Уровень техники
Производные сложных эфиров 2-оксибензойной кислоты являются полезными исходными соединениями для синтеза природных продуктов, см., например, F.M.Hauser и др., Synthesis, 1980 г., стр.72, или для производства фунгицидных безнофенонов, описанных, например, в патенте США №5, 773, 663. Известны способы получения таких сложных эфиров 2-оксибензойной кислоты, см., например, G.Schill и др., Synthesis, 1980 г., стр.814, или Y.Hamada и др., Tetrahedron, том 47, 1991 г., стр.8635. Однако данные известные способы требуют несколько стадий, в них используют неустойчивые или токсичные реагенты, и они не пригодны для
осуществления в крупном масштабе или в условиях промышленного производства.
В двухстадийных способах, описанных в вышеуказанных источниках Synthesis и Tetrahedron, необходимо выделение промежуточных продуктов, что приводит к чрезмерной нагрузке окружающей среды отходящим растворителем. Далее, в данных способах используют газообразный хлористый водород и требуется отдельной стадии окисления с применением окислителей, таких как Вr2 или CuCl2.
Таким образом, задачей изобретения является разработка эффективного и производительного одностадийного способа получения сложных эфиров 5- и/или 6-замещенной 2-оксибензойной кислоты, пригодного для осуществления в крупном масштабе и в условиях промышленного производства.
Другой задачей изобретения является разработка эффективного способа получения сложного эфира замещенной 2-оксибензойной кислоты с высоким выходом при относительно умеренных условиях реакции с использованием легко доступных исходных соединений и реагентов.
Дальнейшей задачей изобретения является разработка возможности щадящего окружающую среду производства сложных эфиров замещенной 2-оксибензойной кислоты, предназначенных для получения важных фитопатогенных фунгицидных веществ и для дальнейшего изучения синтеза природных продуктов. Указанные и еще дальнейшие задачи и признаки изобретения более подробно описаны в нижеследующем.
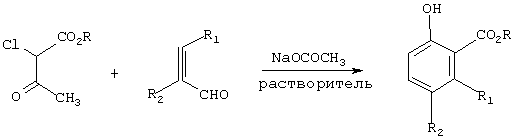
Настоящее изобретение касается одностадийного способа получения соединения формулы (I)
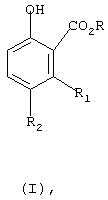
где
R означает С1-С6-алкил, и
R1 и R2 независимо означают водород или С1-С4-алкил,
который отличается тем, что соединение формулы (II)
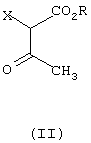
где R означает C1-C6-алкил,и
Х галоген или группа ОСОСН3,
подвергают взаимодействию с соединением формулы (III)
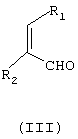
где
R1 и R2 независимо означают водород или С1-С4-алкил,
причем соединение формулы (III) является цис- или транс-изомером или их смесью, в присутствии соли С1-С4-карбоновой кислоты и в среде полярного растворителя.
Объектом изобретения являетя также применение соединения формулы (I) для получения фунгицидного бензофенонового соединения.
Сложные эфиры замещенной 2-оксибензойной кислоты формулы (I) являются ценными исходными соединениями при синтезе природных продуктов и для получения важных бензофеноновых фунгицидных агентов. Последние способствуют получению высококачественных продуктов питания и снабжению потребителей в США и во всем мире зерном. Практически все семена кукурузы и пшеницы и около одной трети соевых бобов в США обрабатывают фунгицидными средствами. По этой причине является крайне желательным иметь в распоряжении высокопроизводительный и щадящий окружающую среду способ получения таких обладающих фунгицидной активностью соединений.
В рамках настоящего изобретения было найдено, что сложные эфиры 5- и/или 6-замещенной 2-оксибензойной кислоты формулы (I) можно получать путем одностадийного способа, используя легко доступные исходные соединения, в относительно умеренных условиях реакции, причем данный способ пригоден для крупномасштабного промышленного производства. Преимущество предлагаемого способа заключается также в том, что не используются неустойчивый газообразный хлористый водород и окислители как, например, Вr2 и CuCl2.
Предпочтительно путем предлагаемого способа получают соединения формулы (I), в которых R1 означает С1-С4-алкил, и R2 означает водород. Особенно предпочтительными являются соединения формулы (I), в которых R1 означает метил и R2 означает водород.
Предпочтительными соединениями формулы (II), используемыми при осуществлении предлагаемого способа, являются соединения, в которых Х означает галоген. Особенно предпочтительными являются те соединения формулы (II), в которых Х означает хлор.
Соединения формулы (III) могут имеется в цис- или транс-конфигурации, или в виде их смеси. В данном описании и в формуле изобретения соединения формулы (III) включают цис- изомер, транс-изомер или их смесь.
Под понятием "галоген" в рамках настоящего описания и формулы изобретения подразумеваются хлор, бром, фтор или йод.
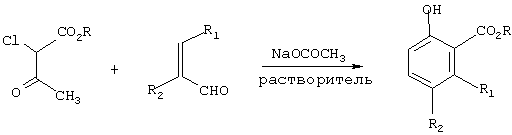
Согласно предлагаемому способу β-кетоэфир формулы (II) подвергают взаимодействию с α,β-ненасыщенным альдегидом формулы (III) в присутствии соли С1-С4-карбоновой кислоты, в количестве предпочтительно примерно от 1,0 до 2,0 мольных эквивалентов, предпочтительно примерно от 1,0 до 1,5 мольных эквивалента, в среде полярного растворителя, представляющего собой предпочтительно C1-C6-алканол, С1-С4-карбоновую кислоту или их смесь, наиболее предпочтительно метанол, этанол, уксусную кислоту или их смесь, для получения соединения формулы (I). Реакция изображена на схеме I, где М означает щелочной или щелочноземельный металл.
Схема I
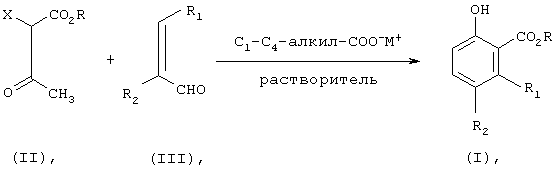
Получаемый сложный эфир оксибензойной кислоты формулы (I) можно выделять известными методами, например путем осаждения, декантирования, фильтрации, экстракции, хроматографического отделения или т.п., предпочтительно путем фильтрации или экстракции.
В предлагаемом способе скорость реакции непосредственно связана с температурой реакции, то есть скорость реакции повышается при повышении температуры. Однако чрезмерно высокая температура реакции может приводить к разложению и образованию нежелательных побочных соединений и, тем самым, к снижению выхода и чистоты. Пригодные температуры реакции для предлагаемого способа лежат между комнатной температурой и температурой флегмы растворителя и составляют предпочтительно примерно от 25°С до 125°С, особенно предпочтительно примерно от 75°С до 120°С.
Пригодными для применения в предлагаемом способе солями кислот являются соли алифатических кислот, предпочтительно соли щелочного или щелочно-земельного металла С1-С4-карбоновой кислоты, более предпочтительно соли щелочного металла уксусной кислоты, как, например, ацетат натрия или калия.
Пригодные растворители для осуществления предлагаемого способа представляют собой полярные растворители, например предпочтительно протонные растворители, как, например, С1-С6-алканолы, С1-С4-карбоновые кислоты или их смесь, более предпочтительно метанол, этанол, уксусная кислота или их смесь.
На практике β-кетоэфир фомрулы (II) и α,β-ненасыщенный альдегид формулы (III) смешивают примерно с 1,2-2,0, предпочтительно примерно 1,0-1,5, более предпочтительно примерно 1,2 мольными эквивалентами соли С1-С4-карбоновой кислоты, предпочтительно соли щелочного или щелочно-земельного металла, более предпочтительно ацетата щелочного металла, в среде полярного растворителя, предпочтительно протонного растворителя, более предпочтительно C1-С4-алканола, С1-С4-карбоновой кислоты или их смеси, при температуре от комнатной до температуры флегмы растворителя, предпочтительно при 25°С-125°С, более предпочтительно 75°С-120°С, причем в итоге получают желаемый сложный эфир 5- и/или 6-замещенной 2-оксибензойной кислоты.
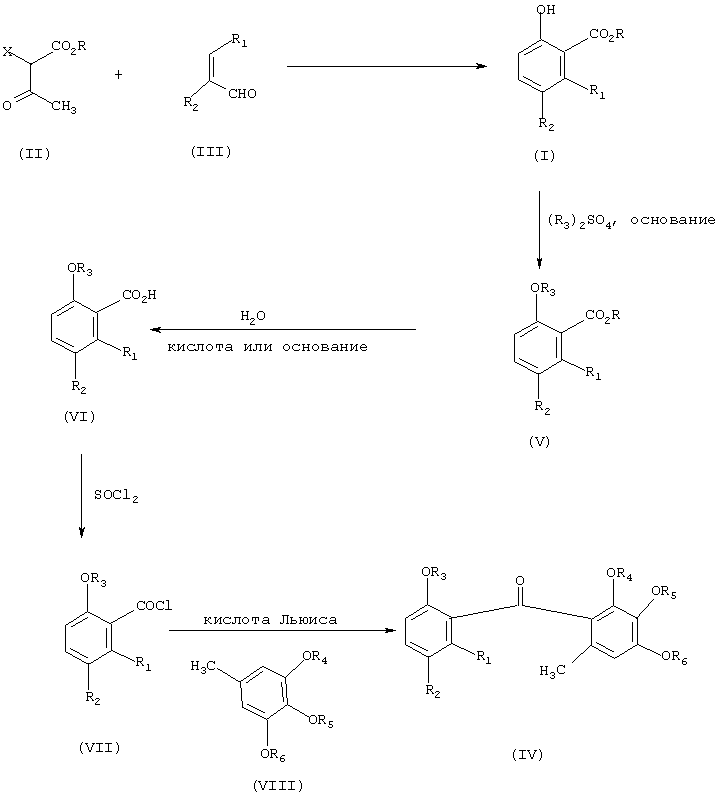
Соединения формулы (I) являются ценными промежуточными соединениями для синтеза природных продуктов и для производства бензофеноновых фунгицидных агентов. Следовательно, согласно одному аспекту изобретения соединение формулы (I), получаемое вышеописанным образом из соединений формулы (II) и (III) путем одностадийного способа, удобным методом можно превращать в бензофеноновое фунгицидное соединение формулы (IV), а именно путем алкилирования соединения формулы (I) ди-(С1-С6-алкил)сульфатом в присутствии основания с получением соответствующего алкоксипроизводного формулы (V); гидролиза производного формулы (V) в присутствии водной кислоты или водного основания с получением соответствующей карбоновой кислоты формулы (VI); взаимодействия соединения формулы (VI) с хлорирующим средством, например SOCl2, с получением хлорангидрида формулы (VII); и взаимодействия соединения формулы (VII) с соединением формулы (VIII) в присутствии Льюисовой кислоты, возможно в среде растворителя, с получением желаемого фунгицидного соединения формулы (IV). Реакция показана на схеме II, на которой R3, R4, R5 и R6 независимо означают C1-C6-алкил.
Соединения формулы (IV), их применение в фунгицидных средствах и способы получения соединений формулы (IV) описаны в патенте США №5, 773, 663 и в патентной заявке США №08/914,966, поданной 20 августа 1997, как ссылки включенных в объем данной заявки. Для лучшего понимания изобретения в нижеследующем приведены примеры. Данные примеры служат лишь для иллюстрации и никаким образом не огранчивают объем или сущность изобретения. ЯМР означает ядерную магнитно-резонансную спектроскопию, где не указано ничего другого, под частями подразумеваются весовые части.
Схема II
Пример 1
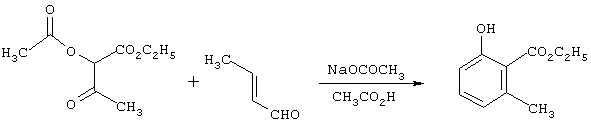
Получение этил-2-окси-6-метилбензоата
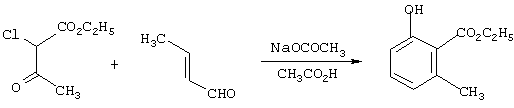
Смесь из 21,0 г (0,30 моль) кротональдегида и 25,0 г (0,30 моль) безводного ацетата натрия в ледяной уксусной кислоте в атмосфере азота нагревают до температуры флегмы, в течение 2,25 ч по каплям добавляют 41,1 г (0,25 моль) 95%-ного этилхлорацетоацетата, держат при температуре флегмы в течение 16 ч, охлаждают до комнатной температуры и концентрируют в вакууме с получением остатка. Последний распределяют между этилацетатом и водой. Органическую фазу разбавляют гексанами, промывают последовательно водой и водным NаНСО3 и концентрируют в вакууме с получением целевого продукта в виде масла чистотой 71,4% с выходом 41,0 г (65%) (анализ ЯМР).
Пример 2
Получение этил-2-окси-5-метилбензоата
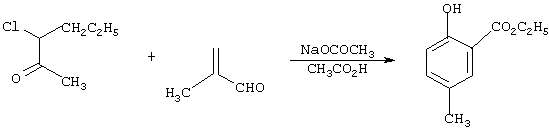
Смесь из 22,8 г (0,30 моль) 92%-ного 2-метилакролеина, 41,1 г (0,25 моль) 95%-ного этил-2-хлорацетоацетата и 24,6 г (0,30 моль) безводного ацетата натрия в уксусной кислоте нагревают с обратным холодильником в атмосфере азота в течение 16 ч, охлаждают до комнатной температуры и концентрируют в вакууме с получением остатка. Последний распределяют между этилацетатом и водой. Органическую фазу концентрируют в вакууме с получением целевого продукта в виде масла чистотой 71,9% с выходом 44,2 г (70,6%) (анализ по ЯМР).
Примеры 3-11
Получение сложных эфиров 5 и/или 6-замещенной 2-оксибензойной кислоты
По методу, подобному методу примеров 1 и 2, и с использованием пригодных кетоэфира и α,β-ненасыщенного альдегида, получают сложные эфиры 2-оксибензойной кислоты, указанные в нижеследующей таблице I.
Пример 12
Получение этил-2-окси-6-метилбензоата через 2-ацетоксиацетоацетат
Согласно данному примеру этил-2-ацетоксиацетоацетат получают путем нагревания с обратным холодильником смеси 95,5 г (0,58 моль) 95%-ного этил-2-хлорацетоацетата и 57,0 г (0,7 моль) ацетата натрия в уксусной кислоте в атмосфере азота в течение 16 ч и концентрации реакционной смеси в вакууме с получением остатка. Остаток распределяют между этилацетатом и водой. Органическую фазу разбавляют гексанами, последовательно промывают водой и водным NaHCO3 и концентрируют в вакууме, в результате чего получают 82 г масла, согласно данным ЯМР и анализа масс-спектра представляющего собой этил-2-ацетоксиацетоацетат.
19,0 г (0,1 моль теории) полученного таким образом этил-2-ацетоксиацетоацетата без дальнейшей очистки смешивают с 10,0 г (0,14 моль) кротональдегида и 3,0 г (0,036 моль) ацетата натрия в уксусной кислоте и нагревают с робратным холодильником в течение 16 ч. Реакционную смесь охлаждают до комнатной температуры и концентрируют в вакууме с получением остатка, который распределяют между этилацетатом и водой. Органическую фазу разбавляют гексанами, последовательно промывают водой и водным NаНСО3 и концентрируют в вакууме, в результате чего получают 16,6 г целевого соединения в виде масла (чистота 41%, выход 37,8%, анализ по ЯМР).
Пример 13
Получение 5-бром-2’,6-диметил-2,4’,5,6’-тетраметоксибензофенона
Стадия а) Этил-2-гидрокси-6-метилбензоат
Указанное в заголовке соединение получают аналогично примеру 1.
Стадия б) Этил-2-метокси-6-метилбензоат
В 10-литровый реакционный сосуд при температуре 10°С загружают 2262,2 г (10,08 молей) 25%-ного гидроксида калия. В течение 2 часов добавляют 1441,6 г (5,04 молей) нагретого до 40°С расплава продукта, полученного на стадии (а) с выходом приблизительно 63%, таким образом, чтобы температура внутри реакционного сосуда не поднималась выше 15°С. После этого в течение 2 часов при температуре 20°С добавляют 889,91 г (7,056 молей) диметилсульфата и затем в течение часа перемешивают при этой же температуре. После этого при температуре 60°С полученную смесь разделяют на фазы. Верхняя фаза содержит 1375,2 г (4,687 молей) указанного в заголовке продукта. Выход 66,2%.
Стадия в) 2-Метокси-6-метилбензойная кислота
В 10-литровый реакционный сосуд загружают 1375,2 г (4,687 молей) продукта, полученного на стадии (б), и при перемешивании добавляют 117,8 г (2,542 молей) этанола. Затем добавляют 1051,98 г (9,374 молей) 50%-ного гидроксида калия и реакционную смесь нагревают с обратным холодильником, после чего перегоняют с приблизительно 1900 г водно-этанольной смеси. Полученный остаток экстрагируют смесью 1406,14 г хлорбензола и 2062,34 г воды при температуре 50°С. Нижнюю фазу отделяют и в течение 3 часов при температуре 60°С добавляют 937,43 г концентрированной соляной кислоты (36-38%), доводя значение рН до приблизительно 1-2. Затем концентрированную кислоту отсасывают и отфильтрованный остаток промывают 585,89 г охлажденного до температуры -5°С хлорбензола и затем сушат при температуре 50°С в вакууме. В результате получают 740 г (4,452 молей) указанного в заголовке чистого продукта.
Стадия в’) 5 -Бром-2-метокси-6-метилбензойная кислота
700 г (4,212 молей) продукта, полученного на стадии (в), суспендируют в 2343,5 г хлорбензола и затем добавляют по каплям 707,2 г (4,423 молей) элементарного брома при постоянной температуре 15°С в течение 3 часов. После этого перемешивают в течение 2 часов при температуре 35°С. Затем перегоняют с 628,7 г хлорбензола при температуре 77-82°С и давлении 200 мбар, при этом избыток брома, а также НВr отгоняются из реакционного сосуда. В результате получают 980,5 г (4,0 моля=выход 95%) суспензии указанного в заголовке продукта в хлорбензоле. Соотношение 5-бром и 3-бромзамещенных производных составляет более 500:1.
Стадия г) 5-Бром-2-метокси-6-метилбензоилхлорид
Суспензию, полученную на стадии (в’), дополнительно разбавляют 754,8 г хлорбензола и охлаждают до температуры 50°С. После этого добавляют 0,95 г (0,013 молей) диметилформамида и затем постепенно в течение 1,5 часов добавляют 528,8 г (4,445 молей) тионилхлорида при температуре 50°С. После этого полученную смесь перемешивают в течение 1,5 часов при температуре 50°С. Затем перегоняют с 754,8 г хлорбензола при температуре 83-90°С и давлении 200 мбар, при этом отгоняются избыток тионилхлорида и остатки соляной кислоты и диоксида серы. В результате получают 1047 г (3,973 молей=выход 99,5%) указанного в заголовке продукта в виде раствора в хлорбензоле.
Стадия д) 5-Бром-2’,6-диметил-2,4’,5’,6’-тетраметоксибензофенон
Раствор 1047 г (3,973 молей) бензоилхлорида в 1715 г хлорбензола (см. стадию /д/) перемешивают с 0,72 г (0,0044 молей) обезвоженного хлорида трехвалентного железа и постепенно в течение 4 часов добавляют к раствору 868,7 г (4,768 молей) 3,4,5-триметокситолуола в 467,8 г хлорбензола при температуре 120°С. Затем полученную смесь перемешивают при этой же температуре в течение 2 часов. Для удаления НСl, образующегося во время добавления реагента и перемешивания, через реакционную смесь постоянно продувают азот. Затем хлорбензол отгоняют при температуре 80-105°С и давлении 80 мбар. Для кристаллизации указанного в заголовке продукта добавляют 4900 г метанола при 50°С и вносят нагретый до 105°С расплав. Затем охлаждают до -5°С и отделяют указанный в заголовке продукт центрифугированием, промывают метанолом и сушат (выход 98,3%).
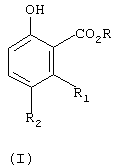
Изобретение относится к усовершенствованному способу получения соединения формулы (I)
где R означает C1-C6-алкил, и R1 и R2 независимо означают водород или С1-С4-алкил, причем соединение формулы (II)
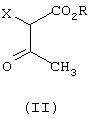
где R означает С1-С6-алкил, и Х - галоген или группа ОСОСН3, подвергают взаимодействию с соединением формулы (III)
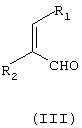
где R1 и R2 независимо означают водород или С1-С4-алкил, в присутствии соли С1-С4-карбоновой кислоты и в среде полярного растворителя. Способ позволяет получить целевое соединение в одну стадию с использованием доступных реагентов. Изобретение относится также к способу получения фунгицидного бензофенонового соединения формулы (IV)
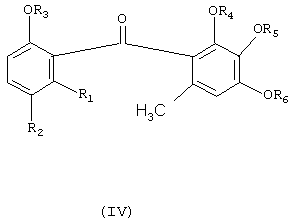
где R1 и R2 независимо означают водород или С1-С4-алкил, и R3, R4, R5 и R6 независимо означают C1-C6-алкил, с использованием соединения формулы (I). 2 н. и 9 з.п. ф-лы, 1 табл.
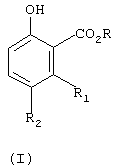
где R означает C1-C6-алкил, и
R1 и R2 независимо означают водород или С1-С4-алкил,
отличающийся тем, что соединение формулы (II)
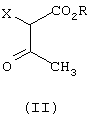
где
R означает С1-С6-алкил, и
Х галоген или группа ОСОСН3,
подвергают взаимодействию с соединением формулы (III)
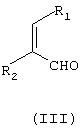
где R1 и R2 независимо означают водород или С1-С4-алкил,
в присутствии соли С1-С4-карбоновой кислоты и в среде полярного растворителя.
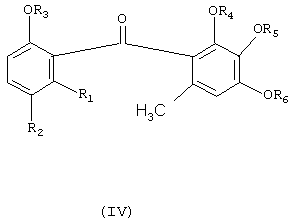
где
R1 и R2 независимо означают водород или С1-С4-алкил, и R3, R4, R5 и R6 независимо означают C1-C6-алкил,
отличающийся тем, что включает следующие стадии:
а) взаимодействие соединения формулы (II)
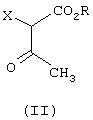
где
R означает С1-С6-алкил, и
Х - галоген или группа ОСОСН3,
с соединением формулы (III)
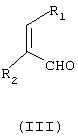
где
R1 и R2 имеют вышеуказанные значения,
в присутствии соли С1-С4-карбоновой кислоты и в среде полярного растворителя,
с получением соединения формулы (I)
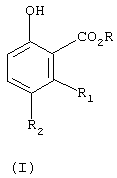
где
R, R1 и R2 имеют вышеуказанное значение;
б) алкилирование соединения формулы (I) с ди(С1-С6-алкил)сульфатом в присутствии основания, с получением соединения формулы (V)
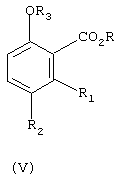
где
R3 означает C1-С6-алкил;
в) гидролиз соединения формулы (V) в водной кислоте или водном основании с получением соединения формулы (VI)
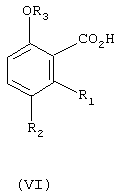
где
R1, R2 и R3 имеют вышеуказанное значение;
г) взаимодействие соединения формулы (VI) с тионилхлоридом с получением соединения формулы (VII)
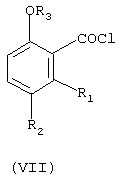
где
R1, R2 и R3 имеют вышеуказанное значение; и
д) взаимодействие соединения формулы (VII), по меньшей мере, с одним мольным эквивалентом соединения формулы (VIII)
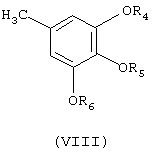
где
R4, R5 и R6 независимо означают C1-С6-алкил, в присутствии кислоты Льюиса, в случае необходимости в среде растворителя, с получением желаемого фунгицидного бензофенонового соединения формулы (IV).
| US 5773663 A, 30.06.1998 | |||
| Способ приготовления сложных эфиров салициловой кислоты | 1924 |
|
SU2744A1 |
| СПОСОБ ПОЛУЧЕНИЯ НОРМАЛЬНОГО И ИЗОАМИЛОВОГО ЭФИРОВ САЛИЦИЛОВОЙкислоты | 0 |
|
SU162122A1 |
| FRANK M | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Поглотительный сосуд для анализа газов | 1923 |
|
SU814A1 |

