных на стадии (4).
Таким образом, в таком способе используется негативная селекция для идентификации микроорганизмов с пониженной способностью к пролиферации в окружающей среде.
Микроорганизм может жить в различных средах и известно, что конкретные гены и их продукты позволяют микроорганизму адаптироваться к окружающей среде обитания. Так, например, для выживания таких патогенных микроорганизмов, как патогенные бактерии или патогенные грибки, в их хозяине требуется продукт одного или более вирулентных генов. Таким образом, в предпочтительном техническом решении изобретения ген микроорганизма, позволяющий ему адаптироваться к конкретной окружающей среде, представляет собой вирулентный ген.
Предпочтительная конкретная окружающая среда представляет собой дифференцированный многоклеточный организм, такой как растение или животное. Известно, что многие бактерии и грибки инфецируют растения, они способны выживать в нем и вызывать заболевание из-за наличия вирулентных генов или их экспрессии. Подходящие микроорганизмы в том случае, когда конкретная окружающая среда представляет собой растение, включают бактерии Agrobacterium tumefaciens, которые образуют опухоли (галлы) особенно на винограде; Erwina amylovara; Pseudomonas solanacearum, которая вызывает увядание большого числа растений; Rhizobium leguminosarum, которая вызывает заболевание бобов; Xanthomonos campestris p.у. citri, которая вызывает язву цитрусовых фруктов; и включают грибок Magnaporthe grisea, который вызывает пирикуляриоз риса; Fusarium spp., который вызывает множество болезней растений; Erisyphe spp., Colletotrichum gloeosporiodes; Gaeumannomyces qraminis, который вызывает заболевания корневой системы и кроны хлебных злаков и трав; Glomus spp., Laccaria spp.; Leptosphaeria maculans; Phoma tracheiphila; Phytophthora spp., Pyrenophora teres; Verticillium alboatrum и V.dahliae; а также Mycosphaerella musicola и M.fijiensis. Как будет подробно описано ниже, в том случае, когда микроорганизм представляет собой грибок, для его жизненного цикла требуется гаплоидная фаза.
Так известно, что многие микроорганизмы, включающие бактерии, грибы, простейшие животные и трипаносомы, инфецируют животных, особенно млекопитающих, включая людей. Выживание микроорганизмов в организме животного и его способность вызывать заболевания в основном связаны с наличием вирулентных генов и их экспрессии. Такие бактерии включают Bordetella spp., особенно В.pertussis, Campylobacter, особенно C.jejuni, Clostridium spp., особенно C.botulinum, Enterococcus spp., особенно E.faecalis, Escherichia spp., особенно E.coli, Haemophilus spp., особенно H.ducreyi и H.influenzae, Helicobacter spp., особенно H.pylori, Klebsiella spp., особенно К.pneumoniae, Legionella spp., особенно L.pneumophila, Listeria spp., особенно L.monocytogenes, Mycobacterium spp., особенно М.smegmatis и M.tuberculosis, Neisseria spp., особенно N.gonorrhoeae и N.meningitidis, Pseudomonas spp., особенно Ps.aeruginosa, Salmonella spp., Shigella spp., Staphylococcus spp., особенно S. aureus, Streptococcus spp., особенно S.pyogenes и pneumoniae, Vibrio spp. и Yersinia spp., особенно Y.pestis. Все такие бактерии вызывают болезни людей и животных. В случае применения бактерий в способе изобретения, конкретной окружающей средой является животное, которое они способны инфицировать и у которого они вызывают болезнь. Так, например, в случае инфицирования мышей Salmonella typhimurium у мышей развивается заболевание, служащее моделью тифоидной лихорадки людей. Staphylococcus aureus вызывает бактериемию и образование ренального абсцесса у мышей (Albus с сотр. (1991) Infect.Immun. 59, 1008-1014) и эндокардиты у кроликов (Perlmann и Freedman (1971) Уа1е J.Biol.Med.44, 206-213).
Необходимо, чтобы грибы или высший эукариотный паразит представляли собой гаплоиды в отдельные периоды его жизненного цикла (например, роста в окружающей среде). Предпочтительно, чтобы имелась ДНК-опосредованная интегрированная трансформационная система, в том случае, когда микроорганизм представляет собой человеческий патоген, удобно, чтобы была доступна животная модель заболевания человека. Грибы, патогенные в отношении людей, включают некоторые Aspergillus spp. (например, A.fumigatus), Cryptococcus neoformans и Histoplasma capsulatum. Очевидно, что указанные выше грибы имеют гаплоидную фазу, и для них имеется в распоряжении ДНК-опосредованная интегрированная трансформационная система. Может также использоваться Тохоplasma, представляющая собой паразит с гаплоидной фазой в ходе инфицирования. Бактерии имеют гаплоидный геном.
Наиболее доступными животными моделями человеческих заболеваний, является мышь, крыса, кролик, собака или обезьяна. Предпочтительно, когда такое животное представляет собой мышь. Вирулентные гены, детектируемые способом настоящего изобретения с использованием животной модели человеческого заболевания, являются генами, определяющими вирулентность микроорганизма в организме человека.
Особенно предпочтительными микроорганизмами для применения в способах настоящего изобретения являются Salmonella typhimurium, Staphylococcus aureus, Streptococcus pneumoniae, Enterococcus faecalis, Pseudomonas aeruginosa и Aspergillus fumigatus.
Далее описывается предпочтительное техническое решение настоящего изобретения.
Нуклеиновую кислоту, содержащую уникальную маркерную последовательность, получают следующим образом. Комплексный пул последовательности двунитевой ДНК "меток" создают с использованием олигонуклеотидного синтеза и полимеразной реакции синтеза цепи (PCR). Каждая ДНК ″метки" имеет последовательность размером в 20-80 п.о., предпочтительно 40 п.о., которая фланкирована последовательностями, "плечами" размером примерно 15-30 п.о., предпочтительно около 20 п.о., которые являются общими для всех "меток". Число пар основания п.о. последовательности является достаточным для создания большого количества (например, > 1010) уникальных последовательностей в ходе беспорядочного олигонуклеотидного синтеза, но не достаточным для образования вторичных структур, которые могут препятствовать PCR. Аналогично, длина последовательности плечей должна быть достаточной для обеспечения эффективного примирования олигонуклеотидов в PCR.
Хорошо известно, что последовательность на 5’-окончании олигонуклеотида не нуждается в совместимости с последовательностью-мишенью для амплифицирования.
Обычно PCR праймеры не содержат каких-либо комплементарных структур, каждая из которых была бы длиннее 2 пар оснований, особенно на их 3’-концах, поскольку это может способствовать образованию искусственных продуктов, называемых "праймерные димеры". В случае гибридизации 3’-концов двух таких праймеров образуется "примированный матричный" комплекс, и праймерная инсерция приводит в результате к образованию короткого дуплексного продукта, называемого "праймерным димером".
Следует избегать образования в праймерах внутренней вторичной структуры. Для симметричной PCR, рекомендуется для обоих праймеров содержание G+C-40-60% с короткой длиной любого основания. Классические расчеты температуры плавления, используемые совместно с исследованиями гибридизации зонда ДНК, часто позволяют предсказать, что данный праймер должен ренатурироваться при конкретной температуре, или, что при повышении температуры до 72°С будет происходить преждевременная диссоциация гибрида праймер/матрица. На практике такие гибриды оказываются более эффективными в PCR процессе, чем это обычно предсказывается простыми расчетами Тm.
Оптимальные температуры отжига могут быть определены эмпирически и могут превышать предположительные значения. При 37-55°С Taq ДНК полимераза является активной, при этом появляется праймерная вставка в ходе стадии отжига и происходит стабилизация гибрида. Концентрации праймеров равны в типичной (симметричной) PCR и, как правило, составляют 0,1-1 мкМ.
"Метки" лигируются в транспозон или транспозон-подобный элемент с образованием нуклеиновой кислоты, содержащей особую маркерную последовательность. В целях удобства транспозон переносится на вектор-"самоубийцу", который сохраняется в виде плазмиды в организме "помощника", но теряется после переноса на микроорганизм, используемый в способе настоящего изобретения. Так, например, организм-"помощник" может представлять собой штамм Escherichia coli, микроорганизм способа может представлять собой Salmonella, а передача происходит в ходе конъюгации. Хотя транспозон может теряться после переноса в определенной части клеток он претерпевает транспозицию даже, если он интегрируется рандомизированно, совместно с его уникальной меткой в геном микроорганизма, используемого в способе изобретения. Наиболее предпочтительно, чтобы транспозон или транспозон-подобный элемент, мог быть подвергнут селекции. Так, например, в случае Salmonella, канамицин-устойчивый ген может присутствовать в транспозоне, при этом эксконъюганты подвергают отбору на среде, содержащей канамицин. Существует также возможность комплементации ауксотрофного маркера в клетке-реципиенте функциональным геном в нуклеиновой кислоте, содержащей уникальный маркер. Такой способ особенно удобен при использовании грибков в способе изобретения.
Предпочтительно, чтобы комплементирующий функциональный ген не был получен из тех же видов микроорганизмов, что и реципиентный микроорганизм иначе могут произойти неслучайные интеграционные события. Так же является предпочтительным перенос транспозона или транспозон-подобного элемента на вектор, который эписомально сохраняется (т.е. не является частью хромосомы) в микроорганизме, используемом в способе по первому аспекту изобретения в начальном заданном состоянии, принимая во внимание, что при его изменении эписом не удерживается в состоянии, при котором возможно осуществлять селекцию клетки, в которой транспозон или транспозон-подобный элемент подвергаются транспозиции, даже если они интегрируются случайным образом, совместно с уникальной меткой, в геном микроорганизма, используемого в способе изобретения. Такое техническое решение является выгодным, поскольку после получения микроорганизма, несущего эписомальный вектор, транспозиционное событие каждый раз отбирается или индуцируется изменением состояния микроорганизма (или его клона) от первого данного состояния во второе данное состояние, и транспозон может интегрироваться на различных сайтах генома микроорганизма. Так, после получения основной коллекции микроорганизмов, каждый член которой содержит особую меченую последовательность в транспозоне или транспозон-подобном элементе, переносимых на эписомальный вектор (в первом данном состоянии), такая коллекция может использоваться повторно для образования пулов рандомизированных инсерционных мутантов, каждый из которых содержит различные меченые последовательности (т.е. особые последовательности в рамках пула). Такое техническое решение особенно полезно поскольку (а) оно уменьшает число и сложность манипуляций, требуемых для создания множества ("пула") независимо мутированных микроорганизмов на стадии (1) настоящего способа; и (б) требуется лишь то же число меток, что и число микроорганизмов во множестве микроорганизмов на стадии (1) способа изобретения. Пункт (а) упрощает такой способ при его использовании в организмах, в которых наиболее трудно осуществить транспозонный мутагенез (например, Staphylococcus aureus), а пункт (б) означает, что могут отбираться меченые последовательности с особенно хорошими гибридизационными характеристиками, в связи с чем облегчается контроль их качества. Как подробно описано ниже, размер "пула" обычно составляет 100 или 200 независимо мутированных микроорганизмов, и поэтому основную коллекцию микроорганизмов удобно хранить на одном или двух 96-луночных титрационных микропланшетах.
В соответствии с особенно предпочтительным техническим решением первое заданное состояние представляет собой первую конкретную температуру или такой температурный интервал, как 25-32°С, наиболее предпочтительно около 30°С, а второе заданное состояние представляет собой вторую конкретную температуру или такой температурный интервал, как 35-45°С, наиболее предпочтительно 42°С. В дополнительных предпочтительных технических решениях первое заданное состояние представляет собой наличие такого антибиотика, как стрептомицин, а второе заданное состояние представляет собой отсутствие указанного антибиотика; либо первое заданное условие представляет собой отсутствие антибиотика, а второе заданное состояние - присутствие указанного антибиотика.
Транспозоны, подходящие для интеграции в геном грамотрицательной бактерии включают Тn5, Тn10 и их производные. Транспозоны, подходящие для интеграции в геном грамположительных бактерий включают Тn916, его производные или аналоги. Транспозоны, особенно подходящие для использования с Staphylococcus aureus, включают Tn917 (Cheung с сотр.) 1992, Proc.Natl.Acad. Sci. США, 89, 6462-6466 (и Tn918) Albus с сотр. (1991) Infect. Immun., 59, 1008-1014).
Особенно предпочтительно, если транспозоны обладают свойствами Tn917 производных, описанных Camilli с сотр. (1990) J.Bacteriol. 172, 3738-3744 и переносятся термочувствительными векторами, такими как рЕ194Т (Villafane с сотр. (1978) J. Bacteriol, 169, 4822-4829).
Следует иметь в виду, что хотя транопозоны и удобны для инсерционной инактивации гена, но также возможно применение любых других способов. Другим удобным способом инсерционной инактивации гена, особенно в некоторых бактериях, таких как Streptococcus, является инсерционно-дупликационный мутагенез, подобный тому, что описан в статье Morrison с сотр. (1984) J.Bacteriol 159, 870 в отношении S.pneumoniae. Общий метод может применяться на других микроорганизмах, особенно на бактериях.
В случае грибков инсерционные мутации создают путем трансформации с использованием фрагментов ДНК или плазмид, несущих "метки" и предпочтительно селектируемый маркер, кодирующий, например, устойчивость к гигромицину В или флеомицину (см. Smith с сотр. (1994) Infect. Immunol. 62, 5247-5254). Неупорядоченная, единичная интеграция фрагментов ДНК, кодирующих устойчивость к гигромицину В, в геном гифомицеты с использованием интеграции под действием рестриктазы (REMI; Schiestl и Petes (1991); Lu с сотр. (1994) Proc.Natl.Acad.Sci. CШA, 91, 12649-12653) является известным явлением.
Простая методика инсерционного мутагенеза гриба описана Schiestl и Petes (1994), на которых ссылаются в настоящем изобретении, и она включает, например, использование Тy элементов и рибосомной ДНК в дрожжах.
Неупорядоченная интеграция транспозона или других последовательностей ДНК позволяет выделить множество независимо мутированных микроорганизмов, в которых различные гены инсерционно инактивированы в каждом мутанте, и каждый мутант содержит маркерную последовательность.
Библиотека таких инсерционных мутантов ранжируется в микротитрационных планшетах так, что каждая лунка содержит различные мутантные микроорганизмы. Аккумулируют ДНК, содержащую уникальную маркерную последовательность от каждого индивидуального мутантного микроорганизма (удобно использовать общую ДНК из клона). Такую операцию удобно проводить путем удаления образца микроорганизма из микротитрационного планшета, нанесения его штрихами на мембрану для гибридизации нуклеиновой кислоты (например, на нитроцеллюлозную или найлоновую мембраны), лизирования микроорганизма в щелочи и фиксации нуклеиновой кислоты на мембране. Таким способом получают копию содержимого лунок.
Из полученных на микротитрационных планшетах пулов микроорганизмов выделяют ДНК. Такую ДНК используют в качестве мишени для PCR с применением праймеров, которые осуществляют ренатурацию с общими участками, фланкирующими "метки", амплифицированную ДНК метят, например, с помощью 32Р. Продукт такой PCR используют для зондирования ДНК, полученной из каждого индивидуального мутанта, с целью обеспечения эталонной гибридизационной карты для реплик микротитрационных планшетов. Такую операцию проводят с целью определения, действительно каждый из индивидуальных микроорганизмов фактически содержит маркерную последовательность и может ли маркерная последовательность быть эффективно амплифицирована и мечена.
Получают пулы транспозонных мутантов для введения в определенную среду. Удобно использовать 96-луночные планшеты для микротитрования, при этом пул содержит 96 транспозонных мутантов. Однако нижний предел размера пула составляет два мутанта; не имеется теоретического верхнего предела размера пула, однако, как обсуждается ниже, верхний предел может быть определен с учетом окружающей среды, в которую введены мутанты.
При введении микроорганизмов в указанную конкретную окружающую среду микроорганизмам создают условия для их роста. Период нахождения микроорганизмов определяется его природой и средой его обитания. По истечении надлежащего времени пребывания микроорганизмы выводят из окружающей среды, выделяют ДНК и такую ДНК используют в качестве матрицы для PCR с применением праймеров, которые осуществляют ренатурацию с "плечами", фланкирующими "метки". Продукт PCR метят, например, с помощью 32Р и используют для зондирования ДНК, запасенной от каждого индивидуального мутанта, реплицированного из микротитрационного планшета. Идентифицировали аккумулированную ДНК, которая слабо гибридизируется или совсем не гибридизируется с зондом, полученным из ДНК, выделенной из микроорганизмов, извлеченных из окружающей среды. Негибридизирующиеся ДНК соответствуют мутантам, адаптация которых в конкретной окружающей среде ослаблена инсерцией транспозона или другой последовательности ДНК.
Наиболее предпочтительно, когда "плечи" не содержат или содержат очень мало меченых атомов по сравнению с "метками".
Так, например, праймеры PCR строят таким образом, чтобы они не содержали или содержали только единственный G-остаток, 32Р-меченый нуклеотид представляет собой dCTP, и в этом случае ни одного или один радиомеченый С-остаток вводится в каждое "плечо", а основное количество радиомеченых С-остатков вводится в "метку". Предпочтительно, чтобы "метка" содержала, по крайней мере, в десять раз большее количество меченых атомов, чем соответствующее количество, введенное в "плечи"; предпочтительно такое количество должно быть больше в двадцать или более раз; более предпочтительно в пятьдесят и более раз. Удобно, чтобы "плечи" могли удаляться из "метки" с использованием соответствующей рестриктазы, сайт для которого может вводиться в праймерную конструкцию.
Как отмечалось выше, наиболее предпочтительно, когда микроорганизм представляет собой патогенный микроорганизм, а конкретная окружающая среда представляет собой животное. В соответствии с таким техническим решением размер пула мутантов, вводимого в животное, определяется (а) числом клеток каждого мутанта, которые вероятно выживают в животном (предполагая, что вирулентный ген не инактивируется), и (б) общим количеством инокулята микроорганизма. Если число по пункту (а) слишком низкое, то могут быть получены ложные положительные результаты, а если число по пункту (б) слишком велико, то животное может погибнуть до того, как достаточное количество мутантов получит возможность для нормального роста. Число клеток по пункту (а) может быть определено для каждого используемого микроорганизма, но предпочтительно оно имеет значение более 50, более предпочтительно выше 100.
Число различных мутантов, которые могут вводиться в одно животное, предпочтительно составляет 50-500, наиболее предпочтительно около 100. Предпочтительно, когда общее количество инокулята не превышает 106 клеток (и предпочтительно, составляет 105 клеток), хотя размер инокулята может быть выше или ниже указанного значения в зависимости от природы микроорганизма и животного.
В соответствии с наиболее удобным методом используют инокулят порядка 105, содержащий 1000 клеток каждого из 100 различных мутантов, в расчете на одно животное. Следует отметить, что согласно такому способу может использоваться одно животное для скрининга 100 мутантов по сравнению с известными способами, в которых требуется, по крайней мере, 100 животных для отбора 100 мутантов.
Однако удобнее всего инокулировать трех животных одним и тем же пулом мутантов так, чтобы, по крайней мере, на двух из них можно было проводить исследования (одно из животных в этом случае служит копией для проверки достоверности способа), в то время как третье животное служит объектом обратного контроля. Несмотря на это, такой способ все еще обеспечивает более, чем 30-кратную экономию числа используемых животных.
Промежуток времени между введением пула мутантов в животное и извлечением микроорганизмов может изменяться в зависимости от природы используемого микроорганизма и животного. Так, например, если животное представляет собой мышь, а микроорганизм представляет собой Salmonella typhimurium, время между инокуляцией и выделением составляет примерно три дня.
Согласно одному из воплощений настоящего изобретения микроорганизмы извлекаются из среды обитания на стадии (5) на сайте, отдаленном от сайта введения на стадии (4) так, что подлежащие исследованию вирулентные гены включают те, что распространились между двумя такими сайтами.
Так, например, в случае растения микроорганизм может вводиться в повреждение на стебле или на один участок листа, а микроорганизм извлекается из другого участка листа, где обнаружено болезненное состояние.
В случае животного микроорганизм может вводиться орально, внутрибрюшинно, внутривенно или интраназально и с течением времени извлекаться из такого внутреннего органа, как селезенка. Может оказаться полезным сравнение вирулентных генов, идентифицированных при оральном применении и тех, что идентифицированы в результате внутрибрюшинного применения, поскольку некоторые гены вызывают заражение одним путем, но не другим. Предпочтительно, чтобы Salmonella вводилась внутрибрюшинно.
Другими предпочтительными окружающими средами, которые могут использоваться для идентификации вирулентных генов, являются животные клетки в культуре (особенно макрофаги и эпителиальные клетки) и растительные клетки в культуре. Хотя использование клеток в культуре полезно само по себе, оно может также дополнять применение целого животного или растения, как это имеет место в определенных случаях, в качестве окружающей среды.
Также предпочтительно, когда окружающая среда представляет собой часть тела животно. В рамках данного взаимодействия хозяин - паразит возможен ряд различных сред обитания, включающих различные органы и ткани, а также их части, например очаг Реуеr.
Число индивидуальных микроорганизмов (т.е. клеток), выделенных из окружающей среды, должно, по крайней мере, в два раза, предпочтительно, по крайней мере, в десять раз, более предпочтительно в 100 раз превышать число различных мутантов, введенных в окружающую среду. Так, например, при инокулировании животного 100 различными мутантами примерно 10000 индивидуальных микроорганизмов должно извлекаться и выделяться их маркерная ДНК.
Дополнительное предпочтительное техническое решение включает стадии:
(1А) удаления ауксотрофов из множества мутантов, продуцированных на стадии:
(1); или
(6А) определения ауксотрофной природы мутанта,отобранного на стадии (6); или
как (1А), так и (6А).
Необходимо различать ауксотрофы (представляющие собой мутантный микроорганизм, требующие наличия факторов роста, не являющихся необходимыми для дикого типа или для прототрофов) и мутантный микроорганизм, в котором ген, обеспечивающий адаптацию такого микроорганизма в конкретной окружающей среде, находится в инактивированном состоянии. Удобно осуществлять такую операцию между стадиями (1) и (2) или после стадии (6).
Предпочтительно, при идентификации вирулентных генов ауксотрофы не удалять.
В соответствии со вторым аспектом настоящего изобретения обеспечивается способ идентификации гена, позволяющего микроорганизму адаптироваться в конкретной окружающей среде, причем такой способ включает способ по первому аспекту изобретения, после которого проводят дополнительную стадию:
(7) выделения инсерционно-инактивированного гена или его части из индивидуального мутанта, отобранного на стадии (6).
Способы выделения гена, содержащего особый маркер, хорошо известны в области молекулярной биологии.
Еще одно предпочтительное техническое решение включает следующую дополнительную стадию:
(8) выделения из микроорганизма дикого типа соответствующего гена дикого типа с использованием инсерционно-инактивированного гена, выделенного на стадии (7) или его части в качестве зонда. Способы генного зондирования хорошо известны в области молекулярной биологии.
Методы молекулярной биологии, подходящие для использования на практике настоящего изобретения, описаны в работе Sambrook с сотр. (1989), на которую ссылаются в тексте.
В том случае, когда микроорганизм представляет собой микроорганизм, патогенный в отношении животного, а ген представляет собой вирулентный ген, транспозон применяют для инсерционной инактивации гена. Удобно клонировать вирулентные гены перевариванием геномной ДНК из индивидуального мутанта, отобранного на стадии (6), с рестриктазой, осуществляющей разрез вне транспозона, лигированием размерно-фракционированной ДНК, содержащей транспозон, в плазмиду. Селекцию плазмидных рекомбинантов осуществляют на основе антибиотической устойчивости, приобретенной от транспозона, а не от плазмиды. Геномную ДНК микроорганизма, примыкающую к транспозону, секвенируют с использованием двух праймеров, которые ренатурируются в терминальные области транспозона, и двух праймеров, которые отжигаются вблизи полилинкерных последовательностей плазмиды. Такие последовательности могут быть подвергнуты исследованию с использованием базы данных ДНК с целью установления влияния транспозона на известный вирулентный ген. Так, например, последовательность, полученную таким способом, сравнивают с последовательностями, присутствующими в таких общедоступных базах данных, как EMBL и GenBank. Наконец, если оказывается, что прерванная последовательность присутствует в новом вирулентном гене, мутацию переносят на новый генетический фон (например, с помощью фаг Р22-управляемой трансдукции в случае Salmonella) и определяют LД50 мутантного штамма с целью подтверждения того, что невирулентный фенотип обусловлен транспозиционным событием, а не вторичной мутацией.
Число индивидуальных мутантов, подвергнутых скринингу для детекции всех вирулентных генов в микроорганизме, зависит от числа генов в геноме микроорганизма. Так, например, вероятно, что 3000-5000 мутантов Salmonella typhimurium необходимо подвергнуть скринингу для детекции основного количества вирулентных генов, тогда как для Aspergillus spp., которая имеет более крупный геном, чем Salmonella, скринингу подвергают около 20000 мутантов. Примерно 4% не являющихся незаменимыми генов S.typhimurium требуется для определения вирулентности (Grossman и Saier, 1990), в этом случае геном S.typhimurium содержит примерно 150 вирулентных генов. Однако способы настоящего изобретения предусматривают более быстрый, более удобный и гораздо более практичный путь идентификации вирулентных генов.
Третий аспект настоящего изобретения обеспечивает микроорганизм, полученный с использованием способа по первому аспекту изобретения.
Значимость таких микроорганизмов заключается в том, что они не адаптируются к выживанию в конкретной окружающей среде.
В соответствии с предпочтительным техническим решением патогенный микроорганизм не адаптируется к выживанию в организме хозяина (среде обитания), и в случае микроорганизма, являющегося патогенным по отношению к животным, особенно млекопитающим, более конкретно к людям, мутант, полученный способом настоящего изобретения, может использоваться в качестве вакцины. Такой мутант не является вирулентным, поэтому его можно использовать для введения пациенту, предполагается, что он является антигенным и вызывает защитный иммунный ответ.
В соответствии с еще одним предпочтительным техническим решением патогенный микроорганизм, не адаптированный на выживание в организме хозяина, полученный способами изобретения, модифицируют предпочтительно путем введения подходящей последовательности ДНК для экспрессии антигенного эпитопа от другого патогена. Такой модифицированный микроорганизм может выполнять функции вакцины для такого другого патогена.
В соответствии с четвертым аспектом изобретения предусматривается микроорганизм, содержащий мутацию в гене, идентифицированном с использованием способа по второму аспекту изобретения.
Таким образом, в микроорганизм по третьему аспекту изобретения специально вводят мутацию в идентифицированный ген. В соответствии с предпочтительным техническим решением, особенно в том случае, когда микроорганизм предназначается для использования в качестве вакцины, мутация в гене представляет собой делению или сдвиг рамки, или какую-либо иную мутацию, которая в значительной степени неспособна к реверсии. Такие ген-специфичные мутации могут создаваться с использованием таких стандартных методов, как введение в микроорганизм копии мутантного гена на автономный репликон (такой как плазмида или вирусный геном) и использование гомологической рекомбинации для введения мутации в копию гена в геноме микроорганизма.
Пятый и шестой аспекты изобретения обеспечивают подходящий микроорганизм, предназначенный для использования в вакцине и вакцину, содержащую подходящий микроорганизм и фармацевтически приемлемый носитель.
Подходящий микроорганизм представляет собой упомянутый выше невирулентный мутант.
Предпочтительна активная иммунизация пациента. В соответствии с таким подходом получают один или более мутантных микроорганизмов в иммуногенной рецептуре, содержащей приемлемые адъюванты и носители, которые известными методами вводят пациенту. Такие адъюванты включают полный или неполный адъюванты Фрейнда, мурамоил дипептид, "Iscoms" из ЕР 109942, ЕР 180564 и ЕР 231039, гидроксид алюминия, сапонин, ДЕАЕ-декстран, нейтральные масла (например, микглиол), растительные масла (например, арахисовое масло), липосомы, полиолы Pluronic или адъювантную систему Ribi (см., например, GB-A-2189141). "Pluronic" представляет собой зарегистрированную торговую марку. Пациент, подвергаемый иммунизации, является пациентом, которому необходима защита от заболевания, вызванного вирулентной формой микроорганизма.
Упомянутые выше авирулентные микроорганизмы изобретения или их рецептуры могут применяться любым традиционным способом, включая оральное применение и парентеральную (например, подкожную или внутримышечную) инъекцию. Лечение может состоять из введения единичной дозы или многократных доз, применяемых в течение определенного времени.
Хотя авирулентный микроорганизм изобретения может применяться сам по себе, предпочитают, чтобы он присутствовал в фармацевтической рецептуре совместно с одним или более приемлемыми носителями. Носитель(ли) должен быть "приемлемым" в смысле совместимости с авирулентным микроорганизмом изобретения без пагубного воздействия на его реципиента. Обычно, в качестве носителей используют воду или физиологический раствор, которые должны быть стерильными и непирогенными.
Следует иметь в виду, что вакцина изобретения в зависимости от входящего в ее состав микроорганизма может использоваться в области медицины человека и ветеринарной медицины.
Болезни, вызываемые микроорганизмами, известны для многих животных, например для домашних животных. Вакцины изобретения, содержащие соответствующий авирулентный микроорганизм, особенно авирулентные бактерии, могут применяться на людях, но также и, например, на коровах, овцах, свиньях, лошадях, собаках и кошках, а также таких домашних птицах, как куры, индейки, утки и гуси.
Седьмой и восьмой аспект изобретения обеспечивает ген, полученный по методу второго аспекта изобретения и закодированный полипептид. Под термином "ген" подразумеваются не только участки ДНК, кодирующие полипептид, но также регуляторные области ДНК, которые регулируют транскрипцию, трансляцию и для некоторых микроорганизмов сплайсинг РНК. Таким образом, такой ген охватывает промоторы, терминаторы, рибосомо-связующие последовательности и, для некоторых организмов, интроны и сайты узнавания сплайсинга.
Обычно получают информацию о последовательности инактивированного гена, полученного на стадии 7. В целях удобства последовательности, близкие к концам транспозона, используются в качестве гибридизационного сайта секвенирующего праймера. Полученная последовательность или рестрикционный фрагмент ДНК, соседствующий с самим инактивированным геном, используется для получения гибридизационного зонда, с помощью которого идентифицируют и выделяют из организма дикого типа соответствующий ген дикого типа.
Предпочтительно осуществлять гибридизационное зондирование в строгих условиях с целью гарантии получения именно целевого гена. Под термином "строгие" подразумевается, что ген гибридизируется зондом при иммобилизации гена на мембране, а зонд (который в таком случае имеет длину > 200 нуклеотидов) находится в растворе, и систему иммобилизованный ген/гибридизированный зонд промывают в 0,l×SSC при 65°С в течение 10 минут. SSC обозначает 0,15 М NaCl/0,015 M цитрата натрия.
Предпочтительные последовательности зонда для клонирования вирулентных генов разновидности Salmonella показаны на фиг.5 и 6 и описаны в примере 2.
В особенно предпочтительном техническом решении вирулентные гены Salmonella содержат последовательность, показанную на фиг.5 и 6 и описанную в примере 2.
Еще более предпочтительные гены, которые содержатся или, по крайней мере, часть которых содержится в последовательностях, показанных на фиг.11 и 12, и которые идентифицированы способом по второму аспекту изобретения. Последовательности, показанные на фиг.11 и 12, представляют собой кластер гена из Salmonella typhimurium, названный вирулентным генным кластером 2 (YGC2). Положение транспозонных инсерций указаны в последовательности, и такие транспозонные инсерций инактивируют вирулентную детерминанту организма. Как более подробно обсуждается ниже и особенно в примере 4, в том случае, когда способ по второму аспекту изобретения используется для идентификации вирулентных генов в Salmonella typhimurium, большинство нуклеиново-кислотных инсерций (и поэтому идентифицированных генов) кластеризуются в относительно небольшой части генома. Такая область, УGС2, содержит другие вирулентные гены, которые, как описано ниже, составляют часть настоящего изобретения.
Ген, выделенный по способу настоящего изобретения, может экспрессироваться в подходящей клетке-хозяине. Так, например, ген (ДНК) в соответствии с известными методиками, может быть соответственно модифицирован в рамках указаний, содержащихся в описании, с целью конструирования вектора экспрессии, который далее используют для трансформации соответствующей клетки-хозяина для экспрессии и продуцирования полипептида настоящего изобретения. Такие методы описаны в патентах США №№4440859, выданном 3 апреля 1984 г. на имя Rutter с сотр., 4530901, выданном 23 июля 1985 г. на имя Weissman, 4582800, выданном 15 апреля 1986 г. на имя Crowl, 4677063, выданном 30 июня 1987 на имя Mark с сотр., 4678751, выданном 7 июля 1987 г. на имя Goeddel, 4704362, выданном 3 ноября 1987 г. на имя Itakura с сотр., 4710463, выданном 1 декабря 1987 г. на имя Murray, 4757006, выданном 12 июля 1988 г. на имя Toole, Jr., с сотр., 4766075, выданном 23 августа 1988 г. на имя Goeddel с сотр., и 4810648, выданном 7 марта 1989 г. на имя Stalker, причем на все эти патенты ссылаются в настоящем описании.
ДНК, кодирующая полипептид настоящего изобретения, может быть соединена с большим числом других последовательностей ДНК для введения в соответствующего хозяина. ДНК-компаньон будет зависеть от природы хозяина, способа введения такой ДНК в организм хозяина и от того, необходимо ли эписомальное содержание или интеграция.
Обычно ДНК встраивают в вектор экспрессии, например в плазмиду, в надлежащей ориентации и правильной рамке считывания для экспрессии. Если в этом есть необходимость, то ДНК может быть связана с соответствующими транскрипционными и трансляционными регуляторными контрольными нуклеотидными последовательностями, распознаваемыми желаемым хозяином, хотя, как правило, такие контроли уже присутствуют в векторе экспрессии. Такой вектор далее вводят в организм хозяина стандартными методами. Как правило, не все хозяева будут трансформироваться таким вектором. Поэтому необходимо проводить селекцию трансформированных клеток-хозяев. Один из приемов такой селекции включает введение в вектор экспрессии последовательности ДНК с какими-либо необходимыми контрольными элементами, которая кодирует селектируемый признак в трансформированной клетке, например устойчивость к антибиотику. С другой стороны, ген для такого отбираемого признака может представлять собой другой вектор, который используется для сотрансформации желаемой клетки-хозяина.
Клетки-хозяева, трансформированные рекомбинантной ДНК настоящего изобретения, затем культивируют в течение достаточного времени, и при соответствующих условиях, известных специалисту в данной области, на основании указаний, приведенных в настоящем описании, с целью осуществления экспрессии полипептида, который затем может быть выделен.
Известны многие экспрессионные системы, включающие бактерии (например, E.coli и Bacillus Subtilis), дрожжи (например, Saccharomyces cerevisiae), гифомицеты (например, Aspergillus), растительные клетки, животные клетки и клетки насекомых.
Векторы включают прокариотный репликон, например, Colelori для экспрессии в прокариоте в других непрокариотных, клеточных типах. Векторы могут также включать прокариотный промотор, способный направлять экспрессию (транскрипцию и трансляцию) генов в таких бактериальных клетках-хозяевах, как E.coli, трансформированных этим геном.
Промотор представляет собой контролирующий экспрессию элемент, образованный последовательностью ДНК, способствующей связыванию РНК-полимеразы и осуществлению транскрипции. Промоторные последовательности, совместимые с упомянутыми бактериальными хозяевами обычно предусматриваются в плазмидных векторах, содержащих удобные рестрикционные сайты, для инсерции сегмента ДНК настоящего изобретения.
Типичные прокариотические векторные плазмиды представляют собой puC18, PuC19, PBR322 и PBR329, приобретенные у Биорад Лабораториз (Ричмонд, КА, США), а также рТrс99А и рКК223-3, приобретенные у Фармация, Пискатавэй, Нью-Джерси, США.
Типичная векторная плазмида клетки млекопитающего представляет собой pSVL, полученная от Фармация, Пискатавэй, Нью-Джерси, США. В таком векторе используется SV 40 поздний промотор для управления экспрессией клонированных генов, причем наивысший уровень экспрессии обнаружен в таких Т-антиген-продуцирующих клетках, как COS-1 клетки.
Примером индуцибельного вектора экспрессии млекопитающего может служить pMSG, также полученный от Фармации. В таком векторе используется глюкокортикоид-индуцибельный промотор длинного концевого повтора вируса опухоли мышиной грудной железы для управления экспрессией клонированного гена.
Используемые дрожжевые плазмидные векторы представляют собой pRS403-406 и pRS413-416, и их обычно получают от Стратаген Клонинг Системс, Ла Йолла, КА 92037, США. Плазмиды pRS403, pRS404, pRS405 и pRS406 представляют собой дрожжевые интегрирующие плазмиды (VIps) и включают дрожжевые селектируемые маркеры HIS3, TRP1, LEU2 и URA3. Плазмиды pRS413-416 представляют собой дрожжевые центромерные плазмиды (VCps).
Известны различные способы для связывания ДНК с векторами через комплементарные липкие концы. Так, например, комплементарные гомополимерные отрезки могут добавляться к сегменту ДНК для ее инсерции в векторную ДНК. Затем такой вектор и сегмент ДНК соединяют через водородную связь между комплементарными гомополимерными хвостами с образованием молекул рекомбинантной ДНК.
Синтетические линкеры, содержащие один или более рестрикционных сайтов, обеспечивают альтернативный способ соединения сегмента ДНК с векторами. Сегмент ДНК, образовавшийся в результате переваривания с рестриктазой, как описано выше, обрабатывают ДНК полимеразой бактериофага Т4 или E.coli ДНК полимеразой 1, ферментами, которые удаляют выступающие 3’-однонитевые окончания с их 3’-5’-экзонуклеолитическими активностями и заполняют рецессивные 3’-окончания с их полимеризующими активностями.
Поэтому комбинация таких активностей приводит к образованию сегментов ДНК с тупыми концами. Затем сегменты с тупыми концами инкубируют с большим молярным избытком линкерных молекул в присутствии фермента, способного катализировать лигирование молекул ДНК с тупыми концами, например, такого, как лигаза ДНК бактериофага Т4. Таким образом, продукты реакции представляют собой сегменты ДНК, несущие на своих концах полимерные линкерные последовательности. Затем такие сегменты ДНК расщепляют с помощью соответствующей рестриктазы и лигируют с вектором экспрессии, который был подвергнут ферментативному расщеплению в результате которого образовались концы, комплементарные концам сегмента ДНК.
Синтетические линкеры, содержащие множество рестрикционных эндонуклеазных сайтов, доступны коммерчески из ряда источников, включающих Интернейшионал Биотекнолоджиз Инк., Нью-Хайвен, ПН, США.
Предпочтительным способом модификации ДНК, кодирующей полипептид изобретения, является использование полимеразной цепьевой реакции в соответствии с описанным Saiki с сотр. (1988) Science 239, 487-491. В таком способе ДНК, подвергающаяся ферментативной амплификации, фланкируется двумя специфическими олигонуклеотидными праймерами, которые сами включаются в амплифицированную ДНК. Указанные специфические праймеры могут содержать сайты распознавания рестрикционной эндонуклеазы, которые могут использоваться для клонирования в векторы экспрессии с использованием методов известных в этой области.
Варианты генов также составляют часть изобретения. Предпочтительно, когда такие варианты имеют, по крайней мере, 70% гомологичность последовательности, более предпочтительно, по крайней мере, 85%, наиболее предпочтительно, по крайней мере, 95% гомологичность последовательности с генами, выделенными способом настоящего изобретения. Разумеется, что допустимы замены, делеции и инсерции. Степень гомологичности между той или иной последовательностями нуклеиновых кислот может быть определена с использованием программы GAP компьютерной группы Висконсинского Университета.
Аналогичным образом, сфера изобретения охватывает варианты белков, закодированные генами.
Под термином "варианты" подразумеваются инсерции, делеции и замены как консервативные, так и неконсервативные, в том случае, когда такие изменения не оказывают существенного влияния на нормальное функционирование белка.
Под термином "консервативные" замены подразумеваются такие комбинации, как Gly, Ala, Val, Ile, Leu, Asp, Glu, Asn, Gln, Ser, Thr, Lys, Arg и Phe, Tyr.
Такие варианты могут быть получены с использованием хорошо известных способов белковой инженерии и сайт-направленного мутагенеза.
Девятый аспект изобретения предусматривает способ идентификации соединения, снижающего способность микроорганизма к адаптации в конкретной окружающей среде, включающий стадии селекции соединения, которое препятствует функционированию (1) гена, полученного способом по второму аспекту изобретения, или (2) полипептида, закодированного таким геном.
Парные скрининги соединений, которые влияют на клетку дикого типа, но не клетку, сверхсинтезирующую ген, выделенный способами настоящего изобретения, составляют часть такого аспекта изобретения.
Так, например, в соответствии с одним из технических решений, одна клетка представляет собой клетку дикого типа, а вторая клетка представляет собой Salmonella, которую получают для сверхсинтеза гена, выделенного способом изобретения. Жизнеспособность и/или рост каждой клетки в конкретной окружающей среде определяется в присутствии соединения, подлежащего тестированию для идентификации соединения, понижающего жизнеспособность или рост клетки дикого типа, но не клетки, сверхсинтезирующей указанный ген.
Предпочтительно, чтобы такой ген представлял собой вирулентный ген.
Так, например, согласно одному из технических решений готовят микроорганизм (например, S.typhimurium) для сверхпродуцирования вирулентного гена, идентифицированного способом по первому аспекту изобретения. Каждый из (а) "сверхпродуцирующего" микроорганизма и (б) эквивалентного микроорганизма (который не способен к сверхсинтезу вирулентного гена) используются для инфицирования клеток в культуре. Способность конкретного испытуемого соединения к селективному ингибированию функции вирулентного гена определяется оценкой количества испытуемого соединения, которое требуется для предотвращения инфицирования клеток-хозяев (а) сверхпродуцирующим микроорганизмом и (б) эквивалентным микроорганизмом (по крайней мере, для некоторых вирулентных генных продуктов предполагается, что испытуемое соединение способно инактивировать их и инактивироваться само по себе в результате связывания с продуктом вирулентного гена). Если значительное количество соединения требуется для предотвращения инфицирования микроорганизмами по (а) и (б), то предполагается, что такое соединение селективно. Предпочтительно, чтобы микроорганизмы (например, Salmonella ) разрушались внеклеточно под действием таких мягких антибиотиков, как гентамицин (который не проникает в клетки-хозяев), и чтобы действие испытуемого соединения по предотвращению инфицирования клетки микроорганизмом заключалось в лизисе указанной клетки и определялось число присутствующих микроорганизмов (например, путем посева на агар).
Парные скрининги и другие скрининги соединений обобщены Kirsch и Di Domenico (1993) в книге "Обнаружение природных продуктов с терапевтическим потенциалом" (изд. Y.P. Gallo), глава 6, стр.177-221, Баттерворс, У.К. (причем на эту работу ссылаются в настоящем описании).
Парные скрининги осуществляют путем сравнения относительной чувствительности соединения, используя два генетически родственных штамма. Если штаммы отличаются друг от друга в одном локусе, то соединение, специфичное в отношении мишени, может идентифицироваться сравнением чувствительности каждого из штаммов к ингибитору. Так, например, ингибиторы, специфичные в отношении мишени, будут более активными в отношении сверхчувствительного испытуемого штамма по сравнению с другим изогенным родственным штаммом. При применении метода диффузии на агаре это определяется измерением диаметра зоны подавления роста тест-организма, окружающей диск или лунку с таким соединением. В процессе диффузии создается непрерывный градиент концентрации соединения, и чувствительность штамма к ингибиторам пропорциональна расстоянию от диска или лунки до края зоны. Обычные антимикробные агенты или антимикробиальные агейты, с отличным способом функционирования, обычно являются агентами, обладающими одинаковыми активностями по отношению к двум таким штаммам.
Другой тип молекулярного генетического скрининга включает пары штаммов, в которых клонированный генный продукт сверхпродуцируется в одном штамме по сравнению с контрольным штаммом. В таком анализе штамм, содержащий повышенное количество белка-мишени, должен быть более устойчивым к ингибиторам, специфичным в отношении клонированного генного продукта, чем изогенный штамм, содержащий обычные количества белка-мишени. При применении метода диффузии на агаре размер зоны подавления роста, окружающей специфическое соединение, будет меньшим у штамма сверхпродуцирующего белок-мишень, чем у другого изогенного штамма.
Дополнительный или альтернативный отбор соединения проводят следующим образом:
1. В качестве контроля используют мутантный микроорганизм, полученный с использованием способа по первому аспекту изобретения. (Он имеет заданный фенотип, например является авиарулентным).
2. Испытуемое соединение смешивают с микроорганизмом дикого типа.
3. Микроорганизм дикого типа вводят в окружающую среду (в присутствии или отсутствие испытуемого соединения).
4. Если микроорганизм дикого типа неспособен адаптироваться в окружающей среде (после обработки соединением или в его присутствии), то такое соединение рассматривается как понижающее способность микроорганизма к адаптации или выживанию в окружающей среде.
Если окружающая среда представляет собой организм животного, а микроорганизм представляет собой патогенный микроорганизм, то соединения, идентифицированные указанным способом, могут использоваться в качестве лекарственного средства, предотвращающего или снижающего заражение микроорганизмом.
В связи с этим, десятый аспект изобретения предусматривает соединение, идентифицируемое с помощью способа по девятому аспекту.
Следует иметь в виду, что применения соединения по десятому аспекту относятся к способу его идентификации, в особенности в отношении хозяина патогенного микроорганизма. Так, например, если соединение идентифицируется с помощью способа, в котором используется вирулентный ген или закодированный им полипептид из бактерии, инфецирующей млекопитающее, такое соединение может использоваться для его лечения.
Аналогичным образом, если соединение идентифицируется способом, в котором используется вирулентный ген или закодированный им полипептид из гриба, который заражает растение, такое соединение может применяться для его лечения.
Согласно одиннадцатому аспекту настоящего изобретения предусматривается молекула, которая селективно взаимодействует и существенно ингибирует функцию гена по седьмому аспекту изобретения или его нуклеиново-кислотного продукта.
Под термином "его нуклеиново-кислотный продукт" подразумевается любая РНК, особенно мРНК, транскрибированная из гена.
Предпочтительно, молекула, селективно взаимодействующая и существенно ингибирующая функцию указанного гена или указанного нуклеиново-кислотного продукта, представляет собой антисмысловую нуклеиновую кислоту или производное нуклеиновой кислоты.
Более предпочтительно, указанная молекула представляет собой антисмысловой олигонуклеотид.
Антисмысловые олигонуклеотиды представляют собой однонитевую нуклеиновую кислоту, которая может специфично связываться с последовательностью комплементарной нуклеиновой кислоты. В результате связывания с последовательностью соответствующей мишени образуются дуплексы РНК-РНК, ДНК-ДНК или РНК-ДНК. Такие нуклеиновые кислоты часто называют "антисмысловыми", поскольку они комплементарны к смысловой или кодирующей нити гена. Недавно удалось получить тройную спираль, в которой олигонуклеотид связан с дуплексом ДНК. Было установлено, что олигонуклеотиды способны распознавать последовательности в главном желобке двойной спирали ДНК. В результате образуется тройная спираль. Это подтверждает тот факт, что существует возможность синтеза молекул, специфичных к последовательности, которые специально связывают двухнитевую ДНК посредством распознавания сайтов связывания водорода в главном желобке.
Очевидно, что последовательность антисмысловой нуклеиновой кислоты или олигонуклеотида может быть легко определена со ссылкой на нуклеотидную последовательность рассматриваемого гена. Так, например, могут быть сконструированы антисмысловая нуклеиновая кислота или олигонуклеотид, которые комплементарны к части последовательности, показанной на фиг.11 или 12, особенно к последовательностям, которые образуют часть вирулентного гена.
Олигонуклеотиды представляют собой объект, который деградируется или инактивируется клеточными эндогенными нуклеазами. Для решения такой проблемы можно использовать модифицированные олигонуклеотиды, например, содержащие измененные интернуклеотидные связи, в которых фосфодиэфирные связи природного происхождения заменены на другую связь. Так, например, Agrawal с сотр. (1988) Proc.Natl.Acad.Sci. США, 85, 7079-7083 продемонстрировали повышенное ингибирование в тканевой культуре НIУ-1 с использованием фосфорамидатов и фосфоротиоатов олигонуклеотида. Sarin с сотр. (1988) в Рroc. Natl. Acad. Sci. США, 85, 7448-7451 продемонстрировали повышенное ингибирование HIY-1 с использованием метилфосфонатов олигонуклеотида. Agrawal с сотр. (1989) в Proc.Natl.Acad.Sci. США, 86, 7790-7794 продемонстрировали ингибирование репликации HIY-1 как в ранее инфицированных, так и в хронически инфицированных клеточных культурах с использованием фосфоротиоатов олигонуклеотида, специфичного к последовательности нуклеотида. Leither с сотр. (1990) в Proc. Natl.Acad.Sci. США, 87, 3430-3434 сообщают об ингибировании в тканевой культуре репликации вируса гриппа с помощью фосфоротиоатов олигонуклеотида. Было показано, что олигонуклеотиды, содержащие искусственные связи, устойчивы к деградации in vivo. Так например, известно из Shaw с сотр. (1991) в Nucleic Acids Res. 19, 747-750, что немодифицированные олигонуклеотиды становятся более устойчивыми к действию нуклеаз in vivo в том случае, когда они блокируются на 3’-конце некоторыми кэппированными структурами, и что некэппированные олигонуклеотидные фосфоротиоаты не деградируются in vivo.
Подробное описание Н-фосфонатного подхода к синтезу олигонуклеозидных фосфоротиоатов приведено Agrawal и Tang (1990) в Tetrahedron Lett. 31, 7541-7544, на которое ссылаются в тексте настоящего описания. Синтезы олигонуклеозидных метилфосфонатов, фосфородитиоатов, фосфорамидатов, сложных фосфатных эфиров, мостиковых фосфорамидатов и мостиковых фосфортиоатов известны в данной области. См., например, статьи Agrawal и Goodchild (1987) Tetrahedron Lett. 28, 3539; Nielsen с сотр. (1988) в Tetrahedron Lett. 29, 2911; Jager с сотр. (1988) Biochemistry 27, 7237; Uznanski с сотр., (1987) в Tetrahedron Lett. 28, 3401; Bannwarth (1988) Helv.Chim.Acta 71, 1517; Crosstick и Vyie (1989) в Tetrahedron Lett. 30, 4693; Agrawal с сотр. (1990) в Proc.Nati.Acad.Sci. США 87, 1401-1405, на содержание которых ссылаются в тексте. Возможны и другие способы получения. В соответствии с предпочтительным техническим решением олигонуклеотид представляет собой дезоксирибонуклеиновую кислоту (ДНК), хотя могут также синтезироваться и применяться последовательности рибонуклеиновой кислоты (РНК).
Олигонуклеотиды, используемые в настоящем изобретении, предпочтительно предназначены для сопротивления деградации под действием эндогенных нуклеолитидных ферментов. Деградация олигонуклеотидов in vivo приводит к образованию продуктов распада олигонуклеотида меньшей длины. Такие продукты распада, по всей вероятности, принимают участие в неспецифической гибридизации, и маловероятно, что они являются эффективными в отношении их копий полной длины. Таким образом, желательно использовать олигонуклеотиды, обладающие устойчивостью к деградации в организме и способные достигать клеток-мишеней. Олигонуклеотидам настоящего изобретения может быть придана повышенная устойчивость к деградации in vivo путем замены нативных фосфодиэфирных связей на одну или более внутренних искусственных интернуклеотидных связей, например, путем замещения в связи фосфата серой. Примерами связей, которые могут использоваться в таких операциях могут служить фосфоротиоаты, метилфосфонаты, сульфон, сульфат, кетил, фосфородитиоаты, различные фосфорамидаты, сложные фосфатные эфиры, мостиковые фосфоротиоаты и мостиковые фосфорамидаты. Приведенные примеры носят иллюстрационный характер, не ограничивающий объем настоящего изобретения. См., например, работу Cohen (1990) в Trends in Biotechnol. Синтез олигонуклеотидов с одной или более таких связей, которыми заменены фосфодиэфирные интернуклеотидные связи, хорошо известен в данной области, и он включает синтетические маршруты получения олигонуклеотидов со смешанными интернуклеотидными связями.
Могут быть получены олигонуклеотиды, устойчивые к удлинению под действием эндогенных ферментов путем "кэппирования", или включения аналогичных групп на 5’ или 3’ терминальных нуклеотидах. Реагент для кэппирования коммерчески доступен в виде препарата Amino-Link 11TM от Эпплайд Биосистемс Инк., Фостер Сити, КА. Способы кэппирования описаны, например, Shaw с сотр. (1991) в Nucleic Acids Res. 19, 747-750 и Agrawal с сотр. (1991) в Proc.Natl.Acad.Sci США 88 (17), 7595-7599, на содержание которых ссылаются в настоящем описании.
Еще один способ получения олигонуклеотидов, устойчивых к воздействию нуклеазы, представляет собой их "самостабилизацию", как описано Tang с сотр. (1993) Nucl.Acids.Res. 21, 2729-2735, причем на содержание этой статьи ссылаются в настоящем описании. Самостабилизированные олигонуклеотиды имеют шпилькообразные петлевые структуры на их 3’-окончаниях и проявляют повышенную устойчивость к деградации под действием фосфодиэстеразы змеиного яда, ДНК полимеразы 1 и фетальной коровьей сыворотки. Самостабилизированная область олигонуклеотида не препятствует гибридизации с комплементарными нуклеиновыми кислотами, а исследования фармакокинетики и стабильности на мышах показало повышение in vivo устойчивости самостабилизированных олигонуклеотидов в отношении их линейных копий.
В соответствии с настоящим изобретением свойственная антисмысловым олигонуклеотидам связующая специфичность, характерная для объединения оснований, усиливается ограничением доступности антисмыслового соединения к его предполагаемому локусу in vivo, что позволяет использовать пониженные дозировки и минимизировать системные эффекты. Так, например, олигонуклеотиды применяют локально для достижения желаемого эффекта. Концентрация олигонуклеотидов в желаемом локусе много выше, чем при системном применении олигонуклеотидов, и терапевтический эффект может достигаться с использованием значительно меньшего общего количества. Высокая локальная концентрация олигонуклеотидов усиливает проникновение клеток-мишеней и эффективно блокирует трансляцию последовательностей нуклеиново-кислотных мишеней.
Олигонуклеотиды могут доставляться в локус с помощью любых средств, подходящих для локализованного применения лекарства. Так, например, раствор олигонуклеотидов может быть инъецирован непосредственно в определенное место или может доставляться вливанием с использованием насоса для вливания. Олигонуклеотиды могут также вводиться в имплантируемое устройство, которое при помещении на желаемый участок обеспечивают выделение олигонуклеотидов в окружающий локус.
Наиболее предпочтительно, вводить такие олигонуклеотиды с помощью гидрогельного материала. Гидрогель является огнестойким и биодеградируемым материалом. В настоящее время известно много таких материалов, включая те, что получают из природных и синтетических полимеров. В соответствии с предпочтительным техническим решением в способе используется гидрогель, представляющий собой жидкость при температуре ниже температуры тела, но загущающийся с образованием сохраняющего форму полутвердого гидрогеля при температуре тела или ниже этой температуры. Предпочтительными гидрогелями являются полимеры из повторяющихся звеньев оксид этилена - оксид пропилена. Свойства такого полимера зависят от его молекулярного веса и относительного содержания в нем полиэтиленоксида и полипропиленоксида. Предпочтительные гидрогели содержат от примерно 10 до примерно 80 вес.% оксида этилена и от примерно 20 до примерно 90 вес.% оксида пропилена. Особенно предпочтительный гидрогель содержит около 70% полиэтиленоксида и 30% полипропиленоксида. Используемые для этих целей гидрогели могут быть получены, например, от BASF Corp., Парсиппани, Нью-Джерси, под торговым наименованием Плюроник (PluronicR)
В соответствии с таким техническим решением гидрогель охлаждают до жидкого состояния и олигонуклеотиды примешивают к жидкости до концентрации около 1 мг олигонуклеотида на грамм гидрогеля. Полученную в результате смесь затем применяют на поверхности подлежащей обработке, например, путем распыления или с помощью мазков в ходе хирургической операции или с использованием катетера или методов эндоскопии. По мере нагревания полимера он загустевает с образованием геля, и олигонуклеотиды диффундируют из такого геля в окружающие клетки в течение периода времени, определяемого точным составом геля.
Олигонуклеотиды могут применяться с помощью других имплантантов, выпускаемых промышленностью или описанных в научной литературе, включающих липосомы, микрокапсулы и имплантируемые устройства. Так, например, имплантанты из таких биодеградируемых материалов, как полиангидриды, полиортоэфиры, полимолочная кислота и полигликолевая кислота, а также их сополимеры, коллаген, белковые полимеры, или такие небиодеградируемые материалы, как этиленвинил ацетат (ЕVАс), поливинилацетат, этилен виниловый спирт и их производные, могут использоваться для локальной доставки олигонуклеотидов. Олигонуклеотиды могут вводиться в материал в ходе его полимеризации или отвердевания с использованием методов испарения расплава или растворителя либо механически смешиваться с материалом. Согласно одному из технических решений олигонуклеотиды примешивают или применяют на покрытиях для имплантируемых средств, таких как шарики из оксида кремния, покрытые декстраном, стенты или катеторы.
Доза олигонуклеотидов зависит от их размера и цели их применения. Как правило, интервал дозировок рассчитывают на основе площади поверхности ткани, подлежащей обработке. Эффективная дозировка олигонуклеотида в некоторой степени зависит от длины и химического состава олигонуклеотида, но, как правило, она составляет 30-3000 мкг на квадратный сантиметр площади поверхности ткани.
Олигонуклеотиды могут применяться на пациенте системно, как в терапевтических, так и в профилактических целях. Олигонуклеотиды могут применяться любым эффективным методом, например парентерально (например, внутривенно, подкожно, внутримышечно) или орально, назально, либо другими методами, которые обеспечивают доступ и циркуляцию олигонуклеотидов в кровеносной системе пациента. Олигонуклеотиды, применяемые системно, предпочтительно дополняют местное применение олигонуклеотидов, однако их можно использовать и без местного применения. Дозировка в интервале примерно 0,1-10 г при применении на взрослом человеке обычно оказывается эффективной для такой цели.
Следует иметь в виду, что молекулы согласно этому аспекту изобретения используются для лечения или профилактики любого заражения, вызываемого микроорганизмом, из которого выделен указанный ген, или он находится в близком родстве с указанным микроорганизмом. Таким образом, указанная молекула представляет собой антибиотик.
Таким образом, двенадцатый аспект настоящего изобретения предусматривает молекулу одиннадцатого аспекта изобретения, предназначенную для применения в медицинских целях.
Тринадцатый аспект настоящего изобретения охватывает способ лечения хозяина, который инфицирован или подвержен инфицированию микроорганизмом, причем такой способ включает применение эффективного количества молекулы в соответствии с одиннадцатым аспектом изобретения, в которой указанный ген присутствует в указанном микроорганизме или является близкородственным указанному микроорганизму.
Под термином "эффективное количество" подразумевается количество, которое в значительное мере предотвращает или улучшает состояние при заражении. Под термином "хозяин" подразумевается любое животное или растение, которые могут инфицироваться микроорганизмом.
Следует принимать во внимание, что фармацевтические рецептуры молекулы по одиннадцатому аспекту изобретения составляют его часть. Такие фармацевтические рецептуры включают указанную молекулу в сочетании с одним или более приемлемых носителей. Носитель должен быть "приемлемым" в том смысле, что он совместим с указанной молекулой изобретения и не оказывает вредного влияния на его реципиента. Обычно в качестве носителей используют воду или физиологический раствор, которые должны быть стерильными и непирогенными.
Как отмечалось выше и более подробно описано ниже в примере 4, было обнаружено, что некоторые вирулентные гены кластеризуются в Salmonella typhimurium в области хромосомы, названной YGC2. ДНК-ДНК гибридизационные эксперименты позволили установить, что последовательности гомологичные, по крайней мере, части УGС2 обнаруживаются во многих видах и штаммах Sаlmonella, но не присутствуют в E.coli и Shigella штаммах, подвергнутых тестированию (см. пример 4). Такие последовательности почти точно соответствуют консервативным генам, по крайней мере, в Salmonella, и лишь некоторые из них являются вирулентными генами. Предполагается, что эквивалентные гены в других разновидностях Salmonella и эквивалентные гены, в случае их наличия, в других кишечных, либо других бактериях также являются вирулентными генами.
Можно легко определить, является ли ген в области VGC2 вирулентным геном. Так, например, те гены в VGC2, которые были идентифицированы способом по второму аспекту изобретения (при использовании Salmonella typhimurium, и в котором окружающая среда представляла собой такое животное, как мышь), являются вирулентными генами. Вирулентные гены также идентифицируются осуществлением мутации в гене (предпочтительно неполярной мутации) и установлением факта авирулентности мутантного штамма. Способы осуществления мутаций в отобранном гене хорошо известны и описаны ниже.
Четырнадцатый аспект изобретения предусматривает VGC2 ДНК Salmonella typhimurium или ее часть, либо вариант указанной ДНК или вариант его части.
VGC2 ДНК Salmonella typhimurium изображена в виде диаграммы на фиг.8, и может быть легко получена из Salmonella typhimurium ATCC 14028 (полученного из коллекции американских типовых культур, 12301 Парлаун Драйв, Роквил, Мэрилэнд 20852, США; и также депонированного в NCTC лаборатория службы здравоохранения, Колиндэйл, Великобритания под каталожным номером NСТС 12021) с использованием информации, предоставленной в примере 4. Так, например, зонды, являющиеся производными последовательностей, показанных на фиг.11 и 12, могут использоваться для идентификации λ клонов из Salmonella typhimurium геномной библиотеки. Стандартные способы скрининга генома могут использоваться для получения всех VGC2 ДНК. Рестрикционная карта, показанная на фиг.8, может использоваться для идентификации и определения местоположения ДНК фрагментов из VGC2.
Под термином "часть VGC2 ДНК Salmonella typhimuriuln" пoдразумевается любая последовательность ДНК, содержащая, по крайней мере, 10 нуклеотидов, предпочтительно, по крайней мере, 20 нуклеотидов, более предпочтительно, по крайней мере, 50 нуклеотидов, и еще более предпочтительно, по крайней мере, 100 нуклеотидов, и наиболее предпочтительно, по крайней мере, 500 нуклеотидов VGC2. Особенно предпочтительной частью VGC2 ДНК является последовательность, показанная на фиг.11, или ее часть. Другой, особенно предпочтительной частью VGC2 ДНК, является последовательность, показанная на фиг.12, или ее часть.
Выгодно, когда часть VGC2 ДНК представляет собой ген или его часть.
Гены могут быть идентифицированы в области VGC2 методами статистического анализа открытых рамок считывания генетической информации с использованием известных компьютерных программ. Если открытая рамка считывания имеет размер более 100 кодонов, то, по всей вероятности, она является геном (хотя известны более мелкие гены). Соответствие открытой рамки считывания полипептид-кодирующей области гена может быть установлено экспериментально. Так, например, часть ДНК, соответствующая открытой рамке считывания, может использоваться в качестве зонда в Нозерн (РНК) блоттинге для установления факта экспрессирования мРНК, которая гибридизируется в указанную ДНК; альтернативно или дополнительно мутация может быть введена в открытую рамку считывания, и может быть определен эффект мутации на фенотип микроорганизма. Если фенотип изменяется, то открытая рамка считывания соответствует гену. Способы идентификации генов в последовательности ДНК известны в этой области.
Под термином "вариант указанной ДНК или вариант ее части" подразумевается любой вариант, подпадающий под термин "вариант" по седьмому аспекту изобретения.
Таким образом, варианты VGC2 ДНК Salmonella typhimurium включают эквивалентные гены или их части из других видов, таких как Salmonella typhi и Salmonella enterica, а также эквивалентные гены или их части из таких других бактерий, как кишечные бактерии.
Под термином "эквивалентный ген" подразумеваются гены, которые функционально эквивалентны, а также те, в которых мутация приводит к аналогичному фенотипу (например, авирулентности). Следует принять во внимание, что до настоящего изобретения VGC2 или содержащиеся в нем гены не были идентифицированы и, разумеется, не охарактеризовывались с точки зрения вирулентности.
Таким образом, дальнейшие аспекты настоящего изобретения предусматривают мутантную бактерию, в которой, если в обычном состоянии содержит ген, являющийся идентичным или эквивалентным гену в VGC2, то указанный ген мутируется или отсутствует в указанной мутантной бактерии; способы получения мутантной бактерии, согласно которым, если бактерия в естественном состоянии содержит ген, идентичный или эквивалентный гену в VGC2, то указанный ген мутируется или отсутствует в указанной мутантной бактерии. Далее описывается предпочтительный способ инактивации VGC2 гена. Вначале проводят субклонирование гена на фрагменте ДНК из библиотеки ДНК Salmonella λ или другой библиотеки ДНК с использованием фрагмента VGC2 в качестве зонда в экспериментах по гибридизации, затем ген картируют относительно сайтов рестрикционного фермента и характеризуют ген секвенированием ДНК в Escherichia coli. С использованием рестрикционных ферментов затем осуществляют введение в кодирующую область гена сегмента ДНК, кодирующего устойчивость к антибиотику (например, к канамицину), возможно после делеции части, кодирующей области клонированного гена с помощью рестрикционных ферментов. Имеются в распоряжении способы и конструкции ДНК, содержащие маркер антибиотической устойчивости, гарантирующие тот факт, что инактивация гена предпочтительно является неполярной, т.е. не оказывает влияния на экспрессию генов ниже по ходу транскрипции от интересующего гена. Затем мутантную версию гена переносят с E.coli на Salmonella typhimurium с использованием трансдукции фага Р22 и трансдуктантов, проверенных Саузерн гибридизацией на гомологическую рекомбинацию мутантного гена в хромосому.
Такой подход обычно используется на Salmonella (и может применяться в S.typhi), и дополнительные подробности могут быть найдены во многих статьях, например в работе Galan с сотр. (1992) 174, 4338-4339.
Еще одним аспектом изобретения является применение указанной мутантной бактерии в вакцине, фармацевтические композиции, содержащие указанную бактерию, и фармацевтически приемлемый носитель, полипептид, закодированный VGC2 ДНК Salmonella typhimurium или ее частью, или вариант его части, способ идентификации соединения, снижающего способность бактерии инфицировать или вызывать заболевание хозяина; соединение, идентифицируемое указанным способом, молекулу, которая селективно взаимодействует и существенно ингибирует функцию гена в VGC2 или его нуклеиновом продукте; а также применение в медицине фармацевтической композиции.
VGC2 содержит гены, которые идентифицированы способами по первому и второму аспектам изобретения, а также гены, которые были идентифицированы по их местоположению (хотя они могут быть идентифицированы способами по первому и второму аспектам изобретения). Эти дополнительные аспекты изобретения весьма близки к четвертому, пятому, шестому, седьмому, восьмому, девятому, десятому, одиннадцатому, двенадцатому и тринадцатому аспектам изобретения, и в соответствии с этим информация, касающаяся этих аспектов, и предпочтения, отмеченые в отношении этих аспектов изобретения, применимы к таким дополнительным аспектам.
Предпочтительно, если используемый ген является геном VGC2 или представляет собой эквивалентный ген из других видов Salmonella, например S.typhi. Предпочтительно, когда мутантная бактерия представляет собой мутант S.typhimurium или мутант других видов Salmonella, например, S.typhi.
Предполагается, что, по крайней мере, некоторые гены из VGC2 обеспечивают способность таких бактерий, как S.typhimurium к проникновению клеток.
Далее настоящее изобретение будет описано со ссылкой на следующие примеры и фигуры, где:
Фиг.1 иллюстрирует диаграмму одного из наиболее предпочтительных способов настоящего изобретения.
Фиг.2 демонстрирует Саузерн гибридизационный анализ ДНК из 12 S.typhimurium эксконъюгантов с последующим перевариванием с EcoRV. Фильтр зондировали с помощью канамицин-устойчивого гена мини-Тn5-транспозона.
На фиг.3 продемонстрирован блоттинг-гибридизационный анализ ДНК на колонии из 48 S.typhimurium эксконъюгантов с половины микротитрационной чаши (А1-Н6). Фильтр гибридизировали с зондом, содержащим меченые амплифицированные метки ДНК, выделенной из пула первых 24 колоний (А1-Д6).
На фиг.4 изображен блоттинг-гибридизационный анализ ДНК на колонии из 95 S.typhimurium эксконъюгантов с микротитрационной чаши (А1-Н11), которые инъецировали в мышей. Репликатные фильтры гибридизировали с мечеными амплифицированными метками из пула (инокулятная структура) или с мечеными амплифицированными метками из ДНК, изолированной из более чем 10000 объединенных колоний, которые выделяли из селезенки инфицированного животного (образец селезенки). Колонии В6, А11 и С8 давали сигналы слабой гибридизации на обеих сериях фильтров. Гибридизационные сигналы от колоний A3, С5, G3 (azOA) и F10 присутствуют в инокулянтном образце, но отсутствуют в образце селезенки.
На фиг.5 изображена последовательность гена Salmonella, выделенного с использованием способа изобретения, и данные сравнения с Escherichia coli cIp протеазным геномом.
Фиг.6 изображает частичные последовательности дополнительного гена Salmonella, выделенного с использованием способа изобретения (SEQ ID №№8-36).
На фиг.7 изображено картирование VGC2 на хромосоме S.typhimurium. (А) ДНК-зонды из трех областей VGC2 использовали в Саузерн гибридизационном анализе лизатов из серии штаммов S. typhimurium, закрепленных в Mud-P22 профагах. Показаны лизаты, которые гибридизировались с Pst1 фрагментом размером в 7,5 т.п.о. (зонд А на фиг.8). Два других используемых зонда гибридизировались с такими же лизатами. Точки инсерции и направления упаковки фага показаны вдоль положения карты в минутах (вариант VIII, ссылка 22 в примере 4). Обозначения фагов соответствуют следующим штаммам: 18Р, ТТ15242; 18Q, 15241; 19Р, ТТ15244; 19Q, ТТ15243; 20Р, ТТ15246 и 20Q, ТТ15245 (см. ссылки в примере 4). Местоположения картированных генов представлены горизонтальными полосами и указаны примерные локализации других генов.
На фиг.8 представлена физическая и генетическая карта VGC2. (А) Положения 16 транспозоновых вставок показаны верхней линией. Протяженность VGC2 указана толстой линией. Положение и направление транскрипции ORF, описанных в тексте примера 4, показаны стрелками, которые находятся ниже линии вместе с названиями аналогичных генов, за исключением ORF 12 и 13, продукты которых аналогичны сенсорным и регуляторным компонентам соответственно, из множества двухкомпонентных регуляторных систем. (В) Местоположение перекрывающихся клонов и EcoRI/XbaI рестрикционного фрагмента из Mud-P22 профагового штамма ТТ15244 показаны жирными линиями. Показаны лишь части λ клонов, которые были картированы, и такие клоны могут распространяться за рамки указанных пределов. (С) Указаны положения рестрикционных сайтов: В, BamHI; E, EcoRI; У, EcoRV; H, Hind III; Р, PstI и X, XbaI. Положения 7,5 т.п.о. PstI фрагмента (зонд А), используемого в качестве зонда на фиг.7 и 2,2 т.п.о PstI/Hind III фрагмента (зонд В), используемого в качестве зонда на фиг.10, указаны внизу рестрикционной карты. Положения последовательности 1 (описанной на фиг.11) и последовательности 2 (описанной на фиг.12) показаны тонкими стрелками (меченые последовательность 1 и последовательность 2).
На фиг.9 представлено картирование границ VGC2. (А) положения картированных генов на 37-38 минутах на E.coli K12 хромосоме приведены в соответстии с соответствующей областью S. typhimurium LT2 хромосомы (минуты 30-31). Расширенная карта VGC2 области показана в виде ДНК фрагментов 11 S.typhimurium, S.t., используемых в качестве зондов (толстые полосы) и рестрикционными сайтами, используемыми для их генерации: В, BamHI; С, ClaI; H, Hind II; К, KpnI; P, PstI; N, NsiI и S, SaiI. Зонды, которые гибридизуются геномной ДНК E.coli K12 (Е.с.) обозначены символом +; зонды, неспособные к гибридизации обозначены символом -.
Фиг.10 показывает, что VGC2 законсервирована и специфична к Salmonella. Геномная ДНК из Salmonella сероваров и других патогенных бактерий подвергалась рестрикции с PstI (A), Hind III или EcoRV (В) и подвергалась Саузерн гибридизационному анализу с использованием 2,2 кb PstI/Hind III фрагмента из λ клона 7 в качестве зонда (зонд В на фиг.2). Фильтры гибридизировали и промывали в строгих (А) и не строгих (В) условиях.
На фиг.11 показана последовательность ДНК "Последовательности 1" VGC2 от центра до левого конца (см. отмеченую стрелкой последовательность 1 на фиг.2). ДНК транслировали во всех шести открытых рамках считывания и указаны стартовые и останавливающие положения предполагаемых генов, а также положения инсерции транспозона для различных мутантов, идентифицированных с помощью STM (SEQ ID №37).
Как общепринято, символ * обозначает стоп-кодон, и, где необходимо, использованы коды нуклеотидной неопределенности.
На фиг.12 показана последовательность ДНК "последовательности 2" VGC2 (кластер С) (см. отмеченую стрелкой последовательность 2 на фиг.2). ДНК транслировали во всех шести рамках считывания и отмечали положения старта и остановки предполагаемых генов, а также положения инсерции транспозона для различных мутантов, идентифицированных STM (SEQ ID №38).
Как общепринято, индекс * обозначает стоп-кодон, и, где необходимо, используются стандартные коды нуклеотидной неопределенности.
Фиг.7-12 большей частью относятся к примеру 4.
ПРИМЕР 1
Идентификация вирулентных генов в Salmonella typhimurium
Материалы и методы
Бактериальные штаммы и плазмиды
Salmonella typhimurium штамм 12023 (эквивалентный штамму 14028 из американской коллекциии типовых культур (АТСС)) был получен от Национальной коллекции типовых культур (NCTC), Сервисной лаборатории здравоохранения, Колиндэйл, Лондон, Великобритания. Спонтанный мутант, устойчивый к налидиксовой кислоте указанного выше штамма (12023 Nalr), был отобран в лаборатории авторов изобретения. Другое производное штамма 12023, CL 1509 (аrОА:Тn10) было получено от Fred Heffron. Escherichia coli штаммы СС118 λ рir(Δ[аrа - leu], araD, Δ lac X74, galE, galK, phoA20, thi-1, rpsE, rpoB, argE/Am/, гесА1, λрir фаговый лизоген/ и S17-1λ pir/Tpr, Smr, recA, thi, pro, hsdR-M+, RP4:2-Tc:Mu:Km Tn7, λpir/ были выделены фирмой Kenneth Timmis. E.coli ДН5 α использовали для размножения puC18 /Гибко-BRL/ и Bluescript (Стратаген) плазмид, содержащих S. typhimurium ДНК. Плазмида риТмини-Тn5Кm2 (de Lorenzo с сотр., 1990) была получена от Kenneth Timmis.
Конструкция меток с полурандомизированными последовательностями и лигирования
Олигонуклеотидный пул RT1 (5’-CTAGGTACCTACAACCTCAAGCTT- /NK/20 - AAGCTTGGTTAGAATGGGTACCATG-3’, //SEQ ID №1) и праймеры Р2 (5’-TACCTACAACCTCAAGCT-3’) (SEQ ID №2), РЗ (5’-CATGGTACССАТТСТААС-3’) (SEQ ID №3), Р4 (5’-TACCCATTCTAACCAAGC-3’) (SEQ ID №4) и Р5 (5’-CTAGGTACCTACAACCTC-3’) (SEQ ID №5) синтезировали на олигонуклеотидном синтезаторе (Эпллайд Биосистемс, модель 380В). Метки двухнитевой ДНК готовили из RTI в 100 мкл объеме PCR, содержащем 1,5 мМ МgС12, 50 мМ КС1 и 10 мМ Трис-Сl (рН 8,0) в присутствии 200 пг RT1 в качестве мишени; по 250 мкМ каждого из dATP, dCTP, dGTP, dTTP; 100 пМ праймеров Р3 и Р5; и 2,5 ед. Ampitaq (Перкин-Элмер Петус). Условия циклической термообработки были следующими: 30 циклов при 95°С в течение 30 с, 50°С в течение 45 с и 72°С в течение 10 с. Продукт PCR очищали на геле (Sambrook с сотр. 1989), пропускали через колонку elutip D (полученную от Schleicher и Schull), расщепляли с помощью KPnl перед лигированием в pUC18 или рUТмини-Тn5Кm2. Для лигирований, плазмиды переваривали с KPnl и дефосфорилировали с телячьей кишечной щелочной фосфатазой (Гибко-BRL). Молекулы линеаризованной плазмиды очищали на геле (Sambrook с сотр., 1989) перед лигированием для удаления любой остаточной неразрезанной в процессе расщепления плазмидной ДНК. Реакционные среды для лигирования содержали примерно 50 нг каждой из плазмид и двухнитевую ДНК-метку в 25 мкл объеме в присутствии 1 единицы Т4 ДНК лигазы (Гибко-BRL) в буфере, содержащим фермент. Лигирования проводили в течение 2 часов при 24°С. Для определения количества бактериальных колоний, возникающих в результате самолигирования плазмидной ДНК, или из неразрезанной плазмидной ДНК, проводили контрольную реакцию, в которой в реакционную среду для лигирования не вводили двухнитевую ДНК-метку. В результате этого не получали ампициллинустойчивых бактериальных колоний после трансформации E.coli CC118 (Sambrook с сотр., 1989) по сравнению с образованием 185 колоний в среде для лигирования, содержащей двухнитевую ДНК-метку.
Бактериальная трансформация и спаривания
Продукты нескольких лигирований между pUT мини-Тn5Кm2 и двухнитевой ДНК-меткой использовали для трансформации E.coli СС118 (Sambrook с сотр., 1989). Составляли пул из примерно 10,300 трансформантов и плазмидную ДНК, экстрагированную из пула, использовали для трансформации E.coli S-17 λ pir (de Lorenzo, Timmis, 1994). Для экспериментов по спариванию пул из примерно 40,000 ампициллинустойчивых E.coli S-17 λ pir трансформантов и S.typhimurium 12023 Nalr индивидуально культивировали до оптической плотности (ОД)580 порядка 1,0. Аликвоты каждой культуры (0,4 мл) смешивали в 5 мл 10 мМ MgSO4 и фильтровали через мембрану Миллипор (с диаметром 0,45 мкм). Такие фильтры помещали на поверхность агара, содержащего М9 соли (de Lorenzo и Timmis, 1994) и инкубировали при 37°С в течение 16 часов. Бактерии были выделены встряхиванием фильтров в жидкой L среде в течение 40 минут при 37°С, и эксконъюганты отобраны путем посева суспензии на LB среду, содержащую 100 мкг мл налидиксовой кислоты (для селекции от донорного штамма) и 50 мкг мл канамицина (для селекции на реципиентный штамм). Каждый эксконъюгант проверяли путем переноса налидиксово-кислотноустойчивых (nalr), канамицин устойчивых (kanr) колоний на МакКонки Лактозную индикаторную среду (для различия между E.coli и S.typhimurium) и на LB среду, содержащую ампициллин. Примерно 90% nalr, kanr колоний оказались чувствительными к ампициллину, что является указанием на их образование в результате аутентичных транспозиционных событий (de Lorenzo, Timmis, 1994). Индивидуальные ампициллин-чувствительные эксконъюганты были помещены на 96-луночные микротитрационные планшеты, содержащие LB среду. При длительном хранении при - 80°С в среду вводили 7% ДМСО или 15% глицерина.
Характеристика фенотипа мутантов
Мутанты пересевали при помощи штампа с микротитрационных планшетов на твердую среду, содержащую М9 соли и 0,4% глюкозы (Sambrook с сотр., 1989) с целью идентификации ауксотрофов. Мутанты, колонии которых имеют ярко выраженную морфологию, определяют путем исследований колоний на агаровых чашках под микроскопом низкого разрешения.
Блоттинги колоний, экстракции ДНК, PCR, мечения ДНК и гибридизации
Для блоттинг-гибридизаций колоний использовали 48-луночный металлический репликатор (Сигма) для переноса эксконъюгантов с микротитрационных планшетов на нейлоновые фильтры Хибанд 11 (Амершам, Великобритания), которые помещали на поверхность LB агара, содержащего 50 мкг мл-1 канамицина. После инкубации в течение ночи при 37°С фильтры с колониями бактерий извлекали и сушили при комнатной температуре в течение 10 минут. Бактерии лизировали 0,4 н. NaOH и фильтры промывали 0,5 н. Трис-Сl рН 7,0 в соответствии с инструкциями производителей фильтров. ДНК бактерии фиксировали на фильтрах путем облучения в УФ-свете от Stratalinker (Стратаген). Гибридизации 32Р-мечеными зондами осуществляли в строгих условиях, как было описано выше (Holden с сотр., 1989). Для выделения ДНК S.tуphimurium транспозоновые мутантные штаммы выращивали в жидкой LB среде в микротитрационных планшетах или повторно суспендировали в LB среде с последующим выращиванием на твердой среде. Общую ДНК получали методом с использованием бромида гексадецилтриметиламмония (СТАВ), согласно методике Ausubel с сотр. (1987). Вкратце, клетки в объеме 150-1000 мкл осаждали центрифугированием и ресуспендировали в 576 мкл ТЕ. К полученной смеси добавляли 15 мкл 20% SDS (додецил сульфат Na) и 3 мкл 20 мг·мл-1 протеиназы К. После инкубирования при 37° в течение 1 часа добавляли 166 мкл 3 М NaCl и смесь тщательно перемешивали, после чего добавляли 80 мкл 10% (вес./об.) СТАВ и 0,7 М NaCl. После тщательного перемешивания раствор инкубировали при 65°С в течение 10 минут. После экстракции фенолом и смесью фенол-хлороформ ДНК осаждали добавлением изопропанола, промывали 70% этанолом и ресуспендировали в ТЕ при концентрации примерно 1 мкг·мкл-1.
Образцы ДНК подвергали двум циклам реакции PCR (полимеразно-цепьевая реакция) с целью получения меченых зондов. Первую PCR проводили в реакционной среде объемом 100 мкл, содержащей 20 мМ Трис-Сl рН 8,3; 50 мМ КС1; 2 мМ MgCl2; 0,01% Твина 80; 200 мкМ каждого из dATP, dCTP, dGTP, dTTP; 2,5 единицы Amlitag полимеразы (Перкин-Элмер Петус); по 770 нг каждого из праймеров Р2 и Р4; и 5 мкг ДНК-мишени. После начальной денатурации в течение 4 минут при 95° проводили циклическую термообработку, включающую 20 циклов в течение 45 секунд при 50°С, 10 с при 72°С и 30 с при 95°С. Продукты PCR экстрагировали смесью хлороформ/изоамиловый спирт (24/1) и осаждали этанолом. ДНК ресуспендировали в 10 мкл ТЕ и продукты PCR очищали электрофорезом через 1,6% Seaplaque (FMC Биопродактс) гель в буфере ТАЕ. Тонкие срезы геля, содержащие фрагменты размером примерно 80 т.п.о. и использовали для второй PCR. Эту реакцию проводили в общем объеме 20 мкл, содержащем 20 мМ Трис-Сl рН 8,3; 50 мМ КС1; 2 мМ МgСl2; 0,01% Твина 80; 50 мкМ каждого из dATP, dTTP, dGTP; 10 мкл 32Р-dCTP (3000 кюри/ммоль, Амершам); по 150 нг каждого из праймеров P2 и Р4; примерно 10 нг ДНК-мишени (1-2 мкл 1,6% Seaplaque агарозы, содержащей продукт первого цикла PCR); 0,5 единиц Amplitag полимеразы. Реакционную смесь покрывали 20 мкл минерального масла и осуществляли циклическую термообработку, как описано выше, количество введенной радиоактивной метки определяли поглощением на Whatman ДЕ81 бумагу (Sambrook с сотр. 1989).
Исследования заражения
Индивидуальные эксконъюганты Salmonella, содержащие меченые транспозоны, выращивали в 2% триптона, 1% дрожжевого экстракта, 0,92% об./об. глицерина, 0,5% Na2PO4, 1% KNO3 (TVGPN среда) (Ausubel с сотр. 1987) в микротитрационных планшетах в течение ночи при 37°С.
Металлический репликатор использовали для переноса небольшого объема, выдержанных в течение ночи, культур на свежий микротитрационный планшет и культуры инкубировали при 37°С до значения ОД580 (оптическая плотность при длине волны 580 нм) (c использованием многоканального спектрофотометра для прочтения микротитрационного планшета Titertec), равного примерно 0,2 в каждой лунке. Затем культуры из отдельных лунок объединяли и с помощью спектрофотометра определяли значение ОД550. Культуру разбавляли стерильным физиологическим раствором до примерно 5×105 КОЕ/мл. Дополнительные разбавления высевали на TVGPN, содержащую налидиксовую кислоту (100 мг×мл-1) и канамицин (50 мг·мл-1) для подтверждения значения КОЕ в инокуляте.
Группам из трех самок BAL В/с мышей (20-25 г) делали внутрибрюшинные инъекции 0,2 мл суспензии бактерий, содержащей примерно 1×105 КОЕ мл-1. Мышей умертвляли через три дня после инокуляции и удаляли их селезенки для выделения бактерий. Половину каждой селезенки гомогенизировали в 1 мл стерильного физиологического раствора в пробирке микрофуги. Клеточному дебрису позволяли осесть и 1 мл физиологического раствора, содержащего клетки все еще в суспендированном состоянии, переносили в свежую пробирку и центрифугировали в течение двух минут на микрофуге. Супернатант отделяли, а осадок после центрифугирования ресуспендировали в 1 мл стерильной дистиллированной воды. Получали серии разбавлении в стерильной дистиллированной воде и 100 мл каждого разбавления помещали на TYGPN агар, содержащий налидиксиновую кислоту (100 uг·мл-1 и канамицин (50 uг·мл-1). Бактерии выделяли с планшетов, содержащих 1000-4000 колоний, и создавали общий пул из примерно 10,000 колоний, выделенных с каждой селезенки и этот пул использовали для получения ДНК для PCR генерации зондов, которые применяли для скрининга пятен колоний.
Клонирование вирулентного гена и ДНК секвенирование
Общую ДНК выделяли из S.typhimurium эксконъюгантов и переваривали по отдельности с SstI, SalI, PstI и SphI. Перевары фракционировали через агароза гели, переносили на Нуbond N+ мембраны (Амершам) и подвергали саузерн-гибридизационному анализу с использованием канамицин устойчивого гена из рuТ мини-Тn5Кm2 в качестве зонда. Зонд метили дигоксигенином (Боэрингер-Маннхейм) и осуществляли хемилюминесцентную детекцию в соответствии с инструкциями производителя. Использовали описанные выше условия гибридизации и промывки. Рестрикционные ферменты, обеспечивающие гибридизационные фрагменты размером 3-5 т.п.о. использовали для переваривания ДНК на препаративном агарозовом геле и фрагменты ДНК, соответствующие размерам сигналов гибридизации, вырезали, очищали и лигировали на pU C18. Реакции лигирования использовали для трансформации E.coli ДН5а в канамициновую устойчивую форму. Плазмиды из канамицин-устойчивых трансформантов очищали пропусканием через колонку clutip D и проверяли путем расщепления рестриктазами. Плазмидные вставки частично секвенировали ди-деокси методом (Sanger с сотр., 1977) с использованием - 40 праймера и обратного секвенирующего праймера (Биохимическая корпорация США), а также праймеров Р6 (5’-CCTAGGCGGCCAGATCTGAT-3’) (SEQ ID №6) и Р7 (5’-GCACTTGTGTGTATAAGAGTCAG-3’) (SEQ ID №7), которые отжигались в 1 и 0 окончания Тn5 соответственно. Нуклеотидные последовательности и выведенные аминокислотные последовательности с использованием метода упаковки по программе Macvector 3,5 на компьютере Макинтош SE/30. Последовательности сравнивали с базами данных EMBL и Genbank ДНК с применением компьютерной системы UNIX/SUN в Исследовательском центре проектирования карты генома человека, Харроу, Великобритания.
РЕЗУЛЬТАТЫ
Строение метки
Структура ДНК-меток показана на фиг.1а. Каждая метка состоит из вариабельной центральной области, фланкированной "плечами" инвариантной последовательности. Последовательность центральной области (/NK/20) конструировалась таким образом, чтобы предотвратить возникновение сайтов для обычно применяемых п.о. распознающих рестрйкционных ферментов, но она была достаточно вариабельной для статистического обеспечения того, что такая же последовательность может возникать лишь раз в 2×10 молекул (ДНК секвенирование 12 случайно отобранных меток показало, что ни одна из них не имеет более чем 50% идентичности в рамках вариабельного участка). (N обозначает любое основание (А, G, С или Т), а К обозначает G или Т). Плечи содержат KpnI сайты вблизи окончаний для облегчения стадии начального клонирования, a HindIII сайты, граничащие с вариабельным участком, используются для выделения радиомеченых вариабельных участков из плеч перед гибридизационным анализом. Кроме этого, плечи конструировали так, что каждый из праймеров Р2 и Р4 содержит лишь один гуаниновый остаток. Поэтому в ходе PCR с использованием таких праймеров лишь один цитозин будет вводиться в каждое вновь синтезированное плечо по сравнению с, в среднем, десятью такими остатками в уникальной последовательности. В том случае, когда радиомеченый dCTP включается в PCR, в среднем, в десять раз большее количество метки будет присутствовать в уникальной последовательности по сравнению с каждым плечом. Целью такой операции является минимизация фоновых гибридизационных сигналов от плеч после их выделения из уникальных последовательностей в результате переваривания с HindIII. Двухнитевые метки лигировали в KpnI сайт мини-Тn5 транспозона Km2, переносимого на плазмиду PUT (de Lorenzo и Timmis 1994). Репликация такой плазмиды зависит от R6К-специфического π продукта pir гена. Он несет oriT последовательность плазмиды RP4, что обеспечивает перенос на множество бактериальных разновидностей (Miller и Mekalanos, 1988), a tnp ген необходим для транспозиции мини-Тn5-элемента. Меченые мини-Тn5 транспозоны переносили на S.typhimurium путем конъюгирования и 288 эксконъюгантов, образовавшихся в результате транспозиционных событий, хранили в лунках микротитрационных планшетов. Общую ДНК, выделенную из 12 таких конъюгантов переваривали с EcoRV и подвергали саузерн-гибридизационному анализу с использованием канамицин-устойчивого гена мини-Тn5 транспозона в качестве зонда. В каждом случае эксконъюгант возникал в результате единичной интеграции транспозона в различные сайты бактериального генома (фиг.2).
Исследования специфичности и чувствительности
Далее определялась эффективность и однородность амплификации ДНК-меток в реакциях PCR с использованием пулов эксконъюгантных ДНК в качестве мишеней реакций. С целью минимизации неравной амплификации меток в PCR, определялось максимальное количество ДНК-мишени, которое следует использовать в 100 мкл реакционной среды и минимальное число циклов PCR, в результате которых образуются продукты, которые могут визуализироваться путем окрашивания бромистым этидием на агарозовом геле (5 мкг ДНК и 20 циклов соответственно).
S. typhimurium эксконъюганты, которые достигли фазы стационарного роста в микротитрационных планшетах объединяли и использовали для экстракции ДНК. Ее подвергали PCR с использованием праймеров Р2 и Р4. PCR продукты размером 80 п.о. подвергали очистке на геле и использовали в качестве мишеней для второй PCR с применением тех же праймеров, но в присутствии 32Р-меченого СТР. В результате этого около 60% радиомеченого dCTP вводилось в PCR продукты. Радиомеченые продукты переваривали с HinIII и использовали в качестве зона для колон-блоттированной ДНК из их соответствующих микротитрационных планшетов. Из 1510 мутантов, тестированных этим методом, 358 оказались неспособными к проявлению отчетливого сигнала на авторадиограмме после ночного облучения пятна колонии. Для такого явления имеется три потенциальных объяснения. Во-первых, возможно, что часть транспозонов не несет меток. Однако при сравнении частот трансформации в результате реакций лигирования с участием транспозонов в присутствии или отсутствие меток представляется маловероятным, что немеченые транспозоны могут составлять более примерно 0,5% от общего их числа (см. Материалы и методы). Более вероятные причины состоят в том, что вариабельная последовательность процессируется в некоторых метках и/или, что некоторые из последовательностей образуют вторичные структуры, прием обе таких причины должны предотвращать амплификацию. Мутанты, не дающие отчетливых сигналов, исключены из дальнейших исследований. Специфичность эффективно амплифицированных меток демонстрировали образованием зонда из 24 колоний микротитрационного планшета и его использованием для зондирования блота из 48 колоний, включающих 24 колонии, которые использовали для образования зонда. Отсутствие какого-либо гибридизационного сигнала от 24 колоний, не использованных для образования зонда (фиг.3), показывает, что используемые условия гибридизации были достаточно строгими для предотвращения кросс-гибридизации среди радиомеченых меток и позволяет предположить, что каждый эксконъюгант не повторяется в микротитрационном планшете.
Имеются дополнительные соображения, касающиеся определения максимального размера пула, который может использоваться в качестве инокулянта в экспериментах на животных. Поскольку количество радиомеченых меток для каждого транспозона обратно пропорционально сложности меченого пула, имеется предел для размера пула, выше которого сигналы гибридизации становятся слишком слабыми для детекции после ночного облучения авторадиограммы. Более важным обстоятельством является то, что поскольку вариабельность пула повышается, должна реализоваться вероятность невозможности присутствия вирулентной составляющей пула в достаточных количествах в селезенке инфицированного животного для того, чтобы получить в достаточной степени меченый зонд. Верхний предел размер пула для использованной модели мышинного сальмонеллоза не определялся, но он должен быть выше 96.
Тесты на вирулентность транспозоновых мутантов
Всего 1152 уникально меченых инсерционных мутантов (с двух микротитрационных планшетов) тестировали на вирулентность у BALB/c мышей в двенадцати пулах, каждый из которых представлял собой 96-луночный микротитрационный планшет. Животным делали внутрибрюшинную инъекцию примерно 103 клеток каждого из 96 транспозоновых мутантов с микротитрационного планшета (всего 105 организмов). Через три дня после инъекции мышей умервляли и бактерии выделяли посевом гомогенатов селезенки на лабораторную среду. Получали пул из примерно 10,000 колоний, выделенных от каждой мыши, и экстрагировали ДНК. Метки, присутствующие в таком образце ДНК, амплифицировали и метили с помощью PCR, а колониевые блоты зондировали и сравнивали с гибридизационным образцом, полученным с использованием меток, амплифицированных из инокулянта (фиг.3). В качестве контроля аrо А мутант S.typhimurium метили и использовали в качестве одного из 96 мутантов в инокулянте. Не следовало ожидать, что такой штамм будет регенерироваться в селезенке поскольку его вирулентность значительно ослаблена (Buch-meier с сотр., 1993). Идентифицировали 41 мутант, ДНК которых была гибридизирована с радиомечеными метками из инокулянта, но не с метками из бактерий, выделенных из селезенки. Эксперимент повторяли и снова идентифицировали аналогичные 41 мутант. Как и ожидалось два из них представляли собой аrоА мутант (один на пул). Другой представлял собой ауксотрофный мутант (он не способен расти на минимальной среде). Все такие мутанты имели нормальную морфологию колоний.
ПРИМЕР 2
Клонирование и частичное охарактеризование последовательностей, фланкирующих транспозон
ДНК экстрагировали из одного из мутантов, описанных в Примере 1 (пул 1, F10), переваривали с SstI и субклонировали на базе канамициновой устойчивости. Последовательность в 450 п.о., фланкирующую один конец транспозона, определяли с использованием праймера Р7. Такая последовательность демонстрировала 80% идентичность E.coli dp (lon) гену, который кодирует терморегулируемую протеазу (фиг.5). Насколько известно, такой ген ранее не рассматривался, как вирулентная детерминанта. Неполные последовательности тринадцати Salmonella typhimurium вирулентных генов показаны на фиг.6 (последовательности А2-А9 и В1-В5). Установленные аминокислотные последовательности Р2D6, S4C3, P3F4, P7G2 и Р9В7 имели подобия семейству секреционно-ассоциированных белков, которые законсервированы бактериальными патогенами животных и растений, и которые известны в Salmonella, как семейство inv. в S. typhimurium inv гены требуются для бактериальной инвазии в кишечную ткань. Вирулентность inv мутантов уменьшается при их инокулировании оральным путем, но не при их внутрибрюшинном применении. Обнаружение inv-родственных генов, которые требуются для вирулентности после внутрибрюшинной инокуляции, предполагает новое секреторное устройство, которое может быть необходимым для инвазии нефагоцитных клеток селезенки и других органов. Продукты таких новых генов могут представлять собой лучшие лекарственные мишени, чем inv белки при лечении установленных инфекций.
Дополнительные характеристики генов идентифицированных в этом примере представлены в примере 4.
ПРИМЕР 3
Определения LД50 и изучение вакцинации мышей
Мутации, идентифицированные способом изобретения, ослабляют вирулентность.
Пять из мутаций в генах, которые ранее не подразумевали вирулентность, переносили Р22-регулируемой трансдукцией на родственный штамм S. typhimurium 12028, чувствительный к налидиксиновой кислоте. Трансдуктанты проверяли рестрикционным картированием и затем внутрибрюшинно инъецировали в группы ВАLВ/с мышей для определения их 50% летальной дозы (LД50). Значения LД50 для мутантов S4C3, P7G2, P3F4 и Р9В7 были на несколько порядков выше соответствующих значений для штамма дикого типа. Различий в значениях LД50 не детектировалось для мутанта P1F10: однако имело место статистически значимое уменьшение количества P1F10 клеток, выделенных из селезенок мышей, получивших инъекции инокулянта, состоящего из равных количеств такого штамма и штамма дикого типа. Это означает, что мутация действительно ослабляет вирулентность, однако степень такого ослабления не детектируется с помощью LД50.
Мутанты P3F4 и Р9В7 также применяли оральным путем при 1 уровне инокулянта порядка 107 клеток/мышь. Ни одна из мышей не заболела, что указывает на то, что оральные LД50 таких мутантов, по крайней мере, на порядок выше соотетствуюших значений для штамма дикого типа.
При исследовании вакцинации мышей группы из пяти самок BALB/c мышей массой 20-25 г, вначале инокулировали орально (р.о.) или внутрибрюшинно (i.p.) серийными десятикратными разбавлениями Salmonella typhimurium мутантных штаммов P3F4 и Р9В7. Затем через четыре недели мышей инокулировали 500 KOE родственного штамма дикого типа. Далее, в течение четырех недель регистрировали смертность.
Группу из двух мышей того же возраста и партии, что и мыши, инокулированные мутантными штаммами, также инокулировали i.p. 500 KOE штамма дикого типа в качестве положительного контроля. Как и ожидалось, обе неиммунизированные мыши погибали в течение четырех недель.
Полученные результаты приведены в таблице.

Из представленных экспериментальных данных можно сделать вывод, что мутант P3F4, по-видимому, обеспечивает некоторую защиту от последующего контрольного заражения дикого типа. Такая защита, по-видимому, выше для мышей, иммунизированных внутрибрюшинно (i.p.)
ПРИМЕР 4
Идентификация вирулентного локуса, кодирующего секреционную систему второго типа III в Salmonella typhimurium
В этом примере используются следующие сокращения: VGC1, вирулентный генный кластер 1; VGC2, вирулентный генный кластер 2.
Объяснение описанных экспериментов
Salmonella typhimurium является главным агентом гастроэнтерита людей и вызывает системные заболевания у мышей, которые служат моделью тифоидной лихорадки людей (1). После оральной инокуляции мышей S.typhimurium бактерии проходят из просвета тонкой кишки через слизистую кишечную оболочку, через энтероциты или М клетки фолликул бляшек Пейера (2). Далее бактерии поражают макрофаги и нейтрофилы, входят в ретикулоэндотелиальную систему и распространяются по другим органам, включая селезенку и печень, дальнейшая репродукция в которых приводит к ярко выраженной и фатальной бактериемии (3). Для поражения клеток-хозяев, выживания и репликации в разнообразных физиологически стрессовых внутриклеточных и клеточных окружающих средах и для преодоления специфических антибактериальных активностей иммунной системы, S.tiphimurium использует сложный набор вирулентных факторов (4).
С целью более полного понимания механизмов вирулентного действия S.typhimurium и других патогенов, в примере 1 описана система мутагенеза транспозона, который для удобства назван "сигнатура-меченым мутагенезом" (STM), который объединяет возможности мутационного анализа со способностью одновременного прослеживания судьбы большого числа различных мутантов в одном животном (ссылка 5 и пример 1; ссылка 5 опубликована после даты приоритета изобретения). С использованием такого подхода авторы изобретения идентифицировали 43 мутанта с пониженной вирулентностью из общего количества в 1152 мутанта, которые были подвергнуты скринингу. Нуклеотидные последовательности ДНК, фланкирующие точки инсерции транспозонов в 5 из таких мутантов показали, что они относятся к генам, кодирующим секреционные системы типа III большого числа бактериальных патогенов (6, 7). Продукты inv/spa генного кластера S.typhimurium (8, 9) представляют собой белки, образующие секреционную систему типа III, требующуюся для сборки поверхностных придатков, обеспечивающей вхождение в эпителиальные клетки (10). Следовательно, вирулентность штаммов, несущих мутации в inv/spa кластере, ослабляется лишь в том случае, когда инокулят применяют орально, а не внутрибрюшинно (8). В отличие от этого 5 мутантов, идентифицированных методом STM, являются авирулентными после внутрибрюшинной инокуляции (5).
В настоящем примере показано, что сайты инсерции транспозона 5 таких мутантов и 11 дополнительных мутантов, идентифицированных с помощью STM, картируются в одну и ту же область S.typhimurium хромосомы. Дополнительный анализ такой области позволил обнаружить дополнительные гены, установленные продукты которых, имеют последовательность, подобную другим компонентам секреционной системы типа III. Такая хромосмальная область, на которую ссылаются как на вирулентный генный кластер 2 (VGC2), отсутствует в ряде других кишечных бактерий и представляет собой важный локус для S.typhimirium вирулентности.
Материалы и методы
Бактериальные штаммы, трансдукция и растительная среда.
Salmonella enterica серотипы 5791 (aberdeen), 423180 (gallinarum), 7101 (cubana) и 12416 (typhimurium LT2) были получены из национальной коллекции типовых культур, Служба лаборатории здравоохранения, Великобритания. Salmonella typhi ВRD 123 геномная ДНК подарена G.Dougan энтерпатогенная Escherichia coli (EPEC), энтерогеморрагическая E.coli (EHEC), Vibro cholera биотип El Tor, Shigella flexneri серотип 2 и Staphylococcus aureus представляли собой клинические изоляты,полученные из департамента инфекционных болезней и бактериологии, Королевская медицинская аспирантура, Великобритания. Геномная ДНК из Yersinia pestis получена J.Heesemann. Однако геномная ДНК может быть выделена стандартными методами. Бактериальные штаммы и способы, применяемые для создания сигнатура-меченых мини-Тn5 транспозоновых мутантов S.typhimurium NCTC штамма 12023, были описаны ранее (5, 11). Общепринятое размножение плазмид осуществляли в E.coli ДН5α. Бактерии выращивали в LB бульоне (12), дополненном соответствующими антибиотиками. Перед оценкой уровней вирулентности индивидуальных мутантных штаммов мутации вначале переносили фаг Р22 - регулируемой трансдукцией (12) на чувствительный к налидиксовой кислоте родительский штамм S. typhimurium 12023. Трансдуктанты анализировали рестрикционным перевариванием и саузерн-гибридизацией перед использованием в качестве инокулянта.
Скрининг лямбда библиотеки. Лямбда (λ) клоны с перекрывающимися ДНК-вставками, покрывающими VGC2, получали стандартными методами (13) из λ 1059 библиотеки (14), содержащей вставки неполного Sau 3А перевара S.typhimurium LT2 геномной ДНК. Библиотеку получали через K.Sanderson из Центра по генетическому семейству Salmonella (SGSC), Калгари, Канада.
Mud-P22 лизогены. Радиомеченые ДНК-зонды гибридизировали Hybond N (Амершам) фильтрами, несущими ДНК, полученную из лизатов ряда S.typhimurium штаммов с якорно-закрепленными Mud-P22 профагами, в известных положениях в S.typhimurium генома. Получение митомицин-индуцированных Mud-P22 лизатов осуществляли, как описано в (12, 15). Набор Mud-P22 профагов был вначале собран Benson и Goldman (16) и был получен из SGSC.
Гель-электрофорез и саузерн-гибридизация. Гель-электрофорез проводили в 1% или 0,6% агарозных гелях в 0,5 × ТВЕ. Гель-фракционированную ДНК переносили на Hybond N или N+ мембраны (Амершам) и гибридизацию в строгих условиях и процедуры промывки (позволяющие проводить гибридизацию между нуклеотидными последовательностями с 10% или менее неправильных спариваний) проводили как описано Holden с сотр. (17). При нестрогих условиях (обеспечивающих гибридизацию между последовательностями с 50% неправильных спариваний) фильтры гидридизировали в течение ночи при 42°С в 10% формамид (0,25 М Na2HPO4/7% SDS и наиболее строгую стадию осуществляли с помощью 20 мМ Na2HPO4/l% SDS при 42°С. ДНК-фрагменты, используемые в качестве зондов, метили с помощью (32P) dCTP с использованием "Radprime" системы (Гибко-BRL ) или с (дигоксигенин-11/ dU TP и детектировали с применением Дигоксигениновой системы (Боэрингер Маннхейм) в соответствии с инструкциями производителей за исключением того, что гибридизацию проводили в том же растворе, что использовался для радиоактивно-меченых зондов. Геномную ДНК получали для саузерн-гибридизации, как описано ранее (13).
Молекулярное клонирование и нуклеотидное секвенирование.
Рестрикционные эндонуклеазы и Т4 ДНК-лигазу получали из Гибко-BRL. Использовали общие методы молекулярной биологии, описанные Sambrook с сотр. (18). Секвенирование нуклеотидов осуществляли с помощью метода дидеоксицепной терминации (19) с использованием Т7 секвенаторного набора (Фармация). Последовательности собирали с помощью программного обеспечения Маc Vector 3,5 или AssemblyLIGN упаковок. Нуклеотидную и соответствующие аминокислотные последовательности сравнивали с образцами из баз данных Европейской лаборатории молекулярной биологии (EMBI) и Swiss Prot с применением программ BLAST и FASTA GCG упаковки из Висконсинского Университа (версия 8) (20) в сетевой службе при Проектно-исследовательском центре картирования человеческого генома, Хинкстон, Великобритания.
Тесты на вирулентность. Группы из пяти самок BALВ/с мышей (20-25 г) инокулировали орально (р.о.) или внутрибрюшинно (i.p.) 10-кратными разбавлениями бактерий, суспендированных в физиологическом растворе. Для получения инокулята бактерии выращивали в течение ночи при 37°С в LB бульоне при встряхивании (50 об/мин) и затем использовали для инокуляции свежей среды в течение различных промежутков времени до достижения оптической плотности (ОД) при 560 нм порядка 0,4-0,6. Для плотностей клеток порядка 5 х 108 колониевообразующих единиц (KOE) на мл и выше, культуры концентрировали центрифугированием и ресуспендировали в физиологическом растворе. Концентрацию коЕ/мл проверяли посевом серий разбавлении инокулята на LB агаровые чашки. Мышей инокулировали i.p. 0,2 мл объемами и р.о. путем проглатывания того же объема инокулята. Значения LД50 рассчитывали через 28 дней по методу Reed и Meunch (21).
Результаты
Локализация транспозоновых инсерций. Образование банка Salmonella typhimurium мини Тn5 транспозоновых мутантов и скрининг, используемый для идентификации 43 мутантов с ослабленной вирулентностью были описаны ранее (5). Транспозоны и фланкирующие ДНК области клонировали из эксконъюгантов путем селекции на устойчивость к канамицину или обратной PCR. Получали нуклеотидные последовательности ДНК, имеющей протяженность 300-600 п.о., фланкирующей транспозоны для 33 мутантов. Сравнение таких последовательностей с соответствующими последовательностями из баз данных по ДНК и белкам показало, что 14 мутантов образовалось в результате инсерций транспозона в ранее известные вирулентные гены, 7 из мутантов образуются в результате инсерций в новые гены, обладающие подобием известным генам энтеробактерий, и 12 образуются в результате инсерций в последовательности, не обладающие подобием элементам ДНК и белковой баз данных (ссылка 5, пример 1 и настоящий пример).
Три основания позволяют предположить, что 16 из 19 транспозоновых инсерций в новые последовательности кластеризуются в трех областях генома, первоначально обозначенных символами А, В и С. Во-первых, сравнение нуклеотидных последовательностей из участков, фланкирующих сайты инсерций транспозонов, друг с другом и с последовательностями из базы данных показывает, что некоторые последовательности перекрываются одна другой или обладают сильным сходством с различными областями того же гена. Во-вторых, саузерн-анализ геномной ДНК, переваренной с несколькими рестрикционными ферментами и зондированной рестрикционными фрагментами, фланкирующими сайты инсерций транспозона, показывает, что некоторые транспозоновые инсерции находятся на одних и тех же рестрикционных фрагментах. В-третьих, в том случае, когда зонды ДНК гибридизированы в бляшки из библиотеки λ ДНК S.typhimiurium, зонды из мутантов, которые, как предполагается на двух предыдущих стадиях, способны связываться, на самом деле гибридизируются с теми же клонами λ ДНК. Таким образом, два мутанта (Р9В7 и P12F5) приписаны кластеру А, пять мутантов (Р2D6, Р9В6, Р11С3, Р11D10 и Р11Н10) - кластеру В, а девять мутантов (P3F4, P4F8, Р7А3, Р7В8, P7G2, P8G12, P9G4, Р10Е11 и Р11В9) приписаны кластеру С (фиг.8).
Гибридизация ДНК-зондов из трех этих кластеров с лизатами из ряда S.typhimurium штаммов с закрепленными замкнутыми Mud-P22 профагами (15, 16) показала, что все три локуса находятся в области 30-31 минут (вариант VIII, ссылка 22) (фиг.7), что является указанием на тот факт, что три этих локуса тесно связаны или составляют один крупный вирулентный локус. Для установления того, что какой-либо из λ клонов, покрывающих кластеры А, В и С, содержит перекрывающиеся ДНК-вставки, фрагменты ДНК из терминальных участков каждого клона использовали в качестве зондов в Саузерн-гибридизационном анализе других λ клонов. Гибридизация фрагментов ДНК показала, что несколько λ клонов перекрываются и что кластеры А, В и С содержат один смежный участок (фиг.8). Затем ДНК фрагменты из концов такого участка использовали для зондирования λ библиотеки с целью идентификации дополнительных клонов, содержащих вставки, представляющих собой соседние участки. Не были идентифицированы λ клоны, которые покрывают крайнее правое окончание локуса, в связи с чем такую область получали клонированием 6,5 т.п.о. EcoRI/XbaI фрагмента лизата Mud-P22 профагового штамма ТТ15244 (16).
Рестрикционное картирование и Саузерн-гибридизационный анализ далее использовали для конструирования физической карты такого локуса (фиг.8). Для того чтобы отличить такой локус от хорошо охарактеризованного inv/spa генного кластера на 63 минуте (вариант VIII, ссылка 22) (8, 9, 23, 24, 25, 26), на последний ссылаются как на вирулентный генный кластер 1 (VGC1), новый вирулентный локус обозначали как VGC2. На фиг.2 показано положение двух частей ДНК с установленной нуклеотидной последовательностью ("последовательность 1" и "последовательность 2"). Нуклеотидная последовательность представлена на фиг.11 и 12.
Картирование границ VGC2 на хромосоме S.typhimurium
Нуклеотидное секвенирование λ клона 7 с левой стороны VGC2 позволило обнаружить наличие открытой рамки считывания (ORF), установленная аминокислотная последовательность которой на примерно 90% идентична полученному продукту сегмента ydhE+ гена E.coli, а секвенирование клонированного фрагмента E.coRI/Xbal из 6,5 т.п.о. с правой стороны VGC2 обнаружило присутствие ORF, что предсказанная аминокислотная последовательность примерно на 90% идентична пируваткиназе 1 E.coli, кодированной pykF геном (27). На E.coli хромосоме ydhE и pykF расположены вблизи друг друга в интервале 37-38 минут (28). Одиннадцать неперекрывающихся ДНК фрагментов, распределенных по длине VGC2, использовали в качестве зондов в нестрогом Саузерн-гибридизационном анализе E.coli и S.typhimurium геномной ДНК. Гибридизация фрагментов ДНК показала, что участок в примерно 40 т.п.о., содержащий VGC2, отсутствует в Е. coli геноме и локализован на границах VGC2 в области 1 т.п.о. (фиг.9). Сравнение местоположения XbaI сайта вблизи с правым концом VGC2 (фиг.8) с картой известных XbaI сайтов (29) в области 30 минут хромосомы (22) позволяет определить позицию карты 30,7 минуты для VGC2.
Структура VGC2. Нуклеотидное секвенирование частей VGC2 обнаружило присутствие 19 ORF (фиг.8). G+C содержание примерно 26 т.п.о. нуклеотидной последовательности в VGC2 составляет 44,6%, по сравнению с 47% для VGC1 (9) и 51-53% установленному для всего Salmonella генома (30).
Полные, установленные аминокислотные последовательности ORF 1-11 аналогичны последовательностям белков секреционных систем типа III (6, 7), которые, как известно, необходимы для экспорта вирулентных детерминант в большое число бактериальных патогенов растений и животных (7). Предсказанные белки ORF 1-8 (фиг.8) аналогичны по организации и последовательности продуктам yscN-U генов Yarsinia pseudotuberculosis (31), invC/spaS кластера inv/spa в VGC1 Salmonella typhimurium (8, 9) и spa 47/spa 40 кластера spa/mxi Shigella flexneri (32, 33, 34, 35). Так, например, предсказанная аминокислотная последовательность ORF 3 (фиг.8) на 50% идентична YscS Y.pseudotuberculosis (31), на 34% идентична spa9 из S.flexneri (35) и на 37% идентична SpaQ VGC1 S.typhimurium (9). Предсказанный белковый продукт ORF9 близкородственен LcrD семейству белков с 43% идентичностью к LcrD Y. enterecolitica (36), 39% идентичностью к MxiA S.flexneri (32) и 40% идентичностью к InvA VGC1 (23). Неполные нуклеотидные последовательности для оставшихся ORF, показанных на фиг.8, указывают на то, что предсказанный белок из ORF10 наиболее близок Y.enterocolitica VscJ (37) липопротеину, расположенному на бактериальной внешней мембране, с ORF11, аналогичной S. typhimurium InvG, члену PulD семейства транслоказ (38). ORF12 и ORF13 демонстрируют значительное подобие сенсорной и регуляторной субединицам соответственно из большого числа белков, содержащих двухкомпонентные регуляторные системы (39). Имеется достаточная кодирующая способность для дополнительных генов между ORF 9 и 10, ORF 10 и 11 и между ORF 19 и правым концом VGC2.
VGC2 законсервирован в и специфичен к Salmonellae
2,2 т.п.о. PstI/HindIII фрагмент, находящийся в центре VGC2 (зонд В, фиг.8), не обладающий подобием последовательности к элементам баз данных ДНК и белков, использовали в качестве зонда в Саузерн-гибридизационном анализе геномной ДНК из Salmonella сероваров и других патогенных бактерий (фиг.10А). Фрагменты ДНК, гибридизирующиеся в нестрогих условиях, показывают, что VGC2 присутствует в S. aberdeen, S.gullinarum, S.cubana, S.typhi и отсутствует в ЕРЕС, ЕНЕС, V.pestis, S.flexneri, V. cholera и S.aureus. Таким образом, VGC2 законсервирован в и, по-видимому, специфичен к Salmonellae. Для установления факта консервации организации локуса среди испытанных Salmonella сероваров проводили Саузерн-гибридизации в строгих условиях геномной ДНК, переваренной с двумя дополнительными рестрикционными ферментами. Гибридизация фрагментов ДНК показала, что имеется некоторая гетерогенность в расположении рестрикционных сайтов между S.typhimurium LT2 и S.gallinarum, S.cubana и S.typhi (фиг.10В). Более того, S.gallinarum и S.typhi содержат дополнительные гибридизирующиеся фрагменты по отношению к тем, что присутствуют в других изученных Salmonellae, что позволяет предположить дупликацию участков VGC2 в таких разновидностях.
VGC2 необходим для вирулентности мышей. Предварительные эксперименты показали, что значения LД50 для i.p. инокуляции транспозоновых мутантов P3F4, P7G2, Р9В7 и Р11С3, по крайней мере, в 100 раз выше, чем для штамма дикого типа (5). Для выяснения важности VGC2 в процессе инфицирования определяли значения LД50 для орального и внутрибрюшинного применений для мутантов P3F4 и Р9В7 (Таблица 1). Оба мутанта демонстрировали понижение вирулентности на, по крайней мере, пять порядков при любом пути инокуляции в сравнении с родительским штаммом. Сильное ослабление вирулентности в результате обоих путей инокуляции демонстрирует тот факт, что VGC2 необходим для событий в инфекционном процессе после проникновения эпителиальной клетки в организм BAL В/с мыши.

Обсуждение
Был идентифицирован ранее неизвестный вирулентный локус в S.typhimurium размером примерно 40 т.п.о., расположенный в области 30,7 минуты на хромосоме путем картирования инсерционных сайтов группы сигнатурно-меченых транспозоновых мутантов с ослабленной вирулентностью. (5). Этот локус отнесен к вирулентному генному кластеру 2 (VGC2) для того, чтобы отличить его от inv/spa вирулентных генов на 63 минуте (вариант VII, ссылка 22), который по предложению авторов был переименован в VGC1. Как VGC1, так и VGC2 кодируют компоненты секреционных систем типа III. Однако такие секреционные системы отличаются друг от друга.
Из 19 мутантов, образующихся в результате инсерций в новые гены (ссылка 5 и настоящий пример), 16 картировались в одинаковый участок хромосомы. Возможно, что мини-Тn5 инсерция осуществляется преимущественно в VGC2. С другой стороны, поскольку негативный отбор, используемый для идентификации мутантов с ослабленной вирулентностью (5) был очень строгим (что отражается на высоких значениях LD50 для VGC2 мутантов), возможно, что среди ранее неизвестных генов лишь мутации в таких VGC2 обеспечивают степень ослабления, достаточную для обнаружения при отборе. Неудача предшествующих поисков S.typhimurium вирулентных детерминант для идентификации VGC2 может быть связана со снижением достоверности анализов на клеточной культуре по сравнению с моделью инфицирования на живом животном. В предыдущем исследовании по идентификации участков S.typhimurium LT2 хромосомы, уникальной в отношении Salmonellae (40), один из таких участков (RF333) был отнесен к области 30,5-32 минут. Поэтому RF333 может соответствовать VGC2, хотя неизвестно, что FF333 принимает участие в определении вирулентности.
Сравнения с секреционными системами типа III, кодированными вирулентными плазмидами Yersinia и Shigella, а также с VGC1 Salmonella показывает, что VGC2 кодирует основные структурные компоненты секреторного устройства. Кроме этого, порядок ORF 1-8 в VGC2 одинаков с генными порядками в гомологиях в Yersinia, Shigella и VGC1 S.typhimurium. Тот факт, что организация и структура VGC2 секреционной системы не более родственна с VGC1, чем с соответствующими генами Yersinia, в совокупности с низким G+C содержанием VGC2, позволяет утверждать, что VGC2 подобно VGC1 (40, 41, 42) независимо приобретается S.typhimurium в результате горизонтальной передачи. Белки, кодированные ORF 12 и 13, обладают сильным сходством с бактериальными двухкомпонентными регуляторами (39) и способны регулировать как ORF 1-11, так и/или секретированные белки такой системы.
Было показано, что многие гены в VGC1 являются важными для проникновения S.typhimurium в эпителиальные клетки. Такой процесс требует бактериального контакта (2) и приводит в результате к цитоскелетным перегруппировкам, приводящим к локализованным активным перемещениям в мембране клетки (43, 44). Роль VGC1 и его рестрикции на этой стадии инфицирования отражается примерно 50-кратным ослаблением вирулентности у BALB/c мышей, инокулированных р.о. мутантами VGC1, и тем фактом, что VGC1 мутанты не демонстрируют потери вирулентности при внутрибрюшинном применении (8). Второе наблюдение также объясняет, почему VGC1 мутанты не получаются в нашем скрининге (5). В отличие от этого, мутанты в VGC2 сильно ослабляются как после р.о., так и i.p. инокуляции. Этот факт показывает, что, в отличие от VGC1, VGC2 необходим для вирулентности у мышей после пенетрации эпителиальной клетки, однако такие факты не исключают роли VGC1 на такой ранней стадии инфицирования.
Таким образом, в резюмирующем картировании инсерционные сайты 16 сигнатура-меченых транспозоновых мутантов на Salmonella typhimurium хромосоме обеспечивают идентификацию 40 т.п.о. вирулентного генного кластера на 30,7 минуте. Такой локус законсервирован среди всех других изученных видов Salmonella, но отсутствует в большом числе других патогенных бактерий или в Escherichia coli K12. Нуклеотидное секвенирование части такого локуса обнаружило 11 открытых рамок считывания, предсказанные белки которых кодируют компоненты секреционной системы типа III. Для проведения различия между ней и секреционной системой типа III, кодированной локусом инвазии inv/spa, на inv/spa локус ссылаются как на вирулентный генный кластер 1 (VGC1), а на новый локус - как на VGC2. VGC2 имеет более низкое G+C содержание, чем у Salmonella генома и фланкирован генами, продукты которых имеют более чем 90% идентичность с продуктами E.coli ydhE и pykF генов. Таким образом, вероятно, что VGC2 приобретается горизонтально путем инсерции в участок, соответствующий области между ydhE и pykF генами E.coli. Исследования вирулентности VGC2 мутантов показали, что она ослабляется, по крайней мере, на пять порядков по сравнению со штаммом дикого типа после оральной или внутрибрюшинной инокуляции.
Литература к этому примеру
1. Carter, P.B. & Collins, F.M. (1974) J. Exp. Med. 139, 1189-1203.
2. Takeuchi, A. (1967) Am. J. Pathol 50, 109-136.
3. Finlay, B.B. (1994) Curr. Top. Microbiol. Immunol. 192, 163-185.
4. Groisman, E.A. & Ochman, H. (1994) Trends Microbiol. 2, 289-293.
5. Hensel, M., Shea, J.E., Gleeson, C., Jones, M.D., Dalton, E. & Holden, D.W. (1995) Science 269, 400-403.
6. Salmond, G.P.C. & Reeves, P.J. (1993) Trends Biochem. Sci. 18, 7-12.
7. Van Gijsegem, F., Genin., S. & Boucher, C. (1993) Trends Microbiol. 1, 175-180.
8. Galan. J.E. & Curtiss, R. (1989) Proc. Natl. Acad. Sci. U.S.A. 86, 6383-6387.
9. Groisman, E.A. & Ochman, H. (1993) EMBO J. 12, 3779-3787.
10. Ginocchio, С.С., Olmsted, S.B., Wells, C.L. & Galan, J.E. (1994) Cell 76. 717-724.
11. de Lorenzo, V. &. Timmis, K.N. (1994) Methods Enzymol. 264, 386-405.
12. Davis, R.H., Botstein, D. & Roth, J.R. (1980) Advanced Bacterial Genetics, Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
13. Ausubel, P.M., Brent, R., Kingston, R.E., Moore, D.D., Seidman, J.G., Smith, J.A. and Struhl, K. (1987) Current Protocols in Molecular Biology Vol 4 John Wiley and Sons, Inc, New York.
14. Maurer, R., Osmond, B.C., Shekhtman, E., Wong, A. & Botstein, D. (1984) Genetics 108, 1-23.
15. Youderain, P., Sugiono, P., Brewer, K.L., Higgins, N.P. & Elliott, T. (1988) Genetics 118, 581-592.
16. "Benson, N.R. & Goldman, B.S. (1992) J. Bacteriol. 174, 1673-1681.
17. Holden, D.W., Kronstad, J.W. & Leong, S. (1989) EMBO J. 8, 1927-1934.
18. Sambrook, J., Fritsch, E.F. & Maniatis, T. (1989) Molecular cloning: a laboratory manual (Cold Spring Harbor Laboratory, Cold Spring Harbor, New York).
19. Sanger, F., Nicklen, S. & Coulson, A.R. (1977) Proc. Natl. Acad. Sci. U.S.A. 74, 5463-5467.
20. Devereux, J., Hearberli, P. & Smithies, O. (1984) Nucl. Acids Res. 12, 387-399.
21. Reed, L.J. & Muench, H. (1938) Am. J. Hyg. 27, 493-497.
22. Sanderson, K.E., Hessel, A. & Rudd, K.E. (1995) Microbiol. Rev. 59, 241-303.
23. Galan, J.E., Ginocchio, С. & Costeas, P. (1992) J. Bacieriol. 174, 4338-4349.
24. Ginocchio, С., Расе, J. & Galan, J.E. (1992) Proc. Natl. Acad. Sci. U.S.A. 89, 5976-5980.
25. Eichelberg, К., Ginocchio, C.C. & Galan, J.E. (1994) J. Bacteriol. 176, 4501-4510.
26. Collazo, C.M., Zierler, M.K. & Galan, J.E. (1995) Mol. Microbiol. 15, 25-38.
27. Ohara, O., Dorit, R.L. & Gilbert, W. (1989) Proc. Nail. Acad. Sci. USA 86, 6883-6887.
28. Bachman, В. (1990) Micro. Rev. 54, 130-197.
29. Liu, S.L., Hessel, A. & Sanderson, K.E. (1993) J. Bacteriol. 175, 4104-4120.
30. Fasman, G.D. (1976) CRC Handbook of Biochemistry and Molecular Biology, CRC Press, Cleveland.
31. Bergman, Т., Erickson, K., Galyov, E., Persson, С. & Wolf Watz, H. (1994) J. Bacteriol. 176, 2619-2626.
32. Andrews, G.P. & Maurelli, A.T. (1992) Infect. Immun. 60, 3287-3295.
33. Allaoui, A., Sansonetti, P.J. & Parsot, C. (1993) Mol. Microbiol. 7, 59-68.
34. Venkatesan, M.. Buysse, J.M. & Oaks, E.V. (1992) J. Bacteriol. 174, 1990-2001.
35. Sasakawa, С., Komatsu, К., Tobe, Т., Suzuki, Т. & Yoshikawa, M. (1993) J. Bacteriol. 175, 2334-2346.
36. Piano, G.V., Barve, S.S. & Straley, S.C. (1991) J. Bacteriol. 173, 7293-7303.
37. Michiels, Т., Vanooteghem, J.C., Lambert de Rouvroit, C., China, В., Gustin, A., Boudry, P. & Cornelis, G.R. (1991) J. Bacteriol. 173, 4994-5009.
38. Kaniga, К., Bossio, J.C. & Galan, J.E. (1994) Mol. Microbiol. 13, 555-568.
39. Ronson, C.W., Nixon, B.T. & Ausubel, F.M. (1987) Cell 49. 579-581.
40. Groisman, E.A., Sturmoski, M.A., Solomon, F.R., Lin, R. & Ochman, H. (1993) Proc. Natl. Acad. Sci. U.S.A. 90, 1033-1037.
41. Altmeyer, R.M., McNern, J.K., Bossio, J.C., Rosenshine, I., Finlay, B.B. & Galan, J.E. (1993) Mol. Microbiol. 7, 89-98.
42. Li, J., Ochman, H., Groisman, E.A., Boyd, E.F., Soloman, F., Nelson, K. & Selander, R.K. (1995) Proc. Nail. Acad. Sci. USA 92, 7252-7256.
43. Finlay, B.B. & Rauschkowski, S. (1991) J. Cell Sci. 99, 283-296.
44. Francis, C.L., Starnbach, M.N. & Falkow, S. (1992) Mol. Microbiol. 6, 3077-3087.
ПРИМЕР 5
Идентификация вирулентных генов в Streptococcus pneumoniae
(а) Мутагенез
В отсутствие удобной транспозоновой системы наиболее эффективный путь создания меченых мутантов Streptococcus pneumoniae заключается в использовании инсерционно-дупликатного мутагенеза (Morrison с сотр. (1984) J.Bacteriol. 159, 870). Случайные S.pneumoniae ДНК фрагменты, размером в 200-400 п.о. могут быть получены перевариванием геномной ДНК рестрикционным ферментом или физическим распределением под действием ультразвука с последующим гель-фракционированием и репарацией конца ДНК с использованием Т4 ДНК полимеразы. Такие фрагменты лигируют в плазмиду pJDC9 (Pearce с сотр. (1993) Mol.Microbiol 9, 1037, которая несет erm ген для селекции эритромицина в E.coli и S.pneumoniae), предварительно модифицированную введением меток последовательности ДНК в один из полилинкерных клонирующих сайтов. Размер клонированной S.pneumoniae ДНК достаточен для обеспечения гомологической рекомбинации и уменьшает возможность возникновения нерепрезентативной библиотеки E.coli (экспрессия S.pneumoniae белков может быть токсичной для E.coli). Имеются в распоряжении альтернативные векторы, несущие селектируемые маркеры, и они могут использоваться вместо pJDC9. Меченые плазмиды, несущие ДНК фрагменты, вводили в соответствующий штамм S.pneumoniae, отобранный на основе серотипа и вирулентности в мышиной модели пневмококковой пневмонии. Регуляция компетентности генетической трансформации в S.pneumoniae управлялась фактором компетентности, пептидом из 17 аминокислот, который недавно был охарактеризован группой Don Morrison в Иллинойском Университете в Чикаго и описан Havarstein, Coomaraswamy и Morrison (1995) Proc.Natl. Acad.Sci. США 92, 11140-11144. Введение малых количеств такого пептида в трансформационные эксперименты приводило к проявлению очень эффективных трансформационных частот в некоторых инкапсулированных клинических изолятах S.pneumoniae. Это позволяет преодолеть главное препятствие в области пневмококковой молекулярной генетики и доступность пептида значительно облегчает конструирование S.pneumoniae мутантных банков и обеспечивает гибкость в выборе штаммов, подлежащих мутации. Определенное количество трансформантов анализировали с целью подтверждения гомологической интеграции плазмидных последовательностей и проверяли на стабильность. Очень низкий уровень реверсии, связанной с мутантами, полученными в результате инсерции-дуприкации, минимизируется благодаря тому факту, что дуплицированные участки являются короткими (200-400 п.о.); однако если уровень реверсии неприемлемо высок, то осуществляют антибиотическую селекцию в ходе роста трансформантов в культуре и в ходе роста в животном.
(b) Животная модель
S.pneumoniae мутантный банк организовывали в пулы для инокуляции в Swiss и/или С57В1/6 мышей. Проводили предварительные эксперименты для определения оптимальной вариабельности пулов и оптимального уровня инокуляции. В одной из моделей использовали инокулят из 105 коE, который орально доставляли в трахею (Veber с сотр. (1993) J.Antimicrobial Chemother. 32, 473). Мыши разновидности Swiss проявляли симптомы острой пневмонии за 3-4 дня, а мыши С57В1/6 демонстрировали подострую пневмонию через 8-10 дней. Такие легочные модели инфицирования обеспечивали 108 cfu/легкое (Veber с сотр. (1993) J.Antimicrobial Chemotherapy 32, 473) ко времени смерти. Если требовалось, то мышей также инъецировали внутрибрюшинно для идентификации генов, требуемых для инфицирования кровеносной системы (Sullivan с сотр. (1993) Antimicrobial Agents and Chemotherapy 37, 234).
(с) Идентификация вирулентного гена
После оптимизации параметров инфекционной модели мутантный банк, состоящий из нескольких тысяч штаммов, подвергали тестам на вирулентность. Мутанты с ослабленной вирулентностью идентифицировали гибридизационным анализом с использованием радиомеченых меток из "начального" и "регенированного" пулов в качестве зондов. Если S.pneumoniae ДНК неспособна к легкому блоттингу колонии, то хромосомальную ДНК высвобождали химически или ферментативно в лунках титрационных микропланшетов перед переносом на найлоновые мембраны с использования устройства для дот-блоттинга. ДНК фланкирующую интегрированную плазмиду клонировали плазмидным "спасением" в E.coli (Morrison с сотр. (1984) J.Bacteriol, 159870) и секвенировали. Библиотеки геномных ДНК конструировали в соответствующих векторах, находящихся либо в E.coli или в грамположительном штамме-хозяине и зондировали рестрикционными фрагментами, фланкирующими интегрированную плазмиду, для выделения клонированных вирулентных генов, которые затем подвергали полному секвенированию и детальному функциональному анализу.
ПРИМЕР 6
Идентификация вирулентных генов в Enterococcus faecalis
(a) Мутагенез
Мутагенез Е.faecalis осуществляли с использованием плазмиды рАТ112 или производного, созданного для этой цели. рАТ112 несет гены для селекции как в грамотрицательных, так и в грамположительных бактериях и att сайт Тn1545. Поэтому она требует присутствия в штамме-хозяине интегразы для транспозиции. Стабильные, однокопиевые инсерции получали в том случае, если хозяин не содержал гена эксцизионной гидролазы (Trieu-Cuot с сотр. (1991) Gene 106, 21). Выделение ДНК, фланкирующей интегрированную плазмиду, осуществляли рестрикционным перевариванием геномной ДНК, внутримолекулярным лигированием и трансформацией E.coli. Присутствие единичных сайтов для рестрикционных ферментов в рАТ112 и ее производных будет (Trieu-Cuot с сотр. (1991) Gene 106, 2l) обеспечивать введение меток последовательности ДНК до переноса на вирулентный штамм Е.faecalis, несущий плазмиду рАТ145 (для обеспечения интегразной функции) в результате конъюгации, электропорации или трансформации (Trieu-Cuot с сотр. (1991) Gene 106, 21; Wirth с сотр. (1986) J.Bacteriol. 165, 831).
(b) Животная модель
Большое число инсерционных мутантов анализировали на рандомизированную интеграцию плазмиды путем выделения ДНК из транципиентов, рестрикционно-энзимного перевара и продуктов Саузерн-гибридизации. Индивидуальные мутанты хранили в лунках микротитрационных планшетов, вариабельность и размер инокулянтных пулов оптимизировали перед скринингом мутантного банка. Использовали две различные модели инфицирования, вызываемого Е.faecalis. Первая из которых представляет собой хорошо установленную модель эндокардита на крысах, предусматривающую инъекцию до 108 коE Е.faecalis в хвостовую вену животных, имеющих катетер, вставленный поперек клапана аорты (Whitman с сотр. (1993) Антимикробные агенты и хемотерапия 37, 1069). Животных умертвляли через различные промежутки времени после инокуляции и бактериальные наросты на клапане аорты вырезали, гомогенизировали и высеивали на культурную среду для выделения бактериальных колоний. Вирулентные бактерии также выделяли из крови через различные промежутки времени после инокуляции. Вторая модель представляла собой перитонит мышей, возникающий после внутрибрюшинной инъекции до 109 коE Е.faecalis/Chenoweth с сотр. (1990) Антимикробные агенты и хемотерапия 34, 1800). Как и в случае модели S.pneumoniae, проводили предварительные эксперименты для установления оптимальной вариабельности пулов и оптимального уровня инокуляции перед скринингом мутантного банка.
(с) Идентификация вирулентного гена
Выделение ДНК, фланкирующей сайт интеграции рАТ112, используя ее E.coli начало репликации, упрощается в связи с отсутствием сайтов большинства рестриктаз, обычно используемых для распознавания 6 п.о. в векторе. В связи с этим, ДНК из интересующих штаммов переваривали с одним из таких ферментов, самолигировали, трансформировали в E.coli и секвенировали с использованием праймеров, базирующихся на последовательностях соседних с att сайтами плазмиды. Геномную ДНК библиотеку Е. faecalis зондировали интересующими последовательностями для идентификации интактных копий вирулентных генов, которые затем секвенировали.
ПРИМЕР 7
Идентификация вирулентных генов в Pseudomonas aeruginosa
(а) Мутагенез
Поскольку транспозон Тn5 использовался другими авторами для мутагенеза Pseudomonas aeruginosa, а мини-Тn5 производное, которое используется для идентификации Salmonella typhimurium вирулентных генов (пример 1), как сообщается, находит широкое применение среди грамотрицательных бактерий, включая некоторые псевдомонады (De Lorenzo и Timaris (1994) Methods Enzym, 264, 386), P.aeruginosa мутантный банк конструировали с использованием существующего пула сигнатура-меченых мини-Тn5 транспозонов путем конъюгального переноса вектора "самоубийцы" на один или более вирулентных (и возможно микоидных) реципиентных штаммов. Такой подход обеспечивает значительную экономию времени. Другие производные Тn5, сконструированные специально для P.aeruginosa мутагенеза (Rella с сотр. (1985) Gene 33, 293), могут альтернативно использоваться с мини-Тn5 транспозонами.
(б) Животная модель и идентификация вирулентного гена
Банк P.aeruginosa инсерционных мутантов подвергали скринингу на ослабленную вирулентность в модели хронической легочной инфекции у крыс. Суспензии P.aeruginosa вводили в бронхи после трахеотомии и заболевание развивалось в течение 30-дневного периода (Woods с сотр. (1982) Infect.Immun.36, 1223). Бактерии выделяли путем посева легочных гомогенатов на лабораторную среду и их меченые последовательности использовали для зондирования бактериальных пятен колоний ДНК, применяемых в качестве инокулянта. Можно также подвергать мутантный банк тестам на вирулентность в модели эндогенной бактериемии (Hirakata с сотр. (1992) Antimicrobial Agents and Chemother. 36, 1198) и циститного фиброза (Davidson с сотр. (1995) Nature Genetics 9, 351) у мышей. Клонирование и секвенирование ДНК, фланкирующую транспозоны, проводили, как описано в примере 1. Библиотеки геномных ДНК для выделения и секвенирования интактных копий генов конструировали в лаборатории стандартными методами.
ПРИМЕР 8
Идентификация вирулентных генов в Aspergillis fumigatus
(а) Мутагенез
Функциональный эквивалент транспозонового мутагенеза в грибах представляет собой регулируемую рестриктазами интеграцию (REMI) трансформирующей ДНК (Schiestl и Petes (1991) Proc.Natl.Acad.Sci. 88, 7585). В таком процессе клетки грибов трансформировали фрагментами ДНК, несущими селектируемый маркер, в присутствии рестрикционного фермента и однокопиевые интеграции осуществляли на различных геномных сайтах, ограниченных последовательностью-мишенью рестрикционного фермента. REMI уже с успехом использовали для выделения вирулентных генов Cochliobolus/Lu с сотр. (1994) Proc.Natl.Acad.Sci США 91, 12649) и Ustilago/Bolker с сотр. (1995) Mol.Gen.Genet. 248, 547) и было показано, что введение активного рестрикционного фермента с плазмидой, кодирующей гигромициновую устойчивость, приводит к единичной и, по-видимому, неупорядоченной интеграции линейной плазмиды в A.fumigatus геном. Меченые последовательности вводили в удобный сайт в одном или двух векторах гигромициновой устойчивости и использовали для трансформации клинического изолята A. fumigatus.
(б) Животная модель и индентификация вирулентного гена
Низкодозовая модель аспергиллеза у нейропениевых мышей особенно близко совместима с ходом легочного заболевания людей (Smith с сотр. (1994) Infect.Immun. 62, 5247). Мышей инокулировали интраназально, введением до 1,000,000 конидиоспор/мышь. Вирулентные мутанты грибов выделяли спустя 7-10 дней с использованием легочных гомогенатов для инокуляции жидкой среды. Через несколько часов собирали гифы, из которых экстрагировали ДНК для амплификации и мечения меток, для зондирования колониевых пятен ДНК из пула трансформантов, включающих инокулят. ДНК из участков, фланкирующих точки REMI инсерции, клонировали перевариванием трансформантной ДНК с рестрикционным ферментом, осуществляющим расщепление вектора вне REMI, самолигированием и трансформацией E.coli. Праймеры на основе известной последовательности плазмиды использовали для определения соседних A.fumigatus ДНК последовательностей. Для доказательства того, что инсерция вектора является причиной возникновения авирулентного фенотипа, выделенную плазмиду повторно расщепляли тем же рестрикционным ферментом, что использовался для клонирования, и подвергали обратной трансформации в родственный штамм A.fumigatus дикого типа. Затем трансформанты, возникшие в результате гомологической рекомбинации, подвергали тестам на вирулентность.



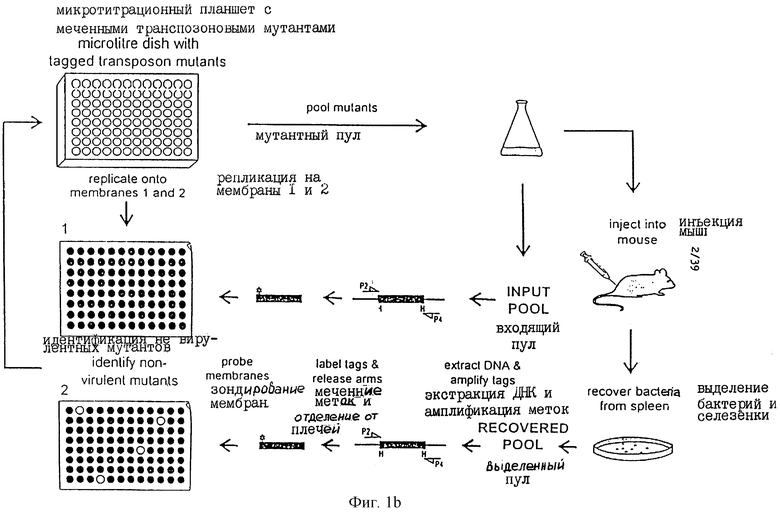
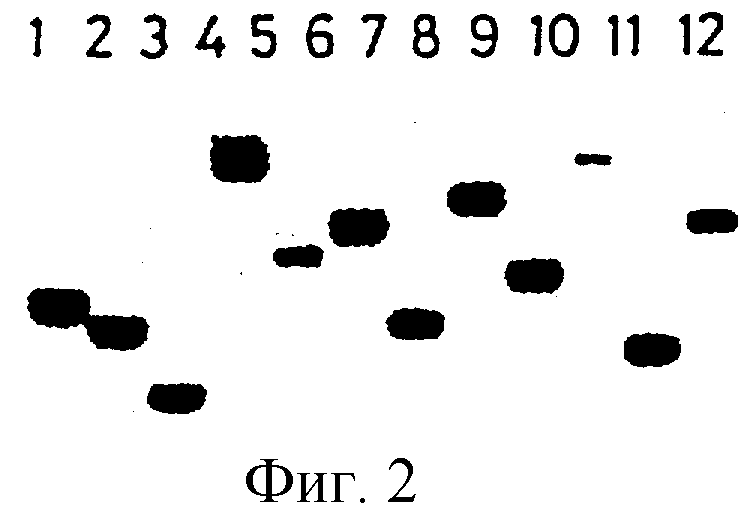

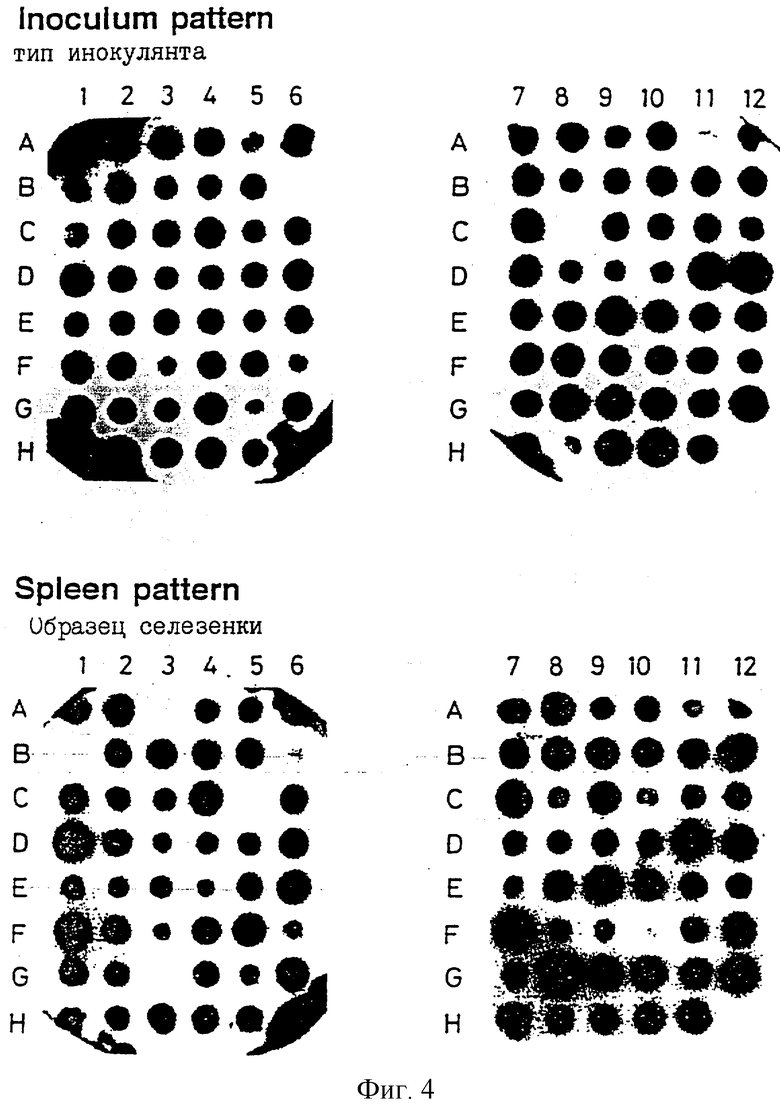




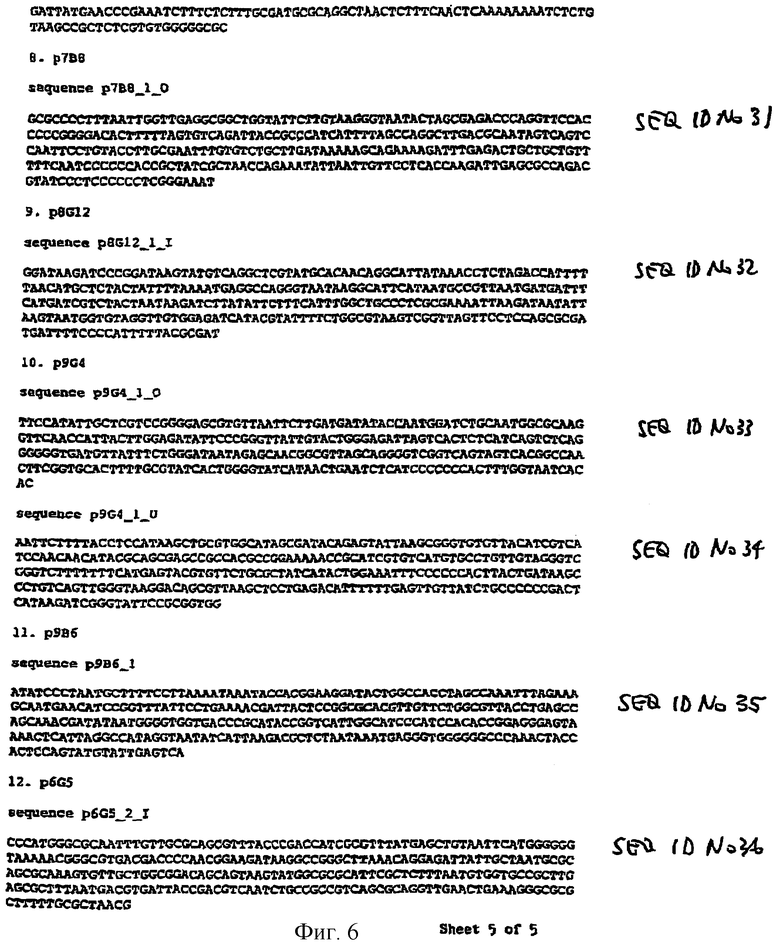
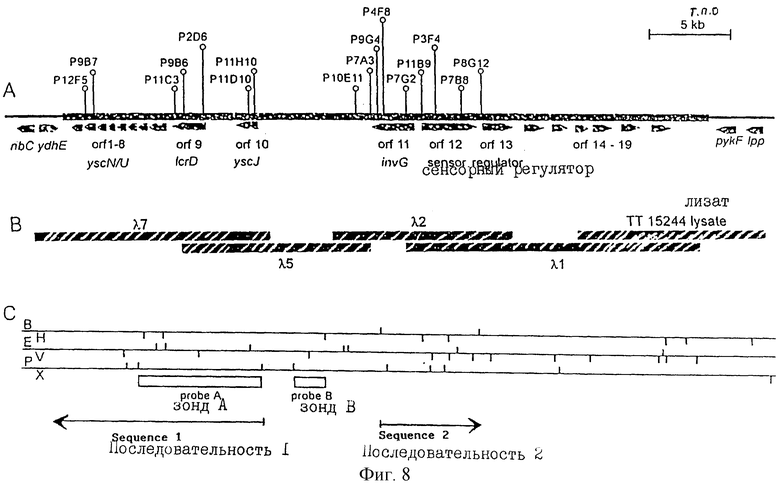
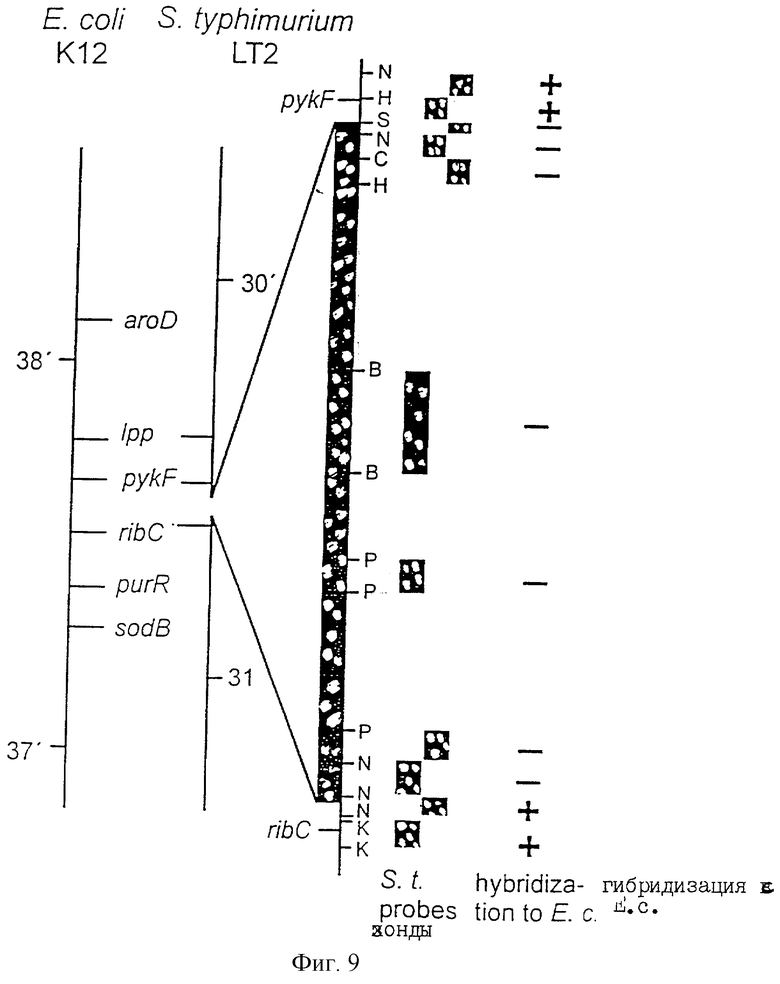
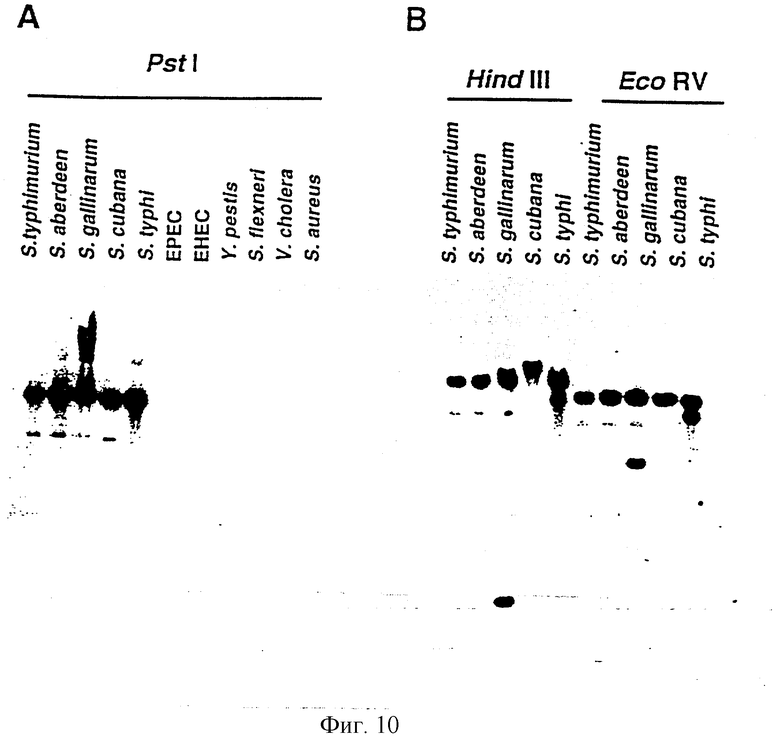

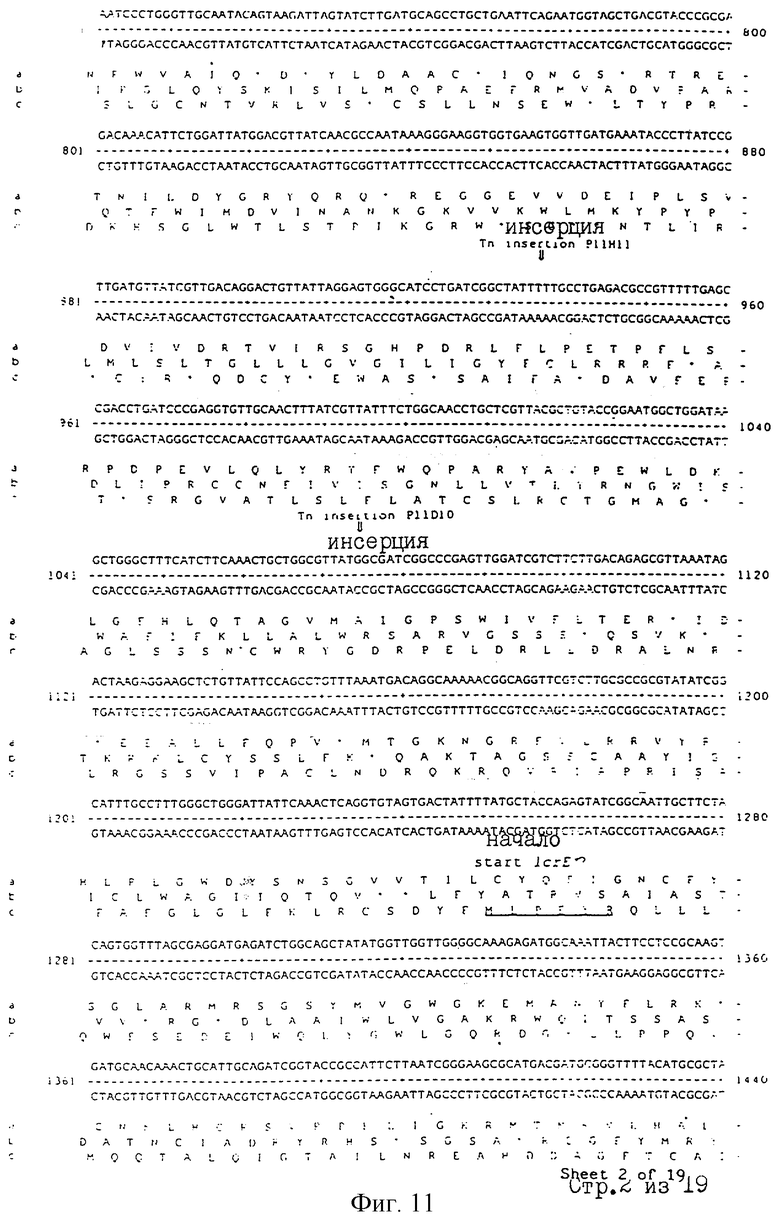
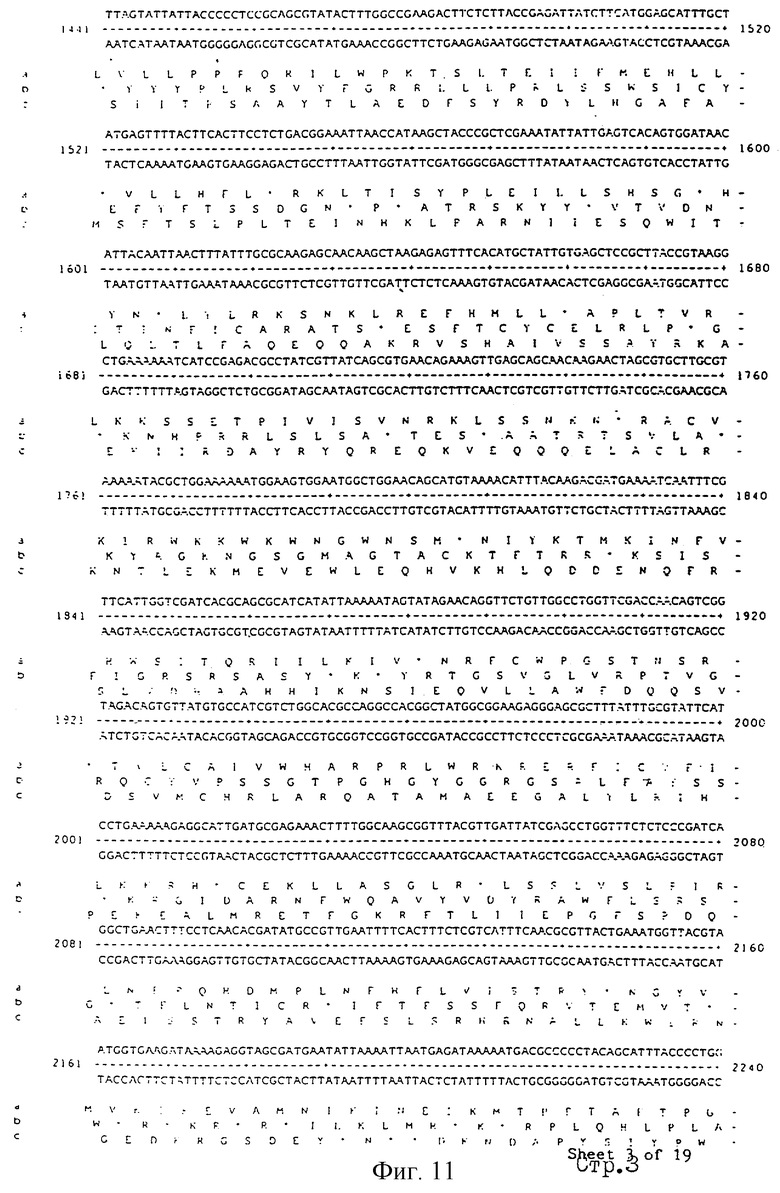


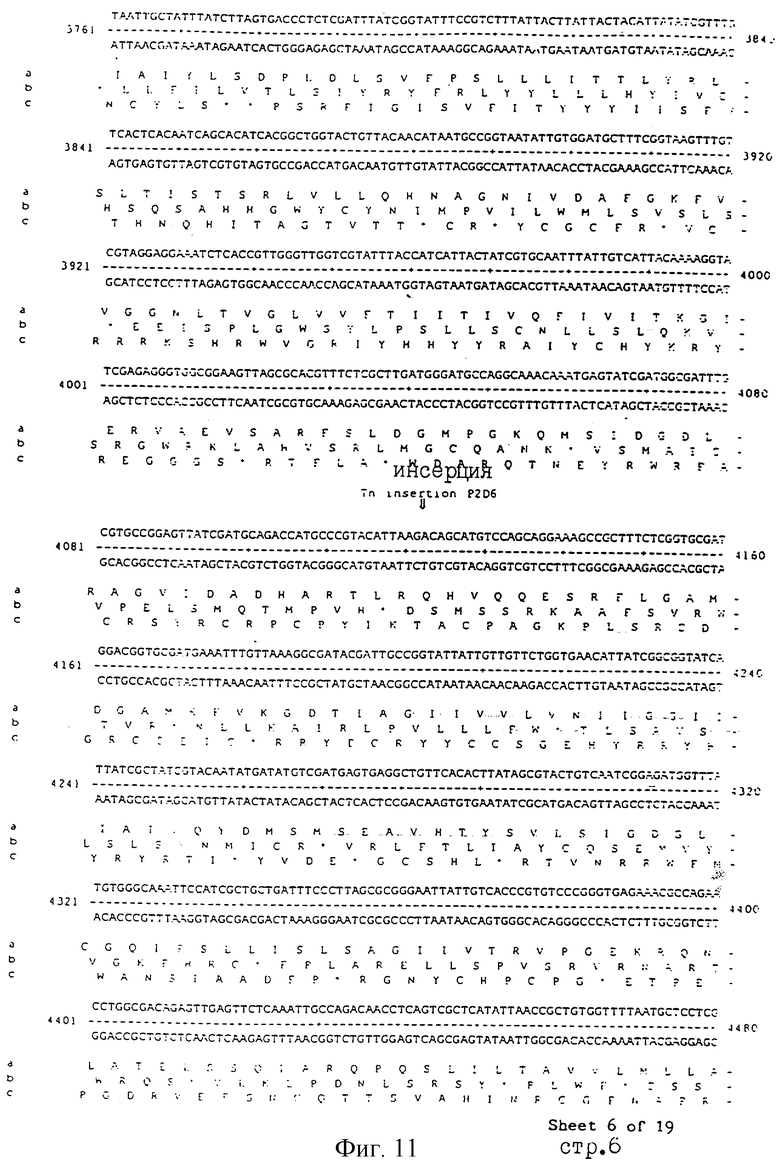
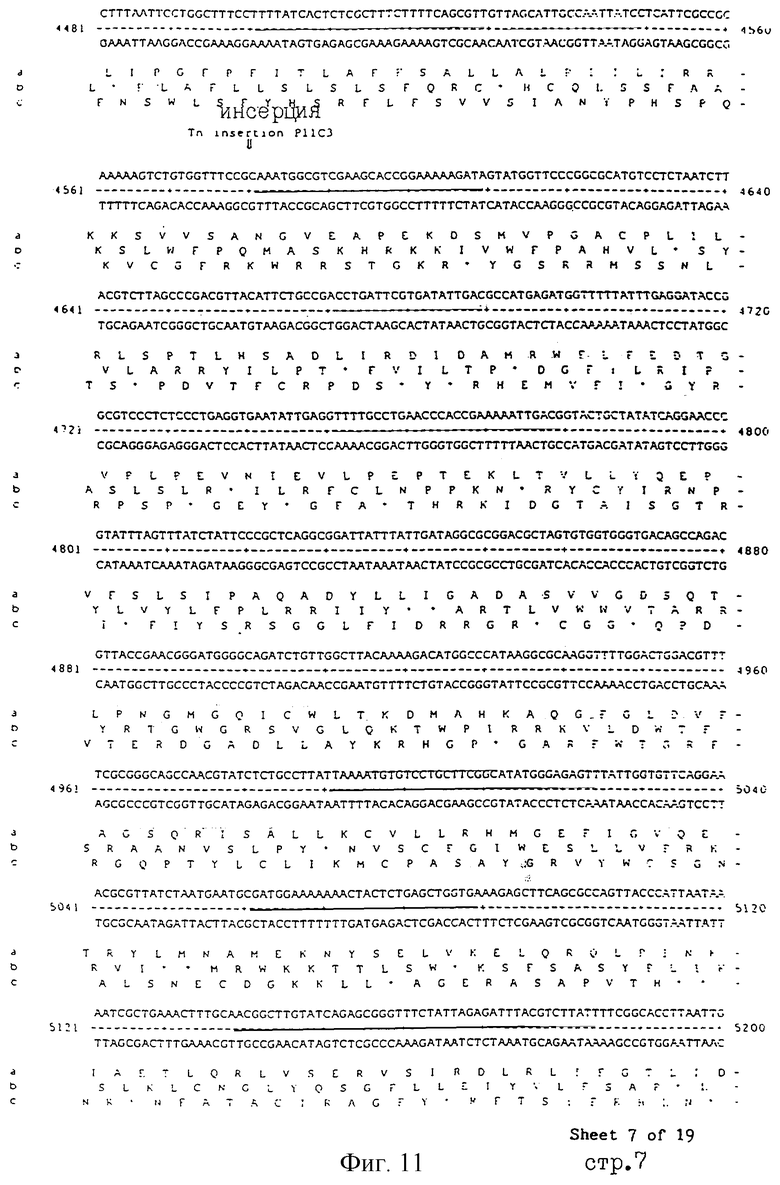
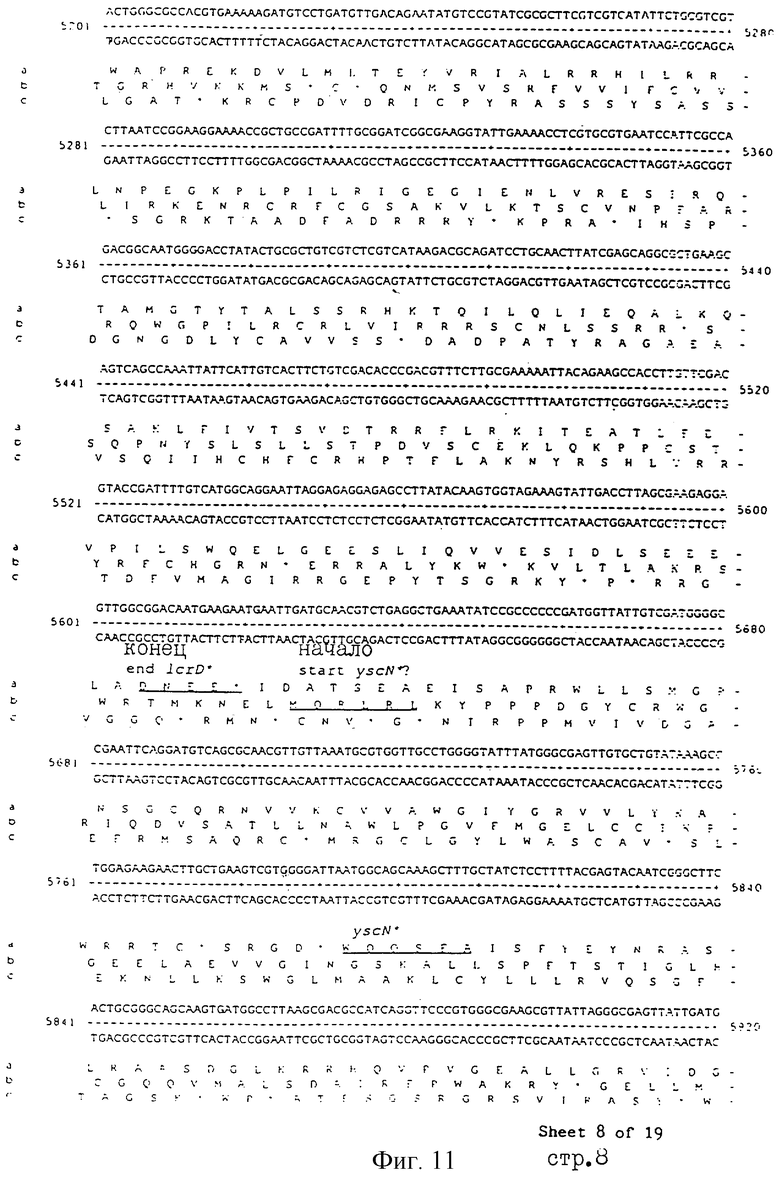
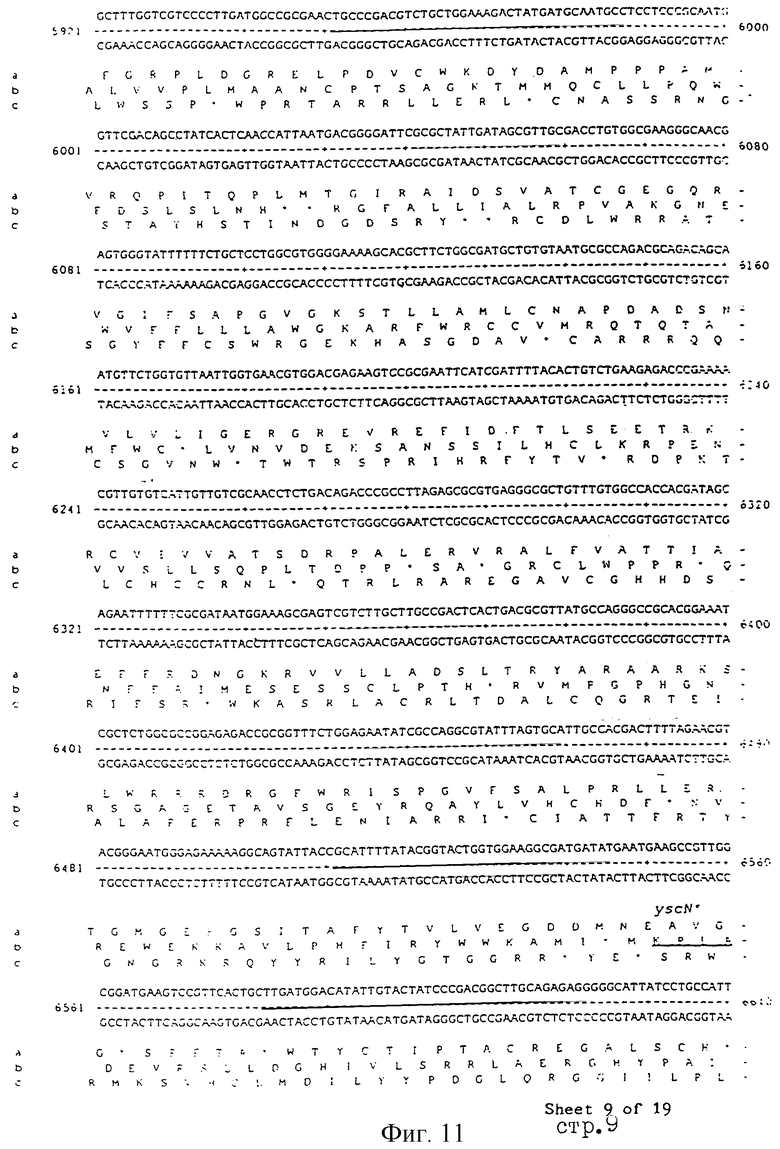


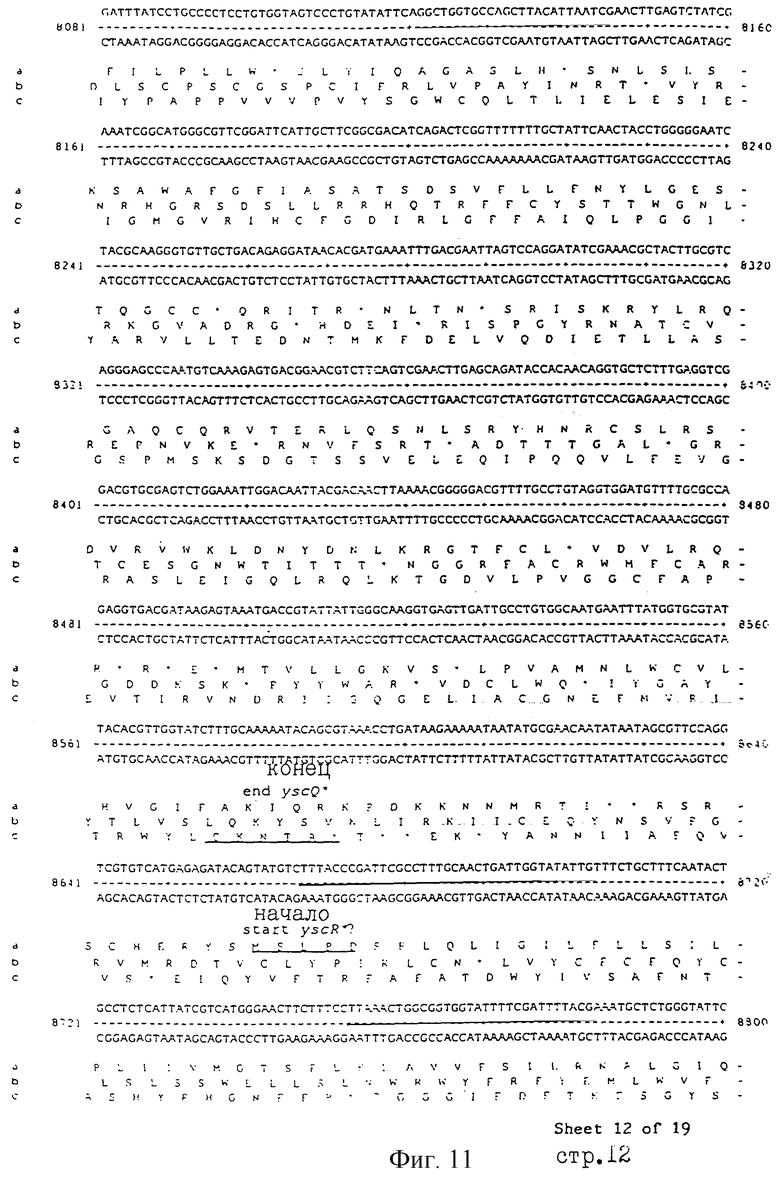

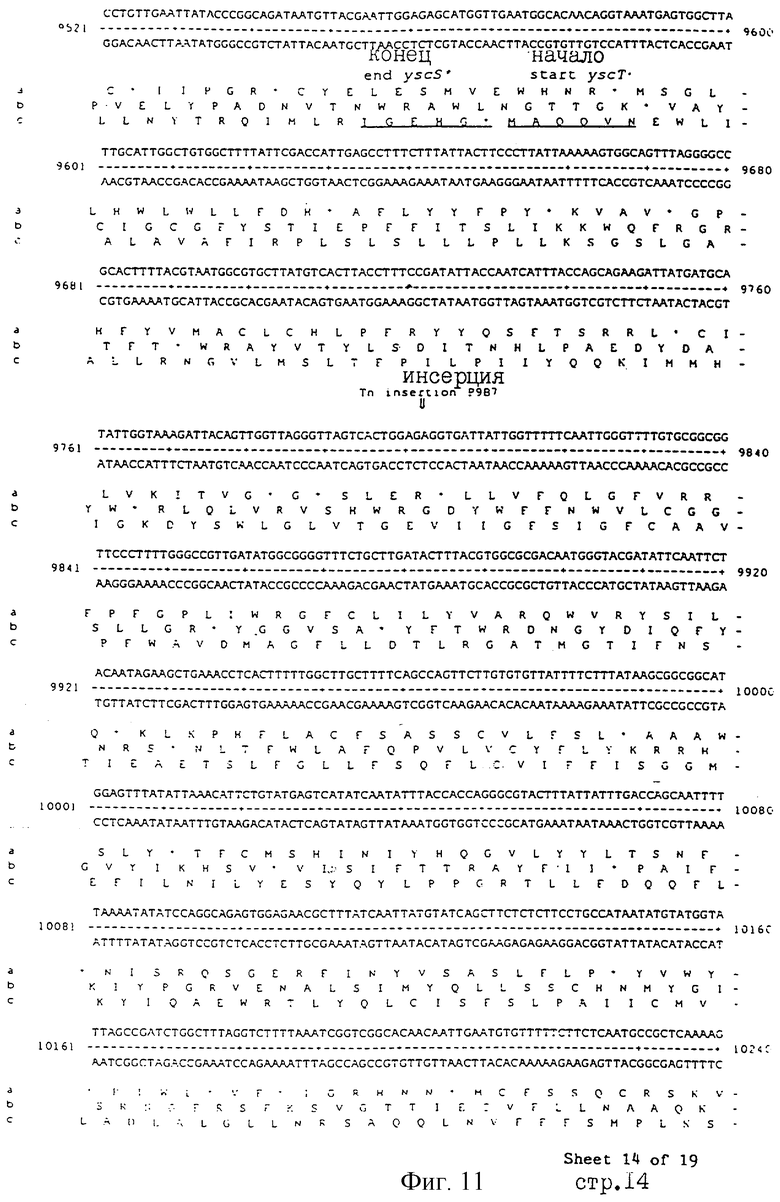
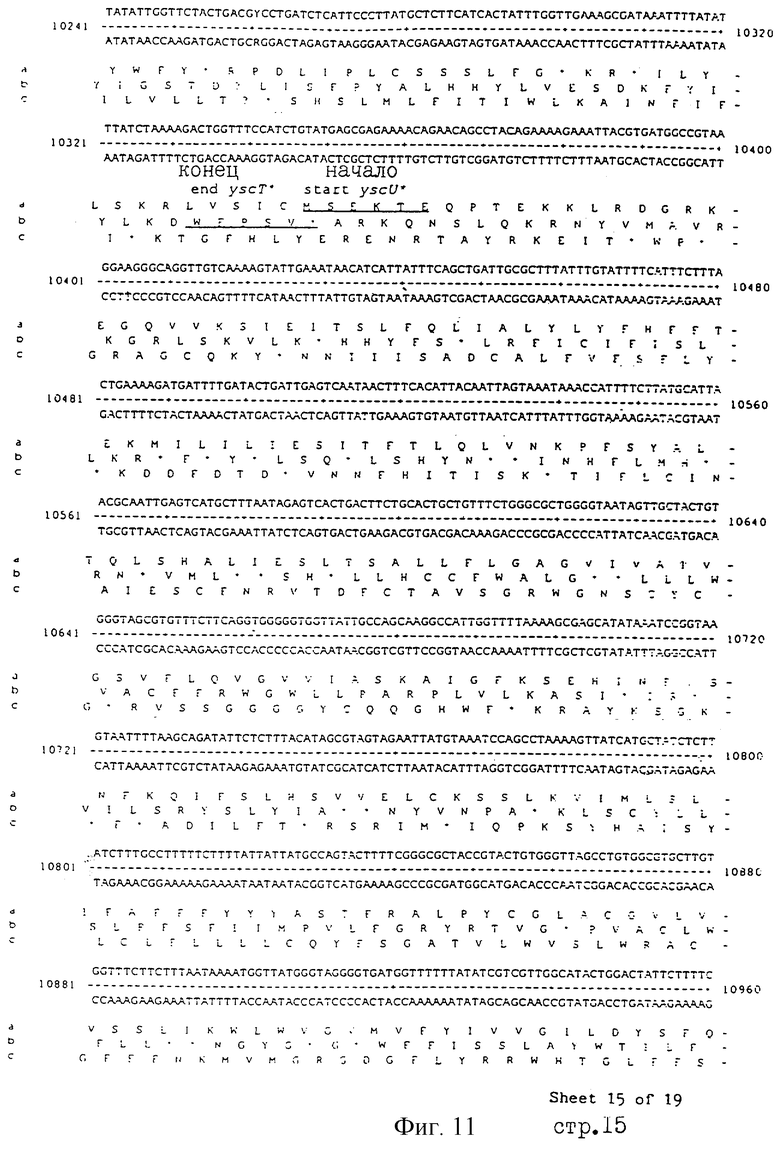
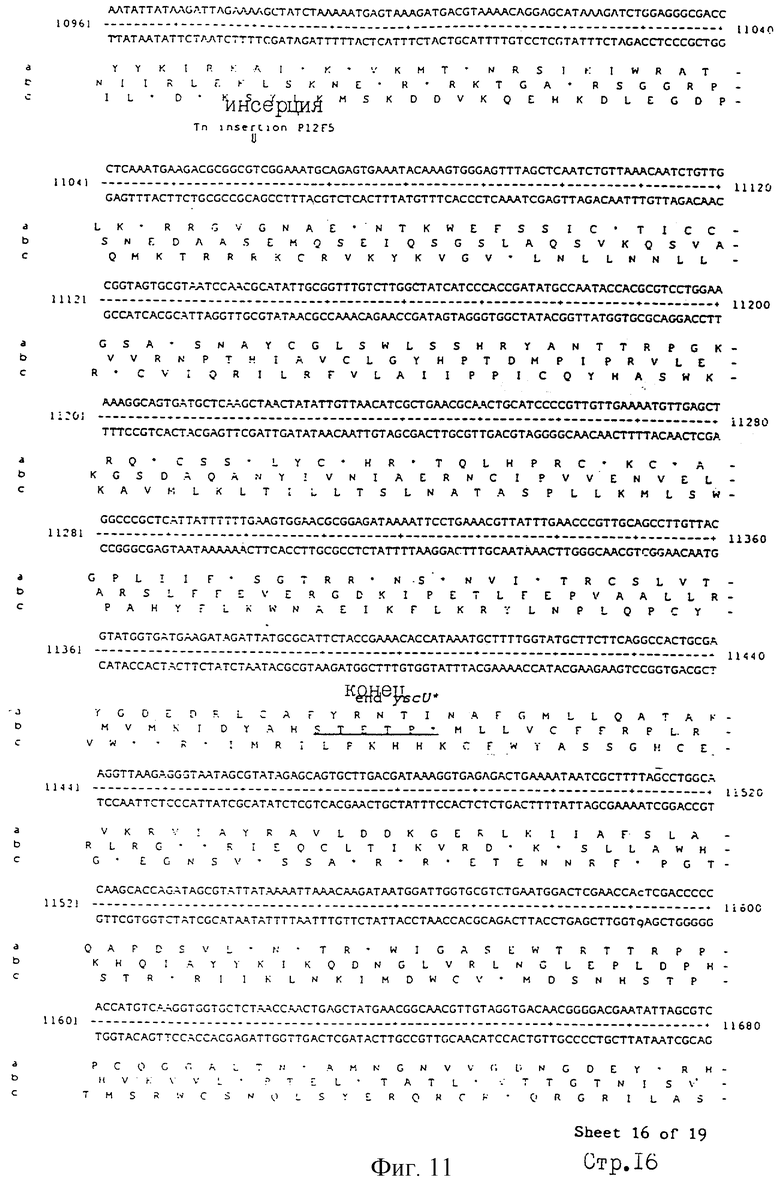
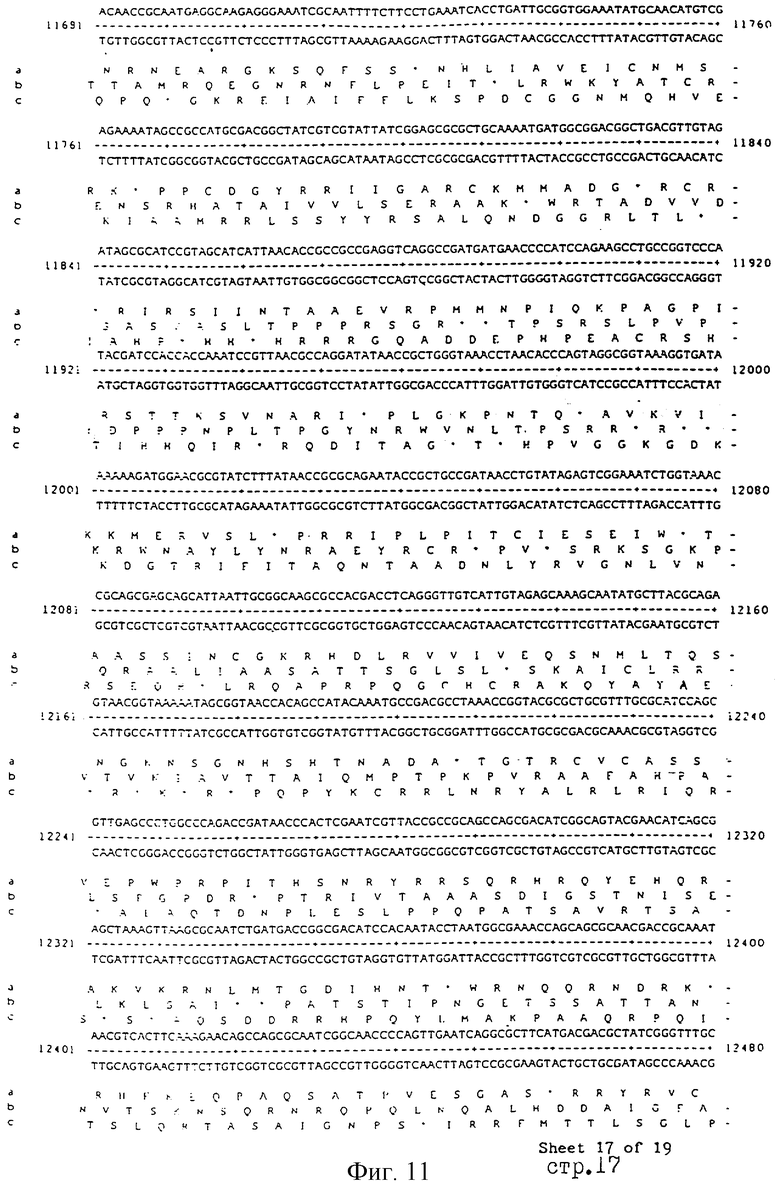
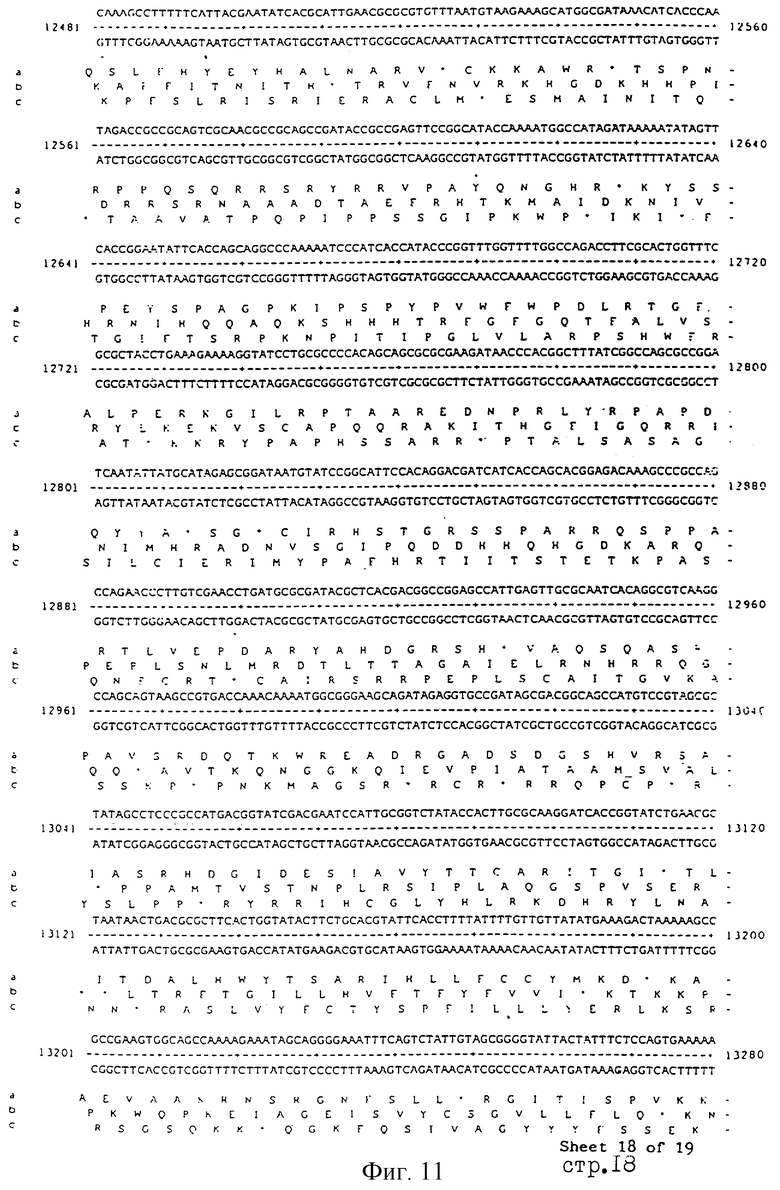
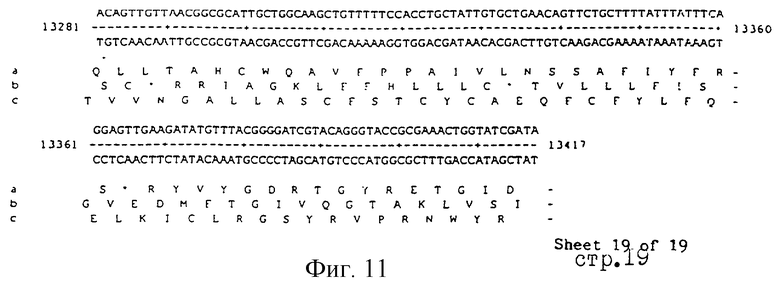




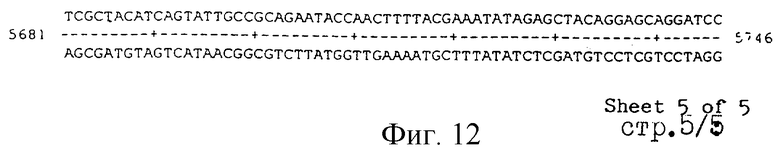
Изобретение относится к генетической инженерии, а в частности способу идентификации генов, участвующих в адаптации организма в среде его обитания. Предложен способ идентификации микроорганизма с пониженной адаптацией к конкретной окружающей среде. Способ предусматривает получение множества микроорганизмов, каждый из которых независимо мутирован инсерционной инактивацией гена нуклеиновой кислотой, содержащей уникальную маркерную последовательность, так, что каждый мутант содержит различные маркерные последовательности, или клонов указанного микроорганизма, стадия (1). Индивидуально хранение образца каждого мутанта, полученного на стадии (1), и индивидуально накопленной нуклеиновой кислоты, содержащей уникальную маркерную последовательность из каждого индивидуального мутанта, стадия (2). Введение множества мутантов, полученных на стадии (1), в указанную конкретную окружающую среду и обеспечение тем микроорганизмам, которые способны к этому, возможности расти в указанной окружающей среде, стадия (3). Извлечение микроорганизмов из указанной окружающей среды или ее выбранной части и выделение нуклеиновой кислоты из этих микроорганизмов, стадия (4). Сравнение любых маркерных последовательностей в нуклеиновой кислоте, выделенной на стадии (4), с уникальной маркерной последовательностью каждого индивидуального мутанта, накопленного на стадии (2), стадия (5). Отбор индивидуального мутанта, который не содержит какой-либо из маркерных последовательностей, выделенных на стадии (4), стадия (6). Предложены также ДНК (варианты), ответственные за снижение адаптации микроорганизма к окружающей среде, выделенные из генома Salmonella, ген вирулентности, содержащий эту ДНК, и полипептид, кодируемый этим геном. Кроме того, предложен способ определения соединения, снижающего адаптацию микроорганизма к окружающей среде. Предложенный способ предусматривает отбор такого соединения, которое препятствует функционированию гена, содержащего ДНК, ответственную за снижение адаптации микроорганизма к окружающей среде. Также предложена антисмысловая нуклеиновая кислота, полученная вышеуказанным способом. Предложенная антисмысловая нуклеиновая кислота используется в способе лечения хозяина, инфицированного или чувствительного к инфицированию микроорганизмом, и входит в фармацевтическую композицию, подавляющую патогенный микроорганизм. 10 н. и 50 з.п.ф-лы, 14 ил., 2 табл.
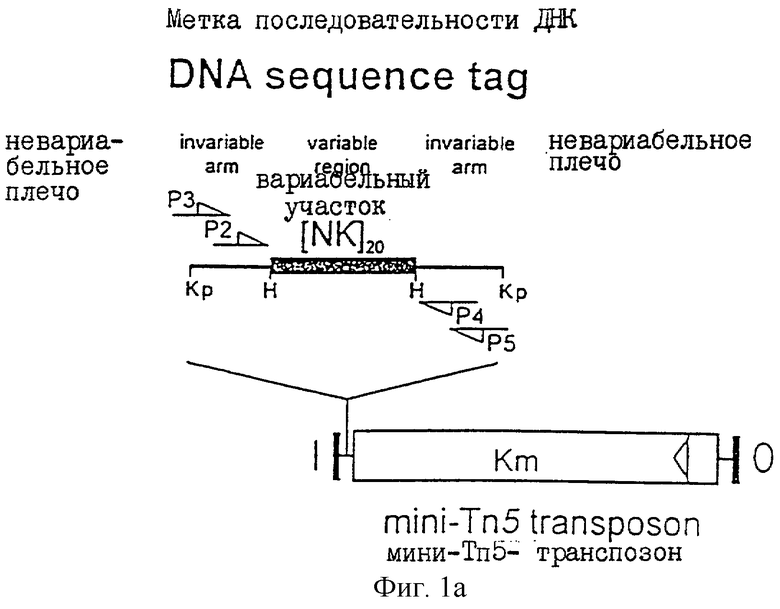
| HENSEL M | |||
| et al | |||
| Simultaneous identification of bacterial virulence genes by negative selection | |||
| Science | |||
| Топка с качающимися колосниковыми элементами | 1921 |
|
SU1995A1 |
| SLAUCH J.M | |||
| et al | |||
| In vivo expression technology for selection of bacterial genes specifically induced in host tissues | |||
| Methods Enzymol | |||
| Прибор для охлаждения жидкостей в зимнее время | 1921 |
|
SU1994A1 |
| PASCOPELLA L | |||
| et al | |||
| Use of in vivo complementation in Mycobacterium tuberculosis to identify a genomic fragment associated with virulence | |||
| Infect Immun | |||
| Прибор для охлаждения жидкостей в зимнее время | 1921 |
|
SU1994A1 |
| US 5397697, 14.03.1995. | |||

