ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Эктеинасцидин (Ecteinascidin) 743 (1, Et 743) является чрезвычайно сильным противоопухолевым агентом1, который получают из морских организмов, в настоящее время он проходит испытания в различных клиниках для лечения людей2. Так как это соединение недостаточно доступно при получении из природного источника, покрытого оболочкой Ecteinascidia turbinata, в промышленности его производят полностью синтетическим путем, описанным в 1996 г.3 Недавно обнаружено, что структурный аналог Et 743, соединение 2 (фталасцидин (phthalascidin), Pt 650) проявляет противоопухолевую активность, по существу не отличимую от активности соединения 14.

Как соединение 1, так и соединение 2 синтезируют из фрагментов 3 и 43 через общее пятициклическое промежуточное соединение 5.

Синтез соединения 5 из фрагментов 3а и 4 впервые был осуществлен способом3, включающим 6 стадий с общим выходом 35% (средний выход на одной стадии составляет примерно 84%). Так как в промышленности соединения 1 и/или 2 из экономических соображений со временем придется получать в количестве многих килограммов, ставилось задачей найти другой, более эффективный и воспроизводимый путь получения соединения 5 из фрагментов 2 и 3.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Один из вариантов воплощения настоящего изобретения таким образом относится к новому способу синтеза промежуточного соединения 5, который проще осуществить, чем первоначальный способ, и который состоит из шести стадий превращения (3b3+4) в 5 с общим выходом 57% (средний выход на одной стадии почти 92%). Этот предпочтительный способ синтеза пятициклического соединения 5 представлен ниже на схеме 15.
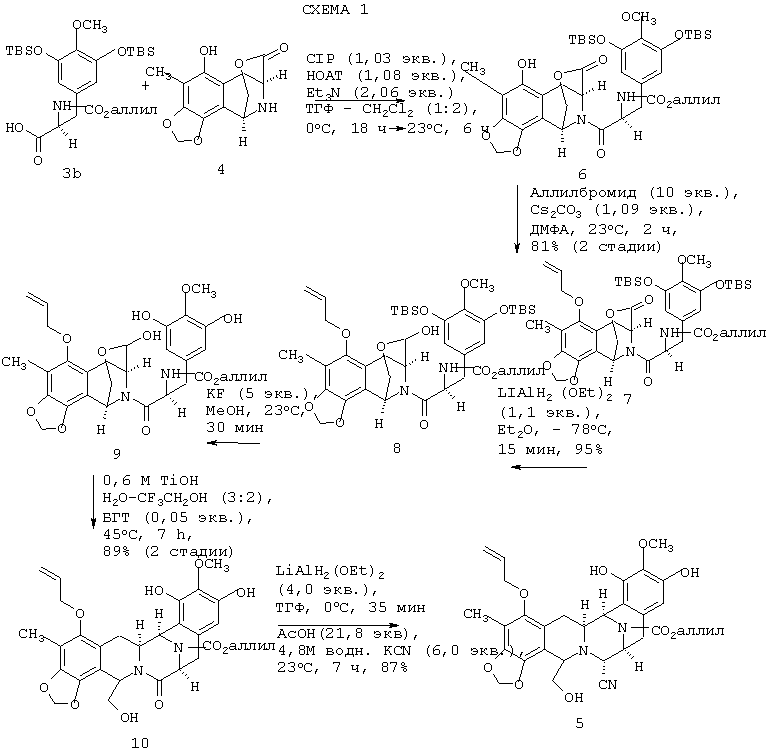
Второй предпочтительный вариант осуществления данного изобретения относится к новому синтетическому способу преобразования пятициклического соединения 5 во фталасцидин 2, которое протекает гладко и с отличным выходом (средний выход на одной стадии составляет 90.8%). Этот способ представлен ниже на Схеме 2.

ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
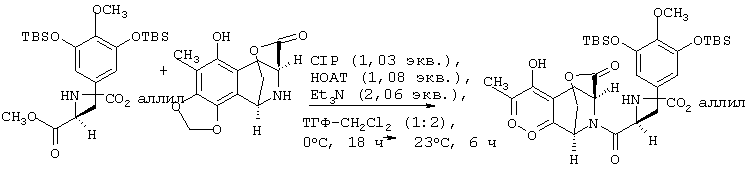
Как показано выше на схеме 1, раствор предварительно азеотропно высушенного (С7Н8-ТГФ) аминолактона 4 в тетрагидрофуране (ТГФ) при 0°С обрабатывают по каплям ацилирующим реагентом, полученным из кислоты 3b3 (1,03 экв.), 1-гидрокси-7-азабензотриазола (НОАТ, 1,08 экв.), 2-хлор-1,3-диметилимидазолидиний гексафторфосфата (CIP, 1,03 экв.) и триэтиламина (2,06 экв.) в растворе СН2Cl2 при 0°С.
Продукт присоединения 6, выделенный экстракцией, аллилируют без дополнительной очистки путем обработки избытком аллилбромида и 1,09 экв. Cs2СО3 в ДМФА при 23°С с получением амида 7 с общим выходом 81% в расчете на 3а и 4 после флеш-хроматографии на силикагеле.
Селективное восстановление лактонной группы соединения 7 в соответствующую лактольную группу с образованием соединения 8 проводят взаимодействием с 1,1 экв. литийдиэтоксиалюминийгидрида (LiAlH2(OEt)2) в эфире при -78°С в течение 15 мин с выходом 95%7,8. Десилилирование соединения 8 с образованием 9 и циклизация 9 (без очистки) с использованием 0,6М трифторметансульфокислоты в смеси 3:2 Н2О-CF3СН2ОН при 45°С в течение 7 ч дают пятициклическое соединение 10 с общим выходом 89% в расчете на лактол 8.
И, наконец, лактамную группу соединения 10 можно чисто восстановить путем обработки 4 экв. (LiAlH2(OEt)2) в ТГФ при 0°С в течение 35 мин до соответствующего циклического аминаля, который при действии HCN дает пятициклический аминонитрил 5 с общим выходом 87% из 10 после флеш-хроматографии на силикагеле9.
Синтез соединения 5, показанный на схеме 1 и описанный выше, имеет преимущества по сравнению с первоначально использовавшимся путем синтеза3 не только из-за существенно более высокого общего выхода (57% по сравнению с 35%), но также из-за простоты и воспроизводимости отдельных стадий, особенно амидного связывания (2а+3→6) и внутримолекулярной циклизации Пикте-Шпенглера (Pictet-Spengler) (9→10).
Кроме того, нет трудностей ни при очистке соединений, ни при увеличении масштаба используемых веществ.
Решающим элементом для успешного осуществления последовательности реакций, показанной на схеме 1, является высокая эффективность и избирательность LiAlH2(OEt)2 на двух стадиях восстановления 8→9 и 10→5, которая позволяет предположить, что этот реагент можно с успехом использовать в синтезе намного чаще, чем он использовался ранее.
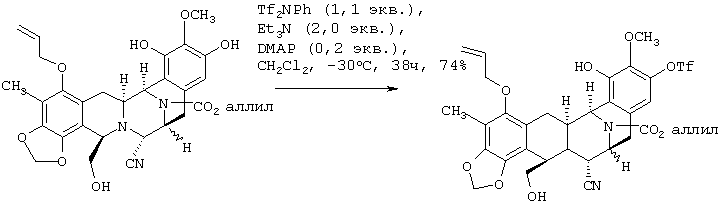
Согласно схеме 2, пятициклический триол 5 сначала преобразуют в фенол монотрифлат 11 (стадия не показана) путем обработки 1,1 экв. с PhNTf2 (реагент McMurry), 2 экв. Et3N и 0,2 экв. 4-диметиламинопиридина в СН2Cl2 при -30°С в течение 38 ч (74%). Преобразование 11 в моно-трет-бутилдиметилсилиловый (TBS) эфир 12 и этерификация с метоксиметилхлоридом (MOMCl) дают соединение 13 с высоким выходом.
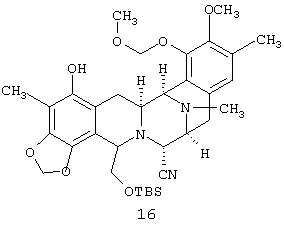
Отщепление N-аллилоксикарбонильной и O-аллильной групп в соединении 13 дает вторичный амин 14 (94%), который N-метилируют до соединения 15 и С-метилируют 15 до 16. Ацетилированием фенола 16 получают соответствующий ацетат 17, который при десилилировании дает первичный спирт 18. Замещение по Мицунобу (Mitsunobu) первичного гидроксила у 18 приводит к фталимиду 19, который при катализируемом кислотой отщеплении метоксиметильного эфира дает чистый фталасцидин 2.
Так как первоначальный путь синтеза Et 743 (1) оказался применимым для крупномасштабных синтезов, можно ожидать, что усовершенствованный способ, описанный в данной заявке, будет даже более полезным, поскольку относится к новому синтезу фталасцидина (2)4. Фталасцидин может оказаться более удобным для практики лечебным средством, чем эстеинасцидин 743, т.к. он более устойчив и его значительно легче получить.
Данное изобретение далее иллюстрируется следующими примерами, которые помогают понять данное изобретение, но которые не должны рассматриваться как ограничивающие. Все процентные количества, указанные в данной заявке, являются массовыми процентами, если не оговорено особо. Все температуры выражены в градусах Цельсия.
Пример 1

Кислоту (224 мг, 0,400 ммоль) растворяют в перегнанной уксусной кислоте (5,0 мл) и 0,2 н HCl10 (1,5 мл) и нагревают до 110°С. Через 5,5 ч реакционную смесь концентрируют в вакууме, сушат повторяющимся азеотропным концентрированием с толуолом в вакууме (3×10 мл) и растворяют в ДМФ (1,0 мл). Прибавляют трет-бутилдиметилсилилхлорид (304 мг, 2,03 ммоль) и имидазол (152 мг, 2,24 ммоль) в виде твердых веществ, и смесь перемешивают при 23°С в течение 2 ч. Реакционную смесь гасят раствором 2:1 уксусной кислоты в воде (1,5 мл) и перемешивают в течение 30 мин. Реакционную смесь выливают в 0,5 М водный раствор щавелевой кислоты (100 мл) и экстрагируют смесью 3:7 этилацетат-гексан (2×100 мл). Объединенные органические слои промывают насыщенным водным раствором хлорида натрия (100 мл), сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Остаток очищают флеш-хроматографией на колонке (100 мл силикагеля, градиентное элюирование смесями 1:1 этилацетат-гексан → 0,1% уксусная кислота-этилацетат) с получением продукта в виде практически чистого прозрачного вязкого масла (204,6 мг, 95%).
Rf 0,10 (этилацетат); 1H ЯМР (400 МГц, CDCl3) δ 10,25 (уш.с, 1H), 6,32 (с, 2Н), 5,90 (ддт, J=17,0, 10,6, 5,4 Гц, 1Н), 5,28 (д, J=17,1 Гц, 1H), 5,20 (д, J=10,4 Гц, 1Н), 5,11 (д, J=8,0 Гц, 1H), 4,61-4,57 (м, 1H), 4,55 (д, J=5,5 Гц, 2Н), 3,70 (с, 3Н), 3,04 (дд, J=14,0, 5,1 Гц, 1H), 2,93 (дд, J=14,0, 6,4 Гц, 1H), 0,99 (с, 18Н), 0,15 (с, 12Н); 13С ЯМР (101 МГц, CDCl3) δ 176,3, 155,7, 149,9, 142,2, 132,5, 130,5, 118,0, 115,6, 66,1, 60,0, 54,5, 37,2, 25,8, 18,4, -4,6. ИК-спектр (с Фурье-преобразованием, записан в тонком слое): 3438 (ср), 3331 (ср), 3088 (ср, оч.ш), 2956 (с), 2931(с), 2894(с), 2863(с), 1719(с), 1578(с), 1496(с), 1435(с), 1361(с), 1253 (с), 1231 (с), 1093 (с), 1010 (ср), 938 (сл), 831 (с) см-1. Анализ методом ВЭЖХ проводят после преобразования в производные с использованием диазометана для получения метилового эфира (ChiralPak AD, 1% изопропанола в гексане, скорость потока элюента: 1,0 мл/мин, λ=226 нм), 96% эи, RT=11,1 мин (осн.), 9,2 мин (незначит.). Масс-спектр высокого разрешения (FAB), [m+H]/z вычислено для C26H46O7NSi2: 540,2813; Найдено: 540,2823; [α]D 23+18,8° (с 1,0, метиленхлорид).
Пример 2
Амин (100.0 мг, 0,380 ммоль) сушат путем азеотропного концентрирования в вакууме со смесью 2:3 ТГФ-толуол (5 мл), растворяют в ТГФ (1,5 мл) и охлаждают до 0°С. Кислоту (211,7 мг, 0,392 ммоль) и 1-гидрокси-7-азабензотриазол (55,8 мг, 0,410 ммоль), помещенные в другую колбу, сушат азеотропным концентрированием в вакууме со смесью ТГФ-толуол (2:3) (5 мл) и растворяют в метиленхлориде (1,5 мл). В эту же колбу добавляют твердый 2-хлор-1,3-диметилимидазолидиний гексафторфосфат (103,9 мг, 0,392 ммоль), шприцем вводят триэтиламин (109 мкл, 0,782 ммоль) и получают прозрачный темно-желтый раствор. Полученную смесь перемешивают при 23°С в течение 3 мин, затем охлаждают до 0°С и переносят с помощью канюли в колбу с амином. Для переноса остатков в колбу используют хлористый метилен (1,5 мл). Золотистый раствор перемешивают при 0°С в течение 18 ч, нагревают до 23°С и дополнительно перемешивают еще в течение 6 ч. Реакционную смесь разбавляют этилацетатом (6 мл) и частично концентрируют в вакууме для удаления метиленхлорида. Полученный раствор выливают в 0,5 М водный раствор уксусной кислоты (100 мл), экстрагируют 3:7 смесью этилацетат-гексан (100 мл) и промывают насыщенным водным раствором бикарбоната натрия (100 мл). Водные слои повторно экстрагируют смесью 3:7 этилацетат-гексан (100 мл) и объединенные органические экстракты сушат над сульфатом натрия, фильтруют и концентрируют в вакууме с получением прозрачной пленки (˜300 мг). Этот остаток далее используют без дополнительной очистки. Продукт можно очистить флеш-хроматографией на колонке (100 мл силикагеля, градиентное элюирование смесями 1:3→2:3 этилацетат-гексан), однако выход в этом случае составляет только 50%, преимущественно из-за того, что силикагель способствует разложению продукта.
Rf 0,36 (2:3 этилацетат-гексан); 1H ЯМР (400 МГц, CDCl3) δ (смесь ротамеров карбамата и амида): 6,35 (с, 1Н), 6,20 (с, 1Н), 5,91-5,81 (м, 3Н), 5,94-5,59 (м, 1,5Н), 5,42 (д, J=3,3 Гц, 0,5Н), 5,30-5,03 (м, 3Н), 4,74-4,63 (м, 1Н), 4,60 (дд, J=10,8, 3,1 Гц, 0,5Н), 4,53 (уш.с, 1Н), 4,45 (д, J=5,1 Гц, 1Н), 4,36 (д, J=10,6 Гц, 0,5Н), 4,19 (д, J=10,6 Гц, 0,5Н), 3,68 (с, 1,5Н), 3,61(с, 1,5Н), 3,56 (д, J=8,4 Гц, 0,5Н), 3,03-2,90 (м, 3Н), 2,79 (дд, J=13,0, 4,6 Гц, 0,5Н), 2,24 (д, J=16,0 Гц, 0,5Н), 2,06 (уш.с, 3Н), 0,99 (с, 9Н), 0,91 (с, 9Н), 0,15 (с, 6Н), 0,06 (с, 6Н); 13С ЯМР (101 МГц, CDCl3) δ (смесь ротамеров карбамата и амида): 169,2, 169,0, 167,9, 167,5, 155,6, 155,2, 150,1, 149,7, 146,9, 146,5, 145,1, 145,0, 142,1, 141,7, 136,8, 136,2, 132,4, 132,3, 130,7, 130,3, 117,9, 117,8, 115,5, 115,3, 111,3, 110,9, 110,5, 108,1, 107,8, 101,4, 73,0, 66,1, 65,9, 60,0, 59,9, 54,7, 52,2, 51,9, 51,0, 47,6, 43,2, 39,6, 38,5, 29,1, 27,4, 25,8, 25,7, 18,4, 18,3, 8,9, -4,53, -4,56, -4,65, -4,74. ИК-спектр (с Фурье-преобразованием, записан в тонком слое): 3406 (сл, ш), 3319 (сл, ш), 2956 (ср), 2931(ср), 2894(сл), 2856(ср), 1725(ср), 1644(ср), 1575 (ср), 1494(ср), 1463(ср), 1431 (с), 1356 (сл), 1231 (с), 1163 (сл), 1094 (с), 1044 (ср), 1013 (ср), 831(с) см-1. Масс-спектр высокого разрешения (ESI), [m+H]/z вычислено для С39Н57O11N2Si2: 785.3501; Найдено: 785.3469; [α]D 24+20.5° (с 1.0, хлороформ).
Пример 3

Фенол (˜300 мг, 0,380 ммоль) сушат в вакууме азеотропным концентрированием с толуолом (5 мл) и растворяют в ДМФ (15 мл). К раствору шприцем прибавляют аллилбромид (330 мкл, 3,82 ммоль) и твердый карбонат цезия (134,7 мг, 0,413 ммоль), осторожно высушенный в вакууме на открытом пламени, и реакционную смесь перемешивают при 23°С в течение 2 ч. Реакционную смесь выливают в воду (300 мл), экстрагируют смесью 1:4 этилацетата и гексана (2×150 мл), промывают насыщенным водным раствором хлорида натрия (100 мл), сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Остаток очищают флеш-хроматографией на колонке (75 мл силикагеля, градиентное элюирование смесями 1:4 → 3:7 этилацетат-гексан,) с получением желаемого продукта в виде практически чистой прозрачной пленки (252,9 мг, общий выход для 2 стадий 81%). Обнаружено, что этот продукт тоже неустойчив по отношению к силикагелю, поэтому решающим фактором для его получения с указанным выходом является быстрое хроматографирование.
Rf 0,47 (2:3 этилацетат-гексан). 1H ЯМР (400 МГц, CDCl3) δ (смесь ротамеров карбамата и амида): 6,35 (с, 1Н), 6,20 (с, 1Н), 6,03-5,78 (м, 5Н), 5,52-5,44 (м, 1,4Н), 5,38-5,33 (м, 1Н), 5,31-5,13 (м, 3,6Н), 4,73-4,59 (м, 1,4Н), 4,55 (д, J=5,1 Гц, 1Н), 4,48 (д, J=5,1 Гц, 1Н), 4,34 (д, J=10,6 Гц, 0,6Н), 4,24-4,04 (м, 3Н), 3,68 (с, 1,5Н), 3,60 (с, 1,5Н), 3,54 (д, J=8,8 Гц, 0,4Н), 3,15-2,90 (м, 2,6Н), 2,77 (дд, J=12,8, 4,8 Гц, 0,6Н), 2,34 (м, 0,4Н), 2,12 (с, 1,5), 2,09 (с, 1,5), 0,99 (с, 9Н), 0,92 (с, 9Н), 0,16 (с, 6Н), 0,07 (с, 3Н), 0,05 (с, 3Н); 13С ЯМР (101 МГц, CDCl3) δ (смесь ротамеров карбамата и амида): 169,2, 168,8, 167,4, 167,1, 155,5, 155,1, 150,2, 150,0, 149,8, 145,5, 145,3, 142,2, 141,9, 139,2, 138,8, 133,4, 133,2, 130,6, 130,2, 118,3, 118,0, 117,9, 117,8, 117,7, 117,2, 115,5, 115,2, 114,3, 113,9, 111,1, 110,9, 101,8, 101,7, 73,8, 73,7, 72,8, 66,1, 65,9, 60,04, 59,99, 54,9, 52,1, 51,9, 51,1, 47,7, 43,3, 39,8, 38,5, 29,6, 27,9, 25,8, 25,7, 18,4, 18,3, 9,6, 9,4, -4,51, -4,54, -4,6, -4,7. ИК-спектр (с Фурье-преобразованием, записан в тонком слое): 3306 (сл, ш), 2956 (ср), 2931(ср), 2898 (ср), 2856(ср), 1750(ср), 1719(ср), 1650 (ср), 1575 (ср), 1494 (ср), 1431(с), 1363 (ср), 1250(ср), 1231 (ср), 1163 (сл), 1094(с), 1044 (ср), 1013 (ср), 944 (сл), 919 (сл), 831 (с) см-1. Масс-спектр высокого разрешения (ESI), [m+H]/z вычислено для C42H61O11N2Si2: 825.3814; Найдено: 825.3788; [α]D 24+21.7° (с 1.0, хлороформ).
Пример 4
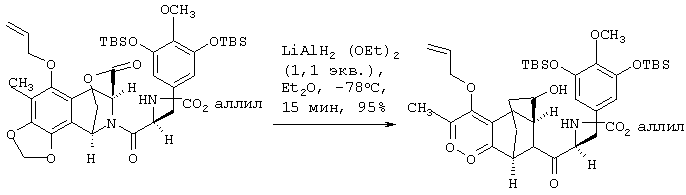
Лактон (354,1 мг, 0,429 ммоль) сушат в вакууме азеотропным концентрированием с толуолом (10 мл), растворяют в диэтиловом эфире (8,0 мл) и охлаждают до -78°С на бане с сухим льдом и ацетоном. 0,10 М Раствор LiAlH2(OEt)2 (4,7 мл, 0,47 ммоль)11 прибавляют по каплям по боковой стенке колбы в течение 2 мин. Реакционную смесь перемешивают при -78°С в течение 15 мин и затем при энергичном перемешивании выливают образующийся светло-желтый раствор в 0,1н раствор HCl (50 мл) при 0°С. Полученный раствор экстрагируют диэтиловым эфиром (2×75 мл), промывают насыщенным водным раствором хлорида натрия (50 мл), сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Остаток очищают флеш-хроматографией на колонке (150 мл силикагеля, градиентное элюирование смесями этилацетат-гексан, 3:7→2:3→1:1) с получением желаемого продукта в виде практически чистой прозрачной пленки (339,0 мг, 95%).
Rf 0,20 (2:3 этилацетат-гексан); 1H ЯМР (400 МГц, CDCl3) δ (смесь аномеров, ротамеров карбамата и амида): 6,43 (с, 0,2Н), 6,37 (с, 0,2Н), 6,16 (с, 1,4Н), 6,15 (с, 0,2Н), 6,05-5,80 (м, 4,4Н), 5,82-5,59 (м, 1,2Н), 5,41-5,14 (м, 3,8Н), 5,07-4,95 (м, 1,5Н), 4,85-4,76 (м, 1,6Н), 4,61-4,46 (м, 2,2Н), 4,26-4,41 (м, 3,8Н), 4,10-3,75 (м, 0,5Н), 3,68 (с, 0,3Н), 3,66 (с, 0,3Н), 3,63 (с, 2,4Н), 3,37 (д, J=11,0 Гц, 0,8Н), 3,33-2,94 (м, 0,7 Н), 2,90-2,65 (м, 2,8 Н), 2,35 (дд, J=17,7, 7,5 Гц, 0,7Н), 2,12 (с, 0,3Н), 2,11(с, 0,3Н), 2,09 (с, 2,4Н), 1,05 (с, 4,5Н), 0,92(с, 13,5Н), 0,16 (с, 3Н), 0,07 (с, 4,5Н), 0,04 (с, 4,5Н); 13С ЯМР (101 МГц, CDCl3) δ (смесь аномеров, ротамеров карбамата и амида): 169,8, 156,1, 149,6, 144,6, 141,7, 138,3, 133,8, 132,2, 130,2, 118,9, 118,0, 116,7, 115,1, 113,4, 112,5, 101,3, 93,4, 73,2, 66,2, 62,4, 60,1, 53,6, 51,4, 44,6, 38,5, 26,5, 25,9, 25,8, 18,5, 18,3, 9,5, -4,56, -4,59. ИК-спектр (с Фурье-преобразованием, записан в тонком слое): 3406 (ср, ш), 3325 (ср, ш), 2956 (ср), 2931(ср), 2894 (ср), 2856(ср), 1714(ср.), 1644(ср), 1578 (ср), 1496(ср), 1433(с), 1360 (ср), 1255(ср), 1234 (ср), 1095 (с), 1044 (ср), 1013 (ср), 941 (сл), 830 (с) см-1. Масс-спектр высокого разрешения (ESI), [m+H]/z вычислено для C42H63O11N2Si2: 827,3970. Найдено: 827,4009; [α]D 25 -1,5°(с 1,0, хлороформ).
Пример 5

Лактол (316,3 мг, 0,382 ммоль) растворяют в метаноле (3,8 мл), который был продут азотом. Безводный фторид калия (110,3 мг, 1,90 ммоль) прибавляют в виде твердого вещества и сосуд насыщают/продувают азотом. Реакционную смесь перемешивают при температуре 23°С в течение 30 мин и полученную светло-розовую смесь разбавляют толуолом (5 мл) и концентрируют в вакууме. Остаток растворяют в 2,2,2-трифторэтаноле (15 мин), который был продут азотом, и прибавляют твердый бутилированный гидрокситолуол (4,3 мг, 0,02 ммоль). В колбу загружают 1,0 М водный раствор трифторметансульфоновой кислоты12 (23 мл), и сосуд снова насыщают/продувают азотом. Раствор перемешивают при 45°С на масляной бане в течение 7 ч. Смесь частично концентрируют в вакууме для удаления спирта и выливают в 80% насыщенный водный раствор хлорида натрия (100 мл), экстрагируют этилацетатом (2×100 мл), промывают насыщенным водным раствором хлорида натрия (50 мл), сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Остаток очищают флеш-хроматографией на колонке (100 мл силикагеля, 5:95 метанол-метиленхлорид) с получением желаемого продукта в виде практически чистого белого твердого вещества (198,5 мг, 89%). Кристаллы получают из толуола.
Т.пл. 130°С (с разл.); Rf 0,11 (5:95 метанол-метиленхлорид); 1Н ЯМР (400 МГц, ацетон-d6) δ (смесь ротамеров карбамата): 8,34 (уш.с, 1Н), 8,32 (уш.с, 1Н), 6,31 (д, J=4, 4 Гц, 1Н), 6,14 (м, 1Н), 5,97 (с, 1Н), 5,97-5,90 (м, 1Н), 5,90 (с, 1Н), 5,68 (м, 1Н), 5,42-5,37 (м, 2Н), 5,31-5,22 (м, 2Н), 5,18-5,12 (м, 1Н), 4,85 (д, J=6, 6 Гц, 1Н), 4,65-4,55 (м, 2Н), 4,38-4,34 (м, 1Н), 4,26-4,22 (м, 1Н), 3,89-3,86 (м, 1Н), 3,77 (с, 3Н), 3,71 (с, 1Н), 3,57 (д, J=15,0 Гц, 1Н), 3,48-3,43 (м, 1Н), 3,25-3,13 (м, 2Н), 3,00 (д, J=16,8 Гц, 1Н), 2,34 (м, 1Н), 2,11 (с, 3Н); 13С ЯМР (101 МГц, ацетон-d6) δ (смесь ротамеров карбамата): 169,4, 169,2, 153,8, 153,7, 150,6, 149,3, 148,2, 148,0, 145,5, 141,0, 135,1, 134,5, 133,9, 130,2, 130,1, 122,4, 117,9, 117,8, 117,7, 117,5, 114,2, 112,7, 111,0, 110,8, 108,4, 108,3, 102,1, 75,4, 66,74, 66,69, 65,6, 61,6, 61,2, 60,9, 54,3, 53,5, 52,9, 50,1, 49,3, 34,1, 33,6, 27,5, 9,7. ИК-спектр (с Фурье-преобразованием, KBr): 3400 (с, ш), 2944 (ср), 2881 (ср), 1700 (с), 1639 (с), 1501 (сл), 1463 (с), 1435 (с), 1356 (ср), 1320 (ср), 1288 (ср), 1269 (ср), 1238 (ср), 1213 (ср), 1166 (ср), 1102 (с), 1065 (с), 1030 (ср), 999 (ср), 938 (ср), 807 (сл) см-1. Масс-спектр высокого разрешения (ESI), [m+H]/z вычислено для C30H33O10N2: 581,2135. Найдено: 581,2112; [α]D 25 - 27,2°(с 0,50, метанол).
Пример 6

Амид (198,0 мг, 0,341 ммоль) сушат путем азеотропного концентрирования в вакууме с толуолом (10 мл), растворяют в ТГФ (10 мл) и охлаждают до 0°С. Прибавляют по каплям 0,20 М раствор LiAlH2(OEt)2 (6,8 мл, 1,36 ммоль)13 в течение 10 мин. Реакционную смесь перемешивают при 0°С в течение 35 мин с получением карбиноламина, Rf 0,59 (4:1 этилацетат-гексан). Прибавляют сначала уксусную кислоту (425 мкл, 17,44 ммоль), чтобы погасить реакцию. Затем прибавляют 4,8 М водный раствор цианида калия (425 мкл, 2,04 ммоль), безводный сульфат натрия (2,5 г, 17,6 ммоль) и Celite® (6 мл) для осуществления преобразования аминонитрила и осаждения солей алюминия. Наблюдают выделение пузырьков и через 5 мин реакционную смесь нагревают до 23°С и перемешивают в течение 7 ч. Суспензию фильтруют через пад из Celite®, элюируя этилацетатом (100 мл). Полученный раствор концентрируют в вакууме и очищают флеш-хроматографией на колонке (100 мл силикагеля, 2:1 этилацетат-гексан) с получением желаемого продукта в виде практически чистой белой пены (175,6 мг, 87%).
Rf 0,31 (4:1 этилацетат-гексан); 1H ЯМР (400 МГц, CDCl3) δ (смесь ротамеров карбамата): 6,43 (уш.с, 0,6Н), 6,26 (с, 0,4), 6,24 (с, 0,6Н), 6,20 (с, 0,4Н), 6,07-6,00 (м, 1Н), 5,97-5,82 (м, 4Н), 5,61 (с, 0,6Н), 5,52 (с, 0,4Н), 5,37-5,17 (м, 3Н), 4,90 (д, J=7,8 Гц, 0,4Н), 4,84 (д, J=8,3 Гц, 0,6Н), 4,73-4,60 (м, 2Н), 4,16-4,08 (м, 2,6Н), 3,97-3,94 (м, 1,4Н), 3,77 (с, 1,2Н), 3,68-3,61 (м, 1Н), 3,62 (с, 1,8Н), 3,49-3,36 (м, 1Н), 3,29-3,19 (м, 3Н), 2,76-2,69 (м, 1Н), 2,11 (с, 1,8Н), 2,08 (с, 1,2Н), 2,00-1,83 (м, 2Н); 13С ЯМР (101 МГц, CDCl3) δ (смесь ротамеров карбамата): 154,3, 153,8, 148,4, 148,34, 148,26, 146,2, 145,9, 144,3, 138,8, 133,62, 133,56, 132,7, 132,2, 130,7, 130,3, 120,5, 120,3, 117,9, 117,8, 117,4, 117,2, 116,3, 112,6, 112,5, 112,1, 111,9, 107,2, 106,4, 101,1, 74,5, 74,0, 66,7, 66,5, 64,5, 64,3, 60,8, 60,5, 59,1, 58,9, 58,0, 56,7, 56,6, 49,9, 49,4, 48,9, 48,7, 31,2, 30,5, 29,7, 25,9, 9,43, 9,35. ИК-спектр (с Фурье-преобразованием, записан в тонком слое): 3369 (ср. ш), 2931 (ср. ш), 1688 (ср), 1500 (сл), 1463 (ср), 1431 (с), 1375 (ср), 1325 (ср), 1294 (ср), 1269 (ср), 1106 (с), 1063 (ср), 994 (ср), 956 (сл) см-1. Масс-спектр высокого разрешения (ESI), [m+H]/z вычислено для С31Н34O9N3: 592,2295. Найдено: 592,2316; [α]D 25+30,4°(с 1,0, хлороформ).
Пример 7
Фенол (170,0 мг, 0,287 ммоль) сушат путем азеотропного концентрирования в вакууме с толуолом (10 мл) и растворяют в метиленхлориде (3,0 мл). Прибавляют триэтиламин (80 мкл, 0,574 ммоль) и 4-диметиламинопиридин (7,0 мг, 0,0574 ммоль) и полученный раствор охлаждают до -30°С на бане из сухого льда - ацетонитрила. Прибавляют твердый N-фенилтрифторметансульфонимид (113,5 мг, 0,318 ммоль), и реакционную смесь перемешивают при -30°С в течение 38 ч на бане Cryobath®. Смесь выливают в смесь 1:1 насыщенного водного раствора бикарбоната натрия и насыщенного водного раствора хлорида натрия (100 мл), экстрагируют метиленхлоридом (2×75 мл), сушат над сульфатом натрия, фильтруют, концентрируют в вакууме. Остаток очищают флеш-хроматографией на колонке (100 мл силикагеля, градиент 2:3→3:4 этилацетат-гексан,) с получением желаемого продукта в виде практически чистой прозрачной пленки (153,4 мг, 74%).
Rf 0,18 (2:3 этилацетат-гексан); 1H ЯМР (400 МГц, CDCl3) δ (смесь ротамеров карбамата): 7,16 (с, 0,6Н), 6,63 (с, 0,4Н), 6,60 (с, 0,6Н), 6,45 (с, 0,4Н), 6,08-5,8 (м, 4Н), 5,74 (м, 0,6Н), 5,59 (м, 0,4Н), 5,40-5,16 (м, 4Н), 4,96-4,89 (м, 1Н), 4,74-4,60 (м, 3Н), 4,26 (м, 1Н), 4,19-4,15 (м, 2Н), 4,00 (м, 1Н), 3,89 (с, 1,2Н), 3,83 (с, 1,8Н), 3,66-3,64 (м, 1Н), 3,39-3,24 (м, 4Н), 2,91-2,83 (м, 1Н), 2,11 (с, 1,2Н), 2,05 (с, 1,8Н), 1,86-1,78 (м, 1Н); 13С ЯМР (101 МГц, CDCl3) δ (смесь ротамеров карбамата):.154,0, 153,9, 148,6, 148,4, 147,3, 146,6, 144,7, 144,5, 141,3, 141,0, 139,1, 138,9, 136,9, 136,7, 133,7, 132,2, 132,1, 131,6, 129,4, 127,0, 123,0, 121,5, 121,3, 119,9, 118,5 (кв, J=321 Гц CF3), 118,2, 117,7, 117,6, 117,4, 116,3, 116,1, 112,6, 112,3, 112,1, 112,0, 101,3, 101,2, 74,5, 66,9, 66,7, 65,7, 65,5, 62,0, 61,9, 59,54, 59,48, 58,6, 56,5, 49,8, 49,3, 49,0, 48,4, 31,0, 30,4, 26,1, 26,0, 9,5, 9,4. 19F ЯМР (376 МГц, BF3·OEt2 стандарт установлен на -153,0 м.д., CDCl3) δ (смесь ротамеров карбамата): -74,02, -74,01. ИК-спектр (с Фурье-преобразованием, записан в тонком слое): 3325 (сл. ш), 2949(сл. ш), 1688 (ср), 1588 (сл), 1500 (ср), 1425 (с), 1319 (ср), 1288 (ср), 1256 (ср), 1213 (с), 1138 (с), 1106 (ср), 1038 (ср), 988 (ср), 875 (сл) см-1. Масс-спектр высокого разрешения (ESI), [m+H]/z вычислено для С32Н33O11N3SF3: 724,1788, Найдено: 724,1803; [α]D 26+34,3°(с 1,0, хлороформ).
Пример 8

Спирт (119,2 мг, 0,165 ммоль) растворяют в ДМФА (1,65 мл) и охлаждают до 0°С. Добавляют твердые трет-бутилдиметилсилилхлорид (32,2 мг, 0,215 ммоль) и имидазол (14,8 мг, 0,218 ммоль), и смесь перемешивают при 0°С в течение 5 часов. Добавляют безводный фторид калия (10,5 мг, 0,181 ммоль) и метанол (1,65 мл), раствор нагревают до 23°С и перемешивают в течение 1 часа. Смесь концентрируют в вакууме с удалением метанола, выливают в воду (50 мл), экстрагируют смесью этилацетат-гексан 1:4 (2×50 мл), промывают водой (25 мл) и насыщенным водным раствором хлорида натрия (25 мл), сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Остаток очищают колоночной флэш-хроматографией (100 мл, силикагель, система этилацетат-гексан 3:7) и получают желаемый продукт в виде прозрачной пленки (122,6 мг, 89%). Rf 0,47 (этилацетат-гексан 2:3).
1Н ЯМР (400 МГц, CDCl3) δ (смесь ротамеров карбоната) 6,92 (с, 0,6Н), 6,58 (с, 0,4Н), 6,54 (с, 0,6Н), 6,17 (с, 0,4Н), 6,14-5,96 (м, 1Н), 5,94 (с, 1Н), 5,93-5,88 (м, 1Н), 5,87 (с, 1Н), 5,70 (ушир. с, 0,6Н), 5,53 (ушир. с, 0,4Н), 5,41-5,14 (м, 4Н), 4,89 (д, J=8,8 Гц, 0,4Н), 4,85 (д, J=8,4 Гц, 0,6Н), 4,71-4,58 (м, 2Н), 4,54 (ушир. с, 0,6Н), 4,51 (ушир.с, 0,4Н), 4,20-4,15 (м, 2Н), 4,00-3,98 (м, 1Н), 3,91 (с, 1,2Н), 3,85 (с, 1,8Н), 3,66-3,64 (м, 1Н), 3,34-3,13 (м, 4Н), 3,01-2,93 (м, 1Н), 2,12 (с, 1,2Н), 2,05 (с, 1,8Н), 1,86-1,80 (м, 1Н), 0,85 (с, 5,4Н), 0,84 (с, 3,6Н), -0,02 (с, 1,8Н), -0,03 (с, 1,2Н), -0,05 (с, 1,8Н), -0,06 (с, 1,2Н); 13С ЯМР (101 МГц, CDCl3) δ (смесь ротамеров карбоната) 154,1, 154,0, 148,6, 148,4, 147,2, 146,4, 144,5, 144,4, 141,1, 140,9, 139,3, 139,1, 136,4, 136,2, 133,7, 133,0, 132,4, 132,3, 122,0, 121,8, 120,5, 120,3, 118,6 (кв, J=321 Гц, CF3), 118,1, 117,7, 117,6, 117,1, 117,0, 116,8, 112,6, 112,5, 112,1, 111,94, 111,88, 101,2, 101,1, 74,5, 69,4, 69,0, 66,8, 66,5, 62,0, 61,9, 60,7, 60,6, 59,1, 56,6, 49,8, 49,3, 49,0, 48,4, 30,7, 30,1, 26,2, 26,1, 26,0, 18,3, 9,5, 9,4, -5,45, -5,48; 19F ЯМР (376 МГц, BF3-Oet2 в качестве стандарта при -153,0 м.д., CDCl3) δ -74,1; FTIR (чистый) 3344 (сл. ушир), 2956 (м), 2931 (м), 2863 (м), 1694 (с), 1588 (ел.), 1500 (м), 1425 (с), 1319 (м), 1256 (м), 1213 (с), 1144 (с), 1106 (с), 1038 (м), 988 (м), 881 (сл.), 838 (м) см-1 ; HRMS (ESI), [m+H]/z вычислено для C38H47O11N3SiSF3: 838,2652, найдено 838,2613; [α]D 25+24,5° (с 1,0, хлороформ).
Пример 9

Фенол (122,6 мг, 0,146 ммоль) растворяют в метиленхлориде (5 мл) и охлаждают до 0°С. При помощи шприца добавляют диизопропилэтиламин (225 мкл, 1,33 ммоль) и хлорметилметиловый эфир (50 мкл, 0,658 ммоль). Реакционную смесь перемешивают при 0°С в течение 10 минут и затем выливают в насыщенный водный раствор бикарбоната натрия (50 мл), экстрагируют метиленхлоридом (3×50 мл), сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Остаток очищают фильтрованием через слой силикагеля, элюируя смесью этилацетат-гексан 1:4, и получают желаемый продукт в виде прозрачной пленки (129,0 мг, 99%). Rf 0,52 (этилацетат-гексан 2:3).
1H ЯМР (400 МГц, CDCl3) δ (смесь ротамеров карбоната) 6,75 (с, 1Н), 6,16-6,06 (м, 1Н), 5,94 (с, 1Н), 5,93-5,88 (м, 1Н), 5,87 (с, 1Н), 5,61-5,13 (м, 7Н), 4,91 (д, J=8,1 Гц, 0,6Н), 4,84 (д, J=8,4 Гц, 0,6Н), 4,71-4,55 (м, 1,8Н), 4,50 (ушир.с, 1Н), 4,20-4,15 (м, 2Н), 3,97 (дд, J=8,1, 1,8 Гц, 1Н), 3,89 (с, 1,8Н), 3,88 (с, 1,2Н), 3,71 (с, 1,8Н), 3,65 (д, J=9,5 Гц, 1Н), 3,62 (с, 1,2Н), 3,33-3,12 (м, 4Н), 2,96 (дд, J=17,6, 12,1 Гц, 1Н), 2,12 (с, 3Н), 1,76 (дд, J=14,8, 12,1 Гц, 1Н), 0,84 (с, 5,4Н), 0,83 (с, 3,6Н), -0,02 (с, 3Н), -0,05 (с, 1,8Н), -0,06 (с, 1,2Н); 13С ЯМР (101 МГц, CDCl3) δ (смесь карбонатных ротамеров) 153,9, 153,8, 148,4, 148,3, 147,53, 147,46, 144,5, 142,4, 142,3, 141,9, 141,8, 139,3, 139,2, 133,6, 132,5, 132,3, 132,1, 128,7, 128,5, 120,1, 118,6 (кв, J=321 Гц, CF3), 118,4, 118,0, 117,51, 117,46, 116,8, 116,7, 116,6, 116,3, 112,5, 112,4, 112,0, 111,9, 101,2, 99,5, 74,3, 69,1, 68,8, 67,0, 66,7, 61,1, 60,6, 60,5, 59,15, 59,09, 58,4, 58,2, 56,4, 50,3, 49,4, 49,2, 48,4, 30,8, 30,2, 26,1, 26,0, 18,3, 9,4, -5,5; 19F ЯМР (376 МГц, BF3OEt2 в качестве стандарта при -153,0 м.д., CDCl3) δ -74,2; FTIR (чистый) 2956 (м), 2931 (м), 2856 (м), 1713 (с), 1581 (сл), 1488 (м), 1425 (с), 1319 (м), 1256 (м), 1213 (м), 1144 (м), 1106 (с), 1019 (м), 988 (м), 925 (м), 843 (м) см-1; HRMS (ESI), [m+H]/z вычислено для C40H51O12N3SiSF3: 882,2915, найдено 882,2886; [α]23 D+49,5° (с 1,0, хлороформ).
Пример 10:

Простой аллиловый эфир (129,0 мг, 0,146 ммоль) растворяют в метиленхлориде (2,5 мл) и к данному раствору добавляют уксусную кислоту (85,0 мкл, 1,49 ммоль), PdCl2 (PPh3)2 (6,4 мг, 0,0091 ммоль) и гидрид трибутилолова (120,0 мкл, 0,445 ммоль). Наблюдают выделение пузырьков, и окраска реакционной смеси изменяется от желтой до темно-оранжевой. Реакционную смесь перемешивают при 23°С в течение 15 минут и затем реакционную смесь непосредственно очищают колоночной флэш-хроматографией (50 мл, силикагель, градиент смеси этилацетат-гексан от 1:1 до этилацетата), получая желаемый продукт в виде прозрачной пленки (104,7 мг, 94%). Rf 0,16 (этилацетат-гексан 1:1).
1H ЯМР (400 МГц, CDCl3) δ 6,76 (с, 1Н), 5,90 (с, 1Н), 5,81 (с, 1H), 5,36 (д, J=5,9 Гц, 1H), 5,21 (д, J=5,9 Гц, 1Н), 4,53 (д, J=1,5 Гц, 1H), 4,45 (д, J=2,6 Гц, 1H), 3,96 (д, J=7,7 Гц, 1H), 3,84 (с, 3Н), 3,68 (с, 3Н), 3,70-3,64 (м, 2Н), 3,23 (д, J=11,4 Гц, 1H), 3,16-3,09 (м, 3Н), 2,95 (д, J=17,6 Гц, 1H), 2,07 (с, 3Н), 1,74-1,66 (м, 1H), 0,84 (с, 9Н), -0,03 (с, 3Н), -0,06 (с, 3Н); 13С ЯМР (101 МГц, CDCl3) δ 148,2, 145,2, 144,5, 141,9, 141,0, 136,6, 133,0, 130,5, 118,6 (кв, J=321 Гц, CF3), 118,1, 116,4, 112,2, 111,7, 106,2, 100,8, 100,0, 69,4, 61,7, 61,3, 59,3, 58,2, 56,7, 49,9, 49,4, 31,1, 26,3, 26,0, 18,3, 8,9, -5,4, -5,5; 19F ЯМР (376 МГц, BF3OEt2 в качестве стандарта при -153,0 м.д., CDCl3) δ -74,2; FTIR (чистый) 3481 (сл ушир), 3325 (сл ушир), 2956 (м), 2931 (м), 2906 (м), 2856 (м), 1656 (сл), 1618 (сл), 1581 (сл), 1488 (м), 1463 (м), 1425 (с), 1344 (сл), 1300 (сл), 1250 (с), 1213 (с), 1144 (с), 1100 (с), 1038 (м), 1013 (м), 981 (м), 919 (м), 837 (м) см-1; HRMS (ESI), [m+H]/z вычислено для С33Н43О10N3SiSF3: 758,2390, найдено 758,2362; [α]D 23-1/6° (с 1,0, хлороформ).
Пример 11

Амин (99,6 мг, 0,131 ммоль) растворяют в ацетонитриле (2,5 мл). Добавляют формалин (0,25 мл, 3,08 ммоль) и твердый цианоборгидрид натрия (31 мг, 0,492 ммоль). К прозрачному желтому раствору добавляют уксусную кислоту (60 мкл, 1,05 ммоль) и образовавшуюся в результате мутную и светло-желтую смесь перемешивают при 0°С в течение 30 минут, затем нейтрализуют насыщенным водным раствором бикарбоната натрия (1 мл). Смесь перемешивают при 23°С в течение 15 минут, выливают в смесь насыщенный водный раствор бикарбоната натрия - насыщенный водный раствор хлорида натрия 5:1 (50 мл), экстрагируют смесью этилацетат-гексан 97:3 (3×50 мл), сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Остаток очищают колоночной флэш-хроматографией (50 мл силикагеля, градиент смеси этилацетат-гексан от 3:7 до 1:1) и получают желаемый продукт в виде прозрачной пленки (92,5 мг, 91%). Rf 0,42 (этилацетат-гексан 1:1).
1H ЯМР (400 МГц, CDCl3) δ 6,75 (с, 1Н), 5,90 (ушир.с, 1Н), 5,82 (ушир.с, 1H), 5,35 (д, J=6,2 Гц, 1Н), 5,21 (д, J=6,2 Гц, 1H), 5,17 (ушир.с, 1H), 4,53 (д, J=2,6 Гц, 1H), 4,23 (д, J=1,8 Гц, 1H), 3,99 (м, 1H), 3,86 (с, 3Н), 3,68 (с, 3Н), 3,68-3,65 (м, 1H), 3,34 (д, J=7,7 Гц, 1H), 3,29 (д, J=9,9 Гц, 1H), 3,14-3,06 (м, 2Н), 3,03 (дд, J=17,9, 8,4 Гц, 1H), 2,76 (д, J=17,9 Гц, 1Н), 2,29 (с, 3Н), 2,09 (с, 3Н), 1,76-1,70 (м, 1H), 0,84 (с, 9Н), -0,03 (с, 3Н), -0,06 (с, 3Н); 13С ЯМР (101 МГц, CDCl3) δ 150,0, 145,2, 144,4, 141,7, 141,4, 136,7, 132,3, 126,7, 118,6 (кв, J=321 Гц, CF3), 118,4, 115,9, 112,3, 112,0, 106,1, 100,8, 99,9, 69,4, 61,8, 61,3, 58,8, 58,2, 57,0, 56,6, 55,1, 48,8, 41,7, 26,0, 25,6, 18,3, 8,9, -5,4, -5,5; 19F ЯМР (376 МГц, BF3OEt2 в качестве стандарта при -153,0 м.д., CDCl3) δ -74,3; FTIR (чистый) 3456 (м ушир), 2965 (м), 2931 (м), 2856 (м), 1719 (сл), 1656 (сл), 1612 (сл), 1581 (сл), 1488 (м), 1425 (с), 1331 (м), 1250 (м), 1213 (с), 1144 (м), 1100 (с), 1063 (м), 1013 (м), 975 (м), 938 (м), 913 (м), 844 (м) см-1; HRMS (ESI), [m+H]/z вычислено для C34H45O10N3SiSF3: 772,2547, найдено 772,2578; [α]D 23-0,6° (с 3,0, хлороформ).
Пример 12

Трифторметансульфонат (97,6 мг, 0,126 ммоль) сушат азеотропным концентрированием в вакууме с толуолом (10 мл) и лиофилизуют из бензола в колбу с объемом 10 мл. Лиофилизат растворяют в 1-метил-2˜пирролидиноне (0,63 мл) и при помощи шприца добавляют уксусную кислоту (0,5 мкл, 0,0087 ммоль). Добавляют твердые PdCl2(PPh3)2 (9,0 мг, 0,0128 ммоль) и высушенный на пламени хлорид лития (27,4 мг, 0,645 ммоль) с получением желтого раствора. Добавляют тетраметилолово (135 мкл, 0,973 ммоль) и емкость закрывают крышкой и перемешивают при 80°С на масляной бане в течение 15 минут. В течение 1 минуты растворяют хлорид лития, и в течение 10 минут раствор становится желто-черным. Реакционную смесь выливают в воду (50 мл), экстрагируют смесью этилацетат-гексан 3:7 (3×50 мл), промывают насыщенным водным раствором хлорида натрия (50 мл), сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Остаток очищают колоночной флэш-хроматографией (50 мл, силикагель, градиент смеси этилацетат-гексан от 3:7 до 2:3) и получают желаемый продукт в виде белого твердого вещества (75,6 мг, 94%). Т.пл.: 144-167°С (с разложением); Rf 0,26 (этилацетат-гексан 2:3).
1H ЯМР (400 МГц, CDCl3) δ 6,64 (с, 1Н), 5,89 (ушир. с, 1Н), 5,79 (ушир. с, 1Н), 5,58 (ушир. с, 1Н), 5,34 (д, J=6,2 Гц, 1Н), 5,15 (д, J=5,9 Гц, 1Н), 4,55 (д, J=2,9 Гц, 1Н), 4,18 (д, J=2,2 Гц, 1Н), 4,00 (д, J=7,3 Гц, 1Н), 3,71 (с, 3Н), 3,70 (с, 3Н), 3,70-3,68 (м, 1Н), 3,30 (м, 1Н), 3,25 (д, J=11,4 Гц, 1Н), 3,20-3,15 (м, 1Н), 3,08 (д, J=14,6 Гц, 1Н), 2,99 (дд, J=17,6, 8,4 Гц, 1Н), 2,69 (д, J=17,6 Гц, 1Н), 2,29 (с, 3Н), 2,23 (с, 3Н), 2,08 (с, 3Н), 1,85-1,79 (м, 1Н), 0,82 (с, 9Н), -0,03 (с, 3Н), -0,08 (с, 3Н); 13С ЯМР (101 МГц, CDCl3) δ 148,8, 147,3, 145,3, 144,3, 136,4, 131,0, 130,4, 124,9, 123,1, 118,7, 112,4, 112,2, 105,9, 100,6, 99,8, 69,1, 61,8, 59,8, 58,8, 57,8, 57,1, 56,5, 55,5, 41,7, 26,2, 26,0, 25,5, 18,3, 15,8, 9,0, -5,4, -5,5; FTIR (чистый) 3469 (м ушир.), 2956 (с), 2931 (с), 2856 (м), 1463 (м), 1438 (м), 1400 (м), 1363 (сл.), 1331 (сл.), 1256 (м), 1238 (м), 1156 (м), 1100 (с), 1006 (м), 969 (м), 919 (м), 838 (м) см-1; HRMS (ESI), [m+H]/z вычислено для C34H48O7N3Si: 638,3261, найдено 638,3233; [α]D 23-17,3° (с 1,0, хлороформ).
Пример 13

Фенол (75,3 мг, 0,118 ммоль) растворяют в метиленхлориде (2,0 мл). К раствору добавляют 4-диметиламинопиридин (1,1 мг, 0,009 ммоль), триэтиламин (75 мкл, 0,538 ммоль) и уксусный ангидрид (50 мкл, 0,529 ммоль) и реакционную смесь перемешивают при 23°С в течение 15 минут. Смесь сразу же очищают колоночной флэш-хроматографией (50 мл, силикагель, этилацетат-гексан 3:7) и получают желаемый продукт в виде прозрачной пленки (75,3 мг, 94%). Rf 0,33 (этилацетат-гексан 2:3).
1H ЯМР (300 МГц, CDCl3) δ 6,64 (с, 1Н), 5,95 (с, 1Н), 5,89 (с, 1Н), 5,13 (д, J=5,7 Гц, 1Н), 5,04 (д, J=5,7 Гц, 1Н), 4,56 (ушир.с, 1Н), 4,14 (д, J=3,1 Гц, 1Н), 3,99 (дд, J=7,9, 1,8 Гц, 1Н), 3,73 (с, 3Н), 3,70 (дд, J=9,9, 2,0 Гц, 1Н), 3,57 (с, 3Н), 3,30 (д, J=7,9 Гц, 1H), 3,25-3,20 (м, 2Н), 2,99 (дд, J=17,6, 7,9 Гц, 1H), 2,78 (д, J=14,5 Гц, 1Н), 2,66 (д, J=17,6 Гц, 1H), 2,32 (с, 3Н), 2,31 (с, 3Н), 2,21 (с, 3Н), 2,00 (с, 3Н), 1,74 (дд, J=15,4, 11,9 Гц, 1H), 0,84 (с, 9Н), -0,03 (с, 3Н), -0,06 (с, 3Н); 13С ЯМР (75 МГц, CDCl3) δ 168,8, 148,2, 148,1, 144,3, 140,9, 140,3, 130,7, 130,5, 125,2, 123,9, 121,1, 118,8, 112,9, 111,4, 101,4, 99,2, 69,0, 61,7, 59,8, 58,6, 57,7, 57,1, 56,5, 55,4, 41,7, 26,4, 25,9, 25,3, 20,2, 18,2, 15,7, 9,3, -5,6; FTIR (чистый) 2931 (с), 2856 (м), 1763 (с), 1463 (м), 1438 (м), 1369 (м), 1325 (сл), 1231 (сл), 1200 (с), 1156 (м), 1106 (с), 1094 (с), 1006 (м), 969 (сл), 931 (сл), 900 (сл), 838 (м) см-1; HRMS (ESI), [m+H]/z вычислено для C36H50O8N3Si: 680,3367, найдено 680,3399; [α]D 26+34,5° (с 1,0, хлороформ).
Пример 14

Силилзащищенный спирт (75,3 мг, 0,111 ммоль) растворяют в ацетонитриле (4,0 мл) в емкости из пластика и добавляют 48%-ный водный раствор фтористоводородной кислоты (0,4 мл). Реакционную смесь перемешивают при 23°С в течение 15 часов и выливают в насыщенный водный раствор бикарбоната натрия (50 мл), экстрагируют этилацетатом (2×75 мл), сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Остаток очищают колоночной флэш-хроматографией (50 мл, силикагель, градиент смеси этилацетат-гексан от 1:1 до 2:1) и получают желаемый продукт в виде прозрачной пленки (62,2 мг, 99%). Rf 0,19 (этилацетат-гексан 1:1).
1H ЯМР (500 МГц, CDCl3) δ 6,70 (с, 1Н), 5,96 (д, J=1,3 Гц, 1Н), 5,90 (д, J=1,4 Гц, 1Н), 5,14 (д, J=5,7 Гц, 1Н), 5,07 (д, J=5,7 Гц, 1Н), 4,21 (д, J=2,3 Гц, 1Н), 4,10 (д, J=1,8 Гц, 1Н), 3,99 (м, 1Н), 3,72 (с, 3Н), 3,66 (д, J=11,1 Гц, 1Н), 3,58 (с, 3Н), 3,49-3,44 (м, 1Н), 3,40-3,32 (м, 2Н), 3,10 (дд, J=18,0, 7,9 Гц, 1Н), 2,79 (д, J=15,7 Гц, 1Н), 2,51 (д, J=18,1 Гц, 1Н), 2,36 (с, 3Н), 2,32 (с, 3Н), 2,21 (с, 3Н), 2,00 (с, 3Н), 1,82-1,70 (м, 2Н); 13С ЯМР (126 МГц, CDCl3) δ 168,5, 148,6, 148,3, 144,5, 140,6, 140,4, 131,3, 129,5, 125,1, 123,6, 120,5, 117,6, 113,2, 111,7, 101,5, 99,2, 63,6, 59,9, 59,8, 58,0, 57,7, 56,9, 56,1, 55,3, 41,6, 26,3, 25,6, 20,1, 15,7, 9,3; FTIR (чистый) 3500 (м ушир), 2935 (с ушир), 2854 (сл), 1760 (с), 1484 (м), 1440 (м), 1434 (м), 1401 (м), 1370 (м), 1341 (сл), 1324 (сл), 1234 (м), 1201 (с), 1158 (м), 1106 (с), 1086 (с), 1075 (с), 1043 (м), 1023 (м), 1000 (м), 961 (м), 912 (м) см-1; HRMS (FAB), [m+Na]/z вычислено для C30H35O8N3Na: 588,2322, найдено 588,2303; [α]D 23+50,2° (с 0,66, метиленхлорид); [α]D 26+50,8° (с 1,0, хлороформ).
Пример 15

Спирт (54,2 мг, 0,0958 ммоль) и фталимид (15,5 мг, 0,105 ммоль) сушат азеотропным концентрированием в вакууме со смесью ТГФ-толуол 1:2 (6 мл), растворяют в ТГФ (3,8 мл) и охлаждают до 0°С. Добавляют трифенилфосфин (37,2 мг, 0,142 ммоль) в виде твердого вещества, затем с помощью шприца по каплям добавляют диэтилазодикарбоксилат (18,0 мкл, 0,114 ммоль). Желтый раствор перемешивают при 0°С в течение 5 минут и затем нагревают до 23°С в течение 2 часов. Избыточные реагенты нейтрализуют уксусной кислотой (0,1 мл) и метанолом (0,1 мл) и реакционную смесь разбавляют толуолом (2 мл). Смесь концентрируют в вакууме и остаток сразу же очищают колоночной флэш-хроматографией (50 мл, силикагель, градиент смеси диэтиловый эфир-гексан от 3:1 до 5:1), получая желаемый продукт в виде белой пены (59,7 мг, 90%). Rf 0,53 (этилацетат-гексан 2:1).
1H ЯМР (400 МГц, CDCl3) δ 7,70-7,63 (м, 4Н), 6,64 (с, 1Н), 5,73 (с, 1Н), 5,50 (с, 1Н), 5,07 (д, J=5,7 Гц, 1Н), 4,98 (д, J=5,7 Гц, 1Н), 4,27 (д, J=2,1 Гц, 1Н), 4,24 (м, 1Н), 4,08 (д, J=2,5 Гц, 1Н), 3,74-3,67 (м, 2Н), 3,53 (с, 3Н), 3,50 (с, 3Н), 3,38 (д, J=7,1 Гц, 1Н), 3,18 (д, J=11,5 Гц, 1Н), 3,02 (дд, J=18,1, 8,1 Гц, 1Н), 2,75 (д, J=16,1 Гц, 2Н), 2,31 (с, 3Н), 2,27 (с, 3Н), 2,18 (с, 3Н), 2,01 (с, 3Н), 1,60 (м, 1Н); 13С ЯМР (101 МГц, CDCl3) δ 168,3, 167,5, 148,1, 147,8, 144,3, 141,2, 140,5, 133,4, 131,8, 130,2, 130,23, 125,3, 123,4, 123,0, 120,8, 118,0, 113,6, 111,7, 101,3, 99,1, 59,8, 59,6, 57,7, 56,7, 56,6, 56,1, 55,4, 41,5, 40,9, 26,7, 25,0, 20,1, 16,0, 9,5; FITR (чистый) 2935 (м ушир), 1764 (м), 1716 (с), 1433 (м ушир), 1394 (м ушир), 1369 (м ушир), 1234 (м), 1198 (с), 1158 (м), 1101 (м ушир), 1072 (м), 1025 (м), 1000 (м), 947 (м), 933 (м) см-1; HRMS (FAB), [m+H]/z вычислено для C38H39O9N4: 695,2717, найдено 695,2744; [α]D 23+21,6° (с 1,0, метиленхлорид); [α]D 23+5,8° (с 1,0, хлороформ).
Пример 16

Метоксиметиловый эфир (59,7 мг, 0,0859 ммоль) растворяют в смеси трифторуксусная кислота-ТГФ-вода 4:1:1 (12,0 мл)1 (1 - важно использовать продутую азотом воду и стабилизированный 0,025% бутилированным гидрокситолуолом ТГФ), и раствор перемешивают при 23°С в течение 9,5 часов. Реакционную смесь разбавляют толуолом (5 мл) и раствор частично концентрируют в вакууме для удаления трифторуксусной кислоты. Смесь выливают в полунасыщенный водный раствор бикарбоната натрия (50 мл), экстрагируют смесью этилацетат-гексан 1:1 (2×50 мл), сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Остаток очищают колоночной флэш-хроматографией (40 мл, силикагель, градиент смеси этилацетат-гексан от 1:1 до 2:1) и получают желаемый продукт в виде прозрачной пленки (46,9 мг, 84%). Rf 0,41 (этилацетат-гексан 2:1).
1H ЯМР (500 МГц, CDCl3) δ 7,73-7,71 (м, 2Н), 7,67-7,65 (м, 2Н), 6,39 (с, 1Н), 5,66 (с, 1Н), 5,59 (с, 1Н), 5,33 (ушир.с, 1Н), 4,25-4,23 (м, 2Н), 4,02 (д, J=2,5 Гц, 1Н), 3,64 (м, 5Н), 3,35 (д, J=8,3 Гц, 1Н), 3,20 (д, J=12,0 Гц, 1Н), 3,02 (дд, J=18,1, 8,1 Гц, 1Н), 2,77 (д, J=14,6 Гц, 1Н), 2,45 (д, J=18,1 Гц, 1Н), 2,29 (с, 6Н), 2,22 (с, 3Н), 1,99 (с, 3Н), 1,73 (м, 1Н);
1H ЯМР (400 МГц, С6D6) δ 7,32 (дд, J=5,4, 3,0 Гц, 2Н), 6,79 (дд, J=5,4, 3,0 Гц, 2Н), 6,38 (с, 1Н), 5,43 (с, 1Н), 5,07 (ушир.с, 1Н), 4,89 (ушир.с, 1Н), 4,50 (дд, J=6,8, 2,8 Гц, 1Н), 4,02 (д, J=2,1 Гц, 1Н), 3,93 (д, J=2,2 Гц, 1Н), 3,68 (дд, J=14,0, 3,1 Гц, 1Н), 3,61 (дд, J=13,8, 7,0 Гц, 1Н), 3,48 (д, J=11,8 Гц, 1Н), 3,05 (с, 3Н), 2,96 (д, J=16,3 Гц, 1Н), 2,82 (д, J=8,0 Гц, 1Н), 2,61 (дд, J=17,9, 7,7 Гц, 1Н), 2,46 (д, J=17,8 Гц, 1Н), 2,09 (с, 6Н), 1,98 (с, 3Н), 1,95-1,90 (м, 1Н), 1,86 (с, 3Н); 13С ЯМР (126 МГц, CDCl3) δ 168,5, 167,7, 146,3, 144,3, 142,6, 141,2, 140,6, 133,5, 131,9, 130,9, 128,3, 123,1, 121,0, 120,9, 118,0, 116,5, 113,7, 111,8, 101,2, 60,5, 60,2, 57,1, 56,4, 55,6, 55,5, 41,8, 41,6, 26,6, 25,3, 20,3, 15,9, 9,6; FTIR (чистый) 3463 (м ушир), 2934 (м ушир), 1764 (м), 1716 (с), 1455 (м ушир), 1433 (м ушир), 1395 (м ушир), 1370 (м), 1233 (м), 1102 (м), 1073 (м) см-1; ВЭЖХ (Columbus, 5 мк, C18, 100 А, 250×4,60 мм, объемная скорость: 1,0 мл/мин, Х=254 нм), RT=13,7 мин (60% СН3CN в воде); HRMS (FAB), [m+H]/z вычислено для С36Н35O8N4: 651,2455, найдено 651,2444; [α]D 23+21,9° (с 1,0, метиленхлорид); [α]D 23+9,7° (с 1,0, хлороформ).
ССЫЛКИ
В перечисленных ниже публикациях содержится информация о предпосылках данного изобретения, и в связи с этим они включены в данную заявку в виде ссылок.
(1) Пионерами исследований в этой области выполнены профессором К.Л.Райнхартом и его группой. См. (a) Rinehart, K.L.; Shield, L.S. in Topics in Pharmaceutical Sciences, eds. Breimer, D.D.; Crommelin, D.J.A.; Midha, K.K. (Amsterdam Medical Press, Noordwijk, The Netherlands), 1989, pp.613. (b) Rinehart, K.L.; Holt, T.G.; Fregeau, N.L.; Keifer, P.A.; Wilson, G.R.; Perun, T.J., Jr.; Sakai, R.; Thompson, A.G.; Stroh, J.G.; Shield, L.S.; Seigler, D.S.; Li, L.H.; Martin, D.G.; Grimmelikhuijzen, C.J.P.; Gäde, G.J. Nat. Prod. 1990, 53, 771. (с) Rinehart, K.L.; Sakai, R.; Holt, T.G.; Fregeau, N.L.; Perun, T.J., Jr.; Seigler, D.S.; Wilson, G.R.; Shield, L.S. Pure Appl. Chem. 1990, 62, 1277. (d) Rinehart, K.L.; Sakai, R.; Holt, T.G.; Fregeau, N.L.; Stroh, J.G.; Keifer, P.A.; Sun, F.; Li, L.H.; Martin, D.G. J. Org. Chem. 1990, 55, 4512. (e) Wright, A.E.; Forleo, D.A.; Gunawardana, G.P.; Gunasekera, S.P.; Koehn, F.E.; McConnel, O.J. J. Org. Chem. 1990, 55, 4508. (f) Sakai, R.; Rinehart, K.L.; Guan, Y.; Wang, H. J. Proc. Natl. Acad. Sci. USA 1992, 89, 11456.
(2) (a) Business Week, 13 September 1999, p.22. (b) Science 1994, 266, 1324.
(3) Corey, E.J.; Gin, D.Y.; Kania, R.J. Am. Chem. Soc. 1996, 118, 9202.
(4) Martinez, E.J.; Owa, Т.; Schreiber, S.L.; Corey, E.J. Proc. Natl. Acad. Sci. USA 1999, 96, 3496.
(5) См. Myers, A.G.; Kung, D.W. J. Am. Chem. Soc. 1999, 121, 10828 для ознакомления с другим подходом к синтезу структур, таких как 5.
(6) Для получения информации о методологии взаимодействия карбоновых кислот и аминов с использованием CIP см.: (a) Akaji, К.; Kuriyama, N.; Kimura, Т.; Fujiwara, Y.; Kiso, Y. Tetrahedron Lett. 1992, 33, 3177. (b) Akaji, К.; Kuriyama, N.; Kiso, Y. Tetrahedron Lett. 1994, 35, 3315. (с) Akaji, К.; Kuriyama, N.; Kiso, Y. J. Org. Chem. 1996, 61, 3350.
(7) Реагент LiAlH2(OEt)2 получают непосредственно перед использованием путем прибавления 1.0 М раствора LiAlH4 в эфире к раствору 1 экв. этилацетата при 0°С и перемешивания при 0°С в течение 2 ч; см.: Brown, H.C.; Tsukamoto, A.J. Am. Chem. Soc. 1964, 86, 1089.
(8) Для знакомства с обзорами по восстановлению лактонов см. следующие публикации: (a) Brown, Н.С.; Krishnamurthy, S.Tetrahedron, 1979, 35, 567. (b) Cha, J.S. Org. Prep. Proc. Int., 1989, 21(4), 451. (с) Seyden-Penne, J. Reduction by the Alumino- and Borohydrides in Organic Synthesis; 2nd Ed.; Wiley-VCH: New York, 1997; Section 3.2.5.
(9) Для получения информации по восстановлению амидов гидридами см. ссылку 7, а также Myers, A.G.; Yang, В.Н.; Chen, Н.; Gleason, J.L. J. Am. Chem. Soc. 1994, 116, 9361.
(10) Получено из воды, через которую пропускают азот.
(11) Реагент приготавливают путем прибавления 1.0 М раствора литийалюминийгидрида в диэтиловом эфире (1 экв.) к раствору 1 экв. этилацетата в Et2O при 0°С. Смесь перемешивают при 0°С в течение 2 ч и порцию этого реагента используют для восстановления лактола. Brown, Н.С.; Tsukamoto, A.J. Am. Chem. Soc. 1964, 86, 1089.
(12) Получено из воды, через которую пропускают азот.
(13) См. ссылку, приведенную в ссылке 11.
Данное изобретение описано подробно, включая предпочтительные варианты его осуществления. Однако должно быть понятно, что изменения и/или улучшения этого изобретения могут быть сделаны специалистами в данной области при рассмотрении материалов данной заявки, при сохранении объема и сущности изобретения, изложенных в формуле изобретения, представленной ниже.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБЫ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ЭСТЕИНАСЦИДИНА, СОЕДИНЕНИЯ, ПРОМЕЖУТОЧНЫЙ ПРОДУКТ | 1997 |
|
RU2194709C2 |
| СИНТЕЗ 4АЛЬФА-АРИЛЭПИКАТЕХИНОВ И ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 2001 |
|
RU2281942C2 |
| ПРЕДШЕСТВЕННИКИ ВИТАМИНА D, СПОСОБ ИХ ПОЛУЧЕНИЯ И ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ | 2000 |
|
RU2247710C2 |
| ИНГИБИТОРЫ КАСПАЗ | 1999 |
|
RU2274642C2 |
| Замещенные 4-арил-гексагидро-7Н-имидазоло[1,5-b][1,2]оксазин-7-оны и способ их получения | 2018 |
|
RU2670097C1 |
| Способ получения (6R,7S,7aS)-6-((R)-1-(3,5-бис(трифторметил)фенил)этокси)-7-(4-фторфенил)гексагидро-3Н-пирролизин-3-она | 2022 |
|
RU2789599C1 |
| АГОНИСТЫ РЕЦЕПТОРА ПЕПТИДА-1, ПОДОБНОГО ГЛЮКАГОНУ, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2003 |
|
RU2342368C2 |
| ВОДОРАСТВОРИМЫЕ ПРОИЗВОДНЫЕ ФЕНИЛПИРИДАЗИНА И СОДЕРЖАЩИЕ ИХ ЛЕКАРСТВЕННЫЕ СРЕДСТВА | 2002 |
|
RU2302413C2 |
| ПРОИЗВОДНОЕ АЛКИНИЛИНДАЗОЛА И ЕГО ПРИМЕНЕНИЕ | 2015 |
|
RU2667486C2 |
| ИНГИБИТОРЫ ВИЧ-ИНТЕГРАЗЫ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2002 |
|
RU2284315C2 |
Изобретение относится к новому способу синтеза соединения 5, ключевого промежуточного соединения в синтезе сильных противоопухолевых агентов эктенасцидина 743 (1) и фталасцидина (2) и легодоступных промежуточных соединений 3b и 4. Технический результат - упрощение процесса. 3 н. и 2 з.п. ф-лы.



включающий стадии:
(а) преобразования соединения 11:
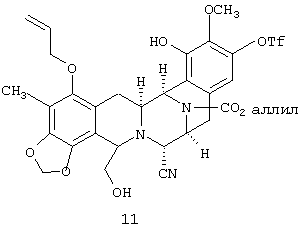
в моно третбутилдиметилсилиловый (ТБС) эфир, соединения 12:

(b) этерификации с помощью метоксиметилхлорида с получением соединения 13:

(с) отщепления N-аллилоксикарбонильной и O-аллильной групп у соединения 13 с получением вторичного амина, соединения 14:

(d) N-метилирования соединения 14 с получением соединения 15:

(е) С-метилирования соединения 15 с получением соединения 16:
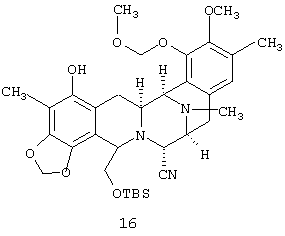
(f) ацетилирования фенола 16 с получением соответствующего ацетата, соединения 17:

(g) десилилирования соединения 17 с получением первичного спирта, соединения 18:
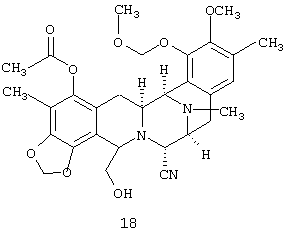
(h) замещения по Мицунобу первичного гидроксила соединения 18 с получением фталимида, соединения 19:

и
(i) кислотно-катализируемого отщепления метоксиметилового эфира у соединения 19.

(а) взаимодействия соединения 3b

с аминолактоном 4:

в присутствии ацилирующего агента с получением продукта присоединения 6:

(b) обработки соединения 6 аллилбромидом с получением амида 7:

(с) восстановления лактона 7 до соответствующего лактола 8:
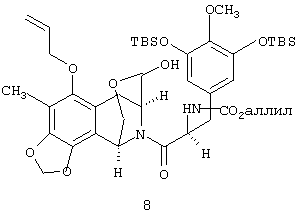
(d) десилилирования соединения 8 с получением соединения 9:

(е) циклизации соединения 9 с получением соединения 10:

и
(f) восстановления лактамной группы в соединении 10 до соответствующего циклического аминаля и обработки HCN.
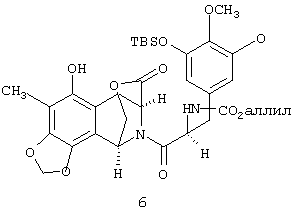
соединение 7:

соединение 8:

соединение 9:
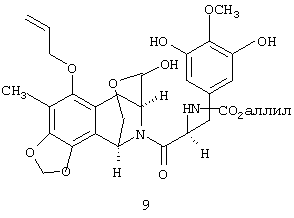
и соединение 10:
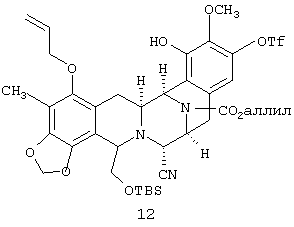
соединение 13:

соединение 14:

соединение 15:

соединение 16:
соединение 17:

соединение 18:
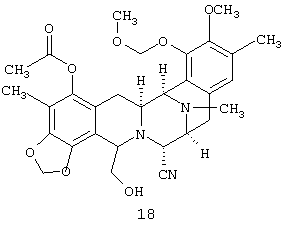
и соединение 19:

| US 5721362 А, 24.02.1998 | |||
| RU 95112485 А1, 20.04.1997 | |||
| RU 96115394 А, 27.12.1998. |

