Изобретение относится к молекулярной биологии, в частности к разработке тестов на обнаружение известных изменений в структуре генов, и может быть использовано для: генодиагностики, в том числе пренатальной, известных наследственных заболеваний; генотипирования известных однонуклеотидных полиморфизмов (SNP); генотипирования вирусов и других микроорганизмов; секвенс-специфичной идентификации ПЦР-фрагментов ДНК.
В настоящее время для определения однонуклеотидных замен существует ряд способов, которые можно подразделить на две главные группы:
I. Способы для выявления неизвестных SNPs ("single-nucleotide polymorphisms") в геноме или его крупных частях;
II. Способы для определения известных SNPs, т.е. для SNP-типирования - в анализе популяций, в поиске "генов-кандидатов" при изучении взаимосвязей генотипа и фенотипа, для детекции известных точечных мутаций и др.
Способы первой и второй групп отличаются принципиально и, как правило, не могут заменять друг друга в решении той или иной задачи.
В зависимости от назначения существующие практические способы II для определения известных однонуклеотидных замен можно подразделить на две подгруппы:
1) Способы для крупномасштабного SNP-типирования - отличаются минимизацией ручного труда, мультиплексными технологиями (типированием по многим SNPs одновременно), автоматическим считыванием и программной обработкой результатов и имеют два основных варианта исполнения - "chip" ("чип") и "microtiter рlаtе" (планшет). По существу - это количественные технологии, т.к. они практически не дают однозначно альтернативных ("да" или "нет"), не требующих статистической обработки прямых ответов.
2) Способы для определения точечных мутаций в целях медицинской диагностики, скрининга популяций или геномов по ограниченному числу определенных SNP-маркеров - допускают технологии с большей долей ручного труда, но требуют необходимым преимуществом простоту считывания результата и относительно низкую стоимость.
Изобретение относится к группе II-2) и позволяет определить однонуклеотидную замену в нуклеиновой кислоте, т.е. установить не только наличие, но и конкретный вариант такой замены. Несмотря на большое разнообразие, другие способы в данной подгруппе предназначены преимущественно для научно-исследовательских лабораторий, т.к. для получения ответа требуют, как правило, или использования радиоактивной метки, или проведения дополнительного электрофоретического анализа в геле, или высокопрофессиональной интерпретации неоднозначных результатов. За исключением таких универсальных способов, как аллель-специфичная гибридизация олигонуклеотидных зондов (ASOH)[1], минисеквенирование (SBE)[2], аллель-специфичная ПНР (PASA)[3] или лигирование олигонуклеотидных зондов (OLA)[4], остальные способы не универсальны в отношении любых нуклеотидных последовательностей и могут использоваться для детекции только ограниченного числа конкретных мутаций. Общим недостатком перечисленных универсальных способов является также их чувствительность к несоблюдению точных условий эксперимента, недостаточно высокий уровень селективности однонуклеотидных замен и в связи с этим необходимость статистической обработки результатов анализа.
Наиболее близким к заявленному способу, прототипом, является способ лигирования олигонуклеотидных зондов, - OLA ("oligonucleotide ligation assay"), - в модификации с высоким уровнем селективности, когда энзиматически лигируют тандем из трех коротких олигонуклеотидов, - тетрамера и двух фланкирующих его олигомеров длиной 8-10 н., заключающийся в следующем [5].
Конструируют три одноцепочечных олигодезоксирибонуклеотидных зонда -тетрануклеотид pN4, 5’-фланкирующий олигонуклеотид pN8-10 и 3’-фланкирующий олигонуклеотид pN*8-10 длиной 8-10 н. - таким образом, чтобы совокупная нуклеотидная последовательность тандема pN8-10+pN4+pN*8-10 была строго комплементарна непрерывной последовательности диагностируемого участка в составе нуклеиновой кислоты, в которой вариантный нуклеотид находится в сайте гибридизации с тетрамером. Для конкретного диагностируемого участка синтезируют по одному варианту последовательностей фланкирующих олигонуклеотидов, но несколько (2-4) вариантов последовательности тетрамера pN4, которые комплементарны разным аллелям предполагаемой однонуклеотидной замены.
В 3’-фланкирующий олигонуклеотид с помощью фермента полинуклеотидкиназы вводят 32Р-радиоактивную метку: (5’)32 pN*8-10.
Указанные наборы из трех тандемных зондов, pN8-10+pN4+32pN*8-10, различающиеся вариантами тетрамера, инкубируют параллельно в гомогенном растворе, содержащем необходимые буферные составляющие, в присутствии фермента ДНК-лигазы и денатурированного ПЦР-фрагмента ДНК, содержащего диагностируемый вариантный нуклеотид.
Между тремя тандемными зондами, находящимися в комплексе с ДНК-матрицей, происходит энзиматическое лигирование:
pN8-10+pN4+32pN*8-10⇒pN8-10-pN4-32pN*8-10, ([32p]-pN20-24)
с образованием 20-24-звенного 32Р-радиоактивного (сигнального) продукта только в той пробе, в которой тетрамерный член тандема полностью комплементарен соответствующему сайту диагностируемой последовательности ДНК. Тандемы в остальных пробах, содержащие любые другие варианты тетрануклеотида, практически не лигируются ДНК-лигазой и не дают сигнального продукта (точнее, сигнального продукта лигирования образуется на два порядка меньше). Сигнальный продукт образуется, если тандемно расположенные гибридизационные комплексы трех зондов с диагностируемой матричной последовательностью не содержат мисматчей в центральном, тетрануклеотидном, дуплексе тандема, и не образуется при наличии любой некомплементарной пары нуклеотидов (мисматча) в какой-либо из четырех нуклеотидных позиций тетрануклеотидного дуплекса тандема. Высокая специфичность лигирования на ДНК-матрице тандемов pN8-10+pN4+pN*8-10 обусловлена как высокой селективностью связывания комплементарного тетрамера с матрицей в присутствии фланкирующих его олигомеров, так и повышенной избирательностью действия фермента, поскольку любой мисматч в тетрануклеотидном дуплексе тандема pN8-10+pN4+pN*8-10 находится вблизи лигируемых сайтов.
По окончании реакции лигирования реакционную смесь прогревают при 100° С в течение 5 минут для инактивации ДНК-лигазы и разрушения водородных связей.
Отличительная особенность сигнального продукта в том, что он имеет размер 20-24 н. и [32р]радиоактивен.
Для отделения сигнального продукта от остаточных зондов-предшественников параллельные постреакционные смеси разделяют электрофорезом в денатурирующем полиакриламидном геле (20%).
Выявление сигнального продукта в геле осуществляют по радиоактивной метке [32р]: после разделения ЭФ-гель ауторадиографируют на рентгеновскую пленку и по ауторадиограмме определяют участки геля, содержащие радиоактивный сигнальный продукт лигирования размером 20-24 н. Детекцию радиоактивности участков геля, содержащих сигнальный продукт, производят в сцинтилляционном счетчике по методу Черенкова. Степень лигирования (%) вычисляют как отношение радиоактивности участка геля, содержащего сигнальный продукт лигирования, к суммарной радиоактивности в соответствующей дорожке.
По наличию радиоактивного сигнального продукта размером 20-24 н. определяют “активный” вариант тетрануклеотида, указывающий на аллель диагностируемой последовательности ДНК.
Недостатками известного способа являются:
1. Лигирование зондов традиционно проводят в гомогенной среде. В буферном растворе присутствуют все необходимые для реакции компоненты и зонды-предшественники, а по окончании лигирования - также продукт реакции. Это вызывает необходимость адекватного анализа сложной постреакционной смеси для выявления продукта лигирования.
2. Для выявления продукта лигирования последний необходимо дифференцировать от зондов-предшественников по принципу, который лежит в основе различий между предшественниками и продуктом лигирования. Радиоактивный продукт лигирования в гомогенной среде интерферирует с радиоактивным остаточным зондом-предшественником. В данном способе единственное отличие продукта лигирования от всех его предшественников - по величине молекулярного веса. Для отделения продукта лигирования по молекулярному весу вынуждены использовать такой продолжительный и трудоемкий метод как разделение олигонуклеотидов электрофорезом в ПАА-геле в денатурирующих условиях.
3. Для детекции сигнального продукта лигирования после его отделения используют радиоактивную метку - изотоп фосфор-32, относящийся к группе В по радиотоксичности и требующий условий работы по классу III радиационной опасности.
4. Детекция радиоактивного продукта лигирования (сигнального продукта) требует количественного подхода и ее осуществляют либо определением радиоактивности соответствующих зон геля в сцинтилляционном счетчике радиоактивности по методу Черенкова, либо сканированием ауторадиографических реплик гелей с последующим компьютерным анализом плотностей почернения рентгеновской пленки в зонах радиоактивных продуктов, т.е. детекция сигнального продукта лигирования нуждается в соответствующем техническом и программном оснащении.
Технической задачей изобретения является упрощение известного способа при сохранении его высокой селективности для применения в широкой практике.
Поставленную техническую задачу решают предлагаемым способом, заключающимся в следующем.
Конструируют три одноцепочечных олигодезоксирибонуклеотидных зонда - тетрануклеотид pN4, 5’-фланкирующий олигонуклеотид pN8-10 и 3’-фланкирующий олигонуклеотид pN*8-10 длиной 8-10 н. - таким образом, чтобы совокупная нуклеотидная последовательность тандема pN8-10+pN4+pN*8-10 была строго комплементарна непрерывной последовательности диагностируемого участка в составе нуклеиновой кислоты, в которой вариантный нуклеотид находится в сайте гибридизации с тетрамером. Для конкретного диагностируемого участка синтезируют по одному варианту последовательностей фланкирующих олигонуклеотидов pN8-10 и pN*8-10, но несколько (2-4) вариантов последовательности тетрамера pN4, которые комплементарны разным аллелям диагностируемой замены.
5’-фланкирующий олигонуклеотид - pN8-10 - ковалентно иммобилизуют через 5’-фосфатный линкер на поверхность твердофазного носителя П:
pN8-10+П⇒ П~pN8-10.
В 3’-фланкирующий олигонуклеотид - pN*8-10 - на его 3’-конец ковалентно вводят репортерную группу (Rep):
pN*8-10+Rep⇒ pN*8-10Rep.
Наборы из трех тандемных зондов, П~pN8-10+pN4+pN*8-10Rep, различающиеся вариантами тетрамера, инкубируют параллельно в лигазном буфере в гетерофазной среде в присутствии фермента ДНК-лигазы и денатурированного ПЦР-фрагмента ДНК, содержащего диагностируемый вариантный нуклеотид. Кроме П~pN8-10, который является твердофазным, все другие составляющие реакционных смесей лигирования - остальные олигонуклеотидные зонды, фермент, ДНК-матрица и составные части лигазного буфера - являются компонентами жидкой фазы.
Между тремя тандемными зондами, находящимися в комплексе с ДНК-матрицей, происходит энзиматическое лигирование
П~pN8-10+pN4+pN*8-10Rep⇒ П~pN8-10-pN4-pN*8-10Rep (П~pN20-24Rep)
с образованием на поверхности твердофазного носителя П (и только там) 20-24-звенного “сигнального” продукта лигирования с репортерной группой (П~pN20-24Rep) только в той пробе, в которой тетрамерный член тандема полностью комплементарен соответствующему сайту диагностируемой последовательности ДНК. Тандемы в остальных пробах, содержащие любые другие варианты тетрануклеотида, не лигируются ДНК-лигазой и не дают 20-24-звенного продукта с репортерной группой (точнее, дают на три порядка меньше) - в таких пробах твердофазный носитель остается без репортерной группы.
По окончании реакции лигирования реакционную смесь прогревают в присутствии 20 мМ ЭДТА-Na при 100° С в течение 5 минут для инактивации ДНК-лигазы и разрушения водородных связей.
Отличительная особенность “сигнального” продукта лигирования в том, что он имеет размер 20-24 н., весь ковалентно иммобилизован на твердофазном носителе и имеет в своем составе репортерную группу.
Для отделения “сигнального” продукта лигирования в составе твердофазного носителя последний отделяют от всех компонентов жидкой фазы интенсивной промывкой, не сохраняющей какие-либо нековалентные связи.
Выявление “сигнального” продукта лигирования на твердофазном носителе осуществляют посредством репортерной группы (Rep), которую сначала преобразуют в ферментную метку (>Е) через аффинный комплекс специфичного к репортерной группе высокоаффинного агента А, конъюгированного с энзиматическим катализатором Е. Для этого отмытый твердофазный носитель выдерживают в гетерогенной среде с раствором, содержащим конъюгат А-Е, для аффинного связывания конъюгата с репортерной группой:
П~pN20-24Rep+A-E⇒ П~pN20-24Rep:A-E (П~pN20-24>E)
В результате “сигнальный” продукт лигирования на твердофазном носителе приобретает ферментную метку (по природе - энзиматический катализатор), которая дает возможность визуализации “сигнального” продукта. Для визуализации ферментную метку “проявляют” - выдерживают в течение 30-60 минут в присутствии специфичных энзимных субстратов, которые под воздействием энзиматического катализатора преобразуются в местах локализации ферментной метки в продукты с сигнальными свойствами, что выражается в окрашивании носителя. Окрашенный носитель детектируют невооруженным глазом или соответствующими сигналу приборами. При необходимости носитель может быть сканирован (сфотографирован).
По наличию окраски твердофазного носителя определяют “активный” вариант тетрануклеотида, указывающий на аллель диагностируемой последовательности ДНК. Если тестируемая ДНК имеет любое комплементарное несоответствие с последовательностью тетрануклеотидного члена тандема, лигирования и специфичной последующей фиксации ферментной метки на твердофазном носителе не происходит - последний не окрашивается. Природу нуклеотидного несоответствия в пределах тетрануклеотидного дуплекса устанавливают параллельным лигированием тандемов с известными различными вариантами pN4-пocлeдoвaтeльнocти; при этом носитель окрашивается только в одном случае - с тем вариантом pN4, который полностью комплементарен соответствующему участку диагностируемой матричной последовательности.
Предложенный способ универсален для любых установленных последовательностей нуклеиновых кислот. Поскольку определение однонуклеотидных замен этим способом осуществляется на основании наличия или отсутствия, - в ответ на аллельные варианты тетрануклеотида, - сигнала, детектируемого визуально (окраска носителя), т.е. способ работает как высокоспецифичный гетерогенный тест, он получил рабочее название “тест-система TSOLA” (далее по тексту -“TSOLA”, от "three short oligonucleotides ligation assay").
Определяющими отличиями от прототипа являются:
1) 5’-фланкирующий олигонуклеотид - pN8-10 - предварительно ковалентно иммобилизуют через 5’-фосфатный линкер на поверхности твердофазного носителя П, в результате чего олигонуклеотид П~pN8-10 становится частью твердой фазы. В качестве твердофазного носителя используют преимущественно производные полимеров метакрилата, акролеина, акриламида или акрилгидразида, а также их сополимеров, капрон (нейлон), лавсан, модифицированное непористое стекло или силикагель - любой формы (гранулы, мембраны, ячейки микроплат, пленки, гели, волокно, пластины).
2) В 3’-фланкирующий олигонуклеотид - pN*8-10-ковалентно вводят на его 3’-конец репортерную группу (Rep) - pN*8-10Rep, что позволяет исключить необходимость использования радиоизотопов.
3) Лигирование зондов проводят не в гомогенном растворе, а в гетерофазной среде, в которой твердой фазой является 5’-фланкирующий олигонуклеотид-предшественник П~pN8-10; а в жидкой фазе присутствуют остальные зонды-предшественники - тетрамер pN4 и 3’-фланкирующий олигонуклеотид pN*8-10Rep, - а также все необходимые для реакции лигирования буферные компоненты, диагностируемая ДНК-матрица и фермент ДНК-лигаза. По окончании лигирования “сигнальный” продукт лигирования с репортерной группой оказывается ковалентно иммобилизованным на твердофазном носителе (П~рN20-24Rер), что позволяет проводить его отделение без какого-либо предварительного анализа и освобождает от необходимости применения электрофореза в ПАА-геле в денатурирующих условиях.
4) Для выявления иммобилизованного продукта лигирования применяют в отличие от радиоактивной метки не представляющую опасности для широкого использования ферментную метку. Последнюю вводят в иммобилизованный “сигнальный” продукт лигирования по месту репортерной группы. Ферментная метка представляет собой энзиматический катализатор Е, конъюгированный со специфичным к репортерной группе высокоаффинным агентом А. В качестве репортерной группы (Rep) и соответствующего ей специфичного высокоаффинного агента (А) используют любые высокоаффинные пары [Rep:A] химических и природных соединений с константой диссоциации аффинного комплекса Кd не более 10-15, преимущественно такие как [биотин: стрептавидин (или ExtrAvidin)], [дигоксигенин: антидигоксигенин-антитело] и другие пары [антиген (гаптен): антитело]. Ферментная метка содержит в качестве энзиматического катализатора Е преимущественно фермент (или его производное) пероксидазу, щелочную фосфатазу, β -D-галактозидазу, или любой другой катализатор, способный генерировать из специфичных субстратов продукты с сигнальными свойствами - хроматическими, люминесцентными, флуоресцентными.
5) В присутствии специфичных субстратов, в ходе процесса под условным названием “проявление”, ферментная метка в составе “сигнального” продукта лигирования развивает цветное, хемилюминесцентное или флуоресцентное окрашивание твердофазного носителя в зонах локализации продукта лигирования, которое является специфичным сигналом наличия продукта. Для “проявления” ферментной метки с визуальной детекцией специфичного сигнала в качестве субстратов используют фермент-специфичные преципитирующие хромогенные субстраты - преимущественно 3,3’-диаминобензидин (DAB), 4-хлор-1-нафтол (4C1N), 5-бром-4-хлор-3-индолилфосфат/р-нитротетразолий-синий (BCIP/NBT); для измерительной детекции используют как преципитирующие, так и непреципитирующие фермент-специфичные субстраты, хромогенные, люминогенные или флюорогенные - преимущественно 3,3’,5,5’-тетраметилбензидин (ТМВ), p-нитрофенилфосфат (pNPP), 5-аминосалициловая кислота (5AS), 4-метилумбеллиферилфосфат (4-MUP).
Кроме этого, отделение продукта лигирования от растворенных остаточных зондов-предшественников в постреакционной смеси осуществляют простой промывкой твердофазного носителя, что позволяет упростить способ. После отмывки на носителе сохраняются только ковалентно иммобилизованные “сигнальный” продукт лигирования (П~рN20-24Reр) и остаточный первый октамер-предшественник (П~pN8-10), который далее никак не интерферирует с “сигнальным” продуктом, т.к. не имеет репортерной группы.
Характер сигнала, генерируемого ферментной меткой (окрашивание носителя), высокая селективность лигирования коротких зондов на твердофазном носителе и связанная с ней однозначность прямых альтернативных ответов (“да”/“нет”) обеспечивают возможность визуальной детекции специфичного сигнала, не требующей в отличие от прототипа обязательного количественного анализа и сопряженного с ним технического и программного оснащения. Все эти параметры предложенного способа позволяют использовать его как простой и надежный тест, доступный для широкого применения.
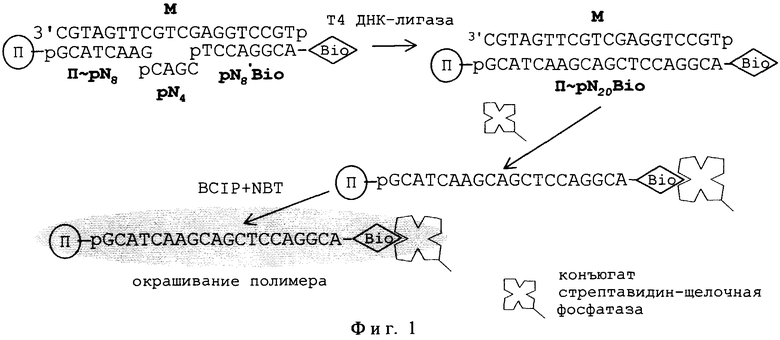
На фиг.1 представлена описанная выше схема лигирования тандема олигонуклеотидов и выявления “сигнального” продукта лигирования в TSOLA, конкретизированная в части длины зондов, репортерной группы (остаток биотина Bio), ферментной метки (конъюгат стрептавидин-щелочная фосфатаза) и хромогенных преципитирующих субстратов (BCIP+NBT), где М - участок диагностируемой матричной ДНК. Остальные обозначения указаны в тексте описания.
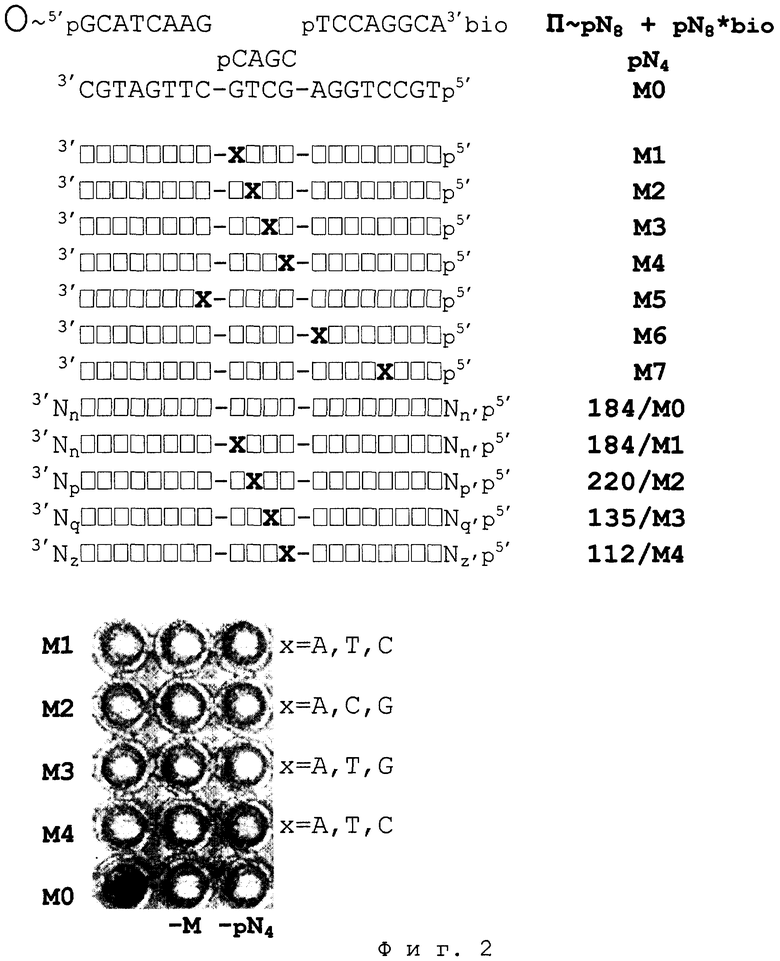
На фиг.2 представлено сканированное изображение планшета, иллюстрирующее результаты TSOLA для примеров 1-13,17,19, где М0 - М7 - модельные одноцепочечные ДНК длиной 20 н.: М0 - правильная ДНК-мишень, не имеющая мисматчей в тандемных гибридизационных комплексах TSOLA; M1-M4 - ДНК-мишени, имеющие в гибридизационных комплексах TSOLA по одному мисматчу - с 1-4-ой соответственно нуклеотидной позицией тетрамера; М5 - ДНК-мишень, имеющая мисматч с 3’-концевым основанием иммобилизованного октамера; М6 - ДНК-мишень, имеющая мисматч с 5’-концевым основанием биотинилированного октамера; М7 - ДНК-мишень, имеющая мисматч с внутренним, - пятым, - основанием биотинилированного октамера; ... /М0 -... /М4 - продукты симметричной ПЦР-амплификации ДНК с разными размерами и последовательностями: 184/М0 - правильная ДНК-мишень длиной 184 п.н., не имеющая мисматчей в тандемных гибридизационных комплексах TSOLA; ... /M1-... /M4 - ДНК-мишени указанной длины, гибридизационные комплексы TSOLA которых имеют один мисматч - с 1-4-ой соответственно нуклеотидной позицией центрального, тетрамерного, члена тандема.  - нуклеотидные позиции, комплементарные тандему TSOLA. Х - некомплементарные позиции.
- нуклеотидные позиции, комплементарные тандему TSOLA. Х - некомплементарные позиции.
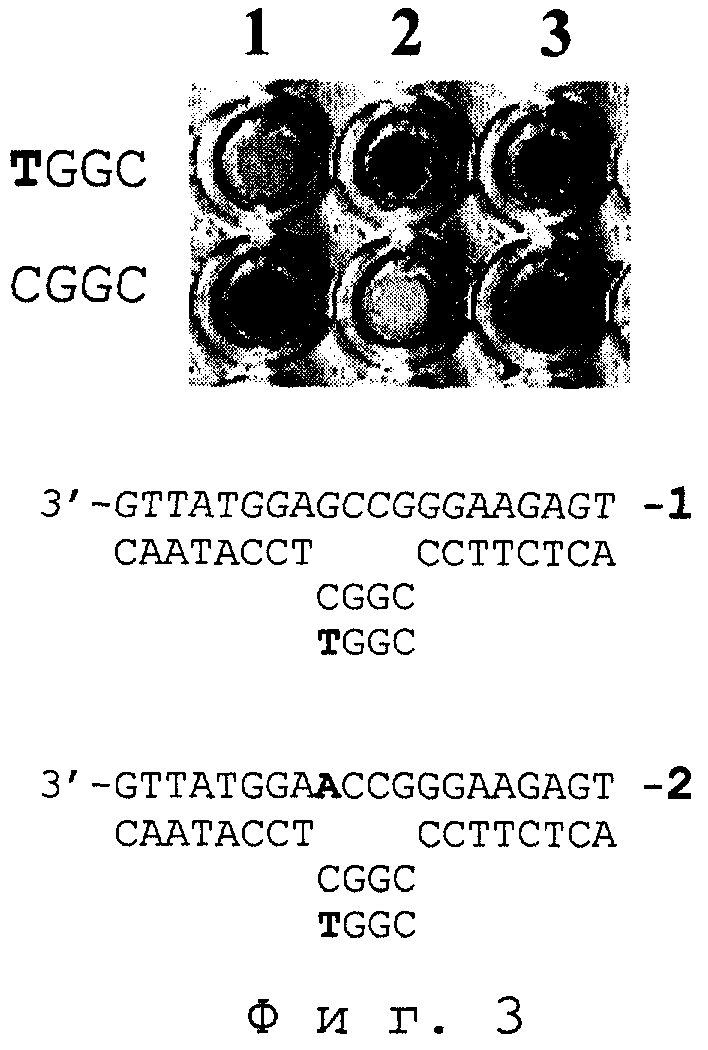
На фиг.3 представлена иллюстрация результата TSOLA-теста на наличие однонуклеотидной замены для трех ПЦР-образцов ДНК размером 184 п.н., один из которых гетерозиготный, а другие - гомозиготные. Тестирование проводили, как описано в примерах 27-30. 1 - гомозиготный образец ДНК, имеет в позиции, гибридизующейся с 1-ым нуклеотидом тетрамерного члена тандема, основание G (примеры 27-28); 2 - гомозиготный образец ДНК, имеет замену G⇒ А в позиции, гибридизующейся с 1-ым нуклеотидом тетрамерного члена тандема (примеры 29-30); 3-гетерозиготный образец. Гомозиготные образцы дают положительный сигнал (окрашивание) только с соответствующим их последовательности тетрануклеотидом, альтернативно. Гетерозиготный образец дает положительный сигнал с каждым из аллельных тетрануклеотидов.
Изобретение иллюстрируется следующими примерами конкретного исполнения изобретения.
Исследовали тест-системы TSOLA с разными последовательностями ДНК-мишеней и олигонуклеотидов. Для моделирования мисматча в определенной позиции комплексов TSOLA вводили однонуклеотидную замену в последовательность матричной ДНК или в последовательность тетрамера.
Пример 1. Твердофазный носитель представлял собой непористые метакрилатные гранулы с иммобилизованным на поверхности 5’-фланкирующим октануклеотидом тандема - П~pN8 (П~pGCATCAAG). Плотность иммобилизации октануклеотида - 1 мкмоль/г. Состав жидкой среды: по 10,0 мкМ тетрануклеотида pN4 (pCAGC) и биотинилированного 3’-фланкирующего октануклеотида pN8*bio (pTCCAGGCAp~bio); 10 нМ ДНК-матрица М0 (фиг.2); 1,5 ед.акт./мкл Т4 ДНК-лигазы, в стандартном лигазном буфере. Проба содержала 0,5 мг сухого веса носителя и 15 мкл жидкой среды.
1) К промытому ТЕ-буфером и хорошо дренированному носителю (0,5 мг сухого веса) добавляли компоненты жидкой среды (общим объемом 15 мкл), смесь взмучивали и инкубировали при 37° С в течение 30 мин.
2) По окончании лигирования пробу разбавляли 20 мМ ЭДТА-Na (рН~8) и прогревали при 100° С в течение 5 мин.
3) Носитель отмывали от биотинилированного октамера-предшественика натрий-цитратным буфером (SSC) путем проточного фильтрования с помощью водоструйного насоса (“Дот”-фильтрация) или 3-4-кратным переосаждением в настольной центрифуге ("Eppendorf").
4) Носитель выдерживали с раствором конъюгата стрептавидина и щелочной фосфатазы в разбавленном SSC-буфере (1 мкг/мл) в течение 30 мин при комнатной температуре и отмывали, как указано для стадии 3.
5) Носитель “проявляли” в течение 60 мин при комнатной температуре в присутствии преципитирующих хромогенных субстратов BCIP/NBT ("Sigma") в стандартных условиях, рекомендуемых фирмой.
Образование сигнального продукта фиксировали визуально по наличию пурпурно-синего окрашивания носителя.
Результат теста положительный: носитель окрашивался; продукт лигирования детектировался. Заключение: последовательность диагностируемого участка ДНК-матрицы комплементарна последовательности использованного варианта тетрануклеотида.
Пример 2. Эксперимент проводили, как описано в примере 1, но в качестве ДНК-матрицы использовали M1 (X=A). Результат теста отрицательный: носитель не окрашивался; продукт лигирования отсутствовал. Заключение: последовательность диагностируемого участка ДНК-матрицы не комплементарна последовательности использованного варианта тетрануклеотида.
Пример 3. Эксперимент проводили, как описано в примере 1, но в качестве ДНК-матрицы использовали M1 (X=T). Результат теста отрицательный: носитель не окрашивался; продукт лигирования отсутствовал. Заключение: последовательность диагностируемого участка ДНК-матрицы не комплементарна последовательности использованного варианта тетрануклеотида.
Пример 4. Эксперимент проводили, как описано в примере 1, но в качестве ДНК-матрицы использовали M1 (X=C). Результат теста отрицательный: носитель не окрашивался; продукт лигирования отсутствовал. Заключение: последовательность диагностируемого участка ДНК-матрицы не комплементарна последовательности использованного варианта тетрануклеотида.
Пример 5. Эксперимент проводили, как описано в примере 1, но в качестве ДНК-матрицы использовали М2 (Х=А). Результат теста отрицательный: носитель не окрашивался; продукт лигирования отсутствовал. Заключение: последовательность диагностируемого участка ДНК-матрицы не комплементарна последовательности использованного варианта тетрануклеотида.
Пример 6. Эксперимент проводили, как описано в примере 1, но в качестве ДНК-матрицы использовали М2 (Х=С). Результат теста отрицательный: носитель не окрашивался; продукт лигирования отсутствовал. Заключение: последовательность диагностируемого участка ДНК-матрицы не комплементарна последовательности использованного варианта тетрануклеотида.
Пример 7. Эксперимент проводили, как описано в примере 1, но в качестве ДНК-матрицы использовали М2 (X=G). Результат теста отрицательный: носитель не окрашивался; продукт лигирования отсутствовал. Заключение: последовательность диагностируемого участка ДНК-матрицы не комплементарна последовательности использованного варианта тетрануклеотида.
Пример 8. Эксперимент проводили, как описано в примере 1, но в качестве ДНК-матрицы использовали М3 (Х=А). Результат теста отрицательный: носитель не окрашивался; продукт лигирования отсутствовал. Заключение: последовательность диагностируемого участка ДНК-матрицы не комплементарна последовательности использованного варианта тетрануклеотида.
Пример 9. Эксперимент проводили, как описано в примере 1, но в качестве ДНК-матрицы использовали М3 (Х=Т). Результат теста отрицательный: носитель не окрашивался; продукт лигирования отсутствовал. Заключение: последовательность диагностируемого участка ДНК-матрицы не комплементарна последовательности использованного варианта тетрануклеотида.
Пример 10. Эксперимент проводили, как описано в примере 1, но в качестве ДНК-матрицы использовали М3 (X=G). Результат теста отрицательный: носитель не окрашивался; продукт лигирования отсутствовал. Заключение: последовательность диагностируемого участка ДНК-матрицы не комплементарна последовательности использованного варианта тетрануклеотида.
Пример 11. Эксперимент проводили, как описано в примере 1, но в качестве ДНК-матрицы использовали М4 (Х=А). Результат теста отрицательный: носитель не окрашивался; продукт лигирования отсутствовал. Заключение: последовательность диагностируемого участка ДНК-матрицы не комплементарна последовательности использованного варианта тетрануклеотида.
Пример 12. Эксперимент проводили, как описано в примере 1, но в качестве ДНК-матрицы использовали М4 (Х=Т). Результат теста отрицательный: носитель не окрашивался; продукт лигирования отсутствовал. Заключение: последовательность диагностируемого участка ДНК-матрицы не комплементарна последовательности использованного варианта тетрануклеотида.
Пример 13. Эксперимент проводили, как описано в примере 1, но в качестве ДНК-матрицы использовали М4 (Х=С). Результат теста отрицательный: носитель не окрашивался; продукт лигирования отсутствовал. Заключение: последовательность диагностируемого участка ДНК-матрицы не комплементарна последовательности использованного варианта тетрануклеотида.
Пример 14. Эксперимент проводили, как описано в примере 1, но в качестве ДНК-матрицы использовали М5 (Х=Т). Результат теста положительный: носитель окрашивался; продукт лигирования детектировался. Заключение: последовательность диагностируемого участка ДНК-матрицы комплементарна последовательности использованного варианта тетрануклеотида.
Пример 15. Эксперимент проводили, как описано в примере 1, но в качестве ДНК-матрицы использовали М6 (Х=Т). Результат теста положительный: носитель окрашивался; продукт лигирования детектировался. Заключение: последовательность диагностируемого участка ДНК-матрицы комплементарна последовательности использованного варианта тетрануклеотида.
Пример 16. Эксперимент проводили, как описано в примере 1, но в качестве ДНК-матрицы использовали М7 (Х=Т). Результат теста положительный: носитель окрашивался; продукт лигирования детектировался. Заключение: последовательность диагностируемого участка ДНК-матрицы комплементарна последовательности использованного варианта тетрануклеотида.
Пример 17. Эксперимент проводили, как описано в примере 1, но отсутствовал тетрануклеотид (-pN4). Результат теста отрицательный: носитель не окрашивался; продукт лигирования отсутствовал. Заключение: неполная тест-система TSOLA.
Пример 18. Эксперимент проводили, как описано в примере 1, но отсутствовала ДНК-лигаза. Результат теста отрицательный: носитель не окрашивался; продукт лигирования отсутствовал. Заключение: неполная тест-система TSOLA.
Пример 19. Эксперимент проводили, как описано в примере 1, но отсутствовала ДНК-мишень (-М). Результат теста отрицательный: носитель не окрашивался; продукт лигирования отсутствовал. Заключение: неполная тест-система TSOLA.
Результаты примеров 1-13,17,19 отражены на фиг.2.
Пример 20. Эксперимент проводили, как описано в примере 1. Состав жидкой среды: 10.0 мкМ биотинилированный октануклеотид pN8*bio; 10 мкМ тетрануклеотид pN4; 100 нМ денатурированная нагреванием ДНК-матрица 184/М0; 3 ед.акт./мкл Т4 ДНК-лигазы, в стандартном лигазном буфере. Результат теста положительный: носитель окрашивался; продукт лигирования детектировался. Заключение: последовательность диагностируемого участка ДНК-матрицы комплементарна последовательности использованного варианта тетрануклеотида.
Пример 21. Эксперимент проводили, как описано в примере 20, но в качестве ДНК-матрицы использовали 220/М2. Результат теста отрицательный: носитель не окрашивался; продукт лигирования отсутствовал. Заключение: последовательность диагностируемого участка ДНК-матрицы не комплементарна последовательности использованного варианта тетрануклеотида.
Пример 22. Эксперимент проводили, как описано в примере 20, но в качестве ДНК-матрицы использовали 135/М3. Результат теста отрицательный: носитель не окрашивался; продукт лигирования отсутствовал. Заключение: последовательность диагностируемого участка ДНК-матрицы не комплементарна последовательности использованного варианта тетрануклеотида.
Пример 23. Эксперимент проводили, как описано в примере 20, но в качестве ДНК-матрицы использовали 112/М4. Результат теста отрицательный: носитель не окрашивался; продукт лигирования отсутствовал. Заключение: последовательность диагностируемого участка ДНК-матрицы не комплементарна последовательности использованного варианта тетрануклеотида.
Пример 24. Эксперимент проводили, как описано в примере 20, но иммобилизованный олигонуклеотид П~pN9 имел длину 9 н., биотинилированный олигонуклеотид pN10*bio - 10 н. и в качестве ДНК-матрицы использовали 341/М0. Результат теста положительный: носитель окрашивался; продукт лигирования детектировался. Заключение: последовательность диагностируемого участка ДНК-матрицы комплементарна последовательности использованного варианта тетрануклеотида.
Пример 25. Эксперимент проводили, как описано в примере 24, но в качестве ДНК-матрицы использовали 341/М1. Результат теста отрицательный: носитель не окрашивался; продукт лигирования отсутствовал. Заключение: последовательность диагностируемого участка ДНК-матрицы не комплементарна последовательности использованного варианта тетрануклеотида.
Пример 26. Эксперимент проводили, как описано в примере 20, но иммобилизованный олигонуклеотид П~pN10 имел длину 10 н., биотинилированный олигонуклеотид pN9*bio - 9 н. и в качестве ДНК-матрицы использовали 341/М0. Результат теста положительный: носитель окрашивался; продукт лигирования детектировался. Заключение: последовательность диагностируемого участка ДНК-матрицы комплементарна последовательности использованного варианта тетрануклеотида.
Пример 27. Твердофазный носитель П представлял собой капроновую мембрану (“Хийу Калур”, Эстония, размер пор - 0.2 мкм) в форме диска диаметром ~3 мм с иммобилизованным на поверхности 5’-фланкирующим октануклеотидом - П~pN8 (77~pCAATACCT). Плотность иммобилизации октануклеотида составляла 5 нмоль/см2. Состав жидкой среды: 10 мкМ тетрануклеотид pN4 (pCGGC); 10 мкМ октануклеотид pN8*Rep с остатком дигоксигенина (гаптен) в качестве репортерной группы (pCCTTCTCA~Dig); 0.1 мкМ денатурированная нагреванием матрица 184/М0 (5’-... TGAGAAGGGCCGAGGTATTG... ); 3 ед.акт./мкл Т4 ДНК-лигазы, в стандартном лигазном буфере.
1) Диски помещали в ячейки микроплаты с ТЕ-буфером для смачивания, промывали 1 раз ТЕ-буфером и тщательно дренировали отсасыванием буфера.
2) На диски в ячейках наносили 30 мкл жидкой среды и инкубировали при 37° С в течение 15 минут.
3) Реакцию останавливали добавлением 150 мкл 25 мМ ЭДТА-Na, рН~8.
4) Диски в ячейках несколько раз промывали горячей (~80-90° С) дистиллированной водой с тщательным дренированием в конце каждой промывки.
5) В ячейки добавляли 150 мкл 0,1 М PBS-T, рН 7,4, содержащего 15 мМ NaN3 и 0,2 мг/мл БСА. Состав 0,1 М PBS-T, рН 7,4 на 1 л: NaCl - 8,0 г, КН2РO4 - 0,2 г; Na2HPO4·12H2O - 2,8 г; КСl - 0,2 г; Твин-20 - 0,05% (v/v). Через 10 мин диски дренировали отсасыванием буфера.
6) Добавляли 30 мкл раствора конъюгата пероксидазы хрена (ПХ) с антителом к дигоксигенину (АнтиDig) в 0,1 М PBS-T, рН 7,4, содержащего 1.0 мкг/мл АнтиDig-ПX; 15 мМ NaN3; 0,2 мг/мл БСА. Инкубировали 1 час при 37° С.
7) Диски в ячейках отмывали 5 раз по 200 мкл 0,1 М PBS-T, рН7.4 с тщательным дренированием.
8) Для проявления в ячейки добавляли по 60 мкл раствора субстратов, содержащего 50 мМ трис-HCl, рН 7,4; 0,5 мг/мл 3,3’-диаминобензидина (DAB); 2 мМ H2O2. Проявление продолжали при комнатной температуре в течение 60 мин и останавливали добавлением 1-2 капли 2 М H2SO4.
Результат теста положительный: носитель окрашивался; продукт лигирования детектировался. Заключение: для ДНК-образца 184/М0 определяется аллель G.
Пример 28. Эксперимент проводили, как описано в примере 27, но состав тетрануклеотида pN4 - pTGGC. Результат теста отрицательный: носитель не окрашивался; продукт лигирования отсутствовал. Заключение: последовательность диагностируемого участка ДНК-матрицы не комплементарна последовательности использованного варианта тетрануклеотида.
Пример 29. Эксперимент проводили, как описано в примере 27, но ДНК-матрица - 184/М1 (5'-... TGAGAAGGGCCAAGGTATTG... ). Результат теста отрицательный: носитель не окрашивался; продукт лигирования отсутствовал. Заключение: последовательность диагностируемого участка ДНК-матрицы не комплементарна последовательности использованного варианта тетрануклеотида.
Пример 30. Эксперимент проводили, как в примере 27, но использовали ДНК-матрицу 184/М1 (пример 30) и тетрануклеотид pTGGC. Результат теста положительный: носитель окрашивался; продукт лигирования детектировался. Заключение: для ДНК-образца 184/М1 определяется аллель А.
Результаты примеров 27-30 отражены на фиг.3 (1, 2).
Таким образом, предлагаемый способ универсален для любых установленных последовательностей нуклеиновых кислот, отличается высокой селективностью и доступен для применения в широкой практике.
Представляем дополнительный пример, иллюстрирующий невоспроизводимость заявленного способа при осуществлении иммобилизации не через 5’-концевой фосфатный линкер 5’-фланкирующего олигонуклеотида тандема, а через 3’-фосфатный линкер 3’-фланкирующего олигонуклеотида тандема:
Rep~pN8-10+pN4+pN*8-10~П
Пример (31).
Твердофазный носитель представлял собой непористые метакрилатные гранулы с иммобилизованным на поверхности через 3’-концевой фосфатный линкер 3’-фланкирующим октануклеотидом тандем -pN8*~П (pTCCAGGCAp~П). Плотность иммобилизации октануклеотида -1 мкмоль/г. Состав жидкой среды: 10.0 мкМ тетрануклеотида pN4 (pCAGC), 10.0 мкМ биотинилированного по 5’-концевой фосфатной группе 5’-фланкирующего октануклеотида Bio~pN8 (Bio~pGCATCAAG); 10 нМ комплементарная ДНК-матрица 5’- TGCCTGGAGCTGCTTGATGC (МО, фиг.2); 1.5 ед.акт./мкл Т4 ДНК-лигазы, в стандартном лигазном буфере. Проба содержала 0.5 мг сухого веса носителя и 15 мкл жидкой среды.
Эксперимент проводили, как описано в примере 1:
1) К промытому ТЕ-буфером и хорошо дренированному носителю (0.5 мг сухого веса) добавляли компоненты жидкой среды (общим объемом 15 мкл), смесь взмучивали и инкубировали при 37° С в течение 30 мин.
2) По окончании лигирования пробу разбавляли 20 мМ ЭДТА-Na (рН~8) и прогревали при 100° С в течение 5 мин.
3) Носитель отмывали от биотинилированного октамера-предшественника натрий-цитратным буфером (SSC) путем проточного фильтрования с помощью водоструйного насоса ("Дот" - фильтрация) или 3-4-кратным переосаждением в настольной центрифуге ("Eppendorf")
4) Носитель выдерживали с раствором конъюгата стрептавидина и щелочной фосфатазы в разбавленном SSC-буфере (1 мкг/мл) в течение 30 мин при комнатной температуре и отмывали, как указано для стадии 3.
5) Носитель "проявляли" в течение 60 мин при комнатной температуре в присутствии преципитирующих хромогенных субстратов BCIP/NBT ("Sigma") в стандартных условиях, рекомендуемых фирмой.
Образование сигнального продукта фиксировали визуально по наличию пурпурно-синего окрашивания носителя.
Результат теста неудовлетворительный: носитель лишь немного изменил оттенок; продукт лигирования достоверно практически не детектируется. Заключение: энзиматического лигирования не происходит по стерическим причинам, технический результат не достигается.
Источники информации
1. Иванов И.Б., Ершов Г.М., Барский В.Е., Бельговский А.И., Кириллов Е.В., Крейндлин Э.Я., Паринов С.В., Мологина Н.В., Мирзабеков А.Д. Диагностика генетических мутаций на олигонуклеотидных микрочипах. // Молекулярная биология. 1997. Т.31. №1. С.159-167.
2. Syvanen AC. From gels to chips: "minisequencing" primer extension for analysis of point mutations and single nucleotide polymorphisms. // Hum Mutat. 1999. Vol.13. №1. P.1-10.
3. Suzuki Y, Sekiya T, Hayashi K. Allele-specific polymerase chain reaction: a method for amplification and sequence determination of a single component among a mixture of sequence variants. // Anal. Biochem. 1991.Vol.192. №1. P.82-84.
4. lannone M.A., Taylor J.D., Chen J., Li M.S., Rivers P., Slentz - Kesler K.A., Weiner M.P. Multiplexed single nucleotide polymorphism genotyping by oligonucleotide ligation and flow cytometry. // Cytometry. 2000. Vol.39. P.131-140.
5. Пышный Д.В., Кривенко А.А., Лохов С.Г., Иванова Е.М., Дымшиц Г.М., Зарытова В.Ф. Взаимодействие производных коротких олигонуклеотидов с нуклеиновыми кислотами. V. Лигирование коротких олигонуклеотидов в тандеме на комплементарной матрице ДНК. VI. Дискриминация комплексов, содержащих мисматч, при лигировании тандема коротких олигонуклеотидов на ДНК-матрице. //Биоорган, химия. 1998. Т.24. №1. С.25-31, 32-37.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ВЫЯВЛЕНИЯ АНАЛИЗИРУЕМОЙ ПОСЛЕДОВАТЕЛЬНОСТИ НУКЛЕИНОВЫХ КИСЛОТ | 1996 |
|
RU2146707C1 |
| СПОСОБ ВЫЯВЛЕНИЯ АНАЛИЗИРУЕМОЙ ПОСЛЕДОВАТЕЛЬНОСТИ ДНК | 2003 |
|
RU2259402C2 |
| Способ определения нуклеотидной последовательности ДНК и устройство для его осуществления | 1991 |
|
SU1794088A3 |
| ДИАГНОСТИЧЕСКИЙ АНАЛИЗ | 2001 |
|
RU2228530C2 |
| РЕАГЕНТ (ВАРИАНТЫ), БИБЛИОТЕКА РЕАГЕНТОВ, СПОСОБЫ ПРОВЕДЕНИЯ АНАЛИЗА | 1994 |
|
RU2158310C2 |
| СПОСОБ ЧАСТИЧНОГО СЕКВЕНИРОВАНИЯ ДНК ДЛЯ ОПРЕДЕЛЕНИЯ МУТАЦИЙ В КОРОТКИХ ФРАГМЕНТАХ ОДНОЦЕПОЧЕЧНОЙ ДНК С ИСПОЛЬЗОВАНИЕМ МИКРОЧИПА | 2001 |
|
RU2206615C1 |
| СПОСОБ СПЕЦИФИЧЕСКОЙ ИДЕНТИФИКАЦИИ ПОСЛЕДОВАТЕЛЬНОСТЕЙ ДНК | 2017 |
|
RU2665631C2 |
| СПОСОБ ИНГИБИРОВАНИЯ РЕПРОДУКЦИИ ВИРУСА ИММУНОДЕФИЦИТА ЧЕЛОВЕКА | 1997 |
|
RU2136752C1 |
| ПРЯМОЙ ЗАХВАТ, АМПЛИФИКАЦИЯ И СЕКВЕНИРОВАНИЕ ДНК-МИШЕНИ С ИСПОЛЬЗОВАНИЕМ ИММОБИЛИЗИРОВАННЫХ ПРАЙМЕРОВ | 2011 |
|
RU2565550C2 |
| МУЛЬТИПЛЕКСНОЕ ДЕТЕКТИРОВАНИЕ НУКЛЕИНОВЫХ КИСЛОТ | 2014 |
|
RU2733887C2 |
Изобретение относится к области молекулярной биологии и может быть использовано в здравоохранении. Предложен способ определения однонуклеотидных замен в известных последовательностях нуклеиновых кислот. Он предусматривает гибридизацию с ПЦР-амплифицированной матричной ДНК и последующее лигирование на ней с помощью ДНК-лигазы тандема, состоящего из тетрануклеотида, содержащего диагностируемую замену, и двух олигонуклеотидов размером 8-10 нуклеотидов, один из которых иммобилизован через 5`-фосфатный линкер на поверхность твердофазного носителя, а второй помечен с 3`-конца биотиновой меткой. Тетрануклеотид гибридизуется с цепью матричной ДНК непосредственно между двумя олигонуклеотидами и может быть лигирован через 5`-конец с иммобилизованным олигонуклеотидом и через 3` -конец - с неиммобилизованным. Продукт лигирования детектируется при помощи преобразования его в ферментную метку через комплекс с высокоаффинным энзиматическим катализатором с последующим проявлением ферментной метки в присутствии хромогенных, люминогенных или флюорогенных субстратов. Применение способа обеспечивает высокоселективное и простое в использовании средство обнаружения известных изменений в структуре генов. 7 з.п. ф-лы, 3 ил.
| КАБИЛОВ М.Р | |||
| и др | |||
| “Молекулярная биология”, 2002, т.36, №3 | |||
| US 5952174, 14.09.1999 | |||
| US 5436327, 25.07.1995 | |||
| US 5800994, 01.09.1998 | |||
| АГРЕГАТ ДЛЯ ТЕРМИЧЕСКОЙ ОБРАБОТКИ ПРОКАТНЫХ ВАЛКОВ, ВОДОСБОРНИК И СПРЕЙЕР ДЛЯ ЭТОГО АГРЕГАТА | 1998 |
|
RU2143009C1 |
| СПОСОБ ВЫЯВЛЕНИЯ АНАЛИЗИРУЕМОЙ ПОСЛЕДОВАТЕЛЬНОСТИ НУКЛЕИНОВЫХ КИСЛОТ | 1996 |
|
RU2146707C1 |

