Предпосылки изобретения
Область техники, к которой относится изобретение
Настоящее изобретение по сути относится к области медицины. Более конкретно настоящее изобретение касается способов получения новых растительных соединений, имеющих терапевтическое применение к млекопитающим.
Состояние проблемы
Растения являются ценным источником для идентификации новых биологически активных молекул. Одним из разнообразных классов молекул, которые были идентифицированы у растений, является класс сапонинов. Сапонины представляют собой высокомолекулярные соединения, являющиеся гликозидами, у которых сахарная составляющая присоединена к тритерпеновому или стероидному агликону. В частности, тритерпеновые сапонины являются объектами более высокого интереса благодаря их биологическим свойствам.
Были исследованы фармакологические и биологические свойства тритерпеновых сапонинов, вырабатываемых различными видами растений, включая фунгицидную, противовирусную, антимутагенную, спермицидную или контрацептивную, сердечно-сосудистую и противовоспалительную активности (Hostettmann et al., 1995). Как известно, в процессе связывания с липидами крови сапонины образуют комплексы с холестерином, тем самым, изменяя динамику его метаболизма (Oakenfull et al., 1983). Тритерпеновые гликозиды, включаемые в пищу, как было показано, снижают содержание холестерина в крови и тканях экспериментальных животных (Cheeke, 1971). Было найдено, что во многих странах сапонины входят в состав лекарственных средств народной медицины и некоторых недавно разработанных фитопрепаратов.
Известно, что тритерпен, глицирретиновая кислота и некоторые его производные обладают противоязвенной, противовоспалительной, противоаллергической, противогепатитной и противовирусной активностью. Например, некоторые производные глицирретиновой кислоты могут быть средствами профилактики или лечения язвы желудка (Doll et al., 1962). Среди таких соединений известными в данной области являются карбеноксолон (патент США №3070623), производные сложного эфира глицирретиновой кислоты, замещенные по 3’-положению (патент США №3070624), аминокислотные соли глицирретиновой кислоты (публикация патента Японии А-44-32798), амидные производные глицирретиновой кислоты (патент Бельгии №753773) и амидные производные 11-дезоксиглицирретиновой кислоты (патент Великобритании №1346871). Было показано, что глицирретиновая кислота ингибирует ферменты, участвующие в биосинтезе лейкотриенов, включая 5-липоксигеназную активность, и, считается, что она ответственна за противовоспалительную активность (Inoue et al., 1986).
Сообщалось, что бетулиновая кислота, пентациклический тритерпен, является избирательным ингибитором роста меланомы человека в ксенотрансплантационных моделях бестимусных мышей, а также, как было показано, является причиной цитотоксичности вследствие индуцирования апоптоза (Pisha et al., 1995). Было продемонстрировано, что тритерпеновый сапонин, происходящий от китайского растения семейства тыквенных, обладает противоопухолевой активностью (Kong et al., 1993). Известно, что моногликозиды тритерпенов показывают сильнодействующую и избирательную цитотоксичность против клеток лейкоза человека MOLT-4 (Kasiwada et al., 1992), а некоторые тритерпеновые гликозиды растений семейства касатиковых подавляют рост опухолей и повышают продолжительность жизни мышей, которым была имплантирована асцитная карцинома Эрлиха (Nagamoto et al., 1988). Препарат сапонина, происходящего от долихоса Dolichos falcatus, относящегося к семейству бобовых, как сообщалось, эффективен в отношении клеток саркомы-37 in vitro и in vivo (Huang et al., 1982). Было показано, что сапонин сои, также представляющей семейство бобовых, эффективно действует на несколько типов опухолей (Tomas-Barbaren et al., 1988). Олеаноловая кислота и гипсогениновые гликозиды, проявляющие гемолитическую и моллюскоцидную активность, были выделены из размельченных плодов бобовых Swartzia madagascariensis (Leguminosae) (Borel & Hostettmann, 1987).
Генистеин, являющийся природным изофлавоноидом, выделенным из продуктов сои, является ингибитором тирозинкиназ, и, как было установлено, ингибирует пролиферацию эстроген-положительных и эстроген-отрицательных линий клеток карциномы молочной железы (Akiyama et al., 1987). Инозитолгексафосфат (фитиновая кислота), широко распространенный в растительном мире и являющийся естественным пищевым компонентом зерновых и бобовых продуктов, как было показано, обуславливает терминальную дифференцировку линии клеток карциномы толстой кишки. Фитиновая кислота также проявляет противоопухолевую активность в отношении экспериментального карциногенеза толстой кишки и молочной железы in vivo (Yang et al., 1995). Также было показано, что некоторые тритерпеновые агликоны обладают цитотоксической или цитостатической активностью: например, было показано, что кора ствола растения Crossopteryx febrifuga (Rubiaceae) обладает цитостатической активностью в отношении линии клеток карциномы толстой кишки человека Со-115 в диапазоне нг/мл (Tomas-Barbaren et al., 1988).
Хотя в предыдущих сообщениях были идентифицированы тритерпеновые соединения, нашедшие широкое применение, до сих пор в данной области техники имеется насущная потребность в идентификации новых биологически активных тритерпеновых соединений. Многие из этих соединений токсичны в отношении нормальных клеток млекопитающих. Кроме того, биологические активности ранее идентифицированных тритерпенов колеблются в широких пределах и многие из них, по-видимому, характеризуются ограниченной или варьирующейся степенью эффективности в лечении любого данного состояния человека или млекопитающего. Большое разнообразие различных тритерпенов, которые были идентифицированы ранее, и широкий диапазон вариабельности и непредсказуемости в проявлении биологических активностей, наблюдаемых даже у близкородственных тритерпеновых соединений, создает трудности, с которыми приходится сталкиваться при получении тритерпенов, которые представляют собой потенциальные терапевтические агенты. Решение трудной задачи идентификации новых тритерпенов с полезной биологической активностью должно открыть новые перспективы в лечении различных заболеваний человека, для которых возможности лечения в настоящее время ограничены.
Краткое содержание изобретения
Настоящее изобретение касается нового применения бобов и корней акации Виктории Acacia victoriae (Benth.) (Leguminosae) для выделения биологически полезных соединений. Семена Acacia victoriae используются в качестве пищевого продукта местными жителями Австралии в течение многих поколений (Lister et al., 1996). Однако, бобы и корни ранее отбрасывались как отходы. Следовательно, заявители настоящего изобретения продемонстрировали наличие новых противоопухолевых и других биологически полезных соединений в тех частях указанного растения, которые ранее не применялись. Например, новые биологически активные сапонины, описанные в настоящем изобретении, часто специфически цитотоксичны в отношении клеток злокачественных опухолей.
В одном из вариантов, настоящее изобретение представляет новые сапониновые соединения и их смеси, которые могут быть выделены из растения Acacia victoriae, и способы их применения. В этом отношении, один из вариантов настоящего изобретения представляет сапониновую композицию, содержащую тритерпен или другую ароматическую терпеноидную композицию. Описанные здесь сапонины также могут включать гликозидную группу.
В предпочтительных вариантах, в которых сапонин включает тритерпеновую составляющую, такая тритерпеновая составляющая обычно является акациевой кислотой или олеаноловой кислотой (карофиллином), или иной структурно сходной тритерпеновой составляющей. Тритерпеновые или тритерпенгликозидные соединения также могут, в основном, включать монотерпеновую(ые) составляющую(ие), и для специалиста в данной области техники должно быть очевидно, что описываемые здесь сапониновые соединения могут быть дополнительно замещены другими химическими функциональными группами. Таким образом, описанные здесь сапониновые соединения могут включать тритерпеновую составляющую, присоединенную, по крайней мере, к одному, предпочтительно к двум, трем и более монотерпеновым составляющим. Когда присутствует более одной монотерпеновой составляющей, то каждая из них может быть присоединена (i) непосредственно к тритерпеновой составляющей; (ii) к сахару или иной связывающей группе, которая присоединена к тритерпеновой составляющей; или (iii) к монотерпеновой составляющей, которая, в свою очередь, присоединена к тритерпеновой составляющей непосредственно или через сахар или другие связывающие группы. Связывающие группы включают сахара, ацильные, амидные, алкокси, кетильные, алкильные, алкиленовые и другие подобные химические группы, которые известны специалистам в данной области техники. Описанные здесь тритерпеновые гликозиды обычно имеют молекулярную массу в диапазоне 1800-2600 а.е.м. или от по крайней мере 1800, 1900, 2000, 2100 до примерно 2200, 2300, 2400 или 2600 а.е.м.
Важным аспектом настоящего изобретения является выделение смеси, содержащей один или большее число выделенных сапонинов или тритерпеновых гликозидов, которые могут быть охарактеризованы следующими свойствами: (а) выделяемостью из тканей Acacia victoriae: (b) наличием, по крайней мере, одного тритерпенового гликозида, имеющего молекулярную массу примерно от 1800 до 2600 а.е.м.; (с) способностью индуцировать цитотоксичность в отношении клетки Jurkat; и (d) способностью индуцировать апоптоз клетки Jurkat.
В конкретных вариантах настоящего изобретения, тритерпеновая композиция может характеризоваться следующими свойствами: способностью индуцировать цитотоксичность в отношении клетки Jurkat при IC50 от примерно 0,12 мкг/мл до примерно 0,40 мкг/мл. В других вариантах настоящего изобретения, апоптоз индуцируют путем введения в клетку Jurkat концентрации примерно от 100 до 400 нг/мл. В других вариантах настоящего изобретения апоптоз индуцируют путем введения в клетки Jurkat концентрации от примерно 200 до примерно 250, 300, 350 или 400 нг/мл или примерно от 300 до примерно 350 или 400 нг/мл.
В следующих вариантах настоящего изобретения уровень апоптоза измеряют по перестройке плазматической мембраны клетки Jurkat по связыванию аннексина. Это можно оценить с применением проточной цитометрии, и уровень индуцированного апоптоза может составлять 16-18%.
В других своих вариантах, настоящее изобретение относится к смеси, содержащей один или большее число выделенных тритерпеновых гликозидов, характеризующихся следующими свойствами: (а) выделяемостью из тканей Acacia victoriae; (b) наличием, по крайней мере, одного тритерпенового гликозида с молекулярной массой от примерно 1800 до примерно 2600 а.е.м.; и (с) способностью индуцировать высвобождение цитохрома-с из митохондрий клеток Jurkat.
В следующих вариантах настоящего изобретения представляется смесь, содержащая один или большее число выделенных тритерпеновых гликозидов, характеризующихся следующими свойствами: (а) выделяемостью из тканей Acacia victoriae; (b) наличием, по крайней мере, одного тритерпенового гликозида с молекулярной массой от примерно 1800 до примерно 2600 а.е.м.; и (с) способностью активировать каспазу-3 в клетке Jurkat, где каспазная активность составляет в пределах примерно от 0,3 до 1,6 единиц флуоресценции/мин/мг.
В других вариантах настоящего изобретения, смесь, содержащая один или большее число выделенных тритерпеновых гликозидов, может характеризоваться следующими свойствами: (а) выделяемостью из тканей Acacia victoriae; (b) наличием, по крайней мере, одного тритерпенового гликозида с молекулярной массой от примерно 1800 до примерно 2600 а.е.м.; и (с) способностью обуславливать расщепление белка PARP в клетке Jurkat.
В других вариантах настоящего изобретения смесь, содержащая один или большее число выделенных тритерпеновых гликозидов, может характеризоваться следующими свойствами: (а) выделяемостью из тканей Acacia victoriae; (b) наличием, по крайней мере, одного тритерпенового гликозида с молекулярной массой от примерно 1800 до примерно 2600 а.е.м.; и (с) способностью подавлять активность киназы PI3 в клетке Jurkat.
В других вариантах настоящего изобретения смесь, содержащая один или большее число выделенных тритерпеновых гликозидов, может характеризоваться следующими свойствами: (а) выделяемостью из тканей Acacia victoriae; и (b) способностью подавлять инициацию и активацию эпителиальных клеток млекопитающего до предопухолевого или злокачественного статуса.
В других вариантах настоящего изобретения, смесь, содержащая один или большее число выделенных тритерпеновых гликозидов, может характеризоваться следующими свойствами: (а) выделяемостью из тканей Acacia victoriae; и (b) способностью индуцировать апоптоз злокачественных клеток у млекопитающих.
В своем важном аспекте настоящее изобретение относится к натуральной фармацевтической композиции, содержащей тритерпеновый гликозид в фармацевтически приемлемой среде, такой как буфер, растворитель, разбавитель, инертный носитель, масло, крем или годный в пищу материал. В одном из вариантов настоящего изобретения нутрицевтическая композиция может содержать высушенные и измельченные корни, бобы Acacia victoriae или их сочетания в фармакологически приемлемой среде. Раскрываемые здесь нутрицевтические композиции могут иметь форму таблетки, капсулы или мази.
В другом своем аспекте, настоящее изобретение относится к способу получения композиции, содержащей смесь одного или большего числа выделенных тритерпеновых гликозидов, включающему: (а) взятие ткани растения Acacia victoriae; (b) экстракцию данной ткани растворителем с получением экстракта; и (с) получение одного или большего числа тритерпеновых гликозидов из данного экстракта. Обычно используемыми в данном способе тканями являются бобы, корни, проростки или их смеси. Растворителем, применяемым для экстракции, может быть любой органический растворитель, пригодный для экстракции, часто путем растворения, нужного сапонинового соединения. Применимыми для экстракции растворителями являются метанол, этанол, изопропиловый спирт, дихлорметан, хлороформ, этилацетат, вода, глицерин и их смеси.
Данный способ может включать дополнительные стадии.
Например, указанный способ может дополнительно включать выделение композиции из багассы фильтрованием после экстракции. В другом варианте данный способ дополнительно включает стадию обезжиривания растительной ткани с использованием органического растворителя перед проведением экстракции. Органическим растворителем может быть любой пригодный для обезжиривания растворитель, такой как гексан, дихлорметан, хлороформ, этилацетат или их смеси. В другом варианте способ выделения дополнительно включает выпаривание растворителя после проведения экстракции.
Данный способ также может включать получение смеси тритерпеновых композиций путем хроматографического выделения, по крайней мере, одного тритерпенового гликозидного соединения. Примерами методов хроматографии являются жидкостная хроматография, ЖХСД или ВЭЖХ. Хотя растворители, которые могут быть использованы для хроматографического выделения, должны быть определены специалистом в данной области техники, примерами таких растворителей являются метанол, ацетонитрил, вода и смеси.
Еще в одном своем аспекте, настоящее изобретение относится к способу получения композиции, содержащей смесь одного или большего числа выделенных тритерпеновых гликозидов, включающему: (а) получение тканевой культуры, включающей клетки растения Acacia victoriae; и (b) экстракцию тритерпеновых композиций из указанных клеток с помощью растворителя, тем самым экстрагируя, по крайней мере, первое тритерпеновое соединение из данной ткани. В одном из аспектов, данная тканевая культура включает культуру волосовидных корней. В другом аспекте настоящего изобретения, тканевую культуру приготавливают путем инфицирования клеток Acacia victoriae клетками Agrobacterium rhizogenes штамма R-1000. В близком аспекте настоящего изобретения, тканевая культура включает культуральную среду, содержащую сахарозу в количестве от примерно 3% до примерно 4% по весу. В другом аспекте настоящего изобретения используемым для экстракции композиции растворителем является метанол, этанол, изопропиловый спирт, дихлорметан, этилацетат, вода или их смеси.
Еще в одном аспекте настоящего изобретения, этот способ, кроме того, включает дополнительные стадии, такие как отфильтровывание растительной багассы из смеси тритерпенов, выделение смешанной тритерпеновой композиции методом жидкостной хроматографии и (или) выпаривание растворителя после экстракции.
В одном из аспектов описывается способ непрерывного воспроизведения тканей Acacia victoriae, из которых можно экстрагировать активные соединения по настоящему изобретению. В одном из вариантов настоящего изобретения, описывается тканевая культура волосовидных корней, включающая клетки Acacia victoriae, которые были инфицированы Agrobacterium rhizogenes R-1000 в соответствующей культуральной среде. В близком варианте эта среда для культивирования тканей содержит от примерно 3% до примерно 4% сахарозы.
В другом аспекте настоящего изобретения, описан способ непрерывного сбора ткани растения Acacia victoriae, включающий: (а) выращивание Acacia victoriae методом гидропоники; и (b) сбор ткани растения от примерно одного до примерно 4 раз в год, причем этот сбор ткани не приводит к гибели растения. В близком варианте настоящего изобретения, системой выращивания является аэропоника. В другом близком варианте настоящего изобретения, используемой для культивирования тканью Acacia victoriae является корневая ткань.
Важным аспектом настоящего изобретения является способ подавления инициации и активации эпителиальных клеток млекопитающего до предопухолевого или злокачественного статуса, включающий введение в клетку млекопитающего терапевтически эффективного количества нутрицевтической композиции, описанной выше. В одном из вариантов, такой эпителиальной клеткой является клетка кожи, клетка толстой кишки, клетка матки, клетка яичника, клетка поджелудочной железы, клетка предстательной железы, почечная клетка, клетка легкого, клетка мочевого пузыря или клетка молочной железы. В близком варианте настоящего изобретения, указанным млекопитающим является человек. Еще в одном близком варианте настоящего изобретения способом введения нутрицевтической композиции является пероральное введение. В альтернативном варианте настоящего изобретения, способом введения нутрицевтической композиции является местное введение.
Настоящее изобретение также относится к способу индукции апоптоза злокачественной клетки млекопитающего, включающему обработку этой клетки терапевтически эффективным количеством нутрицевтической композиции, описанной выше. В одном варианте, указанной клеткой является клетка кожи, клетка толстой кишки, клетка матки, клетка яичника, клетка поджелудочной железы, клетка предстательной железы, почечная клетка, клетка легкого, клетка мочевого пузыря или клетка молочной железы. В близком варианте млекопитающим является человек. Еще в одном близком варианте способом введения нутрицевтической композиции является пероральное введение. В альтернативном варианте настоящего изобретения, способом введения нутрицевтической композиции является местное введение.
Настоящее изобретение также относится к способу предотвращения аномальной пролиферации эпителиальных клеток млекопитающих in vitro или в организме млекопитающего, включающему обработку клетки млекопитающего или введение этому млекопитающему терапевтически эффективного количества нутрицевтических композиций, описанных выше. В одном из аспектов настоящего изобретения эпителиальными клетками являются клетки кишечных крипт. В другом аспекте настоящего изобретения, эпителиальными клетками являются клетки толстой кишки. В близком варианте настоящего изобретения млекопитающим является человек. Еще в одном близком варианте настоящего изобретения, способом введения нутрицевтической композиции in vivo является пероральное введение.
Настоящее изобретение также представляет способ лечения млекопитающего от воспаления, включающий введение этому млекопитающему терапевтически эффективного количества нутрицевтической композиции, описанной выше. В близком варианте настоящего изобретения, таким млекопитающим является человек.
Настоящее изобретение также представляет очищенное тритерпеновое соединение, включающее тритерпеновую составляющую, соединенную с монотерпеновой составляющей следующей формулы:
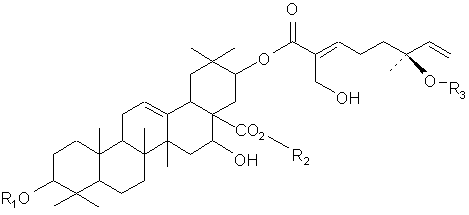
или его фармацевтический препарат, где (a) R1 и R2 выбирают из группы, состоящей из водорода, C1-5-алкила или олигосахарида;
(b) R3 выбирают из группы, состоящей из водорода, гидроксила, C1-5-алкила, C1-5-алкилена, C1-5-алкилкарбонила, сахара и монотерпеновой группы; и (с) данная формула дополнительно включает R4, где R4 выбирают из группы, состоящей из водорода, гидроксила, C1-5-алкила, C1-5-алкилена, C1-5-алкилкарбонила, сахара, C1-5-алкилового сложного эфира и монотерпеновой группы, и где R4 может быть соединен с тритерпеновой составляющей или монотерпеновой составляющей. Настоящее изобретение также относится к соединению, в котором R3 представляет сахар. В близких вариантах настоящего изобретения, сахар выбирают из группы, состоящей из глюкозы, фукозы, рамнозы, арабинозы, ксилозы, хиновозы, мальтозы, глюкуроновой кислоты, рибозы, N-ацетилглюкозамина и галактозы. В других близких вариантах настоящего изобретения соединение дополнительно включает монотерпеновую составляющую, соединенную с сахаром. Настоящее изобретение представляет соединение, в котором R3 имеет формулу:
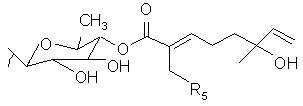
где R5 выбирают из группы, состоящей из водорода, гидроксила, C1-5-алкила, C1-5-алкилена, C1-5-алкилкарбонила, сахара, C1-5-алкилового сложного эфира и монотерпеновой группы.
В одном из вариантов настоящего изобретения, R5 представляет водород или гидроксил. В другом варианте настоящего изобретения, каждый из R1 и R2 включает олигосахарид. Еще в одном варианте настоящего изобретения, каждый из R1 и R2 включает моносахарид, дисахарид, трисахарид или тетрасахарид. В близких вариантах настоящего изобретения, каждый из R1 и R2 включает сахара, которые отдельно и независимо друг от друга выбирают из группы, состоящей из глюкозы, фукозы, рамнозы, арабинозы, ксилозы, мальтозы, хиновозы, глюкуроновой кислоты, рибозы, N-ацетилглюкозамина и галактозы. В других аспектах настоящего изобретения, по крайней мере, один из сахаров метилирован.
В одном из вариантов настоящего изобретения, R4 соединен с тритерпеновой составляющей посредством одного из углеродов метилена, соединенного с тритерпеновой составляющей. В другом варианте настоящего изобретения, тритерпеновой составляющей является олеаноловая кислота вместо акациевой кислоты.
В другом своем варианте, настоящее изобретение относится к композиции, содержащей тритерпеновый гликозид следующей формулы:
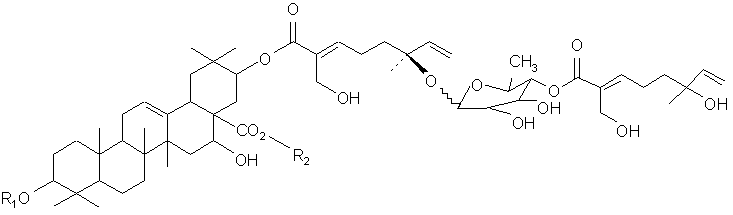
или к ее фармацевтическому препарату, где (а) R1 представляет олигосахарид, включающий N-ацетилглюкозамин, фукозу и ксилозу;
и (b) R2 представляет олигосахарид, включающий глюкозу, арабинозу и рамнозу. В своем близком варианте, настоящее изобретение относится к соединению формулы:
 или его фармацевтическому препарату.
или его фармацевтическому препарату.
В другом своем аспекте, настоящее изобретение относится к очистке композиции, содержащей тритерпеновый гликозид, имеющий молекулярную формулу:
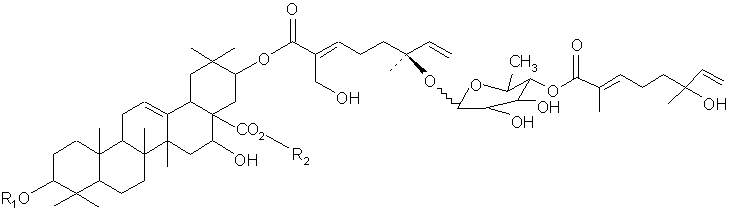
или ее фармацевтического препарата, где (a) R1 представляет олигосахарид, включающий N-ацетилглюкозамин, фукозу и ксилозу; и (b) R2 представляет олигосахарид, включающий глюкозу, арабинозу и рамнозу. В своем близком аспекте, настоящее изобретение относится к очистке и характеризации соединения формулы:
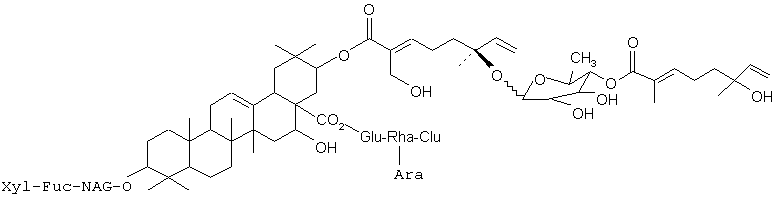
или его фармацевтического препарата.
Еще в одном своем аспекте, настоящее изобретение относится к очистке композиции, содержащей тритерпеновый гликозид, имеющий молекулярную формулу:
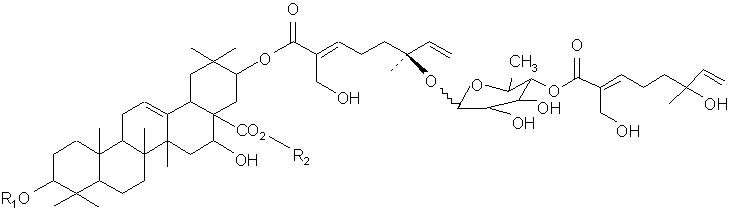
или ее фармацевтического препарата, где (a) R1 представляет олигосахарид, включающий N-ацетилглюкозамин, глюкозу, фукозу и ксилозу; и (b) R2 представляет олигосахарид, включающий глюкозу, арабинозу и рамнозу. В своем близком аспекте, настоящее изобретение относится к очистке и характеризации соединения, имеющего молекулярную формулу:
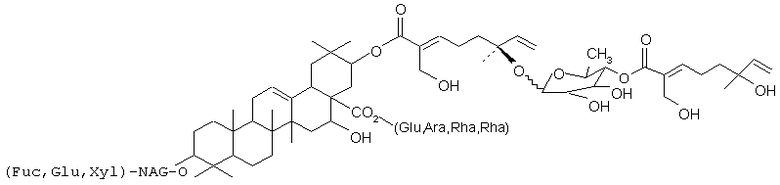 В другом своем аспекте, настоящее изобретение касается соединения, включающего тритерпеновую составляющую, олигосахарид и три монотерпеновые единицы. В одном из вариантов, тритерпеновой составляющей является акациевая кислота или олеаноловая кислота.
В другом своем аспекте, настоящее изобретение касается соединения, включающего тритерпеновую составляющую, олигосахарид и три монотерпеновые единицы. В одном из вариантов, тритерпеновой составляющей является акациевая кислота или олеаноловая кислота.
Важным аспектом настоящего изобретения является получение фармацевтических препаратов, содержащих очищенные и охарактеризованные соединения. В одном из вариантов, фармацевтический препарат представляет собой фармакологически приемлемую среду, включающую буфер, растворитель, разбавитель, инертный носитель, масло, крем или годный в пищу материал. В некоторых аспектах настоящего изобретения, рассматриваемая фармацевтическая композиция, кроме того, содержит агент для направленной доставки. В близких аспектах настоящего изобретения, такой агент направленной доставки способен осуществлять прямую доставку фармацевтической композиции к эпителиальной клетке. В близком варианте настоящего изобретения, таким агентом направленной доставки является антитело, связывающееся с данной эпителиальной клеткой.А
В некоторых вариантах настоящего изобретения, фармацевтическая композиция содержит, по крайней мере, еще одно соединение, которое может уничтожать эпителиальную клетку.
Для соединений по настоящему изобретению был продемонстрирован хемопротективный эффект у мышей, на которых воздействовали канцерогеном DMBA. Следовательно, настоящее изобретение относится к способу подавления инициации и активации эпителиальной клетки млекопитающего до предопухолевого или злокачественного статуса у млекопитающего, включающему введение данному млекопитающему терапевтически эффективного количества фармацевтических композиций, описанных выше. В одном из вариантов настоящего изобретения, эпителиальная клетка является клеткой кожи, клеткой толстой кишки, клеткой матки, клеткой яичника, клеткой поджелудочной железы, клеткой легкого, клеткой мочевого пузыря, клеткой предстательной железы, почечной клеткой или клеткой молочной железы. В близком варианте настоящего изобретения, млекопитающим является человек. Еще в одном близком варианте настоящего изобретения, способом введения фармацевтической композиции является пероральное введение. И еще в одном близком варианте настоящего изобретения способом введения фармацевтической композиции является местное введение. Еще в одном варианте настоящего изобретения, способом введения фармацевтической композиции является внутриопухолевая инъекция. В другом варианте настоящего изобретения, способом введения фармацевтической композиции является внутривенное введение. И еще в одном варианте изобретения, способом введения фармацевтической композиции является ингаляция аэрозолем.
Настоящее изобретение также относится к введению фармацевтических композиций по настоящему изобретению в сочетании с другими способами лечения. В одном из вариантов, такими другими способами лечения являются рентгеновское, ультрафиолетовое, γ- или микроволновое облучение эпителиальной клетки.
Настоящее изобретение также относится к способу стимулирования апоптоза злокачественной клетки млекопитающего, предусматривающему введение этому млекопитающему терапевтически эффективного количества фармацевтических композиций, описанных в данной заявке. В одном из вариантов настоящего изобретения, такой клеткой является клетка кожи, клетка толстой кишки, клетка матки, клетка яичника, клетка поджелудочной железы, клетка легкого, клетка мочевого пузыря, клетка предстательной железы, почечная клетка или клетка молочной железы.
В одном из своих важных аспектов, настоящее изобретение относится к способу предотвращения аномальной пролиферации эпителиальной клетки у млекопитающего, предусматривающему введение этому млекопитающему терапевтически эффективного количества фармацевтических композиций, описанных выше. В одном варианте, такой эпителиальной клеткой является клетка кишечной крипты. В другом варианте настоящего изобретения, такой эпителиальной клеткой является клетка толстой кишки. В близком варианте настоящего изобретения, таким млекопитающим является человек. Еще в одном близком варианте настоящего изобретения, способом введения фармацевтической композиции является пероральное введение. Еще в одном варианте настоящего изобретения, способом введения является местное введение. Еще в одном варианте настоящего изобретения, способом введения является внутриопухолевая инъекция. В другом варианте настоящего изобретения, способом введения фармацевтической композиции является внутривенное введение. В следующем варианте настоящего изобретения, способом введения фармацевтической композиции является ингаляция аэрозолем. Настоящее изобретение также относится к использованию фармацевтических композиций по настоящему изобретению в сочетании с другими способами лечения. В одном из вариантов, такими другими способами лечения являются рентгеновское, ультрафиолетовое, γ- или микроволновое облучение эпителиальной клетки.
Настоящее изобретение также относится к способу лечения млекопитающего от воспаления, включающему введение этому млекопитающему терапевтически эффективного количества описанной здесь фармацевтической композиции, содержащей тритерпеновые соединения. В близком варианте настоящего изобретения, таким млекопитающим является человек. Еще в одном варианте настоящего изобретения, способом введения фармацевтической композиции является пероральное введение. Еще в одном варианте настоящего изобретения, способом введения является местное введение. В другом варианте настоящего изобретения, способом введения фармацевтической композиции является ингаляция аэрозолем.
Другим важным аспектом настоящего изобретения является способ регуляции ангиогенеза у млекопитающего, предусматривающий введение этому млекопитающему терапевтически эффективного количества описанных здесь фармацевтических композиций. Этот способ может быть применен, если этим млекопитающим является человек.
Хотя некоторые из описываемых здесь способов применяются in vitro, однако, предполагается, что и in vivo тритерпеновые гликозидные соединения будут проявлять аналогичные эффекты.
В дополнение к представляемым способам профилактики и лечения раковых опухолей с использованием соединений по настоящему изобретению заявители представляют ряд других путей применения соединений по настоящему изобретению. В частности, соединения по настоящему изобретению могут быть использованы в качестве растворителей, антиоксидантов, противогрибковых и противовирусных агентов, ихтиоцидов или моллюскоцидов, контрацептивов, противогельминтных средств, регуляторов ангиогенеза, средств для защиты от УФ-излучения, отхаркивающих средств, мочегонных средств, противовоспалительных средств, регуляторов метаболизма холестерина, сердечно-сосудистых эффекторов, противоязвенных средств, болеутоляющих средств, седативных средств, иммуномодуляторов, жаропонижающих средств, средств против ломкости капилляров, средств против проявлений старения, средств для повышения содержания коллагена в коже, средств для усиления мужской половой функции и средств для улучшения познавательной способности и памяти.
Краткое описание чертежей
Следующие чертежи являются частью описания настоящего изобретения и включены для дополнительной иллюстрации некоторых его аспектов. Настоящее изобретение может быть лучше понято с помощью отсылки к одному или нескольким чертежам в сочетании с подробным описанием конкретных вариантов.
Фиг.1. Влияние UA-BRF-004-DELEP-F001 на линии опухолевых клеток человека. На фиг.1 проиллюстрировано подавление роста клеточных линий яичников (SK-OV-3, HEY, OVCAR-3), молочной железы (MDA-468), меланомы (А375-М, Hs294t) и эпидермоидных клеток человека (А431), обработанных неочищенным экстрактом бобового растения.
Фиг.2. Влияние UA-BRF-004-DELEP-F023 (фракция 23) на трансформированные и нетрансформированные клеточные линии. На фиг.2 проиллюстрирована цитотоксичность, проявляемая фракцией 23, в отношении клеток яичников (SK-OV-3, OCC1, HEY, OVCAR-3), клетки Т-клеточного лейкоза (Jurkat), простаты (LNCaP), первичных клеток опухоли яичника человека (FTC), фибробластов человека (FS) и эндотелиальных клеток (HUVEC). Уровень цитотоксичности в нетрансформированных клетках составил лишь 15-17%, в то время как в отношении опухолевых клеток она составила 50-95%.
Фиг.3. Влияние фракции 35 (“UA-BRF-004-DELEP-F035” или “F035”) на линии опухолевых клеток человека. На фиг.3 показана цитотоксичность, проявляемая обработанными фракцией 35 клетками яичника (HEY, OVCAR-3, C-1, SK-OV-3), поджелудочной железы (PANC-1) и почек (769-Р, 786-О, А498) человека. Величины IC50 для этих клеточных линий варьировались в пределах 1-6 мкг/мл.
Фиг.4. Влияние фракции 35 на линии лейкозных клеток. На фиг.4 показано, что фракция 35 проявляет сильную цитотоксичность в отношении клеток Jurkat (Т-клеточный лейкоз) при IC50 130 нг/мл и IC50 для клеток REH, KG-1 и NALM-6 (В-клеточные лейкозы) в диапазоне 1-3 мкг/мл.
Фиг.5. Влияние фракции 35 на пролиферацию эндотелиальных клеток. На фиг.5 показано, что фракция 35 является сильным ингибитором пролиферации эндотелиальных клеток при стимуляции bFGF или без таковой.
Фиг.6. Влияние фракции 35 на миграцию эндотелиальных клеток капилляров. На фиг.6 показано отсутствие влияния на миграцию эндотелиальных клеток капилляров, что подтверждает отсутствие токсичности.
Фиг.7. Показаны результаты тонкослойной хроматографии экстрактов проростков и каллюсов. Дорожка 1 - стеблевые каллюсы, сформированные в среде BA-IAA; дорожка 2 - корневые каллюсы, сформированные в среде BA-IAA; дорожка 3 - каллюс из клеток гипокотиля; дорожка 4 - проросток, обработанный метилжасмонатом (100 мкМ) в полутвердой среде; дорожка 5 - контрольный проросток, выращенный в полутвердой среде; дорожка 6 - стандарт F023; дорожка 7 - побег, выращенный в среде ВА; дорожка 8 - проросток, обработанный 50 мкМ метилжасмоната; дорожка 9 - проросток, обработанный 100 мкМ метилжасмоната; дорожка 10 - проросток, обработанный 200 мкМ метилжасмоната; дорожка 11 - контрольный проросток; и дорожка 12 - стандарт F023.
Фиг.8. Приведена фотография мыши SENCAR (слева) и гибридной мыши SENCARxC57B1 (справа), Обеим мышам повторно вводили дозы по 100 нмоль DMBA в течение 8 недель. На 15-й неделе у обеих мышей отмечено развитие множественных папиллом, однако у гибридных мышей SENCAR×C57B1 папилломы были мельче и их число было меньше. Линия С57В1 резистентна к канцерогенезу и у нее не развиваются опухоли.
Фиг.9A-F. Показаны срезы эпидермиса мышей, обработанных ацетоном, DMBA или DMBA + UA-BRF-004-DELEP-F035. Фиг.9А: обработка ацетоном на 4-й неделе. Фиг.9В: обработка ацетоном на 8-й неделе. Фиг.9С: обработка DMBA на 4-й неделе. Фиг.9D: обработка DMBA на 8-й неделе. Фиг.9Е: обработка DMBA + UA-BRF-004-DELEP-F035 на 4-й неделе. Фиг.9F: обработка DMBA + UA-BRF-004-DELEP-F035 на 8-й неделе.
Фиг.10А-В. Продемонстрирован антиоксидантный эффект UA-BRF-004-DELEP-F035 в отношении ДНК через 4 недели. Фиг.10А: показан антиоксидантный эффект после обработки низкой концентрацией UA-BRF-004-DELEP-F035 (0,1 мг на 0,2 мл). Фиг.10В: показан антиоксидантный эффект после обработки высокой концентрацией UA-BRF-004-DELEP-F035 (0,3 мг на 0,2 мл).
Фиг.11А-В. Показана толщина эпидермиса через 4 недели после обработки DMBA и UA-BRF-004-DELEP-F035. Фиг.11А: показано влияние обработки низкой концентрацией UA-BRF-004-DELEP-F035 (0,1 мг на 0,2 мл) на толщину эпидермиса. Фиг.11В: показано влияние обработки высокой концентрацией UA-BRF-004-DELEP-F035 (0,3 мг на 0,2 мл) на толщину эпидермиса.
Фиг.12. Показана степень (в процентах) изменения толщины эпидермиса через 4 недели после обработки DMBA при низкой (0,1 мг на 0,2 мл) или высокой (0,3 мг на 0,2 мл) концентрации UA-BRF-004-DELEP-F035.
Фиг.13. Показана степень (в процентах) уменьшения папиллом через 8 недель после обработки DMBA при низкой (0,1 мг на 0,2 мл) или высокой (0,3 мг на 0,2 мл) концентрации UA-BRF-004-DELEP-F035.
Фиг.14. Показана авторадиограмма реакции ПЦР, иллюстрирующая амплификацию мышиного гена H-ras, мутантного по 61-му кодону.
Фиг.15. Показана исходная стратегия, применяемая для очистки и выделения биологически активных тритерпеновых соединений из Acacia victoriae.
Фиг.16. Показана общая улучшенная схема очистки, выделения и характеризации активных компонентов Acacia victoriae.
Фиг.17А-В. Фиг.17А: показан ВЭЖХ-спектр ацетилированных сахаров, выделенных из гидролизованных активных компонентов, найденных во фракции 94 (“UA-BRF-004Pod-DELEP-F094” или “F094”). Фиг.17В: показан ВЭЖХ-спектр ацетилированных сахаров, выделенных из гидролизованных активных компонентов, обнаруженных в F094.
Фиг.18A-F. Фиг.18А: показаны ВЭЖХ-спектры UA-BRF-004-DELEP-F035 и F035-B2. Фиг.18В: показаны ВЭЖХ-спектры UA-BRF-004Pod-DELEP-F094. Фиг.18С: показаны ВЭЖХ-спектры F140. Фиг.18D: показаны ВЭЖХ-спектры F142. Фиг.18Е: показаны ВЭЖХ-спектры F144. Фиг.18F: показаны ВЭЖХ-спектры F145.
Фиг.19А-В. Анализ клеточного цикла клеток OVCAR-3 до и после обработки (48 часов) фракцией 35. На фигуре показано примерно 8%-ное увеличение числа клеток на стадии G1 и примерно 10%-ное уменьшение числа клеток на S-стадии клеточного цикла после обработки фракцией 35, что свидетельствует о блокировке на G1-стадии. Фиг.19А: анализ клеточного цикла у необработанных клеток OVCAR-3. Фиг.19В: анализ клеточного цикла у опухолевых клеток OVCAR-3, обработанных фракцией 35.
Фиг.20. Данные EMSA показывают заметное ингибирование TNF-активации NF-кВ в результате обработки клеток UA-BRF-004-DELEP-F035 и UA-BRF-004Pod-DELEP-F094. Параметры обработки были такими: дорожка 1 - без обработки; дорожка 2 - TNF (100 пкМ); дорожка 3 - UA-BRF-004-DELEP-F035 (1 мкг/мл); дорожка 4 - TNF + F035 (1 мкг/мл); дорожка 5 - F035 (2 мкг/мл); дорожка 6 - TNF + F035 (2 мкг/мл); дорожка 7 - F094 (1 мкг/мл); дорожка 8 - TNF + F094 (1 мкг/мл); дорожка 9 - F094 (2 мкг/мл); дорожка 10 - TNF + F094 (2 мкг/мл).
Фиг.21. Липидкиназный тест, демонстрирующий подавление киназы PI3 действием UA-BRF-004-DELEP-F035 и вортманнином.
Фиг.22. Анализ геля ДСН-ПААГ методом Вестерн-блоттинга с хемолюминесценцией с использованием антител, специфичных в отношении фосфорилированной АКТ и общей АКТ. После обработки клеток 1 и 2 мкг/мл UA-BRF-004-DELEP-F035 было отмечено заметное подавление АКТ-фосфорилирования (активной АКТ), уровень которого был сходным с уровнем, наблюдаемым после 2-часовой обработки клеток 1 мкМ вортманнина.
Фиг.23. Представлены результаты ПЦР-амплификации части гена го1В из четырех независимо трансформированных корневых клонов (дорожки, слева направо: 1 - шкала т.п.о.; 2 - позитивный контроль (плазмидная ДНК штамма R1000); 3 - негативный контроль (ДНК нетрансформированных корней); 4-7 - четыре независимо трансформированных корневых клона. Следует отметить амплификацию фрагмента в 645 п.о. на материале позитивного контроля и трансформированных корней.
Фиг.24. Структура эллиптозида-А и эллиптозида-Е (Beutler, 1997).
Фиг.25. ВЭЖХ-разделение компонентов F094.
Фиг.26. ВЭЖХ-разделение компонентов F035.
Фиг.27. Первое фракционирование F094 полупрепаративной ВЭЖХ.
Фиг.28. Второе фракционирование F094 с помощью полупрепаративной ВЭЖХ.
Фиг.29. Препаративное фракционирование F094.
Фиг.30. Анализ препаративной фракции D.
Фиг.31. Анализ препаративной фракции G/H.
Фиг.32. Соединение G1 после второй очистки на PFP-колонке.
Фиг.33. Соединение G1 после окончательной очистки на колонке С-18.
Фиг.34. Соединение D1 после очистки на колонке Waters С-18.
Фиг.35. Соединение D1 после окончательной очистки на колонке C-18-Aq.
Фиг.36. Показаны соединения после расщепления соединения D1.
Фиг.37. Показаны соединения после расщепления соединения G1.
Фиг.38. Показаны соединения после расщепления соединения В1.
Фиг.39. Структура тритерпенового гликозида D1.
Фиг.40. Структура тритерпенового гликозида G1.
Фиг.41. Структура тритерпенового гликозида В1.
Фиг.42. Влияние смеси тритерпеновых гликозидов (F035) на линии раковых и нормальных клеток: F035 оценивали на цитотоксичность в соответствии с процедурами, описанными в примерах. Активность F035 оценивали на панели линий раковых и нормальных клеток, как показано на этой фигуре. Для раковых клеток величина IC50 варьировалась в пределах 0,2-5,8 мкг/мл. Не было установлено существенной цитотоксичности (IC50 - от 15 мкг/мл до более чем 25 мкг/мл) по отношению к нормальным и иммортализованным клеточным линиям.
Фиг.43. Профили цитотоксичности очищенных тритерпеновых гликозидов D1 и G1 в отношении линий раковых клеток человека. Очищенные экстракты оценивали на их активность по отношению к следующим линиям раковых клеток человека: Jurkat (Т-клеточный лейкоз), вариант С-2 HEY (яичник), 769-Р (почки), MDA-MB-231, MDA-MB-453 (молочная железа). Полученные результаты представлены как средние значения ± стандартное отклонение.
Фиг.44А-Е. Влияние очищенных соединений D1 и G1 и смеси тритерпеновых гликозидов (F035) на апоптоз. Уровень апоптоза определяли с использованием теста на связывание аннексина-V, в котором клетки окрашивали аннексином V-ФИТЦ, а содержание ДНК измеряли с использованием иодида пропидия (PI) и анализировали методом проточной цитометрии. Клетки инкубировали в течение 16 часов в присутствии 0,5-1,0 мкг/мл экстракта. Через 16 часов обработки наблюдалось 3 популяции клеток. Клетки, которые погибли или находились на поздней стадии апоптоза (позитивные по ФИТЦ-аннексину-V и PI), претерпевающие апоптоз клетки (позитивные по ФИТЦ-аннексину-V и негативные по PI) и жизнеспособные клетки, не подвергшиеся апоптозу (негативные и по ФИТЦ-аннексину-V, и по PI: нижний левый график).
Фиг.45А-В. Подавление Р13-киназной активности и АКТ-фосфорилирования. Способность фосфорилировать фосфатидил-инозитол (ФИ) определяли для иммунопреципитатов белка р85 из клеточных лизатов. Авторадиографически тестировали результаты киназного теста in vitro после разделения методом тонкослойной хроматографии иммунопреципитатов р85 на материале клеток Jurkat. Фиг.45В: Подавление фосфорилирования АКТ по остаткам Ser-473 и Thr-308 очищенными и неочищенными тритерпеновыми гликозидами. Клетки Jurkat инкубировали с неочищенными (F035) и очищенными экстрактами D1 и G1 в течение 16 часов при 37°С. Клеточные лизаты разгоняли методом электрофореза в 9%-ном ДСН-ПААГ и анализировали методом Вестерн-блоттинга-ECL с использованием в качестве зондов антител к Ser-473, Thr-408 и к общей АКТ.
Фиг.46A-D. Подавление TNF-индуцированного фактора NF-кВ и индукция iNOS тритерпеновыми гликозидами. Клетки Jurkat подвергали обработке различными концентрациями F035 (1-4 мкг/мл; фиг.46А) и 2 мкг/мл очищенных экстрактов (D1 и G1; фиг.46В) в течение 16 часов, а NF-кВ активировали 100 пкМ TNF в течение 15 минут при 37°С. ДНК-белковый комплекс разделяли в 7,5%-ном нативном полиакриламидном геле и изотопно меченые полосы визуализовали и тестировали количественно с помощью Phospholmager. NOS индуцировали в клетках U-937 (фиг.46С) и Jurkat (фиг.46D), как описано в разделе “Методы”. Клеточный белок разделяли методом электрофореза в ДСН-ПААГ и тестировали методом Вестерн-блоттинга-ECL с использованием антитела против iNOS.
Фиг.47. Влияние F035 и D1 на расщепление PARP в клетках Jurkat.
Фиг.48. Влияние z-vad fmk на F035-индуцированное расщепление PARP в клетках Jurkat.
Фиг.49. Влияние F035, F094, D1 и G1 на активность каспазы в клетках Jurkat.
Фиг.50. Влияние F035 на высвобождение цитохрома из митохондрий клеток Jurkat.
Подробное описание изобретения
Настоящее изобретение позволяет преодолеть ограничения, имевшиеся в данной области техники, путем представления новых биологически активных тритерпеновых гликозидов. В частности, заявителями были идентифицированы и очищены тритерпеновые соединения растения Acacia victoriae. Идентифицированные соединения обладают сильной противоопухолевой активностью в концентрациях, при которых имеется низкая или вообще отсутствует цитотоксичность в отношении нормальных клеток человека.
Тритерпеновые соединения по настоящему изобретению были идентифицированы в результате целевого скрининга экстрактов из 60 растений, выбранных из видов бобовых, произрастающих в засушливых и полузасушливых регионах. При начальном поиске один из экстрактов, обозначенный как UA-BRF-004-DELEP-F001 и выделенный из тканей акации Acacia victoriae (Benth.) (Leguminosae), обнаруживал сильную противоопухолевую активность в отношении различных линий опухолевых клеток человека. Затем, этот экстракт был дополнительно очищен с получением серии фракций. В двух раундах такой очистки был идентифицирован экстракт, содержавший данные противоопухолевые соединения в чистом виде. Этот экстракт был идентифицирован как содержащий сапонины типа тритерпеновых гликозидов. После этого разработали процедуру эффективного выделения активных компонентов.
Дальнейшее тестирование более очищенного экстракта дополнительно позволило выявить биологические активности этого экстракта. Очищенный экстракт проявлял повышенную противоопухолевую активность по сравнению с неочищенным экстрактом при концентрациях, которые обнаруживали слабую токсичность или вообще не обнаруживали токсичность в отношении нормальных клеток человека. Кроме того, было показано, что этот экстракт обладает хемопротективным эффектом у мышей, подвергнутых воздействию канцерогенов.
Растение, из которого был выделен данный экстракт, Acacia victoriae, было выбрано исходя из таких факторов, как природная среда его обитания и слабая изученность этого вида растений. Acacia victoriae происходит из Австралии, однако как садовая культура этот вид был распространен по всему миру и широко известен как колючая акация или изящная акация. Деревья растут со скоростью от 60 до 120 см в год, характеризуются поздней листопадностью в засушливый сезон и выдерживают понижение температуры до -15°С. Взрослые растения растут до 30-50 метров в высоту и характеризуются голубовато-зелеными двоякоперистыми листьями. На юго-западе США, эта акация обычно цветет с апреля по май, а бобы созревают в июне. Acacia victoriae находит различные применения в сельском хозяйстве, выключая использование в качестве ветрозащитных насаждений, полезащитных насаждений, в качестве пищевого продукта, для стабилизации почвы критических районов, и как декоративное растение, характеризующееся низким уровнем потребления воды. Местное население Австралии в течение многих поколений употребляет в пищу семена различных видов акаций (Lister et al., 1996). Среди акаций Acacia victoriae является наиболее обычным и широко распространенным видом, произрастающим по всей Австралии: поэтому она является наиболее “употребляемым” в пищу видом. Семена акций, обычно называемые “wattleseed”, пользуются большим спросом как натуральный компонент кондитерских изделий и хлеба, а также как ароматический компонент для десерта, особенно для мороженого. Их также используют для приготовления высококачественного кофейного суррогата, а среди видов акаций, Acacia victoriae (Benth.) обычно считается обладателем наиболее приятного аромата (Lister et al., 1996). Однако, нигде не сообщается об использовании бобов и корней этого растения.
Настоящее изобретение касается нового применения бобов и корней Acacia victoriae для целей выделения биологически активных соединений. Заявители настоящего изобретения продемонстрировали присутствие новых противоопухолевых и других биологически активных соединений в тех частях данного вида растений, которые ранее не использовались.
II. Очистка и идентификация тритерпенов по настоящему изобретению
Важный аспект применения растительных экстрактов в качестве фармацевтических композиций связан с характеристикой и определением их отдельных активных компонентов. То же самое относится и к препаратам тритерпеновых сапонинов, что часто требует достаточно сложных способов выделения, распознавания структуры и анализа их компонентов и гликозидов. Для проведения биологического тестирования очищенных соединений, необходимо выделить их в достаточном количестве и при достаточном уровне чистоты.
Поскольку тритерпены и другие родственные сапонины имеют относительно высокую молекулярную массу, а также высокую полярность, может быть оказаться необходимым их выделение. Проблемой, связанной с выделением очищенных сапонинов, является присутствие сложных смесей близкородственных соединений, незначительно различающихся либо по природе их агликона, либо по сахарной составляющей (природа, число, положение и хиральность присоединенных моносахаридов). Трудности также возникают в связи с присутствием лабильных заместителей, таких как сложные эфиры. Например, основной соевый сапонин в чистом виде, являющийся γ-пироновым производным (BOA), экстрагируется исключительно водным раствором этанола при комнатной температуре. Экстракция с нагреванием (до 80°С) приводит к расщеплению сложноэфирной составляющей и образованию соевого сапонина-1 (Bb) (Kudou et al., 1992). У растений, сапонины сопровождаются высокополярными соединениями, такими как сахариды и окрашивающее вещество, включая фенольные соединения и т.п., а поэтому они трудно кристаллизуются и могут быть гигроскопичными, что еще больше затрудняет получение кристаллов.
Характеризация очищенных сапонинов также необходима из-за отсутствия кристаллического материала. Точки плавления не имеют точного значения и часто плавление связано с разложением. Следовательно, определение чистоты образца не может быть в принципе основано только на определении точки плавления, константе оптического вращения или других физических показателях. Более эффективный тест на чистоту сапонина может быть разработан на основе ТСХ или ВЭЖХ, и если это возможно, на основе одновременной хроматографии с аутентичным образцом. Окраска пятен на ТСХ-пластинах после распыления подходящих реагентов является дополнительным индикатором предполагаемых отдельных компонентов. Например, один из тритерпеновых гликозидов по настоящему изобретению (D1) имеет время удерживания в ВЭЖХ 15,2 минуты. Это значение отличается от значения для другого близкого соединения, эллиптозида-Е, выделенного из Archidendron ellipticum (J.Beutler et al., 1997), который имеет время удержания в ВЭЖХ 12,5 минут. Дальнейшая характеризация тритерпенов по настоящему изобретению показала, что такое различие во времени удержания обусловлено, по крайней мере, различием в хиральности и двойных связях в D1 по сравнению с данными, опубликованными для эллиптозида-Е.
(1) Химическая очистка
Методы химической очистки хорошо известны специалистам в данной области техники. Эти методы включают фракционирование неочищенного растительного экстракта на тритерпеновые гликозидные соединения, описываемые в настоящей заявке. После базового разделения соединений из растительного материала по настоящему изобретению, представляющие интерес тритерпеновые гликозиды могут быть дополнительно очищены с использованием описанных здесь способов, например, методом хроматографии, с целью достижений частичной или полной очистки (или очистки до гомогенности). Аналитические способы, конкретно применимые для получения очищенного тритерпенового гликозидного соединения, описаны в данном тексте ниже.
Некоторые аспекты настоящего изобретения касаются очистки, а в конкретном варианте, существенной очистки тритерпеновых гликозидов из растительного материала. В предпочтительном варианте настоящего изобретения тритерпеновые гликозиды очищают из растения, относящегося к семейству бобовых, или, что более предпочтительно, из растения рода акаций, а наиболее предпочтительно, из растения вида Acacia victoriae, а именно вида Acacia victoriae (Benth.). Термин “выделенный тритерпеновый гликозид”, используемый в данном тексте, означает композицию, отделенную от других компонентов, где эта композиция очищена до любой степени по отношению к его встречающемуся в природе состоянию.
Вообще говоря, термин “выделенный” относится к органической молекуле или группе аналогичных молекул, которые были подвергнуты фракционированию с удалением различных других компонентов, причем, эта композиция, в основном, сохраняет свою биологическую активность. Используемый термин “очищен в существенной степени” относится к композиции, в которой тритерпеновые гликозиды образуют главный компонент композиции, так, что он составляет примерно 50%, примерно 60%, примерно 70%, примерно 80%, примерно 90%, примерно 95% или более молекул данной композиции.
В принципе, отсутствует необходимость в том, чтобы тритерпеновые композиции по настоящему изобретению всегда были выделены и представлены в своем наиболее очищенном состоянии. Действительно, предполагается, что в меньшей степени очищенные продукты найдут свое применение в некоторых вариантах. Например, заявители рассматривают применение высушенных корней и бобов Acacia victoriae и их экстрактов в качестве нутрицевтических композиций. По определению, нутрицевтические композиции содержат смесь различных биологически активных соединений, которые оказывают благоприятное синергическое действие на организм. Нутрицевтические композиции по настоящему изобретению могут находиться в форме таблеток или капсул и могут быть введены перорально, или, альтернативно, они могут содержать экстракты по настоящему изобретению в составе мази, которую можно применять местно. Частичная очистка может быть достигнута с использованием меньшего числа стадий очистки в сочетании с (или с использованием только) различными формами той же самой общей схемы очистки. Например, понятно, что хроматография на катионообменных колонках, осуществляемая на аппарате для ВЭЖХ, в целом, позволяет осуществлять очистку более высокого уровня по сравнению с тем же способом, использующим систему хроматографии низкого давления. Способы, обеспечивающие низкую степень относительной очистки, могут иметь некоторые преимущества в общем количестве выделяемого продукта или в сохранении биологической активности тритерпеновых соединений.
(ii) Экстракция и предварительная очистка
Процедуры экстракции должны быть мягкими настолько, насколько это возможно, поскольку некоторые сапонины претерпевают изменения, включая каталитический гидролиз во время водной экстракции, этерификацию кислых сапонинов при спиртовой экстракции, гидролиз лабильных сложноэфирных групп и переацилирование. Следовательно, необходима аккуратность в проведении отдельных стадий процедуры выделения, например, при проведении тонкослойной хроматографии.
Хотя возможны многочисленные изменения, известные общие процедуры получения неочищенных смесей сапонинов обычно основаны на экстракции с использованием метанола, этанола, воды или водно-спиртового раствора; проведении перед экстракцией или в отношении самого экстракта стадии обезжиривания, обычно с использованием петролейного эфира; растворении или суспендировании полученного экстракта в воде; встряхивании или промывке указанного раствора или суспензии насыщенным водным н-бутанолом; и осаждении (что необязательно) сапонинов диэтиловым эфиром или ацетоном. Также может быть включена стадия диализа с целью удаления небольших водорастворимых молекул, таких как сахара (см., например, Zhou et al., 1981; Massiot et al., 1988).
Наиболее эффективная экстракция высушенного растительного материала достигается с использованием метанола или водного раствора метанола. Метанол также используется в случае свежего растительного материала. Хотя вода является, обычно, менее эффективным растворителем для экстракции сапонинов (за исключением конкретной целесообразности получения водорастворимых гликозидов), она имеет ряд преимуществ благодаря более легкой лиофилизуемости и получению более чистого экстракта. В зависимости от количества воды, используемой для экстракции, могут быть получены либо монодесмозидные, либо бидесмозидные сапонины (Domon and Hostettmann, 1984; Kawamura et al., 1988). Свежий растительный материал содержит активные ферменты (эстеразы), которые в случае гомогенизации растворителем способны конвертировать бидесмозиды в монодесмозиды. Даже в сухом материале могут находиться эстеразы, активирующиеся в присутствии воды. В случае момордина- I (монодесмозидный сапонин с олеаноловой кислотой) было установлено, что конверсия в момордин-II (соответствующий бидесмозид) происходит в воде и в 30%-ном и 60%-ном растворах метанола, но не в 80%-ном его растворе и 100%-ном метаноле. В отличие от этого, гомогенаты свежих корней в метаноле сохраняют каталитическую активность. Однако, ферменты могут быть инактивированы сначала путем замачивания свежих корней в 4%-ном растворе соляной кислоты, после чего, как было показано, бидесмозид становится основным компонентом. Поэтому очевидно, что правильный выбор процедуры экстракции является исключительно важным.
Методы, обычно применяемые для очистки белков, такие как диализ, ионообменная хроматография и гель-хроматография, применимы для частичного отделения сапонинов в водном растворе от несапониновых компонентов, но, обычно, они неэффективны в разделении отдельных сапонинов из-за тенденции сапонинов образовывать смешанные мицеллы. Следовательно, обычно эффективное разделение требует использования органических растворителей или систем “вода-растворитель”, которые бы солюбилизировали амфифильные сапонины в виде мономеров, что обеспечивало бы отсутствие помех разделению, обуславливаемых образованием смешанных мицелл.
Распространенной проблемой, связанной с фуростанольными сапонинами, является образование производных 22-ОСН3 в процессе экстракции метанолом. Однако, исходные 22-гидроксифуростанолы могут быть получены либо путем экстракции другим растворителем (например, пиридином), либо путем обработки метоксилированных артефактов кипящим водным ацетоном (Konishi & Shoji, 1979).
(iii) Тонкослойная хроматография (ТСХ)
Качественный анализ тритерпеновых сапонинов с помощью ТСХ важен со всех точек зрения изучения сапонинов. Пластины для ТСХ (обычно из силикагеля) удобны как для очищенных сапонинов, так и для неочищенных экстрактов, недороги, могут быть использованы быстро и эффективно и не требуют специального оборудования. Для распыления на этих пластинах имеется ряд доступных реагентов для визуализации (табл. 2). Существуют следующие методы приготовления наиболее распространенных реагентов:
- Ванилин-серная кислота (реагент Годена). 1%-ный раствор ванилина в этаноле смешивают в соотношении 1:1 с 3% раствором перхлорной кислоты в воде и распыляют на ТСХ-пластину. После этого добавляют 10%-ный раствор серной кислоты в этаноле и нагревают до 110°С.
- Реагент Либермана-Бурхарда. Концентрированную серную кислоту (1 мл) смешивают с уксусным ангидридом (20 мл) и хлороформом (50 мл). В результате нагревания до 85-90°С достигается требуемая окраска ТСХ-пластины.
- Хлорид сурьмы (III). На ТСХ-пластину напыляют 10%-ный раствор хлорида сурьмы в хлороформе и нагревают до 100°С.
- Анисовый альдегид-серная кислота. Анисовый альдегид (0,5 мл) смешивают с ледяной уксусной кислотой (10 мл), метанолом (85 мл) и концентрированной серной кислотой (5 мл). Этот раствор напыляют на ТСХ-пластину, которую после этого нагревают до 100°С.
Распыление ванилин-серной кислоты в присутствии этанола и перхлорной кислоты, например, дает синюю или фиолетовую окраску тритерпеновых сапонинов. Использование анисового альдегида-серной кислоты при нагревании ТСХ-пластины приводит к развитию синей или сине-фиолетовой окраски. Напыление ТСХ-пластин раствором сульфата церия в серной кислоте дает фиолетово-красные, синие или зеленые флуоресцирующие участки при УФ-освещении 365 нм (Kitagawa et al., 1984b). В некоторых случаях, просто распыления воды на ТСХ-пластины достаточно для выявления присутствующих сапонинов. Дополнительное описание напыляемых реагентов можно найти, например, у Stahl (1969).
Наиболее часто используемым в ТСХ растворителем является смесь хлороформ-метанол-вода (65:35:10), хотя могут быть использованы и другие растворители. Смесь растворителей н-бутанол-этанол-аммиак (7:2:5) применима, в частности, для гликозидов, содержащих остатки уроновых кислот, т.е. она применима для высокополярных смесей. Другие широко используемые растворители включают смеси н-бутанол-уксусная кислота-вода (4:1:5; верхний слой) или хлороформ-метанол-уксусная кислота-вода (60:32:12:8).
Системы, применяемые для ТСХ гликоалкалоидов, обычно включают смесь этилацетат-пиридин-вода (30:10:30; верхняя фаза). Визуализацию проводят с использованием стероидных реагентов (анисовый альдегид-серная кислота) или алкалоидных реагентов (реагент Драгендорфа, сульфат церия IV). Другие растворители и визуализующие реагенты для ТСХ описаны у Jadhav et al., 1981 и Baerheim Svendsen & Verpoorte, 1983.
При проведении ТСХ можно осуществлять количественный анализ различного типа. Например, плотность точек, получаемых при распылении подходящего реагента, может быть непосредственно определена с помощью денситометра. Альтернативно, количественные оценки возможны путем проведения ТСХ-разделения, соскоба интересующей полосы с пластин (помещенных, например, в пары иода), элюирования сапонина и измерения величины УФ-поглощения после добавления соответствующего реагента (например, концентрированной серной кислоты).
Пластины для ТСХ с обращенными фазами являются коммерчески доступными и представляют точный аналитический метод исследования сапонинов, который дополняет метод ТСХ на силикагеле. Для проявления обращенно-фазовых пластин используют почти исключительно только смеси метанол-вода и ацетонитрил-вода (например, пластины Merck RP-8 или RP-18 для высокоэффективной ТСХ). Альтернативно, могут быть использованы пластины на стеклянной основе DIOL HPTLC. Для них могут быть использованы растворители для обычной ТСХ на силикагеле или метанол-водный и ацетонитрил-водный растворители, используемые в ОФ-ТСХ.
Примеры реагентов для детекции методом ТСХ и для спектрометрического и колориметрического анализа сапонинов приведены ниже в табл.2.
1. Тонкослойная хроматография с центрифугированием (ТСХЦ)
Метод ТСХЦ является планарным методом, близким к препаративной тонкослойной хроматографии (ТСХ), однако в нем отсутствует необходимость соскоба полос с ТСХ-пластин (Hostettmann et al., 1980). ТСХЦ основана на действии центробежной силы для ускорения перемещения подвижной фазы через кольцевые ТСХ-пластины. Пластину, покрытую подходящим сорбентом (при толщине 1, 2 или 4 мм), вращают со скоростью примерно 800 об/мин с помощью электромотора, образец вносят в центр пластины и элюент подают насосом через сорбент. Элюирование растворителя приводит к образованию концентрических полос на пластине. Их снимают с краев и собирают для ТСХ-анализа. На 2-мм слое сорбента возможным является разделение смеси в количестве 50-500 мг.
Сочетание ТСХЦ со смесью хлороформ-метанол-вода (100:30:3) и колоночной хроматографии было описано при выделении гинзенозидов (гликозидов женьшеня) (Hostettmann et al., 1980). Сапонины также были получены с использованием смеси хлороформ-метанол-вода на силикагелевых пластинах. Два гликозида протопримулагенина-А из корней Eleutherococcus sentlcosus (семейство аралиевых, Araliaceae) были очищены методом ТСХЦ (хлороформ-метанол-вода, 65:35:7) после колоночной хроматографии на силикагеле и гель-фильтрации на сефадексе LH-20 (Segiet-Kujawa & Kaloga, 1991). Для выделения циклоартановых гликозидов пассифлоры Passiflora quadrangularis (Passifloraceae), систему растворителей этилацетат-этанол-вода (8:2:1 или 16:3:2) использовали при скорости потока 1 мл/мин (Orsini et al., 1987) или 1,5 мл/мин (Orsini & Verotta, 1985).
Центрифужный жидкостной хроматограф фирмы Хитачи, модель CLC-5, был описан как применимый для разделения сапонинов. Хроматографию на этом приборе проводят с использованием силикагелевых пластин, а в качестве элюента берут смесь хлороформ-метанол-вода (7:3:1 (нижняя фаза) → 65:35:10 (нижняя фаза)). С использованием этого метода, в целом 1 г полуочищенной сапониновой фракции был подвергнут хроматографии на кольцевой пластине (Kitagawa et al., 1988; Taniyama et al., 1988).
(iv) Хроматография на открытой колонке
Ранее был описан ряд классических систем растворителей для хроматографии сапонинов на колонке с силикагелем, что можно найти, например, у Woitke et al., 1970 и Adler & Hiller, 1985. Хроматографию на открытой колонке часто используют в качестве начального этапа фракционирования неочищенной смеси сапонинов, хотя в некоторых случаях, с ее помощью можно получить чистые продукты. В целом, однако, разделение не бывает высоким, и сложные смеси разделяются только частично. Другие проблемы связаны с потерей материала вследствие необратимой адсорбции и длительным временем, необходимым для проведения таких разделений.
Хроматография на силикагеле со смесью хлороформ-метанол-вода в качестве элюента является одним из самых распространенных методов. При использовании двухфазовой системы, элюентом служит насыщенный раствор хлороформа в воде. Следовательно, градиент смеси хлороформ-метанол-вода (например, 65:35:5→65:40:10) может быть использован для исходного разделения на силикагеле метанольного экстракта растительной ткани. Дальнейшая хроматография на колонках при низком давлении может быть использована для получения, например, монодесмозидного моллюскоцидного сапонина, в то время как бидесмозидный сапонин может быть выделен методом хроматографии на колонках с силикагелем с системой растворителей в виде смеси ацетон-н-пропанол-вода (35:35:5) (Borel et al., 1987).
Комплексная смесь тритерпеновых гликозидов была выделена из луковиц ирисового растения Crocosmia crocosmiiflora (Iridaceae). Три из них, содержащие 2,9,16-тригидроксипальмитиновую кислоту гликозиды полигалактовой кислоты, выделяли с помощью стратегии, включающей хроматографию неочищенной смеси сапонинов на открытой колонке с силикагелем-60 (60-230 мкм) с использованием смеси н-бутанол-этанол-вода (5:1:4, верхний слой) и смеси хлороформ-метанол-вода (60:29:6) в качестве элюентов. Для конечной очистки применяли ВЭЖХ (Asada et al., 1989).
Широкое использование хроматографии на силикагеле также позволило разделить даммарановые гликозиды, актиностеммозиды A-D, тыквенного растения Actinostemma lobatum (Cucurbitaceae). После использования полистирольной гелевой колонки MCI (Mitsubishi Chemical Industries) соответствующие фракции подвергали хроматографии с различными растворителями: хлороформ-метанол-вода (7:3:0,5; 32:8:1), хлороформ-метанол (9:1; 1,1), хлороформ-этанол (17:3), этилацетат-метанол (4:1) и хлороформ-метанол-этилацетат-вода (3:3:4:1,5; нижний слой). Таким способом был получен чистый актиностеммозид С, тогда как для выделения актиностеммозидов А и В требовался дополнительный этап хроматографии при низком давлении, а для актиностеммозида D было необходимо окончательное разделение на колонке С-18 с элюированием 70%-ным метанолом (Iwamoto et al., 1987).
Некоторые сложноэфирные сапонины были очищены хроматографически на силикагеле, пропитанном 2% борной кислотой (Srivastava & Kulshreshtha, 1986, 1988).
В настоящее время в дополнение к стандартному силикагелю при выделении сапонинов с помощью хроматографии на открытой колонке используют крупнофракционные сорбенты обращенной фазы. Из-за неоднородности гранулярного состава и не слишком большого размера колонки пригодными могут оказаться гравитационные колонки. Хроматография с обращенной фазой в принципе используется после начального этапа разделения на силикагеле и позволяет изменить избирательность в отношении разделяемых веществ. Другой возможностью является включение обращенно-фазового разделения после этапа КПТХ (DCCC, Higuchi et al., 1988).
1. Хроматография на открытой колонке с полимерными сорбентами
В течение многих лет в хроматографической практике применялись декстрановые носители, например, в насадках колонок Sephadex. Наибольшее распространение получил сефадекс LH-20, хотя небезынтересна и серия полимеров “G”.
В недавних работах по выделению сапонинов было использовано новое поколение полимеров, в частности, в Японии. Например, полимер Diaion HP-20 (Mitsubishi Chemical Industries, Токио) является высокопористым полимером, который широко применяется на начальных этапах очистки.
Обычно полимерные носители промывают водой после загрузки образца с целью элюирования моносахаридов, небольших заряженных молекул, таких как аминокислоты, и других веществ, характеризующихся высокой растворимостью в воде. Элюирование градиентом смеси вода-метанол (или только метанола) затем позволяет получить фракции сапонинов. Другие хроматографические методы применяются для выделения уже чистых сапонинов.
Также сообщалось об элюировании гелей HP-20 смесями ацетон-вода. Например, при выделении бидесмозидных гликозидов квилаховой кислоты из клубней тыквенного растения Thladiantha dubia (Cucurbitaceae) метанольные экстракты пропускали через колонку Diaion CHP-20P и промывали водой. Неочищенные сапонины элюировали 40%-ным ацетоном. Дальнейшее разделение проводили с помощью хроматографии на силикагеле (с элюированием смесью этилацетат-метанол-вода (6:2:1)) и с помощью ВЭЖХ (Nagao et al., 1990).
Для выделения фибринолитических сапонинов из семян мочалки-люфы Luffa cyilndrica (Cucurbitaceae), водный экстракт подвергали хроматографии на колонке Amberlite XAD-2 с элюированием метанолом с последующим элюированием второй колонки XAD-2 40-70%-ным метанолом. Активные частицы были получены в очищенном состоянии после хроматографии на колонке с силикагелем с элюированием смесью хлороформ-метанол-вода (65:35:10, нижний слой → 65:40:10) (Yoshikawa et al., 1991).
(v) Жидкостная хроматография при среднем давлении (ЖХСД)
Когда требуются относительно большие количества очищенных сапонинов, эффективной может быть ЖХСД. В отличие от коммерчески доступного оборудования для жидкостной хроматографии высокого давления (ЖХВД), граммовые количества образца могут быть загружены на колонки, а разделение осуществлено при давлении до 40 бар. Гранулярный состав такого носителя, обычно, составляет 25-40 мкм, и разделение проходит быстро, для чего требуется существенно меньше времени, чем при хроматографии на открытой колонке. Прямой перенос условий разделения аналитической ВЭЖХ на ЖХСД может быть достигнут с использованием обращенно-фазовых носителей, что облегчает выбор растворителя (Hostettmann et al., 1986).
Например, моллюскоцидные сапонины куссонии колосовидной Cussonia spicata (Araliaceae) были получены в достаточных количествах для биологического тестирования методом ЖХСД на сорбенте С-8 с элюированием смесью метанол-вода (2:1) (Gunzinger et al., 1986). Действительно, при таком подходе для выделения сапонинов из бутанольного экстракта стеблевой коры требуются 2 этапа (один на силикагеле, другой на ОФ-материале).
Выделение сапонинов также может быть достигнуто сочетанием ЖХСД, например, на колонке LiChroprep RP-8 (25-40 мкм, 46×2,6 см) со смесями метанол - вода, с ротационной локулярной противоточной хроматографией (РЛПХ) (Dorsaz & Hostettmann, 1986). В другом варианте в ЖХСД используют колонки с аксиальной компрессией Jobin-Yvon (Elias et al., 1991).
Примеры комбинаций носителей и растворителей, которые применялись для выделения тритерпенов из растительных экстрактов, приведены ниже в табл. 1.
Применение метода ЖХСД для разделения тритерпеновых сапонинов
(vi) Высокоэффективная жидкостная хроматография (ВЭЖХ)
ВЭЖХ является мощным методом с точки зрения получения мультиграммовых количеств из смесей близкородственных соединений и в этом смысле он часто применяется в качестве заключительного этапа очистки. В то время как в методе ЖХСД используются относительно крупные частицы (25-100 мкм), гранулярные фракции для полупрепаративной ВЭЖХ составляют 5-30 мкм, что, следовательно, обеспечивает более высокую степень разделения.
Полупрепаративная ВЭЖХ применялась для разделения тригликозидов олеаноловой кислоты из продуктов их частичного гидролиза. Это было необходимо для того, чтобы определить, присоединена ли галактозная составляющая по положению остатка глюкозы С-3 или С-4. Выделение изомерных сапонинов было осуществлено на колонке LiChrosorb RP-8 (7 мкм; 250×16 мм) с элюированием смесью ацетонитрил-вода (38:62) при скорости потока 10 мл/мин. Детекцию проводили при 206 нм и из 50 мг смеси (Decosterd et al., 1987).
Крупномасштабное разделение сайкосапонинов а, с и d из корней володушки Bupleurum falcatum (семейство зонтичных, Umbrellifeae) было достигнуто на колонках с аксиальной компрессией размером 100х11 см (внутр. диаметр). Предварительную очистку метанольного экстракта проводили путем распределения растворителя и с помощью хроматографии на полимере HP-20. Для препаративной ВЭЖХ колонку упаковывали силикагелем С-18 (размер частиц 20 мкм; 5 кг) и элюировали при скорости потока 210 мл/мин постадийно градиентом водного ацетонитрила. Загрузка образца весом 10 г была достаточной для выделения 400 мг сайкосапонина с, 1200 мг сайкосапонина а и 1600 мг сайкосапонина d (Sakuma & Motomura, 1987).
Гинзенозиды были выделены из женьшеня Рапах trifolius (Araliaceae) с помощью двухстадийной процедуры, включая хроматографию в системе Waters Prep 500 (колонки с радиальной компрессией) с тремя последовательно расположенными картриджами с силикагелем (размером 300×57 мм). Элюентом являлась верхняя фаза смеси н-бутанол-этилацетат-вода (4:1:5), и размер инъецируемого заряда составлял 4 г. Для окончательной очистки использовали полупрепаративную ВЭЖХ с углеводородной колонкой (Waters, 300×7,8 мм) с элюированием смесью ацетонитрил-вода (86:14 или 80:20) при скорости потока 2 мл/мин (Lee & der Marderosian, 1988).
Единственной серьезной проблемой при детекции компонентов в составе элюента при ВЭЖХ является отсутствие подходящего хромофора для УФ-детекции большинства сапонинов, хотя это обычно можно преодолеть с помощью таких методов, как определение показателя преломления, определение молекулярной массы и дериватизация.
Однако, учитывая небольшие изменения градиента, в принципе может быть применена УФ-детекция при длинах волн 203-210 нм с подходящими чистыми растворителями. Успешные разделения также были проведены с использованием градиентов смеси ацетонитрил-вода при УФ-детекции. Ацетонитрил является предпочтительнее метанола при низких значениях длины волны благодаря меньшему поглощению им ультрафиолета. Если в серии тестируемых сапонинов различия в полярности не слишком велики (например, из-за небольших различий в сахарной цепи), возможно изократическое элюирование.
Используемый способ разделения смесей сапонинов включает разделение на октилсвязанной колонке (С8) с использованием для элюирования градиента водного ацетонитрила. Количество ацетонитрила увеличивается с 30% до 40% в течение 20 минут, в результате чего происходит относительно слабый сдвиг базовой величины УФ-поглощения. Более полярные бидесмозидные сапонины обычно элюируются быстрее, чем монодесмозидные сапонины, а глюкурониды удерживаются меньше времени, чем другие гликозиды. Неполярный октилсилильный носитель может быть использован для отбора липофильной составляющей сапонинов. С использованием этого метода гликозиды хедерагенина были элюированы раньше таких же гликозидов менее полярной олеаноловой кислоты (Domon et al., 1984).
Применение дериватизованных тритерпенов
Детекция на коротких длинах волн, связанная с проблемой нестабильности фоновых значений из-за интерференции, обуславливаемой присутствием следовых количеств высоко-УФ-активного материала, может быть улучшена путем ВЭЖХ-анализа дериватизованных тритерпенов. Одним из таких возможных методов является функционализация свободных карбоксильных групп, найденных в сапонине, как было установлено в ходе количественного определения монодесмозидных сапонинов. Обработка гликозидов олеаноловой кислоты 4-бромфенацилбромидом в присутствии бикарбоната калия и краун-эфира приводит к образованию бромфенацильных производных. Эти 4-бромфенацильные производные поглощаются при 254 нм, и детекцию можно проводить при этой длине волны в отсутствие обуславливаемой растворителем интерференции (Slacanin et al., 1988). Такая дериватизация показана ниже:

Альтернативный способ анализа связан с получением флуоресцентных производных кумарина путем образования сложного эфира по остатку карбоновой кислоты. Таким образом, сапонины сои были проанализированы и количественно определены для различных сортов и различных органов растений сои с использованием антрацена в качестве внутреннего стандарта (Kitagawa et al., 1984а; Tani et al., 1985).
2. Очистка образца
С целью удаления постороннего материала, который часто характеризуется высоким уровнем поглощения ультрафиолета, может оказаться необходимым проведение стадии предварительной очистки. Этого можно достичь, например, путем использования картриджей Sep-Pak® C18 (Guedon et al., 1989) или Extrelut® (Sollorz, 1985).
При анализе ионных соединений, таких как соединения, содержащие свободную карбоксильную группу в агликоне или остатки глюкуроновой кислоты, необходимо применение какого-либо метода подавления ионизации, чтобы добиться предотвращения расширения пика. Этого можно достичь добавлением в поток кислоты с низким УФ-поглощением, такой как фосфорная кислота или трифторуксусная кислота. Другим возможным способом является применение ионопарной ВЭЖХ, в которой в подвижную фазу добавляют противоион. Эффективность оценки ионных соединений увеличивается благодаря образованию ионных комплексов с ионопарными реагентами. Дериватизация карбоксильных групп (как указано выше) является альтернативным способом в отношении добавок в подвижную фазу, в результате чего достигается существенное улучшение разрешения пика.
Преимущество количественной ВЭЖХ перед фотометрическими методами состоит в том, что в этом случае могут быть определены количества отдельных сапонинов в смеси или экстракте. Во многих примерах при ВЭЖХ получают лучшие результаты, чем при колориметрии, газовой хроматографии или ТСХ-флуориметрии.
В случаях, когда разрешение пика смесей сапонинов на колонках ВЭЖХ с обращенной фазой недостаточно, может быть использован ряд методов, включая использование колонок с гидроксиапатитом, химически модифицированных колонок из пористого стекла, колонок с силикагелем и ВЭЖХ с использованием боратных комплексов.
3. Гидроксиапатит
Гидроксиапатит (Са10[РO4]6[ОН]2) является более гидрофильным по сравнению с силикагелем и может быть использован вместе с простыми бинарными системами водных растворителей, что способствует облегчению УФ-детекции. Он стабилен в нейтральных и щелочных средах. Недавно были получены твердые частицы гидроксиапатита, устойчивые к высокому давлению (до 150 кг/см2), что расширило сферу применения ВЭЖХ. Сапонины, различающиеся только по конечному пентозному звену и не разделяющиеся методами ОФ-ВЭЖХ, могут быть разделены указанным способом (Kasai et al., 1987b). Разделение гинзенозидов женьшеня Раnах ginseng (Araliaceae) было достигнуто изократически (ацетонитрил-вода, 80:20) или, что дало лучшие результаты, в линейном градиенте смеси ацетонитрил-вода (70:30→90:10) (Kasai et al., 1987b). Как было установлено с использованием силикагеля, гликозиды элюируются по мере возрастания их полярности, т.е. в противоположном наблюдаемому при ОФ-ВЭЖХ направлении.
4. Боратная ионообменная ВЭЖХ
Этот способ находит свое применение в анализе моно- и олигосахаридов. Наилучшие результаты в этом случае получают на анионообменной колонке, например, колонке Asahipak ES-502N™ (100×7,6 мм) фирмы Asahi Kasei Kogyo Co., с 0,4 М Н3ВО3 в 20%-ном (об/об) ацетонитриле (рН 8) при 75°С. Условия хроматографии зависят от образования боратных комплексов с цис-диолами в сахаридной составляющей. После разделения борат может быть удален в виде летучего метилбората путем повторного проведения совместной перегонки полученного элюата с метанолом.
5. Химически модифицированное пористое стекло
Микропористое стекло (МПС) характеризуется высокой химической стойкостью и стабильно в диапазоне рН 2-12. Октадецильное пористое стекло (МПС-ОД) было получено в качестве насадки для ВЭЖХ с обращенной фазой и использовано для быстрого и эффективного разделения сапонинов. Например, как гинзенозиды, так и сайкосапонины можно выделить одновременно из экстрактов комбинации лекарственных средств, содержащих корень женьшеня и володушки, с использованием для разделения смеси ацетонитрил-вода (25,5:74,5) (Kanazawa et al., 1990a). Сравнение МПС-ОД и ОД-силикагелевых колонок, используемых в ВЭЖХ для экстракта женьшеня и смеси гинзенозидов, показало, что характер удерживания был сходным, однако коэффициент производительности колонки МПС-ОД была меньше. Разрешение некоторых пар гинзенозидов было более высоким на колонках МПС-ОД (Kanazawa et al., 1993).
6. Силикагель
Использование подвижных фаз, содержащих воду, часто неизбежно при разделении сапонинов, поэтому ВЭЖХ с силикагелем обычно не пригодна для таких элюентов. Однако модификация упаковки колонки делает возможным разделение водорастворимых гликозидов без износа колонки. Такая процедура включает вначале промывку колонки метанолом, затем смесью хлороформ-метанол-этанол-вода (62:16:16:6) и, наконец, той системой растворителей, которая будет использована для разделения (Kaizuka & Takahashi, 1983). С использованием, например, 5-мкм колонок с силикагелем и водосодержащим элюентом гексан-этанол-вода (8:2:0,5) может быть проведен эффективный анализ сапонинов женьшеня и сайкосапонинов володушки Bupleurum falcatum.
(vii) Другие хроматографические методы
Выделение чистых сапонинов требует одного или, что более обычно, нескольких этапов хроматографического разделения с целью удаления других полярных компонентов из спиртовых или водных растительных экстрактов.
Был описан ряд методов разделения, которые могут быть использованы для разделения тритерпеновых сапонинов, включая флэш-хроматографию, КПТХ, жидкостную хроматографию низкого давления (ЖХНД), жидкостную хроматографию среднего давления (ЖХСД), ВЭЖХ и стандартную хроматографию на открытой колонке (см., например, Hostettmann et al., 1986, 1991; Marston & Hostettmann, 1991b). Информация об условиях разделения, системах растворителей и т.п.хорошо известны специалистам в данной области техники в связи с описываемым изобретением. Наилучшие результаты обычно достигаются с применением стратегий, основанных на комбинации методов, таких как методы, конкретно описанные ниже.
Поскольку ряд сапонинов являются кислотными, могут быть получены их соли, а по завершении хроматографической очистки может оказаться необходимой обработка ионообменной смолой с целью выделения свободного сапонина. Примерами подходящих смол являются Dowex 50Wx8 (Н+-форма) (Kitagawa et al., 1988; Yoshikawa et al., 1991), Amberlite IRC84 (Okabe et al., 1989; Nagao et al., 1990) и Amberlite MB-3 (Mizutani et al., 1984). Однако, если для предотвращения разложения необходима нейтральность или тщательный контроль рН, то этапы фильтрации на ионообменных смолах должны быть исключены.
В некоторых случаях, неочищенные фракции сапонинов были метилированы (в предположении о присутствии свободных карбоксильных групп) с целью достижения удовлетворительного разделения близкородственных сапонинов (Okabe et al., 1989; Nagao et al., 1989, 1990).
1. Флэш-хроматография
Флэш-хроматография представляет собой метод препаративной жидкостной хроматографии под давлением, который позволяет существенно экономить время по сравнению со стандартной хроматографией на открытой колонке. При этом используются обычные стеклянные колонки, однако элюент направляют через сорбент сжатым воздухом или азотом, достигая максимального давления примерно в 2 бар в верхней части колонки. Размер частиц сорбента в некоторой степени снижена благодаря тому, что растворитель подается под давлением, соответственно, разрешение повышается.
Флэш-хроматография может быть использована в качестве быстрого альтернативного метода по отношению к методам хроматографии на открытой колонке, применяемым для предварительного фракционирования. С использованием данного метода разделение образца размером от 10 мг до 10 г может быть осуществлено всего за 10 минут. Например, этим методом были выделены хедерагениновые, байогениновые и медикагениновые глюкозиды из корней Dolichos kilimandscharicus (Leguminosae), обладающие моллюскоцидной и фунгицидной активностью. Метанольный экстракт (3,3 г) был фракционирован на силикагеле (гранулометрия 63-200 мкм) в колонке размером 60×4 см с системой растворителей “хлороформ-метанол-вода” (50:10:1) при скорости потока 15 мл/мин. Этого было достаточно для удаления загрязняющих материалов и получения двух фракций, богатых сапонинами. Очищенные тритерпеновые гликозиды получали путем комбинирования методов КПТХ и ЖХНД на С8-носителях (Marston et al., 1988a).
Хотя в большинстве применений используются силикагелевые сорбенты, имеется отчетливая тенденция к использованию материалов для обращенно-фазовой хроматографии. Флэш-хроматография с обращенной фазой обеспечивает отделение сапонинов от других, более полярных компонентов, таких как олигосахариды.
2. Жидкостная хроматография низкого давления (ЖХНД)
Метод ЖХНД применяется для выделения очищенных сапонинов благодаря высокой скорости разделения и простоте работы. В ЖХНД используются колонки, сорбенты которых характеризуются размером частиц 40-60 мкм. При давлении до 10 бар возможны высокие скорости потока и обычно колонки делают стеклянными. Коммерчески доступные, заранее упакованные колонки различного размера (например, серия "Lobar" фирмы Merck) являются идеальными для препаративной хроматографии сапонинов при объеме образца в 50-500 мг. Высокая и однородная плотность упаковки колонок гарантирует высокую эффективность разделения. Кроме того, условия аналитической ВЭЖХ могут быть относительно легко использованы для разделения методом ЖХНД с учетом того, что химия сорбентов сходна (Marston & Hostettmann, 1991b).
Большинство методов осуществляют на обращенно-фазовых сорбентах с элюированием смесью метанол - вода. В этом случае обычно вносят только предварительно очищенные образцы. Хорошим примером ЖХНД является выделение моллюскоцидных и гемолитических гликозидов олеаноловой кислоты и гипсогенина из Swartzia madagascariensis (Leguminosae). Высушенные измельченные бобы экстрагировали водой, а полученный экстракт фракционировали между н-бутанолом и водой. После разделения органической фазы методом хроматографии на открытой колонке сапонины были разделены на колонке Lobar LiChroprep C-8 (40-63 мкм; 27×2,5 см) с элюированием смесью метанол-вода (75:25) (Borel & Hostettmann, 1987).
Объединение колонок для ЖХНД в ряд позволяет увеличить объем загрузок и (или) мощность разделения. Данный подход был использован в ходе выделения даммарановых гликозидов из Actinostemma lobata (Cucurbitaceae), где три колонки Lobar размером 27×2,5 см были объединены друг с другом. Также элюент содержал небольшое количество воды (смесь этилацетат-н-пропанол-вода, 20:3:0,3) (Iwamoto et al., 1987).
3. Противоточная хроматография
Методы разделения в системе “жидкость-жидкость”, как было подтверждено, является идеальным для применения его в отношении сапонинов. Высокополярные сапонины особенно пригодны для противоточного хроматографического разделения потому, что в этом случае отсутствует потеря материала из-за необратимой адсорбции на материале упаковки колонки. Это может быть существенным аргументом в пользу использования данного подхода для прямого фракционирования неочищенных экстрактов.
4. Капельная противоточная хроматография (КПТХ)
Метод КПТХ основан на непрерывном пропускании капелек подвижной фазы через несмешиваемую жидкую стационарную фазу, содержащуюся в большом количестве вертикальных стеклянных трубок. Сорбат (растворенное вещество) подвергается непрерывному распределению между двумя фазами. В зависимости от того, сверху или снизу была внесена подвижная фаза в указанные трубки, хроматография определяется как, соответственно, “нисходящая” или “восходящая”. С помощью метода КПТХ можно разделить близкородственные сапонины и даже выделить чистые продукты (Hostettmann et al., 1984). Действительно, некоторые варианты разделения, не удававшиеся с помощью жидкотвердой хроматографии, могут быть осуществлены с применением указанного способа. В методе КПТХ удалось разделить изомеры сапонинов, различающиеся только по положению замены ацетатных групп в сахарных остатках (Ishii et al., 1984).
Ряд систем растворителей применяли для разделения сапонинов методом КПТХ (см., например, Hostettmann et al., 1986), и среди них, наибольшее применение нашла смесь хлороформ-метанол-вода (7:13:8). Смеси хлороформ-метанол-вода можно использовать либо для восходящей хроматографии высокополярных сапонинов, либо для нисходящей хроматографии сапонинов с одним или двумя сахарами и несколькими свободными гидроксильными группами.
Была описана крупномасштабная процедура КПТХ, предназначенная для предварительной очистки, на 18 колонках (300×10 мм, внутренний диаметр) с использованием в качестве стационарной фазы насыщенного водой н-бутанола, а в качестве подвижной фазы насыщенной н-бутанолом воды (Komori et al., 1983). Иногда для получения чистых сапонинов проводят два (или больше) этапа КПТХ.
5. Распределительная хроматография с центрифугированием (РХЦ)
Недавно разработанный метод РХЦ оказался многообещающим благодаря его скорости и универсальности (Marston et al., 1990). Метод РХЦ основан на удерживании стационарной фазы полем центробежных сил при вращении 800-2000 или более об/мин, а не гравитационным полем. Принципом данного метода является непрерывный процесс неравновесного распределения сорбата между двумя несмешиваемыми фазами, содержащимися во вращающихся змеевиках или картриджах.
В аппаратах, конструкция которых включает вращающиеся змеевики, может использоваться как орбитальное, так и неорбитальное движение вокруг центральной оси. В одном из таких аппаратов, высокоскоростном противоточном хроматографе, имеется тефлоновая трубка внутреннего диаметра 1,6 или 2,6 мм, намотанная в виде спирали вокруг бобины. Сердечник аппарата состоит из одной, двух или трех таких бобин. В случае кассетной конструкции аппарата картриджи расположены по периферии ротора так, что их продольные оси параллельны направлению центробежной силы. Число и объем картриджей могут варьироваться в зависимости от конкретного использования данного аппарата. По сравнению с КПТХ и ЖПТХ, при которых разделение может продолжаться 2 дня или дольше, в методе РХЦ те же результаты могут быть получены в течение нескольких часов. Емкость аппаратов со змеевиками и картриджами составляет в пределах граммовых количеств. Аппарат с многослойной орбитальной серией змеевиков использовали, например, для предварительной очистки циклоартановых гликозидов абруса Abrus frutlculosus (Leguminosae) (Fullas et al., 1990). Моллюскоцидные тритерпеновые гликозиды были выделены из плюща Hedera helix (Araliaceae) на другом аппарате, хроматографе Sanki LLN (6 картриджей при общем объеме 125 мл). Метанольный экстракт плодов распределяли между н-бутанолом и водой. Бутанольную фракцию вводили непосредственно в аппарат в количествах по 100 мг с использованием в качестве подвижной фазы нижнего слоя смеси растворителей хлороформ-метанол-вода (7:13:8).
Два основных сапонина, азиатикозид и мадекассозид, были выделены из центеллы азиатской Centella asiatica (Umbrellifeae) с использованием сепаратора-экстрактора с многослойными змеевиками Ito (P.С.Inc.), снабженного колонкой размером 66 м на 2,6 мм (внутренний диаметр) (емкостью 350 мл), вращаемой при 800 об/мин. Образец размером 400 мг может быть разделен с использованием системы растворителей хлороформ-метанол-2-бутанол-вода (7:6:3:4; подвижной фазой является ее нижний слой). Для детекции применяли ТСХ (Diallo et al., 1991). Тот же аппарат использовали в ходе выделения тритерпендисахарида из кунжута Sesamum alatum (Pedaliaceae). Нижний слой смеси хлороформ-метанол-изопропанол-вода (5:6:1:4) был выбран в качестве подвижной фазы, а загрузка составляла 1,25 г (Potterat et al., 1992).
6. Комбинации методов
Редко удается за одну стадию хроматографии выделить из экстракта чистый сапонин. Как правило, для получения желаемого продукта требуется использование нескольких препаративных методов. Использование комбинации классических методов (таких как хроматография на открытой колонке) и современных высокоразрешающих технологий (таких как ВЭЖХ), как подтверждается, является достаточным для разделения большинства сапонинов.
Примером является комбинация ЖХСД на силикагеле с ОФ-материалом, ЖХНД и ТСХ с центрифугированием для разделения сапонинов (Hamburger & Hostettmann, 1986). Аналогичным образом, выделение пяти тритерпеновых сапонинов из Swartzia madagascariensis (Leguminosae) потребовало применения хроматографии на открытой колонке, ЖХНД и ЖХСД (Borel & Hostettmann, 1987).
РХЦ была использована в комбинации с флэш-хроматографией и OPLC для выделения тритерпеновых гликозидов из Abrus fruticulosus (Leguminosae). Аппарат со змеевиками, уложенными в несколько слоев (растворители хлороформ-метанол-вода 7:13:8, подвижной фазой является нижний слой), использовали для исходной очистки, в то время как флэш-хроматография и OPLC являются эффективными для получения чистых веществ (Fullas et al., 1990).
Иногда для очистки сапонинов может быть достаточным прямое сочетание флэш-хроматографии на немодифицированном силикагеле с флэш-хроматографией или хроматографией на открытой колонке с обращенными фазами (Schopke et al., 1991).
Другая стратегия основана на пропускании экстрактов (после предварительного разделения) через высокопористые полимеры с последующим фракционированием неочищенных сапониновых смесей. Такой подход был использован для выделения гликозидов 3β-гидроксиолеан-12-ен-28,29-диоевой кислоты из Nothopanax delavayi (Araliaceae). Метанольный экстракт из листьев и стеблей распределяли между гексаном и водой. Водный слой хроматографировали на колонке Diaion HP-20 и элюировали водой, 15% метанолом, 50% метанолом, 80% метанолом, абсолютным метанолом и хлороформом. Гликозиды получали путем последовательной колоночной хроматографии элюата в 80%-ном метаноле с использованием силикагеля и смеси этилацетат-этанол-вода (7:2:1) (Kasai et al., 1987a). Для выделения тритерпеновых и нетритерпеновых сапонинов Acanthopanax senticosus (Araliaceae) данную процедуру начинали с фракционирования метанольного экстракта листьев на полимере Diaion HP-20. Элюированную метанолом фракцию хроматографировали на силикагеле (хлороформ-метанол-вода, 30:10:1) и все полученные в результате фракции подвергали хроматографии на колонках LiChroprep RP-8. Окончательную очистку проводили методом ВЭЖХ на TSK-GEL ODS-120T (300×21 мм; метанол-вода,70:30; 6 мл/мин; радиоиммунная детекция) или хроматографии на колонке с гидроксиапатитом (ацетонитрил-вода, 85:15) (Shao et al., 1988).
В методе разделения гликозидов олеаноловой кислоты использовали комбинацию Сефадекса LH-20 (метанол), КПТХ (хлороформ-метанол-вода, 7:13:8) и ВЭЖХ (С-18; метанол-вода, 65:35) (De Tommasi et al., 1991).
(viii) Реакции окрашивания
Реакции тритерпенов с любым из ряда агентов можно использовать для получения окрашенных соединений с целью количественного или качественного определения этих тритерпенов. Например, ароматические альдегиды, такие как анисовый альдегид или ванилин в сильной минеральной кислоте, например, серной, фосфорной и перхлорной кислотах, взаимодействуя с агликонами, дают окрашенные продукты с максимальным поглощением при длинах волн 510 и 620 нм. В этих реакциях, как считается, происходит дегидратация, в результате чего образуются ненасыщенные метиленовые группы, которые дают окрашенные продукты конденсации с альдегидами. При использовании ванилина с серной кислотой тритерпеновые сапонины с гидроксильной группой в положении С-23 дают пик поглощения между 460-485 нм (Hiai et al., 1976).
Ненасыщенные и гидроксилированные тритерпены и стероиды дают красное, синее или зеленое окрашивание с уксусным ангидридом и серной кислотой (Abisch & Reichstein, 1960). Поскольку тритерпеновые сапонины имеют тенденцию давать розовую или пурпурную окраску, а стероидные сапонины - сине-зеленую окраску, то эти классы сапонинов могут быть идентифицированы этим методом.
Для детекции тритерпенов может быть использовано большое число других реагентов, включая: сульфат церия (IV) или соли железа (III) и неорганические кислоты, такие как серная кислота, которые дают фиолетово-красное окрашивание раствора; 30%-ный раствор хлорида сурьмы (III) в смеси уксусный ангидрид - уксусная кислота, который дает реакции окрашивания с гидрокситритерпенами и гидроксистероидами; хлорид сурьмы (III) в смеси нитробензол-метанол, который может быть использован для выявления 5,6-дигидропроизводных стероидных гликозидов (диосгениновые и соласодиновые гликозиды) и 5α- или 5β-H-производных (например, томатина); и карбазол, который в присутствии бората и концентрированной серной кислоты позволяет выявлять наличие уроновых кислот (Bitter & Muir, 1962).
Примеры реагентов, предназначенных для детекции и спектрофотометрического и колориметрического выявления сапонинов, приведены ниже в табл.2.
Реагенты для визуализации тритерпеновых сапонинов
(ix) Выделение тритерпеновых гликозидов из Acacia victonae
Экстракты бобов были приготовлены методом экстракции смесью хлороформ-метанол или дихлорметан-хлороформ в ун-те штата Аризона в Таксоне (Tucson, AZ). Заявители выделили смеси тритерпеновых гликозидов из акации Виктории Acacia victoriae (Benth.) (Leguminosae). Первый сбор UA-BRF-004-DELEP-F001 был получен следующим образом: (1) измельчение в ступке Вилея до размера частиц 3 мм; (2) расфасовка в двухлитровые емкости для перколяции; (3) экстракция измельченной биомассы смесью дихлорметан-метанол (1:1) в течение 4 часов с последующим отстаиванием в течение ночи, после чего объединенные фракции сушили в вакууме с получением UA-BRF-004-DELEP-F001 (52 г). Далее F001 (51,5 г) экстрагировали этилацетатом с получением активного нерастворимого материала (34,7 г), обозначенного F004. Флэш-хроматографию на силикагеле (1,7 кг; Merck; размер частиц 23-220 мкм) применяли для фракционирования F004 (34,2 г), после чего 51 фракцию по 670 мл элюировали смесью дихлорметан-метанол (ступенчатый градиент от 95% до 0% дихлорметана и от 5% до 100% метанола). Колонку промывали 9 л метанола, затем 6 л смеси метанол - вода (80:20) и, наконец, 6 л того же элюента с добавлением 1% муравьиной кислоты. ТСХ-фракции 23-34 и 39-40 были объединены в 17,2 г препарата F023. Жидкостную хроматографию среднего давления (ЖХСД; система Buchi-632) применяли дважды, каждый раз с 8 г F023, на колонке размером 14,9×46 см, упакованной сорбентом LiChropep C18 (размер частиц 15-25 мкм) с использованием градиента смеси ацетонитрил - вода (0, 10, 20, 30, 50% ацетонитрила в воде) с последующей промывкой абсолютным метанолом. Из 16 г 0-20% ацетонитрила получали семь граммов фракции F027, которая оказалась неактивной. Остальной материал объединяли и подвергали повторной ЖХСД в той же системе с использованием 30-40% ацетонитрила с целью минимизации перекрываний и с получением фракций F028-F036. Хотя большинство этих фракций проявляли противоопухолевую активность, для дальнейшего анализа и оценки выбрали F035 (фракцию 35) (максимальный выход 2,19 г).
III. Структурная характеристика тритерпенов
Для количественного и качественного анализа тритерпенов и их активности может быть использован ряд методов, включая: анализ ихтиоцидной активности, гравиметрический анализ, спектрофотометрию, ТСХ, ГХ, ВЭЖХ, HMQC, HMBC, NOESY, COSY, ЯМР, рентгенокристаллографию и т.п. Анализ на основе классических свойств тритерпеновых сапонинов (поверхностная активность, токсичность для рыб) в основном заменяется фотометрическими методами, такими как денситометрия, колориметрия производных, а в последнее время ГХ, ВЭЖХ и, в частности, ЯМР. Спектрофотометрические методы характеризуются очень высокой чувствительностью, но обычно непригодны для анализа тритерпенов в составе неочищенных растительных экстрактов, поскольку соответствующие реакции неспецифичны и окрашенные продукты могут образовываться соединениями, сопровождающими тритерпены, такими как фитостеролы и флавоноиды. Другая проблема, обычно возникающая при анализе сапонинов, связана с их неполной экстракцией из растительного материала. Однако имеется широкий ряд методов, пригодных для количественного анализа тритерпенов.
При определении структуры сапонинов необходимо решить ряд базовых задач: определить структуру природного агликона; установить структуру и последовательность моносахаридов в углеводной составляющей; определить тип связей моносахаридных звеньев между собой; определить аномерную конфигурацию каждого гликозид-связанного моносахаридного звена; определить положение углеводной составляющей в агликоне.
Для окончательного вывода о структуре необходимо использовать различные методы. Структурный анализ обычно имеет ступенчатый характер, в котором сапонины постепенно разделяют на более мелкие фрагменты, становящиеся объектами отдельного спектрометрического анализа. Путем беспристрастной обработки данных по отдельным фрагментам можно выстроить основную структуру сапонина.
Как правило, количество очищенных сапонинов мало, поэтому при определении их структуры предпочтительным является использование высокочувствительных, высокоразрешающих и, если это возможно, неповреждающих методов. Модификации методов спектроскопии ЯМР и масс-спектроскопии (МС) предоставляют такие необходимые возможности для анализа сложных сапонинов. Путем комбинации этих и других методов может быть осуществлен эффективный структурный анализ. Например, методом FAB-MS получают информацию о молекулярной массе, а во многих случаях, о последовательности сахаров, в то время как методы одно- и двухмерного ЯМР позволяют картировать сахарные связи и помогают в определении структуры агликона. Такой структурный анализ и химический анализ подробно обсуждается в обзоре Tanaka & Kasai, 1984.
(i) Ядерный магнитный резонанс (ЯМР)
Из всех современных методов анализа структуры олигосахаридов и гликозидов ЯМР-спектроскопия позволяет получать наиболее полную информацию, независимо от наличия или отсутствия предварительных сведений о структуре (Agrawal, 1992). Это единственный подход, который, в принципе, может обеспечить определение полной структуры без применения какого-либо дополнительного метода.
1. 13С-Ядерный магнитный резонанс
13С-ЯМР-спектроскопия, широко применяемая в настоящее время для определения структуры сапонинов, является быстрым и недеструктивным методом, хотя и требует достаточно большого количества образца (миллиграммовых количеств). Анализ получаемых спектров позволяет делать заключения о положениях, в которых гликозидные цепи присоединяются к агликону; о последовательности, природе и числе моносахаридов; о конфигурации и конформации межгликозидных связей; о присутствии ацилгликозидов в цепочках; о природе агликона и о структуре присоединенных сложных эфиров кислот.
Для определения химических сдвигов полезно сравнить полученные данные с данными, опубликованными для стандартов и родственных соединений. В качестве эталона величин химических сдвигов в спектрах 13С-ЯМР для тритерпенового сапонина можно использовать данные, полученные для гликозида байогенина (Domon & Hostettmann, 1984). Кроме того, были собраны данные (Nakanishi et al., 1983) по 13C-ЯMP-сигналам, установленным для олеанана (Patra et al., 1981; Agrawal & Jain, 1992), урсана, лупана (Wenkert et al., 1978; Sholichin et al., 1980), хопана (Wenkert et al., 1978; Wilkins et al., 1987) и ланостана (Parrilli et al., 1979). Аналогичные данные для даммарановых гликозидов систематизированы в обзоре (Tanaka & Kasai, 1984); а также описана 13С-ЯМР-спектроскопия сайкогенинов (Tori et аl., 1976а) и сайкосапонинов (Tori et al., 1976b). Были исследованы сапогенины женьшеня и родственные даммарановые гликозиды (Asakawa et al., 1977). Также описана 13С-ЯМР-спектроскопия акациевой кислоты (Kinjo et al., 1992).
При 13С-ЯМР в случае дериватизации гидроксильных групп, т.е. их гликозилирования, метилирования (или ацетилирования), α- и β-атомы углерода как в сахаре, так и в агликоне проявляют характерные сдвиги. Например, α-СН-сигналы сдвинуты в сторону слабого поля, в то время как сигналы β-С сдвинуты в сторону сильного поля, причем сдвиг является результатом общего γ-сильнопольного сдвига. Таким образом, гликозилирование агликона дает сдвиг в сторону слабого поля γ-С и сильнопольный сдвиг (сдвиги гликозидирования) соседних атомов углерода (Tori et al., 1976b; Kasai et al., 1977). В олеанинах гликозидирование группы 3β-ОН дают сдвиг С-3 в сторону слабого поля примерно на 8,0-11,5 м.д., атомов С-2 и С-4 на +0,9 или от -0,9 до -1,9 м.д., атома С-23 в сильное поле на 0,5-5,1 м.д. и атома С-24 от -0,2 до +1,6 м.д. Гликозидирование группы 28-СООН дает сдвиг углерода карбоксильной группы в сторону сильного поля (на 2,5-5,0 м.д.), а сигнала С-17 в сторону слабого поля (на 1,0-2,5 м.д.) (Agrawal & Jain, 1992). Следовательно, сравнение данных 13С-ЯМР агликона и сапонина позволяет определить место присоединения сахара (Seo et al., 1978; Tanaka, 1985).
Аналогичным образом, данные 13С-ЯМР позволяют определить (в более простых сапонинах) положение межгликозидных связей на основе сравнения смещения химических сдвигов с данными для модельных соединений (Konishi et al., 1978). Данные 13С-ЯМР для метил-β-В-фукопиранозида были приведены у Seo et al., 1978, в то время как 13С-ЯМР-сигналы для более сложных сахаров имеются у Gorin & Mazurek (1975) и Dorman & Roberts (1970). Апиоза дает характерные 13С-ЯМР-данные, и эти данные были зарегистрированы (Sakuma & Shoji, 1982; Adinolfi et al., 1987; Reznicek et al., 1990).
2. 1H-Ядерный магнитный резонанс
Хотя анализ спектров 13С-ЯМР и оценка сигналов в них является особенно эффективным методом определения структуры сапонинов, однако, полная расшифровка спектров 1H-ЯМР проводится редко. Протонные 1H-ЯМР-спектры сложны и утомительны для анализа. Подавляющее большинство протонных резонансов углеводных составляющих находится в очень узком спектральном диапазоне 3,0-4,2 м.д., что создает серьезную проблему их перекрывания. Это обуславливается общей массой неаномерных метиновых и метиленовых протонов сахаров, которые характеризуются очень похожими величинами химического сдвига в различных моносахаридных остатках.
Однако, метильные пики тритерпенов легко различимы, поэтому большинство протонных резонансов в структурах олеанена, урсена и близких структурах были определены после 1960 года с применением различных методов (Kojima & Ogura, 1989). Например, полные расшифровки спектров 1H и 13C-ЯМР для сапогенола-В сои (33) и конфигурации его гидроксиметильного заместителя по С-4 были установлены путем комбинации методов 13C-DEPT, 13C-АРТ, двумерной корреляционной спектрометрии (COSY: 1H/13C-COSY; 1H/1H-COSY) и 1H/1H-ROESY (двумерное ядерное усиление по Оверхаузеру во вращающейся системе координат) (Baxter et al., 1990). Результаты расшифровки четвертичных углеродных резонансов у данного сапогенина подтверждены с применением 1H-гетероядерной спектроскопии с множественными (НМВС) и одиночными связями (HMQC) (Massiot et al., 1991b). Также была достигнута полная интерпретация спектров 1H-ЯМР диосгенина и соласодина (Puri et al., 1993).
Некоторые полезные данные могут быть получены из спектров 1H-ЯМР для аномерных конфигураций и связей сахарной цепи. Например, константа спин-спинового взаимодействия протона С-1 для α-связанных глюкозных звеньев составляет примерно 3 Гц, в то время как β-связанные звенья имеют константу взаимодействия 6-7 Гц. Другие подробности о константах взаимодействия протонов в аномерных сахарах можно найти в различных источниках (Lemiex et al., 1958; Capon & Thacker, 1964; Kizu & Tomimori, 1982).
При возникновении трудностей в определении конфигурации гидроксильных групп у С-2, С-3 и С-23, С-24 олеаненовых и урсеновых тритерпенов, ценную информацию можно получить в анализе пиков 1H-ЯМР-сигналов от протонов кислород-несущих атомов углерода (Kojima & Ogura, 1989).
(ii) Методы одномерного и двумерного ЯМР
На практике некоторые спектры 1H- и 13C-ЯМР могут быть идентифицированы и расшифрованы на основе параметров сдвигов, однако для высокоточной интерпретации результатов ЯМР-исследований необходима однозначная расшифровка спектра ЯМР, которая обеспечит установление связи отдельных пиков с соответствующими атомами углерода и (или) водорода в структуре. Во многих случаях такую информацию нельзя получить на материале одномерных спектров 1H и 13C-ЯМР, но можно установить при помощи двумерного исследования. Такие исследования упрощают спектральный анализ за счет распределения информации по двум частотным диапазонам и определения взаимодействий между ядрами. Несмотря на то, что механизмы формирования различных последовательностей импульсов могут быть весьма запутанными, интерпретация двумерных спектров ЯМР обычно весьма проста. С целью установления химических структур было проведено значительное количество исследований с помощью двумерных спектров ЯМР. Примеры таких методов, а также других ЯМР-методов, конкретно рассматриваемых заявителями в связи с использованием для химического анализа тритерпеновых сапонинов по настоящему изобретению, описаны ниже и в табл.3.
1. НМВС, HMQC
Использование спектров многоквантовой когерентности 13C HMQC и НМВС информативно не только в связи с идентификацией агликона, но и для установления деталей последовательности сахаров. Применение методов HMQC и НМВС аналогично гетероядерной корреляционной спектроскопии 13C-1H (HETCOR), только вместо наблюдения 13С наблюдается более обильный 1H. Например, в случае с беллисапонинами, выделенних из маргаритки Bellis perennis (Asteraceae), оказалось возможным определить все химические сдвиги для спектров 1H-ЯМР путем рассмотрения данных 13C-ЯМР-спектра в комбинации с двумерными 1H-детектируемыми спектрами HMQC и НМВС. Почти для всех возможных корреляций в данной молекуле наблюдались перекрестные пики, соответствующие взаимодействию двух или трех связей. Аналогичным образом, широкомасштабные корреляции 1H-13C в спектрах HMQC и НМВС могут быть использованы для определения последовательности и положений присоединения сахарных остатков ( et al., 1991).
et al., 1991).
2. Двумерный NOESY
Данный метод был применен, например, для определения сахарной последовательности в гликозидах цикломеритина-А (ардисиакриспины А и В) (Jansakul et al., 1987) и последовательности моносахаридов в саксифрагифолине-А, выделенных из проломника Androsace saxifragifolia (Primulaceae) (Waltho et al., 1986). Положение рамнозильных и глюкозильных связей на арабинозной составляющей калопанакссапонина-С подтвердили с помощью NOESY на основе анализа сахарной последовательности перметилированного сапонина. Наблюдались конкретные пики для Н-1 рамнозильной составляющей и Н-2 арабинозильной составляющей, а также для Н-1 глюкозильной составляющей и Н-3 арабинозильной составляющей (Shao et al., 1989b). Структуру сахарных составляющих фуростанольных сапонинов, выделенных из Balanites aegyptiaca (Balanitaceae), определяли с применением двумерного NOESY на ЯМР-спектрометре с частотой 400 МГц (Kamel et al., 1991).
Согласованное применение методов двумерного ЯМР позволило полностью проинтерпретировать спектры 13C и 1H для олигосахаридного сегмента сарсапогенинового гликозида 3-O-[{α-L-рамнопиранозил (1→4)}{β-D-глюкопиранозил (1→2)}-β-D-глюкопиранозил]-(255)-5β-спиростан-3β-ола. Комбинация методов DEPT, HETCOR, HETCOR дальнего действия, различных гомоядерных методов, NOESY и INEPT была использована для структурного анализа с целью разрешения проблем, связанных с чрезмерной перенасыщенностью протонного спектра (Pant et al., 1988d).
Идентификация и определение последовательности сахаров в пентасахаридном сапонине 3-O-[β-D-ксилопиранозил (1→3)-α-L-арабинопиранозил] (1→4)-β-D-глюкопиранозил (1β3)-α-L-рамнопиранозил (1→2)-α-L-арабинопиранозил] гедерагенина, выделенного из растения Blighia welwitschii (Sapindaceae), оказались возможными при использовании только методов ЯМР с использованием спектрометра с частотой 500 МГц. Вначале сапонин ацетилировали, а последующий анализ спектров DQF-COSY, NOESY и ROESY позволил определить структуру. Наиболее ценная информация для установления последовательности сахаров была получена с использованием NOE (Penders et al., 1989).
Сапонин, включающий 10 сахарных остатков, выделенный из золотарника гигантского Solidago gigantea (Asteraceae), был идентифицирован методами ЯМР, основанными на многостадийном RCT-анализе. Этот анализ включал методы COSY, гетероядерного COSY, Н-Н-С-когерентного переноса COSY-типа и двумерного NOESY. Таким образом, оказался исключенным анализ, сопряженный со значительными химическими разрушениями молекулы, а для установления структуры потребовалось всего 30 мг продукта (Reznicek et al., 1989a, 1989b). Аналогичные методы использовали в структурном анализе четырех других гликозидов, гигантеасапонинов 1-4 (бидесмозиды байогенина, включающие 9-10 сахарных звеньев), вырабатываемых тем же видом растений (Reznicek et al., 1990а).
Комбинация ЯМР-методов двумерного COSY, HMBC и ROESY была эффективна для установления последовательности и сайтов связей в гексасахариде, входящем в состав мимонозида-А, являющегося сапонином олеаноловой кислоты, выделенным из мимозы мелкоцветковой Mimosa tenuiflora (Leguminosae) (Jiang et al., 1991).
Метод двумерного ЯМР перацетилированного и недериватизованного хризантеллина-А позволил картировать протоны и определить последовательность сахаров. Как было показано, этерифицированный остаток ксилозы находится в β-форме и имеет 1C4-конформацию. В отношении перацетилированного производного применяли методы HMQC, НМВС и ROESY (или, что более точно, метод CAMELSPIN (поперечная релаксация в минимолекулах, эмулированная локализованными спинами)), а в отношении нативного сапонина применяли методы НОНАНА и TOCSY. В частности, для определения последовательности сахаров применяли метод ROESY (Massiot et al., 1991a).
Последовательности сахаров и межгликозидные связи тритерпеновых гликозидов морских организмов были установлены по данным NT1 и в анализе методом NOESY (Miyamoto et al., 1990), однако такой подход оказался ограниченным из-за сложности полученных спектров 1Н-ЯМР в диапазоне 3-5 м.д., что обычно мешает определению NOE при большом числе протонов. Однако, комбинация методов COSY, NOESY, прямого ЯМР и XHCORR-ЯМР позволила полностью расшифровать сигналы и определить структуру пентасахаридных тритерпеновых сапонинов, выделенных из голотурии Holothuria forskalii (Rodriguez et al., 1991).
В структурном анализе сантиагозида, являющегося астеросапонином, выделенным из антарктической морской звезды Neosmilaster georyianus, применяли методы COSY, TOCSY, HMQC и ROESY ЯМР, причем данные метода ROESY использовали для определения точной последовательности углеводов, места их соединения и стереохимической структуры (Vazquez et al., 1992).
3. COSY
Известно два фундаментальных типа двумерной спектроскопии ЯМР: J-разрешающая спектроскопия, при которой одна частотная ось характеризует спиновое сопряжение (J), а другая несет информацию о химическом сдвиге; и корреляционная спектроскопия, при которой обе частотные оси характеризуют химические сдвиги (величины δ) (Agrawal, 1992). Одно из основных преимуществ двумерного анализа заключается в том, что он дает возможность преодолеть проблему переполнения спектров. Это особенно важно в отношении диапазона 2,5-4,0 м.д. при анализе сильнопольного 1H-COSY, т.к. позволяет упростить картирование протонов сахаров. При благоприятных условиях могут быть идентифицированы все протоны, имеющиеся в составе данного углеводного остатка.
На основании спектров COSY можно сделать ряд общих выводов. Например, положения заместителей моносахаридных единиц можно определить по присутствию или отсутствию соответствующих гидроксильных протонов; размер кольца моносахарида можно определить непосредственно; а природа соответствующих друг другу пиков отражает мультиплетность перекрывающихся пиков, что дает возможность оценить константу спин-спинового взаимодействия.
В некоторых случаях определение структуры сапонина, наряду с последовательностью его сахаров, может быть проведено только с помощью двумерной и одномерной спектроскопии 1H-ЯМР (Massiot еt аl., 1986, 1988b). Сначала сапонин перацетилируют и при достаточной напряженности поля (>300 МГц) спектр протонного резонанса сахаров расщепляется на две области: одна в диапазоне 4,75-5,40 м.д., соответствующая СНОАс, и другая в диапазоне 3,0-4,3 м.д., соответствующая СН2OАс, CHOR и CH2OR. Аномерные протоны находятся между этими двумя областями в случае наличия эфирных связей, а при превышении 5,5 м.д. в случае сложноэфирной связи.
При перацетилировании также получают производные, которые растворимы в хлороформе, бензоле или ацетоне. В эквивалентных пердейтерированных растворителях подвижность молекул такова, что сигналы выявляются более отчетливо и константы взаимодействия могут быть определены с высокой точностью. Анализ ацетилированного сапонина корней люцерны методами COSY и COSY дальнего действия оказался достаточным для идентификации его структуры, Аrа-2Glc-2Аrа-3хедерагенин28-Glс (Massiot et al., 1986).
Структуры дополнительно перацетилированных сапонинов, выделенных из листьев люцерны посевной Medicago sativa (Leguminosae) и из Tridesmostemon claessenssi (Sapotaceae), были определены с помощью аналогичных методов, о которых говорилось выше. Подтверждение положения и последовательности сахаров получили с помощью HMQC (для 1J-сопряжений) и НМВС (для 2J- и 3J-сопряжений) и гомоядерных ретранслированных по Хартманну-Хану (НОНАНА) методов COSY и ROSEY (Massiot et al., 1990, 1991b). Сложноэфирные сахарные цепочки сапонинов Т.claessenssi включают остаток β-D-ксилозы в необычной конфигурации lC4 (все заместители аксиальные). При 600 МГц спектр 1H-ЯМР может быть эффективно разрешен с определением химических сдвигов всех протонов без перацетилирования (Schopke et al., 1991).
4. COSY дальнего действия
Этот метод использовали для картирования протонов сахара в стероидном сапонине, выделенном из лука Allium vineale (Liliaceae) (Chen & Snyder, 1987, 1989). Метод 1H-13C COSY дальнего действия также использовали для анализа структуры агликона в циклоастрогенольном сапонине (Wang et al., 1989b) и для определения положения межсахарного 4J-сопряжения между аномерными протонами во внутреннем остатке глюкозы и Н-2 внутреннего остатка арабинозы в описанном выше сапонине Medicago sativa (Massiot et al., 1986).
5. Фазово-чувствительный COSY с двухквантовой фильтрацией (DQF-COSY, DQ-COSY)
Этот метод был применен для анализа протонов сахара в стероидных сапонинах луков рода Allium (Chen & Snyder, 1987, 1989) и определения протонных химических сдвигов у гликозидов 16α-гидроксипротобассоевой кислоты из корней Crossopteryx febrlfuga (Rubiaceae) (Gariboldi et al., 1990). Такой же метод применяли для полного анализа протонов сахарида в ацетилированном производном хедерагенина из плодов сапинда Sapindus rarak (Sapindaceae) (Hamburger et al., 1991). Для анализа межгликозидных связей использовали NOE-спектроскопию (Hamburger et al., 1992).
6. НОНАНА
Система протонных сопряжений в агликонах гликозидов гипсогенина и квилаховой кислоты была полностью расшифрована с использованием метода НОНАНА. Он аналогичен методу COSY (и близок к методу полнокорреляционной спектроскопии - TOCSY), за исключением того, что наблюдаемые соответствующие друг другу корреляционные пики находятся в одной и той же фазе, что тем самым предотвращает внезапное обнуление перекрывающихся пиков. Для определения углеводных цепей константы вицинального сопряжения, полученные в методе НОНАНА, позволяют оценить относительную стереохимию каждого асимметрического центра, тем самым обеспечивая идентификацию отдельных моносахаридов. Гетероядерный 1H-13C-корреляционный анализ можно использовать для анализа резонанса 13С в сахарных составляющих и сахарных связях, определенных по спектрам НМВС (Frechet et al., 1991).
7. FLOCK, COLOC и NOE
Гетероядерную корреляционную спектроскопию дальнего действия, включающую билинейные спин-развязывающие импульсы (FLOCK), использовали для анализа удаленных сопряжений 1H-13C в алатозиде-А, выделенных из кунжута Sesamum alatum (Pedaliaceae). Так, было установлено взаимодействие протона при С-18 и углеродов С-13, С-17 и С-28. В сочетании с гетероядерным 13C-1H корреляционным анализом дальнего действия (XHCORR) наибольшая информация была получена о структуре нового секоурсенового агликона (Potterat et al., 1992).
Примером применения двумерной корреляционной спектроскопии 13C-1H (COLOC) для анализа дальнодействующих сопряжений (2JCH и 3JCH) является установление структуры сапонинов, выделенных из Crossopteryx febrifuga (Rubiaceae) (Gariboldi et al., 1990).
NOE находит широкое применение в анализе структуры сапонинов, например, при картировании сахаридных протонов и определении последовательности сахаров луперозида-I (Okabe et аl., 1989) и камеллидинов-I и II (Nishino et al., 1986). NOE между Н-2 арабинозы и аномерным протоном рамнозы помог подтвердить наличие дисахаридной связи Rha-2Ara в зизифине (Yoshikawa et al., 1991b). Данный метод находит широкое применение благодаря частому обнаружению связности между протоном агликона и аномерным протоном по сайту связывания. Отрицательный NOE наблюдался между протоном при С-3 и аномерным протоном 3-O-гликозидного остатка циклоастрагенола и других сапонинов (Wang et al., 1989b).



(iii) Спектроскопия и другие методы структурного анализа
Структурный анализ сапонинов и соответствующих агликонов основывается не только на химических методах, но и на спектроскопических и близких методах, например, инфракрасном, ультрафиолетовом, ЯМР, масс-спектроскопическом (МС), рентгенографическом анализах, анализе оптической ротационной дисперсии (ORD) и кругового дихроизма (CD). Современные достижения в этих методах, особенно заметные в спектроскопии ЯМР и масс-спектроскопии, облегчают задачу анализа сапонинов и их соответствующих фрагментов после реакций расщепления, и тем самым полученная информация может быть обобщена и могут быть определены соответствующие структуры. Более того, ЯМР-спектроскопия является недеструктивным методом, а ЯМР и МС позволяют анализировать интактный сапонин.
Необходим интегральный подход к установлению структуры сапонинов с включением различных спектроскопических методов, каждый из которых вносит свой вклад в получаемые данные.
1. Масс-спектроскопия (МС)
Выбор метода ионизации для МС зависит от полярности, доступности и молекулярной массы анализируемого соединения. Принципиальным является то, что т.н. “мягкие” методы ионизации, такие как FAB и десорбционная химическая ионизация (DCI), применяются для получения данных о последовательности сахаров в молекулярной массе природных гликозидов (Wolfender et al., 1992). Это позволяет исследовать гликозиды без их дериватизации. В некоторых случаях происходит фрагментация агликонов, а для этой цели более применимы масс-спектры электронного удара (EI-MS).
2. МС с бомбардировкой быстрыми атомами (FAB-MS)
В FAB-анализе пучок ускоренных атомов (или ионов) выпускается из ионной пушки по направлению к мишени, которая была предварительно загружена в вязкую жидкость (т.н. “матрикс”, обычно глицерин или 1-триглицерин), содержащую анализируемый образец (Barber et al., 1981, 1982). Когда пучок атомов сталкивается с матриксом, его кинетическая энергия передается поверхностным молекулам, значительное число которых разбрызгивается из жидкости в высокий вакуум, становясь источниками ионов. Ионизация многих из этих молекул происходит в процессе разбрызгивания, в результате чего образуются положительные и отрицательные ионы. Любой из них может быть зарегистрирован путем выбора подходящих параметров устройства, хотя, как было показано, отрицательные ионы являются, в целом, более применимыми при работе с сапонинами.
3. Масс-спектрометрия вторичных ионов (SIMS)
Это еще один метод десорбции, вызванной ускоренными частицами: в нем кЭВ-ионы, попадающие на поверхность тонкой пленки биологических молекул, индуцируют такую же десорбционную ионизацию, как и в методе PD-MC (Benninghoven & Sichtermann, 1978). Была подтверждена применимость данного метода в структурном анализе трех новых бидесмозидов, ацетилсапонинов A1, A2 и А3 сои, выделенных из семян сои Glycine max (Leguminosae). Ионные пики основных фрагментов дали информацию о характере ацетилирования моносахаридных звеньев, а также о последовательности расположения указанных звеньев (Kitagawa et al., 1988).
4. Лазерная десорбция (LD)
При LD было показано, что возбуждение короткими лазерными импульсами (<10 нc) дает картину десорбированных молекулярных ионов, сходную с картиной, продуцируемой при PD и SIMS. Масс-спектрометрия с лазерной десорбцией и Фурье-преобразованием (LD/FTMS), метод, который также применим для анализа сложных гликозидов, позволяет получать спектры, которые отличаются от спектров FAB-MC, но комплементарны им.
5. МС метод полевой десорбции (FDMS)
Данный метод применим для определения молекулярных масс сапонинов наряду с определением количества, природы и последовательности сахарных остатков (Komori et al., 1985).
Однако, экспериментальная сложность FDMS и тот факт, что спектры FAB-MC требуют более длительного времени для измерения, определили снижение в последнее время популярности метода FDMS. Масс-спектры FD характеризуются дополнительным недостатком, связанным с их усложнением в присутствии катионизованных фрагментов, что существенно затрудняет интерпретацию результатов. В то же время метод FDMS был с большим успехом применен в структурном анализе сапонинов (Hostettmann, 1980).
(iv) Жидкостная хроматография с маcс-спектрометрией (ЖХ-МС)
В настоящее время разработано несколько типов эффективных интерфейсов (соединяющих устройств) для прямой или непрямой подачи эффлюента с ВЭЖХ-колонки для масс-спектрометрии. Например, качественный анализ фракций неочищенных сапонинов осуществляли путем комбинации полумикро-ВЭЖХ с интерфейсом для бомбардировки быстрыми атомами (FRIT-FAB) (Hattori et al., 1988). Для данного анализа используют NН2-колонку (например, колонку μS-Finepak SIL NH2, Jasco; 25 см × 1,5 мм внутренний диаметр), что лучше С18 колонки с октадецилсиликагелем при отношении деления выходящего потока 1:20 (т.е. 100 мкл/мин → 5 мкл/мин). Элюирование в линейном градиенте ацетонитрила и воды, содержащих 1% глицерина, должно в норме обеспечивать лучшую четкость пиков по сравнению с изократическим элюированием. Масс-спектры отрицательных FAB регистрировали для сапонинов с молекулярной массой до 1235 а.е.м. С применением данного метода выявляли псевдомолекулярные ионы [М-1]-, а также фрагментные ионы, связываемые с расщеплением сахарных составляющих (Hattori et al., 1988).
Также была описана система FRIT-FAB ЖХ-МС, предназначенная для разделения смеси изомеров сапонинов, розамультина и арьюнетина (молекулярные массы обоих составляют 650 а.е.м.), выделенных из шиповника Rоsа rugosa (Rosaceae). И розамультин (гликозид урсана), и арьюнетин (гликозид олеанана) включают единственный остаток глюкозы при С-28: их исследовали в отрицательном и положительном типах FAB с инертным газом ксеноном. ВЭЖХ проводили на колонках с октадецилсиликагелем (250×1,5 мм) со смесью ацетонитрил-вода (7:3, содержащей 0,5% глицерина) в качестве растворителя при скорости потока 1 мл/мин. Были выявлены псевдомолекулярные ионы [М+1]- и [М+1]+, наряду с мощными пиками, обусловливаемыми исходными агликонами в масс-спектрах с отрицательными FAB (Young et al., 1988).
Также возможно детектировать сапонины методом динамической масс-спектроскопии вторичных ионов (SIMS), методом, сходным с динамическим FAB, в котором элюент непосредственно запускают в источник ионов. Так, комбинацию ВЭЖХ с УФ- (206 нм) и S IMS-анализом применяли для анализа смеси одного монодесмозидного и двух бидесмозидных тритерпеновых гликозидов (Marston et al., 1991).
Недостатком применения методов FRIT-FAB и CF-FAB является необходимость обеспечения низкой скорости потока (примерно 1-5 мкл/мин). После ВЭЖХ-разделения необходимо проводить разделение выходящего потока. Однако интерфейс в виде термораспылителя (TSP) (Blackley & Vestal, 1983) достаточно прост и способен работать со скоростями потока 1-2 мл/мин. Это делает данный метод более привлекательным для решения задач, связанных с анализом растительных компонентов. Сущностью метода TSP является мягкая ионизация молекул, сходная с химической ионизацией при МС. Это позволяет проводить анализ нелетучих и термолабильных моно-, ди- и даже тригликозидов. При этом получают информацию о молекулярной массе сапонина, природе и последовательности цепей сахаров. Метод TSP ЖХ-МС применяли в исследовании моллюскоцидных сапонинов, экстрагированных метанолом из плодов Tetrapleura tetraptera (Leguminosae) (Maillard & Hostettmann, 1993). В послеколоночный элюент добавляли 0,5 М ацетат аммония (0,2 мл/мин) для получения летучего буфера для ионизации путем ионного выпаривания: общий поток ионов в TSP ЖХ-МС (массой в диапазоне 450-1000 а.е.м.) хорошо соответствовал тому, что наблюдается при анализах ВЭЖХ-УФ при 206 нм. Следы ионов при m/z 660, 676, 880 и 822 давали сигналы, представляющие псевдомолекулярные ионы [М+Н]+ основных сапонинов. Масс-спектры TSP, полученные для каждого сапонина в данном экстракте, давали основной пик для псевдомолекулярного иона [М+Н]+. Фрагментация углеводных остатков наблюдалась в анализе моллюскоцидного сапонина ариданина, у которого утрата N-ацетилгликозильной составляющей привела к образованию пика [А+Н]+, соответствующего агликону (Maillrad & Hostettmann, 1993).
Метод ЖХ-МС, применяемый для анализа сапонинов, перспективен, поскольку метод ГХ-МС имеет небольшое практическое применение, а в ВЭЖХ, используемой отдельно, идентичность пиков может быть подтверждена только по времени удерживания. Метод ЖХ-МС не только применим для анализа тритерпеновых гликозидов в растительных экстрактах, но также может быть ценен (как метод МС-МС) для структурного анализа отдельных сапонинов в этих экстрактах.
(v) Инфракрасная спектроскопия (ИКС)
Помимо обычного применения ИКС, она имеет одну или две особенности, которые являются важными для структурного анализа сапонинов. ИКС применима для изучения стероидных сапогенинов благодаря тому, что наличие нескольких полос высокой интенсивности между частотами 1350 и 875 см-1 является диагностическим для спирокетальной боковой цепи (Jones et al., 1953). Четыре полосы, 980 (А-полоса), 920 (В-полоса), 900 (С-полоса) и 860 см-1 (D-полоса), были определены как характеристические полосы для колец Е и F. В случае с 25R-сапогенинами В-полоса характеризуется более сильным поглощением, чем С-полоса, в то время как в 25R-сериях это соотношение обратное. У сапогенинов, имеющих заместители у кислорода в кольцах Е и F или по положению 27, параметры этих полос в существенной степени изменены (Takeda, 1972).
Присутствие ионизованных карбоксильных групп в сапонинах можно выявить по полосам в ИК-спектре при 1610 и 1390 см-1 (Numata etc al., 1987). Такая информация применима в процессе выделения, когда важным является выяснение того, ионизованы ли карбоксильные группы в этой молекуле.
(vi) Рентгеновская кристаллография
Рентгеновская кристаллография была использована для изучения молекулярной геометрии трисахаридного тритерпенового азиатикозида, выделенного из Centella asiatica (Umbrelliferae). Кристаллизацию проводили из диоксана (Mahato et. al., 1987). Анализ рентгеновской дифракции также оказался успешным с точки зрения подтверждения структуры 3-β-D-глюкозида моллевой кислоты (Pegel & Rogers, 1985).
Рентгеновская кристаллография особенно эффективна при изучении структуры агликонов. Ценную информацию для установления структуры агликона алатозида-А, выделенного из Sesamum alatum (Pedaliaceae), получили методом дифракции рентгеновских лучей кристаллического триацетата артефакта, образующегося после кислотного гидролиза (Potterat et al., 1992). Рентгенокристаллографический анализ исходного агликона 3-O-гликозида медикагеновой кислоты, выделенного из клубней Dolichos killmandscharicus (Leguminosae), показал, что эта молекула имела цис-конденсированные кольца D и Е. Кольцо С характеризовалось слабо нарушенной конформацией “софа”, в то время как кольца А, В, D и Е имели конформацию “кресло” (Stoeckti-Evans, 1989).
(vii) Реакции расщепления
Тритерпеновые сапонины являются гликозидами, в которых гидроксиполуацетальные группы сахаридов в их циклической пиранозной или фуранозной формах формируют ацетали с тритерпеновым или стероидным остатком. Эфирная связь между гидроксиполуацеталем и тритерпеном или стероидом известна как гликозидная связь. Моносахаридные компоненты олигосахаридов также связаны эфирными связями (межгликозидные связи).
По завершении гидролиза гликозида, гликозидная связь расщепляется с высвобождением составляющих моносахаридов и неуглеводной составляющей (агликона или генина). Неуглеводная часть после гидролиза сапонинов обозначается как “сапогенол” или “сапогенин”. Все известные сапонины являются O-гликозидами с эфирными или сложноэфирными связями.
Многочисленные химические реакции и методы были использованы для разделения сапонинов на меньшие компоненты, необходимые для проведения более точного анализа (см., например, Kitagawa, 1981). Такие методы будут находить конкретное применение в структурном анализе тритерпеновых сапонинов.
1. Кислотный гидролиз
Кислотный гидролиз может быть осуществлен путем нагревания с обратным холодильником сапонина в кислоте в течение определенного периода времени, например, в течение 4 часов в 2-4 М соляной кислоте. Остающийся после гидролиза водный раствор экстрагируют диэтиловым эфиром, хлороформом или этилацетатом с получением агликона. Экстракцию сахаров из водного слоя проводят пиридином после нейтрализации раствора (щелочью или основной ионообменной смолой) (Tschesche & Forstmann, 1957; Sanberg & Michel, 1962) и упаривания досуха. С использованием данного метода сапонины полностью расщепляются на компоненты, что дает информацию, позволяющую идентифицировать агликон, а также определить количество и природу присутствующих моносахаридов. Если кислотному гидролизу подвергнут просапогенин (полученный после расщепления сложноэфирной связи при щелочном гидролизе), то можно установить природу сахарных цепей, которые присоединены к агликону эфирными связями. Водная среда реакции может быть заменена спиртом или диоксаном.
Помимо соляной кислоты, для проведения гидролиза сапонинов также можно использовать серную кислоту. При использовании серной кислоты меньше шансов разрушить или перестроить данную молекулу, однако расщепление эфирных связей оказывается не столь эффективным. Стандартный метод получения гипсогеновой кислоты из сапонинов гвоздик рода Dianthus, например, включает гидролиз 1 М серной кислоты в диоксане (Oshima et al., 1984). Сравнительный анализ гидролитических условий для соляной кислоты и серной кислоты в воде и смеси вода - этанол показал, что наилучшее выделение сахаридов достигается при нагревании сапонина в течение 2 часов в растворе 5% серная кислота - вода в запаянной в вакууме ампуле (Kikuchi et al., 1987). В некоторой степени более мягкий гидролиз может быть проведен с использованием трифторуксусной кислоты, например, при нагревании с обратным холодильником в течение 3 часов в 1 М трифторуксусной кислоте.
Альтернативой гидролизу сапонинов в растворе является их гидролиз непосредственно на пластине для ТСХ путем обработки парами соляной кислоты. После упаривания кислоты проводят стандартное элюирование ТСХ-растворителем с целью идентификации имеющихся моносахаридов (Kartnig & Wegschaider, 1971; Не, 1987). В этом методе, концевые сахара, ксилозу и галактозу идентифицировали после частичного гидролиза агавезида-В. ТСХ-пластины проявляли смесью хлороформ-метанол-вода (8:5:1) и для детекции использовали смесь анилин-дифениламин-Н3РO4-метанол (1:1:5:48) (Uniyal et al., 1990).
2. Основный гидролиз
Расщепление О-ацилгликозидных сахарных цепей проводят в условиях основного гидролиза, обычно путем нагревания с обратным холодильником в 0,5 М гидроксиде калия. Альтернативно, можно использовать растворы 1-20% гидроксида калия в этаноле или метаноле, хотя при этом существует риск метилирования, особенно по карбоксильной группе тритерпеновых кислот. Ионообменные смолы, такие как Dowex-I, обеспечивают мягкие условия основного гидролиза (Bukharov & Karlin, 1970). Другой метод предусматривает использование иодида лития в коллидине (Kochetkov et al., 1964).
Путем тщательного контроля условий реакции возможно избирательное расщепление различных сложноэфирных составляющих. Например, гидролиз сапонина кизута К11 при нагревании с обратным холодильником в 0,5 М гидроксиде калия в течение получаса приводил к удалению сахара при С-28 у бидесмозида. Однако, перемешивание этого сапонина в течение 20 часов в 0,1 М гидроксиде калия при комнатной температуре избирательно удаляет ацетатную группу при С-28 сложноэфирной гликозидной цепи (Kizu et al., 1985b).
3. Частичный гидролиз
В некоторых случаях, когда сапонины характеризуются значительно разветвленными или длинными сахарными цепями, необходимой является процедура частичного гидролиза, имеющая целью получение фрагментов, более пригодных для структурного анализа. Этого можно достичь с использованием кислоты или, при необходимости, ферментов. Олигосахарид и/или остальные части сапонина выделяют и затем их характеризуют.
Например, сапонин, выделенный из Phytolacca dodecandra (Phytolaccaceae), гидролизовали 0,1 М соляной кислотой в течение 45 минут с получением смеси трех продуктов. Эти соединения разделяли методом ОФ-ЖХНД и последовательности их сахаров определяли методом МС, 13C-ЯМР и ГХ-МС алдитолацетатов. Обобщение всей полученной информации позволило установить химическую формулу данного соединения, как производного олеаноловой кислоты (Dorsaz & Hostettmann, 1986).
Гидролиз в диоксане обеспечивает более мягкие условия, и поэтому возможен частичный гидролиз. В этом примере сапонин нагревали с обратным холодильником в течение 6 часов в диоксане с 0,1 М соляной кислотой (1:3) (Ikram et al., 1981). Другим методом частичного гидролиза сапонинов является обработка раствора тритерпенового гликозида в спирте щелочным металлом (натрием или калием), а затем добавлением следовых количеств воды (Ogihara & Nose, 1986).
4. Гидротермолиз
Гидротермолиз тритерпеновых гликозидов приводит к образованию соответствующих агликонов и тем самым способствует облегчению определения структуры. Этот метод включает нагревание гликозида с водой или со смесью воды и диоксана при 100-140°С в течение 10-140 часов в зависимости от конкретного образца. Например, Гидротермолиз тритерпеновых 3,28-O-бисгликозидов приводит к образованию соответствующих 3-O-гликозидов (Kim еt аl., 1992).
5. Ферментативный гидролиз
Очень эффективным и мягким методом отщепления сахарных остатков от сапонинов без образования артефактов является ферментативный гидролиз. Хотя соответствующие гидролазы для всех сахаров не являются коммерчески доступными, отщепление остатков β-глюкозы с помощью β-глюкозидазы является в настоящее время перспективным. Дополнительное преимущество расщепления с помощью конкретных ферментов связано с тем, что аномерная конфигурация сахарной составляющей подтверждается автоматически. Некоторые ферменты, которые, в частности, применимы для использования в гидролизе тритерпеновых гликозидов, представляют собой гидролазы β-галактозидазы, целлюлазу, неочищенную хесперидиназу, пектиназу и нарингиназу.
Систематизированный анализ неочищенных препаратов хесперидиназы, нарингиназы, пектиназы, целлюлазы, амилазы и эмульсина показал, что хесперидиназа, нарингиназа и пектиназа являются наиболее эффективными в гидролизе гинзенозидов (Kohda & Tanaka, 1975).
(viii) Анализ агликонов после гидролиза
По завершении гидролиза, агликоны могут быть отделены от гидролизата либо путем простой фильтрации, либо путем рапределения в смеси вода - органический растворитель и проанализированы на известные тритерпены. Наиболее известным методом является ТСХ с использованием такого растворителя, как смесь диизопропиловый эфир - ацетон (75:30). Напыляющими реагентами служат те реагенты, которые обычно используются для анализа сапонинов (см. табл.2).
Для газо-жидкостной хроматографии необходима дериватизация тритерпенов. Например, метиловые сложные эфиры олеаноловой и урсоловой кислот были разделены с помощью ГХ на стеклянной колонке, упакованной 30% OV-17 или SE-30 (Fokina, 1979). Тритерпены могут быть определены методом ГХ после дериватизации с использованием N,O-бис(триметилсилил)ацетамида и хлортриметилсилана, как, например, в случае с сапогенинами А-Е сои или медикагеновой кислоты люцерны (Jurzysta & Jurzysta, 1978).
Метод ГХ-МС также имеет ценность с точки зрения характеристики сапогенинов. Обычно получают триметилсилильные производные, а затем их анализируют в спектрометре. Примером является применение этого метода для изучения тритерпенов олеананового и урсанового ряда. Девять силилированных тритерпенов разделяли методом ГХ на колонке с OV-101 и анализировали параметры их масс-спектров: те из них, которые включали 12-еновую двойную связь, проходили типичную ретрореакцию Дильса-Альдера (Burnouf-Radosevich et al., 1985). Этот метод также использовали для определения тритерпенов, выделенных из солодки (Bombardelli et al., 1979).
ВЭЖХ-анализ не требует дериватизации и характеризуется высокой воспроизводимостью результатов и чувствительностью в анализе тритерпенов. Может быть использована ВЭЖХ с обычной фазой (анализ сапогенинов квинои Burnouf-Radosevich & Delfel, 1984) и с обращенной фазой (Lin et al., 1981), однако недостаток метода ОФ-ВЭЖХ связан с тем, что соединения имеют тенденцию осаждаться в подвижных водных фазах.
(ix) Анализ сахаров после гидролиза
Анализ моносахаридов может быть осуществлен методом ТСХ, например, на силикагелевых пластинах с такими растворителями, как смесь этилацетат-метанол-вода-уксусная кислота (65:25:15:20) и н-бутанол-этилацетат-изопропанол-уксусная кислота-вода (35:100:60:35:30) (Shiraiwa et al., 1991). Детекцию обычно проводят с фталатом п-анизидина, нафторезорцином, тимолсерной кислотой (Kartnig & Wegschaider, 1971) или хлоридом трифенилтетразолия (Wallenfels, 1950; Kamel et al., 1991). Альтернативно, количественный анализ моносахаридов можно проводить методами ГХ или ВЭЖХ.
Ряд методов ВЭЖХ был описан в связи с анализом сахаров, включая: анализ на NH2-связанных колонках с элюированием смесью ацетонитрил-вода (75:25) (Glombitza & Kurth, 1987); анализ на колонках С-18 (ацетонитрил-вода, 4:1) с определением индекса преломления, необходимого для целей количественного анализа, путем сравнения интегрированных ВЭЖХ-пиков со стандартами (Adinolfi et al., 1987); анализ на ион-эксклюзионной колонке Aminex HPX-87H (BioRad) с элюированием раствором 0,005 М серной кислоты (0,4 мл/мин) (Adinolfi et al., 1990); и анализ п-бромбензоатов сахарного ряда (образующихся при метанолизе сапонина смесью 5%-ная соляная кислота-метанол и последующем п-бромбензоилировании метилированных сахаров) методом ВЭЖХ и идентификация путем сравнения с аутентичными производными (Kawai et al., 1988; Sakamoto et al., 1992).
В методе ГХ используются персилилированные сахара (Wulff, 1965), или же проводят ГХ-МС алдитолацетатных производных. Альтернативной процедурой является анализ соответствующим образом дериватизованных моносахаридов комбинированным методом ГХ-Фурье-преобразованый ИКС (FTIR) (Chen & Snyder, 1989).
Наиболее часто обнаруживаемыми сахарами являются D-глюкоза, D-галактоза, L-арабиноза, D-ксилоза, D-фукоза, L-рамноза, D-хиновоза, D-глюкуроновая кислота и D-рибоза.
IV. Производные соединений по настоящему изобретению
Как подробно описано в данной заявке, считается, что ряда преимуществ можно достичь путем манипуляции с тритерпеновыми гликозидами с приданием им новых свойств, более продолжительного времени полураспада in vivo или других предпочтительных свойств. Такими методами являются, но не ограничиваясь ими, манипуляция или модифицирование смесей тритерпеновых гликозидов или их отдельных тритерпеновых молекул, модифицирование или удаление сахаров и конъюгирование тритерпеновых соединений с инертными носителями, такими как различные белки или небелковые компоненты, включая иммуноглобулины или Fc-фрагменты. Следует отметить, что большая продолжительность полураспада не соответствует продолжительности “медленного высвобождения” фармацевтических композиций. Препараты медленного высвобождения обычно создают таким образом, чтобы они обеспечивали доставку постоянного уровня лекарственного средства в течение более длительного периода времени. Увеличение времени полураспада лекарственного средства, такого как тритерпеновый гликозид по настоящему изобретению, дает более высокие плазматические уровни после его введения, причем, эти уровни поддерживаются на протяжении длительного времени, но при этом характер снижения этих уровней зависит от фармакокинетических параметров данного соединения.
(i) Конъюгаты тритерпенов и связанных молекул
Как было описано выше, тритерпеновые соединения по настоящему изобретению, идентифицированные здесь, могут быть соединены с конкретными молекулами с целью повышения эффективности тритерпеновых гликозидов при лечении пациентов, страдающих от любого заболевания, которое может быть вылечено с использованием соединений по настоящему изобретению.
Иллюстративные варианты таких молекул включают контролирующие агенты и агенты, которые должны увеличивать время полураспада тритерпеновых соединений in vivo. Тритерпеновые соединения могут быть соединены с такими вторичными молекулами любым функциональным путем, для того, чтобы он обеспечил каждому сегменту выполнение присущих ему функций без существенного нарушения биологической активности, например, противоопухолевой активности заявляемых здесь соединений.
Тритерпеновые композиции по настоящему изобретению могут быть непосредственно соединены со вторым соединением или они могут быть соединены через линкерную группу. Термин “линкер (линкерная группа)” обозначает одну или несколько бифункциональных молекул, которые могут быть использованы для ковалентного присоединения тритерпеновых соединений или смесей тритерпенов к агенту и которые не будут нарушать биологическую активность указанных тритерпеновых соединений. Линкер может быть присоединен к любой части тритерпена, при условии, что участок присоединения не нарушает биологическую активность, например, противоопухолевую активность соединений по настоящему изобретению.
Характерный вариант для присоединения через линкер тритерпеновых соединений по настоящему изобретению ко второму агенту представляет получение активного сложного эфира тритерпена с последующей реакцией указанного сложного эфира с нуклеофильной функциональной группой того агента, присоединение к которому проводится. Активные сложные эфиры могут быть получены, например, посредством реакции карбоксильной группы тритерпена со спиртом в присутствии дегидратирующего агента, такого как дициклогексилкарбодиимид (DCC), 1-(3-диметиламинопропил)-3-этилкарбодиимид (EDC) и 1-(3-диметиламинопропил)-3-этилкарбодиимидметиодид (EDCI). Использование EDC для получения конъюгатов описано в патенте США №4526714, в патентной заявке РСТ WO 91/01750 и у Arnon et al., 1989: эти источники в полном их объеме приведены здесь для сведения в виде библиографических ссылок. Агент для присоединения к тритерпену, например, специфичное для опухоли антитело, затем смешивают с активированным сложным эфиром в водном растворе с получением данного конъюгата.
Когда линкер между тритерпеном и агентом является желательным, активный сложный эфир тритерпенового гликозида может быть получен в соответствии с описанным выше и подвергнут реакции с линкерной группой, например, 2-аминоэтанолом, алкилендиамином, аминокислотой, такой как глицин, или защищенной по карбоксильной группе аминокислотой, такой как трет-бутиловый сложный эфир глицина. Если в составе линкера имеется защищенная карбоксильная группа, то защитную группу удаляют и получают активный сложный эфир линкера (в соответствии с описанным выше). Активный сложный эфир затем подвергают реакции со второй молекулой с получением конъюгата. Альтернативно, второй агент может быть дериватизирован янтарным ангидридом с получением конъюгата “агент-сукцинат”, который может быть конденсирован в присутствии EDC или EDCI с тритерпен-линкерным производным, в составе которого линкер имеет свободную амино- или гидроксильную группу (см., например, патентную заявку РСТ WO 91/01750, полное описание которой приведено здесь в качестве ссылки).
Также можно получить конъюгат тритерпенового гликозида, включающий линкер со свободной аминогруппой, и перекрестно сшить его со свободной аминогруппой гетеробифункционального кросслинкера, такого как сульфосукцинимидил-4-(N-малеимидоциклогексан)-1-карбоксилат, который будет реагировать со свободными сульфгидрильными группами белковых антигенов.
Тритерпеновый гликозид также можно присоединить к линкерной группе реакцией альдегидной группы с аминолинкером с образованием промежуточного иминного конъюгата с последующим восстановлением борогидридом натрия или цианоборогидридом натрия. Примеры таких линкеров включают аминоспирты, такие как 2-аминоэтанол, и диамино-соединения, такие как этилендиамин, 1,2-пропилендиамин, 1,5-пентандиамин, 1,6-гександиамин и подобные. Тритерпеновый гликозид затем может быть соединен с линкером с получением сначала сукцинированного производного с использованием янтарного ангидрида с последующей конденсацией конъюгата тритерпенового гликозида-линкера с DCC, EDC или EDCI.
Кроме того, тритерпеновый гликозид или агликон могут быть окислены перйодатом, а полученный из него диальдегид можно конденсировать с перечисленными выше аминоспиртами или диаминосоединениями. Свободная гидроксильная или аминогруппа на линкере может быть конденсирована с сукцинатным производным антигена в присутствии DCC, EDC или EDCI. В данной области техники известно большое число типов линкеров, которые можно использовать для получения тритерпеновых конъюгатов. Перечень характерных линкеров для использования в настоящем изобретении приведен ниже в табл.4.
Гетеробифункциональные кросс-линкеры


(ii) Рационализация создания лекарственных средств
Целью способов рационального создания лекарственных средств является получение структурных аналогов биологически активных соединений. Создавая такие аналоги, можно получать лекарства, которые более активны или стабильны по сравнению с природными молекулами, характеризующимися различной чувствительностью к изменениям или способностью нарушать функции различных других молекул. В одном из подходов можно определить пространственную структуру тритерпеновых соединений по настоящему изобретению или их фрагментов. Для этого можно использовать данные рентгеновской кристаллографии, компьютерное моделирование или сочетание обоих этих методов. Другим методом является рандомизированное замещение функциональных групп в молекуле тритерпена и полученное в результате воздействие на функциональную группу.
Также возможно выделение антитела, специфичного в отношении тритерпенового соединения и выбранного методом функционального анализа с последующим определением его кристаллической структуры. В принципе этот метод позволяет получить сердцевину фармацевтического препарата, на основе которого можно создать новое лекарство. Можно вполне избежать кристаллографического анализа белка путем создания антиидиотипических антител, специфичных в отношении функционального фармакологически активного антитела. Используя ситуацию “двойного зеркального отражения”, сайты связывания антиидиотипического антитела, как можно ожидать, будут аналогами сайтов исходного антигена. Антиидиотипическое антитело затем можно использовать для идентификации и выделения пептидов из банков пептидов, полученных химическим или биологическим путем. Отобранные пептиды затем могут служить в качестве сердцевины фармакологического препарата. Антиидиотипические антитела могут быть получены методами, описанными здесь для получения антител, с использованием антитела в качестве антигена.
Таким образом, можно создавать лекарственные средства, обладающие улучшенной биологической активностью, например, противоопухолевой активностью, по сравнению с исходным тритерпеновым соединением. С помощью процедур химического выделения и приводимых здесь описаний, значительные количества тритерпеновых соединений по настоящему изобретению могут быть получены с целью проведения кристаллографического анализа. Кроме того, установление химических свойств этих соединений позволит проводить компьютерное прогнозирование структурно-функциональных взаимосвязей.
V. Лечение раковых опухолей с использованием тритерпеновых соединений по настоящему изобретению
В процессе развития раковой опухоли, клетки млекопитающего претерпевают ряд генетически детерминированных изменений, которые приводят к их аномальной пролиферации. Это может происходить, обычно, в несколько следующих стадий: (1) инициация: внешний агент или стимул индуцирует генетическое изменение в одной или нескольких клетках; и (2) активация: подключение других генетических и метаболических механизмов, одним из которых может быть воспаление. На стадии “активации” в клетке начинаются метаболические преобразования с переходом в стадию роста, в которой блокируется апоптоз.
Раковые клетки характеризуются утратой апоптотического контроля в дополнение к потере контроля за регуляторными этапами клеточного цикла. Раковые клетки (злокачественные клетки) лишены регуляторных механизмов нормального роста вследствие ряда метаболических нарушений, происходящих на стадиях инициации и активации на раннем этапе малигнизации. Эти нарушения являются следствиями генетических изменений в клетке. Данные генетические изменения могут включать: (i) активирующие мутации и/или усиление экспрессии протоонкогенов; и/или (ii) инактивирующие мутации и/или ослабление экспрессии одного или нескольких опухоль-супрессорных генов. Большинство продуктов, кодируемых онкогенами и опухоль-супрессорными генами, являются компонентами путей передачи сигнала, которые контролируют начало или завершение клеточного цикла, активацию дифференцировки, повреждение смысловой ДНК и инициацию механизмов репарации ДНК, и/или регуляцию программированной клеточной гибели. Почти все опухоли имеют мутации во множестве онкогенов и опухоль-супрессорных генов. Можно заключить, что в клетках работают множественные “параллельные” механизмы контроля клеточного роста, дифференцировки, репарации ДНК и апоптоза.
Тритерпеновые соединения по настоящему изобретению могут быть введены нуждающемуся в этом пациенту в целях профилактики ракового заболевания или лечения после диагностирования такого заболевания. Для подавления инициации и активации рака, для уничтожения раковых/злокачественных клеток, для подавления клеточного роста, для индукции апоптоза, для подавления метастазирования, для снижения размера опухоли и для других путей реверсии или редукции проявлений опухолевыми клетками злокачественного фенотипа, с использованием способов и композиций по настоящему изобретению можно обеспечить контакт клетки-“мишени” с описанными здесь тритерпеновыми соединениями. Этого можно достичь путем контакта опухоли или опухолевой клетки с отдельным соединением или фармацевтической композицией, которая содержит тритерпеновые соединения по настоящему изобретению, или путем контакта опухоли или опухолевой клетки более чем с одним соединением или фармацевтическими композициями одновременно, где одна композиция содержит тритерпен по настоящему изобретению, а другая содержит второй агент.
Предпочтительными с точки зрения воздействия по настоящему изобретению раковыми клетками являются клетки эпителиальных карцином, таких как опухолевые клетки кожи, толстой кишки, матки, яичников, поджелудочной железы, легких, мочевого пузыря, молочной железы, почек и предстательной железы. Другими раковыми клетками-мишенями являются клетки опухолей мозга, печени, желудка, пищевода, головы и шеи, яичек, шейки матки, лимфатической системы, гортани, пищевода, околоушных желез, желчных протоков, прямой кишки, матки, эндометрия, почек, мочевого пузыря и щитовидной железы; включая плоскоклеточные карциномы (эпидермиса), аденокарциномы, мелкоклеточные карциномы, глиомы, нейробластомы и т.п. Однако, данный перечень имеет исключительно иллюстративный, но не ограничивающий характер, поскольку потенциально любая опухолевая клетка может быть подвергнута лечению с использованием тритерпеновых соединений по настоящему изобретению. Методы тестирования относительной эффективности соединений по настоящему изобретению с точки зрения воздействия на перечисленные выше опухолевые клетки и другие опухолевые клетки конкретно представлены в данном описании и будут ясны специалистам в данной области техники в свете изложения настоящей заявки.
Соединения по настоящему изобретению предпочтительно вводят в виде нутрицевтической композиции или фармацевтической композиции, содержащей фармацевтически или фармакологически приемлемый растворитель или носитель. Природа носителя зависит от химических свойств используемого(ых) соединения(и), включая параметры растворимости, и/или от способа введения. Например, если желательным является пероральное введение, то может быть выбран твердый носитель, тогда как для внутривенного введения может быть использован жидкий солевой раствор.
Выражение “фармацевтически или фармакологически приемлемый” обозначает молекулярные компоненты и композиции, которые не вызывают негативных, аллергических или иных нежелательных реакций при введении их животному или человеку. Используемыми в данном тексте “фармацевтически приемлемыми носителями” являются любые и все растворители, диспергирующие среды, покрытия, противобактериальные и противогрибковые агенты, изотонические и замедляющие абсорбцию агенты и т.п. Использование таких сред и агентов для фармацевтически активных веществ хорошо известно в данной области техники. Они могут быть использованы в составе терапевтических композиций, за исключением случаев несовместимости любого из этих стандартных сред или агентов с данным активным компонентом. В состав таких композиций могут быть также включены дополнительные активные компоненты.
а. Нутрицевтические композиции
Нутрицевтические композиции являются препаратами природных ингредиентов, которые представляют собой многокомпонентные системы, содержащие предпочтительно синергистически действующие природные продукты и добавки, способствующие оздоровлению. Нутрицевтические композиции могут быть приготовлены из лекарственных растений. Информацию о многочисленных растениях и травах, применяемых для приготовления нутрицевтических композиций, можно получить из научных публикаций, включая "German Commission E Monographs", "Botanical Safety Handbook" и "HerbalGram", ежеквартальное издание Американского Ботанического совета, в котором описываются результаты многочисленных клинических испытаний нутрицевтических композиций.
Информация об описаниях и компонентах, современном применении, дозировках (в зависимости от форм), действии, противопоказаниях, побочных эффектах, взаимодействиях со стандартными лекарствами, путях введения, длительности применения, официальном статусе, рейтинге ботанической безопасности Агентства АНРА США и комментариях доступна для различных растений, включая, помимо прочего, чернику, жостер, питеколобиум, кошачий коготь, стручковый перец, клюкву, лютик полевой, китайский дягель (dong quai) Angelica sinensis, зубровку, масло ослинника, гваюлу, чеснок, имбирь, гингко, женьшень китайский, женьшень сибирский, желтокорень (“золотая печать”), колу, семена винограда, зеленый чай, боярышник, торрейю, солодку, чертополох морской, пальма сереноа, зверобой продырявленный и валериану.
Действие таких нутрицевтических композиций может быть быстрым и/или кратковременным, или может способствовать улучшению здоровья на длительное время. Настоящее изобретение сфокусировано на фитопрепаратах, полученных из акации Виктории Acacia victoriaе. Изобретение относится к нутрицевтическим композициям, содержащим высушенные и измельченные корни и бобы Acacia victoriae или экстракты этих тканей в фармакологически приемлемой среде, предназначенные в качестве природных средств, помимо прочего, для профилактики и лечения раковых заболеваний. Нутрицевтическая композиция может быть использована для предотвращения инициации и активации канцерогенеза, а также для индукции апоптоза злокачественных раковых клеток. Заявляемые здесь нутрицевтические композиции также могут быть использованы в качестве противовоспалительных, антифунгицидных, противовирусных, антимутагенных, спермицидных или контрацептивных, сердечно-сосудистых и контролирующих метаболизм холестерина средств. Нутрицевтические композиции могут содержаться в такой среде, как буфер, растворитель, разбавитель, инертный носитель, масло, крем или годный в пищу материал.
Нутрицевтическая композиция может быть введена перорально и может быть приготовлена в форме таблетки или капсулы. Пероральное введение может быть предпочтительным для лечения рака толстой кишки и других опухолей внутренних органов.
Альтернативно, нутрицевтическая композиция может быть приготовлена в форме мази, содержащей экстракты корней или бобов Acacia victoriae в масле или креме, предназначенной для местного нанесения на кожу. Такая форма нутрицевтической композиции может быть использована для предотвращения инициации раковых опухолей кожи. Применение данных нутрицевтических композиций предоставляет способ подавления инициации и активации эпителиальных клеток млекопитающего в предзлокачественное или злокачественное состояние путем введения в данную клетку млекопитающего терапевтически эффективного количества нутрицевтической композиции. Это особенно применимо в случаях рака эпителиальных клеток, такого как рак кожи.
b. Фармацевтические композиции
Настоящее изобретение, кроме того, относится к выделенным композициям Acacia victoriae, которые частично или полностью очищены и охарактеризованы по их структуре. Очистка и анализ этих тритерпеновых гликозидных соединений подробно описаны в примерах. D1, G1 и В1 представляют собой три композиции, которые были полностью очищены и их структура была почти полностью охарактеризована (фиг.39, 40 и 41). В биологических тестах на активность этих соединений в отношении линий раковых клеток было продемонстрировано подавление клеточного роста и индукция апоптоза злокачественных клеток (фиг.43, 44А-Е). Более того, частично очищенные композиции этих сапонинов, выделенных из Acacia victoriae, также проявляют хемопротективный эффект у мышей, которые были подвергнуты воздействию канцерогеном DMBA (фиг.8, 9, 11, 12 и 13). Таким образом, данные композиции обладают противораковой активностью и действует ряд механизмов, индуцирующих апоптоз раковых клеток. Фармацевтические композиции на основе этих соединений представляются как мощные химиотерапевтические средства, которые могут быть применены сами по себе или в сочетании с другими формами противораковой терапии, такой как химиотерапия, радиотерапия, хирургическое вмешательство, генная терапия и иммунотерапия. Эта комплексная терапия подробно описана далее. Специалист в данной области техники сможет определить эффективные дозы и режимы комплексной терапии.
с. Способы введения
(i) Парентеральное введение
В одном из своих вариантов, настоящее изобретение относится к препаратам для парентерального введения тритерпеновых композиций, например, приготовленных для инъекций внутривенным, внутримышечным, подкожным или иным подобным путем, включая прямую доставку в опухоль или в пораженную область. Исходя из настоящего описания, специалист в данной области может приготовить водные композиции, которые содержат тритерпеновое соединение. Обычно такие композиции могут быть получены в виде жидких растворов или суспензий для инъекций; также могут быть получены твердые формы, пригодные для получения растворов или суспензий путем добавления жидкости перед инъекцией; и эти препараты также могут быть эмульгированы.
Растворы активных соединений в виде свободного основания или фармакологически приемлемых солей могут быть приготовлены в воде, соответствующим образом смешанной с поверхностно-активным веществом, таким как гидроксипропилцеллюлоза. Дисперсии могут быть также приготовлены в глицерине, жидких полиэтиленгликолях и в их смесях, а также в масле. В стандартных условиях хранения и применения, данные препараты содержат консервант, необходимый для предотвращения роста микроорганизмов.
Фармацевтические формы, пригодные для инъекций, включают стерильные водные растворы или дисперсии; препараты, содержащие кунжутное масло, арахисовое масло или водный пропиленгликоль; и стерильные порошки в целях приготовления инъецируемых растворов или дисперсий для немедленного приема. Во всех случаях лекарственная форма должна быть стерильной и должна быть настолько жидкой, чтобы легко проходить через шприц. Она должна быть стабильной в условиях промышленного производства и хранения и должна быть защищена от загрязняющего действия микроорганизмов, таких как бактерии и грибы.
Тритерпеновые соединения могут быть включены в состав композиции в нейтральной форме или в виде солей. Фармацевтически приемлемыми солями являются аддитивные соли кислоты (образуемые свободными аминогруппами белка), образуемые неорганическими кислотами, такими как, например, соляная или фосфорная кислоты, или органическими кислотами, такими как уксусная, щавелевая, винная, миндальная кислоты и подобными. Соли, образованные свободными карбоксильными группами, также могут быть образованы неорганическими основаниями, такими как, например, гидроксиды натрия, калия, аммония, кальция или трехвалентного железа, и органическими основаниями, такими как изопропиламин, триметиламин, гистидин, прокаин и подобными.
Носитель также может быть растворителем или дисперсионной средой, содержащей, например, воду, этанол, полиол (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль и т.п.), их подходящие смеси и растительные масла. Достаточная текучесть может обеспечиваться, например, путем использования покрытий, таких как лецитин, путем поддерживания требуемого размера частиц в случае суспендирования, и путем использования поверхностно-активных веществ. Для предотвращения действия микроорганизмов можно использовать различные противобактериальные и противогрибковые агенты, например, парабены, хлорбутанол, фенол, сорбиновую кислоту, тимерозаль и подобные.
Часто предпочтительным является включение изотонических агентов, например, сахаров или хлорида натрия. Пролонгированная абсорбция инъецируемых композиций может быть достигнута использованием в этих композициях агентов, замедляющих поглощение, например, моностеарата алюминия и желатина.
Стерильные растворы для инъекций приготавливают путем внесения активных соединений в необходимом количестве в подходящем растворителе с различными другими ингредиентами, перечисленными выше, если они необходимы, с последующим стерильным фильтрованием. В целом, дисперсии приготавливают внесением различных стерилизованных активных ингредиентов в стерильный носитель, который содержит основную дисперсионную среду и необходимые другие ингредиенты из тех, которые перечислены выше. В случае стерильных порошков, предназначенных для приготовления стерильных растворов для инъекций, предпочтительными методами их получения являются сушка в вакууме и сушка вымораживанием, дающие активный компонент в виде порошка плюс дополнительные желательные компоненты из предварительно стерильно отфильтрованного их раствора.
(ii) Другие способы введения
Другие способы введения также найдут применение в связи с настоящим изобретением. Например, тритерпеновые соединения по настоящему изобретению могут быть приготовлены в виде суппозиториев, а в некоторых случаях в виде аэрозолей и интраназальных композиций. Для суппозиториев, наполнитель композиции должен включать традиционные связывающие вещества и носители, такие как полиалкиленгликоли или триглицериды. Такие суппозитории могут быть приготовлены из смесей, содержащих активный компонент в количестве от примерно 0,5% до примерно 10% (по массе), предпочтительно от примерно 1% до примерно 2%.
Композиции для перорального введения могут быть приготовлены в форме растворов, суспензий, таблеток, пилюль, капсул, препаратов пролонгированного действия или порошков. Такие композиции могут быть введены, например, путем проглатывания или вдыхания. Если фармацевтическая композиция предназначена для ингаляции, то она предпочтительно должна быть аэрозольной. Примеры процедур для приготовления водных аэрозолей для использования в соответствии с настоящим изобретением можно найти в патенте США №5049388, полное содержание которого вводится в настоящее описание посредством ссылки. Приготовление сухих аэрозольных препаратов описано, например, в патенте США №5607915, полное содержание которого вводится в настоящее описание посредством ссылки.
Также применимо введение соединений по настоящему изобретению непосредственно в виде трансдермальных препаратов, содержащих усилители проницаемости, такие как ДМСО. Эти композиции также могут содержать любые другие подходящие носители, наполнители или разбавители. Для лечения некоторых симптомов заболевания могут быть введены другие препараты для местного применения. Например, могут быть приготовлены интраназальные препараты, которые содержат наполнители, не раздражающие слизистую носовой полости и, в основном, не нарушающие циллиарной функции. В связи с настоящим изобретением могут быть использованы разбавители, такие как вода, водно-солевой раствор или другие известные вещества. Препараты для введения в нос также могут содержать консерванты, такие как, не ограничиваясь ими, хлорбутанол и бензалконийхлорид. Может присутствовать поверхностно-активное вещество, усиливающее поглощение данных соединений слизистой носа.
(iii) Приготовление и введение
После приготовления растворы должны быть введены путем, совместимым с готовой лекарственной формой и в таком количестве, которое является терапевтически эффективным. Выбранный способ приготовления может быть осуществлен с использованием различных наполнителей, включая, например, маннит фармацевтической чистоты, лактозу, крахмал, стеарат магния, натрий-содержащую сахаринцеллюлозу, карбонат магния и т.п.
Обычно соединения по настоящему изобретению должны содержать от менее чем 1% до примерно 95% активного компонента, предпочтительно от примерно 10% до примерно 50%. Предпочтительно пациенту вводят от примерно 10 мг/кг веса тела в день до примерно 25 мг/кг веса тела в день. Частота введения должна быть определена врачом на основе наблюдений за реакцией пациента. Другие эффективные дозы могут быть легко определены специалистом в данной области с помощью стандартных испытаний путем построения кривой зависимости “доза-ответ”.
Независимо от пути введения, подходящие фармацевтические композиции в соответствии с настоящим изобретением должны в принципе включать количество тритерпеновой композиции, смешанной с подходящим фармацевтическим разбавителем или наполнителем, таким как стерильный водный раствор, с получением конечных концентраций в соответствующем диапазоне в зависимости от цели применения. Методы приготовления лекарственных препаратов в целом хорошо известны специалистам и их примеры можно найти в руководстве: Remington’s Pharmaceutical Sciences, 16th ed.. Mack Publ. Co., 1980, полное содержание которого вводится в настоящее описание посредством ссылки. Следует отметить, что загрязнение эндотоксинами должно быть на минимально безопасном уровне, например, менее 0,5 нг/мг белка. Более того, предназначенные для введения человеку препараты должны отвечать требованиям стерильности, пирогенности, общей безопасности и чистоты в соответствии со стандартами, принятыми Федеральным департаментом биологических стандартов США.
Терапевтически эффективные дозы легко определяются с использованием животной модели в соответствии с исследованиями, подробно описанными здесь. Например, часто экспериментальных животных с твердыми опухолями используют для оптимизации подходящих терапевтических доз перед использованием их в клинической практике. Известно, что такие модели являются достаточно надежными с точки зрения определения эффективных противоопухолевых стратегий.
В некоторых вариантах может быть желательным обеспечение непрерывной доставки терапевтических композиций пациенту. При внутривенном и внутриартериальном введении это осуществляется с помощью капельниц. Для местного применения должно быть осуществлено повторное введение. Для различных целей могут быть использованы препараты пролонгированного высвобождения, которые выделяют небольшие, но постоянные количества терапевтического агента на протяжении длительного периода времени. Для внутреннего применения предпочтительной может быть непрерывная перфузия интересующего участка. Для этого может быть осуществлена катетеризация, в некоторых случаях после операции с последующим непрерывным введением терапевтического средства. Продолжительность перфузии должна определяться лечащим врачом для конкретного пациента и конкретного случая, и это время должно варьироваться от примерно 1-2 часов до примерно 2-6 часов, до примерно 6-10 часов, до примерно 10-24 часов, до примерно 1-2 дней, до примерно 1-2 недель или более. В целом, доза терапевтической композиции при непрерывной перфузии должна быть эквивалента дозе, вводимой при однократной или многократных инъекциях, рассчитанной для того периода времени, за который такие инъекции проводят. Считается, однако, что при перфузии могут быть использованы более высокие дозы.
1. Протокол лечения
Заявители настоящего изобретения рассматривают два основных способа использования тритерпеновых соединений по настоящему изобретению как по отдельности, так и в комбинированной терапии. Первый из них направлен на метастазирующий рак у пациентов, которым до этого не проводилась химио-, радио- или биотерапия, или у пациентов, получавших предварительно такое лечение. Таким больным проводится системное введение путем внутривенного, подкожного, перорального введения или инъекций непосредственно в опухоль. Вводимыми фармацевтическими дозами предпочтительно являются 10-25 мг тритерпеновых соединений по настоящему изобретению на 1 кг веса тела больного в день, включая 13, 16, 19 и 22 мг/кг в день. Альтернативно, для лечения больного могут быть использованы фармацевтические композиции, содержащие от примерно 1 мг/кг в день тритерпеновых композиций по настоящему изобретению до примерно 100 мг/кг в день, включая 3, 6, 9, 12, 15, 18, 21, 28, 30, 40, 50, 60, 70, 80 и 90 мг/кг в день тритерпеновых композиций по настоящему изобретению.
Курс лечения обычно включает ежесуточное введение, по крайней мере, в течение восьми недель или еженедельную инъекцию, по крайней мере, в течение восьми недель. В соответствии с назначением лечащего врача, курс лечения может быть продолжен по той же схеме до тех пор, пока будет наблюдаться рост опухоли или пока не прекратится реакция на лечение.
Другое применение соединений по настоящему изобретению связано с лечением больных, у которых заболевания были вылечены хирургически, с помощью химиотерапии и (или) радиотерапии. Вспомогательная терапия должна проводиться в том же режиме, который описан выше, в течение по крайней мере одного года с целью предотвращения рецидива заболевания.
2. Профилактика ракового заболевания с использованием соединений по настоящему изобретению
Другое применение соединений и смесей по настоящему изобретению связано с профилактикой рака в группах повышенного риска. Таким пациентам (например, тем, у которых имеется генетическая предрасположенность к таким опухолям, как рак молочной железы, рак толстой кишки, рак кожи и другие) введение должно осуществляться через рот (опухоли желудочно-кишечного тракта), путем локального нанесения на кожу (кожные опухоли) или системно в течение, по крайней мере, одного года, а, вероятно, и более продолжительное время с целью профилактики ракового заболевания. Такой метод использования может быть применен к пациентам и точно установленным предраковым повреждениям, таким как полипы прямой кишки и другие предзлокачественные поражения кожи, молочной железы, легких или других органов.
3. Клинический протокол
Клинический протокол был рассчитан заявителями таким образом, чтобы обеспечить лечение рака с использованием тритерпеновых соединений по настоящему изобретению. В соответствии с таким протоколом отбирают больных, для которых гистологическими методами подтвержден диагноз ракового заболевания, например, рака яичника, рака поджелудочной железы, рака почек, рака предстательной железы, рака легких или рака мочевого пузыря. К больным могла ранее применяться (хотя это и не обязательно) химио-, радио- и генная терапия. Оптимально, чтобы у таких больных сохранялась нормальная функция костного мозга (определяемая по абсолютному числу периферических гранулоцитов >2000 на 1 мм3, и количеству тромбоцитов 100000 на 1 мм3), адекватная функция печени (билирубин ≤1,5 мг/дл) и почек (креатинин <1,5 мг/дл).
Указанный протокол предусматривает введение путем внутриопухолевой инъекции фармацевтической композиции, содержащей примерно 10-25 мг тритерпеновых соединений по настоящему изобретению на 1 кг веса тела пациента. Для опухоли размером ≥4 см вводимый объем должен составлять 4-10 мл (предпочтительно 10 мл), в то время как для меньших опухолей <4 см должен использоваться объем в 1-3 мл (предпочтительно 3 мл). Многократные инъекции, выстроенные на основе объема разовой дозы в 0,1-0,5 мл, должны проводиться на расстоянии 1 см или больше.
Курс лечения включает примерно 6 доз, вводимых в течение двух недель. По назначению лечащего врача этот курс может быть продлен: по 6 доз каждые две недели или с меньшей частотой (в месяц, за 2 месяца, в квартал и т.п.).
Если для больных показано хирургическое вмешательство, то опухоль у них должна обрабатываться так же, как описывалось выше, в течение, по крайней мере, двух последовательных двухнедельных курсов лечения. Через неделю после завершения второго курса (или большего, например, третьего, четвертого, пятого, шестого, седьмого, восьмого и т.п.) больному должна быть проведена хирургическая операция. Перед зашиванием операционного разреза 10 мл фармацевтической композиции, содержащей тритерпеновые соединения по настоящему изобретению, должно быть нанесено на место операции (операционное ложе), контакт с которым должен продолжаться, по крайней мере, 60 минут. После этого рану закрывают, оставляя в ней катетер или дренажную трубку. На 3-й день после операции дополнительные 10 мл фармацевтической композиции вводят через эту трубку и оставляют в контакте с операционным ложем, по крайней мере, в течение 2 часов. Затем ее удаляют отсасыванием и вынимают катетер в клинически подходящее время.
4. Лечение искусственных и естественных полостей тела
Одним из основных источников рецидивов опухоли являются остаточные микроскопические поражения, которые после ее удаления остаются в месте первичной опухоли, а также локальные и периферические поражения. Кроме того, аналогичные ситуации имеют место тогда, когда естественные полости тела загрязняются микроскопическими опухолевыми клетками. Эффективное лечение такого микроскопического поражения должно иметь существенные преимущества перед терапевтическими схемами лечения.
Так, в некоторых вариантах раковая опухоль может быть удалена хирургически, в результате чего образуется “полость”. И в процессе хирургического вмешательства, и после него (периодически или непрерывно) терапевтическую композицию по настоящему изобретению вводят в полость тела. По сути это является “местным” применением по отношению к поверхности такой полости. Количество композиции должно быть достаточным для того, чтобы вся поверхность полости проконтактировала с вводимой композицией.
В одном из вариантов, введение должно осуществляться путем простой инъекции терапевтической композиции в полость, образованную удаленной опухолью. В другом варианте желательным может быть применение с помощью механического предмета: губки, тампона и т.п. Любой из данных подходов может быть использован после удаления опухоли, а также и в ходе первой хирургической операции. Еще в одном варианте катетер вводят в полость до закрытия хирургического разреза. Затем полость может быть непрерывно перфузирована в течение желательного периода времени.
При другом способе лечения “местное” применение терапевтической композиции направлено на естественную полость тела, такую как полости рта, глотки, пищевода, гортани, трахеи, грудная полость, брюшная полость или полости полых органов, включая мочевой пузырь, толстую кишку и другие внутренние органы. В этом случае в данной полости первичная опухоль может присутствовать или отсутствовать. Это лечение нацелено на микроскопические поражения в такой полости, но иногда оно также может воздействовать на массу первичной опухоли, если она не была перед этим удалена, или предопухолевые повреждения, которые могут присутствовать в такой полости. И в этом случае, ряд методов может быть применен для “местного” применения в отношении этих внутренних органов или полостных поверхностей. Например, полость глотки просто может быть обработана путем промывки и полоскания. Однако, для местной обработки гортани и трахеи может потребоваться эндоскопия и локальная доставка терапевтической композиции. В отношении внутренних органов, таких как слизистые мочевого пузыря или толстой кишки, может потребоваться внутренняя катетеризация для инфузии через катетер или прямая визуализация с помощью цистоскопа или иного эндоскопического прибора. Такие полости, как грудная полость или брюшная полость, можно обрабатывать с помощью внутренних катетеров или хирургическими методами, которые обеспечивают доступ в эти области.
(iv) Терапевтические наборы.
Настоящее изобретение также относится к терапевтическим наборам, включающим описываемые здесь тритерпеновые композиции. Данные наборы в целом содержат фармацевтически приемлемый препарат, по крайней мере, одного тритерпенового соединения по настоящему изобретению, размещенный в подходящем контейнере. Данные наборы также включают другие фармацевтически приемлемые препараты, такие как препараты, содержащие компоненты для доставки тритерпенового соединения в конкретные участки тела пациента по необходимости их лечения или любые одно или несколько лекарственных средств, например, химиотерапевтических средств, которые могут действовать с тритерпеновыми соединениями согласованно.
Эти наборы могут содержать единую упаковку, которая содержит тритерпеновые соединения с любыми дополнительными компонентами или без них, или они могут содержать отдельный контейнер для каждого желательного агента. Если компоненты набора представляются в виде одного или нескольких жидких растворов, то такой жидкий раствор является водным раствором, причем, особенно предпочтительным является стерильный водный раствор. Однако, компоненты данного набора могут присутствовать в виде сухого(их) порошка(ов). Если реагенты или компоненты присутствуют в виде сухого порошка, то этот порошок может быть разведен путем добавления подходящего растворителя. Предусматривается, что растворитель также может находиться в отдельном контейнере. Контейнером в составе данного набора, в принципе, может быть, по крайней мере, одна ампула, тест-пробирка, колба, пузырек, шприц или контейнер иной формы, в который может быть помещен тритерпеновый гликозид и любой другой желательный агент, предпочтительно в виде аликвот. В случае включения дополнительных компонентов, этот набор может также содержать вторую ампулу или иной контейнер, в который эти компоненты помещают, что позволяет вводить отдельно приготовленные дозы. Эти наборы могут также включать второй/третий контейнер для стерильного фармацевтически приемлемого буфера или иного разбавителя.
Данные наборы также могут включать приспособление, с помощью которого тритерпеновые композиции вводят животному или человеку, например, одну или большее число игл или шприцев, или глазную капельницу, пипетку или иное подобное устройство, с помощью которого конкретный препарат может быть введен животному или применен на желательную область тела. Наборы по настоящему изобретению также обычно включают приспособление для установки ампул или т.п.и другие компоненты, с учетом ограничений, накладываемых условиями коммерческой реализации, такие как, например, инъекционные или ингаляционные пластиковые емкости, в которых размещают и сохраняют желательные ампулы и другие устройства.
VI. Химиотерапевтические комбинации и лечение
В некоторых вариантах настоящего изобретения желательным может быть введение тритерпеновых композиций по настоящему изобретению с одним или большим числом других агентов, обладающих противоопухолевой активностью, включая химиотерапевтические и радиотерапевтические средства, а также терапевтические белки или гены. Это может усиливать общую противоопухолевую активность, достигаемую путем терапии с использованием только соединений по настоящему изобретению, или это можно использовать для предотвращения или подавления резистентности опухолей к нескольким лекарственным средствам.
Использование настоящего изобретения в сочетании с введением второго химиотерапевтического агента предусматривает просто введение животному тритерпеновой композиции вместе со вторым химиотерапевтическим агентом таким образом, чтобы это было эффективно для достижения их комбинированного противоопухолевого воздействия у этого животного. Следовательно, эти агенты должны быть представлены в эффективном количестве и в течение периода времени, эффективного для достижения их совместного присутствия в опухолевой ткани и их совместного воздействия на среду этой опухоли. Для достижения этой цели тритерпеновая композиция и химиотерапевтические агенты могут быть введены животному одновременно как в составе общей композиции, так и в виде двух раздельных композиций с использованием разных путей введения.
Альтернативно, введение тритерпеновой композиции может предвосхищать применение химиотерапевтического агента, радиотерапии или белковой или генной терапии или следовать за ним с интервалом от минут до недель. В вариантах, в которых второй агент и тритерпеновую композицию вводят животному раздельно, в целом, нужно обеспечить, чтобы интервал времени между каждым из введений был не слишком большим для того, чтобы дополнительный агент и тритерпеновая композиция не утратили способность проявить преимущественный совместный эффект в отношении данной опухоли. В этих случаях предусматривается, чтобы контакт данной опухоли с каждым из обоих агентов протекал в течение примерно от 5 минут до примерно одной недели для каждого из них или, что более предпочтительно, в течение примерно 12-72 часов для каждого, при времени задержки примерно 24-48 часов. В некоторых случаях может оказаться желательным существенное увеличение времени лечения, где перерывы между соответствующими введениями составляют несколько дней (2, 3, 4, 5, 6 или 7) или даже несколько недель (1, 2, 3, 4, 5, 6, 7 и 8). Также предполагается, что желательным может быть неоднократное введение либо тритерпенового гликозида, либо второго агента. Для достижения регрессии опухолей, оба агента доставляют в общем количестве, эффективном для подавления их роста, независимо от сроков введения.
Ряд агентов применим для использования в описываемых здесь способах комбинированной терапии. Химиотерапевтическими агентами, приводимыми в качестве примеров, являются, например, этопозид (VP-16), адриамицин, 5-фторурацил (5-FU), камптотецин, актиномицин-D, митомицин-С и цисплатин (CDDP).
Как должно быть понятно специалистам в данной области, подходящие дозы химиотерапевтических агентов, в целом, должны находиться в диапазонах, уже применяемых в клинических способах лечения, в которых химиотерапевтические средства вводят отдельно или в комбинации с другими химиотерапевтическими средствами. В качестве примера таких агентов можно привести цисплатин; могут быть также использованы другие ДНК-алкилирующие агенты. Цисплатин широко применяется для лечения рака, а эффективные дозы, используемые в клинической практике, составляют 20 мг/м2 в течение 5 дней каждые три недели на протяжении трех курсов. Цисплатин не абсорбируется при пероральном введении, а следовательно, его вводят путем внутривенных, подкожных, внутриопухолевых или внутрибрюшинных инъекций.
Дополнительными применимыми агентами являются соединения, которые препятствуют репликации ДНК, митозу и сегрегации хромосом. Таким химиотерапевтическим соединением является адриамицин, а также известны доксорубицин, этопозид, верапамил, подофиллотоксин и т.п. Будучи широко применяемыми в клинической практике для лечения опухолей, эти соединения вводят путем болюсных внутривенных инъекций при дозах в диапазоне от 25-75 мг/м2 с 21-дневным интервалом для адриамицина до 35-50 мг/м2 для этопозида, с удвоением дозы при пероральном введении.
Также могут быть использованы агенты, которые нарушают синтез и соответствие полинуклеотидных предшественников, В частности, применимыми являются агенты, которые проходят интенсивное тестирование и которые легко доступны. С учетом этого, такие агенты, как 5-фторурацил (5-FU), предпочтительно используются опухолевой тканью, что делает этот агент особенно подходящим для направленной доставки к опухолевым клеткам. Хотя 5-FU является токсичным, он может быть использован в составе широкого круга носителей, включая местное введение, причем, чаще всего осуществляют внутривенное введение дозами в пределах 3-15 мг/кг в день.
Примеры химиотерапевтических агентов, которые применяются в связи с комбинированной терапией, перечислены в табл.5.
Каждый из перечисленных в ней агентов приведен в качестве примера и не является ограничивающим. В связи с этим, можно отослать специалиста к справочнику "Remington’s Pharmaceutical Sciences", 15th ed., глава 33, в частности, стр.624-652. Может оказаться необходимым некоторое варьирование дозировок в зависимости от состояния конкретного больного, лечение которого проводится. В любом случае ответственное за применение лицо должно установить подходящую дозу для каждого конкретного пациента. Более того, стерильность, пирогенность, общие уровни безопасности и чистоты препаратов для введения человеку должны удовлетворять стандартным требованиям, предъявляемым Федеральным департаментом биологических стандартов США.
Химиотерапевтические агенты, применяемые в отношении раковых заболеваний
Другими факторами, вызывающими повреждения ДНК, являются широко применяемые γ-облучение, рентгеновское облучение и (или) направленная доставка радиоактивных изотопов в опухолевые клетки. Другими факторами, вызывающими повреждения ДНК, являются микроволны и ультрафиолетовое излучение. Наиболее вероятно, что все эти факторы вызывают широкий спектр повреждений ДНК, ДНК-предшественников, нарушение процессов репликации и репарации ДНК, сборки и стабильности хромосом. Дозы рентгеновского облучения варьируются от 50 до 200. Рентген в сутки при продолжительных периодах времени (3-4 недели), а разовые дозы составляют 2000-6000 Рентген. Диапазоны доз для радиоактивных изотопов варьируются в широких пределах, что определяется временем полураспада изотопа, силой и типом испускаемого излучения и поглощаемостью их раковыми клетками.
VII. Направленная терапия раковых опухолей
Описываемые здесь тритерпеновые соединения могут быть связаны с одной или несколькими молекулами, которые направляют соединения непосредственно в раковые клетки. Такая направленная доставка имеет то преимущество, что она может использоваться для повышения общей концентрации лекарственного средства непосредственно в месте лечения, например, в опухоли, минимизируя тем самым системный “расход” лекарственного средства. Как и в случае с обсуждавшимися выше химиотерапевтическими агентами, направленное тритерпеновое соединение может быть использовано в комбинации со вторым агентом, таким как химиотерапевтический агент. И сам тритерпен, и второй агент могут быть направлены на одну или разные мишени в опухолевой ткани. В этом случае может быть достигнут суммарный, превышающий суммарный или даже явный синергитический эффект.
Примерами направляющих агентов, используемых в комбинации с тритерпеновыми соединениями по настоящему изобретению, будут те направляющие агенты, которые способны доставлять тритерпеновые соединения в область опухоли, т.е. способны локализоваться в месте ее расположения. Также желательными являются агенты, направленные на сосудистую систему опухоли. В частности, следует учесть, что направленная доставка тритерпеновых гликозидных соединений позволяет использовать более эффективные концентрации в области опухоли без побочных эффектов или с минимальными побочными эффектами, которые могут наблюдаться при более широком или системном распределении тритерпеновых соединений. В частности, мишенью направляющего агента могут быть: компоненты опухолевых клеток; компоненты опухолевой сосудистой системы; компоненты, связывающиеся или обычно ассоциированные с опухолью; компоненты, связывающиеся или обычно ассоциированные с сосудистой системой опухоли; компоненты внеклеточного матрикса опухоли или стремы, или связанные с ними, и даже клетки таких типов, которые обнаруживаются в сосудистой системе опухоли.
(i) Опухолевые клетки-мишени и антитела
Злокачественные клетки, составляющие опухоль, могут быть помечены с использованием биспецифичного антитела, которое включает участок связывания с относительно специфичным маркером или антигеном такой опухолевой клетки. Например, специфическое ингибирование или уничтожение опухолевых клеток может быть достигнуто путем связывания конъюгата “антитело-тритерпеновая композиция” с опухолевой клеткой-мишенью.
Описано множество т.н. “опухолевых антигенов”, любой из которых может быть выбран в качестве мишени в связи с направляющим мечением по настоящему изобретению. Здесь далее перечислено значительное число антигенов, ассоциированных с твердыми опухолями. Получение и применение антител против таких антигенов хорошо известно специалистам в данной области техники и, в частности, описано здесь. Примерами антител являются антитела к опухолям женской половой сферы (см., например, каталог коллекции АТСС): ОС 125; ОС 133; OMI; Mo v1; Мо v2; 3C2; 4С7; ID3; DU-PAN-2; F 36/22; 4F7/7A10; OV-TL3; В72.3; DF3; 2C8/2F7; MF 116; Mov18; CEA 11-H5; СА 19-9 (1116NS 19-9); Н17-Е2; 791Т/36; NDOG2; H317; 4D5, 3H4, 7C2, 6E9, 2C4, 7F3, 2H11, 3E8, 5B8, 7D3, SB8; HMFG2; 3.14.A3; опухолям молочной железы: DF3; NCRC-11; 3C6F9; МВЕ6; CLNH5; MAC 40/43; EMA; HMFG1; HFMG2; 3.15.C3; M3, M8, М24; М18; 67-D-11; D547Sp, D75P3, H222; анти-EGF; LR-3; TA1; H59; 10-3D-2; HmAB1,2; MBR 1,2·3; 24·17·1; 24·17·2 (3E1·2); F36/22.M7/105; С11, G3, H7; В6·2; B1·1; Cam 17·1; SM3; SM4; C-Mul (566); 4D5 3H4, 7C2, 6E9, 2С4, 7F3, 2Н11, 3Е8, 5В8, 7D3, 5В8; ОС 125; МО v2; DU-PAN-2; 4F7/7A10; DF3; B72·3; cccccCEA 11; H17·E2; 3·14·A3; FO23C5; опухолям толстой и прямой кишки: B72·3; (17·1А) 1083·17·1А; С017·1А; ZCE·025; AB2; НТ·29·15; 250·30.6; 44Х14; А7; GA73·3; 791Т/36; 28А32; 28.19.8; X ММСО-791; DU-PAN-2; ID3; CEA 11-H5; 2С8/2F7; СА-19-9 (1116NS 19·9); PR5C5; PR4D2; PR4D1; меланомам: 4·1; 8·2 M17; 96·5; 118·1; 133·2, (113·2); L1, L10, R10(R19); I12; K5; 6·1; R24; 5·1; 225.28S; 465.12S; 92·27; F11; 376.96S; 465.12S; 15·75; 15·95; Mel-14; Mel-12; Me3-TB7; 225.28SD; 763.24TS; 705F6; 436910; М148; опухолям желудочно-кишечного тракта: ID3; DU-PAN-2; OV-TL3; B72-3; CEA 11-H5; 3-14-A3; С COLI; СА-19-9 (1116NS 19-9) и СА50; ОС125; опухолям легкого: 4D5 3Н4, 7С2, 6Е9, 2С4, 7F3, 2Н11, ЗЕ8, 5В8, 7D3, SB8; МО v2; B72-3; DU-PAN-2; CEA 11-H5; MUC 8-22; MUC 2-63; MUC 2-39; MUC 7-39; и неоднородным типам опухолей: РАb 240; PAb 246; РАb 1801; ERIC-1; M148; FMH25; 6-1; CA1; 3F8; 4F7/7А10; 2C8/2F7; CEA 11-H5.
Другой способ определения и мечения опухоли связан с непосредственными характеристиками самой опухоли, а не с биохимическими свойствами антигена, экспрессируемого опухолевой клеткой. Известен ряд линий опухолевых клеток, которые могут быть использованы для получения направляющих агентов. Например, интактные клетки или клеточные гомогенаты, полученные от известных клеточных линий, могут быть использованы для получения противоопухолевых антител, метящих соответствующие типы опухолей. Аналогичным образом, такие линии опухолевых клеток могут найти применение при проведении различных тестов in vitro. В этом случае специалист может обратиться к каталогу АТСС с целью выбора характерных линий опухолевых клеток человека, к которым имеется открытый доступ (из каталога АТСС). Примерами таких линий являются: J82; RT4; ScaBER; T24; TCCSUP; 5637; SK-N-MC; SK-N-SH; SW 1088; SW 1783; U-87 MG; U-118 MG; U-138 MG; U-373 MG; Y79; BT-20; BT-474; MCF7; MDA-MB-134-VI; MDA-MD-157; MDA-MB-175-VII; MDA-MB-361; SK-BR-3; C-33 A; HT-3; ME-180; MS751; SiHa; JEG-3; Caco-2; HT-29; SK-CO-1; HuTu 80; A-253; FaDu; A-498; A-704; Caki-1; Caki-2; SK-NEP-1; SW 839; SK-HEP-1; A-427; Calu-1; Calu-3; Calu-6; SK-LU-1; SK-MES-1; SW 900; EB1; EB2; P3HR-1; HT-144; Malme-3М; RPMI-7951; SK-MEL-1; SK-MEL-2; SK-MEL-3; SK-MEL-5; SK-MEL-24; SK-MEL-28; SK-MEL-31; Caov-3; Caov-4; SK-OV-3; SW 626; Capan-1; Capan-2; DU 145; A-204; Saos-2; SK-ES-1; SK-LMS-1; SW 684; SW 872; SW 982; SW 1353; U-2 OS; Malme-3; KATO III; Cate-1B; Tera-1; Tera-2; SW 579; AN3 CA; HEC-1-A; HEC-1-B; SK-UT-1; SK-UT-1B; SW 954; SW 962; NCI-H69; NCI-H128; BT-483; BT-549; DU4475; HBL-100; Hs 578Bst; Hs 578T; MDA-MB-330; MDA-MB-415; MDA-MB-435S; MDA-MB-436; MDA-MB-453; MDA-MB-468; T-47D; Hs 766T; Hs 746T; Hs 695T; Hs 683; Hs 294T; Hs 602; JAR; Hs 445; Hs 700T; H4; Hs 696; Hs 913T; Hs 729; FHs 738Lu; FHs 173We; FHs 738B1; NIH:OVCAR-3; Hs 67; RD-ES; ChaGo K-1; WERI-Rb-1; NCI-H446; NCI-H209; NCI-H146; NCI-H441; NCI-H82; H9; NCI-H460; NCI-H596; NCI-H676B; NCI-H345; NCI-H820; NCI-Н520; NCI-H661; NCI-H510A; D283 Med; Daoy; D341 Med; AML-193 и MV4-11.
Можно ознакомиться с каталогом АТСС за любой последующий год с целью идентификации других подходящих клеточных линий. Также, если желательным является конкретный тип клеток типа, то каждому специалисту известны способы получения таких клеток и/или их непосредственный источник. Поэтому при анализе научной литературы можно легко выбрать источник опухолевой клетки любого типа, который желательно наметить как мишень.
Как объяснялось выше, антитела представляют собой прямое средство распознавания опухолевого антигена-мишени. Известно, что очень большое количество антител направлено против твердых опухолей. Некоторые из применимых противоопухолевых антител были перечислены выше. Однако, как должно быть известно специалистам в данной области техники, некоторые из перечисленных антител не обладают желательными биохимическими свойствами или недостаточно специфичны в отношении опухоли, чтобы их можно было использовать как терапевтические средства. Примером является антитело MUC8-22, распознающее цитоплазматический антиген. Использование подобных антител может представлять лишь экспериментальный интерес, например, в модельных системах или в скрининг-тестах.
В целом, антитела для применения в данных аспектах настоящего изобретения должны распознавать антигены, которые представлены на поверхности клеток и которые предпочтительно или даже специфически экспрессируются опухолевыми клетками.
Также такие антитела предпочтительно должны характеризоваться высокой аффинностью, например, Кd<200 нМ, а предпочтительно, Kd<100 нМ, и не должны проявлять значимой реактивности в отношении жизненно важных нормальных тканей, таких как одна или большее число тканей, выбираемых из тканей сердца, почек, головного мозга, печени, костного мозга, толстой кишки, молочной железы, предстательной железы, щитовидной железы, желчного пузыря, легких, надпочечников, скелетных мышц, нервных волокон, поджелудочной железы, кожи или иных жизненно важных органов или тканей в организме человека. Среди “жизненно важных” тканей наиболее важными тканями с точки зрения целей настоящего изобретения и исходя из условий низкой в их отношении реактивности являются ткани сердца, почек, центральной и периферической нервной системы и печени. Используемый в данном тексте термин “отсутствие значимой реактивности” характеризует антитело или фрагмент антитела в том, что, в случае их применения в отношении конкретной ткани в подходящих для иммуногистохимического анализа условиях, не будет вызвано окрашивание вовсе или будет обусловлено едва заметное окрашивание по отдельным позитивным клеткам, рассеянным в массе подавляющего большинства негативных клеток.
Конкретно перспективными антителами, представляемыми для использования в настоящем изобретении, являются те антитела, которые селективны в отношении твердых опухолей. Например, антитела, связывающиеся с протоонкогенами TAG72 и HER-2, которые избирательно обнаруживаются на поверхности многих раковых опухолей молочной железы, легких и толстой кишки (Thor et al., 1986; Colcher et al., 1987; Shepard et al., 1991); MOv18 и OV-TL3 и антитела, которые связываются с ядром муцинового белка молока и жировыми шариками молока человека (Miotti etc al., 1985; Burchell et al., 1983); и антитело 9.2.27, которое связывается с антигенами меланомы с высокой Mr (Reisfeld et al., 1982). Также применимыми являются антитела против фолат-связывающего белка, которые, как известно, гомогенно экспрессируются почти во всех карциномах яичников; антитела против онкогенов семейства erb, которые сверхэкспрессируются в плоскоклеточных опухолях и в большинстве глиом; и другие антитела, известные как объекты преклинического и клинического тестирования.
Антитела В3, KSI/4, CC49, 260F9, ХММСО-791, D612 и SM3, как считается, пригодны для использования в клинической практике после стандартного преклинического тестирования, обычно проводимого в клинике. Антитело В3 (патент США №5242813; Brinkmann et al., 1991) имеет депозитарный №HB10573 в АТСС; антитело KSI/4 может быть получено в соответствии с описанным в патенте США №4975369; и антитело D612 (патент США №5183756) имеет депозитарный №НВ9796 в АТСС.
Другой способ определения ассоциированной с опухолью мишени связан с непосредственными характеристиками самой опухоли, а не с определением биохимических свойств антигена, экспрессируемого опухолевой клеткой. Следовательно, заявители считают, что любое антитело, которое предпочтительно связывается с опухолевой клеткой, может быть использовано в качестве направляющего компонента в составе конъюгата с тритерпеновым соединением. Предпочтительное связывание с опухолевой клеткой основывается на антителе, которое обладает высокой степенью аффинности в отношении этой опухолевой клетки и не проявляет значимой реактивности в отношении жизненно важных нормальных клеток или тканей, которые были определены выше.
Также настоящее изобретение представляет ряд способов получения антитела для использования в целях доставки тритерпеновых гликозидов к опухолевым клеткам в соответствии с описанным в данном тексте. Для получения специфичного для опухолевой клетки антитела можно иммунизировать животное композицией, содержащей антиген опухолевой клетки, и отобрать (что будет подробнее описано далее) образующееся в результате антитело с подходящей специфичностью. Эта иммунизирующая композиция может содержать полностью или частично очищенный препарат любого из перечисленных выше антигенов; она может быть такой композицией, как мембранный препарат, обогащенный любыми из перечисленных выше антигенов; любыми клетками, перечисленными выше; или содержать смесь или популяцию клеток, включая любые типы клеток, перечисленных выше.
Очевидно, что независимо от источника антитела, при практическом применении настоящего изобретения в медицине может быть отдано предпочтение предварительной уверенности в том, что являющаяся клиническим объектом опухоль экспрессирует тот антиген, который в конечном счете отобран. Этого можно достичь путем прямого теста, включающего тестирование на антигенность образца опухолевой ткани, взятой, например, путем хирургической биопсии, или, возможно, тестирование циркулирующего антигена. Это может быть легко осуществлено путем иммунологического скрининга, например, методом ELISA (твердофазный иммуноферментный анализ), в котором антитела из гибридомного банка тестируют на аффинность связывания в отношении данной опухоли. Затем антитела, обладающие соответствующей селективностью и аффинностью в отношении опухоли, отбирают для получения биспецифичных антител по настоящему изобретению.
Благодаря хорошо известному явлению перекрестной реактивности, предполагается, что используемые антитела, могут быть получены в соответствии со схемой иммунизации, где используемые исходные антигены происходят от животного, например, мыши или примата, в дополнение к тем иммунизациям, для которых используют исходные антигены клетки человека. При использовании происходящих от человека антигенов, они могут быть получены от линии опухолевых клеток человека, либо они могут быть получены путем взятия биопробы от конкретного обследуемого больного. Действительно, известны способы получения антител, являющихся для опухоли конкретного больного “скроенными на заказ” (Stevenson et al., 1990), и они представляются в связи с настоящим изобретением.
1. Способы получения антител
Как отмечалось выше, антитела могут найти применение в конкретных вариантах настоящего изобретения. Например, могут быть получены антитела, специфичные в отношении конкретного участка в организме пациента или конкретного типа ткани. Такие антитела затем могут быть присоединены к тритерпеновому соединению по настоящему изобретению, что тем самым позволит специфическим образом направить (“прицелить”) данные тритерпеновые соединения на ткань, в отношении которой специфично данное антитело. Примером такого антитела является антитело, которое связывается с опухолевой клеткой. В предпочтительном варианте настоящего изобретения антитело является моноклональным антителом. Способы получения моноклональных и поликлональных антител хорошо известны в данной области техники и, в частности, описаны здесь далее (см., например, Howell & Lane, 1988).
Кратко, поликлональное антитело получают путем иммунизации животного иммуногеном, включающим желательный антиген, и взятия антисыворотки от такого иммунизированного животного. Для получения антисывороток может быть использован широкий круг видов животных. Обычно животным, используемым для получения антисывороток, служит не являющееся человеком животное, включая кроликов, мышей, крыс, хомячков, свиней или лошадей. Благодаря относительно большому количеству крови, предпочтительным выбором для получения поликлональных антител являются кролики.
Как поликлональные, так и моноклональные антитела, специфичные в отношении изоформ антигена, могут быть получены с использованием стандартных методов иммунизации, что в целом хорошо известно в данной области техники. Композицию, содержащую антигенные эпитопы конкретных типов клеток, или, с другой стороны, соединения по настоящему изобретению, можно использовать для иммунизации одного или большего числа животных, таких как кролик или мышь, которых затем используют для получения антител, специфичных в отношении данных антигенов. Поликлональные антисыворотки можно получить после определенного промежутка времени, необходимого для формирования антител, просто путем взятия крови у этих животных и получения образцов сыворотки на материале цельной крови.
Можно считать, что моноклональные антитела по настоящему изобретению найдут практическое применение в иммунохимических процедурах, которые могут быть использованы для тестирования на присутствие тритерпеновых соединений по настоящему изобретению у других видов растений, помимо Acacia victoriae, или в других процедурах, в которых используются антитела, специфичные для конкретных антигенов. Как обсуждалось выше, примером использования антител в связи с настоящим изобретением является получение антител, специфичных в отношении опухолевых антигенов, связывание антител с тритерпеновыми соединениями по настоящему изобретению и лечение больных с использованием конъюгата “антитело-тритерпен”, благодаря чему тритерпеновые соединения специфическим образом доставляются к опухолевым клеткам или другим клеткам, вовлеченным в то состояние, лечение которого может быть осуществлено с использованием тритерпеновых соединений по настоящему изобретению. В целом, в различных вариантах настоящего изобретения могут быть использованы и поликлональные, и моноклональные антитела к различным антигенам. Например, их можно использовать для очистки тритерпеновых соединений на аффинных колонках. Способы получения и характеризации таких антител хорошо известны в данной области и изложены, например, у Harlow & Lane, 1988; полное содержание этого руководства включено в настоящее описание посредством ссылки.
Как хорошо известно в данной области, иммуногенность конкретного соединения может варьироваться. Поэтому часто бывает необходимым дополнительно простимулировать иммунную систему реципиента, чего можно достичь посредством связывания пептидного или полипептидного иммуногена с носителем. Примерами и предпочтительными носителями являются гемоцианин лимфы улитки (KLH) и бычий сывороточный альбумин (БСА). В качестве носителей могут быть также использованы другие альбумины, такие как овальбумин, мышиный сывороточный альбумин или кроличий сывороточный альбумин. Способы конъюгирования полипептида с белком-носителем хорошо известны в данной области и включают использование глутарового альдегида, сложного эфира м-малеимидобенкоил-N-гидроксисукцинимида, карбодиимида и бис-биазотированного бензидина.
Также, как хорошо известно в данной области, иммуногенность конкретной иммуногенной композиции может быть усилена за счет использования неспецифических стимуляторов иммунного ответа, называемых адъювантами. Примеры и предпочтительные адъюванты включают полный адъювант Фрейнда (неспецифический стимулятор иммунного ответа, содержащий убитые туберкулезные микобактерии Mycobacterium tuberculosis), неполный адъювант Фрейнда и гидроксид алюминия.
Количество иммуногенной композиции, используемой для получения поликлональных антител, варьируется в зависимости от природы иммуногена, а также вида используемого для иммунизации животного. Могут быть использованы различные пути введения иммуногена (подкожный, внутримышечный, внутрикожный, внутривенный и внутрибрюшинный). Выработку поликлональных антител можно контролировать путем взятия проб крови у иммунизированного животного через различные промежутки времени после иммунизации. Также может быть проведена вторая, бустер-инъекция. Бустинг и титрование антител продолжают до тех пор, пока не будет достигнут желательный титр. По достижении желательного уровня иммуногенности, иммунизированное животное может быть умерщвлено, а сыворотка выделена и сохранена, и/или данное животное может быть использовано для генерирования моноклональных антител (mAbs).
Моноклональные антитела могут быть легко получены с применением хорошо известных технологий, таких как описано в патенте США №4196265, полное содержание которого включено в настоящее описание посредством ссылки. Обычно данная технология включает иммунизацию подходящего животного выбранной иммуногенной композицией, например, очищенным или частично очищенным опухолевым антигеном, полипептидом или пептидом, или опухолевой клеткой. Иммунизирующую композицию вводят так, чтобы обеспечить эффективную стимуляцию клеток, вырабатывающих антитела. Предпочтительными животными являются грызуны, такие как мыши и крысы, однако также возможно использование клеток кроликов, овец или лягушек. Использование крыс имеет некоторые преимущества (Coding, 1986), однако мыши предпочтительнее, а наиболее предпочтительными являются мыши линии BALB/c, поскольку это наиболее широко используемая стандартная линия, дающая высокий процент стабильных клеточных гибридов.
После иммунизации, соматические клетки, обладающие потенциалом по выработке антител, в частности, В-лимфоциты (В-клетки), отбирают для использования в соответствии с протоколом получения моноклональных антител. Эти клетки могут быть взяты путем биопсии селезенки, миндалин или лимфатических узлов или из пробы периферической крови. Предпочтительны спленоциты и клетки крови: первые являются богатым источником вырабатывающих антитела клеток, находящихся на стадии делящихся лимфобластов, а вторые предпочтительны благодаря легкодоступности проб крови. Часто должна быть проиммунизирована группа животных, чтобы выделить селезенку у животного, характеризующегося наивысшим титром антител, после чего лимфоциты селезенки получают путем гомогенизации селезенки шприцем. Обычно в селезенке иммунизированной мыши содержится приблизительно 5×107-2×108 лимфоцитов.
Затем В-лимфоциты иммунизированного животного, вырабатывающие антитела, гибридизуют с клетками иммортализованной миеломной линии, обычно от того же вида животных, особи которого были иммунизированы. Предпочтительными линиями миеломных клеток, пригодными для гибридизации с получением гибридом, являются клетки, которые не вырабатывают антител, характеризуются высокой гибридизационной способностью и дефицитны по ферментам, что делает их неспособными расти в некоторых селективных средах, которые поддерживают рост только желательных гибридных клеток (гибридом).
Могут быть использованы любые из ряда миеломных клеток, которые хорошо известны в данной области техники. Например, для иммунизации мышей можно использовать клетки Р3-Х63/Аg8, Р3-Х63-Аg8.653, NS1/1.Ag-4 1, Sp210-Ag14, FO, NSO/U, MPC-11, MPC-11-X45-GTG 1.7 и S194/5XXO Bul; для крыс можно использовать клетки R210.RCY3, Y3-Ag 1.2.3, IR983F и 4В210; и во всех случаях, для слияния клеток можно использовать клетки U-266, GM-1500-GRG2, LICR-LON-HMy2 и UC729-6 (см., например, Goding, 1986; Campbell, 1984; каталог АТСС).
Способы получения гибридов между вырабатывающими антитела клетками селезенки или лимфатических узлов и миеломными клетками обычно включает смешивание соматических клеток с миеломными клетками в соотношении 2:1, хотя это отношение может варьироваться от примерно 20:1 до примерно 1:1, соответственно в присутствии агента или агентов (химических или электрических), способствующих слиянию клеточных мембран. Были описаны методы гибридизации с использованием вируса Сендай (Kohler & Milstein, 1975, 1976) и с использованием полиэтиленгликоля (ПЭГ), например, 37%-ного ПЭГ (о/о) (Gefter et al., 1977). Подходящими методами также являются электрически индуцированные слияния (Coding, 1986).
В результате процедур слияния, жизнеспособные гибриды обычно образуются с низкой частотой, примерно от 1×10-6 до 1×10-8. Однако, это не создает проблемы потому, что жизнеспособные, т.е. слитые гибриды дифференцируются от родительских неслитых клеток (в частности, от неслитых миеломных клеток, которые обычно должны продолжать неограниченно делиться) при культивировании в селективной среде. Селективной средой в принципе является среда, содержащая агент, который блокирует de novo синтез нуклеотидов в культуральной среде. Предпочтительными примерами таких агентов являются аминоптерин, метотрексат и азасерин. Аминоптерин и метотрексат блокируют синтез de novo как пуриновых, так и пиримидиновых оснований, в то время как азасерин блокирует лишь пуриновый синтез. При использовании аминоптерина или метотрексата в среду включают гипоксантин и тимидин в качестве источников нуклеотидов (т.н. НАТ-среда). При использовании азасерина в культуральную среду добавляют гипоксантин.
Предпочтительной селективной средой является НАТ-среда. Только те клетки, которые способны действовать по пути “спасения” нуклеотидов, способны выживать в НАТ-среде. Миеломные клетки дефектны по ключевым ферментам этого механизма, например, по гипоксантинфосфорибозилтрансферазе (HPTR), поэтому они выживать не могут. В-клетки могут действовать по этому механизму, но они характеризуются ограниченной продолжительностью жизни в культуре и обычно погибают в течение 2 недель. Поэтому в селективных средах могут выживать только клетки-гибриды миеломных и В-клеток.
В результате такого культивирования получают популяцию гибридом, из которой отбирают специфичные гибридомы. Обычно отбор гибридом проводят путем культивирования клеток в моноклональном разведении на микротировальных планшетах с последующим тестированием супернатантов отдельных клонов (примерно через 2-3 недели) на искомую активность. Такой тест должен быть чувствительным, простым и оперативным, таким как радиоиммунологический тест, ферментный иммунологический тест, тест на цитотоксичность, тест на образование бляшек, дот-блот-иммуноанализ и т.п.
Отобранные гибридомы затем должны быть подвергнуты серийному разведению и клонированы в отдельные линии антителопродуцирующих клеток, а затем эти клоны неограниченно воспроизводят с получением моноклональных антител (mAbs). Клеточные линии можно использовать для получения mAb двумя основными способами. Образец гибридомных клеток может быть инъецирован (обычно в брюшную полость) гистосовместимому животному того вида, который был использован как источник соматических и миеломных клеток для исходной гибридизации. У инъецированного животного развиваются опухоли, секретирующие специфическое моноклональное антитело, продуцируемое слитыми гибридными клетками. Затем физиологические жидкости этого животного, такие как сыворотка и асцитная жидкость, могут быть отобраны для получения из них mAbs в высокой концентрации. Отдельные клеточные линии могут быть также культивированы in vitro так, что mAbs будут естественным путем выделяться в культуральную среду, из которой их можно легко извлечь в высоких концентрациях. mAbs, полученные любым из этих способов, могут быть дополнительно очищены, если это желательно, с применением фильтрации, центрифугирования и различных хроматографических методов, таких как ВЭЖХ или аффинная хроматография.
(iii) Дополнительные мишени опухолевых клеток и связывающиеся лиганды
Помимо антител для доставки тритерпеновых соединений по настоящему изобретению к месту локализации опухоли за счет связывания с антигеном опухолевых клеток, могут быть использованы и другие лиганды. В случае опухолевых антигенов, являющихся сверхэкспресированными рецепторами (например, рецептором эстрогенов, EGF-рецептором) или мутантными рецепторами, направляющими агентами могут служить соответствующие лиганды.
Аналогично лигандам рецепторов эндотелиальных клеток, могут существовать компоненты, которые специфически или предпочтительно связываются опухолевыми клетками. Например, если опухолевый антиген является сверхэпрессированным рецептором, то опухолевая клетка может быть покрыта специфичным лигандом in vivo. Следовательно, затем такой лиганд может быть помечен либо специфичным для него антителом, либо с помощью самого рецептора. Конкретными примерами такого типа метящих (направляющих) агентов являются антитела против лигандов TIE-1 или TIE-2, антитела против тромбоцитарного фактора-4 и белка адгезии лейкоцитов.
(iv) Токсины
В некоторых вариантах предусматривается, что вторые терапевтические агенты, применяемые в сочетании с тритерпеновыми соединениями, описываемыми в данной заявке, должны быть фармакологическими агентами, соединенными с антителами или факторами роста, в частности, цитотоксическими или обладающими иной противоклеточной активностью агентами, способными уничтожать, подавлять рост или деление эндотелиальных клеток. В общих чертах, настоящее изобретение относится к применению любого фармакологического агента, вместе с и в дополнение к описываемым здесь тритерпеновым соединениям, который может быть конъюгирован с направляющим агентом, предпочтительно, с антителом, и доставлен в активной форме к опухолевым клеткам-мишеням. Примерами антиклеточных агентов являются химиотерапевтические агенты, радиоактивные изотопы, а также цитотоксины. В случае химиотерапевтических агентов заявители считают, что особенно предпочтительными являются такие агенты, как стероидные гормоны; антиметаболит, такой как цитозинарабинозид, фторурацил, метотрексат или аминоптерин; антрациклин; митомицин-С; алкалоиды барвинка;
демекальцин; этопозид; митрамицин; или противоопухолевый алкилирующий агент, такой как хлорамбуцил или мелфалан. Другие варианты могут включать такие агенты, как цитокин, фактор роста, бактериальный эндотоксин или А-липидная составляющая бактериального эндотоксина. В любом случае считается, что такие агенты, как перечисленные выше, если это желательно, могут вместе с тритерпеновыми соединениями по настоящему изобретению с успехом связываться с направляющими агентами, предпочтительно с антителом, таким образом, чтобы обеспечивалась их направленная доставка, интернализация, секреция или презентация компонентам крови в месте локализации клеток-мишеней (см., например, Ghose et al., 1983; Ghose et al., 1987).
Ряд химиотерапевтических и других фармакологических агентов в настоящее время успешно конъюгируют с антителами с подтверждением их фармакологической функциональности (см., например, Vaickus et al., 1991). Примерами противоопухолевых агентов, которые были в этом отношении исследованы, являются доксорубицин, дауномицин, метотрексат, винбластин и другие (Dillman et al., 1988; Pietersz et al., 1988). Более того, было описано присоединение других агентов, таких как неокарциностатин (Kimura et al., 1983), макромицин (Manabe et al., 1984), тренимон (Ghose, 1982) и α-аманитин (Davis & Preston, 1981). Конкретные способы получения конъюгатов тритерпеновых соединений по настоящему изобретению с подходящими направляющими молекулами конкретно описаны выше.
VII. Другие способы использования соединений по настоящему изобретению
Заявителями конкретно рассматривается использование соединений по настоящему изобретению для ряда других применений помимо лечения или профилактики раковых заболеваний. В частности, заявители рассматривают использование тритерпеновых соединений по настоящему изобретению в качестве растворителей, противогрибковых и противовирусных агентов, ихтиоцидов или моллюскоцидов, контрацептивов, противогельминтозных средств, средств защиты от УФ-облучения, отхаркивающих средств, мочегонных средств, противовоспалительных агентов, регуляторов метаболизма холестерина, эффекторов сердечно-сосудистой системы, противоязвенных средств, болеутоляющих средств, седативных средств, иммуномодуляторов, жаропонижающих средств, регуляторов ангиогенеза, в качестве агентов для снижения ломкости капилляров, в качестве агентов для борьбы с проявлениями старения и в качестве агентов для улучшения познавательной способности и памяти.
Соединения по настоящему изобретению играют определенную роль в регуляции ангиогенеза. Ангиогенез или неоваскуляризация определяется как образование новых кровеносных сосудов. Опухоли и рак индуцируют ангиогенез с целью обеспечения жизнедеятельности опухоли за счет снабжения их кислородом и питательными веществами. Развитие новых кровеносных сосудов также предоставляет раковым клеткам возможность выхода в другие части тела. Следовательно, подавление ангиогенеза должно иметь позитивный эффект для раковых больных. С другой стороны, иногда ангиогенез бывает необходим, например, при заживлении ран. Подобными повреждениями могут быть наружные раны или раны внутренних органов, причинами которых являются несчастные случаи, ожоги, травмы и хирургическое вмешательство. Таким образом, агенты, стимулирующие ангиогенез, имеют важное значение для заживления ран.
Рассматривается также применение соединений по настоящему изобретению для модуляции метаболизма холестерина. В частности, соединения и нутрицевтические композиции по настоящему изобретению могут быть использованы для снижения уровней холестерина в сыворотке у больных. Следовательно, при лечении больных с использованием тритерпеновых композиций по настоящему изобретению либо при пероральном, либо при внутривенном введении можно надеяться, что смертность, ассоциированная с повышенным уровнем холестерина и связанными с этим сердечно-сосудистыми заболеваниями, будет снижена.
Считается, что для лечения сердечно-сосудистых заболеваний, соединения по настоящему изобретению можно использовать для лечения аритмии и также в качестве сосудистых релаксантов, обусловливающих гипотензивную активность.
Другим особенно важным применением соединений по настоящему изобретению является их использование в качестве противовоспалительного агента. Было показано, что активные тритерпеновые соединения по настоящему изобретению являются сильными ингибиторами транскрипционного фактора NF-кВ, который играет важную роль в формировании воспалительного ответа. Этот факт имеет особо важное значение, если учесть все возрастающее количество данных, свидетельствующих о центральной роли воспалительного ответа в процессах канцерогенеза. Лечение больных с использованием тритерпеновых соединений, заявляемых здесь, может, следовательно, облегчать широкий круг болезненных состояний, связанных с воспалениями, включая образование опухолей и повреждения тканей.
Начальные фазы воспалительного ответа характеризуются повышением проницаемости кровеносных сосудов и высвобождением (экссудацией) гистамина, серотонина и основных полипептидов и белков. Это сопровождается гиперемией и образованием отеков. После этого происходит клеточная инфильтрация и формирование новой соединительной ткани. Считается, что применение соединений по настоящему изобретению может ограничить эти ранние фазы воспаления, и тем самым уменьшить отрицательные эффекты, связанные с воспалительным состоянием.
Вид растений, из которого были выделены соединения по настоящему изобретению, Acacia victoriae, был выбран, в частности, потому, что природной областью его произрастания являются засушливые зоны. Важной функцией метаболизма растений в этих районах является продуцирование соединений, которые защищают клетки от ультрафиолетового облучения. Заявители, в частности, считают, что тритерпеновые соединения по настоящему изобретению могут служить в качестве таких средств защиты от УФ-излучения. Следовательно, можно считать, что соединения по настоящему изобретению найдут широкое применение в случаях, когда желательной является защита от ультрафиолетового облучения. Например, такое применение предусматривает использование тритерпеновых соединений по настоящему изобретению в качестве компонентов средств против загара и других подобных лосьонов, предназначенных для нанесения на кожу человека.
Потенциальное достоинство такой композиции определяется хемопротективными эффектами благодаря соединениям по настоящему изобретению. Следовательно, лосьоны и средства от загара, содержащие тритерпеновые соединения по настоящему изобретению, в частности, будут целесообразны для применения индивидуумами, характеризующимися предрасположенностью к раку кожи. Примерами таких лиц являются люди с чувствительной кожей или индивидуумы с генетической предрасположенностью к раку кожи. Формы такой предрасположенности включают наследуемые мутации в онкогенах и нарушения клеточных механизмов, опосредующих репарацию ДНК после УФ-индуцированных повреждений. В частности, существенны мутации в генах, регулирующих процессы репарации, например, вырезание УФ-индуцированных тимин-тиминовых димеров. Аналогичным образом, соединения по настоящему изобретению могут быть добавлены в состав любой композиции, для которой желательна улучшенная защита от УФ-излучения, и эти соединения применимы для любого живого или неодушевленного объекта, для которого предусматривается защита от УФ.
Другими возможными применениями тритерпеновых соединений по настоящему изобретению являются защита от повреждений центральной нервной системы, например, при потере памяти или для повышения познавательной способности, использование в качестве антиоксиданта (для контроля уровней окислителей в крови) или повышение количества оксида азота (NO), необходимого при лечении гипертензии или атеросклероза. Кроме того, заявители, в частности, рассматривают местное применение тритерпеновых соединений по настоящему изобретению для усиления половой функции. Также рассматривается местное применение соединений по настоящему изобретению для повышения содержания кожного коллагена, что связано с преодолением эффектов старения кожи.
IX. Тесты и способы скрининга активных соединений
Специалистам в данной области техники известен ряд тестов, которые могут быть использованы для дальнейшей характеризации тритерпеновых соединений по настоящему изобретению. Они включают тесты на биологическую активность, а также тесты на химические свойства. Результаты этих тестов позволяют сделать важные выводы о свойствах соединений, а также об их возможном применении для лечения человека и других млекопитающих. Тесты, предположительно применимые для этих целей, включают скрининг биологической активности in vivo и in vitro и иммунологические тесты.
(i) Тесты in vivo
Настоящее изобретение предусматривает использование различных животных с моделью заболевания. В данном случае идентичность, имеющаяся между мышью и человеком, представляет отличную возможность протестировать функцию потенциального терапевтического агента, например, тритерпенового соединения по настоящему изобретению. Можно использовать опухолевые мышиные модели, которые характеризуются хорошей предсказательностью для раковых опухолей у человека и других млекопитающих. В этих моделях можно проводить ортотопическое или системное введение опухолевых клеток с целью имитации первичного и/или метастазирующего рака. Альтернативно, раковые опухоли можно индуцировать у животных, воздействуя на них агентами, о которых известно, что они участвуют в процессах злокачественной трансформации и/или развития опухоли.
Лечение животных с использованием тестируемых соединений предусматривает введение этого соединения данному животному в подходящей форме. Введение должно осуществляться любым из путей, которые могут использоваться для клинических или внеклинических целей, включая, но не ограничиваясь ими, пероральное, интраназальное, трансбуккальное, ректальное, вагинальное или местное введение. Альтернативно, введение может быть осуществлено путем внутритрахейной или бронхиальной инсталляции (закапывания), внутрикожной, подкожной, внутримышечной, внутрибрюшинной или внутривенной инъекции. В частности, предусматривается внутривенная системная инъекция, введение в конкретный участок через кровь или лимфу и внутриопухолевая инъекция.
Определение эффективности соединения in vivo может базироваться на различных критериях. Такими критериями являются, но не ограничиваются ими, выживаемость, снижение предрасположенности к опухоли или ее массы, прекращение или замедление развития опухоли, рассасывание опухолей, подавление или предотвращение метастазов, повышение уровня активности, улучшение эффекторной иммунологической функции и улучшение усвоения пищи.
Одним из конкретно применимых тестов in vivo на противоопухолевую активность является использование мышиной кожной модели. Мышиная кожная модель, которая представляет собой одну из наиболее хорошо изученных экспериментальных моделей многостадийного канцерогенеза, позволяет выделить три отчетливые фазы развития рака: инициацию, активацию и развитие (прогрессирование). В настоящее время известно, что клеточная эволюция малигнизации включает последовательные альтерации протоонкогенов и/или опухоль-супрессорных генов, продукты которых участвуют в главных механизмах передачи сигналов и/или регуляции экспрессии генов. Фазы активации и развития кожных опухолей характеризуются избирательной и постоянной гиперплазией, изменениями дифференцировки и генетической нестабильностью, приводящей к специфической пролиферации инициированных клеток с образованием папиллом и карцином. Было показано, что индукция стойкой гиперплазии отчетливо коррелирует с активностью различных агентов, стимулирующей развитие опухолей кожи, таких как форболовые сложные эфиры, некоторые перекиси и хризаробин. Было показано, что в мышиной кожной модели все известные канцерогены и опухолевые активаторы вызывают стойкую гиперплазию эпидермиса. В целом, это предвосхищается развитием воспалительного ответа.
Обширные данные указывают на четкую корреляцию канцерогенности и мутагенности. Многие опухоль-инициирующие агенты либо генерируют, либо метаболически преобразуются в элекрофильные агенты, способные ковалентно связываться с клеточной ДНК. Некоторые свободные радикалы и модифицированные основания ДНК, являющиеся свободными радикалами, участвуют в инициации опухоли и/или активации опухоли в стадиях канцерогенеза. Было доказано, что активация гена Ha-ras происходит на ранних стадиях кожного канцерогенеза у мышей, что, по-видимому, эквивалентно событию инициации. Например, было показано, что присутствие активированного гена c-Ha-ras в кожных папилломах мыши, вызванных действием 7,12-диметилбенз[а]антрацена, ассоциировано с высокой частотой трансверсий А→Т в 61-м кодоне. Последующие исследования показали, что тип этой мутации зависит от природы химического инициатора и не зависит от активатора, что позволяет предположить о наличии прямого влияния индуктора на ген с-На-ras. Более того, инфицирование мышиной кожи геном На-rаs, активированным вирусом (v-Ha-ras), может служить в качестве инициирующего события в двухстадийном канцерогенезе. Необходимо отметить, что все кожные химические канцерогены и индукторы опухолей кожи, как было показано, продуцируют мутации в онкогене H-ras. Однако, активаторы кожных опухолей не приводят к мутации гена На-rаs.
(ii) Подтверждающие тесты In vivo и клинический анализ
Для специалиста в данной области техники должно быть понятно, что химиотерапевтические агенты, включая тритерпеновые соединения по настоящему изобретению, или их комбинации с дополнительными агентами должны быть в целом протестированы в моделях in vivo перед введением человеку. Такое преклиническое испытание на животном является стандартным методом в данной области. Для проведения таких подтверждающих тестов, все что требуется - это подходящее с научной точки зрения животное с моделью анализируемого заболевания, такое как животное, имеющее твердую опухоль. В данном контексте может быть использовано любое животное, такое как, например, мышь, крыса, морская свинка, хомячок, кролик, собака, шимпанзе и т.д. С точки зрения лечения раковых заболеваний, исследования с использованием мелких животных, таких как мыши, широко распространены с точки зрения предсказуемости клинической эффективности по отношению к человеку, а, следовательно, такие животные модели являются предпочтительными с точки зрения настоящего изобретения, поскольку они легкодоступны и относительно недороги, по крайней мере, по сравнению с другими экспериментальными животными.
Способ проведения теста на экспериментальном животном может быть разработан самим специалистом в данной области техники. Все что необходимо для проведения такого теста, это создать эквивалентно обрабатываемые группы и ввести тестируемые соединения одной группе при параллельном проведении различных форм контроля на эквивалентных животных в другой группе (группах). В ходе исследования осуществляют непрерывный мониторинг животных, после чего животных умерщвляют для анализа эффекта обработки.
Одним из главных применений настоящего изобретения является лечение рака. Следовательно, противоопухолевые тесты могут быть проведены с целью определения конкретных эффектов в отношении сосудистой системы опухолей и общих противоопухолевых эффектов. В качестве части таких исследований, необходимо также проводить мониторинг на специфичность таких эффектов, включая общее состояние этих животных.
Что касается лечения твердых опухолей, то считается, что эффективные количества тритерпеновых соединений по настоящему изобретению должны быть такими, чтобы они обеспечивали гибель или апоптоз, по крайней мере, 10% клеток в опухоли. Предпочтительно должно быть уничтожено, по крайней мере, примерно 20%, примерно 30%, примерно 40% или примерно 50% клеток в конкретном участке локализации опухоли. Наиболее предпочтительно должно быть уничтожено 100% клеток в участке локализации опухоли.
Уровень гибели клеток в опухоли оценивают по отношению к уровню сохранения здоровых тканей во всех участках тела. Предпочтительным должно быть использование доз соединений по настоящему изобретению, способных индуцировать некроз опухоли, по крайней мере, примерно на 60%, примерно на 79%, примерно на 80%, примерно на 85%, примерно на 90%, примерно на 95% и включая 100%, причем, используемые дозы не должны вызывать значительных побочных эффектов или иных неблагоприятных воздействий на животное. Все такие определения могут быть легко осуществлены и точно оценены специалистом в данной области техники. Например, медперсонал, ученые и врачи могут использовать такие данные, полученные на экспериментальных животных для оптимизации доз, походящих для лечения человека. У больных с запущенным заболеванием возникновение побочных эффектов на определенном уровне можно не принимать во внимание. Однако, для лечения больных на ранних стадиях заболевания можно применять более умеренные дозы с целью достижения значимого терапевтического эффекта при отсутствии побочных эффектов. Результаты таких исследований на экспериментальных животных должны быть статистически значимыми по сравнению с контрольными уровнями и должны быть воспроизводимыми в разных анализах.
Кроме того, для специалистов в данной области техники должно быть понятно, что комбинации и дозы соединений по настоящему изобретению, которые вызывают специфический некроз опухоли, в соответствии с настоящим изобретением могут быть использованы, начиная от нижней границы эффективного диапазона. Например, в вариантах, в которых предусматривается непрерывное применение активных агентов, исходная доза, которая приводит примерно лишь к 10%-ному уровню некроза, тем не менее применима, в частности, потому, что часто отмечается свойство такой исходной дозы стимулировать деструктивные процессы в опухоли, усиливаемые при последующем возобновлении лечения. В любом случае, даже если в конечном счете и не достигается примерно 40%-ное подавление опухоли, тем не менее очевидно, что может быть использована любая индукция тромбоза и некроза, если только она обеспечивает улучшение состояния пациентов перед проведением лечения. Более того, предполагается, что доза соединений по настоящему изобретению, которая предотвращает или уменьшает вероятность либо метастазирования, либо канцерогенеза de novo, также должна оказывать терапевтическое благоприятное воздействие на пациента, проходящего курс лечения.
Как обсуждалось выше в связи с тест-системой in vitro, очевидно, что комбинации агентов, предназначенные для совместного применения, должны быть протестированы и оптимизированы. Соединения по настоящему изобретению могут быть непосредственно проанализированы в комбинации с одним или несколькими химиотерапевтическими агентами, иммунотоксинами, коагулянтами и т.п. Должен быть проведен анализ комбинированного действия таких агентов и его результаты должны быть оценены в соответствии с приведенными выше описаниями.
(iii) Тесты in vitro
В одном из вариантов настоящего изобретения, скрининг растительных экстрактов проводят in vitro с целью идентификации тех соединений, которые способны подавлять рост или уничтожать опухолевые клетки. Уничтожение опухолевых клеток, или цитотоксичность, обычно происходит благодаря некрозу или апоптозу. Некроз является достаточно обычным механизмом, “запускаемым” внешними сигналами. В этом процессе утрачивается целостность клеточных мембран и клеточных компартментов. С другой стороны, апоптоз, или генетически запрограммированная гибель клеток, является высокоорганизованным процессом, включающим морфологические события, которые синхронизованы активацией и дезактивацией специфических генов (Thompson et al., 1992; Wyllie, 1985).
Эффективный способ тестирования цитотоксичности in vitro включает систематизированное воздействие отобранных растительных экстрактов на панель опухолевых клеток. Такие тесты и линии опухолевых клеток, пригодные для проведения таких тестов, хорошо известны специалистам в данной области техники. Конкретно предпочтительными линиями опухолевых клеток человека для применения в тестах in vitro на противоопухолевую активность, являются линии клеток рака яичника человека SKOV-3, HEY, OCC1 и OVCAR-3; Т-лейкозные клетки Jurkat; линия опухоли молочной железы человека MDA-468; клетки рака предстательной железы человека LNCaP; линии меланомных клеток человека А375-М и Hs294t; и клетки рака почки человека 769-Р, 786-0, А498. Предпочтительным типом нормальных клеток для использования в качестве контроля являются клеточные линии фибробластов крайней плоти человека FS или Hs27.
Анализ in vitro эффективности соединения по уничтожению опухолевых клеток может быть проведен, например, с помощью тестов на экспрессию и индукцию различных генов, участвующих в блокировке клеточных циклов (р21; р27; ингибиторы циклин-зависимых киназ) и в апоптозе (bcl-2, bcl-XL и bax). Для проведения такого теста клетки обрабатывают тестируемым соединением, лизируют, выделяют белки и затем анализируют методом электрофореза в ДСН-ПААГ, а связанные в геле белки переносят на нитроцеллюлозные мембраны. Вначале эти мембраны зондируют с использованием первичных антител (например, антител к белкам р21, р27, bax, bcl-2 и bcl-ХL, и т.д.) и затем детектируют с помощью разведенных вторичных антител, конъюгированных с пероксидазой хрена, и эти мембраны экспонируют хемолюминесцентным (ЭХЛ) детекционным реагентом, с последующей визуализацией на ЭХЛ-фотопленке. Посредством анализа на относительное содержание данных белков можно оценить процент клеток, находящихся на данной стадии клеточного цикла, например, стадии G0/G1, стадии S или стадии G2/M.
Цитотоксичность соединения в отношении раковых клеток также может быть эффективно оценена in vitro с использованием окрашивания МТТ или кристаллическим фиолетовым. В этом методе клетки высевают, обрабатывают различными концентрациями тестируемых соединений, инкубируют и окрашивают либо МТТ, (3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолийбромид, Sigma Chemical Co.), либо кристаллическим фиолетовым. На обработанные МТТ чашки наносят лизирующий буфер (20% додецилсульфат натрия в 50% DMF) и проводят дополнительное инкубирование с последующим считыванием оптической плотности (ОП) при 570 нм. Окрашенные кристаллическим фиолетовым чашки промывают для удаления красителя в буфере Соренсена (0,1 М цитрата натрия, рН 4,2; 50% этанол по объему) и считывают при 570-600 нм (Mujoo et al., 1996). Уровни относительных значений оптической плотности являются показателем суммарной цитотоксичности.
(iv) Иммунологические тесты
Иммунологические тесты могут найти применение в связи с настоящим изобретением, например, для скрининга экстрактов растений других видов, не являющихся акацией Виктория Acacia victoriae, на присутствие тритерпеновых соединений по настоящему изобретению. Охватываемые настоящим изобретением иммунологические тесты включают, не ограничиваясь ими, те тесты, которые были описаны в патенте США №4367110 (“сэндвич”-тест с двумя моноклональными антителами) и в патенте США №4452901 (Вестерн-блоттинг). Другими тестами являются иммунопреципитация меченых лигандов и иммуноцитохимические методы, как in vitro, так и in vivo.
В самом общем и прямом смысле, иммунологические тесты являются тестами на связывание. Некоторыми предпочтительными иммунологическими тестами являются различные варианты твердофазных иммуноферментных анализов (ELISA) и радиоиммунологических анализов (РИА), известные специалистам. В частности, также эффективными являются иммуноцитохимические методы детекции с использованием гистологических препаратов.
В одном характерном варианте ELISA антитритерпеновые антитела иммобилизуют на выбранную подложку, обладающую аффинностью в отношении белков, такую как лунка полистиролового микротитровального планшета. Затем в лунку добавляют тестируемую композицию, предположительно содержащую тритерпеновые соединения по настоящему изобретению, такую как экстракт растения, родственного Acacia victoriae. После связывания и промывки с целью удаления неспецифических иммунных комплексов связанный антиген может быть детектирован. В принципе, детекцию проводят путем добавления другого антитела, специфичного в отношении желательного антигена и присоединенного к детектируемой метке. Такой вариант ELISA представляет собой простой “сэндвич-ELISA””. Детекция может быть также осуществлена путем добавления второго антитела, специфичного в отношении желаемого антигена с последующим добавлением третьего антитела, которое аффинно по связыванию со вторым антителом, причем, третье антитело соединено с детектируемой меткой.
Модификации методов ELISA известны специалистам в данной области техники. В одном таком варианте образцы, проверяемые на содержание желаемого антигена, иммобилизуют на поверхности лунки и затем приводят в контакт с препаратом антител. После связывания и подходящей промывки детектируют образовавшиеся иммунные комплексы. Если исходные антиген-специфичные антитела соединены с детектируемой меткой, то иммунные комплексы могут быть детектированы прямым методом. И в этом случае, иммунные комплексы могут быть детектированы с использованием второго антитела, которое обладает аффинностью в отношении первого антиген-специфичного антитела, причем, данное второе антитело соединено с детектируемой меткой.
Также могут быть использованы конкурентные ELISA: в них тестируемые образцы конкурируют по связыванию с известными количествами меченых антигенов или антител. Это количество реактивных молекул в неизвестном образце определяют путем смешивания этого образца с известным количеством меченых молекул до или в процессе инкубации в сенсибилизированных лунках. Присутствие молекул реактивного вида в данном образце обусловливает снижение количества меченых молекул, связанных с лункой, что в конечном счете приводит к ослаблению конечного сигнала.
Независимо от применяемого формата, для ELISA характерен ряд общих стадий, таких как сенсибилизация, инкубирование или связывание, промывка с целью удаления не специфически связанных молекул и детекция связанных иммунных комплексов. Эти стадии охарактеризованы ниже.
Антиген или антитела также могут быть присоединены к твердой подложке, такой как планшет, шарики, палочка, мембрана или колоночный носитель, а предназначенный для анализа образец наносят на иммобилизованный антиген или антитело. Для сенсибилизации планшета либо антигеном, либо антителом, обычно, лунки этого планшета инкубируют с раствором антигена или антитела либо в течение ночи, либо в течение определенного периода времени. Затем лунки планшета надо промыть с целью удаления не полностью адсорбировавшегося материала. Затем любые оставшиеся после этого доступные поверхности на лунках “покрывают” неспецифическим белком, являющимся антигенно нейтральным по отношению к анализируемым антисывороткам. Таковыми являются бычий сывороточный альбумин (БСА), казеин и растворы сухого молока. Это покрытие обеспечивает блокировку сайтов неспецифического связывания на иммобилизованной поверхности, что тем самым снижает фон, обуславливаемый неспецифическим связыванием антисыворотки на данной поверхности.
При проведении ELISA, по-видимому, более привычно использовать способы вторичной или третичной детекции в отличие от прямого способа. Так, после связывания антигена или антитела на лунке, покрытия нереактивным материалом с целью подавления фона и промывки с целью удаления несвязанного материала, иммобилизирующую поверхность подвергают контакту с анализируемым клиническим или биологическим образцом в условиях, эффективных для обеспечения образования иммунного комплекса (антиген/антитело). Затем, для детекции иммунного комплекса необходимо использовать меченые вторичносвязывающийся лиганд или антитело, или вторично связывающийся лиганд или антитело, конъюгированные с меченым “третичным” антителом или третьим связывающимся лигандом.
Выражение “в условиях, эффективных для образования иммунного комплекса (антиген/антитело)” означает, что предпочтительны такие условия, которые включают разведение антигенов и антител такими растворами, как растворы БСА, бычьего гамма-глобулина (БГГ) или забуференного фосфатом физиологического раствора (ЗФР)/Твин. Такие добавленные агенты также способствуют снижению уровня неспецифического фона.
Подходящие условия также означают, что инкубацию проводят при такой температуре и в течение такого периода времени, которые достаточны для обеспечения эффективного связывания. Стадии инкубации обычно продолжаются примерно от 1-2 до 4 часов, температура предпочтительно составляет порядка 25-27°С, или они могут быть проведены в течение ночи при примерно 4°С или т.п.
После всех стадий инкубации в ELISA, проконтактировавшую поверхность промывают с целью удаления не образовавшего комплекс материала. Обычно для промывки используют раствор ЗФР/Твин или боратный буфер. После образования специфичных иммунных комплексов между анализируемым образцом и исходным связанным материалом и последующей промывки может быть выявлено даже очень небольшое количество иммунных комплексов.
Для проведения детекции, ко второму или третьему антителу должна быть присоединена метка. Предпочтительно это должен быть фермент, который способствует развитию окраски в результате инкубации с соответствующим хромогенным субстратом. Так, например, первый или второй иммунные комплексы могут быть подвергнуты контакту и инкубации с антителом, конъюгированным с уреазой, глюкозоксидазой, щелочной фосфатазой или гидропероксидазой в течение определенного периода времени и в условиях, которые благоприятствуют образованию следующего иммунного комплекса, например, путем инкубации в течение 2 часов при комнатной температуре в ЗФР-содержащем растворе, таком как ЗФР/Твин.
После инкубации с меченым антителом и промывки с целью удаления несвязанного материала проводят количественную оценку метки, например, путем инкубации с хромогенным субстратом, таким как мочевина и бромкрезоловый пурпурный или 2,2’-азино-ди-3-этилбензтиазолин-6-сульфоновая кислота (ABTS) и Н2О2, если ферментной меткой является пероксидаза. Количественную оценку затем осуществляют по степени окрашивания, например, используя спектрофотометр в видимом спектре. Альтернативно, эта метка может быть хемолюминесцентной. Применение таких меток описано в патентах США №5310687, 5238808 и 5221605.
Методы анализа in vitro и in situ хорошо известны и включают оценку связывания антиген-специфических антител с тканями, клетками или клеточными экстрактами. Специалистам в данной области техники известны стандартные методы подобного типа. Например, антитела, специфичные в отношении опухолевых антигенов, могут быть использованы в сочетании как со свежезамороженными, так и фиксированными в формалине, залитыми в парафин тканевыми блоками, приготовленными для иммуноцитохимического анализа (ИЦХА). Каждый тканевый блок может включать 50 мг остаточной “рассеянной” опухоли. Способ приготовления тканевых блоков, полученных из образцов, состоящих их частиц, ранее был уже успешно применен в ИЦХА для различных прогностических оценок, например, в случаях опухоли молочной железы, т.е. он хорошо известен специалистам в данной области техники.
Кратко, замороженные срезы могут быть приготовлены путем замачивания 50 нг замороженной рассеянной опухоли при комнатной температуре в ЗФР в небольших пластиковых капсулах; осаждения частиц путем центрифугирования; их ресуспендирования в вязкой заливочной среде; выворачивания капсулы и повторного осаждения путем центрифугирования; мгновенного замораживания в изопентане при -70°С; отрезания пластиковой капсулы и выделения замороженной ткани в форме цилиндра; закрепления этого цилиндра в патроне криостатного микротома; и изготовления 25-50 последовательных срезов, в каждый из которых в среднем попадает примерно по 500 явно интактных опухолевых клеток.
Непрерывные срезы могут быть приготовлены аналогичным образом, включая повторное гидратирование 50 мг образца в пластиковой микроцентрифужной пробирке; центрифугирование; ресуспендирование в 10%-ном формалине с 4-часовой фиксацией; промывку/центрифугирование; ресуспендирование в теплом 2,5%-ном агаре; центрифугирование; охлаждение в ледяной воде для отверждения агара; выделение тканево-агарового блока из пробирки; процеживание блока и заливку его в парафин; и приготовление до 50 последовательно-перманентных срезов.
В свете настоящего изложения можно использовать скрининг-тесты, направленные на идентификацию соединений, обладающих по сути одинаковыми химическими характеристиками и биологической активностью по сравнению с тем, что описано в данном тексте. В частности, в настоящей заявке можно использовать тесты на биологическую активность тритерпеновых гликозидов тех растений, которые близкородственны Acacia victoriae, например, представителей рода акаций, Acacia. Применение этих тестов может иметь различные форматы, что может определяться тем типом активности, скрининг которой осуществляется. Предпочтительными являются те тесты, в которых исследуется противоопухолевая активность, такая, как была описана здесь для экстрактов Acacia victoriae. Используемый в настоящем описании термин “противоопухолевая активность” означает подавление межклеточных сигналов, роста, метастазов, деления клеток, миграции клеток, формирования колоний в мягком агаре, подавление контактов, инвазионности, ангиогенеза, развития опухоли или другого злокачественного фенотипа или индукцию апоптоза. В частности, рассматриваются функциональные тесты, в которых проводят анализ на использование соединений по настоящему изобретению в качестве противогрибковых и противовирусных агентов, ихтиоцидов или моллюскоцидов, контрацептивов, противогельминтных средств, средств для защиты от УФ-излучения, отхаркивающих средств, мочегонных средств, противовоспалительных средств, регуляторов метаболизма холестерина, сердечно-сосудистых эффекторов, противоязвенных средств, болеутоляющих средств, седативных средств, иммуномодуляторов, жаропонижающих средств, регуляторов ангиогенеза и средств, снижающих ломкость капилляров. В свете настоящего описания подобные тесты хорошо известны специалистам в данной области техники. Как и при прямом тесте на активности in vitro и in vivo, данные тесты могут включать анализ подавления связывания с субстратом, лигандом, рецептором или другими партнерами по связыванию с соединением по настоящему изобретению.
X. Рост и тканевые культуры Acacia victoriae
Важным аспектом получения соединений по настоящему изобретению является доступность ткани Acacia victoriae. Т.к. заявителями было показано, что соединения по настоящему изобретению концентрируются в корнях и бобах Acacia victoriae, то доступность именно этих тканей является наиболее важной.
Также заявителями было показано, что другим источником для выделения соединений по настоящему изобретению являются молодые проростки. Acacia victoriae растет на юго-западе США и в Австралии, т.е. растительный материал доступен людям. Кроме того, примерно 2500 семян Acacia victoriae было 7 мая 1998 г. внесено заявителями в Американскую коллекцию типовых культур (АТСС: 10801 University blvd., Manassas, VA 20110-2209, США). Этим депонированным семенам присвоен номер доступа в АТСС №209835. Они были депонированы в соответствии со сроками и условиями, определяемыми Будапештским соглашением о депонировании микроорганизмов, на срок, по крайней мере, тридцать (30) лет и на срок, по крайней мере, пять (05) лет после самого последнего запроса на предоставление депонированного образца, полученного данным депозитарием, или на срок действия настоящего патента, независимо от его продолжительности, причем этот материал подлежит замене в случае, если он потеряет свою жизнеспособность в течение указанного периода.
Таким образом, в свете настоящей заявки специалист в данной области техники может высадить семена из указанного депозитария, вырастить из них растения и выделить ткань из полученных растений с целью получения тритерпеновых соединений и нутрицевтических композиций по настоящему изобретению. Также можно выделить ткань из растений природных популяций Acacia victoriae. Однако получение ткани для выделения соединений по настоящему изобретению может быть достигнуто более легким путем, если для воспроизведения ткани Acacia victoriae будет применена подходящая технология выращивания. Одним из условий получения такой ткани должно быть крупномасштабное выращивание данного вида. Однако, более предпочтительным выбором являются тканевые культуры Acacia victoriae и использование системы аэропоники для выращивания.
(i) Выращивание методами аэропоники
Ряд преимуществ может быть достигнут в случае применения методов аэропоники для выращивания Acacia victoriae. Во-первых, в этом случае, темп роста растений примерно в два раза превышает темпы роста, достигаемые при стандартных методах выращивания. Во-вторых, при необходимости, корни могут быть легко собраны без повреждения растения. Более того, отрезание корней приводит к интенсивному боковому росту мочковатой части корня. Следовательно, корни можно собирать по несколько раз за год. В популяциях Acacia victoriae дикого типа сбор бобов ограничен несколькими неделями в году, а сбор корней без повреждения или уничтожения растения весьма затруднен.
Аэропоническая система выращивания является закрытой системой, в которой корни растения находятся на воздухе и опрыскиваются полноценной питательной средой. Корни закрывают в водонепроницаемой коробке и через определенные периоды времени опрыскивают питательной средой. Питательный раствор содержит все необходимые компоненты, обеспечивающие этим растениям полный жизненный цикл. Несмотря на то, что для различных растений требуются разные уровни и составы для достижения оптимального роста, в целом, однократно сбалансированный питательный раствор дает удовлетворительные результаты.
(ii) Тканевые культуры Acacia victoriae
Получение тканевых культур представляют собой другой путь выращивания Acacia victoriae. Для формирования тканевых культур, семена Acacia victoriae тщательно промывают водопроводной водой с противомикробным мылом и обрабатывают 20%-ным раствором бытовой хлорной извести в течение 15 минут. После повторной промывки деионизованной водой, семена обрабатывают кипятком в целях стимуляции прорастания и инкубируют в течение ночи. На следующее утро семена вновь дезинфицируют бытовой хлорной известью и промывают 2-3 раза стерильной деионизованной водой. Затем обработанные семена культивируют в среде Мурасиге-Скоога (MS) (Murashige et al., 1962), дополненной MS-витаминами и 2% сахарозы (для культур эксплантатов используют добавление 3% сахарозы) и среду отверждают путем добавления 0,7% агара или 0,2% гельрита.
Используемые для культивирования эксплантаты представляют собой практически любую ткань, включая кончики побегов, сегменты узлов, гипокотили и сегменты корней. Обычно эксплантаты культивируют в чистой среде MS или в MS, дополненной регуляторами роста, такими как IAA, NAA, IBA, 2,4-D и ВАР (как по отдельности, так и в сочетании). Обычно культуры поддерживают при 25±2°С при светопериоде 16 часов с интенсивностью освещения 1000 люкс: для освещения использовали белые флуоресцентные лампы холодного свечения. Полученные проростки выдерживают в теплице при опрыскивании в течение 1 месяца до уплотнения ткани с последующим высаживанием в самой теплице, в открытый грунт или внесением в систему аэропоники.
В настоящем изобретении были сформированы культуры волосовидных корней Acacia victoriae. Развитие волосовидных корней обусловливается заражением растения клубеньковой бактерией Agrobacterium rhizogenes штамма R-1000, что приводит к интеграции в геном растения и экспрессии Т-ДНК. Культуры волосовидных корней отличаются быстрым ростом, плагиотропны и характеризуются интенсивным ветвлением корней в безгормональной среде, а также проявляют высокий уровень генетической стабильности (Aird et al., 1988). Генетическая трансформация и индукция волосовидных корней у Acacia victoriae и оптимальные условия для роста подробно описаны в одном из разделов в примерах. Культуры волосовидных корней обеспечивают быстрый крупномасштабный рост ткани, которая может быть использована для выделения тритерпеновых соединений по настоящему изобретению.
Достоинство тканевой культуры состоит в том, что могут быть сформированы клональные культуры, которые экспрессируют соединения по настоящему изобретению. Такие культуры могут быть выращены в большом объеме и могут быть размножены до промышленного масштаба для растительной ткани в целях выделения тритерпеновых соединений. Кроме того, регенерированные из этих культур растения обычно характеризуются существенной изменчивостью. Следовательно, с использованием этих культур могут быть получены клональные клеточные линии или регенерированные из таких культур растения, которые будут “элитными” с точки зрения получения из них тритерпеновых соединений по настоящему изобретению. Полученные растения могут быть воспроизведены путем самоопыления и отобраны в каждом поколении селекции с получением настоящих отселекционированных элитных линий.
Элитные сорта не обязательно должны быть сформированы на материале тканевых культур, однако, благодаря существенной генетической изменчивости, они существуют и в диких популяциях Acacia victoriae. Следовательно, заявители считают, что обнаруживаемая в природных популяциях Acacia victoriae генетическая изменчивость включает и изменчивость генов, контролирующих выработку эндогенных тритерпеновых соединений. Если так, то возможным является выявление в популяциях Acacia victoriae особей, которые характеризуются повышенными уровнями выработки тритерпенов по сравнению с другими особями этих природных популяций, а также возможен отбор таких линий для использования в системах выращивания, направленных на получение ткани для выделения тритерпеновых соединений по настоящему изобретению. Системой выращивания может являться, например, стандартное сельскохозяйственное выращивание, аэропоника, тканевое культивирование или любой подходящий метод воспроизводства ткани Acacia victoriae. Помимо того, эти растения могут быть отобраны для использования в селекционных мероприятиях для получения сортов, являющихся более элитными и также воспроизводимыми как гомозиготы.
XI. Определения
Артикль “а” означает “один или более”. Таким образом, слово “составляющая” (“a moiety”) может относиться к одной, двум, трем или большему числу составляющих.
“Активные компоненты” обозначают наиболее чистый экстракт, сохраняющий активность. В связи с настоящим изобретением термины “активный компонент” или “активное соединение” обозначают активные тритерпеновые соединения, идентифицированные авторами настоящего изобретения. Эти соединения были очищены и идентифицированы, например, во фракции UA-BRF-004-DELEP-F094.
“Бобы” обозначают плоды Acacia victoriae типа бобов.
Термин “цитотоксический” определяет гибель клеток, в то время как термин “цитостатический” определяет подавление роста и/или пролиферацию клеток.
“Апоптоз” определяется как нормальный физиологический процесс запрограммированной гибели клеток, который происходит в ходе эмбриогенеза и в ходе поддержания тканевого гомеостаза. Процесс апоптоза может быть подразделен на ряд метаболических преобразований в апоптотических клетках. Отдельные ферментативные каскады реакции в различных регуляторных механизмах или механизмах передачи сигнала могут быть определены в связи с выявлением апоптоза в клетке или клеточной популяции или определены в связи с утратой способности к апоптозу у раковых клеток. Также апоптотическая программа отражается на морфологии клетки, что включает изменения плазматической мембраны (например, утрату ее асимметричности), конденсацию цитоплазмы и ядра и межнуклеосомное расщепление ДНК (т.н. “пульверизация” хроматина). В результате происходит гибель клеток, превращающихся в т.н. “апоптотические тельца”.
Методы оценки различных ферментативных процессов и процессов передачи сигналов, участвующих в апоптозе, разработаны в виде стандартных протоколов многофакторного изучения апоптоза. Примером одного из маркеров ранней стадии апоптоза является высвобождение из митохондрий цитохрома-с с последующей активацией каспазного-3 пути (PharMingen, San Diego, CA). Индукция каспаз (группы цитоплазматических протеаз) является одним из наиболее существенных свойств апоптоза. В частности, каспаза-3 играет ключевую роль в данном процессе. После активации каспазы расщепляют белки-мишени:
одним из самых важных из них является RARP, поли-АДФ-рибозополимераза, локализованная в ядре. Следовательно, эффективное определение апоптоза может быть осуществлено в тестах, направленных на детекцию высвобождения цитохрома-с, детекцию каспазы-3 и детекцию разрушения RARP.
Более того, агенты, которые вызывают высвобождение цитохрома-с из митохондрий злокачественных клеток, могут быть определены как потенциальные лекарственные средства, нацеленные, по крайней мере, на некоторые аспекты клеточного контроля запрограммированной клеточной гибели.
Другим тестом на апоптоз является детекция аннексина-V (BioWhitaker, Walkerville, MD). Обычно фосфатидилсерин (ФС) расположен на внутренней поверхности плазматической мембраны. Однако, на ранних стадиях апоптоза происходит экстернализация ФС. Аннексин-V представляет собой кальций-связывающий белок, который связывается с ФС и может быть выявлен после окрашивания комплекса аннексин-У-ФИТЦ методом проточной цитометрии (Martin et al., 1995). Способность клеток, обработанных описываемыми в настоящем изобретении соединениями Acacia victoriae, связывать аннексин-V, принимается в качестве индикатора вступления клеток в апоптоз.
В других примерах заявителями был использован тест на киназу PI-3 и для выявления апоптотической активности клеток, обработанных смесями обладающих противоопухолевой активностью соединений, выделенных из Acacia victoriae. Фосфоинозитид-3-кинаэа (PI3K), являющаяся ассоциированным с клеточной мембраной ферментом, способна фосфорилировать инозитное кольцо фосфатидилинозитола по атому в 3-положении, что тем самым определяет новый липидный сигнальный путь в тех клетках, в которых активна PI3K. В случае активности Р13-киназы, другая киназа, обозначаемая АКТ, рекрутируется на мембрану клетки. АКТ кодируется онкогеном и каталитически активируется после присоединения к мембране. Полностью активированная киназа АКТ играет ключевую роль в выживании клеток. Взаимодействие PI3K/AKT представляет собой механизм, по которому клетки избегают апоптоза. Таким образом, способ подавления PI3K в клетках злокачественных опухолей обуславливает вероятное терапевтические средство для восстановления, по крайней мере, некоторых элементов клеточного контроля апоптоза.
Выражение “аномальная пролиферация” указывает на ряд генетически детерминированных изменений, которые происходят в клетках при патологическом состоянии, известное как раковое заболевание. В конце концов, данный процесс приводит к потере контроля за апоптозом в раковых клетках. Это может происходить постадийно и обычно обозначается как: (1) “инициация”, что определяет стадию, в которой внешний агент или стимул обуславливает генетическое изменение в одной или нескольких клетках; и (2) активация, определяемая как стадия, приводящая к дальнейшим генетическим и метаболическим изменениям, способным вызвать воспаление. В “стадии активации” клетки начинают “метаболический переход” в стадию клеточного роста, в которой апоптоз заблокирован.
“Злокачественные клетки” определяются как раковые клетки, лишенные нормальных механизмов контроля клеточного роста в результате серии метаболических изменений, происходящих в процессе инициации и активации стадий начала малигнизации. Эти изменения являются следствием генетических изменений в клетках (либо активирующие мутации и/или усиление экспрессии протоонкогенов, либо инактивирующие мутации и/или ослабление экспрессии одного или нескольких опухоль-супрессорных генов). Продукты большинства онкогенов и опухоль-супрессорных генов являются компонентами каскадов реакций передачи сигнала, которые контролируют вступление в клеточные деления или выход из них, дифференцировку, повреждения кодирующей ДНК и инициацию репарационных механизмов, и/или регулируют программы клеточной гибели. В клетках работают множественные параллельные механизмы регуляции роста, дифференцировки клеток, контроля повреждений ДНК и апоптоза. Практически все опухолевые и злокачественные клетки имеют мутации во множестве онкогенов и опухоль-супрессорных генов.
“Экстракт” или “фракция” обозначают последовательные образцы, получаемые различными способами из тканей. Такие “экстракты” или “фракции” могут быть проанализированы на желательную противоопухолевую активность, а также дополнительно “экстрагированы” или “фракционированы” для получения более чистых компонентов, соответствующих активному компоненту.
“Тритерпен” или “тритерпеновый гликозид” обозначает новые и/или биологически активные сапониновые соединения, идентифицированные здесь из Acacia victoriae. Тритерпены или тритерпеновые гликозиды не обязательно должны быть выделены из Acacia victoriae, т.к. любой специалист в данной области техники исходя из данных настоящего изобретения может выделить соединения из родственных видов или химически синтезировать аналоги тритерпенов и тритерпеновых гликозидов, описанных здесь. “Тритерпены” по настоящему изобретению включают сапониновые соединения, описанные в данном тексте, которые включают, по крайней мере, тритерпеновое(ые) звено(ья) и, в случае тритерпеновых гликозидов, также сахар или сахарид. Также данные термины охватывают соединения, включающие дополнительные составляющие или химические функциональные группы, включая, но тем самым, не ограничиваясь ими, монотерпеновые звенья, на что будет указано в другой части данной заявки. Таким образом, тритерпены по настоящему изобретению также включают агликоны, образующиеся в результате гидролиза сахарных составляющих, а также включают другие модификации тритерпеновых соединений, если только такие модификации не нарушают биологическую активность этих соединений.
XII. Примеры
Нижеследующие примеры приводятся для иллюстрации предпочтительных вариантов настоящего изобретения. Специалисту в данной области техники должно быть понятно, что способы, описанные в нижеследующих примерах, являются способами, позволяющими успешно осуществить настоящее изобретение, а следовательно, они могут рассматриваться как предпочтительные модели для его осуществления. Однако, исходя из настоящего изобретения, каждому специалисту в данной области техники должно быть понятно, что в конкретные варианты могут быть внесены различные изменения, которые дадут подобные или сходные результаты, не выходящие за рамки идеи, существа и объема настоящего изобретения. Более конкретно, должно быть ясно, что различные химически и физиологически родственные агенты могут быть использованы для замены описанных здесь агентов, если при этом будут достигнуты такие же или аналогичные результаты. Все такие подобные замены и модификации, очевидные каждому специалисту, рассматриваются как отвечающие сущности, объему и идее настоящего изобретения, сформулированного в прилагаемой формуле изобретения.
ПРИМЕР 1
Предварительный скрининг и очистка активных противоопухолевых компонентов из Acacia victoriae
Шестьдесят видов растений были отобраны при осуществлении исследовательской программы DELEP (изучение бобовых растений пустынной зоны) с целью идентифицировать новые соединения, обладающие привлекательными биологическими свойствами. Программа DELEP (университет штата Аризона в Таксоне) включает коллекцию пустынных видов бобовых растений, создаваемую при сотрудничестве университета Аризоны и Юго-Западного дендрария им. Б.Томпсона. Экспериментальные полевые образцы были собраны от каждого вида растений, высушены на воздухе в течение 3-4 дней, измельчены на частицы размером 3 мм в мельнице Уилли (экран с отверстиями 3 мм) и экстрагированы 2 или 3 раза методом перколяции смесью 1:1 дихлорметана (DCM) и метанола (МеОН). Каждую перколяционную экстракцию проводили в течение, по крайней мере, 5 часов и часто продолжали в течение ночи. Основную массу экстракта получали после первых двух этапов перколяционной очистки. Затем полученную биомассу промывали объемом метанола, равным половине незанятого объема с выделением неочищенного экстракта, содержащегося в метанольных аликвотах. Обычно образцы выделяли и готовили для биотестов удалением метанола в вакууме, пропусканием водной фазы через частицы RP-C18, возвращением активных компонентов в МеОН и затем роторным выпариванием МеОН с получением экстракта в виде твердого вещества. Неочищенный экстракт затем ресуспендировали в воде, ДМСО или их смесях (менее полярные соединения ресуспендировались в ДМСО, в то время как более полярные соединения ресуспендировались в воде или в смеси воды и ДМСО; агликоны ресуспендировались в ДМСО).
Каждый из экстрактов затем подвергали скринингу в отношении панели опухолевых и неопухолевых клеток человека, включая линии раковых клеток человека, Т-лейкозные клетки, эпидермоидные клетки человека, клетки рака молочной железы человека, клетки рака предстательной железы человека, фибробласты крайней плоти человека, эндотелиальные клетки человека и клетки рака почек человека. Вначале клетки высевали в 96-луночные планшеты на 18-24 часа при 37°С. Затем клетки обрабатывали различными концентрациями растительных экстрактов и инкубировали в течение 72 часов при 37°С и окрашивали либо 3-(4,5-диметилтиазол-2-ил)-2,5-дифенилтетразолийбромидом (МТТ) (Sigma Chemical Co.) в течение 4 часов, либо кристаллическим фиолетовым (Sigma Chemical Co.) в течение 20 минут при комнатной температуре. На МТТ-окрашенные планшеты наносили литический буфер (20% додецилсульфата натрия в 50%-ном DMF) и инкубировали еще в течение 6 часов с последующим считыванием оптической плотности при 570 нм. Окрашенные кристаллическим фиолетовым планшеты промывали, краситель экстрагировали в течение 3-4 часов буфером Соренсена (0,1 М цитрат натрия, рН 4,2; 50% этанол по объему) и планшеты считывали при 570-600 нм (Mujoo et al., 1996). Цитотоксичность протестированных экстрактов определяли, сравнивая данные считывания ОП между “чистой” обработанной средой и клетками, обработанными растительным экстрактом. Уровень цитотоксичности в процентах определяли, вычитая из 100 процентов контроля, где процент контроля = [((ОП клеток, обработанных растительным экстрактом (обработанный образец))/(ОП клеток, обработанных “пустой” средой (необработанный образец)))×100].
В ходе исходного скрининга для одного растительного экстракта было продемонстрировано сильное подавление роста раковых клеток при слабой токсичности в отношении нормальных фибробластов человека. Этот экстракт, обозначенный UA-BRF-004-DELEP-F001, был выделен из бобового растения Acacia victoriae. Этот экстракт проявлял IC50 на уровне приблизительно 12 мкг/мл (SKOV-3), 26 мкг/мл (OVCAR-3) и 13 мкг/мл (HEY) при тестировании линий клеток рака яичника человека; более чем 50 мкг/мл (А375-М) и примерно 38 мкг/мл (HS294T) в отношении меланомных клеток человека; примерно 15 мкг/мл для эпидермоидных клеток человека (А431); и свыше 50 мкг/мл для линии клеток рака молочной железы MDA-468 (фиг.1) (см. пример 13, в котором описаны эти клеточные линии). В отношении нормальных фибробластов крайней плоти человека (FS) и фибробластов мыши (L929), обработанных тем же самым экстрактом, цитотоксичность выявлена не была.
По данным ТСХ этот экстракт содержал смесь большого числа компонентов. Следовательно, предварительные усилия были сконцентрированы на очистке данного экстракта с целью выделения активных компонентов, ответственных за избирательную цитотоксичность. Хроматографические фракции, обогащенные активными компонентами, были выделены из исходного экстракта в соответствии с базовой схемой, показанной на фиг.15.
Исходный экстракт, UA-BRF-004-DELEP-F001, был приготовлен из 538 г растительного материала Acacia victoriae методом перколяции (двукратной), как описано выше. Затем экстракт сушили в вакууме с получением примерно 52,0 г порошка. После этого 51,5 г сухого материала трижды обрабатывали 1 л этилацетата (EtOAc). Приблизительно 15,75 г растворенного в EtOAc материала подвергали колоночной хроматографии на силикагеле (1,5 кг). 54 субфракции по 670 мл элюировали с использованием смесей с увеличивающейся полярностью гексана, EtOAc и МеОН. 54 субфракции были отобраны в 13 отдельных фракций, обозначенных от UA-BRF-004-DFELEP-F006 до UA-BRF-004-DELEP-F018. Затем эти фракции тестировали на противоопухолевую активность с использованием описанной выше процедуры. Ни одна из тестированных фракций не продемонстрировала противоопухолевой активности, выявленной для UA-BRF-004-DELEP-F001.
Нерастворимый в EtOAc материал (приблизительно 34,7 г) также хроматографировали на силикагеле (1,7 кг). 51 субфракцию по 670 мл и 3 дополнительные субфракции общим объемом 21 л элюировали смесями с увеличивающейся полярностью DCM, МеОН и воды. Эти субфракции собирали в 8 отдельных фракций, обозначенных от UA-BRF-004-DELEP-F019 до UA-BRF-004-DELEP-F026, в соответствии с табл. 6.
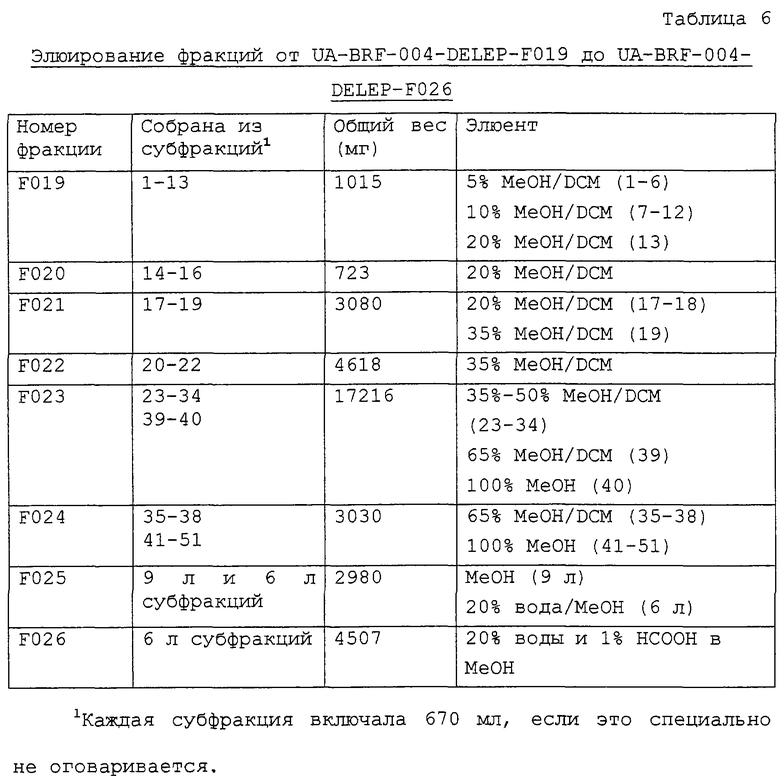
Затем каждую фракцию подвергали скринингу на противоопухолевую активность в отношении панели опухолевых клеток, как описано выше для неочищенного экстракта. Одна из этих фракций, UA-BRF-004-DELEP-F023, проявляла противоопухолевую активность, более высокую по сравнению с UA-BRF-004-DELEP-F001. Полученные результаты показали, что 6 мкг/мл фракции UA-BRF-004-DELEP-F023 проявляли цитотоксичность на уровне 50% (ОСС1), 63% (SKOV-3), 85% (HEY) и 48% (OVCAR-3) в отношении клеток рака яичника человека; цитотоксичность приблизительно 60% в отношении клеток рака предстательной железы человека (LNCaP); цитотоксичность примерно 92% в отношении лейкозных клеток (Jurkat) и цитоксичность приблизительно 73% в отношении свежевыделенных клеток рака яичника человека из проб асцитной жидкости больных (FTC). В биотестах в отношении нормальных клеток были определены величины IC50: 10,6 мкг/мл для клеток FS и 23 мкг/мл для клеток HUVEC (фиг.2).
Далее, очистку биологически активных компонентов из UA-BRF-004-DELEP-F023 осуществляли путем разделения методом жидкостной хроматографии при среднем давлении (ЖХСД) с многократно обращенной фазой с целью выделения и характеризации активного(ых) компонента(ов). Образцы элюировали из дегазированных смесей при повышающейся концентрации ацетонитрила (ACN) в воде в 4 л с шагом 10% при следующих этапах: 0%, 10%, 20%, 30%, 40% ацетонитрила в воде. Затем 2-4-литровые фракции элюировали метанолом. 10 фракций были отобраны после повторных проходов и помечены от UA-BRF-004-DELEP-F027 до UA-BRF-004-DELEP-F036 в соответствии с табл. 7.
Элюирование фракций от UA-BRF-004-DELEP-F027 до UA-BRF-004-DELEP-F036
Некоторые из этих фракций аналогичным образом были выделены методом ТСХ. Сильное проявление противоопухолевой активности было установлено для фракции (35), UA-BRF-004-DELEP-F035, характеризующейся одним из наиболее высоких выходов.
Скрининг UA-BRF-004-DELEP-F035 на противоопухолевую активность определил IC50 на уровне, соответственно, 3,0, 1,2, 2,0 и 3,5 мкг/мл в отношении линий клеток опухоли яичников HEY, SKOV-3, OVCAR-3 и С-1 (резистентная к цисплатину сублиния OVCAR-3); IС50=2,4 мкг/мл в отношении клеток опухоли поджелудочной железы (Рапс-1); IС50=1,2 мкг/мл, 3,0 мкг/мл и 3,7 мкг/мл, соответственно, для линий клеток почечной опухоли 769-Р, 786-0 и А498; IC50=130 нг/мл для Т-лейкозных клеток Jurkat; и ICso в диапазоне 1-3 мкг/мл в отношении В-лейкозных клеток линий KG1, REH и NALM-6 (фиг.3, 4). Как показано в табл.8, очистка сырого растительного экстракта приводит к резкому возрастанию биологической активности.
Цитотоксичность неочищенного экстракта в сопоставлении с UA-BRF-004-DELEP-F035
Фракция 35 обнаруживает IC50 на уровне приблизительно 4,7 мкг/мп в отношении нормальных фибробластов человека и IC50 приблизительно 13,3 мкг/мл в отношении нормальных клеток Нs27 человека. Когда влияние фракции 35 (F035) оценивали в отношении нормальных популяций эритроидной и миелоидной линий (клетки, выделенные из костного мозга) при концентрации 3,0 мкг/мл, уровень ингибирования составил 12-18% (табл.9).
Влияние фракции 35 на эритроидные и миелоидные колонии клеток
Из приведенных выше данных по сильной противоопухолевой активности фракции 35 был проведен биотест, соответствующий описанному выше, с использованием концентрации фракции 35 вплоть до 0,095 мкг/мл. В этом анализе, различные концентрации фракции 35 были применены к расширенной панели линий опухолевых клеток. Полученные в этом тесте результаты показывают, что фракция 35 даже в концентрации 1,56 мкг/мл проявляет противоопухолевую активность в отношении многих клеточных линий (табл.10).
Цитотоксичность различных концентраций UA-BRF-004-DELEP-F035 в отношении разных линий опухолевых клеток
Пример 2
Способы выделения активных компонентов из Acacia victoriae
Был разработан способ прямого получения фракций, содержащих активные компоненты, входящие в UA-BRF-004-DELEP-F035, выделенную в ходе предварительной очистки, подробно описанной в примере 1. Приблизительно 9665 г свежесобранной ткани бобов Acacia victoriae измельчали в молотковой мельнице с 3-мм ситом и затем экстрагировали 80%-ным МеОН в воде (трижды) с последующей фильтрацией. 8200 г выжимок отбрасывали. Результаты трех промывок собирали по отдельности, обозначая их как следующие фракции: F068 в 21,5 л (первая промывка); F069 в 24 л (вторая промывка); и F070 в 34,3 л (третья промывка). Фракцию F068 далее очищали разделением на 1-литровые аликвоты, добавляя по 400 мл воды к каждой аликвоте и промывая хлороформом (2×250 мл). Объединенные полярные фазы (28,5 л) обозначали как фракцию F078, а объединенные органические фазы обозначали как фракцию F079 (выход 42 г после удаления органического растворителя на роторном испарителе).
МеОН удаляли из F078 в вакууме и далее F078 фракционировали методом ОФ-ЖХСД с использованием колонки размером 29×460 мм, загруженной 530 г восстановленного Bakerbond RP-C18, размер частиц 40 мкм. По 500 мл водного раствора аспирировали в колонку и фракции отбирали в соответствии с табл.11.
Элюирование фракций от F091 до F094
Затем каждую фракцию сушили удалением метанола, пропуская через частицы С-18, возвращали в метанол и выделяли в вакууме в виде твердого вещества. Твердое вещество ресуспендировали в воде и тестировали на противоопухолевую активность (для менее полярных фракций в воду добавляли ДМСО; агликоны ресуспендировали в ДМСО). Полученные результаты показали, что биологическая активность метанольного (100%) элюента, обозначенного как фракция F094, по сути эквивалента активности, определенной для фракции UA-BRF-004-DELEP-F035 (табл.12). Активные компоненты также содержались во фракции F093. Химическое сходство фракций F094 и F035 подтверждали с помощью методов ТСХ и ВЭЖХ, хотя F094, по-видимому, содержит дополнительные компоненты.
Цитотоксичность различных концентраций F094 в отношении различных линий опухолевых клеток
F094 была дополнительно фракционирована в соответствии с табл. 13 и проанализирована методом ТСХ и в биотестах с целью выделения очищенного(ых) активного(ых) компонента(ов). Результаты биотестов, проведенных на различных количествах полученных фракций (F138-F147), приведены в табл.14.
Элюирование фракций F138-F147
Биологическое тестирование фракций F137-F147
Процент подавления роста
Хотя описанные выше процедуры имели целью выделение активных компонентов из бобов Acacia victoriae, также активные компоненты могут быть экстрагированы из корней. В этом случае корни перемалывают в течение получаса и заливают 100% метанолом. Затем эту смесь отфильтровывают и разводят 80%-ным МеОН в воде. Если необходимо провести экстракцию большого количества корней, предпочтительным затем может быть проведение экстракции путем перколяции, что было описано выше. Обоснованием такого разграничения процедур экстракции является то, что корни обычно экстрагируют свежесобранными, а бобы, как правило, перед экстракцией высушивают.
Пример 3
Ступенчатая препаративная процедура получения активных компонентов из фракции UA-BRF-004-DELEP-F094
Модифицированная процедура экстракции/разделения была использована для поэтапного (ступенчатого) получения смесей активных компонентов из состава фракции UA-BRF-004-DELEP(бобы)-F094 (F094). Эту процедуру повторяли многократно, постепенно получая высокоактивные фракции. Обычно 20-25 г F094 или ее эквивалента растворяли в 150-175 мл 50% МеОН в воде, после чего аспирировали в колонку: (26 мм × 460 мм)+(70 мм × 460 мм), RP-C18, 40 мкм, 1200 г, уравновешенную 60% МеОН/Н2О. Фракции элюировали поэтапно 8 л 60% МеОН/Н2О; 7,5 л 70% МеОН/Н2O; и 2 л чистого МеОН и полученные фракции обозначали в соответствии с табл. 15. Фракция F035-B2 содержит смесь активных компонентов, имеющихся в F094, F133-136 (выделенных из F093) и F138-147 (выделенных из F094), как показано на фиг.18A-F. F094 является приемлемым заменителем F035 при 1-2-кратном снижении эффективности, а фракция F035-B2 имеет меньшую эффективность, чем F094.
Выделение F035-B2
Для дальнейшей очистки активных компонентов фракции F035-B2, заявителями была разработана процедура получения фракций, обладающих аналитическими ВЭЖХ-характеристиками фракции UA-BRF-004-DELEP-F035. Эта процедура заключалась в следующем: препаративную ВЭЖХ осуществляли на материале фракции F035-B2 с использованием колонки для обращенно-фазовой хроматографии с частицами 10 мкм с целью элюирования градиентными смесями ацетонитрила и воды, начиная с 26% ацетонитрила в воде до 40% ацетонитрила в воде с шагом градиента в 1-2%, с последующими промывкой 100% ацетонитрилом и промывкой 100% метанолом, в результате чего достигается уникальное разделение F035-B2 на несколько фракций, содержащих 0-20 минутных пиков (в расчете на стандартный 6-мкм аналитический метод ВЭЖХ с RP-C18), которые не выявлялись в исходной фракции F035 (фиг.18А). Остальные полученные фракции соответствовали 1-3-компонентному фракционированию F035. Как было установлено в описанном выше анализе, фракции F139-F147 сходны с этими фракциями, проявляя в некоторой степени более высокую противоопухолевую активность по отношению к одним линиям опухолевых клеток по сравнению с другими. Уникальная смесь активных компонентов, присутствующих в составе фракции F035, может быть получена, в целом, путем модифицирования исходной композиции переднего ряда растворителей от 16% до 26% ацетонитрила в воде с последующей очисткой методом ЖХСД с получением мультиграммовых количеств эквивалента UA-BRF-004-DELEP-F035.
Дальнейшее совершенствование описанной выше процедуры экстракции, а также других процедур экстракции, описываемых в данном тексте, может быть осуществлено путем использования смесей трех растворителей, ацетонитрила, метанола и воды. Их процентные соотношения могут динамично расти и могут быть оптимизированы специалистом в области стандартного хроматографического разделения. Таким же образом связанные на силикагеле фазы могут варьироваться в зависимости от использования сочетания комбинированных обращенно-фазовых систем, включая, но не ограничиваясь ими, С-8, CN, диметилдиол и С-18. На заключительных этапах даже силикагель с нормальной фазой может быть использован для процедуры окончательной очистки.
Пример 4
Альтернативные процедуры выделения активных компонентов из Acacia victoriae
Фракции F094 (250 г), F035 (50 мг) и измельченные бобы Acacia victoriae (1 кг) (т.е. “плодо-семенной” порошок) были получены, как описано выше. В этом примере описывается аналитическая процедура анализа фракции F094 и последующего фракционирования F094.
4.1. Аналитическая процедура
Ряд методов, включая разнообразные С8 и С18 колонки в градиентных и изократических условиях, были применены для расщепления фракции F094. Мониторинг предусматривал анализ УФ-поглощения при 220 нм и детекцию рассеяния света при выпаривании (ELSD). Наилучшее разрешение пиков имело место при включении в подвижные фазы трифторуксусной кислоты (ТФУ). Ниже описан способ, получивший здесь название “акация-257”. Этот способ обеспечивает разрешение хорошего уровня при коротком времени фракционирования.
ВЭЖХроматограф был оборудован детектором с диодной матрицей (DAD) или детектором с переменной длиной волны и колонкой 4,6×150 мм Inertsil C18, 3 мк (MetaChem). Детектор был настроен на длину волны 220 нм.
Использовали следующие градиенты.
На фиг.25 показана хроматограмма F094, полученная данным способом. F094 состоит из трех групп или семейств с наличием в каждом из них по несколько пиков. Группа-1 (8-20 минут: пики A-D), группа-2 (22-35 минут; пики Е-Н) и группа-3 (36-47 минут; пики I-L). Фракция F035 была также проанализирована данным способом: соответствующая хроматограмма показана на фиг.26. Пики второй группы более многочисленны в случае с F035 по сравнению с F094, где наиболее многочисленны пики 1-й группы.
4.2. Фракционирование
4.2.1. Начальное фракционирование
Начальное фракционирование было сосредоточено на пиках группы-1. Полупрепаративную колонку Symmetry C8 (Waters) (7,8×300 мм, 7 мк) использовали для этой цели с параметрами градиента элюирования, показанными ниже. Семь субфракций выделяли в соответствии с показанным на фиг.27. Последняя фракция (№2160-007-31) включает все пики группы-2 и группы-3. Эти фракции, а также исходный материал (F094) были включены в биологическое тестирование.
4.2.2. Второе фракционирование
Целью данного этапа фракционирования было разделение пиков соединений группы-2. Для этого использовали ту же полупрепаративную колонку с С8. Подвижная фаза была изократической: 32% ацетонитрила в воде + 0,1% ТФУ. Хроматографические отметки, указывающие на разделение 7 фракций, показаны на фиг.28. Первая отделенная фракция в данном случае охватывает все пики группы-1.
4.2.3. Биологические тесты
Результаты биотестирования субфракций, полученных при первом и втором фракционировании, показаны, соответственно, в табл.16 и 17.
Цитотоксичность субфракций первого фракционирования в отношении клеток Jurkat
Цитотоксичность субфракций второго фракционирования в отношении клеток Jurkat
Два очищенных тритерпеновых гликозида, обозначенные как D1 и G1, были получены из фракции F094 Acacia. В результате кислотного гидролиза D1 образуется агликон.
4.4. Ступенчатое препаративное фракционирование с получением пиков областей D и G/H
F094 (2,3 г) фракционировали на колонке для ВЭЖХ Prep PFP (пентафторфенил) (50×250 мм, 10 мкм). Подвижная фаза: ацетонитрил / вода с 0,1% трифторуксусной кислотой (ТФУ) при градиенте ацетонитрила от 27% до 32%, время 38 минут. Как показано на фиг.29, в этом способе были разделены фракции, содержащие пики D и G/H. Выделенные при данном препаративном разделении фракции подвергали биологическому тестированию. Результаты аналитических тестов D и G/H показаны на фиг.30 и 31. В этом случае применяли способ “акация-257”, описанный в этом разделе выше.
Фракцию G/H дополнительно очищали на колонке PFP Prep с целью получения G1 с хроматографической чистотой 68%: этот материал далее очищали на полупрепаративной колонке С18 с получением чистого G1.
Примерно 100 мг смеси G/H загружали на ту же самую колонку PFP, которая описана выше. Использовали следующий градиент.
Были получены пять фракций (G1, G2, G3, H1 и Н2). Аналитические данные для G1 (фиг.32) указали на 68%-ную хроматографическую чистоту. Эту фракцию дополнительно очищали на полупрепаративной колонке.
Использовали колонку YMC C18-Aq (10×250 мм, 5 мкм). Подвижная фаза: 31% ацетонитрила в воде с 0,1% ТФУ. Конечный продукт G1 характеризовался хроматографической чистотой 100% (фиг.33).
Фракция D (45% D1) с колонки PFP Prep вначале была фракционирована с использованием колонки Waters C-18 (25×100 мм). Подвижная фаза: 61% метанол в воде с 0,1% ТФУ. ВЭЖХ-анализ показал, что D1 характеризуется чистотой 78% (фиг.34) и содержит дополнительный пик (обозначенный D1.5). Образец D1 с хроматографической чистотой 100% (фиг.35) был получен путем дополнительного фракционирования неочищенного D1 на колонке YMC C18-Aq смесью 33% ацетонитрил - вода с 0,1% ТФУ в качестве подвижной фазы. Было показано, что D1 более стабильна в разбавленных растворах кислот, чем в воде при 40°С. С учетом этого, в состав растворителей в ходе очистки D1 была включена ТФУ (0,1%).
4.4.1. Биологические тесты
Биотесты были проведены с использованием линий клеток Jurkat, и влияние субфракций и чистых D1 и G1 показаны, соответственно, в табл. 18, 19 и 20. D1 и G1 тестировали при двух разных величинах рН. Полученные данные указывают на немного более высокую активность при рН 6,5, нежели при рН 7,5. Однако, рост клеток подавлялся примерно на 40% при меньших величинах рН.
Цитотоксичность фракций с колонки PFP Prep
Цитотоксичность фракций, полученных при фракционировании G/H
Цитотоксичность D1 и G1 при рН=6,5 и 7,5
4.4.2. Кислотный гидролиз D1
Сапонин D1 в этаноле гидролизовали 3 N соляной кислотой в течение 3 часов при 100°С. Полученный агликон очищали методом ВЭЖХ. По данным масс-спектроскопии, молекулярная масса агликона составила 652.
Пример 5
Структура D1, G1 и В1
5.1. Структура D1
D1 является основным компонентом бобов Acacia victoriae. При тестировании этого соединения была установлена его значительная биологическая активность.
5.1.1. Полная молекула D1
D1 выделяли в виде бесцветного аморфного твердого вещества из частично очищенного экстракта F094, полученного с использованием препаративного ВЭЖХ-разделения в соответствии с вышеописанными примерами. При масс-спектроскопии MALDI ее молекулярная масса составила 2104 а.е.м., а за вычетом иона натрия реальная молекулярная масса составила 2081. В высокоразрешающем методе масс-спектроскопии FAB была подтверждена эта молекулярная масса и определена молекулярная формула C98H155NO46. Такая молекула слишком велика для определения ее структуры только спектроскопическим методом; в связи с этим использовали программу ее разделения, показанную на схеме 1 фиг.36. D1 на фиг.36 помечена как структура "(1)".
Протонный и углеродный ЯМР молекулы D1 показали присутствие тритерпена, двух монотерпенов и приблизительно восьми сахаров (см. в табл.21 полученные показатели 13C-ЯМР в столбце (1)).
Параметры 13C-ЯМР для D1 (1), G1 (14), B1 (21), агликона (2) и акациевой кислоты (3): номера в круглых скобках 1, 14, 21, 2 и 3 соответствуют структурам D1, G1, B1, агликона и акациевой кислоты, изображенным на фиг.36, фиг.37 и фиг.38, соответственно.
5.1.2. Сильный кислотный гидролиз D1
В результате гидролиза Dl 3 н НС1 при 100°С в течение 2 часов был получен “агликон D1”, показанный как (2) на фиг.36;
и как показала масс-спектроскопия, имел молекулярную массу 652. По данным ЯМР в агликоне Dl имеется тритерпен и модифицированный монотерпен, а сахара отсутствуют. Путем омыления (1,3 н NaOH при 100°С в течение 30 минут в метаноле) этот продукт расщепляли дополнительно, выделив следующее.
5.1.2.а. Тритерпен. Данные 13C-ЯМР идентичны данным, полученным ранее для акациевой кислоты (см. фиг.36, структура (3), а также см. табл. 21, данные 13C-ЯМР в столбце (3)), а установленная молекулярная масса 488 согласуется с его структурой.
5.1.2.b. Циклизованный монотерпен. Молекулярная масса и ЯМР данного соединения указывали на присутствие карбоновой кислоты, двух метильных групп, присоединенных к двойной связи, и двух винильных протонов, обуславливающих наличие пирановой структуры. Хотя эта структурная единица, обозначенная на фиг.36 как (4), также присутствует в “агликоне D1”, она отсутствует в исходном D1. Этот D1 включает ациклический монотерпен, обозначенный на фиг.36 как (5), и эта структура циклизуется в ходе кислотного гидролиза в соответствии с показанным ниже:
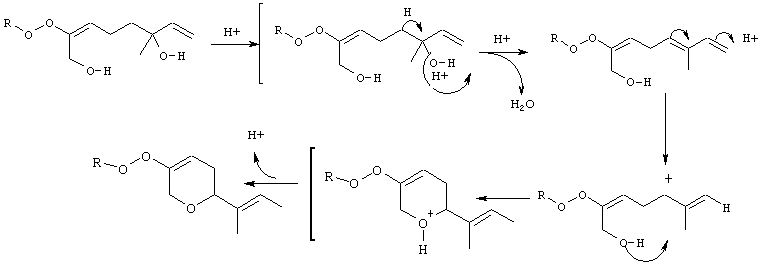 Эти структуры наряду с параметрами молекулярной массы и спектральных свойств D1, хорошо согласуются со структурой агликона D1, показанной на фиг.36 под меткой (2). Выбранные параметры 13C-ЯМР показаны в табл.21 в столбце (2).
Эти структуры наряду с параметрами молекулярной массы и спектральных свойств D1, хорошо согласуются со структурой агликона D1, показанной на фиг.36 под меткой (2). Выбранные параметры 13C-ЯМР показаны в табл.21 в столбце (2).
5.1.3. Слабое омыление D1
При обработке D1 0,5 н NH4OH при комнатной температуре в течение 1 часа происходит полная конверсия в два новых соединения.
5.1.3.а. Монотерпен. Эта молекула имеет молекулярную массу 200, а параметры ЯМР указывают на наличие у нее структуры ациклического монотерпена, что подтверждает предполагаемое расщепление. Данная структура показана на фиг.36 как (5).
5.1.3.b. Тритерпеновый/монотерпеновый олигосахарид. Это соединение более полярно, чем D1, а данные ЯМР согласуются с присутствием акациевой кислоты, одного монотерпена и нескольких моносахаридов. Эта структура показана на 36 как (6).
5.1.4. Анализ сахаров D1
С применением сильного кислотного гидролиза D1 (2 н НСl при 100°С в течение 2 часов) с последующей дериватизацией (получением триметилсилильных сложных эфиров) и применением метода ГХ/МС было подтверждено присутствие восьми сахарных остатков в исходной молекуле: арабинозы, рамнозы, фукозы, ксилозы, 6-дезоксиглюкозы (= хиновозы), N-ацетилглюкозамина и двух молекул глюкозы.
5.1.5. Более активное омыление тритерпенового/монотерпенового олигосахарида
В результате обработки тритерпенового/монотерпенового олигосахарида 0,3 н NaOH в течение 1 часа при 60°С были получены три соединения.
5.1.5.а. Олигосахарид. Выделение и анализ данного высокополярного фрагмента подтвердил, что он является олигосахаридом. Анализ на сахар методом кислотного гидролиза (2 н НСl в течение 2 часов при 100°С) и данные анализа ГХ/МС в отношении триметилсилильных эфиров моносахаридов подтвердили, что данный олигосахарид является тетрасахаридом, составленным из двух молекул глюкозы и по одной каждой из молекул арабинозы и рамнозы.
5.1.5.b. Монотерпеновый гликозид.
По данным ЯМР, этот компонент соответствует структуре (8), показанной на фиг.36. Результаты кислотного гидролиза (2 н НСl в течение 2 часов при 100°С) идентифицировали сахар как 6-дезоксиглюкозу. Обработка монотерпенового гликозида β-глюкозидазой привела к получению монотерпена со структурой (9), показанной на фиг.36, данные ЯМР которого соответствуют структуре транс-2-гидроксиметил-6-гидрокси-6-метил-2,7-октадиеновой кислоты. Гидролиз этой связи под действием β-глюкозидазы указывает на то, что связь между этими двумя группами является β-связыо.
5.1.5.с. Тритерпеновый гликозид.
Это соединение имеет молекулярную массу 951, а по данным ЯМР, соответствует лактону акациевой кислоты, содержащему трисахарид по положению С-3, что показано как (10b) на фиг.36. При кислотном гидролизе (2 н НСl в течение 2 часов при 100°С) этого соединения, входящие в его состав сахара идентифицируются как N-ацетилглюкозамин, фукоза и ксилоза в виде их триметилсилильных производных в методе ГХ/МС. Эта молекула была выявлена и в открытой форме кислоты/спирта, что обозначено на фиг.36 как (10а), и в закрытой лактонной форме, что показано на фиг.36 как (10b).
Анализ сахаров и молекулярной массы фрагментов по сравнению с интактной молекулой D1 подтвердил, что все части D1 соответствуют фрагментам, структуры которых на фиг.36 обозначены как (5), (7), (8) и (10а).
5.1.6. Слабый кислотный гидролиз D1
Слабый кислотный гидролиз D1 (1 н НСl в течение 16 часов при 25°С) позволил получить две новые молекулы.
5.1.6.а. Монотерпеновый сахар.
Молекулярная масса, спектры ЯМР и анализ сахаров соответствуют структуре монотерпен-6-дезоксиглюкозы. Структура этой молекулы изображена как (11) на фиг.36.
5.1.6.b. Тритерпен-монотерпен-гликозид.
Вторая молекула была идентифицирована как тритерпенмонотерпеновый гликозид, а структура этой молекулы показана на фиг.36 как (12).
5.1.7. Соединение подгрупп в D1
По данным ЯМР, карбоновая кислота внешнего монотерпена образует сложный эфир по атому С-4 в кольце 6-дезоксиглюкозы (хиновозы). Данные ЯМР и гидролиза показали, что аномерный углерод хиновозы присоединен к гидроксильной группе при атоме С-6 внутреннего монотерпена. Стереохимические параметры аномерного углерода в хиновозе указывают на то, что это β-связь.
Проведение гидролиза (2 н НСl в течение 2 часов при 100°С) и анализ изомеризации сахаров показывают, что мономерами данного тетрасахарида являются две молекулы глюкозы и по одной молекуле арабинозы и рамнозы. Тетрасахарид непосредственно проэтерефицирован по атому С-28 карбоновой кислоты тритерпена, как показано на фиг.39. Анализ методом масс-спектроскопии с ионной ловушкой показывает, что тетрасахаридная структура имеет два остатка глюкозы и арабинозы, присоединенных к занимающему центральное положение остатку рамнозы, как это показано на фиг.39. Типы связей этих сахаров еще не известны.
По данным ЯМР установлено, что N-ацетилглюкозамин (NAG) присоединен непосредственно к атому С-3 тритерпена. Остальными элементами последовательности сахаров являются фукоза в середине и ксилоза на конце, на что указывают данные ЖХ/МС продуктов частичного гидролиза (1 н НСl в течение 1 часа при 60°С в 50% метаноле). Тип связи между этими сахарами еще не известен.
5.1.8. Эллиптозид-Е
В составе D1 имеются тритерпен и два монотерпена, обычно обнаруживаемые в сапонинах, описанных для других видов растений, включая другие представители рода Acacia. Хотя структура D1 сходна с эллиптозидом-Е (фиг.24), описанным для Archidendron ellipticum (Beutler et al., 1997), в настоящем изобретении удельное вращение D1 составило [α]D=-30,0°, что не совпадает со значением -24,3°, приведенным для эллиптозида-Е.
Описанный ранее эллиптозид-Е (Beutler et al., 1997) и D1 характеризуются различным временем удерживания в ВЭЖХ (15,2 минут для D1 и 12,5 минут для эллиптозида-Е). Следовательно, эти две молекулы должны иметь определенные различия, такие как конкретное место присоединения их субъединиц или присутствие оптических или структурных изомеров.
Заявителями было установлено, что удельное вращение внутреннего монотерпена, показанного на фиг.36 как (9), составляет +11,2° в метаноле и +16° в хлороформе. Для аналогичного фрагмента эллиптозида-Е было установлено -9,1° в хлороформе. Более того, только для одного хирального центра во внутреннем монотерпене D1 была установлена конфигурация "S", что прямо противоположно найденной конфигурации у эллиптозида-Е. Удельное вращение внешнего монотерпена D1 находится на стадии изучения. Более того, по данным протонного ЯМР, двойные связи в монотерпенах D1 имеют “транс”-конфигурацию, в то время как двойные связи монотерпенов эллиптозида-Е находятся в “цис”-конфигурации (Beutler et al., 1997). Эти два признака определяют первое структурное отличие, обнаруженное между D1 и эллиптозидом-Е. Ферментативный каталитический гидролиз отдельных сахаров показал, что расположение сахаров такое же, как и в эллиптозиде-Е.
5.2. Структура G1
Биологические тесты этого материала показали, что G1 биологически более активен по сравнению с D1.
5.2.1. Интактная молекула G1 (14)
Второй структурой, установленной в настоящем изобретении, является G1. Ее также выделяли из фракции F094 путем препаративной ВЭЖХ, но при меньшем выходе соединения. G1 характеризуется меньшей полярностью, чем D1. Методом масс-спектрометрии с лазерной десорбцией (MALDI) определяли молекулярную массу 2065, что на 16 а.е.м. меньше, чем у D1. Удельное вращение G1 составило -26,9° (в метаноле). По данным протонного ЯМР G1 также является сапонином, очень похожим на D1, но отличается от D1 отсутствием одного атома кислорода во внешнем монотерпене, который, соответственно, является Транс-2,6-диметил-6-гидрокси-2,7-октадиеновой кислотой: см. (14) на фиг.37 и отдельные параметры спектра 13C-ЯМР в табл. 21 в столбце (14). Разрушение G1 изображено на схеме 2 (фиг.37).
5.2.2. Слабое омыление G1
При обработке G1 раствором 0,5 н NH4OH при комнатной температуре даже в течение нескольких минут происходит полная конверсия в более полярный продукт слабого омыления и монотерпен.
5.2.2.а. Монотерпен. Молекулярная масса и спектры ЯМР данного материала указывают на наличие метильной группы у атома С-2, у которого имеется гидроксиметил. Это показано под меткой (15) на фиг.37.
5.2.2.b. Тритерпен-монотерпеновый олигосахарид.
По данным ЯМР этого соединения оно идентично по времени удерживания в ВЭЖХ и по спектрам протонного ЯМР его структура (16) на фиг.37 сходна со структурой (6) на фиг.36, происходящей от D1, и оно включает акациевую кислоту, один монотерпен и восемь моносахаридов, что было показано и для D1. Сходство структур (16) и (6) указывает на сходство стереохимических параметров, полученных для внутреннего монотерпена D1.
5.2.3. Анализ сахаров G1
При сильном кислотном гидролизе G1 (2 н НСl в течение 2 часов при 100°С) получали те же моносахаридные остатки, что и в D1: арабинозу, рамнозу, фукозу, ксилозу, 6-дезоксиглюкозу, N-ацетилглюкозамин и две молекулы глюкозы.
5.2.4. Кислотный гидролиз G1
Кислотный гидролиз продукта слабого омыления позволил выделить три молекулы, как и в случае с D1. Для каждой из них был проведен ЯМР и анализ на сахар (2 н НСl в течение 2 часов при 100°С). Результат показан на фиг.37 как структура (16).
5.2.4.а. Олигосахарид включает две молекулы глюкозы и по одной молекуле арабинозы и рамнозы, как показано на фиг.37 под меткой (17).
5.2.4.b. Монотерпеновый гликозид включает ациклический монотерпен, показанный на фиг.37 как (15), и 6-дезоксиглюкозу, а его полная структура обозначена на фиг.37 как (18).
5.2.4.с. Тритерпеновый гликозид включает акациевую кислоту и по одной молекуле N-ацетилглюкозамина, фукозы и ксилозы. Порядок этих сахаров такой же, как и в D1. Данная структура на фиг.37 обозначена как (19).
5.2.5. Эллиптозид-А
Состав терпенов и сахаров в G1 такой же, как и у эллиптозида-А (см. фиг.24 и Beutler, 1997). Однако, как было установлено, эллиптозид-А заметно отличается по времени удерживания в ВЭЖХ (G1 29,09 минут; эллиптозид-А 26,04 минут): это указывает на то, что эти две молекулы должны в определенной степени различаться, например, конкретным порядком соединения их частей или присутствием оптических изомеров, или и тем и другим одновременно. Также были выявлены различия по величинам химических сдвигов в спектрах 1H и 13C-ЯМР G1 и эллиптозида-А. Предполагается, что удельные вращения внутренних и внешних монотерпенов в составе этих соединений также могут различаться. На фиг.37 структура (14) представляет G1.
5.3. Структура В1
По данным биологических тестов В1 в существенной степени менее активна по сравнению с D1 или G1.
5.3.1. Интактная молекула В1 (21)
Выделение В1 было осуществлено путем получения растительного экстракта и флэш-хроматографии с С-18 с последующей препаративной и полупрепаративной хроматографией с С-18. По данным ЯМР В1 является типичной для семейства сапонинов с тритерпен-монотерпен-хиновоза-монотерпен структурой. Также данные ЯМР указывают на присутствие четырех дезоксисахаров и одной N-ацетильной группы: т.е. эта молекула должна отличаться от D1 по своим сахарным составляющим. Конкретные параметры 13C-ЯМР показаны в табл.21 в столбце (21). Расщепление этой молекулы отображено на фиг.38.
5.3.2. Анализ сахаров В1
Данные ЯМР указывают на присутствие более одной копии одного остатка 6-дезоксиметилсахаров (т.е. фукозы, рамнозы, 6-дезоксиглюкозы). Анализ сахарного состава полной молекулы после гидролиза (2 н НСl в течение 2 часов при 100°С) показал наличие девяти сахаров: по одной молекуле фукозы, арабинозы, ксилозы, хиновозы и глюкозамина и двух молекул глюкозы и рамнозы. В исходной молекуле присутствует глюкозамин, являющийся “остатком” от N-ацетилглюкозамина. Структура В1 показана на фиг.38 под меткой (21).
5.3.3. Слабое омыление В1
При обработке В1 раствором 0,5 н NH4ОH при комнатной температуре даже в течение нескольких минут происходит полная конверсия в более полярное соединение, продукт слабого омыления и монотерпен.
5.3.3.а. Монотерпен. По молекулярной массе и данным ЯМР этот материал имеет ту же структуру, что и монотерпен из состава D1, показанный на фиг.37 под меткой (5). На фиг.38 он показан под меткой (22).
5.3.3.b. Тритерпен/монотерпеновый олигосахарид. Данные ЯМР этого соединения указывают на то, что оно включает акациевую кислоту, один монотерпен и несколько моносахаридов. Это показано на фиг.38 как (23).
5.3.4. Более активное омыление тритерпен/монотерпенового олигосахарида
Более активное омыление (0,3 н NaOH в течение 1 часа при 6°С) продукта слабого омыления позволило выделить три молекулы, как это ранее было получено для D1 и G1. Были получены данные ЯМР и анализа сахаров для каждого из них.
5.3.4.а. Олигосахарид включает глюкозу, арабинозу и две молекулы рамнозы. Это показано на фиг.38 под меткой (24).
5.3.4.b. Монотерпеновый гликозид включает 6-дезоксиглюкозу и монотерпен. Это показано на фиг.38 под меткой (25).
5.3.4.с. Тритерпеновый гликозид включает акациевую кислоту с присоединенным тетрасахаридом в С-3-положении. Этот тетрасахарид включает по одной молекуле N-ацетилглюкозамина, фукозы, глюкозы и ксилозы. Это показано на фиг.38 как структура (26).
Пример 6
Де-этерификация тритерпеновых соединения по настоящему изобретению
Проводили деэтерификацию F094 и продукты деэтерификации тестировали с целью поиска активных компонентов. 1,00 г F094 растворяли в 100 мл воды с последующим добавлением 1 г КОН и нагреванием с обратным холодильником в течение полутора часов. Раствор оставляли охлаждаться до комнатной температуры и его рН доводили до 7 с помощью 1 н НСl и затем промывали гексаном (2×50 мл). Затем водный раствор подвергали дальнейшей поэтапной экстракции с получением фракций 159-162. Например, раствор вначале экстрагировали н-бутанолом (2×50 мл) с получением 0,127 г растворимого в органических растворителях твердого вещества (фракция F159) после сушки в вакууме. Водную фазу подкисляли до рН 5 с использованием 1 н НСl и экстрагировали EtOAc (2×50 мл) с получением 0,397 г растворимого в EtOAc твердого вещества (F160) и затем н-бутанолом (2×50 л) с получением 0,338 г твердого вещества (F161) после удаления органических растворителей. И наконец, водный слой нейтрализовали до рН 7 1 н NaOH. Из конечного водного слоя было выделено 1,808 г твердого вещества (F162).
Биологические тесты показали, что продукты деэтерификации обладают слабой активностью или лишены ее вовсе. Была протестирована цитотоксичность F159-162 в отношении клеточных линий 769-Р, Panc-1, HEY, MDA-MB-453 и Jurkat. Активность выявили только для F161, которая проявляла цитотоксичность в отношении MDA-MB-453 на уровне 1,6% при 50 мкг/мл, а также для F159 в отношении клеток Jurkat на уровне 15,50%, 6,60% и 3,80%, соответственно, при 50 мкг/мл, 25 мкг/мл и 12,5 мкг/мл. Эти данные указывают на то, что для проявления биологической активности наличие боковой сложноэфирной цепи необходимо. Считается, что боковая сложноэфирная цепь в соединениях по настоящему изобретению обеспечивает противоопухолевую активность и/или согласованно функционирует вместе с тритерпеновым скелетом данных соединений по настоящему изобретению с получением проявляемого ими сильного противоопухолевого эффекта.
Пример 7
Гидролиз сахаров в соединениях по настоящему изобретению
Входящие в состав F094 сахара также были гидролизованы с целью характеризации активных компонентов. 12 г F094 растворяли в 400 мл 2 н Н2SO4 и нагревали с обратным холодильником в течение 75 минут: в это время происходило образование нерастворимого материала. Раствор охлаждали и отфильтровывали через спекшееся пористое стекло. Остаток промывали водой, получая 4,8 г агликона (F191), что подтверждается ТСХ-анализом. Фильтрат темно-янтарного цвета нейтрализовали КОН или NaOH. Собирали образующийся осадок белого цвета. Растворитель удаляли в вакууме и продукт ресуспендировали в метаноле, в результате чего получали осадок белого цвета, который, по-видимому, соответствует солям сульфата, на что указывают данные теста на окрашивание пламени. Почти прозрачный фильтрат сушили в вакууме, а остаток ацетилировали и анализировали методом ВЭЖХ, по данным которой он включает смесь сахаров, что показано на фиг.17А и 17В. Данная смесь предположительно содержит, по крайней мере, пять сахаридов. Эти сахара могут быть дополнительно охарактеризованы выделением тетраметилсилильных производных для характеристики с помощью ГХ/МС; хроматографией на бумаге; выделением бензильных производных методом ВЭЖХ разделения или ЯМР DEPT; или 13C-ЯМР, что в полном объеме было описано выше. С применением стандартной одно- и двумерной целлюлозной, бумажной и нормальнофазовой тонкослойной хроматографии, основные сахара были идентифицированы как глюкоза, ксилоза, рамноза и арабиноза, наряду с некоторым количеством второстепенного дополнительного сахара, вероятно являющегося источником аминогликозида, в частности, ацетамидозамещенного сахара.
Количество различных сахаров, по-видимому, объясняет образование сложных ВЭЖХ-спектров, которые указывают на присутствие дюжин близкородственных соединений. В частности, некоторые активные компоненты предположительно гликозилированы по двум различным сайтам (сайт спирта и сайт карбоновой кислоты). Различные комбинации шести сахаров, присоединенных по указанным двум сайтам, видимо, дают большое число близкородственных соединений, разделение которых весьма затруднительно.
Альтернативно, при более слабом гидролизе с перегонкой со 100% этанолом (азеотропная смесь с 5% воды) и с 0,1-2,0 н H2SO4 (при том, что дальнейшая обработка не изменяется) при слабом нагревании до точки дефлегмации, но без жесткой дефлегмации, образуется смесь агликонов. Некоторые компоненты утрачиваются; это указывает на определенную степень изомеризации при более жестких условиях.
Затем агликон(ы) F191 (1 г) метилировали нагреванием с обратным холодильником в течение 5-7 часов с метилиодидом (1 мл) и безводным К2СО3 (1 г) в безводном ацетоне (10 мл). В результате получали 0,315 г нерастворимого материала и 0,54 г сложных метиловых эфиров, обозначенных F197. 500 мг F197 далее разделяли методом ЖХСД, используя колонки 15×460 мм (45 г SiO2, 15-25 мкм), в которых образец предварительно адсорбировали на 1,5 г SiO2 (15-25 мкм). Соединения элюировали 790 мл 7%-ного изопропилового спирта (ИПС) в гексанах (субфракции 1-10), 470 мл 10%-ного ИПС в гексанах (субфракции 1-14), 275 мл смеси 20%-ный ИПС - гексаны (субфракции 14-15), 200 мл дихлорметана и 100 мл смеси DCM/MeOH (1:1), в в соответствии с табл.22.
Фракционирование F197 на фракции от F198 до F208
Биологическое тестирование F191 и F197 показало наличие цитотоксичности в отношении клеток опухоли яичника (линия HEY), клеток рака почек (линия 769-Р), клеток рака поджелудочной железы (линия Раnс-1), Т-лейкозных клеток Jurkat и клеток рака молочной железы MDA-MB-453 в концентрациях, соответствующих показанному в табл.23.
Биологическое тестирование фракций F191 и F197
F199 (116 мг) дополнительно фракционировали с использованием той же колонки, которая была использована при фракционировании F191, и элюировали 100 мл смеси 2% ИПС - гексаны, 525 мл смеси 4% ИПС - гексаны и 250 мл смеси 10% ИПС - гексаны в соответствии с табл.24.
Фракционирование F199 на фракции от F209 до F214
F212 (85 мг) дополнительно фракционировали методом ВЭЖХ на хроматографе Waters Prep LC-4000 на колонке Alltech Econosil C18 (22×500 мм; 10 мкм; уравновешена смесью 75% ацетонитрил - вода), элюировали смесью 80% ацетонитрил - вода и промывали ацетонитрилом при скорости потока 40 мл/мин с установкой детектора на 220 нм и отбором субфракций каждый 30 секунд (20 мл) в соответствии с табл.25.
Фракционирование F212 на фракции от F220 до F228
F223 сначала была очищена в виде метилового сложноэфирного производного с получением С-191. С-191 подвергали классической процедуре ацетилирования. В частности, С-191 (47 мг) перемешивали в течение ночи при комнатной температуре со смесью с уксусного ангидрида и пиридина в соотношении 2:1. Реакцию гасили водой и раствор распределяли между диэтиловым эфиром и 5 н соляной кислотой. Затем органическую фазу промывали до нейтральности, выпаривали на роторном испарителе и остаток подвергали ПТСХ: препаративную пластину размером 20×20 см элюировали смесью гексан:изопропиловый спирт (90:10) с последующим ПТСХ-элюированием смесью дихлорметан:метанол (98:2) с получением С191-ацетата (F229, которая далее была обозначена как С-194).
F201 (85 мг) также дополнительно фракционировали методом ЖХСД на той же колонке, на которой разделяли F199, элюировали и собирали в смесь 2% ИПС-гексаны (120 мл), в смесь 4% ИПС-гексаны (330 мл), в смесь 7% ИПС-гексаны (460 мл), в смесь 20% ИПС-гексаны (150 мл), в дихлорметан (50 мл) и в смесь дихлорметан/МеОН (1:1) (70 мл) в соответствии с табл.26.
Фракционирование F201 на фракции от F215 до F219
Пример 8
Биологические характеристики активных тритерпенов по настоящему изобретению
Ангиогенез или образование новых кровеносных сосудов представляет собой процесс, в котором клетки рекрутируются фактором(ами), образуемым(ми) опухолью с целью обеспечения самой себя питающей сосудистой системой. Подавление ангиогенеза замедляет разрастание опухоли за счет ограничения притока крови к данной опухоли. Эту функцию анализировали с использованием теста на пролиферацию эндотелиальных клеток капилляров быка, в которых клетки обрабатывали фракцией 35 (UA-BRF-004-DELEP-F035). Тест осуществляли следующим образом:
эндотелиальные клетки капилляров быка получали и культивировали стандартным образом (Folkman et al., 1979). Эти клетки промывали ЗФР и суспендировали в растворе трипсина (0,05%). Суспензию клеток (25000 клеток на 1 мл) формировали в культуральной среде DMEM + 10% БСА + 1% GPS, высевали на желатинизированные 24-луночные культуральные планшеты (0,5 мл на лунку) и суспензию инкубировали в течение 24 часов при 37°С. Культуральную среду заменяли на 0,25 мл DMEM + 5% БСА + 1% GPS и использовали различные концентрации UA-BRF-004-DELEP-F035. После 20-минутной инкубации добавляли культуральную среду и FGF-β, достигая конечного объема 0,5 мл DMEM + 5% БСА + 1% GPS + 1 нг/мл FGF-β. Через 72 часа клетки диспергировали в трипсине, ресуспендировали в Hematall (Fischer Scientific, Pittsburgh, PA) и подсчитывали на кольтер-счетчике, измеряющем электрическое сопротивление непроводящих частиц, суспендируемых в электролите (т.н. “принцип Coulter”) (O’Reilly et al., 1997).
По результатам данного теста было показано значимое подавление пролиферации эндотелиальных клеток как в присутствии основного фактора роста фибробластов, так и без него (фиг.5). Эти данные показывают, что активные компоненты растительного экстракта являются сильными ингибиторами пролиферации эндотелиальных клеток и часто являются предиктором in vivo подавления ангиогенеза. Кроме того, данная фракция не влияет на миграцию эндотелиальных клеток капилляров, что свидетельствует об отсутствии токсичности в отношении нормальных клеток (фиг.6).
Общей проблемой, связанной со стероидными сапонинами (например, дигитонином и генин-диосгенином, выделенных из ямса), является лизис эритроцитов. С использованием простого анализа крови в культуральной пробирке было показано, что после обработки 1 мг/мл UA-BRF-004-DELEP-F035, наблюдался лишь очень слабый лизис. Напротив, обработка 10-25 мкг/мл дигитонином дает 100%-ный лизис крови в пробирке.
Кроме того, с целью дальнейшего изучения механизма, по которому происходит ингибирование опухолевых клеток активными компонентами, индуцируемую TNF-α активацию транскрипционного фактора NF-кВ анализировали в клетках Jurkat (3·106), которые обрабатывали 1-2 мкг/мл UA-BRF-004-DELEP-F035 и UA-BRF-004-(бoб)DELEP-F094. Данный анализ проводили следующим образом: клетки Jurkat предварительно обрабатывали 1-2 мкг/мл F035 или F094 в течение 15 часов при 37°С. Клетки собирали и ресуспендировали в 1 мл культуральной среды RPMI и обрабатывали 100 пкМ TNF-α в течение 30 минут при 37°С. После обработки TNF-α, ядерные экстракты приготавливали в соответствии с Schreiber et al. (1989). Кратко, клетки промывали охлажденным на льду ЗФР и суспендировали в 0,4 мл лизис буфера (10 мМ HEPES, рН 7,9; 10 мМ КСl, 0,1 мМ ЭДТА, 0,1 Мм EGTA, 1 мМ дитиотреитола, 0,5 мМ PMSF, 2 мкг/мл лейпептина, 2 мкг/мл апротинина и 0,5 мг/мл бензамидина). Клетки оставляли на льду на 15 минут и к ним добавляли по 25 мкл 10% Nonidet-40. Пробирки перемешивали на мешалке и микроцентрифугировали в течение 30 секунд. Ядерный осадок ресуспендировали в 25 мкл охлажденного на льду ядерно-экстракционного буфера (20 мМ HEPES, рН 7,9; 0,4 М Nad, 1 мМ ЭДТА, 1 мМ EGTA, 1 мМ дитиотреитола, 1 мМ PMSF, 2 мкг/мл лейпептина, 2 мкг/мл апротинина и 0,5 мг/мл бензамидина) и пробирки инкубировали на льду с непостоянным перемешиванием. Ядерный экстракт микроцентрифугировали в 5 этапов при 4°С и полученные супернатанты хранили при -70°С.
Тест на изменение электрофоретической подвижности (ТИЭП) осуществляли путем инкубации ядерных экстрактов (7 мкг белка) с 32Р-мечеными олигонуклеотидами гена NF-кВ (SEQ ID NO 1; консенсусный олигонуклеотид NF-кВ; Santa Cruz Biotechnology) в присутствии 0,5 мкг поли-dI-dC в связывающем буфере (25 мМ HEPES, рН 7,9; 0,5 мМ ЭДТА, 0,5 мМ дитиотреитола, 1% Nonidet Р-40, 5% глицерина и 50 мМ NaCl) в течение 20 минут при 37°С (Nabel & Baltimore, 1987; Collart et al., 1990; Hassanain et al., 1993). Образованный ДНК-белковый комплекс отделяли от свободного олигонуклеотида в 5%-ном нативном полиакриламидном геле с использованием буфера, содержащего 50 мМ Трис, 200 мМ глицина и 1 мМ ЭДТА. Этот гель фиксировали в 10%-ной уксусной кислоте и сушили, а полосы визуализовали авторадиографически с использованием усиливающего экрана при -70°С.
Результаты ТИЭП показывают, что в необработанных клетках низок базальный уровень фактора NF-кВ, который активируется TNF (фиг.20: дорожки 1 и 2). Предварительная обработка клеток 1 мкг/мл F035 или F094 с последующей активацией TNF (фиг.20: дорожки 4 и 8) приводила к отсутствию подавления активации NF-кВ. Когда клетки обрабатывали 2 мкг/мл UA-BRF-004-DELEP-F035 или UA-BRF-004(бобы)-DELEP-F094 (фиг.20: дорожки 6 и 10), обнаруживали отчетливое подавление активации NF-кВ под влиянием TNF. Результаты этого анализа показывают, что и F035, и F094 способны индуцировать мощный противовоспалительный ответ. Помимо подтверждения потенциальной противовоспалительной активности тритерпеновых соединений, полученные результаты, в частности, являются существенным дополнительным аргументом, показывающим ключевую роль воспаления в канцерогенезе (Sieweke et al., 1990; Prehn, 1997; Schuh et al., 1990).
Пример 9
Анализ механизма передачи сигнала F035
С целью дальнейшего выяснения молекулярных мишеней для активных соединений экстракта растений Acacia victoriae был проведен анализ влияния F035 на активность фосфатидилинозитол-3-киназы (PI3K), а также на активность АКТ (протеинкиназа-В, являющаяся серин-треониновой киназой), негативного эффектора киназы PI3. Киназа PI3 является ферментом, который участвует в сигнальных путях факторов роста за счет связывания с рецепторными и нерецепторными тирозинкиназами. Известны два ингибитора киназы PI3: вортманнин, являющийся грибковым метаболитом, и LY294002, являющийся синтетическим соединением, которое структурно сходно с растительным биофлавоноидом кверцетином.
Данный тест проводили следующим образом: клетки Jurkat (1х107), голодавшие в течение ночи, обрабатывали различными концентрациями (1-8 мкг/мл, в зависимости от клеточной линии) фракции F035 в течение различного времени (2-16 часов) при 37°С. Через разные промежутки времени клетки отбирали и промывали ЗФР при 2000 об/мин в течение 10 минут. Клетки лизировали в лизис буфере с 1% NP-40 в течение 30 минут при 4°С и полученные лизаты выделяли центрифугированием в течение 5 минут при 15000 об/мин при 4°С. С целью проведения иммунопреципитации киназы PI3, 5 мкл кроличьего анти-р85 антитела (р85 - адаптерный белок тирозинкиназного рецептора: Upstate Biotechnology Inc.) инкубировали с 1 мл клеточного лизата в течение 90 минут при 4°С. Иммунные комплексы выделяли с использованием 100 мкл шариков с 20% белка-А/сефарозы в течение еще 90 минут при 4°С. Полученные иммунопреципитаты последовательно промывали: (а) ЗФР, 100 мМ Nа3VO4, 1% Тритон Х-100; (b) 100 мМ Трис, рН 7,6, 0,5 мМ LiCl, 100 мМ Na3VO4; (с) 100 мМ Трис, рН 7,6, 100 мМ NaCl, 1 мМ ЭДТА, 100 мМ Nа3VO4; и (d) 20 мМ HEPES, рН 7,5, 50 мМ NaCl, 5 мМ ЭДТА, 30 мМ NaPPi, 200 мМ Nа3VO4, 1 мМ PMSF, 0,03% Тритон Х-100. Затем иммунопреципитаты ресуспендировали в 30 мкл киназо-реактивного буфера (33 мМ Трис, рН 7,6, 125 мМ NaCl, 15 мМ MgCl2, 200 мМ аденозина, 20 мМ АТФ, 30 мкКи γ-32Р-АТФ). Фосфатидилинозит (PI: 50 мкл) сушили в атмосфере азота и ресуспендировали в 20 мМ HEPES, рН 7,5, 2 мг/мл, и обрабатывали ультразвуком на льду в течение 10 минут. Р13-киназную реакцию инициировали добавлением 10 мкл суспензии PI и 10 мкл γ-меченого АТФ. Реакционную смесь оставляли на полчаса при комнатной температуре с последующей остановкой реакции путем добавления 100 мкл 1 н НСl. Липиды экстрагировали в 600 мкл смеси хлороформ:метанол (1:1) и разделяли на силикагеле (G60) методом тонкослойной хроматографии (ТСХ) с элюированием смесью хлороформ:метанол:NH4OH:H2O (60:47:2:11,3). Радиоактивно меченый фосфатидилинозитфосфат визуализовали авторадиографически и уровень ингибирования определяли количественно с использованием прибора Storm (Okada et al., 1994; Vlahos et al., 1994). Полученные результаты (фиг.21) показывают, что через 2 и 6 часов после обработки F035 (4 мкг/мл) происходило подавление активности киназы PI3. Аналогично, когда клетки обрабатывали 2 мкг/мл F035 в течение 15 часов, уровень наблюдаемого подавления составил 95%, что сходно с действием вортманнина (грибковый метаболит, являющийся известным ингибитором киназы PI3) на клетки Jurkat.
Далее, было изучено влияние F035 на АКТ, являющуюся негативным эффектором киназы PI3. АКТ, также известная как протеинкиназа-В, является клеточным гомологом вирусного онкогена v-akt, активируется рядом фактором роста и функционирует в составе механизма, элементом которого является активация PI3K, чувствительной к действию вортманнина. Ген АКТ кодирует серин-треониновую протеинкиназу: для этого гена была продемонстрирована амплифицируемость в 12,1% случаев карцином яичников и 2,8% случаев раковых опухолей молочной железы. АКТ участвует в антиапоптотическом механизме через фосфорилирование Bad - антиапоптической молекулы (фактора преодоления апоптоза). Больные с опухолью яичников с изменениями АКТ отличаются неблагоприятным прогнозом (Bellacosa et al., 1995). Как было показано, АКТ активно блокирует апоптоз, в частности, за счет активации киназы p70S6 (Kennedy et al., 1997). Киназа p70S6 является активируемой митогенами серин-треониновой протеинкиназой, необходимой для клеточного роста и развития в фазе G1 клеточного цикла (Спои & Blenis, 1996). Активность киназы p70S6 находится под контролем множественных событий фосфорилирования на участке, расположенном в этом полипептиде в пределах каталитического и псевдосубстратного доменов (Cheatham et al., 1995; Weng et al., 1995).
Влияние F035 на фосфорилирование АКТ анализировали следующим образом. Клетки Jurkat (5·106) находились в бессывороточной среде и их обрабатывали F035 в течение 15 часов и 2 часов с вортманнином при 37°С. Клетки индуцировали cd3XL (поперечно сшитый CD3) в течение 10 минут при 37°С или не индуцировали вовсе и лизировали в АКТ-лизис буфере, а белки разделяли электрофоретически в 8%-ном ДСН-ПААГ и анализировали методом Вестерн-блоттинга с использованием антител к фосфорилированной по серину-473 АКТ (New England Biolabs) и к общей АКТ. Тест на влияние F035 на киназу р70S6 можно осуществить аналогичным образом, лишь используя набор антител к киназе p70S6 Phosphoplus (New England Biolabs), предназначенный для анализа фосфорилирования киназы p70S6 (по серину-411, по треонину-421/серину-424). Полученные результаты изучения АКТ (фиг.22) показали, что перекрестно сшитый cd3 индуцирует слабое фосфорилирование АКТ. Последующая обработка клеток 1 и 2 мкг/мл F035 обуславливала отчетливое подавление фосфорилирования АКТ (активацию АКТ), что аналогично 2-часовой обработке клеток 1 мкМ вортманнином. Однако, различий в общем уровне экспрессии АКТ не было. Аналогичное подавление фосфорилирования АКТ также было продемонстрировано для клеток опухоли яичников OVCAR-3 и С-2 (вариант линии HEY) и клеток Jurkat, обработанных 2-4 мкг/мл F094. Полученные данные показывают, что F035 подавляет фосфорилирование киназы АКТ в клетках Jurkat и клетках рака яичников. Это убедительно подтверждает то, что сигнальный механизм с участием киназ Р13/АКТ, как это было показано, передает антиапоптотический сигнал (Kennedy et al., 1997). Полученные результаты подтверждают, что F035 и F094 опосредуют апоптоз опухолевых клеток за счет подавления сигнального механизма киназы PI3.
Пример 10
Выявление апоптоза в анализе клеточного цикла и тесте на связывание аннексина-V
С целью дальнейшей характеризации механизма подавления роста и цитотоксичности, проявляемых активными соединениями из данного растительного экстракта, приблизительно 1×106 опухолевых клеток линии OVCAR-3 высевали на 60-мм3 чашки, обрабатывали различными концентрациями UA-BRF-004-DELEP-F035 и инкубировали в течение 18-24 часов при 37°С. Клетки собирали, дважды промывали ЗФР и ресуспендировали в концентрации 1×106 клеток на 1 мл. Параформальдегид (в конечной концентрации 1%) добавляли по каплям к аккуратно перемешиваемым клеткам. Клетки вновь промывали ЗФР после 15-минутной инкубации на льду и осадок ресуспендировали в 70%-ном этаноле, охлажденном на льду, и инкубировали в течение ночи при -20°С. Наконец, этанол один раз отмывали ЗФР и клетки ресуспендировали в 10 мкг/мл йодистого пропидия (Sigma Chemical Co.) с 0,1% РНКазой (RNase). Клетки снова однократно инкубировали при комнатной температуре в течение 30 минут, а затем переносили в 4°С и анализировали через 18 часов методом проточной цитометрии (Pallavicini, 1987). Полученные результаты показывают, что перед обработкой клеток UA-BRF-004-DELEP-F035, 48% клеток находились в фазе G0/G1, 36% клеток находились в фазе S и 7% клеток находились в фазе G2/M. Однако, через 48 часов после обработки клеток OVCAR-3 фракцией F035 примерно 58% клеток находились в фазе G1 и только 27% клеток находились в фазе S клеточного цикла: т.е. на 8% увеличивается доля клеток в G1 и примерно на 10% уменьшается число клеток в фазе S клеточного цикла (фиг.19А, В). Полученные результаты указывают на явную блокировку клеток опухоли яичников человека OVCAR-3 в фазе G1.
Также было изучено влияние F035 на профиль клеточного цикла клеток рака молочной железы человека. Клетки рака молочной железы MDA-MB-435 и MDA-MB-453 обрабатывали различными концентрациями F035 и анализировали через 72 часа, как описано выше. Полученные результаты показывают, что F035 индуцирует апоптоз клеток MDA-MB-435, о чем свидетельствует появление пика, соответствующего фазе Sub.G0 (табл.27). Кроме того, регуляция клеточного цикла также наблюдалась по снижению процента клеток, находящихся в фазах клеточного цикла S и G2/M.
Анализ клеточных циклов у клеток опухоли молочной железы MDA-МВ-435 после обработки F035
С использованием клеток MDA-MB-435 полученные результаты показывают, что F035 индуцирует блокировку клеточного цикла с увеличением примерно на 10% числа клеток в фазе G1 и уменьшение на 4-10% клеток в фазе S клеточного цикла через 72 часа после обработки F035 (табл.28). Эти данные указывают на блокировку клеточного цикла и апоптоз опухолевых клеток, индуцируемые данным растительным экстрактом.
Анализ клеточных циклов у клеток опухоли молочной железы MDA-МВ-453
Клетки Jurkat (1×106) обрабатывали различными концентрациями UA-BRF-004-DELEP-F035 (50-1000 нг/мл) в течение 18 часов при 37°С. Клетки однократно промывали ЗФР, ресуспендировали в связывающем буфере (10 мМ HEPES/NaOH, 140 мМ NaCl, 2 мМ CaCl2), содержащем 5 мкл конъюгата аннексина-V-ФИТЦ (Biowhittaker, Walkersville, MD), и инкубировали в течение 10 минут в темноте. Клетки промывали и ресуспендировали в связывающем буфере, содержащем 10 мкл 20 мкг/мл йодистого пропидия (Sigma Chemical Co.), и анализировали с применением клеточного сортера с возбуждаемой флуоресценцией (FACS) (Martin et al., 1995).
Полученные результаты показывают, что очищенные активные компоненты были способны вызывать апоптоз клеток Jurkat. Это наблюдение дополнительно подтверждается способностью обработанных клеток связывать аннексин-V, что является показателем вступления клеток в апоптоз (табл. 29). Обычно, фосфотидилсерин (ФС) локализуется на внутренней поверхности плазматической мембраны. Однако, на ранних стадиях апоптоза происходит выход ФС на наружную поверхность плазматической мембраны. Аннексин-V является кальций-связывающим белком, который связывается с ФС и может быть выявлен методом проточной цитометрии с окрашиванием аннексином-V-ФИТЦ (Martin et al., 1995).
Анализ апоптоза по параметрам связывания с аннексином-V в клетках Jurkat, обработанных различными концентрациями UA-BRF-004-DELEP-F035
Пример 11
UA-BRF-004-DELEP-F035 как хемопротективный агент
Влияние UA-BRF-004-DELEP-F035 было исследовано на модели многостадийного кожного канцерогенеза у мышей SENCAR. Кожу этих животных обрабатывали (“разрисовывали”) ацетоном, канцерогеном DMBA (7,12-диметилбенз[а]антрацен), DMBA + UA-BRF-004-DELEP-F035 и DMBA + фракция 60 (негативный контроль) при низкой (100 мкг UA-BRF-004-DELEP-F035 или фракции 60 на 1 животное) и высокой (500 мкг UA-BRF-004-DELEP-F035 или фракции 60 на 1 животное) дозах растительного экстракта, применявшихся два раза в неделю на протяжении 4 недель. UA-BRF-004-DELEP-F035 или контроль наносили на кожу мышей за 5 минут до применения DMBA. У животных наблюдали образование папиллом и после этого их умерщвляли и ткани анализировали гистологически (фиг.9A-F). Результаты проведенного анализа систематизированы на фиг.10А, фиг.10В, фиг.11А, фиг.11В, фиг.12 и фиг.13.
Через 8 недель проведения этих экспериментов в группе мышей, обработанных DMBA, отмечено по 8 папиллом на 1 животное, в то время как в группах с совместным воздействием DMBA и UA-BRF-004-DELEP-F035 было по 0,66 папиллом на 1 животное; в группе с обработкой DMBA и фракцией 60 (негативный контроль) имелось по 6,9 папиллом на 1 животное. Эти результаты указывают на значительный протективный эффект UA-BRF-004-DELEP-F035 в отношении опухолей, в то время как фракция 60, в основном, не обладала протективным эффектом.
В дальнейших исследованиях на мышах in vivo было показано, что UA-BRF-004-DELEP-F035 является хемопротективным агентом в случаях с опухолями, индуцированными канцерогенами, за счет предотвращения мутаций в онкогене ras. Стадию инициации канцерогенеза в коже мышей обеспечивали путем прямого воздействия канцерогена (например, DMBA) и, в основном, она являлась необратимой. Подавление канцерогенеза определяли через 8 недель по снижению интенсивности образования папиллом, индуцируемых DMBA. Молекулярный анализ обработанной кожи показал, что UA-BRF-004-DELEP-F035 предотвращает способность DMBA индуцировать мутации в онкогене ras (см. примеры 14, 15 и 16 ниже).
Пример 12
Процедура выявления активных тритерпеноидов у Acacia victoriae
Была использована процедура, позволяющая эффективно выявлять активные тритерпены в образце ткани растения. Эту процедуру осуществляли следующим образом. Приблизительно 5 г листьев и веточек разрезали на небольшие кусочки ножницами или, альтернативно, образцы корней разрезали ножом на мелкие кусочки. Растительный материал обрабатывали в небольшом смесителе, объединяли примерно с 25 мл 80%-ного (об/об) метанола и оставляли, по крайней мере, на 2 часа при встряхивании каждый час. Нерастворимый материал удаляли центрифугированием при 10000 g. Затем экстракт использовали для проведения тонкослойной хроматографии на обращенно-фазовых пластинах (алюминиевые ТСХ-пластины, RP-C18 F254s) с 40% ацетонитрила (об/об). После обработки ТСХ-пластин смесью 0,1% ванилин (4-гидрокси-3-метоксибензальдегид/Н2SO4 и подсушивания при 70°С в течение 15-30 минут, активные тритерпеноидные соединения визуализуются в виде буровато-красных пятен (Rf=0,2-0,3).
Пример 13
Локализация тритерпеновых соединений в растениях Acacia
victoriae
На начальных этапах исследований, высушенные надземные части растения собирали в начале лета для получения экстрактов. Последующие повторные сборы осенью оказались неактивными. После этого был проведен систематический анализ с целью определения относительных уровней присутствия или отсутствия активных тритерпеновых соединений в различных частях растений Acacia victoriae. После мониторинга химии растения было установлено, что, в основном, все активные компоненты надземной части данного растения сконцентрированы в бобах и проростках, в то время как они в основном или полностью отсутствуют в ветках, коре ствола, листьях и семенах. Следовательно, активный сбор материала ограничен примерно тремя неделями от начала формирования бобов до их раскрытия. Также было установлено, что корни растения вырабатывают такой же активный материал при колеблющемся соотношении сахаров и активных компонентов. Последние параметры указывают на то, что аэропоника, при которой наблюдается мощный корневой рост при сохранении нормального развития растений, может быть пригоден для Acacia victoriae.
Пример 14
Линии опухолевых клеток и их рост
Следующие линии опухолевых клеток человека были получены из Американской коллекции типовых культур (АТСС: Rockville, MD): клеточные линии SK-OV-3 и OVCAR-3 (яичник), Jurkat (Т-клеточный лейкоз), U-937 (гистиоцитарная лимфома), MDA-MB-468, MDA-MB-453, MDA-MB-435, SK-BP-3, MCF-7, MDA-MB-231, ВТ-20 (молочная железа), LNCaP, PC-3, DU145 (предстательная железа), 769-Р, 786-0, А-498 (почки) и PANC-1 (поджелудочная железа). Клеточные линии HEY и Dov-13 (яичники) были получены из Онкологического центра M.D.Anderson. Следующие нетрансформированные клеточные линии MCF-10A и 10F (эпителий молочной железы) были получены из Онкологического центра M.D.Anderson. Линии Hs-27 (фибробласты крайней плоти человека) и L929 (фибробласты мыши) были получены из АТСС. Линии SK-OV-3, MDA-MB-468, Hs-27, L929 культивировали в минимальной поддерживающей среде. Линии OVCAR-3, Jurkat, U-937, LNCaP, DU-145, PC-3, HEY, Dov-13, PANC-1, MCF-10A, MCF-10F и остальные клеточные линии рака молочной железы культивировали в среде RPMI-1640, а культуральную среду F-12 использовали для роста клеток 769-Р, 786-0 и А-498. Все используемые культуральные среды дополняли 10% фетальной телячьей сывороткой, 200 мМ глутамина и 0,05% гентамицина.
Пример 15
Амплификация мутантного по 61-му кодону САА→СТА гена Ha-ras мыши с использованием специфичных для этой мутации праймеров (MSP)
Данный протокол использован в соответствии с Nelson et al., (1992). Обратный праймер, обозначенный как 3MSP61mut, конструировали так, чтобы 3’-концевой нуклеотид (А) спаривался со средним нуклеотидом (подчеркнут) в кодонах 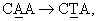 представляющих трансверсию 61-го кодона гена Ha-ras, и обеспечивал избирательную амплификацию мутантной ДНК в условиях, описанных ниже. Тест основывается на том, что Taq-полимераза лишена 3’-экзонуклеазной активности и поэтому не способна исправить ошибку по 3'-концу отжигаемого праймера. Условия теста зависят от обратного праймера, не способного в сколько-нибудь существенной степени отжигаться на последовательность дикого типа, в результате чего достройки цепи не происходит. С использованием того же прямого праймера одна реакция проходит с обратным включающим ошибку праймером (3MSP61mut), а другая реакция проходит с обратным праймером, соответствующим дикому типу (3MP61wt). В этом протоколе выявляется только трансверсия
представляющих трансверсию 61-го кодона гена Ha-ras, и обеспечивал избирательную амплификацию мутантной ДНК в условиях, описанных ниже. Тест основывается на том, что Taq-полимераза лишена 3’-экзонуклеазной активности и поэтому не способна исправить ошибку по 3'-концу отжигаемого праймера. Условия теста зависят от обратного праймера, не способного в сколько-нибудь существенной степени отжигаться на последовательность дикого типа, в результате чего достройки цепи не происходит. С использованием того же прямого праймера одна реакция проходит с обратным включающим ошибку праймером (3MSP61mut), а другая реакция проходит с обратным праймером, соответствующим дикому типу (3MP61wt). В этом протоколе выявляется только трансверсия 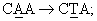 однако, именно эти мутации являются, в основном, преобладающими среди мутаций, затрагивающих 61-й кодон. В результате этой трансверсии образуется ПДРФ-сайт, разрезаемый рестриктазой Xbal
однако, именно эти мутации являются, в основном, преобладающими среди мутаций, затрагивающих 61-й кодон. В результате этой трансверсии образуется ПДРФ-сайт, разрезаемый рестриктазой Xbal  . Мутации можно проверить путем обработки амплифицированной ДНК рестриктазой Xbal или путем прямого секвенирования ДНК с использованием 32Р-концевого мечения праймера 5MSP61. Реакции, включающие продукты с мутацией, могут быть подвергнуты электрофорезу в 2%-ной легкоплавкой агарозе для последующих очистки и секвенирования. Последовательности использованных праймеров, 5MSP61, 3MSP61mut и 3MSP61wt, приведены ниже: и соответственно, в SEQ ID NO 2, SEQ ID NO 3 и SEQ ID NO 4.
. Мутации можно проверить путем обработки амплифицированной ДНК рестриктазой Xbal или путем прямого секвенирования ДНК с использованием 32Р-концевого мечения праймера 5MSP61. Реакции, включающие продукты с мутацией, могут быть подвергнуты электрофорезу в 2%-ной легкоплавкой агарозе для последующих очистки и секвенирования. Последовательности использованных праймеров, 5MSP61, 3MSP61mut и 3MSP61wt, приведены ниже: и соответственно, в SEQ ID NO 2, SEQ ID NO 3 и SEQ ID NO 4.
5MSP61 (23-мерный): 5’-CTA AGC CTG TTG TTT TGC AGG AC-3’ (SEQ ID NO 2);
3MSP61mut (20-мерный): 5’-CAT GGC ACT ATA CTC TTC TA-3’ (SEQ ID NO 3);
3MSP61wt (20-мерный): 5’-CAT GGC ACT ATA CTC TTC TT-3’ (SEQ ID NO 4).
Последовательность 3MSP61wt включает 2 или 3 ошибки по сравнению с последовательностями, соответственно, N-ras и К-ras, размер фрагмента составляет 110 пар нуклеотидов. ДНК-матрица и реактивы для амплификации следующие:
ДНК (позитивный контроль) OR 1,0 мкг
ДНК (негативный контроль, т.е. дикий тип) OR 1,0 мкг
ДНК (образец, т.е. парафинизированный блок) OR 5,0 мкл
Без ДНК (т.е. Н2О) 5,0 мкл
Буфер Rxn (10x) (10x = 500 мМ КСl,
100 мМ Трис, рН=8,3; 15 мМ MgCl2) 5,0 мкл
Смесь dNTP, по 500 мкМ каждого
(конечная концентрация =20 мкМ) 2,0 мкл
[32P]dCTP, 3000 Ки/ммоль, 5 мкМи,
1,7 пикомоль, 0,034 мкМ 0,50 мкл
5’-праймер (10 пкмоль/мкл), 7,5 пкмоль
(конечная концентрация 0,15 мкМ) 0,75 мкл
3’-праймер (10 пкмоль/мкл), 7,5 пкмоль
(конечная концентрация 0,15 мкМ) 0,75 мкл
Taq-полимераза (5 ед./мкл, 3,0 ед.) 0,60 мкл
Н2O до 50,0 мкл 50 мкл
Минеральное масло 2 капли
Условия цикла амплификации при использовании термоячейки фирмы Perkin-Elmer были следующими:
Проверку теста осуществляли обработкой следующих контролей: дикий тип (WT), мутации дикого типа (MUT) и негативный контроль (H2O). В качестве позитивного контроля использовали мутантную (МОТ) ДНК из плазмиды pHras61mut. Плазмида pHras61 включает клонированную ДНК-последовательность 2-го экзона гена Ha-ras из клеток мышиной опухоли Sencar. Правильность клонированной мутации подтверждали путем секвенирования ДНК. Этой мутацией является трансверсия САА→СТА в кодоне 61 (находится во 2-м экзоне) гена Ha-ras мыши, являющегося образцом ДНК из опухоли аденокарциномы, содержащей мутацию гена Ha-ras в 61-м кодоне (см. фиг.14).
Пример 16
Тест с ПЦР™ “с горячим стартом” на ПДРФ-мутации в кодонах 12/13 гена Н-ras мыши
Данный тест основывается на разрушении нативного рестрикционного MnlI-сайта, охватывающего все 3 нуклеотида 12-го кодона и первый нуклеотид 13-го кодона (GGA GGC, нуклеотиды 34-37 в кодирующей последовательности генов Ha-ras крысы и мыши). Распознаванием сайта MnlI является N7GGAG. Мутации в любом из этих четырех сайтов будут приводить к неспособности Mnll разрезать ПЦР-фрагмент, в составе которого имеется эта область. Недостатком данного теста является то, что иногда происходит неполное расщепление с помощью MnlI. Этот факт затрудняет разграничение между очень малым процентом устойчивой к расщеплению ДНК дикого типа и характеризующейся низким уровнем истинной мутации. Это иногда наблюдается тогда, когда источник ДНК включает смесь ДНК дикого типа и ДНК с мутацией и когда для повышения чувствительности используется 32Р-меченый фрагмент. Набор праймеров для ПЦР™, используемый в данном тесте, приведен ниже и в последовательностях SEQ ID NO 5 и SEQ ID NO 6. Размер амплифицированного продукта гена Ha-ras 12 составляет 214 пар нуклеотидов.
Праймер #3 (5’): 5’-ССТ TGG СТА AGT GTG СТТ СТС АТТ GG-3’ (SEQ ID NO: 5);
Праймер #6 (3’): 5’-АСА GCC САС СТС TGG CAG GTA GG-3’ (SEQ ID NO: 6).
Праймер #6 используют для секвенирования при 55°С при следующих условиях реакции:
Буфер Rxn (10x) (10x = 500 мМ KCl,
100 мМ Трис, рН=8,3; 15 мМ MgCl2) 1,0 мкл
Смесь dNTP, по 0,5 мМ каждого нуклеотида 1,0 мкл
Праймер (5’) 6 пкмол
Праймер (3’) 6 пкмол
32P-dCTP, 3000 Ки/ммоль 0,5 мкл
Taq-полимераза (5 ед./мкл, 0,65 ед.) 0,13 мкл
Н2О до 10 мкл
ДНК (позитивный контроль) >200,0 нг
ДНК (негативный контроль, т.е. дикий тип) >200,0 нг
ДНК (образец, т.е. парафинизированный блок) 5,0 мкл
Без ДНК (т.е. Н2О) 5,0 мкл
Минеральное масло 2 капли
Условия амплификации при использовании набора реактивов
для ПЦР Perkin-Elmer PCR™ были следующими:
Пример 17
Результаты тестирования: выявление мутаций в гене c-Ha-ras
Через 4 дня после последнего введения DMBA, растительного экстракта и контроля, выделенную из свежезамороженных тканей ДНК (по 5 мышей из каждой группы) анализировали методом ПЦР™ на наличие мутаций в кодонах 12 и 13 и в кодоне 61 гена c-Ha-ras. Заявителями были использованы образцы 4-дневной выдержки, поскольку некоторые 2-дневные образцы ДНК разрушились и не были пригодны для анализа Ha-ras. В кодонах 12 и 13 находится рестрикционный MnlI-сайт, охватывающий 3 нуклеотида 12-го кодона и первый нуклеотид 13-го кодона в последовательности дикого типа. Мутация любого из них приводила в результате к утрате MnlI-сайта. На материале геномной ДНК был амплифицирован 1-й экзон (в котором находятся 12-й и 13-й кодоны) гена c-Ha-ras, для чего использовали термоячейку фирмы Perkin-Elmer. Реакционную смесь экстрагировали смесью фенол-СНСl3 и ДНК осаждали этанолом. Затем эту ДНК ресуспендировали в ферментном буфере и ПЦР-продукт рестриктировали MnlI с последующим электрофорезом полученного продукта в 8%-ном не денатурирующем полиакриламидном геле. В тестируемом материале не было выявлено потери рестрикционного MnlI-сайта и было сделано заключение о том, что в кодонах 12 и 13 мутации отсутствуют. ДНК для анализа гена Ha-ras также была выделена из залитых в парафин гистологических срезов толщиной 8 мкм, полученных через 2 дня после последней обработки. По 25 срезов из каждого запарафинированного блока помещали в микроцентрифужную пробирку, депарафинизировали ксилолом и этанолом, центрифугировали и ресуспендировали в 5% Chelex® с протеиназой-К.
Первая процедура, связанная с 61-м кодоном гена Ha-ras, разработана Nelson et al., 1992 (см. выше пример 15). С использованием одного и того же прямого праймера, одна реакция проходит с обратным “ошибочным” праймером (3MSP61mut), a другая реакция с обратным праймером, соответствующим дикому типу (3MSP61wt). В этом методе выявляется только мутация типа трансверсии 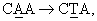 которая является наиболее преобладающей точечной мутацией в 61-м кодоне. В результате этой трансверсии создается ПДРФ по XbaI-сайту
которая является наиболее преобладающей точечной мутацией в 61-м кодоне. В результате этой трансверсии создается ПДРФ по XbaI-сайту 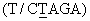 . Реакции, включающие продукт с мутацией, подвергают электрофорезу в 2%-ной легкоплавкой агарозе для последующей очистки и секвенирования. Соотношение разрезанной (ДНК дикого типа) и неразрезанной (мутантной ДНК) определяют количественно путем оценки интенсивности окрашивания этидийбромидом или с помощью 32Р-мечения. В качестве позитивного контроля используют ДНК плазмиды pHras61mut. Плазмида рНrаs61 включает клонированную ДНК 2-го экзона гена Ha-ras опухоли мышей Sencar. Правильность клонированной мутации подтверждали путем секвенирования ДНК. Мутацией является трансверсия САА→СТА в кодоне 61 (находящимся в последовательности 2-го экзона) гена Ha-ras мыши. Условия реакции описаны в примере 15.
. Реакции, включающие продукт с мутацией, подвергают электрофорезу в 2%-ной легкоплавкой агарозе для последующей очистки и секвенирования. Соотношение разрезанной (ДНК дикого типа) и неразрезанной (мутантной ДНК) определяют количественно путем оценки интенсивности окрашивания этидийбромидом или с помощью 32Р-мечения. В качестве позитивного контроля используют ДНК плазмиды pHras61mut. Плазмида рНrаs61 включает клонированную ДНК 2-го экзона гена Ha-ras опухоли мышей Sencar. Правильность клонированной мутации подтверждали путем секвенирования ДНК. Мутацией является трансверсия САА→СТА в кодоне 61 (находящимся в последовательности 2-го экзона) гена Ha-ras мыши. Условия реакции описаны в примере 15.
Пример 18
Влияние F035 на возникновение аномальных крипт у крыс F344, подвергавшихся воздействию азоксиметана
Самцы крыс линии Fischer-344 (F344) были получены из лаборатории Charles River (Raleigh, NC) в возрасте 6 недель. Крыс кормили без ограничений рационом AIN-76A, закупленным у Dyets Inc. (Bethlehem, PA). В состав рациона входит казеин (20%), DL-метионин (0,3%), кукурузный крахмал (15%), сахароза (50%), кукурузное масло (5%), целлюлоза (5%), солевая смесь AIN-76 (3,5%), витаминная смесь AIN-76 (1%) и битартрат холина (0,2%). Также животным без ограничений давали питьевую воду. Азоксиметан (АОМ), являющийся индуктором аномальных крипт кишечника крыс, был приобретен у Sigma Chemical Co. (St.Louis, МО). Животных выдерживали на 3-дневном карантине с “крысиной пищевой смесью”, а затем выкармливали рационом AIN-76A до возраста 7 недель. После этого животных для обработки случайным образом разделяли на 3 группы (10 животных/группу). Животных 1-й группы выкармливали только рационом AIN-76A, 2-я группа получала рацион AIN-76A с добавлением 5 мг/мг F035, а 3-я группа - AIN-76A и 10 мг/кг F035 (табл.30).
Экспериментальные группы по изучению влияния F035 на индукцию аномальных крипт у крыс F344 под действием азоксиметана
После такого кормления в течение недели, всем животным внутрибрюшинно вводили АОМ (15 мг/кг веса тела). Еще одну инъекцию АОМ проводили через неделю после первой. В течение всего эксперимента, животных еженедельно взвешивали. Выкармливание животных проводили на протяжении 4 недель. Через 31 день животных умерщвляли с помощью углекислотной асфиксии. Затем удаляли толстую кишку, промывали ее холодным ЗФР, разрезали вдоль средней продольной оси, помещали на фильтровальную бумагу и фиксировали 70%-ным спиртом в течение, по крайней мере, 24 часов. Ткани толстой кишки окрашивали метиленовым синим (0,25% в ЗФР) в течение примерно 1 минуты. Аномальные крипты подсчитывали под микроскопом с увеличением 20х. Аномальные крипты отличаются от окружающих их нормальных крипт по увеличенному размеру, резко увеличенному расстоянию от верхушки складки (просвета) до основания лакуны клеток и увеличенной перикриптальной зоной. Все животные получили условные метки и подсчет проводили вдвоем “вслепую”. Статистическую достоверность результатов при сравнении групп между собой определяли с помощью одномерного дисперсионного анализа ANOVA. При обнаружении различий использовали критерий Стьюдента (t) с поправкой Бонферрони для оценки множественных сравнений обеих доз F035 с контрольной группой. Обсчет проб толстой кишки проводили вдвоем и “вслепую”, т.е. не зная, обсчет какой экспериментальной группы проводится. Между данными, полученными двумя “счетчиками”, имелось хорошее соответствие.
Было установлено, что F035 достоверно снижает общее число очагов аномальных крипт при включении в рацион в количестве 10 мг на 1 кг корма, что примерно эквивалентно суточной дозе 1 мг F035 на 1 кг веса тела (табл.31, табл.33). Та же доза также в существенной степени снижает число аномальных крипт в синглетных и дублетных (1 и 2 аномальные крипты на очаг) категориях (табл.32, табл.34). Нижняя доза F035, 5 мг на 1 кг корма (что примерно эквивалентно суточной дозе 500 мг F035 на 1 кг веса тела), приводит к снижению общего числа аномальных крипт, числа их синглетных и дублетных очагов, однако уровень различий по сравнению с контролем статистически недостоверен (табл.32, табл.34). На протяжении всего эксперимента достоверные отличия по весовому приросту между экспериментальными и контрольной группами отсутствовали (табл.35, табл.36, табл.37).
Влияние F035 на число аномальных крипт в толстой кишке у крыс, подвергавшихся воздействию АОМ
“+” - достоверно по сравнению с контролем (Р<0,05);
С - заключительное исследование
Влияние F035 на число аномальных крипт в очаге у крыс, подвергавшихся воздействию АОМ
Обобщение предварительных данных анализа влияния F035 на среднее число аномальных крипт на толстую кишку у крыс, подвергавшихся воздействию АОМ
b - среднее по АОМ-инъецированным крысам (N=10) за 4 недели;
с - достоверно по сравнению с контролем (Р<0,05)
Обобщение предварительных данных о влиянии F035 на число аномальных крипт в очаге у крыс, подвергавшихся воздействию АОМ
Вес тела крыс, которым вводили АОМ и скармливали F035 из расчета 5 мг на 1 кг веса тела
Вес тела крыс, которым вводили АОМ и скармливали F035 из расчета 10 мг на 1 кг веса тела
Вес тела крыс, которым вводили АОМ
Пример 19
Противоопухолевая активность агликонов
Проведенные исследования подтвердили важность сахаров для проявления биологической активности, поскольку удаление сахаров из ядра тритерпеновой молекулы приводит в результате к значительной потере биологической активности. Как показано в табл. 38, фракция UA-BRF-004(бобы)-DELEP-F164 (генерированная путем гидролиза Сахаров из UA-BRF-004(бобы)-DELEP-F094 с присоединенными сложными эфирами) и UA-BRF-004(бобы)-DELEР-F245 (смесь метиловых сложных эфиров продуктов гидролиза UA-BRF-004(бобы)-DELEP-F094) обнаруживает отчетливую утрату противоопухолевой активности в отношении панели линий опухолевых клеток. Аналогично, UA-BRF-004(бобы)-DELEP-C194 (очищенный ацетат агликона 1) не проявляет существенной противоопухолевой активности в отношении панели линий опухолевых клеток по сравнению с данными, полученными для тритерпенового гликозида фракции 35. Следовательно, имеется значительная потеря биологической активности после гидролиза сахарной составляющей заявляемого здесь тритерпенового гликозида.
Биологическое тестирование фракций F164, F245 и С194
Пример 20
Анализ влияния F035 на метаболизм холестерина
Целью данного анализа является изучение влияния биологически активных тритерпеновых гликозидов по настоящему изобретению на профилактику сердечно-сосудистых заболеваний. Непосредственным объектом данного анализа является установление того факта, что тритерпеновые соединения, вносимые в рацион млекопитающих, включая человека, будут снижать уровень сывороточного холестерина. Отобранной для исследований модельной системой линии хомячков с гиперлипидемией является модель грызунов, которая, в отличие от крысиной модели, очень точно имитируют изменения уровней в плазме рецептора LDL и липопротеина у человека в зависимости от уровня холестерина (Spady et al., 1993).
Тритерпеновый гликозид вводят в двух различных концентрациях в очищенный корм для хомячков без какого-либо изменения в содержании кальция, калия, фосфора и других необходимых элементов рациона. Для того, чтобы продемонстрировать зависимость от дозы, использовали два различных уровня добавляемого тритерпенового гликозида. Животным обеспечивали свободный доступ к очищенному корму для хомячков Dyets, приготовленному в соответствии с рекомендациями NRC (Reeves et al., 1993) с добавлением 1% холестерина или без него (Davis et al., 1989). Очищенный корм для хомячков Dyets, содержащий холестерин, модифицировали внесением тритерпенового гликозида в концентрациях, обозначенных ниже. Экспериментальный гранулированный корм и контрольный рацион были приготовлены фирмой Dyets Inc. (Bethlehem, PA) без изменений в содержании кальция, фосфора или любого другого важного пищевого микрокомпонента. Животных контролировали на усвояемость корма и прирост веса тела еженедельно. Используемыми животными были золотистые (сирийские) хомячки аутбредной линии в возрасте 4 недель, не содержащие вирусных инфекций (Charles River Lab., Wilmington, MA). Животных разделяли на группы, помещая по 3 особи на садок случайным образом с использованием генератора случайных чисел программы Statview: садки размещали в комнате с освещением 12 часов в сутки и поддержанием температуры 22±1,0°С.
Через 0, 4 и 8 недель выкармливания на соответствующих рационах, включающих или не включающих тритерпеновый гликозид, по 12 животных в каждой группе отбирают случайным образом и умерщвляют в 9-11 часов утра. Затем удаляют печень и почки, взвешивают, обрабатывают и хранят при -70°С для дальнейшего анализа. В момент умерщвления берут кровь путем пункции сердца перед удалением печени и почек, и тестируют липидную картину крови. Проводят сравнение липидных профилей сыворотки крови у экспериментальной и контрольной группы животных. Было исследовано две контрольные группы (группы 1 и 2) и две экспериментальные группы (группы 3 и 4). Все эти группы выкармливают на рационе NRC в ходе 2-недельного карантина. Группа 1 продолжает получать рацион NRC на протяжении всего периода эксперимента. Группы 2-4 кормят рационом NRC с добавлением 1% холестерина в течение еще 2 недель с целью индуцирования гиперхолестеринемии. Затем группа 2 продолжает получать этот рацион до конца эксперимента, в то время как группы 3 и 4 кормят тем же рационом, дополненным тритерпеновым гликозидом (например, F035 или F094). Обобщение параметров экспериментальных групп дано ниже в табл.39.
Схема модификации рационов
b - Ход = холестерин;
с - ТГ = тритерпеновый гликозид
Через 0 (только контрольная группа), 4 и 8 недель кормления соответствующим рационом, включающим или не включающим тритерпеновый гликозид, по 12 особей из каждой группы отбирают случайным образом и умерщвляют в 9-11 часов утра. Печень и почки хомячков удаляли, взвешивали и анализировали на возможные аномалии. Фрагмент каждого органа, характеризующегося какими-либо аномалиями, приготавливали для гистологического исследования, т.е. замораживали парафиновые срезы, которые окрашивали гематоксилином и эозином. Кровь брали в момент умерщвления путем пункции сердца перед хирургическим удалением печени и почек. Затем приготавливали сыворотку и выдерживали ее при -20°С для анализа липидного профиля крови. Перед умерщвлением хомячки голодали в течение ночи. Полученные данные показаны в табл.40.
Образцы крови, собранные в ходе данного эксперимента, используют для определения уровня общего холестерина, триглицеридов, HDL-холестерина и LDL-холестерина, а также VLDL-холестерина (Mackness & Durrington, 1992) в лаборатории Roche Biomedical Lab., Burlington, NC. Статистическую обработку полученных данных проводили на компьютере “Макинтош-9600” с программой для дисперсионного анализа, определения уровней значимости и линейной регрессии (Armitage, 1971). В частности, данные анализа липидных профилей в каждой из экспериментальных групп проводили методом анализа дисперсий с использованием критерия Ньюмена-Кейлсa (Steel & Torrie, 1980).
Влияние непрерывного скармливания хомячкам тритерпенового гликозида (ТГ) в течение 6 недель
по 12 хомячков в группе получали очищенный хомячковый рацион с 1% холестерина
Пример 21
Анализ профилактики УФВ-индуцированного канцерогенеза с использованием фракции 35
Данный анализ был сосредоточен на профилактике канцерогенеза, вызываемого ультрафиолетом-В, в модели кожи мышей с использованием тритерпеновых гликозидов по настоящему изобретению. Непосредственным объектом данного исследования является выявление того факта, что в модели кожи мышей тритерпеновые гликозиды будут предотвращать повреждения, вызываемые УФ-В. Экспериментальную мышиную модель использовали потому, что она наиболее точно отображает аналогичную ситуацию у людей. В данном анализе планируется продемонстрировать, что местное применение активных тритерпеновых соединений по настоящему изобретению в ацетоне на кожу спины бесшерстных мышей линии SKH-1, облученных УФ-В, будет предотвращать повреждения кожи, вызываемые таким облучением.
В данном анализе, бесшерстных мышей SKH-1 облучают ультрафиолетом-В дозой 1,8 кДж/м2 в течение 15 минут. Предварительно, мышей подвергают воздействию двух различных доз F035 (2 мг и 4 мг в одну дозу), а также ставят негативный контроль (F060 или только ацетон). Считается, что для получения статистически значимых результатов, в каждой группе необходимо иметь минимум по 10 мышей. Каждое тестируемое соединение применяют местно за 10 минут до облучения трижды в неделю в течение 6-10 недель. Исследования проводят в течение короткого периода времени с целью оценки профилактического эффекта данных соединений. Увидеть сформированные, визуально различимые опухоли не ожидали, даже при воздействии только УФ-В, учитывая в данных временных рамках только повреждения кожи. Может быть выявлена слабая эритема (небольшое покраснение кожи), которая исчезала на следующий день после облучения.
В качестве источника УФ использовали прибор с 8 УФ-лампами Westinghouse FS40, радиометр/фотометр IL-1400A и присоединенный к фототерапевтическому УФВ-радиометру IL-1403 детектор SEL-240/UVB-1/TD. В срединной части этого прибора имеется несколько камер, в каждую из которых помещали по 1 мыши. По бокам камер имеются отверстия, обеспечивающие вентиляцию в процессе облучения мышей. Во время облучения камеры вращались в горизонтальной плоскости, что обеспечивало равномерное воздействие УФ-В на мышей. В устройстве имеются дверцы, которые должны быть закрыты во время включения УФВ-лампы, и поэтому весь ультрафиолет остается внутри устройства. Для измерения уровня УФ-В используется УФВ-радиометр. Мыши не должны находиться в камерах более 10-15 минут.
Целью данного анализа было продемонстрировать светозащитное действие против индуцируемых УФ-В повреждений кожи мышей. УФ-В поглощается непосредственно клеточной ДНК и вызывает повреждения, которые могут индуцировать мутации в генах-мишенях, в конечном счете приводящих к раку. Раннее выявление таких повреждений и предотвращение таких повреждений может указывать на наличие хемопротективных эффектов (Berton et al., 1997; Chatterjee et al., 1996; Youn et al., 1997; Shirazi et al., 1996; Baba et al., 1996; Takema et al., 1996).
Режимы облучения УФ-В
Экспериментальные группы для исследования описаны в табл. 41. Предполагается, что размер этих групп достаточен для того, чтобы проконтролировать изменчивость гиперплазии кожи и воспаления кожи в данной группе, межиндивидуальную изменчивость толщины эпидермиса и воспаление кожи у животных одного и того же возраста и стадии развития. Основными параметрами, которые регистрируют в данном анализе, являются гиперплазия и кожные воспаления. Оставшуюся кожу сохраняют для измерения других биологических маркеров, таких как модифицированные нуклеотиды в ДНК (8-OH-dG) и экспрессия онкогена (онкоген Ha-ras).
Животные получают свободный доступ к гранулированному корму и питьевой воде на протяжении всего эксперимента. Животных контролируют по усвояемости пищи и приросту веса тела еженедельно. Используют мышей бесшерстной линии SKH-1 аутбредные самки в возрасте 7 недель, не содержащие вирусных инфекций (Charles River Lab., Wilmington, МА). Животных разделяют на группы, помещая по 5 особей на садок случайным образом с использованием генератора случайных чисел программы Statview: садки размещают в комнате с освещением 12 часов в сутки и поддержанием температуры 22±1,0°С.
Статистическую обработку полученных данных проводят на компьютере “Макинтош-G3” с оригинальной программой для дисперсионного анализа, определения уровней значимости и линейной регрессии (Armitage, 1971). В частности, данные анализа по толщине эпидермиса в каждой группе лекарственного вещества оценивают с помощью дисперсионного анализа (Armitage, 1971).
Пример 22
Влияние биологически активных тритерпенов на экспрессию белков, участвующих в блокировке клеточного цикла и апоптозе
Апоптоз определяется как нормальный физиологический процесс запрограммированной гибели клеток, который происходит в ходе эмбрионального развития и при поддержании тканевого гомеостаза. Процесс апоптоза может быть подразделен на ряд метаболических изменений в апоптотической клетке. Отдельные ферментативные стадии нескольких регуляторных или сигнальных механизмов могут быть проанализированы для того, чтобы продемонстрировать ход апоптоза в клетке или клеточной популяции или же чтобы выявить нарушение процесса клеточной гибели в раковых клетках. Апоптотическая программа также наблюдается по морфологическим признакам, которые включают изменения в плазматической мембране (такие как утрата асимметричности), конденсацию цитоплазмы и ядра и межнуклеосомное расщепление ДНК. Все это завершается гибелью клетки, которая, разрушаясь, превращается в “апоптотические тельца”.
Методы анализа некоторых ферментативных реакций и процессов передачи сигналов, участвующих в апоптозе, были разработаны в качестве стандартных протоколов, применяемых в мультифакторном изучении апоптоза. Одним из примеров маркеров раннего этапа апоптоза является высвобождение цитохрома-с из митохондрий с последующей активацией каспазного-3 пути (PharMingen, San Diego, CA). Индукция каспаз (семейство цитоплазматических протеаз) является одним из наиболее часто наблюдаемых признаков апоптоза. В частности, каспаза-3 играет ключевую роль в данном процессе. При активации каспазы расщепляются белки-мишени, а одним из основных таких белков является белок PARP (ядерный белок). Следовательно, эффективными с точки зрения идентификации апоптоза являются тесты на выявление высвобождения цитохрома-с, выявление активности каспазы-3 и выявление расщепления белка PARP.
Более того, агенты, которые обуславливают высвобождение цитохрома-с из митохондрий опухолевых клеток, могут рассматриваться как вероятные терапевтические средства, восстанавливающие, по крайней мере, один из аспектов механизма клеточного контроля запрограммированной гибели клеток.
Другой тест на апоптоз основан на выявлении аннексина-V (BioWhitaker, Walkerville, MD). Обычно, фосфотидилсерин (ФС) находится на внутренней поверхности клеточной мембраны.
Однако, на ранних этапах апоптоза происходит экстернализация (перемещение на внешнюю поверхность) ФС. Аннексин-V представляет собой кальций-связывающийся белок, который связывается с ФС и может быть выявлен методом проточной цитометрии с ФИТЦ-окрашиванием аннексина-V (Martin et al., 1995). Способность клеток, обработанных соединениями Acacia victoriae, описываемыми в настоящем изобретении, связывать аннексин-V, рассматривается как индикатор того, что эти клетки подвержены апоптозу.
В других примерах заявителями был использован тест с киназой PI3, направленный на оценку апоптотической активности в клетках, обработанных противораковыми соединениями, выделенными из Acacia victoriae. Фосфоинозитид-3-киназа (PI3K), фермент, ассоциированный с клеточной мембраной, способна фосфорилировать по 3-му положению инозитольное кольцо фосфатидилинозита, тем самым определяя новый липидный сигнальный механизм в тех клетках, в которых киназа PI3 активна. Если PI3K активна, то на клеточную мембрану рекрутируется киназа, обозначаемая АКТ. АКТ является продуктом онкогена, который каталитически активируется после рекрутирования на мембрану. Полностью активированная АКТ играет ключевую роль в выживании клеток. Механизм, вовлекающий PI3K/AKT, соответствует механизму избегания клеткой апоптоза. Таким образом, способ подавления PI3K в опухолевых клеток, очевидно, может явиться терапевтическим методом восстановления, по крайней мере, некоторых аспектов механизма клеточного контроля апоптоза.
Пример 23
Анализ клеточных циклов
Для анализа клеточных циклов применяли стандартные методы проточной цитофотометрии с некоторыми модификациями. Кратко, 1×106 клеток высевали в 60-мм чашки и обрабатывали различными концентрациями F035 в течение 72 часов при 37°С. Клетки промывали ЗФР и ресуспендировали в концентрации 1×106 клеток в 1 мл. Клетки фиксировали сначала 1%-ным параформальдегидом и затем 70%-ным этанолом, охлажденным на льду. Затем клетки окрашивали йодистым пропидием (10 мкг/мл; Sigma Chemical Co.; St.Louis, МО), содержащим 0,1% РНКазы (Sigma), в течение 30 минут при комнатной температуре и анализировали на проточном цитометре FAC SCAN фирмы Beckton-Dickinson.
Пример 24
Тест на связывание аннексина-V-флуоресцеинизотиоцианата (ФИТЦ)
Индукцию апоптоза в опухолевых клетках изучали в тесте на связывание аннексина-V-ФИТЦ. Клетки Jurkat (1×106) обрабатывали различными концентрациями смеси тритерпеновых гликозидов (F035) и очищенными экстрактами D1 и G1 (0,5-2,0 мкг/мл) в течение 18 часов при 37°°С. После промывки клеток ЗФР их ресуспендировали в буфере для связывания (10 мМ HEPES + NaOH, 140 мМ NaCl, 2 мМ CaCl2), содержащем 5 мкл конъюгата аннексина-V-ФИТЦ (BioWhittaker, Walkersville, MD), и инкубировали в течение 10 минут в темноте. Затем клетки окрашивали йодистым пропидием (20 мкг/мл) и анализировали методом проточной цитометрии (Martin et al., 1995).
Пример 25
Тест с фосфатидилинозитол-3-киназой (киназой PI3)
Клетки в бессывороточной среде Jurkat обрабатывали 2 мкг/мл F035 в течение 2-15 часов или вортманнином в течение 0,5 час при 37°С. Активность киназы PI3 определяли, как описано ранее (Whitman et al., 1985; Royal & Park, 1995). Киназу PI3 иммунопреципитировали из 1 мг клеточного белка с использованием 5 мкл кроличьей анти-р85 антисыворотки при 4°С в течение 90 минут. Иммунные комплексы собирали на шариках сефарозы с 20% белка-А в течение 90 минут при 4°С. Затем полученные иммунопреципитаты ресуспендировали в 30 мкл буфера для киназной реакции (33 мМ Трис, рН=7,6, 125 мМ NaCl, 15 мМ MgCl2, 200 мМ аденозина, 20 мМ АТФ, 30 мкКи γ-32Р-АТФ). Реакцию киназы PI3 инициировали добавлением 10 мкл суспензии PI и 10 мкл γ-АТФ и оставляли на 30 минут при комнатной температуре. Реакцию останавливали добавлением 100 мкл 1 н НСl. Липиды экстрагировали из реакционной смеси смесью хлороформ:метанол (1:1) и разделяли методом тонкослойной хроматографии (ТСХ) с элюированием смесью хлороформ:метанол:NH4OH: Н2О (60:47:2:11,3) на силикагелевых пластинах G60. Радиоактивно меченый фосфатидилинозитфосфат визуализовали методом авторадиографии и ингибирование количественно оценивали с использованием системы Storm-860 (Molecular Dynamics).
Пример 26
Анализ общей и фосфорилированной форм АКТ
Экспрессию общей и фосфорилированной форм АКТ определяли методом Вестерн-блоттинга. Клетки линии Jurkat культивировали в среде, содержащей 0,5% ПТС (FBS), и обрабатывали F035 и очищенными экстрактами D1 и G1 (2,0 мкг/мл) в течение 15 минут при 37°С. Клетки лизировали в АКТ-лизис буфере (20 мМ Трис-НСl, 150 мМ NaCl, 1 мМ ЭДТА, 1мМ EGTA, 1% Тритон Х-100, 2,5 мМ пирофосфата натрия, 1 мМ 8-глицерофосфата, 1 мМ Na3VO4, 1 мл лейпептина, 1 мМ PMSF, рН=7,5). Клеточный белок (40 мкг) разделяли путем электрофореза в 8% ДСН-ПААГ и переносили на нитроцеллюлозную мембрану. Эти мембраны тестировали с использованием антитела, специфичного в отношении фосфорилированной (по серину-473) или общей АКТ с последующей обработкой козьим антикроличьим антителом, конъюгированным с пероксидазой хрена. Белки детектировали хемолюминесцентным методом (ECL, Amersham, Arlington Heights, IL).
Пример 27
Тест на изменение электрофоретической подвижности (ТИЭП)
В соответствии с описанным выше тест ТИЭП был проведен с целью изучения влияния неочищенного (F035) и очищенных экстрактов D1 и G1 на фактор NF-кВ, индуцированный TNF (Genetech Inc.). Клетки линии Jurkat (1×106 на 1 мл) обрабатывали различными концентрациями неочищенных и очищенных экстрактов в течение 15 минут при 37°С. Затем клетки подвергали воздействию 100 пкМ TNF в течение 15 минут при 37°С. Ядерные экстракты были приготовлены в соответствии с описанным выше. Ядерные экстракты инкубировали с 16 фкмоль 45-мерного двухцепочечного олигонуклеотида NF-кВ, несущего концевую 32Р-метку и происходящего от длинного концевого повтора вируса иммунодефицита человека,
5’-TTGTTACAAGGGACTTTCCGCTGGGGACTTTCCAGGGAGGCTGG-3’ (SEQ ID NO:9),
в течение 15 минут при 37°С в присутствии 2 мкг поли-(dI-dC). ДНК-белковый комплекс отделяли от свободного олигонуклеотида в нативном 7,5%-ном полиакриламидном геле. Радиоактивные полосы из высушенных гелей визуализовали и количественно оценивали с применением Phospholmager (Molecular Dynamics, Sunnyvale, CA) и компьютерной программы ImageQuant.
Пример 28
Индукция и анализ индуцибельной синтазы окиси азота (iNOS)
Клетки линий U-937 и Jurkat использовали для анализа фермента iNOS. Клетки U-937 дифференцировались в макрофаги при культивировании их с РМА (100 нМ) в течение 72 часов при 37°С. Дифференцированные клетки обрабатывали F035 (2 мкг/мл) в течение 15 часов с последующей 4-часовой обработкой ЛПС (10 мкг/мл) с целью индукции iNOS. В клетках Jurkat iNOS индуцировали воздействием на 0,5×106 клеток ФГА (10 мкг/мл) и РМА (10 мл) в течение 24 часов при 37°С. Клеточные лизаты готовили повторным замораживанием-оттаиванием в буфере RIPA (1% NP-40, 0,5% дезоксихолата натрия, 0,1% ДСН в ЗФР). Клеточный белок (200 мкг) разделяли электрофоретически в 7,5%-ном ДСН-ПААГ, переносили на нитроцеллюлозную мембрану, тестировали с использованием кроличьего антитела к iNOS, a затем козьего антикроличьего антитела, конъюгированного с пероксидазой хрена. Белки детектировали хемолюминесцентным методом (ECL, Amersham, Arlington Heights, IL).
Пример 29
Индукция цитотоксичности в отношении опухолевых клеток смесью и очищенными тритерпеновыми гликозидами
Влияние смеси тритерпеновых гликозидов (F035) на жизнеспособность панели раковых и нетрансформированных клеток исследовали в соответствии с описанными методами. Как показано на фиг.42, (Т-лейкозные) клетки Jurkat были высокочувствительными к F035 при IС50=0,2 мкг/мл. Аналогичным образом F035 подавляла рост ряда линий раковых клеток при ингибирующей концентрации IС50 в диапазоне 1,7-2,8 (клетки рака яичников), 2,0-3,3 (рак почек), 0,93 (рак поджелудочной железы), 1,2-6,5 (рак предстательной железы) и 0,72-4,0 мкг/мл (некоторые клетки рака молочной железы). Однако, другие линии клеток молочной железы были устойчивы к цитотоксическому действию F035. Как видно по четырем последним блокам на фиг.42, для уничтожения половины нетрансформированных клеток (фибробласты человека и мыши и иммортализованные клетки эпителия молочной железы) необходимо более чем 25 мкг/мл F035: это подтверждает, что F035 специфически цитотоксична в отношении опухолевых клеток.
Кроме того, два очищенных тритерпеновых гликозида, D1 и G1, были протестированы на их цитотоксичность в отношении 5 клеточных линий. На фиг.43 видно, что величина IC50 для D1 сопоставима с F035 для 3 клеточных линий (769-Р, MDA-MB-453 и MDA-MB-231). В отношении клеток С-2 (вариант линии HEY) и Jurkat D1 в два раза более активен, чем с F035. Однако, G1 в существенно большей степени цитотоксичен по сравнению с F035 и D1 в отношении большинства протестированных клеточных линий: это может объясняться меньшей полярностью G1 по сравнению с экстрактом D1 (фиг.43).
Пример 30
Блокировка клеточного цикла и индукция апоптоза смесью тритерпеновых гликозидов
Для анализа влияния F035 на клеточный цикл линии опухолевых клеток MDA-MB-453 и MDA-MB-435 обрабатывали различными концентрациями F035. В табл.42 показано увеличение числа клеток в фазе G1 (7-10%) и соответствующее уменьшение процента клеток в фазе S (6-10%): это подтверждает наличие блокировки клеточного цикла клеток MDA-MB-453 в фазе G1. Кроме того, через 72 часа после воздействия F035 16% клеток MDA-MB-435 (другая линия клеток рака молочной железы) находится в Фазе Sub.Go клеточного цикла (табл.42), что позволяет предположить, что клетки вступили в апоптоз. Данное наблюдение дополнительно было подтверждено при анализе апоптоза в тесте TUNEL.
Анализ клеточных циклов в клетках, обработанных F035
Клетки MDA-MB-453 и MDA-MB-435 обрабатывали различными концентрациями F035 в течение 72 часов при 37°С. Анализ клеточных циклов проведен после окрашивания йодистым пропидием в соответствии с описанным в разделе “Методы”.
Для выяснения механизма, лежащего в основе F035-индуцированной гибели клеток, заявителями был осуществлен тест на связывание аннексина-V-ФИТЦ с использованием обработки F035, D1 и G1 клеток линии Jurkat. В табл. 43 показано связывание аннексина-V с клетками, обработанными 1 мл F035, D1 и G1 (15-17%), что указывает на апоптотический механизм, приводящий к гибели клеток.
Тест на 72-часовой анализ цитотоксичности в отношении Т-лейкозных клеток Jurkat с использованием D1-контроля, D1-агликона, D1 без монотерпенов и монотерпеновых углеводов
Пример 31
Подавление активности киназы Р13 смесью тритерпеновых гликозидов
С целью выявления молекулярных мишеней F035, заявители исследовали механизм передачи сигнала киназы PI3. Результаты иммунопреципитации с анти-р85 (адаптерный белок) антителом и последующим липидкиназным тестом показали, что F035 подавляет активность киназы PI3 в клетках линии Jurkat. На фиг.45А показано, что после 2-часовой предварительной обработки F035 активность киназы PI3 подавляется на 50-70%. При 6-часовой предварительной обработке наблюдалось подавление активности киназы PI3 на 92-95%, которое сохранялось вплоть до 15-го часа после обработки. Аналогичное подавление киназной активности в клетках Jurkat было установлено при действии вортманнина (1 мкМ, 30 минут после обработки), являющегося известным ингибитором киназы PI3 (фиг.45А).
Пример 32
Подавление фосфорилирования АКТ смесью тритерпеновых гликозидов, D1 и G1
Авторы настоящего изобретения определили влияние F035 и очищенных экстрактов на АКТ, являющуюся серин-треониновой киназой и негативным эффектором Р13-киназного сигнального механизма. В противоположность быстрому подавлению активности киназы PI3, подавления фосфорилирования АКТ не происходило вплоть до 15-го часа после обработки. Обработка клеток Jurkat фракцией F035 (2 мл) в течение 15 часов приводила к снижению уровня фосфорилирования АКТ. Однако, эта обработка также приводила к снижению общего количества белка АКТ, как это можно видеть на фиг.45В. Заявители подтвердили, что очищенные тритерпеновые гликозиды подавляли активность АКТ. Очищенные тритерпеновые гликозиды D1 и G1 (2 мкг/мл) также подавляли фосфорилирование АКТ и общий уровень экспрессии белка АКТ (фиг.45В). Обработка клеток Jurkat вортманнином и LY294002 (известные ингибиторы РТЗ-киназы) выявила подавление фосфорилирования АКТ.
Пример 33
Подавление TNF-индуцированного NF-кВ смесью тритерпеновых гликозидов, D1 и G1
С целью дальнейшего установления медиаторов апоптотического механизма, заявители оценили влияние F035, D1 и G1 на транскрипционный фактор NF-кВ, который, как было показано, вовлечен в апоптоз. Приведенные на фиг.46А результаты показывают, что в клетках Jurkat F035 подавляет индуцированную фактором TNF активацию NF-кВ в дозо-зависимом режиме. В необработанных клетках и в клетках, обработанных только F035, активации NF-кВ не происходит. Также аналогичные результаты были получены для очищенных экстрактов D1 и G1. Предварительная обработка клеток 2 мл G1 и D1 приводила к снижению на 54% и 87% уровней NF-кВ, соответственно (фиг.46В). В клетках, обработанных D1 или G1, активация NF-кВ не выявляется (фиг.46В). Поскольку недавно было показано, что киназа PI3 участвует в регуляции NF-кВ, то предварительная обработка клеток вортманнином (1 мкМ) приводит к почти полному подавлению TNF-индуцированного NF-кВ.
Пример 34
Подавление iNOS фракцией F035
Поскольку транскрипция гена iNOS регулируется фактором NF-кВ, авторы настоящего изобретения исследовали влияние F035 на индукцию фермента iNOS. В клетках U-937, которые были дифференцированы в макрофаги, iNOS индуцировался в ответ на обработку липополисахаридом (фиг.46С). Предварительная обработка этих клеток F035 (1 мкг/мл) полностью блокирует индукцию iNOS. Вортманнин также обладает аналогичным эффектом на ЛПС-индуцированную iNOS в этих клетках.
Также было исследовано влияние F035 на индукцию iNOS в клетках Jurkat. iNOS индуцировали с использованием ФГА и РМА, как описано в разделе “Методы”. Полученные результаты показали, что предварительная обработка клеток Jurkat фракцией F035 блокировала индукцию iNOS (фиг.46D).
Пример 35
Анализ разрушения PARP методом иммуноблоттинга
Апоптоз, индуцированный F035 и D1, тестировали по протеолитическому расщеплению поли-АДФ-рибозополимеразы (белок PARP). Клетки Jurkat (2×106 на 1 мл) обрабатывали F035 (2 мкг/мл) и D1 (2 мкг/мл) в разные промежутки времени. Клеточные лизаты приготавливали в буфере, содержащем 20 мМ HEPES, 250 мМ NaCl, 2 мМ ЭДТА, 0,1% NP-40, 2 мкг/мл лейпептина, 2 мкг/мл апротинина, 0,5 мкг/мл бензамидина, 1 мМ дитиотреитола и 1 мМ PMSF. Клеточные белки (60 мкг/мл) разделяли путем электрофореза в 7,5%-ном ДСН- полиакриламидном геле и переносили на нитроцеллюлозную мембрану с помощью электроблоттинга. Эту мембрану тестировали с использованием вначале моноклонального антитела к PARP (Pharmagen), а затем антимышиного антитела, конъюгированного с пероксидазой хрена. Белковые полосы детектировали хемолюминесцентным путем (ECL, Amersham). В качестве маркера апоптоза использовали степень расщепленности белка PARP с молекулярной массой 116 кД на пептидные фрагменты 85 кД и 41 кД (Tewari et al., 1995).
Пример 36
Тест на каспазу-3 протеазы
Активность каспазы-3 измеряли, как описано ранее (Enari et al., 1995) с некоторыми изменениями. Кратко, клетки Jurkat (1×106 на 1 мл) обрабатывали F035, D1 и G1 в различные промежутки времени. Цитоплазматические экстракты приготавливали путем повторного замораживания-оттаивания в 300 мкл экстракционного буфера (12,5 мМ Трис, рН=7,0, 1 мМ дитиотреитола (DTT), 0,125 мМ ЭДТА, 5% глицерина, 1 мкМ PMSF, 1 мкг/мл лейпептина, 1 мкг/мл пепстатина и 1 мкг/мл апротинина). Клеточные лизаты разводили 1:2 буфером ICE (50 мМ Трис, рН=7,0, 0,5 мМ ЭДТА, 4 мМ дитиотреитола и 20% глицерина) и инкубировали при 37°С с 20 мкМ субстрата каспазы-3 (ацетил-Asp-Glu-Val-Asp-аминометилкумарина). Активность каспазы-3 прослеживали на образование флуоресцирующего аминометил-кумарина на сканере Fluoroscan-II с возбуждением при 355 нм и поглощением при 460 нм (Labsystems, Helsinki, Финляндия).
Пример 37
Выявление высвобождения цитохрома-с из митохондрий
Высвобождение цитохрома-с из митохондрий в ответ на обработку F035 определяли методом Вестерн-блоттинга. Клетки Jurkat (10×106) обрабатывали 2 мкг/мл F035 в течение 4 и 6 часов при 37°С. Клеточные осадки промывали сахарозным буфером (0,25 М сахарозы, 30 мМ Трис, рН=7,7, 1 мМ ЭДТА). К этим клеточным осадкам добавляли 20 мкл сахарозного буфера, содержащего 1 мкМ PMSF, 1 мкг/мл лейпептина, 1 мкг/мл пепстатина и 1 мкг/мл апротинина. Клетки разрывали путем 120-кратного продавливания через 0,3-мл “даунсер” с пестиком “В” (“мягким”) (Kontes Glass Company). Клеточный белок (60 мкг) растворяли в 15%-ном ДСН-полиакриламидном геле и электротрансферировали на нитроцеллюлозную мембрану. Эту мембрану тестировали с использованием сначала моноклонального антитела, специфичного в отношении цитохрома-с (Pharmagen), a затем антимышиного антитела, конъюгированного с пероксидазой хрена. Белковые полосы детектировали хемолюминесцентным путем (ECL, Amersham).
Пример 38
Индукция расщепления PARP действием F035 и D1
F035 и D1 индуцировали расщепление PARP в клетках Jurkat в зависимости от продолжительности действия. Данные, приведенные на фиг.47, показывают, что за 4 часа и F035, и D1 начинают индуцировать расщепление PARP, и завершение расщепления происходит за 6-8 часов. Это указывает на действенную роль в этом каспаз и тем самым, в апоптозе, т.е. механизме вовлечения в клеточную гибель, индуцируемую F035 и D1.
Пример 39
Влияние z-vad fmk на F035-индуцированную гибель клеток
Для дальнейшего подтверждения роли каспаз в F035-опосредованной гибели клеток исследовали влияние z-vad fmk, являющегося ингибитором каспаз, на клетки, обработанные F035.
Предварительная обработка клеток Jurkat 100 мкМ z-vad fmk в течение 1 часа при 37°С приводит к полной реверсии F035-индуцированного расщепления PARP (фиг.48).
Пример 40
Индукция активации каспазы-3 действием F035
Полученные заявителями результаты отчетливо подтверждают роль каспаз в апоптозе, индуцируемом действием F035. Были проведены дополнительные исследования активации каспазы-3 в клетках, обработанных F035, F094, D1 и G1, потому что эта протеаза во временной динамике располагается непосредственно перед PARP в каспазе-3 (фиг.49). Активация начинается через 4 часа после обработки во всех случаях, пик достигается на 6-8-й час и затем снижается.
Пример 41
Высвобождение цитохрома-с из митохондрий под действием F035
Как считается, высвобождение цитохрома-с в некоторых апоптотических механизмах является причиной активации каспазы-3. Для анализа справедливости этого предположения, при индукции апоптоза действием F035 определяли уровни цитохрома-с в цитоплазматических экстрактах клеток, обработанных F035. Авторами настоящего изобретения было установлено высвобождение цитохрома-с из митохондрий этих клеток в зависимости от продолжительности действия (фиг.50). Высвобождение цитохрома-с выявляется через 4 часа после обработки F035, что согласуется с моментом времени, когда происходит активация каспазы-3 и начинается расщепление PARP. Необходимо исследовать более ранние моменты времени, чтобы понять, предшествует ли высвобождение цитохрома-с активации каспазы-3.
Пример 42
Аэропоническая система выращивания
В результате обнаружения того факта, что тритерпеновые соединения по настоящему изобретению концентрируются в корнях и бобах растений Acacia victoriae, желательно разработать способ воспроизведения необходимых тканей, из которых могут быть выделены данные соединения. Для решения этой задачи была разработана аэропоническая система выращивания корней Acacia victoriae. Аэропоника представляет собой закрытую систему, в которой корни растения находятся на воздухе и опрыскиваются полноценной питательной средой. Ящик размером 8×4×3,5 футов был сделан из 3/4-дюймовых фанерных листов, скрепленных шурупами, и выстлан листовым оргстеклом, чтобы получить водонепроницаемый объем. Верх ящика покрывали двумя (2×8 футов) листами полистирола, в котором были сделаны 12 круглых сквозных отверстий, хотя новая конструкция, основанная на ПВХ-облицованной проволочной крышке для птичьих клеток, закрытой непрозрачной спрессованной черной полиэтиленовой пленкой, выкрашенной в белый цвет, рассматривается как крышка камеры для будущей работы. Для опрыскивания корней в течение 12 секунд каждые 4,5 минут использовали электронный таймер.
Вертикальная конструкция камеры для аэропоники представляет собой закрытую систему, собранную из 3/4-дюймовых листов ПВХ с шестью полыми струйными коническими полипропиленовыми распылительными соплами. На дне камеры устанавливали 720-литровый резервуар с питательным раствором, который распыляли на корни растений снизу вверх с помощью внешнего насоса. Использовали насос Little Giant 4-MD, 3250 об/мин, мощностью 1/12 л.с.
Работа насоса находилась под контролем таймера Tork, установленного на интервалы в 30 секунд для распыления каждые 4,5 минуты. Температуру контролировали с помощью двухиндикаторного термометра Tork, показывающего температуру внутри и за пределами камеры; температуру регистрировали вручную. В зимние месяцы для нагревания питательного раствора использовали два полупогружных аквариумных водонагревателя Visi-therm мощностью 300 Вт; этого было достаточно для поддержания активного роста растений без прогрева окружающего воздуха при отсутствии отопления и охлаждения под крытым навесом вне помещения в условиях Таксона, штат Аризона.
Питательный раствор содержал все необходимые элементы, необходимые для полного жизненного цикла растения. Несмотря на то, что различным растениям требуются разные количества и составы для достижения оптимального роста, в целом, удовлетворительные результаты были получены при использовании однократно сбалансированного раствора. Состав этого раствора приведен ниже в табл.44.
Питательный раствор для аэропоники
Затем семена Acacia victoriae скарифицировали и замачивали в не содержащей почвы смеси сфагнума (50%) и вермикулита (50%). Проростки смачивали два раза в день и удобряли одной дозой осмакота (гранулированное удобрение на основе костной муки). По достижении проростком длины 15-20 см, что обычно достигается за 3-4 месяца выращивания, корневые системы тщательно промывали с целью удаления любых остатков сфагнума и вермикулита. Затем корни просовывали через отверстия в полистироловых крышках и верхнюю часть проростков фиксировали с помощью веревок, закрепленных за фрагменты конструкции теплицы. Цилиндрическими кусками пены размером 7 см окутывали верхнюю часть проростков, чтобы не допустить попадания распыляемого раствора на листья и в окружающее пространство. После этого ящик заполняли питательным раствором, слоем примерно 30 см, и включали насос.
После размещения проростков на месте и начала их опрыскивания, контроль за ними ограничивался обеспечением роста проростков вдоль подвязок, для чего использовали пластмассовые прищепки, а также пополнением питательного раствора после того, как его уровень снижался до 10 см и менее. До тех пор, пока проростки росли внутри теплицы, температурный контроль за питательным раствором не являлся необходимым. Однако, если ящик для аэропоники подвергается воздействию открытой среды (т.е. находится вне помещения), рекомендуется повысить температуру питательного раствора до 21°С, для того, чтобы растения не “засыпали” в течение зимних месяцев.
Для сбора корней, корневую массу отдельного растения промывают водой непосредственно в ящике для аэропоники и затем отрезают эту корневую массу ножницами, отступая на несколько дюймов выше распылителя. Избыток воды удаляют путем обсушивания бумажными полотенцами с последующим взвешиванием образца. Затем корневую массу измельчают ножницами на кусочки по 3-4 дюйма и подвергают химической экстракции, как описано выше. Альтернативно, для обеспечения непрерывного сбора корней, насос выключают и корни отщепляют от растущей корневой массы. Затем эти корни измельчают на кусочки размером 3-4 дюйма и экстрагируют, как описано. При этом требуется аккуратность, чтобы не повредить несобираемые корни, которые продолжают расти.
При выращивании растений в аэропонике достигается ряд преимуществ. Во-первых, темп роста примерно вдвое выше по сравнению со стандартными способами выращивания. Во-вторых, при необходимости, корни могут быть легко собраны, не причиняя вреда растению. Такое срезание корней дополнительно приводит к интенсивному нарастанию боковых отростков по типу мочковатого корня. Следовательно, корни можно собирать по несколько раз в год. Кроме того, выращенные в аэропонике растения цветут в первый год жизни, а первое цветение растений открытого грунта происходит на 3-5-й год.
Пример 43
Культивирование тканей и проращивание Acacia victoriae
Семена/субстрат. Семена собирали на растениях Сельскохозяйственного центра университета штата Аризона в Таксоне. Семена тщательно промывали водопроводной водой, используя антимикробное мыло (Vionex, Viro Res. Intern. Inc., Durango, Colorado), затем обрабатывали 20%-ной (об/об) бытовой хлорной известью в течение 15 минут. После повторной промывки деионизованной водой семена обрабатывали кипятком (примерно 200 мл на 100 семян) и оставляли остывать на ночь. Затем их обрабатывали 20%-ной (об/об) бытовой хлорной известью, 2-3 раза промывали в стерильной деионизованной воде и культивировали в полной среде Мурасиге-Скоога (MS) (Murashige & Skoog, 1962) и в среде MS половинной силы. Эту культуральную среду дополняли витаминами MS, 2% (в/о) сахарозы и для отверждения использовали либо 0,7% агар, либо 0,2% гельрит. В одном из экспериментов семена скарифицировали концентрированной серной кислотой, промывали в стерильной воде и культивировали в среде. Все культуральные среды автоклавировали при 121°С в течение 15 минут. Культуры поддерживали при 25±2°С при светопериоде 16 часов при 1000 люкс с использованием флуоресцентных ламп холодного свечения. Каждый анализ воспроизводили 18 раз.
Размножение. Кончики побегов и сегменты узлов, отрезанные от 3-недельных проростков, культивировали только в среде MS и также в среде MS, дополненной 0,1 мг/л ауксинов (IAA, NAA или IBA) и ВАР (0,1; 0,3; 0,5; 1,0 и 1,3 мг/л) как по отдельности, так и в комбинациях. Для анализа образования корней на побегах тестировали IAA (0,1 мг/л), IBA (0,1 и 0,6 мг/л) и NAA (0,1 и 0,2 мг/л). Для переноса в почву, проростки вынимали из культуральных пробирок, корни промывали водопроводной водой для удаления приклеившихся к корням фрагментов питательного субстрата и переносили в горшки, заполненные почвой аридного типа. Растения укрывали коробками Magenta для поддержания влажности и выдерживали при опрыскивании и низкой освещенности в течение 3 недель. Через 3 недели, коробки Magenta убирали и растения переносили из опрыскиваемой зоны в освещенные участки теплицы.
Индукция каллюсов. Образование каллюсов индуцировали на материале гипокотилей и корневых сегментов, отрезанных у 3-недельных проростков, проращенных in vitro. Эксплантаты культивировали в среде MS, дополненной 2,4-D (1 мг/л), NAA (0,5 и 1 мг/л), IAA (0,2 и 1 мг/л), тидиазуроном (0,2 мг/л), дикамбой (0,2 и 2 мг/л), ВАР (0,3 мг/л) и KN (0,5 и 3 мг/л) как по отдельности, так и в комбинациях.
Проращивание семян. Семена, обработанные горячей водой, прорастали с появлением корня через 3-4 дня и образованием полного проростка за 1 неделю. Семена, не обработанные горячей водой, не прорастали вовсе. Большее число семян прорастало на среде, уплотненной с использованием гельрита (0,2%) по сравнению с агаром (0,7%). Максимальный уровень всхожести достиг 88,7%, что было отмечено в среде MS с половинной концентрацией, уплотненной гельритом. Данные по прорастанию в различных средах обобщены в табл.45.
Прорастание семян Acacia victoriae
Пригодные для пересадки проростки были получены через 3-4 недели. Семена Acacia victoriae в естественных условиях характеризуются плохой всхожестью из-за высокой частоты нахождения в диапаузе (Kaul & Ganguly, 1965; Grice & Westoby, 1987). Для преодоления диапаузы необходимо удалить внешний покров семени путем либо соскабливания с помощью острого инструмента, либо кислотной скарификации, либо обработки кипятком с последующим остыванием в воде в течение ночи. Заявителями было обнаружено, что уровень всхожести может быть увеличен до 88,7% с использованием обработки кипятком и последующим культивированием семян в культуральной среде MS половинной концентрации, уплотненной гельритом (0,2%). В соответствии с Larsen 1964, всхожесть семян A. victoriae в условиях in vivo в случае обработки кипятком может быть увеличена до 36%. Без предварительной обработки, всхожесть составляет 19,4% (Kaul & Ganguly, 1965). Кроме того, в этом случае на развитие корня уходит 12 дней, а полные проростки образуются за 79 дней. В используемом заявителями способе всхожесть увеличена (до 88,7%), и пригодные для пересадки проростки могут быть получены за 3-4 недели.
Культуры кончиков побегов: Для изучения размножения побегов, их кончики (примерно 1,0 см в длину) культивировали либо в чистой среде MS, либо в среде MS, дополненной ВА или ВА в сочетании с IAA. В чистой среде MS мощность побегов была низкой при слабом росте корешков (1-3 корешка на культуру). В среде, содержащей ВА (1,3 мг/л), на кончиках побегов образовывались многочисленные побеги (в среднем 3,94 побега на культуру). Среди многочисленных побегов один или два побега удлинялись и достигали за 4 недели длины 8,6 см. Комбинации ВА и IAA также способствовали образованию множественных побегов.
Эти результаты систематизированы в табл.46.
Влияние различных уровней ВА и IAA (0,2 мг/л) на образование множественных побегов у Acacia victoriae
При более высоких концентрациях ВА (1,0 и 1,3 мг/л), число побегов увеличивается. Как было установлено, комбинация ВА (1 мг/л) и IAA (0,2 мг/л) улучшает размножение побегов. Каллюсы получали на материале обрезков кончиков при всех комбинациях BA-IAA. Был подтвержден (Kaur et al., 1998) синергизм ВА-NAA в отношении индукции развития почек на побегах Acacia catechu, в то время как при более высоких концентрациях NAA (1-2 мг/л) такого эффекта не наблюдалось. Также утверждалось, что IAA неэффективен по интенсификации образования почек на побегах; тем не менее на материале таких эксплантатов каллюсы были получены.
Для изучения образования корешков, сформированные in vitro побеги отрезали и переносили в культуральную среду, содержащую IAA, NAA или IBA. Полученные данные систематизированы в табл.47. Среди исследованных вариантов наилучшие параметры образования корешков имели место в среде MS половинной концентрации с NAA (0,2 мг/л). Корешки образовывались почти на 100% побегах. Побеги достигали высоты 9-11 см за 4 недели. Сообщалось (Kaur et al., 1998), что у Acacia catechu непостоянное развитие каллюсов происходит в местах сочленения побегов и корешков, а для контроля развития каллюсов концентрацию сахарозы снижали с 3% до 1,5%. Аналогичные данные были получены для феронии лимонной Feronia limonia (Purohit & Tak, 1992) и акации Acacia auriculiformis (Das et al., 1993). В данном исследовании слабое развитие каллюсов было также выявлено при 3% сахарозы, и оно сводилось до минимум 2% сахарозы. Побеги с образовавшимися корешками переносили в теплицу. Выживаемость после переноса составила 100%. Рассаду акклиматизировали при опрыскивании в течение 3 недель, а затем проростки выращивали в условиях обычной теплицы.
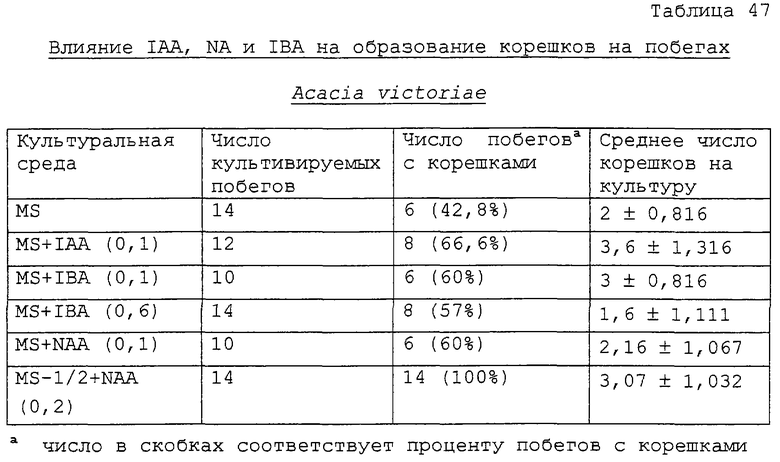
Культуры сегментов узлов. Сегменты узлов (семядольные узлы), отрезанные от проращенных in vitro проростков, культивировали в среде MS/ дополненной 0,1 мг/л IAA, NAA или IBA. На одном эксплантате развивалось по одному или два пазушных побегов. Однако, эти побеги характеризовались слабым ростом. Поэтому узелковые эксплантаты не брали для дальнейшего использования.
Индукция каллюсов на материале гипокотилей и сегментов корней. Каллюсы индуцировали на материале сегментов гипокотилей, отрезанных от 3-недельных проростков, проращенных in vitro. Каллюс, сформированный в присутствии 2,4-D (1 мг/л), тидиазурона (0,2 мг/л) и дикамбы (0,2 мг/л), был зеленоватым, компактным и твердым. Количество образующихся каллюсов было умеренным при большинстве испытуемых концентраций (табл.48). Интенсивное образование каллюсов выявили в среде MS с добавлением 2,4-D (4 мг/л) + IAA (1 мг/л) + NAA (1 мг/л).
Сегменты корней, отрезанные от 3-недельных проростков, полученных in vitro, культивировали в среде MS только с 2,4-D (1 мг/л) и с 2,4-D в сочетании с кинезином (0,5 мг/л), и на них отмечено образование светло-желтоватых рыхлых каллюсов, причем, на них образовывалось небольшое число корешков. Образование каллюсов имело место в 100% культур. Обильное образование беловатых, рыхлых, ломких каллюсов выявили на сегментах корней, культивируемых в среде с добавлением ВА (0,3 мг/л) и IAA (0,2 мг/л). Образование многочисленных светло-желтоватых каллюсов отметили на сегментах корней, культивировавшихся в среде с добавлением 2,4-D (4 мг/л) в сочетании с IAA и NAA (по 1 мг/л). Аналогичный тип каллюсов был выявлен в присутствии тидиазурона (0,2 мг/л), дикамбы (2 мг/л) и IAA (0,1 мг/л). На сегментах корней, культивировавшихся в среде с дикамбой (2 мг/л) и IAA (0,1 мг/л), образовывались светло-зеленые компактные твердые каллюсы. Попытки регенерировать проростки на материале этих каллюсов оказались неудачными. Для некоторых древесных пород растений, например, для Albiizzia lebbeck (Lakshmana Rao & De, 1987) и Lonicera japonica (Georges et al., 1993), была установлена изменчивость типов эксплантатов в связи с развитием каллюсов. В исследованиях заявителей также было обнаружено, что у развивающихся в идентичной культуральной среде каллюсов, образованных гипокотилями и корнями, имеется различие. Каллюсы, сформировавшиеся из ткани гипокотиля при комбинации BA-IAA, были светло-зеленоватыми, компактными и твердыми, в то время как каллюсы корневых сегментов были беловатыми, мягкими (рыхлыми), ломкими; также было показано, что в некоторых вариантах на каллюсах образовывались корешки. Каллюсы Dalbergia latifolia при помещении их в регенерирующую среду становились компактными, твердыми и темно-зелеными, и на них дифференцировались почки побегов (Pradhan et al., 1998). В исследованиях заявителей был отмечен аналогичный характер развития каллюсов, однако такие каллюсы не обладали способностью к регенерации. В этом анализе заявители продемонстрировали, что A.victoriae может быть воспроизведена in vitro на материале кончиков побегов. Стандартизованный способ применим для микровоспроизведения элитных особей, выявленных в гетерогенной популяции проростков, и для поддержания элитных линий для дальнейших исследований.
Развитие каллюсов на материале гипокотилей и сегментов корней Acacia victoriae
Пример 44
Индукция образования волосовидных корней Acacia victoriae с целью выработки противоопухолевых соединений
Заражение растительного материала Acacia victoriae бактерией Agrobacterium rhizogenes приводит к интеграции в состав генома растения и экспрессии Т-ДНК, что обуславливает развитие фенотипа “волосовидные корни” (Grant et al., 1991). Культуры волосовидных корней характеризуются быстрым ростом, ассоциируются с плагиотропным корневым ростом и интенсивно ветвятся в безгормональной среде. Волосовидные корни также характеризуются высоким уровнем генетической стабильности (Aird et al., 1988). Многие виды двудольных растений восприимчивы к заражению A. rhizogenes, и для многих видов была проведена регенерация растений из культур волосовидных корней (Christey, 1997).
Генетическая трансформация и индукция волосовидных корней были осуществлены авторами настоящего изобретения в качестве способа выработки активных тритерпенов из А. victoriae. Естественная способность почвенной бактерии Agrobacterium rhizogenes трансформировать гены в геноме организма-реципиента приводит к тому, что участки корней, в которых произошло такое заражение, можно использовать для получения культур волосовидных корней. Волосовидные корни характеризуются очень быстрым ростом, интенсивным ветвлением вспомогательных корней, которые продолжают расти in vitro в безгормональной среде.
Заявителями была продемонстрирована индукция волосовидных корней Acacia victoriae с использованием Agrobacterium rhizogenes штамма R1000 (генно-инженерный штамм Agrobacterium tumefaciens, в состав которого была внесена плазмида pRiA4 Agrobacterium rhizogenes - АТСС №43056). Продуцирование нужного соединения волосовидными корнями была подтверждена методом ВЭЖХ. Индукцию развития волосовидных корней осуществляли следующим образом. Сначала семена Acacia victoriae были собраны с растений, выращенных в открытом грунте в Таксоне, штат Аризона. Эти семена обрабатывали кипятком и оставляли на ночь замоченными в этой воде, которая в это время остывала, а поверхность семян стерилизовали с использованием 15%-ной бытовой хлорной извести в течение 30 минут. После повторной промывки в стерильной воде семена культивировали в жидкой среде MS (Murashige & Skoog, 1962), дополненной витаминами MS и 2% сахарозы, в 250-мл конических колбах с 50 мл среды в каждой из них. Культуры поддерживали во вращающемся шейкере в культуральном боксе при 25±2°С в темноте. Через 4 дня культивирования, зародыши отрезали от проростков и использовали для заражения бактерией.
Перед использованием для заражения, Agrobacterium rhizogenes, штамм R1000 культивировали в течение ночи в среде YENB. Среда YENB была приготовлена путем добавления дрожжевого экстракта (7,5 г/л) и питательного бульона (8 г/л) (Difco Lab., Detroit, MI). Зародыши эксплантатов инфицировали с использованием тонкой иглы из нержавеющей стали, которую перед этим окунали в раствор с бактериями. После инфицирования каплю бактериальной суспензии (1:20 в среде MS) наносили на поверхность эксплантата. Затем эксплантаты переносили в среду MS и в среду MS с добавлением ацетосирингона (100 мкМ) (3,5-диметокси-4-гидроксиацетофенон) (Aldrich Chem. Co., Milwaukee, WI) для совместного культивирования. Совместное культивирование проводили в течение 3 дней в темноте. Через 3 дня совместного культивирования, эксплантаты переносили в среду MS с добавлением цефотаксима (500 мг/л) (Agri-Bio. North Miami, FL) с целью предотвращения избыточного роста бактерий. Инициация образования корней была отмечена в месте инфицирования, в основном на материале молодых развивающихся листьев зародыша спустя 3-4 недели. Через 4 недели, эксплантаты наряду с корешками переносили в чистую среду MS и продолжали инкубацию в темноте с целью достижения развития волосовидных корней. Развитие волосовидных корней было отмечено еще через 8 недель. Образованные таким образом волосовидные корни размножали стандартным путем в среде MS методом пересевов. Трансгенную природу волосовидных корней подтверждали методом ПЦР с использованием набора праймеров для амплификации участка гена го1В. Эти праймеры были следующими:
1) 5’-GAGGGGATCCGATTTGCTTTTC-3’ (SEQ ID NO 7);
2) 5’-CTGATCAGGCCCCGAGAGTC-3’ (SEQ ID NO 8).
50 мкл реакционной смеси ПЦР содержали праймеры (конечная концентрация каждого из них 1 мкМ), Тaq-полимеразу (1,0 ед.), 125 мкМ каждого из нуклеотидов dNTP, 1x ПЦР-реакционный буфер, 1,5 мМ MgCl2, 300 нг изолированной ДНК. Использовались такие условия ПЦР: исходная денатурация в течение 5 минут при 92°С, затем проводили 35 циклов по 50 секунд при 92°С, 1 минуте при 55°С (отжиг), 1,5 минут при 72°С (достройка цепи) и в конце 1 минут при 72°С (окончательная достройка). Был амплифицирован фрагмент длиной 645 пар нуклеотидов.
Культуры волосовидных корней в жидкой среде. Для оптимизации условий роста, волосовидные корни, растущие в полужидкой среде MS, отрезали и культивировали в жидкой среде MS в колбах разной емкости (125, 250, 500 и 1000 мл), заливая в них, соответственно, по 20, 50, 100 и 400 мл среды. Размер исходного инокулята волосовидных корней составил 6 г/л. Рост волосовидных корней также был протестирован в следующих базовых средах: MS, среде N&N (Nitsch & Nitsch, 1969), среде Шенка-Хильдербрандта (SH) (Schenk & Hilderbrandt, 1972) и среде для древесных пород растений WPM (Woody Plant Medium) (Lloyd & McCown, 1981). Для тестирования влияния различных источников углерода на рост волосовидных корней, в среду MS добавляли по 2% (масс/об) следующих источников: сахароза, глюкоза, фруктоза и манноза. Эффект гибберелловой кислоты (0,1, 0,5 и 1 мг/л) на рост волосовидных корней тестировали путем добавления стерилизованного фильтрованием раствора в среду MS после ее автоклавирования.
Инициация образования корешков в месте инфицирования была отмечена через 3-4 недели. Четыре независимых трансформированных клона волосовидных корней были сформированы на материале зародышей, инфицированных штаммом R1000 в присутствии ацетосирингона (100 мкМ). Зародыши, культивировавшиеся вместе с A. rhizogenes без ацетосирингона, не образовывали волосовидных корней (табл. 49). Было установлено, что 3-дневное совместное культивирование в присутствии ацетосирингона является оптимальным для индукции волосовидных корней. Активирующий эффект ацетосирингона был описан для шалфея Salvia militiorrhiza (Нu & Alfermann, 1993). Полученные результаты показали, что ацетосирингон, являющийся активатором генов vir Agrobacterium, повышает частоту трансформации. Аналогично, в проведенном здесь исследовании, ацетосирингон является необходимым для индукции волосовидных корней.
Трансформированную природу таких корней подтверждали методом ПЦР-амплификации с использованием набора праймеров, обеспечивающих амплифицирование фрагмента гена rо1В. Диагностический фрагмент длиной 645 пар нуклеотидов был амплифицирован во всех четырех протестированных клонах волосовидных корней.
Растущие в жидкой среде волосовидные корни характеризуются мощным развитием. Среди протестированных исходных сред, наилучшие показатели в связи с ростом волосовидных корней были определены для среды MS (табл.51). В 125-мл колбах отмечалось 268-кратное усиление роста за 4 недели. При использовании сред WPM и N&N, кратность увеличения составила, соответственно, 254 и 196. Среды B5 и SH оказались не оптимальными для роста волосовидных корней. На этих двух средах наблюдалось замедленное потемнение волосовидных корней. В одном эксперименте, волосовидные корни выращивали в различных по емкости колбах (125, 250, 500 и 1000 мл) с 20, 50, 100 и 400 мл среды MS, соответственно. Параметры динамики роста обобщены в табл. 50. Вначале рост волосовидных корней был мощным и достигал 25,77-кратного увеличения за 4 недели в 125-мл колбах при величине исходного инокулята 150 мг. По мере увеличения емкости колб, интенсивность роста корней медленно снижалась.
Рост волосовидных корней может быть чувствителен к составу культуральной среды, особенно к минеральным ионам и источнику углерода (Wysokinska & Chmiel, 1997). Для Acacia victoriae, пять различных исходных сред (MS, N&N, SH, WPM и B5) были протестированы по влиянию на рост волосовидных корней. Наилучшей для роста оказалась среда MS. По данным сравнения роста волосовидных корней Coleus forskohlii в различных питательных средах (Sasaki et al., 1998) наилучшей для роста волосовидных корней была среда WPM.
В проведенном исследовании, сахароза способствовала росту волосовидных корней по сравнению с другими источниками углерода (фруктозой, глюкозой и маннозой). Наилучший рост (24,52-кратное увеличение) был обнаружен в культуральной среде, содержащей сахарозу. Среда, содержащая глюкозу, в небольшой степени подавляла рост, а манноза рост подавляла полностью (табл.52). У Catharanthus roseus выработка катарантина может быть удвоена путем использования фруктозы в качестве источника углерода в культуральной среде. Однако, сообщалось, что использование фруктозы давало приблизительно 40%-ное снижение роста по сравнению с сахарозой (Jung et al., 1992).
Для роста волосовидных корней необязательно добавлять экзогенные факторы роста, поскольку гены, обеспечивающие повышенную восприимчивость к ауксинам, имеются и в составе плазмиды Ri (Wysokinska & Chmiel, 1997). Однако, известны сообщения, согласно которым экзогенные гормоны стимулируют рост. Заявителями был исследован эффект гибберелловой кислоты (0,1, 0,5 и 1,0 мг/л) на рост волосовидных корней. Рост волосовидных корней был наилучшим в среде, лишенной GА3, по сравнению со средой, содержащей GА3 (15,77-кратное увеличение). Различные уровни GA3 существенно не влияют на рост (табл.53). У полыни рода Artemisia, GА3 не усиливает накопление биомассы в целом, но облегчает более быстрое достижение стационарной фазы роста по сравнению с культурами, выращенными на среде без GА3 (Smith et al., 1997). Было установлено (Rhodes et al., 1994), что реакция волосовидных корней белой горчицы Brassica Candida на GA3 в значительной степени зависит от анализируемого клона. Однако, было выявлено, что в целом GА3 проявляет положительный эффект на рост и снижение уровня накопления алкалоидов наряду с изменениями условий их продуцирования. Сообщалось (Ohkawa et al., 1989), что GA3 в концентрациях 10 нг/л и 1 мг/л ускоряет рост, усиливает удлинение и способствует увеличению боковых ветвлений волосовидных корней дурмана Datura innoxia. Было подтверждено (Zobel, 1989), что GА3 действует в отношении роста корней как аналог CO2. В отношении волосовидных корней Acacia victoriae, GА3 не усиливает рост, что может указывать на неодинаковую реакцию на нее различных генотипов.
Использование культур волосовидных корней Acacia victoriae предоставляет эффективный способ получения однородной культуры растительной ткани, из которой могут быть выделены тритерпеновые гликозиды по настоящему изобретению, включая выделение смесей или отельных очищенных соединений.

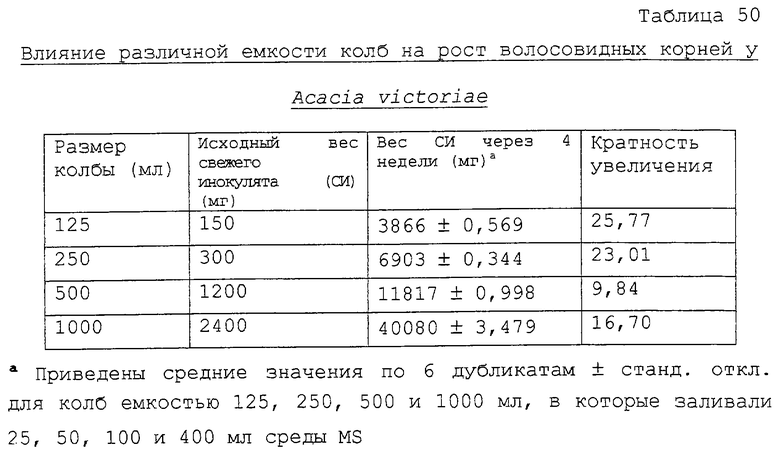
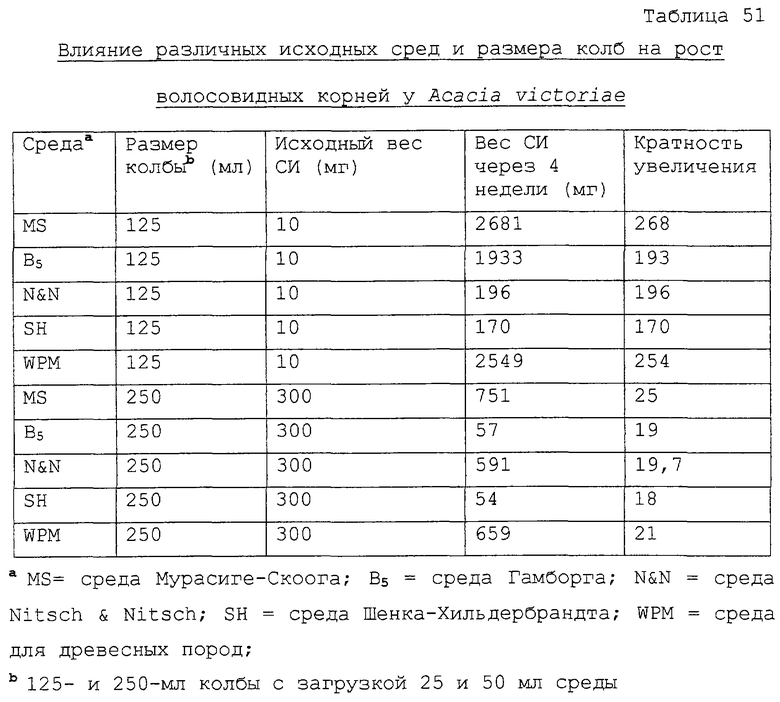
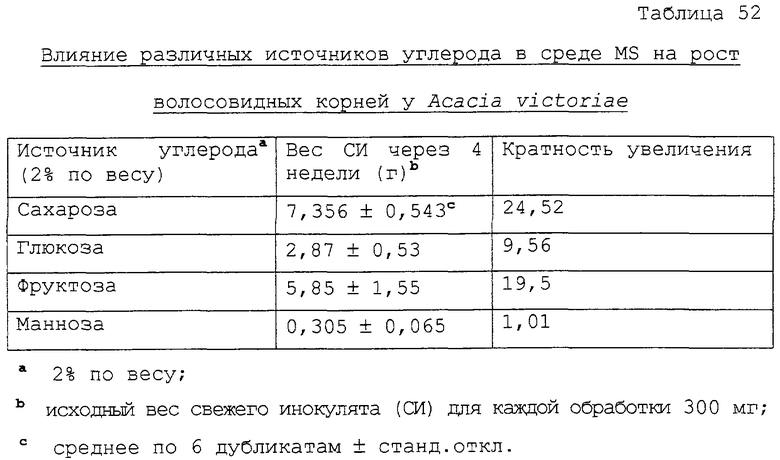
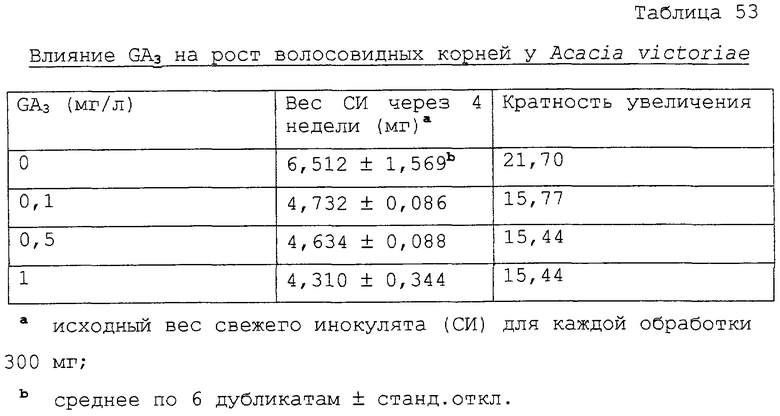
Различные среды тестировали по росту в них волосовидных корней. Наилучший рост был выявлен в среде MS, содержащей 2% сахарозы. Исследовали влияние различной емкости колб и гибберелловой кислоты на рост волосовидных корней. Также волосовидные корни культивировали в жидкой среде MS на вращающемся шейкере в 125-мл конической колбе с внесением 20 мл среды. За 4 недели был отмечен 84-кратный прирост. Продуцирование тритерпеновых сапонинов, соответствующих тем, которые идентифицировали во фракции F035, была подтверждена с применением ВЭЖХ с использованием стандартного аутентичного образца.
Все композиции и способы, описанные и заявленные в данном тексте, могут быть выполнены и применены без дополнительных экспериментов в свете настоящей заявки. Хотя композиции и способы по настоящему изобретению описаны в виде предпочтительных вариантов, специалисту в данной области техники должно быть понятно, что в эти композиции и способы могут быть внесены модификации, равно как и в отдельные этапы и последовательность этапов этих способов, описанных в данном тексте, без отклонения от идеи, существа и объема настоящего изобретения. Более конкретно должно быть понятно, что некоторые агенты, которые близки и химически, и физиологически, могут быть использованы для замены агентов, описанных здесь, если при этом будут достигнуты такие же или аналогичные результаты. Все такие аналогичные замены и модификации, очевидные для специалиста в данной области техники, рассматриваются как соответствующие существу, объему и идее настоящего изобретения, определенного формулой изобретения.
БИБЛИОГРАФИЯ
Нижеследующие библиографические ссылки в той степени, в которой они призваны проиллюстрировать процедуры или иные подробности, дополняющие заявляемое в данном тексте, включены здесь для сведения в виде библиографии.
Aerts et al., Plant J., 5:635-643, 1994.
Agrawal, "NMR spectroscopy in the structural elucidation of oligosaccharides and glycosides," Phytochemistry, 31:3307-3330, 1992.
Aird, Hamill, Rhodes, "Cytogenetic analysis of hairy root cultures from a number of species transformed by Agrobacterium rhizogenes," Plant Cell Tissue Organ Cult., 15:47-57; 1988.
Akiyama et al., J. Blol. Chem., 262:5592-5595, 1987.
Alien et al., "Leguminosae, A source book of characteristics uses and nodulation," The University of Wisconsin Press, Madison, Wisconsin, 1981.
Armitage, Jn: Statistical Methods in Medical Research, Wiley and Sons, New York, NY.p 205, 1971.
Amon, R., et al., Proc. Natl. Acad. Sci. (USA) 77: 6769-6772, 1980.
Baba, Hanada, Hashimoto, "The study of ultraviolet B-induced apoptosis in cultured mouse keratinocytes and in mouse skin," J. Dermatol. Sci., 12:18-23, 1996.
Baxter, Price, Fenwick, "Sapogenin structure: analysis of the
13C- and 1H-NMR spectra of soyasapogenol b," J. Nat. Prod., 53:298-302, 1990.
Bellacosa, Feo, Godwin, Bell, Cheng, et al., Int. J. Cancer, 64:280-285, 1995.
Berton, Mitchell, Fischer, Locniskar, "Epidermal proliferation but not the quantity of DNA photodamage is correlated with UV-induced mouse skin carcinogenesis," Invest.
Dermatol., 109:340-347, 1997.
Beutler, Kashman, Pannell, Cardellina, Alexander, Balaschak, Prather, Shoemaker, Boyd, Bioorganic and Medicinal Chemistry, 5:1509-1517, (1997).
Boll and von Philipshom, "NMR studies and the absolute configuration of Solarium alkaloids (spiroaminoketal-alkaloids), Acta Chem. Scand., 19: 1365-1370, 1965.
Brinkmann et al., Proc. Natl. Acad. Sci., USA, 88(19):8616-8620, 1991.
Burchell et al., J. Immunol.. 131 (1): 508-513, 1983.
Campbell. in Monoclonal Antibody Technology, Laboratory
Techniques in Biochemistry and Molecular Biology Vol.13,
Burden and Von Knippenberg, Eds. pp.75-83, Amsterdam,
Elseview, 1984.
Capaidi et al., Blochem. Biophys. Res. Comm., 76:425, 1977. Capon and Thacker, "The nuclear magnetic resonance spectra of some aldofuranosides and acyclic aldose acetals," Proc.
Chem. Soc. bond., 369, 1964. Chatterjее, Agarwal, Muhtar, "Ultraviolet В radiation-induced
DNA lesions in mouse epidermis," Biochem. Blophys. Res.
Commun., 229:590-595, 1996.
Cheatham et al., Proc. Natl. Acad. Sci., 92: 11696-11700, 1995.
Cheeke, Can. J. Animal Sci., 51: 621-632, 1971.
Chen and Snyder, "Diosgenin-bearing, molluscicidal saponins from Allium vineale: an NMR approach for the structural assignment of oligosaccharide units," J. Org. Chem.,
54: 3679-3689, 1989.
Chen and Snyder, "Molluscicidal saponions form Allium vineale," Tetrahedron Lett., 28:5603-5606, 1987.
Cho, Widholm, Tanaka, Nakanishi, Murooka, "Agrobacterlum
rhizogenes-mediated transformation and regeneration of the legume Astragalus sinicus (Chinese milk vetch),"Riant Science, 138: 53-65;1998.
Chou and Blems, Cell, 85:573-583, 1996.
Christey, "Transgenic crop plants using Agrobacterium rhizogenes-mediated transformation," Doran, P.M., (ed.)
Hairy roots: Culture and applications, Harwood,
Amsterdam, 99-111, 1997.
Colchere et al., Cancer Res., 47: 1185 and 4218, 1987.
Cohart, Baeuerle, Vassalli, Mol. Cell. Biol., 10: 1498-1506, 1990.
Creeimane et al., Proc. Natl, Acad. Sci. USA, 89: 4938-4941, 1992.
Davis & Preston Analytical Biochemistry, 116(2):402-407, 1981.
Davis, Sinensky, Junker, Pharmac. Ther., 43: 221-36, 1989.
Defago, Bеr. Schweiz. Bot. Ges., 87:79-132, 1977.
Dillman et al., Antibody Immunocon. Radiopharm., 1:65-77,1988.
Doll, R. et ai., Lancet 1: 793, 1962.
Enari. Hug, Nagata, Nature, 375: 78-81, 1995.
Folkman, Haudenschild, Zetter, Proc. Natl. Acad. Sci.,
76: 5217-5221, 1979.
Franceschi et al., Proc. Nati. Acad. Sci. USA, 88: 6745-6749, 1991.
Frechet, Christ, du Sorbier, Fischer, Vuilhorgne, "Four triterpenoid saponins from dried roots of Gypsophlla species," Phytochemistry, 30:927-931, 1991.
Gamborg, Miller, Ojima, "Nutrient requirements of suspension cultures of soybean root cells," Exp. Cell Res., 50: 151-158;
1968. Gariboldi, Verotta, Gabetta, "Saponins from Crossopteryx
febrlfuga, Phyto chemistry, 29:2629-2635, 1990.
Gefter et al., Somatic Cell Genet., 3: 231-236, 1977. Ghose et al., CRC Critical Reviews In Therapeutic Drug Carrier
Systems, 3: 262-359, 1987.
Ghose, et al., Meth. Enzymology, 93: 280-333, 1983. Coding, 1986, In: Monoclonal Antibodies: Principles and.
Practice, 2d ed., Academic Press, Orlando, Fla., pp.60-61, and 71-74, 1986.
Grant, Dommisse, Christey, Conner, "Gene transfer to plants using Agrobacterium," In: Murray, D.R., (ed.) Advanced methods in plant breeding and biotechnology, CAB
International, Wallingford, 1991:50-73. Gundalche et al., Proc. Natl. Acad. Sci. USA, 89: 2389-2393, 1992.
Hamburger, Slacanin, Hostettmann, Dyatmiko, Sutarjadi,
"Acetylated saponins with molluscicidal activity from
Saplndus rarak: unambiguous structure determination by proton nuclear magnetic resonance and quantitative analysis," Phytochem. Anal., 3: 231-237, 1992.
Hansen, Nielsen, Berg, J. Immunologlcal Methods, 119: 203-210, 1989.
Harlow and Lane, Antibodies: A Laboratory manual. Cold Spring
Harbor Laboratory, 1988.
Harwood, Chandler, Pellarin, Bangerter, Wilkins, Long, Cosgrove, Malinow, Marzetta, Pettini, Savoy, Mayne,
"Pharmacologic consequences of cholesterol absorption inhibition: alteration in cholesterol metabolism and reduction in plasma cholesterol concentration induced by the synthetic saponin p-tigogenin cellobioside (CP-88,818; tiqueside), J. Lipid. Res. 34:377-395, 1993.
Hassanain, Dai, Gupta, Anal. Biochem., 213:162-167, 1993.
Hostettmann et al., "Chemistry and pharmacology of natural products," In Saponins, Cambridge University Press, pp.1-548, 1995.
Hu, Alfermann, "Diterpenoid production in hairy root cultures of Salvia’ miltlorrhlza," Phy to chemistry, 32 (3): 699-703; 1993.
Huang et al., Zhongueo Yaoil XuebaOr Chemical abstract No.98100885, 3: 286-288, 1982.
Ikeda, Fujiwara, Kinjo, Nohara, Ida, Shoji, Shingu, Isobe,
Kajimoto, Bull. Chem. Soc. Jpn., 68:3483-3490 (1995).
Inoue, H., et al., Chem. Pharm. Bull. 6) 2:897-901, 1986.
Jansakul, Baumann, Kenne, Samuelsson, "Ardisiacrispin A and B, two utero-contracting saponins from Ardlsia crispa,"
Planta Medico, 53: 405-409, 1987.
Jiang, Massiot, Lavaud, et al., "Triterpenoid glycosides from
the bark of Mimosa tenuiflora, Phytochemlstry, 30: 2357-2360, 1991.
Jung, Kwak, Kirn, Lee, Choi, Lin, "Improvement of the catharanthine productivity in hairy root cultures of Catharanthus roseus by using monosaccharides as a carbon source," Blotech. Lett., 14: 695-700; 1992.
Kamel, Ohtani, Kurokawa, et al., "Studies on Balanltes aegyptiaca fruits, an antidiabetic Egyptian folk medicine," Chem. Pharm. Bull., 39: 1229-1233, 1991.
Kasiwada et al., J. Org. Chem., 57: 6946-6953, 1992.
Kelly and Tsai, "Effect of pectin, gum arable and agar on cholesterol absorption, synthesis and turnover in rats,"
J. Nutr., 108:630-639, 1978.
Kennedy, Wagner, Conzen, Jordan, Bellacosa, Tsichlis, Nissam,
Genes and Dev., 11: 701-713, 1997.
Kimura et al., Immunogenetics., 11: 373-381, 1983. Kinjo, Araki, Fukui, Higuchi, Iceda, Nohara, Ida, Takemoto,
Miyakoshi, Shoji, Chem. Pharm. Bull. 40(12); 3269-3273 (1992).
Kizu and Tomimori, "Studies on the constituents of Clematis species. V. On the saponins of the root of Clematis chinensis OSBECK," Chem. Pharm. Bull., 30: 3340-3346, 1982.
Kohler and Milstein, Eur. J. Immunol., 6: 511-519,1976.
Kohler and Milstem, Nature, 256: 495-497, 1975.
Kojima and Ogura. "Configurational studies on hydroxy groups at C-2, 3 and 23 or 24 of oleanene and ursene-type triterpenes by NMR spectroscopy," Phytochemistry, 28: 1703-1710, 1989.
Kong et al., Phyto chemistry. 33: 427-430, 1993.
Konoshima and Sawada, Chem. Pharm. Bull., 30: 2747-2760, 1982.
Kutney, "Nuclear magnetic resonance (N.M.R.) study in the steroidal sapogenin series. Stereochemistry of the spiro ketal system," Steroids, 2: 225-235, 1963.
Lemieux, Kullnig, Bernstein, Schneider, "Configurational effects on the proton magnetic resonance spectra of six-membered ring compounds," J. Am. Chem. Soc., 80: 6098-6105, 1958.
Lister, P.P.., P. Holford, T. Haigh, and D.A. Morrison. Acacia in Australia: Ethnobotany and potential food crop. p.228-236. In: J. Janick (ed.). Progress in new crops. ASHS Press, Alexandria, VA, 1996.
Lloyd, McCown, "Commercially feasible micropropagation of mountain laurel, Kalmia latifolia by use of shoot tip culture," Comb. Proc. Inti. Plant Prop. Soc., 30: 421-427; 1981.
Mackness, Durrington, Converse, Skinner (Eds.), Jn:
Iiipoprotein Analysis: A Practical Approach, Oxford University Press, Oxford, p 1, 1992.
Mahato, Pal, Nandy, Tetrahedron, 48: 6717-6728 (1992).
Manabe et al, J. Lab. Clin. Med., 104 (3):445-454, 1984.
Martin et al., J. Exp. Med., 182:1545-1556, 1995.
Martin, Reueelingsperger, McGahon, Rader, van Schie, Laface, Green, J. Exp.Med., 182:1545-1556, 1995.
Massiot, Lavaud, Besson, Le Men-Olivier, van Binst, "Saponins from aerial parts of alfalfa (Medicago sativa)," J. Agric. Food Chem., 39: 78-82, 1991b.
Massiot, Lavaud, Delaude, van Binst, Miller, Fales, "Saponins from Tridesmostemon claessenssi," Phy to chemistry, 29:3291-3298, 1990.
Massiot, Lavaud, Guillaume, Le Men-Olivier, van Binst, "Identification and sequencing of sugars in saponins using 2D 1H NMR spectroscopy," J. Chem. Soc., Chem. Common., 1485-1487, 1986.
Massiot, Lavaud, Le Men-Olivier, van Binst, Miller, Fales, "Structural elucidation of alfalfa root saponins by mass spectrometry and nuclear magnetic resonance analysis,". J. Chem. Soc., Perkin Trans., 1:3071-3079, 1988.
Massiot, Lavaud, Nuzillard. "Revision des structures des chrysantellines par resonance magnetique nucleaire," Bull. Soc. Chim. Fr., 127: 100-107, 1991a.
Miotti et al.. Cancer Res., 65: 826, 1985.
Miyamoto, Togawa, Higuchi. Komori, Sasaki, "Six newly identified biologically active triterpenoid glycoside sulphates from the sea cucumber," Cucumana echinata. Annalen, 453-460, 1990.
Monk, "Variegation in epigenetic inheritance", TIG, 6: 110-114, 1990.
Mujoo, Maneval, Anderson, Gutterman, Oncogene, 12: 1617-1623, 1996.
Murashige, Skoog, "A revised medium for rapid growth and bioassay of tobacco tissue culture," Physiol. Plant, 15: 473-482; 1962.
Murashige, Т and Skoog, F. "A revised medium for rapid growth and bio-assays with tobacco tissue cultures," Physlologia
Plantarum 15: 473-497, 1962.
Nabel and Baltimore, Nature 326:711-713, 1987.
Nagamoto et a2., Planta Medica., 54:305-307, 1988.
Nagao, Hachiyama, Oka, Yamauchi, "Studies on the constituents of Aster tatarlcus L. f. П.Structures of aster saponins isolated from the root," Chem. Pharm. Bull., 37:1977-1983, 1989.
Nelson, Futscher, Kinsella, Wymer, Bowden, "Detection of mutant Ha-ras genes in chemically initiated mouse skin epidermis before the development of benign tumors," Proc.
Natl. Acad. Sci. USA, (14):6398-6402, 1992.
Nishino, Manabe, Enoki, Nagata, Tsushida, Hamaya, "The structure of the tetrasaccharide unit of camellidins, saponins, possessing antifungal activity," J. Chem. Soc.,
Chem. Commun., 720-723, 1986. Nitsch, Nitsch, "Haploid plants from pollen grains," Science, 163:85-87, 1969.
O’Reilly, Boehm, Shing, Fukai, Vasios, Lane, Flynn, Birkhead, Olsen, Folkman, Cell, 88: 277-285, 1997.
Oakenfull et al., Athe-rosc-Iero.s-LS, 48: 301 (1983).
Ohkawa, Kamada, Sudo, Harada, "Effects of gibberellic acid on hairy root growth in Datura innoxia," J. Plant Physiol., 134:633-636; 1989.
Okabe, Nagao, Hachiyama, Yamauchi, "Studies on the constituents of Luff a operculata cogn. П.Isolation and structure elucidation of saponins in the herb," Chezn.
Pharm. Bull., 37: 895-900, 1989.
Okada, Koyama, Takahashi, Okuyama. Shibata. Planta Med. 40: 185-192, (1980).
Okada, Sakuma. Fukui, Hazeki, Ui, J. Blo. Chem., 269: 3563-3567, 1994.
Pallavicini, Jn: Techniques In Cell Cycle Analysis, Gray and Parzynkiewicz (Eds.), Humana Press Inc., Clifton, NJ, pp.139, 1987.
Pant, Panwar, Negi, Rawat, Morris, Thompson, "Structure elucidation of a spirostanol glycoside from. Asparagus offlclnalis fruits by concerted use of two-dimensional
NMR techniques," Mag. Reson. Chem., 26: 911-918, 1988.
Fenders, Delaude, Pepermans, van Binst, "Identification and sequencing of sugars in an acetylated saponin of Bllghia welwltschil by N.M.R. spectroscopy," Carbohyd. Res., 190: 109-120, 1989.
Pietenpol et al.. Cancer Res., 55: 1206-1210, 1995.
Pieterez et al.. Antibody Inmiunoconj. Radlopharm., 1: 79-103, 35, 1988.
Pisha et al., Mature Medicine, 1: 1046-1051, 1995.
Polyak et al.. Genes Dev., 8: 9-22, 1994.
Potterat, Hostettmann, Stoeckli-Evans, Saadou, "Saponins with an unusual secoursene skeleton from Sesamum alatum
THONN., Helv. Chlm. Ada, 75:833-841, 1992.
Prehn, "Regeneration versus neoplastic growth,"
Carcinogenesis, 18(8): 1439-1444, 1997.
Puri, Wong, Puri, "Solasodine and diosgenin: 1H and 13C assignments by two-dimensional NMR spectroscopy," Mag.
Res. Chem., 31: 278-282, 1993. Reeves, Nielson, Fahey, Am. Inst. Nutr., 1939, 1993.
Reisfeld et al., Melanoma Antigens and Antibodies, p.317, 1982.
Reznicek, Jurenitsch, Kubelka, Michi, Korhammer, Haslinger, "Isolierung und Struktur der vier Hauptsaponine aus Solldago gigantea var. serotlna," Annalen, 989-994, 1990.
Reznicek, Jurenitsch, Michi, Haslinger, "The first structurally confirmed saponin from Solldago gigantea: structure elucidation by modern NMR techniques," Tetrahedron Lett., 30: 4697-4100. 1989b.
Reznicek, Jurenitsch, Robien, Kubelka, "Saponins in Cyclamen species: configuration of cyclamiretin С and structure of isocyclamin," Phyto chemistry, 28: 825-828.1989a.
Rhodes, et al., "Influence of exogenous hormones on the growth and secondary metabolite formation in transformed root cultures," Plant Cell Tissue Organ Culture, 3 8: 143-151; 1994.
Rodriguez, Castro, Riguera, "Holothurinosides: new antitumour non sulphated triterpenoid glycosides from the sea
cucumber Holothrula forskalii, " Tetrahedron, 47: 4753-4762, 1991.
Royal I and Park M, J. Biol. Chem. 270: 27780-27787, 1995.
Sasaki, Udagawa, Ishimaru, Hayashi, Alfermann, Nakanishi,
Shimomura, "High forskolin production in hairy roots of
Coleus forskohlll" Plant Cell Reports 17: 457-459, 1998.
Sashida, Kawashima, Mimaki, "Novel polyhydroxylated steroidal
saponins from Allium giganteum," Chem. Pharm. Bull, 39: 698-703, 1991.
Schenk, Hilderbrandt, "Medium and techniques for induction and
growth of monocotyledonous and dicotyledonous plant cell cultures," Can. J. Bot., 50: 199-204; 1972.
Schopke, Wray, Rzazewska, Hiller, "Bellissaponins BAi and BAa, acylated saponins from Bellis perennis," Phytochemlstry, 30: 627-631, 1991.
Schreiber, Matthias, Muller, Schafmer, Nucleic Acids -Res., 17: 6419, 1989.
Schuh et al., "Obligatory wounding requirement for tumorigenes is in v-jun transgenic mice," Nature, 346:756-760, 1990.
Shao, Kasai, Xu, Tanaka, "Saponins from roots of Kalopanax septemlobus. (THUNB.) KOIDZ., Ciqiu: structures of kalopanaxsaponins С, D, E and F, " CAem. Pha-nn. Bull., 37: 311-314, 1989.
Shayesteh, Lu, Kuo, Baldocchi, Godfrey, Collins, Pinkel,
Powell, Mills, Grey, Nat. Gent., 21: 99-102, 1999.
Shepard et al., J. Clln. Jmmunol., 11: 117-127, 1991.
Shirazi, Liu, Trott, "Exposure to ultraviolet В radiation
increases the tolerance of mouse skin to daily X-radiation," Pad. Res., 145: 768-775, 1996.
Sieweke et al., "Mediation of wound-related rous sarcoma virus
tumorigenesis by TGF-P; Science, 248: 1656-1660, 1990.
Smith, Weathers, Cheetham, "Effects of gibberellic acid on hairy root cultures of Artemlsia annua: growth and artemisinin production," In Vitro Cell Dev. Blol., 33: 75-79; 1997.
Spady, Wollett, Dietschy, Annu. Rev. Nutr., 13:355, 1993. Steel and Torrie, In: Principals and Procedures of Statistics, 2nd Ed., McGraw-Hill, New York, p.383, 1980.
Stevenson et al., CAem. Immunol., 48:126-166, 1990.
Takema, Fujimura, Ohsu, Imokawa, "Unusual wrinkle formation after temporary skin fixation followed by UVB irradiation in hairless mouse skin." Exp. Dermatol., 5:145-149, 1996.
Tewari, Quan, Rourke, Zeng, Beidler, Salvesan, Dixit, "Yaina/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly (ADP-ribose) polymerase," Cell, 81: 801, 1995.
Thompson et al., Cancer Epidemiol. Biomarker Prevent., 1: 597-602, 1992.
Thor et al.. Cancer Res., 46:3118, 1986.
Tomas-Barbaren et al., Planta Medica., 54: 266-267 (1988).
Tori and Aono, Ann. jRept. Shionogi Res. Lab., 14: 136, 1964.
Vaickus et al.. Cancer Invest., 9: 195-209, 1991. Vazquez, Quinoa, Riguera, San Martin, Darias, "Santiagoside, the first asterosaponin from an Antarctic starfish {Neosmllaster georgianus}" Tetrahedron, 48: 6739-6746, 1992.
Vlahos and Matter, FEBS Lett., 309: 242-248, 1992.
Vlahos,Matter, Hui, Brown, J. Bio. Chem., 269: 5241-5248, 1994.
Waltho, Williams, Mahato, Pal, Barna, "Structure elucidation of two triterpenoid tetrasaccharides from Androsace saxliragifolia," J. Chem. Soc., Perkin 1: 1527-1531, 1986.
Wang, He, Ling, Li, "Chemical study of Astragalus plant. П.Structures of asernestioside A and B, isolated from Astragalus ernest-ii COMB. Huaxue Xuebao, 47: 583-587,
Chem. Abstr., 1989.
Wens et al., Proc. Natl. Acad. Sci., 92: 5744-5748, 1995.
White, Genes Dev., 10: 1-15, 1996. Whitman M, Kaplan D.R.. Schatthausen B, Cantley L.C. and Roberts, Т.М. Nature, 315: 239-242, 1985.
Willker and Leibfritz, "Complete assignment and conformational studies of tomatine and tomatidine," Mag. Res. Chem., 30: 645-650, 1992.
Wyllie, Anticancer Res., 5: 131-136, 1985.
Wysokinska, Chmiel, "Transformed root cultures for biotechnology," Ada Blotechnol., 17: 131-159; 1997.
Yang et al., Anticancer Res., 15: 2479-2488, 1995.
Yoshikawa, Shimono, Arihara, "Antisweet substances, jujubasaponins I-III from Zizyphus jujuba,. Revised structure of ziziphin," Tetrahedron Lett., 32:7059-7062, 1991.
Yoshikawa, Suzaki, Tanaka, Arihara, Nigam, J. Wat. Prod., 60: 1269-1274 (1997).
Youn, Park, Chung, Lee, PAotodermatoi PAoto-Lmmuno-I. PAotomed., 13: 109-114, 1997.
Yukimune et al.. Nature Biotech., 14: 1129-1132, 1996.
Zobel, "Study-state control and investigation of root system morphology," In: Torrey J.G., Winship, L.J. (eds.) Applications of continuous and. steady-state methods to root biology, Kluwer, Amsterdam, 165-182, 1989.



| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИРОВАНИЕ NF-κB КОМПОЗИЦИЯМИ ТРИТЕРПЕНОВ | 2001 |
|
RU2288706C2 |
| АНТИТЕЛО, СЕЛЕКТИВНОЕ В ОТНОШЕНИИ РЕЦЕПТОРА ЛИГАНДА, ИНДУЦИРУЮЩЕГО АПОПТОЗ И СВЯЗАННОГО С ФАКТОРОМ НЕКРОЗА ОПУХОЛИ, И ЕГО ПРИМЕНЕНИЕ | 2002 |
|
RU2313537C2 |
| КОМБИНАЦИИ АНТИТЕЛ, ОБЛАДАЮЩИХ СЕЛЕКТИВНОСТЬЮ ПО ОТНОШЕНИЮ К РЕЦЕПТОРУ ЛИГАНДА, ИНДУЦИРУЮЩЕМУ АПОПТОЗ, АССОЦИИРОВАННЫЙ С ФАКТОРОМ НЕКРОЗА ОПУХОЛИ, И ДРУГИХ ТЕРАПЕВТИЧЕСКИХ СРЕДСТВ | 2002 |
|
RU2313368C2 |
| АНТИТЕЛО, ОБЛАДАЮЩЕЕ СЕЛЕКТИВНОСТЬЮ ПО ОТНОШЕНИЮ К РЕЦЕПТОРУ ЛИГАНДА, ИНДУЦИРУЮЩЕМУ АПОПТОЗ, АССОЦИИРОВАННЫЙ С ФАКТОРОМ НЕКРОЗА ОПУХОЛИ, И ЕГО ИСПОЛЬЗОВАНИЕ | 2001 |
|
RU2298013C2 |
| АНАЛИЗЫ И СПОСОБЫ ПРИМЕНЕНИЯ БИОМАРКЕРОВ | 2006 |
|
RU2409817C2 |
| СПОСОБЫ ПРИМЕНЕНИЯ АГОНИСТОВ АРО-2L-РЕЦЕПТОРОВ И АКТИВАТОРОВ NK-КЛЕТОК | 2005 |
|
RU2395294C2 |
| АПОПТОТИЧЕСКАЯ ЧУВСТВИТЕЛЬНОСТЬ К Apo2L/TRAIL ПУТЕМ ТЕСТИРОВАНИЯ ЭКСПРЕССИИ GalNac-T14 В КЛЕТКАХ/ТКАНЯХ | 2006 |
|
RU2416097C2 |
| СПОСОБ ПОЛУЧЕНИЯ ФРОНДОЗИДА А И СПОСОБ СТИМУЛИРОВАНИЯ ИММУННОЙ СИСТЕМЫ МЛЕКОПИТАЮЩИХ | 2005 |
|
RU2339644C2 |
| БИОЛОГИЧЕСКИЕ МАТЕРИАЛЫ И ИХ ПРИМЕНЕНИЕ | 2006 |
|
RU2432363C9 |
| СРЕДСТВО, СТИМУЛИРУЮЩЕЕ АПОПТОЗ КЛЕТОК ЛЕЙКЕМИИ ЧЕЛОВЕКА | 2007 |
|
RU2360692C1 |

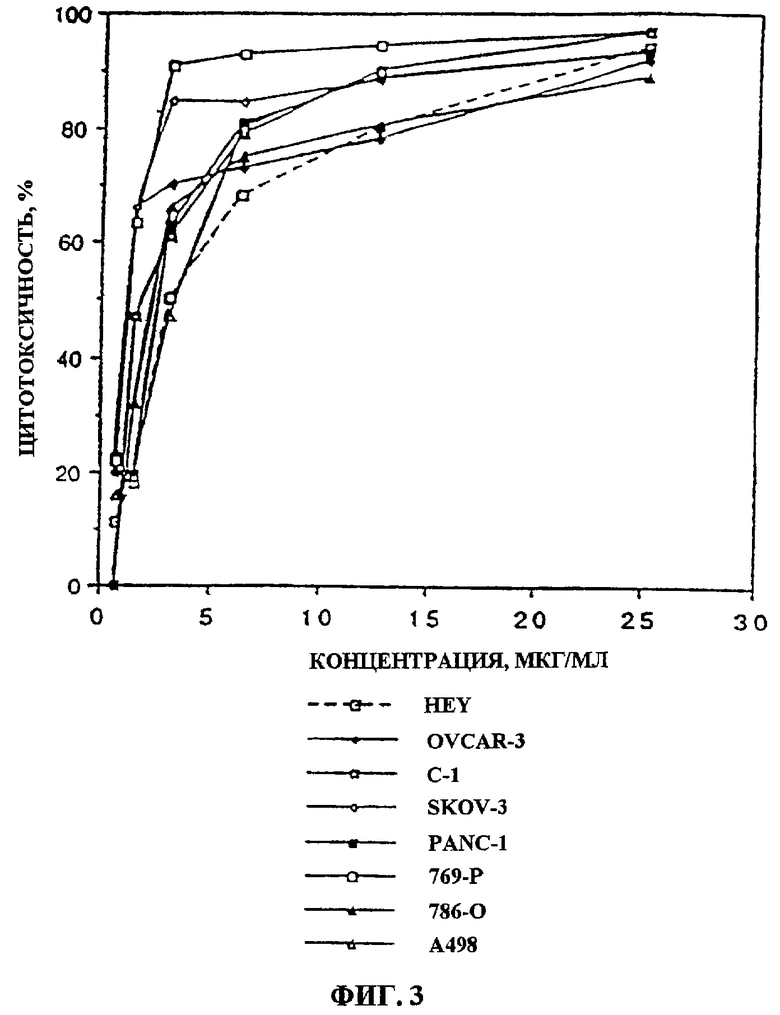
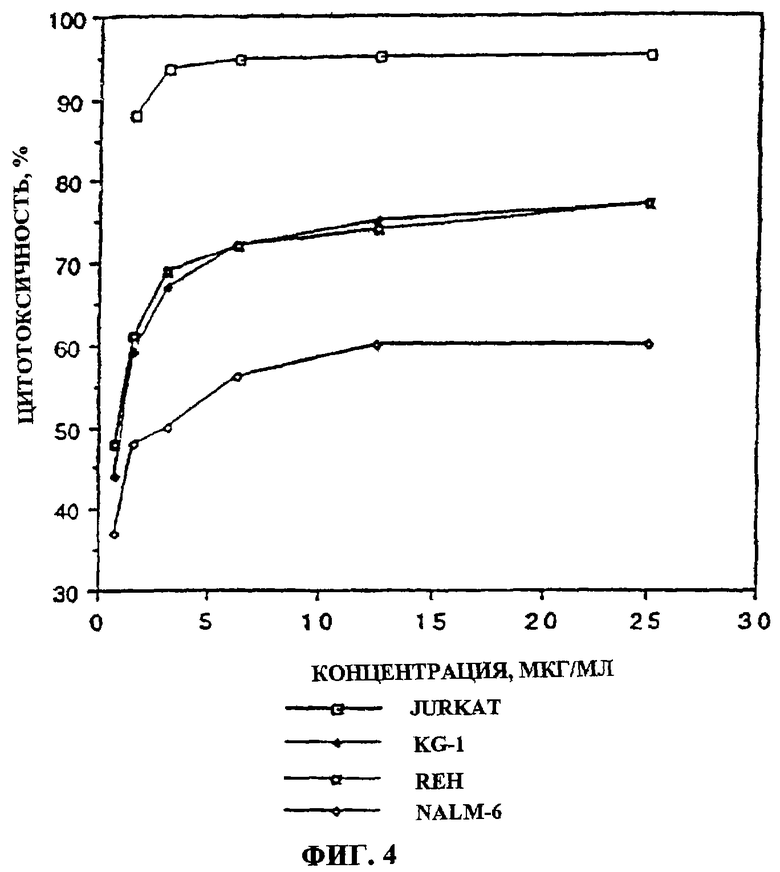
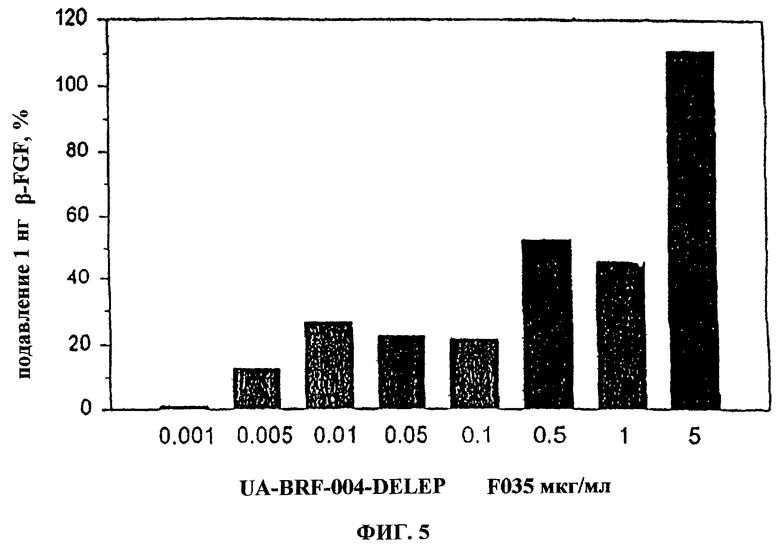
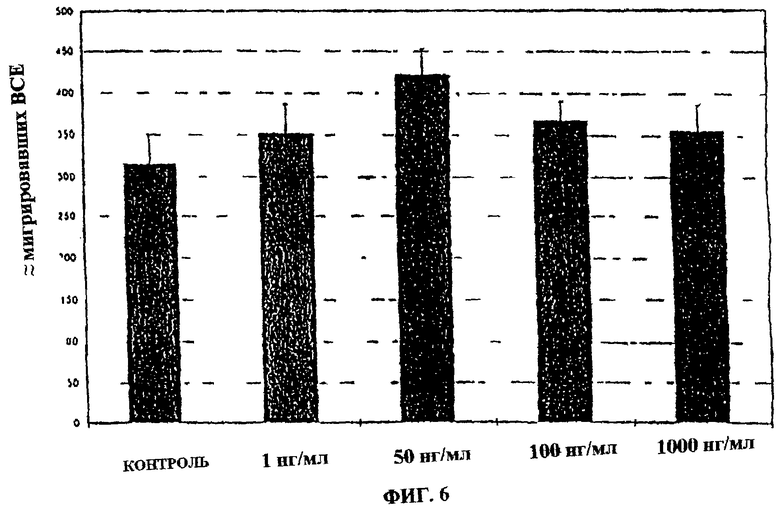
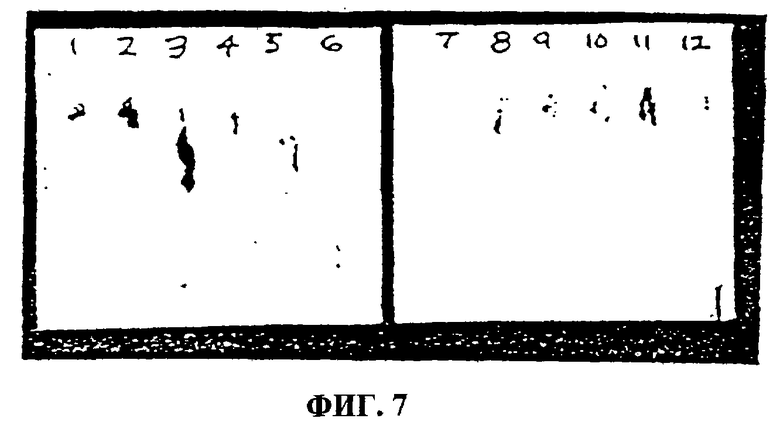

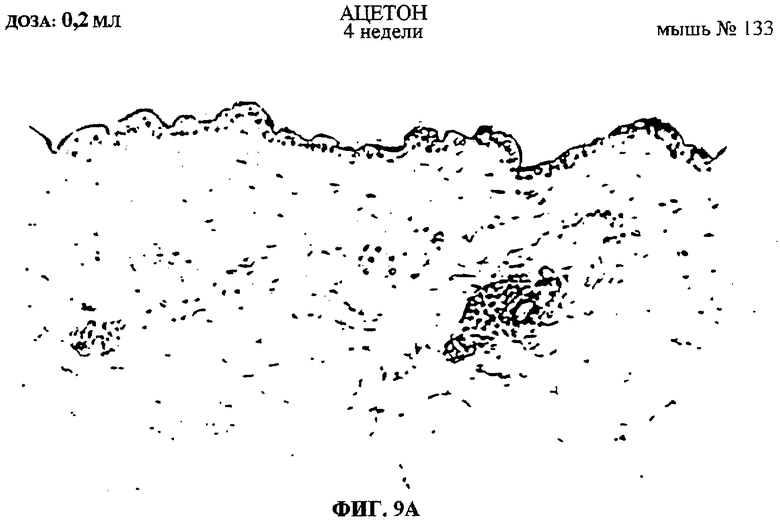
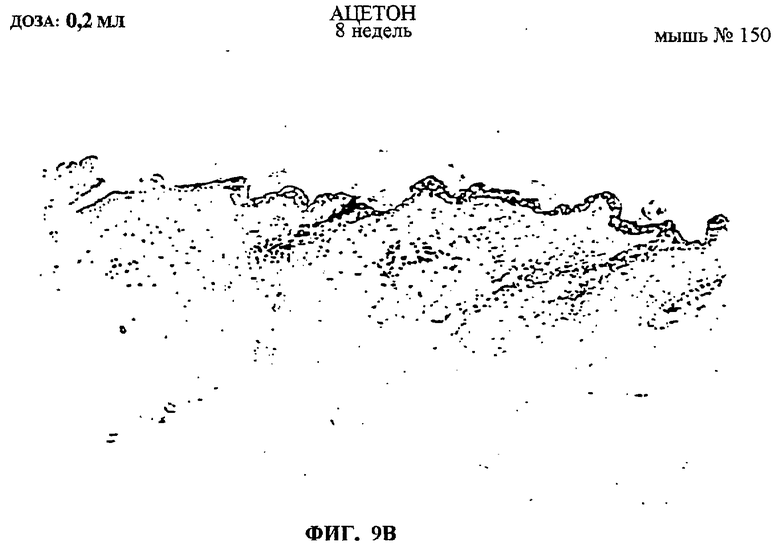

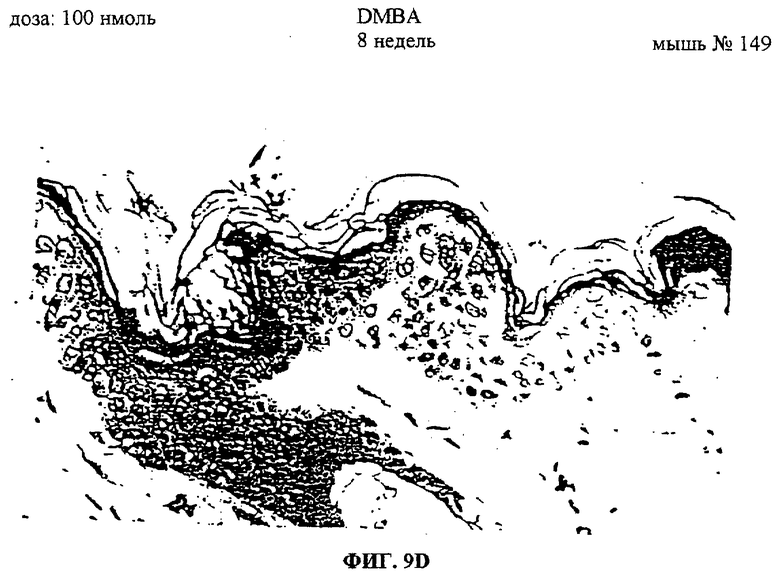

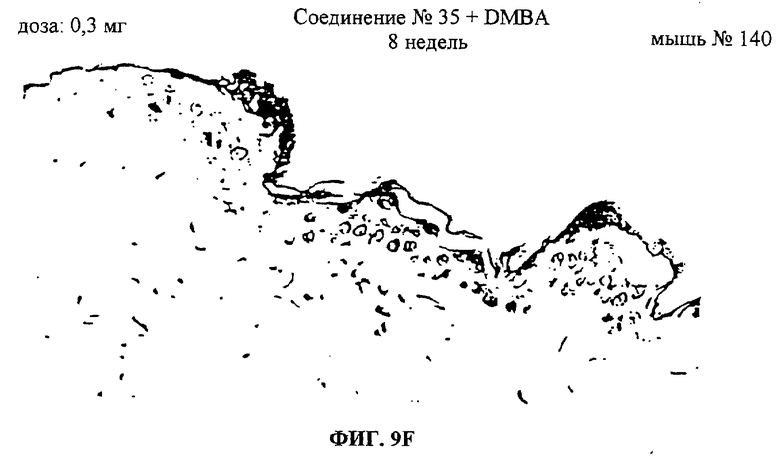
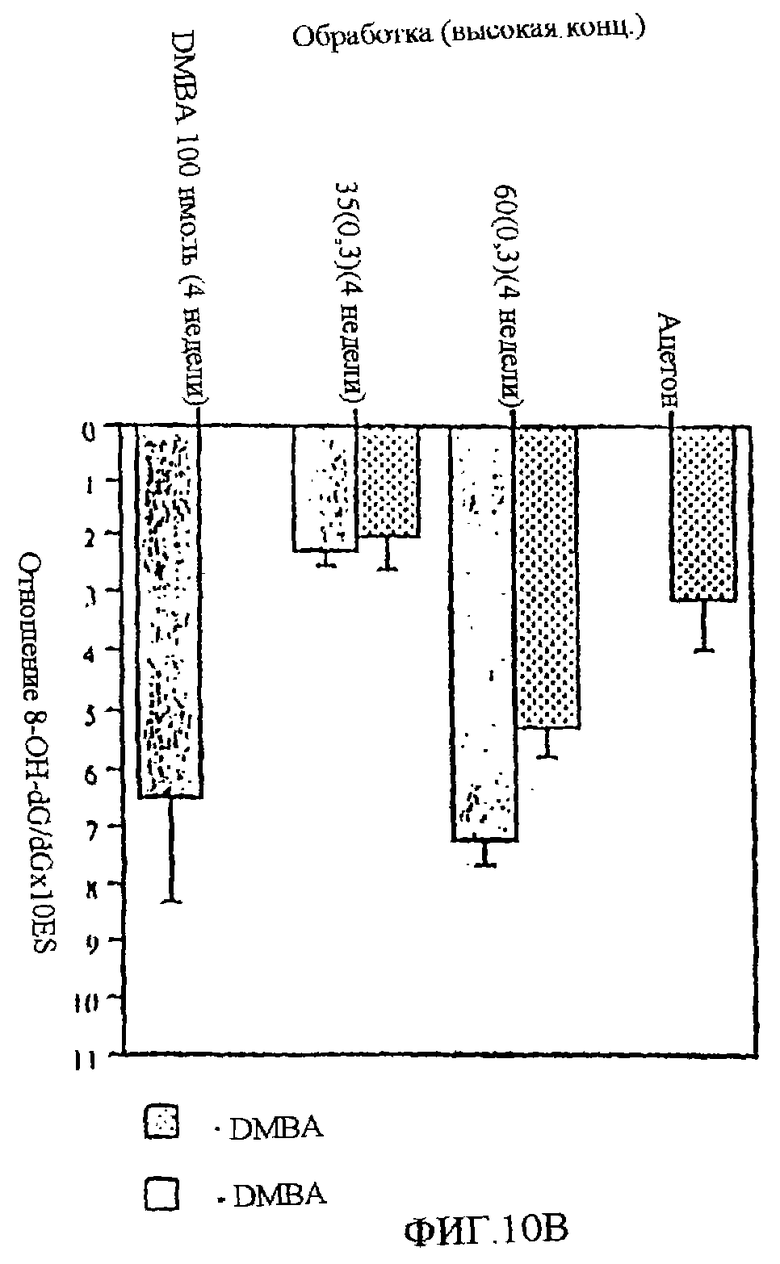
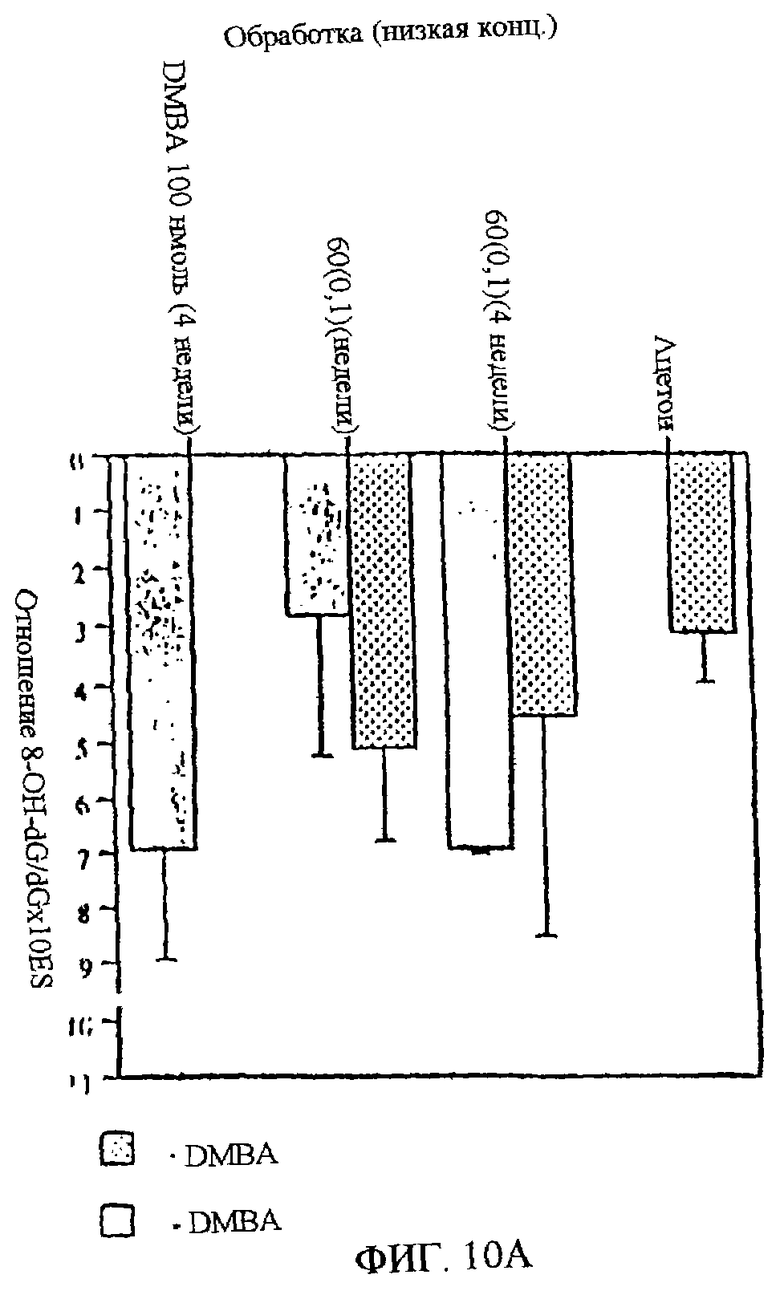

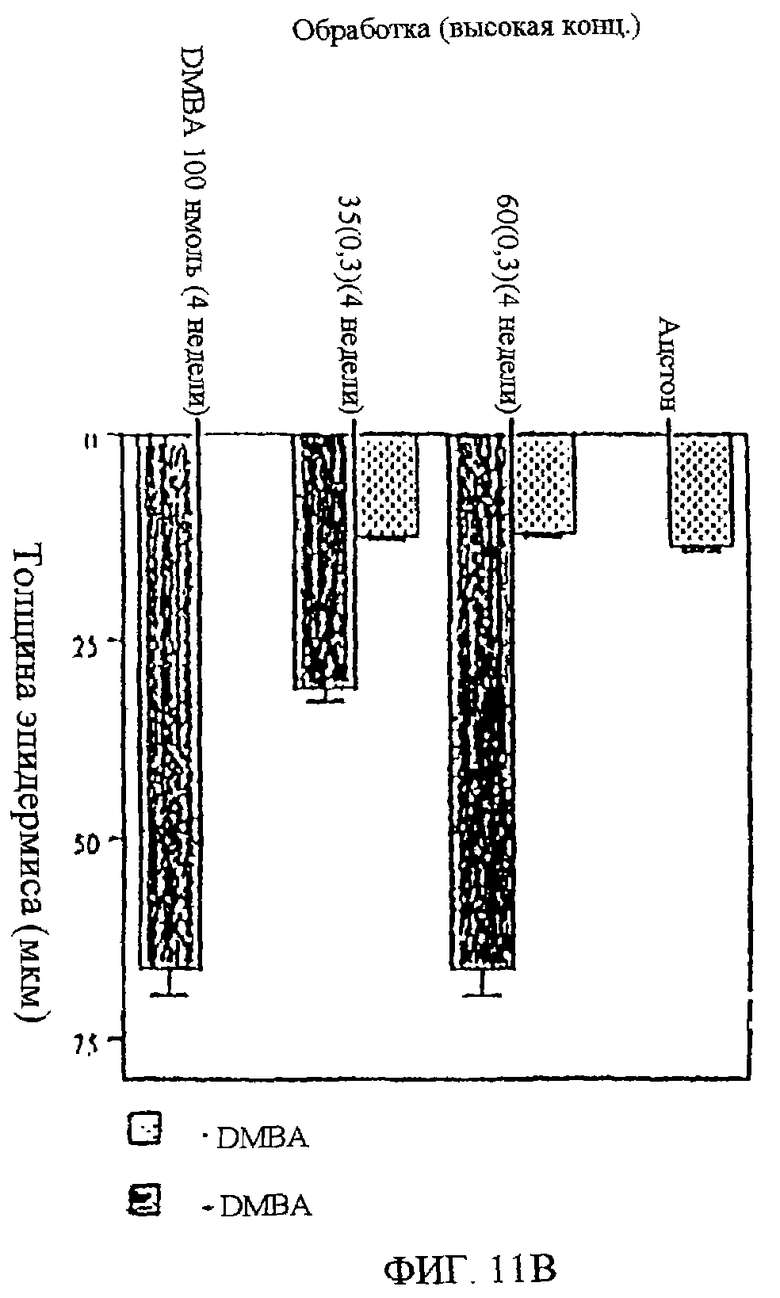
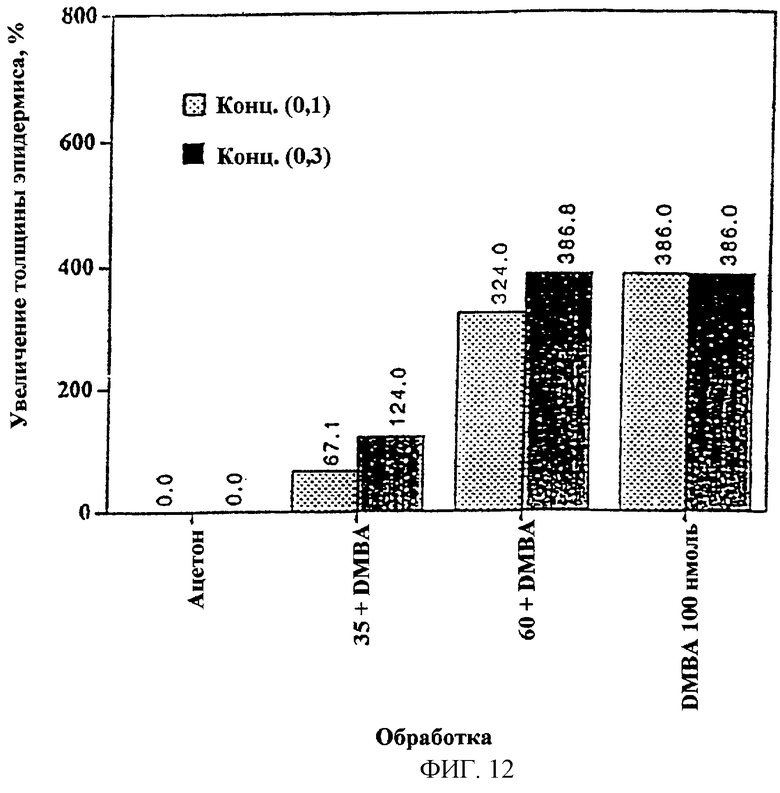
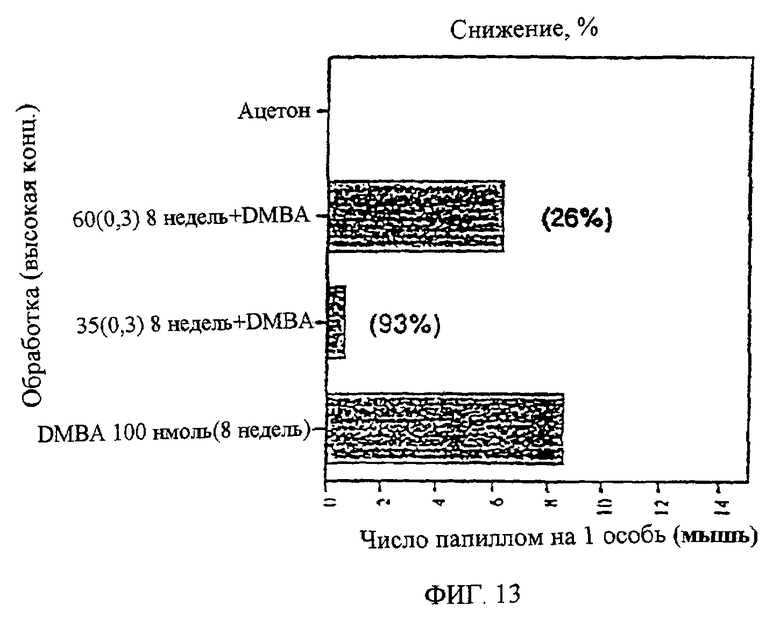
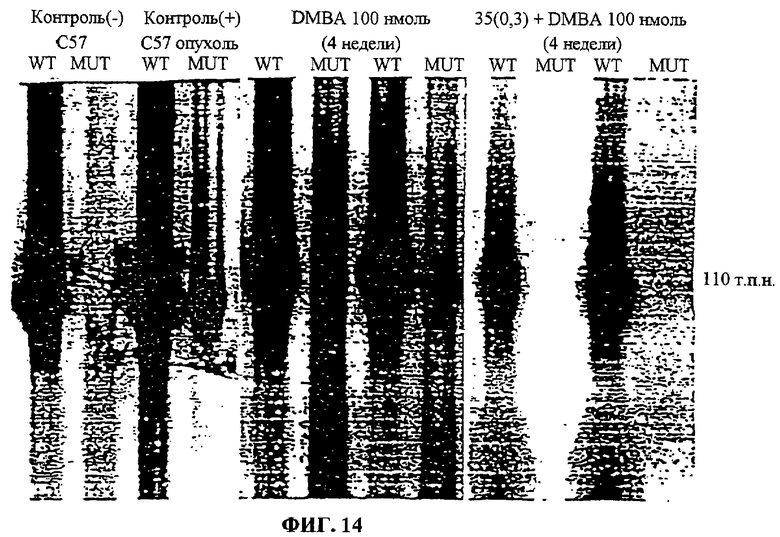
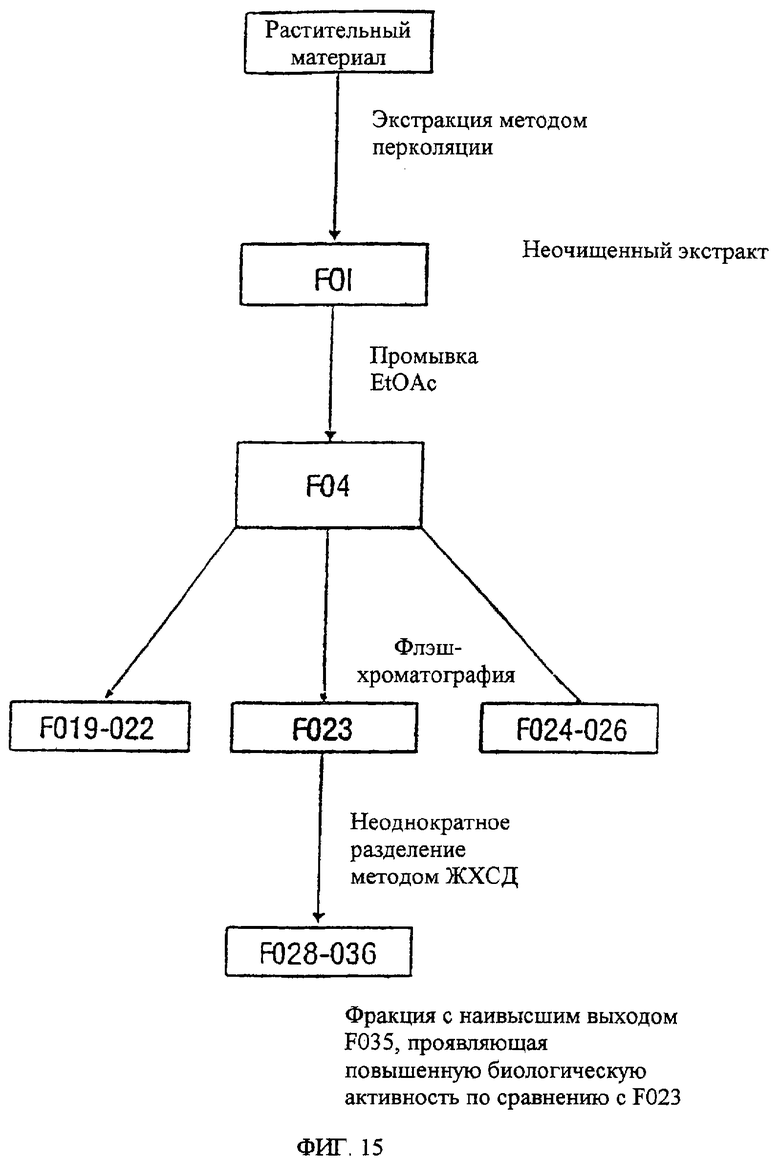
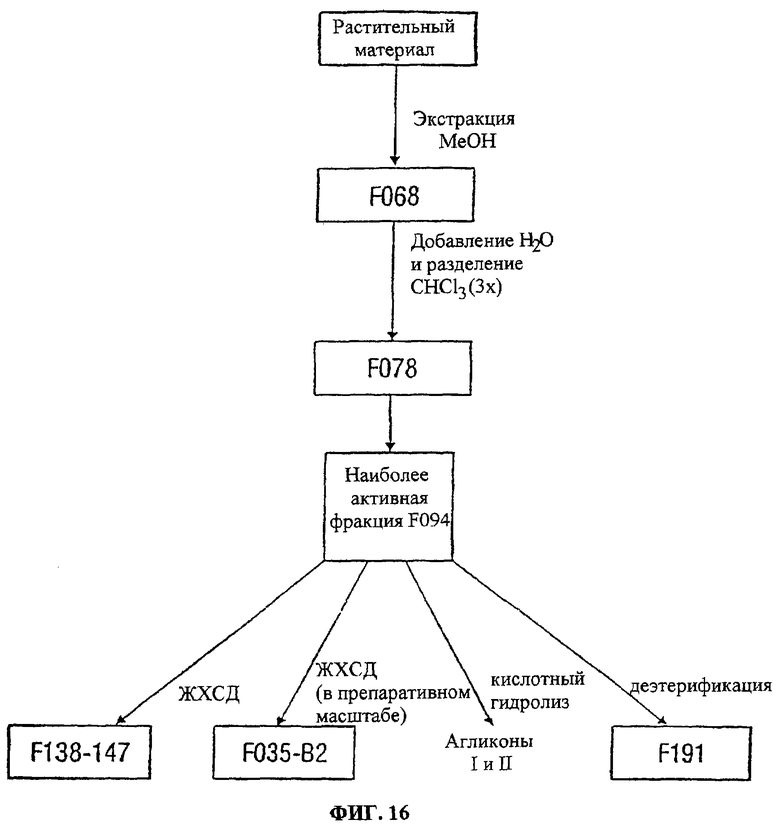
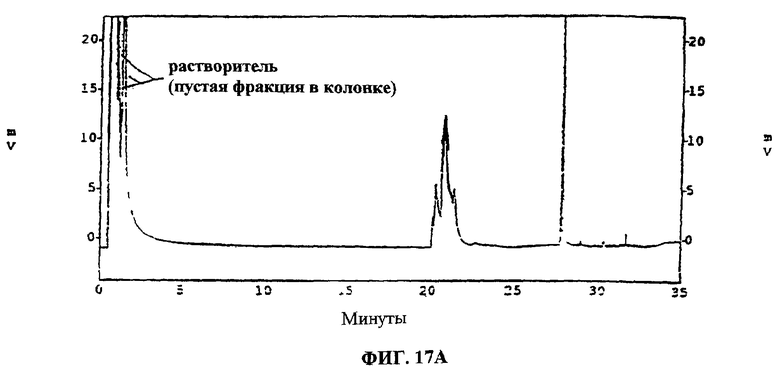
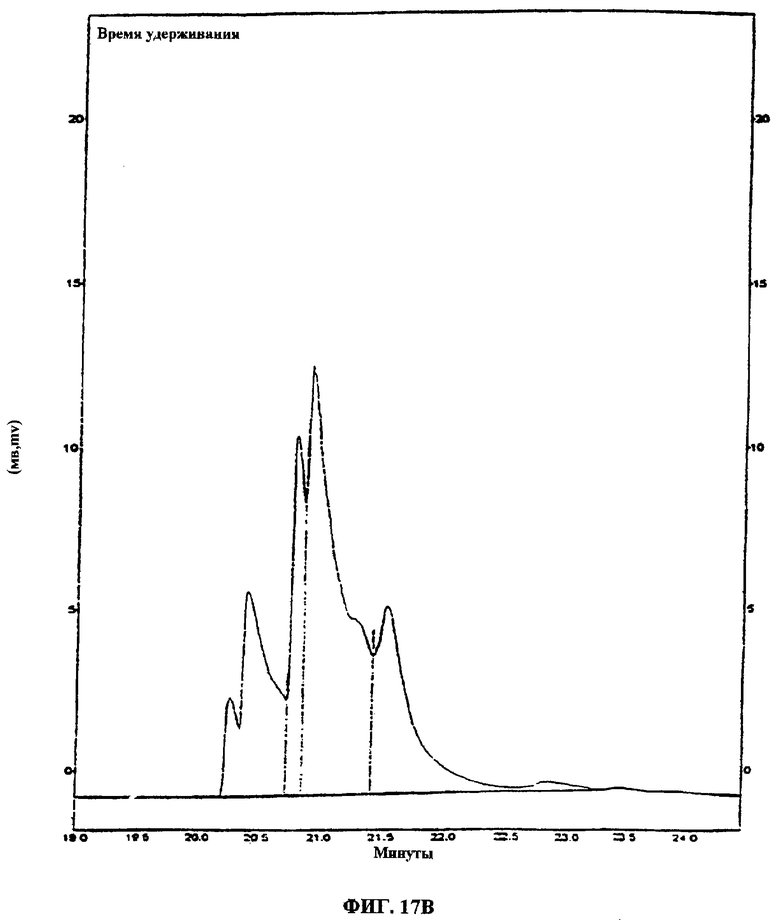
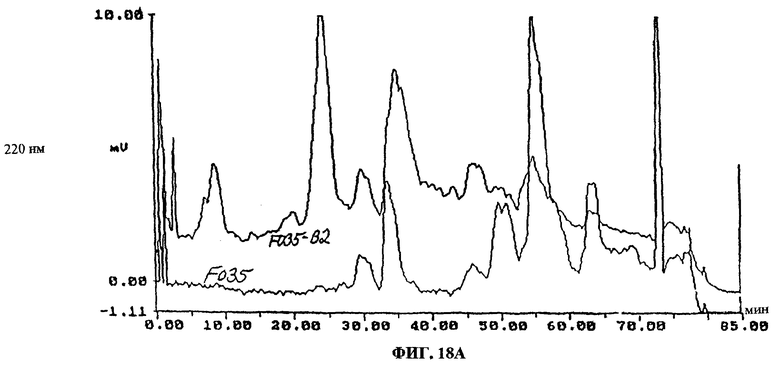
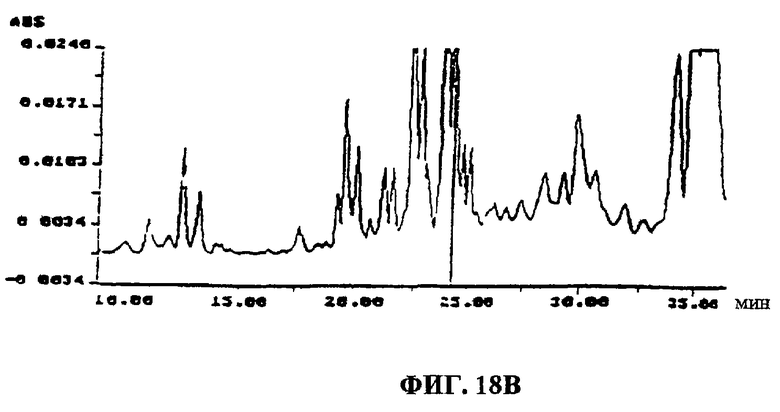

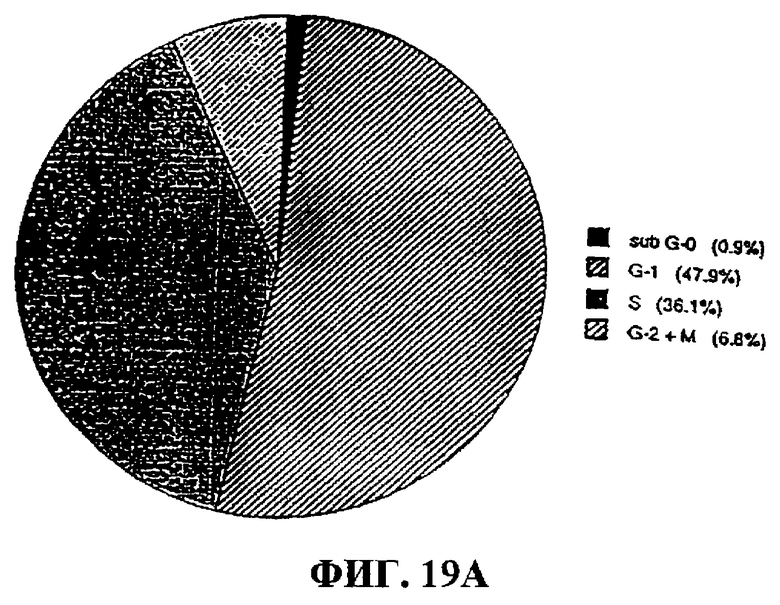
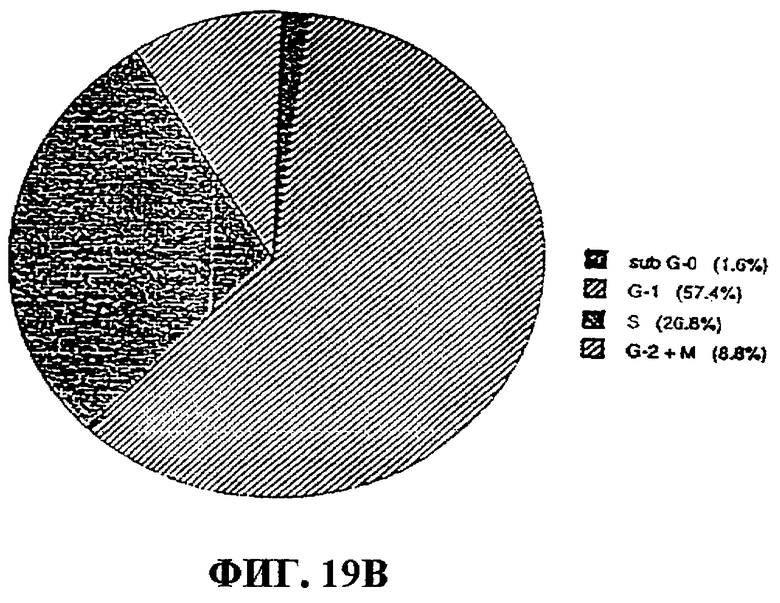
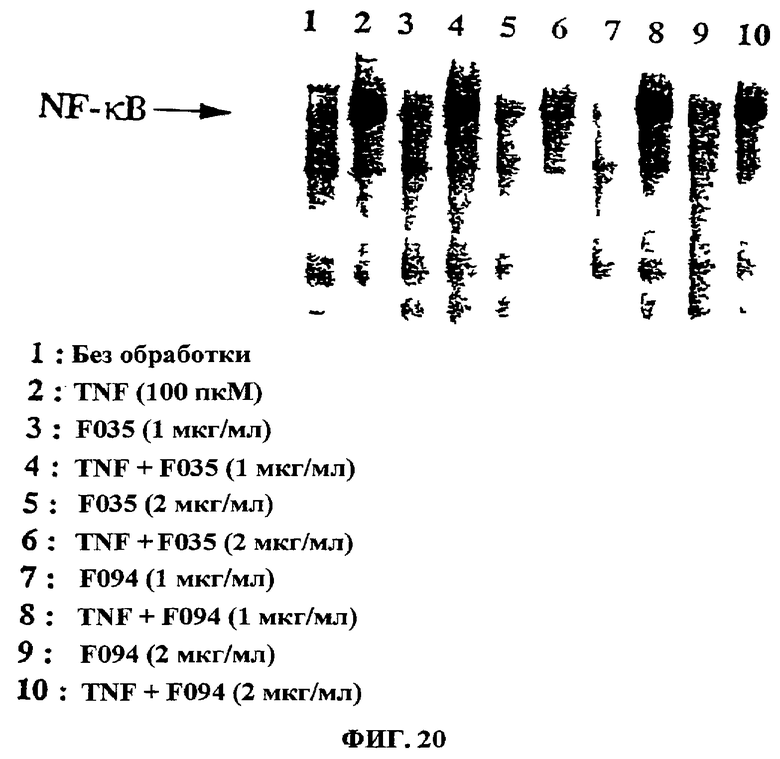
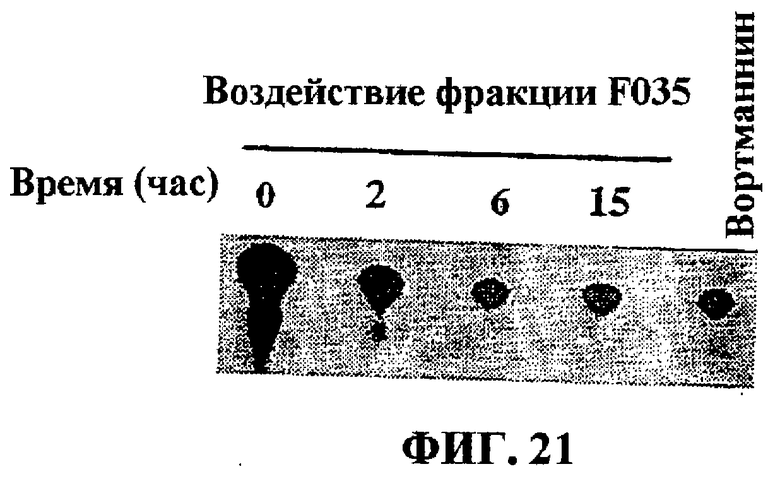
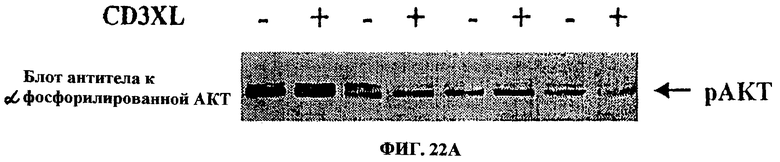
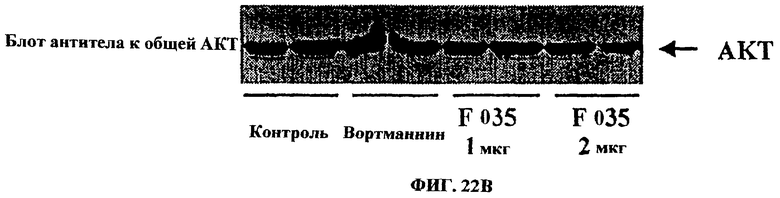

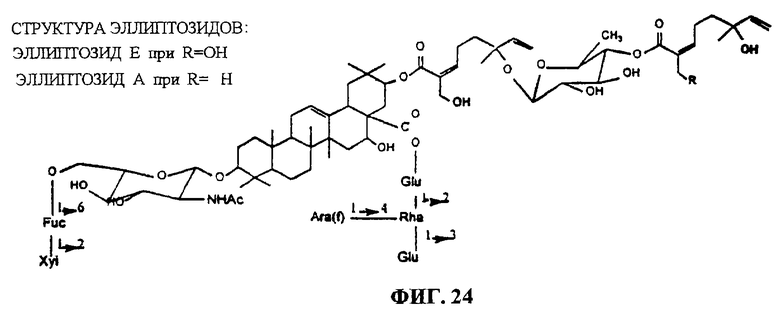
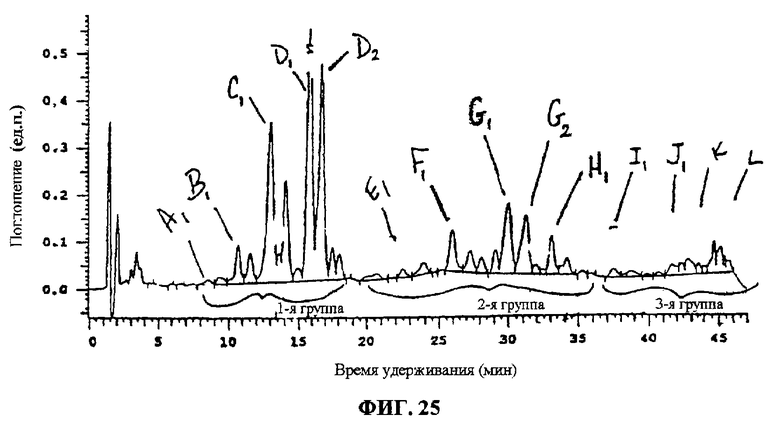
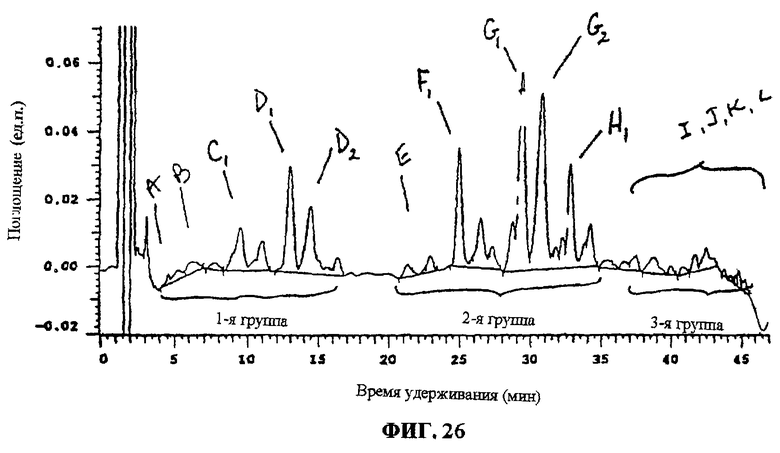
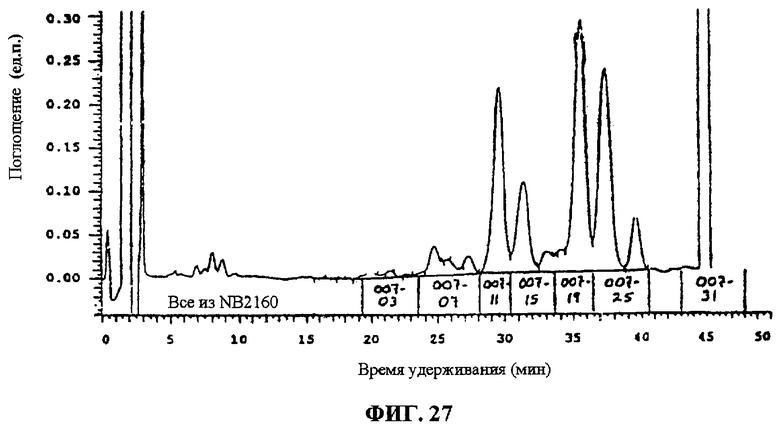

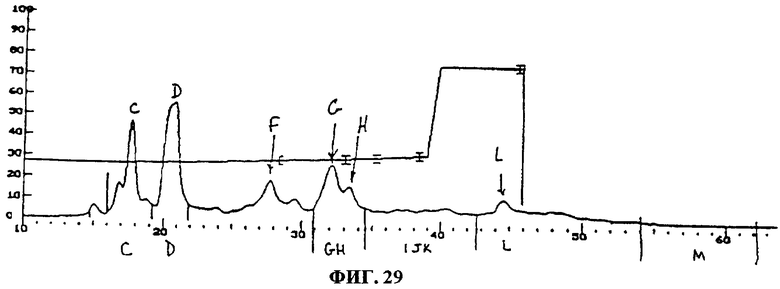
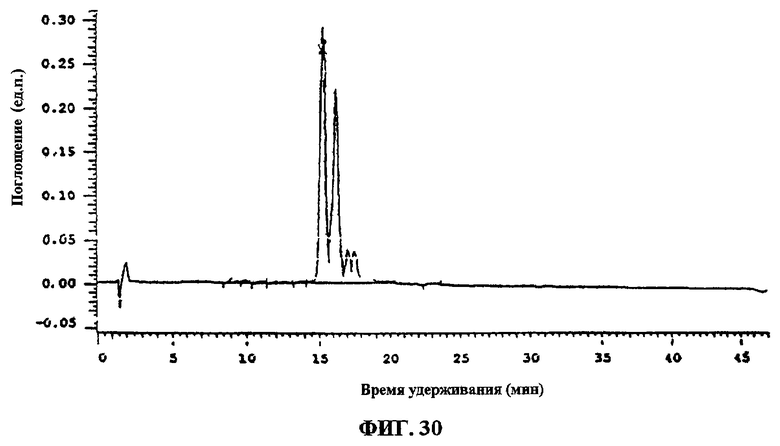
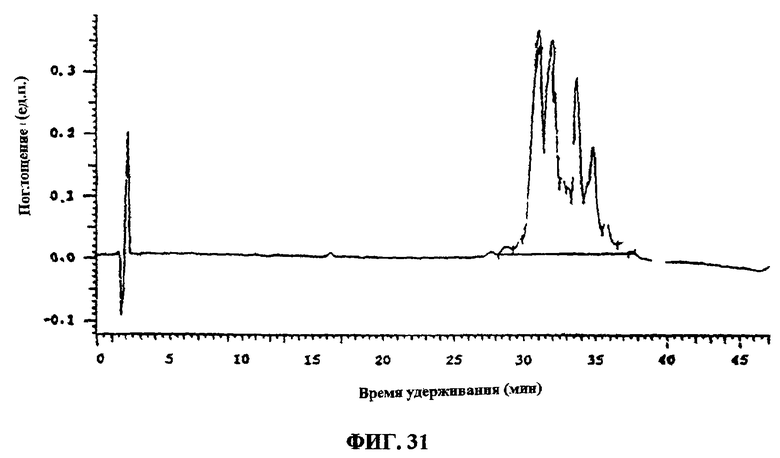

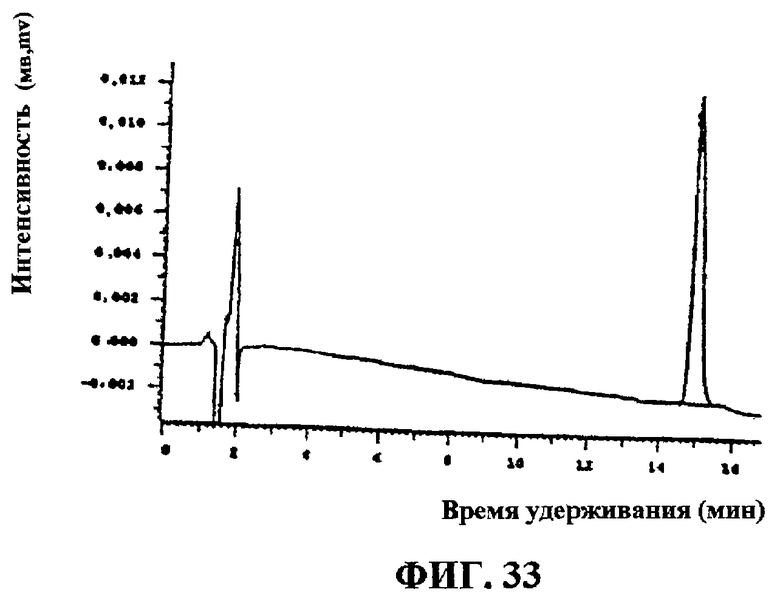
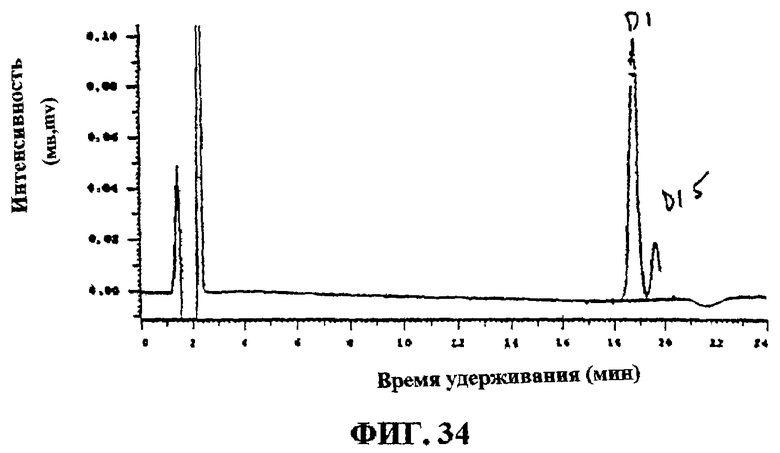
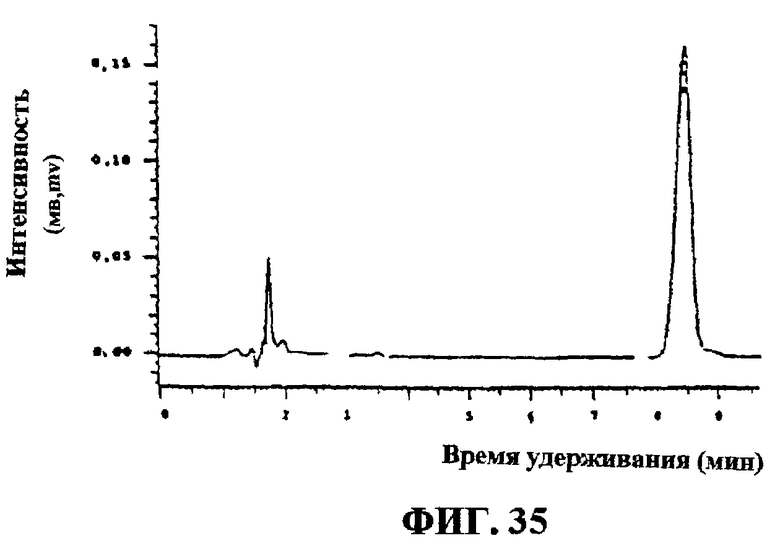
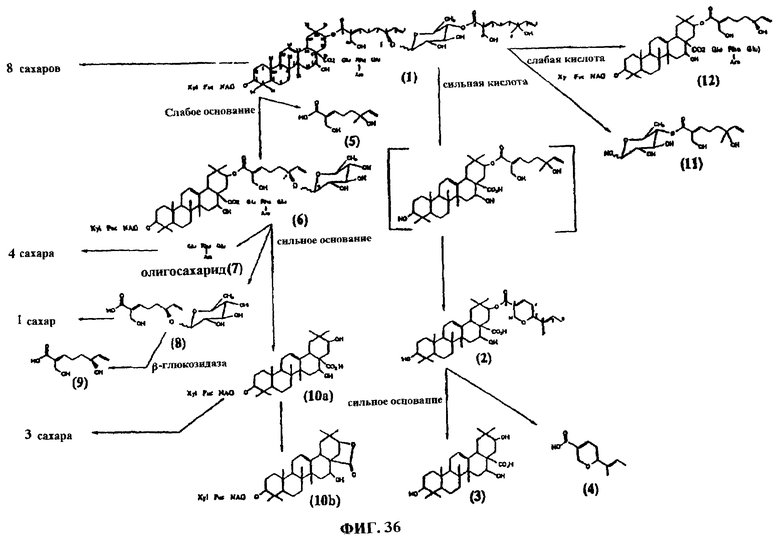
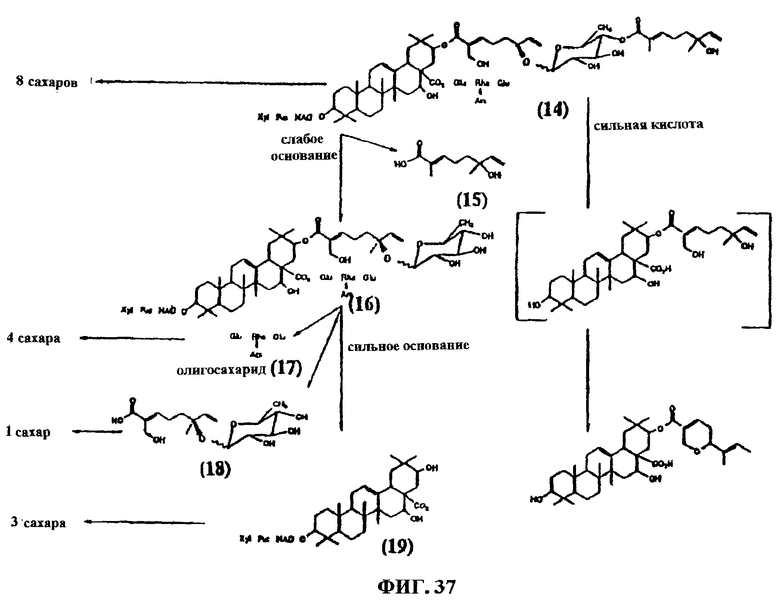

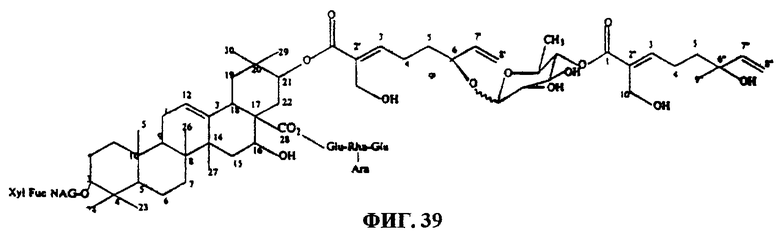
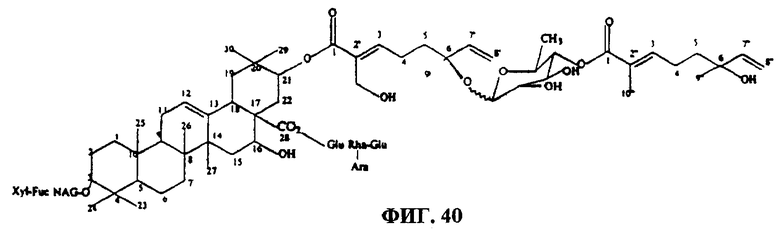
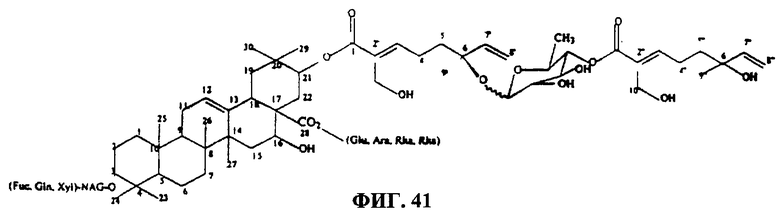

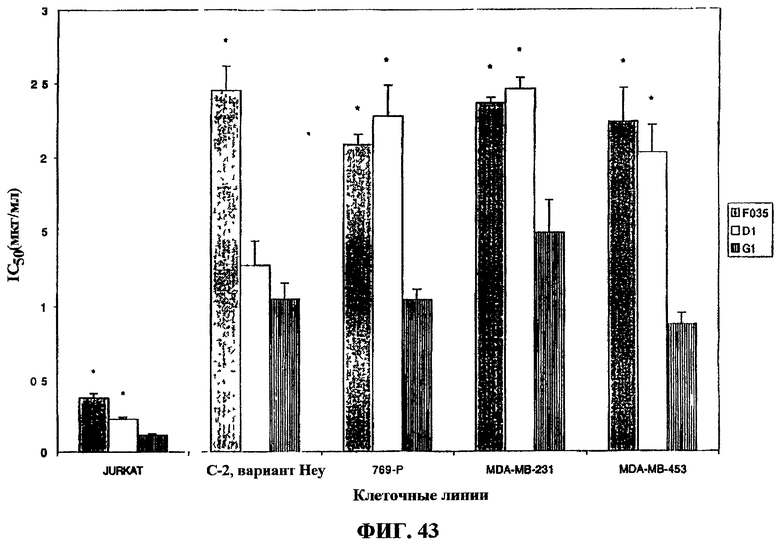

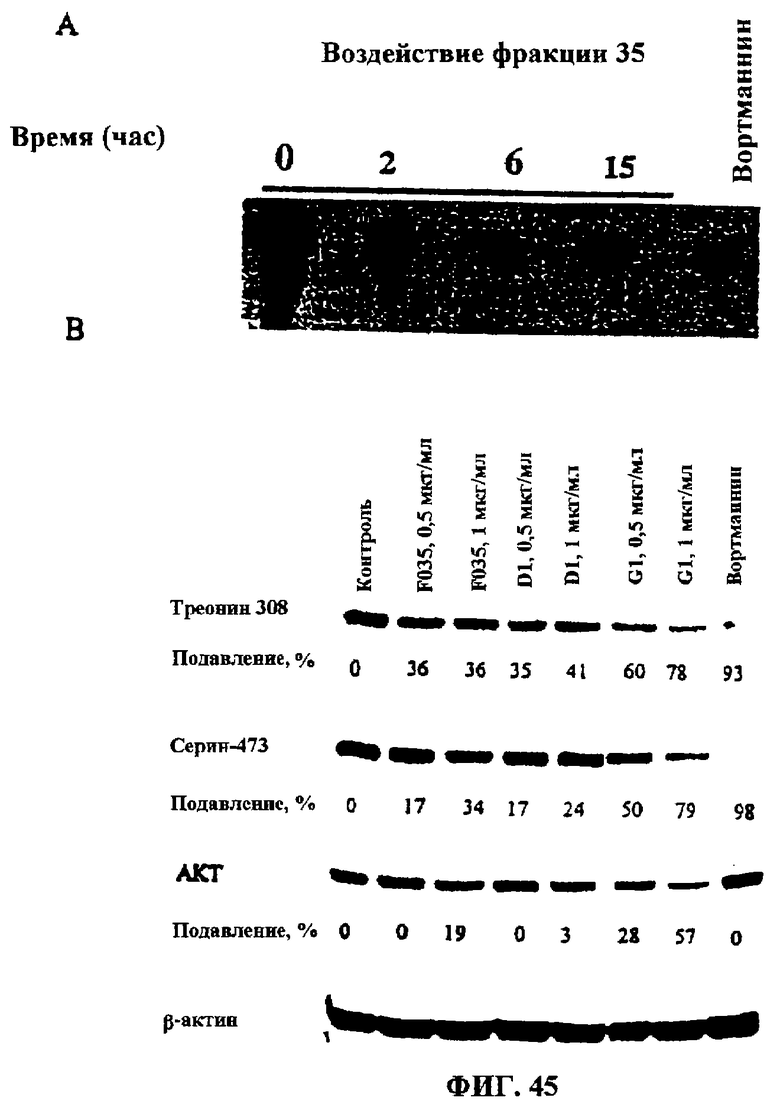
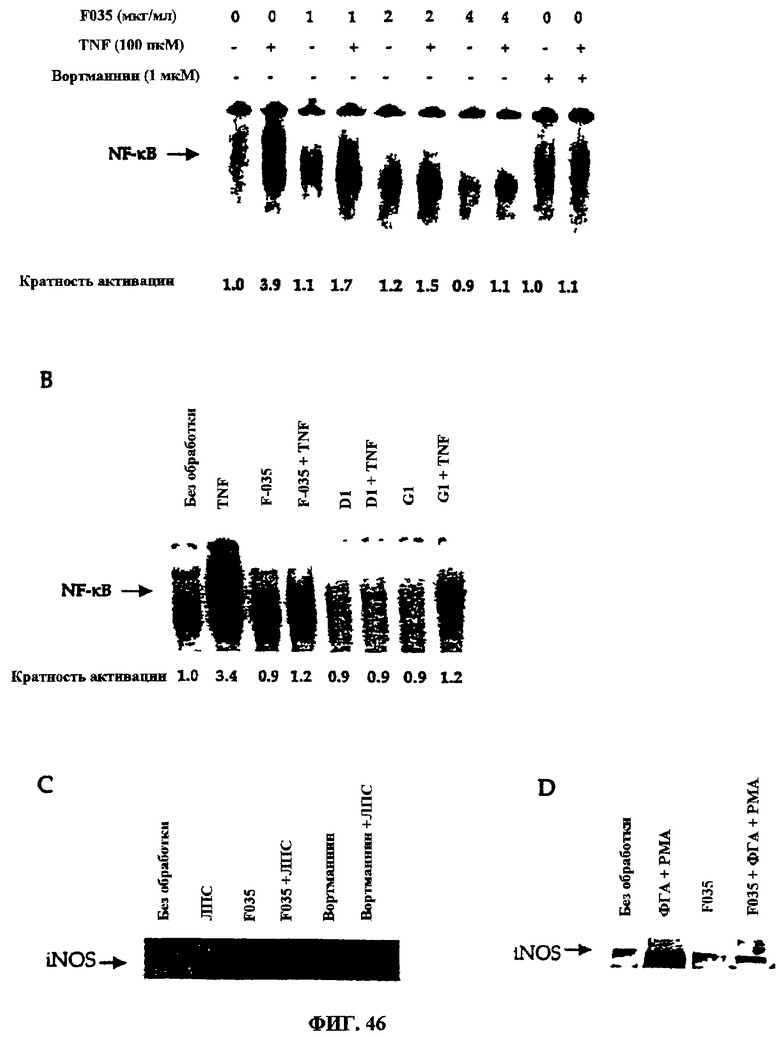


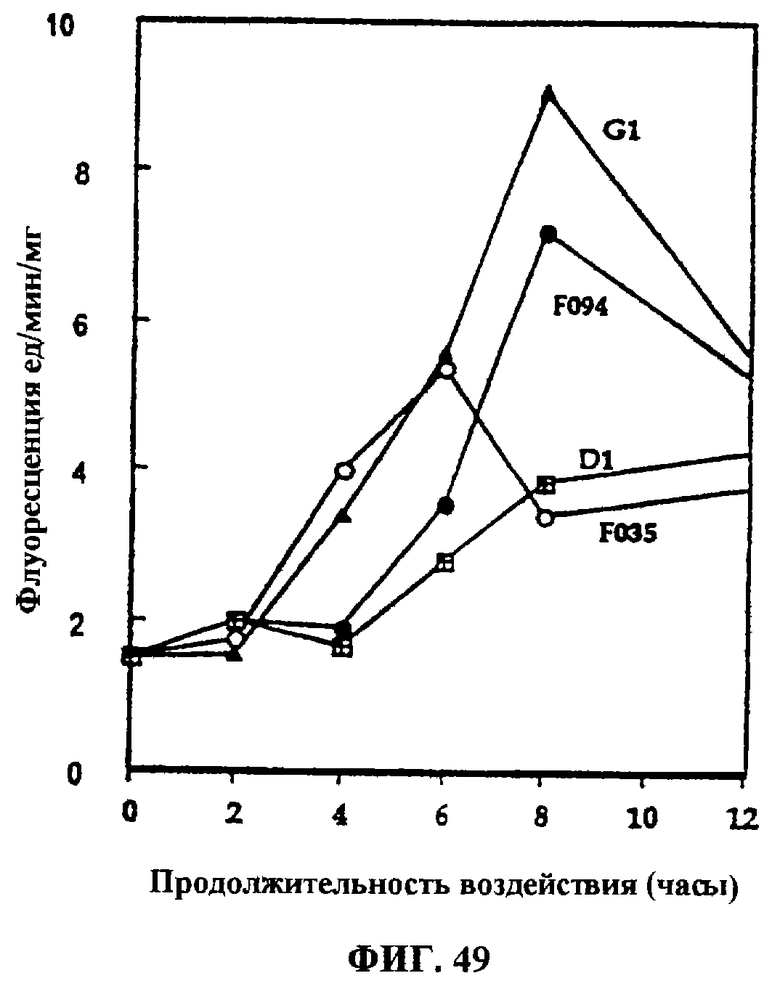
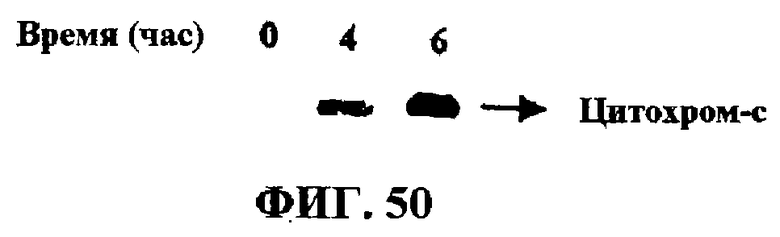
Изобретение представляет собой новые сапониновые смеси для ингибирования инициации и активации эпителиальной клетки млекопитающего в предзлокачественном или злокачественном состоянии, стимулирования апоптоза злокачественной клетки млекопитающего, предотвращения аномальной пролиферации эпителиальной клетки млекопитающего, лечения воспаления и регуляции ангиогенеза у млекопитающего, которые выделены из растений вида Acacia victoriae, и способы их применения. Эти соединения могут включать тритерпеновую составляющую, такую как акациевая или олеаноловая кислоты, к которой присоединены олигосахариды и монотерпеноидные составляющие. Технический результат: смеси и соединения обладают свойствами, связанными с регуляцией апоптоза и цитотоксичности клеток, и обладают сильным противоопухолевым действием по отношению к различным опухолевым клеткам. 17 н. и 26 з.п. ф-лы, 53 табл., 50 ил.
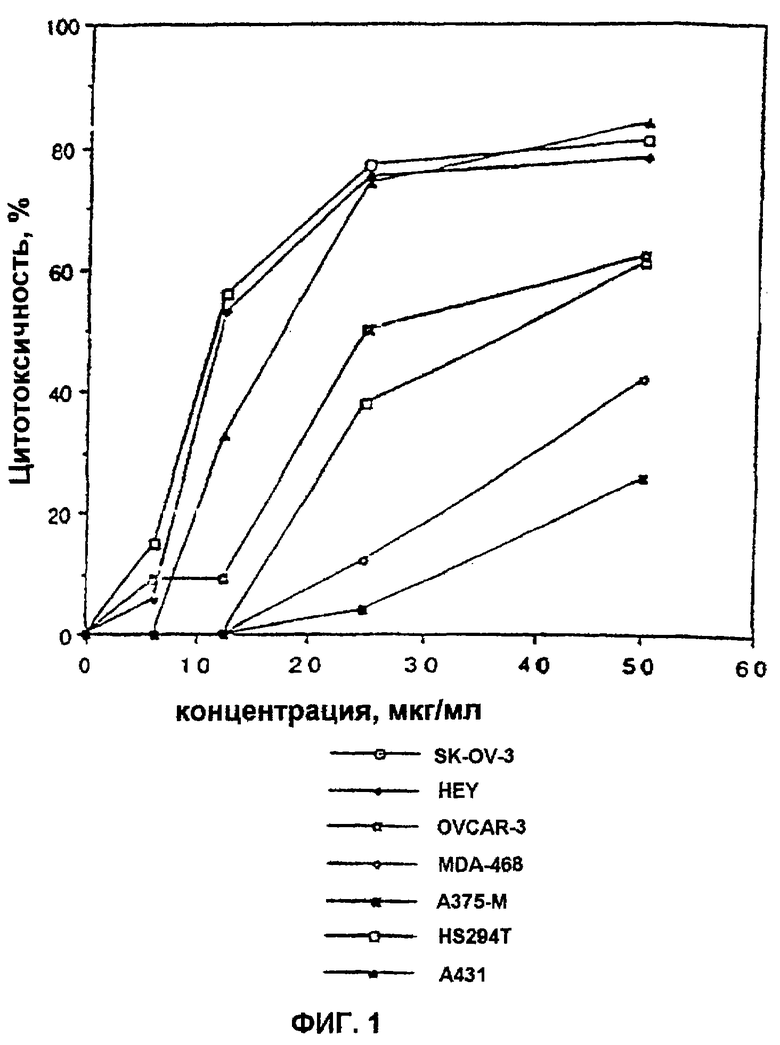
а) выделяемостью из тканей бобов или проростков Acacia victoriae;
b) содержанием, по крайней мере, одного тритерпенового гликозида, имеющего молекулярной массу примерно 1800 - 2600 а.е.м.;
с) способностью индуцировать цитотоксичность клетки Jurkat и
d) способностью индуцировать апоптоз клетки Jurkat.
а) выделяемостью из тканей бобов и проростков Acacia victoriae;
b) содержанием, по крайней мере, одного тритерпенового гликозида, имеющего молекулярной массу примерно 1800 - 2600 а.е.м. и
с) способность индуцировать высвобождение цитохрома-с из митохондрий клетки Jurkat.
а) приготовление тканевой культуры волосовидных корней, включающую клетки растения Acacia victoriae, которые были инфицированы Agrobacterium rhizogenes в соответствующей культуральной среде, и
b) экстракцию указанной композиции тритерпеновых гликозидов из указанной культуры растворителем, тем самым экстрагируя тритерпеновое гликозидное соединение, в котором тритерпеновая составляющая является акациевой кислотой, олеаноловой кислотой или иной структурно сходной тритерпеновой составляющей, имеющей формулу:
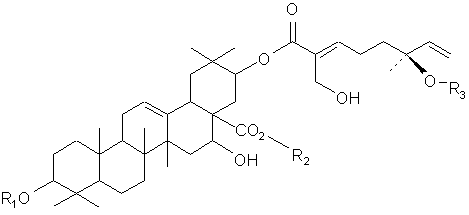 ,
,
где
а) R1 и R2 выбирают из группы, состоящей из водорода, С1-5-алкила и олигосахарида;
b) R3 выбирают из группы, состоящей из водорода, гидроксила, С1-5-алкила, С1-5-алкилена, С1-5-алкилкарбонила, сахара и монотерпеновой группы и
с) формула дополнительно включает R4, где R4 выбирают из группы, состоящей из водорода, гидроксила, С1-5-алкила, С1-5-алкилена, С1-5-алкилкарбонила, сахара, С1-5-алкилового сложного эфира и монотерпеновой группы, и где R4 может быть присоединен к тритерпеновой составляющей или монотерпеновой составляющей.
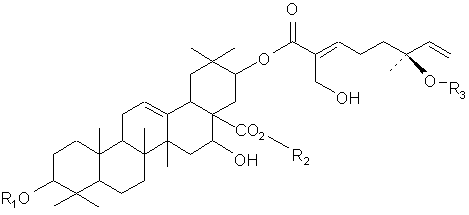 ,
,
где
а) R1 и R2 выбирают из группы, состоящей из водорода, С1-5-алкила и олигосахарида;
b) R3 выбирают из группы, состоящей из водорода, гидроксила, С1-5-алкила, С1-5-алкилена, С1-5-алкилкарбонила, сахара и монотерпеновой группы и
с) формула дополнительно включает R4, где R4 выбирают из группы, состоящей из водорода, гидроксила, С1-5-алкила, С1-5-алкилена, С1-5-алкилкарбонила, сахара, С1-5-алкилового сложного эфира и монотерпеновой группы, и где R4 может быть присоединен к тритерпеновой составляющей или монотерпеновой составляющей.
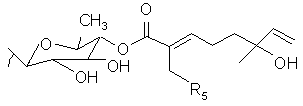
R5 выбирают из группы, состоящей из водорода, гидроксила, С1-5-алкила, С1-5-алкилена, С1-5-алкилкарбонила, сахара, С1-5-алкилового сложного эфира и монотерпеновой группы.
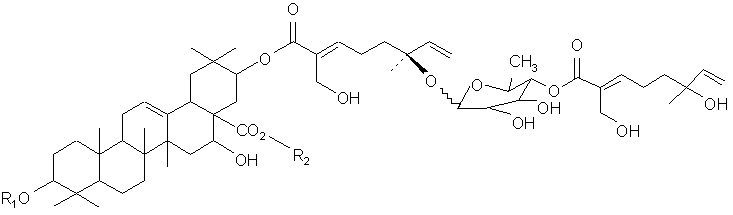
где
а) R1 представляет олигосахарид, включающий N-ацетилглюкозамин, фукозу и ксилозу, и
b) R2 представляет олигосахарид, включающий глюкозу, арабинозу и рамнозу.
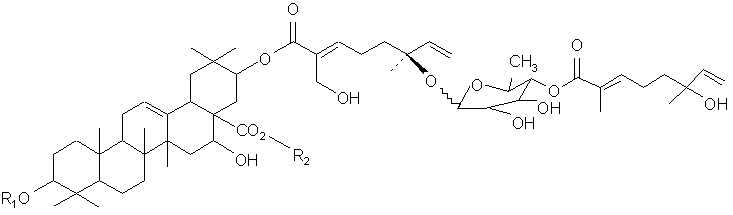
где
а) R1 представляет олигосахарид, включающий N-ацетилглюкозамин, фукозу и ксилозу, и
b) R2 представляет олигосахарид, включающий глюкозу, арабинозу и рамнозу.

где
а) R1 представляет олигосахарид, включающий N-ацетилглюкозамин, глюкозу, фукозу и ксилозу, и
b) R2 представляет олигосахарид, включающий глюкозу, арабинозу и рамнозу.
| WO 9701334 А1, 16.01.1997 | |||
| СРЕДСТВО ДЛЯ ПРОФИЛАКТИКИ И ПОСЛЕОПЕРАЦИОННОГО ЛЕЧЕНИЯ ЗЛОКАЧЕСТВЕННЫХ НОВООБРАЗОВАНИЙ (ЕГО ВАРИАНТЫ) | 1993 |
|
RU2027441C1 |
| WO 8800194 А1, 14.01.1988 | |||
| Устройство для испытаний образцов на изгиб при высоких температурах | 1982 |
|
SU1017960A1 |
| JP 6073084 А1, 15.03.1994 | |||
| Способ изготовления электрических сопротивлений посредством осаждения слоя проводника на поверхности изолятора | 1921 |
|
SU19A1 |
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |

